

Abstract
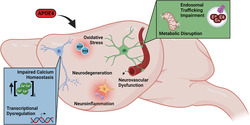
The APOE gene encoding the Apolipoprotein E protein is the single most significant genetic risk factor for late‐onset Alzheimer's disease. The APOE4 genotype confers a significantly increased risk relative to the other two common genotypes APOE3 and APOE2. Intriguingly, APOE4 has been associated with neuropathological and cognitive deficits in the absence of Alzheimer's disease‐related amyloid or tau pathology. Here, we review the extensive literature surrounding the impact of APOE genotype on central nervous system dysfunction, focussing on preclinical model systems and comparison of APOE3 and APOE4, given the low global prevalence of APOE2. A multi‐hit hypothesis is proposed to explain how APOE4 shifts cerebral physiology towards pathophysiology through interconnected hits. These hits include the following: neurodegeneration, neurovascular dysfunction, neuroinflammation, oxidative stress, endosomal trafficking impairments, lipid and cellular metabolism disruption, impaired calcium homeostasis and altered transcriptional regulation. The hits, individually and in combination, leave the APOE4 brain in a vulnerable state where further cumulative insults will exacerbate degeneration and lead to cognitive deficits in the absence of Alzheimer's disease pathology and also a state in which such pathology may more easily take hold. We conclude that current evidence supports an APOE4 multi‐hit hypothesis, which contributes to an APOE4 pathophysiological state. We highlight key areas where further study is required to elucidate the complex interplay between these individual mechanisms and downstream consequences, helping to frame the current landscape of existing APOE‐centric literature.
Keywords: ageing, APOE, multihit hypothesis, pathophysiology, review
Evidence is presented for a multihit hypothesis of APOE4 pathophysiology whereby multiple hits are exacerbated by cellular stressors and exhibit crosstalk between pathways with APOE4 placing the system in a state of vulnerability. Assertions of the hypothesis, potential interactions and implications are discussed aiming to frame existing APOE‐centric literature.
List of Abbreviations
- ABCA1
ATP binding cassette transporter 1
- AD
Alzheimer's disease
- Akt
AKT serine/threonine kinase 1
- AMPAR
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor
- AP‐1
Activator protein 1
- ApoE
Apolipoprotein E
- APOE‐KO
Apolipoprotein E knock‐out
- APOER2
APOE receptor 2
- APOE‐TR
APOE targeted replacement
- ATP
Adenosine triphosphate
- Aβ
Beta‐amyloid
- BBB
Blood–brain barrier
- CBF
Cerebral blood flow
- CBV
Cerebral blood volume
- CNS
Central nervous system
- COX2
Prostaglandin‐endoperoxide synthase 2
- COXIV
Cytochrome oxidase 4
- CSF
Cerebrospinal fluid
- CypA
Cyclophilin A
- DG
Dentate gyrus
- eEF2
Eukaryotic elongation factor 2
- ER
Endoplasmic reticulum
- ERK1/2
Extracellular signal regulated kinase 1/2
- ETC
Electron transport chain
- GABA
Gamma aminobutyric acid
- GABA‐INs
GABAergic interneurons
- GLUT1
Glucose transporter 1
- GLUT4
Glucose transporter 4
- HDAC4
Histone deacetylase 4
- HDL
High‐density lipoprotein
- hIPSC
human‐induced pluripotent stem cells
- HNE
4‐hydroynonenal
- HSP
Heat shock protein
- HSPG
Heparan‐sulphate proteoglycans
- IR
Insulin receptor
- JNK
c‐Jun N‐terminal kinase
- LDL
Low‐density lipoprotein
- LDLR
Low‐density lipoprotein receptor
- LOAD
Late onset Alzheimer's disease
- LRP
Low‐density lipoprotein receptor‐related protein
- LRP1
low‐density lipoprotein receptor‐related protein 1
- miR
Micro‐RNA
- MMP9
Matrix metalloprotease 9
- NFAT
Nuclear factor of activated T cells
- NF‐ĸB
Nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- NHE
Na/H + exchangers
- NMDAR
N‐methyl‐D‐aspartate receptor
- NO
Nitric oxide
- p38 MAPK
p38 mitogen activated protein kinase
- PPARɣ
Peroxisome proliferator‐activated receptor gamma
- PSD‐95
Post‐synaptic density 95
- RNA
ribonucleic acid
- ROS
Reactive oxygen species
- SERPINA3
Serpin family member 3
- SIRT1
Sirtuin 1
- SNP
Single nucleotide polymorphisms
- SOD2
Superoxide dismutase 2
- Src
SRC non‐receptor tyrosine kinase
- TBI
Traumatic brain injury
- TCA
Tricarboxylic acid cycle
- TLR
Toll‐like receptor
- TREM2
Triggering receptor on myeloid 2 cells
- UPR
Unfolded protein response
- vATPase
Vacuolar‐type ATPase
- VLDL
Very low‐density lipoprotein
- ZO‐1
Zonula occludens 1
1. INTRODUCTION
Apolipoprotein E (ApoE) is a 34 kDa glycoprotein (299 amino acids) encoded by the ~3.6 kb gene on chromosome 19, APOE. The primary physiological role of ApoE is in lipoprotein homeostasis, though is implicated in multiple diverse roles in both the central nervous system (CNS) and periphery (Mahley, 1988; Martínez‐Martínez et al., 2020). In the CNS, ApoE is the primary apolipoprotein with expression predominantly driven by astrocytes (Boyles et al., 1985; Grehan et al., 2001; Pitas et al., 1987) and also within microglia (Nakai et al., 1996; Xu et al., 1999), the neurovasculature (Majacek et al., 1988) and neurons under conditions of stress (Xu et al., 1996). Typically, ApoE is secreted from astrocytes and lipidated via members of the adenosine triphosphate (ATP) cassette binding transporter (ABC) family, such as ABCA1. ApoE mediates the transport of cholesterol and various lipids in lipoprotein complexes to neurons for internalisation by interacting with ApoE receptors (ApoER2/LRP8), members of the low‐density lipoprotein receptor (LDLR) and LDLR‐related protein (LRP) families as well as heparan sulphate proteoglycans (HSPGs) (Bu, 2009; DeMattos et al., 2001; Hirota et al., 2015; Mahley et al., 2009; Strasser et al., 2004).
APOE exists as three common allelic isoforms: APOE2, APOE3 and APOE4 (encoding proteins ApoE2, ApoE3 and ApoE4), which arise from single nucleotide polymorphisms (SNPs) conferring single amino acid substitutions. Relative to ApoE3 (cysteine 112, arginine 158), these SNPs result in C112R substitutions in ApoE4 and R158C in ApoE2 (Mahley & Rall, 2000; Nickerson et al., 2000; Zannis & Breslow, 1981; Zannis et al., 1982). Debate concerning the structural consequence of APOE isoform is still ongoing, with data suggesting either an ApoE4‐specific conformational C‐N terminal domain interaction or aggregation and subtle structural differences (Dong et al., 1994; Dong & Weisgraber, 1996; Raffaï et al., 2001; Raulin et al., 2019; Weisgraber, 1994; Xu et al., 2004). Ultimately, APOE isoform profoundly impacts lipoprotein transport and metabolism. ApoE4 exhibits a preference for larger, triglyceride rich very low‐density lipoprotein (VLDL) particles in the periphery (Mahley, 1988; Mahley & Huang, 1999; Mahley & Rall, 2000), instead of the smaller phospholipid rich high‐density lipoprotein (HDL) particles preferred by ApoE2 and ApoE3 (Dong et al., 1994; Dong & Weisgraber, 1996). In the cerebrospinal fluid (CSF) however, ApoE4 is associated with smaller and less lipid‐rich lipoprotein particles (Heinsinger et al., 2016; Lanfranco et al., 2020; Mahley, 2016). Furthermore, circulating levels of ApoE4 in the CNS are the lowest of the three main isoforms (Riddell et al., 2008; Ulrich et al., 2013), likely as a consequence of increased endosomal accumulation (Chen et al., 2010; Morrow et al., 2002; Xian et al., 2018) and proteolytic degradation (Brecht et al., 2004; Harris et al., 2003; Huang et al., 2001).
At a population level, APOE3 has an allelic frequency of ~77%, APOE4 of ~15% and APOE2 of ~7% (Huang & Mahley, 2014). APOE4 is the single strongest genetic predictor for late onset (LO) non‐familial Alzheimer's disease (AD, LOAD), the most common form of AD. APOE4 increases LOAD risk by ~2 to ~8 times for heterozygotic and homozygotic carriers, relative to APOE3‐carriers, respectively (Corder et al., 1993, 1994; Gureje et al., 2006; Mayeux et al., 1998; Raber et al., 2004; Saunders et al., 1993). The primary pathological hallmarks of AD include extracellular senile plaques comprising complex aggregations of beta‐amyloid (Aβ) species and intraneuronal neurofibrillary tangles formed of the microtubule associated protein tau.
Crucially, a growing body of evidence points towards an emerging APOE4 phenotype, in which APOE4 can both interact with these AD‐dependent Aβ and tau‐related processes while also independently contributing to cellular pathophysiology (e.g., Dose et al., 2016; Flowers & Rebeck, 2020; Huang, 2010; Huang & Mahley, 2014). Much of the supporting work has been performed in cell and animal model systems such as the targeted replacement (TR) mice, in which the mouse Apoe gene is replaced with human APOE (Knouff et al., 1999; Sullivan et al., 1997, 1998, 2004). Importantly, classic AD pathology is absent in many of these animals, allowing the study of APOE function in isolation. Evidence from these systems is converging on a series of APOE4‐dependent ‘hits’ that we propose are interlinked to induce an APOE4‐mediated pathophysiological state, independent of AD pathology.
This review focuses on the preclinical evidence supporting individual APOE4 hits and their underpinning mechanisms, before considering how these functional disruptions converge to place the CNS in a state vulnerable to cellular stressors that further increase LOAD risk. We focus on comparing APOE4 function to the population norm, APOE3, primarily in preclinical systems due to the challenge of studying these pathways in humans. However, where appropriate, references are made to supporting or contradicting human literature. We then suggest how future research should aim to investigate the mechanistic interplay between these hits in combination rather than in isolation to understand systems‐level interactions. Specifically, we describe eight key APOE4 hits: neuronal maturation and neurodegeneration, neurovascular integrity and function, inflammation and glial function, oxidative stress, endosomal trafficking, lipid and cellular metabolism, calcium homeostasis and transcriptional dysregulation. Throughout the described hits, the influence of contextual factors including ageing, biological sex and injury are considered. Ultimately, we suggest that a multi‐hit hypothesis promotes understanding of a complex APOE4‐pathophysiology and emphasises the importance of cross modal approaches to study life‐long CNS vulnerability which may initially arise independent of, but subsequently contribute to, AD‐pathology.
2. HIT 1: NEURODEGENERATION AND NEURODEVELOPMENT
Neuronal cell death and loss of neuronal structural integrity are typical pathologies of neurodegenerative disorders. However, the events preceding gross cell loss including subtle changes to neuronal morphology and synaptic density as well as susceptibility of specific neuronal subtypes have been implicated in the aetiology of multiple neurodegenerative conditions (Najm et al., 2019; Stranahan & Mattson, 2010). ApoE is recognised to have a key role in the regulation of neuronal morphology and response to CNS injury (Boyles et al., 1989; Mahley, 1988; Nathan et al., 1995). However, an extensive body of evidence implicates APOE4 in the dysfunction of these pathways, promoting age‐dependent gross neurodegeneration within the hippocampus and cortex in E4‐TR mice (Speidell et al., 2019; Yin et al., 2011, 2014). Here we summarise the broad evidence that APOE4 plays a major role in driving neurodegeneration, altering proliferation, cell fate and damage repair processes with particular focus on the susceptibility of inhibitory interneurons as the first major hit in a multihit hypothesis.
2.1. APOE4‐mediated GABA‐interneuron selective cell loss
Multiple lines of evidence suggest that APOE4 drives a sex‐dependent neurodegenerative phenotype in dentate gyrus (DG) hilar gamma aminobuyric acid (GABA)‐ergic interneurons (GABA‐INs) without necessitating pre‐existing AD‐related pathology. Firstly, female E4‐TR mice exhibit reductions in GABAergic terminals in the DG, followed by significant loss of DG hilar GABA‐INs across the lifespan, suggesting an excitatory: inhibitory imbalance which correlated with spatial learning and memory defects (Andrews‐Zwilling et al., 2010; Jones et al., 2019; Knoferle et al., 2014; Leung et al., 2012; Li et al., 2009; Tong et al., 2016). Interestingly, these deficits were not detected in male E4‐TR mice, although GABA‐IN loss was inducible following cumulative environmental stress (Lin et al., 2016; Zhang et al., 2021) but not following toxic lead exposure (Engstrom et al., 2017). This likely suggests a general increased susceptibility of GABA‐INs to APOE4‐mediated neurodegeneration. Importantly, the GABAergic and spatial memory phenotypes were rescued via restoring GABAergic signalling through multiple methods (Andrews‐Zwilling et al., 2010; Knoferle et al., 2014; Tong et al., 2016) including via knock out of APOE4 specifically within all neurons or interneurons alone (Knoferle et al., 2014). This evidence suggests a gain of toxic function role for APOE4 in driving GABAergic‐specific neurodegeneration, mediated by an unclear sex‐dependent mechanism.
Strong evidence supports a causative role for ApoE4 proteolytic fragmentation within neurons and subsequent neurotoxicity, particularly within GABAergic interneurons (Figure 1, Path A). Elevated neuronal ApoE4, but not ApoE3, fragmentation is exacerbated by injury and ageing (Brecht et al., 2004), is associated with tau inclusions (Brecht et al., 2004; Harris, Brecht, et al., 2004) and tau‐dependent GABAergic cell death in vitro and in vivo (Andrews‐Zwilling et al., 2010; Li et al., 2009; Wang et al., 2018). ApoE4 exhibits enhanced mitochondrial and ER sequestration, while ApoE4 fragments cause neuronal mitochondrial dysfunction and astrocytic ER stress in vitro (Hit 6; Brodbeck et al., 2011; Chang et al., 2005; Chen et al., 2011; Huang & Mahley, 2014). The ApoE4 fragmentation process is likely regulated by a putative C‐N terminal domain interaction between Glu255 and Arg61 (Dong & Weisgraber, 1996; Huang, 2010; Wang et al., 2018; Xu et al., 2004). Importantly, preventing the ApoE4 domain interaction via pharmacological inhibition (Brodbeck et al., 2011; Wang et al., 2018) or gene editing (Chen et al., 2011; Wang et al., 2018) rescued ApoE4 trafficking, reduced neurotoxic ApoE4 fragmentation, mitochondrial dysfunction and tau‐associated GABA‐IN degeneration. Similarly, knockout of endogenous tau rescued GABA‐IN loss in vivo in an ApoE4 fragment mouse model (Andrews‐Zwilling et al., 2010). Thus, evidence strongly suggests that ApoE4 fragments generated in neurons are key mediators of hippocampal GABA‐IN degeneration via a tau‐dependent process and of disruption of mitochondrial function which is exacerbated by exogenous stressors.
FIGURE 1.
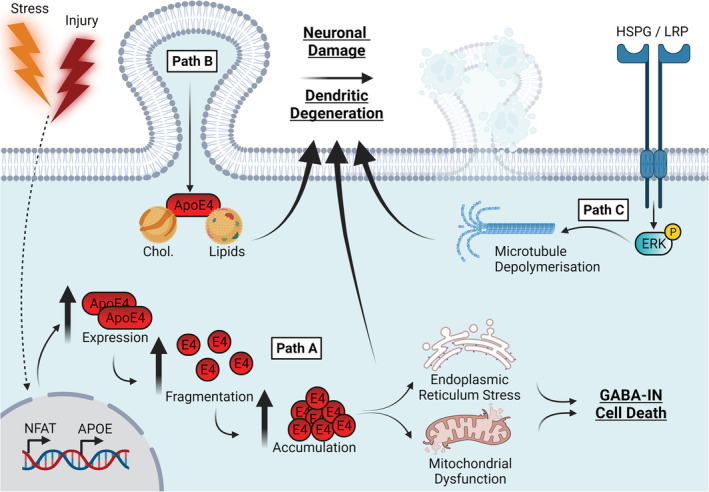
APOE4 drives a neurodegenerative phenotype via fragmentation, microtubule depolymerisation and somal ApoE4 sequestration. Path A: Under conditions of cellular stress and injury, neuronal ApoE expression is upregulated, and there is an increased propensity of ApoE4 to undergo proteolytic fragmentation which results in enhanced intracellular ApoE4 fragment accumulation. ApoE4 fragments disrupt mitochondrial function and induce ER stress, to which, GABA‐Ins are particularly sensitive and exhibit exacerbated cell death. Path B: Somal sequestration of ApoE4 and its associated lipoproteins alongside away from the dendritic cell surface poorer lipid availability inhibits effective dendritic repair, remodelling and maintenance. Path C: In addition, through action at surface LRP/HSPGs ApoE4 enhances microtubule depolymerisation in a mechanism thought to be driven by enhanced ERK signalling. The culmination of these pathways is a heightened state of neuronal vulnerability and dendritic degeneration, and thus a neurodegenerative phenotype. Abbreviations: ApoE4, apolipoprotein E4; ER, endoplasmic reticulum; ERK, extracellular signal‐related kinase; GABA‐INs, gamma aminobutyric acid (GABA) interneurons; LRP‐HSPG, low‐density lipoprotein receptor‐related protein;. Created with BioRender.com
2.2. APOE4 disrupts maintenance of neuronal morphology
In addition to neuronal cell loss, there is a clear link between APOE genotype and both the establishment and maintenance of neuronal morphology. Decreases in dendritic spine and neurite density have been observed across the lifespan in the cortex layers II/III, hippocampus, entorhinal cortex and amygdala of E4‐TR and other APOE4 mouse models (Bell et al., 2012; Dumanis et al., 2009; Jain et al., 2013 Ji et al., 2003; Neustadtl et al., 2017; Rodriguez et al., 2013; Sun et al., 2017; Veinbergs et al., 1999; Wang et al., 2005; Xu et al., 1996), although there are some reports of no changes in these measures (Dumanis et al., 2009; Jain et al., 2013; Jones et al., 2021; Klein et al., 2014). These differences are exacerbated in multiple APOE4 mouse models following excitotoxic and deafferentation injury, with increased neuronal ApoE expression (Xu et al., 1996), impaired post‐injury sprouting responses and reduced neuronal integrity relative to APOE3 controls (Bott et al., 2016; Buttini et al., 1999; White et al., 2001). Other sources of cellular stress such as environmental stress, chronic high‐fat diet, environmental toxicants and neuroinflammation appear to have their effects exacerbated in E4‐TR mice, including reducing dendritic complexity (Engstrom et al., 2017), glutamatergic synaptic integrity (Lin et al., 2016) and neurotrophic BDNF signalling (Maioli et al., 2012). As observed for GABA‐INs, evidence suggests that morphological impairments are most aggressive when ApoE4 expression occurs in neurons themselves (e.g., Jain et al., 2013), particularly following injury (Buttini et al., 1999, 2010).
Evidence points to several mechanisms by which ApoE4 may impair the development and maintenance of neuronal integrity. First, a reduction in the total or membrane associated levels of ApoE4 (DiBattista et al., 2016; Nathan et al., 1995) and reduced lipidation state of ApoE4 (Heinsinger et al., 2016; Hu et al., 2015; Lanfranco et al., 2020) may impair its ability to effectively mediate trafficking of lipids required to support neurite remodelling as shown in vitro (DeMattos et al., 1998; Holtzman et al., 1995; Mahley, 2016; Nathan et al., 1994, 1995; Figure 1, Path B). Second, evidence suggests that ApoE4 exhibits reduced microtubule binding and impaired microtubule polymerisation (Nathan et al., 1995) and contributed to via aberrant HSPG/LRP signalling, ERK1/2 (Extracellular signal regulated kinase) activation and downstream tau phosphorylation (Brecht et al., 2004; Harris, Brecht, et al., 2004, Figure 1, Path C). Reduced binding may also be contributed to via increased somal ApoE4 sequestration (Harris, Tesseur, et al., 2004; Nathan et al., 1995; Figure 1, Path B). Third, neuronal ApoE4 expression (Dumanis et al., 2009; Teter et al., 2002) and neurotoxic proteolytic fragmentation (Figure 1, Path A; Brodbeck et al., 2008) impair neuronal sprouting and are partially rescued by targeting the ApoE4 domain interaction pharmacologically (Brodbeck et al., 2011). ApoE4 fragmentation is suggested to be regulated by serine and chymotrypsin‐like proteases (Chu et al., 2016; Harris et al., 2003; Muñoz et al., 2018; Tamboli et al., 2014), and expression of a chymotrypsin inhibitor SERPINA3 (α1 antichymotrypsin) was recently shown to be upregulated in the E4‐TR mouse brain in vivo (J. Zhao et al., 2020). This may represent an attempted compensatory response to ameliorate elevated ApoE4 fragmentation. Finally, ApoE4 may also enhance calcineurin‐mediated spine collapse, which was rescued in vivo in E4‐TR mice via calcineurin inhibition (Neustadtl et al., 2017).
Therefore, evidence to date supports the view that ApoE4 in its basal state mediates disruption of neuronal architecture and is exacerbated when combined with stressors. However, while this evidence provides compelling explanatory mechanisms, the relative physiological contribution of each of these pathways during ageing remains to be fully understood.
2.3. APOE4 alters synaptic integrity and synaptogenesis
Synapse loss is a salient marker of disease progression that precedes gross degeneration and predicts impaired neuronal function in multiple neurodegenerative conditions, including AD (LeBlanc, 2005; Clare et al., 2010). APOE4 may contribute to synapse degradation prior to larger‐scale neurodegeneration, although evidence is contentious (e.g., see Tzioras et al., 2019).
Some evidence suggests an age‐dependent reduction in pre‐ or post‐synaptic proteins in multiple APOE4 mouse models, at a young age in cortex and hippocampus (Bell et al., 2012; Liu et al., 2015; Zhu et al., 2012) and at late age in the hippocampus (Zalocusky et al., 2021) accompanied by reduced synapse number and increased synaptic size (Cambon et al., 2000). In contrast, no basal changes were detected in synaptic markers including synaptophysin at mid‐age across APOE models (Buttini et al., 1999, 2010; Chouinard‐Watkins et al., 2017; Nichol et al., 2009; Veinbergs et al., 1999). However, as with overall morphology, APOE4‐related synaptic protein loss is exacerbated by stressors, injury, and neuronal ApoE4 expression (Buttini et al., 1999, 2000, 2010; Lin et al., 2016; Zhang et al., 2021). Similarly, in vitro studies suggest a possible loss of excitatory and inhibitory synapses and impaired maturational rate via glial‐derived ApoE4 (Konings et al., 2021; Nwabuisi‐Heath et al., 2013), supporting the importance of both glial and neuronal ApoE in synapse formation and maintenance.
Conversely, other studies using human‐induced pluripotent stem cell (hiPSC)‐derived neuron systems suggest that ApoE4 may, in fact, exert an acute synaptogenic influence, without affecting gross dendritic morphology or necessitating AD pathology. This was suggested to occur via an ApoE receptor to CREB‐dependent (cAMP response element binding protein) signalling pathway (Huang et al., 2017, 2019; Lin et al., 2018). However, another study found only synaptic protein reductions in APOE4‐carrier AD patient derived‐, but not healthy APOE4‐carrier derived‐, hIPSC‐derived organoids (N. Zhao et al., 2018). Ultimately, the reason for this conflict between an inhibitory and stimulatory effect of ApoE4 on synaptogenesis is unclear. It possibly reflects a dissociation between acute and chronic ApoE‐mediated signalling or variation between model systems and the techniques resolution (Tzioras et al., 2019). More in‐depth research is needed to tease apart physiological effects of acute and chronic ApoE signalling on synaptic structural dynamics.
2.4. APOE4 alters neurogenesis and maturation
ApoE also influences neurogenesis and newborn neuron maturation into existing neuronal circuitry. ApoE appears to promote neuronal proliferation by negatively regulating astrogenesis cell fate and maintaining progenitor cell pools in the DG subgranular zone via a Noggin‐dependent signalling pathway (Li et al., 2009; Yang et al., 2011). The directionality of an APOE isoform difference remains uncertain, however. An increase (Li et al., 2009; Rijpma et al., 2013), decrease (Koutseff et al., 2014) or no change (Tensaouti et al., 2018) in newborn neuron proliferation in the DG has been reported in young or adult E4‐TR mice compared with E3‐TRs. However, dendritic maturation in newborn neurons (reflecting circuit integration) was generally diminished by APOE4 in females with or without toxicant stress (Engstrom et al., 2017; Li et al., 2009) but was also observed in both sexes following injury (Tensaouti et al., 2020).
Mechanistically, ApoE4 may impair newborn neuron maturation via disruption of GABAergic network function and is rescued by GABAA receptor potentiation in female E4‐TR mice (Li et al., 2009). The role of GABA‐INs in neurogenesis also hints at involvement of ApoE4 fragmentation (Huang & Mahley, 2014; Li et al., 2009) and supports a circuit level mechanism by which ApoE4 can indirectly influence neurogenesis via loss of GABAergic control (Section 2.2; Figure 1, Path A). Together, these data suggest that ApoE4 mediates a dissociation between enhancement of neuronal proliferation and disruption of effective circuit maturation.
Evidence from human APOE4‐carriers somewhat supports these findings. Gross anatomical magnetic resonance imaging data suggest that APOE4 is associated with late‐age reductions in regional brain volume in the hippocampus and medial temporal lobe, although this is not always replicated and the influence of APOE isoform at younger age remains unclear (e.g., Bussy et al., 2019; Cherbuin et al., 2007; Evans et al., 2020; Flowers & Rebeck, 2020). Additionally, reductions in white matter integrity (Heise et al., 2011) and changes in microstructure (Slattery et al., 2017; Westlye et al., 2012) have been reported in APOE4‐carriers but again not consistently (e.g., Evans et al., 2020). Whether APOE4 affects synaptic and GABAergic function in humans is also unclear, due in part to the difficulty in dissociating even low‐level AD pathological load (e.g., Kanekiyo et al., 2014). Some evidence suggests that APOE4 is associated with dysregulation of the synaptic proteome (Hesse et al., 2019), excitatory and inhibitory synapse number (Koffie et al., 2012; Kurucu et al., 2021), possible loss of GABA‐synthesising enzymes (Grouselle et al., 1998) and elevated APOE4 fragmentation (Huang et al., 2001) in the post‐mortem AD patient brain. Further, poorer neurological outcomes for APOE4‐carriers after traumatic brain injury (TBI) are consistent with findings reported in animal models (e.g., Buttini et al., 1999, 2010; White et al., 2001), although again between studies results vary (e.g., Kassam et al., 2016; Lawrence et al., 2015).
Overall, an expansive set of literature suggests that ApoE4 disrupts neuronal development and maintenance in an age‐dependent manner, which appears to push cells towards a degeneration‐prone state and is accelerated by exogenous stressors. This likely involves impaired GABAergic signalling, altered ApoE availability, toxic fragmentation, mitochondrial dysfunction and microtubule dynamics. While neurodegeneration is likely consequent to APOE4 disrupting other cellular processes (Hits 2 to 8), we posit that APOE4 also uniquely contributes to disrupting neuronal integrity via overlapping pathways within a multihit hypothesis.
3. HIT 2: NEUROVASCULAR DYSFUNCTION
Neuronal circuitry is reliant on the provision of adequate blood supply to meet its high metabolic demands. This delivery and facilitation is mediated via the process of neurovascular coupling (Hall et al., 2014; Iadecola, 2017), whereby active neurons signal to dilate local blood vessels. Increasingly, compromised brain perfusion, neurovascular coupling and blood–brain barrier (BBB) integrity are observed as key pathological features of disease progression in multiple neurodegenerative conditions, including LOAD (Bell & Zlokovic, 2009; Erickson & Banks, 2013; Huang et al., 2020). Additionally, vascular damage may be induced by both acute and chronic activation of the neuroinflammatory and oxidative stress response, driving neurodegeneration and poorer outcomes during ageing (Cechetto et al., 2008; Grammas, 2011; Zhang & Jiang, 2014). APOE4 is a risk factor for peripheral cardiovascular complications (Belloy et al., 2019; Lahoz et al., 2001) alongside having a direct impact on the structural and functional integrity of the BBB in clinical populations. Preclinical studies have started to untangle the mechanisms through which APOE4 impacts the structure and function of the cerebral vasculature, which we review here.
