

Abstract
Cryopreservation of chimeric antigen receptor (CAR) T cells facilitates shipment, timing of infusions, and storage of subsequent doses. However, reports on the impact of cryopreservation on CAR T cell efficacy have been mixed. We retrospectively compared clinical outcomes between patients who received cryopreserved versus fresh CAR T cells for treatment of B cell leukemia across two cohorts of pediatric and young adult patients: those who received anti-CD22 CAR T cells and those who received bispecific anti-CD19/22 CAR T cells. Manufacturing methods were consistent within each trial but differed between the two trials, allowing for exploration of cryopreservation within different manufacturing platforms. Among 40 patients who received anti-CD22 CAR T cells (21 cryopreserved cells and 19 fresh), there were no differences in in vivo expansion, persistence, incidence of toxicities, or disease response between groups with cryopreserved and fresh CAR T cells. Among 19 patients who received anti-CD19/22 CAR T cells (11 cryopreserved and 8 fresh), patients with cryopreserved cells had similar expansion, toxicity incidence, and disease response, with decreased CAR T cell persistence. Overall, our data demonstrate efficacy of cryopreserved CAR T cells as comparable to fresh infusions, supporting cryopreservation, which will be crucial for advancing the field of cell therapy.
Keywords: cell therapy, CAR T, clinical outcomes, cryopreservation, CAR T manufacturing, cytokine release syndrome, CAR T expansion
Graphical abstract
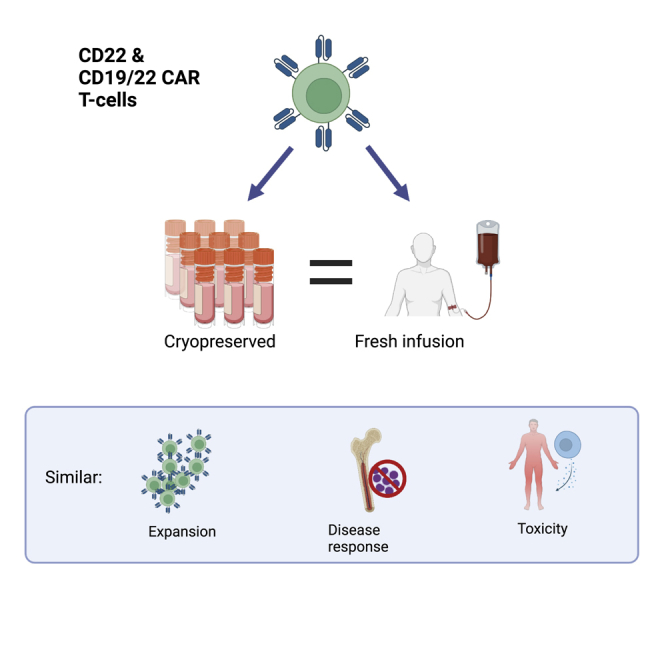
Cryopreservation of CAR T cell therapy products is necessary for expanding production and access to CAR T therapies; however, there have been mixed data on the effect of cryopreservation on CAR T cell function. Dr. Shah and colleagues report comparable clinical outcomes after infusion of fresh and frozen CD22 and CD19/22 CAR T cells.
Introduction
Chimeric antigen receptor (CAR) T cell therapy has shown remarkable efficacy for treatment of relapsed/refractory B cell malignancies1,2 and is a rapidly expanding field of research, with ongoing efforts to both improve access and extend the therapeutic index across a wide range of targets, including hematologic and solid tumors.3,4 As the use and effectiveness of CAR T cell therapy increases, it is important to optimize both its efficacy and availability.
Cryopreservation of cellular therapy products allows for storage of cellular products for shipment, preparation of products for potential subsequent infusions, and coordination of care for patients traveling for therapy, potentially improving access to care.5,6,7 It also allows optimal timing of both cell collection and final product infusion in the context of patients’ clinical status. Furthermore, cryopreservation improves CAR T cell safety since all lot-release testing can be completed prior to giving cells to the patient.
In CAR T cell manufacturing, the cells used to begin the manufacturing process may be cryopreserved immediately after they are collected by leukapheresis or later in the course of CAR T cell production. Cryopreservation of the leukapheresis product has become a standard methodology in the manufacture of CD19 CAR T cells and has resulted in similar clinical outcomes when compared with fresh infusions.8 Cryopreservation of T cells is also well established based on historical use of donor lymphocyte infusions.9,10 Thus, the majority of leukapheresis products are cryopreserved prior to CAR T cell manufacture. Subsequently, depending on the clinical situation, CAR T cell products in early phase studies have been given either as a fresh infusion or cryopreserved.11 Products in later stages of development, including FDA-approved products (e.g., tisagenlecleucel, axicabtagene ciloleucel, and lisocabtagene maraleucel), were cryopreserved in the initial studies that led to approval and are all cryopreserved prior to shipment.1,12,13,14,15,16 Thus, reported efficacy data are based on infusion of cryopreserved products. While the cryopreservation of CAR T cell products is routinely utilized, it has not been extensively studied.
The primary concern with cryopreservation of CAR T cells is loss of efficacy or increased toxicity as a result of cells undergoing the freeze-thaw cycle. Some preclinical data have suggested differences in cytokine production between fresh and cryopreserved anti-B cell maturation antigen (BCMA) CAR T cells in mouse models of lymphoma; however, overall tumor response in this study remained the same.17 Frozen CAR T cells also exhibit different gene expression, with increased expression of genes associated with apoptosis and cell cycle damage compared with fresh cells.18 Furthermore, it is known that different lymphocyte subsets have varying tolerance to the freeze-thaw process,19,20,21 so results from one cell type cannot necessarily be extrapolated to others. The added delay in CAR T cell infusion when cells need to be cryopreserved and shipped may also lead to disease progression and subsequently worse outcomes, but it is likely necessary to facilitate the commercial development of this novel treatment approach.
One recent study describing 22 patients treated with bispecific anti-CD19/20 CAR T cells manufactured using a self-contained, automated closed-process system (CliniMACs Prodigy)22 reported a significantly higher rate of disease response in the 15 patients who received freshly infused cells, compared with those seven who received cryopreserved cells.23 Of note, in this study, fresh infusion was intended for all patients, with cryopreservation only performed for clinical complications such as infection precluding a planned fresh infusion. Conversely, a study by Panch et al. described six different CAR T cell types across 145 patients that were cryopreserved and showed that they had similar in vitro characteristics to fresh cells, including fold expansion, transduction efficiency, and CD3 percent.11 Although patient numbers did not allow for clinical outcome comparisons for all cell types in this study, cryopreserved anti-CD19 CAR T cells showed similar persistence, clinical response, and rate of cytokine release syndrome when compared with freshly infused cells. For the 16 anti-CD22 CAR T cells evaluated, frozen products led to lower in vivo expansion but similar overall persistence and clinical response. Another recent study of cryopreserved CD19 CAR T cells demonstrated phenotypic differences between fresh and cryopreserved products but comparable anti-tumor activity based on in vitro assays.24
As applications of CAR T cell therapy have increased, novel automated manufacturing methods have been introduced, which may lead to subtle changes in CAR T cell characteristics.25,26 CAR T cells produced in automated systems may respond differently to the freeze-thaw process. In order to ship and transport these important products, understanding how cryopreservation plays a role in outcomes remains critical.
