

Abstract
To date, the use of immune checkpoint inhibitors has proven largely ineffective in patients with advanced pancreatic ductal adenocarcinoma. A combination of low tumor antigenicity, deficits in immune activation along with an exclusive and suppressive tumor microenvironment result in resistance to host defensives. However, a deepening understanding of these immune escape and suppressive mechanisms has led to the discovery of novel molecular targets and treatment strategies that may hold the key to a long-awaited therapeutic breakthrough. In this review, we describe the tumor-intrinsic and microenvironmental barriers to modern immunotherapy, examine novel immune-based and targeted modalities, summarize relevant pre-clinical findings and human experience, and, finally, discuss novel synergistic approaches to overcome immune-resistance in pancreatic cancer. Beyond checkpoint inhibition, immune agonists and anti-tumor vaccines represent promising strategies to stimulate host response via activation and expansion of anti-tumor immune effectors. Off-the-shelf natural killer cell therapies may offer an effective method for bypassing downregulated tumor antigen presentation. In parallel with this, sophisticated targeting of crosstalk between tumor and tumor-associated immune cells may lead to enhanced immune infiltration and survival of anti-tumor lymphocytes. A future multimodal treatment strategy involving immune priming/activation, tumor microenvironment reprogramming, and immune checkpoint blockade may help transform pancreatic cancer into an immunogenic tumor.
Keywords: Immunotherapy, Pancreatic Cancer, Review, Tumor Microenvironment, Targeted Therapy, Developmental Therapeutics
1. Introduction:
Pancreatic ductal adenocarcinoma (PDAC) has the highest case-fatality rate of any solid tumor, and, is projected to surpass colorectal cancer as the 2nd leading cause of cancer-related mortality nationally in the next 5 years [1,2]. Even for the 15–20% of patients who are eligible for curative resection at diagnosis, their 5-yr overall survival remains discouraging low at 20% with >80% of cases recurring just two years post-surgical intervention [3]. The current mainstay of treatment for advanced PDAC continues to be limited to largely two cytotoxic chemotherapy regimens: 1) fluorouracil, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX); 2) gemcitabine (Gem) and nanoparticle albumin-bound paclitaxel (NP) [4]–6]. While immune checkpoint blockade (ICB), namely inhibitors of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1) T-cell checkpoints, has dramatically changed frontline therapy and survival outcomes in an impressive breadth of solid tumors, it has proven, largely, ineffective in patients with PDAC. With the exception of mismatch repair deficient/microsatellite instability high (dMMR/MSI-H) tumors, which account for <1% of all PDAC tumors, there are currently no approved immunotherapy (IO) regimens for PDAC [7,8]. However, IO remains an area ripe for novel targets and strategies that could lead to a therapeutic breakthroughs in PDAC. In this review, we will describe tumor-intrinsic and microenvironmental barriers to effective IO in PDAC, examine novel IO and targeted modalities, summarize relevant pre-clinical findings and clinical trial experience, and discuss novel synergistic approaches to overcome the intrinsic immune-resistance in PDAC.
2. Contemporary IO and PDAC
2.1. CTLA-4 and PD-[L]1 Checkpoint Inhibition:
Unrivaled in clinical experience and commercial ubiquity, inhibitors of CTLA-4 and PD-[L]1 T-cell checkpoints form the cornerstone of modern immunotherapy. Briefly, the binding of CTLA-4 on activated T-cells by its ligands B7–1|2 prevents CD28 co-stimulation and leads to T-cell anergy/apoptosis [9]. Generally, while CTLA-4 functions during central T-cell priming, PD-1 signaling functions during the T-cell effector phase in the peripheral tissues. The binding of PD-1 on T-cells with its ligand, PD-L1, inhibits T-cell activation, and leads to T-cell exhaustion. Unlike CTLA-4 which is T-cell exclusive, PD-1 is expressed on activated T-cells, B cells, and myeloid cells [9]. Cancer cells & tumor-recruited immune cells overexpress PD-L1 as a means to blunt and escape T-cell mediated immune response [10]. Though the expression of both CTLA4 and PD-L1 are upregulated and associated with poor outcomes in PDAC [11,12], the targeting of these checkpoints has not improved upon outcomes with current standard of care (SOC). In early phase clinical trials, the anti-CTLA-4 antibodies, ipililumab and tremelimumab were ineffective as monotherapies and demonstrated no additive benefit when added to a Gem-backbone in patients with previously treated PDAC [13]–15]. In KEYNOTE-028, objective response rate (ORR) among PDAC patients treated with pembrolizumab monotherapy (aPD-1 mAb) was 0% and median progression-free survival (mPFS0 was just 1.7 months [16]. Durvalulmab (aPD-L1 mAb) also proved ineffective both as a monotherapy (0% ORR) and when combined with CTLA-4 blockade (3.1% ORR and 22% incidence of grade 3+ toxicity) in patients advanced PDAC [17].
2.2. IO-responsive PDAC:
While conventional ICB has disappointed in PDAC, overall, there are rare, but notable exceptions. Patients with mutations in homologous recombination repair genes may lead to increased genomic instability and IO-sensitivity but evidence remains anecdotal. In small case series of patients with refractory mPDAC harboring either germline BRCA or RAD51 mutations, one patient (gBRCA1 mutated) achieved a durable complete response while another (gRAD51C mutated) had an ongoing PR when treated with combination ipililumab and nivolumab [18]. More robust data in ICB-responsive PDAC has been observed in patients with tumors characterized as dMMR/MSI-H. KEYNOTE-158 included 22 patients with dMMR/MSI-H advanced, heavily-pretreated, PDAC who had an observed 18% ORR (1 CR, 3 PR) and a median duration of response (DOR) of 13.2 months with pembrolizumab monotherapy. It should be noted that mPFS and mOS for patients with this rare genotype was still only 2.0 and 4.0 months, respectively. When comparing across dMMR/MSI-H GI primaries, the 18% ORR of PDAC underperforms compared to cholangiocarcinoma (40.9%), small intestine (42.1%), gastric (45.8%), or colorectal adenocarcinomas (53%) [8,19]. While this remains a promising treatment signal, the noticeable disparity in therapeutic response reinforces the need for novel combinatorial strategies to achieve a breakthrough in IO and improve survival in PDAC, regardless of MMR status.
2.3. Intrinsic Immune-Resistance:
An anti-tumor immune response requires tumor recognition, immune cell activation, tumor bed infiltration, and immune-mediated cytolysis (Fig. 1). PDAC has adapted well to evade immune surveillance and suppress immune response at each of these levels: a combination of low tumor antigenicity, deficits in immune activation along with an exclusive and suppressive tumor microenvironment all act as individual roadblocks to effective IO in PDAC (Fig. 2). Strategic targeting of these immunologic barriers may optimize the anti-tumor potential of IO-based therapies in PDAC.
Fig. 1: Adaptive Immune Response in Cancer.
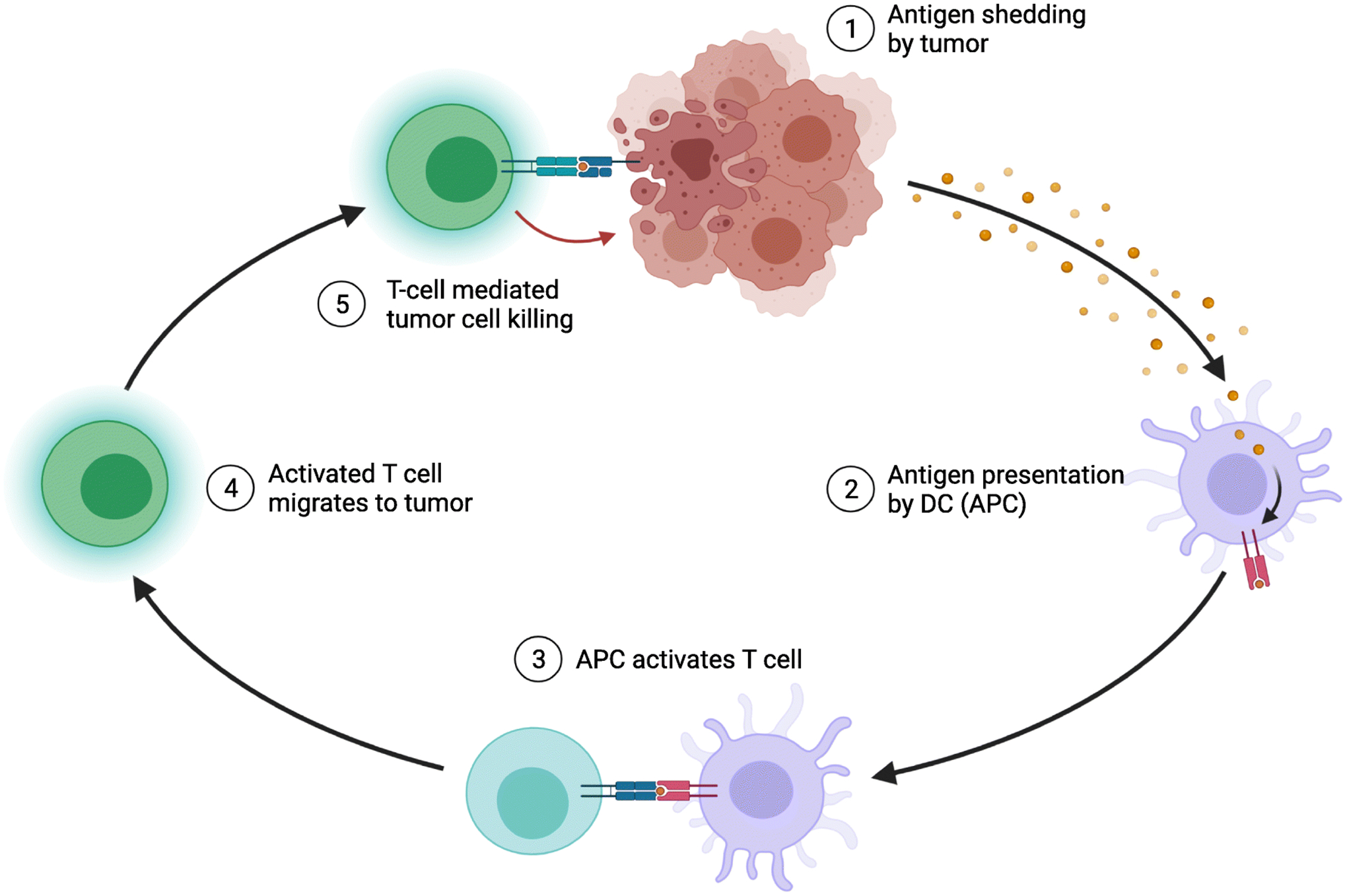
Immunotherapy relies on the ability of host immune cells to recognize cancer as cells that need to be eliminated. For this to happen, a tumor cell needs to alert surveilling antigen presenting cells (APCs), such as dendritic cells (DC), that they are abnormal by expressing antigens on their cell surface. When APCs recognize this abnormal signal, they take up the antigen, traffic to lymph nodes to mature and, in turn, activate cytotoxic and helper T cells with T cell receptors (TCR) specific to that tumor antigen. Once these T cells are activated, they should further differentiate, expand, and migrate to the tumor bed and kill that tumor cell. Adapted from “Antigen Presentation in Cancer”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
Fig. 2: Mechanisms of Immune Resistance in Pancreatic Cancer.