3.1. APOE4 disrupts microvascular integrity and induces BBB degeneration
The maintenance of the BBB is vital for protecting the brain from toxic blood‐borne molecules while supplying it with required nutrients and clearing waste products. It comprises a semipermeable border of capillary endothelial cells, pericytes and astrocyte endfeet that allow for regulation and movement of molecules, ions and cells between the blood, CSF and central nervous system (Zlokovic, 2008). Ageing and disease can reduce BBB integrity and nutrient transporters, including GLUT1 (glucose transporter 1) and tight junction proteins such as ZO‐1 (Zonula Occludens 1; Sweeney et al., 2018; Montagne et al., 2021).
APOE4 appears to contribute to both developmental and age‐dependent microvascular and BBB defects. E4‐TR mice exhibit decreased microvascular length and density, as well as BBB degeneration from 2 weeks of age linearly declining across lifespan and precedes gross neurodegeneration (Bell et al., 2012; Koizumi et al., 2018; Montagne et al., 2021). Enhanced BBB permeability and a consequent infiltration of blood‐borne proteins (e.g., haemosiderin; Bell et al., 2012) in APOE4 mouse models are likely linked to loss of tight junction and basement membrane proteins, critical for regulating BBB permeability (Alata et al., 2015; Montagne et al., 2021). Similarly, BBB degeneration has been observed in APOE‐knock‐out (APOE‐KO) mice (Soto et al., 2015) or at late age in E4‐TR mice with cerebral Aβ challenge (Hawkes et al., 2012). Thus, there is evidence of life‐long, progressive BBB degeneration linked to APOE4, which may be mechanistically driven by a loss of function.
Inflammatory mechanisms are thought to be the primary pathways through which APOE4 disrupts vascular and BBB integrity. In pericytes in vitro, APOE4 expression and calcineurin‐nuclear factor of activated T cells (NFAT) signalling are both specifically upregulated, leading to pericyte degeneration (as illustrated in Figure 2, Path A) and cerebral amyloid angiopathy. Critically, cerebral amyloid angiopathy, and both NFAT and APOE4 upregulation in pericytes, were rescued by calcineurin inhibition via the shared Cyclophilin A (CypA) inhibitor, cyclosporine (Blanchard et al., 2020). Upregulated calcineurin activity in E4‐TR mice has also been linked to dendritic abnormalities (Section 1; Neustadtl et al., 2017), suggesting a possible conserved target in vascular and neuronal degeneration.
FIGURE 2.
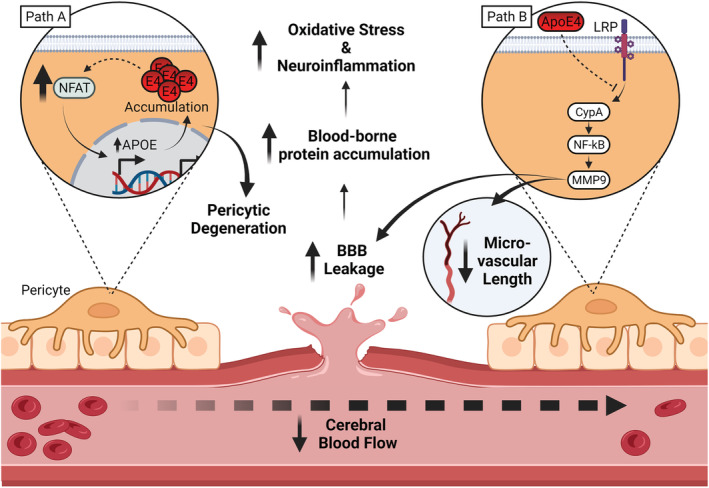
ApoE4 drives neurovascular impairment. Path A: Increased NFAT drives APOE expression in pericytes, ultimately resulting in the accumulation of ApoE within pericytes, promoting pericyte degeneration. Path B: A reduced ability of ApoE4 to associate with LRP results in impaired suppression of CypA activity. Disinhibited CypA drives MMP9 activity through an NF‐kB dependant pathway. Elevated MMP9 activity has been directly linked to decreased microvascular length and degeneration of the blood brain barrier. Combined, these pathways represent significantly reduced maintenance of both the wider neurovascular system and result in a ‘leaky BBB’ phenotype. This leaky BBB in turn causes an increase in blood born proteins in the CSF, increasing neuroinflammation and oxidative stress. In addition, net cerebral blood flow and volume reductions have been observed likely linked to impaired oxidative responsivity and microvascular integrity Abbreviations: ApoE4, apolipoprotein E4; BBB, blood brain barrier; CNS, central nervous system; CSF, cerebrospinal fluid; CypA, cyclophilin A; LRP, low‐density lipoprotein receptor; MMP9, matrix metalloproteinase 9; NFAT, nuclear factor of activated T cells; NF‐kB, nuclear factor kappa B. Created with BioRender.com
Degeneration of the BBB observed in E4‐TR mice has been attributed to pro‐inflammatory CypA activating an NF‐ĸB‐dependent (nuclear factor kappa‐light‐chain‐enhancer of activated B cells) pathway, culminating in elevation of matrix metalloproteinases (MMP) 9 in brain microvessels (Bell et al., 2012; Montagne et al., 2021). ApoE3 is thought to signal via its receptor, LRP1 (LDLR‐related protein‐1) to suppress the NF‐ĸB pathway, whereas ApoE4 was shown to exhibit reduced LRP1 binding in pericytes and thus reduced pathway suppression (Bell et al., 2012). The failure to suppress CypA inflammatory activation in brain microvessels correlates with tight junction degeneration and subsequent neurodegeneration in ageing E4‐TR mice, which is partially rescued by CypA inhibition (Montagne et al., 2021). This provides compelling evidence that ApoE4 loss of function promotes an inflammatory cascade of neurovascular degeneration (Montagne et al., 2021; Main et al., 2018; Figure 2, Path B).
3.2. APOE4 impairs neurovascular function
Another key function of the cerebral microvasculature is the control of blood flow, to match energy supply with demand. This is achieved through the coordinated responses of pericytes and smooth muscle to signals from active neurons and astrocytes to spread dilation signals through the endothelium (Howarth et al., 2021). As APOE4 impairs BBB integrity and microvascular density (see Section 3.1), it might be expected that cerebral blood flow and volume (CBF and CBV, respectively) would also be impacted by APOE isoform.
However, there is some conflict in the effect of APOE4 in the extent and age of onset for changes in basal or stimulus‐evoked CBF/CBV. Reductions in CBF in APOE4 mouse models have been reported across the lifespan, from as early as 2 weeks (Bell et al., 2012), to adult maturity (Koizumi et al., 2018) and late age, although it is unclear whether these late changes vary by brain region (Montagne et al., 2021; Wiesmann et al., 2016; Zerbi et al., 2014). Additionally, evidence is more equivocal in APOE4 mouse models at mid age, with demonstrations of a decrease (Bell et al., 2012) or no isoform differences (Wiesmann et al., 2016; Zerbi et al., 2014). Interestingly, more subtle changes were reported in a recent preprint, including decreased arteriole vasomotion and responsivity to sensory stimulation in young to mid‐aged E4‐TR mice (Bonnar et al., 2021, Preprint). Finally, one study also demonstrated enhanced CBV in the entorhinal cortex of late‐age E4‐TR mice, associated with neuronal hyperactivity, highlighting a complication in dissociating the effects of APOE4 on neuronal and neurovascular function (Nuriel, Angulo, et al., 2017). This evidence suggests that APOE4 may be associated with subtle early life impairments in vascular function while more global changes emerge at late age.
APOE4 may induce reductions in CBF/CBV by increasing oxidative stress leading to endothelial dysfunction, rather than aberrant neuronal activity or vasodilatory responsivity, with CBF rescue achieved with an ROS scavenger in vivo (Koizumi et al., 2018). Further, recent evidence has shown a strong correlation between reductions in capillary length and reduced CBF in old‐aged E4‐TR mice, independent of Aβ deposition. This suggests a link between APOE4‐induced microvascular loss and subsequent ineffective CBF regulation (Montagne et al., 2021). However, it is unclear what may drive discrepancies in the effect of APOE4 on CBF/CBV at mid‐age and further lifespan characterisation is needed. Finally, E4‐TR mice exhibited impaired recovery of neurovascular, cognitive and myelination deficits following hypoperfusive injury, indicating an interaction between APOE4 and severity of neurovascular insult (Koizumi et al., 2018).
Evidence from human studies appears to support these observations. Firstly, multiple reports highlight reductions in regional CBF in APOE4‐carriers (e.g., Filippini et al., 2011; Rhea et al., 2020; Thambisetty et al., 2010). However, debate surrounding potential age‐related compensation in cerebrovascular reactivity is ongoing (e.g., Tai et al., 2016; Trachtenberg et al., 2012). Additionally, recent evidence linked elevated BBB permeability in the hippocampal complex to exacerbated cognitive decline in older APOE4‐carriers. This association proved independent of Aβ and tau accumulation, supporting a plausible interspecies conservation of BBB disruption by APOE4 (Montagne et al., 2020). Further, elevated CSF levels of the putative pericyte marker sPDGFRβ correlate with cognitive decline in APOE4 but not E3‐carriers, suggesting that degenerative loss of pericytes is a conserved early pathological event independent of pre‐existing AD pathology (Montagne et al., 2020; Nation et al., 2019). Strikingly, neurovascular aberrations have been noted as some of the earliest to emerge during the development of LOAD (Iturria‐Medina et al., 2016), highlighting the importance in identifying neurovascular phenotypes associated with APOE4.
Together this evidence places neurovascular dysfunction as a central hit of the multihit hypothesis. This dysfunction appears early, with life‐long BBB degeneration which appears driven by pro‐inflammatory action within the vasculature. Additionally, while APOE4‐associated BBB degeneration and neurovascular coupling likely interact (Figure 2, Path B), impairments in ROS handling and hypoxic stress sensitivity may contribute to disruptions of the haemodynamic response and warrants further investigation.
4. HIT 3: NEUROINFLAMMATION
Pro‐ and anti‐inflammatory signalling and activation of glia are critical components of the immune response within the CNS, playing important roles in responding to pathogenic stimuli. These responses mediate both neuronal apoptotic and phagocytic processes, as well as reparative roles of glia. The delicate balance of these processes is crucial for normal immune function, while dysregulation is associated with multiple neurodegenerative disorders (Schain & Kreisl, 2017; Shabab et al., 2016). Evidence has outlined multiple roles for APOE in regulating immune responses under basal and immune‐stimulated conditions in both the periphery and CNS (Kloske & Wilcock, 2020). This section of the review will focus on the evidence of isoform‐dependent effects of APOE on immune function, its associated pathways and consequences for normal CNS function.
4.1. APOE4 impacts the homeostatic control of the inflammatory state
The role of ApoE in the regulation of immune responses is generally thought to involve anti‐inflammatory action via suppression of pro‐inflammatory signalling pathways (Figure 2, Path B and Figure 3, Path A). ApoE4, however, demonstrates a loss of anti‐inflammatory function. Specifically, ApoE4 fails to suppress the basal activation of macrophages and microglia and the response to pro‐inflammatory stimuli including lipopolysaccharide, resulting in stimulation of JNK (c‐Jun N‐terminal kinase), p38 MAPK (p38 mitogen activated protein kinase) (Maezawa, Nivison, et al., 2006; Cash et al., 2012) and NF‐ĸB signalling (Jofre‐Monseny et al., 2007). Subsequently, ApoE4 promotes upregulation of pro‐inflammatory cytokines, nitric oxide (NO) release and macrophage cell death in vitro (Brown et al., 2002; Colton et al., 2002, 2004; Grainger et al., 2004; Jofre‐Monseny et al., 2007; Laskowitz et al., 2001; Lynch et al., 2003, 2005; Pocivavsek, Burns, & Rebeck, 2009; Zhu et al., 2010). ApoE4 was also associated with opposing cell‐specific effects in pro‐inflammatory cytokine release, increasing in microglia and decreasing in astrocytes (Maezawa, Maeda, et al., 2006; Maezawa, Nivison, et al., 2006). Similarly, in APOE4 and APOE‐KO mouse models, increased pro‐inflammatory cytokine release, NF‐ĸB signalling and astrogliosis are observed across age and may precede neurodegeneration (Maezawa, Nivison, et al., 2006; Maezawa, Zaja‐Milatovic, et al., 2006; Ophir et al., 2005; Vitek et al., 2009; Yin et al., 2011; Zhu et al., 2012).
FIGURE 3.
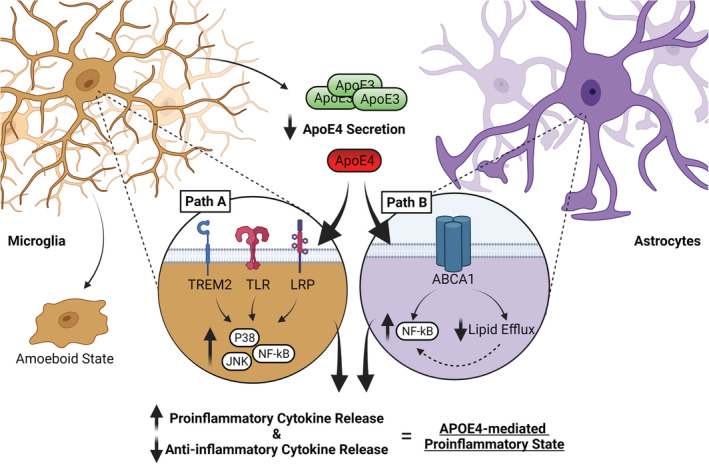
An elevated pro‐inflammatory state is caused by ApoE4. Path A: ApoE4 fails to suppress pro‐inflammatory activation via a loss of activation of LDLR/LRP family signalling, resulting in chronic activation of JNK and P38 culminating in activation of the NF‐kB pathway, driving the release of pro‐inflammatory factors such as TNFa, while reducing anti‐inflammatory cytokine release. ApoE also interacts with TREM2 to induce a phagocytic state, although the isoform interaction is unclear. Path B: ApoE4 drives activation of lipid‐sensitive inflammatory pathways. L/RXR can supress NF‐kB activation, while promoting lipid efflux via increasing APOE and ABCA1 expression upon lipid loading. ApoE4 impairs lipid efflux and excessive lipid accumulation may subsequently increase activation of inflammatory factors including NF‐kB. TLR signalling is similarly sensitive to lipid loading, with the net effect being elevated NF‐kB. Impaired lipid trafficking likely mediated in part by endosomal accumulation as discussed in hit 5 of the review. These imbalances result in an overall pro‐inflammatory state that is also associated with aberrant glial behaviour including reduced clearance of cellular debris and microglia entering an aberrant amoeboid state. Abbreviations: ABCA1, adenosine triphosphate (ATP)‐binding membrane cassette transporter A1; ApoE4, apolipoprotein E4; JNK, c‐Jun N‐terminal kinase; L/RXR, liver/retinoid X receptors; LRP, low‐density lipoprotein receptor‐related protein; NF‐kB, nuclear factor kappa B; TLR, toll‐like receptor; TREM2, triggering receptor expressed on myeloid cells 2. Created with BioRender.com
Impaired action of ApoE4 at LDL family receptors, particularly LRP1, appears primarily responsible for the loss of anti‐inflammatory signalling (Figure 3, Path A, Baitsch et al., 2011; Laskowitz et al., 2001; Lynch et al., 2005; Maezawa, Nivison, et al., 2006; Mikhailenko, et al., 2009; Pocivavsek, Burns, & Rebeck, 2009; Pocivavsek, Vitek et al., 2009; Zhu et al., 2010). Moreover, agonism of PPARɣ/COX2 (peroxisome proliferator‐activated receptor gamma; cyclooxygenase 2), indirectly inhibiting NF‐ĸB, rescued abnormal ApoE4 cellular distribution and dendritic degeneration in adult E4‐TR mice (DiBattista et al., 2016). This may point to a mechanistic bridge between chronic dysregulation of a pro‐inflammatory NF‐ĸB pathway, ApoE4 somal sequestration and aberrant neuronal morphology.
An alternate mechanism may exist via activation of lipid‐sensitive inflammatory pathways. Cholesterol accumulation in lipid rafts is crucial for toll‐like receptor (TLR) inflammatory signalling, converging on activation of NF‐ĸB (Fessler & Parks, 2011; Gale et al., 2014; Ito et al., 2015; Zhu et al., 2010). Additionally, nuclear liver/retinoid X receptors normally suppress NF‐ĸB transcription and can upregulate both ApoE and ABCA1 expression upon lipid loading to promote lipid efflux (Geyeregger et al., 2006; Liang et al., 2004; Yassine & Finch, 2020). However, ApoE4 impairs cholesterol efflux (Lin et al., 2018; Okoro et al., 2012; Rebeck, 2017) which may therefore indirectly contribute to enhanced inflammatory NF‐ĸB activation. Thus, ApoE4 may contribute to toxic feedback via loss of function in acute inflammatory suppression (Figure 3, Path A) and activation of lipid‐sensitive inflammatory responses (Figure 3, Path B; see Section 7.2).
4.2. APOE4 causes dysfunctional immune cell behaviour
In addition to altering acute inflammatory signalling, APOE4 is associated with abnormal immune cell behaviour. APOE4‐expressing macrophages and microglia exhibit impaired migration, phagocytosis and clearance of cellular debris including Aβ, increased amoeboid structure and enhanced risk of cell death and promote neurotoxicity (Cash et al., 2012; Cudaback et al., 2015; Grainger et al., 2004; Lin et al., 2018; Muth et al., 2019; Vitek et al., 2009). APOE4 also appears to impair astrocytic support for neurite regrowth (Maezawa, Maeda, et al., 2006). Recent evidence further suggests that ApoE4 undergoes differential post‐translational modifications in astroglia, with impaired ApoE4 secretion from microglia following TLR stimulation. This suggests that impaired ApoE4 circulation may contribute to deficient immunomodulation (Figure 3, Path A; Lanfranco et al., 2021; Rebeck, 2017). Thus, ApoE4 impairs normal astroglial behaviour including debris clearance and phagocytosis while impairing support for neuronal repair from inflammatory damage.
4.3. APOE interacts with TREM2 and regulates disease‐associated microglial state
APOE has also been specifically linked to regulation of the disease/damage‐associated microglia (DAM) state. DAM activation is regulated by the triggering receptor on myeloid cells 2 (TREM2) receptor, subsequent ApoE upregulation and activation of a ‘DAM state’ transcriptional response (Castranio et al., 2017; Keren‐Shaul et al., 2017; Kloske & Wilcock, 2020; Krasemann et al., 2017; Tay et al., 2018). Loss of APOE or TREM2 in microglia reduces neuronal apoptosis in injury models, although it is unclear whether this interaction is dependent on APOE isoform (Atagi et al., 2015; Bailey et al., 2015; Kloske & Wilcock, 2020; Wolfe et al., 2019). For example, ApoE4 potentiated signalling downstream of TREM2, without a consequent change in phagocytic activity in vitro (Yao et al., 2019), while transcriptomic studies show discrepancies in the relationship between APOE4 and TREM2 (Lin et al., 2018; Shi & Holtzman, 2018; J. Zhao et al., 2020). However, lipid metabolism may link APOE and TREM2 function, as loss of either gene (Damisah et al., 2020; Nugent et al., 2020) or expression of APOE4 (Lin et al., 2018) results in increased glial cholesterol accumulation, including following injury. These data highlight that there may be an indirect effect of ApoE4 on TREM2 signalling to support neuronal repair via lipid efflux, but the mechanistic underpinning remains unresolved.
There is evidence for an immunomodulatory role of APOE in human studies, with APOE4‐carrier AD‐patients exhibiting elevated gliosis in post‐mortem brain tissue and shifts in expression of microglial markers (Egensperger et al., 1998; Jofre‐Monseny et al., 2008; Minett et al., 2016; Tai et al., 2015; Tzioras et al., 2019). Further, peripheral inflammatory studies similarly suggest a pro‐inflammatory influence of APOE4, although debate remains over the definition and the consequences of ‘inflammatory profile phenotypes’ (Gale et al., 2014; Tai et al., 2015). One key limitation is the debated animal‐to‐human translatability of glial transcriptional signatures such as DAM state (Serrano‐Pozo et al., 2021; Srinivasan et al., 2020). Such pathways require thorough investigation using a combination of human and preclinical models to assess conservation of APOE‐dependent immunoregulation.
In summary, ApoE has a complex role in modulating immune homeostasis and neuroinflammation, summarised in Figure 3. ApoE4 displays loss of anti‐inflammatory function, likely via impaired receptor‐mediated signalling, and promotes inflammatory glial cell behaviour via common regulatory targets such as NF‐ĸB. Additionally, an APOE‐TREM2 pathway regulates the microglial DAM state and phagocytic microglial activity, although an APOE isoform‐specific role is unclear. Ultimately, APOE4 disruption of immunomodulation represents a pivotal hit of the multihit hypothesis, in which chronic immune stimulation may ‘set the stage’ for neuronal and neurovascular degeneration.
5. HIT 4: OXIDATIVE STRESS
Oxidative stress occurs when the production of oxidants outweighs the antioxidant defence system of the cell. Overproduction of reactive oxygen species (ROS) and nitrogen species can lead to extensive oxidation of biomolecules including lipids, proteins and DNA. The brain is particularly vulnerable to oxidative damage as it is rich in highly oxidizable lipids and has a high oxygen consumption rate alongside relatively low levels of antioxidants (Salim, 2017). ApoE has been linked to the oxidative stress lifecycle (Butterfield & Mattson, 2020), and evidence for an APOE isoform‐specific role in oxidative stress as a hit to cellular function will be discussed.
5.1. APOE4 alters basal and induced antioxidant responses
Firstly, evidence from multiple in vitro studies suggests that ApoE4 regulates the oxidative stress response. ApoE4 expression or acute application causes an upregulation of NO and subsequent cell death in neuronal cell lines compared with ApoE3 (Dose et al., 2016; Miyata & Smith, 1996; Pocernich et al., 2004), while no effect was observed in macrophages (Jofre‐Monseny et al., 2007, 2008). ApoE4 may interact with sex in effecting oxidative stress, with elevated NO only in male E4‐TR‐derived macrophages but comparable NO elevation of E4‐TR microglia (Brown et al., 2002). Further, proteomic profiling in female E4‐TR synaptosomes revealed a reduction in oxidative stress response proteins including SOD (Superoxide dismutase) and HSP (heat‐shock protein) families, which was greater in synaptosomal than somal compartment mitochondria (Shi et al., 2014).
In vivo evidence also suggests that basal antioxidant protein levels are reduced by APOE4, with E4‐TR mice demonstrating downregulation of cortical and hippocampal anti‐oxidative proteins including nuclear factor erythroid 2‐related factor 2, thioredoxin (Persson et al., 2017), SOD2, cytochrome oxidase 4 (COXIV) and glutathione levels suggesting a global enhancement of oxidative stress (Figure 4, Path A; Shi et al., 2014). Some effects were also exacerbated in female E4‐TR mice, again suggesting sex‐dependent modulation of oxidative risk (Shi et al., 2014). In contrast, responses to environmental oxidative challenge are more mixed. Reductions in TBI‐induced SOD2 expression were reported in E4‐TR mice (Ferguson et al., 2010). Paradoxically, however, multiple studies show environmental oxidative challenge elicits and enhanced redox protein response, decreased astrogliosis, synaptic integrity and attenuated memory impairment in E4‐TR mice (Basaure et al., 2018; Jiang et al., 2019; Peris‐Sampedro et al., 2015; Villasana et al., 2016; Yeiser et al., 2013), although with some inconsistency (Basaure et al., 2019).
FIGURE 4.
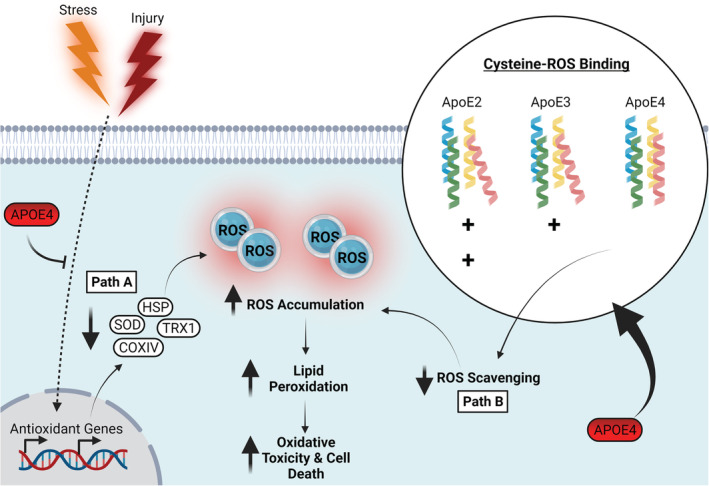
ApoE4 associated oxidative stress. Path A: The presence of APOE4 impairs the basal expression of antioxidant proteins such as SOD, HSP, TRX1 and COXIV and may impair ability to buffer oxidative challenge. Path B: ApoE4 exhibits a reduced affinity for reactive oxygen species and lipid peroxidation byproducts, likely due to a decreased number of ROS binding cysteine residues relative to that of ApoE2 and ApoE3. Combined, these pathways result in elevated ROS accumulation causing lipid peroxidation enhanced oxidative toxicity, ultimately increasing susceptibility to cell death. Abbreviations: ApoE4, apolipoprotein E4; COXIV, cytochrome oxidase subunit 4; HSP, heat shock protein; SOD, superoxide dismutase; TRX, thioredoxin; ROS, reactive oxygen species. Created with BioRender.com
These data suggest that APOE4 downregulates critical antioxidant proteins likely in multiple cell types (Figure 4, Path A). However, conflicting evidence surrounding the effect of APOE4 on the response to oxidative challenge may hint at a potential compensatory mechanism for basal impairments in ROS handling capabilities (Persson et al., 2017; Shea et al., 2002).
5.2. ApoE4 structure confers reduced intrinsic antioxidant function
Aside from altering the basal and stress‐induced antioxidant responses, ApoE may also impact oxidative stress via its own intrinsic antioxidant activity. An early observation that APOE‐KO mice had increased lipid peroxidation linked ApoE to oxidative resistance (Hayek et al., 1994), and further studies have investigated the antioxidant potential of individual isoforms. ApoE protected against hydrogen peroxide cytotoxicity and showed antioxidant activity in the order ApoE2 > ApoE3 > ApoE4 (Figure 4, Path B; Jolivalt et al., 2000; Miyata & Smith, 1996). How exactly ApoE exerts its direct antioxidant effects remains unclear, although there are several proposed mechanisms. One such mechanism is through the binding and sequestration of metals, with ApoE4 having the lowest efficiency for inhibition of copper‐mediated lipoprotein oxidation (Miyata & Smith, 1996). ApoE may also act directly as an ROS scavenger. 4‐hydroxynonenal (HNE) is a by‐product of lipid peroxidation which can cross‐link more effectively with ApoE3 than ApoE, with HNE cross‐linking protecting against cell death (Jiang et al., 2019; Montine et al., 1996; Pedersen et al., 2000). Further, direct oxidative modifications to ApoE can decrease its hydrophobicity and reduce binding efficacy to phospholipids, indicating that both physiological structure and function of ApoE would be affected by oxidation (Jolivalt et al., 2000). All APOE isoforms possess potential antioxidant activity, owing to possession of seven methionine residues open to oxidation (Levine et al., 1999), particularly within the receptor binding domain (Pham et al., 2005). However, the increased cysteine residues because of the corresponding allelic SNP sites in ApoE2 and ApoE3 may confer ApoE4 with poorer antioxidant capacity (Figure 4, Path B; Jolivalt et al., 2000; Miyata & Smith, 1996; Pham et al., 2005).