To more closely evaluate the effect of cryopreservation on CAR T cell therapy, we examined clinical outcomes and toxicities in a larger cohort of patients who received anti-CD22 CAR T cells as well as a second cohort of patients who received bispecific CD19/22 CAR T cells. The effect of cryopreservation on these bispecific CAR T cells, which were manufactured using the CliniMACs Prodigy system, has not previously been described. By studying the impact of cryopreservation across a similar patient population using two different constructs and manufacturing methodologies, our results provide further insight into the feasibility of cryopreserving CAR T cells and maintaining clinical outcomes.
Results
40 patients who received anti-CD22 CAR T cell therapy at a uniform dose and 19 patients who received bispecific CD19/22 CAR T cell therapy at a uniform dose were included in our analysis (Figure 1). Manufacturing methods were consistent within each cohort, with CD22 CAR T cells being manufactured using bag culture and bispecific CD19/22 CAR T cells manufactured using the Prodigy system. In the CD22 cohort, 21 patients received cryopreserved cells and 19 received fresh infusions. In the bispecific CD19/22 CAR T cell cohort, 11 received cryopreserved cells and eight received fresh infusions. Throughout the treatment period, although fresh infusions were generally considered the preferred method, cells were cryopreserved primarily for logistical and scheduling considerations.
Figure 1.

Diagram of patients included in the study
Our study included patients from the expansion doses of two CAR T cell dose-escalation trials: one involving anti-CD22 CAR T cells and one involving bispecific anti-CD19/CD22 CAR T cells. (A) depicts patients in the CD22 cohort and (B) depicts patients in the CD19/22 cohort.
Baseline patient characteristics
Baseline characteristics, including demographics, disease burden, and prior therapies were compared between the cryopreserved and freshly infused groups (Table 1). In the CD22 cohort, no significant differences were found in patient demographics or baseline disease status, including disease burden, presence of central nervous system (CNS) disease, or presence of extramedullary disease. Patients were generally heavily pretreated, with an average of six prior lines of therapy. The majority, 65%, had previously received CAR T cell therapy, and almost half of patients had previously had a stem cell transplant, but there was no difference in prior therapies between the fresh and cryopreserved groups.
Table 1.
Baseline characteristics of patients from both CD22 and bispecific CD19/22 CAR T cell cohorts included in the study
| Characteristic | CD22 CAR T cells (n = 40) |
CD19/22 CAR T cells (n = 15) |
||||
|---|---|---|---|---|---|---|
| Cryopreserved | Fresh | p value | Cryopreserved | Fresh | p value | |
| n | 21 | 19 | 11 | 8 | ||
| Gender (%male) | 57% | 84% | 0.09 | 45% | 38% | 1 |
| Median age (yrs) | 13 | 12 | 0.96 | 28 | 17 | 0.41 |
| Median weight (kg) | 47 | 50 | 0.94 | 67 | 52 | 0.12 |
| Race | 67% White | 63% White | 1 | 54% White | 63% White | 1 |
| 19% Asian | 5% Asian | 0.35 | 18% Black | 0% Black | 0.49 | |
| 5% multiple | 11% multiple | 0.60 | 9% Asian | 0% Asian | 1 | |
| 10% unknown | 21% unknown | 0.40 | 18% unknown | 38% unknown | 0.60 | |
| Ethnicity | 29% Hispanic | 39% Hispanic | 0.52 | 27% Hispanic | 75% Hispanic | 0.07 |
| Diagnosis | 100% ALL | 89% ALL, 6% CML w/ALL blast crisis, 6% B-LLy | 1 | 100% ALL | 100% ALL | 1 |
| Median disease burden (percent MNCs in BM) | 48% | 40% | 0.93 | 0.70% | 25.15% | 0.03 |
| Disease burden M | M1 24%, M2 9.5%, M3 67% | M1 26%, M2 16%, M3 58% | 0.79 | M1 73%, M2 9%, M3 18% | M1 38%, M2 13%, M3 50% | 0.28 |
| CNS status | 95% CNS1, 5% CNS2 | 100% CNS1 | 1 | 100% CNS1 | 100% CNS1 | 1 |
| Extramedullary disease | 5/21 (24%) | 3/19 (16%) | 0.70 | 4/11 (36%) | 1/8 (13%) | 0.34 |
| Prior HSCT | 10/21 (48%) | 9/19 (47%) | 0.99 | 7/11 (64%) | 3/8 (38%) | 0.37 |
| Prior CAR T therapy | 12/21 (57%) | 14/19 (74%) | 0.33 | 2/11 (18%) | 2/8 (25%) | 1 |
| Median number of prior treatments | 6 | 6 | 0.41 | 3 | 4.5 | 0.16 |
MNC = mononuclear cell, BM = bone marrow, HSCT = hematopoietic stem cell transplant, ALL = acute lymphoblastic leukemia, CML = chronic myelogenous leukemia, BLLy = B-lymphoblastic lymphoma, CNS = central nervous system.
Statistically significant p-values are shown in bold.
In the bispecific CD19/22 cohort, there were no significant differences in demographics. Importantly, the baseline disease burden differed significantly, with median baseline disease burden of 0.7% blasts on bone marrow flow cytometry in the cryopreserved group versus 25% in the fresh infusion group. Other baseline characteristics, including incidence of CNS disease, extramedullary disease, and prior lines of therapy, were similar between groups.
Baseline cell characteristics
Baseline CAR T cell product characteristics were compared at the end of the manufacturing process (Table 2). In some cases, the mononuclear cell (MNC) leukapheresis products were cryopreserved prior to CAR T cell production, and the proportion of patients whose MNCs were cryopreserved did not differ between groups. Viability, transduction efficiency, CD4:8 ratio, and fold expansion were evaluated on the day of CAR T cell harvest, and there were no baseline differences between fresh and cryopreserved groups.
Table 2.