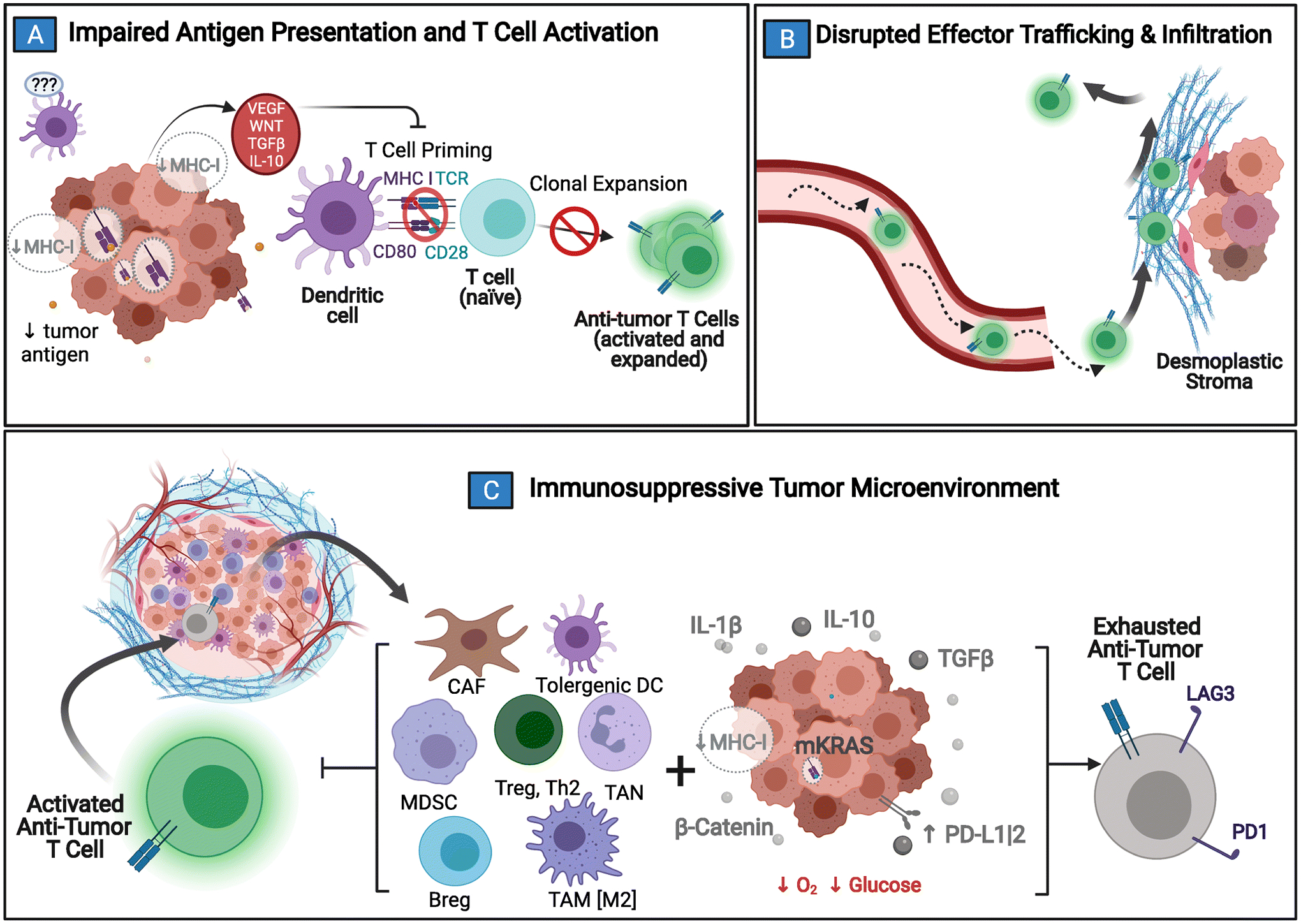
Pancreatic cancer (PDAC) has several protections against the generation and execution of an anti-tumor host-immune response. A) PDAC’s low antigen burden, sequestration of MHC/HLA-I molecules, and local tolerogenic TME signaling all contribute to poor immune surveillance and stunt normal dendritic cell maturation. Without normal antigen processing and presentation, anti-tumor effectors are not activated. B) If activation and clonal expansion of effector T cells still does manage to occur, trafficking to the tumor bed is complicated by disrupted chemokines gradients, abnormal vasculature, and the desmoplastic tumor stroma that acts as both a physical & chemical barrier to entry. C) Anti-tumor immune cells that are able to penetrate the TME are quickly exhausted by local metabolic conditions combined with immunosuppressive molecular crosstalk (e.g. upregulated interleukin 1 beta [IL-1β], transforming growth factor beta [TGFβ], interleukin 10 [IL-10], and beta-catenin [β-catenin]) between tumor and tumor-coopted immune cell populations (cancer-associated fibroblasts [CAF]; tumor associated macrophages [TAM]; tumor associated neutrophils [TAN]; tolerogenic dendritic cells [DC]; myeloid derived suppressor cells [MDSC]; type II T helper cells [Th2]; T regulatory cells [Treg]; B regulatory cells [Breg]). Adapted from “Challenges for CAR T-Cell Immunotherapy in Solid Tumors” and “Tumor Extracellular Matrix Reduces Therapeutic Efficiency in Solid Tumors”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
3. [IO Barrier] Impaired Tumor Recognition and Blunted Host Immune Response:
The process of adaptive immune recognition should be initiated when tumor antigens are processed and displayed onto major histocompatibility complexes (MHC/HLA) by nucleated cells (class I) and antigen presenting cells (class II) [20] Once these antigenic flags are taken up by antigen presenting cells (APCs), such as dendritic cells (DCs), these immunostimulatory cells then traffic to lymph nodes and secondary immune organs to mature and present their antigen-MHC complex to previously-quiescent naïve T-cells [20]. Following T cell receptor (TCR) binding with the antigen-MHC (or antigen-HLA) complex, along with a co-stimulatory signal, the T cell undergoes clonal expansion and differentiation towards effector (CD8+ cytotoxic T lymphocytes [CTL] and CD4+ helper T cells [Th]) and memory functions (central [Tcm], tissue resident [Trm]& effector [Tem] subtypes) [21]. There are three main areas where PDAC hampers this process: 1) antigenic load; 2) tumor cell MHC presentation; 3) APC dysfunction.
3.1. [IO Barrier] Low Tumor Neoantigen Burden:
Although the proteins encoded by cancer genes are intracellular, short peptides (epitopes) derived through proteolytic processing within the tumor cell are presented on the cell surface by MHC (HLA) molecules for recognition as self/foreign antigen by surveilling immune cells. There are three broad categories of tumor antigens:
Tumor-associated antigens (TAA) - normal host proteins that demonstrate distinct expression profiles between host and tumor cells. Though the best characterized, TAAs induce limited endogenous T cell responses due to central tolerance and/or lower TCR-MHC binding affinity due to similarity to self-peptides[22].
Cancer-germline or cancer/testis antigens (CTA) - CTAs can be expressed in testes, fetal ovaries, or trophoblasts, but are otherwise absent in healthy somatic cells [22]. Although CTA targets are also shared between tumors, they are known to be expressed in only a limited number of tumor types, potentially reducing the applicability of CTA-targeted immunotherapy [23].
Tumor-specific antigens (TSA) - these cancer-restricted antigens are generated from tumor-specific mutations (neoantigens) and oncogenic viral proteins (e.g. HPV, EBV). Since neoantigens arise from non–synonymous somatic mutations during oncogenesis, they are uniquely expressed on tumors, and T cells involved in their recognition are less likely to be eliminated by central tolerance [23]. For the purposes of this review, antigens in the context of “antigen burden” or “tumor antigenicity” will be in reference specifically to tumor neoantigens (TSAs).
Tumor mutational burden (TMB), a predictor of ICB response [24], strongly correlates neoantigen load [25]. As compared to PDAC, ICB-responsive tumors such melanoma, non small cell lung cancer (NSCLC), and dMMR tumors, generally have significantly higher TMB indices and neoantigen loads (1000s-10,000s) [26]. In comparison, PDAC expresses an average of 30–50 neoantigens per tumor [27]. Among those small numbers, only a select few immunogenic neoantigens can trigger T cell activation and expansion. Consistent with this, data shows that tumors with both abundant CD8+ T cell infiltrates and a high volume of neoantigens—but neither alone—are associated with the longest survival in patients with PDA [28].
3.1.1. [IO Strategy] Increase the Tumor Antigen/Neoepitope Pool with Radiation and DNA Damage Repair Inhibition
Radiation:
Combining localized radiation therapy (RT, SBRT) with ICB may provide a boost to what is known as the “Abscopal Effect” – an observation of focal intervention inducing a systemic antitumor response in sites outside the treatment-area [29]. This is presumably due to RT-induced tumor cell injury leading production of additional tumor antigens which are then engulfed by APCs [30,31]. In addition, RT can diversify the TCR repertoire of intratumoral & peripheral T-cells and can synergize with ICB through non-redundant immune activation mechanisms [32]–34]. Preliminary results from a phase II study examining use of dual CTLA-4 and PD-1 blockade with RT (NCT03104439) has shown promising results as a strategy to stimulate immune response in patients with pretreated metastatic PDAC. Among 22 patients enrolled, all pMMR tumors, disease control rate (DCR) was 27% and ORR was 14% including 1 CR. Notably, all objective responses occurred outside of the radiation field[35]. There are several other ongoing clinical trials looking at RT in combination with several classes of IO and TME modifying therapies (e.g. NCT03599362, NCT02648282, NCT04327986).
