

Abstract
Chromatin dysfunction has been implicated in a growing number of cancers especially in children and young adults. In addition to chromatin modifying and remodeling enzymes, mutations in histone genes are linked to human cancers. Since the first reports of hotspot missense mutations affecting key residues at histone H3 tail, studies have revealed how these so-called “oncohistones” dominantly (H3K27M and H3K36M) or locally (H3.3G34R/W) inhibit corresponding histone methyltransferases and misregulate epigenome and transcriptome to promote tumorigenesis. More recently, widespread mutations in all four core histones are identified in diverse cancer types. Furthermore, an “oncohistone-like” protein EZHIP has been implicated in driving childhood ependymomas through a mechanism highly reminiscent of H3K27M mutation. We will review recent progresses on understanding the biochemical, molecular and biological mechanisms underlying the canonical and novel histone mutations. Importantly, these mechanistic insights have identified therapeutic opportunities for oncohistone-driven tumors.
Keywords: oncohistones, histone mutations, chromatin, EZHIP, cancer epigenetics, histone methylation
1. Introduction
Nucleosomes, the basic repeating units of eukaryotic genomes, are made of an octamer of histones H2A, H2B, H3 and H4 wrapped around ~150bp of DNA. Nucleosomes and the linker histone H1 are instrumental in genome packaging. Furthermore, the composition, positioning and post-translational modifications (PTMs) of nucleosomes and linker histones play critical regulatory functions in transcription, genome integrity and replication. Indeed, combinatorial patterns of histone PTMs are demonstrated to act both in cis and in trans to modulate various DNA-templated programs in a highly specific manner, as predicted by the histone code hypothesis (Jenuwein & Allis 2001).
Owing to their importance, histone genes are highly conserved throughout evolution and exist in multiple copies, many of which are closely clustered in the genome. The redundancy and complexity of histone genes, especially in mammalian systems, present significant challenges to dissect the function of histone residues and PTMs using genetic approaches. The discovery and characterization of cancer-associated histone mutations over the past decade, however, has challenged this notion (Behjati et al. 2013; Nacev et al. 2019; Schwartzentruber et al. 2012; Wu et al. 2012). These so-called “oncohistone” mutations are heterozygous and affect only one of the many histone genes, yet often producing dominant effects on the landscape of histone PTMs and gene expression programs.
The first reported histone mutations are highly specific missense mutations affecting select residues of the histone H3 tail (H3K27, H3G34 and H3K36) (Behjati et al. 2013; Schwartzentruber et al. 2012; Wu et al. 2012). These “canonical” oncohistone mutations also exhibit a remarkable tissue specificity. More recently, a plethora of pan-cancer-associated “non-canonical” histone mutations occurring in the tails and globular domains of core histones were identified (Nacev et al. 2019). Furthermore, “oncohistone-like” protein and linker histone H1 mutations have been linked to human cancers (Li et al. 2014; Pajtler et al. 2018; Yusufova et al. 2021). In this review, we will focus on the recent exciting advances in our understanding of the biochemical, molecular and biological mechanisms by which histone mutations reprogram cellular epigenome to promote tumor development.
2. H3K27M mutation and “H3K27M-like” EZHIP
2.1. H3K27M mutations are associated with pediatric midline gliomas
As one of the first reported oncohistone mutations, H3K27M was found in the majority (~70%) of diffuse intrinsic pontine gliomas (DIPG) – pediatric brainstem gliomas with dismal prognosis (Schwartzentruber et al. 2012; Wu et al. 2012). Additional midline pediatric high-grade gliomas (pHGG) (eg. thalamus and spinal cord) also harbor recurrent H3K27M mutations (Sturm et al. 2012) (Figure 1). While we refer the readers to excellent reviews on the clinical characteristics of H3K27M-mutant midline gliomas (Buczkowicz & Hawkins 2015; Funato & Tabar 2018), here we highlight two intriguing features with potential mechanistic implications. First, both canonical histone H3.1/2 and variant histone H3.3 can carry K27M mutations, yet with a number of distinctions. H3.1K27M-mutant tumors are found in younger patients with secondary mutations such as activating mutations in ACVR1. In contrast, patients with H3.3K27M-mutant gliomas are older and H3.3K27M mutations tend to co-occur with PDGFRA amplification and mutations in TP53 and ATRX (Castel et al. 2015, 2018). Second, as described below, H3K27M mutations dominantly inhibit the H3K27 methyltransferase complex PRC2. However, inactivating mutations in PRC2 members are rare in pediatric gliomas and conversely H3K27M mutations are not found in cancer types where PRC2 is recurrently mutated or deleted (Lee et al. 2014). Together, these correlative observations suggest that (1) there may be subtle isoform-specific effects of H3K27M mutations and (2) H3K27M oncohistone may not be functionally equivalent to PRC2 loss.
Figure 1:
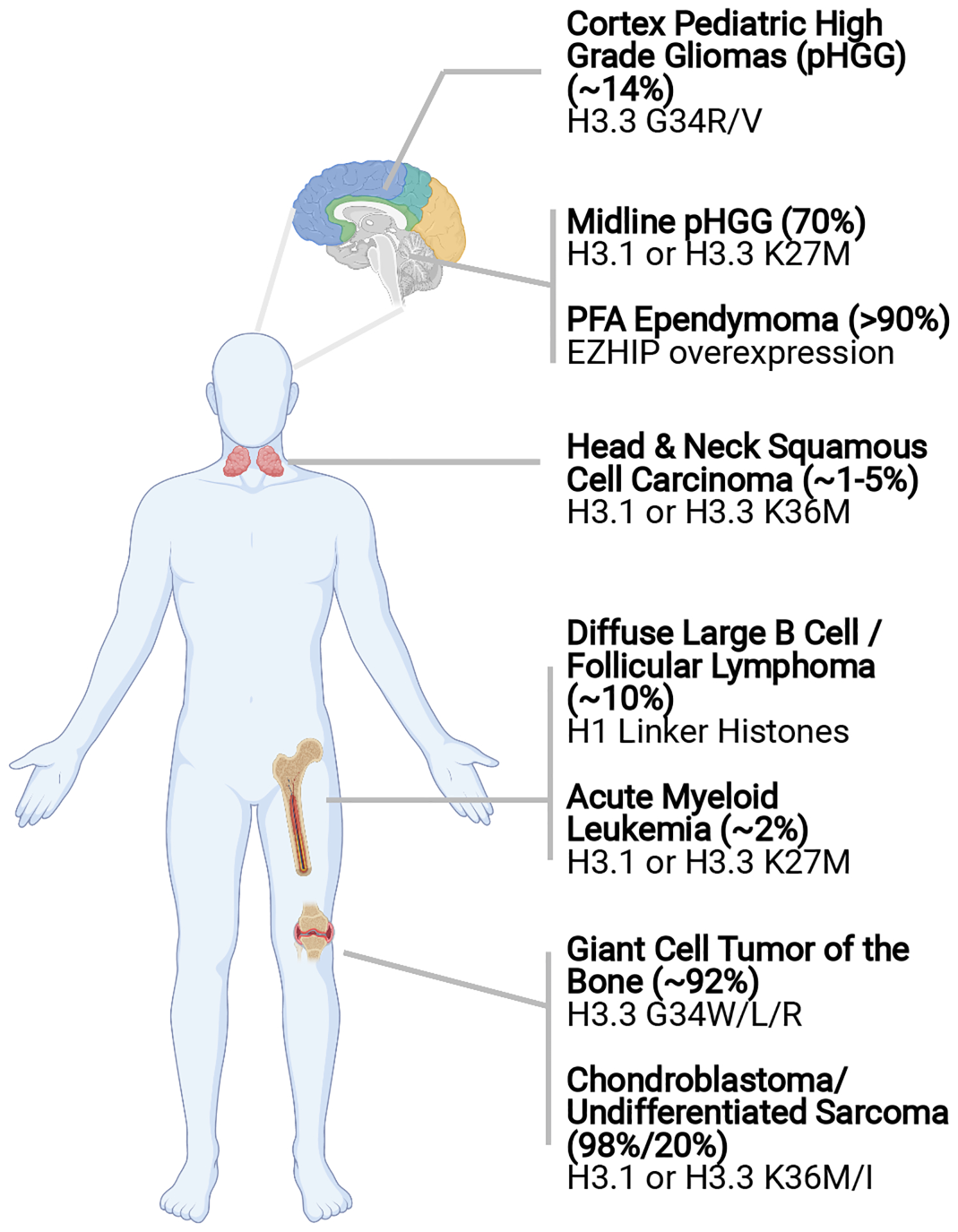
Summary of cancer-associated histone mutations and their frequencies within disease subtypes.
