

Summary
Lsd1/Kdm1a functions both as a histone demethylase enzyme and as a scaffold for assembling chromatin modifier and transcription factor complexes to regulate gene expression. The relative contributions of Lsd1’s demethylase and scaffolding functions during embryogenesis are not known. Here, we analyze two independent zebrafish lsd1/kdm1a mutant lines and show Lsd1 is required to repress primitive hematopoietic stem cell gene expression. Lsd1 rescue constructs containing point mutations that selectively abrogate its demethylase or scaffolding capacity demonstrate the scaffolding function of Lsd1, not its demethylase activity, is required for repression of gene expression in vivo. Lsd1’s SNAG-binding domain mediates its scaffolding function and reinforces a negative feedback loop to repress the expression of SNAG-domain-containing genes during embryogenesis, including gfi1 and snai1/2. Our findings reveal a model in which the SNAG-binding and scaffolding function of Lsd1, and its associated negative feedback loop, provide transient and reversible regulation of gene expression during hematopoietic development.
Subject areas: Molecular biology, Molecular interaction, Cell biology, Stem cells research
Graphical abstract
Highlights
-
•
LSD1 controls embryonic gene expression by a catalysis-independent scaffold function
-
•
SNAG-domain binding to LSD1 is critical for its scaffold function
-
•
LSD1-GFI1 generates a negative feedback loop to repress gfi1 expression in vivo
Molecular biology; Molecular interaction; Cell biology; Stem cells research
Introduction
Lysine (K)-specific demethylase 1A (LSD1, also known as KDM1A) is a well-established and highly conserved flavin adenine dinucleotide (FAD)-dependent demethylase that controls gene expression during differentiation and development. LSD1 functions through interaction with multiprotein complexes, such as BHC/CoREST (BRAF-histone deacetylase complex/Corepressor for element-1-silencing transcription factor), NuRD (nucleosome remodeling and deacetylase), and CtBP (C-terminal binding protein), to remove mono- and di-methyl marks from histone 3 lysine 4 (H3K4) and histone 3 lysine 9 (H3K9).1,2 LSD1 also demethylates transcription factors such as TP53 and E2F1 to modulate transcriptional programs.3,4,5 Evidence from multiple malignancy models indicates that LSD1 may also have non-catalytic functions.6,7,8,9,10,11 For example, in acute myeloid leukemia (AML) cells, LSD1 enzymatic activity is not required for cell survival or differentiation. Instead, LSD1 acts as a scaffold for recruiting the growth factor independence 1 protein (GFI1) to enhancer regions located adjacent to genes that regulate myeloid differentiation and survival.6,7,9 The relative contribution of LSD1’s catalytic and non-catalytic functions during tumorigenesis and normal development, however, are not well understood. Such knowledge will be essential for understanding and manipulating LSD1-dependent gene expression programs for therapeutic purposes.
Hematopoiesis in vertebrates is characterized by two separate developmental programs called the primitive and definitive waves.12,13,14 The primitive wave primarily gives rise to early erythrocytes and macrophages in the developing embryo. Immediately following the primitive wave, the definitive wave generates hematopoietic stem cells (HSCs) that differentiate into all blood lineages of the adult organism.12,13,14 LSD1 is essential for the development of HSCs and granulocytes, and for erythroid differentiation.15,16,17,18,19,20 Germline deletion of Lsd1 in mice is embryonic lethal by day E6.5, and conditional Lsd1 deletion in hematopoietic stem cells and erythroid cells causes severe anemia.17,21,22 Similarly, germline mutations in lsd1 in zebrafish result in embryonic lethality, failure to generate primitive erythrocytes19 and loss of expression of essential erythrocyte genes such as gata1.19,20 Of interest, recent studies suggest that the primitive hematopoietic defects in zebrafish lsd1 mutants are demethylase-independent.20 However, the basis for assigning a demethylase-independent role for Lsd1 was derived from the use of a presumed catalytically dead lsd1 rescue construct,20 which has subsequently been shown to retain up to 20% normal Lsd1 activity.23 Thus, a demethylase-independent role for Lsd1 during hematopoietic differentiation remains debatable. More importantly, the mechanism underlying Lsd1's repression of gene expression during primitive hematopoiesis in vivo remains unknown.
Both the catalytic and scaffolding functions of LSD1 are thought to require a hydrophobic pocket within the LSD1 protein that enables substrate binding.24,25 The structure of LSD1 includes an N-terminal amino-oxidase-like domain (AOL), a central tower domain, and a C-terminal AOL domain. The AOL domains fold around the tower domain to form the substrate-binding hydrophobic pocket that contains an FAD cofactor essential for LSD1’s catalytic activity.24,25 The binding of histone tails to the hydrophobic pocket is required for their demethylation by LSD1. The non-catalytic or scaffolding functions of LSD1 appear to be mediated through direct binding to transcription factors that contain a highly conserved, 21-amino-acid, N-terminal domain called the SNAIL/GFI (“SNAG”) domain, which resembles the structure of the tail region of histone H3 and binds to the same hydrophobic pocket in LSD1.26,27
Transcriptional repression mediated by either GFI- or SNAIL-family proteins is critical for normal embryonic development.28,29 The SNAIL family of zinc-finger transcription factors direct many cellular processes, including the formation of mesoderm and neural crest cells, and the epithelial-to-mesenchymal transition (EMT) in both development and malignancy.29 GFI1 and GFI1B are important drivers of both hematopoietic stem cell (HSC) differentiation and leukemogenesis.18,30,31 GFI1 promotes normal lymphoid and myeloid development whereas GFI1B promotes the generation of erythrocytes and megakaryocytes.31,32 A common attribute of these SNAG-domain-containing transcription factors is their ability to negatively regulate their own gene expression and that of other members of their gene family.29,33,34,35,36,37,38,39 The SNAG-dependent transcriptional feedback loops as well as the GFI- and SNAIL-mediated developmental programs are dependent on interactions between these transcription factors and LSD1 in vitro26,40; however, whether such interactions also mediate feedbacks loops during embryogenesis remains unknown.
The mechanistic basis for LSD1-mediated primitive hematopoiesis has not yet been investigated in vivo. Here, we performed RNA-Sequencing (RNA-Seq) to analyze the transcriptional program regulated by Lsd1 during primitive hematopoiesis using two previously undescribed lsd1 zebrafish mutants. Surprisingly, only a limited number of genes were upregulated in lsd1 mutants and were enriched for snail1/2 and gfi family genes, suggesting that Lsd1 normally functions to selectively repress SNAG-domain-containing genes during embryogenesis. As expected,20 we found that loss of lsd1 impaired erythrocyte development in vivo and that both wild-type Lsd1 and the Lsd1-K661A catalytic mutant rescued defective primitive hematopoiesis in lsd1-mutant embryos. Therefore, to determine whether Lsd1 binding to SNAG-domain-containing proteins is needed for Lsd1-mediated hematopoiesis, we analyzed the structure of SNAG-domain-bound Lsd1 and mutated amino acid residues that were likely critical for Lsd1 binding to SNAG-domain-containing proteins but not its catalyic activity. None of these SNAG-binding-compromised Lsd1 mutant proteins were able to rescue defective hematopoiesis in the lsd1 mutants. Moreover, injection of embryos with an exogenous SNAG peptide inhibited the ability of wild-type Lsd1 to rescue defective hematopoiesis in the lsd1 mutants. These experiments show that LSD1 controls gene expression during primitive hematopoiesis primarily via its scaffolding function, which requires binding to SNAG-domain-containing proteins. In addition, we demonstrate that SNAG-dependent binding to LSD1 represses the expression of SNAG-containing genes themselves, thereby reinforcing a negative feedback loop to control gene expression during embryogenesis.
Results
Newly identified zebrafish lsd1 alleles confirm that Lsd1 function is required for erythrocyte development in vivo
The zebrafish genome contains one copy of the lsd1 gene, which encodes for a protein that is highly identical (>90%) to human LSD1 (Figure 1A). Zebrafish lsd1 was previously analyzed using a mutant allele, lsd1it627, that was identified through an N-ethyl-N-nitrosourea (ENU) mutagenesis screen and found to contain a premature stop codon (replacing Gln609) that leads to a truncation of the Lsd1 protein within the C-terminal amine-oxidase-like (AOL) domain19. This mutation was reported to promote endothelial cell differentiation at the expense of erythroid cell development as early as 12.5 h post-fertilization (hpf), and to cause lethality by 10 days post-fertilization (dpf).19 To verify that Lsd1 is required for erythroid cell development in zebrafish, we analyzed two previously uncharacterized ENU-generated lsd1 alleles called kdm1a(sa23095) and kdm1a(sa36433). The kdm1a(23095) allele contains a donor-splice-site mutation at the C-terminus of exon 19, and kdm1a(36433) contains a premature stop mutation in exon 17 that replaces Gln632 (Figure 1A).41 Zebrafish that are homozygous mutant for either allele fail to survive to adulthood. Embryos derived from an incross between either kdm1a(23095) or kdm1a(36433) were grown to 4 dpf, and the developing cardiac region was examined for red cells (erythrocytes). Consistent with lsd1it627 mutants, a quarter of the embryos derived from either incross exhibited a lack of red cells, suggesting that kdm1a(23095) and kdm1a(36433) homozygous mutants either fail to generate erythrocytes or their erythrocytes have a reduction or loss of hemoglobin (known as “pale erythrocytes”, Figures 1B and 1C). To better understand the effect of the kdm1a(23095) and kdm1a(36433) mutations on their respective mRNA expression levels, we performed RNA-Seq and found that lsd1 mRNA levels were similar between kdm1a(23095) mutants and their wild-type siblings, whereas kdm1a(36433) mRNA was largely undetectable, indicating that it is likely degraded through nonsense-mediated decay (Figure 1D). Examination of the mutant kdm1a(23095) allele with the Integrative Genomics Viewer revealed that it leads to lsd1 mRNA transcripts containing either an excision of exon 19 or a retention of the intron between exon 19 and 20 (Figure S1A). Both scenarios are predicted to produce a non-functional protein because they would disrupt the C-terminal AOL domain, which is critical for forming the substrate- and FAD-binding subdomains. Finally, to determine if the kdm1a(23095) and kdm1a(36433) mutations disrupt hematopoietic gene expression programs during primitive hematopoiesis similar to the lsd1it627 mutants,19 we analyzed the expression of erythrocyte (gata1) and endothelial-cell (etv2 and fli1a) markers by whole-mount in situ hybridization (WISH). Indeed, kdm1a(23095) and kdm1a(36433) homozygous mutants showed diminished gata1 expression (Figures S1B and S1E) and elevated fli1a (Figures S1C and S1F) and etv2 (Figures S1D and S1G) expression. The reduction in primitive erythrocytes did not appear to be driven by an overall increase in apoptotic cells or decrease in cellular proliferation in the tail region encompassing the intermediate cell mass (ICM), because there were no significant changes in activated-Caspase-3-positive cells or phospho-histone-H3-positive cells, respectively, in lsd1 mutants in this region (see Figure S2 and STAR Methods). Thus, a phenotypic analysis of three different zebrafish lsd1 mutant alleles from two different laboratories demonstrates highly reproducible and robust primitive hematopoietic defects in vivo that support a model in which loss of LSD1 function is required for the differentiation of primitive hematopoietic cell types, such as erythrocytes, during embryonic development.
