

Abstract
Aims
To improve the tolerability and therapeutic application of histone deacetylase inhibitors (HDACi), by application of an esterase‐sensitive motif (ESM), to target pharmacological activity directly to mononuclear myeloid cells expressing the processing enzyme carboxylesterase‐1 (CES1).
Methods
This first‐in‐human study comprised single and multiple ascending dose cohorts to determine safety and tolerability. Pharmacodynamic parameters included acetylation, cytokine inhibition and intracellular concentrations of processed acid metabolite in isolated monocytes. Mechanistic work was conducted in vitro and in a CES1/Es1e lo mouse strain.
Results
ESM‐HDAC391 showed transient systemic exposure (plasma half‐life of 21–30 min) but selective retention of processed acid for at least 12 hours, resulting in robust targeted mechanistic engagement (increased acetylation in monocytes plus inhibition of ex vivo stimulated cytokine production). ESM‐HDAC391 was well tolerated and clinical toxicities common to non‐targeted HDACi were not observed. ESM‐HDAC391 treatment was accompanied by the novel finding of a dose‐dependent monocyte depletion that was transient and reversible and which plateaued at 0.06 × 109 monocytes/L after repeat dosing with 20 or 40 mg. Characterisation of monocyte depletion in transgenic mice (CES1/Es1e lo ) suggested that colony stimulating factor 1 receptor loss on circulating cells contributed to ESM‐HDAC‐mediated depletion. Further mechanistic investigations using human monocytes in vitro demonstrated HDACi‐mediated change in myeloid fate through modulation of colony stimulating factor 1 receptor and downstream effects on cell differentiation.
Conclusion
These findings demonstrate selective targeting of monocytes in humans using the ESM approach and identify monocytopaenia as a novel outcome of ESM‐HDACi treatment, with implications for potential benefit of these molecules in myeloid‐driven diseases.
Keywords: ESM‐HDAC391, histone deacetylase inhibitor, monocyte, monocytopaenia, myeloid, pharmacodynamics, pharmacokinetics
What is already known about this subject
Histone deacetylase (HDAC) inhibitors are potent anti‐inflammatory molecules but their clinical application has been restricted by dose‐limiting toxicities.
Esterase‐sensitive motif (ESM) conjugation enables targeting to mononuclear myeloid cells.
What this study adds
ESM‐HDAC391 is a novel targeted inhibitor demonstrating sustained potent mechanistic engagement and anti‐inflammatory activity.
ESM‐HDAC391 mitigates common conventional HDAC toxicities such as neutropaenia and thrombocytopaenia.
ESM‐HDAC391 causes reversible monocytopaenia in volunteers; this has been further characterised in a novel transgenic mouse model and is driven by downregulation of colony stimulating factor 1 receptor.
1. INTRODUCTION
Histone deacetylases (HDACs), as pivotal enzymes in cellular physiology and pathology, represent impactful drug discovery targets. 1 , 2 Originally defined epigenetically via their catalytic role in the removal of accessible acetyl groups on histones to control gene expression, additional activity against non‐histone substrates and mechanisms has become evident. 3 , 4 Histone hypoacetylation is causative in the aetiology of various clinical disorders 5 and studies demonstrate HDAC inhibitory efficacy in treating inflammatory diseases and modulating immune endpoints. 6 , 7
Several first‐generation, conventional HDAC inhibitors (HDACi) have been approved as therapeutic agents or are in development, primarily for the treatment of cancer. 8 , 9 , 10 These molecules have restricted clinical utility beyond oncology studies due to toxicities including thrombocytopaenia, neutropaenia, cardiovascular effects, gastrointestinal disturbances and fatigue. 11 However, there is also wider interest in areas such as heart disease, 12 diabetes, 13 neurological indications, 14 and infectious and inflammatory diseases. 15 Obtaining therapeutic windows sufficient to allow extensive use of HDACi for chronic inflammatory disorders therefore requires more selective or targeted approaches. 16
Esterase‐sensitive motif (ESM) technology allows the specific targeting of small molecule inhibitors to immune cells which express human carboxylesterase‐1 (CES1), primarily monocytes and macrophages. Selective hydrolysis of a dosed ester moiety by CES1 yields an acid that, being less permeable, is retained within CES1‐expressing cells for significantly longer over other cell types that cannot process the ester to retained acid (Figure 1A). Targeted and prolonged pharmacodynamic (PD) effects specifically in CES1+ cells, combined with lower systemic compound exposure, therefore result in a greater therapeutic window. The utility of ESM technology has been evaluated preclinically by conjugation of CES1 substrates onto various small molecule inhibitors, including p38 MAP kinase, HSP‐90, IκB kinase 2, as well as HDACs, 17 , 18 , 19 and also clinically by the ESM‐HDAC inhibitor tefinostat 20 and an ESM‐BET inhibitor. 21
FIGURE 1.
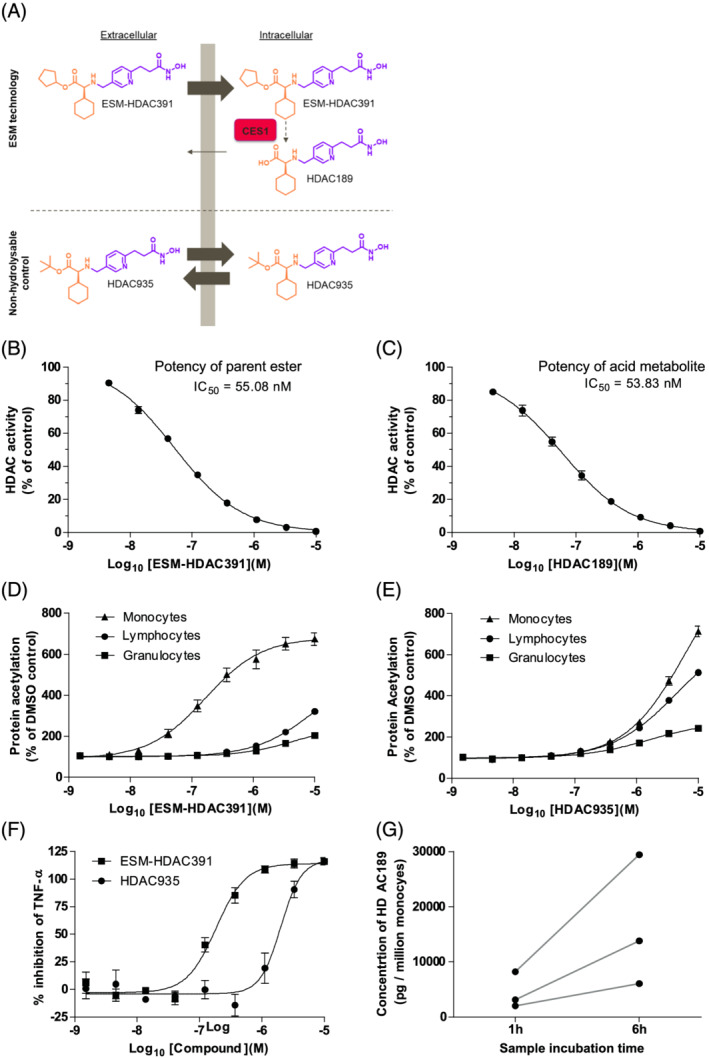
ESM‐HDAC391 demonstrates myeloid‐targeted histone deacetylase (HDAC) inhibitory effects in vitro. (A) Schematic of ESM targeting, showing conversion of ester ESM‐HDAC391 to acid HDAC189 in carboxylesterase‐1‐expressing cells in comparison to the non‐hydrolysable control molecule HDAC935. The HDACi pharmacophore is shown in purple and the linked ester/acid moieties are shown in orange. (B, C) Inhibition of HeLa nuclear extract HDAC activity by ESM‐HDAC391 (n = 6) and HDAC189 (n = 2), respectively. (D, E) Acetylation of leucocyte populations in human whole blood following 4 hours incubation with ESM‐HDAC391 (n = 13) and HDAC935 (n = 4), respectively. (F) Inhibition of Tumour Necrosis Factor‐α (TNF‐α) production in lipopolysaccharide‐stimulated blood by ESM‐HDAC391 and HDAC935 (n = 4). (G) Quantification of HDAC189 levels in isolated monocytes following incubation of blood with ESM‐HDAC391 for 1 or 6 hours (n = 3), with each donor plotted separately
ESM‐HDAC391 (also known as GSK3117391 and CHR‐5154) is a novel ESM‐conjugated HDACi suitable for oral administration (Figure 1A). Both the parent ester (ESM‐HDAC391) and its acid metabolite (HDAC189, also known as GSK3339189) have sub‐micromolar HDACi activity. The matched control ester HDAC935, which is not a substrate for CES1, acts as a non‐targeted HDACi. This paper describes the myeloid‐specific targeting of ESM‐HDAC391 both preclinically and in a Phase I study evaluating its safety, tolerability, pharmacokinetics (PK) and PD after single and multiple doses in healthy male volunteers. In addition to predicted monocyte‐specific outcomes (compound retention, prolonged acetylation and ex vivo cytokine inhibition), transient and reversible monocytopaenia was an unexpected PD observation. Through further mechanistic work, we provide evidence that ESM‐HDAC inhibitor‐induced monocytopaenia is linked to loss of the key myeloid fate regulator colony stimulating factor 1 receptor (CSF1R). Together, our data support the application of ESM‐HDAC inhibitors as important orally active agents for the treatment of diseases beyond oncology.
2. METHODS
2.1. Clinical study design
Study NCT01934101 (https://clinicaltrials.gov/ct2/show/NCT01934101) was conducted at Biokinetic Europe (Belfast, NI). All participants provided written, informed consent in accordance with the Declaration of Helsinki. Approval by a national ethics committee (NRES Committee East of England, Nottingham, NG1 6FS, UK; approval number 13/EE/0200) was granted for the study, which was conducted in accordance with Good Clinical Practice from 12th August 2013–13th June 2014.
The primary objective of the study was to determine the safety and tolerability of single‐ and multiple‐ascending doses (SAD and MAD) of ESM‐HDAC391 in healthy volunteers. Secondary objectives were focussed on investigating PK profiles after single/repeat doses while exploratory objectives assessed PD endpoints (ex vivo lipopolysaccharide [LPS] stimulation, cellular acetylation) and the intracellular quantification of HDAC189 in monocytes.