2.2. H3K27M/I mutations dominantly inhibit PRC2 methyltransferase activity
Remarkably, expression of H3K27M mutant histones caused a global reduction of H3K27 tri-methylation (H3K27me3) (Chan et al. 2013; Lewis et al. 2013; Venneti et al. 2013), despite that at the protein level, K27M mutant H3 accounted for a small pool of the total H3 (Lewis et al. 2013). Biochemical studies found that H3K27M mutant is a potent and competitive inhibitor for PRC2 methyltransferase complex that catalyzes H3K27 methylation (Bender et al. 2013; Brown et al. 2014; Lewis et al. 2013). Although PRC2 inhibition by H3K27M has been well-established, the exact molecular basis is still an active area of investigation. H3K27M substitution occupies the same position as K27 in the active site of EZH2 (Justin et al. 2016), but the linear and hydrophobic side chain markedly increases the mutant’s affinity to EZH2 (Brown et al. 2014; Justin et al. 2016; Lewis et al. 2013). Indeed, substituting K27 with isoleucine, another residue with unbranched hydrophobic side chain, inhibited PRC2 in vitro and reduced H3K27me3 in cells (Brown et al. 2014; Lewis et al. 2013). H3K27I mutation was also identified in DIPG (Castel et al. 2015). Furthermore, co-immunoprecipitation showed an increased enrichment of PRC2 on H3K27M mutant nucleosomes (Chan et al. 2013; Lewis et al. 2013). These findings have led to the proposed “sequestration” model, in which H3K27M mutant “traps” PRC2 and prevents its availability to wildtype histones. More recent single-molecule studies lend support to this model by demonstrating that H3K27M stabilizes the engagement of PRC2 with nucleosomes and increases its residence time (Leicher et al. 2020; Tatavosian et al. 2018). At the same time, findings that are seemingly incompatible with the “sequestration” model also emerged and helped reveal important mechanistic insights. For example, increased interaction between H3K27M and PRC2 requires the presence of the co-factor S-adenosyl-methionine (Diehl et al. 2019; Wang et al. 2017). Furthermore, the “sequestration” is likely a highly dynamic and transient event, which may explain why less sensitive methods likes ChIP-seq has failed to detect genome-wide co-localization between H3K27M mutant histone and PRC2 (Mohammad et al. 2017; Piunti et al. 2017).
Recent studies have identified additional mechanisms contributing to the inhibition of PRC2 by H3K27M. PRC2 complex purified from H3K27M-mutant cells appeared to be less active, suggesting that binding to H3K27M alters the activity and conformation of PRC2 in a long-lasting manner (Stafford et al. 2018). Indeed, H3K27M was found to impair the automethylation of EZH1/2 and SUZ12 which is critical to PRC2’s catalytic function (Lee et al. 2019). It was also reported that allosterically activated PRC2 was more sensitive to H3K27M inhibition (Stafford et al. 2018), consistent with the finding that H3K27M-H3K27me3 di-nucleosome led to a greater inhibition of PRC2 activity (Diehl et al. 2019). Therefore, H3K27M mutation can block multiple steps that are key to the positive feedback loop required for PRC2 chromatin propagation and establishment of H3K27me3 domains.
2.3. Epigenome reprogramming by H3K27M
Despite the global loss of H3K27 methylation, genome-wide analyses suggest that the impact of H3K27M on H3K27me3 distribution is not uniform (Figure 2). Strong PRC2 target genes such as p16Ink4A, preferentially retained high levels of H3K27me3 (Chan et al. 2013; Mohammad et al. 2017). More recent studies demonstrated that H3K27M mutation had little impact on PRC2 recruitment and initial deposition of H3K27me3 around the PRC2 nucleation sites, yet markedly inhibited the spread of H3K27me3 (Harutyunyan et al. 2019, 2020). These observations are consistent with in vitro evidence that H3K27M impedes the propagation of PRC2 activity along chromatin and offered an explanation for why H3K27me3 levels are less affected at strong PRC2 binding sites. Interestingly, a “step down” model was described in which H3K27M limited the enrichment patterns of H3K27me1 and H3K27me2 to that of H3K27me2 and H3K27me3 in wild-type cells, respectively (Harutyunyan et al. 2020). The function of H3K27me2/1 in gene regulation is poorly understood, and it is not entirely clear what is the impact of this progressive restriction of H3K27 methylation on gene expression in H3K27M mutant cells. It has been proposed, however, that persisting levels of H3K27me3 led to enhanced silencing of strong PRC2 target genes and represented a vulnerability of H3K27M mutant gliomas (Mohammad et al. 2017).
Figure 2:
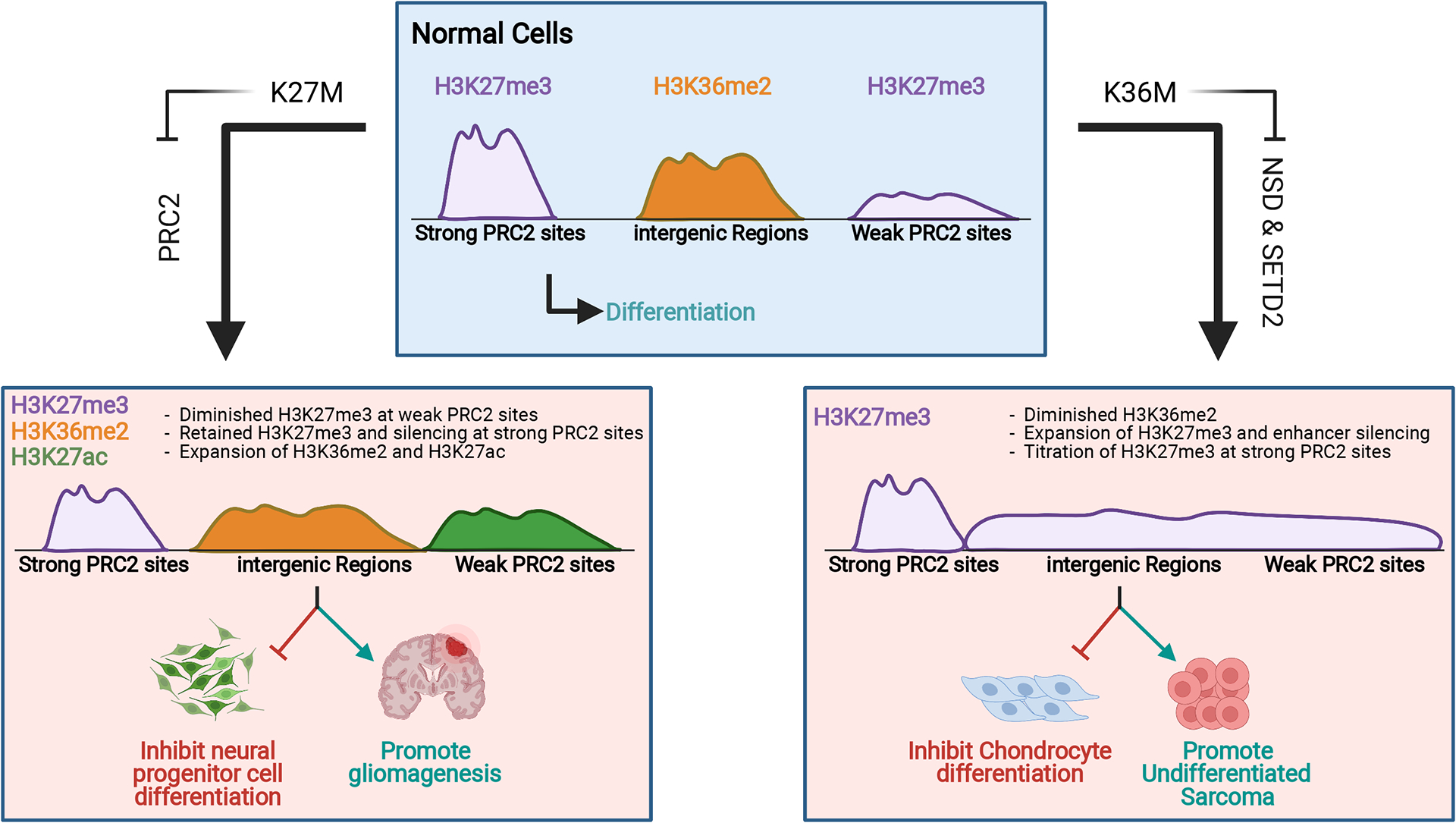
Epigenome reprogramming by “K-to-M” H3 mutations. In H3K27M mutant cells (left), inhibition of PRC2 leads to diminished H3K27me3 and increased expression of weak PRC2 target genes. Strong PRC2 target genes retain H3K27me3 and remain silenced. Loss of global H3K27me3 is also accompanied by expansion of H3K36me2 domains and pervasive increases in H3K27ac. In H3K36M mutant cells (right), inhibition of NSD family enzymes leads to diminished H3K36me2. Spreading of H3K27me3 into H3K36me2 domains results in enhancer silencing. The genome-wide H3K27me3 increase also has a “diluting” effect on strong PRC2 target genes, leading to their de-repression.
As described above, although H3.3K27M and H3.1K27M mutations are both identified in DIPG, they are associated with distinct secondary mutations and patient demographics (Castel et al. 2015, 2018). The enhancer landscape between H3.3K27M versus H3.1K27M mutant tumors is also different, suggesting that there is some degree of variant-specific effect of H3K27M mutation (Nagaraja et al. 2019). ChIP-seq of H3.1K27M and H3.3K27M showed that the oncohistones follow the known deposition patterns of H3 isoforms, where H3.3 is enriched at enhancers and gene bodies and H3.1 is distributed across the genome (Fang et al. 2018a; Nagaraja et al. 2019; Sarthy et al. 2020). Accordingly, H3.1K27M had a stronger effect on genome-wide decreases in H3K27me3, whereas H3K27me3 depletion correlated with levels of H3.3K27M, indicating local inhibition of PRC2 activity (Nagaraja et al. 2019; Sarthy et al. 2020). Furthermore, the enrichment of H3.3K27M at poised enhancers correlated with enhanced binding of EZH2, and the relative depletion of H3.3K27M at PRC2-targted CpG islands may facilitate the local retention of H3K27me3 (Fang et al. 2018a; Sarthy et al. 2020). These features, together with the disparate cell-of-origin and mutational background in which K27M mutation occurs, argue that H3.1K27M and H3.3K27M mark distinct subgroups of DIPG which may require specific clinical management and therapeutic strategy.