Figure 1.

Mutant lsd1 alleles in zebrafish
(A) Schematic showing the percent amino acid identity between human and zebrafish Lsd1 as well as the locations of the mutations in the C-terminal AOL region that are relevant to this study.
(B and C) Brightfield images of 4-dpf embryos derived from an incross between either kdm1a(23095) heterozygotes (B) or kdm1a(36433) heterozygotes (C). Arrowheads indicate erythrocytes in the heart. The number of animals with a similar phenotype to the representative image, out of the total number of animals visualized, is shown in the top right to indicate Mendelian ratio. Scale bar, 200 μm.
(D) The relative mRNA expression levels of kdm1a in mutants and wild-type siblings are shown in the graph.
gfi1 hematopoietic transcriptional regulators are de-repressed in lsd1 mutants
We next questioned whether the lsd1-mutant primitive hematopoietic phenotypes were because of a block in differentiation via mis-regulation of hematopoietic stem cell transcription factors. The GFI family of transcription factors, GFI1 and GFI1B, were likely candidates because they both require LSD1 to mediate gene repression for lineage-specific differentiation in hematopoiesis.6,9,18,40 Both GFI1 and GFI1B can autoregulate as well as cross-regulate expression from their respective genes to modulate hematopoietic-lineage differentiation.33,34,35,36,37 This autoregulation is dependent on functional SNAG and DNA-binding domains within the GFI1 and GFI1B proteins.26,33,34,36,42 The zebrafish genome contains three gfi family members. The gfi1aa and gfi1ab genes are homologous to human GFI1, whereas gfi1b is homologous to human GFI1B.43 We performed WISH in 24-hpf lsd1 mutants and siblings and found that gfi1aa expression is normally observed exclusively in the ICM and is upregulated in lsd1 mutants (Figures 2A and 2D). Expression of the gfi1ab gene is normally restricted to hair cells of the developing ear at 24 hpf44 (Figures 2B and 2E); however, in lsd1 mutants gfi1ab expression is ectopically induced in the anterior lateral plate mesoderm by 14 hpf (Figures S3A and S3B), as well as the ICM at 24 hpf (Figures 2B and 2E), and remains elevated in the caudal hematopoietic tissue (CHT) at 3 dpf (Figure S3C). The gfi1b gene is normally expressed in the ICM at 24 hpf, and WISH analysis showed no qualitative difference in gfi1b ICM expression between lsd1 mutants and siblings at this stage (Figures 2C and 2F). However, analysis of gfi1b expression by WISH at 3 dpf showed elevated gfi1b in the CHT (Figure S3D). These data indicate that Lsd1 is required to repress gfi1 paralog expression during zebrafish primitive hematopoiesis.
Figure 2.

Expression of GFI-family transcription factors are dysregulated in lsd1 mutants
(A–F) Whole-mount in situ hybridization (WISH) of embryos derived from an incross of either kdm1a(23095) heterozygotes (A–C) or kdm1a(36433) heterozygotes (D–F) using probes to detect gfi1aa (A and D), gfi1ab (B and E), or gfi1b (C and F), as indicated. Representative images are shown with the number of animals with similar staining patterns for the indicated genotype shown in the upper right corner of each image. The lower left corner indicates the approximate stage of the animal at the time of fixation. Embryos were imaged, blindly scored by expression levels and then genotyped by HRMA. Carons indicate ICM expression; closed arrowheads indicate posterior blood island/caudal hematopoietic tissue; narrow triangles indicate inner-hair-cell expression. Scale bar, 200 μm.
Genetic mutants in gfi1/1b phenocopy lsd1 mutant hematopoietic defects
Gfi1/1b family members control the differentiation of stem/progenitor cells into different lineages during hematopoiesis,18,30,31 in part through repressing their own expression during lineage commitment.33,34,35,36,37 Our WISH analysis of gfi1/1b genes in lsd1 mutants suggested that Lsd1 is required for Gfi1 and Gfi1b to repress their own expression in vivo, particularly gfi1ab. Thus, we next hypothesized that the loss of differentiated hematopoietic cell types in lsd1 mutants is because of loss of Gfi1/1b function. To test this possibility, we analyzed primitive hematopoiesis in gfi1aa; gfi1ab; gfi1b triple mutants (Figure S4). We found that loss of gfi1/1b genes in zebrafish caused loss of primitive hematopoietic markers, including gata1 expression at 24 hpf in the ICM (Figure S4B) as well as other morphological abnormalities (Figure S4C). Indeed, during the course of this study, another group reported similar results with gfi1/1b triple mutants.45 We also analyzed individual mutants in the Gfi1 family and found that, similar to Wu et al.,45 loss of gfi1aa is largely sufficient to phenocopy the lsd1 mutant hematopoietic phenotypes, with a minor contribution by loss of gfi1ab (data not shown). Together, these studies support a model in which Lsd1 is required for Gfi1aa to repress the expression of itself and other family members, particularly gfi1ab, which in turn is required for the generation of differentiated primitive hematopoietic cell types, such as gata1-expressing erythrocytes.
Lsd1 broadly regulates the mRNA expression of SNAG domain transcription factors
As a histone demethylase, LSD1 is predicted to impact the expression of multiple genes during embryogenesis, mainly through transcriptional repression by demethylation of H3K4. To better understand how LSD1 controls gene expression during embryogenesis, we used a scalpel to cut 24-hpf embryos derived from an incross of kdm1a(23095) or kdm1a(36433) heterozygotes just posterior to the yolk sac (Figure 3A). The head was used to genotype the embryo and the tail was preserved in RNA stabilization solution. On completion of genotyping, five tails from homozygous lsd1 mutants or wild-type siblings were pooled, and RNA-Seq was performed on the pooled RNA (Figure 3B, Table S4). Despite Lsd1’s established role as an epigenetic modifier, we found that the expression of only a small subset of genes was significantly affected by loss of lsd1 at this stage of embryogenesis. Of 21,497 genes, 409 were upregulated and 524 were downregulated (p<0.05, Figure 3B, Table S4). Examination of the most differentially expressed genes in lsd1 mutants revealed three important findings: (1) An enrichment among the upregulated genes for SNAG-domain-containing transcription factors (Figure 3B), including both gfi and snail family genes, (2) elevated levels of endothelial/hematopoietic stem or progenitor genes, including tbx16, etv2, lmo2, gfi1aa and tal1 (Figure S5), and (3) decreased levels of differentiated hematopoietic cell markers, such as lyz and hbbe1.3 (Figure S5). These findings were consistent with the WISH analysis above showing differential effects on gfi1 and gata1 gene expression in lsd1 mutants. In addition, they show Lsd1 is normally required to repress hematopoietic/endothelial stem cell genes during embryogenesis, including all gfi1/1b genes.
Figure 3.

SNAG-domain proteins are dysregulated in lsd1 mutants
(A) Schematic of the sample preparation workflow.
(B) Heatmap of the top 40 differentially expressed genes in kdm1a(23095) and kdm1a(36433) mutants compared to wild-type siblings. SNAG-domain proteins are boxed.
(C–H) WISH of embryos from an incross of kdm1a(23095) heterozygotes for snail1a, snail1b, and snail2 at 14 hpf (C–E), lateral (left) and dorsal (right) views) and 24 hpf (F–H). Representative images are shown with the number of animals with similar staining patterns for the indicated genotype shown in the upper right corner of each image. The lower left corner indicates the approximate stage of the animal at the time of fixation. Embryos were imaged, blindly scored by expression and then genotyped by HRMA. Carons indicate somite expression; narrow triangles indicate tail bud; double carons indicate adaxial cell expression; arrows indicate neural plate border; closed arrowheads indicate trunk neural crest cells (snail1b) or ventral mesoderm (snai2). Scale bar, 200 μm.
The RNA-Seq analysis suggests that Lsd1 function is required to regulate a limited number of genes during early embryogenesis, and that SNAG-domain-containing genes as a group are targeted by Lsd1 for repression. To determine the extent to which Lsd1 is required to repress the expression of SNAG-domain-containing genes in other embryonic tissues, WISH experiments were performed on embryos from a lsd1-heterozygous mutant incross at 14 and 24 hpf (Figures 3C–3H). We observed the upregulation of snail1a, snail1b and snai2 in a number of different cell types, including muscle, lateral plate mesoderm, neural crest and tailbud. These data show that LSD1 functions in a number of embryonic tissues to selectively repress the expression of SNAG-domain-containing genes.
Wild-type Lsd1 restores normal expression levels of primitive hematopoietic transcription factors in lsd1 mutants
To determine if expression of lsd1 mRNA can repress expression of gfi1aa and gfi1ab in kdm1a(23095) and kdm1a(36433) mutants, we injected embryos derived from a heterozygous in-cross of each mutant line with mRNA encoding either wild-type zebrafish lsd1 (zlsd1) or human LSD1 (hLSD1). We then confirmed the level of protein expression via western blot (Figure S6) and performed WISH. Indeed, the injection of either wild-type zlsd1 mRNA or wild-type hLSD1 mRNA was sufficient to repress gfi1aa and gfi1ab expression, and to restore gata1 expression, in both kdm1a(23095) and kdm1a(36433) embryos (Figures 4A–4F and S7A–S7F). Moreover, the injection of mRNA encoding zlsd1-ΔQ632, which mimics the predicted kdm1a(36433) protein product (a premature stop codon substituted at Gln632), and mRNA encoding lsd1 lacking the C-terminal AOL domain failed to rescue changes in the transcriptional program (Figures S8 and S9). Although our data is consistent with previous reports showing conservation of Lsd1 function with respect to restoring gata1 expression,20 it shows that the ability of LSD1 to repress expression of gfi1aa/ab genes during primitive hematopoiesis is also conserved. As Lsd1 is largely associated with the repression of gene expression, such findings allowed us to design assays to interrogate the mechanism(s) required for Lsd1’s repressive function in vivo.
Figure 4.

Wild-type lsd1 rescues hematopoietic transcription factor expression
(A–F) Single-cell embryos from a kdm1a(23095) heterozygous incross were injected with either zlsd1 (A–C) or hLSD1 mRNA (D–F). At 24 hpf, WISH was performed. Representative images are shown with the number of animals with similar staining patterns for the indicated genotype shown in the upper right corner. The lower right corner indicates the mRNA injection conditions. Uninjected mutant embryos are shown in the middle column for comparison. Embryos were imaged, blindly scored by expression and then genotyped by HRMA. Carons indicate ICM expression; closed arrowheads indicate posterior blood island/caudal hematopoietic tissue; narrow triangles indicate inner-hair-cell expression. Scale bar, 200 μm.