Reductions in multiple antioxidant proteins, including SOD and glutathione peroxidase, have also been noted in APOE4‐carrier brain tissue from mixed AD, MCI and healthy patient cohorts (Kharrazi et al., 2008; Ramassamy et al., 2000; Yin et al., 2020), alongside enhanced lipid peroxidation (Jofre‐Monseny et al., 2008; Ramassamy et al., 2000). Post‐mortem cortical tissue from APOE4‐carrier AD patients also showed dysregulation of multiple antioxidant and mitochondrial function genes, alongside lipid homeostasis and inflammatory pathways including NF‐ĸB (Caberlotto et al., 2016). Further, a cooperative enhanced risk of AD diagnosis has been shown in APOE4 carriers possessing mutations within genes involved in the oxidative stress response, including the uncoupling protein family (UCPs, Montesanto et al., 2016; Butterfield & Mattson, 2020). This suggests that APOE4 may similarly impair antioxidant protein levels and induce dysregulation of interconnected inflammatory and oxidative stress pathways as observed in preclinical systems. However, more research is needed to establish the extent of oxidative stress load conferred by APOE4 across the lifespan.
Overall, accumulating evidence supports a role of ApoE4 in oxidative stress and increasing vulnerability to cellular lipid peroxidation. ApoE4 may act both directly by sequestering oxidative derivatives and indirectly through altering activity of endogenous antioxidant systems. However, discrepancies in the literature remain unexplained and appear modulated by both sex and oxidative challenge. Therefore, further work is needed to understand these complex interactions and interplay with other cellular hits. For example, ApoE4 elevation of inflammatory NF‐ĸB is a redox‐sensitive transcription factor, while oxidative stress can accumulate as a by‐product of respiratory dysfunction (Hit 6; Dose et al., 2016; Singh et al., 2019).
6. HIT 5: ENDOSOMAL TRAFFICKING IMPAIRMENTS
Endosomal trafficking in the CNS facilitates receptor recycling, processing of neurotoxic protein fragments and the integration of phospholipids amongst other functions (Elkin et al., 2016). When ApoE binds to its receptor, ApoER2, the resulting complex is endocytosed into an early endosome where sorting is thought to occur (Jovic et al., 2010), before the receptor complex is recycled. This recycling is necessary to maintain levels of ApoER2 on the cell surface for further activation and threatens a cycle of impaired signalling when disrupted.
Recent evidence suggests that ApoE4 disrupts endosomal trafficking within the CNS, with predominant mechanisms thought to involve changes in endosomal pH (Figure 5). The additional Arginine in ApoE4 (Arg‐112) relative to ApoE3 raises its isoelectric point from 6.1 to 6.4. The isoelectric point of ApoE4 is thus closer to the pH of the early endosome (Xian et al., 2018), increasing the propensity of internalised ApoE4 to deform into a ‘molten globule’ state (Morrow et al., 2002). This state then prevents the efficient recycling of ApoE receptor complexes evidenced by the reduced surface expression of ApoER2 with ApoE4 (Chen et al., 2010; Feng et al., 2020; Xian et al., 2018). The reduction in surface ApoER2 consequently limits the ability of its alternative ligand, Reelin, to enhance long‐term potentiation of excitatory synaptic transmission (Chen et al., 2010). Surface expression of other membrane receptors, important for processes including cell signalling and synaptic transmission (Figure 5, path A), is also reported to be directly impaired by ApoE4‐mediated endosomal trafficking defects. Specifically, surface levels of both AMPAR (α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor) and NMDARs (N‐methyl‐D‐aspartate receptor) are reduced rapidly following exposure to ApoE4 (Chen et al., 2010), while total levels of these protein levels are unaffected. In addition to glutamatergic synaptic receptors, neuronal insulin receptors (IR) (N. Zhao et al., 2017), astrocytic ABCA1 receptor (Rawat et al., 2019) and ApoER2 itself (Chen et al., 2010; Feng et al., 2020; Xian et al., 2018) have all also been shown to exhibit reduced surface expression levels when exposed to ApoE4. Crucially, this suggests that ApoE4 can induce a multifaceted disruption of cell signalling function and subsequent intracellular trafficking.
FIGURE 5.
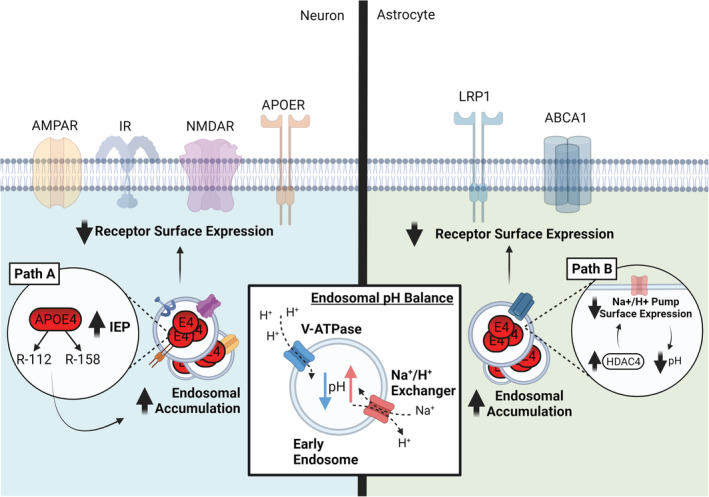
ApoE4 impairs endosomal trafficking. In both neurons and astrocytes, normal endosomal trafficking is stalled by accumulation of ApoE4 in endosomes, preventing not only their recycling but also the surface expression of multiple receptors including AMPAR, NMDAR, IR, APOER and ABCA1. However, cell‐specific differences are evident as described below; Path A: In neurons, endosomal pH is thought to be close to the altered isoelectric point of ApoE4 resulting in denaturation to a molten globule state promoting subsequent accumulation. Path B: In astrocytes, excessive HDAC4 activity prevents expression of the Na+/H + exchanger NHE6, lowering the endosomal pH and resulting in similar endosomal accumulation as seen in neurons. Inset box: Endosomal pH is regulated by the opposing actions of Na+/H + exchangers and V‐ATPases. Abbreviations: ABCA1, adenosine triphosphate binding cassette transporter A1; AMPAR, alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole propionic acid receptor; APOER, ApoE receptor; ApoE4, apolipoprotein E4; HDAC4, histone deacetylase 4; IR, insulin receptor; NHE6, Na+/H + exchanger 6; NMDAR, N‐methyl‐D‐aspartate receptor. Created with BioRender.com
Strategies to alleviate the ApoE4‐mediated deficits in endosomal trafficking have centred around altering early endosome pH away from the isoelectric point of ApoE4 to prevent aggregation of ApoE4‐receptor complexes (Xian et al., 2018). Endosomal pH is regulated by the balance of pH lowering proton pumps and the pH raising Na/H + exchangers (NHEs) (Figure 5, inset box; Fuster & Alexander, 2014). NHE6 disruption has previously been shown to lower endosomal pH (Brett et al., 2002), while proton pump inhibition is associated with impaired lipid release from endosomes (Goldstein et al., 1985). Targeted inhibition of NHE6 rescued surface ApoER2 levels following ApoE4‐mediated trapping, while increasing endosomal pH impaired ApoER2 surface trafficking, again illustrating the pH sensitivity of the endosomal pathway and the inherent importance of NHE6. It has been suggested that following aggregation, ApoE4 would not travel past the early endosome where NHE6 is predominantly localised. Crucially, it was also noted that pharmacological rescue of normal endosomal trafficking in ApoE4‐treated neurons restored aberrant surface expression of glutamatergic receptors and purportedly relieved the previously described ApoE4‐mediated insensitivity of long‐term potentiation to Reelin (Xian et al., 2018).
Intriguingly, a conflicting study in the same year suggests an alternate process operating in astrocytes raising possible cell type‐specific mechanisms. APOE4‐expressing cultured astrocytes had higher levels of the histone deacetylase HDAC4, which was linked to significantly reduced expression of NHE6 (Figure 5, Path B; Prasad & Rao, 2018). Consequently, hyperacidification of astrocytic endosomes was observed, leading to decreased LRP1 surface expression and impaired Aβ clearance, which proved rescuable by HDAC4 inhibition. Recent evidence suggested that not only is endosomal acidification able to restore receptor trafficking and synaptic deficits but also improve Aβ clearance in brain sections from E4‐TR mice, linking Aβ‐related and unrelated pathways (Pohlkamp et al., 2021). These differences between findings could be accounted for both by cell type‐specific processes (Wong, 2020) and different model systems (APOE overexpressing astrocyte cultures vs tissue from APOE‐TR mice). However, combined these studies highlight the importance of APOE isoform interactions with endosomal compartments and highlight how subtle manipulations to pH balance can alter trafficking efficiency. It is likely that an optimal window exists for endosomal pH and perturbation too far in either direction from this window can stall endosomal trafficking, particularly in the case of ApoE4 (Prasad & Rao, 2018; Xian et al., 2018). Interestingly, carnosic acid treatment can reduce ApoE4‐mediated receptor trafficking and Reelin signalling defects pH‐independently by promoting cell surface LDLR localisation via sorting nexin 17 (Feng et al., 2020; Stockinger et al., 2002).
Finally, ApoE4 has been suggested to perturb the formation and secretion of exosomes, vesicles produced from late‐endocytic multivesicular bodies (Peng et al., 2019). Exosomes are reported to be critical for cellular clearance of TDP‐43 (Iguchi et al., 2016), a pathological protein related to frontotemporal dementia (Tan et al., 2015), and likely other forms of neurotoxic material. This impairment in normal exosome trafficking thus posits another risk to protein aggregation and cell death (see Hit 1).
While the study of endosomal trafficking mechanisms in humans is challenging due to technical limitations, some studies provide supporting evidence for its involvement in pathophysiology linked to APOE4. In post‐mortem brain tissue of AD patients, carrying one or two copies of APOE4 was associated with endosomal enlargement, which preceded Aβ‐associated neuropathology (Cataldo et al., 2000). This may be due to the accumulation of ApoE4 and subsequent stalling of the endosomal recycling pathway (Nixon & Yang, 2011; Small et al., 2017). Further, APOE4 was associated with impaired endocytic trafficking in hiPSC‐derived astrocytes, which proved rescuable with elevated expression of PICALM, a modifier of APOE4‐associated defects (Narayan et al., 2020). Taken together, the limited human‐specific evidence should motivate further work to establish the role of APOE4 in endosomal trafficking and on the therapeutic efficacy of manipulating endosomal pH.
ApoE4 driven impairments to endosomal trafficking represent a significant factor in the multihit hypothesis that may drive some of the other hits described in this review. For example, surface receptor depletion will result in deficits to synaptic transmission, insulin signalling and lipoprotein trafficking (Hit 6). Subsequent intracellular accumulation of receptors and ApoE will stall the clearance of toxic Aβ fragments and cellular waste and may increase neurodegenerative vulnerability (Hit 1). However, further work is needed to understand the interactions between endosomal trafficking and other APOE4‐related cellular hits.
7. HIT 6: LIPID AND CELLULAR METABOLISM DISRUPTION
The homeostatic regulation of cellular lipid content and the metabolic processes underpinning cellular respiratory pathways are critical to ensure the energetic demands of the cellular environment are met (Martínez‐Reyes & Chandel, 2020). Glucose uptake and metabolism, mitochondrial function and lipid homeostasis are essential components of cellular health and allow the maintenance of optimal concentrations of metabolic respiratory precursors, ATP synthesis and supporting cellular membrane synthesis, amongst many other functions (Martínez‐Reyes & Chandel, 2020; Reddy, 2009). APOE is primarily implicated in lipoprotein trafficking but has been demonstrated to play a role in each of these cellular processes, with isoform specific differences. This section of the review will focus on the evidence for these isoform disparities in lipid and cellular metabolism.
7.1. APOE4 disrupts normal glucose metabolism
Preclinical evidence suggests that APOE isoform modulates cerebral glucose uptake but with unclear directionality. Reports of increased brain glucose uptake (Venzi et al., 2017) are contradicted by decreased CNS‐wide glucose uptake following dietary/cognitive challenge (Johnson et al., 2019) or environmental stress in aged E4‐TR mice (Fang et al., 2021). Further, young E4‐TR mice demonstrated impaired brain glucose metabolic rate which was rescuable with rapamycin treatment, which also reduced CypA expression (Lin et al., 2017), hinting at a potential conserved target between the APOE4‐mediated vascular impairment (Hit 2), neuroinflammation (Hit 3) and metabolic disruption (Hit 6). Cell and region‐specific glucose modulation may contribute to these conflicting observations, with some studies demonstrating decreased glucose uptake specifically in astrocytes (Williams et al., 2020; Zhong et al., 2009) or neurons (Li et al., 2021) in vitro. While further investigation is needed, these studies trend towards an APOE4 decrease in glucose utilisation.
The mechanism by which APOE genotype modulates glucose utilisation within the CNS is likely insulin‐mediated. Insulin is critical for promoting glucose transport, metabolism and glycolysis (Petersen & Shulman, 2018). Insulin is maintained at physiological levels in part by the insulin degrading enzyme (IDE), disruption of which leads to chronically elevated insulin, receptor internalisation and insulin resistance (Hulse et al., 2009; Petersen & Shulman, 2018). Neuronal IDE expression was significantly impaired in the presence of ApoE4 (Figure 6, path A; Du et al., 2009; Keeney et al., 2015) or APOE‐KO which was rescued by ApoE3 but not ApoE4 exposure (Du et al., 2009). This effect is likely mediated by an LDLR family dependent signalling pathway. In E4‐TR mice, LDLR family receptors, as well as the insulin receptor itself (N. Zhao et al., 2017), exhibit lower neuronal surface levels due to ApoE4 disrupting endosomal trafficking (Hit 5; Chen et al., 2010; Nuriel, Angulo, et al., 2017; Xian et al., 2018). In addition, adult E4‐TR female mice show reduced expression of multiple genes involved in glucose and insulin regulation including glucose transporter 4 (GLUT4) and PPARɣ than their E3‐TR counterparts (Keeney et al., 2015). Combined, these results suggest that ApoE4 has a complex but disruptive relationship with glucose metabolism, likely via altered glucose uptake and use caused by impaired insulin signalling (Figure 6, Path A). However, mixed evidence as to the direction of these effects highlights the need for further investigation and consideration of diet appears an important factor (Yassine & Finch, 2020). Glucose availability is critically linked to efficient mitochondrial respiratory function (Section 7.3) and as such, ApoE4‐mediated disruptions of glucose availability may subsequently exacerbate shifts in energy supply–demand dynamics.
FIGURE 6.
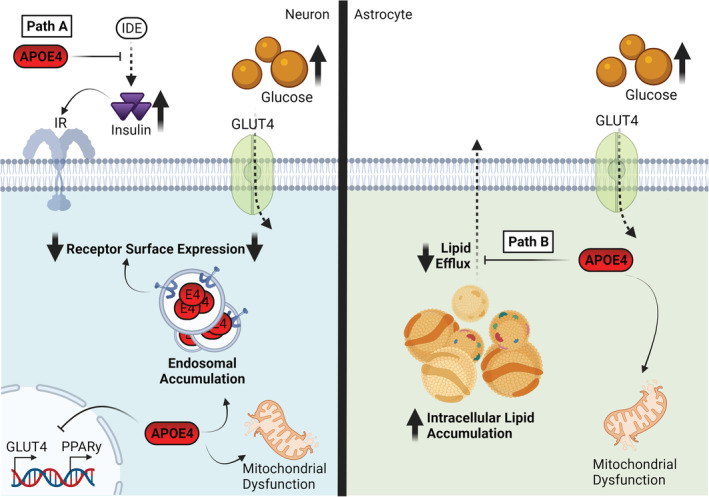
Metabolic dysfunction caused by ApoE4. Path A: ApoE4 impairs the activity of IDE, resulting in elevated extracellular levels of insulin. This causes receptor internalisation, that is, exacerbated by the previously described endosomal trafficking defects and leads to reduced surface expression of GLUT4 and impaired glucose uptake. This is likely exacerbated by reduced PPARy and GLUT4 expression, associated with ApoE4. Path B: ApoE4 shifts the mitochondrial metabolome away from effective oxidative phosphorylation while promoting intracellular lipid accumulation and lipid droplet formation through impaired lipid efflux. Path C: ApoE4 escaping the secretory pathway within neurons undergo proteolytic fragmentation. ApoE4 fragments accumulate in the mitochondria and ER, impairing mitochondrial complex function and inducing cellular toxicity. Abbreviations: ApoE4, apolipoprotein E4; GLUT4, glucose transporter 4; IDE, insulin degrading enzyme; PPARy, peroxisome proliferator‐activated receptor y.Created with BioRender.com
7.2. APOE4 impairs lipid homeostasis and metabolism
Intracellular lipid efflux, sequestration and accumulation in the CNS are critically dependent on APOE isoform (Flowers & Rebeck, 2020; Huang & Mahley, 2014). In vitro evidence suggests that ApoE4 displays a reduced affinity for phospholipids (Xu et al., 2004) and a reduced lipidation state, which may be due to reduced affinity for HDL, resulting in smaller lipid complexes in the CNS (Hatters et al., 2006; Heinsinger et al., 2016; Hu et al., 2015; Lanfranco et al., 2020; Rawat et al., 2019). This shifts towards an unlipidated ApoE4 pool which is more prone to aggregation and exhibits neurotoxicity, particularly following fragmentation (Figure 6, Path C; Flowers & Rebeck, 2020; Hatters et al., 2006; Hubin et al., 2019; Raulin et al., 2019; Rawat et al., 2019). In addition, ApoE4 appears to impact effective lipid handling and efflux. ApoE4 increases cholesterol accumulation and impairs lipid sensitivity in primary E4‐TR or APOE4 hIPSC‐derived astrocytes (Larramona‐Arcas et al., 2020; Lin et al., 2018) and primary neurons (Gong et al., 2007; Michikawa et al., 2000). This is likely mediated by impaired cholesterol efflux (Figure 6, Path B), although some contradiction is present with either no difference or even an enhancement of cholesterol efflux by ApoE4 observed dependent on cell type (Lanfranco et al., 2020; Lee et al., 2021; Michikawa et al., 2000). Structural analyses of ApoE4 have demonstrated that the putative C‐N domain interaction, the amino terminal region or lack of self‐dimerization relative to ApoE3 may all contribute a loss of lipid efflux function (Flowers & Rebeck, 2020; Frieden et al., 2017; Minagawa et al., 2009).
Further, intracellular lipid stores known as lipid droplets are also implicated in ApoE4 metabolic dysfunction. Lipid droplets serve as energy dense reserves for membrane synthesis and metabolic processes including mitochondrial beta‐oxidation (Walther & Farese, 2012; Welte & Gould, 2017). However, excessive lipid droplet formation has been associated with multiple neurodegenerative conditions, enhances the risk of insulin resistance and can stimulate pro‐inflammatory signalling such as the complement cascade (Yin et al., 2019; Farmer et al., 2020). Importantly, E4‐TR astrocytes exhibit greater lipid droplet number but smaller size following lipid‐rich challenge, alongside an increase in endogenous: exogenous fatty acid oxidation (Farmer et al., 2019). Overall, this evidence converges on a failure of ApoE4‐mediated glial lipid efflux, decreased lipoprotein complex size, lipid sensitivity, and disrupted lipid storage. This predicts a multilevelled impairment of normal lipid handling and delivery to neurons, while promoting lipid‐sensitive pro‐inflammatory responses (Hit 3; Martínez‐Martínez et al., 2020; Rebeck, 2017).
7.3. APOE4 induces mitochondrial dysfunction
In conjunction with lipid homeostasis and glucose handling, experimental evidence implicates APOE isoform in mitochondrial dysfunction; similarly, a common target of neurodegenerative disease (Cenini & Voos, 2019). The association between ApoE and mitochondrial dysfunction appears regulated both via its role in lipid homeostasis, crucial for metabolite synthesis (Lanfranco et al., 2020; Huang & Mahley, 2014), and via directly inducing mitochondrial toxicity (Huang & Mahley, 2014; Liu et al., 2013). Indeed, transcriptional profiling studies in various models demonstrate APOE4‐mediated dysregulation of genes involved in cholesterol biosynthesis (Zalocusky et al., 2021; J. Zhao et al., 2021), lipid transport (Lin et al., 2018), the electron transport chain (Area‐Gomez et al., 2020; Nuriel, Angulo, et al., 2017) and oxidative phosphorylation (Farmer et al., 2021). However, recent metabolomic and respiratory function analyses are more conflicted. Astrocytes from E4‐TR mice exhibited elevated lactate, decreased oxidative tricarboxylic acid (TCA)‐cycle activity (Williams et al., 2020) and decreased exogenous fatty acid oxidation (Farmer et al., 2019). However, entorhinal cortex samples from aged E4‐TR mice instead showed increased oxidative phosphorylation, decreased free fatty acids and an opposing sex‐isoform effects in state 3 respiration (Area‐Gomez et al., 2020; Nuriel, Peng, et al., 2017). Therefore, interactions between sex hormones and ApoE4 may drive separable metabolic profiles, with ApoE4 contributing to unstable respiratory function in hyper or hypometabolic states (Arnold et al., 2020; Farmer et al., 2019).
The effect of ApoE4 on mitochondrial function may be mediated by mitochondrial toxicity and ER stress. Neuronally expressed ApoE4 and its proteolytic fragments (see Hit 1) escape the secretory pathway, accumulate at the mitochondria and ER and disrupt mitochondrial complex function inducing neurotoxicity (Brodbeck et al., 2011; Chang et al., 2005; Chen et al., 2011; Nakamura et al., 2009; Figure 6, Path C). Importantly, the complexity of ApoE4 fragmentation extends beyond this and has been reviewed elsewhere (Muñoz et al., 2018). ER stress also is elevated in E4‐TR mouse brains (Segev et al., 2013) and predominately in astrocytes and macrophages in vitro (Brodbeck et al., 2011; Cash et al., 2012; Zhong et al., 2009). Chronic ER stress, including via calcium accumulation, activates the unfolded protein response (UPR). The UPR can reduce ER protein folding load by inhibiting global protein translation, but chronic activation can stimulate pro‐inflammatory signalling converging on NF‐ĸB and apoptosis (Dose et al., 2016). Interestingly, UPR activation has been linked to ApoE4 expression in E4‐TR mouse brain (Zalocusky et al., 2021; N. Zhao et al., 2020) and ApoE4 fragment accumulation in vitro (Zhong et al., 2009), while in parallel ApoE4 may suppress global translation via prolonged neuronal calcium influx (Ramakrishna et al., 2021). Speculatively, ApoE4 may converge on ER stress and mitochondrial dysfunction via direct ApoE4 fragment accumulation and indirectly via chronically elevated intracellular calcium levels (Hit 7).
Evidence for metabolic disturbance associated with APOE4 in patient populations is growing. Disrupted brain glucose metabolism has been reported in asymptomatic young and old APOE4‐carriers (Jagust & Landau, 2012; Reiman et al., 1996, 2004; Small et al., 1995) and is associated with both LOAD and cognitive decline (Arnáiz et al., 2001; Dong et al., 2021; Ouchi et al., 1998). While difficult to dissect, these findings may indicate impaired glucose utilisation and consequent mitochondrial dysfunction. Additionally, case studies discussing the consequences of an altered ApoE‐insulin‐glucose axis are beginning to appear (Stoykovich and Gibas, 2019), and insulin delivery has shown promise in cognitive outcomes for APOE4 LOAD patients (Claxton et al., 2013, 2015; Reger et al., 2006). Young female APOE4‐carriers may also exhibit impaired glucose oxidation, hinting at an APOE4–sex interaction in energy expenditure (Farmer et al., 2021). Finally, ApoE lipid particle size in CSF is reportedly reduced in APOE4‐carriers (Heinsinger et al., 2016) alongside increased fractions of lipid‐depleted ApoE4 (Hanson et al., 2013) supporting observations of impaired ApoE4 lipidation in preclinical models.
Together evidence suggests a complex, multifaceted role for ApoE4 in driving metabolic dysfunction at the level of glucose metabolism, lipid handling and mitochondrial function. The effects of ApoE4 include impaired lipid binding and efflux; shifts in glucose flux and insulin signalling; defects in respiratory function, mitochondrial and ER toxicity caused by ApoE4 fragmentation; and possible UPR activation. Ultimately, these events may impair the ability of the cell to effectively respond to metabolic demands such as sustained synaptic function, while increasing vulnerability to oxidative stress (Hit 4) and chronic metabolic inefficiency. However, conflicts between studies around the effects of cell type, sex, age and brain region remain unresolved and require thorough investigation.
8. HIT 7: CALCIUM DYSHOMEOSTASIS
Homeostatic control of cellular calcium levels is crucial for regulating neuronal activity and a myriad of processes including cell signalling cascades, organelle function and gene expression (Foster, 2007; Lane‐Donovan et al., 2014; Lane‐Donovan & Herz, 2017). Numerous studies have implicated ApoE in the regulation of calcium homeostasis, and here we review the evidence of an APOE isoform‐dependent effect on these processes.
Early observations demonstrated that the direct application of full length or truncated ApoE to neuronal cells resulted in an increase in intracellular calcium (Ohkubo et al., 2001; Qiu et al., 2004; Tolar et al., 1999; Wang & Gruenstein, 1997) and calcium bursting responses (Konings et al., 2021). Further, ApoE4 specifically elevated intracellular calcium levels relative to ApoE3 and was shown to be likely mediated by ApoE receptor activation of L‐type voltage gated calcium channels (VGCCs) and NMDARs, which may involve prolonged calcium influx dynamics (Ohkubo et al., 2001; Qiu et al., 2003; Ramakrishna et al., 2021; Veinbergs et al., 2002; Figure 7, Path A). Further, NMDAR‐dependent accumulation of intracellular calcium and induced cell death was also shown to be exacerbated by injury in the presence of APOE4 or APOE‐KO but was rescued by APOE3 in vitro (Jiang et al., 2015). This suggests that ApoE4 has impaired ability to suppress both basal and injury induced calcium accumulation.
FIGURE 7.
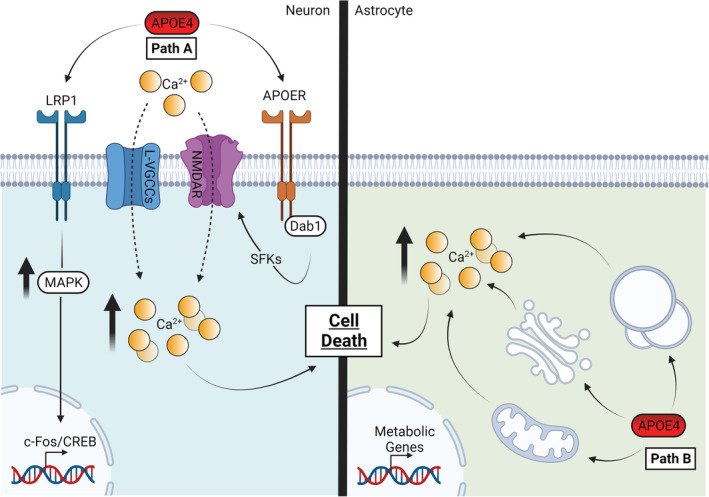
ApoE4 mediated impaired calcium homeostasis. Path A: Elevated calcium influx through L‐VGCCs and NMDARs results in an elevated level of intracellular calcium. A Dab1/SFK mediated pathway downstream of ApoE binding to APOERs promotes NMDAR mediated calcium influx. ApoE4 binding to LDLR/LRP family receptors upregulates MAPK signalling activation and is linked to activation of cFos/CREB synaptogenic signalling which may be chronically dysregulated. Path B: In astrocytes ApoE4 has been shown to induce the release of calcium from intracellular stores such as the mitochondria and endo/lysosomal compartments. The net effect of these pathways is a prolonged increase in intracellular calcium concentration, calcium signalling and subsequent cell death. Abbreviations: APOER, apolipoprotein E receptor; ApoE4, apolipoprotein E4; cFos, FBJ osteosarcoma oncogene; CREB, cyclic adenosine monophosphate (cAMP) response element binding protein; Dab1, disabled 1; L‐VGCCs, L type voltage gated calcium channels; LRP1, Lowdensity lipoprotein receptor‐related protein 1; MAPK, mitogen activated protein kinase; NMDAR, N‐methyl‐daspartate receptor; SFK, serine family kinases. Created with BioRender.com
A recent study also suggested that ApoE4 impaired calcium homeostasis in astrocytes via an alternative mechanism. Immortalised astrocytes from E4‐TR mice exhibited elevated spontaneous and ATP‐induced intracellular calcium levels, while in vivo male, but not female, E4‐TR astrocytes showed increased calcium event frequency and amplitude (Larramona‐Arcas et al., 2020). Furthermore, elevated lysosomal vATPase (Vacuolar‐type ATPase) activity was associated with increased basal calcium in APOE4‐expressing astrocytes, indicating that enhanced calcium efflux from organelle stores is another likely mechanism (Larramona‐Arcas et al., 2020; Figure 7, Path B). This was also suggested to reflect greater calcium accumulation in the endolysosomal compartments driven by ApoE4. Finally, whereas ApoE3 was shown to increase astrocytic calcium influx in lipid‐poor conditions, ApoE4 retained enhanced calcium influx during both lipid‐rich and poor conditions, while increasing perinuclear lysosome accumulation (Larramona‐Arcas et al., 2020).
