

Abstract
According to the Developmental Origins of Health and Disease (DOHaD) hypothesis, the intrauterine environment influences fetal programming and development, affecting offspring disease susceptibility in adulthood. In recent years, therapeutic use of the Type 2 diabetes drug metformin has expanded to the treatment of pre-diabetes, polycystic ovarian syndrome, and gestational diabetes. Because metformin both undergoes renal excretion and binds to receptors on the placenta, the fetus receives equivalent maternal dosing. Although no teratogenic nor short-term harmful fetal impact of metformin is known to occur, the effects of metformin exposure on longer-range offspring development have not yet been fully elucidated. This review encapsulates the (albeit limited) existing knowledge regarding the potential longer-term impact of intrauterine metformin exposure on the development of key organs including the liver, central nervous system, heart, gut, and endocrine pancreas in animal models and humans. We discuss molecular and cellular mechanisms that would be altered in response to treatment and describe the potential consequences of these developmental changes on postnatal health. Further studies regarding the influence of metformin exposure on fetal programming and adult metabolic health will provide necessary insight to its long-term risks, benefits, and limitations in order to guide decisions for use of metformin during pregnancy.
Keywords: metformin, pregnancy, gestational diabetes, preeclampsia, polycystic ovarian syndrome, fetal growth restriction
INTRODUCTION
Inspired by epidemiological findings in England and Wales that demonstrated a correlation between living in poorer areas and increased prevalence of ischemic heart disease [1], Dr. David Barker and colleagues analyzed health outcomes in the offspring of pregnant women who experienced the Dutch Hunger Winter of November 1944 through April 1945. The findings of these studies revealed that children exposed to undernutrition in utero were prone to the development of cardiometabolic disease and impaired glucose tolerance in adulthood, while offspring born to the same parents outside of the period of famine did not show these tendencies [2–4]. Thus, the Developmental Origins of Health and Disease (DOHaD) hypothesis was born, which postulates that the intrauterine environment alters fetal developmental programming in a way that influences susceptibility to metabolic disease in adulthood [5].
More recently, the increased incidence of obesity in women of childbearing age has spurred research that seeks to understand the influence of maternal overnutrition on the metabolic health of offspring. A meta-analysis of 23 published studies examining a combined total of over 1.3 million women across multiple ethnicities revealed that women in the United States and Europe have higher pre-pregnancy body mass index (BMI) and gain more weight during pregnancy than women of Asian descent, leading to an increased number of offspring that are large for gestational age in these populations [6]. Likewise, a study from Portugal revealed that offspring exposed to maternal overnutrition and obesity have increased BMI from infancy to two years of age [7]. Children with obesity are more likely to be obese as adults, which is associated with impaired glucose tolerance and increased mortality [8–10].
In non-human primate (NHP) offspring, a maternal calorically dense, high fat or Western style diet (WSD) results in reduced oxidative capacity in fetal skeletal muscle and impaired insulin-induced glucose uptake in the skeletal muscle of juvenile offspring, even in the absence of maternal obesity [11, 12]. In this same model, maternal WSD also alters the ratio of insulin-producing beta cells to glucagon-producing alpha cells in pancreatic islets, with a decrease in alpha cell number leading to an increased proportion of beta cells. Islets from NHP offspring exposed to overnutrition during development also show an over-secretion of insulin in response to a rise in glucose [13]. Of note, NHP offspring exposed to maternal WSD during development demonstrate increased reactive and ritualized anxiety, demonstrating an effect of maternal diet on offspring behavior in addition to physiology [14]. At a mechanistic level, we have shown that this largely occurs in humans and primates via two overarching means: 1) altered offspring gene expression driven by epigenetic changes to the early life “histone code” [15–21] and 2) alterations in the developing microbiome [22–24]. Taken together, these studies demonstrate that maternal WSD consumption, in both the absence and presence of obesity, impacts the development of various organ systems in offspring. The sum of the changes in fetal development predisposes offspring to metabolic disease and potentially to mental health issues later in life. Thus, lifestyle changes and treatments that reduce maternal obesity and overnutrition are predicted to have great impact on subsequent generations [25, 26].
In this review, we discuss what is known about metformin—a commonly used therapeutic for the treatment of Type 2 diabetes (T2D) whose use has recently expanded to include treatment of pre-diabetes, gestational diabetes mellitus (GDM), and polycystic ovarian syndrome (PCOS)—and its effects on fetal development and postnatal physiology. Additionally, we will explore how the mechanisms of action of metformin might influence the development of the fetal liver, brain, heart, gut (microbiome) and pancreas. Finally, we review clinical outcomes from studies of women who received metformin in pregnancy and their offspring, which suggest that the cellular processes by which maternal metformin acts may be disruptive to normal fetal growth and may ultimately result in childhood metabolic disease.
Metformin use in pregnancy
Women who are overweight or obese are prone to diseases associated with metabolic dysfunction, including PCOS, pre-diabetes, T2D, and GDM [27, 28]. These conditions are associated with impairments in insulin secretion and glucose homeostasis, introducing concerns regarding exposure of the fetus to hyperglycemia during pregnancy. Offspring of women with poorly managed GDM have increased adiposity, are large for gestational age, and have increased cardiometabolic risk [29, 30]. Long-term studies reveal that glycemic disorders in pregnant women predispose offspring to insulin resistance and T2D. These studies substantiate the notion that poor management of the maternal metabolic environment during pregnancy programs poor metabolic compensation in offspring later in life. Thus, it is imperative that maternal dysglycemia be appropriately managed and treated for the benefit of both the mother and her offspring.
Approved in 1994 by the Food and Drug Administration (FDA), metformin is the frontline therapeutic for T2D due to its low cost, oral delivery, and efficiency in reducing hyperglycemia [31]. Usual dosing of metformin starts at 500 mg nightly for one week, and increases to 500 mg twice daily up to a maximum dose of 2000 mg per day [32]. After being absorbed in the jejunum via organic cation transporters, metformin targets the liver, where it reduces gluconeogenesis and glycogenolysis, thus helping to lower blood glucose. Unlike most other medications, metformin is excreted by the kidneys without first pass hepatic metabolism [33, 34]. Metformin also acts on adipose tissue and skeletal muscle to improve glucose uptake and insulin sensitivity by increasing translocation of the glucose transporter, GLUT4 to the plasma membrane [35, 36]. Well-tolerated by most people, metformin has been deemed a “miracle drug” for its anti-hyperglycemic and anti-obesity effects. However, 30–40% of patients experience clinically significant gastrointestinal (GI) side effects, including diarrhea, vomiting and bloating, and elevations of hepatic transaminases. These common GI side effects are due to two mechanisms: first, the high turnover of glucose and bile acids in the intestine as metformin accumulates; and second, the serious albeit rare side effect of lactic acidosis due to increased anaerobic metabolism of glucose secondary to increased glucose uptake [37]. Forslund et al. (2015) demonstrated that metformin-driven gut dysbiosis resulted from aberrant microbial-mediated fatty acid, butyrate, and tryptophan degradation [38].
Given its success in effectively treating hyperglycemia, therapeutic use of metformin has been expanded for the treatment of GDM, pregestational diabetes, PCOS, weight management, cancer, and infertility in patients undergoing in vitro fertilization (Figure 1) [39–41]. Metformin has also been trialed in the context of maternal obesity and pre-eclampsia to improve maternal and fetal outcomes [32, 42]. Use of metformin in the management of GDM occurs despite the lack of approval by the FDA for this clinical indication [43]. In light of this increased use among women who are pregnant, it is worthwhile recalling that metformin readily crosses the placental barrier through organic cation transporters, resulting in the developing fetus experiencing the full dose of metformin as administered to mothers during pregnancy [44]. There is also documented potential for increased bioavailability of metformin during pregnancy [45]. However, it is important to note that congenital anomalies associated with first trimester metformin treatment are rare [46].
Figure 1. Reasons for metformin use in pregnancy.