Lsd1 variants that impair catalytic activity have differential effects on restoring gene expression during primitive hematopoiesis
LSD1 catalytic activity occurs through an FAD cofactor intermediate.24,25 In the LSD1 hydrophobic pocket, the H3K4 residue from the bound histone H3 tail is located in close proximity to the N5 atom of FAD,46 which enables FAD to oxidize the methylated lysine. This converts the methylated lysine to an intermediate imine molecule and a demethylated side chain.47 The imine is then hydrolyzed to formaldehyde, and the flavin cofactor is reoxidized by molecular oxygen to complete the enzymatic cycle.47 Several residues are required for LSD1 demethylase activity on in vitro substrates such as dimethylated histone H3 peptides or reconstituted nucleosomes.23,24,25 In general, there are two groups of mutations that impair LSD1 catalytic activity on histone substrates: (1) Mutations that interfere with stabilization and subsequent oxidization of the reduced flavin, such as the LSD1 K661A allele and (2) mutations located in the hydrophobic pocket that are required for the binding of LSD1 to the histone H3 tail, such as the LSD1 A539E allele.23,25,27 Notably, because the LSD1 K661A allele does not demethylate histone peptides, it has been widely used to infer catalysis-independent functions of LSD1 in different cell- and animal-based processes, including zebrafish primitive hematopoiesis.20 However, recent studies showed that the LSD1 K661A allele retains approximately 20% of its wild-type histone demethylase activity when combined with reconstituted nucleosome substrates.23 In contrast, the demethylase activity of the LSD1 A539E allele is significantly impaired using both peptide and nucleosome substrates, retaining only ∼5% of wild-type LSD1 activity. We therefore determined the extent to which these catalytically impaired LSD1 mutations could restore primitive hematopoietic gene expression programs in vivo. Similar to wild-type zlsd1 mRNA, injection of zlsd1K661A mRNA into both kdm1a(23095) and kdm1a(36433) mutants rescued normal expression patterns of gfi1ab and gata1 expression during primitive hematopoiesis (Figures 5 and S10).20 In contrast, injection of mRNA encoding zlsd1A539E failed to repress gfi1ab expression or restore gata1 levels (Figures S11A and S11B) despite robust Lsd1 A539E protein expression (Figure S6). These data show that catalytically impaired Lsd1 alleles have differential outcomes with respect to rescue of gene expression programs, revealing that either a threshold level of LSD1 nucleosome-based catalytic activity (between 5 and 20%), or integrity of the substrate binding domain of LSD1, or both, are required to control gene expression programs during primitive hematopoiesis in vivo.
Figure 5.

Demethylase-deficient lsd1 rescues hematopoietic transcription factor gene expression
(A and B) Single-cell zebrafish embryos from a kdm1a(23095) heterozygous incross were injected with zebrafish zlsd1-K661A. WISH was then performed at 24 hpf for gata1 (A) and gfi1ab (B). Representative images are shown with the number of animals with similar staining patterns for the indicated genotype shown in the upper right corner. The lower right corner indicates the mRNA injection conditions. Uninjected mutant embryos are shown in the middle column for comparison. Embryos were imaged, blindly scored by expression and then genotyped by HRMA. Carons indicate ICM expression; closed arrowheads indicate posterior blood island/caudal hematopoietic tissue; narrow triangles indicate inner-hair-cell expression. Scale bar, 200 μm.
SNAG-domain binding to Lsd1 is required for Lsd1-mediated regulation of primitive hematopoiesis
To determine whether SNAG-domain binding is specifically required for LSD1-mediated transcription during primitive hematopoiesis, we sought to design SNAG-binding mutations in LSD1. We analyzed the crystal structure of LSD1 in complex with an N-terminal SNAIL SNAG peptide27 and identified LSD1 residues in the hydrophobic pocket that were within 4 Å of the first six amino acids of the peptide (Table S1). To interfere with SNAG-dependent binding of Lsd1 without significantly impacting its FAD-dependent catalytic activity, we chose to analyze the Asp555 residue, which is conserved from humans to zebrafish (D555, Figure 6A) and located within 4 Å of the proline-2 residue of the SNAG domain that is essential for its binding to LSD1 (Figure 6B, top panel).40,42 We mutated the negatively charged Asp555 to a positively charged Lys (D555K, Figure 6B, bottom panel). We found that, similar to injection of substrate-binding-mutant zlsd1A539E mRNA, injection of zlsd1D555K mRNA into lsd1 mutants was unable to restore gata1 (Figure 6C) or repress gfi1ab (Figure 6D) gene expression. As the LSD1 D555K mutant protein retains more LSD1 demethylase activity than the LSD1 K661A mutant protein on histone peptides,24,25 our data supports a model in which LSD1’s regulation of primitive hematopoietic gene expression programs is largely catalysis-independent.
Figure 6.

SNAG-binding-deficient lsd1 does not rescue gene expression
(A) Schematic of the zebrafish Lsd1 C-terminal AOL domain with exon locations of predicted SNAG-binding-deficient mutations indicated.
(B) Top panel: the structure of LSD1 (green) with the FAD moiety (red) and the SNAG domain of SNAIL (yellow) is shown. LSD1 D555 is highlighted in teal. The second proline residue of the SNAG domain is highlighted in white to provide orientation. Bottom panel: the PyMOL mutagenesis feature (PyMOL Molecular Graphics System, Version 2.3.4 Schrodinger, LLC) was utilized to change Asp 555 to Lys. The atom providing positive charge is indicated with a (+) symbol.
(C and D) Single-cell zebrafish embryos from a kdm1a(23095) heterozygous incross were injected with mRNA encoding zlsd1-D555K (C and D). WISH was then performed on 24-hpf embryos for gata1 (C) and gfi1ab (D) expression. Representative images are shown with the number of animals with similar staining patterns for the indicated genotype shown in the upper right corner. The lower right corner indicates the mRNA injection conditions. Embryos were imaged, blindly scored by expression and then genotyped by HRMA. Carons indicate ICM expression; closed arrowheads indicate posterior blood island/caudal hematopoietic tissue; narrow triangles indicate inner-hair-cell expression. Scale bar, 200 μm.
To our knowledge, the LSD1 K661A mutation represents the most well-characterized catalytically-impaired molecule that does not interfere with substrate binding.23,24,25 Mutations at other LSD1 residues, such as D555 and A539, have been shown both to impair catalytic activity and interfere with substrate binding,23,24,25 and we found no published LSD1 mutants that block substrate binding without also negatively impacting LSD1 catalytic activity. Therefore, to test whether blocking substrate binding to LSD1 is sufficient to disrupt gene expression in vivo, we synthesized a peptide representing the SNAG domain (Figure 7A), which we and others have previously shown to bind to LSD1 in vitro.6,26,27,48,49 We injected the SNAG-domain peptide with wild-type lsd1 mRNA into one-cell-stage zebrafish embryos derived from an incross between heterozygous lsd1 mutants and analyzed gata1 and gfi1ab expression by WISH. We found that the SNAG-domain peptides blocked the ability of wild-type Lsd1 to both restore normal gata1 expression and repress gfi1ab expression in lsd1 mutants (Figures 7B and 7C). These data further support a model in which LSD1’s regulation of gene expression during primitive hematopoiesis in vivo relies primarily on scaffolding and SNAG-domain transcription factor binding, rather than its demethylase activity.
Figure 7.

The SNAG-LSD1 interaction is critical for the regulation of primitive hematopoiesis
(A) Sequence of the SNAG domain peptide with dimethylated Lys 8 indicated in red.
(B and C) Single-cell zebrafish embryos from a kdm1a(23095) heterozygous incross were injected with both zlsd1-3XFLAG mRNA and the SNAG peptide. WISH was then performed at 24 hpf for gata1 (B) and gfi1ab (C) expression.
(D and E) Single-cell zebrafish embryos from a kdm1a(23095) heterozygous incross were injected with zlsd1-ΔTower mRNA. WISH was then performed at 24 hpf for gata1 (D) and gfi1ab (E) expression. Representative images are shown with the number of animals with similar staining patterns for the indicated genotype shown in the upper right corner. The lower right corner indicates the mRNA injection conditions. Embryos were imaged, blindly scored by expression and then genotyped by HRMA. Carons indicate ICM expression; closed arrowheads indicate posterior blood island/caudal hematopoietic tissue; narrow triangles indicate inner-hair-cell expression. Scale bar, 200 μm.
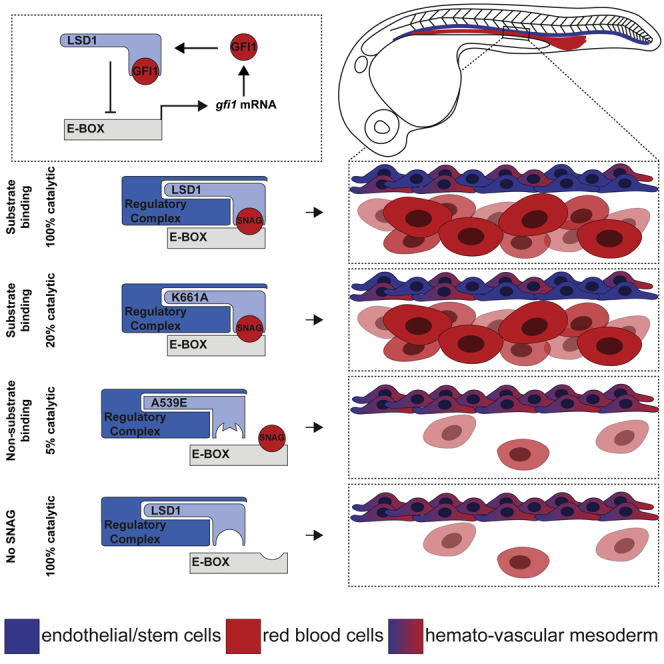
(F) Model: LSD1-dependent hematopoietic phenotypes require SNAG-domain binding. Under normal conditions, a SNAG-domain protein recruits LSD1 and a regulatory complex (e.g., CoREST) to target specific loci to silence the endothelial/stem program and promote hematopoietic cell differentiation. (II) In the absence of LSD1, SNAG-target loci are not transcriptionally repressed and an endothelial/stem cell fate is permitted. (III) An SNAG-domain protein is still able to recruit a demethylase-deficient LSD1 (K661A) and a regulatory complex to target the genetic loci that enable primitive hematopoietic gene expression. (IV) LSD1 SNAG-domain-binding mutations, such as A539E or D555K, prevent LSD1 from binding SNAG-domain-containing proteins. These LSD1 mutants are therefore not recruited to SNAG-protein target genes, such as endothelial/stem genes, which remain expressed and permit an endothelial/stem cell fate.
The Lsd1 tower domain is required for Lsd1-mediated regulation of primitive hematopoiesis
Once LSD1 is recruited by SNAG-domain-containing transcription factors to specific genomic locations, it can form a variety of different multiprotein complexes, including BHC/CoREST, NuRD, and CtBP, to regulate gene transcription.1,5,50 The tower domain of LSD1 is its most well-established protein-protein interaction domain and is critical for binding RCOR1, MTA1, MTA2, and SIN3A.24,50,51 To determine whether the SNAG-dependent function of Lsd1 requires the tower domain for multiprotein-complex recruitment and subsequent transcriptional regulation, we injected mRNA encoding a zebrafish lsd1 allele with deletion of the tower domain (zlsd1-ΔTower) into embryos derived from an incross between kdm1a(23095) heterozygotes and examined gene expression by WISH. Indeed, the zlsd1-ΔTower mRNA was unable to restore normal gata1 or gfi1ab expression (Figures 7D and 7E), showing that the LSD1 scaffolding function is critical for LSD1-mediated transcriptional regulation during primitive hematopoiesis.