Baseline characteristics of CD22 and bispecific CD19/22 CAR T cells
| Characteristic | CD22 CAR T cells (n = 40) |
CD19/22 CAR T cells (n = 15) |
||||
|---|---|---|---|---|---|---|
| Cryopreserved | Fresh | p value | Cryopreserved | Fresh | p value | |
| n | 21 | 19 | 11 | 8 | ||
| CAR T cells manufactured using cryopreserved MNCs | 15/21 (71%) | 16/19 (84%) | 0.46 | 7/11 (64%) | 3/8 (38%) | 0.37 |
| Median CAR T cell viability at harvest | 97% | 97.5% | 0.59 | 95% | 97.7% | 0.05 |
| Median CAR T cell transfection efficiency (percent Protein L) | 44% | 34% | 0.08 | 70% | 63% | 0.35 |
| Median CAR T cell in vitro fold expansion | 3.02 | 3.405 | 0.99 | 15.01 | 14.07 | 0.49 |
| CAR T cell median CD4:8 ratio | 1.670 | 2.056 | 0.98 | 1.67 | 2.18 | 0.78 |
| Median days of cryopreservation (range) | 24 (6–108) | n/a | n/a | 20 (3–44) | n/a | n/a |
| Median CAR T cell viability at Infusion | 91% | 97.5% | 0.0001 | 86% | 97.7% | <0.0001 |
MNC = mononuclear cell.
Statistically significant p-values are shown in bold.
Viability was re-checked for cryopreserved samples at the time of infusion. The percent cell viability at the time of infusion was notably decreased in cryopreserved cells compared with freshly infused cells for both the CD22 and the bispecific CD19/22 cohorts. For the CD22 cohort, the median time of cryopreservation was 24 days, with a range of 6–108 days. For the CD19/22 cohort, the median time of cryopreservation was 20 days, with a range from 3 to 44. There was no correlation between final viability and the number of days of cryopreservation. Of note, cell doses for infusion were calculated based on numbers of viable cells, so differences in percent viability did not affect the absolute numbers of viable cells infused.
For select cryopreserved samples, post-thaw viability, CD3 percent, transduction efficiency, and CD4:8 ratios were measured as part of quality assurance. This included 7 samples from the CD22 cohort and all 11 samples from the CD19/22 cohort. Grouped pre- and post-thaw measurements are depicted in Figure 2, with the baseline measurements in freshly infused cells shown for comparison.
Figure 2.

Manufacturing characteristics of CAR T cells before and after cryopreservation
Viability, CD3 percent, transduction efficiency (based on Protein L percent), and CD4:8 ratios are shown for pre-cryopreservation, post-cryopreservation, and fresh samples. Viability was tested for all samples prior to infusion. Post-thaw CD3 percent, Protein L percent, and CD4:8 ratios were only tested in a subset of anti-CD22 CAR T cells (n = 7) and for all cryopreserved anti-CD19/22 CAR T cells (n = 11). The results of testing anti-CD22-CAR T cell products are shown in (A)–(D), and those of anti-CD19/CD22 bispecific CAR T cell products are shown in (E)–(H). (A) and (E) show the viability, (B) and (F) show CD3 percent, (C) and (G) the percent of cells expressing Protein L, and (D) and (H) the ratio of CD4:CD8-expressing cells. p values represent a comparison between freshly infused and post-cryopreservation products. For pre- and post-cryopreservation comparisons, please refer to Figure S2.
Paired pre- and post-thaw samples are shown in Figure S1. Viability was decreased post-thaw, as expected (p < 0.01). In addition, the CD4:8 ratio was decreased in the post-thaw samples (p = 0.02 for CD22 cohort, p < 0.01 for CD19/22 cohort), although the baseline CD4:8 ratios were similar between fresh and cryopreserved groups. We also noted that the Protein L percent (transduction efficiency) was higher in the cryopreserved samples post-thaw in the CD22 cohort (p = 0.02), but not in the CD19/22 cohort.
In vivo expansion and persistence
In vivo CAR T cell expansion was compared among cryopreserved and fresh CAR T cell products, based on peak expansion in the peripheral blood (Figure 3). In the CD22 cohort, there was no difference in expansion between cryopreserved and freshly infused cells, with median expansion of 82.5% CAR T cells and 76.6%, respectively (p = 0.34). Peak expansion was not different between groups whether measured by percent CAR T cells in peripheral blood or absolute number of CAR T cells per microliter. The proportion of patients who had low expansion (<10% of T cells expressing the CAR construct) did not differ between groups. The timing of peak expansion also did not differ, with median peak at day 15 for both cryopreserved and fresh cells. A detailed illustration of CAR T cell expansion over time can be found in Figure S2.
Figure 3.

Peak expansion and day 28 persistence in patients who received cryopreserved versus fresh CAR T cells
The results of testing for peak expansion and persistence of cryopreserved and freshly infused anti-C22 CAR T cells are shown in (A)–(D) and of anti-CD19/CD22 bispecific CAR T cells in (E)–(H). (A) and (E) show peak in vivo expansion in peripheral blood expressed as the percentage of T cells expressing the CAR construct. (B) and (F) show peak in vivo expansion in peripheral blood expressed as the absolute number of CAR T cells per microliter. Please note different y-axes used in these panels due to vastly different expansion between the two CAR constructs. (C) and (G) show day 28 CAR T cell persistence in bone marrow expressed as percentage of T cells expressing the CAR construct. Anti-CD22 CAR T cells were measured by the expression of the CD22 CAR and anti-CD19/CD22 CAR T cells by the expression of the CD19 CAR. All patients receiving anti-CD22 CAR T cells (n = 40) and anti-CD19/22 CAR T cells (n = 19) were evaluated for peak expansion and day 28 (+/− 4 days) bone marrow persistence. In addition, (D) and (H) show peak expansion with patients grouped by baseline disease burden in the CD22 and the CD19/22 cohorts, respectively, demonstrating the relationship between expansion and disease burden especially in the CD19/22 cohort.
There was no difference in CD22 CAR T cell persistence based on bone marrow biopsies on day 28. For those patients who had bone marrow biopsy data after day 28 and prior to transplant, the majority did have detectable CAR T cells, but there was no difference between cryopreserved (median 40.9% CAR T cells in bone marrow) and fresh infusion (median 30.5% CAR T cells in bone marrow) groups (p = 0.22). Among patients with available bone marrow data after 28 days, 9 of 10 patients who received fresh infusions and 11 of 13 patients who received cryopreserved cells had detectable CAR T cells.
In the bispecific CAR T cell cohort, there were some differences noted between fresh and cryopreserved cells after infusion. There was a trend toward greater expansion in the fresh infusion group, with median peak expansion of 50% for freshly infused cells and 10% for cryopreserved cells (p = 0.06). Expansion over the first month is shown in Figure S2. Persistence at 28 days was higher in patients who received fresh infusions compared with those who received cryopreserved cells, but overall persistence of this product was low, with a median of 4.5% CAR T cells in bone marrow in the fresh infusion group and 0.3% in the cryopreserved group (p = 0.01). There were insufficient data to compare persistence beyond 28 days, particularly because of the limited persistence of this CAR T cell construct overall.
