

Abstract
The cochlea uses two types of mechanosensory cell to detect sounds. A single row of inner hair cells (IHCs) synapse onto neurons to transmit sensory information to the brain, and three rows of outer hair cells (OHCs) selectively amplify auditory inputs1. So far, two transcription factors have been implicated in the specific differentiation of OHCs, whereas, to our knowledge, none has been identified in the differentiation of IHCs2–4. One such transcription factor for OHCs, INSM1, acts during a crucial embryonic period to consolidate the OHC fate, preventing OHCs from transdifferentiating into IHCs2. In the absence of INSM1, embryonic OHCs misexpress a core set of IHC-specific genes, which we predict are involved in IHC differentiation. Here we find that one of these genes, Tbx2, is a master regulator of IHC versus OHC differentiation in mice. Ablation of Tbx2 in embryonic IHCs results in their development as OHCs, expressing early OHC markers such as Insm1 and eventually becoming completely mature OHCs in the position of IHCs. Furthermore, Tbx2 is epistatic to Insm1: in the absence of both genes, cochleae generate only OHCs, which suggests that TBX2 is necessary for the abnormal transdifferentiation of INSM1-deficient OHCs into IHCs, as well as for normal IHC differentiation. Ablation of Tbx2 in postnatal, largely differentiated IHCs makes them transdifferentiate directly into OHCs, replacing IHC features with those of mature and not embryonic OHCs. Finally, ectopic expression of Tbx2 in OHCs results in their transdifferentiation into IHCs. Hence, Tbx2 is both necessary and sufficient to make IHCs distinct from OHCs and maintain this difference throughout development.
With the goal of identifying a gene driving inner hair cell (IHC)-specific differentiation, we surmised that such a gene would (1) encode a gene regulator such as a transcription factor, (2) be expressed during normal development in IHCs but not outer hair cells (OHCs) and (3) be expressed during abnormal development (that of Insm1 mutants) in those OHCs that transdifferentiate into IHCs. One such gene is Tbx2.
By in situ hybridization, we found that Tbx2 mRNA is expressed in IHCs and other epithelial cell types lining the cochlear scala media in mice, including supporting cells of the inner compartment of the organ of Corti (inner border and phalangeal), Kölliker’s organ, interdental cells of the spiral limbus, Reissner’s membrane, stria vascularis, spiral prominence and Claudius’ cells. By contrast, we detected little or no Tbx2 mRNA in cells of the outer compartment of the organ of Corti (OHCs and supporting pillar, Deiters’ and Hensen’s cells) (Extended Data Fig. 1a, d, f). Despite this broad expression pattern and an early role for Tbx2 information of the inner ear from the otocyst5, we suspected that Tbx2 has a specific role in IHCs because, in the absence of INSM1, embryonic OHCs express a small set of IHC-specific genes that includes Tbx2 and nearly half these cells transdifferentiate into IHCs2. We reasoned that, among these genes, those misexpressed in all INSM1-deficient OHCs would render the cells prone to transdifferentiation into IHCs, whereas those solely expressed in transdifferentiating hair cells could have an early role in IHC differentiation. We found that Tbx2 was not misexpressed in all INSM1-lacking OHCs (OHCs of Insm1 mutants) but only in those that transdifferentiated into IHCs (Extended Data Fig. 1b, c, e, g). Early in development, all INSM1-lacking OHCs that expressed Tbx2 also expressed the functional IHC marker Vglut3 (50/114 at postnatal day (P) 0 (P0), 14/30 at P2 and 9/21 at P4; Extended Data Fig. 1f–h); however, no hair cell expressed either gene alone. On the basis of this complete correlation between early Tbx2 expression and transdifferentiation into IHCs and because TBX2 is a transcriptional regulator involved in differentiation of various cell types6–8, we postulated that TBX2 would be a critical factor in regulating IHC differentiation.
Conditional ablation of Tbx2 (using a loxP-flanked allele9) with Atoh1cre in nascent hair cells and most supporting cells at about embryonic day (E) 13.5 (E13.5)10, before their differentiation, resulted in embryos in which all of the hair cells in the position of IHCs expressed the early OHC markers Insm1 and Bcl11b and downregulated the early IHC markers Fgf8, Brip1 and Msx1 (Fig. 1a–e′). Mature mice in which Tbx2 had been conditionally ablated as above with Atoh1cre or with Gfi1cre (expressed in all hair cells at E15.5; ref. 11) were deaf and lacked auditory brainstem responses (ABRs, for which IHCs are essential), as expected for mice with no functional IHCs (Fig. 1f). In these mice, the cochlear hair cells in the position of IHCs (those in the inner, or medial, compartment of the organ of Corti) displayed all examined molecular, anatomical and physiological features of OHCs but none of the features of IHCs. These cells expressed OHC markers such as the electromotile protein prestin, the calcium buffer oncomodulin, the potassium channel KCNQ4 and, in their stereocilia, the calcium pump PMCA2. However, they lacked IHC markers such as the vesicular glutamate transporter VGLUT3, the calcium buffer CALB2, the big potassium (BK) channel and nuclear CtBP2 (Fig. 1g–p′ and Extended Data Table 1). Anatomically, these cells resembled OHCs in having a cylindrical rather than flask shape, stereocilia with a less intense phalloidin label and tight bundling rather than a fanned-out appearance like IHCs12,13 (Fig. 1o–p′), although their alignment was not as precisely arcuate as that of OHCs. Furthermore, these cells had smaller nuclei positioned at the base as opposed to larger nuclei near the centre of the hair cell (the nuclear size and position for IHCs) (Extended Data Fig. 2) and reduced numbers of synaptic ribbons labelled with anti-CtBP2 antibodies (Fig. 1n, n′ and Extended Data Table 1). Although these hair cells were in the normal position for IHCs (the inner compartment), they molecularly and anatomically appeared as bona fide OHCs; hence, we termed them ic-OHCs. We conclude that, in the absence of TBX2, hair cells born in the position of IHCs instead proceed to differentiate as OHCs (Extended Data Fig. 5), which suggests that TBX2 is required for IHCs to develop as distinct cells from OHCs.
Fig. 1 |. Early ablation of Tbx2 results in the generation of OHCs in the position of IHCs.
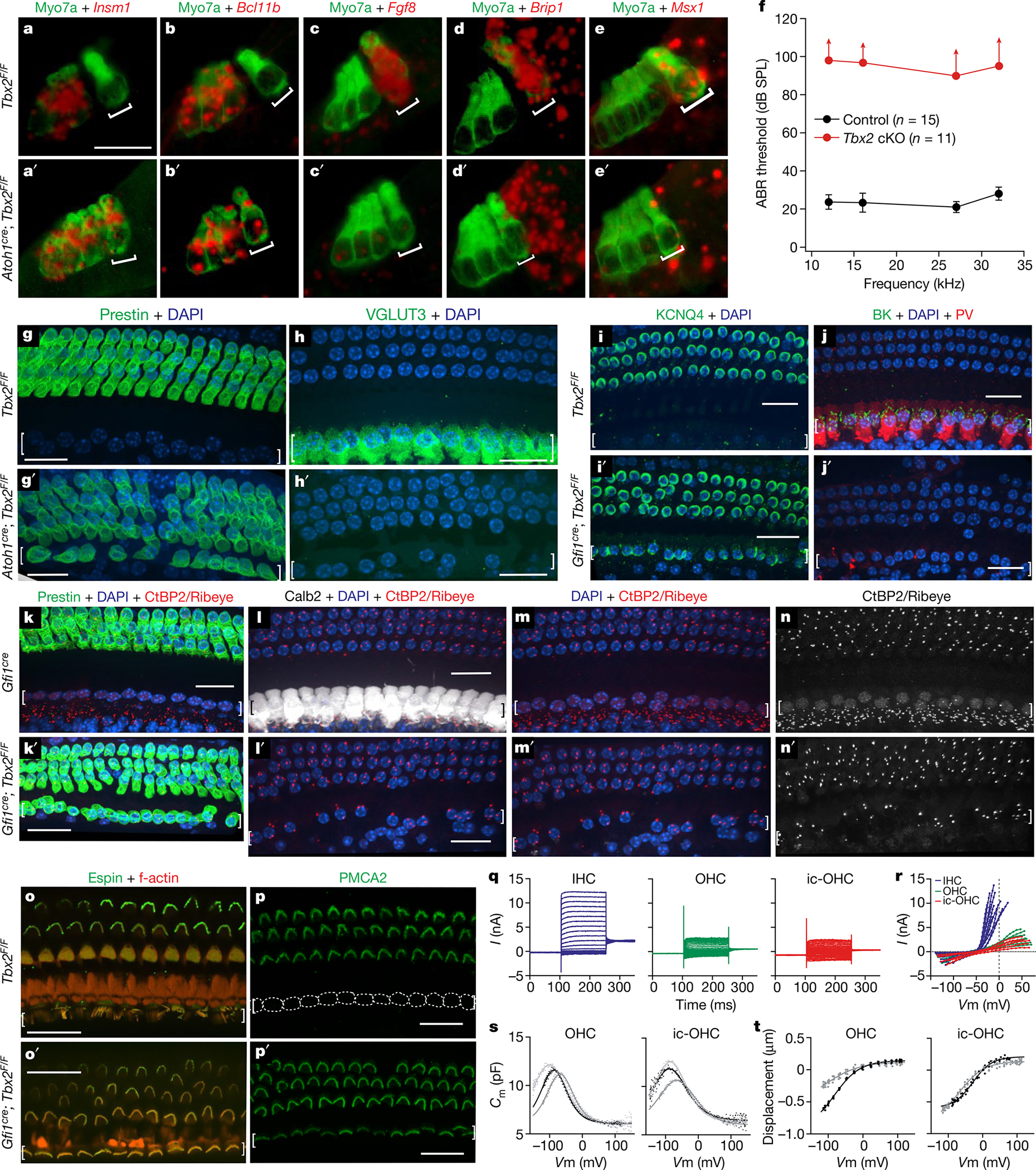
a–e′, In situ hybridizations (red) with immunohistochemistry for hair cells using myosin VIIa (green) on E17.5 cochleae after conditional Tbx2 deletion. In mutants, embryonic hair cells in the position of IHCs (square brackets) express markers of developing OHCs (a–b′) but not those of IHCs (c–e′). Expression of Brip1 and Msx1 in supporting cells near IHCs is unaltered in conditional knockouts (cKOs) (n ≥ 3 biologically independent samples). f, ABRs are absent (up-facing arrows) from mature or weaned (P26–P39) mice with embryonic ablation of Tbx2 (red; n = 11: 5 Atoh1cre/+; Tbx2F/F plus 6 Gfi1cre/+; Tbx2F/F). Littermate controls (black; n = 15: Tbx2F/F, Atoh1cre/+; Tbx2F/+, Gfi1cre/+; Tbx2F/+ and Gfi1cre/+) had normal ABRs. Error bars, s.d. g–p′, Immunohistochemistry of mature cochleae indicating that, after embryonic Tbx2 ablation, all hair cells in the position of IHCs (square brackets) displayed all examined features of OHCs but none for IHCs. These features include expression of prestin (g, g′, k, k′), KCNQ4 (i, i′) and PMCA2 (p, p′); lack of VGLUT3 (h, h′), BK, parvalbumin (j, j′), CALB2 (l, l′) and nuclear CtBP2 (m–n′); few synaptic ribbons (Ribeye+ puncta; k–n′); small nuclei (DAPI+; m, m′) located at the base and not the middle of the hair cell (h, i′, j); and shorter stereocilia tightly bundled as in OHCs rather than the long and fanning pattern for IHCs (o–p′). Dotted lines (p) delineate IHCs, which lack PMCA2 (n ≥ 3 biologically independent samples). q–t, Whole-cell currents from ic-OHCs (of Fgf8creER; Tbx2F/F; R26LSL-tdTomato/+ mice treated with tamoxifen at birth; tdTomato+) and from control IHCs and OHCs. q, r, Current responses to voltage steps (−140 mV to +80 mV) in ic-OHCs (n = 7) were characteristic of OHCs (n = 7) but not IHCs (n = 12). s, t, Electromotility assessment from nonlinear capacitance (s) and video recordings (t). Two-state Boltzmann fits. Similar to control OHCs (n = 9), ic-OHCs (n = 10) were electromotile. Hair cells originated from the apical one-fourth of the cochlea at P25–P29. The cells were from ≥3 animals. Scale bars, 20 μm.