We propose a model in which the SNAG domains of SNAG-domain-containing proteins recruit LSD1, which acts as a scaffold to assemble chromatin-modifying complexes (such as BHC/CoREST) to repress early developmental programs (such as primitive hematopoiesis, Figure 7F, panel I). In the absence of LSD1, repressive complexes are not recruited to SNAG-bound targets, preventing the down-regulation of endothelial/hematopoietic stem cell programs required to initiate primitive myeloid and erythroid differentiation (Figure 7F, panel II). A catalytically impaired form of LSD1 (K661A) is still able to bind SNAG-domain proteins and repress target genes (Figure 7F, panel III); however, SNAG-binding-impaired LSD1 proteins (e.g., D555K) are unable to repress gene expression (Figure 7F, panel IV). As gfi1/1b genes are themselves a target of the LSD1-GFI repressive complex, the scaffolding function of LSD1 also controls a negative feedback loop with GFI1/1B. Thus, the LSD1-GFI1 complex would repress stem and/or endothelial genes to promote the initial wave of primitive hematopoietic differentiation, however the subsequent down-regulation of LSD1 scaffold function (through repression of Gfi1/Gfi1b levels) would re-establish the hematopoietic stem cell program to drive definitive hematopoiesis. The catalysis-independent function of LSD1 therefore creates a dynamic system that allows for reversible control of gene expression programs in a progenitor population at different developmental time points, and obviates the need for the reversal of epigenetic/histone events to mediate hematopoietic program switching.
Discussion
LSD1 regulates a diverse array of biological processes through both catalytic and non-catalytic mechanisms that involve interactions with a wide variety of transcription factors and multiprotein complexes. Thus, the inherent complexity of LSD1-mediated gene regulation presents a significant challenge to understanding how LSD1 orchestrates distinct biological outcomes. In this study, we sought to understand the mechanism driving LSD1-mediated transcriptional regulation during primitive hematopoiesis. The catalytically impaired LSD1 K661A mutant protein has been used in a number of studies to identify potential non-catalytic functions both in vitro and in vivo.6,7,8,9,10,11,20 For example, a recent study concluded that LSD1 drives primitive hematopoiesis in a demethylase-independent manner because zlsd1K661A was able to restore primitive hematopoiesis in lsd1-mutant zebrafish.20 However, it was subsequently shown that the LSD1 K661A mutant protein retains 20% of its catalytic activity in nucleosome assays.23 In light of this data, no conclusions can yet be made about whether LSD1’s catalytic activity is required for primitive hematopoiesis. Moreover, because all previous reports using lsd1-mutant alleles have proven capable of rescuing primitive hematopoiesis in lsd1-mutant zebrafish, the domains within LSD1 that are required for LSD1-mediated regulation of primitive hematopoiesis have remained elusive. Here, we use multiple lsd1-mutant alleles that have similar levels of impaired catalytic activity but differential effects on rescuing lsd1 mutant primitive hematopoiesis, with SNAG peptides to interfere with the LSD1 substrate-binding pocket, to show that (1) Lsd1 represses hematopoietic gene expression in a catalysis-independent manner, (2) the interaction between Lsd1 and Gfi1 creates a negative feedback loop that represses gfi1 mRNA expression, and (3) the Lsd1 SNAG-binding and tower domains are critical for Lsd1-mediated transcriptional regulation during primitive hematopoiesis in vivo.
In this study, we show that loss of lsd1 results in an upregulation of gfi1ab and a downregulation of gata1, similar to what we and others have observed with loss of gfi1aa.42,52,53 These data suggest a model whereby Gfi1aa normally recruits Lsd1 to repress the expression of stem cell genes, such as scl1, lmo2, tbx16 and etv2, in a common hemato-vascular mesoderm (HVM) cell, thereby providing a permissive landscape for driving cells toward primitive hematopoietic cell fates. Loss of, or interference with, either Lsd1 or Gfi1aa would then lead to elevated levels of stem cell genes in HVM cells, promoting a shift toward endothelial and/or HVM cell fates at the expense of primitive erythrocyte and myeloid cells. In this model, the reversible nature of SNAG-binding to LSD1 would allow for the repression of HVM stem cell genes to occur transiently, thereby ensuring such programs can be reactivated later in development.
An LSD1-SNAG-dependent negative feedback loop would represent an ideal mechanism for controlling oscillating gene expression programs in HVM cells. Indeed, it is known that SNAG family proteins negatively regulate their own gene expression, as well as expression of other members of their gene family, by unknown mechanisms.33,34,35,36,37,38,39 In lsd1-mutant zebrafish, we found that Lsd1 negatively regulates GFI-family genes in the ICM, where primitive hematopoiesis takes place in the developing embryo. We also observed the upregulation of SNAIL-family genes in neural-crest-related tissues in lsd1 zebrafish mutants, suggesting that LSD1 may also regulate SNAIL-mediated processes, such as neural crest development. This latter possibility was suggested in a recent study that showed upregulation of SNAI2 following LSD1 inhibition in epidermal progenitor cells.54 LSD1 is also recruited by SNAIL1 and SNAIL2 to prompt the epithelial-to-mesenchymal transition (EMT), a process critical for both development and cancer progression.49,55 Thus, LSD1 is likely to be intricately linked with diverse biological programs through its interactions with SNAG-family proteins, particularly during embryogenesis. It is likely that the design of LSD1 mutant alleles that can effectively uncouple LSD1 demethylase activity from its substrate binding capacity will be necessary for understanding the role of LSD1 in controlling these feedback loops.
A model is emerging in which LSD1, GFI-family proteins and the BHC/CoREST complex function together to regulate transcriptional programs that drive hematopoiesis. We and others found that the BHC/CoREST complex is critical for GFI-family-mediated regulation of the hematopoietic lineage,5,40 and the GFI-LSD1 interaction is required for AML cell survival.6,7,8,9 In this study, we found that the LSD1 tower domain, which is important for binding to RCOR1 (a member of the BHC/CoREST and CtBP complexes), MTA1 and MTA2 (members of the NuRD complex), and SIN3A,24,50,51 is critical for maintaining early hematopoietic programs. Although the BHC/CoREST complex is widely associated with the GFI family and hematopoietic lineage regulation,5,6,7,8,18,40,56,57,58,59,60 links to the NuRD, CtBP, and Sin3A complexes have also been reported.30,61,62,63 Future studies involving the use of LSD1-protein-partner mutants will clarify the complexes that are necessary for LSD1-mediated transcriptional programs that drive primitive hematopoiesis.
LSD1 has become a therapeutic target-of-interest because of its elevated expression in malignancies such as neuroblastoma, small-cell lung cancer, and leukemia.64,65,66 Like-wise, SNAG-domain transcription factors, many whose functions in transcriptional control depend on LSD1, are emerging as critical determinants of malignant behavior. For example, GFI1 has pro-survival functions in both myeloid and lymphoid leukemias, in medulloblastoma and in the pre-malignant condition, severe congenital neutropenia.67,68,69,70 Similarly, SNAIL family members figure prominently in the development and progression of breast cancer.71,72 Inhibitors of LSD1 could therefore have broad reaching implications for cancer treatment. The first available inhibitors of LSD1 included tranylcypromine derivatives that irreversibly inactivate LSD1 through covalent modification of FAD.73 However, disrupting interactions between LSD1 and its protein partners may be a more effective therapeutic strategy than inhibiting its catalytic activity. Indeed, uncoupling LSD1 from GFI1 or GFI1B alters cellular differentiation in cancer.6,8,9,74,75 A better understanding of the catalysis-dependent and-independent functions of LSD1, the specific multiprotein complexes with which LSD1 interacts, the post-translational modifications of histones and other transcription factors governed by LSD1, the changes brought on by LSD1 inhibition, and the loci that LSD1 regulates will better inform the design of effective therapeutic strategies targeting LSD1 and its SNAG domain transcription factor partners for cancer treatment.