To elucidate the role of baseline disease burden as a confounder, we compared peak expansion between patients with different levels of baseline disease. We found that in the CD19/22 cohort, in vivo CAR T cell expansion correlated with baseline disease burden (p < 0.01). Peak expansion grouped by baseline disease is illustrated in Figures 3D and 3H and is shown for cryopreserved and freshly infused cells separately in Figure S3. Expansion in proportion to baseline disease is an important contributor to adequate disease response to CAR T cell therapy. Day 28 persistence also correlated with baseline disease burden in the CD19/22 cohort (p < 0.01). Interestingly, in the CD22 cohort, where much greater absolute expansion was observed, the relationship between expansion and disease burden was more variable.
Toxicities
Incidence of CAR T cell-associated toxicities as well as peak levels of ferritin and C-reactive protein (CRP) are shown in Figure 4. The incidences of cytokine release syndrome (CRS), neurotoxicity, and lymphohistiocytosis (HLH)-like toxicities were similar between groups for both the CD22 and CD19/22 cohorts. In the CD22 cohort, the grade of CRS was similar, with a median grade of 2 for cryopreserved and 1 for freshly infused cells. The proportion of patients who developed severe CRS (grade 3 or higher) was also similar between groups (p = 0.7). In the CD19/22 cohort, higher grade CRS occurred in the patients who were infused with fresh cells, with median CRS grade 1.5, compared with median CRS grade 1 in patients who received cryopreserved cells (p = 0.03). The proportion of patients with severe CRS did not differ significantly between groups (p = 0.42).
Figure 4.

CAR T cell-associated toxicities for patients receiving cryopreserved versus fresh CAR T cells
CAR T cell-associated toxicities in patients who received anti-CD22 CAR T cells are shown in (A) and for those who received anti-CD19/CD22 CAR T cells in (D). The percentage of patients experiencing any grade of CRS, severe CRS defined as grades 3 or 4, neurotoxicity, or HLH are shown. The brackets show the p values for comparison of patients receiving cryopreserved and fresh products. Following the infusion of CAR T cells, ferritin and C-reactive protein (CRP) were measured for all patients. (B) shows peak ferritin levels and (C) shows peak CRP levels in patients receiving anti-CD22 CAR T cells (n = 40). For patients receiving anti-CD19/CD22 CAR T cells (n = 19), peak ferritin levels are shown in (E) and peak CRP levels in (F). Please note that scales on y axes differ between different cohorts in order to optimize viewing of individual data points.
The peak levels of ferritin and peak levels of CRP after CAR T cell infusion were similar for patients who received cryopreserved versus freshly infused cells in both cohorts. The median days at which peak CRP and peak ferritin levels were observed were also similar between groups. CRP peaked on day 12 for both fresh and cryopreserved groups in the CD22 cohort. Ferritin peaked at day 13 for fresh and day 14 for cryopreserved products. Similarly, in the CD19/22 cohort, CRP peaked at day 4 for fresh infusions and day 5 for cryopreserved products. Ferritin peak was at day 5 and day 4 for fresh and cryopreserved products, respectively.
Disease response
Importantly, for both CD22 and CD19/22 cohorts, disease response, based on presence of complete response (CR) as best overall response after infusion, did not differ between patients who received freshly infused cells and those who received cryopreserved cells (Figure 5).
Figure 5.

Disease response in patients who received cryopreserved versus fresh CAR T cells
The number of patients whose best clinical outcome was a complete response (CR) after infusion of cryopreserved versus fresh cells is shown for those receiving CD22 CAR T cells in (A) and CD19/CD22 CAR T cells in (B).
Plots showing more long-term follow-up information for patients in both cohorts are included in Figure S4.
Outcomes as a function of viability
Since the percent viability at infusion was a major difference between fresh and cryopreserved cells, we also examined the relationship between viability and clinical outcomes. In both cohorts, patients with low cell viability at infusion did tend to have lower peak expansion in vivo, but this did not lead to a significant correlation. In the CD19/22 cohort, lower viability did correlate with lower day 28 persistence (Figure S5). With the cohorts split into two groups by percent viability at infusion (above or below 90% for CD22 and above or below 70% for CD19/22), there was no difference in rate of complete response at 28 days, rate of CRS, grade of CRS, rate of neurotoxicity, or rate of HLH between the high and low viability groups.
Discussion
This study compared cell characteristics and clinical outcomes for two cohorts of patients who received either cryopreserved or freshly infused CAR T cells across two different constructs and using two different manufacturing platforms. We include a large cohort of patients who were treated with uniform dosing of anti-CD22 CAR T cells, which were manufactured using CD4/CD8 antibody selected T cells and bag culture. We also report data on patients who received bispecific anti-CD19/22 CAR T cells, which were manufactured using CD4/CD8 antibody selected T cells and the Prodigy system. We compare detailed clinical outcomes for both cohorts, including comparisons of baseline disease status and CAR T cell-associated toxicities.
Our results showed no significant difference between freshly infused or cryopreserved cells for anti-CD22 or bispecific CD19/22 CAR T cells in patients with B cell leukemia, with similar expansion, toxicity profiles, and disease response. Our observed difference in day 28 persistence in the CD19/22 cohort (4.5% CAR T cells in bone marrow for the fresh infusion group versus 0.3% in cryopreserved group), while statistically significant, may not be clinically significant. Importantly, of the two patients in the CD19/22 cohort who did not achieve a CR following infusion of a cryopreserved product, one had a CR to a second infusion, which was a cryopreserved product from the original manufacturing. The one patient in this cohort who failed to achieve a CR after a fresh infusion did have full clearance of his B-ALL but emerged with lineage switch and extramedullary disease, demonstrating CAR efficacy with immune escape. Overall, our results support the use of both CD22 and bispecific CD19/CD22 CAR T cryopreserved cells without compromising safety or efficacy.
Among patients in the CD19/22 cohort, those who received fresh infusions had higher baseline disease burden compared with those who received cryopreserved cells, which may account for the observed differences in day 28 persistence and CRS grade. High disease burden is generally associated with worse clinical outcomes and increased rates of CRS.27,28 While the relationship between disease burden and expansion has been variable in the literature,29,30,31 the results from our CD19/22 cohort show correlations between disease burden and expansion (p < 0.01) as well as day 28 persistence (p < 0.01). This is consistent with prior studies that have shown an association between high baseline disease burden and greater expansion of anti-CD19 CAR T cells.32,33,34
We also examined whether lower percent viability, which was noted in a larger proportion of cryopreserved samples, was related to clinical outcomes. In our patients, cell dose is calculated based on absolute number of viable cells at infusion, so the percent viability from the thawed samples should not alter the number of viable cells infused. However, we did find a correlation between viability and persistence for the CD19/22 cohort. In this group, as discussed above, there was overlap between the cryopreserved samples with lower viability and patients who had lower baseline disease, which may account for the low persistence. It is also possible that among cryopreserved samples, cells that are viable at the time of thaw undergo delayed apoptosis after infusion or have decreased in vivo activation or proliferation.35 Importantly, even with these potential differences, we did not observe different rates of toxicity or disease response.