DNA Damage Repair Modulators:
Defective DNA repair response (DDR) pathway mutations - such as BRCA1, BRCA2, ATM and PALB2 - occur frequently in both inherited and sporadic PDAC [36]. Germline DDR mutations are found in 5–10% of PDAC patients. Additionally, a quarter of PDAC tumors carry somatic mutations in either BRCA1, BRCA2 and other “BRCA-like” DNA repair genes (e.g. ATM, ATR, BARD1, BRIP1, CHK1, CHK2, PALB2, RAD51, and FANC) [37]. These mutations may predict increased sensitivity to platinum-based chemotherapy and could be therapeutically targetable 34.. Poly ADP-ribose polymerase inhibitors (PARPi) were welcomed into the arsenal of approved pancreatic cancer treatments after maintenance olaparib demonstrated a significant PFS & OS benefit of in advanced PDAC patients harboring germline BRCA mutations who had disease control with first-line metastatic therapy [38]. Inhibiting DDR with PARPi may lead tumor cells to develop a more immunogenic repertoire of antigens and cause release interferons to serve as signals for immune response that can be further augmented with ICB [39]. Preclinical data have shown that treatment with PARP inhibition is associated with upregulated PD-L1 expression in the TME [40]. Consistent with this observation, synergy between PARPi and ICB has been demonstrated in mouse models of BRCA1-deficient ovarian and breast cancers [41,42]. Current ongoing studies BRCA-mutated PDAC patients include neratinib (PARPi) in combination with dostarlimab (anti-PD1) (NCT04493060), dostarlimab plus RT (NCT04409002), nivolumab, or ipilimumab (anti-CTLA-4) (NCT03404960). Outside of PARPi, ATM and ATR inhibitors are in the early stages of clinical development in patients with solid tumors, but preclinical data has suggested potential therapeutic synergy between these inhibitors, chemotherapy, ICB, and RT [43]–46].
3.1.2. [IO Strategy] Administering Anti-Cancer Vaccines & Immune Adjuvants to Optimize Immune Priming:
To overcome the issue of low immune recognition of PDACs, several anti-cancer vaccine platforms have been developed to generate a novel or boost existing immune responses [47]. Several vaccination strategies have demonstrated antigen-specific immunological responses in patients with PDAC [48]–50].
GVAX:
The most extensively studied vaccine in PDAC is the allogenic, whole cell vaccine, GVAX. GVAX is composed of two human PDAC cell lines modified to express granulocyte-macrophage colony stimulating factor (GM-CSF) to activate T-cells against a variety of tumor antigens. This whole cell, allogeneic vaccine is less specific, but offers the advantages of being standardized, and faster to give off-the-shelf to patients. This is as opposed to autologous, personalized vaccines that use the patient’s own tumor as an antigen source [51]. In clinical trials, GVAX combined with chemoRT in the adjuvant setting for resected PDAC revealed an expansion of mesothelin-specific CD8+ T cells. Mesothelin, a TAA, has relatively high specificity for PDAC and has been implicated as mediator of tumor progression/metastasis [52,53]. This led to the development of a live-attenuated, mesothelin-expressing, Listeria monocytogenes vaccine (CRS-207) added as a method to prime-boost response to GVAX. Despite promising phase I results in a metastatic PDAC population [54], this combination, ultimately, failed to improve OS compared physician’s choice chemotherapy in the 2nd & 3rd line metastatic settings [55]. Notably, on correlative tissue analysis, vaccination-induced increases in tumor infiltrating (TILs) CD8+ Teff cells were met with an upregulation of PDL1 and other immunosuppressive markers suggesting potential synergy with ICB [56]. In line with this hypothesis, a phase Ib study in pretreated mPDAC patients showed that combining GVAX and Ipilimumab (anti-CTLA4) was associated with improved overall survival compared to treatment with ipilimumab alone (5.7 vs. 3.6 mo) [57]. However, in a recent phase II trial, the addition of nivolumab to a backbone of a combined GVAX+CRS-207 vaccine regimen provided no additional benefit to OS compared to vaccination alone in mPDAC patients [58]. This has prompted investigation into combining anti-tumor vaccination with immunomodulators beyond conventional aCTLA-4/aPD-1 targets. Combinatorial GVAX + Nivolumab + an anti-CD137 immune agonist, administered to 10 resectable PDAC patients prior surgery, resulted in three moderate pathologic responses on resected tumor surgical specimens. Additional doses of this regimen were given prior to and continued (every 4 weeks) after completion of standard adjuvant chemotherapy. At a median 12-month follow up, 9 of 10 enrolled patients remained disease-free. Correlative studies notably showed treatment-related expansion of activated, cytolytic Granzyme B+ CD8 TILs [59].
Additional TAA-targeted vaccines:
As with GVAX and CRS-207, other TAA-targeting vaccines using various platforms, including dendritic cells [60], viruses [61], and peptides [62], have yielded mixed results thus far. A notable phase II study tested GV1001, a human telomerase reverse transcriptase catalytic subunit (hTERT) peptide vaccine, along with GM-CSF adjuvant, demonstrated an objective immune responses in 63% participants. Those same patients also experienced significantly longer OS when compared to the patients who did not have an immune response to GV1001[50]. However, this may have been more a function of the tumor/host-immune substrate rather than due to the intrinsic-efficacy of the vaccine. In a follow up phase III trial, the addition of GV1001 (with GM-CSF) to adjuvant gemcitabine and capecitabine failed increase OS compared to the chemotherapy alone[63]. BSK01, an intra-dermal, autologous DC vaccine (ex vivo primed & matured DCs pulsed with TAAs - SP17, AKAP4, PTTG1, Ropporin-1, Span-Xb) demonstrated immunogenicity and clinical response in a patient with refractory, metastatic pancreatic cancer, but has not yet moved onto a phase II trial [64]. Another vaccine, BN-CV301, a genetically modified poxvirus expressing TAAs carcinoembryonic antigen (CEA) and mucin-1 (MUC-1) along with three costimulatory molecules (B7.1, ICAM-1 and LFA-3) is currently in trial with durvalumab (anti-PDL1) and maintenance capecitabine in the advanced PDAC patients who are responsive to first-line chemotherapy (NCT03376659). Overall, TAA-based vaccination strategies remain limited in generating robust host immune responses due to variable immunogenicity and presence/expression of selected TAAs across PDAC patients and within heterogeneous tumors. The expression of TAAs by normal cells risks further damping of response due to negative selection of TAA-specific T-cells and/or off-target autoimmune toxicity[51].
TSA-Targeted Vaccines:
Targeting TSAs, as opposed to TAAs, may offer a solution to the above limitations since it targets antigen unique solely to the tumor. However, these biologic/immunologic benefit currently comes with a high technologic and economic cost: neoantigens often being unique to each individual patient and thus typically require personalized vaccines [65]. Generating a neoantigen vaccine first must begin with neoantigen identification. Generally, this starts with tumor and host genomic and whole exome sequencing to identify unique neoepitopes produced by the tumor. Next, these antigens are assessed for immunogenicity via factors such as variant allele frequency, MHC-binding avidity predictions [66] and similarity to self. Those neoantigens identified as most likely to trigger a robust immune response are synthesized into long chain peptides and combined with an adjuvant to induce innate immune response (e.g. GM-CSF, polyinosinic-polycytidylic acid [poly-ICLC], a synthetic RNA TLR3 agonist, etc.,). This technological workflow has already been successfully carried out in human trials.
Pilot studies in melanoma, glioblastoma, and other solid tumors have shown that the generation/administration of personalized neoantigen peptide vaccines is not only feasible but also induces neoantigen–specific CD4+ and CD8+ T-cells in peripheral blood in response to vaccines targets [67]–72]. Ott and colleagues generated and administered personalized neoantigen peptide vaccines (n=13–21, 21-mer synthetic long chain peptides) with poly-ICLC adjuvant in 8 melanoma patients following definitive resection; six of whom demonstrated peripheral T cell responses [67]. Thirty-two months post-vaccination, half of participants remained disease free, with the 2 patients recurring, who had peripheral T cell response, going on to achieve complete responses after treatment with subsequent anti-PD-1 therapy [67]. Pilot studies in glioblastoma, a low TMB tumor similar to pMMR PDAC, have shown that neoantigen vaccines can induce peripheral immune response and effector T-cell infiltration [68,69]. In a very recent combinatorial protocol [73], personalized neoantigen vaccines were added to on-going nivolumab (anti-PD1) treatment in patients with high–TMB tumors (e.g. melanoma, NSCLC, and bladder cancer). The addition of the neoantigen vaccine to ongoing PD1 blockade resulted in robust neoantigen–specific T cell immunity an led to vaccine-induced direct tumor cell killing. Taken together, the pre-clinical and human pilot study data supports the therapeutic synergy of increasing immune activation/expansion with an anti-tumor vaccine and optimizing effector response/countering immunoregulatory signaling with ICB.
With regards to neoantigen vaccine use in PDAC, specifically, a pre-clinical proof of concept study was successfully conducted by Zaidi and colleagues, who generated and administered a neoantigen-targeted long peptide vaccine (PancVAX), with STING adjuvant (ADU-V16), in Panc02 mouse model [74]. Encouragingly, this vaccine led to neoepitope-specific T-cell responses and transient tumor regression that was noted to be significantly more durable when combined with immune checkpoint modulators (e.g. aPD-1 and agonist OX40 mAbs) [74]. These findings formed the basis of two clinical trials. The first, opening later this year, will seek to generate and administer a personalized neoantigen peptide vaccine with poly-ICLC adjuvant along with MGA-012 (aPD-1) therapy to metastatic PDAC patients in the maintenance setting. The second, is an ongoing phase 1 clinical trial (NCT04117087) testing the safety and immunogenicity of a administering dual ICB (nivolumab and ipilimumab) with a mutant KRAS “off-the shelf” neoantigen vaccine - consisting of six SLPs corresponding to KRAS G12C, G12D, G12V, G12R, G12A, G13D mutations, respectively, with poly-ICLC adjuvant - in patients with resected mKRAS PDAC following adjuvant chemotherapy. Overall, while currently carrying a high technologic and economic cost to produce, the growing ubiquity & standardization of high-throughput next generation sequencing and immunogenicity algorithms, neoantigen-targeted vaccination may soon have the feasibility to realize its therapeutic potential in PDAC and other immunologically “cold” tumors types.