Limitations of the study
LSD1/KDM1A is a multifaceted protein because of its ability to demethylate substrates (histones and transcription factors) and provide a scaffold to recruit various chromatin-modifying complexes. Unraveling the different functions of LSD1 will require methods that uncouple its enzymatic and structural functions for testing in both biochemical and in vivo assays. Previous studies have relied heavily on the use of one point mutation that abolishes LSD1’s demethylase activity on mono- or di-methylated histone peptide substrates in vitro, called the K661A mutation. However, recent studies showed that the K661A mutation still retains 20% activity when reconstituted nucleosomes are used as substrate, limiting our ability to interpret LSD1’s potential non-catalytic functions in prior cell- or animal-based experiments. Indeed, to our knowledge, there are no described LSD1 point mutations that completely uncouple its catalytic activity from its substrate binding capacity. The A539E point mutation used in this study was recently shown to have less than 5% catalytic activity on nucleosomes; however, this is likely because of its inability to effectively bind substrates because the hydrophobic pocket is disrupted by a charged residue. Therefore, based on LSD1 activity toward nucleosome substrates alone, our data could support the conclusion that LSD1 catalytic activity is required for controlling gene expression in vivo, as the K661A allele (20% activity) rescued Lsd1-mediated gene regulation but the A539E allele (5% activity) did not. For this reason, we also analyzed the ability of the D555K point mutation to rescue lsd1 mutants, as D555K is located in the hydrophobic pocket and was reported to exhibit higher catalytic activity (>20%) than K661A on histone peptides,23,24,25 although its activity on reconstituted nucleosomes is not known. The inability of D555K to restore lsd1 mutant phenotypes bolsters the conclusion that the substrate-binding property of LSD1 is more critical than its catalytic activity during primitive hematopoietic development. This conclusion was further strengthened by the use of exogenous SNAG peptides to block wild-type LSD1 function in vivo. However, at this time our data cannot rule out an alternative model in which both substrate binding and a threshold level of LSD1 catalytic activity (between 5 and 20%) is required for control of gene expression programs in vivo. To address this possibility, biochemical assays using nucleosomes as substrates will be required in combination with new LSD1 mutants that can clearly uncouple catalytic activity from substrate binding. Nonetheless, our studies show that LSD1 substrate binding, particularly to SNAG-domain-containing transcription factors, is a prerequisite for mediating LSD1’s function in vivo, regardless of whether LSD1 catalytic activity is required.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Caspase 3 | BD Bioscience | RRID: AB_397274; Cat#559565 |
| Donkey Anti-Rabbit Alexa Fluor 568 | Life Technologies | RRID: AB_2534017; Cat#A10042 |
| Anti-Digoxigenin | Sigma Aldrich | RRID: AB_2734716; Cat#11093274910 |
| Anti-Flag M2 | Sigma Aldrich | RRID: AB_259529; Cat#F3165 |
| HRP-Donkey Anti-Mouse IgG | Jackson Immunoresearch | RRID: AB_2340770; Cat#715-035-150 |
| Anti-Phospho-Histone H3 | Cell Signaling Technology | RRID: AB_331535; Cat#9701 |
| Chemicals, peptides, and recombinant proteins | ||
| SNAG peptide | GenScript | N/A |
| Proteinase K | Sigma Aldrich | Cat#RPROTK-RO |
| Ambion SP6 RNA Polymerase | Invitrogen | Cat#AM2071 |
| Ambion T7 RNA Polymerase | Invitrogen | Cat#AM2082 |
| Ribonucleic Acid Type VI | Sigma Aldrich | Cat#R6625 |
| Heparin | Sigma Aldrich | Cat#H3393 |
| Maleic Acid | Sigma Aldrich | Cat#M0375 |
| Blocking Reagent | Roche | Cat#11096176001 |
| Fetal Bovine Serum, Heat Inactivated | Gibco | Cat#10438026 |
| RNAlater | Qiagen | Cat#76104 |
| Phusion High-Fidelity DNA Polymerase | NEB | Cat#M0530S |
| Protease Inhibitor Cocktail | Sigma Aldrich | Cat#P8340 |
| gfi1b sgRNA | This paper | N/A |
| Critical commercial assays | ||
| OneStep RT-PCR | Qiagen | Cat#210210 |
| QuikChange II Site-Directed Mutagenesis Kit | Agilent | Cat#200523 |
| QIAquick Gel Extraction Kit | Qiagen | Cat#28704 |
| pGEM-T-Easy Vector Systems | Promega | Cat#A1360 |
| LightScanner Master Mix | BioFire Defense | Cat#HRLS-ASY-0003 |
| mMessage mMachine SP6 Transcription Kit | Invitrogen | Cat#AM1340 |
| NucAway Spin Columns | Thermo Fisher | Cat#AM10070 |
| DIG RNA Labeling Mix | Sigma Aldrich | Cat#11277073910 |
| BCIP/NBT Substrate Kit | Vector Laboratories | Cat#SK5400 |
| PureLink RNA MicroKit | Invitrogen | Cat#12183016 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher | Cat#34095 |
| Cas9 | IDT | Cat#1081058 |
| HiScribe T7 Quick High Yield RNA Synthesis RNA Kit | NEB | Cat#E2050S |
| RNeasy Plus Mini Kit | Qiagen | Cat#74134 |
| Protease inhibitor cocktail | Sigma Aldrich | Cat#P8340 |
| PVDF membrane | Millipore | Cat#IPVH00010 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher | Cat#34095 |
| Autoradiography film | Genesee Scientific | Cat#30-507 |
| Deposited data | ||
| Raw data | This paper | GEO: GSE198476 |
| Experimental models: Organisms/strains | ||
| Zebrafish: kdm1a 23095 | ZFIN: ZDB-ALT-131217-16908 | |
| Zebrafish: kdm1a 36433 | ZFIN: ZDB-ALT-161003-15414 | |
| Zebrafish: gfi1aa 16850 | ZFIN: ZDB-ALT-131217-13025 | |
| Zebrafish: gfi1ab 40706 | ZFIN: ZDB-ALT-160601-6870 | |
| Zebrafish: plrg1 hi3174aTg | ZFIN: ZDB-ALT-040826-6 | |
| Zebrafish: gfi1b Δ17 | This paper | N/A |
| Oligonucleotides | ||
| Primers for genotyping, see Table S2 | This paper | N/A |
| Primers for cDNA amplification, see Table S2 | This paper | N/A |
| Primers for site directed mutagenesis, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pUC57-zlsd1-3XFLAG | This paper | N/A |
| pUC57-zlsd1-K661A | This paper | N/A |
| pUC57-zlsd1-ΔQ632-3XFLAG | This paper | N/A |
| pUC57-zlsd1-D555K-3XFLAG | This paper | N/A |
| pUC57-zlsd1-A539E-3XFLAG | This paper | N/A |
| pUC57-zlsd1-ΔTower-3XFLAG | This paper | N/A |
| pUC57-zlsd1-ΔAOLC | This paper | N/A |
| pUC57-hLsd1-3XFLAG | This paper | N/A |
| pUC57-hLsd1-ΔAOLC | This paper | N/A |
| pGEM-T-Easy-gfi1b | This paper | N/A |
| pGEM-T-Easy-gfi1aa | This paper | N/A |
| pGEM-T-Easy-gfi1ab | This paper | N/A |
| pGEM-T-Easy-fli1a | This paper | N/A |
| pBluescript-gata1 | Detrich et al.76 | N/A |
| pUC57-etv2 | Sumanas et al.77 | N/A |
| pBK-snail1a | Thisse et al.78 | N/A |
| pBK-snail1b | Thisse et al.79 | N/A |
| pBS-IISK-snai2 | Thisse et al.80 | ZFIN: ZDB-EST-030328-40 |
| Software and algorithms | ||
| ImageJ | ||
| LightScanner Call-IT | Idaho Technology Inc. | |
| Adobe Illustrator | Adobe | |
| Adobe Photoshop | Adobe | |
| Integrated Genomics Viewer | Broad Institute | |
| RStudio | RStudio | |
| Pymol | Schrodinger Inc. | |
| GraphPad Prism | Dotmatics | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to Rodney Stewart (rodney.stewart@utah.edu).
Materials availability
Plasmids generated in this study are available upon request from the lead contact.
Experimental model and subject details
Ethics statement
All experiments involving zebrafish conformed to the regulatory standards and guidelines of the University of Utah Institutional Animal Care and Use Committee.
Zebrafish husbandry
Zebrafish were bred and maintained as described.81 The kdm1a(23095), kdm1a(36433), gfi1aa(16850), gfi1ab(40706), and plrg1(hi3174aTg) lines were obtained from the Zebrafish International Resource Center (http://www.zebrafish.org/home/guide.php). The gfi1b(Δ17) line was generated as described below.
Method details
Molecular biology and cloning
The entire cDNA of zlsd1 (GRCz11 transcript 201) and hLSD1 (GRCh38 transcript 201) was synthesized by Genewiz into the pUC57 backbone with flanking SP6 and T7 promoter sequences. pUC57-etv2 was synthesized from Genewiz as described by Sumanas et al.77 Site-directed mutagenesis (Agilent 200523) was performed on pUC57-zlsd1 to generate the K661A, ΔQ632, D555K, and A539E mutations. The reaction was carried out for 5 min at 95°C, 16 cycles of 30 s at 95°C, 30 s at 55°C, 12 min at 68°C, ending with 15 min at 68°C and cooling to 4°C. An annealing temperature of 62°C was used to generate the A539E mutation. Mutagenesis was confirmed by Sanger sequencing. pUC57-zlsd1 was used as a template to generate zlsd1-ΔTower using Phusion DNA Polymerase (NEB M0530S). The reaction was carried out for 30 s at 98°C, 35 cycles of 10 s at 98°C, 30 s at 68.6°C, 45 s at 72°C, ending with 10 min at 72°C and cooling to 4°C. Deletion from Gln399 to Ser502 was confirmed by BsaI digest and Sanger sequencing.
The zebrafish gfi1aa, gfi1ab, gfi1b, and fli1a cDNAs were amplified from 24-hpf zebrafish RNA using the One-Step RT-PCR kit (Qiagen 210,210). The reaction was carried out for 30 min at 50°C, 15 min at 95°C, 35 cycles of 30 s at 94°C, 30 s at 55°C (gfi1aa, gfi1ab, gfi1b) or 60°C (fli1a), 80 s at 72°C, ending with 10 min at 72°C and cooling to 4°C. The amplicon was purified via gel extraction (Qiagen 28,704) and ligated to pGEM via the pGEM-T-Easy Vector System (Fisher Scientific PRA1360). To determine the orientation of each cDNA, pGEM-gfi1aa and pGEM-gfi1ab were digested with SacI, pGEM-gfi1b was digested with NdeI, and pGEM-fli1a was digested with AccI. Sanger sequencing was performed to validate results.
Genomic DNA extraction and high-melt-resolution analysis (HRMA)
Genomic DNA was extracted from adult zebrafish by fin clipping. Fins were lysed in alkaline lysis solution (25 mM NaOH, 0.2 mM EDTA) at 95°C for 2 h and the solution was neutralized with 40 mM Tris pH 5.0. Isolation of genomic DNA from embryo heads or whole embryos following WISH was modified from Draper et al.82 Embryos were placed in HRMA lysis buffer (20 mM Tris pH 8.0, 50 mM KCl, 0.3% Tween 20, 0.3% NP-40, 0.5 mg/mL proteinase K (Sigma Aldrich RPROTK-RO)), incubated at 95°C for 20 min, 55°C for 75 min, and ending with 20 min at 95°C.
Genotyping was performed by HRMA to amplify a 96-bp and 86-bp product for kdm1a(23095) and kdm1a(36433), respectively. Similarly, HRMA was used to amplify a 91-bp product for gfi1aa(16850), a 98-bp product for gfi1ab(40706), and a 106-bp product for gfi1b(Δ17). Briefly, 20 μL of mineral oil was added to each well of a 96-well plate (BioRad HSP9665). Each reaction contained 2 μL of LightScanner master mix (Idaho Technologies HRLS-ASY-0003), 0.5 μM of each primer, and 1 μL of genomic DNA. The plate was covered with an optically transparent seal, centrifuged at 1800 rcf for 2 min, and cycled with the following conditions: denaturation at 96°C for 5 min; 55 cycles of 30 s at 96°C, 30 s at 68°C, 30 s at 72°C, ending with 3 min at 95°C and cooled to 4°C. Genotyping data were collected on a LightScanner (Idaho Technology) and analyzed with the LightScanner Call-IT Software as previously described.83,84 Homozygous kdm1a mutants were distinguished from wild-type animals, as described by Boer et al.85 All samples that grouped with known wild-type samples were mixed with 1–4 μL of wild-type reaction, heated to 95°C for 5 min, and reanalyzed on the LightScanner. HRMA genotyping results were validated by standard polymerase chain reaction (PCR) of genomic DNA and Sanger sequencing.
Microinjection
To generate capped sense mRNA, the zlsd1-ΔAOLC mRNA and the hLSD1-ΔAOLC plasmids were digested with EcoRV. The pUC57-zlsd1 and pUC57-hLSD1 plasmids were digested with StuI. All other constructs were linearized with NotI. Linearized plasmids (400 ng) were in vitro transcribed using the SP6 mMESSAGE mMACHINE kit (Life Technologies AM1340). mRNA was purified using NucAway spin columns (Life Technologies AM10070) and microinjected into the single cell of zebrafish embryos at a dosage of 50–65 ng.
SNAG domain peptides were synthesized by GenScript to >85% purity. Peptides were reconstituted according to manufacturer’s instructions in water at a stock concentration of 1 mg/mL. Equal concentrations of peptide and zlsd1-3XFLAG mRNA were microinjected into the single cell of zebrafish embryos at a dosage of 50–65 ng.
Whole-mount in situ hybridization (WISH)
To generate antisense RNA probes, pGEM-fli1a, pGEM-gfi1b, pGEM-gfi1aa,pGEM-gfi1ab were digested with SacII. pBluescript-gata1 was digested with XbaI,76 pUC57-etv2 with KpnI, pBK-snail1a with BamHI,78snail1b with XbaI,79 and pUC57-snai2 with NotI.80 A total of 2 ug of digested plasmid was in-vitro transcribed with either SP6 (Invitrogen AM2071) (fli1a, gfi1b, gfi1aa, gfi1ab, etv2) or T7 (Invitrogen AM2082) (gata1, snail1a, snail1b, snai2) polymerase and labeled with digoxigenin (Sigma Aldrich 11277073910). Probes were purified on NucAway spin columns (Life Technologies AM10070).