Metformin is increasingly prescribed beyond the treatment of Type 2 diabetes and gestational diabetes. Created with Biorender.com
Due to low rates of aerobic as compared to aerobic metabolism during early pregnancy, the embryo has few, immature mitochondria [47]. As such, in the first trimester, the embryo expresses very low levels of organic cation transporters which are responsible for transporting metformin into cells [47]. For this reason, metformin is likely safe in the first trimester. However, in the second and third trimesters, the placenta and fetus both express metformin transporters, exhibit high rates of aerobic metabolism, and are dependent on the activity of mature mitochondria [47]. Given metformin’s inhibitory effects on the mitochondrial pathways, there is concern that metformin could adversely affect the function, growth, or differentiation of fetal or placental tissues into which metformin is transported [48]. These properties raise the possibility that metformin might induce fetal programming that may be maladaptive to metabolic health later in adulthood. Characterization of the cellular processes by which metformin acts on the developing fetus and placenta during pregnancy is crucial to understanding how this medication has the potential to induce developmental programming of metabolic disease in offspring and will be discussed in subsequent sections.
Mechanisms of metformin action
Intracellularly, metformin is thought to act mainly by altering the redox state of the cytoplasm to activate 5’ adenosine monophosphate activated protein kinase (AMPK), a nutrient-sensing kinase that regulates cellular growth and metabolism. AMPK activation occurs via direct inhibition of Complex I of the mitochondrial electron transport chain, raising the intracellular AMP:ATP ratio while also increasing the presence of reactive oxygen species (ROS) [49]. Additionally, some studies suggest that AMPK is activated via direct phosphorylation by intracellular kinases that respond to the cellular redox status such as liver kinase B (LKB/STK11) and ATM via an indirect mechanism [50]. Interference with mitochondrial respiration increases glycolytic flux, resulting in the buildup of lactate, which accounts for the increased incidence of lactic acidosis observed in patients prescribed metformin [51, 52]. A study that analyzed effects of in vitro treatment of embryonic day (e) 8.5 mouse embryos with metformin for 6, 12, and 24 hours found increased glucose uptake and glycolysis in response as well as high lactate, indicating that metformin also induces high levels of lactate in embryos [53].
Another outcome of AMPK activation is the inhibition of protein synthesis regulated by the mammalian target of rapamycin (mTOR) complex, which is critical for fetal growth (Figure 2). The mTOR signaling pathway is a major nutrient-sensing mechanism in the placenta [47]. Consequently, by inhibiting the mTOR pathway, metformin could restrict nutrients like glucose and amino acids from reaching the feto-placental unit [54, 55]. In a study of late gestation mouse embryos (e18.5), offspring of dams that consumed a high fat diet (HFD) containing 60% kcal from fat and treated with a metformin via the drinking water unexpectedly weighed less than offspring of dams that consumed a control diet while treated with the same dose of metformin. Placental evaluation revealed a decrease in phosphorylated S6, a target of mTORC1 signaling [56], suggesting that metformin exposure, coupled with maternal HFD consumption, has inhibitory effects on the mTOR pathway. Nutrient restriction during fetal development has been previously established as a progenitor of adult diseases like T2D, obesity, and cardiovascular disease [57]. Thus, the effects of metformin on AMPK and the mTOR pathway and subsequent nutrient restriction could conceivably result in adult metabolic disease in offspring exposed to metformin in utero.
Figure 2. Intracellular actions of metformin.
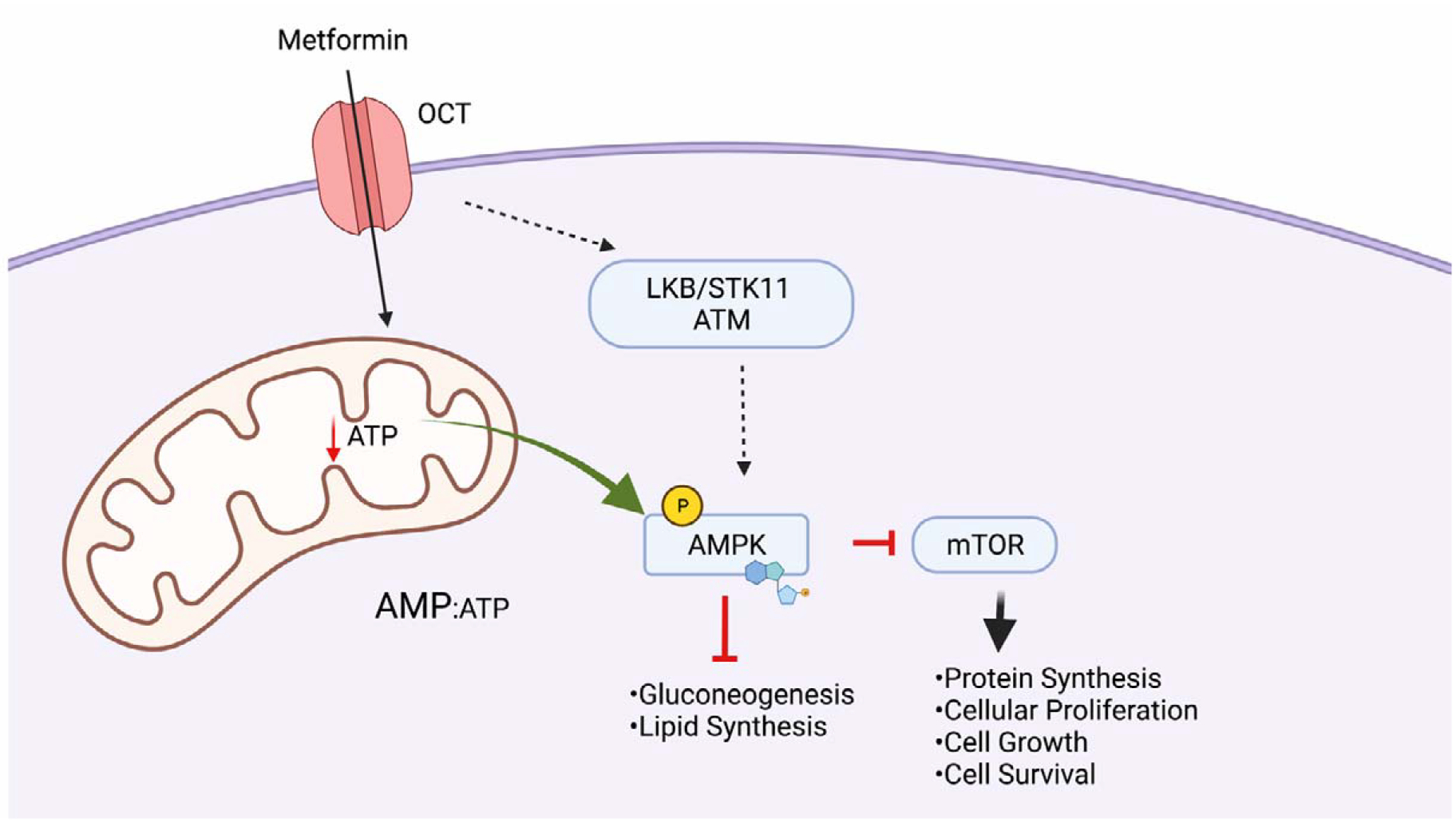
Metformin enters the cell via organic cation transporters (OCT). Following entry, the cellular redox status results in phosphorylation of AMPK by intracellular kinases LKB/ATM/ STK11. Reduced ATP production by metformin-mediated Complex I inhibition in mitochondria also results in activation of AMPK via increased AMP:ATP ration. Activation of AMPK reduces anabolism in cells via downstream targets, ultimately inhibiting cell growth. Created with Biorender.com
Overall impact of developmental metformin exposure
While initiation of pharmacological treatment of GDM is important to prevent neonatal complications like macrosomia, it is important to consider the underlying molecular mechanisms driving the fetal growth effects of hyperglycemia and the potential long-term cost of specific treatments. Long-term studies of children that were prenatally exposed to metformin reveal that as neonates they have lower birthweight and are small for gestational age but have increased BMI at 18 months of age and in early childhood due to catch-up growth [58–61]. Low birthweight is hypothesized to predispose offspring to glucose intolerance and insulin resistance in adulthood according to the thrifty phenotype hypothesis [62]. The MiTY study has demonstrated definitive concern for maternal metformin use resulting in low birthweight with disrupted post-natal growth velocity [63]. Thus, what could be interpreted as a metformin-driven “favorable” birthweight reduction in association with improved maternal glycemic control could instead be partly attributable to detrimental growth inhibition of the feto-placental unit by metformin with unknown long-term consequences [47].