To determine whether ic-OHCs display the physiological features of OHCs but not IHCs, we generated Fgf8creER; Tbx2F/F; R26LSL-tdTomato/+ mice. The Fgf8creER allele14 expresses the tamoxifen-inducible CreER recombinase in IHCs from their earliest differentiation in the embryo until about P9, after which expression subsides first in basal IHCs and later in apical IHCs. Tamoxifen administration at birth ablated Tbx2 in IHCs and labelled them with permanent expression of the live reporter tdTomato (Extended Data Fig. 3a, a′). Following maturation (P25–P29), we dissociated fluorescently labelled ic-OHCs from these mice as well as from IHCs and OHCs obtained from control mice and then performed whole-cell patch-clamp recordings. We found that ic-OHCs displayed basolateral currents characteristic of OHCs (KCNQ4-mediated IK,n) but not those of IHCs (BK-mediated IK,f) (Fig. 1q, r). Notably, voltage changes revealed that ic-OHCs were electromotile (as assessed both through changes in capacitance and by visual observations of changes in cell length), which is the most defining property of OHCs (Fig. 1s, t and Extended Data Fig. 3b). We conclude that the ic-OHCs generated by ablation of Tbx2 are functional OHCs.
We have shown that, in the absence of TBX2, embryonic IHCs express INSM1 and differentiate as OHCs (Fig. 1), whereas in the absence of INSM1 many embryonic OHCs express TBX2 and transdifferentiate into IHCs2,15 (Extended Data Table 1 and Extended Data Fig. 5). We wondered whether INSM1 expression is required for IHC-to-OHC conversion and whether TBX2 is required for OHC-to-IHC conversion. We generated mice lacking both TBX2 and INSM1 in embryonic IHCs and OHCs (Atoh1cre; Tbx2F/F; Insm1F/F) and found that Tbx2 is epistatic to Insm1: none of the OHCs transdifferentiated into IHCs, and all of the IHCs transdifferentiated into OHCs (Fig. 2, Extended Data Table 1 and Extended Data Figs. 4 and 5). This result is consistent with the dispensable nature of INSM1 in OHC differentiation: whereas removal of INSM1 renders OHCs susceptible to becoming IHCs, over half of these cells differentiate into mature OHCs2. Although INSM1 is thought to prevent embryonic OHCs from responding to an IHC-inducing signal, it is not essential for OHC differentiation. By contrast, our results demonstrate that TBX2 is necessary for IHC differentiation during normal IHC formation and through abnormal transdifferentiation from INSM1-lacking OHCs (Extended Data Fig. 5).
Fig. 2 |. Tbx2 is epistatic to Insm1.
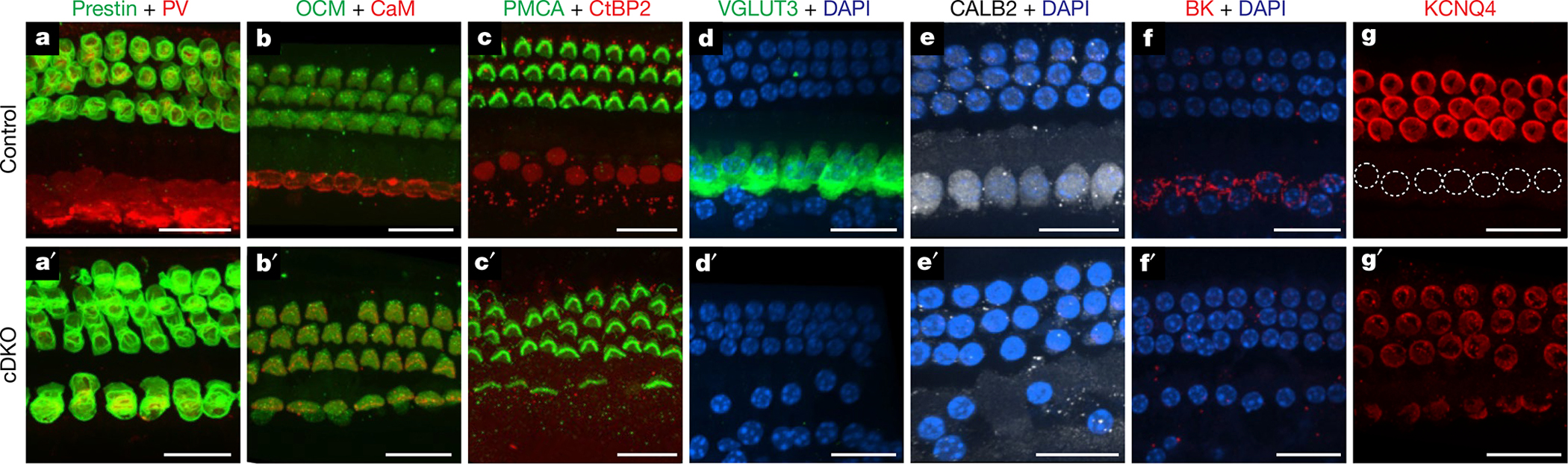
Immunohistochemistry in cochleae of mice (P20 in d, d′; P29 in f–g′; P26 in all other panels) in which both Tbx2 and Insm1 were conditionally ablated (cDKOs: Atoh1cre/+; Insm1F/F; Tbx2F/F; a′–g′) or in control littermates in which both genes were functional (Insm1F/F; Tbx2F/F; a–g). Staining with antibodies to prestin and parvalbumin (PV; a, a′), oncomodulin and calmodulin (b, b′), PMCA2 and CtBP2 (Ribeye and nuclear isoforms; c, c′), VGLUT3 (d, d′), CALB2 (e, e′), BK (f, f′) and KCNQ4 (g, g′) is shown. Nuclei are labelled with DAPI (d–f′) or are outlined with a dotted white line (IHCs in g). In cDKOs, the phenotype is exactly as in Tbx2-knockout mice (both IHCs and OHCs express OHC markers). By contrast, cDKOs do not display the Insm1-knockout phenotype, in which nearly half of the hair cells in the position of OHCs expressing IHC markers, as previously shown2,15. Expression of prestin, oncomodulin and PMCA2, few CtPB2+ synaptic ribbons and smaller nuclei at the basal end of the hair cell are all features of normal OHCs and are also displayed by all of the examined cells in the position of IHCs in cDKO mice. Expression of calmodulin, CALB2 and nuclear CtBP2, numerous CtPB2+ synaptic ribbons and larger nuclei located in the middle of the hair cell are features of normal IHCs that are missing in the cDKO cells in the position of IHCs. All images are from middle portions of the cochlea (spanning approximately 31–82% of the cochlear length, from base to apex) except those in f–g′, which are more basal (approximately 10–31%). The images shown are examples of results obtained with three separate tissue samples (n = 3 biologically independent samples). Scale bars, 20 μm.
At birth, IHCs are already at an advanced stage of differentiation, expressing markers of functionally mature IHCs such as VGLUT3. The fact that removal of TBX2 after birth (Fig. 1q–t and Extended Data Fig. 3) caused these cells to become OHCs indicates that, for their differentiation to proceed and to be maintained, TBX2 is required not only at the initiation of IHC differentiation but also subsequently for their differentiation to proceed and be maintained. Removal of TBX2 in partially differentiated IHCs by tamoxifen injection at P0 in Fgf8creER; Tbx2F/F; R26LSL-tdTomato/+ pups resulted in Cre-mediated deletion by P1, as evidenced by tdTomato expression (Extended Data Fig. 4d), and in the eventual conversion of these cells into ic-OHCs (Fig. 3). Postnatal IHCs that lose TBX2 either dedifferentiate into early cochlear hair cells that then recapitulate OHC differentiation, as normally occurs during embryogenesis, or transdifferentiate directly into postnatal OHCs. In situ hybridization of cochlear sections, with each transecting the organ of Corti at three to four positions from the more mature base to the less differentiated apex, at each postnatal day between P1 and P7–P8 revealed that most of the IHCs converting into OHCs did not express the early OHC markers Insm1 and Bcl11b2 (Extended Data Tables 1 and 2 and Extended Data Fig. 4e–g). Moreover, these transdifferentiating cells did not express SOX2, which is expressed in supporting cells and also transiently expressed in nascent hair cells produced during normal embryogenesis16,17 as well as in those that can transdifferentiate postnatally from supporting cells18–20. Furthermore, immunohistochemistry at the same stages revealed that, for several days after ablation of Tbx2 (at P0), IHCs converting into OHCs expressed the OHC markers PMCA2 beginning at P1 and prestin beginning at P4 while still expressing the IHC marker VGLUT3 until P8 (Extended Data Fig. 4a–c, g). Hence, unlike IHCs missing TBX2 embryonically, which differentiated as OHCs (Fig. 1 and Extended Data Table 2), these transdifferentiating cells switched from IHCs to OHCs without recapitulating OHC differentiation (Extended Data Fig. 5).
Fig. 3 |. Late or postnatal ablation of Tbx2 results in the transdifferentiation of IHCs into OHCs.