While calcium dyshomeostasis is a persistent hypothesis surrounding the aetiology of AD (Guan et al., 2021; Magi et al., 2016; Puzianowska‐Kuznicka & Kuznicki, 2009), evidence for a direct association between APOE isoform and calcium homeostasis in humans is limited. A demonstration of elevated calcium serum levels in APOE4‐carriers, correlating with impaired cognitive performance (Van Vliet et al., 2009), supports a role for APOE4 in disrupting calcium homeostasis. Aβ‐dependent dysregulation of calcium homeostasis has been linked to the induction of ER stress and cell death in multiple models in vitro (Magi et al., 2016), but there is growing interest in the amyloid‐dependent role of APOE in calcium homeostasis (Guan et al., 2021). However, further investigation is needed during ageing in both healthy and AD states to validate preclinical findings.
In summary, calcium dysregulation is apparent across cell types in the presence of ApoE4, largely thought to be through VGCC and NMDAR mediated responses in neurons while intracellular calcium stores appear perturbed in astrocytes. Elevated intracellular calcium poses as threat to general organelle function and enhances the risk of cell death, thus forming another APOE4‐dependent hit. Future studies should replicate and expand these findings to clarify the extent to which APOE isoforms influence calcium accumulation across cell types and the mechanisms that may underpin interactions with lipid handling.
9. HIT 8: ALTERED TRANSCRIPTIONAL REGULATION
There is mounting evidence of APOE isoform specific modulation of both direct and indirect transcriptional regulation thus providing another hit to cellular function. These pathways appear to be mediated by both calcium‐dependent and independent pathways, including a putative direct DNA binding role for APOE in mediating transcriptional control.
9.1. APOE4‐associated transcriptomic signatures
Transcriptomic studies are elucidating the extent to which transcriptional dysregulation is induced by APOE4. Both bulk brain tissue and single cell ribonucleic acid (RNA) sequencing from E4‐TR mice and hIPSC‐derived cells are converging on a dysregulated signature of gene expression in multiple cellular pathways including immune responses, synaptic function, metabolic function, lysosomal processing and deoxyribonucleic acid (DNA)/RNA processing (Lin et al., 2016; Nuriel, Peng, et al., 2017; N. Zhao, et al., 2020). Levels of neuronal APOE expression have isoform‐independently been correlated with a similar signature, specifically within DG‐CA1 pyramidal neurons and interneurons but not astroglia (Zalocusky et al., 2021). Further, a conserved signature of dysregulated synaptic and immune function genes in hIPSC‐derived neurons and glia from both APOE4‐carrier AD and healthy patients correlated with Aβ plaque density and clinical dementia score. Importantly, this may hint at an APOE4‐specific transcriptomic phenotype occurring prior to AD‐pathology (Lin et al., 2016). This evidence suggests not only that APOE4 modulates the transcriptomic signatures of numerous cellular processes but that these processes may be directly influenced as a function of the levels of neuronal APOE expression itself.
9.2. APOE4 influences calcium‐sensitive transcriptional control
APOE isoform has been further associated with dysregulation of calcium‐dependent intracellular signalling cascades involved in transcriptional regulation. Key signalling proteins elevated downstream of ApoE‐ApoER interactions include ERK1/2, Akt (AKT serine/threonine kinase 1), Src (SRC non‐receptor tyrosine kinase) and eEF2 (Eukaryotic elongation factor 2) (Huang et al., 2017, 2019; Korwek et al., 2009; Ohkubo et al., 2001; Yong et al., 2014), although one study demonstrated an ApoE4‐dependent decrease in signalling activation (Hoe et al., 2005). These proteins are central regulators of the activity‐dependent transcription response and are involved in downstream synaptogenic signalling. Recent in vitro evidence in hIPSC‐derived neurons also provided direct evidence for ApoE4‐ApoER‐mediated potentiation of a Fos/CREB synaptogenic signalling pathway (see also Hit 1), although the physiological relevance of this is unclear (Huang et al., 2017, 2019). Intriguingly, one study suggested that ageing may interact with APOE4 and signalling, with an initial upregulation during adulthood (including pCREB and pERK) before pathological downregulation in old age in female E4‐TR mice (Yong et al., 2014).
The mechanisms underpinning these perturbations in signalling may fall downstream of elevated intracellular calcium influx by ApoE4 (Hit 7). Indeed, ApoE4 application to cultured neurons led to sustained calcium influx both basally and with stimulation, followed by a greater p‐eEF2‐mediated inhibition of global translation but preferential upregulation of multiple synaptic gene targets (Ramakrishna et al., 2021). As discussed in ‘Hit 1’, it is unclear what may mediate acute synaptogenic to chronic synaptotoxic signalling involving APOE4, although speculatively this may involve gradual compensatory changes in kinase‐phosphatase activation.
9.3. A role for APOE in microRNA‐mediated and direct transcriptional control
Some limited evidence also suggests that ApoE may directly regulate gene expression via action as a transcription factor or by modulating pre‐/post‐transcriptional machinery. In culture, it was demonstrated that ApoE3 and ApoE4 binds dsDNA at low nanomolar affinities. However, ApoE4 bound diverse gene promoter sites in the order of twofold that of ApoE3. Validation demonstrated that the ApoE4 interaction with multiple gene promoters led to dysregulation of genes including SIRT1/2 (Sirtuin 1/2) and MADD (MAPK activating death domain; Theendakara et al., 2016), important for cell survival (Theendakara et al., 2013). This suggests that transcription‐like activity of ApoE4 could be an important mechanism in neurodegenerative risk (Theendakara et al., 2016, 2018).
One recent study also suggested that astrocytic ApoE‐bound lipoproteins deliver micro‐RNAs (miRs) targeted to downregulating cholesterol biosynthesis in neurons. The miR‐mediated downregulation of cholesterol synthesis was associated with increased histone acetylation and both immediate‐early and synaptic gene expression. Crucially each component of the pathway was attenuated by ApoE4 both in vitro and in E4‐TR mice (Li et al., 2021). This supports an indirect role for ApoE4 in transcriptional control via impaired lipoprotein miRNA delivery, contributing to cholesterol accumulation (Hit 6), but again, these novel findings require further validation.
In humans, data from post‐mortem AD brain samples provide some convincing support for APOE4‐mediated disruption of transcriptional regulation. Studies have shown differential expression of genes involved in processes including metabolism, immunity, DNA damage, synaptic integrity, cell signalling and transcription associated with APOE4 (Bossers et al., 2010; Panitch et al., 2022; Simpson et al., 2011). Further, the association between elevated neuronal APOE expression and a unique transcriptional signature observed E4‐TR mice (outlined in Section 9.1) was replicated in both AD and healthy aged patient brains (Zalocusky et al., 2021). However, the mechanistic origins of these transcriptomic changes remain unknown.
In summary, ApoE likely regulates transcription indirectly through receptor‐mediated activation of calcium signalling and possible direct DNA binding activity. Evidence from both pathways converge to suggest that APOE4 expression may induce dysregulation of transcriptional signatures for numerous cellular processes, including immune function, metabolism, RNA processing, UPR, endo/lysosomal trafficking and neuronal function. This dysregulation poses a hit to multiple pathways, and future studies should investigate mechanisms by which ApoE action on pre‐/post‐transcriptional regulation may drive both AD‐dependent and AD‐independent processes under physiological circumstances.
10. DISCUSSION
Predominantly focussing on preclinical models, in this review, we have highlighted evidence associating APOE4 with the disruption of multiple cellular processes that converge on a pathophysiological state independent of typical AD‐pathology. Next, we will summarise and predict the consequences of the APOE4 multihit hypothesis, explain how it can be used to frame future research and consider its implications for understanding APOE4 as a risk factor for LOAD and other neurodegenerative disorders. Importantly, we stress that this includes predictions of the directionality of interactions between hits which will require further validation.
10.1. The APOE4 multihit hypothesis
In summary, APOE4 in its basal state promotes cellular ‘susceptibility’ with initially low‐level dysfunction that accumulates with age. Further, this state is exacerbated by multiple sources of endogenous and exogenous stress including physical injury, pro‐inflammatory and oxidative challenge and metabolic disturbances. This susceptibility appears to be determined consequent to structural and regulatory features of ApoE4. Generally, ApoE4 exhibits reduced expression, impaired oxidative scavenging capabilities, intracellular sequestration, impaired lipid efflux and trafficking, an increased propensity for fragmentation, reduced lipid binding affinity, less efficient receptor‐ligand interactions and altered DNA binding activity. These features ultimately result in global loss of normal and gain of toxic functions for ApoE4 across multiple cellular processes, or ‘hits’. The ways we envisage these multiple hits converge, and predictions of their functional consequences are outlined below and illustrated in Figure 8.
FIGURE 8.
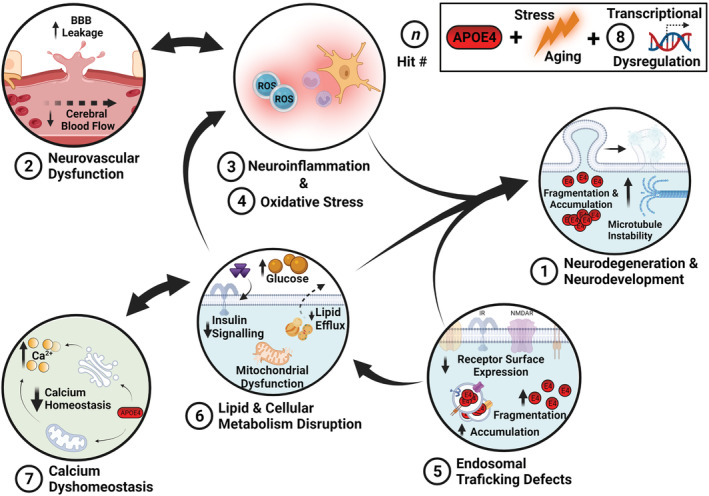
The multihit hypothesis. APOE4 genotype promotes a pathophysiological state of vulnerability to further damage through multiple interacting hits. Cellular stress, ageing and an altered transcriptional landscape act as key moderators of many of the hits described and are thus highlighted in their own box (top right). There are multiple points of crosstalk between APOE4‐mediated hits (as described in Section 10). Including the following: lipid and metabolic homeostasis and neuronal maintenance; calcium homeostasis and transcriptional regulation; neuroinflammation, oxidative stress and neurovascular dysfunction; and endosomal trafficking, lipid homeostasis and neurodegeneration. Abbreviations: ApoE4, apolipoprotein E4; CNS, central nervous system. Created with BioRender.com
10.2. Predictions of the APOE4 multihit hypothesis
As the primary role of ApoE is in lipoprotein trafficking, reduced ApoE4 lipidation and lipid efflux will enhance astrocytic lipid loading, lipid droplet formation and impair lipid delivery capacity from glia to neurons. This failure to meet the lipid‐rich demands of neuronal membrane remodelling will impair neurite growth and enhance sensitivity to injury‐induced neurodegeneration. Additionally, somal/endosomal sequestration of ApoE4 away from neurites will similarly reduce effective repair and microtubule dynamics. Importantly, the developmental consequences of this are likely minor in the absence of exogenous stressors but will accumulate with age.
Reductions in effective ApoE4 interaction with its cognate receptors will result in loss of anti‐inflammatory signalling including enhancing NF‐ĸB signalling, while lipid loading will contribute to the lipid‐sensitive glial inflammatory responses such as priming TLR activation. Additionally, reduced antioxidant functions of ApoE4 will promote lipid peroxidation and act in concert with lower ApoE4 anti‐inflammatory function to generate an environment of chronic low‐level inflammatory stress. Inflammatory and oxidative stress pathways will subsequently promote chronic blood vessel and BBB degeneration via loss of supporting tight junction proteins. BBB breakdown across age will also be accelerated by peripheral pro‐inflammatory factor infiltration and impaired clearance of toxic products, generating a toxic feedback loop between ApoE4‐induced inflammation and BBB degeneration. Cumulative action of this inflammatory state will also crosstalk with neuronal integrity, promoting synaptic engulfment and impairment of overall morphology, reflected during adulthood and ageing. Larger‐scale inflammatory disruption of the neurovascular unit will interact with oxidative stress disruption of neurovascular signalling, contributing to a neuronal energy supply–demand mismatch, resulting in impaired metabolic neuronal support, synaptic dysfunction and further degenerative risk.
In parallel, disruptions to intracellular trafficking pathways and toxic ApoE4 fragmentation will contribute to multiple downstream impairments. Firstly, enhanced endosomal sequestration of ApoE4, ApoE receptors and synaptic receptors will prevent continual receptor recycling, effective signalling, and promote synaptic dysfunction. Second, reductions in neuronal lipid availability and enhanced endosomal sequestration of the insulin receptor will impair glucose handling and induce oxidative phosphorylation supply–demand deficits. In concert, the intracellular accumulation of ApoE4 and its toxic truncated fragments, particularly within neurons, will disrupt critical mitochondrial complex function and stimulate ER/UPR stress responses. Disruption of mitochondrial complex function will then alter metabolite utilisation via electron transport chain/tricarboxylic acid cycle flux, shifting away from effective oxidative phosphorylation. These metabolic shifts and disruption of mitochondrial function will be exacerbated by ROS by‐product accumulation and elevated endogenous oxidative stress. Thus, chronic ApoE4 fragmentation will promote neuronal cell death via accumulation of these disruptions.
Additionally, aberrant ApoE4 signalling will promote activation of calcium channels and organelle store release to induce chronic intracellular calcium accumulation, contributing to both mitochondrial and ER/UPR dysfunction. Elevated calcium will also stimulate calcium‐dependent signalling pathways, activating transcription factors such as AP‐1 and CREB, of which chronic activation may lead to a compensatory shift towards synaptotoxic signalling. Finally, both via signalling and possible direct DNA interactions, ApoE4 will induce transcriptional dysregulation which will contribute to multiple hits through altered expression of genes related to cellular trafficking, metabolism and immune function amongst others.
Ultimately, these parallel hits promote convergence on a chronic state of cellular vulnerability, increasing degenerative risk that acts cumulatively with age, stressors or injury. These features will be modified by systemic contextual factors such as biological sex, cell type and brain region, with regions (e.g., the hippocampal complex) and cell types (e.g., GABAergic interneurons) particularly vulnerable to pathology at higher risk of dysfunction. Finally, the multihit hypothesis predicts that the convergence of individual hits onto common pathways will manifest in disruptions of higher‐order processing, which in some cases results in top‐down systems‐level dysfunction. For example, ApoE4 fragment‐induced mitochondrial dysfunction is linked to GABAergic degeneration, which subsequently leads to a systems‐level defect in GABAergic signalling, disrupting neuronal maturation.
10.3. Utility of the APOE4 multihit hypothesis and its relation to APOE4 as a risk factor for neurodegenerative disease
The APOE4 multihit hypothesis provides a framework to understand the temporality and interaction between the APOE4 hits and environmental variables. The interplay between the different APOE4 hits is increasingly more pertinent in the era of multi‐omics, with recent studies yielding associations between APOE and simultaneous dysregulation of multiple pathways (Lin et al., 2018; Nuriel, Peng, et al., 2017; Zalocusky et al., 2021; N. Zhao et al., 2020). There is a need to consider these complex interactions as we generate models of APOE4‐mediated pathophysiology, independent of, and also in the context of causing an increased risk of LOAD (Lewandowski et al., 2020).
While we have explicitly presented evidence of AD pathology‐independent roles for APOE4, we emphasise that within the multihit hypothesis, pathological load (e.g., Aβ accumulation, tauopathy) can be viewed as sources of cellular stress that would be predicted to cooperatively exacerbate an APOE4‐related pathophysiological state. We therefore argue that this does not conflict with the well‐established mechanisms by which ApoE4 can interact with AD pathology directly (e.g., enhanced amyloid beta aggregation, tau phosphorylation). That APOE4 has also been implicated as a risk factor for multiple neurological disorders including Parkinson's disease, frontotemporal dementia, as well as LOAD suggesting a diverse involvement in the aetiology of disease‐related CNS dysfunction (Forero et al., 2018; Huang & Mahley, 2014).
Previous thematic reviews and hypotheses have placed APOE at the interface between insult and inflammation, neuronal dysfunction, degeneration and gene–environment interactions, noting APOE as a risk or ‘homeostatic frailty’ gene (Bu, 2009; Gerdes et al., 2000; Huang & Mahley, 2014; Liu et al., 2013; Mahley & Huang, 2012; Najm et al., 2019; Tzioras et al., 2019; Yassine & Finch, 2020). The multihit hypothesis is congruent with these views but places emphasis on the accumulation and interplay between multiple disruptions of cellular physiology without necessitating AD pathology. It is perhaps less compatible with the theory of antagonistic pleiotropy, which posits age‐dependent detrimental but beneficial early and latest‐age roles for APOE4 (Han & Bondi, 2008; Han & Tuminello, 2011). The multihit hypothesis predominantly predicts a trajectory of cumulative APOE4 burden supported by much of the preclinical evidence base reviewed here, but we are unable to discount the possibility of some systems‐level benefits of APOE4 under specific circumstances. Potentially, antagonistic pleiotropy could be accommodated as a mechanism for accelerating late‐life vulnerability and multihit cascade. We acknowledge, however, the common criticism of the inconsistency with which APOE4 is associated with a clear cognitive phenotype. We posit that the multihit hypothesis is better suited to deal with this limitation with injury and stressors as conditional modifiers for neurocognitive risk rather than placing specific ageing constraints on expected cognitive outcome.
Finally, some crucial limitations. There is an absolute necessity to compare findings from preclinical model systems with human studies and neurodegenerative conditions. This review has focussed on preclinical data to derive APOE4 ‘hits’ and provided some specific examples of where these media are or are not congruent, but we must be wary of overinterpretation in the absence of direct evidence of pathway conservation. Alignment between the questions and aims using preclinical and human systems is essential to improve understanding of APOE and the translational value of our research. Further, while we have discussed the modifying role of sex in APOE4 pathophysiology, the influence of ethnicity has received attention in human studies but cannot be addressed by animal models. Ethnicity has been suggested to be a mediator in the effect of APOE isoform on multiple outcomes, including neurocognitive decline (Makkar et al., 2020), AD (Belloy et al., 2019; Farrer et al., 1998) and modifying environmental risk factors (Trumble & Finch, 2019; Yassine & Finch, 2020). How the multihit hypothesis could account for this is not clear, and valid criticisms may be raised concerning the bias towards interpretation of APOE4 phenotype on a Western genetic background. Speculatively, if considering translation, protective genetic variations associated with ethnicity may offer resilience against APOE4 pathophysiology and may help offset a multihit cascade. It is difficult to study such effects in preclinical models, but novel hIPSC‐based techniques modelling ethnic genetic diversity will be useful going forward.
11. CONCLUSION
In this review, we have presented evidence to support a multihit model of APOE4 pathophysiology, whereby basal alterations in ApoE4 function contribute to aberrant activation of multiple cellular pathways including neuronal maintenance, neurovascular function, inflammation and oxidative stress, lipid and cellular metabolism, endosomal trafficking, calcium homeostasis and transcriptional regulation. Cumulative perturbation in, and interactions between, these pathways converge on age‐dependent synaptic dysfunction, neurovascular impairment, neurodegeneration and presumptive eventual cognitive impairment. We have highlighted the critical role of contextual biological variables including cellular stressors, ageing and sex. We have also described briefly how the multihit hypothesis may compare to some key existing hypotheses of APOE function. Future work is required to better understand the physiological relevance, temporality, and crosstalk between these pathways. Indeed, studies demonstrating mutual exacerbation between the described APOE4‐dependent hits are emerging, supporting crosstalk between inflammation, lipid metabolism and neurotoxicity (e.g., de Leeuw et al., 2022; Zalocusky et al., 2021). We propose that some of the key outstanding questions to understand the role of APOE from low‐level cellular dysfunction to neurodegenerative disease are ‘how’ and ‘to what extent’ do APOE‐mediated pathways contribute and interact to form an APOE4 pathophysiological state.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTION
OGS and ACS equally contributed to the text, multihit hypothesis and created the figures. OGS and ACS contributed equally and should be cited as “Steele & Stuart et al. (2022)” where possible. LM drafted Hits 3 and 4, provided thoughtful discussion and aided in overall editing. KS, OB, SA and CH co‐drafted Hit 2. ACP, JR, LS and CH were involved in the final edits. SK was heavily involved in overall drafting, editing and structuring of the review.
NOMENCLATURE
In all cases, APOE refers to the Apolipoprotein E gene, while ApoE refers to the functional protein. APOE gene isoform is denoted by APOE2/3/4, while ApoE protein is represented as ApoE2/3/4.
12.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/ejn.15685.
ACKNOWLEDGEMENTS
Funding for this review was received from the Alzheimer's Society, specifically the Alzheimer's Society Doctoral Training Centre Grant (AS‐DTC‐2014‐003) awarded to LS, JR, SK, CH and ACP and supporting OGS, OB and SA. Funding was also received from Sussex Neuroscience which supports ACS. LM is supported by the University of Sussex School of Life Sciences doctoral studentship, while KS is supported by MRC Discovery Award (MC_PC_15071) and Project Grant (MR/S026495/1) held by CH. No funding providers had any input on the scope, structure or content of the review. Figures were created with BioRender.com and referenced appropriately throughout.
Steele, O. G. , Stuart, A. C. , Minkley, L. , Shaw, K. , Bonnar, O. , Anderle, S. , Penn, A. C. , Rusted, J. , Serpell, L. , Hall, C. , & King, S. (2022). A multi‐hit hypothesis for an APOE4‐dependent pathophysiological state. European Journal of Neuroscience, 56(9), 5476–5515. 10.1111/ejn.15685
Funding information Alzheimer's Society Grant Code: AS‐DTC‐2014‐003; Sussex Neuroscience
Edited by: Tara Spires‐Jones
Steele, O.G. and Stuart, A.C. contributed equally to the generation of this manuscript and are listed as, and should be cited as, co‐first authors (Steele & Stuart et al., 2022).
DATA AVAILABILITY STATEMENT
This review does not include any data.