Metformin has also been shown to lead to deficiencies in maternal vitamin B12 and folate, both of which are critical for regulating one-carbon metabolism for nucleotide, lipid, and amino acid synthesis [64, 65]. In development, folate and B12 are important for the prevention of neural tube defects via regulation of DNA methylation. Vitamin B12 deficiency during fetal development has been suggested to result in insulin resistance and increased adiposity in children [66]. Thus, in utero metformin exposure could alter growth via both metabolic and epigenetic mechanisms in both fetal and childhood development. Additionally, metformin inhibits the production of tricarboxylic acid (TCA)-derived metabolites needed for biosynthesis and growth while also impeding thiamine transport [67–69]. As such, the aforementioned anti-growth properties of metformin make this drug a good anti-cancer therapeutic; it has been evaluated in breast cancer, hepatocellular carcinoma, and gastric cancer [70–72]. However, these same anti-growth properties may not be beneficial in the context of the rapid growth that occurs during embryogenesis.
Given the antiproliferative and mitochondrial suppressive effects of metformin described above, some have postulated that metformin may be used to prevent or lessen the severity of pre-eclampsia, a hypertensive disease of pregnancy. While the pathophysiology of pre-eclampsia is still not well understood, metformin could prevent pre-eclampsia through its effects on cell metabolism, the anti-angiogenic state, and other processes associated with pre-eclampsia [73]. Cluver et al. (2021) performed a randomized control trial in 180 women who were pregnant (gravidae) with pre-term pre-eclampsia and found that the metformin-exposed cohort experienced a non-significant 7.6 day pregnancy prolongation compared to the placebo cohort [74]. Unfortunately, this study is likely underpowered for secondary analyses aimed at measuring the potential for longer-term harm in the offspring [63, 75]. This would have been a key outcome in this trial, given that any condition associated with placental insufficiency is a relative contraindication for its use due to concerns of lactic acidosis in the setting of the decreased placental perfusion which accompanies placental insufficiency [75]. As noted above, the MiTY trial noted a 2-fold increased risk of being born small for gestational age in the metformin arm of this T2D trial [63]. Before moving forward with prescribing metformin for any theoretical effects it may have on the treatment of pre-eclampsia, it is crucial to collect long-term safety data and fully explore any adverse childhood and/or adulthood metabolic effects associated with the drug.
In summary, metformin demonstrates some degree of effectiveness among gravidae in the management of glucose tolerance and possibly pre-eclampsia. However, these benefits may be accompanied by risk of longer-term harm to the metabolic health of the offspring as a result of fetal exposure to maternal metformin concentrations effectively restricting relative fetal growth and modifying fetal programming to predispose offspring to maladaptive effects later in life. Whether there is further additive or synergistic effects of a calorically-dense maternal or post-weaning diet remains unexplored, but is a key area of active investigation.
Effects of metformin on pancreas development and function
Detailed reviews of pancreas development can be found in [25, 76]. In brief, the pancreas forms as an evagination of the posterior foregut endoderm epithelium. Glucose homeostasis is primarily driven by the secretion of the endocrine hormones glucagon and insulin by the alpha and beta cells of the pancreatic islets of Langerhans, respectively, which make up approximately 2% of the total adult pancreas volume. The remaining 98% of the adult pancreas is comprised of exocrine tissue (acinar and ductal cells) that release and transport digestive enzymes. Both endocrine and exocrine cells are derived from the endodermal epithelium. Insulin, produced by pancreatic beta cells, encourages the uptake and utilization of glucose in peripheral tissues, while glucagon produced by alpha cells stimulates liver glycogenolysis and gluconeogenesis to increase blood glucose levels during times of fasting. New alpha and beta cells form from neogenesis from endocrine progenitors within the ductal epithelium during development. Just prior to birth in the mouse, neogenesis decreases and further increases in alpha and beta cell mass are due mainly to proliferation continuing into the postnatal period. Beta cell proliferation slows as individuals reach young adulthood and is very low in adult humans. However, beta cell proliferation can increase in response to proliferative stimuli such as weight gain and pregnancy. The number of beta cells at birth and the effectiveness of postnatal beta cell mass expansion contribute to the overall size of the beta cell pool in adulthood. Perturbations to the beta cell differentiation program by the intrauterine and postnatal environments could alter the size of the beta cell pool available to maintain glucose homeostasis in adulthood. In the setting of obesity, beta cell compensation leads to an increase in insulin production and secretion to counteract insulin resistance and hyperglycemia. Mechanisms of compensation include increased beta cell size and proliferation, as well as increased insulin transcription. A smaller initial beta cell pool might contribute to beta cell failure and T2D.
Data evaluating the effect of developmental metformin exposure on beta cell development and function is conflicting. Studies by Gregg et al. using mouse models in which dams were administered metformin via the drinking water revealed an increase in multipotent pancreatic progenitor cells, endocrine progenitors, and an overall increase in pancreatic bud size in embryos analyzed at e14.0 [77]. This finding was consistent in pancreatic rudiments from e13.0 embryos cultured in metformin for 72 hours, though the number of endocrine pancreatic progenitors was decreased at the end of in vitro culture. The authors postulated that increased cell number could be due to increased signaling downstream of mTORC1 secondary to AMPK activation. This was supported by increased phosphorylated S6 and phosphorylated acetyl CoA carboxylase (p-ACC), a target of AMPK. At birth, offspring exposed to metformin had an increased beta cell fraction, indicating a positive effect of metformin on increasing the size of the beta cell pool at birth [77]. A later study by the same group observed that adult male offspring at six and nine weeks of age exposed to metformin in utero have improved glucose tolerance and insulin secretion due to increased L-type calcium channel transcription leading to a more robust calcium response in islets despite having similar beta cell mass to that of control offspring [78]. In the context of this study, intrauterine metformin exposure increases glucose-stimulated insulin secretion via proposed epigenetic modification to L-type calcium channels, though the increased beta cell fraction present in early postnatal life is no longer observed in offspring in adulthood.
Zebrafish embryos expressing cyan florescent protein (CFP) under control of the insulin promoter treated in vitro with metformin daily at 4–6 days post fertilization (dpf) have increased expression of the transcript for the hormone somatostatin, which is secreted by delta cells within the islets. Somatostatin is a counterregulatory hormone that reduces the secretion of insulin and glucagon. The increase in somatostatin production in response to metformin suggests that exposure may attenuate insulin secretion in zebrafish embryos, although functional studies evaluating insulin secretion would be required to support this idea. At six dpf, embryos exposed to metformin had increased beta cell number with no increase in insulin mRNA. The authors suggested that increased cell number in response to metformin is counterbalanced by attenuation of insulin production and secretion [79].
A recent study evaluating the consequence of in vitro treatment of cultured H9 human embryonic stem cells (hESCs) with metformin during a beta-like cell differentiation protocol demonstrates similar findings to those in zebrafish. Metformin treatment during pancreas/beta cell differentiation resulted in smaller clusters of beta-like cells and reduced expression of all hormone transcripts (insulin, glucagon, somatostatin, and chromogranin A). RNA sequencing of beta-like clusters from this study revealed that metformin treatment downregulated genes responsible for hormone production and secretion. Furthermore, metformin resulted in an increase in transcripts associated with mitochondrial respiration. Treatment of the immortalized human beta cell line, EndoC-BH1, with metformin had negative effects on mitochondrial function including decreased basal respiration, maximal respiration, spare respiratory capacity, and ATP production. Further, metformin treatment decreased glycolytic function in EndoC-BH1 cells. Therefore, in addition to reducing beta-cell number and expression of hormones, in vitro metformin treatment decreases mitochondrial respiration, an essential process for the ATP production needed to trigger insulin secretion [80].
Taken together, more studies evaluating the developmental effects of exposure to metformin on pancreas and islet development and function are necessary to elucidate whether metformin treatment is maladaptive or beneficial to the offspring pancreas. The differences observed in hormone regulation, beta-cell number, and glucose homeostasis in mice relative to human-derived cells demonstrate a need for studies in animal models more evolutionarily related to humans, such as the NHP. Islet architecture, proportion of different islet cell types, and expression of some islet transcription factors are more similar between humans and NHP than they are between humans and rodents [81–84]. Furthermore, NHP demonstrate heterogeneity in their response to diets high in fat and carbohydrates resulting in varying susceptibility to metabolic disease, in a similar manner to humans [85]. Future studies in ex vivo islets and in vivo studies of glucose tolerance in offspring will provide more evidence as to the effect of in utero metformin exposure on the endocrine pancreas of the offspring.