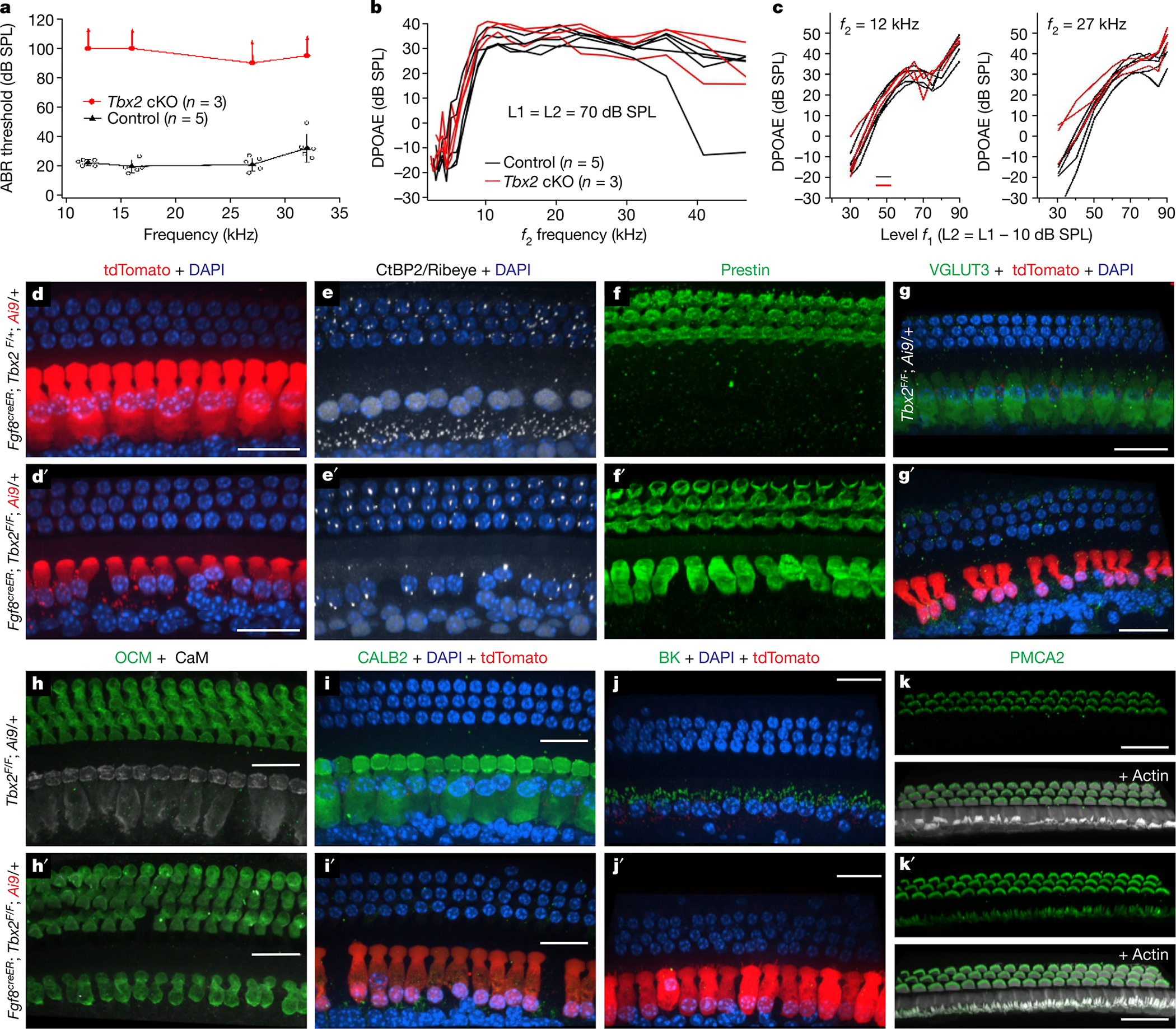
a–c, Hearing tests in post-weaning, mature mice (P31) in which Tbx2 was ablated in IHCs with tamoxifen at P7 or P9 (Fgf8creER/+; Tbx2F/F; R26LSL-tdTomato/+, with Fgf8creER/+; Tbx2F/+; R26LSL-tdTomato/+ and Tbx2F/F; R26LSL-tdTomato/+ littermates as controls). a, ABRs were absent in tamoxifen-treated mutants (up-facing arrows), whereas littermate controls had normal ABRs. Error bars, s.d.; centre symbols (triangles and ovals) indicate averages. b, c, Distortion product otoacoustic emissions (DPOAEs): iso-input functions (b) and input–output functions (c). The DPOAEs, which indicate OHC function, were similar to those of littermate controls, as expected for a phenotype affecting IHCs and not OHCs. d–k′, Immunohistochemistry on the mature cochleae of mice (P31 in d–i′; P27 in h, h′, k, k′; P28 in j, j′) in which Tbx2 was ablated in IHCs by tamoxifen administration at P7 (d′–g′) or P9–P11 (h′–k′). Control littermates are shown in d–k. d–f and d′–f′ depict the same cells labelled with various markers, whereas every other panel depicts a cochlear portion from a different animal. All examined cells in the position of IHCs displayed features of OHCs but not those of IHCs. These features include a cylindrical rather than flask shape; a small nucleus at the cell base rather than a large nucleus at the middle; few ribbons; and expression of prestin, oncomodulin and stereocilliary PMCA2 but not that of VGLUT3, CALB2, BK and nuclear CtBP2; the exception was stereocilia arrangement (lower panels in k, k′). The red fluorophore tdTomato results from Cre-dependent recombination of the Ai9 allele (R26LSL-tdTomato), thereby demonstrating recombination in the labelled cells (d, d′, g′, i′, j′). All images are confocal projections of apical (82–100% of the cochlear length; j, j′), mid-apical (approximately 57–82%; i, i′, k, k′) and mid-basal (approximately 31–57%; d–h′) portions of the cochlea. The images shown are examples of results obtained with at least three separate tissue samples (n = 3 biologically independent samples). Scale bars, 20 μm.
Because TBX2 expression in IHCs is not transient but is permanent21,22, we wondered whether it was still needed at increasingly later stages of IHC differentiation. IHCs begin to differentiate in embryos at about E15.5 at the base of the cochlea and continue differentiating after birth. A single administration of tamoxifen to Fgf8creER; Tbx2F/F; R26LSL-tdTomato/+ mice on P0, P3, P7 and P9 resulted in Cre-dependent expression of tdTomato in all IHCs starting 1 day later. Administration up to P11 caused recombination in more apical IHCs, presumably those in which Fgf8creER expression persisted. With each of these treatments, ablation of Tbx2 resulted in almost complete transdifferentiation of IHCs into OHCs. Specifically, cells lost expression of the IHC markers VGLUT3, CALB2, BK and nuclear CtBP2; gained expression of the OHC markers prestin, oncomodulin and PMCA2; had few CtBP2+ synaptic ribbons like OHCs and not the larger number present in IHCs; lacked the flask shape characteristic of IHCs; and displayed small nuclei positioned at the hair cell basal pole as in OHCs and not larger nuclei in the middle of the cell, which is characteristic of IHCs (Fig 3d–k′, Extended Data Fig. 3 and Extended Data Table 1). These postnatally transdifferentiated hair cells, however, were not like OHCs in one respect: their stereocilia, despite expressing the OHC marker PMCA2, had the overall appearance of IHC stereocilia (Extended Data Table 1 and Fig. 3k, k′). This may be attributed to the irreversibility of stereocilia shape and arrangement once stereocilia are formed. In mouse cochleae, hair cell stereocilia develop during a critical embryonic-to-early postnatal period12 and are not reformed if subsequently lost23,24. Hence, cochlear hair cells probably cannot alter their stereocilia size and bundling arrangement once these stereocilia are formed, in keeping with the lack of plasticity of this organelle even if cellular identities switch from IHCs to OHCs, as otherwise occurs by ablating Tbx2 in IHCs. Notably, however, all examined IHC and OHC features already present by the time of Tbx2 ablation were respectively lost and gained, such as expression of VGLUT3, which starts in IHCs by birth, and that of PMCA2, prestin and oncomodulin, which is detectable in OHCs by P1, P4 and P6, respectively (Fig. 3 and Extended Data Fig. 5). Therefore, it appears that, in addition to being necessary for initiating the specific differentiation of IHCs during embryogenesis, TBX2 is also required throughout the postnatal period (at least until P9) for IHCs to continue expressing their already acquired features and to keep them from expressing those of OHCs; that is, TBX2 is required for IHCs to maintain their differentiating state.
Although we have demonstrated that removal of TBX2 from IHCs converts them into OHCs and that removal of TBX2 from OHCs lacking INSM1 prevents them from converting into IHCs, we wondered whether the addition of TBX2 to OHCs would make them become IHCs. We engineered an adeno-associated virus (AAV; Anc80 serotype, which infects hair cells25) expressing TBX2-IRES-mCherry, which is the coding sequences of Tbx2 and mCherry separated by an internal ribosome entry site for expression of the red fluorescent reporter, and used it to ectopically express TBX2 in cultures of developing cochleae. To confirm that ectopically expressed TBX2 was functional, we transfected some IHCs at the same time that we removed endogenous TBX2 from all IHCs. In these experiments, we established cultures from Fgf8creER; Tbx2F/F mice at P0, administered 4-hydroxytamoxifen plus AAVs after 1 day in vitro (DIV) and examined the hair cells after 7 DIV. Whereas non-transfected IHCs lacking TBX2 transdifferentiated into OHCs and expressed prestin, transfected IHCs, identified by expression of mCherry, did not appear to transdifferentiate as they did not express prestin (Fig. 4a, a′). Hence, ectopic TBX2 compensated for the removal of endogenous TBX2 from IHCs. We then proceeded to transfect wild-type cochlear explants as described above and examined hair cells in the outer compartment. We found that OHCs expressing mCherry, and hence TBX2, also expressed the IHC marker VGLUT3 (Fig. 4b, b′) and downregulated the OHC markers prestin (Fig. 4c, c′) and PMCA2 (Fig. 4d). Hence, ectopic expression of TBX2 in perinatal OHCs caused their transdifferentiation into IHCs (at least to the extent that we were able to assess this in the timespan of these cultures) (Extended Data Fig. 5). Together, our results reveal that TBX2 is both necessary and sufficient to cause a cochlear hair cell to differentiate into and remain differentiated as an IHC and not an OHC. On the basis of these properties, we conclude that TBX2 is a master regulator of IHC versus OHC differentiation.
Fig. 4 |. Ectopic expression of TBX2 in OHCs results in their conversion into IHCs.
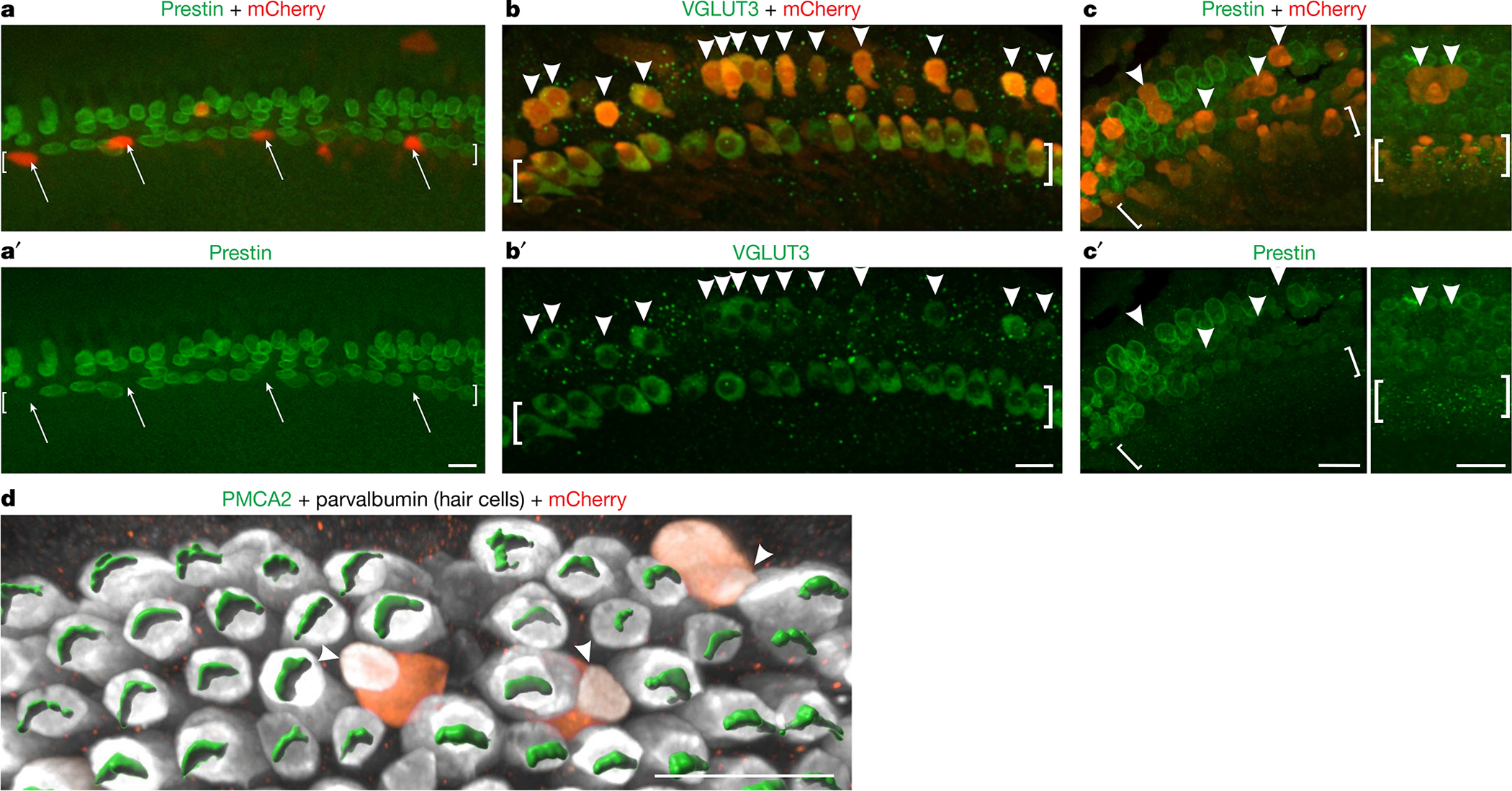
a, a′, Organ of Corti explants from neonatal Fgf8creER/+; Tbx2F/F mice were established at P0, exposed to Anc80 AAVs expressing TBX2-IRES-mCherry and 4-hydroxytamoxifen (to ablate Tbx2) from 1 DIV to 3 DIV, fixed at 7 DIV and immunostained to detect the OHC marker prestin. Untransfected IHCs expressed prestin, as expected, owing to their loss of TBX2. However, transfected IHCs (mCherry+, arrows) showed no prestin expression, demonstrating that ectopic TBX2 compensated for the loss of endogenous TBX2. b–d, Organ of Corti explants from wild-type mice were established at E17.5, exposed to Anc80 AAVs expressing TBX2-IRES-mCherry from 1 DIV to 3 DIV and fixed at 6 DIV (b–c′) or 7 DIV (d). Cells in the position of OHCs that were transfected (mCherry+, arrowheads) expressed the IHC marker VGLUT3 (b, b′) but not the OHC markers prestin (c, c′) or PMCA2 (d), as visualized with Imaris software. The image in d shows only the outer compartment. Square brackets surround the IHCs. The images shown are examples of results obtained with at least three separate cultures (n = 3 biologically independent samples). Scale bars, 20 μm.