For the CAR T cells that were cryopreserved, it is also interesting to note the pre- and post-thaw differences in cell characteristics. The patterns were similar for both CD22 and bispecific CAR T cells, with higher percentage of cells reacting with Protein L and lower CD4:8 ratios post-thaw. Since there was no notable difference in CD3 percent, this suggests that the cells that do survive the freeze-thaw cycle are more likely to be those that were transduced. Since dosing is based on the pre-cryopreservation flow measurements, this also implies that patients who received cryopreserved cells may have received a higher proportion of transduced cells. The difference in CD4:8 ratio similarly suggests that CD8 cells are more likely to tolerate the freeze-thaw cycle. Others have reported that naive, central memory, and CD4+CD25+ (Treg) cells are more affected by cryopreservation than effector memory cells,36,37 leading to higher proportions of effector cells in thawed products. This may explain our observed change in CD4:8 ratios.
Since manufacturing methods, such as use of different costimulatory molecules or cell selection procedures, can affect CAR T cell function,38,39 it is worth nothing the differences in methods used to produce our CD22 and bispecific CAR T cells. Both the CD22 and CD19/CD22 vectors use the 4-1BB costimulatory molecule. Both of the apheresis products also underwent CD4/8 selection. Notably, the CD22 CAR T cells were produced using bag cultures, while bispecific cells were made using a closed system in the CliniMACS Prodigy. These systems involve different T cell activation methods and different culture conditions. We have previously shown that different activation methods affect the proportions of T cell subtypes.40 We are currently investigating how cells produced in the Prodigy system may differ from CAR T cells produced in bag culture, which could provide insights into whether certain manufacturing platforms are predisposed to a greater sensitivity to cryopreservation. Preliminary results show that cells produced in the Prodigy have a more naive memory cell phenotype,41 which may be more sensitive to cryopreservation compared with effector memory cells.36,37
Among prior publications, cells produced in an automated system have been infused fresh42 as well as thawed.43 A study by Maschan et al. describes the clinical efficacy of freshly infused CD19-targeted CAR T cells produced using the Prodigy, but it also includes preclinical data showing that freshly infused cells had increased anti-tumor activity compared with cryopreserved cells in a mouse lymphoma model.44 The publication by Shah et al.,23 which showed inferior outcomes with cryopreserved CAR T cells, is the only previous direct clinical comparison between fresh and cryopreserved cells manufactured in the Prodigy system. A limitation of the Shah et al. study is the notably prolonged culture time, with a 14-day culture instead of a 7-day culture. Longer culture periods may be associated with T cell differentiation,45 so it is possible that the prolonged culture time also yields a different T cell phenotype, more sensitive to cryopreservation.46 Furthermore, prolonged culture has also been associated with impaired anti-tumor efficacy, which confounds the attribution of worse outcomes solely to cryopreservation.47
While the aim of this study was not to compare anti-CD22 and bispecific CD19/22 CAR T cells, we observed some differences in clinical response between the two cohorts, with overall lower peak expansion, ferritin, and CRP levels and lower incidence of toxicities in the bispecific cohort compared with the CD22 cohort. These differences are described in the previous publication reporting results of the CD19/22 CAR T cell phase I trial.48
There are several limitations to this study. Most importantly, this is a retrospective analysis, and the decision to cryopreserve CAR T cells was not randomly assigned. While the baseline characteristics of patients receiving cryopreserved and freshly infused cells were comparable in the CD22 cohort, the difference in baseline disease burden in our bispecific cohort makes it difficult to interpret differences in outcomes related to expansion and persistence. Treatment for patients with greater disease burden was preferentially expedited, so they were more likely to be given fresh cells, which is particularly evident in the CD19/22 cohort. Nonetheless, one patient who received cryopreserved CD19/22 CAR T cells had a baseline disease burden of >90% and, accordingly, had high peak expansion and CAR T cell persistence. Despite lack of randomization, the relatively large sample size and homogeneous population in the CD22 group provides convincing support for the safety and efficacy of cryopreserved CAR T cells. The results of the CD19/22 cohort also support the efficacy of cryopreserved CAR T cells for patients with both high and low disease burden. A larger sample size will be needed to demonstrate a consistent response to cryopreserved CD19/22 CAR T cells in patients with high baseline disease. Additional limitations include lack of post-thaw cell characterization data for all patients. As more CAR T cell products are being cryopreserved, our center has now started routinely measuring post-thaw CD3 percent, CD4:8 ratios, and Protein L percent on all samples, so this information will be available for future analyses. Furthermore, we do not have more detailed flow cytometry data for phenotypic characterization of CAR T cells, which may elucidate more subtle differences in the future. We also excluded data for the second infusion among several patients who received repeated infusions of CAR T cells. Finally, long-term follow-up data are limited especially in the bispecific cohort, which includes more recently treated patients. Future work may include long-term follow-up of both cohorts of patients as well as comparison of outcomes from cell products that were infused first fresh and then reinfused after cryopreservation.
In conclusion, cryopreserved CAR T cells produced in bag cultures and in the Prodigy closed system have demonstrated the ability to expand and to effectively target leukemia in vivo. This study, in combination with previously reported and ongoing trials that use cryopreserved CAR T cells, adds to the body of literature supporting use of cryopreservation routinely in CAR T cell manufacture.
Materials and methods
We retrospectively reviewed two cohorts of pediatric and young adult patients with relapsed/refractory B cell leukemia or lymphoma who were treated with CAR T cell therapy at the NIH Clinical Center. Patients received either anti-CD22 CAR T or bispecific anti-C19/22 CAR T cells as part of phase I/II dose-escalation trials for these CAR T cell therapies (NCT02315612, NCT03448393). Trial protocols were approved by the National Cancer Institute institutional review board.
Patients who received CAR T cell therapy prior to October 1, 2022, were screened for inclusion. Only patients who received CD4/8 selected cells were included. The patient cohort included in this study, which included only patients who had CAR T cells manufactured from CD4/8 selected cells and infused at the final expansion dose, did not overlap with the 16 patients reported previously by Panch et al. Only patients who received the final dose chosen for enrollment expansion were included. For the CD22 CAR T cell cohort, these were patients who received dose level 1, 0.3 × 106 cells/kg. For the bispecific CAR T cell cohort, the expansion dose was dose level 3, 3 × 106 cells/kg. For patients who received more than one cell infusion, only data from the first infusion were included. Figure 1 summarizes the patient selection.