Effect of metformin on hepatic cell growth and viability
The primary functional cells of the liver are derived from endodermal cells in the anterior foregut in response to inductive signals such as fibroblast growth factor (FGF) from the adjacent cardiac mesoderm. The endoderm undergoes extensive branching and growth into the mesoderm of the septum transversum, which contributes the mesenchymal component of the mature organ. Importantly, prior to formation of the bone marrow the liver is the site of fetal hematopoiesis, and thus, a smaller liver results in defects in blood development. A detailed review of the liver including developmental origins, cell types, and metabolism can be found in [86].
The consensus in the field is that in adults, metformin primarily acts on hepatocytes to reduce gluconeogenesis and glycogenolysis. Different mechanisms for this response have been proposed, including reduced cAMP response after interaction of glucagon with its receptor, inhibition of mitochondrial glycerol phosphate dehydrogenase (mGPD), and activation of AMPK [87–89]. Nevertheless, because many studies evaluate the effect of metformin on primary hepatocytes from adult animals, little data exists on the influence of metformin on the developmental program of the liver.
One study evaluating the effect of intrauterine metformin exposure in mouse embryos isolated at e18.5 revealed that metformin directly increases the expression of Hnf4a, a transcription factor important for hepatocyte differentiation and activation of the gluconeogenic program. This effect was correlated with decreased methylation in the Hnf4a promoter, mediated by increased expression of the long noncoding RNA, H19. Notably, metformin also decreased the expression of transcripts encoding the insulin receptor in these embryos as well [90]. Therefore, metformin has been shown to influence the developmental program of the liver via epigenetic modification of important transcription factor genes. The fact that these mice were born at lower birthweights than the non-exposed controls but then had a higher weight gain postnatally with consumption of a WSD is clinically concerning. In the offspring of gravidae treated with metformin, there is an increased risk in some cohorts for higher BMI during childhood when eating an ad lib diet [59, 91, 92]. These findings suggest that the effects of metformin on the hepatic epigenome (or gut microbiome) could result in long-term adverse hepatic function and metabolism.
While prenatal exposure of mice to metformin resulted in smaller birthweights compared to non-exposed mice, the metformin-exposed offspring fed a WSD post-weaning demonstrated an accelerated weight gain, greater measures of mesenteric fat, and more frequent occurrence of hepatomegaly [93]. The male mice exposed to metformin in this study later demonstrated impaired glucose tolerance and fasting hyperglycemia. In addition to these metabolic changes, the noted hepatomegaly is of clinical concern as there is a high risk of nonalcoholic fatty liver disease (NAFLD) in obese children [94]. NAFLD can progress into nonalcoholic steatohepatitis (NASH), one of the leading causes for liver transplant worldwide. Studies have shown that newborns from gravidae with both obesity and GDM have 68% increased hepatic fat measured by Magnetic Resonance Imaging/Magnetic Resonance Spectroscopy (MRI/MRS) when compared to infants born to gravidae with neither GDM nor obesity [95], which may serve as a “first hit” and increase their risk of developing NAFLD in the future [47].
Studies assessing the effects of metformin in ex vivo primary hepatocytes and in vivo liver function can be evaluated to speculate about potential effects of metformin on the developmental program of the liver. Livers isolated from adult male mice treated with metformin after a fasting/refeeding paradigm showed inhibited activation of mTORC1 as measured by a reduction in phosphorylated S6K1, phosphorylated S6, and phosphorylated 4EBP1. This effect was reversed in mice lacking both alleles of AMPKa1 and AMPKa2, suggesting that the effect of mTORC inhibition is a result of AMPK activation in response to metformin. Further evaluation revealed reduced protein translation in response to metformin [96]. Reduction in mTORC signaling as the liver develops could result in growth restriction of the liver. Fetal hepatic metabolism is unique as the fetus develops in a low-oxygen environment with limited capacities for lipid and amino acid oxidation until birth [97]. The fetal liver has little or no gluconeogenesis, fewer mitochondria, and lower activity of carnitine palmitoyl-CoA transferase-1 (CPT1, the enzyme responsible for mitochondrial fatty acid transport) when compared to adult livers [97]. The developing and adult liver secrete insulin-like growth factor (IGF) which influences the development of other organ systems including the reproductive organs and the central nervous system. A reduction in the size of the liver and the attenuation of protein synthesis in hepatocytes would reduce the bioavailability of important growth factors for fetal organogenesis. This possibility needs to be directly evaluated in preclinical studies.
While the aforementioned studies suggest that metformin could interfere with normal cellular and organ growth during development, metformin is also protective against hepatic cell death in response to lipotoxicity. Immortalized human hepatocytes (HepG2 cells) and primary rat hepatocytes treated with metformin prior to exposure to the fatty acid palmitate were protected from palmitate-induced necrotic cell death. The authors proposed that metformin mediates these protective effects by reducing ROS accumulation via increasing transcription of the anti-oxidant enzyme superoxide dismutase 2 (SOD2), and mild inhibition of respiratory Complex I independent of AMPK activation [98]. During development, maternal HFD consumption is expected to increase the presence of fatty acids such as palmitate in the fetal circulation; therefore, metformin may protect developing hepatocytes from lipotoxicity induced by maternal overnutrition as well.
Taken together, these studies highlight the consideration that metformin can maintain cellular viability during stress; however, the anti-growth properties of metformin in primary hepatocytes from adult animals warrant the consideration of the role of metformin in fetal growth restriction. Moreover, the combined effects of metformin and WSD may have unintended long-lasting consequences on hepatic function.
Risk of neural tube defects in response to metformin exposure
The neural tube forms from folding of the ectodermal neural plate epithelium during development, eventually giving rise to the central nervous system (CNS; brain and spinal cord). Neural tube closure along the dorsal midline does not occur simultaneously along the entire anterior-posterior axis; the neural tube remains open for an extended time in both the anterior-most and posterior regions (called the anterior and posterior neuropores, respectively) [99]. Defects in completion of neural tube closure are some of the most common birth defects. Folate and vitamin B12 are critical nutritional cofactors for proper neural tube closure and women are recommended to consume folate supplements during pregnancy. Children born to mothers with reduced folate levels show a persistent reduction in total brain volume from the third trimester up to 10 years of age [100].
Individuals with T2D that take metformin exhibit deficiencies in vitamin B12 and folate due to reduced absorption from the gut [101–103]. Vitamin B12 is beneficial for hematopoiesis, nervous system maintenance, and methionine metabolism. In addition to reduction of serum B12 levels during pregnancy resulting from increased micronutrient demands and dilution, metformin intake may also reduce circulating B12 and its bioavailability to the fetus. A study of 120 Egyptian graviade, in which offspring were monitored for neural tube defects (NTD), and levels of serum homocysteine, methyl malonic acid, and vitamin B12 were measured, found a correlation between low serum vitamin B12 and increased incidence of NTD such as anencephaly, encephalocele, and spina bifida [104]. Thus, folate levels in gravidae exposed to metformin should be monitored for the benefit of offspring CNS development.
Mouse models have also been used to investigate whether metformin could contribute to risk of occurrence of NTD. In an early study, delays in neural tube closure increased in a dose-dependent manner in e9.0 embryos cultured for 24–48 hours in metformin [105]. Importantly, metformin did not induce gross neural defects. The relevance of delayed neural tube closure in embryos in this study is unknown. Another study by the Loeken group revealed that neural tube defects are not observed in response to metformin culture [106]. In a mouse model of T2D in which dams were fed a HFD, administration of metformin reduced oxidative stress, ER stress, apoptosis, and the incidence of NTD in e8.5 embryos [107]. Hyperglycemia induced by T2D is known to increase the incidence of NTD, but metformin treatment reduces the incidence of NTD in this setting [108–110]. Therefore, the extent to which metformin delays neural tube closure or reduces risk of NTD should be considered in the context of the metabolic status of the mother.
In summary, metformin has not been associated with overt NTD; however, the finding that metformin causes delays in neural tube closure at a dose observed in the therapeutic range experienced in humans warrants more investigation into consequences of such delays. One should also consider the metabolic status of the mother, as the effects of fetal exposure to maternal diabetes can be attenuated by metformin, while different effects may be observed in the offspring of mothers consuming a healthy diet.