H3K27 methylation is known to influence the levels of other histone modifications such as H3K27 acetylation (H3K27ac) and H3K36 methylation (H3K36me2/3). As expected, the contraction of H3K27 methylation domains by H3K27M was followed by increases in H3K27ac and H3K36me2 (Krug et al. 2019; Stafford et al. 2018). Interestingly, the elevated H3K27ac in H3K27M mutant cells was not shown as increased peaks, but rather as diffuse distribution across the genome. This led to increased expression of retrotransposon elements and a “viral mimicry” effect, making H3K27M mutant gliomas potentially more immunogenic.
2.4. Modeling H3K27M mutations in DIPG
There have been considerable efforts to develop faithful pre-clinical models of H3K27M-driven gliomas. In neural progenitor cells (NPCs) derived from human embryonic stem cells, expression of H3.3K27M together with p53 deletion and PDGFRA activation resulted in low-grade brainstem gliomas (Funato et al. 2014). Similarly, co-expression of H3.3K27M and PDGFB in mouse NPCs accelerated tumor growth and the resulting brainstem gliomas shared gene expression profiles with human DIPG (Mohammad et al. 2017). The synergy between H3.3K27M, p53 loss and PDGF signaling was further demonstrated by three independent models. Introduction of p53 knockout and co-expression of H3.3K27M and PDGFB using the RCAS/tv-a retroviral system into neonatal NPCs induced aggressive brain tumors which showed similar radioimaging characteristics to human DIPG (Cordero et al. 2017; Subashi et al. 2016). Using an in utero electroporation system, mosaic H3.3K27M expression and CRISPR-Cas9-mediated Trp53 deletion in NPCs of both cortex and hindbrain are sufficient for tumorigenesis, which can be furthered enhanced by PDFGRA overexpression (Pathania et al. 2017). Finally, endogenous expression of H3.3K27M using an inducible knock-in model demonstrated that the mutation could accelerate spontaneous DIPG formation from postnatal NPCs together with p53 deletion and PDGFRα activating mutation (Larson et al. 2019). Therefore, H3K27M alone appears insufficient for glioma initiation and requires cooperating mutations. Further supporting this notion and in keeping with the mutational patterns observed in human DIPG (Buczkowicz et al. 2014; Taylor et al. 2014; Wu et al. 2014), ACVR1 activating mutations cooperate with H3.1K27M mutation to drive diffuse gliomagenesis in two independent mouse models (Fortin et al. 2020; Hoeman et al. 2019).
Recent studies have also investigated the developmental context underlying the neoplastic transformation by H3K27M. In a human induced pluripotent stem cell-derived model, DIPG-like tumor formation was only observed upon inducible knock-in of H3.3K27M with p53 inactivation in neural stem cells (NSCs) but not in more differentiated oligodendrocyte progenitor cells (Haag et al. 2021). Intriguingly, ectopic expression of H3.3K27M from the Fabp7 promoter, which is active in both NPCs and other tissue progenitors, was sufficient to increase the frequency and decrease the latency in the development of multiple tumor types including lymphomas and carcinomas, although high-grade gliomas were only observed in p53 knockout background (Pajovic et al. 2020).
Lastly, either RNAi-mediated suppression or CRISPR-Cas9-mediated knockout of H3.3K27M mutant allele could delay the growth of human DIPG cell lines in vitro and in vivo (Harutyunyan et al. 2019; Silveira et al. 2019). Therefore, H3K27M mutation is required for tumor maintenance, providing important rationale for developing targeted therapeutic strategies.
2.5. Therapeutic strategies targeting H3K27M mutation
The discovery of H3K27M mutations in DIPG has attracted strong interests to develop targeted epigenetic therapies for these deadly tumors with currently limited treatment options. To counteract the global depletion of H3K27 methylation, GSK-J4 – a pharmacologic inhibitor of H3K27 demethylases (UTX and JMJD3) – was tested and demonstrated to exhibit antitumor activity and radiosensitize H3K27M mutant DIPG cells (Hashizume et al. 2014; Katagi et al. 2019). Conversely, inhibitors of EZH2 also delayed the growth of DIPG cells (Mohammad et al. 2017). While this may seem counterintuitive, the therapeutic benefit of EZH2 inhibition is consistent with the notion that the persisting H3K27me3 levels at strong PRC2 target genes are critical for tumor maintenance (Chan et al. 2013; Mohammad et al. 2017). Consistently, several reports have found that BMI-1, a member of the H3K27me3 “reader” PRC1 complex, was upregulated and represents a viable therapeutic target for H3K27M mutant DIPG (Balakrishnan et al. 2020; Filbin et al. 2018; Kumar et al. 2017).
Beyond targeting H3K27 methylation, a drug screen found that multiple histone deacetylase inhibitors (HDACi) exhibited therapeutic efficacy in pre-clinical DIPG models (Grasso et al. 2015). Based on these results, several clinical trials are ongoing to evaluate the clinical benefits of HDACi panobinostat in DIPG patients. It has been demonstrated that poly-acetylated H3K27M mutant histones are poor inhibitors of PRC2 (Brown et al. 2014). Therefore, HDAC inhibition may elevate global histone acetylation to “detoxify” H3K27M oncohistone. Furthermore, H3K27M mutant cells already show increased H3K27ac and a propensity to de-silencing retroviral elements, which may be further augmented by HDAC inhibition to elicit anti-tumor immune response (Krug et al. 2019). Several additional epigenetic compounds show promise in treating DIPG tumors, including inhibitors targeting BRD4, CDK7, CDK9, and combined inhibitions of LSD1/HDAC and FACT/HDAC (Anastas et al. 2019; Dahl et al. 2020; Ehteda et al. 2021; Nagaraja et al. 2017; Piunti et al. 2017). However, it should be noted that the therapeutic effects of these compounds apply to both H3K27M mutant and H3 wildtype tumors, suggesting that chromatin and transcriptional dysregulation is a universal druggable dependency for DIPG.
Finally, H3K27M oncohistone was recently shown to extensively reprogram cellular metabolic network, and this metabolism-chromatin crosstalk can be therapeutically targeted by the inhibition of metabolic enzymes including HK2, IDH1, and GDH (Chung et al. 2020). H3K27M DIPG also uniformly express high levels of the disialoganglioside GD2, which can be targeted by chimeric antigen receptor (CAR)-expressing T cells. Indeed, treatment of GD2-targeted CAR T cells demonstrated marked anti-tumor effects in H3K27M mutant DIPG xenograft models (Mount et al. 2018). Taken together, the plethora of epigenetic, metabolic, and immune therapies under preclinical and clinical development offer hope to improve the outcome of patients carrying H3K27M mutant DIPG.
2.6. Ependymoma-associated protein EZHIP exhibits H3K27M-like activity
Ependymoma arises from the ependymal cells and is another type of rare brain tumor affecting young children (Pajtler et al. 2015). A subtype of hindbrain ependymomas, known as posterior fossa group A (PFA) ependymomas, exhibit distinct gene expression profiles and bear a worse prognosis (Mack et al. 2014). Interestingly, H3K27me3 levels are globally reduced in PFA ependymomas and its genome-wide distribution pattern mimics that of H3K27M mutant DIPG (Bayliss et al. 2016), even though H3K27M mutation is rare in ependymomas. Instead, a germ cell-specific gene, EZHIP/CXorf67, was found to be highly expressed in PFA ependymomas and, when introduced, was able to bind to PRC2 and inhibit H3K27me3 in cells (Pajtler et al. 2018). Shortly after this discovery, multiple independent studies have demonstrated that EZHIP competitively inhibits PRC2 via a H3K27M-like mechanism (Hübner et al. 2019; Jain et al. 2019; Piunti et al. 2019; Ragazzini et al. 2019). Remarkably, a short (<15aa) and highly conserved amino acid sequence at the C-terminal region of EZHIP shares similarity to the H3K27M mutant histone tail, with the Methionine 27 corresponding to the Methionine 406 of EZHIP. Indeed, both H3K27M mutant histone and EZHIP preferentially bind to allosterically active PRC2 and inhibit its chromatin spreading but not recruitment (Jain et al. 2020b; Ragazzini et al. 2019). A subset of H3 wild-type DIPG overexpress EZHIP (Castel et al. 2020), further supporting its functional similarity to H3K27M mutation. Therefore, it would be interesting to test if therapeutic strategies targeting H3K27M mutant DIPG could be repurposed to treat EZHIP-overexpressing PFA ependymomas.