The study comprised 2 parts: Part A was an SAD design with a separate food effect arm and Part B was an MAD design (once daily dosing for 7 days). Forty‐four participants were randomised in Part A (6 participants in cohorts 1 and 6, and 8 participants in cohorts 2–5) to receive ESM‐HDAC391 (range 5–60 mg) or placebo (2 in each cohort). Food effects were also investigated: participants from cohorts 2 and 4 returned to the study unit following a 2‐week washout period to receive the same dose of ESM‐HDAC391 administered during their first in‐house stay (20 or 40 mg), but in the fed (fatty breakfast) state. In Part B, 20 participants were randomised to receive 20 mg, 40 mg or placebo. Out of the 10 participants in each cohort, 8 received ESM‐HDAC391 and 2 received placebo. Based on the food effect results in Part A, the study drug was administered in the fasted state in Part B.
Safety assessments included electrocardiography, vital signs, serum chemistry, haematology and urinalysis. Treatment‐emergent adverse events (TEAEs) were recorded from the first drug administration. Adverse events were coded using the MedDRA Dictionary, Version 17.0. If a subject experienced >1 TEAE, the subject was counted once for each system organ class and once for each preferred term, each ordered in decreasing frequency.
2.2. Dose selection
Starting doses were selected based on 2 complementary approaches. Firstly, the no adverse event level (NOAEL) from the most sensitive preclinical toxicology species (monkey), was converted to a human dose of 60 mg, and a factor of 12 was applied to give the initial starting dose of 5 mg. Subsequent escalation in later dosing cohorts was allowed as indicated by the clinical safety monitoring data, providing that the resultant systemic exposures did not exceed this NOAEL level.
Secondly, an estimation of the minimum anticipated biological effect level came from a comparison with Chroma Therapeutic's first macrophage‐targeted HDAC inhibitor, tefinostat (CHR‐2845). In its Phase I study in cancer patients, 40 mg was the lowest dose that induced monocyte histone acetylation 20 and applying a 6–7‐fold correction factor for the enhanced in vitro potency of ESM‐HDAC391 also supported the 5 mg starting dose.
2.3. PK assessments
Levels of ESM‐HDAC391 and HDAC189 were measured in plasma from trial participants by liquid chromatography–tandem mass spectrometry (LC–MS/MS) following protein precipitation; 3 quality control samples were included and the lower limits of quantification were 1.00 and 10.00 ng/mL, respectively. To confirm intracellular levels of ester and acid, analogous LC–MS/MS assays were run on lysates from isolated monocytes; the lower limit of quantification was 1.00 pg/mL for both compounds in this matrix.
2.4. PD assessments
PD samples were obtained pretreatment, 1, 4 and 12 hours post‐treatment on the day of dosing and, in the repeat dose cohorts, on the final (seventh) day of dosing. Measurement of cytokines (tumour necrosis factor [TNF]α, interleukin [IL]‐6, IL‐10, IL‐12 and interferon [IFN]γ) after ex vivo LPS stimulation was initiated no later than 2 hours post‐blood draw (Randox Testing Services). Levels of lysine acetylation in monocytes, granulocytes and lymphocytes were determined by flow cytometry in blood fixed no later than 30 minutes after collection (Java Clinical Research). Peripheral blood mononuclear cells from 8 mL of blood were obtained by Ficoll gradient and monocytes were isolated by negative selection (EasySep) with the remaining cells (CD2+, CD3+, CD19+, CD20+, CD56+, CD66b+, CD123+ and glycophorin A+) constituting the non‐monocyte population. Cells were counted, lysates frozen and analysed by LC–MS (Quotient Bio Analytics Sciences) within 2 months.
For Part A, all 44 subjects were included in the PD analysis. For Part B, the PD subset consisted of n = 3 treated and n = 1 placebo for Cohort 1 (20 mg) and n = 7 treated with n = 2 placebo for Cohort 2. Seven subjects were excluded from the PD analysis set (5 subjects in Cohort 1 who received 20 mg, 1 subject in Cohort 1 who received placebo and 1 subject in Cohort 2 who received 40 mg) due to major protocol deviations. In Part B, 20 subjects reported 45 minor protocol deviations, and 7 major deviations. The major deviations were all related to incorrectly processed laboratory samples, of which 6 out of the 7 were concerned with 4‐hour LPS samples.
2.5. Preclinical assays
2.5.1. HDAC fluorometric activity
HDAC activity in HeLa nuclear extracts was measured using the Fluor de Lys assay kit (Enzo Life Sciences, Inc., New Jersey) according to manufacturer's instructions.
2.5.2. HDAC selectivity assays
Recombinant HDAC enzymes were incubated with fluorogenic substrate containing an acetylated lysine side chain and ESM‐HDAC391 or HDAC189. Trichostatin A and TMP269 were used as assay controls. Assays were carried out by Reaction Biology Corp (Malvern, Pennsylvania).
2.6. Inhibition of LPS‐stimulated TNFα production in human blood
Compounds (10 μM – 1.5 nM in 0.7% DMSO) were added to heparinised blood from healthy volunteers for 30 min prior to stimulation with 200 ng/mL LPS. After overnight incubation, TNFα plasma levels were determined by MesoScale Discovery technology.
2.7. Quantification of acid metabolite HDAC189 in monocytes
Heparinised blood was incubated with 10 μM ESM‐HDAC391 (0.1% DMSO) for 1 or 6 hours at 37°C. CD14+ monocytes were isolated using StraightFrom Whole Blood CD14 microbeads (Miltenyi Biotec) and lysates were snap‐frozen. Concentrations of ESM‐HDAC391 and HDAC189 were quantified by reverse‐phase LC–MS/MS.
2.8. Human flow cytometry
Compounds (10 μM–1.5 nM in 0.7% DMSO) were added to heparinised blood for indicated times. To measure intracellular acetylation, samples were fixed and lysed using FACS lysing solution (BD Pharmingen), Fc receptors were blocked with human IgG (Sigma) and populations stained with fluorescently‐labelled antibodies (listed in Table S1) before permeabilisation and staining with anti‐Ac‐K‐PE. To measure CSF1R and CCR2, samples were stained with cell surface markers prior to fixation with BD Phosflow Lyse/Fix buffer. Data were acquired using a BD FACS Canto II Flow Cytometer with FACS Diva software (BD BioSciences) and recorded as median fluorescence intensity.
2.9. Monocyte differentiation
Positively isolated human CD14+ monocytes were incubated with compound or vehicle control, with or without 100 ng/mL macrophage colony‐stimulating factor (M‐CSF). Compound was removed by washing after 4 or 20 hours and cells cultured with fresh media ± M‐CSF. At 48 hours post‐plating, cells were fixed with 10% formalin and stained with Hoechst 33342 and Alexa Fluor 488‐phalloidin. Plates were imaged on a Cellomics ArrayScan VTI HCS Reader and 32 fields per well were recorded. Data were normalised to the mean intensity of the positive control (monocytes + M‐CSF).
2.10. CES1/Es1e lo murine model
The CES1/Es1e lo mouse strain was designed by Chroma Therapeutics and generated by Genoway (Lyon, France) using Quick Knock‐In technology. Expression of the CES1 transgene is driven by the human CD68 promoter. 22 , 23 Mice were cross‐bred with a plasma esterase‐low (Es1elo) mouse (Jackson Labs USA: strain 000785 ‐ B6;D2‐a Ces1ce/EiJ) at Charles River (Margate, UK). For in vivo studies, ESM‐HDAC391, HDAC935 or vehicle (5% v/v DMSO: 10% w/v kleptose in saline) were administered intraperitoneally at 10 mg/kg unless specified. Male age‐matched mice were used in all experiments.
Animal studies were ethically reviewed and carried out in accordance with the UK Animals (Scientific Procedures) Act 1986 and GSK Policy on Care, Welfare and Treatment of Animals.
2.11. Murine flow cytometry
Murine blood was collected by cardiac puncture into EDTA‐coated tubes and, where indicated, incubated with inhibitors (10 μM–1 nM in 0.7% DMSO) for 3 hours, stained with the fluorescently‐labelled antibodies listed in Table S1 and fixed using FACS Lysing Solution (BD Biosciences). To measure intracellular acetylation, cells were fixed with BD Phosflow Lyse/Fix Buffer prior to nuclear permeabilisation buffer (Biolegend) and stained with anti‐acetylated lysine‐PE. Data were acquired on a BD FACS Canto II Flow Cytometer, analysed using BD FACSDiva software (BD Biosciences) or FlowJo v10 and recorded as the median fluorescence intensity.
2.12. Statistical analysis
Data were analysed using Microsoft Excel 2007 and GraphPad Prism v5.04. Data are represented as mean ± standard error of the mean unless stated. Preclinical data were fitted using a nonlinear regression curve fit (log [inhibitor] vs. response analysis‐variable slope [4 parameters]). Details of statistical analyses are indicated in legends. Significance is illustrated as follows; * P ≤ .05, ** P ≤ .01, *** P ≤ .001.
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019 a,b). 24
3. RESULTS
3.1. ESM‐HDAC391 is a myeloid‐targeted HDAC inhibitor
The HDAC selectivity profile, myeloid selective effects on acetylation and potency increase following the addition of ESM technology were demonstrated preclinically. Both ESM‐HDAC391 and its acid metabolite HDAC189 were confirmed as potent HDACi in a HeLa cell nuclear extract fluorometric assay with mean 50% inhibitory concentration (IC50) values of 55 and 54 nM, respectively (Figure 1B–C). Selectivity of ESM‐HDAC391 across the HDAC family was investigated using a panel of recombinant HDACs (Table S2). Both ESM‐HDAC391 and HDAC189 potently inhibited Class I, Class IIb and Class IV members (HDAC 1/2/3/8, HDAC 6/10, and HDAC 11, respectively), with Class IIa enzymes (HDAC 4/5/7/9) unaffected.
Myeloid‐selective increases in protein acetylation by ESM‐HDAC391 were demonstrated in human whole blood (Figure 1D), where increased global acetylation in monocytes (EC50 158 nM) indicated marked selectivity over other leucocyte populations. In comparison, the matched t‐butyl control ester HDAC935 (not a substrate for CES1 so acting as a non‐targeted HDACi) elevated global acetylation less potently (EC50 6.31 μM) and lacked selectivity for monocytes (Figure 1E). Furthermore, ESM‐HDAC391 inhibited TNFα production in LPS‐stimulated blood (IC50 174 nM) whereas HDAC935 was markedly less potent (IC50 1.66 μM; Figure 1F). Following incubation of blood with ESM‐HDAC391 for 1 or 6 hours, increasing concentrations of HDAC189 were subsequently detected within isolated monocytes (Figure 1G), whereas ESM‐HDAC391 itself was undetectable, indicative of rapid intracellular conversion from CES1‐labile ester to acid metabolite in these cells.