Effect of metformin on the developmental program of the heart
In adults, metformin protects against T2D-associated cardiovascular complications by reducing inflammation and by inducing autophagy to prevent cardiac hypertrophy [111, 112]. The extent to which this protection would be extended to the fetus is less known. Randomized, controlled studies in women that took metformin while pregnant suggest that intrauterine exposure to metformin does not result in gross effects on cardiac development and morphogenesis in offspring when assessed in early childhood. Four- and six-year-old offspring of obese pregnant women (in the absence of diabetes) exposed to metformin during gestation demonstrate improved cardiac function and hemodynamics in early childhood [113, 114]. Meanwhile, studies of offspring from women with GDM and PCOS that consumed metformin during pregnancy concluded that increased BMI in early childhood would predispose offspring to inferior cardiometabolic health as adults [60, 61]. The influence of metformin exposure during fetal development on cardiovascular disease susceptibility in adulthood is not known. Zebrafish embryos exposed to metformin displayed dysregulation of transcripts associated with cardiovascular development, dilation, enlargement, and function, in addition to cardiac edema when introduced from four hours post fertilization to five days post fertilization, a critical window of cardiac development [115].
As metformin is typically prescribed to women to manage gestational diabetes in pregnancy, one must consider the effect of hyperglycemia on cardiovascular development in the fetus. Exposure to maternal hyperglycemia and diabetes in utero has been associated with increased susceptibility to congenital heart defects and increased incidence of cardiovascular disease in offspring [116–120]. The mechanisms responsible for congenital heart defects in offspring of dams with Type 1 and Type 2 Diabetes include increased apoptosis, stress-induced oxidative stress, and endoplasmic reticulum stress [107]. Mouse models of gestational diabetes reveal that perturbed maternal glycemia results in cardiovascular defects due to the downregulation of genes responsible for cardiac neural crest development when insulin-producing beta cells are ablated with streptozotocin mid pregnancy [121]. Therefore, poor management of maternal diabetes makes the fetus susceptible to stress-induced changes to genes responsible for cardiovascular development. The glucose-lowering effect of metformin would be beneficial for evading diabetes induced teratogenicity and embryopathy in offspring.
Long-term physiological/metabolic consequences of developmental metformin exposure
There is evidence that metformin places exposed fetuses at risk for developmental programming and lifetime alterations that may enable an obesogenic phenotype, particularly in a postnatal environment of nutritional and caloric excess [122, 123]. Several recent randomized clinical trials in women with GDM or PCOS suggest that exposure to metformin in utero results in a metabolic phenotype that increases offspring weight in childhood [59, 60, 91]. These studies, which are some of the first to examine the long-term consequences of fetal exposure to metformin, found that children whose mothers received metformin during pregnancy weigh more and manifest larger waist circumferences and higher fat mass at four and nine years of age [59, 60, 91]. In the PregMet study, mothers with PCOS were randomized to either metformin or placebo during pregnancy and the effects of metformin on offspring growth were determined for up to four years of age [91]. The 5–10 year follow-up of the PregMet study (the PedMed study) found that these same children (mean = 7.5 years) exposed to metformin in utero had significantly higher BMI, waist-to-height ratio, and waist circumference z-scores with borderline significantly higher body fat than their control peers [60]. A small follow-up study of offspring of women with PCOS who had been randomized to metformin or placebo demonstrated that although the children exposed to in utero metformin had significantly higher fasting blood glucose levels, they had lower low-density lipoprotein at eight years of age than the children exposed to the placebo [124]. These findings suggest that the actions of metformin in utero may ultimately worsen some long-term metabolic outcomes.
Similarly, the MiG trial randomized mothers with GDM to metformin (46% of whom also received insulin to achieve glycemic targets) versus insulin alone to examine the effects of in utero metformin exposure on fetal and childhood development [75]. In the follow-up to the MiG trial (MiG Tofu trial), children exposed to metformin had larger measures of subcutaneous fat without evidence of a decrease in visceral fat at age two [58]. These same children were assessed at nine years of age, and the metformin-exposed offspring were still noted to be statistically significantly larger on several measures including weight, mid-upper arm circumference, waist circumference, and waist-to-height ratio [59]. Thus, these randomized trials in women with PCOS and GDM provide evidence that is concurrent with animal studies and that supports the notion that metformin exposure in utero may ultimately result in or contribute to a metabolic phenotype that increases childhood weight or obesity. Further studies are needed to follow the offspring long-term for metabolic risk.
CONCLUSIONS
Recommendations for metformin use in pregnancy are growing in prevalence for the treatment of pregestational diabetes, GDM, PCOS, and preeclampsia. Metformin ameliorates the teratogenic effects of poor glycemic management in pregnancy as seen in the case of neural tube defects and cardiovascular defects in response to maternal diabetes. These recommendations are made based on findings from follow up studies in human trials that show that metformin does not cause gross anatomical abnormalities in offspring (for example, neural tube and cardiac defects).
Nevertheless, metformin alters the metabolic program of the fetus during development such that changes in the expression of genes responsible for the development of the pancreas, liver, central nervous system, and heart are observed. Maternal metformin use inhibits cellular processes that are crucial for normal fetal growth. When accompanied by maternal and/or postnatal nutrient excess, clinically apparent childhood metabolic disease ensues, as recent clinical studies suggest. The subtle changes to the epigenetic landscape of organs in development by metformin exposure combined with environmental exposures following birth could reasonably contribute to disease susceptibility in adulthood. Additionally, metformin inhibits cellular growth in disease, though this effect has not been thoroughly evaluated enough in fetal tissue after in utero exposure. Additional follow-up studies into adulthood of offspring whose mothers consumed metformin during pregnancy are imperative so that more data on long-term metabolic health is available. Animal studies should consider the timepoint at which metformin is introduced to dams, as critical periods of development differ for major organs. Future studies should follow up on the observation that metformin-exposed offspring have been shown to have low birthweight and increased BMI in early childhood (Figure 3). Fetal exposure to metformin may influence offspring metabolism to a similar degree as observed in response to maternal overnutrition and undernutrition. These studies highlight the necessity of not only considering the diet consumed by women while pregnant, but also the therapeutics that are employed to combat metabolic disease in pregnant women.
Figure 3. Effects of metformin on offspring health following intrauterine exposure.
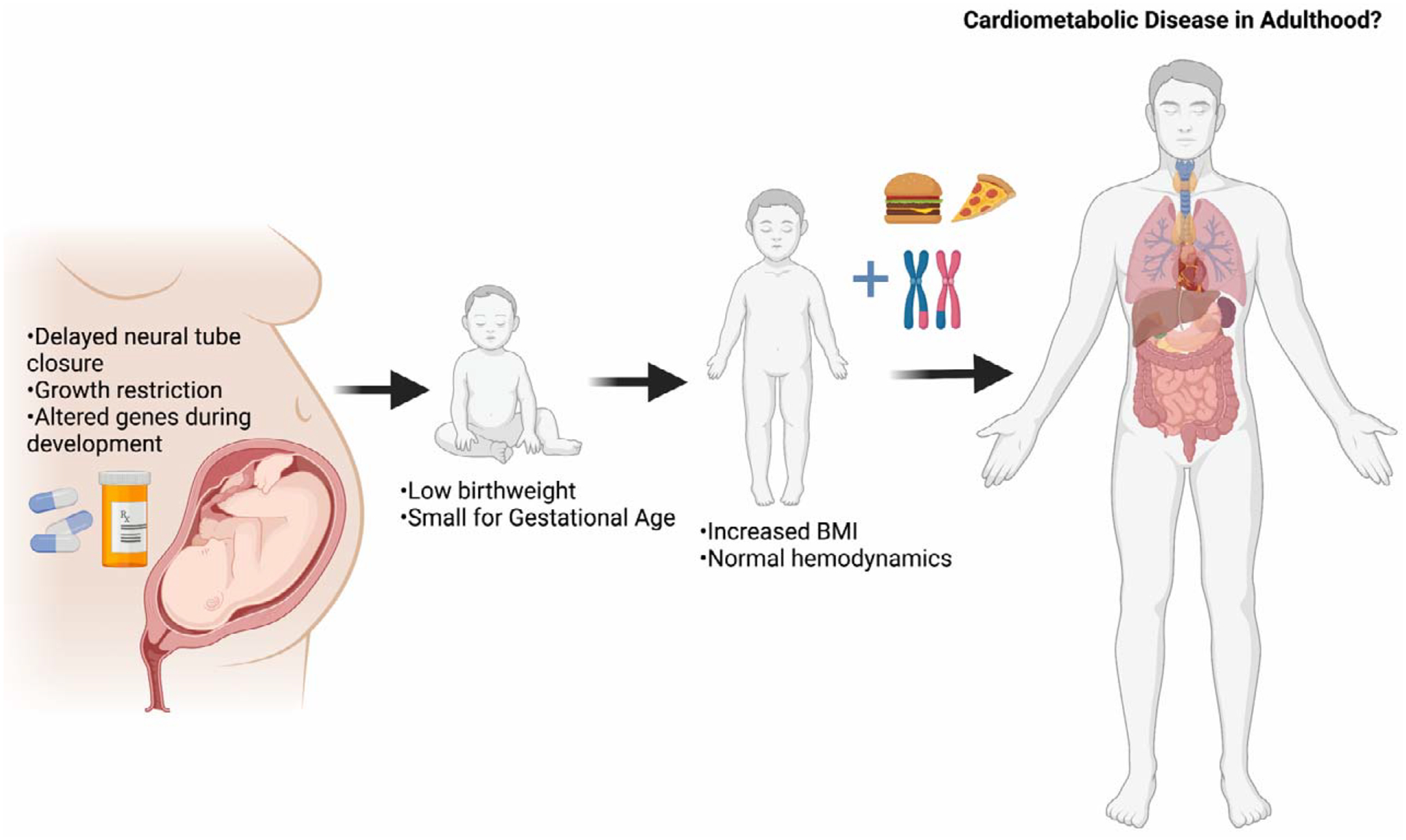
Exposure to metformin results in delayed neural tube closure, restriction of growth via inhibition of the mTOR pathway, and alteration of genes involved in development of the pancreas, liver, heart, and CNS. Human follow-up studies show that offspring are born small for gestational age with low birthweight in some women that take metformin while pregnant. Studies also show that offspring have increased BMI z-scores from 18 months of age into early childhood. The combination of intrauterine exposure to metformin, genetic predisposition, and offspring diet will influence the susceptibility of offspring to cardiometabolic disease in adulthood. Created with Biorender.com
ACKNOWLEDGEMENTS
We would like to thank Juliann Burkett for advice on figures. D.T.C. was supported in part by the Vanderbilt University Training Program in Molecular Endocrinology (5T32 DK7563-30). K.M.A. and M.G. were supported by the NIH/NIDDK (R24DK090964-06, R01DK089201 and 1R01 DK128187-01A1). AS is supported by the WRHR (K12 HD103087). M.G. was also supported by VA Merit awards (I01 BX003744-01 and I01 BX005399).