3.2. [IO Barrier] Sequestration of Tumor MHC/HLA I Molecules:
Effector T-cells directly identify tumor cells as foreign via antigen presentation by MHC (HLA) class I molecules expressed on the surface of tumor cells. However, when examining the tumors of PDAC patients, the locus-specific expression of HLA I was found to be reduced in 61% of PDAC primary tumors and in 93% of tumor metastases [75]. Recently, Yamamoto and colleagues, using a mouse-model of PDAC, observed that, instead of being located of the cell surface, most PDAC antigen-MHC I complexes were sequestered inside the tumor cell within autophagosomes and autolysosomes [76]
3.2.1. [IO Strategy] Increase Tumor HLA Class 1 Expression by inhibiting Autophagy:
PDAC cells exhibit constitutive elevated basal autophagy which plays multiple pro-tumorigenic roles, including promoting immune evasion and supporting tumor metabolic demand in a nutrient-deprived microenvironment [77,78]. In the same preclinical study mentioned above, the investigators showed that inhibiting an autophagy mediator, NRB1 with chloroquine (CQ) induced an influx of CTLs to the TME and generated robust antitumor clinical responses[76]. While encouraging, this synergy between IO and CQ/hydroxychloroquine (HCQ) has not proven consistent with other preclinical studies suggesting CQ/HCQ attenuates IO response [79]. Clinically, HCQ has not shown significant anti-tumor benefit as a single-agent [80] or in combination with standard chemotherapy in PDAC [81,82]. This may be, in part, due to off-target systemic immunosuppressive effects of HCQ/CQ’s unselective inhibition of endosomal degradation and vesicular trafficking [83]–85]. Given this, the future role of autophagy inhibitors in IO-based regimens will be dependent on the discovery and exploitation of tumor-specific autophagy/metabolic pathways.
3.2.2. [IO Strategy] Target KRAS signaling to increase sensitivity to autophagy inhibition and IO:
Oncogenic KRAS (mKRAS), mutated in 95% of PDAC, has recently been implicated as a mediator of both antitumor immune response and autophagy influx. In models of mKRAS PDAC, inhibition of signaling downstream of RAS (e.g. MEK/ERK) increased tumor cell metabolic reliance on autophagy [86]. Notably, autophagic signaling and transcription of autophagy-associated genes was increased in cells with suppressed mKRAS but not in PDAC cells with suppressed wild-type (WT) KRAS. This could represent a therapeutic vulnerability for combinatorial targeted therapy, autophagy inhibition, and IO in mKRAS tumors [87]. Based on these preclinical results, a phase I/II (NCT04214418) is underway evaluating combination of cobimetinib (MEK inhibitor), atezolizumab (aPDL-1), and HCQ (autophagy inhibitor) in KRAS-mutated advanced malignancies, including PDAC [88].
Furthermore, promising early phase trial results of the first-in-class, KRAS Gly12Cys inhibitor, AMG510, in KRAS G12C-mutated advanced solid tumors[89], particularly in NSCLC [90], shows it is possible to directly target mKRAS. While KRAS G12C mutations are only present in ~2% of PDAC, inhibitors of more common KRAS mutations (e.g. G12A, G12V, or G12A) may soon follow suit. This would have important implications beyond single agent anti-cancer activity since inhibiting mKRAS may be also enhance IO response. In a mouse model of mKRAS PDAC, tumors were unable to evade host immune response after KRAS inactivation. The ongoing phase 1b CodeBreak 101 study (NCT04185883) is examining several combinatorial AMG510-based regimens, including with ICB, in advanced solid tumors harboring mKRAS G12C [91].
3.2.3. [IO Strategy] Bypass Absent Antigen Presentation with NK-Cells:
PDAC is known to suppress signaling needed to recruit and activate natural killer (NK) cells, and rightly so [92]–96]. The innate ability of NK cells to recognize the absence of MHC-proteins and respond rapidly - independent of transcription or proliferation – makes them promising cellular agents to target tumors evading T-cell immunity via mechanisms such as disruption of INF-γ signaling or downregulating MHC-I presentation [97]. NK-cell based strategies for PDAC include receptor-mediated activation and ex vivo expansion of NK cells, chimeric antigen receptor engineering (CAR-NK), adoptive immunotherapy using donor-transformed NK cells, and increasing antibody-dependent cellular cytotoxicity (ADCC) [98].
NK Cell Checkpoint Inhibitors:
Similar to ICBs, mAbs have been developed to target inhibitory receptors on the surface of NK cells such as the killer-cell Ig-like receptors (KIRs) and Natural Killer Group 2A receptors (NKG2A). Lirilumab (IPH2102/BMS-986015), which targets several inhibitory KIRs, unfortunately has yet to demonstrate a definite improvement in survival outcomes in human trials [99]. IPH2201, a first in class mAb targeting NKG2a was shown to be well-tolerated with durvalulmab (aPDL-1) in advanced solid tumors (NCT02671435) and demonstrated encouraging preliminary outcomes when add to first line SOC therapy (mFOLFOX6 + Bevacizumab) in pMMR mCRC patients [100].
CAR-NK Cell Therapy:
While T cells equipped with chimeric antigen receptors (CAR) have proven highly beneficial in B cell lymphoma and ALL, the required time and difficulty in generating a therapeutic dose of autologous CAR-T cells combined with lack of surface targets in PDAC limits CAR-T cell therapeutic utility. Here, allogenic CAR-NK cells may be a more effective, and more readily available alternative. A novel combinatorial immunotherapy protocol of 1) reduced dose metronomic chemotherapy; 2) SBRT; 3) Avelumab (aPD-L1); 4) N803 (IgG1 Fc-engineered IL-15-fusion protein), 5) off-the-shelf Allogeneic (PDL1-targted) allogeneic high-affinity NK cell infusion (haNK) cells resulted in the first reported complete response (CR) to an NK-based immunotherapy combination in advanced PDAC [101]. QUILT-88 (NCT04390399), an ongoing phase 2, randomized, three-cohort study (2 randomized, 1 single-arm) is evaluating PD-L1 targeted-haNK cells as part of multimodal treatment regimen combining IO (IL-15 agonist [N-803] and PDL1 t-haNK), SBRT, and SOC chemo in PDAC. Cohort A will test above experimental combinatorial regimen against gem-NP in the first line maintenance setting, cohort B will randomize patients to either the experimental regimen or SOC 5-FU + liposomal irinotecan in the second line setting, and cohort C will be a single arm examining the study therapy in the 3rd line or later setting (total goal accrual of 250 patients across all cohorts and arms) [102]. Finally, CAR-NK-cell tumor infiltration could be further optimized with an NK cell-recruiting protein-conjugated antibody (NRP-body) that has been shown to enhance tumor-infiltration of ex vivo expanded NK cells (via CXCL16 gradient) and increase survival in mouse-models of PDAC [98].
3.3. [IO Barrier] Paucity of Mature Dendritic Cells:
A recent study by Hedge and colleagues (2020) demonstrated that a strongly-matched pair of an immunogenic neoantigen-MHC complex and CD8+ TCR was not enough to mount an anti-tumor response without functioning conventional dendritic cells (cDC) required for antigen presentation and T-cell priming. In mice bearing PDAC, the rare DCs present in the TME were observed to be phenotypically immature and dysfunctional both in terms of both antigen sampling and trafficking to draining lymph nodes [103]. DC dysfunction is mediated, in part, by dysregulating cytokines (e.g. IL-10, TGFβ) secreted directly by the tumor or tumor-recruited immune subsets [104].
3.3.1. [IO strategy] Optimize dendritic cell recruitment and maturation
CD40 Agonism:
In addition to blockade of negative immune checkpoints (e.g. CTLA-4, PD1-PDL1 axis), immune agonism (e.g. co-stimulatory mAbs targeting CD27, CD137, and CD40) is an area of growing clinical inquiry. Anti-CD40 agonism, to date, has garnered the most translational experience in PDAC. Preclinically, CD40 activation on DCs and monocytes upregulates the expression of other costimulatory molecules (e.g. CD80 and CD86), enhance antigen presentation, licenses DCs, and activates effector T cells [105]–108]. The CD40 agonistic monoclonal antibody, APX005M, and SOC chemotherapy (Gem-NP) with or without nivolumab (aPD-1) was recently examined in the first-line setting for advanced PDAC (NCT03214250). Fourteen of the 24 patients enrolled in the initial study phase showed a partial response (58%), and 8 (33%) had stable disease. Compared to a historical mOS of between 8.5–12.1 mo for Gem-NP alone [5,109], mOS for total study cohort was an impressive 20.1 mo (12·7 months for cohort B1[0·1 mg/kg APX005M], 20·1 months for cohort B2 [0·3 mg/kg APX005M], 15·9 months for cohort C1 [0·1 mg/kg APX005M + aPD-1], and not evaluable for cohort C2 [0·3 mg/kg APX005M dose + aPD-1]) [110]. In a follow up phase II, presented at ASCO 2021, the primary endpoint of 1-year OS > 35% (historical OS rate for Gem-NP) was met when combining chemo with either aPD-1 (1-year OS 57%, mOS 16.7 mo, mPFS 4.8 mo) or APX005M (1-year OS 51%, mOS 14.5 mo, mPFS 5.5 mo); however, not with the combination of aCD40 + aPD-1 [1-year OS 41%, mOS 10.7 mo, mPFS 6.7 mo] [111]. Detailed multi-omic immune and tumor biomarker analyses are underway to identify patient subsets that benefit most from these combinations [111]. While these results do temper expectations, aCD40 agonism, along with other immune co-stimulants, are still likely to play a key role in a future effective IO-based regimen(s) for PDAC.
Recombinant FLT3L:
Recombinant FLT3L has been shown to increase the number of DC precursors and DCs in blood and tissue in vivo[112], while CD40 agonism can promote DC maturation [113]. Interim results from a phase I study (NCT03329950) shows that the combination of CDX-1140 (aCD40 mAb agonist) with CDX-301 (recombinant FLT3L) is well-tolerated and in patients with treatment-refractory solid tumors. Additionally, pretreatment of patients with recombinant FLT3L greatly increasing the number of circulating and aCD40-responsive DCs[114].