Initially, we proposed that TBX2 would be necessary for IHC differentiation. However, in its absence, IHCs did not simply lose their specific features and markers; they also acquired those of OHCs. The default state for an IHC not expressing TBX2 is not an undifferentiated cochlear hair cell but a fully differentiated OHC. This finding is contrary to common opinion regarding how cochlear IHCs and OHCs evolved and develop. From an evolutionary perspective, OHCs appear to be a novel cell type because they differ the most from other hair cells and other sensory cells in their acquisition of electromotility instead of robust synaptic transmission in afferents26. However, although evolutionarily the features of OHCs may be more recent than those of IHCs, the developmental programme that distinguishes the two cell types from each other operates such that TBX2 prevents IHCs from acquiring and maintaining a default OHC fate.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-04668-3.
Methods
Animals
All animal care and procedures were in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and were approved by Northwestern University’s Institutional Animal Care and Use Committee (Animal Study Protocols IS00006235 and IS00000593). Mice were group housed with food and water provided ad libitum under a 12-h light/12-h dark cycle and temperatures of 18–23 °C with 40–60% humidity. The loxP-flanked Tbx2F mice9, provided by V. Christoffels (Amsterdam University Medical Center), were bred in the FVB background. The loxP-flanked Insm1F mice2 were bred in the C57BL/6J background. The Atoh1cre knock-in mouse line2 was bred from a mixed background of CD1 and C57BL/6. The Gfi1cre knock-in mouse line11 was bred from a C57BL/6J background. The Fgf8creER knock-in mouse line14, provided by A. Moon and M. Deans (University of Utah), was bred from a mixed 129SV and C57BL/6J background. For the conditional ablation of Tbx2, we injected Fgf8creER; Tbx2F/F; R26LSL-tdTomato/+ and Tbx2F/F; R26LSL-tdTomato/+ littermate controls intraperitoneally (i.p.) at P0, P3, P7, P9 or P11 with a single dose of tamoxifen (80 mg kg−1; Sigma, T5648) freshly solubilized in corn oil (20 mg ml−1 stock) using a Hamilton syringe and a 27-gauge needle. We confirmed activation of the tamoxifen-inducible CreER in IHCs by their expression of tdTomato from the recombined R26LSL-tdTomato (Ai9) allele27.
All mice used in this study were generated by intercrossing the above mouse lines. Similar numbers of male and female animals were used for all analyses at the ages indicated in the figure legends. Tests were performed with randomly selected littermate control mice.
Mouse tissue preparation and RNA in situ hybridization with immunohistochemistry
After fixation (4% paraformaldehyde for 24 h) and cryoprotection in sucrose solutions (5%, 10% and 20%), cochleae were mounted in optimal cutting temperature compound (Fisher) and frozen on dry ice. Samples were sectioned at 12-μm thickness, mounted on SuperFrost Plus slides (Fisher) and dried briefly at room temperature before storage at −80 °C. The manufacturer’s standard protocol for chromogenic in situ hybridization (RNAscope 2.5 HD Red Assay, Advanced Cell Diagnostics) was followed. Probes obtained from Advanced Cell Diagnostics were used for the following mouse genes: Insm1 (430621), Tbx2 (448991), Bcl11b (570631), Fgf8 (313411), Msx1 (421841) and Brip1 (529199). In situ hybridization was followed by haematoxylin and eosin staining according to the manufacturer’s instructions or by immunohistochemistry. In brief, sections were washed with PBS and incubated for 1 h in blocking solution (1× PBS, 3% BSA, 3% normal donkey serum). Sections were incubated for 1 h at room temperature in blocking solution containing anti-myosin VIIa antibody (Proteus). Sections were washed three times with 1× PBS followed by incubation in Alexa Fluor 488 donkey anti-rabbit antibody for 1 h. After four washes with 1× PBS, sections were mounted with Prolong Gold mounting medium (ThermoFisher).
Mouse tissue preparation and immunohistochemistry of cochlear whole mounts
Tissue was collected and immunostained using one of three methods—standard, freeze–thaw or antigen retrieval—as detailed in refs. 2,15. The respective primary antibodies, working dilutions and immunohistochemical methods used were as follows: mouse anti-CtBP2 (BD Transduction Laboratories, 612044), 1:100, standard; rabbit anti-BK (Alomone Labs, APC-021), 1:500, standard; rabbit anti-KCNQ4 (gift from B. Kachar, National Institute of Health–National Institute on Deafness and Other Communication Disorders), 1:500, standard; mouse anti-parvalbumin (Millipore, MAB1572), 1:250, freeze–thaw; rabbit anti-oncomodulin (Novusbio, NBP2–14568), 1:100, standard; goat anti-VAChT (Millipore, ABN100), 1:200, standard; rabbit anti-calretinin/Calb2 (Invitrogen, MA5–14540) 1:100, freeze–thaw; rabbit anti-prestin (gift from J. Zheng, Northwestern University), 1:1,000, standard; rabbit anti-Vglut3 (Synaptic Systems, 135 203), 1:500, standard; mouse anti-calmodulin (Sigma, C3545), 1:200, standard; rat anti-Ctip2/BCL11b (Abcam, 18465), 1:200, antigen retrieval; goat anti-parvalbumin (Swant, PVG-213), 1:2,000, freeze–thaw; rabbit anti-glutamate receptor 2 (Millipore, MAB397), 1:1,000, freeze–thaw; rabbit anti-myosin VIIa (Proteus BioSciences, 25–6790), 1:800, standard; mouse anti-β-tubulin (Millipore, T6199), 1:400, freeze–thaw; rabbit anti-espin (gift from J. Bartles, Northwestern University), 1:80, freeze–thaw; goat anti-SOX2 (Santa Cruz, sc-17320, lot H0516), 1:500, standard; rabbit anti-PMCA2 (Invitrogen, PA1-915, lot VL309644), 1:800, freeze–thaw; chicken anti-mCherry (Abcam, 205402), 1:100, standard.
Secondary antibodies and working dilutions were as follows: Alexa Fluor 488 donkey anti-rabbit (Jackson ImmunoResearch, 711-545-152), 1:100; Alexa Fluor 488 donkey anti-mouse (Jackson ImmunoResearch, 711-545-151), 1:100; AlexaFluor 488 donkey anti-goat (Jackson ImmunoResearch, 711-545-147), 1:100; Alexa Fluor 488 donkey anti-guinea pig (Jackson ImmunoResearch, 711-545-148), 1:100; Alexa Fluor 594 donkey anti-rabbit (Jackson ImmunoResearch, 711-585-152), 1:100; Alexa Fluor 594 donkey anti-mouse (Jackson ImmunoResearch, 711-585-151), 1:100; Alexa Fluor 594 donkey anti-chicken (Jackson ImmunoResearch, 711-585-155), 1:200; Alexa Fluor 594 donkey anti-rat (Jackson ImmunoResearch, 711-585-153), 1:100; Alexa Fluor 647 donkey anti-rabbit (Jackson ImmunoResearch, 711-605-152), 1:100; Alexa Fluor 647 donkey anti-rat (Jackson ImmunoResearch, 711-605-153), 1:100; Alexa Fluor 647 donkey anti-mouse (Jackson ImmunoResearch, 711-605-151), 1:100.
Image acquisition and analysis
Images of cochlear whole mounts and organotypic cultures were acquired using either a Yokogawa CSU-W1 spinning disk on a Nikon Ti2 microscope with a Hamamatsu Flash 4.0 V3 camera operated by NIS-Elements or a Nikon A1 confocal microscope, using ×100 or ×60 lenses. Exposure times were set to ensure high signal-to-noise ratio and no saturation in the image. Gain and offset adjusting were performed to ensure that no saturated or undersaturated pixels were present. Identical capture and analysis conditions were used for each experimental and control tissue.
Images were processed and three-dimensional renderings were generated using NIS-Elements and Imaris. Nuclear volumes were measured on the basis of DAPI fluorescence using Imaris, as detailed in ref. 2.
Cochlear explant cultures and AAV transfections
For explant cultures, cochleae were isolated from either wild-type CD1 mice at E17.5 or Fgf8creER/+; Tbx2F/F mice at P0. The cochlear roof and any excess connective tissue were removed to expose the sensory epithelium, as described in refs. 28, which was then transferred to a plastic culture dish containing fresh DMEM supplemented with 1% foetal bovine serum, 1% N2 supplement and ampicillin at 50 μg ml−1. After 1 day, each culture was surrounded by a cloning cylinder 8 mm in diameter (Sigma, C1059-1EA) to reduce the culture volume to approximately 100 μl. To this, we added AAV (2.5 μl of 2.59 × 1012 genome copies of AAV per millilitre) Anc80 serotype25 expressing TBX2-IRES-mCherry under the CMV promoter, custom generated by BrainVTA. The mixture was kept for 2 days (1 DIV to 3 DIV), at which time the cloning cylinder was removed and the culture medium was replaced with fresh medium. For Fgf8creER/+; Tbx2F/F cultures, AAV was added together with 4-hydroxytamoxifen (at a concentration of 10 μM), to ablate endogenous Tbx2 concurrently with the transfection of exogenous Tbx2. The cultures were kept for a total of 6 or 7 days in vitro (6 DIV or 7 DIV) at 37 °C with 5% CO2. Following the culture period, we washed the explants ten times in PBS, fixed them in 4% paraformaldehyde in PBS for 30 min at room temperature, and then processed them for immunostaining and confocal microscopy.