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts to report.
REFERENCES
- 1.Barker DJ and Osmond C 1986, Lancet, 1(8489), 1077–81. [DOI] [PubMed] [Google Scholar]
- 2.Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C and Winter PD 1991, BMJ, 303(6809), 1019–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martyn CN and Barker DJ 1994, Health Rep, 6(1), 45–53. [PubMed] [Google Scholar]
- 4.Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN and Bleker OP 1998, Lancet, 351(9097), 173–7. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman DJ, Reynolds RM and Hardy DB 2017, Nutrition Reviews, 75(12), 951–970. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein RF, Abell SK, Ranasinha S, Misso ML, Boyle JA, Harrison CL, Black MH, Li N, Hu G, Corrado F, Hegaard H, Kim YJ, Haugen M, Song WO, Kim MH, Bogaerts A, Devlieger R, Chung JH and Teede H 2018, J. BMC Med, 16(1), 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nazareth M, Pinto E, Severo M, Lopes C and Rego C 2021, Public Health Nutrition, 24(10), 2798–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon-Larsen P, The NS and Adair LS 2010, Obesity (Silver Spring), 18(9), 1801–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H, Cupples A, Stokes A and Liu C-T. 2018, JAMA Network Open, 1(7), e184587–e184587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benestad B, Juliusson PB, Siegfried W, Lekhal S, Smastuen MC, Hertel JK, Agosti F, Marazzi N, Hjelmesaeth J and Sartorio A 2019, J. Acta Paediatrica, 108(3), 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCurdy CE, Schenk S, Hetrick B, Houck J, Drew BG, Kaye S, Lashbrook M, Bergman BC, Takahashi DL, Dean TA, Nemkov T, Gertsman I, Hansen KC, Philip A, Hevener AL, Chicco AJ, Aagaard KM, Grove KL and Friedman JE 2016, JCI insight, 1(16), e86612–e86612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campodonico-Burnett W, Hetrick B, Wesolowski SR, Schenk S, Takahashi DL, Dean TA, Sullivan EL, Kievit P, Gannon M, Aagaard K, Friedman JE and McCurdy CE 2020, Diabetes, db191218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elsakr JM, Dunn JC, Tennant K, Zhao SK, Kroeten K, Pasek RC, Takahashi DL, Dean TA, Velez Edwards DR, McCurdy CE, Aagaard KM, Powers AC, Friedman JE, Kievit P and Gannon M 2019, Molecular Metabolism, 25, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson JR, Gustafsson HC, DeCapo M, Takahashi DL, Bagley JL, Dean TA, Kievit P, Fair DA and Sullivan EL 2018, Frontiers in Endocrinology, 9, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R and Lane RH 2008, J. Mol. Endocrinol, 41(2), 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox J, Williams S, Grove K, Lane RH and Aagaard-Tillery KM 2009, Am. J. Obstet Gynecol, 201(3), 281.e1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris RA, Alcott CE, Sullivan EL, Takahashi D, McCurdy CE, Comstock S, Baquero K, Blundell P, Frias AE, Kahr M, Suter M, Wesolowski S, Friedman JE, Grove KL and Aagaard KM 2016, Scientific Reports, 6(1), 36123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suter M, Bocock P, Showalter L, Hu M, Shope C, McNight R, Grove K, Lane R and Aagaard-Tillery K 2011, FASEB J, 25(2), 714–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suter MA, Chen A, Burdine MS, Choudhury M, Harris RA, Lane RH, Friedman JE, Grove KL, Tackett AJ and Aagaard KM 2012, FASEB J, 26(12), 5106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suter MA, Sangi-Haghpeykar H, Showalter L, Shope C, Hu M, Brown K, Williams S, Harris RA, Grove KL, Lane RH and Aagaard KM 2012, Mol. Endocrinol, 26(12), 2071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suter MA, Takahashi D, Grove KL and Aagaard KM 2013, Pediatr. Res, 74(3), 252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma J, Prince AL, Bader D, Hu M, Ganu R, Baquero K, Blundell P, Harris RA, Frias AE, Grove KL and Aagaard KM 2014, Nat. Commun, 5, 3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pace RM, Prince AL, Ma J, Belfort BDW, Harvey AS, Hu M, Baquero K, Blundell P, Takahashi D, Dean T, Kievit P, Sullivan EL, Friedman JE, Grove K and Aagaard KM 2018, BMC Microbiol, 18(1), 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prince AL, Pace RM, Dean T, Takahashi D, Kievit P, Friedman JE and Aagaard KM 2019, Am. J. Primatol, 81(10–11), e22980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elsakr JM and Gannon M 2017, Trends in Developmental Biology, 10, 79–95. [PMC free article] [PubMed] [Google Scholar]
- 26.Elsakr JM, Zhao SK, Ricciardi V, Dean TA, Takahashi DL, Sullivan E, Wesolowski SR, McCurdy CE, Kievit P, Friedman JE, Aagaard KM, Velez Edwards DR and Gannon M 2021, Sci. Rep, 11(1), 12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber TM, McCarthy MI, Wass JA and Franks S 2006, Clinical Endocrinology, 65(2), 137–145. [DOI] [PubMed] [Google Scholar]
- 28.Chu SY, Callaghan WM, Kim SY, Schmid CH, Lau J, England LJ and Dietz PM 2007, Diabetes Care, 30(8), 2070–6. [DOI] [PubMed] [Google Scholar]
- 29.Bianco ME and Josefson JL 2019, Curr. Diab. Rep, 19(12), 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perng W, Kelsey MM, Sauder KA, and Dabelea D 2021, Diabetologia, 64(10), 2237–2246. [DOI] [PubMed] [Google Scholar]
- 31.Bailey CJ 2017, Diabetologia, 60(9), 1566–1576. [DOI] [PubMed] [Google Scholar]
- 32.ACOG Practice Bulletin No. 190: Gestational Diabetes Mellitus. 2018, Obstetrics & Gynecology, 131(2), e49–e64. [DOI] [PubMed] [Google Scholar]
- 33.Thomas I and Gregg B 2017, Pediatric Diabetes, 18(1), 10–16. [DOI] [PubMed] [Google Scholar]
- 34.LaMoia TE and Shulman GI 2021, Endocr. Rev, 42(1), 77–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matthaei S, Reibold JP, Hamann A, Benecke H, Haring HU, Greten H and Klein HH 1993, Endocrinology, 133(1), 304–11. [DOI] [PubMed] [Google Scholar]
- 36.Fischer Y, Thomas J, Rosen P and Kammermeier H 1995, Endocrinology, 136(2), 412–20. [DOI] [PubMed] [Google Scholar]
- 37.Bailey CJ, Wilcock C and Scarpello JH 2008, Diabetologia, 51(8), 1552–3. [DOI] [PubMed] [Google Scholar]
- 38.Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Pedersen HK, Arumugam M, Kristiansen K, Voigt AY, Vestergaard H, Hercog R, Costea PI, Kultima JR, Li J, Jørgensen T, Levenez F, Dore J, MetaHIT consortium, Nielsen HB, Brunak S, Raes J, Hansen T, Wang J, Ehrlich SD, Bork P and Pedersen O 2015, Nature, 528(7581), 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jinno M, Kondou K and Teruya K 2010, Hormones (Athens), 9(2), 161–70. [DOI] [PubMed] [Google Scholar]