CAR T cell manufacturing
Anti-CD22 and bispecific anti-CD19/CD22 CAR T cells were manufactured at the Center for Cellular Engineering, NIH Clinical Center. Anti-CD22 CAR T cells were manufactured as previously described.48 Briefly, for anti-CD22 CAR T cells, the mononuclear cell (MNC) leukapheresis product used to begin manufacturing was either freshly collected or cryopreserved and thawed. The target for cell collection is 3–6 x 109 CD3+ cells. The MNCs underwent CD4/8 selection using magnetic bead separation in the Miltenyi CliniMACS system. Culture was then initiated with 1.5–2 x 106 cells/mL in bags with volume ranging from 20 to 325 mL depending on the required cell dose. Culture medium was supplemented with GlutaMAX and IL-2. CD3/CD28 Dynabeads were used for activation. Lentiviral transduction with a vector containing CD22 single-chain variable fragments (scFvs) with 41BB and CD3zeta costimulatory domains was conducted on day 2. Cells then expanded in culture for 9 days, with transduction efficiency being measured on day 7 using flow cytometry to determine the percent of cells reacting with Protein L. At the time of harvest on day 9, evaluation of cells following expansion included flow cytometry for CD3, CD4, and CD8 percentage. Cells were then either used for fresh infusion or cryopreserved, depending on clinical scenario.
Leukapheresis and CD4/8 selection was similarly conducted for bispecific CD19/22 CAR T cells. The cell manufacturing was completed as previously described.49 Culture was initiated with cell counts ranging from 110–132 × 106 cells, in medium supplemented with IL-2. TransAct CD3/CD28 beads were used for activation. Transduction and expansion were performed using the CliniMACS Prodigy enclosed CAR T manufacturing system. Cells were transduced with a lentiviral vector containing two distinct scFv recognizing CD19 and CD22 proteins as well as 41BB and CD3zeta costimulatory domains. The cells were transduced after 1 day of culture and were harvested after 7 days of culture. Cell counts and viabilities were monitored on day 5. After harvest, cells were evaluated by flow cytometry as above.
Cryopreservation
Products were cryopreserved using a controlled rate freezer. Clinical aliquots were preserved at 1.3–2x the required volume. Cryopreservation was done either in cryovials (1.8–4.5 mL) or cryobags (25–50 mL), depending on the required total volume. Cell concentration ranged from 1–100 × 106 cells/mL for vials or 20–300 × 106 cells/mL for bags. Two different cryoprotectants were used. For earlier patients, cells were preserved in 5% dimethyl sulfoxide (DMSO), 6% pentastarch, and 4% human serum albumin. We then transitioned to using CryoStor, a pre-made cryopreservation media from Sigma-Aldrich that contains 10% DMSO. This transition affected one of the reported anti-CD22 CAR T cell products and six of the reported bispecific CAR T cell products.
Thaw of cryopreserved products
Thawing was performed in a water bath at 37°C. Immediately after thawing, products were diluted to a final required volume with Plasma-Lyte, containing 10 u/mL preservative-free heparin. Post-thaw quality control was completed on all samples, including WBC count, Trypan-blue viability, and sterility testing. Flow cytometry post-thaw was only conducted for selected products.
Cell infusions
Anti-CD22 CAR T cells were infused at a dose of 0.3 × 106 cells/kg. Bispecific CD19/22 CAR T cells were infused at a dose level of 3 × 106 cells/kg. The final cell doses were calculated as a product of total cell count, percent viability, percent CD3, and transduction efficiency. Importantly, these calculations are completed at the end of manufacture, prior to cryopreservation. Patients were treated with a lymphodepleting regimen including fludarabine and cyclophosphamide.
Cell manufacturing characteristics
Data on the following cell manufacturing characteristics were collected at the time of harvest from cell culture: percent viability, fold expansion, transduction efficiency (measured by Protein L percent), as well as CD3, CD4, and CD8 percent. Fold expansion was calculated from day 1 to day of harvest (day 9 for CD22 CAR T cells and day 7 for Bispecific CD19/22 cells). For cells that were cryopreserved, the number of days of cryopreservation was documented. Furthermore, post-thaw viability was also measured. For some cell products, post-thaw data on CD3, CD4, and CD8 percent and transduction efficiency was also available. These baseline cell characteristics were compared between freshly infused and cryopreserved groups using Mann-Whitney U tests. Paired comparisons for pre- and post-thaw products were conducted using Wilcoxon signed rank tests.
Clinical characteristics and outcomes
Data on the following baseline characteristics were collected: demographics, disease burden prior to cell infusion, presence of CNS disease, presence of extramedullary disease, and number of lines of prior treatment, specifying prior CAR T cell or stem cell transplant therapy. Clinical outcomes included in vivo CAR T cell expansion and persistence, incidence, severity, and day of onset for CAR T cell associated toxicities (CRS, neurotoxicity, and CAR T cell-associated hemophagocytic HLH), and best overall disease response. Cell expansion was measured by both percent of T cells that expressed CAR T cell targets (CD22 for the first cohort and CD19 for the bispecific cohort) and absolute number of CAR T cells in peripheral blood. CAR T cell persistence was reported based on percentage of CAR T cells identified by flow cytometry in the day 28 bone marrow sample. Since some of these patients were treated prior to utilization of the immune effector cell-associated neurotoxicity syndrome scoring system,50 neurotoxicity was more broadly defined and included any neurologic symptoms, including headache, documented in the post-infusion period.
Continuous variables were compared between patients in the fresh infusion and cryopreserved groups using Mann-Whitney U tests, and categorical variables were compared using chi squared, Fisher’s exact, and Wilcoxon rank-sum tests. Correlations between continuous variables were calculated using Spearman’s correlation. Comparisons between multiple groups were done using Kruskal-Wallis tests. Statistical analyses were performed using GraphPad Prism software.
Acknowledgments
We gratefully acknowledge the study participants and their families, referring medical care teams, the faculty and staff of the NIH Clinical Center who provided their expertise in the management of the study participants, and the data managers and research nurses and patient care coordinators involved with this work. We also acknowledge the cell manufacturing team at the NIH Center for Cell Engineering who produced the CAR T cell products and provided the manufacturing data analyzed in this study.
This work was supported in part by the Intramural Research Program, Center of Cancer Research, National Cancer Institute, and NIH Clinical Center, National Institutes of Health (ZIA BC 011823, N.N.S).
The graphical abstract was made using Biorender.com.
Author contributions
A.D., S.P., N.N.S., and D.S. contributed to the conceptualization of the study. A.D., B.Y., H.S., and N.N.S. contributed to data curation. A.D. completed data analysis and wrote the original draft. D.S. and N.N.S. provided supervision. All authors contributed to review and editing of the manuscript.
Declaration of interests
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. The authors have no conflicts of interest to disclose.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.12.004.
Contributor Information
David Stroncek, Email: dstroncek@mail.cc.nih.gov.