WISH was performed in either biological duplicates or triplicates, as previously described.78 Briefly, embryos were grown to the desired stage, dechorionated, and fixed overnight in 4% paraformaldehyde at 4°C. The following day embryos were rinsed three times in PBST (1X PBS with 0.1% Tween 20) and dehydrated overnight at −20°C in methanol. To rehydrate, embryos were washed three times in PBST and placed in prewarmed HYB(−) solution (50% formamide, 5X SSC, 0.1% Tween 20) for 15 min at 68-70°C while gently rocking. The HYB(−) solution was replaced with HYB(+) solution (HYB(−), 5 mg/mL Torula RNA Type VI (Sigma Aldrich R6625), 50 ug/ml heparin (Sigma Aldrich H3393)) and rocked for 1 h at 68-70°C. The RNA probe was added and hybridization was performed overnight at 68-70°C. Embryos were washed at 68-70°C in 2X SSCT (2X SSC, 0.1% Tween 20) with 50% formamide two times for 30 min each, one time for 15 min in 2X SSCT, and two times for 30 min each in 0.2X SSCT. Embryos were transferred to room temperature and washed three times for 5 min each in 1X MABT pH 7.5 (100 mM maleic acid (Sigma Aldrich M0375), 150 mM NaCl, 100 mM Tris pH 9.5, 0.1% Tween 20) and subsequently placed in blocking solution (MABT, 2% BMB blocking reagent (Sigma Aldrich 11096176001), 10% heat-inactivated fetal bovine serum (Fisher Scientific 10438026) for 1 h. Anti-digoxygenin FAB fragment antibody (Sigma Aldrich 11093274910) was added at a 1:5000 final dilution and rocked overnight at 4°C. The next day embryos were washed at room temperature in blocking solution for 30 min, 1X MABT for 1 h, 1X MABT for 30 min, and 0.1M Tris pH 9.5 three times for 5 min each. BCIP/NBT substrate (Vector Laboratories SK-5400) was added, and embryos were covered with aluminum foil and rocked at room temperature until stained. Following staining, embryos were rinsed in PBST two times for 5 min each to stop the reaction, gradually transferred to 80% glycerol and imaged. Embryos older than 30 hpf were initially treated with 10 μg/mL of proteinase K, rinsed three times with PBST and fixed again in 4% paraformaldehyde overnight at 4°C before beginning the dehydration step.
RNA sequencing
Embryos from a heterozygous incross of kdm1a(23095) or kdm1a(36433) were dissected at 24 hpf just posterior to the yolk sac. The head was used for genomic DNA extraction and HRMA while the tail was placed in RNA stabilization solution (QIAGEN 76104) until genotyping was completed. Three independent clutches were collected and five tails from homozygous lsd1 mutants or wild-type siblings were combined per clutch. Tails were rinsed in 1X PBS and pelleted. Tissue homogenization and RNA isolation was performed using the PureLink RNA Micro kit (Invitrogen 12183016) according to manufacturer’s instructions with the following exceptions. Tails were only homogenized with an 18.5-gauge needle 10 times before proceeding directly to the binding, washing and elution steps. Carrier RNA was not utilized.
RNA quality was assessed using the Agilent ScreenTape Assay (Agilent 5067-5579 and 5067-5580) and the Illumina TruSeq Stranded Total RNA kit with Ribo-Zero Gold (Illumina 20020598) was used for library preparation. Sequencing libraries were chemically denatured and applied to an Illumina NovaSeq flow cells using the NovaSeq XP chemistry workflow (Illumina 20021665). The flow cell was transferred to an Illumina NovaSeq instrument and a 2 x 150 cycle paired-end sequence run with 100 M reads was performed using a NovaSeq S2 reagent kit (Illumina 20012860). DESeq2 was used to perform differential gene expression analysis.86 A heatmap of the top 40 differentially regulated genes was generated using RStudio. RNA sequences were visualized using integrated genomics viewer (IGV version 2.4.9).87
Whole-mount antibody staining
Immunofluorescence for activated Caspase-3 and phospho-histone H3 was performed as described.88 Briefly, dechorionated, fixed and dehydrated 24-hpf embryos were washed at room temperature in 1X PDT (1X PBS, 0.1% Tween 20, 0.3% Triton X-, 1% DMSO) two times for 30 min and then transferred to blocking buffer (1X PBS, 0.1% Tween 20, 10% heat-inactivated fetal bovine serum, 2% BSA) for 1 h. Anti-activated Caspase 3 antibody (BD Biosciences 559,565) or anti-phospho-histone H3 antibody (Cell Signaling Technology 9701) was added at a 1:500 or 1:200 dilution for 2 h at room temperature, respectively. Embryos were washed in 1X PDT overnight at 4°C and transferred to blocking buffer the next day for 1 h at room temperature. Secondary anti-rabbit Alexa 568 antibody (Life Technologies A10042) was added at a 1:200 dilution for 2 h at room temperature; embryos were washed in 1X PDT overnight at 4°C and imaged the following day. Mutant plrg1(hi3174aTg) embryos display severe developmental cell death and were used as a positive control for the activated Caspase-3 assay.89
Protein lysates and western blotting
Wild-type embryos were microinjected with 3XFLAG-tagged mRNA constructs as described above. Ten embryos per condition were pooled at 24 hpf in 20 μL of RIPA buffer (150 mM NaCl, 50 mM Tris pH 8.0, 0.5% Deoxycholate, 1% NP-40, 0.1% SDS) supplemented with 0.7 mM PMSF and 1:100 of protease inhibitor cocktail (Sigma Aldrich P8340). Samples were homogenized and denatured at 95°C with 10 μL of 2X LSB/DTT (0.125 M Tris pH 6.8, 4% SDS, 20% glycerol, 0.005% bromophenol blue, 20 mM DTT). Samples were separated by SDS-PAGE, transferred to PVDF membranes (Millipore IPVH00010) using transfer buffer (1X Tris-Glycine buffer, 20% methanol, 0.1% SDS) for 1.5 h at 4°C. Membranes were blocked with 5% milk in PBST (1X PBS, 0.05% Tween 20) for 1 h at room temperature. Anti-Flag M2 (Sigma Aldrich F3165) primary antibody was added at 1:1000 and incubated overnight at 4°C. Membranes were washed 3X for 5 min each with PBST and incubated with anti-mouse-HRP (Jackson Immunoresearch 715-035-150) secondary antibody at 1:10,000 for 1 h at room temperature. Membranes were washed 3X for 5 min each with PBST and proteins were visualized by chemiluminescence detection (Thermo Fisher 34,095) using autoradiography film (Genesee Scientific 30-507).
CRISPR/Cas9
A sgRNA was designed against exon 2 of gfi1b (GRCz10 transcript 201) (agaatcacggaatcatgccacgg). sgRNA was made using the HiScribe T7 Quick High Yield RNA Synthesis RNA kit (NEB E2050S) and purified using the RNeasy Plus Mini kit (Qiagen 74,134) for small RNAs. sgRNA (400 ng/μL) and Cas9 (1000 ng/μL, IDT 1081058) were combined and injected into the single cell of zebrafish embryos. Injected embryos were grown to adults and genotyped to identify individuals with potential gfi1b exon 2 mutations. These adults were outcrossed to wild-type AB animals and progeny examined for transmission of gfi1b mutations. Adult F1 individuals were then outcrossed again to establish an F2 generation containing a deletion of 17 bp in exon 2 (Chr 21: 17295972-17295988) which was confirmed by DNA and RNA sequencing.
Image acquisition and processing
Both brightfield and fluorescent images were acquired on an Olympus SZX16 microscope with an aperture of 0.3 and configured with an Olympus DP74-CU camera. Embryos were imaged in 80% glycerol (WISH) or 4% methylcellulose (whole-mount antibody staining) on a 35-mm glass-bottom dish (Mattek Corporation P35G-1.0 20-C) at 4 to 5× magnification. Olympus cellSens Entry software was used for image acquisition. Adobe Illustrator CC 2017 and Adobe Photoshop CC 2017 were used to generate figures. The brightness/contrast of images was adjusted as indicated in the figure legends using Adobe Photoshop CC 2017.
Statistical analysis
The Wald test was used to determine statistical significance and the Benjamini-Hochberg test for false discovery rate in DESeq2.86 An unpaired t-test with Welch’s correction was performed using GraphPad Prism to determine if the number of phospho-histone H3 cells in the tail were significantly different between lsd1 siblings.
Acknowledgments
We thank Katherine E. Varley for critical reading of this manuscript, the H. Joseph Yost laboratory for reagents, David J. Grunwald for reagents and technical support, David McClellan for sgRNA design assistance, and Chris Stubben for help with RNA sequencing analysis. Research reported in this publication utilized the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at the University of Utah Huntsman Cancer Institute, which is supported by the National Cancer Institute (P30CA042014) as well as the University of Utah Mutation Generation and Detection Core. We thank current and former members of the RAS and MEE laboratories for thoughtful discussions, and the Huntsman Cancer Institute/University of Utah Zebrafish Facility for providing animal husbandry. This work was supported by funding from the National Institutes of Health (R01NS106527, P30CA042014) (RAS), (R01CA201235) (MEE), T32 DK007115 (MJC) as well as the Huntsman Cancer Foundation and the Hyundai Hope on Wheels Foundation.
Author contributions
This study was conceived and designed by M.J.C., R.A.S., and M.E.E. M.J.C. and A.M.C. performed the experiments. M.J.C., A.M.C., A.V.T., and R.A.S. analyzed the data. The manuscript was prepared by M.J.C., C.A.J., and R.A.S. with input from all authors.
Declaration of interests
The authors declare no conflicts of interest.
Published: January 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105737.
Contributor Information
Michael E. Engel, Email: mee2mj@hscmail.mcc.virginia.edu.
Rodney A. Stewart, Email: rodney.stewart@utah.edu.
Supplemental information
Genes were filtered to include those that were protein-coding and with padj<0.05.