- 40.Seifarth C, Schehler B and Schneider HJ 2013, Exp. Clin. Endocrinol. Diabetes, 121(1), 27–31. [DOI] [PubMed] [Google Scholar]
- 41.Kasznicki J, Sliwinska A and Drzewoski J 2014, Ann. Transl. Med, 2(6), 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo L, Ma J, Tang J, Hu D, Zhang W and Zhao X 2019, J. Diabetes Res, 2019, 9804708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.American Diabetes Association. 2004, Diabetes Care, 27(suppl. 1), s88.14693936 [Google Scholar]
- 44.Lee N, Herbert MF, Wagner DJ, Easterling TR, Liang CJ, Rice K and Wang J 2018, Molecular Pharmacology, 94(4), 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao MZ, Flood Nichols SK, Ahmed M, Clark S, Hankins GD, Caritis S, Venkataramanan R, Haas D, Quinney SK, Haneline LS, Tita AT, Manuck T, Wang J, Thummel KE, Morris Brown L, Ren Z, Easterling TR and Hebert. MF 2020, Drug Metabolism and Disposition, 48(4), 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Given JE, Loane M, Garne E, Addor M-C, Bakker M, Bertaut-Nativel B, Gatt M, Klungsoyr K, Lelong N, Morgan M, Neville AJ, Pierini A, Rissmann A and Dolk H 2018, BMJ, 361, k2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbour LA, Scifres C, Valent AM, Friedman JE, Buchanan TA, Coustan D, Aagaard K, Thornburg KL, Catalano PM, Galan HL, Hay WW, Frias AE, Shankar K, Simmons RA, Moses RG, Sacks DA and Loeken MR 2018, Am. J. Obstet. Gynecol, 219(4), 367.e1–367.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heckman-Stoddard BM, DeCensi A, Sahasrabuddhe AA and Ford LO 2017, Diabetologia, 60(9), 1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bridges HR, Jones AJY, Pollak MN and Hirst J 2014, Biochem. J, 462(3), 475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong L, Goswami S, Giacomini KM, Altman RB and Klein TE 2012, Pharmacogenet Genomics, 22(11), 820–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scarpello JH and Howlett HC 2008, Diab. Vasc. Dis. Res, 5(3), 157–67. [DOI] [PubMed] [Google Scholar]
- 52.Sanchez-Rangel E and Inzucchi SE 2017, Diabetologia, 60(9), 1586–1593. [DOI] [PubMed] [Google Scholar]
- 53.Smoak IW 1999, Toxicology in Vitro, 13(1), 27–33. [DOI] [PubMed] [Google Scholar]
- 54.Jansson T, Aye IL and Goberdhan DC 2012, Placenta, 33(Suppl. 2), e23–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, Jansson T and Powell TL 2013, J. Clin. Endocrinol. Metab, 98(1), 105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grace MR, Dotters-Katz SK, Zhou C, Manuck T, Boggess K and Bae-Jump V 2019, AJP Rep, 9(2), e138–e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Portha B, Chavey A and Movassat J 2011, Exp. Diabetes Res, 2011, 105076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rowan JA, Rush EC, Obolonkin V, Battin M, Wouldes T and Hague WM 2011, Diabetes Care, 34(10), 2279–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rowan JA, Rush EC, Plank LD, Lu J, Obolonkin V, Coat S and Hague WM 2018, BMJ Open Diabetes Res. Care, 6(1), e000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanem LGE, Salvesen O, Juliusson PB, Carlsen SM, Nossum MCF, Vaage MO, Odegard R and Vanky E 2019, Lancet Child Adolesc Health, 3(3), 166–174. [DOI] [PubMed] [Google Scholar]
- 61.Tarry-Adkins JL, Aiken CE and Ozanne SE 2019, PLoS Med, 16(8), e1002848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hales CN and Barker DJ 2001, Br. Med. Bull, 60, 5–20. [DOI] [PubMed] [Google Scholar]
- 63.Feig DS, Donovan LE, Zinman B, Sanchez JJ, Asztalos E, Ryan EA, Fantus IG, Hutton E, Armson AB, Lipscombe LL, Simmons D, Barrett JFR, Karanicolas PJ, Tobin S, McIntyre HD, Tian SY, Tomlinson G, Murphy KE, and MiTy Collaborative Group. 2020, Lancet Diabetes Endocrinol,, 8(10), 834–844. [DOI] [PubMed] [Google Scholar]
- 64.Niafar M, Hai F, Porhomayon J and Nader ND 2015, Intern. Emerg. Med, 10(1), 93–102. [DOI] [PubMed] [Google Scholar]
- 65.Owen MD, Baker BC, Scott EM and Forbes K 2021, Int. J. Mol. Sci, 22(11), 5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yajnik CS, Deshpande SS, Jackson AA, Refsum H, Rao S, Fisher DJ, Bhat DS, Naik SS, Coyaji KJ, Joglekar CV, Joshi N, Lubree HG, Deshpande VU, Rege SS and Fall CHD 2008, Diabetologia, 51(1), 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindsay RS and Loeken MR 2017, Diabetologia, 60(9), 1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen L, Shu Y, Liang X, Chen EC, Yee SW, Zur AA, Li S, Xu L, Keshari KR, Lin MJ, Chien H-C, Zhang Y, Morrisey KM, Liu J, Ostrem J, Younger NS, Kurhanewicz J, Skokat KM, Ashrafi K and Giacomini KM 2014, Proc. Natl. Acad. Sci. USA, 111(27), 9983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, Viollet B, DeBerardins RJ and Jones RG 2015, PLoS Biol, 13(12), e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Park D-B 2015, Diabetes Metabolism Journal, 39(6), 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sekino N, Kano M, Matsumoto Y, Sakata H, Murakami K, Toyozumi T, Otsuka R, Yokoyama M, Shiraishi T, Okada K, Kamata T, Ryuzaki T and Maysubara H 2018, Anticancer Res, 38(11), 6263–6269. [DOI] [PubMed] [Google Scholar]
- 72.Zhang ZJ, Yuan J, Bi Y, Wang C and Liu Y 2019, Pharmacol. Res, 141, 551–555. [DOI] [PubMed] [Google Scholar]
- 73.Brownfoot FC, Hastie R, Hannan NJ, Cannon P, Tuohey L, Parry LJ, Senadheera S, Illanes SE, Kaitu’u-Lino TJ and Tong S 2016, Am. J. Obstet. Gynecol, 214(3), 356.e1–356.e15. [DOI] [PubMed] [Google Scholar]
- 74.Cluver CA, Hiscock R, Decloedt EH, Hall DR, Schell S, Mol BW, Brownfoot F, Kaitu’u-Lino TJ, Walker SP and Tong S 2021, BMJ, 374, n2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rowan JA, Hague WM Gao W, Battin MR, Moore MP and MiG Trial Investigators. 2008, N. Engl. J. Med, 358(19), 2003–15. [DOI] [PubMed] [Google Scholar]
- 76.Kropp PA and Gannon M 2016, Trends Dev Biol, 9, 43–57. [PMC free article] [PubMed] [Google Scholar]
- 77.Gregg B, Elghazi L, Alejandro EU, Smith MR, Blandino-Rosano M, El-Gabri D, Cras-Meneur C and Bernal-Mizrachi E 2014, Diabetlogia, 57(12), 2566–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gregg BE, Botezatu N, Brill JD, Hafner H, Vadrevu S, Satin LS, Alejandro EU and Bernal-Mizrachi E 2018, Sci. Rep, 8(1), 5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wyett G, Gibert Y, Ellis M, Castillo HA, Kaslin J and Aston-Mourney K 2018, Endocrine United States, 419–425. [DOI] [PubMed] [Google Scholar]
- 80.Nguyen L, Lim LY, Ding SSL, Amirruddin NS, Hoon S, Chan S-Y and Keong Teo AK Diabetes, 2021, 70(8), 1689–1702. [DOI] [PubMed] [Google Scholar]
- 81.Plentz RR, Palagani V, Wiedemann A, Diekmann U, Glage S, Naujok O, Jorns A and Muller T 2012, Islets, 4(2), 123–129. [DOI] [PubMed] [Google Scholar]