Hearing tests
DPOAEs and ABRs were measured in mice 3–8 weeks of age, as previously described2. After checking for a pinna reflex, each animal was weighed and anaesthetized with 100 mg kg−1 ketamine and 10 mg kg−1 xylazine given i.p. In brief, the ABR thresholds were determined by using three subdermal needle electrodes with one at the vertex, one at the mastoid and the reference electrode on the opposite shoulder. The signals were amplified (1,000×), bandpass filtered (0.3–3 kHz) and averaged (3,000 samples). The stimuli were 10-ms sinusoids including 1-ms rise and fall times and were presented by a speaker (Super Tweeter, RadioShack) fitted with a speculum. The latter was positioned at the entrance to the ear canal. The stimuli were calibrated using real-head calibration29. DPOAEs were acquired using a microphone (ER-10B+ Low Noise DPOAE Microphone, Etymotic Research) coupled to a custom emission probe designed and fabricated by J. H. Siegel (Northwestern University) that fit securely in the mouse’s ear canal. Calibrations were determined for each individual mouse by measuring the stimulus pressures in the ear canal using the emission microphone. DPOAEs were acquired using a two-tone input with a frequency ratio of f2/f1 = 1.2. Responses were analysed using Emission Averager30. These results are provided as iso-input functions, in which the parameter is stimulus frequency, and as input–output or growth functions, in which the parameter is signal level. For the latter results, the level of f1 was 10 dB greater than that for f2.
Whole-cell patch-clamp recordings
Whole-cell recordings were performed at room temperature using an Axopatch 200B amplifier (Molecular Devices) with a 10-kHz low-pass filter. Recording pipettes were pulled from borosilicate glass to achieve initial bath resistances averaging 3 MΩ. Pipettes were filled with an intracellular solution containing (in mM) 140 KCl, 2 MgCl2, 10 EGTA and 10 HEPES (pH 7.3). Cells were bathed in Hanks’ balanced salt solution (14025, ThermoFisher Scientific). After establishing the whole-cell configuration, the intracellular pressure was not adjusted. Command voltages were constant step functions of 150 ms in duration (from −140 mV to +80 mV, 10-mV step). Current data were collected by jClamp (SciSoft Company). For cell membrane electric capacitance (Cm) measurements, recording pipettes were filled with an intracellular solution containing (in mM) 140 CsCl, 2 MgCl2, 10 EGTA and 10 HEPES (pH 7.4). Cells were bathed in an extracellular solution containing (in mM) 120 NaCl, 20 TEA-Cl, 2 CoCl2, 2 MgCl2 and 10 HEPES (pH 7.4). Osmolality was adjusted with glucose to 320 mOsm per kilogram. The electric current response to a sinusoidal voltage stimulus (2.5 Hz, 120 mV or 150 mV in amplitude) superimposed with two higher-frequency stimuli (390.6 Hz and 781.2 Hz, 10 mV in amplitude) was recorded. Cm was determined from this current response by a fast Fourier transform-based admittance analysis31. OHC electromotility was captured using a WV-CD22 camera (Panasonic), and the obtained sequential images were analysed using ImageJ, as described previously32.
Nonlinear capacitance and electromotility data analysis
Voltage-dependent Cm data were analysed using the following two-state Boltzmann equation:
where α is the slope factor of the voltage dependence of charge transfer, Qmax is the maximum charge transferred, Vm is the membrane potential, Vpk is the voltage at which the maximum charge movement is attained and Clin is the linear capacitance. Electromotility data were analysed using the following equation:
where Dmax is the maximum cell length change and D0 is a base reference point at which an OHC or ic-OHC shows zero cell contraction (at infinitely hyperpolarized membrane potential).
Extended Data
Extended Data Fig. 1 |. Expression of Tbx2 mRNA in cochlea.
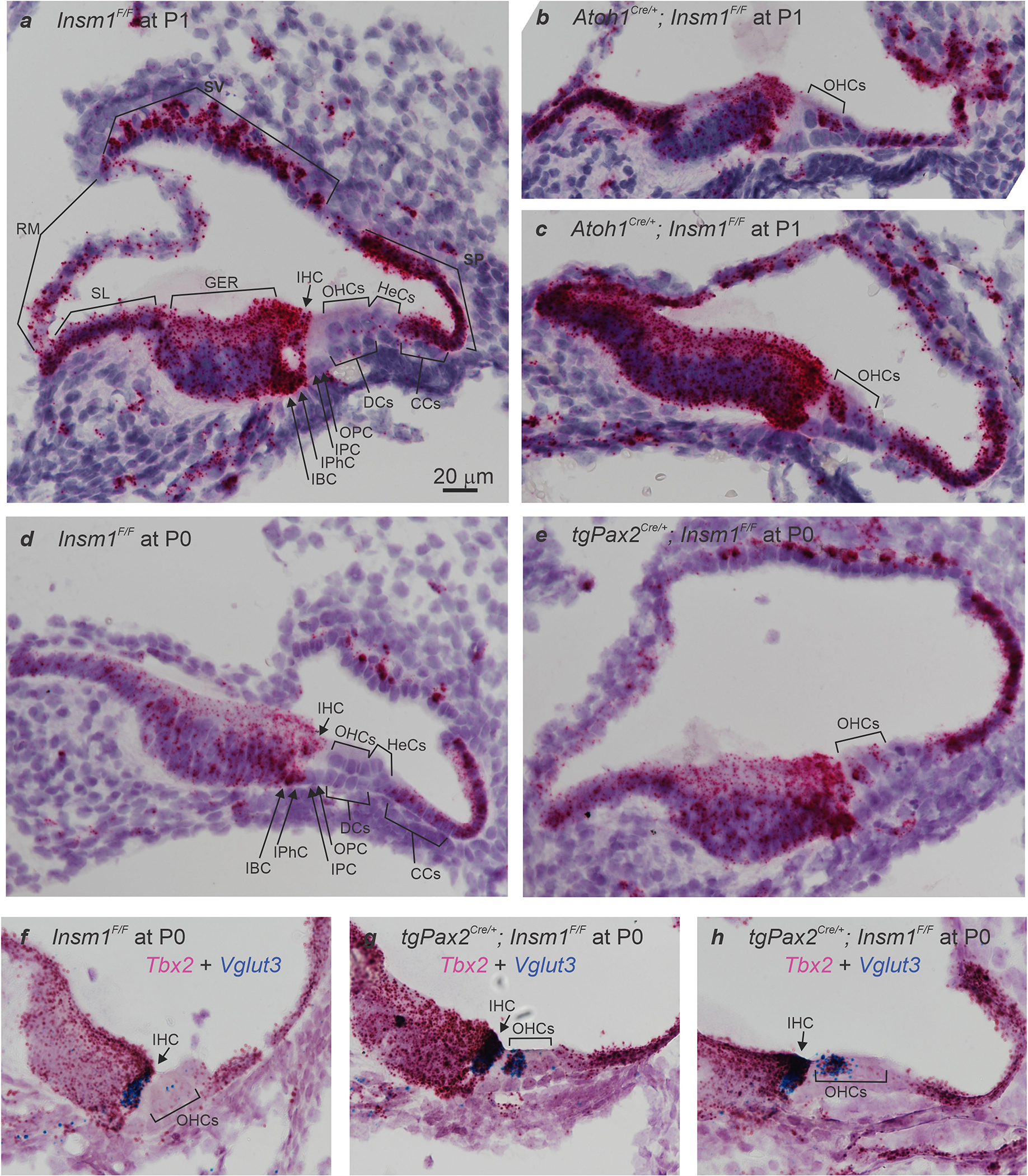
(a,d) In situ hybridizations in sections of neonatal (P0 and P1) cochleae from control mice (Insm1F/F) reveal Tbx2 expression in most cells lining the scala media including IHCs, Inner Phalangeal Cells (IPhC), Inner Border Cells (IBC), Greater Epithelial Ridge (GER), interdental cells of the Spiral Limbus (SL), Reissner’s Membrane (RM), some cells of the Stria Vascularis (SV), Spiral Prominence (SP), and Claudius’ Cells (CCs); but little or no expression (above background levels) of Tbx2 in OHCs or other cells of the outer compartment, namely Hensen’s Cells (HeCs), Deiters’ Cells (DCs), and Outer and Inner Pillar cells (OPC and IPC). (b,c,e) In situ hybridizations in sections of developing Insm1 mutant cochleae (Atoh1Cre/+; Insm1F/F in b and c; TgPax2Cre/+; Insm1F/F in e) reveal that Tbx2 is expressed in about half of the OHCs, presumably those transdifferentiating into IHCs. In controls, none of the OHCs (0/117 from N=4 Insm1F/F mice at E16.5, P0 and P1) expressed Tbx2, whereas in Insm1 mutants, in which nearly half of the OHCs transdifferentiate into IHCs (46.0±5.6%)2, an equivalent fraction of hair cells in the position of the OHCs expressed Tbx2 (46.6%; 62/133 cells from N=4 TgPax2Cre/+; Insm1F/F or Atoh1Cre/+; Insm1F/F mice at E16.5, P0 and P1). (f-h) Double in situ hybridizations in sections of neonatal (P0) cochleae for detection of Tbx2 (magenta) and Vglut3 (blue), the earliest marker of functional, mature IHCs. (f) In Insm1F/F controls, only IHCs express high levels of Vglut3 (a few dots label OHCs, perhaps representing a much lower level of expression). (g,h) In TgPax2Cre/+; Insm1F/F cochleae, only OHCs misexpressing Tbx2 (50/114 at P0, 14/30 at P2 and 9/21 at P4) also misexpressed high levels (those characteristic of IHCs) of Vglut3, whereas none of the OHCs misexpressed either gene alone (at IHC-like levels). Hence, early expression of Tbx2 in OHCs lacking INSM1 appears to correlate perfectly with their transdifferentiating into IHCs. Images shown are examples of results obtained in three separate tissue samples (n=3 biologically independent samples). Scale bar of 20 μm in (a) applies to all other panels.
Extended Data Fig. 2 |. Nuclear volumes of hair cells in the inner or outer compartment of controls and in various Tbx2 conditional KOs.