3.2. ESM‐HDAC391 demonstrates selective myeloid‐targeting in a phase I study
The clinical potential of ESM‐HDAC391 was evaluated in a Phase I healthy volunteer study consisting of SAD cohorts ranging from 5 to 60 mg (Part A) and MADs of 20 or 40 mg (Part B). The participant disposition of the study and its cohorts is illustrated in Figure 2. Subject demographics are summarised in Tables 1 and 2 for Parts A/B respectively.
FIGURE 2.
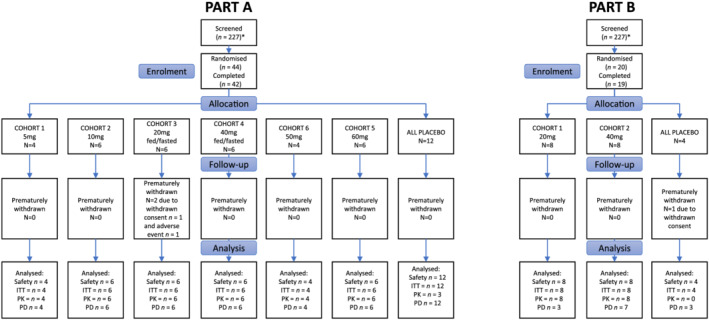
Participant disposition for Part A and Part B. In Part A, 44 participants were randomised and 42 completed the study. In Part B, 20 patients were randomised and 19 completed the study. Reasons for discontinuation are shown in the diagram
TABLE 1.
Subject demographics (Part A)
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 6 | Cohort 5 | All placebo | Overall | |
|---|---|---|---|---|---|---|---|---|
| PART A | 5 mg | 10 mg | 20 mg | 40 mg | 50 mg | 60 mg | (part A) | (part A) |
| Fasted/fed | Fasted/fed | |||||||
| (n = 4) | (n = 6) | (n = 6) | (n = 6) | (n = 4) | (n = 6) | (n = 12) | (n = 44) | |
| Age (y) | ||||||||
| Mean | 27 | 29.3 | 26.5 | 29 | 31.5 | 26.7 | 25.2 | 27.4 |
| (SD) | 10.71 | 8.78 | 8.43 | 9.63 | 8.66 | 4.89 | 5.91 | 7.55 |
| Minimum | 21 | 21 | 21 | 18 | 20 | 19 | 19 | 18 |
| Median | 22 | 27.5 | 23.5 | 28.5 | 32.5 | 27 | 23.5 | 25 |
| Maximum | 43 | 41 | 43 | 40 | 41 | 34 | 41 | 43 |
| BMI (kg/m 2 ) | ||||||||
| Mean | 24.63 | 25.7 | 24.93 | 23.98 | 28.18 | 26.58 | 23.78 | 25.09 |
| (SD) | 6.129 | 2.937 | 3.56 | 3.305 | 3.762 | 2.4 | 2.102 | 3.328 |
| Minimum | 18.4 | 21 | 19.9 | 20.4 | 24.2 | 22.8 | 20.2 | 18.4 |
| Median | 24.5 | 26.05 | 25 | 23.65 | 28.25 | 27.2 | 24.45 | 24.65 |
| Maximum | 31.1 | 29.1 | 30 | 30 | 32 | 28.9 | 27.3 | 32 |
| Height (cm) | ||||||||
| Mean | 177.13 | 175.8 | 180.92 | 178.55 | 175.88 | 178.37 | 177.25 | 177.75 |
| (SD) | 10.664 | 4.195 | 2.498 | 7.117 | 6.738 | 6.038 | 5.231 | 5.789 |
| Minimum | 170 | 171 | 177.5 | 170.8 | 169 | 167.5 | 169.1 | 167.5 |
| Median | 172.75 | 175 | 180.5 | 176.25 | 174.75 | 181.3 | 177 | 177.25 |
| Maximum | 193 | 183 | 185 | 191 | 185 | 183.1 | 187.9 | 193 |
| Weight (kg) | ||||||||
| Mean | 78.55 | 79.32 | 81.78 | 76.27 | 87.5 | 84.37 | 74.92 | 79.4 |
| (SD) | 27.477 | 7.72 | 13.398 | 9.811 | 15.601 | 5.71 | 9.234 | 12.314 |
| Minimum | 55 | 64.4 | 64.6 | 67.5 | 73.8 | 76.6 | 61.2 | 55 |
| Median | 71.7 | 81.8 | 80.75 | 72.55 | 85.6 | 84.6 | 77.3 | 79.8 |
| Maximum | 115.8 | 85.2 | 102.6 | 94.6 | 105 | 91.5 | 90 | 115.8 |
| Ethnicity, n (%) | ||||||||
| Hispanic or Latino: | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Not Hispanic or Latino: | 4 | 6 | 6 | 6 | 4 | 6 | 12 | 44 |
| Race, n (%) | ||||||||
| Black or African American | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 1 (2.3) |
| American Indian or Alaskan native | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Asian | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Native Hawaiian or other Pacific islander | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| White | 4 (100.0) | 5 (83.3) | 6 (100.0) | 6 (100.0) | 4 (100.0) | 6 (100.0) | 12 (100.0) | 43 (97.7) |
| Other | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Abbreviation: SD, standard deviation.
TABLE 2.
Subject demographics (Part B)
| Cohort 1 | Cohort 2 | All placebo | Overall | |
|---|---|---|---|---|
| PART B | 20 mg | 40 mg | (part B) | (part B) |
| (n = 8) | (n = 8) | (n = 4) | (n = 20) | |
| Age (y) | ||||
| Mean | 28 | 23.4 | 27.3 | 26 |
| (SD) | 6.93 | 5.13 | 5.91 | 6.15 |
| Minimum | 19 | 19 | 19 | 19 |
| Median | 29.5 | 21 | 29 | 25 |
| Maximum | 35 | 34 | 32 | 35 |
| BMI (kg/m 2 ) | ||||
| Mean | 24.83 | 24.86 | 25.3 | 24.94 |
| (SD) | 2.636 | 3.158 | 3.542 | 2.872 |
| Minimum | 19.5 | 19.5 | 21.4 | 19.5 |
| Median | 24.7 | 25.2 | 25.3 | 24.95 |
| Maximum | 27.9 | 29.1 | 29.2 | 29.2 |
| Height (cm) | ||||
| Mean | 175.08 | 183.99 | 175.23 | 178.67 |
| (SD) | 7.186 | 4.634 | 3.706 | 6.997 |
| Minimum | 165.5 | 175 | 171 | 165.5 |
| Median | 175.05 | 185 | 174.95 | 179.2 |
| Maximum | 185 | 189.2 | 180 | 189.2 |
| Weight (kg) | ||||
| Mean | 76.08 | 84.21 | 77.93 | 79.7 |
| (SD) | 9.022 | 11.588 | 12.883 | 10.974 |
| Minimum | 57.7 | 62.9 | 62.5 | 57.7 |
| Median | 76.25 | 85.65 | 80.15 | 80.45 |
| Maximum | 89.3 | 102.4 | 88.9 | 102.4 |
| Ethnicity, n (%) | ||||
| Hispanic or Latino: | 0 | 0 | 0 | 0 |
| Not Hispanic or Latino: | 8 (100.0) | 8 (100.0) | 4 (100.0) | 20 (100.0) |
| Race, n (%) | ||||
| Black or African American | 0 | 0 | 0 | 0 |
| American Indian or Alaskan native | 0 | 0 | 0 | 0 |
| Asian | 0 | 0 | 0 | 0 |
| Native Hawaiian or other Pacific islander | 0 | 0 | 0 | 0 |
| White | 8 (100.0) | 8 (100.0) | 4 (100.0) | 20 (100.0) |
| Other | 0 | 0 | 0 | 0 |
Abbreviation: SD, standard deviation.
ESM‐HDAC391 was well tolerated with a low rate of TEAEs across groups (Table 3). All TEAEs were mild or moderate in severity, with headache most commonly reported. One TEAE (increased blood bilirubin) led to treatment withdrawal in 1 subject from single‐dose cohort 3 after receiving 20 mg ESM‐HDAC391 under fasting conditions. In Part B, greater numbers of TEAEs were reported in cohort 2 (40 mg) compared with cohort 1 (20 mg).
TABLE 3.
Summary of treatment‐emergent adverse events (TEAEs; Part A and B). E = events; S = subjects
| Cohort 1 | Cohort 2 | Cohort 3 (20 mg) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PART A | 5 mg, n = 4 | 10 mg, n = 6 | Fasted (n = 6) | Fed (n = 5) | ||||||
| E | S (%) | E | S (%) | E | S (%) | E | S (%) | |||
| Total number of TEAEs | 0 | 0 | 4 | 3 (50.0) | 2 | 2 (33.3) | 0 | 0 | ||
| Headache | 0 | 0 | 3 | 2 (33.3) | 1 | 1 (16.7) | 0 | 0 | ||
| Syncope | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Nausea | 0 | 0 | 1 | 1 (16.7) | 0 | 0 | 0 | 0 | ||
| Cheilitis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Vessel puncture site bruise | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Contusion | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Blood bilirubin increased | 0 | 0 | 0 | 0 | 1 | 1 (16.7) | 0 | 0 | ||
| Cohort 4 (40 mg) | Cohort 6 | Cohort 5 | All placebo | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PART A | Fasted (n = 6) | Fed (n = 5) | 50 mg, n = 4 | 60 mg, n = 6 | n = 12 | |||||
| E | S (%) | E | S (%) | E | S (%) | E | S (%) | E | S (%) | |
| Total number of TEAEs | 4 | 2 (33.3) | 3 | 3 (60.0) | 1 | 1 (25.0) | 1 | 1 (16.7) | 1 | 1 (8.3) |
| Headache | 1 | 1 (16.7) | 3 | 3 (60.0) | 1 | 1 (25.0) | 0 | 0 | 0 | 0 |
| Syncope | 1 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nausea | 1 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cheilitis | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 (16.7) | 0 | 0 |
| Vessel puncture site bruise | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 (8.3) |
| Contusion | 1 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Blood bilirubin increased | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cohort 1 | Cohort 2 | All placebo | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PART B | 20 mg, n = 8 | 40 mg, n = 8 | n = 4 | |||||||
| E | S (%) | E | S (%) | E | S (%) | |||||
| Total number of TEAEs | 2 | 2 (25.0) | 10 | 5 (62.5) | 1 | 1 (25.0) | ||||
| Headache | 1 | 1 (12.5) | 4 | 3 (37.5) 0 0 | 0 | 0 | ||||
| Dizziness | 0 | 0 | 1 | 1 (12.5) | 0 | 0 | ||||
| Nausea | 0 | 0 | 2 | 2 (25.0) | 0 | 0 | ||||
| Abdominal pain | 0 | 0 | 2 | 2 (25.0) | 0 | 0 | ||||
| Abdominal pain upper | 0 | 0 | 0 | 0 | 1 | 1 (25.0) | ||||
| Constipation | 0 | 0 | 1 | 1 (12.5) | 0 | 0 | ||||
| Application site pruritus | 1 | 1 (12.5) | 0 | 0 | 0 | 0 | ||||
PK analyses of ESM‐HDAC391 and HDAC189 were undertaken (Tables 4 and 5). After single dosing, ESM‐HDAC391 was rapidly absorbed and eliminated from the body with a short plasma half‐life of 21–30 minutes and a Tmax of ~30 minutes. Plasma exposure of ESM‐HDAC391 (Cmax and AUC) increased with dose and HDAC189 showed similar kinetics (plasma half‐life 36–46 min). Transient kinetics were confirmed in the multiple dose cohorts (plasma half‐life and Tmax for ESM‐HDAC391 and HDAC189 <1 h) and small, although variable, degrees of accumulation (indicated by the AUC metric Rac and the Cmax metric RCmax) were generally observed for ESM‐HDAC391 when comparing Day 7 levels with Day 1 (Table 5). Comparison of the fed vs. fasted state for the 20 and 40 mg single dose cohorts (Table S3) confirmed modest but complex food effects on both ESM‐HDAC391 and HDAC189, with a tendency to reduced Cmax but mixed effects on AUC0‐t (increased at 20 mg, decreased at 40 mg). Part B was conducted in the fasted state.