3. H3K36M mutation
3.1. H3K36M mutations are oncogenic and dysregulate cell differentiation
H3K36M is another “K-to-M” missense mutation that has emerged as a driver of oncogenesis (Figure 1). Behjati et al. in 2013 showed that H3F3B, a gene encoding histone H3.3, was mutated in 95% cases of chondroblastomas – a type of benign cartilage tumors affecting young adults (Behjati et al. 2013). The mutation invariably results in substitution of K36 with methionine. Since then, multiple reports have detected H3K36M mutations in head and neck squamous cell carcinomas and undifferentiated sarcomas, as well as in rare cases of congenital soft-tissue neoplasm and histiocytic tumor of the skull (Kernohan et al. 2017; Papillon-Cavanagh et al. 2017; Snuderl et al. 2019).
Analogous to H3K27M, H3K36M was hypothesized to act as oncogene and exerts dominant effects on H3K36 methylation. Indeed, functional studies demonstrated that ectopic expression of H3K36M mutant histone impaired the differentiation of immortalized mesenchymal progenitor cells (Lu et al. 2016). Knock-in of H3.3K36M mutation to the endogenous H3F3B gene in immortalized human chondrocytes also promoted malignant features including increased colony formation and resistance to apoptosis (Fang et al. 2016). Furthermore, H3K36M-expressing mesenchymal progenitor cells generated undifferentiated sarcomas in nude mice (Lu et al. 2016). This was also seen for H3K36I, which mirrors the H3K27I mutation found in pediatric gliomas. More recently, a knock-in mouse model has been developed where H3.3K36M can be inducibly expressed at the endogenous H3f3b locus. By crossing with Prx1-Cre mice, H3.3K36M was conditionally expressed in the chondrogenic progenitor cells, which led to delayed limb development and impaired maturation of chondrocytes. However, these mice did not develop chondroblastomas, suggesting that additional co-operating mutations may be required (Abe et al. 2020).
Interestingly, the impact of H3K36M mutation on cellular differentiation extends beyond the cartilage compartment. Brumbaugh et al. found that induced H3K36M transgene expression in mice resulted in testicular atrophy and an absence of goblet and paneth cells in the intestine, indicating impaired specifications of spermatocyte and intestinal epithelium, respectively. In addition, H3K36M mutation promoted aberrant transcriptional programs in hematopoietic progenitor and stems cells, leading to a failure in stem cell maintenance, defective erythropoiesis and severe anemia (Brumbaugh et al. 2019). Using an independent transgenic mouse model to examine the effect of H3K36M on adipogenesis, Zhuang et al. observed impaired development of brown adipose tissue and muscle development (Zhuang et al. 2018). In Arabidopsis thaliana, introduction of the H3K36M mutation induced a range of developmental defects from increased branching to early flowering by altering the expression of development- and metabolism-associated genes (Lin et al. 2018; Sanders et al. 2017).
3.2. Inhibition of H3K36 methyltransferases by H3K36M mutation
H3K36M mutation markedly reduces levels of H3K36 di- and tri-methylation when expressed in cells. H3K36M mutant peptides and nucleosomes also dominantly inhibit the catalytic activities of H3K36 methyltransferases such as NSD2 and SETD2 (Fang et al. 2016; Lu et al. 2016). Structural studies of the SETD2 catalytic domain bound to H3K36M or H3K36I revealed that the methionine or isoleucine was positioned in a newly formed substrate channel (Yang et al. 2016). Additional analysis showed that H3G34 and H3P38 residues play an interesting role in SETD2 engagement, since when either was mutated, it alleviated the inhibitory effects of H3K36M on H3K36 methylation (Zhang et al. 2017). Co-immunoprecipitation experiments demonstrated that H3K36M mutant histone appeared to interact more strongly with NSD1/2 in mesenchymal progenitor cells (Lu et al. 2016) and in tissue lysates derived from H3.3K36M knock-in mice (Abe et al. 2020). In addition, H3K36M stabilizes the interaction between histone and SDG8, Arabidopsis homolog of mammalian SETD2, and alters SDG8’s subcellular localization (Lin et al. 2018). Therefore, while more studies are needed, it seems that H3K36M mutant nucleosomes “trap” H3K36 methyltransferases through a mechanism reminiscent of H3K27M.
Unlike H3K27, where a single methyltransferase complex PRC2 catalyzes mono-, di- and tri-methylation, methylation states of H3K36 are regulated by distinct enzymes. ASH1L and NSD family enzymes catalyze H3K36me1/2, whereas SETD2 is the only enzyme responsible for generating H3K36me3 (Wagner & Carpenter 2012). Although both can be inhibited by H3K36M mutant nucleosomes in vitro, recent evidence suggests that the impaired activity of NSD1/2, and thus the depletion of H3K36me2, are critical mediators of H3K36M mutation. With individual or combined knockout of H3K36 methyltransferases, it was shown that the loss of H3K36me2 could largely recapitulate H3K36M’s effect on gene expression, chondrocyte differentiation and drug response in mesenchymal progenitor cells (Rajagopalan et al. 2021). The impaired adipogenesis induced by H3K36M can be phenocopied by knockdown of NSD2 in preadipocytes (Zhuang et al. 2018). Finally, expression of H3K36M in a fibrosarcoma line HT1080 led to reduced proliferation, which can be mirrored by knockdown of NSD2 but not SETD2 (Sankaran & Gozani 2017). These results are consistent with the finding that H3K36M mutations are mutually exclusive with NSD1 deletions and loss-of-function mutations in head and neck cancers (Papillon-Cavanagh et al. 2017). However, given H3K36M mutation’s broad impact on lineage specification, additional tissue/cell types need to be examined to fully understand the context-dependent contribution of various H3K36 methylation states to H3K36M’s mechanism of action.
3.3. Impact of H3K36 methylation loss on epigenome reprogramming
H3K36 methylation is implicated in several pathways involved in transcription and genome integrity. H3K36me3 is found at gene bodies of actively transcribed genes, and has been linked to transcriptional elongation, RNA processing and DNA-damage induced homologous recombination (Carvalho et al. 2014; Kuo et al. 2011; Simon et al. 2014; Wen et al. 2014). Consistently, the developmental defects observed with H3K36M mutation in Arabidopsis thaliana agrees with the role of H3K36me3 in regulating the expression of genes involved in development-related pathways like brassinosteroid signaling pathways (Lin et al. 2018; Sanders et al. 2017). In contrast, H3K36me2 diffusely marks intergenic regions, and its function is less well understood. Both H3K36me2 and H3K36me3 are required for the deposition of DNA methylation via the recruitment of DNMT3A and DNMT3B, respectively (Baubec et al. 2015; Weinberg et al. 2019). Furthermore, nucleosomes marked by H3K36me2/3 are poor substrates for PRC2 and therefore H3K36 methylation can serve to oppose the establishment and spreading of H3K27me3 (Finogenova et al. 2020; Jani et al. 2019; Yuan et al. 2011). Indeed, decreased DNA methylation and increased H3K27me3 are observed in cells and patient samples carrying H3K36M mutations (Lu et al. 2016; Papillon-Cavanagh et al. 2017; Rajagopalan et al. 2021).
The global increase of H3K27me3 appears to play an important role in shaping the transcriptome of H3K36M mutant cells (Figure 2). On one hand, active genes and enhancers embedded within H3K36 methylation domains became silenced following the increase in H3K27me3 (Rajagopalan et al. 2021). For example, the adipogenesis blockade effect of H3K36M was linked to a decreased ratio of H3K36me2/H3K27me3 that inhibited the expression of adipocyte master regulators like C/EBPα and PPAR-γ (Zhuang et al. 2018). Unexpectedly, genome-wide gain of H3K27me3 also abrogates the silencing of strong polycomb target genes. For example, a collapse of normal H3K27me3 distribution at HoxA gene clusters resulted in their mis-regulated expression, which likely contributed to the delayed limb development phenotype of H3K36M mutant mice (Abe et al. 2020). One possible mechanism is that the pervasive spreading of H3K27me3 causes a “titration” of the H3K27me3 reader complex PRC1 at strong polycomb target genes, which in turn decreases chromatin compaction and transcriptional repression (Lu et al. 2016). It would be interesting to know if restoration of the H3K27 methylation landscape could reverse the developmental abnormality and oncogenesis driven by H3K36M mutation.
The variant-specific effect of H3K36M mutation remains incompletely understood. A recent report examined the context specific role of H3.1K36M versus H3.3K36M in driving chondroblastoma (Zhang & Fang 2021). H3.3K36M but not H3.1K36M mutant chondrocytes showed increased colony formation and defects in differentiation. Locus-specific deposition of K36M mutant H3.1 or H3.3 resulted in local inhibition of H3K36 methylation and differential epigenetic and transcriptomic reprogramming. These results are consistent with the observation that only H3.3K36M mutations are found in human chondroblastomas.
4. H3.3G34 mutations
4.1. Mutational patterns of H3G34 in cancers
Histone H3G34 mutations were reported in ~20% of pHGGs of the forebrain cerebral cortex (Schwartzentruber et al. 2012), ~90% of giant cell tumors of the bone (GCTB) and more rarely in other bone neoplasms (Behjati et al. 2013; Koelsche et al. 2017) (Figure 1). Two interesting features distinguish the patterns of H3G34 mutations from H3 “K-to-M” mutations. First, H3G34 mutations are exclusively found in the variant histone H3.3, whereas both H3.3 and H3.1/2 can carry “K-to-M” mutations. Since H3.3 is deposited in more defined regions of the genome including promoters, enhancers and repetitive elements, H3.3G34 mutations likely exert a local effect on chromatin. Second, there is a high degree of tissue specificity in the substitution of G34 mutations. In pHGGs, G34 is frequently converted to arginine and less commonly to valine (G34R/V). In GCTB, however, the majority of mutations are G34 to tryptophan followed by leucine (G34W/L). It remains unclear if such a specificity reflects the different mutational processes between cancer types or implies a functional distinction among G34 substitutions.