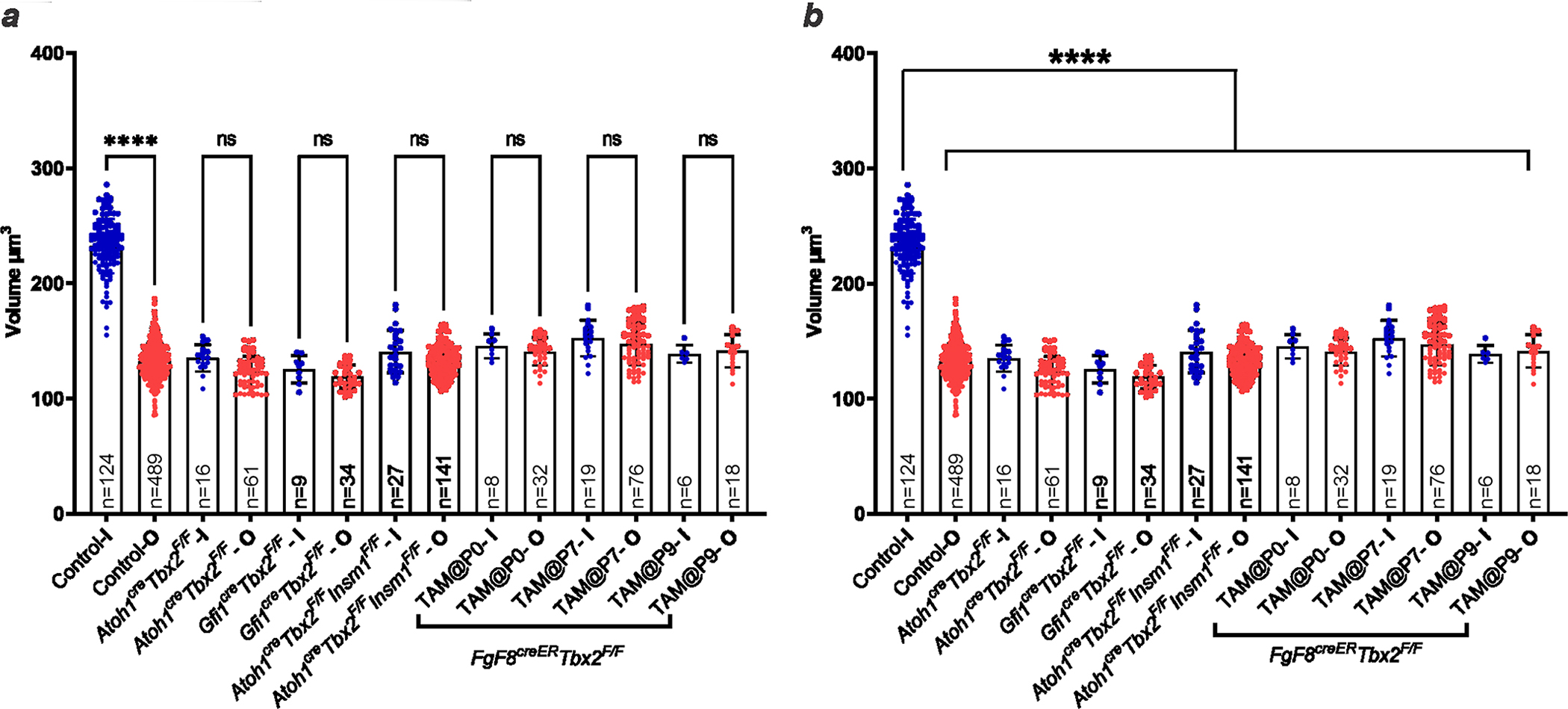
Blue dots represent nuclear volumes of hair cells from the inner compartment (I in the column labels), whether IHCs (controls) or ic-HCs (mutants). Red dots represent nuclear volumes of hair cells from the outer compartment (OHCs; O in the column labels). Sample size (n) is indicated in each column. The time of tamoxifen administration to Fgf8CreER; Tbx2F/F pups is indicated in the column labels as TAM@P0 to P9. (a,b) While in controls the nuclei of IHCs are statistically larger than those of OHCs, in the mutants (all Tbx2 cKOs as well as the double cKO of Tbx2 and Insm1) the nuclei of the inner compartment hair cells (ic-HCs) are statistically smaller than those of control IHCs (b) and indistinguishable from those of OHCs (a). Error bars indicate SD and column tops the averages. Statistical comparisons were performed by one-way ANOVA with a Bonferroni post test. **** indicates P<0.0001, while ns (not significant) indicates P>0.05 (for panel a, from left to right, P=0.0792 for Atoh1CreTbx2F/F; P>0.9999 for Gfi1CreTbx2F/F; P=0.1285 for Atoh1CreGfi1CreTbx2F/F; P>0.9999 for Fgf8CreERTbx2F/F + Tamoxifen at P0, P7 and P9).
Extended Data Fig. 3 |. Non-linear capacitance parameters evince electromotility of ic-OHCs.
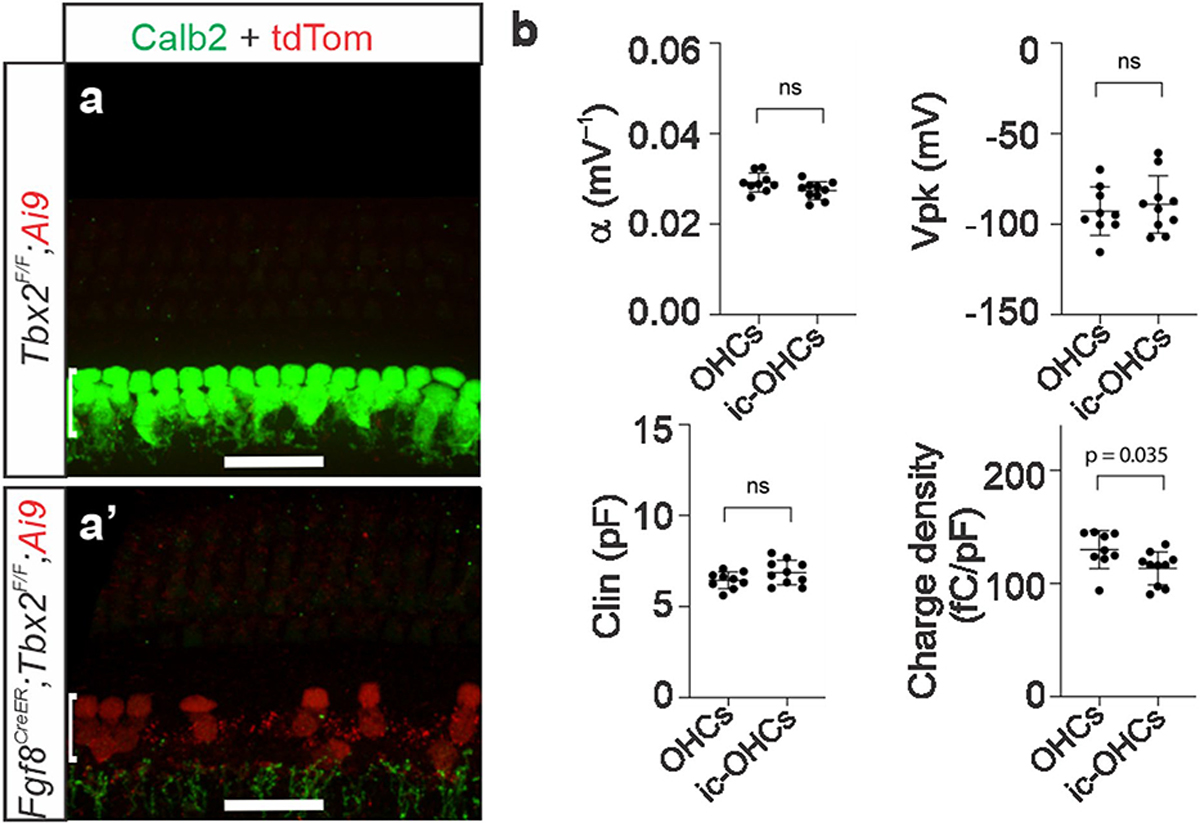
(a,a’) Hair cells in the position of IHCs (ic-OHCs)that lacked TBX2 (from Fgf8CreER; Tbx2F/F; R26LSL-tdTomato/+ treated with tamoxifen at birth) were identified by their red fluorescence (tdTomato+; a’), whereas control IHCs and OHCs were obtained from equally treated littermates that lacked Fgf8CreER (Tbx2F/F; R26LSL-tdTomato/+), so that their IHCs did not transdifferentiate into ic-OHCs, expressing IHC markers like Calb2 and not tdTomato (a). These control IHC and OHC were distinguished as before15. Images shown are examples of results obtained in at least three separate tissue samples (n=3 biologically independent samples). (b) Summary of the non-linear capacitance (NLC) parameters of ic-OHCs lacking TBX2 and control OHCs (including those examined in Fig. 1s, t). Like control OHCs (n=9), all examined ic-OHCs (n=10) were electromotile. Samples of each cell type were obtained from ≥ 3 separate animals. Horizontal bars indicate the means and the standard deviations. Statistical comparisons were performed by unpaired t-tests (two-tailed). “ns” indicates not significant (p > 0.05).
Extended Data Fig. 4 |. Cochlear hair cells transdifferentiating postnatally transiently co-express markers of mature IHCs (VGLUT3) and OHCs (Prestin and PMCA2), but not of nascent hair cells (SOX2) nor of differentiating OHCs (Insm1 and Bcl11b).
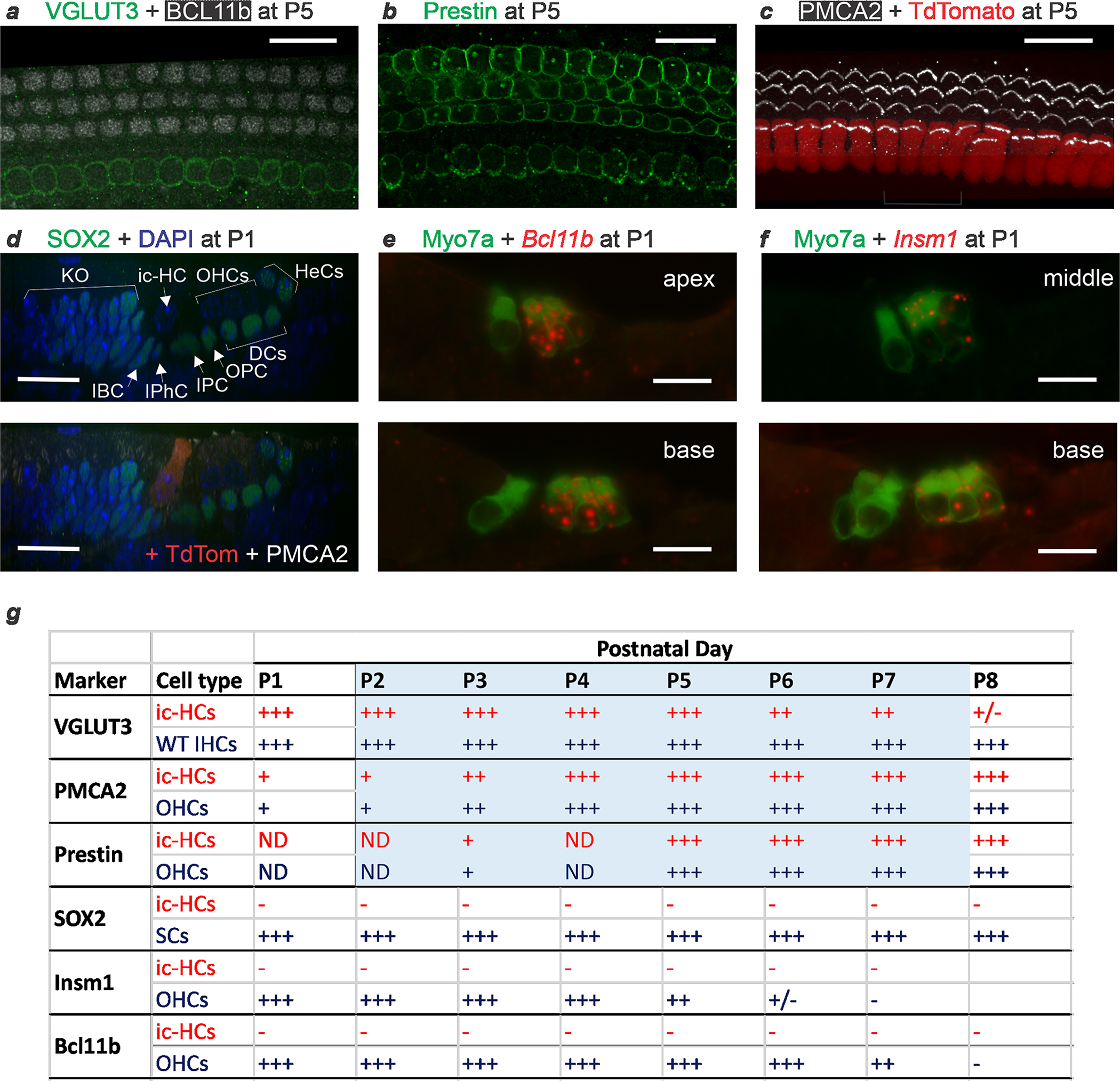
Fgf8CreER; Tbx2F/F; R26LSL-tdTomato/+ newborn (P0) pups were treated with tamoxifen at P0 and collected for examination at each subsequent day from P1 to P8. (a-c) Immunohistochemistry at P5 reveals that hair cells in the position of the IHCs express the IHC functional marker VGLUT3 (and not the OHC developmental market BCL11b) (a; confocal optical section), but also the OHC functional markers Prestin (b; confocal optical section) and PMCA2 (c; confocal projection). In (c), former IHCs are identified by the expression of TdTomato. Hence, at this time transdifferentiating hair cells display features of both IHCs and OHCs. (d) Immunohistochemistry at P1, displayed as a confocal radial optical section, reveals that cells transdifferentiating from IHCs to OHCs (at this point ambiguously termed ic-HCs; TdTomato+ in the lower panel) do not express SOX2, a nuclear marker of both supporting cells and nascent hair cells, and are beginning to express PMCA2 in their stereocilia (bottom panel). For a better visualization of SOX2, the top panel displays its immunoreactivity only with the nuclear counterstain DAPI. (e,f) In situ hybridization combined with immunohistochemistry (for Myosin VIIa, to label IHCs and OHCs) on cryostat sections from P1 cochleae reveal that ic-HCs transdifferentiating from IHCs to ic-OHCs do not express Bcl11b (e) or Insm1 (f) mRNAs, which are expressed by OHCs during their early (embryonic to postnatal) differentiation. Images were taken at apical, middle and basal cochlear positions at P1 and subsequent postnatal days. Images shown are examples of results obtained in at least three separate tissue samples (n=3 biologically independent samples). All scale bars are 20 μm. (g) Schematic summary of all the stainings performed on mice at P1 to P8 in which Tbx2 ablation had been induced by tamoxifen administration at P0. Blue shading denotes the period (P1 to P7) during which transdifferentiating ic-HCs co-express VGLUT3 with OHC markers PMCA and/or Prestin. At no point from P1 to P8 do ic-HCs express the nascent HC marker SOX2 or the OHC differentiation markers Bcl11b (mRNA or protein) or Insm1. Expression levels are subjectively categorized as strong (+++) to undetectable above background (−). Expression in ic-HCs (red font) is provided by comparison to expression in the following control cells (blue font) in the same tissues or stages: Wild type (WT) IHCs for VGLUT3; adjacent OHCs for Prestin, PMCA2, Bcl11b and Insm1; and Supporting Cells (SC) for SOX2. Supporting and other cells labeled in (d) as expressing SOX2 are epithelial cells of Kölliker’s Organ (KO), Inner Border (IBC), Inner Phalangeal (IPhC), Inner Pillar (IPC), Outer Pillar (OPC), Deiters’ (DC) and Hensen’s (HeC) cells.