TABLE 4.
Pharmacokinetic parameters for ESM‐HDAC391 and HDAC189 (Part A)
| ESM‐HDAC391 (n = 6 unless otherwise stated) | ||||||
|---|---|---|---|---|---|---|
| Part A | Cohort 1 b | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 6 d | Cohort 5 |
| 5 mg | 10 mg | 20 mg | 40 mg | 50 mg | 60 mg | |
| Cmax | 16.53 | 42.82 | 47.38 | 209.99 | 256.47 | 287.97 |
| (ng/mL) | (81.5) | (45.7) | (72.8) | (17.8) | (31.4) | (38.4) |
| Tmax a | 0.75 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| (h) | (0.50–1.00) | (0.50–1.00) | (0.50–4.00) | (0.50–0.75) | (0.50–0.75) | (0.50–1.00) |
| AUC0‐t | 13.95 | 32.308 | 39.419 | 151.509 | 186.026 | 244.011 |
| (ng.h/mL) | (39.1) | (24.9) | (15.8) | (11.4) | (39.1) | (40.4) |
| AUC0–24 | NC | NC | 40.109 b | 156.821 | 189.255 | 246.834 |
| (ng.h/mL) | (18.4) | (11.7) | (38) | (39.8) | ||
| AUC0‐∞ | NC | NC | 40.109 b | 156.82 | 189.258 | 246.855 |
| (ng.h/mL) | (18.4) | (11.7) | (17) | (39.8) | ||
| t½ | NC | NC | 0.343 b | 0.429 | 0.366 | 0.5 |
| (h) | (11.5) | (25) | (17) | (56.4) | ||
| CL/F | NC | NC | 498.623 b | 255.067 | 264.195 | 243.057 |
| (L/h) | (18.3) | (11.7) | (38) | (39.8) | ||
| Vz/F | NC | NC | 245.398 b | 157.777 | 140.073 | 176.016 |
| (L) | (27) | (30.1) | (37.7) | (32.2) | ||
| Part A | HDAC189 (n = 6 unless otherwise stated) | |||||
|---|---|---|---|---|---|---|
| Cohort 1 d | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 6 | Cohort 5 | |
| 5 mg | 10 mg | 20 mg | 40 mg | 50 mg | 60 mg | |
| Cmax | 18.99 | 28.44 | 47.30 | 154.57 | 206.8 b | 213.23 |
| (ng/mL) | (13.0) | (16.0) | (62.2) | (23.0) | (26.3) | (119.5) |
| Tmax a | 1.000 | 1.000 | 0.760 | 0.875 | 1.00 b | 1.01 |
| (h) | (1.00–1.00) | (1.00–2.00) | (0.75–4.00) | (0.75–1.00) | (1.00–1.00) | (0.75–1.25) |
| AUC0‐t | 11.741 | 37.208 | 61.488 | 189.109 | 289.224 b | 280.364 |
| (ng.h/mL) | (31.9) | (16.8) | (34.3) | (16.3) | (41.3) | (114.4) |
| AUC0–24 | NC | NC | 87.070 b | 250.956 b | 362.960 d | 437.829 b |
| (ng.h/mL) | (23.9) | (9.6) | (17.0) | (25.8) | ||
| AUC0‐∞ | NC | NC | 65.480 c | 243.317 d | 362.961 d | 437.822 b |
| (ng.h/mL) | (NC) | (8.9) | (17.0) | (25.8) | ||
| t½ | NC | NC | 0.767 b | 0.723 b | 0.709 d | 0.604 b |
| (h) | (39.9) | (30.3) | (6.0) | (29.2) | ||
Note: Date are represented as geometric mean (% coefficient of variation geometric mean) unless otherwise stated.
Abbreviations: AUC, area under the plasma drug concentration–time curve, as defined from 0 h to the last time of quantifiable concentration (t), to 24 h, or extrapolated to infinity time (∞). CL/F, apparent clearance; Cmax, maximum serum concentration; F, oral bioavailability; Tmax, time to maximum observed plasma drug concentration; t1/2, terminal half‐life; Vz/F, apparent volume of distribution.
n = the number of subjects with a measurement, NC: not calculated.
median and range presented;
n = 4;
n = 1;
n = 3.
TABLE 5.
Pharmacokinetic parameters for ESM‐HDAC391 and HDAC189 (Part B)
| Part B | ESM‐HDAC391 (n = 6 unless otherwise stated) | |||
|---|---|---|---|---|
| Cohort 1 (20 mg) | Cohort 2 (40 mg) | |||
| Day 1 | Day 7 e | Day 1 | Day 7 h | |
| Cmax | 102.54 | 108.81 | 165.5 | 283.84 |
| (ng/mL) | (51.3) | (40.8) | (70.7) | (30.3) |
| Tmax g | 0.500 | 0.625 | 0.625 | 0.625 |
| (h) | (0.25–1.25) | (0.50–2.00) | (0.50–2.00) | (0.50–1.00) |
| AUC0‐t | 76.695 | 94.351 | 137.736 | 228.723 |
| (ng.h/mL) | (39.5) | (31.4) | (41.7) | (27.3) |
| AUCtau | 85.654 b | 98.844 a | 159.639 a | 251.381 b |
| (ng.h/mL) |
(43.8) |
(36.9) | (36.1) | (17.4) |
| t½ | NC | 0.368 a | NC | 0.408 b |
| (h) | (24.1) | (17.6) | ||
| CL/F | NC | 0.723 a | NC | 159.118 b |
| (L/h) | (30.3) | (17.4) | ||
| Vz/F | NC | 107.083 a | NC | 93.470 b |
| (L) | (43.6) | (20.7) | ||
| Rac g | NC | 1.140 c | NC | 1.410 c |
| (0.99–1.57) | (1.22–2.45) | |||
| Rcmax g | NC | 0.9 | NC | 1.775 |
| (0.53–2.48) | (0.61–4.78) | |||
| Part B | HDAC189 (n = 8 unless otherwise stated) | |||
|---|---|---|---|---|
| Cohort 1 (20 mg) | Cohort 2 (40 mg) | |||
| Day 1 | Day 7 | Day 1 | Day 7 | |
| Cmax | 78.01 | 69.47 | 151.46 | 180.98 |
| (ng/mL) | (35.6) | (39.6) | (33.1) | (31.9) |
| Tmax g | 1.000 | 1.125 | 1.015 | 1.125 |
| (h) | (0.75–2.00) | (1.00–2.00) | (0.75–2.00) | (0.75–2.00) |
| AUC0‐t | 97.850 | 89.376 | 221.401 | 316.144 |
| (ng.h/mL) | (36.6) | (24.3) | (36.3) | (21.4) |
| AUC0‐tau | 136.873 g | NC | 307.974 d | 380.624 e |
| (ng.h/mL) | (1.4) | (0.3) | (14.5) | |
| t½ | NC | NC | NC | 0.754 e |
| (h) | (11.7) | |||
| Rac g | NC | NC | NC | 1.020 f |
| Rcmax g | NC |
0.835 (0.60–1.51) |
NC |
1.120 (0.67–2.85) |
Note: Date are represented as geometric mean (% coefficient of variation geometric mean) unless otherwise stated.
Abbreviations: AUC, area under the plasma drug concentration–time curve, as defined from 0 h to the last time of quantifiable concentration (t), to 24 h, or extrapolated to infinity time (∞). CL/F, apparent clearance; Cmax, maximum serum concentration; F, oral bioavailability; Rac, accumulation ratio for AUC; Rcmax, accumulation ratio for Cmax; Tmax, time to maximum observed plasma drug concentration; t1/2, terminal half‐life; Vz/F, apparent volume of distribution.
n = 6;
n = 7;
n = 5;
n = 2;
n = 4;
n = 1;
median and range presented;
at steady state; NC: not calculable.
Despite transient systemic PK, measurement of intracellular HDAC189 in monocytes confirmed lasting target engagement and selective, dose‐dependent retention of acid in isolated monocytes relative to non‐monocytes (Figures 3A and 4A,B). Importantly, sustained average intracellular concentrations of HDAC189 within monocytes were maintained above its in vitro IC50 (18.05 ng/mL, equivalent to the HeLa cell lysate IC50 of 53.83 nM in Figure 1C) at 12 hours post‐dose with 10 mg or higher. Furthermore, intracellular acid levels in monocytes were above the IC90 for at least 4 hours following doses ≥40 mg.
FIGURE 3.