Nirali N. Shah, Email: nirali.shah@nih.gov.
Supplemental information
Data availability
The data that support the findings in this study are stored in a controlled access repository due to sensitive contents involving protected health information. Deidentified data can be available from the corresponding author upon reasonable request.
References
- 1.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. JULIET Investigators Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 3.Martinez M., Moon E.K. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin Y.J., Mashouf L.A., Lim M. CAR T cell therapy in primary brain tumors: current investigations and the future. Front. Immunol. 2022;13:817296. doi: 10.3389/fimmu.2022.817296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li R., Johnson R., Yu G., McKenna D.H., Hubel A. Preservation of cell-based immunotherapies for clinical trials. Cytotherapy. 2019;21:943–957. doi: 10.1016/j.jcyt.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meneghel J., Kilbride P., Morris G.J. Cryopreservation as a key element in the successful delivery of cell-based therapies-A review. Front. Med. 2020;7:592242. doi: 10.3389/fmed.2020.592242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holland E.M., Molina J.C., Dede K., Moyer D., Zhou T., Yuan C.M., Wang H.W., Stetler-Stevenson M., Mackall C., Fry T.J., et al. Efficacy of second CAR-T (CART2) infusion limited by poor CART expansion and antigen modulation. J. Immunother. Cancer. 2022;10:e004483. doi: 10.1136/jitc-2021-004483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyagarajan S., Schmitt D., Acker C., Rutjens E. Autologous cryopreserved leukapheresis cellular material for chimeric antigen receptor-T cell manufacture. Cytotherapy. 2019;21:1198–1205. doi: 10.1016/j.jcyt.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Stroncek D.F., Xing L., Chau Q., Zia N., McKelvy A., Pracht L., Sabatino M., Jin P. Stability of cryopreservedwhite blood cells (WBCs) prepared for donor WBC infusions. Transfusion. 2011;51:2647–2655. doi: 10.1111/j.1537-2995.2011.03210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bashey A., Manion K.L., Smith P., Mccollum J., Holland H.K., Morris L.E., Solomon S.R. Cryopreserved G-CSF mobilized donor lymphocyte infusion (DLI) without withdrawal of immunosuppression is safe and highly effective for treating isolated poor donor T-cell chimerism following reduced intensity allogeneic transplantation (RICT) Blood. 2009;114:3308. [Google Scholar]
- 11.Panch S.R., Srivastava S.K., Elavia N., McManus A., Liu S., Jin P., Highfill S.L., Li X., Dagur P., Kochenderfer J.N., et al. Effect of cryopreservation on autologous chimeric antigen receptor T cell characteristics. Mol. Ther. 2019;27:1275–1285. doi: 10.1016/j.ymthe.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kymriah [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2020.
- 13.Yescarta [package insert]. Santa Monica, CA: Kite Pharma, Inc.; 2022.
- 14.Breyanzi [package insert]. Bothel, WA: Juno therapeutics, Inc.; 2021.
- 15.Locke F.L., Neelapu S.S., Bartlett N.L., Siddiqi T., Chavez J.C., Hosing C.M., Ghobadi A., Budde L.E., Bot A., Rossi J.M., et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol. Ther. 2017;25:285–295. doi: 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abramson J.S., Palomba M.L., Gordon L.I., Lunning M.A., Wang M., Arnason J., Mehta A., Purev E., Maloney D.G., Andreadis C., et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–852. doi: 10.1016/S0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 17.Xu H., Cao W., Huang L., Xiao M., Cao Y., Zhao L., Wang N., Zhou J. Effects of cryopreservation on chimeric antigen receptor T cell functions. Cryobiology. 2018;83:40–47. doi: 10.1016/j.cryobiol.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Berens C., Heine A., Müller J., Held S.A.E., Mayer K., Brossart P., Oldenburg J., Pötzsch B., Wolf D., Rühl H. Variable resistance to freezing and thawing of CD34-positive stem cells and lymphocyte subpopulations in leukapheresis products. Cytotherapy. 2016;18:1325–1331. doi: 10.1016/j.jcyt.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 19.Pi C.H., Hornberger K., Dosa P., Hubel A. Understanding the freezing responses of T cells and other subsets of human peripheral blood mononuclear cells using DSMO-free cryoprotectants. Cytotherapy. 2020;22:291–300. doi: 10.1016/j.jcyt.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haider P., Hoberstorfer T., Salzmann M., Fischer M.B., Speidl W.S., Wojta J., Hohensinner P.J. Quantitative and functional assessment of the influence of routinely used cryopreservation media on mononuclear leukocytes for medical research. Int. J. Mol. Sci. 2022;23:1881. doi: 10.3390/ijms23031881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tompa A., Nilsson-Bowers A., Faresjö M. Subsets of CD4+, CD8+, and CD25hi lymphocytes are in general not influenced by isolation and long-term cryopreservation. J. Immunol. 2018;201:1799–1809. doi: 10.4049/jimmunol.1701409. [DOI] [PubMed] [Google Scholar]
- 22.Zhu F., Shah N., Xu H., Schneider D., Orentas R., Dropulic B., Hari P., Keever-Taylor C.A. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy. 2018;20:394–406. doi: 10.1016/j.jcyt.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Shah N.N., Johnson B.D., Schneider D., Zhu F., Szabo A., Keever-Taylor C.A., Krueger W., Worden A.A., Kadan M.J., Yim S., et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat. Med. 2020;26:1569–1575. doi: 10.1038/s41591-020-1081-3. [DOI] [PubMed] [Google Scholar]
- 24.Brezinger-Dayan K., Itzhaki O., Melnichenko J., Kubi A., Zeltzer L.A., Jacoby E., Avigdor A., Shapira Frommer R., Besser M.J. Impact of cryopreservation on CAR T production and clinical response. Front. Oncol. 2022;12:1024362. doi: 10.3389/fonc.2022.1024362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blaeschke F., Stenger D., Kaeuferle T., Willier S., Lotfi R., Kaiser A.D., Assenmacher M., Döring M., Feucht J., Feuchtinger T. Induction of a central memory and stem cell memory phenotype in functionally active CD4+ and CD8+ CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19+ acute lymphoblastic leukemia. Cancer Immunol. Immunother. 