Extended Data Fig. 5 |. Schematic illustration of the various forms of transdifferentiation between IHCs and OHCs.
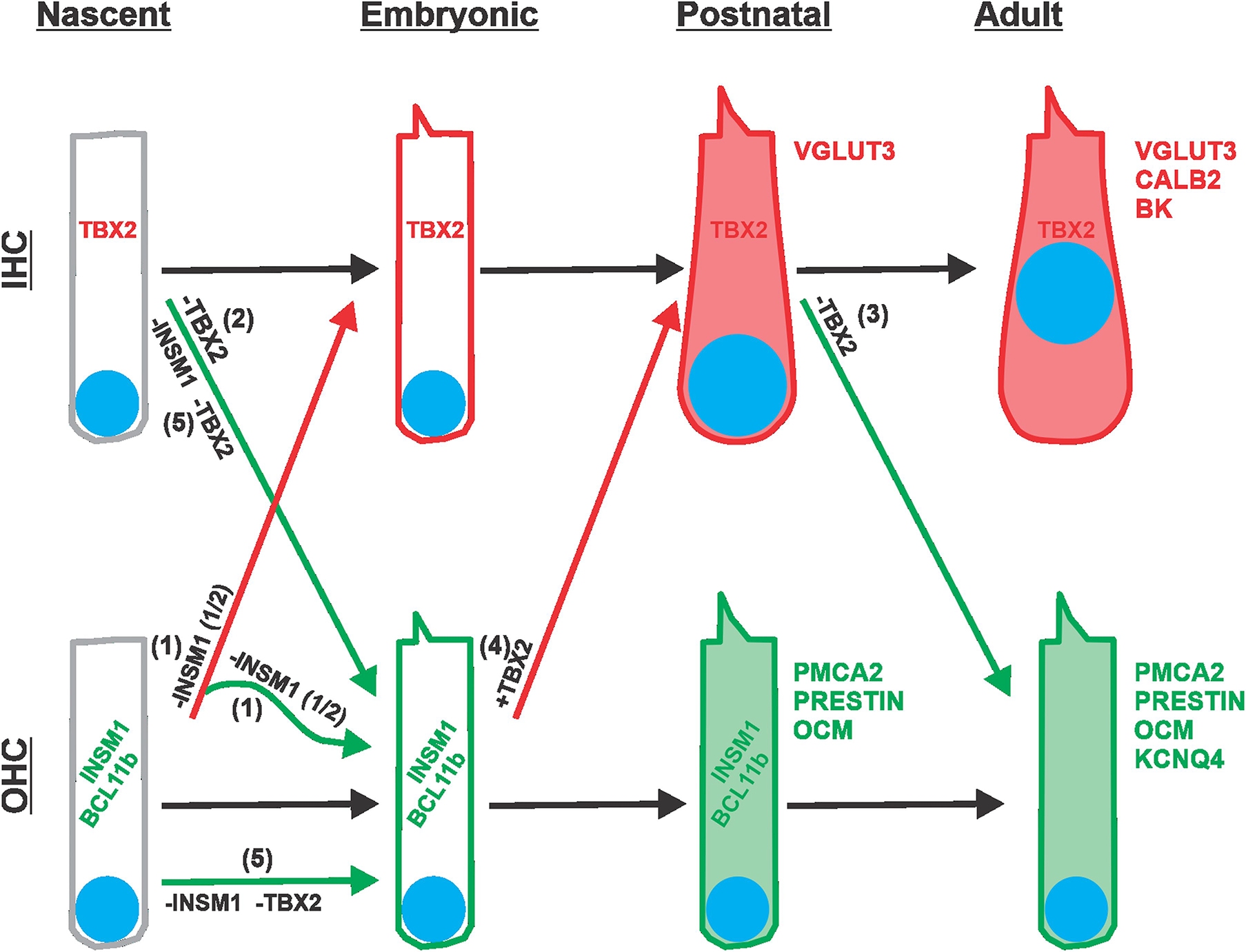
IHCs and OHCs are depicted from their onset after proliferation of progenitors has ceased (nascent, ~E14.5), through embryonic (E15.5 to E20.5) and postnatal (P0 to P9) stages, and into their adult form. As differentiation proceeds, hair cells acquire increasing numbers of characteristic markers (represented in red for IHCs and green for OHCs; nuclei are represented as blue disks). The course of normal development is indicated with black arrows. IHCs express TBX2 from their onset and throughout life. OHCs express INSM1 and BCL11b transiently during embryonic and early postnatal stages. Transitions due to genetic manipulations are indicated by colored arrows: red if leading to an IHC-like differentiation, and green if leading to an OHC-like differentiation. (1) Removal of INSM1 from nascent OHCs (−INSM1) results in about half of them expressing TBX2 and transdifferentiating into early IHCs (red arrow), which then proceed to differentiate into mature IHCs. The other half of INSM1 lacking OHCs do not express TBX2 and proceed to differentiate into mature OHCs (green). Note that, subsequent to its deletion, there is no expression of INSM1 (whose lable has been kept in the illustration for simplicity). (2) Removal of TBX2 from the onset of IHC formation (−TBX2) results in its switching to differentiate like an OHC, transiently expressing early markers INSM1 and BCL11b, and eventually maturing into OHCs like those of adults (albeit in the position of the IHCs, and hence termed ic-OHCs). (3) Removal of TBX2 from a postnatal (up to P9) IHC (−TBX2) results in its direct transdifferentiation into a differentiated OHC, without expressing early OHC markers INSM1 and BCL11b, and transiently co-expressing markers of both mature IHCs (VGLUT3) and OHCs (PMCA2 and Prestin), before proceeding to becoming OHC-like in most respects. (4) Ectopic expression of TBX2 in late embryonic OHCs (+TBX2) results in their transdifferentiation into IHCs (expressing VGLUT3 but not PMCA2 or Prestin). (5) In the absence of both INSM1 and TBX2, nascent IHCs (−INSM1 −TBX2) transdifferentiate into OHCs whereas nascent OHCs do not transdifferentiate into IHCs. In other words, Tbx2 is epistatic to Insm1.
Extended Data Table 1 |.
Summary of OHC and IHC specific features displayed by hair cells depending on position and genotype
| Genotype | HC type | Prestin | OCM | KCNQ4 | BK | VGLUT3 | CaM | Calb2 | CtBP2+ Ribbon # | Nuclear CtBP2 | Nuclear Size | Nuclear Possition | Stereocilia | Transient Insm1 | Transient Bcl11b | Transient Fgf8 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | OHCs | Yes | Yes | Yes | No | No | Low | No | Few | No | Small | Basal | Small | Yes(E15.5–P5 or P8) | Yes(E15.5–P7 or P8) | No |
| Control | IHCs | No | No | No | Yes | Yes | High | Yes | Many | Yes | Large | Apical | Large | No | No | Yes (E14.5-P11) |
| Atoh1 Cre ; Tbx2 F/F | ic-HCs | Yes | Yes | ND | ND | No | ND | No | Few | No | Small | Basal | Small | Yes (E17.5) | Yes (E17.5) | No (E17.5) |
| Gfi1 Cre ; Tbx2 F/F | ic-HCs | Yes | ND | Yes | No | No | ND | No | Few | No | Small | Basal | Small | ND | ND | ND |
| Fgf8CreER; Tbx2F/F + TAM at P0 | ic-HCs | Yes | ND | ND | ND | No | ND | No | ND | ND | Small | Basal | Large | No (P1–8) | No (P1–8) | ND |
| Fgf8CFeER; Tbx2F/F + TAM at P3 | ic-HCs | Yes | ND | ND | ND | No | ND | No | ND | ND | Small | Basal | Large | ND | ND | ND |
| Fgf8CneER; Tbx2F/F + TAM at P7 | ic-HCs | Yes | Yes | ND | ND | No | Low | No | Few | No | Small | Basal | Large | ND | ND | ND |
| Fgf8CreER; Tbx2F,F + TAM at P9 | ic-HCs | Yes | ND | ND | ND | ND | ND | ND | Few | No | Small | Basal | Large | ND | ND | ND |
| Atoh1Cre; Insm1 F/F | oc-IHCs | No | No | No | Yes | Yes | High | Yes | Interm | Yes | Large | Apical | Large | NA | Lost ~E17.5 | Yes (since E17.5) |
| Atoh1 Cre; Tbx2 F/F; Insml F/F | ic-HCs | Yes | Yes | Yes | No | No | Low | No | Few | No | Small | Basal | Small | NA | ND | ND |
| Atoh1 Cre; Tbx2 F/F; Insm1 F/F | oc-HCs | Yes | Yes | Yes | No | No | Low | No | Few | No | Small | Basal | Small | NA | ND | ND |
Features characteristic of control OHCs are in blue font, those of control IHCs in red font, and those of intermediate magnitude in purple font. ND stands for not determined and NA for not applicable. Hair cells in the position of IHCs on Tbx2 mutant backgrounds are termed ic-HCs (inner compartment hair cells). Outer compartment hair cells that have transdifferentiated into IHCs in Insm1 mutants are termed oc-IHCs. Hair cells in Tbx2 and Insm1 double mutants, all of which exhibit the features of OHCs, are termed depending on location: ic-HCs if in the inner compartment and oc-HCs if in the outer compartment. Columns 3 (Prestin) to 16 (Stereocilia) represent features of mature hair cells. Stereocilia are categorized as “OHC-Like” if relatively shorter, less intensely labelled with filamentous-actin-marker phalloidin, and arranged in a somewhat compact V or W-shape bundle; or as “IHC-Like” if larger, more intensely labelled with phalloidin, and arranged in somewhat fanning-out straight line bundle. See examples in Fig. 1o, o’ and Fig. 3k, k’. Columns 17 to 19 represent transiently expressed markers: Insm1, Bcl11b and Fgf8 mRNAs. Rows 6 to 9 represent ic-HCs from Fgf8CreEr/+; Tbx2F/F; R26LSL-tdTom3to/+ to which tamoxifen was administered at P0, P3, P7, and P9, respectively. All data were derived from this manuscript except that of row 10 (Atoh1Cre/+; Insm1F/F), which are summarized from prior publications (Wiwatpanit et al. 2 Webber et al., 2021).