IL-10 modulation:
The anti-tumor and systemic immune effects of IL-10 signaling, as is the case with other direct interleukin modulatory treatments, remain complex and, at times, even contradictory. For example, IL-10 agonism and antagonism have been observed to enhance anti-tumor outcomes, preclinically [115,116]. Despite promising early phase results, IL-10 agonist, pegilodecakin, failed to enhance response to FOLFOX in the 2nd line for mPDAC [117] and led to increased toxicity without enhancing survival when paired with aPD-1 therapy in NSCLC[118]. Human trials with IL-10 inhibition are limited. A phase 1 study (NCT02731742) examining the combination of an IL-10 inhibitor (MK-1966) with a TLR9 agonist (SD-101) was recently halted due to lack of efficacy [119]. More investigation is required to examine context-dependent IL-10 signaling, specifically the effects of blockade on peripheral and tumor-infiltrating T cell activation and expansion [120].
4. Impaired Anti-Tumor Immune Cell Trafficking to Tumor Bed:
Once primed, activated T cells, primarily the effector subtypes, should traffic to the tumor site via chemotaxis, crossing vasculature, while being maintained by pro-survival growth factors signaling [121]. However, upon examination of the PDAC TME, we instead observe an “immune desert” with only a small and impotent T cell infiltrate.
4.1. [IO Barrier] Desmoplastic Stroma & Dysregulated T cell recruitment:
A major factor contributing to this immune exclusion is the dense stromal compartment of PDAC. This a collagenous, desmoplastic hypo-vascular matrix, produced by cancer-induced pancreatic stellate cells and accounts for the vast majority of tumor cellularity [122]. The resultant desmoplasia is known to be responsible for creating a mechanical barrier around the tumor cells, preventing appropriate vascularization and thus limiting exposure to chemotherapy and leading to poor immune cell infiltration. In addition to a physical boundary, PDAC sets up a chemical barrier via disrupted chemokine expression to deregulate signaling involved in effector T-cell recruitment resulting in significantly reduced a CD8+ T-cell infiltrate. Intratumoral TGFβ signaling via cancer-associated fibroblasts (CAF) appears critical in counteracting anti-tumor immunity via restricting movement of CTLs in the TME. TGFβ signaling also mediates recruitment of immune suppressive cells (e.g. regulatory T cell [Tregs] & myeloid-derived suppressor cell [MDSC]) via downstream signaling targets (e.g. VEGF) further establishing an unfavorable environment for Teff-infiltration [122,123].
4.1.1. [IO Strategy] Disrupting the extracellular matrix (ECM):
Non-specific targeting of the ECM with matrix metalloproteinases (MMP) inhibitors such as marimastat and tanomastat failed to demonstrate significant clinical activity in patients with advanced-stage pancreatic cancer [124,125]. A more specific approach to weakening ECM-scaffolding is targeting hyaluronan (HA), a major component of the PDAC ECM with high levels associated with poor prognosis [126]. In preclinical models, decreasing the hydrostatic pressure in the dense PDAC stroma with PEGPH20, a pegylated hyaluronidase (HA), was shown to increase vascular permeability, increase drug delivery when used in combination with chemotherapy [127] Paradoxically, a phase 1b/II trial of adding PEGPH20 to mFOLFIRINOX resulted in significantly inferior OS, PFS, and ORR with compared to chemotherapy alone [128]. In contrast, PEGPH20 appeared to add a PFS benefit to Gem-NP [129]. However, a follow up phase III trial (HALO 109–301), PEGPH20 in combination with chemotherapy failed to improve PFS or OS in metastatic PDAC patients, even among those with HA-high tumor expression [130]. Since it has been observed that PEGPH20 enhances GVAX-induced CD4+ and CD8+ T-cell TME infiltration in a preclinical PDAC model [131], it is worth considering whether adding an immunomodulator/ICB to the studied regimen would have led to a change in outcome [132]. Along these lines, the combination of atezolizumab with PEGPH20 is currently being tested in pre-treated advanced PDAC population (NCT03214250).
4.2. [IO Strategy] Eliminating Cancer-Associated Fibroblasts:
Fibroblast Activation Protein (FAP) is found ~90% CAFs and higher levels of tumor expression are associated with poor clinical outcomes in PDAC. Despite preclinical models showing anti-tumor effects of FAP-targeting/ablative strategies[133]–135], a FAP antagonist mAb (sibrotuzumab) and small molecule inhibitors (talabostat) have not been able to translate this meaningful benefit to the clinical trial setting [136,137]. Recently, there has been a growing appreciation for the heterogeneity and plasticity of CAFs and their associated pro- and anti-tumorigenic functioning [137]. This phenotypic complexity may underly the preclinical observations that show depletion of CAFs paradoxically accelerates tumor progression [138,139]. The next generation of therapies targeting stromal resistance mechanisms in PDAC must account for the functional heterogeneity of CAFs.
4.2.1. [IO Strategy] Reprogramming Cancer-Associated Fibroblasts:
Instead of simply eliminating the stromal fibroblasts from the TME, a more sophisticated approach might be to disrupt crosstalk between tumor-stroma-immune compartments. An example of this is blocking CAF-mediated pro-tolerogenic signaling (e.g. CXCL12/CXCR4) or disrupting CAF–cancer cell immunosuppressive communication loops through blockade of TGFβ and IL-1β.
Targeting CXCL12/CXCR4 Chemokine Axis:
CXCL12, produced by CAFs, misdirects effector T cells to the extratumoral stroma, preventing them from entering the tumor [140]. Pre-clinically, disrupting the binding of CXCL12 with its receptor CXCR4 improves the anti-tumor activity of ICB and the combination has been shown to reactivate endogenous TILs in PDAC models [141,142]. On the basis of this, AMD3100, a small molecule inhibitor of CXCR4, is now being studied in patients with metastatic pancreatic cancer in combination with Cemiplimab (aPD-1) in metastatic PDAC (NCT04177810). BL-8040 another CXCR4 antagonist, is being examined in combination with pembrolizumab and chemotherapy for 2nd or 3rd line treatment for metastatic PDAC. Preliminary outcomes from the expansion cohort of the COMBAT trial (NCT02826486) showed that the BL-8040 and pembrolizumab added to the NAPOLI-1 regimen (liposomal irinotecan, fluorouracil and leucovorin) resulted in an encouraging ORR of 32% and a DCR of 77% in PDAC patients following 1st-line Gem-based treatment [143]. These results compared favorably with the NAPOLI-1’s ORR of 17% and DCR of 52% [144] suggesting BL-8040 and pembrolizumab may expand the benefit of chemotherapy in PDAC [143,144].
TGFβ Inhibition:
Transforming growth factor beta (TGFβ), released by PDAC tumor cells, stromal fibroblasts and other cell types in the TME contributes to the tolerogenic architecture of the TME and suppresses the cytotoxic activities of anti-tumor immune cells [145]. T-cells with constitutively active TGFβ signals remain refractory to full activation [146]. Serum TGFβ have correlated with poor prognosis in pancreatic cancer [147]. Several preclinical models have demonstrated therapeutic synergy of TGFβ signaling inhibition and immunomodulatory agents [148]–150]. The combination of gemcitabine and Galunisertib, a small molecule inhibitor of TGFβ, enhanced OS compared to chemo alone in first line treatment in patients with mPDAC (mOS 8.9 and 7.1 months for galunisertib and placebo, respectively [HR = 0.79, 95% CI: 0.59–1.09, posterior probability HR < 1 = 0.93]) [151]. Galunisterib with durvalumab (aPD-L1) was shown to be well-tolerated but had limited in clinical efficacy when used the 3rd or later line for patients with mPDAC [152]. More recently, a bifunctional checkpoint inhibitor comprised of an aPD-L1 mAb fused with a TGFβ ligand trap (M7824, Bintrafusp Alpha) was tested in the phase I setting in a cohort treatment-refractory advanced solid tumor patients. Of the 5 PDAC patients included, there was 1 PR but this patient also had dMMR disease [153]. Expectations for the dual-targeting fusion protein have been further tempered since M7824 fell short of its primary endpoint for second line treatment in advanced biliary tract cancers (BTC) [154]. However, there are pending results of an ongoing phase II/III of M7824 combined with chemo in the 1st-line for advanced BTC (NCT04066491), and a similar approach may warrant consideration in PDAC.
IL-1β antagonism:
High stromal IL-1β expression is associated with poor overall survival of PDAC patients [155]. CAFs are reprogrammed by tumor-derived IL-1β to produce cytokines and chemokines that subvert anti-tumor immunity and further promote cancer growth [155,156]. Preclinically, the addition of anti-IL-1β antibody significantly enhanced the anti-tumor activity of aPD-1 mAb in the variety of tumor models, including PDAC, and resulted in increased tumor infiltration of CD8+ T cells [156]. In human experience, combination of pembrolizumab and the aIL-1β antagonist, canakinumab, has demonstrated safety in metastatic solid tumor patients[157]. A phase III study (NCT03631199) evaluating the additive impact of canakinumab to SOC first-line immunotherapy and chemotherapy in NSCLC is expected to report final results before the end of the year. A phase II trial of examining dual IL-1β and PD1 blockade in patients resectable PDAC is in development.
FAK Inhibition:
Another stromal target is focal adhesion kinase-1 (FAK1), a tyrosine kinase expressed on both tumor and stromal cells that has been implicated in driving stromal desmoplasia. Pre-clinically, FAK inhibition decreased tumor cell proliferation, reduced tumor-associated macrophages and CAFs, increased CD8+ T cell infiltration, sensitized tumors to RT[158,159]. A phase I trial of defactinib, an oral FAK1 TKI, in combination pembrolizumab and gemcitabine (NCT02546531) showed modest but encouraging efficacy signals in both maintenance and treatment refractory mPDAC settings [160]. Correlative studies with paired biopsies in PDAC patients show increased proliferating CD8+ T cells, while Tregs, macrophages, and stromal density decreased with treatment[160]. FAK inhibition’s limited clinical efficacy may be in part related to a resistance mechanism via STAT3 upregulation triggered by FAK-induced stromal depletion; arguing for a combinational approach with direct/indirect STAT3 inhibitors [161,162].
5. [IO Barrier] Immunosuppressive Cellular TME:
Even with sufficient effector T-cell priming, infiltration through the desmoplastic ECM, anti-tumor immune cells are still left to deal with local immunosuppressive cell populations that further dampen and exhaust effector functions. The PDAC TME is populated by multiple types of immunosuppressive cells, including regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and tumor-associate-neutrophils (TANs) [163].