Data and code availability
-
•
Raw RNA sequencing data is publicly available and has been deposited at GEO under the accession number GEO: GSE198476. Analyzed RNA sequencing data is available via Table S4.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Shi Y., Lan F., Matson C., Mulligan P., Whetstine J.R., Cole P.A., Casero R.A., Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Metzger E., Wissmann M., Yin N., Müller J.M., Schneider R., Peters A.H.F.M., Günther T., Buettner R., Schüle R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 3.Huang J., Sengupta R., Espejo A.B., Lee M.G., Dorsey J.A., Richter M., Opravil S., Shiekhattar R., Bedford M.T., Jenuwein T., Berger S.L. P53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 4.Kontaki H., Talianidis I. Lysine methylate regulates E2F1-induced cell death. Mol. Cell. 2010;39:152–160. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 5.McClellan D., Casey M.J., Bareyan D., Lucente H., Ours C., Velinder M., Singer J., Lone M.D., Sun W., Coria Y., et al. Growth factor independence 1B-mediated transcriptional repression and lineage allocation require lysine-specific demethylase 1-dependent recruitment of the BHC complex. Mol. Cell Biol. 2019;39:000200–e119. doi: 10.1128/MCB.00020-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maiques-Diaz A., Spencer G.J., Lynch J.T., Ciceri F., Williams E.L., Amaral F.M.R., Wiseman D.H., Harris W.J., Li Y., Sahoo S., et al. Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 2018;22:3641–3659. doi: 10.1016/j.celrep.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vinyard M.E., Su C., Siegenfeld A.P., Waterbury A.L., Freedy A.M., Gosavi P.M., Park Y., Kwan E.E., Senzer B.D., Doench J.G., et al. CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol. 2019;15:529–539. doi: 10.1038/s41589-019-0263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto R., Kawahara M., Ito S., Satoh J., Tatsumi G., Hishizawa M., Suzuki T., Andoh A. Selective dissociation between LSD1 and GFI1B by a LSD1 inhibitior NCD38 induces the activation of ERG super-enhancer in erythroleukemia cells. Oncotarget. 2018;9:21007–21021. doi: 10.18632/oncotarget.24774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barth J., Abou-El-Ardat K., Dalic D., Kurrle N., Maier A.M., Mohr S., Schütte J., Vassen L., Greve G., Schulz-Fincke J., et al. LSD1 inhibition by tranylcypromine derivatives interferes with GFI1-mediated repression of PU.1 target genes and induces differentiation in AML. Leukemia. 2019;33:1411–1426. doi: 10.1038/s41375-018-0375-7. [DOI] [PubMed] [Google Scholar]
- 10.Carnesecchi J., Cerutti C., Vanacker J.M., Forcet C. ERRα protein is stabilized by LSD1 in a demethylation-independent manner. PLoS One. 2017;12:e0188871. doi: 10.1371/journal.pone.0188871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sehrawat A., Gao L., Wang Y., Bankhead A., 3rd, McWeeney S.K., King C.J., Schwartzman J., Urrutia J., Bisson W.H., Coleman D.J., et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA. 2018;115:4179–4188. doi: 10.1073/pnas.1719168115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Jong J.L.O., Zon L.I. Use of the zebrafish system to study primitive and definitive hematopoiesis. Annu. Rev. Genet. 2005;39:481–501. doi: 10.1146/annurev.genet.39.073003.095931. [DOI] [PubMed] [Google Scholar]
- 13.Stachura D.L., Traver D. Cellular dissection of zebrafish hematopoiesis. Methods Cell Biol. 2016;133:11–53. doi: 10.1016/bs.mcb.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 14.Kwan W., North T.E. Netting novel regulators of hematopoiesis and hematologic malignancies in zebrafish. Curr.Top. Dev. Biol. 2017;124:125–160. doi: 10.1016/bs.ctdb.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Maiques-Diaz A., Somervaille T.C. LSD1: biologic roles and therapeutic targeting. Epigenomics. 2016;8:1103–1116. doi: 10.2217/epi-2016-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sprüssel A., Schulte J.H., Weber S., Necke M., Händschke K., Thor T., Pajtler K.W., Schramm A., König K., Diehl L., et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia. 2012;26:2039–2051. doi: 10.1038/leu.2012.157. [DOI] [PubMed] [Google Scholar]
- 17.Kerenyi M.A., Shao Z., Hsu Y.J., Guo G., Luc S., O’Brien K., Fujiwara Y., Peng C., Nguyen M., Orkin S.H. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife. 2013;2:e00633. doi: 10.7554/eLife.00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thambyrajah R., Mazan M., Patel R., Moignard V., Stefanska M., Marinopoulou E., Li Y., Lancrin C., Clapes T., Möröy T., et al. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol. 2016;18:21–32. doi: 10.1038/ncb3276. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi M., Fuse Y., Watanabe M., Andrea C.S., Takeuchi M., Nakajima H., Ohashi K., Kaneko H., Kobayashi-Osaki M., Yamamoto M., Kobayashi M. LSD1/KDM1A promotes hematopoietic commitment of hemangioblasts through downregulation of Etv2. Proc. Natl. Acad. Sci. USA. 2015;112:13922–13927. doi: 10.1073/pnas.1517326112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamaoki J., Takeuchi M., Abe R., Kaneko H., Wada T., Hino S., Nakao M., Furukawa Y., Kobayashi M. Splicing- and demethylase-independent functions of LSD1 in zebrafish primitive hematopoiesis. Sci. Rep. 2020;10:8521. doi: 10.1038/s41598-020-65428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J., Scully K., Zhu X., Cai L., Zhang J., Prefontaine G.G., Krones A., Ohgi K.A., Zhu P., Garcia-Bassets I., et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–887. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 22.Wang J., Hevi S., Kurash J.K., Lei H., Gay F., Bajko J., Su H., Sun W., Chang H., Xu G., et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 23.Kim S.A., Zhu J., Yennawar N., Eek P., Tan S. Crystal structure of the LSD1/CoREST histone demethylase bound to its nucleosome substrate. Mol. Cell. 2020;78:903–914.e4. doi: 10.1016/j.molcel.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y., Yang Y., Wang F., Wan K., Yamane K., Zhang Y., Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc. Natl. Acad. Sci. USA. 2006;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stavropoulos P., Blobel G., Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat. Struct. Mol. Biol. 2006;13:626–632. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- 26.Lin Y., Wu Y., Li J., Dong C., Ye X., Chi Y.I., Evers B.M., Zhou B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010;29:1803–1816. doi: 10.1038/emboj.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baron R., Binda C., Tortorici M., McCammon J.A., Mattevi A. Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure. 2011;19:212–220. doi: 10.1016/j.str.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beauchemin H., Möröy T. Multifaceted actions of GFI1 and GFI1B in hematopoietic stem cell self-renewal and lineage commitment. Front. Genet. 2020;11:591099. doi: 10.3389/fgene.2020.591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nieto M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002;3:155–166. doi: 10.1038/nrm757. [DOI] [PubMed] [Google Scholar]
- 30.Shooshtarizadeh P., Helness A., Vadnais C., Brouwer N., Beauchemin H., Chen R., Bagci H., Staal F.J.T., Coté J.F., Möröy T. Gfi1b regulates the level of Wnt/β-catenin signaling in hematopoietic stem cells and megakaryocytes. Nat. Commun. 2019;10:1270. doi: 10.1038/s41467-019-09273-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Möröy T., Vassen L., Wilkes B., Khandanpour C. From cytopenia to leukemia: the role of Gfi1 and Gfi1b in blood formation. Blood. 2015;126:2561–2569. doi: 10.1182/blood-2015-06-655043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Meer L.T., Jansen J.H., van der Reijden B.A. Gfi1 and Gfi1b: key regulators of hematopoiesis. Leukemia. 2010;24:1834–1843. doi: 10.1038/leu.2010.195. [DOI] [PubMed] [Google Scholar]
- 33.Doan L.L., Porter S.D., Duan Z., Flubacher M.M., Montoya D., Tsichlis P.N., Horwitz M., Gilks C.B., Grimes H.L. Targeted transcriptional repression of Gfi1 by GFI1 and GFI1B in lymphoid cells. Nucleic Acids Res. 2004;32:2508–2519. doi: 10.1093/nar/gkh570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yücel R., Kosan C., Heyd F., Möröy T. Gfi1:Green fluorescent protein knock-in mutant reveals different expression and autoregulation of the growth factor independence 1 (Gfi1) gene during lymphocyte development. J. Biol. Chem. 2004;279:40906–40917. doi: 10.1074/jbc.M400808200. [DOI] [PubMed] [Google Scholar]
- 35.Huang D.Y., Kuo Y.Y., Chang Z.F. GATA-1 mediates auto-regulation of Gfi-1B transcription in K562 cells. Nucleic Acids Res. 2005;33:5331–5342. doi: 10.1093/nar/gki838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vassen L., Fiolka K., Mahlmann S., Möröy T. Direct transcriptional repression of genes encoding the zinc-finger proteins Gfi1b and Gfi1 by Gfi1b. Nucleic Acids Res. 2005;33:987–998. doi: 10.1093/nar/gki243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anguita E., Villegas A., Iborra F., Hernández A. GFI1B controls its own expression binding to multiple sites. Haematologica. 2010;95:36–46. doi: 10.3324/haematol.2009.012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boettiger A.N., Levine M. Rapid transcription kinetics foster coordinate snail expression in the Drosophila embryo. Cell Rep. 2013;3:8–15. doi: 10.1016/j.celrep.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sundararajan V., Tan M., Tan T.Z., Ye J., Thiery J.P., Huang R.Y.J. SNAI1 recruits HDAC1 to suppress SNAI2 transcription during epithelial to mesenchymal transition. Sci. Rep. 2019;9:8295. doi: 10.1038/s41598-019-44826-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saleque S., Kim J., Rooke H.M., Orkin S.H. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell. 2007;27:562–572. doi: 10.1016/j.molcel.2007.06.039. [DOI] [PubMed] [Google Scholar]
- 41.Busch-Nentwich E., Kettleborough R., Dooley C.M., Scahill C., Sealy I., White R., Herd C., Mehroke S., Wali N., Carruthers S., et al. ZFIN Direct Data Submission; 2013. Sanger Institute Zebrafish Mutation Project Mutant Data Submission. [Google Scholar]
- 42.Grimes H.L., Chan T.O., Zweidler-McKay P.A., Tong B., Tsichlis P.N. The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol. Cell Biol. 1996;16:6263–6272. doi: 10.1128/mcb.16.11.6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooney J.D., Hildick-Smith G.J., Shafizadeh E., McBride P.F., Carroll K.J., Anderson H., Shaw G.C., Tamplin O.J., Branco D.S., Dalton A.J., et al. Teleost growth factor independence (gfi) genes differentially regulate successive waves of hematopoiesis. Dev. Biol. 2013;373:431–441. doi: 10.1016/j.ydbio.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dufourcq P., Rastegar S., Strähle U., Blader P. Parapineal specific expression of gfi1 in the zebrafish epithalamus. Gene Expr. Patterns. 2004;4:53–57. doi: 10.1016/s1567-133x(03)00148-0. [DOI] [PubMed] [Google Scholar]
- 45.Wu M., Chen Q., Li J., Xu Y., Lian J., Liu Y., Meng P., Zhang Y. Gfi1aa/Lsd1 facilitates hemangioblast differentiation into primitive erythrocytes by targeting etv2 and sox7 in zebrafish. Front. Cell Dev. Biol. 2021;9:786426. doi: 10.3389/fcell.2021.786426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fraaije M.W., Mattevi A. Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem. Sci. 2000;25:126–132. doi: 10.1016/s0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- 47.Forneris F., Binda C., Vanoni M.A., Mattevi A., Battaglioli E. Histone demethylation catalyzed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005;579:2203–2207. doi: 10.1016/j.febslet.2005.03.0. [DOI] [PubMed] [Google Scholar]