REFERENCES
- Alata, W. , Ye, Y. , St‐Amour, I. , Vandal, M. , & Calon, F. (2015). Human apolipoprotein E ε4 expression impairs cerebral vascularization and blood‐brain barrier function in mice. Journal of Cerebral Blood Flow and Metabolism, 35(1), 86–94. 10.1038/jcbfm.2014.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews‐Zwilling, Y. , Bien‐Ly, N. , Xu, Q. , Li, G. , Bernardo, A. , Yoon, S. Y. , Zwilling, D. , Yan, T. X. , Chen, L. , & Huang, Y. (2010). Apolipoprotein E4 causes age‐ and Tau‐dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. Journal of Neuroscience, 30(41), 13707–13717. 10.1523/JNEUROSCI.4040-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area‐Gomez, E. , Larrea, D. , Pera, M. , Agrawal, R. R. , Guilfoyle, D. N. , Pirhaji, L. , Shannon, K. , Arain, H. A. , Ashok, A. , Chen, Q. , Dillman, A. A. , Figueroa, H. Y. , Cookson, M. R. , Gross, S. S. , Fraenkel, E. , Duff, K. E. , & Nuriel, T. (2020). APOE4 is associated with differential regional vulnerability to bioenergetic deficits in aged APOE mice. Scientific Reports, 10(1), 1–20. 10.1038/s41598-020-61142-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnáiz, E. , Jelic, V. , Almkvist, O. , Wahlund, L. O. , Winblad, B. , Valind, S. , & Nordberg, A. (2001). Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. Neuroreport, 12(4), 851–855. 10.1097/00001756-200103260-00045 [DOI] [PubMed] [Google Scholar]
- Arnold, M. , Nho, K. , Kueider‐Paisley, A. , Massaro, T. , Huynh, K. , Brauner, B. , MahmoudianDehkordi, S. , Louie, G. , Moseley, M. A. , Thompson, J. W. , John‐Williams, L. S. , Tenenbaum, J. D. , Blach, C. , Chang, R. , Brinton, R. D. , Baillie, R. , Han, X. , Trojanowski, J. Q. , Shaw, L. M. , … Kastenmüller, G. (2020). Sex and APOE ε4 genotype modify the Alzheimer's disease serum metabolome. Nature Communications, 11(1), 1148. https://www.nature.com/articles/s41467-020-14959-w, 10.1038/s41467-020-14959-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atagi, Y. , Liu, C. C. , Painter, M. M. , Chen, X. F. , Verbeeck, C. , Zheng, H. , Li, X. , Rademakers, R. , Kang, S. S. , Xu, H. , Younkin, S. , Das, P. , Fryer, J. D. , & Bu, G. (2015). Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). Journal of Biological Chemistry, 290(43), 26043–26050. 10.1074/jbc.M115.679043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, C. C. , Devaux, L. B. , & Farzan, M. (2015). The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. Journal of Biological Chemistry, 290(43), 26033–26042. 10.1074/jbc.M115.677286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitsch, D. , Bock, H. H. , Engel, T. , Telgmann, R. , Müller‐Tidow, C. , Varga, G. , Bot, M. , Herz, J. , Robenek, H. , Von Eckardstein, A. , & Nofer, J. R. (2011). Apolipoprotein e induces antiinflammatory phenotype in macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology, 31(5), 1160–1168. 10.1161/ATVBAHA.111.222745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basaure, P. , Guardia‐Escote, L. , Cabré, M. , Peris‐Sampedro, F. , Sánchez‐Santed, F. , Domingo, J. L. , & Colomina, M. T. (2018). Postnatal chlorpyrifos exposure and apolipoprotein E (APOE) genotype differentially affect cholinergic expression and developmental parameters in transgenic mice. Food and Chemical Toxicology, 118, 42–52. 10.1016/j.fct.2018.04.065 [DOI] [PubMed] [Google Scholar]
- Basaure, P. , Guardia‐Escote, L. , Cabré, M. , Peris‐Sampedro, F. , Sánchez‐Santed, F. , Domingo, J. L. , & Colomina, M. T. (2019). Learning, memory and the expression of cholinergic components in mice are modulated by the pesticide chlorpyrifos depending upon age at exposure and apolipoprotein E (APOE) genotype. Archives of Toxicology, 93(3), 693–707. 10.1007/s00204-019-02387-9 [DOI] [PubMed] [Google Scholar]
- Bell, R. D. , Winkler, E. A. , Singh, I. , Sagare, A. P. , Deane, R. , Wu, Z. , Holtzman, D. M. , Betsholtz, C. , Armulik, A. , Sallstrom, J. , Berk, B. C. , & Zlokovic, B. V. (2012). Apolipoprotein e controls cerebrovascular integrity via cyclophilin a. Nature, 485(7399), 512–516. 10.1038/nature11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, R. D. , & Zlokovic, B. V. (2009). Neurovascular mechanisms and blood‐brain barrier disorder in Alzheimers disease. Acta Neuropathologica, 118(1), 103–113. 10.1007/s00401-009-0522-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloy, M. E. , Napolioni, V. , & Greicius, M. D. (2019). A quarter century of APOE and Alzheimers disease: Progress to date and the path forward. Neuron, 101(5), 820–838. 10.1016/j.neuron.2019.01.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard, J. W. , Bula, M. , Davila‐Velderrain, J. , Akay, L. A. , Zhu, L. , Frank, A. , Victor, M. B. , Bonner, J. M. , Mathys, H. , Lin, Y. T. , Ko, T. , Bennett, D. A. , Cam, H. P. , Kellis, M. , & Tsai, L. H. (2020). Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nature Medicine, 26(6), 952–963. 10.1038/s41591-020-0886-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnar, O. , Shaw, K. , Grijseels, D. M. , Clarke, D. , Bell, L. , Anderle, S. … & Hall, C. N. (2021). APOE4 genotype increases neuronal calcium signals and decreases pial arteriole responsivity and vasomotion in visual cortex of awake mice. bioRxiv. Preprint
- Bossers, K. , Wirz, K. T. S. , Meerhoff, G. F. , Essing, A. H. W. , Van Dongen, J. W. , Houba, P. , Kruse, C. G. , Verhaagen, J. , & Swaab, D. F. (2010). Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimers disease. Brain, 133(12), 3699–3723. 10.1093/brain/awq258 [DOI] [PubMed] [Google Scholar]
- Bott, J. B. , Héraud, C. , Cosquer, B. , Herbeaux, K. , Aubert, J. , Sartori, M. , Goutagny, R. , & Mathis, C. (2016). APOE‐sensitive cholinergic sprouting compensates for hippocampal dysfunctions due to reduced entorhinal input. Journal of Neuroscience, 36(40), 10472–10486. 10.1523/JNEUROSCI.1174-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyles, J. K. , Pitas, R. E. , Wilson, E. , Mahley, R. W. , & Taylor, J. M. (1985). Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. Journal of Clinical Investigation, 76(4), 1501–1513. 10.1172/JCI112130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyles, J. K. , Zoellner, C. D. , Anderson, L. J. , Kosik, L. M. , Pitas, R. E. , Weisgraber, K. H. , Hui, D. Y. , Mahley, R. W. , Gebicke‐Haerter, P. J. , Ignatius, M. J. , & Shooter, E. M. (1989). A role for apolipoprotein E, apolipoprotein A‐I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. Journal of Clinical Investigation, 83(3), 1015–1031. 10.1172/JCI113943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht, W. J. , Harris, F. M. , Chang, S. , Tesseur, I. , Yu, G. Q. , Xu, Q. , Fish, J. D. , Wyss‐Coray, T. , Buttini, M. , Mucke, L. , Mahley, R. W. , & Huang, Y. (2004). Neuron‐specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. Journal of Neuroscience, 24(10), 2527–2534. 10.1523/JNEUROSCI.4315-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett, C. L. , Wei, Y. , Donowitz, M. , & Rao, R. (2002). Human Na+/H+ exchanger isoform 6 is found in recycling endosomes of cells, not in mitochondria. American Journal of Physiology ‐ Cell Physiology, 282(5 51–5), 51–55. 10.1152/ajpcell.00420.2001 [DOI] [PubMed] [Google Scholar]
- Brodbeck, J. , Balestra, M. E. , Saunders, A. M. , Roses, A. D. , Mahley, R. W. , & Huang, Y. (2008). Rosiglitazone increases dendritic spine density and rescues spine loss caused by apolipoprotein E4 in primary cortical neurons. Proceedings of the National Academy of Sciences of the United States of America, 105(4), 1343–1346. 10.1073/pnas.0709906104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbeck, J. , McGuire, J. , Liu, Z. , Meyer‐Franke, A. , Balestra, M. E. , Jeong, D. E. , Pleiss, M. , McComas, C. , Hess, F. , Witter, D. , Peterson, S. , Childers, M. , Goulet, M. , Liverton, N. , Hargreaves, R. , Freedman, S. , Weisgraber, K. H. , Mahley, R. W. , & Huang, Y. (2011). Structure‐dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small‐molecule structure correctors. Journal of Biological Chemistry, 286(19), 17217–17226. 10.1074/jbc.M110.217380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, C. M. , Wright, E. , Colton, C. A. , Sullivan, P. M. , Laskowitz, D. T. , & Vitek, M. P. (2002). Apolipoprotein E isoform mediated regulation of nitric oxide release. Free Radical Biology and Medicine, 32(11), 1071–1075. 10.1016/S0891-5849(02)00803-1 [DOI] [PubMed] [Google Scholar]
- Bu, G. (2009). Apolipoprotein e and its receptors in Alzheimers disease: Pathways, pathogenesis and therapy. Nature Reviews Neuroscience, 10(5), 333–344. 10.1038/nrn2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussy, A. , Snider, B. J. , Coble, D. , Xiong, C. , Fagan, A. M. , Cruchaga, C. , Benzinger, T. L. S. , Gordon, B. A. , Hassenstab, J. , Bateman, R. J. , & Morris, J. C. (2019). Effect of apolipoprotein E4 on clinical, neuroimaging, and biomarker measures in noncarrier participants in the dominantly inherited Alzheimer network. Neurobiology of Aging, 75, 42–50. 10.1016/j.neurobiolaging.2018.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield, D. A. , & Mattson, M. P. (2020). Apolipoprotein E and oxidative stress in brain with relevance to Alzheimers disease. Neurobiology of Disease, 138, 104795. 10.1016/j.nbd.2020.104795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini, M. , Akeefe, H. , Lin, C. , Mahley, R. W. , Pitas, R. E. , Wyss‐Coray, T. , & Mucke, L. (2000). Dominant negative effects of apolipoprotein E4 revealed in transgenic models of neurodegenerative disease. Neuroscience, 97(2), 207–210. 10.1016/S0306-4522(00)00069-5 [DOI] [PubMed] [Google Scholar]
- Buttini, M. , Masliah, E. , Yu, G. Q. , Palop, J. J. , Chang, S. , Bernardo, A. , Lin, C. , Wyss‐Coray, T. , Huang, Y. , & Mucke, L. (2010). Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. American Journal of Pathology, 177(2), 563–569. 10.2353/ajpath.2010.090973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini, M. , Orth, M. , Bellosta, S. , Akeefe, H. , Pitas, R. E. , Wyss‐Coray, T. , Mucke, L. , & Mahley, R. W. (1999). Expression of human apolipoprotein E3 or E4 in the brains of Apoe(−/−) mice: Isoform‐specific effects on neurodegeneration. Journal of Neuroscience, 19(12), 4867–4880). Isoform‐Specific Effects on Neurodegeneration. 10.1523/jneurosci.19-12-04867.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caberlotto, L. , Marchetti, L. , Lauria, M. , Scotti, M. , & Parolo, S. (2016). Integration of transcriptomic and genomic data suggests candidate mechanisms for APOE4‐mediated pathogenic action in Alzheimers disease. Scientific Reports, 6(1), 1–10. 10.1038/srep32583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambon, K. , Davies, H. A. , & Stewart, M. G. (2000). Synaptic loss is accompanied by an increase in synaptic area in the dentate gyrus of aged human apolipoprotein E4 transgenic mice. Neuroscience, 97(4), 685–692. 10.1016/S0306-4522(00)00065-8 [DOI] [PubMed] [Google Scholar]
- Cash, J. G. , Kuhel, D. G. , Basford, J. E. , Jaeschke, A. , Chatterjee, T. K. , Weintraub, N. L. , & Hui, D. Y. (2012). Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. Journal of Biological Chemistry, 287(33), 27876–27884. 10.1074/jbc.M112.377549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castranio, E. L. , Mounier, A. , Wolfe, C. M. , Nam, K. N. , Fitz, N. F. , Letronne, F. , Schug, J. , Koldamova, R. , & Lefterov, I. (2017). Gene co‐expression networks identify Trem2 and Tyrobp as major hubs in human APOE expressing mice following traumatic brain injury. Neurobiology of Disease, 105, 1–14. 10.1016/j.nbd.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo, A. M. , Peterhoff, C. M. , Troncoso, J. C. , Gomez‐Isla, T. , Hyman, B. T. , & Nixon, R. A. (2000). Endocytic pathway abnormalities precede amyloid β deposition in sporadic alzheimers disease and down syndrome: Differential effects of APOE genotype and presenilin mutations. American Journal of Pathology, 157(1), 277–286. 10.1016/S0002-9440(10)64538-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cechetto, D. F. , Hachinski, V. , & Whitehead, S. N. (2008). Vascular risk factors and Alzheimers disease. Expert Review of Neurotherapeutics, 8(5), 743–750. 10.1586/14737175.8.5.743 [DOI] [PubMed] [Google Scholar]
- Cenini, G. , & Voos, W. (2019). Mitochondria as potential targets in Alzheimer disease therapy: An update. Frontiers in Pharmacology, 10, 902. 10.3389/fphar.2019.00902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, S. , Ma, T. R. , Miranda, R. D. , Balestra, M. E. , Mahley, R. W. , & Huang, Y. (2005). Lipid‐ and receptor‐binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America, 102(51), 18694–18699. 10.1073/pnas.0508254102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. K. , Ji, Z. S. , Dodson, S. E. , Miranda, R. D. , Rosenblum, C. I. , Reynolds, I. J. , Freedman, S. B. , Weisgraber, K. H. , Huang, Y. , & Mahley, R. W. (2011). Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for alzheimer disease. Journal of Biological Chemistry, 286(7), 5215–5221. 10.1074/jbc.M110.151084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Durakoglugil, M. S. , Xian, X. , & Herz, J. (2010). ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proceedings of the National Academy of Sciences of the United States of America, 107(26), 12011–12016. 10.1073/pnas.0914984107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbuin, N. , Leach, L. S. , Christensen, H. , & Anstey, K. J. (2007). Neuroimaging and APOE genotype: A systematic qualitative review. Dementia and Geriatric Cognitive Disorders, 24(5), 348–362. 10.1159/000109150 [DOI] [PubMed] [Google Scholar]
- Chouinard‐Watkins, R. , Vandal, M. , Léveillé, P. , Pinçon, A. , Calon, F. , & Plourde, M. (2017). Docosahexaenoic acid prevents cognitive deficits in human apolipoprotein E epsilon 4‐targeted replacement mice. Neurobiology of Aging, 57, 28–35. 10.1016/j.neurobiolaging.2017.05.003 [DOI] [PubMed] [Google Scholar]
- Chu, Q. , Diedrich, J. K. , Vaughan, J. M. , Donaldson, C. J. , Nunn, M. F. , Lee, K. F. , & Saghatelian, A. (2016). HtrA1 proteolysis of ApoE in vitro is allele selective. Journal of the American Chemical Society, 138(30), 9473–9478. 10.1021/jacs.6b03463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clare, R. , King, V. G. , Wirenfeldt, M. , & Vinters, H. V. (2010). Synapse loss in dementias. Journal of Neuroscience Research, 88(10), 2083–2090. 10.1002/jnr.22392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton, A. , Baker, L. D. , Hanson, A. , Trittschuh, E. H. , Cholerton, B. , Morgan, A. , Callaghan, M. , Arbuckle, M. , Behl, C. , & Craft, S. (2015). Long‐acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early‐stage Alzheimers disease dementia. Journal of Alzheimers Disease, 44(3), 897–906. 10.3233/JAD-141791 [DOI] [PubMed] [Google Scholar]
- Claxton, A. , Baker, L. D. , Wilkinson, C. W. , Trittschuh, E. H. , Chapman, D. , Watson, G. S. , Cholerton, B. , Plymate, S. R. , Arbuckle, M. , & Craft, S. (2013). Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or alzheimers disease. Journal of Alzheimers Disease, 35(4), 789–797. 10.3233/JAD-122308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton, C. A. , Brown, C. M. , Cook, D. , Needham, L. K. , Xu, Q. , Czapiga, M. , Saunders, A. M. , Schmechel, D. E. , Rasheed, K. , & Vitek, M. P. (2002). APOE and the regulation of microglial nitric oxide production: A link between genetic risk and oxidative stress. Neurobiology of Aging, 23(5), 777–785. 10.1016/S0197-4580(02)00016-7 [DOI] [PubMed] [Google Scholar]
- Colton, C. A. , Needham, L. K. , Brown, C. , Cook, D. , Rasheed, K. , Burke, J. R. , Strittmatter, W. J. , Schmechel, D. E. , & Vitek, M. P. (2004). APOE genotype‐specific differences in human and mouse macrophage nitric oxide production. Journal of Neuroimmunology, 147(1–2), 62–67. 10.1016/j.jneuroim.2003.10.015 [DOI] [PubMed] [Google Scholar]
- Corder, E. H. , Saunders, A. M. , Risch, N. J. , Strittmatter, W. J. , Schmechel, D. E. , Gaskell, P. C. , Rimmler, J. B. , Locke, P. A. , Conneally, P. M. , Schmader, K. E. , Small, G. W. , Roses, A. D. , Haines, J. L. , & Pericak‐Vance, M. A. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nature Genetics, 7(2), 180–184. 10.1038/ng0694-180 [DOI] [PubMed] [Google Scholar]
- Corder, E. H. , Saunders, A. M. , Strittmatter, W. J. , Schmechel, D. E. , Gaskell, P. C. , Small, G. W. , Roses, A. D. , Haines, J. L. , & Pericak‐Vance, M. A. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimers disease in late onset families. Science, 261(5123), 921–923. 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- Cudaback, E. , Yang, Y. , Montine, T. J. , & Keene, C. D. (2015). APOE genotype‐dependent modulation of astrocyte chemokine CCL3 production. Glia, 63(1), 51–65. 10.1002/glia.22732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damisah, E. C. , Rai, A. , & Grutzendler, J. (2020). TREM2: Modulator of lipid metabolism in microglia. Neuron, 105(5), 759–761. 10.1016/j.neuron.2020.02.008 [DOI] [PubMed] [Google Scholar]
- de Leeuw, S. M. , Kirschner, A. W. T. , Lindner, K. , Rust, R. , Budny, V. , Wolski, W. E. , Gavin, A. C. , Nitsch, R. M. , & Tackenberg, C. (2022). APOE2, E3, and E4 differentially modulate cellular homeostasis, cholesterol metabolism, and inflammatory response in isogenic iPSC‐derived astrocytes. Stem Cell Reports, 17(1), 110–126. 10.1016/J.STEMCR.2021.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos, R. B. , Brendza, R. P. , Heuser, J. E. , Kierson, M. , Cirrito, J. R. , Fryer, J. , Sullivan, P. M. , Fagan, A. M. , Han, X. , & Holtzman, D. M. (2001). Purification and characterization of astrocyte‐secreted apolipoprotein E and J‐containing lipoproteins from wild‐type and human apoE transgenic mice. Neurochemistry International, 39(5–6), 415–425. 10.1016/S0197-0186(01)00049-3 [DOI] [PubMed] [Google Scholar]
- DeMattos, R. B. , Curtiss, L. K. , & Williams, D. L. (1998). A minimally lipidated form of cell‐derived apolipoprotein E exhibits isoform‐specific stimulation of neurite outgrowth in the absence of exogenous lipids or lipoproteins. Journal of Biological Chemistry, 273(7), 4206–4212. 10.1074/jbc.273.7.4206 [DOI] [PubMed] [Google Scholar]
- DiBattista, A. M. , Dumanis, S. B. , Newman, J. , & Rebeck, G. W. (2016). Identification and modification of amyloid‐independent phenotypes of APOE4 mice. Experimental Neurology, 280, 97–105. 10.1016/j.expneurol.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, L. M. , & Weisgraber, K. H. (1996). Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. Journal of Biological Chemistry, 271(32), 19053–19057. 10.1074/jbc.271.32.19053 [DOI] [PubMed] [Google Scholar]
- Dong, L. M. , Wilson, C. , Wardell, M. R. , Simmons, T. , Mahley, R. W. , Weisgraber, K. H. , & Agard, D. A. (1994). Human apolipoprotein E. role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. Journal of Biological Chemistry, 269(35), 22358–22365. 10.1016/s0021-9258(17)31797-0 [DOI] [PubMed] [Google Scholar]
- Dong, Q. Y. , Li, T. R. , Jiang, X. Y. , Wang, X. N. , Han, Y. , & Jiang, J. H. (2021). Glucose metabolism in the right middle temporal gyrus could be a potential biomarker for subjective cognitive decline: A study of a Han population. Alzheimers Research and Therapy, 13(1), 74. 10.1186/s13195-021-00811-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dose, J. , Huebbe, P. , Nebel, A. , & Rimbach, G. (2016). APOE genotype and stress response ‐ a mini review. Lipids in Health and Disease, 15(1), 121. 10.1186/s12944-016-0288-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, J. , Chang, J. , Guo, S. , Zhang, Q. , & Wang, Z. (2009). ApoE 4 reduces the expression of Aβ degrading enzyme IDE by activating the NMDA receptor in hippocampal neurons. Neuroscience Letters, 464(2), 140–145. 10.1016/j.neulet.2009.07.032 [DOI] [PubMed] [Google Scholar]
- Dumanis, S. B. , Tesoriero, J. A. , Babus, L. W. , Nguyen, M. T. , Trotter, J. H. , Ladu, M. J. , Weeber, E. J. , Turner, R. S. , Xu, B. , Rebeck, G. W. , & Hoe, H. S. (2009). ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. Journal of Neuroscience, 29(48), 15317–15322. 10.1523/JNEUROSCI.4026-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egensperger, R. , Kösel, S. , Von Eitzen, U. , & Graeber, M. B. (1998). Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathology, 8(3), 439–447. 10.1111/j.1750-3639.1998.tb00166.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkin, S. R. , Lakoduk, A. M. , & Schmid, S. L. (2016). Endocytic pathways and endosomal trafficking: A primer. Wiener Medizinische Wochenschrift, 166(7–8), 196–204. 10.1007/s10354-016-0432-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom, A. K. , Snyder, J. M. , Maeda, N. , & Xia, Z. (2017). Gene‐environment interaction between lead and apolipoprotein E4 causes cognitive behavior deficits in mice. Molecular Neurodegeneration, 12(1), 1–23. 10.1186/s13024-017-0155-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, M. A. , & Banks, W. A. (2013). Blood‐brain barrier dysfunction as a cause and consequence of Alzheimers disease. Journal of Cerebral Blood Flow and Metabolism, 33(10), 1500–1513. 10.1038/jcbfm.2013.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, S. L. , Dowell, N. G. , Prowse, F. , Tabet, N. , King, S. L. , & Rusted, J. M. (2020). Mid age APOE ε4 carriers show memory‐related functional differences and disrupted structure‐function relationships in hippocampal regions. Scientific Reports, 10(1), 1–11. 10.1038/s41598-020-59272-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, W. , Xiao, N. , Zeng, G. , Bi, D. , Dai, X. , Mi, X. , Ye, Q. , Chen, X. , & Zhang, J. (2021). APOE4 genotype exacerbates the depression‐like behavior of mice during aging through ATP decline. Translational Psychiatry, 11(1), 1–9. 10.1038/s41398-021-01631-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, B. , Kluemper, J. , & Johnson, L. (2019). Apolipoprotein E4 alters astrocyte fatty acid metabolism and lipid droplet formation. Cell, 8(2), 182. 10.3390/cells8020182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, B. C. , Walsh, A. E. , Kluemper, J. C. , & Johnson, L. A. (2020). Lipid Droplets in Neurodegenerative Disorders. Frontiers in Neuroscience, 14, 742. 10.3389/fnins.2020.00742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, B. C. , Williams, H. C. , Devanney, N. A. , Piron, M. A. , Nation, G. K. , Carter, D. J. , Walsh, A. E. , Khanal, R. , Young, L. E. A. , Kluemper, J. C. , Hernandez, G. , Allenger, E. J. , Mooney, R. , Golden, L. R. , Smith, C. T. , Brandon, J. A. , Gupta, V. A. , Kern, P. A. , Gentry, M. S. , … Johnson, L. A. (2021). APOΕ4 lowers energy expenditure in females and impairs glucose oxidation by increasing flux through aerobic glycolysis. Molecular Neurodegeneration, 16(1), 1–18. 10.1186/s13024-021-00483-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer, L. A. , Cupples, L. A. , Myers, R. H. , van Duijn, C. M. , Mayeux, R. , Haines, J. L. , Kukull, W. A. , Hyman, B. , Pericak‐Vance, M. A. , & Risch, N. (1998). Effects of age and ethnicity on the link between APOE ϵ4 and Alzheimer disease—Reply. Jama, 279(8), 581‐582. 10.1001/jama.279.8.581a 9486747 [DOI] [Google Scholar]
- Feng, M. , Cui, D. , Li, Y. , Shi, J. , Xiang, L. , Bian, H. , Ma, Z. , Xia, W. , & Wei, G. (2020). Carnosic acid reverses the inhibition of ApoE4 on cell surface level of ApoER2 and Reelin signaling pathway. Journal of Alzheimers Disease, 73(2), 517–528. 10.3233/JAD-190914 [DOI] [PubMed] [Google Scholar]
- Ferguson, S. , Mouzon, B. , Kayihan, G. , Wood, M. , Poon, F. , Doore, S. , Mathura, V. , Humphrey, J. , OSteen, B. , Hayes, R. , Roses, A. , Mullan, M. , & Crawford, F. (2010). Apolipoprotein E genotype and oxidative stress response to traumatic brain injury. Neuroscience, 168(3), 811–819. 10.1016/j.neuroscience.2010.01.031 [DOI] [PubMed] [Google Scholar]
- Fessler, M. B. , & Parks, J. S. (2011). Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. The Journal of Immunology, 187(4), 1529–1535. 10.4049/jimmunol.1100253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini, N. , Ebmeier, K. P. , MacIntosh, B. J. , Trachtenberg, A. J. , Frisoni, G. B. , Wilcock, G. K. , Beckmann, C. F. , Smith, S. M. , Matthews, P. M. , & Mackay, C. E. (2011). Differential effects of the APOE genotype on brain function across the lifespan. NeuroImage, 54(1), 602–610. 10.1016/j.neuroimage.2010.08.009 [DOI] [PubMed] [Google Scholar]
- Flowers, S. A. , & Rebeck, G. W. (2020). APOE in the normal brain. In Neurobiology of disease (Vol. 136) (p. 104724). Academic Press Inc. 10.1016/j.nbd.2019.104724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forero, D. A. , López‐León, S. , González‐Giraldo, Y. , Dries, D. R. , Pereira‐Morales, A. J. , Jiménez, K. M. , & Franco‐Restrepo, J. E. (2018). APOE gene and neuropsychiatric disorders and endophenotypes: A comprehensive review. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 177(2), 126–142. 10.1002/ajmg.b.32516 [DOI] [PubMed] [Google Scholar]
- Foster, T. C. (2007). Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell, 6(3), 319–325. 10.1111/j.1474-9726.2007.00283.x [DOI] [PubMed] [Google Scholar]
- Frieden, C. , Wang, H. , & Ho, C. M. W. (2017). A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain‐domain interactions. Proceedings of the National Academy of Sciences of the United States of America, 114(24), 6292–6297. 10.1073/pnas.1705080114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster, D. G. , & Alexander, R. T. (2014). Traditional and emerging roles for the SLC9 Na+/H+ exchangers. Pflugers Archiv European Journal of Physiology, 466(1), 61–76. 10.1007/s00424-013-1408-8 [DOI] [PubMed] [Google Scholar]
- Gale, S. C. , Gao, L. , Mikacenic, C. , Coyle, S. M. , Rafaels, N. , Murray Dudenkov, T. , Madenspacher, J. H. , Draper, D. W. , Ge, W. , Aloor, J. J. , Azzam, K. M. , Lai, L. , Blackshear, P. J. , Calvano, S. E. , Barnes, K. C. , Lowry, S. F. , Corbett, S. , Wurfel, M. M. , & Fessler, M. B. (2014). APOε4 is associated with enhanced in vivo innate immune responses in human subjects. Journal of Allergy and Clinical Immunology, 134(1), 127–134.e9. 10.1016/j.jaci.2014.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes, L. U. , Jeune, B. , Ranberg, K. A. , Nybo, H. , & Vaupel, J. W. (2000). Estimation of apolipoprotein E genotype‐specific relative mortality risks from the distribution of genotypes in centenarians and middle‐aged men: Apolipoprotein E gene is a “frailty gene,” not a “longevity gene.”. Genetic Epidemiology, 19(3), 202–210. [DOI] [PubMed] [Google Scholar]