Tumor-associated macrophages:
Tumor-associated macrophages (TAMs) are one of the most abundant immune cell populations in the pancreatic tumor stroma due in part to recruitment by oncogenic KRAS [164]. Either via direct tumor-signaling or via tumor-CAF crosstalk, TAMs are generally polarized to the immunosuppressive phenotype (M2) defined by Csf1R, CD206, and IL-10 expression along with reduced expression of MHC class II and Ly6C [156,165]. These M2 TAMs support tumorigenesis, immune escape (e.g. promoting Th2 cell differentiation), metastasis, and chemotherapeutic resistance [166].
Myeloid-derived suppressor cells:
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that accumulate in the TME, reducing anti-tumor T cell activity and promoting tumor immune escape. These cells can be divided into two general groups termed granulocytic (G-MDSCs)and monocytic (M-MDSCs), phenotypically and morphologically similar to neutrophils and monocytes, respectively. MDSCs suppress effector TILs by depleting metabolites critical for T cell function (via upregulation of arginases, and IDO-mediated sequestration of cysteine), blocking T cell homing (via nitric oxide-mediated downregulation L-selectin expression in T cells), induction of other immunosuppressive cells (e.g. FoxP3+ T reg and M2 TAMs), and upregulating expression of PDL1 to induce T cell anergy [167].
Tumor-associated neutrophils:
More recently, tumor-associated neutrophils (TANs) have become increasingly recognized for their ability to promote tumor progression, mediate resistance to therapy, regulate immunosuppression, and correlate with poor prognosis numerous cancers in a variety of cancers including PDAC [168]–170]. TAN-recruitment to the TME appears to be mediated via the chemokine CXCR2.
5.1. [IO Strategy] Reprogramming TME Myeloid Cells:
5.1.1. CD40 agonism:
As discussed earlier, CD40, is a key mediator of adaptative immune response. Additionally, it may also help reprogram the TME reprograming via effects on myeloid cell populations. Specifically, CD40 agonism potentiates the immunogenic profile of DCs, converts TAMs from a tumor permissive (M2) to a tumoricidal phenotype (M1), and may help reverse stromal desmoplasia [171,172]. A summary of the current clinical trial data with CD40 agonism can found in section 3.3.1.
5.1.2. CD11b modulation:
CD11b is almost universally expressed on myeloid cells in PDAC and is an important mediator of myeloid cell migration and functional phenotype. Panni and colleagues developed a small molecule, allosteric agonist of CD11b (ADH-503) that resulted in a partially active CD11b that suppressed myeloid cell infiltration by increasing adhesion to the endothelium, repolarized TAMs to M1 phenotypes, and lead to tumor regression and long-term immune memory when combined with ICB [173,174]. These preclinical findings are the basis for an early phase clinical trial (NCT04060342) examining safety and tolerance of GB1275, a first-in-class CD11b modulator, alone and in combination with pembrolizumab. Preliminary results from the trial suggest the combination is well-tolerated but responses in PDAC patients included were limited [175].
5.1.3. BTK Inhibition:
Activation of Bruton’s Tyrosine Kinase (BTK), mediated by PI3Kδ protein kinase, has been linked to M2 macrophages polarization. Blockade of the signaling pathway using BTK inhibitors has resulted in decreased tumor growth, reduced M2-TAMs & B reg TME populations, and increased CD8+ TILs in preclinical models of PDAC. [176,177]. On the basis of these findings, the BTK inhibitor, acalabrutinib, was studied in a phase II trial as monotherapy and in conjunction pembrolizumab in pretreated advanced PDAC patients (NCT02362048). Though Acalabrutinib treatment was associated with reduced circulating MDSCs and the combination induced activation of CD4 and CD8 memory T cells detected in peripheral blood, BTKi plus aPD-1 did not improve PFS/OS [178]. Due to limited on-treatment tumor biopsies, it was not possible to assess treatment-related TME changes [178].
5.1.4. CSF1/CSF-1R Inhibition:
The colony stimulating factor 1 and colony stimulating factor 1 receptor (CSF-1/CSF-1R) pathway is crucial for the differentiation and survival of pro-tolerogenic M2 macrophages and represents a potential target of reprogramming TAMs from M2 to M1 type [179]. Preclinically, blockade of CSF-1/CSF-1R pathway has shown promising results in repolarizing TAM towards M1 type and induced distinct TME remodeling [180]–182]. Although early studies showed potential synergy of cabiralizumab, a humanized IgG4 CSF-1R mAb, with PD1 blockade, the combination when added to SOC chemotherapy-backbone did not improve PFS compared to SOC chemotherapy alone in advanced pretreated PDAC [183]. A small molecule inhibitor of CSF1R, pexidartinib, is being examined in combination with durvalumab in patients with metastatic PDAC or CRC (NCT02777710). Preliminary results from the study showed an ORR of 0% and DCR of 21% with 4 patients achieving SD, two of which happened to be dMMR CRC patients[184].
5.1.5. CCL2/CCR2 Inhibition:
In mouse models of PDAC, small molecule inhibitors of CCR2 decreased TAM infiltration, increased TIL CD8+Teff to CD4+Treg ratios, and enhanced response to ICB [185,186]. This led to phase I study of PF-04136309, an oral inhibitor of CCR2, in combination with Gem-NP in patients with mPDAC (NCT02732938). The trial concluded that PF-04136309 was associated with worse pulmonary toxicities and added no clinical benefit compared to gemcitabine and nab-paclitaxel alone [187].
5.1.6. CXCR2 Inhibition:
Preclinically, inhibiting CXCR2 decreases TAN infiltration, abrogates PDAC metastasis, augments T cell infiltration and synergizes with aPD-1 & chemotherapy to extend survival [188,189]. A phase Ib/II clinical trial (NCT02583477) investigating AZD5069, an oral small molecule inhibitor of CXCR2, in combination with aPD-L1, durvalumab, demonstrated limited efficacy in advanced PDAC patients with 8 (40%) of patients experiencing serious adverse events causally related to treatment (3 [15% of patients] requiring study discontinuation, but no reported deaths related to therapy) [190,191].
5.1.7. Epigenetic Therapy:
Epigenetic changes are an integral part of PDAC’s development, progression, intratumoral heterogeneity, TME makeup, immune escape, and chemoresistance [192]. Revising dynamic epigenetic changes with DNA methyltransferases (DNMT) or histone deacetylases (HDAC) inhibitors may augment immunotherapy [193] by preventing/reversing immune exhaustion [194], increase tumor antigens and MHC 1 expression [195], and reprogramming local stromal and immune cells to create a more immune permissive TME [196,197]. Recently, epigenetic regulation of the biologic behavior of MDSCs has emerged as a promising tool in PDAC therapy. In a murine model of PDAC, use of the HDACi, entinostat, changed the polarity of immunosuppressive MDSCs to a nonfunctional phenotype. Additionally, the combination of HDACi plus aPD-1 significantly improved survival as compared with mice treated with either agent [196]. On the basis of these findings, entinostat and nivolumab are under current clinical trial investigation (NCT03250273) for use in pre-treated advanced PDAC patients with preliminary outcomes expected to be reported soon.
5.2. [IO Strategy] Additional TME-Targeted Approaches
5.2.1. Reducing T regulatory cells:
CD4+CD25+FOXP3+ Tregs, in the context of PDAC, can limit anti-tumor immune responses and are associated with poor prognosis [198]. However, depletion of Tregs in a preclinical PDAC model failed to relieve immunosuppression and paradoxically led to accelerated tumor progression. Clinically, targeting CCR4, highly expressed on Tregs with Mogamulizumab, reduced intratumoral Tregs, but did not enhance responses to ICB [199].
5.2.2. MEK Inhibition:
MEK inhibitors have shown immunomodulatory effects and efficacy when combined with PD-(L)1 inhibitors in multiple preclinical models via a variety mechanisms including enhanced MHC I/II expression, increased CD8+ TIL infiltration and survival, reduced recruitment of mMDSCs, M2 TAMs, and B regs [200]–207]. Unfortunately, the combination of a MEK inhibitor (cobimetinib) plus a aPD-L1 inhibitor (atezolizumab) failed to show compelling immune activity in a large phase III clinical trial in colon cancer [208]. However, a phase II trial of this same combination demonstrated a beneficial PFS therapeutic signal in advanced pretreated BTC patients [209]. One possible explanation for the discrepancy between preclinical and clinical outcomes is that systemic MEK inhibition may impair T cell priming and activation [200]. Dovetailing off of this, multiple pre-clinical models, including PDAC and mKRAS cell lines, have demonstrated synergy between MEKi and immune agonists [203,210,211]. A clinical trial combining MEKi, ICB, and an aCD27 immune agonism is in development for advanced BTC patients (NCT04941287), and could also represent a future immunotherapeutic strategy in PDAC should it prove successful.
5.2.3. Targeting B-cells:
While contemporary IO is firmly grounded in optimizing cellular immunity, mainly T cell function, the anti-tumor potential of humoral immunity and B cell modulation is a relatively new area of exploration. In addition to producing antibodies and secreting cytokines, B cells are able to recognize antigens, regulate antigen processing and presentation, and modulate cellular immune response [212]. The latter has been shown clinically in treatment of autoimmune processes. For example, in multiple sclerosis and rheumatoid arthritis, conditions characterized by pathogenic T-cell function, selective B-cell depletion with aCD20 antagonizing mAbs improves symptoms, lowers disease activity, and increases time between disease flares [213,214]. As with other cellular counterparts, B cell pro and anti-tumorigenic functions are context-dependent and influenced by the TME milieu. The use of CD20-targeted B cell therapy in patients with advanced solid tumors patients has shown mixed-to-no clinical benefit [215,216]. Several strategies to antagonize immunosuppressive B-regs have shown promise in the preclinical setting but their clinical utility has yet to be established [217]. Therapies activating of B cells are also under investigation. Beyond optimizing DC function, IL-10 inhibition and CD40 agonism may sustain anti-tumor B cells in the TME as well as promote B-cell mediated T-cell activation and memory formation [218,219]. Additionally, B cell–activating factor (BAFF, BLyS), a B cell–activating cytokine, has been shown to activate B cells expressing high levels of costimulatory molecules (CD40, ICOSL), augment antigen presentation to CD4+ T cells through increased expression of MHC-II, and increases IL-12 expression to promote the differentiation of Th1 cells and T memory cells [220]. BAFF/BLyS was safely tolerated in a clinical trial for treatment of IgA deficiency (NCT00024934) may be valuable adjuvant for CD40 targeted and/or vaccine-based regimens [220,221].