Extended Data Table 2 |.
Quantification of in situ hybridization (ISH) results revealing expression of transient OHC markers Insm1 and Bcl11b on cochlear hair cells after induction of IHC to OHC transdifferentiation
| cKO driver | Stage | Control OHCs | Control IHCs | cKOOHCs | cKO ic-HCs | ||||||||
| Insm1+ | Total | % Insm1+ | Insm1+ | Total | % Insm1+ | Insm1+ | Total | % Insm1+ | Insm1+ | Total | % Insm1+ | ||
| Atohl Cre | E17.5 | 63 | 63 | 100.0% | 0 | 21 | 0.0% | 45 | 45 | 100.0% | 15 | 15 | 100.0% |
| Fgf8CreER + TAM at PO | P1 | 67 | 72 | 93.1% | 0 | 24 | 0.0% | 33 | 36 | 91.7% | 0 | 12 | 0.0% |
| Fgf8CreER + TAM at PO | P2 | 33 | 33 | 100.0% | 0 | 11 | 0.0% | 75 | 75 | 100.0% | 0 | 26 | 0.0% |
| Fgf8CreER + TAM at PO | P3 | 46 | 48 | 95.8% | 0 | 16 | 0.0% | 36 | 36 | 100.0% | 0 | 11 | 0.0% |
| Fgf8CreER + TAM at PO | P4 | 40 | 42 | 95.2% | 0 | 14 | 0.0% | 40 | 57 | 70.2% | 1 | 19 | 5.3% |
| Fgf8CreER +TAM at PO | P5 | 38 | 48 | 79.2% | 0 | 16 | 0.0% | 27 | 51 | 52.9% | 0 | 17 | 0.0% |
| Fgf8CreER + TAM at PO | P6 | 11 | 46 | 23.9% | 0 | 16 | 0.0% | ||||||
| Fgf8CreER +TAM at PO | P7 | 6 | 54 | 11.1% | 0 | 18 | 0.0% | ||||||
| Fgf8CreER + TAM at PO | P8 | 32 | 45 | 71.1% | 0 | 18 | 0.0% | ||||||
| Bcl11b+ | Total | % Bcl11b+ | Bcl11b+ | Total | % Bcl11b+ | Bcl11b+ | Total | % Bel11b+ | Bcl11b+ | Total | % Bcl11b+ | ||
| Atohl Cre | E17.5 | 54 | 54 | 100.0% | 0 | 18 | 0.0% | 54 | 54 | 100.0% | 15 | 18 | 83.3% |
| Fgf8CreER + TAM at PO | PI | 81 | 81 | 100.0% | 0 | 27 | 0.0% | 51 | 51 | 100.0% | 0 | 17 | 0.0% |
| Fgf8CreER + TAM at PO | P2 | 42 | 42 | 100.0% | 0 | 14 | 0.0% | 45 | 45 | 100.0% | 0 | 15 | 0.0% |
| Fgf8c,eER + TAM at PO | P3 | 45 | 45 | 100.0% | 0 | 15 | 0.0% | 42 | 42 | 100.0% | 0 | 14 | 0.0% |
| Fgf8CreER +TAM at PO | P4 | 59 | 60 | 98.3% | 0 | 20 | 0.0% | 60 | 60 | 100.0% | 2 | 19 | 10.5% |
| Fgf8CreER + TAM at PO | P5 | 47 | 48 | 97.9% | 2 | 15 | 13.3% | 38 | 45 | 84.4% | 1 | 15 | 6.7% |
| Fgf8CreER +TAM at PO | P6 | 45 | 45 | 100.0% | 1 | 15 | 6.7% | ||||||
| Fgf8CreER + TAM at PO | P7 | 28 | 45 | 62.2% | 0 | 18 | 0.0% | ||||||
| Fgf8CreER + TAM at PO | P8 | 9 | 45 | 20.0% | 0 | 15 | 0.0% | ||||||
Cochleae examined at E17.5 were from Atoh1Cre/+; Tbx2F/F and Tbx2F/F littermate controls. While control IHCs did not express high levels of Insm1 and Bcl11b mRNAs, ic-HCs of conditional KOs, in which Tbx2 was ablated embryonically (at or after E13.5), expressed these two OHC markers. Postnatal ablation of Tbx2 was induced by the administration of tamoxifen at P0 to Fgf8CreER/+; Tbx2F/F; R26Ai9/+ mice. Controls were equally treated Tbx2F/F; R26Al9/+ littermates as well as untreated Tbx2F/+ mice. We collected cochleae every subsequent day from P1 to P8, by which time hair cells in the inner compartment (ic-HCs) expressed the mature OHC markers Prestin and Oncomodulin. In experimental animals, cells in the position of IHCs are termed ic-HCs as they are transitioning from displaying features of IHCs to those of OHCs, despite their position. ISH revealed Insm1 and Bcl11b mRNAs in nearly all OHCs at early stages, before slowly declining at P5 and P7, respectively. In contrast, most ic-HCs in postnatally-treated cKOs, like most IHCs in controls but unlike the ic-HCs transdifferentiating embryonically, did not express these mRNAs. Each examined section displayed hair cells at 3 or 4 cochlear locations: apical, mid-apical, mid-basal and basal.
Supplementary Material
Acknowledgements
NUcore facilities used were the Transgenic and Targeted Mutagenesis Laboratory and the Center for Advanced Microscopy (partially supported by P30-CA060553 to the Robert H. Lurie Comprehensive Cancer Center). This study was supported by National Institutes of Health grants R01-DC015903, R01-DC019834 and R01-DC017482. The expertise of J.H. Siegel regarding hearing tests is appreciated.
Footnotes
Competing interests The authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Peer review information Nature thanks Alan Cheng, Oliver Hobert and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Reprints and permissions information is available at http://www.nature.com/reprints.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-04668-3.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information. Source data are provided with this paper.
References
- 1.Dallos P In Proceedings of the 9th International Symposium on Hearing (eds Cazals Y et al. ) 3–17 (Elsevier, 1992). [Google Scholar]
- 2.Wiwatpanit T et al. Trans-differentiation of outer hair cells into inner hair cells in the absence of INSM1. Nature 563, 691–695 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chessum L et al. Helios is a key transcriptional regulator of outer hair cell maturation. Nature 563, 696–700 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzen SM, Duggan A, Osipovich AB, Magnuson MA & García-Añoveros J Insm1 promotes neurogenic proliferation in delaminated otic progenitors. Mech. Dev. 138, 233–245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaiser M et al. Regulation of otocyst patterning by Tbx2 and Tbx3 is required for inner ear morphogenesis in the mouse. Development 148, dev195651 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Chen Y et al. The transcription factor TBX2 regulates melanogenesis in melanocytes by repressing Oca2. Mol. Cell. Biochem. 415, 103–109 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Aydoğdu N et al. TBX2 and TBX3 act downstream of canonical WNT signaling in patterning and differentiation of the mouse ureteric mesenchyme. Development 145, dev171827 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Wojahn I, Lüdtke TH, Christoffels VM, Trowe M-O & Kispert A TBX2-positive cells represent a multi-potent mesenchymal progenitor pool in the developing lung. Respir. Res. 20, 292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wakker V et al. Generation of mice with a conditional null allele for Tbx2. Genesis 48, 195–199 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Yang H, Xie X, Deng M, Chen X & Gan L Generation and characterization of Atoh1-Cre knock-in mouse line. Genesis 48, 407–413 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang H et al. Gfi1-Cre knock-in mouse line: a tool for inner ear hair cell-specific gene deletion. Genesis 48, 400–406 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekerková G, Richter C-P & Bartles JR Roles of the espin actin-bundling proteins in the morphogenesis and stabilization of hair cell stereocilia revealed in CBA/CaJ congenic jerker mice. PLoS Genet. 7, e1002032 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen DW & Slepecky N Stereocilia. in Neurobiology of Hearing: The Cochlea (eds Altschuler RA et al. ) 23–46 (Raven Press, 1986). [Google Scholar]
- 14.Ratzan EM, Moon AM & Deans MR Fgf8 genetic labeling reveals the early specification of vestibular hair cell type in mouse utricle. Development 147, dev192849 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Webber JL et al. Axodendritic versus axosomatic cochlear efferent termination is determined by afferent type in a hierarchical logic of circuit formation. Sci. Adv. 7, eabd8637 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dabdoub A et al. Sox2 signaling in prosensory domain specification and subsequent hair cell differentiation in the developing cochlea. Proc. Natl Acad. Sci. USA 105, 18396–18401 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kempfle JS, Turban JL & Edge ASB Sox2 in the differentiation of cochlear progenitor cells. Sci. Rep. 6, 23293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox BC et al. Spontaneous hair cell regeneration in the neonatal mouse cochlea in vivo. Development 141, 816–829 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu L et al. Diphtheria toxin-induced cell death triggers Wnt-dependent hair cell regeneration in neonatal mice. J. Neurosci. 36, 9479–9489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bramhall NF, Shi F, Arnold K, Hochedlinger K & Edge ASB Lgr5-positive supporting cells generate new hair cells in the postnatal cochlea. Stem Cell Rep. 2, 311–322 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H et al. Cell-specific transcriptome analysis shows that adult pillar and Deiters’ cells express genes encoding machinery for specializations of cochlear hair cells. Front. Mol. Neurosci. 11, 356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H et al. Characterization of transcriptomes of cochlear inner and outer hair cells. J. Neurosci. 34, 11085–11095 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia S, Yang S, Guo W & He DZZ Fate of mammalian cochlear hair cells and stereocilia after loss of the stereocilia. J. Neurosci. 29, 15277–15285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Hirose K & Liberman MC Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J. Assoc. Res. Otolaryngol. 3, 248–268 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landegger LD et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 35, 280–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coffin A, Kelley M, Manley GA & Popper AN Evolution of sensory hair cells. in Evolution of the Vertebrate Auditory System, Vol. 22 (eds Manley GA et al. ) 55–94 (Springer, 2004). [Google Scholar]
- 27.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haque KD, Pandey AK, Kelley MW & Puligilla C Culture of embryonic mouse cochlear explants and gene transfer by electroporation. J. Vis. Exp. 12, 52260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pearce M, Richter C-P & Cheatham MA A reconsideration of sound calibration in the mouse. J. Neurosci. Meth. 106, 57–67 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Neely ST & Liu Z EMAV: Otoacoustic Emission Averager. Technical memo no. 17 (Boy’s Town National Research Hospital, 1994). [Google Scholar]
- 31.Santos-Sacchi J, Kakehata S & Takahashi S Effects of membrane potential on the voltage dependence of motility-related charge in outer hair cells of the guinea-pig. J. Physiol. 510, 225–235 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Homma K & Dallos P Evidence that prestin has at least two voltage-dependent steps. J. Biol. Chem. 286, 2297–2307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its Supplementary Information. Source data are provided with this paper.