4.2. Effects of H3.3G34 mutations on writer, eraser and reader enzymes
Unlike “K-to-M” mutations, H3.3G34 mutations do not alter global levels of histone modifications. Instead, they appear to act in cis through affecting the local binding and activity of chromatin enzymes and reader proteins. It was shown that in vitro G34R/V/W mutant nucleosomes block the methyltransferase activity of SETD2 towards the adjacent K36 on the same H3 tail (Fang et al. 2018b; Lewis et al. 2013). Consistently, ectopically expressed tagged G34 mutant histones, but not endogenous wildtype histones, displayed low levels of H3K36me3. Structural studies suggest that substitution of G34 with more bulky residues causes steric hindrance with the surrounding narrow tunnel formed by Y1604, F1668 and Y1671 of SETD2, inhibiting the enzyme’s access to mutant histone (Fang et al. 2018b; Yang et al. 2016; Zhang et al. 2017). Interestingly, these residues are not conserved in NSD family enzymes, suggesting that they may be able to tolerate G34 mutations. Indeed, H3.3G34 mutant nucleosome does not inhibit NSD2 activity and retains H3K36me2 levels (Jain et al. 2020a).
Nucleosomes carrying H3K36 methylation can inhibit H3K27 methylation by blocking PRC2 activity. Accordingly, H3.3G34 mutant histones exhibit increased levels of H3K27me3 (Jain et al. 2020a; Shi et al. 2018) and there is a genome-wide co-localization between H3.3G34 mutant histones and increased H3K27me3 (Huang et al. 2020; Jain et al. 2020a; Shi et al. 2018). Interestingly, point mutations affecting other residues (eg. G33, K36 and K37), while similarly inhibiting H3K36me3, do not alter H3K27me3 levels in cis, since these mutations prohibit the engagement and/or activity of PRC2 (Jain et al. 2020a). H3.3G33/K36/K37 mutations are also rarely found in human cancers, which strongly implies that the gain of H3K27me3 – rather than the mere loss of H3K36me3 – is a critical consequence of H3.3G34 mutations. Indeed, the effects of H3.3G34W mutation on gene expression were markedly rescued by substituting K27 to arginine (Jain et al. 2020a). Beyond the crosstalk with H3K27me3, H3K36 methylation facilitates the recruitment of PWWP domain-containing proteins such as the mismatch repair factor MutSα and the de novo DNA methyltransferases DNMT3A/B (Baubec et al. 2015; Li et al. 2013; Weinberg et al. 2019). Consistently, cells harboring H3G34 mutations demonstrate a weak mutator phenotype (Fang et al. 2018b). H3.3G34 mutations are also associated with a distinct DNA methylation signature that is conserved across different cancer types and can be recapitulated in an H3.3G34W mutant isogenic system (Lutsik et al. 2020; Sangatsuda et al. 2020). Future work is required to dissect the context-specific contribution of these (epi)genomic alterations to the biology of H3.3G34 mutant tumors.
H3.3G34 mutations may also affect the chromatin landscape in an H3K36 methylation-independent manner. H3.3G34R was shown to preferentially bind to and inhibit the activities of the KDM4 family histone demethylases, effectively “sequestering” these enzymes and causing genome-wide changes to H3K36 and H3K9 methylation (Voon et al. 2018). Supporting this mechanism, the epigenomic and transcriptomic effects of H3.3G34R can be phenocopied by triple knockout of KDM4A-C. Furthermore, RACK7 (ZMYND8), which normally recognizes H3K4me1/H3K14ac, was found to interact with H3.3G34R mutant nucleosomes in vitro and localize to and suppress the expression of H3.3G34R-marked genes in glioma cells (Jiao et al. 2020). A potential role of H3.3G34 mutations in the aberrant recruitment of chromatin complexes is further supported by a recent interactome study, which found that splicing factors such as hnRNPs preferentially bound to H3.3G34W mutant histones (Lim et al. 2017). These pleiotropic effects downstream of H3.3G34 mutations may explain how a single mutation could be sufficient for the development of human cancers like GCTB.
4.3. Modeling the oncogenic function of H3.3G34 mutations
In fission yeast, histone H3 is encoded by only three genes, making this organism an attractive model to study histone genetics. Yadav et al. modeled G34R mutation by deleting two histone H3 genes and introduced the mutation to the remaining single H3 gene (Yadav et al. 2017). Despite reduced global H3K36me3, H3K36me2 levels were maintained in G34R mutant strain. Since fission yeast only has one H3K36 methyltransferase Set2, this result suggests that G34R mutation partially inhibits or alters the activity of Set2. Indeed, G34R mutation had a more limited impact on gene expression compared to Set2 deletion. Furthermore, G34R mutant strain exhibited defects in maintaining genome integrity, including increased replication stress and impaired homologous recombination-mediated DNA repair. These features were missing in Set2 deletion strain, suggesting the involvement of H3K36 methylation-independent mechanisms. More recently, additional G34 mutations were modeled using fission yeast (Lowe et al. 2021). Surprisingly, the effects of G34R on genome instability were not recapitulated by G34V or G34W mutations, and H3K36me3 levels were retained in G34W mutant strain. These results point to the complex and subtle differences between G34 substitutions, which could underlie their high degree of cancer type specificity.
Modeling of GCTB-associated H3.3G34W mutation suggests that it plays a sufficient and necessary role in tumor development. Ectopic expression of H3.3G34W mutant in an osteosarcoma cell line increased proliferation compared to H3.3 wildtype-expressing cells (Lim et al. 2017). Implantation of mouse mesenchymal progenitor cells transduced with H3.3G34W mutation led to shortened survival time compared to wildtype and H3.3G34W/K27R double mutant cells, reinforcing the notion that H3K27 methylation is a key mediator of G34W mutation’s oncogenic potential (Jain et al. 2020a). On the other hand, knockdown of H3.3G34W mutant histone impaired the in vitro and in vivo growth of GCTB-derived cell lines (Fellenberg et al. 2019). This is consistent with the recent report that deletion or correction of the H3.3G34W mutant allele impaired GCTB tumor growth (Khazaei et al. 2020). This study further revealed that H3.3G34W mutant cells resembled developmentally stalled osteoblast-like cells, which not only promoted sustained proliferation but also facilitated the recruitment of multinucleated osteoclasts and in turn contributed to bone destruction. Importantly, H3.3G34W mutant cells retain the capacity to undergo enforced terminal maturation, suggesting that differentiation therapy may be an effective treatment strategy for GCTB.
Early analysis using an H3.3G34V-mutuant pediatric glioma line (KNS42) linked the histone mutation to the upregulation of MYCN oncogene (Bjerke et al. 2013). Several recent reports, however, revealed that the oncogenic mechanisms by H3.3G34 mutations in gliomas are complex and highly context-dependent. Comparing to transcriptome atlases of forebrain development, Chen et al. found that H3.3G34R/V gliomas shared gene expression programs with GSX2/DLX+ progenitors of the prenatal interneuron lineage (Chen et al. 2020). H3.3G34R/V oncohistones appeared to inhibit terminal neuronal differentiation, thus “locking” cells in a specific differentiation and chromatin state. This enables the stabilization of an otherwise transient interaction between PDGFRA – a gene commonly co-mutated with H3.3G34R/V - and GSX2 regulatory elements, leading to PDGFRA overexpression. The exquisite spatial-temporal window required for H3.3G34R/V-mediated glioma initiation was also elegantly demonstrated by studies showing robust oncogenic transformation of the forebrain but not hindbrain neural stem cells by H3.3G34R (Bressan et al. 2021; Funato et al. 2021). Both studies also discovered that H3.3G34R could lead to aberrant expression and/or splicing of forebrain master transcription regulators (eg. FOXG1) and signaling pathways (eg. NOTCH2NL). These transcriptional abnormalities may be attributed to diminished binding of ZMYND11 – a “reader” of H3.3K36me3 involved in transcription elongation and splicing (Wen et al. 2014) – to the H3.3G34R mutant nucleosomes. These valuable mechanistic insights and pre-clinical models will pave the way for discovering therapies targeting H3.3G34 oncohistones.
5. Non-canonical oncohistones
5.1. Core histone globular domain mutations are found in human cancers
Numerous cancer-associated mutations in all four core histones, affecting both the tails and globular domains, were recently reported and need further investigation to determine their role in promoting and/or sustaining tumorigenesis. Using a computational approach to comprehensively catalog the landscape of histone mutations across various cancers, Nacev et al. documented a total of 4,205 histone missense mutations in 3,143 samples from 3,074 unique patients across 183 specific tumor types (Nacev et al. 2019). Another recent study incorporated a DNA-barcoded mononucleosome library and a humanized yeast library to profile the biochemical and cellular effects of 150 histone mutations, highlighting that many of the globular domain mutations were lethal in yeast, upregulated cancer-associated gene pathways and inhibited cellular differentiation by altering the expression of lineage-specific transcription factors (Bagert et al. 2021; Jain & Strahl 2021). These “non-canonical” histone mutations collectively occur at a higher frequency than the canonical H3 tail mutations and appear to affect gene expression by locally altering the nucleosome stability, positioning and dynamics. Together these two datasets provide a library of testable hypotheses about the functional roles of non-canonical histone mutations in oncogenesis. For example, some histone mutations may disrupt higher order chromatin structure and override the need of SWI/SNF complex to aberrantly activate gene expression, whereas other mutations lead to swapped amino acid charges, thus possibly causing nucleosome instability in a dominant fashion.