- Geyeregger, R. , Zeyda, M. , & Stulnig, T. M. (2006). Liver X receptors in cardiovascular and metabolic disease. Cellular and Molecular Life Sciences, 63(5), 524–539. 10.1007/s00018-005-5398-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, J. L. , Brown, M. S. , Anderson, R. G. , Russell, D. W. , & Schneider, W. J. (1985). Receptor‐mediated endocytosis: Concepts emerging from the LDL receptor system. Annual Review of Cell Biology, 1, 1–39. 10.1146/annurev.cb.01.110185.000245 [DOI] [PubMed] [Google Scholar]
- Gong, J. S. , Morita, S. Y. , Kobayashi, M. , Handa, T. , Fujita, S. C. , Yanagisawa, K. , & Michikawa, M. (2007). Novel action of apolipoprotein e (ApoE): ApoE isoform specifically inhibits lipid‐particle‐mediated cholesterol release from neurons. Molecular Neurodegeneration, 2(1), 1–11. 10.1186/1750-1326-2-9/FIGURES/7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger, D. J. , Reckless, J. , & McKilligin, E. (2004). Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic Proinflammatory state in apolipoprotein E‐deficient mice. The Journal of Immunology, 173(10), 6366–6375. 10.4049/jimmunol.173.10.6366 [DOI] [PubMed] [Google Scholar]
- Grammas, P. (2011). Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimers disease. Journal of Neuroinflammation, 8(1), 1–12. 10.1186/1742-2094-8-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grehan, S. , Tse, E. , & Taylor, J. M. (2001). Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. Journal of Neuroscience, 21(3), 812–822. 10.1523/jneurosci.21-03-00812.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grouselle, D. , Winsky‐Sommerer, R. , David, J. P. , Delacourte, A. , Dournaud, P. , & Epelbaum, J. (1998). Loss of somatostatin‐like immunoreactivity in the frontal cortex of Alzheimer patients carrying the apolipoprotein epsilon 4 allele. Neuroscience Letters, 255(1), 21–24. 10.1016/S0304-3940(98)00698-3 [DOI] [PubMed] [Google Scholar]
- Guan, P. P. , Cao, L. L. , & Wang, P. (2021). Elevating the levels of calcium ions exacerbate alzheimers disease via inducing the production and aggregation of β‐amyloid protein and phosphorylated tau. International Journal of Molecular Sciences, 22(11), 5900. 10.3390/ijms22115900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureje, O. , Ogunniyi, A. , Baiyewu, O. , Price, B. , Unverzagt, F. W. , Evans, R. M. , Smith‐Gamble, V. , Lane, K. A. , Gao, S. , Hall, K. S. , Hendrie, H. C. , & Murrell, J. R. (2006). APOE ε4 is not associated with Alzheimers disease in elderly Nigerians. Annals of Neurology, 59(1), 182–185. 10.1002/ana.20694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, C. N. , Reynell, C. , Gesslein, B. , Hamilton, N. B. , Mishra, A. , Sutherland, B. A. , Oâ Farrell, F. M. , Buchan, A. M. , Lauritzen, M. , & Attwell, D. (2014). Capillary pericytes regulate cerebral blood flow in health and disease. Nature, 508(1), 55–60. 10.1038/nature13165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, S. D. , & Bondi, M. W. (2008). Revision of the apolipoprotein E compensatory mechanism recruitment hypothesis. Alzheimers and Dementia, 4(4), 251–254. 10.1016/j.jalz.2008.02.006 [DOI] [PubMed] [Google Scholar]
- Han, S. D. , & Tuminello, E. R. (2011). The apolipoprotein e antagonistic pleiotropy hypothesis: Review and recommendations. International Journal of Alzheimers Disease, 2011, 726197. 10.4061/2011/726197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, A. J. , Bayer‐Carter, J. L. , Green, P. S. , Montine, T. J. , Wilkinson, C. W. , Baker, L. D. , Watson, G. S. , Bonner, L. M. , Callaghan, M. , Leverenz, J. B. , Tsai, E. , Postupna, N. , Zhang, J. , Lampe, J. , & Craft, S. (2013). Effect of apolipoprotein e genotype and diet on apolipoprotein e lipidation and amyloid peptides randomized clinical trial. JAMA Neurology, 70(8), 972–980. 10.1001/jamaneurol.2013.396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, F. M. , Brecht, W. J. , Xu, Q. , Mahley, R. W. , & Huang, Y. (2004). Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal‐regulated kinase. Modulation by Zinc. Journal of Biological Chemistry, 279(43), 44795–44801. 10.1074/jbc. M408127200 [DOI] [PubMed] [Google Scholar]
- Harris, F. M. , Brecht, W. J. , Xu, Q. , Tesseur, I. , Kekonius, L. , Wyss‐Coray, T. , Fish, J. D. , Masliah, E. , Hopkins, P. C. , Scearce‐Levie, K. , Weisgraber, K. H. , Mucke, L. , Mahley, R. W. , & Huang, Y. (2003). Carboxyl‐terminal‐truncated apolipoprotein E4 causes Alzheimers disease‐like neurodegeneration and behavioral deficits in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America, 100(19), 10966–10971. 10.1073/pnas.1434398100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, F. M. , Tesseur, I. , Brecht, W. J. , Xu, Q. , Mullendorff, K. , Chang, S. , Wyss‐Coray, T. , Mahley, R. W. , & Huang, Y. (2004). Astroglial regulation of apolipoprotein E expression in neuronal cells: Implications for Alzheimers disease. Journal of Biological Chemistry, 279(5), 3862–3868. 10.1074/jbc. M309475200 [DOI] [PubMed] [Google Scholar]
- Hatters, D. M. , Peters‐Libeu, C. A. , & Weisgraber, K. H. (2006). Apolipoprotein E structure: Insights into function. Trends in Biochemical Sciences, 31(8), 445–454. 10.1016/j.tibs.2006.06.008 [DOI] [PubMed] [Google Scholar]
- Hawkes, C. A. , Sullivan, P. M. , Hands, S. , Weller, R. O. , Nicoll, J. A. R. , & Carare, R. O. (2012). Disruption of arterial perivascular drainage of amyloid‐β from the brains of mice expressing the human APOE ε4 allele. PLoS ONE, 7(7), e41636. 10.1371/journal.pone.0041636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayek, T. , Oiknine, J. , Brook, J. G. , & Aviram, M. (1994). Increased plasma and lipoprotein lipid peroxidation in apo E‐deficient mice. Biochemical and Biophysical Research Communications, 201(3), 1567–1574. 10.1006/BBRC.1994.1883 [DOI] [PubMed] [Google Scholar]
- Heinsinger, N. M. , Gachechiladze, M. A. , & Rebeck, G. W. (2016). Apolipoprotein e genotype affects size of ApoE complexes in cerebrospinal fluid. Journal of Neuropathology and Experimental Neurology, 75(10), 918–924. 10.1093/jnen/nlw067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise, V. , Filippini, N. , Ebmeier, K. P. , & Mackay, C. E. (2011). The APOE ɛ4 allele modulates brain white matter integrity in healthy adults. Molecular Psychiatry, 16(9), 908–916. 10.1038/mp.2010.90 [DOI] [PubMed] [Google Scholar]
- Hesse, R. , Hurtado, M. L. , Jackson, R. J. , Eaton, S. L. , Herrmann, A. G. , Colom‐Cadena, M. , Tzioras, M. , King, D. , Rose, J. , Tulloch, J. , McKenzie, C. A. , Smith, C. , Henstridge, C. M. , Lamont, D. , Wishart, T. M. , & Spires‐Jones, T. L. (2019). Comparative profiling of the synaptic proteome from Alzheimers disease patients with focus on the APOE genotype. Acta Neuropathologica Communications, 7(1), 1–18. 10.1186/s40478-019-0847-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota, Y. , Kubo, K. i. , Katayama, K. i. , Honda, T. , Fujino, T. , Yamamoto, T. T. , & Nakajima, K. (2015). Reelin receptors ApoER2 and VLDLR are expressed in distinct spatiotemporal patterns in developing mouse cerebral cortex. Journal of Comparative Neurology, 523(3), 463–478. 10.1002/cne.23691 [DOI] [PubMed] [Google Scholar]
- Hoe, H. S. , Harris, D. C. , & Rebeck, G. W. (2005). Multiple pathways of apolipoprotein E signaling in primary neurons. Journal of Neurochemistry, 93(1), 145–155. 10.1111/j.1471-4159.2004.03007.x [DOI] [PubMed] [Google Scholar]
- Holtzman, D. M. , Pitas, R. E. , Kilbridge, J. , Nathan, B. , Mahley, R. W. , Bu, G. , & Schwartz, A. L. (1995). Low density lipoprotein receptor‐related protein mediates apolipoprotein E‐dependent neurite outgrowth in a central nervous system‐derived neuronal cell line. Proceedings of the National Academy of Sciences of the United States of America, 92(21), 9480–9484. 10.1073/pnas.92.21.9480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth, C. , Mishra, A. , & Hall, C. N. (2021). More than just summed neuronal activity: How multiple cell types shape the BOLD response. Philosophical Transactions of the Royal Society B: Biological Sciences, 376(1815), 20190630. 10.1098/rstb.2019.0630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, J. , Liu, C. C. , Chen, X. F. , Zhang, Y. W. , Xu, H. , & Bu, G. (2015). Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Aβ metabolism in apoE4‐targeted replacement mice. Molecular Neurodegeneration, 10(1), 6. 10.1186/s13024-015-0001-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. (2010). Aβ‐independent roles of apolipoprotein E4 in the pathogenesis of Alzheimers disease. Trends in Molecular Medicine, 16(6), 287–294. 10.1016/j.molmed.2010.04.004 [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Liu, X. Q. , Wyss‐Coray, T. , Brecht, W. J. , Sanan, D. A. , & Mahley, R. W. (2001). Apolipoprotein E fragments present in Alzheimers disease brains induce neurofibrillary tangle‐like intracellular inclusions in neurons. Proceedings of the National Academy of Sciences of the United States of America, 98(15), 8838–8843. 10.1073/pnas.151254698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. , & Mahley, R. W. (2014). Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimers diseases. Neurobiology of Disease, 72(Part A), 3–12. 10.1016/j.nbd.2014.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. W. A. , Zhou, B. , Nabet, A. M. , Wernig, M. , & Südhof, T. C. (2019). Differential signaling mediated by ApoE2, ApoE3, and ApoE4 in human neurons parallels Alzheimers disease risk. Journal of Neuroscience, 39(37), 7408–7427. 10.1523/JNEUROSCI.2994-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. W. A. , Zhou, B. , Wernig, M. , & Südhof, T. C. (2017). ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell, 168(3), 427–441, e21. 10.1016/j.cell.2016.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Z. , Wong, L. W. , Su, Y. , Huang, X. , Wang, N. , Chen, H. , & Yi, C. (2020). Blood‐brain barrier integrity in the pathogenesis of Alzheimers disease. Frontiers in Neuroendocrinology, 59, 100857. 10.1016/j.yfrne.2020.100857 [DOI] [PubMed] [Google Scholar]
- Hubin, E. , Verghese, P. B. , van Nuland, N. , & Broersen, K. (2019). Apolipoprotein E associated with reconstituted high‐density lipoprotein‐like particles is protected from aggregation. FEBS Letters, 593(11), 1144–1153. 10.1002/1873-3468.13428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulse, R. E. , Ralat, L. A. , & Wei‐Jen, T. (2009). Chapter 22 structure, function, and regulation of insulin‐degrading enzyme. Vitamins and Hormones, 80(C), 635–648. 10.1016/S0083-6729(08)00622-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola, C. (2017). The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron, 96(1), 17–42. 10.1016/j.neuron.2017.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi, Y. , Eid, L. , Parent, M. , Soucy, G. , Bareil, C. , Riku, Y. , Kawai, K. , Takagi, S. , Yoshida, M. , Katsuno, M. , Sobue, G. , & Julien, J. P. (2016). Exosome secretion is a key pathway for clearance of pathological TDP‐43. Brain, 139(12), 3187–3201. 10.1093/brain/aww237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, A. , Hong, C. , Rong, X. , Zhu, X. , Tarling, E. J. , Hedde, P. N. , Gratton, E. , Parks, J. , & Tontonoz, P. (2015). LXRs link metabolism to inflammation through Abca1‐dependent regulation of membrane composition and TLR signaling. eLife, 4, e08009. 10.7554/eLife.08009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturria‐Medina, Y. , Sotero, R. C. , Toussaint, P. J. , Mateos‐Pérez, J. M. , Evans, A. C. , Weiner, M. W. , Aisen, P. , Petersen, R. , Jack, C. R. , Jagust, W. , Trojanowki, J. Q. , Toga, A. W. , Beckett, L. , Green, R. C. , Saykin, A. J. , Morris, J. , Shaw, L. M. , Khachaturian, Z. , Sorensen, G. , … Furst, A. J. (2016). Early role of vascular dysregulation on late‐onset Alzheimers disease based on multifactorial data‐driven analysis. Nature Communications, 7(1), 1–14. 10.1038/ncomms11934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust, W. J. , & Landau, S. M. (2012). Apolipoprotein E, not fibrillar β‐amyloid, reduces cerebral glucose metabolism in normal aging. Journal of Neuroscience, 32(50), 18227–18233. 10.1523/JNEUROSCI.3266-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, S. , Yoon, S. Y. , Leung, L. , Knoferle, J. , & Huang, Y. (2013). Cellular source‐specific effects of apolipoprotein (Apo) E4 on dendrite Arborization and dendritic spine development. PLoS ONE, 8(3), e59478. 10.1371/journal.pone.0059478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Y. , Gong, Y. , Gan, W. , Beach, T. , Holtzman, D. M. , & Wisniewski, T. (2003). Apolipoprotein E isoform‐specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimers disease patients. Neuroscience, 122(2), 305–315. 10.1016/j.neuroscience.2003.08.007 [DOI] [PubMed] [Google Scholar]
- Jiang, C. , Stewart, L. T. , Kuo, H. C. , McGilberry, W. , Wall, S. B. , Liang, B. , van Groen, T. , Bailey, S. M. , Kim, Y. i. , Tipple, T. E. , Jones, D. P. , McMahon, L. L. , & Liu, R. M. (2019). Cyclic O3 exposure synergizes with aging leading to memory impairment in male APOE ε3, but not APOE ε4, targeted replacement mice. Neurobiology of Aging, 81, 9–21. 10.1016/j.neurobiolaging.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, L. , Zhong, J. , Dou, X. , Cheng, C. , Huang, Z. , & Sun, X. (2015). Effects of ApoE on intracellular calcium levels and apoptosis of neurons after mechanical injury. Neuroscience, 301, 375–383. 10.1016/j.neuroscience.2015.06.005 [DOI] [PubMed] [Google Scholar]
- Jofre‐Monseny, L. , de Pascual‐Teresa, S. , Plonka, E. , Huebbe, P. , Boesch‐Saadatmandi, C. , Minihane, A. M. , & Rimbach, G. (2007). Differential effects of apolipoprotein E3 and E4 on markers of oxidative status in macrophages. British Journal of Nutrition, 97(5), 864–871. 10.1017/S0007114507669219 [DOI] [PubMed] [Google Scholar]
- Jofre‐Monseny, L. , Minihane, A. M. , & Rimbach, G. (2008). Impact of apoE genotype on oxidative stress, inflammation and disease risk. Molecular Nutrition and Food Research, 52(1), 131–145. 10.1002/mnfr.200700322 [DOI] [PubMed] [Google Scholar]
- Johnson, L. A. , Torres, E. R. , Weber Boutros, S. , Patel, E. , Akinyeke, T. , Alkayed, N. J. , & Raber, J. (2019). Apolipoprotein E4 mediates insulin resistance‐associated cerebrovascular dysfunction and the post‐prandial response. Journal of Cerebral Blood Flow and Metabolism, 39(5), 770–781. 10.1177/0271678X17746186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt, C. , Leininger‐Muller, B. , Bertrand, P. , Herber, R. , Christen, Y. , & Siest, G. (2000). Differential oxidation of apolipoprotein E isoforms and interaction with phospholipids. Free Radical Biology and Medicine, 28(1), 129–140. 10.1016/S0891-5849(99)00232-4 [DOI] [PubMed] [Google Scholar]
- Jones, E. A. , Gillespie, A. K. , Yoon, S. Y. , Frank, L. M. , & Huang, Y. (2019). Early hippocampal sharp‐wave ripple deficits predict later learning and memory impairments in an Alzheimers disease mouse model. Cell Reports, 29(8), 2123, e4–2133. 10.1016/J.CELREP.2019.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, N. S. , Watson, K. Q. , & Rebeck, G. W. (2021). High‐fat diet increases gliosis and immediate early gene expression in APOE 3 mice, but not APOE4 mice. Journal of Neuroinflammation, 18(1), 1–13. 10.1186/s12974-021-02256-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovic, M. , Sharma, M. , Rahajeng, J. , & Caplan, S. (2010). The early endosome: a busy sorting station for proteins at the crossroads. Histology and Histopathology, 25(1), 99–112. 10.14670/HH-25.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo, T. , Xu, H. , & Bu, G. (2014). ApoE and Aβ in Alzheimers disease: Accidental encounters or partners? Neuron, 81(4), 740–754. 10.1016/j.neuron.2014.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassam, I. , Gagnon, F. , & Cusimano, M. D. (2016). Association of the APOE‐ϵ4 allele with outcome of traumatic brain injury in children and youth: A meta‐analysis and meta‐regression. Journal of Neurology, Neurosurgery and Psychiatry, 87(4), 433–440. 10.1136/jnnp-2015-310500 [DOI] [PubMed] [Google Scholar]
- Keeney, J. T. R. , Ibrahimi, S. , & Zhao, L. (2015). Human ApoE isoforms differentially modulate glucose and amyloid metabolic pathways in female brain: Evidence of the mechanism of neuroprotection by ApoE2 and implications for Alzheimers disease prevention and early intervention. Journal of Alzheimers Disease, 48(2), 411–424. 10.3233/JAD-150348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren‐Shaul, H. , Spinrad, A. , Weiner, A. , Matcovitch‐Natan, O. , Dvir‐Szternfeld, R. , Ulland, T. K. , David, E. , Baruch, K. , Lara‐Astaiso, D. , Toth, B. , Itzkovitz, S. , Colonna, M. , Schwartz, M. , & Amit, I. (2017). A unique microglia type associated with restricting development of Alzheimers disease. Cell, 169(7), 1276, e17–1290. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Kharrazi, H. , Vaisi‐Raygani, A. , Rahimi, Z. , Tavilani, H. , Aminian, M. , & Pourmotabbed, T. (2008). Association between enzymatic and non‐enzymatic antioxidant defense mechanism with apolipoprotein E genotypes in Alzheimer disease. Clinical Biochemistry, 41(12), 932–936. 10.1016/j.clinbiochem.2008.05.001 [DOI] [PubMed] [Google Scholar]
- Klein, R. C. , Acheson, S. K. , Mace, B. E. , Sullivan, P. M. , & Moore, S. D. (2014). Altered neurotransmission in the lateral amygdala in aged human apoE4 targeted replacement mice. Neurobiology of Aging, 35(9), 2046–2052. 10.1016/j.neurobiolaging.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloske, C. M. , & Wilcock, D. M. (2020). The important interface between apolipoprotein E and neuroinflammation in Alzheimers disease. Frontiers in Immunology, 11, 754. 10.3389/fimmu.2020.00754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoferle, J. , Yoon, S. Y. , Walker, D. , Leung, L. , Gillespie, A. K. , Tong, L. M. , Bien‐Ly, N. , & Huang, Y. (2014). Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. Journal of Neuroscience, 34(42), 14069–14078. 10.1523/JNEUROSCI.2281-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouff, C. , Hinsdale, M. E. , Mezdour, H. , Altenburg, M. K. , Watanabe, M. , Quarfordt, S. H. , Sullivan, P. M. , & Maeda, N. (1999). Apo E structure determines VLDL clearance and atherosclerosis risk in mice. Journal of Clinical Investigation, 103(11), 1579–1586. 10.1172/JCI6172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie, R. M. , Hashimoto, T. , Tai, H. C. , Kay, K. R. , Serrano‐Pozo, A. , Joyner, D. , Hou, S. , Kopeikina, K. J. , Frosch, M. P. , Lee, V. M. , Holtzman, D. M. , Hyman, B. T. , & Spires‐Jones, T. L. (2012). Apolipoprotein E4 effects in Alzheimers disease are mediated by synaptotoxic oligomeric amyloid‐β. Brain, 135(7), 2155–2168. 10.1093/brain/aws127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi, K. , Hattori, Y. , Ahn, S. J. , Buendia, I. , Ciacciarelli, A. , Uekawa, K. , Wang, G. , Hiller, A. , Zhao, L. , Voss, H. U. , Paul, S. M. , Schaffer, C. , Park, L. , & Iadecola, C. (2018). Apoε4 disrupts neurovascular regulation and undermines white matter integrity and cognitive function. Nature Communications, 9(1), 3816. 10.1038/s41467-018-06301-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konings, S. C. , Torres‐Garcia, L. , Martinsson, I. , & Gouras, G. K. (2021). Astrocytic and neuronal apolipoprotein E isoforms differentially affect neuronal excitability. Frontiers in Neuroscience, 15. 10.3389/fnins.2021.734001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korwek, K. M. , Trotter, J. H. , Ladu, M. J. , Sullivan, P. M. , & Weeber, E. J. (2009). Apoe isoform‐dependent changes in hippocampal synaptic function. Molecular Neurodegeneration, 4(1), 1–18. 10.1186/1750-1326-4-21/FIGURES/10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutseff, A. , Mittelhaeuser, C. , Essabri, K. , Auwerx, J. , & Meziane, H. (2014). Impact of the apolipoprotein e polymorphism, age and sex on neurogenesis in mice: Pathophysiological relevance for Alzheimers disease? Brain Research, 1542, 32–40. 10.1016/j.brainres.2013.10.003 [DOI] [PubMed] [Google Scholar]
- Krasemann, S. , Madore, C. , Cialic, R. , Baufeld, C. , Calcagno, N. , El Fatimy, R. , Beckers, L. , OLoughlin, E. , Xu, Y. , Fanek, Z. , Greco, D. J. , Smith, S. T. , Tweet, G. , Humulock, Z. , Zrzavy, T. , Conde‐Sanroman, P. , Gacias, M. , Weng, Z. , Chen, H. , … Butovsky, O. (2017). The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity, 47(3), 566, e9–581. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurucu, H. , Colom‐Cadena, M. , Davies, C. , Wilkins, L. , King, D. , Rose, J. , Tzioras, M. , Tulloch, J. H. , Smith, C. , & Spires‐Jones, T. L. (2021). Inhibitory synapse loss and accumulation of amyloid beta in inhibitory presynaptic terminals in Alzheimers disease. European Journal of Neurology, 00, 1–13. 10.1111/ene.15043 [DOI] [PubMed] [Google Scholar]
- Lahoz, C. , Schaefer, E. J. , Cupples, L. A. , Wilson, P. W. F. , Levy, D. , Osgood, D. , Parpos, S. , Pedro‐Botet, J. , Daly, J. A. , & Ordovas, J. M. (2001). Apolipoprotein E genotype and cardiovascular disease in the Framingham heart study. Atherosclerosis, 154(3), 529–537. 10.1016/S0021-9150(00)00570-0 [DOI] [PubMed] [Google Scholar]
- Lane‐Donovan, C. , & Herz, J. (2017). ApoE, ApoE receptors, and the synapse in Alzheimers disease. Trends in Endocrinology and Metabolism, 28(4), 273–284. 10.1016/j.tem.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane‐Donovan, C. , Philips, G. T. , & Herz, J. (2014). More than cholesterol transporters: Lipoprotein receptors in CNS function and neurodegeneration. In Neuron (Vol. 83, Issue 4, pp. 771–787). Cell Press. 10.1016/j.neuron.2014.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranco, M. F. , Ng, C. A. , & Rebeck, G. W. (2020). ApoE lipidation as a therapeutic target in Alzheimers disease. International Journal of Molecular Sciences, 21(17), 1–19. 10.3390/ijms21176336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranco, M. F. , Sepulveda, J. , Kopetsky, G. , & Rebeck, G. W. (2021). Expression and secretion of apoE isoforms in astrocytes and microglia during inflammation. Glia, 69(6), 1478–1493. 10.1002/glia.23974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larramona‐Arcas, R. , González‐Arias, C. , Perea, G. , Gutiérrez, A. , Vitorica, J. , García‐Barrera, T. , Gómez‐Ariza, J. L. , Pascua‐Maestro, R. , Ganfornina, M. D. , Kara, E. , Hudry, E. , Martinez‐Vicente, M. , Vila, M. , Galea, E. , & Masgrau, R. (2020). Sex‐dependent calcium hyperactivity due to lysosomal‐related dysfunction in astrocytes from APOE4 versus APOE3 gene targeted replacement mice. Molecular Neurodegeneration, 15(1), 1–23. 10.1186/s13024-020-00382-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowitz, D. T. , Thekdi, A. D. , Thekdi, S. D. , Han, S. K. D. , Myers, J. K. , Pizzo, S. V. , & Bennett, E. R. (2001). Downregulation of microglial activation by apolipoprotein E and apoE‐mimetic peptides. Experimental Neurology, 167(1), 74–85. 10.1006/exnr.2001.7541 [DOI] [PubMed] [Google Scholar]
- Lawrence, D. W. , Comper, P. , Hutchison, M. G. , & Sharma, B. (2015). The role of apolipoprotein E episilon (ε)‐4 allele on outcome following traumatic brain injury: A systematic review. Brain Injury, 29(9), 1018–1031. 10.3109/02699052.2015.1005131 [DOI] [PubMed] [Google Scholar]
- LeBlanc, A. (2005). The role of apoptotic pathways in Alzheimers disease neurodegeneration and cell death. Current Alzheimer Research, 2(4), 389–402. 10.2174/156720505774330573 [DOI] [PubMed] [Google Scholar]
- Lee, S. I. , Jeong, W. , Lim, H. , Cho, S. , Lee, H. , Jang, Y. , Cho, J. , Bae, S. , Lin, Y. T. , Tsai, L. H. , Moon, D. W. , & Seo, J. (2021). APOE4‐carrying human astrocytes oversupply cholesterol to promote neuronal lipid raft expansion and Aβ generation. Stem Cell Reports, 16(9), 2128–2137. 10.1016/j.stemcr.2021.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, L. , Andrews‐Zwilling, Y. , Yoon, S. Y. , Jain, S. , Ring, K. , Dai, J. , Wang, M. M. , Tong, L. , Walker, D. , & Huang, Y. (2012). Apolipoprotein E4 causes age‐ and sex‐dependent impairments of hilar GABAergic interneurons and learning and memory deficits in mice. PLoS ONE, 7(12), e53569. 10.1371/journal.pone.0053569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, R. L. , Berlett, B. S. , Moskovitz, J. , Mosoni, L. , & Stadtman, E. R. (1999). Methionine residues may protect proteins from critical oxidative damage. Mechanisms of Ageing and Development, 107(3), 323–332. 10.1016/S0047-6374(98)00152-3 [DOI] [PubMed] [Google Scholar]
- Lewandowski, C. T. , Maldonado Weng, J. , & LaDu, M. J. (2020). Alzheimers disease pathology in APOE transgenic mouse models: The who, what, when, where, why, and how. Neurobiology of Disease, 139, 104811. 10.1016/j.nbd.2020.104811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. , Bien‐Ly, N. , Andrews‐Zwilling, Y. , Xu, Q. , Bernardo, A. , Ring, K. , Halabisky, B. , Deng, C. , Mahley, R. W. , & Huang, Y. (2009). GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 Knockin mice. Cell Stem Cell, 5(6), 634–645. 10.1016/j.stem.2009.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Zhang, J. , Li, D. , He, C. , He, K. , Xue, T. , Wan, L. , Zhang, C. , & Liu, Q. (2021). Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone‐acetylation‐mediated memory. Neuron, 109(6), 957, e8–970. 10.1016/j.neuron.2021.01.005 [DOI] [PubMed] [Google Scholar]
- Liang, Y. , Lin, S. , Beyer, T. P. , Zhang, Y. , Wu, X. , Bales, K. R. , DeMattos, R. B. , May, P. C. , Li, S. D. , Jiang, X. C. , Eacho, P. I. , Cao, G. , & Paul, S. M. (2004). A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. Journal of Neurochemistry, 88(3), 623–634. 10.1111/j.1471-4159.2004.02183.x [DOI] [PubMed] [Google Scholar]