6. Conclusions and Perspectives
Pancreatic adenocarcinoma has multiple layers of immune-resistance to overcome, but it is not an immune-refractory tumor[222]. A growing understanding of its resistance mechanisms and intricate tumor microenvironment dynamics are revealing novel IO therapeutic targets and combinatorial approaches (see Table 1 for a selection ongoing clinical trials examining novel IO-based regimens). Specifically, immune agonists, such as aCD40 agonism, and anti-tumor vaccines represent promising strategies to stimulate host immune response via activation and expansion of anti-tumor effector cells. With the advancement of high-throughput multi-omics technologies accurately characterizing tumor-specific antigens and their respective immunogenicity, neoantigen-targeting strategies may enhance the effectiveness of current systemic immunotherapies, minimize off-target toxicities, and lead to formation of long-term anti-tumor memory. Furthermore, off-the-shelf NK-cell therapies may be an effective method for bypassing downregulated antigen-MHC complex presentation on PDAC cells. Optimization of anti-tumor lymphocyte activation will need to be coupled with strategies that disrupt tumor-myeloid immunosuppressive communication loops within the TME to promote anti-tumor effector infiltration and prevent immune exhaustion. This will require a sophisticated and nuanced approach to targeting the TME that focuses more on reprogramming (e.g. inhibiting CXCL12/CXCR4 chemokine signaling, or blocking tumor-CAF pro-tolerogenic crosstalk mediated via TGFβ and IL-1β) rather than simply depleting cellular targets.
Table 1:
Selected Clinical Trials Examining Novel Immunotherapy Regimens in Patients with Pancreatic Ductal Adenocarcinoma
| Study NCT Registry Number | Study Agent(s) | Standard Agent(s) | Subjects | Treatment Setting | Phase | Status/Results |
|---|---|---|---|---|---|---|
| Multimodal IO Regimens | ||||||
|
NCT03329248
NCT03586869 |
Metronomic chemo (aldoxorubicin, gem, NP, oxal, 5FU, and cy) haNK aPD-L1 PD-L1 t-haNK |
SBRT | mPDAC | Metastatic ≤2nd line |
Ib/II | Closed mOS 8.0 mo mPFS 6.6 mo 1 CR (treated with PD-L1 t-haNK) |
| NCT04390399 | Aldoxorubicin N-803 (IL-15) PD-L1 t-haNK |
Chemo (gem+NP [1st line] or NAPOLI-1 [2nd line]) SBRT |
mPDAC | Metastatic Maintenance (group A) 2nd line (group B) ≤3rd line (group C) |
II | Open |
| NCT03767582 | CCR2/CCR5i aPD-1 ± GVAX |
Chemo SBRT |
LA-PDAC | Neoadjuvant & Adjuvant | II | Open |
| NCT03153410 | GVAX aPD1 CSF1Ri |
Chemo (neoadj) SBRT (neoadj) |
BR-PDAC | Neoadjuvant & Adjuvant | Pilot | Open |
| NCT03006302 | CRS-207 IDOi aPD-1 ± GVAX |
mPDAC | Metastatic ≤2nd line |
II | Open | |
| Anti-Tumor Vaccines | ||||||
| NCT04853017 | Peptide Vaccine (Shared Neoantigen [KRAS], adj AMP-CpG) | rPDAC [KRAS/N RAS mut] |
NED Maintenance | I/II | Open | |
| NCT03948763 | mRNA Vaccine (Shared Neoantigen [KRAS], lipid-based) ± aPD-1 |
mPDAC [mKRAS] | Not specified | I | Open | |
| NCT04799431 | Peptide vaccine (Personalized Neoantigen, adj pICLC) aPD-1 |
mPDAC | Maintenance | Pilot/I | Not yet Open | |
| NCT02600949 | Peptide vaccine (Personalized Neoantigen, adj imiquimod) | mPDAC | Maintenance ≤2nd Line |
Pilot | Open | |
| NCT03468244 | mRNA Vaccine (Personalized Neoantigen) | Advanced PDAC | ≤2nd Line | Pilot | Open | |
| NCT03592888 | Autologous DC Vaccine, (Shared KRAS Neoantigens) | rPDAC [mKRAS] | Adjuvant | I | Open | |
| NCT04627246 | Autologous DC Vaccine, (Personalized Neoantigen) aPD-1 |
Chemo | rPDAC | Adjuvant | I | Open |
| NCT04157127 | Autologous DC Vaccine* (Personalized Tumor Lysate, adj peg-IFN) *perinodal injection |
Chemo | rPDAC | Adjuvant | I | Open |
| NCT04111172 | TAA-encoding Viral Vaccine (Shared, adj il-PADRE [TH1 epitope]) | Chemo | rPDAC | Adjuvant | II | Open |
| NCT03806309 | Peptide vaccine (Shared TAA/TSA, adj il-PADRE [TH1 epitope) ± aPD-1 |
± FOLFIRI | LA or mPDAC | Maintenance | II | Open |
| NCT03376659 | TAA-encoding Viral Vaccine (Shared, adj TRICOM) + aPD-L1 |
Maintenance Chemo | mPDAC Maintenan ce |
Maintenance | II | Enrolled Prelim results expected soon |
| NCT03161379 | GVAX aPD1 |
Chemo SBRT |
BR-PDAC | Neoadjuvant | II | Open |
| NCT03190265 | CRS-207 aPD-1 aCTLA4 ± GVAX |
Chemo SBRT |
mPDAC | ≤2nd line | II | Open |
| NCT02451982 | GVAX a4-1BB aPD-1 |
Chemo SBRT |
rPDAC | Neoadjuvant & adjuvant | II | Open Prelim results expected soon |
| Immune Agonists and Adjuvants | ||||||
| NCT04807972 | aCD40 agonist ± aPD-1 |
Chemo | mPDAC | 1st Line | II | Open |
| NCT03329950 | aCD40 agonist ± aPD-1 ± FLT3L |
±Chemo | mPDAC | 1st line (aCD40 + Chemo arm) Refectory (other groups) |
I/II | Open |
| NCT03214250 | aCD40 agonist ± aPD-1 |
Chemo | mPDAC | 1st line | II | [Prelim] No improvement in PFS/OS/ORR when aCD40 added to SOC chemo + PD-1 |
| NCT03193190 | aCD40 agonist aPD-L1 |
Chemo | mPDAC | 2nd line | I/II | Open |
| NCT02376699 | aCD40 agonist aPD-1 |
Chemo | mPDAC | 1st Line | II | Enrolled Prelim results expected soon |
| NCT04387071 | aOX40 agonist + TLR9 adjuvant (intratumoral) | LA or mPDAC | 2nd Line | II | Not Yet Open | |
| NCT04612530 | Electroporation CpG adj aPD-1 |
mPDAC (hepatic mets) | Maintenance | I | Open | |
| NCT04050085 | TLR9 agonist (intratumoral) aPD-1 |
SBRT | mPDAC | Refractory | I | Open |
| NCT03983954 | Superantigen fAb aPD-L1 |
mPDAC | Refractory | I | Open | |
| NCT03225989 | TMZ-CD40L and 4–1BBL-encoded Virus | Chemo | LA or mPDAC | ≤1st line | I/II | Open |
| TME Cellular/Signaling Modulators | ||||||
| NCT04390763 | TGFβ antagonist ± aPD-1 |
Chemo | mPDAC | 1st line | II | Open |
| NCT03821935 | GARP-TGFβ1 antagonist ± aPD-1 |
LA or mPDAC | Refractory | I | Open | |
| NCT04581343 | aIL-1β antagonist aPD-1 |
Chemo | mPDAC | 1st line | I | Open |
| NCT04543071 | CXCR4i aPD-1 |
aPD1 Chemo | mPDAC | 1st line | II | Open |
| NCT02826486 | CXCR4i aPD-1 |
± Chemo | mPDAC | ≥2nd line | I/II | Enrolled [Prelim] DCR 77%, ORR 32% w/chemo DCR 34%, ORR 3% w/o chemo |
| NCT03193190 | CXCR4i aPD-L1 |
mPDAC | ≥2nd line | II | ||
| NCT04477343 | CXCR1|2i aPD-1 |
mPDAC | Maintenance | I | Open | |
| NCT03257761 | Epigenetic modifier (DNMTi) aPD-L1 |
mPDAC | ≥2nd line | Ib | Open | |
| NCT03250273 | Epigenetic modifier (HDACi) aPD-1 |
mPDAC | ≥2nd line | II | Enrolled Interim results expected soon |
|
| NCT04060342 | CD11b modulator ± aPD-1 |
± Chemo | mPDAC | 1st line (for chemo combo) | I/II | Open |
| NCT03193190 | aIL-6 antagonist aPD-L1 |
Chemo | mPDAC | 1st line | II | Open |
| NCT03727880 | FAKi aPD-L1 |
Chemo | rPDAC | Neoadjuvant & Adjuvant | II | Open |
| NCT03496662 | CCR2/CCR5i aPD-1 |
Chemo | BR or LA-PDAC | Neoadjuvant 1st line |
I/II | Open |
Key (selected terms): adj – vaccine/immune adjuvant; AMP-CpG – amphiphilic-CpG (pathogen-associated molecular pattern); BR-PDAC – borderline resectable pancreatic ductal adenocarcinoma; CRS-207 – live, attenuated double-deleted Listeria vaccine targeting mesothelin; CSF-1R, colony-stimulating factor-1 receptor; Cy – cyclophosphamide; DC – dendritic cell; DCR – disease control rate; DNMTi - DNA methyltransferase inhibitor; FAKi – focal adhesion kinase inhibitor; Gem – gemcitabine; GVAX - GM-CSF secreting allogeneic pancreatic tumor cell vaccine; haNK – high affinity natural killer cells; HDACi - histone deacetylase inhibitor; IDOi - indolamine 2,3-dioxygenase inhibitor; IO – immunotherapy, immuno-oncology; LA-PDAC – locally advanced pancreatic ductal adenocarcinoma; mPDAC – metastatic pancreatic ductal adenocarcinoma; NAPOLI-1- 5FU and leucovorin plus liposomal irinotecan; NP – nab-paclitaxel; ORR – objective responsive rate; OS – overall survival; Oxal – oxaliplatin; PD-L1 t-haNK – PD-L1 targeted high affinity natural killer cells; PFS – progression-free survival; pICLC - polyinosinic-polycytidylic acid; rPDAC– resected pancreatic ductal adenocarcinoma; SOC – standard of care; TAA – tumor-associated antigen; TLR – toll-like receptor; TME – Tumor Microenvironment; TSA – tumor-specific antigen or neoantigen.