In addition to these systematic analyses, several recent studies have also focused on select histone mutations to understand their roles in chromatin regulation and tumor biology. In carcinosarcomas of the ovary, neoplasms containing both carcinomatous and sarcomatous elements, whole-exome sequencing identified mutations in genes encoding histone H2A and H2B. Introduction of these mutations to USC-ARK2 cells led to a decreased expression of epithelial markers and increased expression of N-cadherin as well as transcription factor SNAI2/Slug, suggesting that these mutations may contribute to the epithelial-mesenchymal transition process (Zhao et al. 2016). Crystal structure analysis of H2BE76K, a charge swap mutation found in patients with bladder and head and neck cancers, showed perturbed interactions between H2B α2 and H4 α3 helices which destabilized the nucleosome (Figure 3). Further functional characterization of this mutation suggested that it could induce oncogenesis in the presence of wildtype H2B by relaxing chromatin and cooperating with oncogenes like PIK3CA and RAS (Bennett et al. 2019). H2BE76K has also been investigated as an oncohistone mutation in breast cancer. In this context, H2BE76K mutation directly regulated the transcription of ADAM19, a metalloproteinase-domain containing protein which is highly expressed in invasive breast carcinomas, by facilitating efficient transcription elongation (Evangeline Kang et al. 2021). In addition, when this mutation was added back into the MDA-MD-231 cells, it promoted colony formation and weakened histone octamer stability. A similar effect of nucleosome destabilization can be seen with short H2A histone variants (eg. H2A.B) that are typically expressed in testes of placental mammals. Analysis of existing genomic datasets identified unexpected aberrant H2A.B upregulation in a broad array of human cancers, which was associated with altered gene splicing patterns (Chew et al. 2021). Finally, another H2B mutation – G53D – has been identified in patients with pancreatic adenocarcinoma, glioblastoma, and lung squamous cell carcinoma (Wan et al. 2020). This mutation is located near the DNA entry/exit site and plays a role in mediating DNA-histone interaction. As a result, knock-in of this mutation in cells led to enhanced transcription elongation (Wan et al. 2020). Future investigations are needed to determine if the nucleosome destabilization by histone globular domain mutations affect other DNA-templated processes such as DNA repair and replication.
Figure 3:
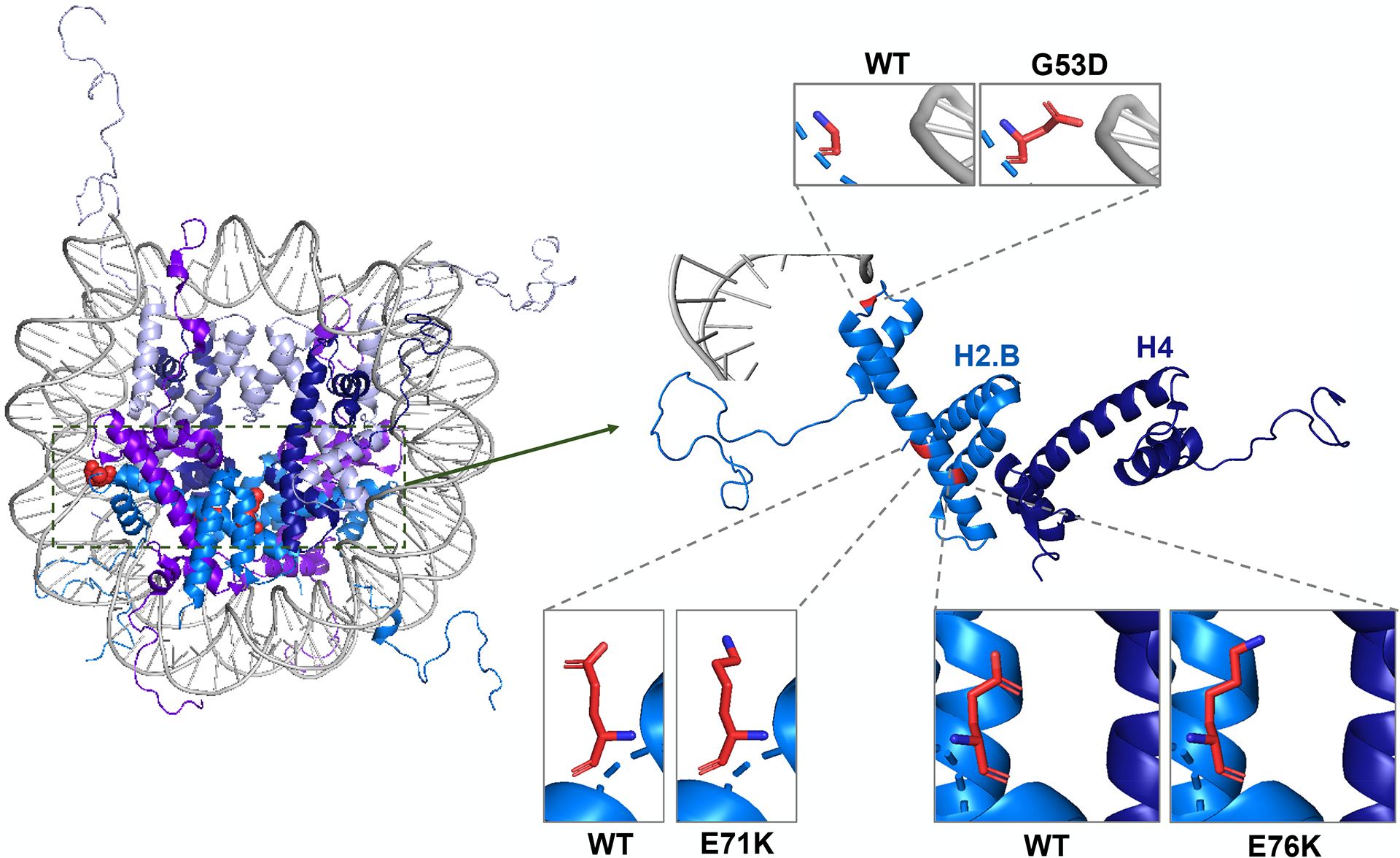
Nucleosome crystal structure with non-canonical histone mutations highlighted in red (Left). (Right) Close up of Histone H2.B (aqua blue) interacting with DNA (Gray) and H4 (Dark Blue). Side by side images of WT and mutated amino acid at reside 53, 71, and 76 (PDB code 1KX5).
5.2. Linker histone H1 mutations in lymphomas
The H1 linker histones are expressed in a replication-dependent manner, regulate nucleosome spacing and coordinate nucleosome arrays for compaction (Yusufova et al. 2021). Whole-genome sequencing of follicular lymphomas revealed clustered missense mutations in the DNA-binding globular domain of at least one histone H1 gene in 28% of samples, with HIST1H1C and HIST1H1E being the most common (Okosun et al. 2014) (Figure 1). To test the functional consequences of these mutations, mouse embryonic stem cells with combined knockout of H1 genes (Hist1h1c, Hist1h1d, Hist1h1e) were rescued with human wildtype or S102F mutant HIST1H1C. It was observed that the mutation impaired H1’s association with chromatin, suggesting that H1 mutations likely lead to a loss-of-function phenotype by reducing the DNA-binding affinity of H1 during chromatin compaction (Okosun et al. 2014). HIST1H1E was furthered categorized as a driver gene from a CRISPR screen in germinal center B cell-like diffuse large B cell lymphomas (GCB-DLBCL) (Reddy et al. 2017). Consistently, genetically engineered mouse models where either H1C and H1E or H1C, H1E, and H1D were knocked out in hematopoietic stem cells demonstrated that loss of H1 protein led to T cell re-activation and reduction in mature B cells and CD4+, CD8+ T cells in the thymus (Willcockson et al. 2021). Furthermore, H1 regulates local nucleosome density, promotes PRC2-mediated H3K27me3 and inhibits NSD2-mediated H3K36me2. The disrupted H3K36-H3K27 methylation balance in H1-deficient lymphomas caused remodeling of nuclear 3D compartmentalization, leading to de-silencing of stemness genes (Yusufova et al. 2021). Therefore, by promoting the transition to a relaxed chromatin conformation and conferring germinal center B cells with enhanced fitness and self-renewal capacity, H1 mutations act as drivers for lymphomas and represent an enticing target for therapeutic intervention.