2018;67:1053–1066. doi: 10.1007/s00262-018-2155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W., Jordan K.R., Schulte B., Purev E. Characterization of clinical grade CD19 chimeric antigen receptor T cells produced using automated CliniMACS prodigy system. Drug Des. Dev. Ther. 2018;12:3343–3356. doi: 10.2147/DDDT.S175113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hay K.A., Hanafi L.A., Li D., Gust J., Liles W.C., Wurfel M.M., López J.A., Chen J., Chung D., Harju-Baker S., et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017;130:2295–2306. doi: 10.1182/blood-2017-06-793141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li M., Xue S.L., Tang X., Xu J., Chen S., Han Y., Qiu H., Miao M., Xu N., Tan J., et al. The differential effects of tumor burdens on predicting the net benefits of ssCART-19 cell treatment on r/r B-ALL patients. Sci. Rep. 2022;12:378. doi: 10.1038/s41598-021-04296-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Talleur A.C., Qudeimat A., Métais J.Y., Langfitt D., Mamcarz E., Crawford J.C., Huang S., Cheng C., Hurley C., Madden R., et al. Preferential expansion of CD8+ CD19-CAR T cells postinfusion and the role of disease burden on outcome in pediatric B-ALL. Blood Adv. 2022;6:5737–5749. doi: 10.1182/bloodadvances.2021006293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng B., Pan J., Liu Z., Liu S., Chen Y., Qu X., Zhang Y., Lin Y., Zhang Y., Yu X., et al. Peripheral leukemia burden at time of apheresis negatively affects the clinical efficacy of CART19 in refractory or relapsed B-ALL. Mol. Ther. Methods Clin. Dev. 2021;23:633–643. doi: 10.1016/j.omtm.2021.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mueller K.T., Maude S.L., Porter D.L., Frey N., Wood P., Han X., Waldron E., Chakraborty A., Awasthi R., Levine B.L., et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130:2317–2325. doi: 10.1182/blood-2017-06-786129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curran K.J., Margossian S.P., Kernan N.A., Silverman L.B., Williams D.A., Shukla N., Kobos R., Forlenza C.J., Steinherz P., Prockop S., et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134:2361–2368. doi: 10.1182/blood.2019001641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finney O.C., Brakke H.M., Rawlings-Rhea S., Hicks R., Doolittle D., Lopez M., Futrell R.B., Orentas R.J., Li D., Gardner R.A., Jensen M.C. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J. Clin. Invest. 2019;129:2123–2132. doi: 10.1172/JCI125423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baust J.G., Snyder K.K., Van Buskirk R., Baust J.M. Integrating molecular control to improve cryopreservation outcome. Biopreserv. Biobanking. 2017;15:134–141. doi: 10.1089/bio.2016.0119. [DOI] [PubMed] [Google Scholar]
- 36.Li B., Yang C., Jia G., Liu Y., Wang N., Yang F., Su R., Shang Y., Han Y. Comprehensive evaluation of the effects of long-term cryopreservation on peripheral blood mononuclear cells using flow cytometry. BMC Immunol. 2022;23:30. doi: 10.1186/s12865-022-00505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg A., Song L.Y., Wilkening C., Sevin A., Blais B., Louzao R., Stein D., Defechereux P., Durand D., Riedel E., et al. Pediatric ACTG Cryopreservation Working Group. Optimization and limitations of use of cryopreserved peripheral blood mononuclear cells for functional and phenotypic T-cell characterization. Clin. Vaccine Immunol. 2009;16:1176–1186. doi: 10.1128/CVI.00342-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savoldo B., Ramos C.A., Liu E., Mims M.P., Keating M.J., Carrum G., Kamble R.T., Bollard C.M., Gee A.P., Mei Z., et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah N.N., Highfill S.L., Shalabi H., Yates B., Jin J., Wolters P.L., Ombrello A., Steinberg S.M., Martin S., Delbrook C., et al. CD4/CD8 T-Cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J. Clin. Oncol. 2020;38:1938–1950. doi: 10.1200/JCO.19.03279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keskar V., Sood A., Loghin E., Kovacs E., Duthie R.S., Liu S., Park J.H., Chadwick C., Smith R., Brown M., et al. Novel DNA-based T-cell activator promotes rapid T-cell activation and expansion. J. Immunother. 2020;43:231–235. doi: 10.1097/CJI.0000000000000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song H.W., Shao L., Prochazkova M., Cheuk A., Jin P., Stroncek D., Khan J., Highfill S. Poster Presentation. Society for ImmunoTherapy of Cancer. 2021. Comparison of CAR-T cell manufacturing platforms reveals distinct phenotypic and transcriptional profiles. [Google Scholar]
- 42.Jackson Z., Roe A., Sharma A.A., Lopes F.B.T.P., Talla A., Kleinsorge-Block S., Zamborsky K., Schiavone J., Manjappa S., Schauner R., et al. Automated manufacture of autologous CD19 CAR T cells for treatment of non-hodgkin lymphoma. Front. Immunol. 2020;11:1941. doi: 10.3389/fimmu.2020.01941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ortíz-Maldonado V., Rives S., Castellà M., Alonso-Saladrigues A., Benítez-Ribas D., Caballero-Baños M., Baumann T., Cid J., Garcia-Rey E., Llanos C., et al. CART19-BE-01: a multicenter trial of ARI-0001 cell therapy in patients with CD19+ relapsed/refractory malignancies. Mol. Ther. 2021;29:636–644. doi: 10.1016/j.ymthe.2020.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maschan M., Caimi P.F., Reese-Koc J., Sanchez G.P., Sharma A.A., Molostova O., Shelikhova L., Pershin D., Stepanov A., Muzalevskii Y., et al. Multiple site place-of-care manufactured anti-CD19 CAR T cells induce high remission rates in B-cell malignancy patients. Nat. Commun. 2021;12:7200. doi: 10.1038/s41467-021-27312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herda S., Heimann A., Obermayer B., Ciraolo E., Althoff S., Ruß J., Grunert C., Busse A., Bullinger L., Pezzutto A., et al. Long-term in vitro expansion ensures increased yield of central memory T cells as perspective for manufacturing challenges. Int. J. Cancer. 2021;148:3097–3110. doi: 10.1002/ijc.33523. [DOI] [PubMed] [Google Scholar]
- 46.Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghassemi S., Nunez-Cruz S., O'Connor R.S., Fraietta J.A., Patel P.R., Scholler J., Barrett D.M., Lundh S.M., Davis M.M., Bedoya F., et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol. Res. 2018;6:1100–1109. doi: 10.1158/2326-6066.CIR-17-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B., et al. CD22-targeted CART cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shalabi H., Qin H., Su A., Yates B., Wolters P.L., Steinberg S.M., Ligon J.A., Silbert S., DéDé K., Benzaoui M., et al. CD19/22 CAR T cells in children and young adults with B-ALL: phase I results and development of a novel bicistronic CAR. Blood. 2022;140:451–463. doi: 10.1182/blood.2022015795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee D.W., Santomasso B.D., Locke F.L., Ghobadi A., Turtle C.J., Brudno J.N., Maus M.V., Park J.H., Mead E., Pavletic S., et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transplant. 2019;25:625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings in this study are stored in a controlled access repository due to sensitive contents involving protected health information. Deidentified data can be available from the corresponding author upon reasonable request.