- 48.Velinder M., Singer J., Bareyan D., Meznarich J., Tracy C.M., Fulcher J.M., McClellan D., Lucente H., Franklin S., Sharma S., Engel M.E. GFI1 functions in transcriptional control and cell fate determination require SNAG domain methylation to recruit LSD1. Biochem. J. 2016;473:3355–3369. doi: 10.1042/BCJ20160558. [DOI] [PubMed] [Google Scholar]
- 49.Ferrari-Amorotti G., Fragliasso V., Esteki R., Prudente Z., Soliera A.R., Cattelani S., Manzotti G., Grisendi G., Dominici M., Pieraccioli M., et al. Inhibiting interactions of lysine demethylase LSD1 with Snail/Slug blocks cancer cell invasion. Cancer Res. 2013;73:235–245. doi: 10.1158/0008-5472.CAN-12-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y., Zhang H., Chen Y., Sun Y., Yang F., Yu W., Liang J., Sun L., Yang X., Shi L., et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell. 2009;138:660–672. doi: 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 51.Yang Y., Huang W., Qiu R., Liu R., Zeng Y., Gao J., Zheng Y., Hou Y., Wang S., Yu W., et al. LSD1 coordinates with the SIN3A/HDAC complex and maintains sensitivity to chemotherapy in breast cancer. J. Mol. Cell Biol. 2018;10:285–301. doi: 10.1093/jmcb/mjy021. [DOI] [PubMed] [Google Scholar]
- 52.Moore C., Richens J.L., Hough Y., Ucanok D., Malla S., Sang F., Chen Y., Elworthy S., Wilkinson R.N., Gering M. Gfi1aa and gfi1b set the pace for primitive erythroblast differentiation from hemangioblasts in the zebrafish embryo. Blood Adv. 2018;2:2589–2606. doi: 10.1182/bloodadvances.2018020156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thambyrajah R., Ucanok D., Jalali M., Hough Y., Wilkinson R.N., McMahon K., Moore C., Gering M. A gene trap transposon eliminates hematopoietic expression of zebrafish Gfi1aa, but does not interfere with hematopoiesis. Dev. Biol. 2016;417:25–39. doi: 10.1016/j.ydbio.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Egolf S., Aubert Y., Doepner M., Anderson A., Maldonado-Lopez A., Pacella G., Lee J., Ko E.K., Zou J., Lan Y., et al. LSD1 inhibition promotes epithelial differentiation through derepression of fate-determining transcription factors. Cell Rep. 2019;28:1981–1992.e7. doi: 10.1016/j.celrep.2019.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin T., Ponn A., Hu X., Law B.K., Lu J. Requirement of the histone demethylase LSD1 in Snail1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene. 2010;29:4896–4904. doi: 10.1038/onc.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laurent B., Randrianarison-Huetz V., Frisan E., Andrieu-Soler C., Soler E., Fontenay M., Dusanter-Fourt I., Duménil D. A short GFI-1B isoform controls erythroid differentiation by recruiting the LSD1-CoREST complex through the dimethylationn of its SNAG domain. J. Cell Sci. 2012;125:993–1002. doi: 10.1242/jcs.095877. [DOI] [PubMed] [Google Scholar]
- 57.Laurent B., Randrianarison-Huetz V., Kadri Z., Roméo P.H., Porteu F., Duménil D. Gfi-1B promoter remains associated with active chromatin marks throughout erythroid differentiation of human primary progenitor cells. Stem Cell. 2009;27:2153–2162. doi: 10.1002/stem.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tatsumi G., Kawahara M., Yamamoto R., Hishizawa M., Kito K., Suzuki T., Takaori-Kondo A., Andoh A. LSD1-mediated repression of GFI1 super-enhancer plays an essential role in erythroleukemia. Leukemia. 2020;34:746–758. doi: 10.1038/s41375-019-0614-6. [DOI] [PubMed] [Google Scholar]
- 59.Chowdhury A.H., Ramroop J.R., Upadhyay G., Sengupta A., Andrzejczyk A., Saleque S. Differential transcriptional regulation of meis1 by Gfi1b and its co-factors LSD1 and CoREST. PLoS One. 2013;8:e53666. doi: 10.1371/journal.pone.0053666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andrade D., Velinder M., Singer J., Maese L., Bareyan D., Nguyen H., Chandrasekharan M.B., Lucente H., McClellan D., Jones D., et al. SUMOylation regulates growth factor independence 1 in transcriptional control and hematopoiesis. Mol. Cell Biol. 2016;36:1438–1450. doi: 10.1128/MCB.01001-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ross J., Mavoungou L., Bresnick E.H., Milot E. GATA-1 utilizes Ikaros and polycomb repressive complex 2 to suppress Hes1 and to promote erythropoiesis. Mol. Cell Biol. 2012;32:3624–3638. doi: 10.1128/MCB.00163-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hamlett I., Draper J., Strouboulis J., Iborra F., Porcher C., Vyas P. Characterization of megakaryocyte GATA1-interacting proteins: the corepressor ETO2 and GATA1 interact to regulate terminal megakaryocyte maturation. Blood. 2008;112:2738–2749. doi: 10.1182/blood-2008-03-146605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang X., Hu C., Arnovitz S., Bugno J., Yu M., Zuo Z., Chen P., Huang H., Ulrich B., Gurbuxani S., et al. miR-22 has a potent anti-tumour role with therapeutic potential inn acute myeloid leukaemia. Nat. Commun. 2016;7:11452. doi: 10.1038/ncomms11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schulte J.H., Lim S., Schramm A., Friedrichs N., Koster J., Versteeg R., Ora I., Pajtler K., Klein-Hitpass L., Kuhfittig-Kulle S., et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 65.Lv T., Yuan D., Miao X., Lv Y., Zhan P., Shen X., Song Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One. 2012;7:e35065. doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harris W.J., Huang X., Lynch J.T., Spencer G.J., Hitchin J.R., Li Y., Ciceri F., Blaser J.G., Greystoke B.F., Jordan A.M., et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 67.Khandanpour C., Thiede C., Valk P.J.M., Sharif-Askari E., Nückel H., Lohmann D., Horsthemke B., Siffert W., Neubauer A., Grzeschik K.H., et al. A variant allele of growth factor independence 1 (GFI1) is associated with acute myeloid leukemia. Blood. 2010;115:2462–2472. doi: 10.1182/blood-2009-08-239822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khandanpour C., Phelan J.D., Vassen L., Schütte J., Chen R., Horman S.R., Gaudreau M.C., Krongold J., Zhu J., Paul W.E., et al. Growth factor independence 1 antagonizes a p53-induced DNA damage response pathway in lymphoblastic leukemia. Cancer Cell. 2013;23:200–214. doi: 10.1016/j.ccr.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Northcott P.A., Lee C., Zichner T., Stütz A.M., Erkek S., Kawauchi D., Shih D.J.H., Hovestadt V., Zapatka M., Sturm D., et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428–434. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Person R.E., Li F.Q., Duan Z., Benson K.F., Wechsler J., Papadaki H.A., Eliopoulos G., Kaufman C., Bertolone S.J., Nakamoto B., et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat. Genet. 2003;34:308–312. doi: 10.1038/ng1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujita N., Jaye D.L., Kajita M., Geigerman C., Moreno C.S., Wade P.A. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207–219. doi: 10.1016/s0092-8674(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 72.Côme C., Magnino F., Bibeau F., De Santa Barbara P., Becker K.F., Theillet C., Savagner P. Snail and slug play distinct roles during breast carcinoma progression. Clin. Cancer Res. 2006;12:5395–5402. doi: 10.1158/1078-0432.CCR-06-0478. [DOI] [PubMed] [Google Scholar]
- 73.Mould D.P., McGonagle A.E., Wiseman D.H., Williams E.L., Jordan A.M. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date. Med. Res. Rev. 2015;35:586–618. doi: 10.1002/med.21334. [DOI] [PubMed] [Google Scholar]
- 74.Fiskus W., Sharma S., Shah B., Portier B.P., Devaraj S.G.T., Liu K., Iyer S.P., Bearss D., Bhalla K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia. 2014;28:2155–2164. doi: 10.1038/leu.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun W., Guo J., McClellan D., Poeschla A., Bareyan D., Casey M.J., Cairns B.R., Tantin D., Engel M.E. GFI1 cooperates with IKZF1/IKAROS to activate gene expression in T-cell acute lymphoblastic leukemia. Mol. Cancer Res. 2022;20:501–514. doi: 10.1158/1541-7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Detrich H.W., Kieran M.W., Chan F.Y., Barone L.M., Yee K., Rundstadler J.A., Pratt S., Ransom D., Zon L.I. Intraembryonic hematopoietic cell migration during vertebrate development. Proc. Natl. Acad. Sci. USA. 1995;92:10713–10717. doi: 10.1073/pnas.92.23.10713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sumanas S., Jorniak T., Lin S. Identification of novel vascular endothelial-specific genes by the microarray analysis of the zebrafish cloche mutants. Blood. 2005;106:534–541. doi: 10.1182/blood-200401204653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thisse C., Thisse B., Schilling T.F., Postlethwait J.H. Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development. 1993;119:1203–1215. doi: 10.1242/dev.119.4.1203. https://doi:10.1242/dev.119.4.1203. [DOI] [PubMed] [Google Scholar]
- 79.Thisse C., Thisse B., Postlethwait J.H. Expression of snail2, a second member of the zebrafish snail family, in cephalic mesendoderm and presumptive neural crest of wild-type and spadetail mutant embryos. Dev. Biol. 1995;172:86–99. doi: 10.1006/dbio.1995.0007. [DOI] [PubMed] [Google Scholar]
- 80.Thisse B., Pflumio S., Furthauer M., Loppin B., Heyer V., Degrave A., Woehl R., Lux A., Steffan T., Charbonnier X.Q., Thisse C. ZFIN Direct Data Submission; 2001. Expression of the Zebrafish Genome during Embryogenesis. [Google Scholar]
- 81.Westerfield M. University of Oregon Press; 1993. The Zebrafish Book. [Google Scholar]
- 82.Draper B.W., McCallum C.M., Moens C.B. Nanos1 is required to maintain oocyte production in adult zebrafish. Dev. Biol. 2007;305:589–598. doi: 10.1016/j.ydbio.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dahlem T.J., Hoshijima K., Jurynec M.J., Gunther D., Starker C.G., Locke A.S., Weis A.M., Voytas D.F., Grunwald D.J. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012;8:e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xing L., Quist T.S., Stevenson T.J., Dahlem T.J., Bonkowsky J.L. Rapid and efficient zebrafish genotyping using PCR with high-resolution melt analysis. J. Vis. Exp. 2014:e51138. doi: 10.3791/51138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boer E.F., Howell E.D., Schilling T.F., Jette C.A., Stewart R.A. Fascin1-dependent filopodia are required for directional migration of a subset of neural crest cells. PLoS Genet. 2015;11:e1004946. doi: 10.1371/journal.pgen.1004946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sorrells S., Toruno C., Stewart R.A., Jette C. Analysis of apoptosis in zebrafish embryos by whole-mount immunofluorescence to detect activated Caspase 3. J. Vis. Exp. 2013:e51060. doi: 10.3791/51060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Amsterdam A., Nissen R.M., Sun Z., Swindell E.C., Farrington S., Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes were filtered to include those that were protein-coding and with padj<0.05.
Data Availability Statement
-
•
Raw RNA sequencing data is publicly available and has been deposited at GEO under the accession number GEO: GSE198476. Analyzed RNA sequencing data is available via Table S4.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.