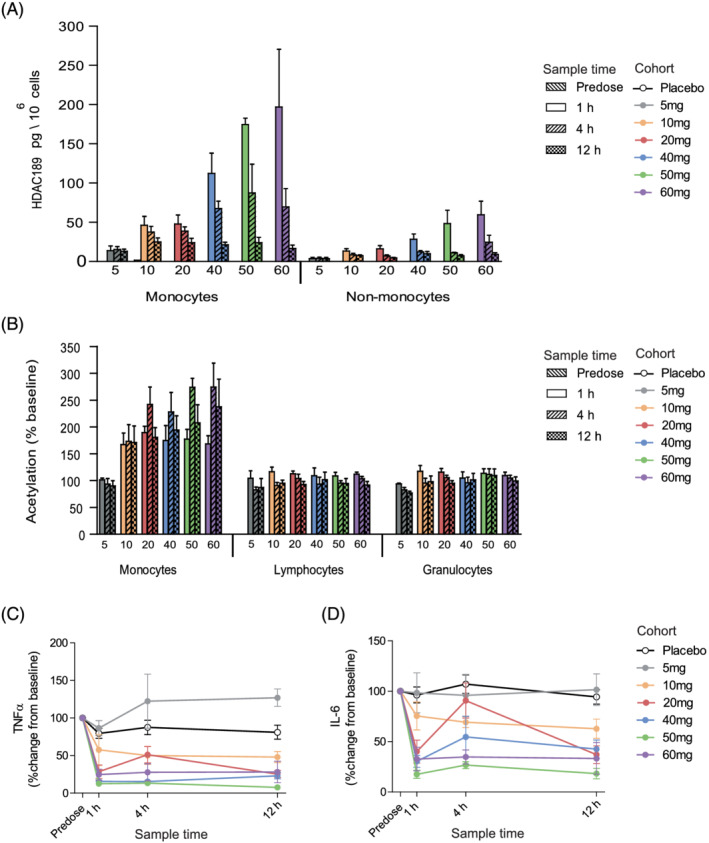
Part A, single‐ascending dose, clinical characterisation of ESM‐HDAC391. (A) Quantification of HDAC189 levels in monocyte and non‐monocyte fractions per dose cohort. (B) Acetylation levels in gated monocyte, lymphocyte and granulocyte populations for each assessed timepoint and dose cohort. (C, D) Time course of inhibition of ex vivo lipopolysaccharide‐stimulated cytokine production (tumour necrosis factor [TNF]α and interleukin [IL]‐6, respectively) per dose cohort. Data are shown as mean and standard error. n = 6 for all treatment cohorts with the exception of 5 mg and 50 mg, where n = 4. For panels C and D, n = 12 placebos from all cohorts were included
FIGURE 4.
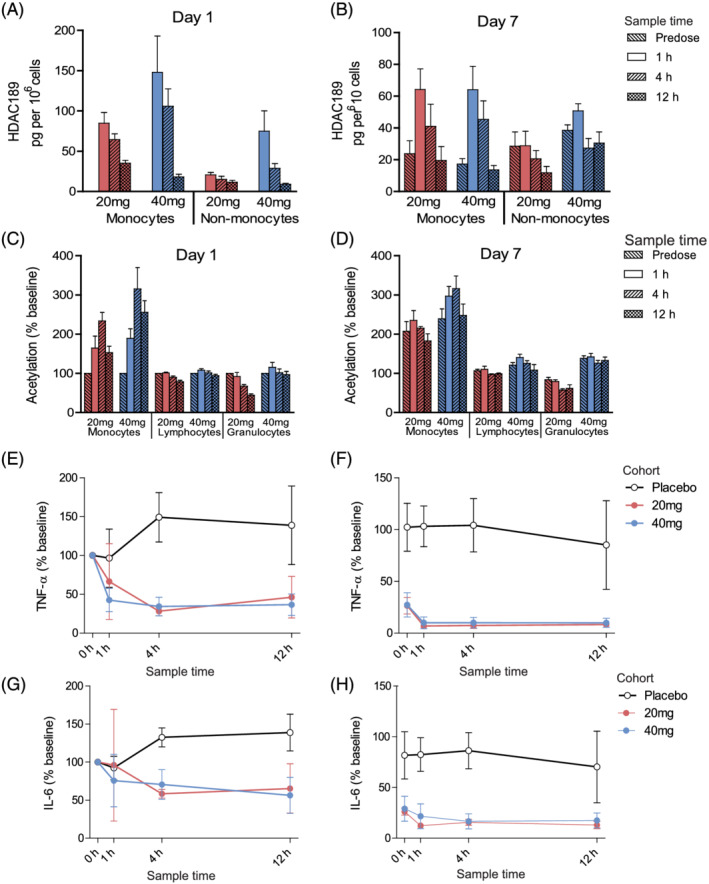
Part B, multiple‐ascending dose, clinical characterisation of ESM‐HDAC391. (A, B) Quantification of HDAC189 levels in monocyte and non‐monocyte fractions per dose cohort. (C, D) Acetylation levels in gated monocyte, lymphocyte and granulocyte populations for each assessed timepoint and dose cohort. (E–H) Time course of inhibition of ex vivo LPS‐stimulated cytokine production (tumour necrosis factor [TNF]α in E, F and interleukin [IL]‐6 in G‐H) per dose cohort. Panels A, C, E, G represent Day 1 data; panels B, D, F, H represent Day 7 data. Data are shown as mean and standard error. For all panels, 20 mg (n = 3) and 40 mg (n = 7) data are included. For panels E‐H, n = 3 placebos (n = 1 from 20 mg and n = 2 from 40 mg cohort) were included
Evidence for mechanistic consequences of preferential retention in monocytes were provided by 2 complementary endpoints. Firstly, clear dose‐dependent increases in global acetylation levels were seen in monocyte populations over lymphocytes and granulocytes in the SAD cohorts and maintained for at least 12 hours (Figure 3B), consistent with the aims of ESM targeting. These were mirrored upon repeat dosing (Figure 4C–D), with selective acetylation of monocytes over other leucocyte populations seen on Day 1 and Day 7. Importantly, the predose analysis on Day 7, 24 hours after the previous dose, demonstrated enhanced monocyte acetylation relative to Day 1 levels, indicating a cumulative, prolonged effect. Secondly, ex vivo LPS stimulation of blood showed inhibition of cytokines in a dose‐dependent manner (exemplified by TNFα and IL‐6 in Figure 3C–D, with similar findings for IL‐1β, IFN‐γ and IL‐10). Mean reductions to <50% of baseline were attained for all cytokines measured, at single doses of 20 mg and above (except IFN‐γ, which achieved this level with 40 mg). With repeat dosing (Figure 4E–H), Day 1 cytokines were similarly inhibited by ESM‐HDAC391 at both doses. The Day 7 data confirmed this reduction, but from markedly reduced t = 0 levels, again demonstrating PD effects persisting from previous doses in the absence of measurable plasma concentrations of parent or acid. This PK‐PD disconnect, with acetylation levels and cytokine inhibition exceeding even the prolonged acid retention observed in targeted monocytes, probably reflects the relative stability/slow turnover of the generated acetylation marks, suggesting further therapeutic impact from targeting HDACs by ESM technology.
3.3. ESM‐HDAC391 causes transient and rapidly reversible monocytopaenia in the absence of neutropaenia or thrombocytopaenia
Haematological analysis revealed that ESM‐HDAC391 led to substantial reduction in the total number of circulating monocytes, which had not been evident in preclinical studies. Throughout Part A, monocytopaenia increased in magnitude and duration in a dose‐dependent manner (Figure S1A). In Part B, decreases were observed on Day 1 (4 h postdose) and reached maximal depletion on Day 2 (24 h; Figure 5A,B). Reductions were maintained after each successive dose, with maximal depletion levels of 0.06 x109 monocytes/L with both 20 and 40 mg doses. Following cessation of dosing, recovery of monocytes was evident on Days 8–9, with normalisation by post‐study visits (4–7 d after last treatment).
FIGURE 5.
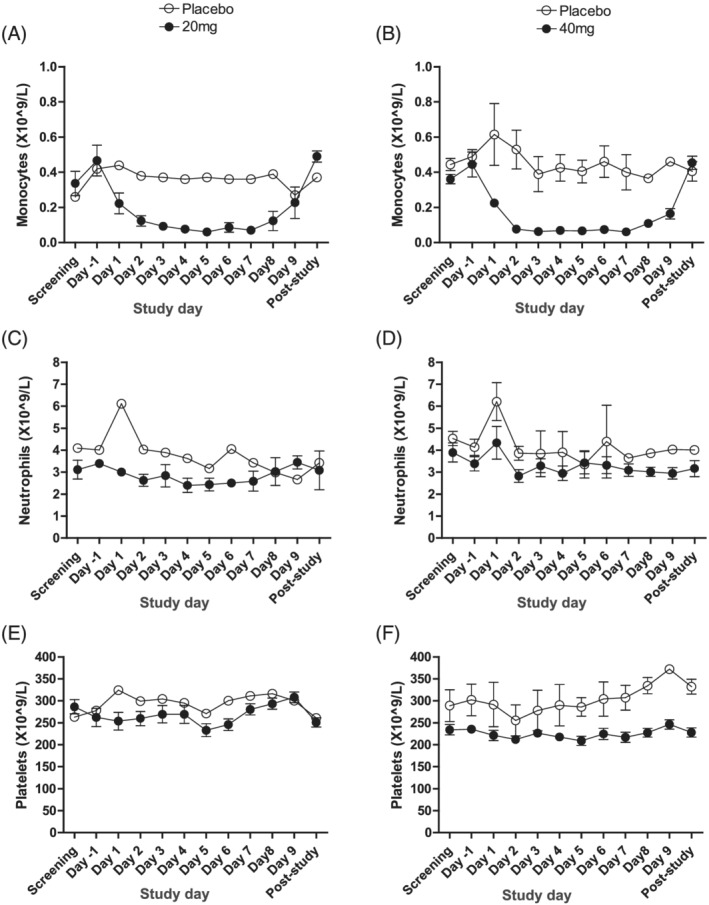
Blood leucocyte counts in subjects dosed with ESM‐HDAC391 vs. placebo in Part B, multiple ascending dose. Levels of monocytes, neutrophils and platelets are indicated in panels (A, B), (C, D) and (E, F), respectively. The 20‐mg repeat dose cohort (n = 3 treated and n = 1 placebo) measurements are shown in panels (A), (C) and (E), and the 40‐mg cohorts (n = 7 treated and n = 2 placebo) in panels (B), (D) and (F). Post‐study visit occurred 4–7 days after last study treatment. Data are shown as mean and standard error
Importantly, parallel assessment of neutrophils and platelets (Figure 5C–F and S1B,C), commonly depleted by non‐targeted HDACi, demonstrated no evidence of neutropaenia or thrombocytopaenia with single or multiple dosing.