6. Concluding remarks
In less than a decade from the initial reports of cancer-associated histone mutations, we have witnessed a remarkable progress in elucidating the function and mechanism of oncohistones. Importantly, mechanistic insights gained from lab investigations have identified therapeutic opportunities and nominated novel drug candidates, some of which have entered clinical testing. However, several key questions remain to be addressed. For the canonical oncohistones, therapeutic strategies targeting H3K36M and H3G34R/V/W mutations are largely lacking. For the non-canonical oncohistones, which lack the exquisite cancer type specificity, identifying the precise context (eg. cell-of-origin and cooperating mutations) that enables these globular domain mutations to activate oncogenic expression programs in cis remains a major challenge. Furthermore, it is perhaps reasonable to speculate that additional “oncohistone-like” proteins exist and remain to be identified. Notably, beyond their roles in cancer, histone mutations have become important tools for probing chromatin complex structures (Justin et al. 2016; Yang et al. 2016) and epigenome dynamics (Brumbaugh et al. 2019; Herz et al. 2014). Therefore, a more comprehensive and in-depth analysis of oncohistones will have a broad and far-reaching impact on chromatin biology in development and human diseases.
Acknowledgements
We apologize to colleagues whose work could not be cited because of space limitations. We thank the Lu lab for critical reading of the manuscript. C.L. is supported by funding from the National Institutes of Health (NIH) Grant P01CA196539 and the Pew-Stewart Scholars for Cancer Research Award.
Footnotes
Conflict of Interests
The authors declare that they have no conflict of interest.
References
- Abe S, Nagatomo H, Sasaki H, Ishiuchi T. 2020. A histone H3.3K36M mutation in mice causes an imbalance of histone modifications and defects in chondrocyte differentiation. Epigenetics. 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas JN, Zee BM, Kalin JH, Kim M, Guo R, et al. 2019. Re-programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell. 36(5):528–544.e10 [DOI] [PubMed] [Google Scholar]
- Bagert JD, Mitchener MM, Patriotis AL, Dul BE, Wojcik F, et al. 2021. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat. Chem. Biol 17(4):403–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan I, Danis E, Pierce A, Madhavan K, Wang D, et al. 2020. Senescence Induced by BMI1 Inhibition Is a Therapeutic Vulnerability in H3K27M-Mutant DIPG. Cell Rep. 33(3):108286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, et al. 2015. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 520(7546):243–47 [DOI] [PubMed] [Google Scholar]
- Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, et al. 2016. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci. Transl. Med 8(366):366ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, et al. 2013. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet 45(12):1479–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, et al. 2013. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 24(5):660–72 [DOI] [PubMed] [Google Scholar]
- Bennett RL, Bele A, Small EC, Will CM, Nabet B, et al. 2019. A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discov. 9(10):1438–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, et al. 2013. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 3(5):512–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan RB, Southgate B, Ferguson KM, Blin C, Grant V, et al. 2021. Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants. Cell Stem Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ZZ, Müller MM, Jain SU, Allis CD, Lewis PW, Muir TW. 2014. Strategy for “detoxification” of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J. Am. Chem. Soc 136(39):13498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumbaugh J, Kim IS, Ji F, Huebner AJ, Di Stefano B, et al. 2019. Inducible histone K-to-M mutations are dynamic tools to probe the physiological role of site-specific histone methylation in vitro and in vivo. Nat. Cell Biol 21(11):1449–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczkowicz P, Hawkins C. 2015. Pathology, Molecular Genetics, and Epigenetics of Diffuse Intrinsic Pontine Glioma. Front. Oncol 5:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, et al. 2014. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet 46(5):451–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho S, Vítor AC, Sridhara SC, Martins FB, Raposo AC, et al. 2014. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife. 3:e02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel D, Kergrohen T, Tauziède-Espariat A, Mackay A, Ghermaoui S, et al. 2020. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. 139(6):1109–13 [DOI] [PubMed] [Google Scholar]
- Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, et al. 2015. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 130(6):815–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel D, Philippe C, Kergrohen T, Sill M, Merlevede J, et al. 2018. Transcriptomic and epigenetic profiling of “diffuse midline gliomas, H3 K27M-mutant” discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun 6(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K-M, Fang D, Gan H, Hashizume R, Yu C, et al. 2013. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 27(9):985–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CCL, Deshmukh S, Jessa S, Hadjadj D, Lisi V, et al. 2020. Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell. 183(6):1617–1633.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew G-L, Bleakley M, Bradley RK, Malik HS, Henikoff S, et al. 2021. Short H2A histone variants are expressed in cancer. Nat. Commun 12(1):490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C, Sweha SR, Pratt D, Tamrazi B, Panwalkar P, et al. 2020. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell. 38(3):334–349.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero FJ, Huang Z, Grenier C, He X, Hu G, et al. 2017. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol. Cancer Res 15(9):1243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl NA, Danis E, Balakrishnan I, Wang D, Pierce A, et al. 2020. Super Elongation Complex as a Targetable Dependency in Diffuse Midline Glioma. Cell Rep. 31(1):107485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl KL, Ge EJ, Weinberg DN, Jani KS, Allis CD, Muir TW. 2019. PRC2 engages a bivalent H3K27M-H3K27me3 dinucleosome inhibitor. Proc. Natl. Acad. Sci. U. S. A 116(44):22152–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehteda A, Simon S, Franshaw L, Giorgi FM, Liu J, et al. 2021. Dual targeting of the epigenome via FACT complex and histone deacetylase is a potent treatment strategy for DIPG. Cell Rep. 35(2):108994. [DOI] [PubMed] [Google Scholar]
- Evangeline Kang TZ, Zhu L, Yang D, Ding D, Zhu X, et al. 2021. The elevated transcription of ADAM19 by the oncohistone H2BE76K contributes to oncogenic properties in breast cancer. J. Biol. Chem 296:100374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Gan H, Cheng L, Lee J-H, Zhou H, et al. 2018a. H3.3K27M mutant proteins reprogram epigenome by sequestering the PRC2 complex to poised enhancers. Elife. 7: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Gan H, Lee J-H, Han J, Wang Z, et al. 2016. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. 352(6291):1344–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Huang Y, Mao G, Yang S, Rennert G, et al. 2018b. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc. Natl. Acad. Sci. U. S. A 115(38):9598–9603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellenberg J, Sähr H, Mancarella D, Plass C, Lindroth AM, et al. 2019. Knock-down of oncohistone H3F3A-G34W counteracts the neoplastic phenotype of giant cell tumor of bone derived stromal cells. Cancer Lett. 448:61–69 [DOI] [PubMed] [Google Scholar]
- Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, et al. 2018. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science. 360(6386):331–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finogenova K, Bonnet J, Poepsel S, Schäfer IB, Finkl K, et al. 2020. Structural basis for PRC2 decoding of active histone methylation marks H3K36me2/3. Elife. 9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J, Tian R, Zarrabi I, Hill G, Williams E, et al. 2020. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell. 37(3):308–323.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. 2014. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 346(6216):1529–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Smith RC, Saito Y, Tabar V. 2021. Dissecting the impact of regional identity and the oncogenic role of human-specific NOTCH2NL in an hESC model of H3.3G34R-mutant glioma. Cell Stem Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Tabar V. 2018. Histone Mutations in Cancer. Annu. Rev. Cancer Biol 2(1):337–51 [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, et al. 2015. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med 21(6):555–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag D, Mack N, Benites Goncalves da Silva P, Statz B, Clark J, et al. 2021. H3.3-K27M drives neural stem cell-specific gliomagenesis in a human iPSC-derived model. Cancer Cell. 39(3):407–422.e13 [DOI] [PubMed] [Google Scholar]
- Harutyunyan AS, Chen H, Lu T, Horth C, Nikbakht H, et al. 2020. H3K27M in Gliomas Causes a One-Step Decrease in H3K27 Methylation and Reduced Spreading within the Constraints of H3K36 Methylation. Cell Rep. 33(7):108390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, et al. 2019. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun 10(1):1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, et al. 2014. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med 20(12):1394–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz H-M, Morgan M, Gao X, Jackson J, Rickels R, et al. 2014. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. 345(6200):1065–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeman CM, Cordero FJ, Hu G, Misuraca K, Romero MM, et al. 2019. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat. Commun 10(1):1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TY-T, Piunti A, Qi J, Morgan M, Bartom E, et al. 2020. Effects of H3.3G34V mutation on genomic H3K36 and H3K27 methylation patterns in isogenic pediatric glioma cells. Acta Neuropathol. Commun 8(1):219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner J-M, Müller T, Papageorgiou DN, Mauermann M, Krijgsveld J, et al. 2019. EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro. Oncol 21(7):878–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain K, Strahl BD. 2021. Oncohistones: corruption at the core. Nat. Chem. Biol 17(4):370–71 [DOI] [PubMed] [Google Scholar]
- Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, et al. 2019. PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun 10(1):2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SU, Khazaei S, Marchione DM, Lundgren SM, Wang X, et al. 2020a. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. U. S. A 117(44):27354–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SU, Rashoff AQ, Krabbenhoft SD, Hoelper D, Do TJ, et al. 2020b. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol. Cell 80(4):726–735.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jani KS, Jain SU, Ge EJ, Diehl KL, Lundgren SM, et al. 2019. Histone H3 tail binds a unique sensing pocket in EZH2 to activate the PRC2 methyltransferase. Proc. Natl. Acad. Sci. U. S. A 116(17):8295–8300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. 2001. Translating the histone code. Science. 293(5532):1074–80 [DOI] [PubMed] [Google Scholar]
- Jiao F, Li Z, He C, Xu W, Yang G, et al. 2020. RACK7 recognizes H3.3G34R mutation to suppress expression of MHC class II complex components and their delivery pathway in pediatric glioblastoma. Sci. Adv 6(29):eaba2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, et al. 2016. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun 7:11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagi H, Louis N, Unruh D, Sasaki T, He X, et al. 2019. Radiosensitization by Histone H3 Demethylase Inhibition in Diffuse Intrinsic Pontine Glioma. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res 25(18):5572–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernohan KD, Grynspan D, Ramphal R, Bareke E, Wang YC, et al. 2017. H3.1 K36M mutation in a congenital-onset soft tissue neoplasm. Pediatr. Blood Cancer 64(12): [DOI] [PubMed] [Google Scholar]