- Lin, L. Y. , Zhang, J. , Dai, X. M. , Xiao, N. A. , Wu, X. L. , Wei, Z. , Fang, W. T. , Zhu, Y. G. , & Chen, X. C. (2016). Early‐life stress leads to impaired spatial learning and memory in middle‐aged ApoE4‐TR mice. Molecular Neurodegeneration, 11(1), 1–16. 10.1186/s13024-016-0107-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. T. , Seo, J. , Gao, F. , Feldman, H. M. , Wen, H. L. , Penney, J. , Cam, H. P. , Gjoneska, E. , Raja, W. K. , Cheng, J. , Rueda, R. , Kritskiy, O. , Abdurrob, F. , Peng, Z. , Milo, B. , Yu, C. J. , Elmsaouri, S. , Dey, D. , Ko, T. , … Tsai, L. H. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimers disease phenotypes in human iPSC‐derived brain cell types. Neuron, 98(6), 1141, e7–1154. 10.1016/j.neuron.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, A. L. , Jahrling, J. B. , Zhang, W. , DeRosa, N. , Bakshi, V. , Romero, P. , Galvan, V. , & Richardson, A. (2017). Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre‐symptomatic Alzheimer's disease. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 37(1), 217–226. 10.1177/0271678X15621575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. C. , Kanekiyo, T. , Xu, H. , & Bu, G. (2013). Apolipoprotein e and Alzheimer disease: Risk, mechanisms and therapy. Nature Reviews Neurology, 9(2), 106–118. 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D. S. , Pan, X. D. , Zhang, J. , Shen, H. , Collins, N. C. , Cole, A. M. , Koster, K. P. , Ben Aissa, M. , Dai, X. M. , Zhou, M. , Tai, L. M. , Zhu, Y. G. , Ladu, M. J. , & Chen, X. C. (2015). APOE4 enhances age‐dependent decline in cognitive function by down‐regulating an NMDA receptor pathway in EFAD‐Tg mice. Molecular Neurodegeneration, 10(1), 1–17. 10.1186/s13024-015-0002-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, J. R. , Tang, W. , Wang, H. , Vitek, M. P. , Bennett, E. R. , Sullivan, P. M. , Warner, D. S. , & Laskowitz, D. T. (2003). APOE genotype and an ApoE‐mimetic peptide modify the systemic and central nervous system inflammatory response. Journal of Biological Chemistry, 278(49), 48529–48533. 10.1074/jbc.M306923200 [DOI] [PubMed] [Google Scholar]
- Lynch, J. R. , Wang, H. , Mace, B. , Leinenweber, S. , Warner, D. S. , Bennett, E. R. , Vitek, M. P. , McKenna, S. , & Laskowitz, D. T. (2005). A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Experimental Neurology, 192(1), 109–116. 10.1016/j.expneurol.2004.11.014 [DOI] [PubMed] [Google Scholar]
- Maezawa, I. , Maeda, N. , Montine, T. J. , & Montine, K. S. (2006). Apolipoprotein E‐specific innate immune response in astrocytes from targeted replacement mice. Journal of Neuroinflammation, 3(1), 10. 10.1186/1742-2094-3-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa, I. , Nivison, M. , Montine, K. S. , Maeda, N. , & Montine, T. J. (2006). Neurotoxicity from innate immune response is greatest with targeted replacement of ε4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. The FASEB Journal, 20(6), 797–799. 10.1096/fj.05-5423fje [DOI] [PubMed] [Google Scholar]
- Maezawa, I. , Zaja‐Milatovic, S. , Milatovic, D. , Stephen, C. , Sokal, I. , Maeda, N. , Montine, T. J. , & Montine, K. S. (2006). Apolipoprotein E isoform‐dependent dendritic recovery of hippocampal neurons following activation of innate immunity. Journal of Neuroinflammation, 3(1), 21. 10.1186/1742-2094-3-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magi, S. , Castaldo, P. , MacRi, M. L. , Maiolino, M. , Matteucci, A. , Bastioli, G. , Gratteri, S. , Amoroso, S. , & Lariccia, V. (2016). Intracellular calcium dysregulation: Implications for Alzheimers disease. BioMed Research International, 2016, 1–14. 10.1155/2016/6701324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley, R. W. (1988). Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science, 240(4852), 622–630. 10.1126/science.3283935 [DOI] [PubMed] [Google Scholar]
- Mahley, R. W. (2016). Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arteriosclerosis, Thrombosis, and Vascular Biology, 36(7), 1305–1315. 10.1161/ATVBAHA.116.307023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley, R. W. , & Huang, Y. (1999). Apolipoprotein E: From atherosclerosis to Alzheimers disease and beyond. Current Opinion in Lipidology, 10(3), 207–217. 10.1097/00041433-199906000-00003 [DOI] [PubMed] [Google Scholar]
- Mahley, R. W. , & Huang, Y. (2012). Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron, 76(5), 871–885. 10.1016/j.neuron.2012.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley, R. W. , & Rall, S. C. (2000). Apolipoprotein E: Far more than a lipid transport protein. Annual Review of Genomics and Human Genetics, 1(2000), 507–537. 10.1146/annurev.genom.1.1.507 [DOI] [PubMed] [Google Scholar]
- Mahley, R. W. , Weisgraber, K. H. , & Huang, Y. (2009). Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimers disease to AIDS. Journal of Lipid Research, 50 (SUPPL.), S183–S188. 10.1194/jlr. R800069‐JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main, B. S. , Villapol, S. , Sloley, S. S. , Barton, D. J. , Parsadanian, M. , Agbaegbu, C. , Stefos, K. , McCann, M. S. , Washington, P. M. , Rodriguez, O. C. , & Burns, M. P. (2018). Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Molecular Neurodegeneration, 13(1), 1–18. 10.1186/s13024-018-0249-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maioli, S. , Puerta, E. , Merino‐Serrais, P. , Fusari, L. , Gil‐Bea, F. , Rimondini, R. , & Cedazo‐Minguez, A. (2012). Combination of apolipoprotein E4 and high carbohydrate diet reduces hippocampal BDNF and arc levels and impairs memory in young mice. Journal of Alzheimers Disease, 32(2), 341–355. 10.3233/JAD-2012-120697 [DOI] [PubMed] [Google Scholar]
- Majacek, R. A. , Castle, C. K. , Goodman, L. V. , Weisgraber, K. H. , Mahley, R. W. , Shooter, E. M. , & Gebicke‐Haerter, P. J. (1988). Expression of apolipoprotein E by cultured vascular smooth muscle cells is controlled by growth state. Journal of Cell Biology, 107(3), 1207–1213. 10.1083/jcb.107.3.1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkar, S. R. , Lipnicki, D. M. , Crawford, J. D. , Kochan, N. A. , Castro‐Costa, E. , Lima‐Costa, M. F. , Diniz, B. S. , Brayne, C. , Stephan, B. , Matthews, F. , Llibre‐Rodriguez, J. J. , Llibre‐Guerra, J. J. , Valhuerdi‐Cepero, A. J. , Lipton, R. B. , Katz, M. J. , Wang, C. , Ritchie, K. , Carles, S. , Carriere, I. , & Sachdev, P. (2020). APOE ε4 and the influence of sex, age, vascular risk factors, and ethnicity on cognitive decline. Journals of Gerontology ‐ Series a Biological Sciences and Medical Sciences, 75(10), 1863–1873. 10.1093/gerona/glaa116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Martínez, A. B. , Torres‐Perez, E. , Devanney, N. , Del Moral, R. , Johnson, L. A. , & Arbones‐Mainar, J. M. (2020). Beyond the CNS: The many peripheral roles of APOE. In Neurobiology of disease (Vol. 138) (p. 104809). Academic Press Inc. 10.1016/j.nbd.2020.104809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Reyes, I. , & Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nature Communications, 11(1), 1–11. 10.1038/s41467-019-13668-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayeux, R. , Saunders, A. M. , Shea, S. , Mirra, S. , Evans, D. , Roses, A. D. , Hyman, B. T. , Crain, B. , Tang, M.‐X. , & Phelps, C. H. (1998). Utility of the apolipoprotein E genotype in the diagnosis of Alzheimers disease. New England Journal of Medicine, 338(8), 506–511. 10.1056/nejm199802193380804 [DOI] [PubMed] [Google Scholar]
- Michikawa, M. , Fan, Q. W. , Isobe, I. , & Yanagisawa, K. (2000). Apolipoprotein E exhibits isoform‐specific promotion of lipid efflux from astrocytes and neurons in culture. Journal of Neurochemistry, 74(3), 1008–1016. 10.1046/j.1471-4159.2000.0741008.x [DOI] [PubMed] [Google Scholar]
- Minagawa, H. , Gong, J. S. , Jung, C. G. , Watanabe, A. , Lund‐Katz, S. , Phillips, M. C. , Saito, H. , & Michikawa, M. (2009). Mechanism underlying apolipoprotein E (ApoE) isoform‐dependent lipid efflux from neural cells in culture. Journal of Neuroscience Research, 87(11), 2498–2508. 10.1002/jnr.22073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett, T. , Classey, J. , Matthews, F. E. , Fahrenhold, M. , Taga, M. , Brayne, C. , Ince, P. G. , Nicoll, J. A. R. , & Boche, D. (2016). Microglial immunophenotype in dementia with Alzheimers pathology. Journal of Neuroinflammation, 13(1), 1–10. 10.1186/s12974-016-0601-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata, M. , & Smith, J. D. (1996). Apolipoprotein E allele‐specific antioxidant activity and effects on cytotoxicity by oxidative insults and β‐amyloid peptides. Nature Genetics, 14(1), 55–61. 10.1038/ng0996-55 [DOI] [PubMed] [Google Scholar]
- Montagne, A. , Nation, D. , Sagare, A. , & Nature, G. B. (2020). APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature, 2020(7806), 71–76. 10.1038/s41586-020-2247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne, A. , Nikolakopoulou, A. M. , Huuskonen, M. T. , Sagare, A. P. , Lawson, E. J. , Lazic, D. , Rege, S. V. , Grond, A. , Zuniga, E. , Barnes, S. R. , Prince, J. , Sagare, M. , Hsu, C.‐J. , LaDu, M. J. , Jacobs, R. E. , & Zlokovic, B. V. (2021). Author correction: APOE4 accelerates advanced‐stage vascular and neurodegenerative disorder in old Alzheimers mice via cyclophilin a independently of amyloid‐β. Nature Aging, 1(7), 624–624. 10.1038/s43587-021-00090-y [DOI] [PubMed] [Google Scholar]
- Montesanto, A. , Crocco, P. , Anfossi, M. , Smirne, N. , Puccio, G. , Colao, R. , Maletta, R. , Passarino, G. , Bruni, A. C. , & Rose, G. (2016). The genetic variability of UCP4 affects the individual susceptibility to late‐onset Alzheimers disease and modifies the diseases risk in apoe‐ ϵ 4 carriers. Journal of Alzheimers Disease, 51(4), 1265–1274. 10.3233/JAD-150993 [DOI] [PubMed] [Google Scholar]
- Montine, T. J. , Huang, D. Y. , Valentine, W. M. , Amarnath, V. , Saunders, A. , Weisgraber, K. H. , Graham, D. G. , & Strittmatter, W. J. (1996). Crosslinking of apolipoprotein E by products of lipid peroxidation. Journal of Neuropathology and Experimental Neurology, 55(2), 202–210. 10.1097/00005072-199602000-00009 [DOI] [PubMed] [Google Scholar]
- Morrow, J. A. , Hatters, D. M. , Lu, B. , Höchtl, P. , Oberg, K. A. , Rupp, B. , & Weisgraber, K. H. (2002). Apolipoprotein E4 forms a molten globule: A potential basis for its association with disease. Journal of Biological Chemistry, 277(52), 50380–50385. 10.1074/jbc.M204898200 [DOI] [PubMed] [Google Scholar]
- Muñoz, S. S. , Li, H. , Ruberu, K. , Chu, Q. , Saghatelian, A. , Ooi, L. , & Garner, B. (2018). The serine protease HtrA1 contributes to the formation of an extracellular 25‐kDa apolipoprotein e fragment that stimulates neuritogenesis. Journal of Biological Chemistry, 293(11), 4071–4084. 10.1074/jbc.RA117.001278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth, C. , Hartmann, A. , Sepulveda‐Falla, D. , Glatzel, M. , & Krasemann, S. (2019). Phagocytosis of apoptotic cells is specifically upregulated in apoe4 expressing microglia in vitro. Frontiers in Cellular Neuroscience, 13, 181. 10.3389/fncel.2019.00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najm, R. , Jones, E. A. , & Huang, Y. (2019). Apolipoprotein E4, inhibitory network dysfunction, and Alzheimers disease. Molecular Neurodegeneration, 14(1), 1–13. 10.1186/s13024-019-0324-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai, M. , Kawamata, T. , Taniguchi, T. , Maeda, K. , & Tanaka, C. (1996). Expression of apolipoprotein E mRNA in rat microglia. Neuroscience Letters, 211(1), 41–44. 10.1016/0304-3940(96)12716-6 [DOI] [PubMed] [Google Scholar]
- Nakamura, T. , Watanabe, A. , Fujino, T. , Hosono, T. , & Michikawa, M. (2009). Apolipoprotein E4 (1‐272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Molecular Neurodegeneration, 4(1), 1–11. 10.1186/1750-1326-4-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan, P. , Sienski, G. , Bonner, J. M. , Lin, Y. T. , Seo, J. , Baru, V. , Haque, A. , Milo, B. , Akay, L. A. , Graziosi, A. , Freyzon, Y. , Landgraf, D. , Hesse, W. R. , Valastyan, J. , Barrasa, M. I. , Tsai, L. H. , & Lindquist, S. (2020). PICALM rescues endocytic defects caused by the Alzheimers disease risk factor APOE4. Cell Reports, 33(1), 108224. 10.1016/j.celrep.2020.108224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan, B. P. , Bellosta, S. , Sanan, D. A. , Weisgraber, K. H. , Mahley, R. W. , & Pitas, R. E. (1994). Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science, 264(5160), 850–852. 10.1126/science.8171342 [DOI] [PubMed] [Google Scholar]
- Nathan, B. P. , Chang, K. C. , Bellosta, S. , Brisch, E. , Ge, N. , Mahley, R. W. , & Pitas, R. E. (1995). The inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule depolymerization. Journal of Biological Chemistry, 270(34), 19791–19799. 10.1074/jbc.270.34.19791 [DOI] [PubMed] [Google Scholar]
- Nation, D. A. , Sweeney, M. D. , Montagne, A. , Sagare, A. P. , DOrazio, L. M. , Pachicano, M. , Sepehrband, F. , Nelson, A. R. , Buennagel, D. P. , Harrington, M. G. , Benzinger, T. L. S. , Fagan, A. M. , Ringman, J. M. , Schneider, L. S. , Morris, J. C. , Chui, H. C. , Law, M. , Toga, A. W. , & Zlokovic, B. V. (2019). Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nature Medicine, 25(2), 270–276. 10.1038/s41591-018-0297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neustadtl, A. L. , Winston, C. N. , Parsadanian, M. , Main, B. S. , Villapol, S. , & Burns, M. P. (2017). Reduced cortical excitatory synapse number in APOE4 mice is associated with increased calcineurin activity. Neuroreport, 28(10), 618–624. 10.1097/WNR.0000000000000811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichol, K. , Deeny, S. P. , Seif, J. , Camaclang, K. , & Cotman, C. W. (2009). Exercise improves cognition and hippocampal plasticity in APOE ϵ4 mice. Alzheimers and Dementia, 5(4), 287–294. 10.1016/j.jalz.2009.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson, D. A. , Taylor, S. L. , Fullerton, S. M. , Weiss, K. M. , Clark, A. G. , Stengård, J. H. , Salomaa, V. , Boerwinkle, E. , & Sing, C. F. (2000). Sequence diversity and large‐scale typing of SNPs in the human apolipoprotein E gene. Genome Research, 10(10), 1532–1545. 10.1101/gr.146900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon, R. A. , & Yang, D. S. (2011). Autophagy failure in Alzheimers disease‐locating the primary defect. Neurobiology of Disease, 43(1), 38–45. 10.1016/j.nbd.2011.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent, A. A. , Lin, K. , van Lengerich, B. , Lianoglou, S. , Przybyla, L. , Davis, S. S. , Llapashtica, C. , Wang, J. , Kim, D. J. , Xia, D. , Lucas, A. , Baskaran, S. , Haddick, P. C. G. , Lenser, M. , Earr, T. K. , Shi, J. , Dugas, J. C. , Andreone, B. J. , Logan, T. , et al. (2020). TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron, 105(5), 837–854. 10.1016/j.neuron.2019.12.007 [DOI] [PubMed] [Google Scholar]
- Nuriel, T. , Angulo, S. L. , Khan, U. , Ashok, A. , Chen, Q. , Figueroa, H. Y. , Emrani, S. , Liu, L. , Herman, M. , Barrett, G. , Savage, V. , Buitrago, L. , Cepeda‐Prado, E. , Fung, C. , Goldberg, E. , Gross, S. S. , Hussaini, S. A. , Moreno, H. , Small, S. A. , & Duff, K. E. (2017). Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimers disease‐like pathology. Nature Communications, 8(1), 1–14. 10.1038/s41467-017-01444-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuriel, T. , Peng, K. Y. , Ashok, A. , Dillman, A. A. , Figueroa, H. Y. , Apuzzo, J. , Ambat, J. , Levy, E. , Cookson, M. R. , Mathews, P. M. , & Duff, K. E. (2017). The endosomal‐lysosomal pathway is dysregulated by APOE4 expression in vivo. Frontiers in Neuroscience, 11, 702. 10.3389/fnins.2017.00702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwabuisi‐Heath, E. , Rebeck, G. W. , LaDu, M. J. , & Yu, C. (2013). ApoE4 delays dendritic spine formation during neuron development and accelerates loss of mature spines in vitro. ASN Neuro, 6(1), AN20130043. 10.1042/an20130043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo, N. , Mitsuda, N. , Tamatani, M. , Yamaguchi, A. , Lee, Y. D. , Ogihara, T. , Vitek, M. P. , & Tohyama, M. (2001). Apolipoprotein E4 stimulates cAMP response element‐binding protein transcriptional activity through the extracellular signal‐regulated kinase pathway. Journal of Biological Chemistry, 276(5), 3046–3053, M005070200. 10.1074/jbc [DOI] [PubMed] [Google Scholar]
- Okoro, E. U. , Zhao, Y. , Guo, Z. M. , Zhou, L. , Lin, X. , & Yang, H. (2012). Apolipoprotein E4 is deficient in inducing macrophage ABCA1 expression and stimulating the Sp1 signaling pathway. PLoS ONE, 7(9), e44430. 10.1371/journal.pone.0044430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophir, G. , Amariglio, N. , Jacob‐Hirsch, J. , Elkon, R. , Rechavi, G. , & Michaelson, D. M. (2005). Apolipoprotein E4 enhances brain inflammation by modulation of the NF‐κB signaling cascade. Neurobiology of Disease, 20(3), 709–718. 10.1016/j.nbd.2005.05.002 [DOI] [PubMed] [Google Scholar]
- Ouchi, Y. , Nobezawa, S. , Okada, H. , Yoshikawa, E. , Futatsubashi, M. , & Kaneko, M. (1998). Altered glucose metabolism in the hippocampal head in memory impairment. Neurology, 51(1), 136–142. 10.1212/wnl.51.1.136 [DOI] [PubMed] [Google Scholar]
- Panitch, R. , Hu, J. , Xia, W. , Bennett, D. A. , Stein, T. D. , Farrer, L. A. , & Jun, G. R. (2022). Blood and brain transcriptome analysis reveals APOE genotype‐mediated and immune‐related pathways involved in Alzheimer disease. Alzheimers Research and Therapy, 14(1), 30. 10.1186/s13195-022-00975-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, W. A. , Chan, S. L. , & Mattson, M. P. (2000). A mechanism for the neuroprotective effect of apolipoprotein E: Isoform‐ specific modification by the lipid peroxidation product 4‐hydroxynonenal. Journal of Neurochemistry, 74(4), 1426–1433. 10.1046/j.1471-4159.2000.0741426.x [DOI] [PubMed] [Google Scholar]
- Peng, K. Y. , Pérez‐González, R. , Alldred, M. J. , Goulbourne, C. N. , Morales‐Corraliza, J. , Saito, M. , Saito, M. , Ginsberg, S. D. , Mathews, P. M. , & Levy, E. (2019). Apolipoprotein E4 genotype compromises brain exosome production. Brain, 142(1), 163–175. 10.1093/brain/awy289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris‐Sampedro, F. , Basaure, P. , Reverte, I. , Cabré, M. , Domingo, J. L. , & Colomina, M. T. (2015). Chronic exposure to chlorpyrifos triggered body weight increase and memory impairment depending on human apoE polymorphisms in a targeted replacement mouse model. Physiology & Behavior, 144, 37–45. 10.1016/j.physbeh.2015.03.006 [DOI] [PubMed] [Google Scholar]
- Persson, T. , Lattanzio, F. , Calvo‐Garrido, J. , Rimondini, R. , Rubio‐Rodrigo, M. , Sundström, E. , Maioli, S. , Sandebring‐Matton, A. , & Cedazo‐Mínguez, Á. (2017). Apolipoprotein E4 elicits lysosomal Cathepsin D release, decreased Thioredoxin‐1 levels, and apoptosis. Journal of Alzheimers Disease, 56(2), 601–617. 10.3233/JAD-150738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, M. C. , & Shulman, G. I. (2018). Mechanisms of insulin action and insulin resistance. Physiological Reviews, 98(4), 2133–2223. 10.1152/physrev.00063.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham, T. , Kodvawala, A. , & Hui, D. Y. (2005). The receptor binding domain of apolipoprotein E is responsible for its antioxidant activity. Biochemistry, 44(20), 7577–7582. 10.1021/bi0472696 [DOI] [PubMed] [Google Scholar]
- Pitas, R. E. , Boyles, J. K. , Lee, S. H. , Hui, D. , & Weisgraber, K. H. (1987). Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E (LDL) receptors in the brain. Journal of Biological Chemistry, 262(29), 14352–14360. 10.1016/s0021-9258(18)47945-8 [DOI] [PubMed] [Google Scholar]
- Pocernich, C. B. , Sultana, R. , Hone, E. , Turchan, J. , Martins, R. N. , Calabrese, V. , Nath, A. , & Butterfield, D. A. (2004). Effects of apolipoprotein E on the human immunodeficiency virus protein tat in neuronal cultures and synaptosomes. Journal of Neuroscience Research, 77(4), 532–539. 10.1002/jnr.20182 [DOI] [PubMed] [Google Scholar]
- Pocivavsek, A. , Burns, M. P. , & Rebeck, G. W. (2009). Low‐density lipoprotein receptors regulate microglial inflammation through c‐Jun N‐terminal kinase. Glia, 57(4), 444–453. 10.1002/glia.20772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocivavsek, A. , Mikhailenko, I. , Strickland, D. K. , & Rebeck, G. W. (2009). Microglial low‐density lipoprotein receptor‐related protein 1 modulates c‐Jun N‐terminal kinase activation. Journal of Neuroimmunology, 214(1–2), 25–32. 10.1016/j.jneuroim.2009.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlkamp, T. , Xian, X. , Wong, C. H. , Durakoglugil, M. S. , Werthmann, G. C. , Saido, T. C. , Evers, B. M. , White, C. L. , Connor, J. , Hammer, R. E. , & Herz, J. (2021). NHE6 depletion corrects ApoE4‐mediated synaptic impairments and reduces amyloid plaque load. eLife, 10, e72034. 10.7554/elife.72034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad, H. , & Rao, R. (2018). Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proceedings of the National Academy of Sciences of the United States of America, 115(28), E6640–E6649. 10.1073/pnas.1801612115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzianowska‐Kuznicka, M. , & Kuznicki, J. (2009). The ER and ageing II: Calcium homeostasis. Ageing Research Reviews, 8(3), 160–172. 10.1016/j.arr.2009.05.002 [DOI] [PubMed] [Google Scholar]
- Qiu, Z. , Crutcher, K. A. , Hyman, B. T. , & Rebeck, G. W. (2003). apoE isoforms affect neuronal N‐methyl‐D‐aspartate calcium responses and toxicity via receptor‐mediated processes. Neuroscience, 122(2), 291–303. 10.1016/j.neuroscience.2003.08.017 [DOI] [PubMed] [Google Scholar]
- Qiu, Z. , Hyman, B. T. , & Rebeck, G. W. (2004). Apolipoprotein E receptors mediate neurite outgrowth through activation of p44/42 mitogen‐activated protein kinase in primary neurons. Journal of Biological Chemistry, 279(33), 34948, M401055200–34956. 10.1074/jbc [DOI] [PubMed] [Google Scholar]
- Raber, J. , Huang, Y. , & Ashford, J. W. (2004). ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiology of Aging, 25(5), 641–650. 10.1016/j.neurobiolaging.2003.12.023 [DOI] [PubMed] [Google Scholar]
- Raffaï, R. L. , Dong, L. M. , Farese, R. V. , & Weisgraber, K. H. (2001). Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proceedings of the National Academy of Sciences of the United States of America, 98(20), 11587–11591. 10.1073/pnas.201279298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna, S. , Jhaveri, V. , Konings, S. C. , Nawalpuri, B. , Chakraborty, S. , Holst, B. , Schmid, B. , Gouras, G. K. , Freude, K. K. , & Muddashetty, R. S. (2021). Apoe4 affects basal and nmdar‐mediated protein synthesis in neurons by perturbing calcium homeostasis. Journal of Neuroscience, 41(42), 8686–8709. 10.1523/JNEUROSCI.0435-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramassamy, C. , Averill, D. , Beffert, U. , Theroux, L. , Lussier‐Cacan, S. , Cohn, J. S. , Christen, Y. , Schoofs, A. , Davignon, J. , & Poirier, J. (2000). Oxidative insults are associated with apolipoprotein E genotype in Alzheimers disease brain. Neurobiology of Disease, 7(1), 23–37. 10.1006/nbdi.1999.0273 [DOI] [PubMed] [Google Scholar]
- Raulin, A. C. , Kraft, L. , Al‐Hilaly, Y. K. , Xue, W. F. , McGeehan, J. E. , Atack, J. R. , & Serpell, L. (2019). The molecular basis for apolipoprotein E4 as the major risk factor for late‐onset Alzheimers disease. Journal of Molecular Biology, 431(12), 2248–2265. 10.1016/j.jmb.2019.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat, V. , Wang, S. , Sima, J. , Bar, R. , Liraz, O. , Gundimeda, U. , Parekh, T. , Chan, J. , Johansson, J. O. , Tang, C. , Chui, H. C. , Harrington, M. G. , Michaelson, D. M. , & Yassine, H. N. (2019). ApoE4 alters ABCA1 membrane trafficking in astrocytes. Journal of Neuroscience, 39(48), 9611–9622. 10.1523/JNEUROSCI.1400-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck, G. W. (2017). The role of APOE on lipid homeostasis and inflammation in normal brains. Journal of Lipid Research, 58(8), 1493, R075408–1499. 10.1194/jlr [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, P. H. (2009). Role of mitochondria in neurodegenerative diseases: Mitochondria as a therapeutic target in Alzheimers disease. CNS Spectrums, 14(8 Suppl 7), 8–13. 10.1017/s1092852900024901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reger, M. A. , Watson, G. S. , Frey, W. H. , Baker, L. D. , Cholerton, B. , Keeling, M. L. , Belongia, D. A. , Fishel, M. A. , Plymate, S. R. , Schellenberg, G. D. , Cherrier, M. M. , & Craft, S. (2006). Effects of intranasal insulin on cognition in memory‐impaired older adults: Modulation by APOE genotype. Neurobiology of Aging, 27(3), 451–458. 10.1016/j.neurobiolaging.2005.03.016 [DOI] [PubMed] [Google Scholar]
- Reiman, E. M. , Caselli, R. J. , Yun, L. S. , Chen, K. , Bandy, D. , Minoshima, S. , Thibodeau, S. N. , & Osborne, D. (1996). Preclinical evidence of Alzheimers disease in persons homozygous for the ε4 allele for apolipoprotein E. New England Journal of Medicine, 334(12), 752–758. 10.1056/nejm199603213341202 [DOI] [PubMed] [Google Scholar]
- Reiman, E. M. , Chen, K. , Alexander, G. E. , Caselli, R. J. , Bandy, D. , Osborne, D. , Saunders, A. M. , & Hardy, J. (2004). Functional brain abnormalities in young adults at genetic risk for late‐onset Alzheimer's dementia. Proceedings of the National Academy of Sciences of the United States of America, 101(1), 284–289. 10.1073/pnas.2635903100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea, E. M. , Raber, J. , & Banks, W. A. (2020). ApoE and cerebral insulin: Trafficking, receptors, and resistance. Neurobiology of Disease, 137, 104755. 10.1016/j.nbd.2020.104755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell, D. R. , Zhou, H. , Atchison, K. , Warwick, H. K. , Atkinson, P. J. , Jefferson, J. , Xu, L. , Aschmies, S. , Kirksey, Y. , Hu, Y. , Wagner, E. , Parratt, A. , Xu, J. , Li, Z. , Zaleska, M. M. , Jacobsen, J. S. , Pangalos, M. N. , & Reinhart, P. H. (2008). Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. Journal of Neuroscience, 28(45), 11445–11453. 10.1523/JNEUROSCI.1972-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijpma, A. , Jansen, D. , Arnoldussen, I. A. C. , Fang, X. T. , Wiesmann, M. , Mutsaers, M. P. C. , Dederen, P. J. , Janssen, C. I. F. , & Kiliaan, A. J. (2013). Sex differences in presynaptic density and neurogenesis in middle‐aged ApoE4 and ApoE knockout mice. Journal of Neurodegenerative Diseases, 2013, 1–9. 10.1155/2013/531326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, G. A. , Burns, M. P. , Weeber, E. J. , & Rebeck, G. W. (2013). Young APOE4 targeted replacement mice exhibit poor spatial learning and memory, with reduced dendritic spine density in the medial entorhinal cortex. Learning and Memory, 20(5), 256–266. 10.1101/lm.030031.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim, S. (2017). Oxidative stress and the central nervous system. Journal of Pharmacology and Experimental Therapeutics, 360(1), 201–205. 10.1124/jpet.116.237503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, A. M. , Strittmatter, W. J. , Schmechel, D. , St. George‐Hyslop, P. H. , Pericak‐Vance, M. A. , Joo, S. H. , Rosi, B. L. , Gusella, J. F. , Crapper‐MacLachlan, D. R. , Alberts, M. J. , Hulette, C. , Crain, B. , Goldgaber, D. , & Roses, A. D. (1993). Association of apolipoprotein E allele 4 with late‐onset familial and sporadic Alzheimer’s disease. Neurology, 43(8), 1467–1467. 10.1212/wnl.43.8.1467 [DOI] [PubMed] [Google Scholar]