3.4. Characterisation of monocytopaenia in a CES1/Es1e lo transgenic mouse model
To investigate the mechanisms underlying the unexpected monocyte depletion, a transgenic CES1/Es1e lo mouse model was used. These mice express human CES1 under the myeloid‐specific promoter CD68 to drive expression in monocytes and macrophages 22 , 23 on a background strain with low plasma esterase (Ces1ce), which is otherwise elevated in rodents, to mimic the lack of plasma CES1 in humans. To confirm that ESM‐directed target engagement occurs in these mice, acetylation levels in leucocyte populations in blood treated ex vivo for 3 hours with ESM‐HDAC391 were determined by flow cytometry. Selective acetylation occurred in monocytes compared with other cell types, with maximal differences occurring at 1 μM ESM‐HDAC391, and an EC50 value of 226 nM in monocytes (Figure 6A).
FIGURE 6.
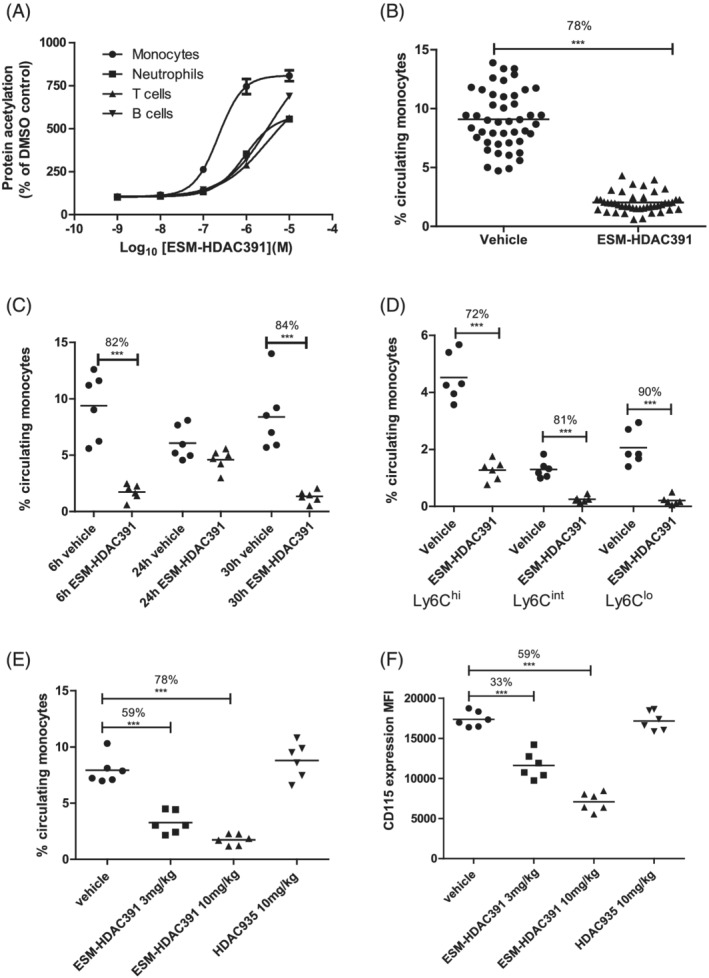
Demonstration of monocytopaenia in CES1/Es1e lo mice. (A) Acetylation of leucocyte populations in mouse whole blood following 3 hours incubation with ESM‐HDAC391 (n = 6). (B) Reduction in total circulating monocyte population 6 hours after intraperitoneal (ip) dosing of CES1/Es1e lo mice with 10 mg/mL ESM‐HDAC391 (n = 46) compared to vehicle controls (n = 45); data pooled across 8 in vivo studies. (C) Time course of reduction in total circulating monocytes in CES1/Es1e lo mice either 6 or 24 hours after a single dose, or 6 hours after a second dose at 24 hours, of 10 mg/mL ESM‐HDAC391 vs. vehicle (ip), all compared with time‐matched vehicle controls; n = 6 mice per group. (D) Analysis of monocyte sub‐populations (Ly6Chi, Ly6Clo and Ly6Cint) 6 hours after dosing CES1/Es1e lo mice with 10 mg/mL ESM‐HDAC391 compared with vehicle controls (n = 6). (E) Reduction in total circulating monocyte population 6 hours after dosing CES1/Es1e lo mice with 3 or 10 mg/mL ESM‐HDAC391 vs. vehicle (ip) compared with 10 mg/kg HDAC935 and matched vehicle controls (n = 6). (F) Levels of CD115 (measured as median fluorescence intensity) on monocyte populations in panel (E). Statistical analysis was carried out by an unpaired, 2‐tailed t‐test (B) or a 1‐way ANOVA with Bonferroni post‐test (C–F)
To replicate monocytopaenia, CES1/Es1e lo mice were dosed intraperitoneally with 10 mg/kg ESM‐HDAC391, modelled to mimic 40 mg dosing for humans. A significant decrease of 78% of circulating monocytes was observed after 6 hours (Figure 6B). This effect was transient (recovering by 24 h) and reproducible, as repeat administration of ESM‐HDAC391 at 24 hours caused a subsequent decrease in total circulating monocytes at 30 hours (Figure 6C). Significant reductions in all monocyte subsets were observed, with a progressive increase in depletion associated with sequential monocyte maturation status: classical Ly6Chi monocytes (72%) < intermediate Ly6Cint population (81%) < non‐classical Ly6Clo cells (90%; Figure 6D). Further investigations into the effects of longer‐term dosing on monocytopaenia were carried out (Figure S2) and confirmed replenishment of circulating monocytes from bone marrow, and no effect on bone marrow progenitor cells, even with 14 days dosing.
To confirm monocyte selectivity and increased potency relative to a non‐targeted HDACi, ESM‐HDAC391 was compared with 10 mg/kg control ester HDAC935 (modelled to match ester blood profiles) and the effects on leucocyte populations in the blood at 6 hours postdose were determined. Both doses of ESM‐HDAC391 caused a significant reduction in circulating monocytes but no effect was observed with HDAC935 (Figure 6E), indicating that monocyte depletion is ESM‐dependent and probably due to specific retention of intracellular acid. Interestingly, a significant reduction in CD115 expression levels (also known as CSF1R, or M‐CSF receptor) was observed on the remaining circulating monocytes from mice treated with ESM‐HDAC391 (Figure 6F). No change in CD115 expression was observed in mice treated with HDAC935.
3.5. Phenotypic changes in CSF1R expression on human monocytes and functional consequences
The reduction in CD115 (CSF1R) expression on mouse monocytes suggested a possible mechanism for the monocytopaenia observed clinically. To determine if ESM‐HDAC391 caused decreased expression of CSF1R on human monocytes, freshly isolated human blood was treated for 1, 4 or 8 hours with ESM‐HDAC391 or HDAC935. As shown in Figure 7A and B, time‐ and concentration‐dependent decreases in expression of CSF1R on monocytes following incubation with ESM‐HDAC391 were observed, whilst HDAC935 treatment only decreased CSF1R expression at much higher compound concentrations. Notably, no downregulation of the chemokine receptor CCR2, critical for monocyte egress from the bone marrow and chemotaxis, 25 was observed with either compound (Figure 7C,D), indicating the effect on CSF1R is unlikely to be due to compromised viability. Further investigations into cell health along with adhesion molecule expression and function extend this observation (Figure S3A–C).
FIGURE 7.
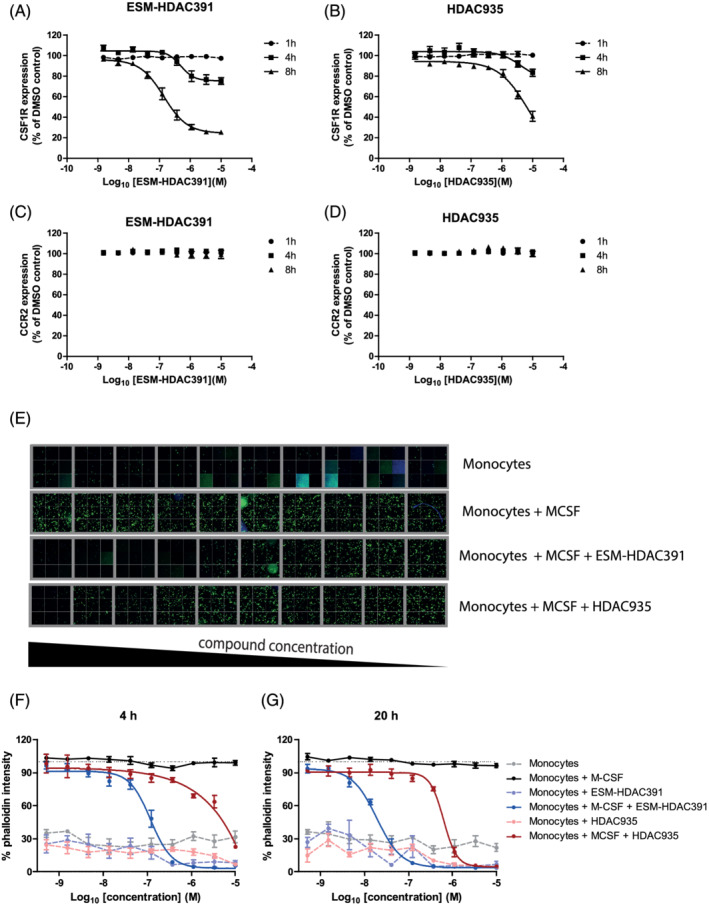
ESM‐HDAC391 downregulates CSF1R and affects monocyte differentiation. (A, B) Concentration‐ and time‐dependent reduction in CSF1R on monocytes incubated with ESM‐HDAC391 and HDAC935, respectively (n = 4). (C, D) Parallel expression of CCR2 levels on monocytes following incubation as above with ESM‐HDAC391 and HDAC935, respectively (n = 4). (E) Cytoskeletal F‐actin staining images following 48 hours differentiation of CD14+ monocytes in the presence of macrophage colony‐stimulating factor (M‐CSF) with an initial 4 hours exposure to ESM‐HDAC391 or HDAC935 (final concentrations titrated from 10 μM to 0.51 nM) followed by compound washout and continued incubation with M‐CSF in fresh media for the remaining 44 hours; representative data of 32 fields of views per well from 1 of the n = 4 replicate donors are shown. (F, G) Normalised quantification of cytoskeletal F‐actin staining following 48 hours differentiation of CD14+ monocytes in the presence of M‐CSF with an initial incubation with ESM‐HDAC391 or HDAC935 for 4 or 20 hours (respectively) followed by washout (n = 4)
To investigate functional consequences of CSF1R downregulation, CD14+ monocytes were incubated for 4 hours with ESM‐HDAC391 or HDAC935 in the presence of M‐CSF, after which cells were washed to remove compound and cultured with M‐CSF. Differentiation was assessed at 48 hours by increase in cytoskeleton as determined by F‐actin staining intensity (Figure 7E). Furthermore, the addition of ESM‐HDAC391 for either 4 or 20 hours of culture markedly inhibited monocyte adherence and differentiation in response to M‐CSF signalling in a concentration‐dependent manner (IC50 118 nM at 4 h, 19 nM at 20 h), confirming that even transient exposure to ESM‐HDAC391 is sufficient to prevent monocytes responding to M‐CSF. In contrast, HDAC935 was markedly less potent, (IC50 > 10 μM at 4 h, 625 nM at 20 h; Figure 7F,G).