- Khazaei S, De Jay N, Deshmukh S, Hendrikse LD, Jawhar W, et al. 2020. H3.3 G34W Promotes Growth and Impedes Differentiation of Osteoblast-Like Mesenchymal Progenitors in Giant Cell Tumor of Bone. Cancer Discov. 10(12):1968–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelsche C, Schrimpf D, Tharun L, Roth E, Sturm D, et al. 2017. Histone 3.3 hotspot mutations in conventional osteosarcomas: a comprehensive clinical and molecular characterization of six H3F3A mutated cases. Clin. Sarcoma Res 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug B, De Jay N, Harutyunyan AS, Deshmukh S, Marchione DM, et al. 2019. Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell. 35(5):782–797.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Sengupta S, Lee K, Hura N, Fuller C, et al. 2017. BMI-1 is a potential therapeutic target in diffuse intrinsic pontine glioma. Oncotarget. 8(38):62962–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, et al. 2011. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44(4):609–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, et al. 2019. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell. 35(1):140–155.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-H, Yu J-R, Granat J, Saldaña-Meyer R, Andrade J, et al. 2019. Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev. 33(19–20):1428–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, et al. 2014. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet 46(11):1227–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leicher R, Ge EJ, Lin X, Reynolds MJ, Xie W, et al. 2020. Single-molecule and in silico dissection of the interaction between Polycomb repressive complex 2 and chromatin. Proc. Natl. Acad. Sci. U. S. A 117(48):30465–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, et al. 2013. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 340(6134):857–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Mao G, Tong D, Huang J, Gu L, et al. 2013. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell. 153(3):590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Kaminski MS, Li Y, Yildiz M, Ouillette P, et al. 2014. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 123(10):1487–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Park JH, Baude A, Yoo Y, Lee YK, et al. 2017. The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing. Sci. Rep 7(1):13459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Zhou Y, Li M, Fang Y. 2018. Histone 3 lysine 36 to methionine mutations stably interact with and sequester SDG8 in Arabidopsis thaliana. Sci. China. Life Sci 61(2):225–34 [DOI] [PubMed] [Google Scholar]
- Lowe BR, Yadav RK, Henry RA, Schreiner P, Matsuda A, et al. 2021. Surprising phenotypic diversity of cancer-associated mutations of Gly 34 in the histone H3 tail. Elife. 10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, et al. 2016. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 352(6287):844–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutsik P, Baude A, Mancarella D, Öz S, Kühn A, et al. 2020. Globally altered epigenetic landscape and delayed osteogenic differentiation in H3.3-G34W-mutant giant cell tumor of bone. Nat. Commun 11(1):5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, et al. 2014. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 506(7489):445–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, et al. 2017. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med 23(4):483–92 [DOI] [PubMed] [Google Scholar]
- Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, et al. 2018. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med 24(5):572–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacev BA, Feng L, Bagert JD, Lemiesz AE, Gao J, et al. 2019. The expanding landscape of “oncohistone” mutations in human cancers. Nature. 567(7749):473–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja S, Quezada MA, Gillespie SM, Arzt M, Lennon JJ, et al. 2019. Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol. Cell 76(6):965–980.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, et al. 2017. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell. 31(5):635–652.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okosun J, Bödör C, Wang J, Araf S, Yang C-Y, et al. 2014. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet 46(2):176–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajovic S, Siddaway R, Bridge T, Sheth J, Rakopoulos P, et al. 2020. Epigenetic activation of a RAS/MYC axis in H3.3K27M-driven cancer. Nat. Commun 11(1):6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Wen J, Sill M, Lin T, Orisme W, et al. 2018. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 136(2):211–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Witt H, Sill M, Jones DTW, Hovestadt V, et al. 2015. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell. 27(5):728–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papillon-Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, et al. 2017. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet 49(2):180–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, et al. 2017. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell. 32(5):684–700.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, et al. 2017. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med 23(4):493–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Smith ER, Morgan MAJ, Ugarenko M, Khaltyan N, et al. 2019. CATACOMB: An endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci. Adv 5(7):eaax2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragazzini R, Pérez-Palacios R, Baymaz IH, Diop S, Ancelin K, et al. 2019. EZHIP constrains Polycomb Repressive Complex 2 activity in germ cells. Nat. Commun 10(1):3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan KN, Chen X, Weinberg DN, Chen H, Majewski J, et al. 2021. Depletion of H3K36me2 recapitulates epigenomic and phenotypic changes induced by the H3.3K36M oncohistone mutation. Proc. Natl. Acad. Sci. U. S. A 118(9): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, et al. 2017. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell. 171(2):481–494.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders D, Qian S, Fieweger R, Lu L, Dowell JA, et al. 2017. Histone Lysine-to-Methionine Mutations Reduce Histone Methylation and Cause Developmental Pleiotropy. Plant Physiol. 173(4):2243–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangatsuda Y, Miura F, Araki H, Mizoguchi M, Hata N, et al. 2020. Base-resolution methylomes of gliomas bearing histone H3.3 mutations reveal a G34 mutant-specific signature shared with bone tumors. Sci. Rep 10(1):16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran SM, Gozani O. 2017. Characterization of H3.3K36M as a tool to study H3K36 methylation in cancer cells. Epigenetics. 12(11):917–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarthy JF, Meers MP, Janssens DH, Henikoff JG, Feldman H, et al. 2020. Histone deposition pathways determine the chromatin landscapes of H3.1 and H3.3 K27M oncohistones. Elife. 9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, et al. 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 482(7384):226–31 [DOI] [PubMed] [Google Scholar]
- Shi L, Shi J, Shi X, Li W, Wen H. 2018. Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis. J. Mol. Biol 430(11):1562–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira AB, Kasper LH, Fan Y, Jin H, Wu G, et al. 2019. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 137(4):637–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, et al. 2014. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 24(2):241–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snuderl M, Dolgalev I, Heguy A, Walsh MF, Benayed R, et al. 2019. Histone H3K36I mutation in a metastatic histiocytic tumor of the skull and response to sarcoma chemotherapy. Cold Spring Harb. Mol. case Stud 5(5): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford JM, Lee C-H, Voigt P, Descostes N, Saldaña-Meyer R, et al. 2018. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv 4(10):eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang D-A, Jones DTW, et al. 2012. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 22(4):425–37 [DOI] [PubMed] [Google Scholar]
- Subashi E, Cordero FJ, Halvorson KG, Qi Y, Nouls JC, et al. 2016. Tumor location, but not H3.3K27M, significantly influences the blood-brain-barrier permeability in a genetic mouse model of pediatric high-grade glioma. J. Neurooncol 126(2):243–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavosian R, Duc HN, Huynh TN, Fang D, Schmitt B, et al. 2018. Live-cell single-molecule dynamics of PcG proteins imposed by the DIPG H3.3K27M mutation. Nat. Commun 9(1):2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, et al. 2014. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet 46(5):457–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, et al. 2013. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 23(5):558–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon HPJ, Udugama M, Lin W, Hii L, Law RHP, et al. 2018. Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun 9(1):3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Carpenter PB. 2012. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol 13(2):115–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan YCE, Leung TCS, Ding D, Sun X, Liu J, et al. 2020. Cancer-associated histone mutation H2BG53D disrupts DNA-histone octamer interaction and promotes oncogenic phenotypes. [DOI] [PMC free article] [PubMed]
- Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, et al. 2017. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat. Struct. Mol. Biol 24(12):1028–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, et al. 2019. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 573(7773):281–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, et al. 2014. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 508(7495):263–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcockson MA, Healton SE, Weiss CN, Bartholdy BA, Botbol Y, et al. 2021. H1 histones control the epigenetic landscape by local chromatin compaction. Nature. 589(7841):293–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, et al. 2012. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet 44(3):251–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, et al. 2014. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet 46(5):444–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav RK, Jablonowski CM, Fernandez AG, Lowe BR, Henry RA, et al. 2017. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. Elife. 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Zheng X, Lu C, Li G-M, Allis CD, Li H. 2016. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 30(14):1611–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. 2011. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem 286(10):7983–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, et al. 2021. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature. 589(7841):299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Fang D. 2021. The incorporation loci of H3.3K36M determine its preferential prevalence in chondroblastomas. Cell Death Dis. 12(4):311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shan C-M, Wang J, Bao K, Tong L, Jia S. 2017. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci. Rep 7:43906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Bellone S, Lopez S, Thakral D, Schwab C, et al. 2016. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. U. S. A 113(43):12238–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang L, Jang Y, Park Y-K, Lee J-E, Jain S, et al. 2018. Depletion of Nsd2-mediated histone H3K36 methylation impairs adipose tissue development and function. Nat. Commun 9(1):1796. [DOI] [PMC free article] [PubMed] [Google Scholar]