- Schain, M. , & Kreisl, W. C. (2017). Neuroinflammation in neurodegenerative disorders—A review. Current Neurology and Neuroscience Reports, 17(3), 25. 10.1007/s11910-017-0733-2 [DOI] [PubMed] [Google Scholar]
- Segev, Y. , Michaelson, D. M. , & Rosenblum, K. (2013). ApoE ε4 is associated with eIF2α phosphorylation and impaired learning in young mice. Neurobiology of Aging, 34(3), 863–872. 10.1016/j.neurobiolaging.2012.06.020 [DOI] [PubMed] [Google Scholar]
- Serrano‐Pozo, A. , Li, Z. , Noori, A. , Nguyen, H. N. , Mezlini, A. , Li, L. , Hudry, E. , Jackson, R. J. , Hyman, B. T. , & Das, S. (2021). Effect of APOE alleles on the glial transcriptome in normal aging and Alzheimers disease. Nature Aging, 1(10), 919–931. 10.1038/s43587-021-00123-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabab, T. , Khanabdali, R. , Moghadamtousi, S. Z. , Kadir, H. A. , & Mohan, G. (2016). Neuroinflammation pathways: A general review. International Journal of Neuroscience, 127(7), 624–633. 10.1080/00207454.2016.1212854 [DOI] [PubMed] [Google Scholar]
- Shea, T. B. , Rogers, E. , Ashline, D. , Ortiz, D. , & Sheu, M. S. (2002). Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radical Biology and Medicine, 33(8), 1115–1120. 10.1016/S0891-5849(02)01001-8 [DOI] [PubMed] [Google Scholar]
- Shi, L. , Du, X. , Zhou, H. , Tao, C. , Liu, Y. , Meng, F. , Wu, G. , Xiong, Y. , Xia, C. , Wang, Y. , Bi, G. , & Zhou, J. N. (2014). Cumulative effects of the ApoE genotype and gender on the synaptic proteome and oxidative stress in the mouse brain. International Journal of Neuropsychopharmacology, 17(11), 1863–1879. 10.1017/S1461145714000601 [DOI] [PubMed] [Google Scholar]
- Shi, Y. , & Holtzman, D. M. (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nature Reviews Immunology, 18, 759–772. 10.1038/s41577-018-0051-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, J. E. , Ince, P. G. , Shaw, P. J. , Heath, P. R. , Raman, R. , Garwood, C. J. , Gelsthorpe, C. , Baxter, L. , Forster, G. , Matthews, F. E. , Brayne, C. , & Wharton, S. B. (2011). Microarray analysis of the astrocyte transcriptome in the aging brain: Relationship to Alzheimers pathology and APOE genotype. Neurobiology of Aging, 32(10), 1795–1807. 10.1016/j.neurobiolaging.2011.04.013 [DOI] [PubMed] [Google Scholar]
- Singh, A. , Kukreti, R. , Saso, L. , & Kukreti, S. (2019). Oxidative stress: A key modulator in neurodegenerative diseases. Molecules, 24(8), 1583. 10.3390/molecules24081583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery, C. F. , Zhang, J. , Paterson, R. W. , Foulkes, A. J. M. , Carton, A. , Macpherson, K. , Mancini, L. , Thomas, D. L. , Modat, M. , Toussaint, N. , Cash, D. M. , Thornton, J. S. , Henley, S. M. D. , Crutch, S. J. , Alexander, D. C. , Ourselin, S. , Fox, N. C. , Zhang, H. , & Schott, J. M. (2017). ApoE influences regional white‐matter axonal density loss in Alzheimers disease. Neurobiology of Aging, 57, 8–17. 10.1016/j.neurobiolaging.2017.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, G. W. , Mazziotta, J. C. , Collins, M. T. , Baxter, L. R. , Phelps, M. E. , Kaplan, A. , Adamson, C. F. , Guze, B. H. , Chang, L. , Mandelkern, M. A. , La Rue, A. , Corder, E. H. , Saunders, A. M. , Pericak Vance, M. A. , Roses, A. D. , & Haines, J. L. (1995). Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA: The Journal of the American Medical Association, 273(12), 942–947. 10.1001/jama.1995.03520360056039 [DOI] [PubMed] [Google Scholar]
- Small, S. A. , Simoes‐Spassov, S. , Mayeux, R. , & Petsko, G. A. (2017). Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimers disease. Trends in Neurosciences, 40(10), 592–602. 10.1016/j.tins.2017.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto, I. , Graham, L. C. , Richter, H. J. , Simeone, S. N. , Radell, J. E. , Grabowska, W. , Funkhouser, W. K. , Howell, M. C. , & Howell, G. R. (2015). APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biology, 13(10), 1–33. 10.1371/journal.pbio.1002279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speidell, A. P. , Demby, T. , Lee, Y. , Rodriguez, O. , Albanese, C. , Mandelblatt, J. , & Rebeck, G. W. (2019). Development of a human APOE Knock‐in mouse model for study of cognitive function after Cancer chemotherapy. Neurotoxicity Research, 35(2), 291–303. 10.1007/s12640-018-9954-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, K. , Friedman, B. A. , Etxeberria, A. , Huntley, M. A. , van der Brug, M. P. , Foreman, O. , Paw, J. S. , Modrusan, Z. , Beach, T. G. , Serrano, G. E. , & Hansen, D. V. (2020). Alzheimers patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Reports, 31(13), 107843. 10.1016/j.celrep.2020.107843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger, W. , Sailler, B. , Strasser, V. , Recheis, B. , Fasching, D. , Kahr, L. , Schneider, W. J. , & Nimpf, J. (2002). The PX‐domain protein SNX17 interacts with members of the LDL receptor family and modulates endocytosis of the LDL receptor. EMBO Journal, 21(16), 4259–4267. 10.1093/emboj/cdf435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoykovich, S. , & Gibas, K. (2019). APOE ε4, the door to insulin‐resistant dyslipidemia and brain fog? A case study. Alzheimers and Dementia: Diagnosis, Assessment and Disease Monitoring, 11, 264–269. 10.1016/j.dadm.2019.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan, A. M. , & Mattson, M. P. (2010). Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimers disease. Neural Plasticity, 2010, 1–8. 10.1155/2010/108190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser, V. , Fasching, D. , Hauser, C. , Mayer, H. , Bock, H. H. , Hiesberger, T. , Herz, J. , Weeber, E. J. , Sweatt, J. D. , Pramatarova, A. , Howell, B. , Schneider, W. J. , & Nimpf, J. (2004). Receptor clustering is involved in Reelin signaling. Molecular and Cellular Biology, 24(3), 1378–1386. 10.1128/mcb.24.3.1378-1386.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, P. M. , Mace, B. E. , Maeda, N. , & Schmechel, D. E. (2004). Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience, 124(4), 725–733. 10.1016/j.neuroscience.2003.10.011 [DOI] [PubMed] [Google Scholar]
- Sullivan, P. M. , Mezdour, H. , Aratani, Y. , Knouff, C. , Najib, J. , Reddick, R. L. , Quarfordt, S. H. , & Maeda, N. (1997). Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet‐induced hypercholesterolemia and atherosclerosis. Journal of Biological Chemistry, 272(29), 17972–17980. 10.1074/jbc.272.29.17972 [DOI] [PubMed] [Google Scholar]
- Sullivan, P. M. , Mezdour, H. , Quarfordt, S. H. , & Maeda, N. (1998). Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human APOE*2. Journal of Clinical Investigation, 102(1), 130–135. 10.1172/JCI2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, G. Z. , He, Y. C. , Ma, X. K. , Li, S. T. , Chen, D. J. , Gao, M. , Qiu, S. F. , Yin, J. X. , Shi, J. , & Wu, J. (2017). Hippocampal synaptic and neural network deficits in young mice carrying the human APOE4 gene. CNS Neuroscience and Therapeutics, 23(9), 748–758. 10.1111/cns.12720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney, M. D. , Sagare, A. P. , & Zlokovic, B. V. (2018). Blood‐brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nature Reviews Neurology, 14(3), 133–150. 10.1038/nrneurol.2017.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, L. M. , Ghura, S. , Koster, K. P. , Liakaite, V. , Maienschein‐Cline, M. , Kanabar, P. , Collins, N. , Ben‐Aissa, M. , Lei, A. Z. , Bahroos, N. , Green, S. J. , Hendrickson, B. , Van Eldik, L. J. , & LaDu, M. J. (2015). APOE‐modulated Aβ‐induced neuroinflammation in Alzheimers disease: Current landscape, novel data, and future perspective. Journal of Neurochemistry, 133(4), 465–488. 10.1111/jnc.13072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai, L. M. , Thomas, R. , Marottoli, F. M. , Koster, K. P. , Kanekiyo, T. , Morris, A. W. J. , & Bu, G. (2016). The role of APOE in cerebrovascular dysfunction. Acta Neuropathologica, 131(5), 709–723. 10.1007/s00401-016-1547-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamboli, I. Y. , Heo, D. , & Rebeck, G. W. (2014). Extracellular proteolysis of apolipoprotein e (apoE) by secreted serine neuronal protease. PLoS ONE, 9(3), 93120. 10.1371/journal.pone.0093120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, R. H. , Kril, J. J. , Fatima, M. , McGeachie, A. , McCann, H. , Shepherd, C. , Forrest, S. L. , Affleck, A. , Kwok, J. B. J. , Hodges, J. R. , Kiernan, M. C. , & Halliday, G. M. (2015). TDP‐43 proteinopathies: Pathological identification of brain regions differentiating clinical phenotypes. Brain, 138(10), 3110–3122. 10.1093/brain/awv220 [DOI] [PubMed] [Google Scholar]
- Tay, T. L. , Sagar, Dautzenberg, J. , Grün, D. , & Prinz, M. (2018). Unique microglia recovery population revealed by single‐cell RNAseq following neurodegeneration. Acta Neuropathologica Communications, 6(1), 87. 10.1186/s40478-018-0584-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tensaouti, Y. , Stephanz, E. P. , Yu, T. S. , & Kernie, S. G. (2018). ApoE regulates the development of adult newborn hippocampal neurons. ENeuro, 5(4), ENEURO.0155–ENEU18.2018. 10.1523/ENEURO.0155-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tensaouti, Y. , Yu, T. S. , & Kernie, S. G. (2020). Apolipoprotein e regulates the maturation of injury‐induced adult‐born hippocampal neurons following traumatic brain injury. PLoS ONE, 15(3), e0229240. 10.1371/journal.pone.0229240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter, B. , Xu, P. T. , Gilbert, J. R. , Roses, A. D. , Galasko, D. , & Cole, G. M. (2002). Defective neuronal sprouting by human apolipoprotein E4 is a gain‐of‐negative function. Journal of Neuroscience Research, 68(3), 331–336. 10.1002/jnr.10221 [DOI] [PubMed] [Google Scholar]
- Thambisetty, M. , Beason‐Held, L. , An, Y. , Kraut, M. A. , & Resnick, S. M. (2010). APOE ε4 genotype and longitudinal changes in cerebral blood flow in normal aging. Archives of Neurology, 67(1), 93–98. 10.1001/archneurol.2009.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theendakara, V. , Patent, A. , Libeu, C. A. P. , Philpot, B. , Flores, S. , Descamps, O. , Poksay, K. S. , Zhang, Q. , Cailing, G. , Hart, M. , John, V. , Rao, R. V. , & Bredesen, D. E. (2013). Neuroprotective Sirtuin ratio reversed by ApoE4. Proceedings of the National Academy of Sciences of the United States of America, 110(45), 18303–18308. 10.1073/pnas.1314145110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theendakara, V. , Peters‐Libeu, C. A. , Bredesen, D. E. , & Rao, R. V. (2018). Transcriptional effects of ApoE4: Relevance to Alzheimers disease. Molecular Neurobiology, 55(6), 5243–5254. 10.1007/s12035-017-0757-2 [DOI] [PubMed] [Google Scholar]
- Theendakara, V. , Peters‐Libeu, C. A. , Spilman, P. , Poksay, K. S. , Bredesen, D. E. , & Rao, R. V. (2016). Direct transcriptional effects of apolipoprotein E. Journal of Neuroscience, 36(3), 685–700. 10.1523/JNEUROSCI.3562-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar, M. , Keller, J. N. , Chan, S. , Mattson, M. P. , Marques, M. A. , & Crutcher, K. A. (1999). Truncated apolipoprotein E (ApoE) causes increased intracellular calcium and may mediate ApoE neurotoxicity. Journal of Neuroscience, 19(16), 7100–7110. 10.1523/jneurosci.19-16-07100.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, L. M. , Yoon, S. Y. , Andrews‐Zwilling, Y. , Yang, A. , Lin, V. , Lei, H. , & Huang, Y. (2016). Enhancing gaba signaling during middle adulthood prevents age‐dependent gabaergic interneuron decline and learning and memory deficits in apoe4 mice. Journal of Neuroscience, 36(7), 2316–2322. 10.1523/JNEUROSCI.3815-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg, A. J. , Filippini, N. , & Mackay, C. E. (2012). The effects of APOE‐ε4 on the BOLD response. Neurobiology of Aging, 33(2), 323–334. 10.1016/j.neurobiolaging.2010.03.009 [DOI] [PubMed] [Google Scholar]
- Trumble, B. C. , & Finch, C. E. (2019). The exposome in human evolution: From dust to diesel. Quarterly Review of Biology, 94(4), 333–394. 10.1086/706768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzioras, M. , Davies, C. , Newman, A. , Jackson, R. , & Spires‐Jones, T. (2019). Invited review: APOE at the interface of inflammation, neurodegeneration and pathological protein spread in Alzheimers disease. Neuropathology and Applied Neurobiology, 45(4), 327–346. 10.1111/nan.12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich, J. D. , Burchett, J. M. , Restivo, J. L. , Schuler, D. R. , Verghese, P. B. , Mahan, T. E. , Landreth, G. E. , Castellano, J. M. , Jiang, H. , Cirrito, J. R. , & Holtzman, D. M. (2013). In vivo measurement of apolipoprotein e from the brain interstitial fluid using microdialysis. Molecular Neurodegeneration, 8(1), 13. 10.1186/1750-1326-8-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vliet, P. , Oleksik, A. M. , Mooijaart, S. P. , De Craen, A. J. M. , & Westendorp, R. G. J. (2009). ApoE genotype modulates the effect of serum calcium levels on cognitive function in old age. Neurology, 72(9), 821–828. 10.1212/01.wnl.0000343852.10018.24 [DOI] [PubMed] [Google Scholar]
- Veinbergs, I. , Everson, A. , Sagara, Y. , & Masliah, E. (2002). Neurotoxic effects of apolipoprotein E4 are mediated via dysregulation of calcium homeostasis. Journal of Neuroscience Research, 67(3), 379–387. 10.1002/jnr.10138 [DOI] [PubMed] [Google Scholar]
- Veinbergs, I. , Mallory, M. , Mante, M. , Rockenstein, E. , Gilbert, J. R. , & Masliah, E. (1999). Differential neurotrophic effects of apolipoprotein E in aged transgenic mice. Neuroscience Letters, 265(3), 218–222. 10.1016/S0304-3940(99)00243-8 [DOI] [PubMed] [Google Scholar]
- Venzi, M. , Tóth, M. , Häggkvist, J. , Bogstedt, A. , Rachalski, A. , Mattsson, A. , Frumento, P. , & Farde, L. (2017). Differential effect of APOE alleles on brain glucose metabolism in targeted replacement mice: An [18F]FDG‐μPET study. Journal of Alzheimers Disease Reports, 1(1), 169–180. 10.3233/adr-170006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasana, L. E. , Weber, S. , Akinyeke, T. , & Raber, J. (2016). Genotype differences in anxiety and fear learning and memory of WT and ApoE4 mice associated with enhanced generation of hippocampal reactive oxygen species. Journal of Neurochemistry, 138(6), 896–908. 10.1111/jnc.13737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek, M. P. , Brown, C. M. , & Colton, C. A. (2009). APOE genotype‐specific differences in the innate immune response. Neurobiology of Aging, 30(9), 1350–1360. 10.1016/j.neurobiolaging.2007.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther, T. C. , & Farese, R. V. (2012). Lipid droplets and cellular lipid metabolism. Annual Review of Biochemistry, 81, 687–714. 10.1146/annurev-biochem-061009-102430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Najm, R. , Xu, Q. , Jeong, D. E. , Walker, D. , Balestra, M. E. , Yoon, S. Y. , Yuan, H. , Li, G. , Miller, Z. A. , Miller, B. L. , Malloy, M. J. , & Huang, Y. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC‐derived neurons is ameliorated by a small‐molecule structure corrector article. Nature Medicine, 24(5), 647–657. 10.1038/s41591-018-0004-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Wilson, W. A. , Moore, S. D. , Mace, B. E. , Maeda, N. , Schmechel, D. E. , & Sullivan, P. M. (2005). Human apoE4‐targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiology of Disease, 18(2), 390–398. 10.1016/j.nbd.2004.10.013 [DOI] [PubMed] [Google Scholar]
- Wang, X. S. , & Gruenstein, E. (1997). Rapid elevation of neuronal cytoplasmic calcium by apolipoprotein E peptide. Journal of Cellular Physiology, 173(1), 73–83. [DOI] [PubMed] [Google Scholar]
- Weisgraber, K. H. (1994). Apolipoprotein E: Structure‐function relationships. Advances in Protein Chemistry, 45, 249–302. 10.1016/s0065-3233(08)60642-7 [DOI] [PubMed] [Google Scholar]
- Welte, M. A. , & Gould, A. P. (2017). Lipid droplet functions beyond energy storage. Biochimica et Biophysica Acta ‐ Molecular and Cell Biology of Lipids, 1862(10), 1260–1272. 10.1016/j.bbalip.2017.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlye, L. T. , Reinvang, I. , Rootwelt, H. , & Espeseth, T. (2012). Effects of APOE on brain white matter microstructure in healthy adults. Neurology, 79(19), 1961–1969. 10.1212/WNL.0b013e3182735c9c [DOI] [PubMed] [Google Scholar]
- White, F. , Nicoll, J. A. R. , & Horsburgh, K. (2001). Alterations in apoE and apoJ in relation to degeneration and regeneration in a mouse model of entorhinal cortex lesion. Experimental Neurology, 169(2), 307–318. 10.1006/exnr.2001.7655 [DOI] [PubMed] [Google Scholar]
- Wiesmann, M. , Zerbi, V. , Jansen, D. , Haast, R. , Lütjohann, D. , Broersen, L. M. , Heerschap, A. , & Kiliaan, A. J. (2016). A dietary treatment improves cerebral blood flow and brain connectivity in aging apoE4 mice. Neural Plasticity, 2016(6846721), 1–15. 10.1155/2016/6846721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, H. C. , Farmer, B. C. , Piron, M. A. , Walsh, A. E. , Bruntz, R. C. , Gentry, M. S. , Sun, R. C. , & Johnson, L. A. (2020). APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiology of Disease, 136, 104742. 10.1016/j.nbd.2020.104742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, C. M. , Fitz, N. F. , Nam, K. N. , Lefterov, I. , & Koldamova, R. (2019). The role of APOE and TREM2 in Alzheimers disease—Current understanding and perspectives. International Journal of Molecular Sciences, 20(1). 10.3390/ijms20010081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, C. O. (2020). Endosomal‐lysosomal processing of neurodegeneration‐associated proteins in astrocytes. International Journal of Molecular Sciences, 21(14), 1–17. 10.3390/ijms21145149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian, X. , Pohlkamp, T. , Durakoglugil, M. S. , Wong, C. H. , Beck, J. K. , Lane‐Donovan, C. , Plattner, F. , & Herz, J. (2018). Reversal of ApoE4‐induced recycling block as a novel prevention approach for Alzheimers disease. eLife, 7, e40048. 10.7554/eLife.40048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Q. , Bernardo, A. , Walker, D. , Kanegawa, T. , Mahley, R. W. , & Huang, Y. (1996). Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. Journal of Neuroscience, 26(19), 4985–4994. 10.1523/JNEUROSCI.5476-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Q. , Brecht, W. J. , Weisgraber, K. H. , Mahley, R. W. , & Huang, Y. (2004). Apolipoprotein E4 domain interaction occurs in living neuronal cells as determined by fluorescence resonance energy transfer. Journal of Biological Chemistry, 279(24), 25511–25516. 10.1074/jbc.M311256200 [DOI] [PubMed] [Google Scholar]
- Xu, P.‐T. , Gilbert, J. R. , Qiu, H.‐L. , Ervin, J. , Rothrock‐Christian, T. R. , Hulette, C. , & Schmechel, D. E. (1999). Specific Regional Transcription of Apolipoprotein E in Human Brain Neurons. The American Journal of Pathology, 154(2), 601–611. 10.1016/s0002-9440(10)65305-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. P. , Gilley, J. A. , Zhang, G. , & Kernie, S. G. (2011). ApoE is required for maintenance of the dentate gyrus neural progenitor pool. Development, 138(20), 4351–4362. 10.1242/dev.065540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, H. , Coppola, K. , Schweig, J. E. , Crawford, F. , Mullan, M. , & Paris, D. (2019). Distinct signaling pathways regulate TREM2 phagocytic and NFκB antagonistic activities. Frontiers in Cellular Neuroscience, 13, 457. 10.3389/fncel.2019.00457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassine, H. N. , & Finch, C. E. (2020). APOE alleles and diet in brain aging and Alzheimers disease. Frontiers in Aging Neuroscience, 12, 150. 10.3389/fnagi.2020.00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeiser, L. A. , Villasana, L. E. , & Raber, J. (2013). ApoE isoform modulates effects of cranial 56Fe irradiation on spatial learning and memory in the water maze. Behavioural Brain Research, 237(1), 207–214. 10.1016/j.bbr.2012.09.029 [DOI] [PubMed] [Google Scholar]
- Yin, C. , Ackermann, S. , Ma, Z. , Mohanta, S. K. , Zhang, C. , Li, Y. , Nietzsche, S. , Westermann, M. , Peng, L. , Hu, D. , Bontha, S. V. , Srikakulapu, P. , Beer, M. , Megens, R. , Steffens, S. , Hildner, M. , Halder, L. D. , Eckstein, H. H. , Pelisek, J. , … Habenicht, A. (2019). ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nature Medicine, 25(3), 496–506. 10.1038/s41591-018-0336-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, J. , Reiman, E. M. , Beach, T. G. , Serrano, G. E. , Sabbagh, M. N. , Nielsen, M. , Caselli, R. J. , & Shi, J. (2020). Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology, 94(23), e2404–e2411. 10.1212/WNL.0000000000009582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, J. , Turner, G. , Coons, S. , Maalouf, M. , Reiman, E. , & Shi, J. (2014). Association of Amyloid Burden, brain atrophy and memory deficits in aged apolipoprotein ε4 mice. Current Alzheimer Research, 11(3), 283–290. 10.2174/156720501103140329220007 [DOI] [PubMed] [Google Scholar]
- Yin, J. X. , Turner, G. H. , Lin, H. J. , Coons, S. W. , & Shi, J. (2011). Deficits in spatial learning and memory is associated with hippocampal volume loss in aged apolipoprotein e4 mice. Journal of Alzheimers Disease, 27(1), 89–98. 10.3233/JAD-2011-110479 [DOI] [PubMed] [Google Scholar]
- Yong, S. M. , Lim, M. L. , Low, C. M. , & Wong, B. S. (2014). Reduced neuronal signaling in the ageing apolipoprotein‐E4 targeted replacement female mice. Scientific Reports, 4(1), 1–9. 10.1038/srep06580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalocusky, K. A. , Najm, R. , Taubes, A. L. , Hao, Y. , Yoon, S. Y. , Koutsodendris, N. , Nelson, M. R. , Rao, A. , Bennett, D. A. , Bant, J. , Amornkul, D. e. J. , Xu, Q. , An, A. , Cisne‐Thomson, O. , & Huang, Y. (2021). Neuronal ApoE upregulates MHC‐I expression to drive selective neurodegeneration in Alzheimers disease. Nature Neuroscience, 24(6), 786–798. 10.1038/s41593-021-00851-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannis, V. I. , & Breslow, J. L. (1981). Human very Low density lipoprotein apolipoprotein E Isoprotein polymorphism is explained by genetic variation and posttranslational modification. Biochemistry, 20(4), 1033–1041. 10.1021/bi00507a059 [DOI] [PubMed] [Google Scholar]
- Zannis, V. I. , Breslow, J. L. , Utermann, G. , Mahley, R. W. , Weisgraber, K. H. , Havel, R. J. , Goldstein, J. L. , Brown, M. S. , Schonfeld, G. , Hazzard, W. R. , & Blum, C. (1982). Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. Journal of Lipid Research, 23(6), 911–914. 10.1016/s0022-2275(20)38094-9 [DOI] [PubMed] [Google Scholar]
- Zerbi, V. , Wiesmann, M. , Emmerzaal, T. L. , Jansen, D. , Beek, M. V. , Mutsaers, M. P. C. , Beckmann, C. F. , Heerschap, A. , & Kiliaan, A. J. (2014). Resting‐state functional connectivity changes in aging apoe4 and apoe‐ko mice. Journal of Neuroscience, 34(42), 13963–13975. 10.1523/JNEUROSCI.0684-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , & Jiang, L. (2014). Neuroinflammation in Alzheimers disease. Neuropsychiatric Disease and Treatment, 11, 243–256. 10.2147/NDT.S75546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Lin, L. , Dai, X. , Xiao, N. , Ye, Q. , & Chen, X. (2021). ApoE4 increases susceptibility to stress‐induced age‐dependent depression‐like behavior and cognitive impairment. Journal of Psychiatric Research, 143, 292–301. 10.1016/j.jpsychires.2021.09.029 [DOI] [PubMed] [Google Scholar]
- Zhao, J. , Fu, Y. , Yamazaki, Y. , Ren, Y. , Davis, M. D. , Liu, C. C. , Lu, W. , Wang, X. , Chen, K. , Cherukuri, Y. , Jia, L. , Martens, Y. A. , Job, L. , Shue, F. , Nguyen, T. T. , Younkin, S. G. , Graff‐Radford, N. R. , Wszolek, Z. K. , Brafman, D. A. , … Bu, G. (2020). APOE4 exacerbates synapse loss and neurodegeneration in Alzheimers disease patient iPSC‐derived cerebral organoids. Nature Communications, 11(1), 5540. 10.1038/s41467-020-19264-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , Lu, W. , Ren, Y. , Fu, Y. , Martens, Y. A. , Shue, F. , Davis, M. D. , Wang, X. , Chen, K. , Li, F. , Liu, C. C. , Graff‐Radford, N. R. , Wszolek, Z. K. , Younkin, S. G. , Brafman, D. A. , Ertekin‐Taner, N. , Asmann, Y. W. , Dickson, D. W. , Xu, Z. , … Bu, G. (2021). Apolipoprotein E regulates lipid metabolism and α‐synuclein pathology in human iPSC‐derived cerebral organoids. Acta Neuropathologica, 142(5), 807–825. 10.1007/s00401-021-02361-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, N. , Liu, C. C. , Qiao, W. , & Bu, G. (2018). Apolipoprotein E, receptors, and modulation of Alzheimers disease. Biological Psychiatry, 83(4), 347–357. 10.1016/j.biopsych.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, N. , Liu, C. C. , Van Ingelgom, A. J. , Martens, Y. A. , Linares, C. , Knight, J. A. , Painter, M. M. , Sullivan, P. M. , & Bu, G. (2017). Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron, 96(1), 115, e5–129. 10.1016/j.neuron.2017.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, N. , Ren, Y. , Yamazaki, Y. , Qiao, W. , Li, F. , Felton, L. M. , Mahmoudiandehkordi, S. , Kueider‐Paisley, A. , Sonoustoun, B. , Arnold, M. , Shue, F. , Zheng, J. , Attrebi, O. N. , Martens, Y. A. , Li, Z. , Bastea, L. , Meneses, A. D. , Chen, K. , Thompson, J. W. , … Bu, G. (2020). Alzheimers risk factors age, APOE genotype, and sex drive distinct molecular pathways. Neuron, 106(5), 727, e6–742. 10.1016/j.neuron.2020.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, N. , Ramaswamy, G. , & Weisgraber, K. H. (2009). Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astrocyte function. Journal of Biological Chemistry, 284(40), 27273–27280. 10.1074/jbc. M109.014464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y. , Kodvawala, A. , & Hui, D. Y. (2010). Apolipoprotein E inhibits toll‐like receptor (TLR)‐3‐ and TLR‐4‐mediated macrophage activation through distinct mechanisms. Biochemical Journal, 428(1), 47–54. 10.1042/BJ20100016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y. , Nwabuisi‐Heath, E. , Dumanis, S. B. , Tai, L. M. , Yu, C. , Rebeck, G. W. , & Ladu, M. J. (2012). APOE genotype alters glial activation and loss of synaptic markers in mice. Glia, 60(4), 559–569. 10.1002/glia.22289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic, B. V. (2008). The blood‐brain barrier in health and chronic neurodegenerative disorders. Neuron, 57(2), 178–201. 10.1016/j.neuron.2008.01.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This review does not include any data.