4. DISCUSSION
Characterisation of ESM‐HDAC391 highlights an opportunity for the specific targeting of HDACi to mononuclear myeloid cells. Such mechanistic engagement, in concert with a favourable safety profile over conventional HDACi, should expand the therapeutic potential for HDACi to target wider inflammatory diseases along with oncology indications (e.g. leukaemias of a monocyte lineage).
Preclinically, ESM‐HDAC391 demonstrates broad activity across Class I, IIb and IV HDACs whilst increasing cellular acetylation levels preferentially in monocytes over other leucocyte populations. In this first clinical trial of ESM‐HDAC391, no safety concerns were identified in healthy male participants up to single oral doses of 60 mg and repeat doses (over 7 days) to 40 mg. Complementing this well‐tolerated profile, ESM‐HDAC391 demonstrated dose‐proportional PK and PD effects. Whereas blood concentration levels of ester and acid were short‐lived systemically, their resulting PD effects were prolonged and demonstrated robust myeloid targeting and mechanistic engagement. In the repeat dose cohorts, increased acetylation and repressed cytokine inhibition in the Day 7 predose samples, 24 hours after prior drug administration, again confirmed sustained pharmacology. Crucially, targeted pharmacology was evident in the absence of common HDAC toxicities (neutropaenia and thrombocytopaenia). These data indicate that ESM‐HDACi should have an increased therapeutic index vs. conventional HDACi. Further clinical studies to evaluate the efficacy of selective targeting of myeloid cells with HDACi will be required to fully understand this potential and to allow these molecules to reach wider applicability.
Unexpectedly, transient drops in circulating monocytes were detected. These reductions were dose‐dependent and reversible upon cessation of dosing. Since not accompanied by clinical signs or symptoms in trial volunteers, monocytopaenia was not recorded as an adverse event. Monocytopaenia was not observed clinically with nontargeted HDACi or previous ESM‐HDACi tefinostat. 20 While the full implications require further study and vigilant monitoring, dosing regimens of ESM‐HDAC391 causing moderate/transient monocytopaenia could bring additional therapeutic benefits for the treatment of chronic inflammatory disorders via depletion of pathogenic myeloid cells. 26 , 27 , 28 , 29 , 30 , 31
Consistent with conventional HDACi, 32 , 33 a dose‐proportional inhibition of stimulated cytokine release was observed in blood samples stimulated ex vivo with LPS. As dose levels demonstrating profound PD coincided with monocytopaenia, this beneficial inhibition probably results from loss of monocytes in concert with direct action of ESM‐HDAC391 on the production of pro‐inflammatory cytokines.
Transgenic mice, in which human CES1 has been inserted under the CD68 promoter to drive expression in monocytes, have previously been used to demonstrate selective targeting and efficacy of an ESM‐HDAC inhibitor in an rheumatoid arthritis model. 17 Here, we further refined this model by crossing with mice deficient in plasma carboxylesterase‐1, 34 which is present in rodents but absent in humans. 35 We confirmed monocyte‐specific targeting, as evidenced by increased acetylation, and successfully recapitulated robust, rapid monocytopaenia, with subsequent recovery of circulating monocytes.
Whilst it was not possible to retrospectively characterise monocyte populations in the circulation of healthy volunteers in study NCT01934101, subset analysis of monocytes in the CES1/Es1e lo model indicated that all subsets were significantly depleted. Furthermore, Ly6Clo monocytes, equivalent to the more mature CD14−CD16+ non‐classical monocytes in humans, were the most significantly reduced population. The order of re‐emergence of monocyte subsets into the circulation after chronic treatment followed the same maturation pathway previously defined in both mice 36 and humans, 37 suggesting reconstitution of circulating monocytes from the bone marrow.
The down‐regulated expression of CD115 (CSF1R) in the CES1/Es1e lo model and specific and functional downregulation of CSF1R in CD14+ human monocytes led to the hypothesis that the mechanism of monocyte depletion may be mediated via disruption of this pathway. The CSF‐CSF1R axis plays a central role in monocyte development, survival and homeostasis of macrophage populations 38 , 39 , 40 and targeting this pathway is of clinical interest. 41 , 42 Pan‐HDACi have previously been shown to rapidly inhibit the transcription factor PU.1, 43 which regulates the expression of CSF1R. 44 , 45 Decreases in the CD14loCD16hi nonclassical monocyte sub‐populations have been noted in several clinical studies targeting the CSF‐CSF1R axis. 46 , 47 , 48 The marginally greater depletion of mature monocyte subsets in the mouse model is consistent with the observation of HDACi‐driven blockade of myeloid differentiation and maturation. 19 , 49 Given the precedence for monocyte depletion in clinical and preclinical settings following the disruption of CSF1R signalling, we propose that monocytopaenia driven by ESM‐HDAC391 is a result of the downregulation of CSF1R observed in monocytes and the previously undocumented effect in classical monocytes is probably due to the enhanced potency and prolonged retention in monocytes with ESM technology.
Together, these findings demonstrate the extensive characterisation of ESM‐HDAC391 as a novel targeted therapeutic approach potentially applicable to a wide range of diseases with unmet medical need. ESM‐HDAC391 demonstrates the effectiveness of ESM targeting (both preclinically and clinically) through selective effects on mononuclear myeloid cells. Clinical target engagement is accompanied by a parallel depletion of circulating monocytes, which has been investigated mechanistically. This phenotype is reversible and linked to a change in the levels of CSF1R, and may contribute to the therapeutic potential of myeloid‐targeted HDACi. Overall, these data provide novel evidence for the pharmacological and biological effects of ESM‐HDACi, highlighting important opportunities for future therapies.
COMPETING INTERESTS
All authors were employees and shareholders of GSK during the generation of these data or drafting of this manuscript. J.M., N.P., D.H., F.A., Y.H. and M.T. are currently employees/shareholders of companies, as indicated by their stated affiliations.
CONTRIBUTORS
Phase I Study conceptualisation, delivery and analysis: J.M., N.P., D.H., R.S., F.A., M.T., H.D.L.; preclinical investigations: R.C.F., A.G., N.C.M., A.K.B., C.S., S.B., R.P.D.; formal analysis for publication: R.C.F., H.D.L.; writing—original draft/review and editing, R.C.F., H.D.L.; visualisation, R.C.F.; project administration, R.C.F., D.F.T., R.K.P., H.D.L. All authors read and approved the final manuscript.
Supporting information
FIGURE S1 Blood leucocyte counts in subjects dosed in Part A with ESM‐HDAC391 vs. placebo.
FIGURE S2 Consequences of 14 days of dosing of ESM‐HDAC391 on circulating monocytes and bone marrow progenitor cells in CES1/Es1elo mice
FIGURE S3 Effects on viability, extracellular markers and adhesion of human monocytes.
TABLE S1 Antibodies used for preclinical flow cytometry
TABLE S2 Selectivity of ESM‐HDAC391 across the HDAC family
TABLE S3 Comparison of fed and fasted phases (Part A, cohorts 3 and 4)
ACKNOWLEDGEMENTS
The authors would like to acknowledge David Bell (BioKinetik Europe) who provided critical input into the clinical study design and was the Principal Investigator. The study originally was sponsored and funded by Chroma Therapeutics and was transferred to GSK during the course of the trial. GSK assumed sponsorship and funding responsibilities.
Furze RC, Molnar J, Parr NJ, et al. Phase 1 and preclinical profiling of ESM‐HDAC391, a myeloid‐targeted histone deacetylase inhibitor, shows enhanced pharmacology and monocytopaenia. Br J Clin Pharmacol. 2022;88(12):5238‐5256. doi: 10.1111/bcp.15428
The authors confirm that the PI for this paper is Prof. David Bell (BioKinetic) and that he had direct clinical responsibility for participants. Prof. Bell declined the invitation to be an author on this manuscript.
DATA AVAILABILITY STATEMENT
Additional details can be found at https://www.gsk-clinicalstudyregister.com/files2/gsk-201302-clinical-study-result-summary.pdf.