- 82.Dolenšek J, Rupnik MS and Stožer A 2015, Islets, 7(1), e1024405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Conrad E, Dai C, Spaeth J, Guo M, Cyphert HA, Scoville D, Carroll J, Yu W-M, Goodrich LV, Harlan DM, Grove KL, Roberts CT, Powers AC, Gu G and Stein R 2016, Am. J. Physiol. Endocrinol. Metab, 310(1), E91–E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsuchitani M, Sato J and Kokoshima H 2016, J. Toxicol. Pathol, 29(3), 147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pound LD, Kievit P and Grove KL 2014, Current Opinion in Endocrinology, Diabetes and Obesity, 21(2), 89–94. [DOI] [PubMed] [Google Scholar]
- 86.Trefts E, Gannon M and Wasserman DH 2017, Curr. Biol, 27(21), R1147–R1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miller RA, Chu Q, Xie J, Foretz M, Viollet B and Birnbaum MJ 2013, Nature, 494(7436), 256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cao J, Meng S, Chang E, Beckwith-Fickas K, Xiong L, Cole RN, Radovick S, Wondisford FE and He L 2014, J. Biol. Chem, 289(30), 20435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Madiraju AK, Erion DM, Rahimi Y, Zhang X-M, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez J-P, Lee H-Y, Cline GW, Samuel VT, Kibbey RG and Shulman GI 2014, Nature, 510(7506), 542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deng J, Mueller M, Geng T, Shen Y, Liu Y, Ramilapalli R, Taylor HS, Paidas M and Huang Y 2017, Cell Death Dis, 8(12), e3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hanem LGE, Stridsklev S, Juliusson PB, Salvesen O, Roelants M, Carlsen SM, Odegard R and Vanky E 2018, J Clin Endocrinol Metab, 103(4), 1612–1621. [DOI] [PubMed] [Google Scholar]
- 92.Carlsen SM, Martinussen MP and Vanky E 2012, Pediatrics, 130(5), e1222–6. [DOI] [PubMed] [Google Scholar]
- 93.Salomäki H, Vahatalo LH, Laurila K, Jappinen NT, Penttinen A-M, Ailanen L, Ilyasizadeh J, Personen U and Koulu M 2013, PLoS One, 8(2), e56594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brumbaugh DE and Friedman JE 2014, Pediatr. Res, 75(1–2), 140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brumbaugh DE, Tearse P, Cree-Green M, Fenton LZ, Brown M, Scherzinger A, Reynolds R, Alston M, Hoffman C, Pan Z, Friedman JE and Barbour LA 2013, J. Pediatr, 162(5), 930–6.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ and Manning BD 2017, Cell Metabolism, 25(2), 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baker PR and Friedman JE 2018, J. Clin. Invest, 128(9), 3692–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Geng Y, Hernandez Villanueva A, Oun A, Buist-Homan M, Blokzijl H, Faber KN, Doga A and Moshage H 2020, Biochimica Et Biophysica Acta-Molecular Basis of Disease, 1866(3), 11. [DOI] [PubMed] [Google Scholar]
- 99.Nikolopoulou E, Galea GL, Rolo A, Greene NDE and Copp AJ 2017, Development, 144(4), 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zou R, Al Marroun H, Cecil C, Jaddoe VWV, Hillegers M, Tiemeier H and White T 2021, Clin. Nutr, 40(5), 3391–3400. [DOI] [PubMed] [Google Scholar]
- 101.Kim J, Ahn CW, Fang S, Lee HS, and Park JS 2019, Medicine (Baltimore), 98(46), e17918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Owhin SO, Adaja TM, Fasipe OJ, Akhideno PE, Kalejaiye OO and Kehinde MO 2019, SAGE Open Med, 7, 2050312119853433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Infante M, Leoni M, Caprio M and Fabbri A 2021, World J Diabetes, 12(7), 916–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Senousy SM, Farag MK, Gouda AS, El Noury MA, Dabbous OA and Gaber KR 2018, J. Obstet Gynaecol. Res, 44(10), 1902–1908. [DOI] [PubMed] [Google Scholar]
- 105.Denno KM and Sadler TW 1994, Teratology, 49(4), 260–6. [DOI] [PubMed] [Google Scholar]
- 106.Lee H-Y, Wei D and Loeken MR 2014, Diabetes/metabolism research and reviews, 30(1), 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu Y, Reece EA, Zhong J, Dong D, Shen W-B, Harman CR and Yang P 2016, Am. J. Obstet. Gynecol, 215(3), 366.e1–366.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Becerra JE, Khoury MJ. Cordero JF and Erickson JD 1990, Pediatrics, 85(1), 1–9. [PubMed] [Google Scholar]
- 109.Ramos-Arroyo MA, Rodriguez-Pinilla E and Cordero JF 1992, Eur. J. Epidemiol, 8(4), 503–8. [DOI] [PubMed] [Google Scholar]
- 110.Reece EA, Wiznitzer A, Homko CJ Hagay Z and Wu YK 1996, Am. J. Obstet. Gynecol, 174(4), 1284–8. [DOI] [PubMed] [Google Scholar]
- 111.Turner RC, Cull CA, Frighi V and Holman. R. R. 1999, JAMA, 281(21), 2005–12. [DOI] [PubMed] [Google Scholar]
- 112.Soraya H, Clanachan AS, Rameshrad M, Maleki-Dizaji N, Ghazi-Khansari M and Garjani A 2014, Eur, J, Pharmacol,, 737, 77–84. [DOI] [PubMed] [Google Scholar]
- 113.Panagiotopoulou O, Syngelaki A, Georgiopoulos G, Simpson J, Akolekar R, Shehata H, Nicolaides K and Charakida M 2020, Am. J. Obstet. Gynecol, 223(2), 246.e1–246.e10. [DOI] [PubMed] [Google Scholar]
- 114.Yang L, Lacey L, Whyte S, Quenby S, Denison FC, Dhaun N, Norman JE, Drake AJ and Reynolds RM 2021, J. Dev. Orig. Health Dis, 1–5. [DOI] [PubMed] [Google Scholar]
- 115.Phillips J, Akemann C, Shields JN, Wu C-C, Meyer DN, Baker BB, Pitts DK and Baker TR 2021, Environ. Toxicol. Pharmacol, 87, 103716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tabib A, Shirzad N, Sheikhbahaei S, Mohammadi S, Qorbani M, Haghpanah V, Abbasi F, Hasani-Ranjbar S and Baghaei-Tehrani R 2013, Iran J. Pediatr, 23(6), 664–8. [PMC free article] [PubMed] [Google Scholar]
- 117.Zhao Z and Reece EA 2013, Clin. Lab. Med, 33(2), 207–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tam WH, Ma RCW, Ozaki R, Li AM, Chan MHM, Yuan LY, Lao TTH, Yang X, Ho CS, Tutino GE and Chan JCN 2017, Diabetes Care, 40(5), 679–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dib A, Payen C, Bourreau J, Munier M, Grimaud L, Fajloun Z, Loufrani L, Henrion D and Fassot C 2018, Front Physiol, 9, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yu Y, Arah OA, Liew Z, Cnattingius S, Olsen J, Sorensen HT, Qin G and Li J 2019, BMJ, 367, 16398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kumar SD, Dheen ST and Tay SS 2007, Cardiovasc. Diabetol, 6, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Catalano PM and Shankar K 2017, BMJ, 356, j1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Friedman JE 2015, Diabetes Care, 38(8), 1402–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rø TB, Ludvigsen HV, Carlsen SM and Vanky E 2012, Scand J. Clin. Lab. Invest, 72(7), 570–5. [DOI] [PubMed] [Google Scholar]