REFERENCES
- 1. New M, Olzscha H, La Thangue NB. HDAC inhibitor‐based therapies: can we interpret the code? Mol Oncol. 2012;6(6):637‐656. doi: 10.1016/j.molonc.2012.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673‐691. doi: 10.1038/nrd4360 [DOI] [PubMed] [Google Scholar]
- 3. Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science. 2009;325(5942):834‐840. doi: 10.1126/science.1175371 [DOI] [PubMed] [Google Scholar]
- 4. Li W, Sun Z. Mechanism of Action for HDAC Inhibitors‐Insights from Omics Approaches. Int J Mol Sci. 2019;20(7):1616. doi: 10.3390/ijms20071616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Qiu X, Xiao X, Li N, Li Y. Histone deacetylases inhibitors (HDACis) as novel therapeutic application in various clinical diseases. Prog Neuropsychopharmacol Biol Psychiatry. 2017;72:60‐72. doi: 10.1016/j.pnpbp.2016.09.002 [DOI] [PubMed] [Google Scholar]
- 6. Dinarello CA, Fossati G, Mascagni P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol Med. 2011;17(5–6):333‐352. doi: 10.2119/molmed.2011.00116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011;32(7):335‐343. doi: 10.1016/j.it.2011.04.001 [DOI] [PubMed] [Google Scholar]
- 8. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci. 2017;18(7):1414. doi: 10.3390/ijms18071414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McClure JJ, Li X, Chou CJ. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv Cancer Res. 2018;138:183‐211. doi: 10.1016/bs.acr.2018.02.006 [DOI] [PubMed] [Google Scholar]
- 10. Banik D, Moufarrij S, Villagra A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int J Mol Sci. 2019;20(9):2241. doi: 10.3390/ijms20092241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shah RR. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019;42(2):235‐245. doi: 10.1007/s40264-018-0773-9 [DOI] [PubMed] [Google Scholar]
- 12. Xie M, Tang Y, Hill JA. HDAC inhibition as a therapeutic strategy in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol. 2019;129:188‐192. doi: 10.1016/j.yjmcc.2019.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Makkar R, Behl T, Arora S. Role of HDAC inhibitors in diabetes mellitus. Curr Res Transl Med. 2019;68(2):45‐50. doi: 10.1016/j.retram.2019.08.001 [DOI] [PubMed] [Google Scholar]
- 14. Shieh PB. Emerging Strategies in the Treatment of Duchenne Muscular Dystrophy. Neurotherapeutics. 2018;15(4):840‐848. doi: 10.1007/s13311-018-00687-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8(1):57‐64. doi: 10.1016/j.coph.2007.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Das Gupta K, Shakespear MR, Iyer A, Fairlie DP, Sweet MJ. Histone deacetylases in monocyte/macrophage development, activation and metabolism: refining HDAC targets for inflammatory and infectious diseases. Clin Transl Immunol. 2016;5(1):e62. doi: 10.1038/cti.2015.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Needham LA, Davidson AH, Bawden LJ, et al. Drug targeting to monocytes and macrophages using esterase‐sensitive chemical motifs. J Pharmacol Exp Ther. 2011;339(1):132‐142. doi: 10.1124/jpet.111.183640 [DOI] [PubMed] [Google Scholar]
- 18. Elfiky AMI, Ghiboub M, Li Yim AYF, et al. Carboxylesterase‐1 assisted targeting of HDAC inhibitors to mononuclear myeloid cells in Inflammatory Bowel Disease. J Crohns Colitis. 2021;16(4):668‐681. doi: 10.1093/ecco-jcc/jjab176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luque‐Martin R, Van den Bossche J, Furze RC, et al. Targeting Histone Deacetylases in Myeloid Cells Inhibits Their Maturation and Inflammatory Function With Limited Effects on Atherosclerosis. Front Pharmacol. 2019;10:1242. doi: 10.3389/fphar.2019.01242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ossenkoppele GJ, Lowenberg B, Zachee P, et al. A phase I first‐in‐human study with tefinostat ‐ a monocyte/macrophage targeted histone deacetylase inhibitor ‐ in patients with advanced haematological malignancies. Br J Haematol. 2013;162(2):191‐201. doi: 10.1111/bjh.12359 [DOI] [PubMed] [Google Scholar]
- 21. Brown JA, Bal J, Simeoni M, et al. A Randomized Study of the Safety and Pharmacokinetics of GSK3358699, a Mononuclear Myeloid‐Targeted Bromodomain and Extra‐Terminal Domain Inhibitor. Br J Clin Pharmacol. 2021;88(5):2140‐2155. doi: 10.1111/bcp.15137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gough PJ, Gordon S, Greaves DR. The use of human CD68 transcriptional regulatory sequences to direct high‐level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology. 2001;103(3):351‐361. doi: 10.1046/j.1365-2567.2001.01256.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iqbal AJ, McNeill E, Kapellos TS, et al. Human CD68 promoter GFP transgenic mice allow analysis of monocyte to macrophage differentiation in vivo. Blood. 2014;124(15):e33‐e44. doi: 10.1182/blood-2014-04-568691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexander SPH, Fabbro D, Kelly E, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176(Suppl 1):S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762‐774. doi: 10.1038/nri3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scott MKD, Quinn K, Li Q, et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: a retrospective, multicentre cohort study. Lancet Respir Med. 2019;7(6):497‐508. doi: 10.1016/S2213-2600(18)30508-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chara L, Sanchez‐Atrio A, Perez A, et al. The number of circulating monocytes as biomarkers of the clinical response to methotrexate in untreated patients with rheumatoid arthritis. J Transl Med. 2015;13(1):2. doi: 10.1186/s12967-014-0375-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krenkel O, Puengel T, Govaere O, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology. 2018;67(4):1270‐1283. doi: 10.1002/hep.29544 [DOI] [PubMed] [Google Scholar]
- 29. Rana AK, Li Y, Dang Q, Yang F. Monocytes in rheumatoid arthritis: Circulating precursors of macrophages and osteoclasts and, their heterogeneity and plasticity role in RA pathogenesis. Int Immunopharmacol. 2018;65:348‐359. doi: 10.1016/j.intimp.2018.10.016 [DOI] [PubMed] [Google Scholar]
- 30. Yamanaka K, Umezawa Y, Yamagiwa A, et al. Biologic therapy improves psoriasis by decreasing the activity of monocytes and neutrophils. J Dermatol. 2014;41(8):679‐685. doi: 10.1111/1346-8138.12560 [DOI] [PubMed] [Google Scholar]
- 31. Gibbons MA, MacKinnon AC, Ramachandran P, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184(5):569‐581. doi: 10.1164/rccm.201010-1719OC [DOI] [PubMed] [Google Scholar]
- 32. Gatla HR, Muniraj N, Thevkar P, Yavvari S, Sukhavasi S, Makena MR. Regulation of Chemokines and Cytokines by Histone Deacetylases and an Update on Histone Decetylase Inhibitors in Human Diseases. Int J Mol Sci. 2019;20(5):1110. doi: 10.3390/ijms20051110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hoeksema MA, de Winther MPJ. Epigenetic Regulation of Monocyte and Macrophage Function. Antioxid Redox Signal. 2016;25(14):758‐774. doi: 10.1089/ars.2016.6695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morton CL, Wierdl M, Oliver L, et al. Activation of CPT‐11 in mice: identification and analysis of a highly effective plasma esterase. Cancer Res. 2000;60(15):4206‐4210. [PubMed] [Google Scholar]
- 35. Bahar FG, Ohura K, Ogihara T, Imai T. Species difference of esterase expression and hydrolase activity in plasma. J Pharm Sci. 2012;101(10):3979‐3988. doi: 10.1002/jps.23258 [DOI] [PubMed] [Google Scholar]
- 36. Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79‐91. doi: 10.1016/j.immuni.2012.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Patel AA, Zhang Y, Fullerton JN, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. 2017;214(7):1913‐1923. doi: 10.1084/jem.20170355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dai X‐M, Ryan GR, Hapel AJ, et al. Targeted disruption of the mouse colony‐stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood. 2002;99(1):111‐120. doi: 10.1182/blood.V99.1.111 [DOI] [PubMed] [Google Scholar]
- 39. Chitu V, Stanley ER. Colony‐stimulating factor‐1 in immunity and inflammation. Curr Opin Immunol. 2006;18(1):39‐48. doi: 10.1016/j.coi.2005.11.006 [DOI] [PubMed] [Google Scholar]
- 40. Sweet MJ, Hume DA. CSF‐1 as a regulator of macrophage activation and immune responses. Arch Immunol Ther Exp (Warsz). 2003;51(3):169‐177. [PubMed] [Google Scholar]
- 41. Kumari A, Silakari O, Singh RK. Recent advances in colony stimulating factor‐1 receptor/c‐FMS as an emerging target for various therapeutic implications. Biomed Pharmacother. 2018;103:662‐679. doi: 10.1016/j.biopha.2018.04.046 [DOI] [PubMed] [Google Scholar]
- 42. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369‐382. doi: 10.1038/s41577-019-0127-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Laribee RN, Klemsz MJ. Loss of PU.1 expression following inhibition of histone deacetylases. J Immunol. 2001;167(9):5160‐5166. doi: 10.4049/jimmunol.167.9.5160 [DOI] [PubMed] [Google Scholar]
- 44. Zhang DE, Hetherington CJ, Chen HM, Tenen DG. The macrophage transcription factor PU.1 directs tissue‐specific expression of the macrophage colony‐stimulating factor receptor. Mol Cell Biol. 1994;14(1):373‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bencheikh L, Diop MK, Riviere J, et al. Dynamic gene regulation by nuclear colony‐stimulating factor 1 receptor in human monocytes and macrophages. Nat Commun. 2019;10(1):1935. doi: 10.1038/s41467-019-09970-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Genovese MC, Hsia E, Belkowski SM, et al. Results from a Phase IIA Parallel Group Study of JNJ‐40346527, an Oral CSF‐1R Inhibitor, in Patients with Active Rheumatoid Arthritis despite Disease‐modifying Antirheumatic Drug Therapy. J Rheumatol. 2015;42(10):1752‐1760. doi: 10.3899/jrheum.141580 [DOI] [PubMed] [Google Scholar]
- 47. Masek‐Hammerman K, Peeva E, Ahmad A, et al. Monoclonal antibody against macrophage colony‐stimulating factor suppresses circulating monocytes and tissue macrophage function but does not alter cell infiltration/activation in cutaneous lesions or clinical outcomes in patients with cutaneous lupus erythematosus. Clin Exp Immunol. 2016;183(2):258‐270. doi: 10.1111/cei.12705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wesolowski R, Sharma N, Reebel L, et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF‐1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther Adv Med Oncol. 2019;11:1758835919854238. doi: 10.1177/1758835919854238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cabanel M, Brand C, Oliveira‐Nunes MC, et al. Epigenetic Control of Macrophage Shape Transition towards an Atypical Elongated Phenotype by Histone Deacetylase Activity. PLoS ONE. 2015;10(7):e0132984. doi: 10.1371/journal.pone.0132984 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Blood leucocyte counts in subjects dosed in Part A with ESM‐HDAC391 vs. placebo.
FIGURE S2 Consequences of 14 days of dosing of ESM‐HDAC391 on circulating monocytes and bone marrow progenitor cells in CES1/Es1elo mice
FIGURE S3 Effects on viability, extracellular markers and adhesion of human monocytes.
TABLE S1 Antibodies used for preclinical flow cytometry
TABLE S2 Selectivity of ESM‐HDAC391 across the HDAC family
TABLE S3 Comparison of fed and fasted phases (Part A, cohorts 3 and 4)
Data Availability Statement
Additional details can be found at https://www.gsk-clinicalstudyregister.com/files2/gsk-201302-clinical-study-result-summary.pdf.