Looking ahead, the template for successful immunotherapy in PDAC will be built on multipronged targeting of immune priming/activation pathways in parallel with TME modulation and immune checkpoint blockade to ensure sustained anti-tumor responses (Fig. 3). Given the numerous barriers preventing a robust, and durable anti-tumor immune response, it is not surprising that the vast majority of the clinical trials discussed in this review have returned largely negative, or, at best, modest outcomes because the IO strategies examined, at their core, are incomplete. An effective IO regimen for pMMR PDAC will require several IO and molecular agents on top of conventional therapies. Standard chemotherapy with combinatorial cocktail of an anti-tumor vaccine, co-stimulatory agent (aCD40), TME crosstalk modulator (CXC4Ri or TGFβ trap), and ICB (aPD-1) is one such potential combination. The IO breakthrough that PDAC needs will take the form of regimen that takes this comprehensive approach to immune response while accounting for compensatory signaling pathways, and context-dependent immunomodulatory roles of therapeutic targets.
Fig. 3: A Framework for Successful IO-based Treatment in PDAC.
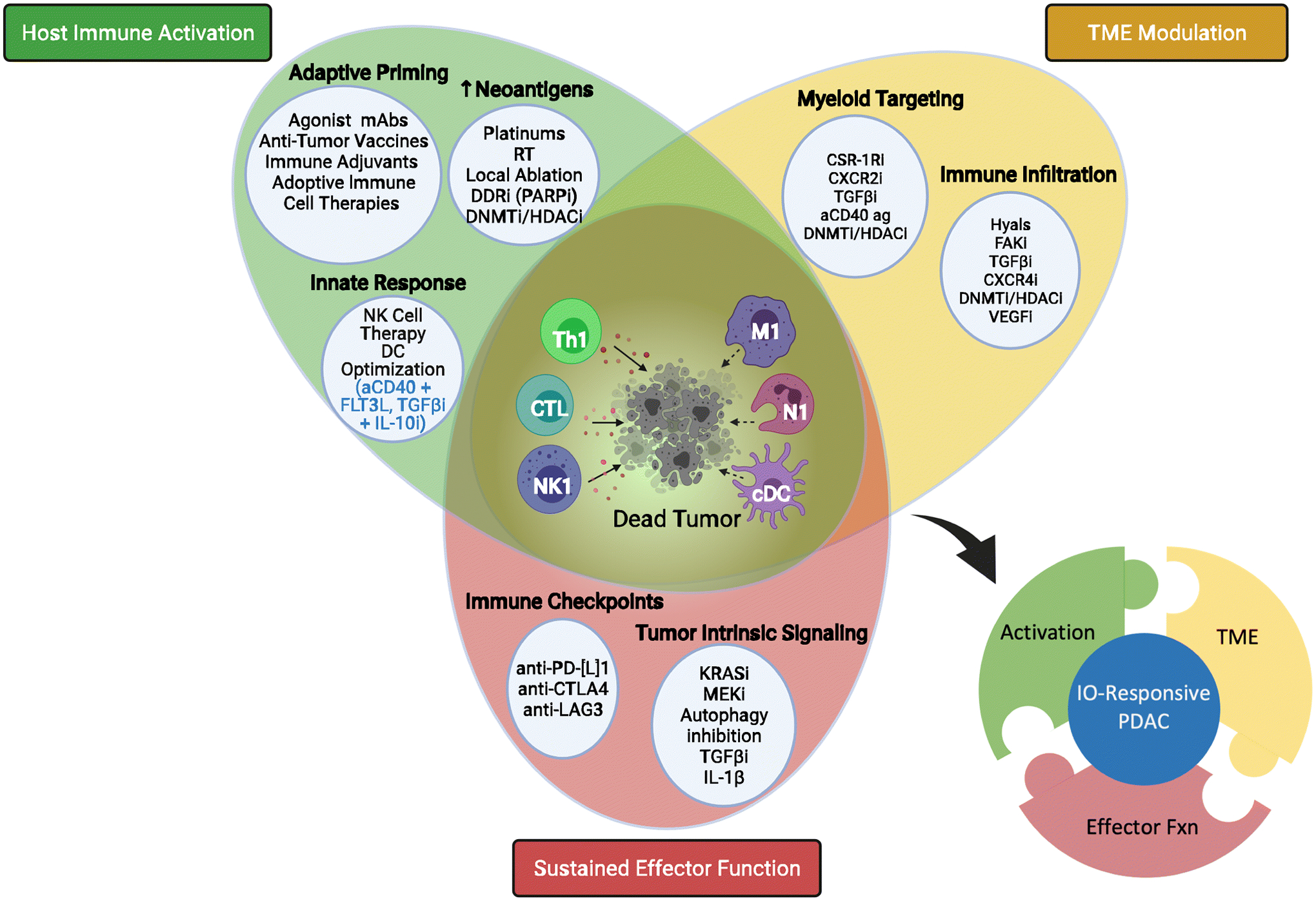
To block immune escape, next generation IO-based approaches for PDAC must be built on targeting immune priming/activation pathways simultaneously with modulating local TME immunosuppression and blocking immune checkpoints blockade for effective sustained anti-tumor immune trafficking and sustained cytotoxic response. This will require several IO and targeted agents on top of conventional therapies. Standard chemotherapy with combinatorial cocktail of an anti-tumor vaccine, co-stimulatory agent (aCD40), TME crosstalk modulator (CXC4Ri or TGFβ trap), and ICB (aPD-1) is one such potential combination. Key (selected terms): [cDC] conventional dendritic cell; [CSF-1Ri] colony-stimulating factor-1 receptor inhibitor; [CTL] cytotoxic T lymphocyte; [CTLA-4] cytotoxic T lymphocyte antigen 4; [DDRi] DNA damage repair inhibitor; [DNMTi] DNA methyltransferase inhibitor; [FAKi] focal adhesion kinase inhibitor; [FLT3L] Fms-related receptor tyrosine kinase 3; [HDACi] histone deacetylase inhibitor; [Hyals] hyalanurinases; [LAG3] lymphocyte activating 3; [M1] tumor associated macrophage, type I phenotype; [mAbs] monoclonal antibodies; [N1] Neutrophil, type 1 phenotype; [NK] – natural killer; [NK1] natural killer cell, type 1 phenotype; [PARPi] Poly (ADP-ribose) polymerase inhibitor; [PD-L1] – programmed death-ligand 1; [RT] radiation therapy; [TGFβ] transforming growth factor beta; [Th1] T helper cell, type 1 phenotype; [VEGFi] Vascular endothelial growth factor inhibitor. Created with BioRender.com.
Acknowledgments
Dr. Azad receives research funding from BMS, Merck, Incyte, Syndax, Intensity, Bayer, EMD Serono. She receives financial compensation as a consultant for AstraZeneca and Merck/EMD Serono.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest Statement:
Dr. Heumann has no conflict of interests.
References
- 1.Rahib L, Wehner MR, Matrisian LM, Nead KT. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw Open. 2021;4(4):e214708. 10.1001/jamanetworkopen.2021.4708. Accessed 4/11/2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians. 2018;68(6):394–424. https://onlinelibrary.wiley.com/doi/abs/10.3322/caac.21492. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Sohal DPS, Kennedy EB, Cinar P, et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. Jco. 2020:JCO.20.01364. 10.1200/JCO.20.01364. [DOI] [PubMed] [Google Scholar]
- 4.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX Versus Gemcitabine for Metastatic Pancreatic Cancer. The New England Journal of Medicine. 2011;364(19):1817–1825. https://search.datacite.org/works/10.1056/nejmoa1011923. 10.1056/nejmoa1011923. [DOI] [PubMed] [Google Scholar]
- 5.Von Hoff DD, Ervin T, Arena FP, et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel Plus Gemcitabine. The New England Journal of Medicine. 2013;369(18):1691–1703. 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX Or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. The New England Journal of Medicine. 2018;379(25):2395–2406. 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 7.Hu ZI, Shia J, Stadler ZK, et al. Evaluating Mismatch Repair Deficiency in Pancreatic Adenocarcinoma: Challenges and Recommendations. Clinical Cancer Research. 2018;24(6):1326–1336. https://www.ncbi.nlm.nih.gov/pubmed/29367431. 10.1158/1078-0432.CCR-17-3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le DT, Durham JN, Smith KN, et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (American Association for the Advancement of Science). 2017;357(6349):409–413. https://search.datacite.org/works/10.1126/science.aan6733. 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of their Inhibition. American Journal of Clinical Oncology. 2016;39(1):98–106. https://www.ncbi.nlm.nih.gov/pubmed/26558876. 10.1097/coc.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD-L1 Expression through 3′-UTR Disruption in Multiple Cancers. Nature (London). 2016;534(7607):402–406. https://www.ncbi.nlm.nih.gov/pubmed/27281199. 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- 11.Yamaki S, Yanagimoto H, Tsuta K, Ryota H, Kon M. PD-L1 Expression in Pancreatic Ductal Adenocarcinoma is a Poor Prognostic Factor in Patients with High CD8+ Tumor-Infiltrating Lymphocytes: Highly Sensitive Detection using Phosphor-Integrated Dot Staining. Int J Clin Oncol. 2017;22(4):726–733. https://www.ncbi.nlm.nih.gov/pubmed/28314962. 10.1007/s10147-017-1112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knudsen ES, Vail P, Balaji U, et al. Stratification of Pancreatic Ductal Adenocarcinoma: Combinatorial Genetic, Stromal, and Immunologic Markers. Clinical Cancer Research. 2017;23(15):4429–4440. https://www.ncbi.nlm.nih.gov/pubmed/28348045. 10.1158/1078-0432.CCR-17-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ROYAL RE, LEVY C, ROSENBERG SA, et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced Or Metastatic Pancreatic Adenocarcinoma. Journal of Immunotherapy (1997). 2010;33(8):828–833. https://www.ncbi.nlm.nih.gov/pubmed/20842054. 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma P, Dirix L, De Vos Filip Yves Francine Leon, et al. Efficacy and Tolerability of Tremelimumab in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Journal of Clinical Oncology. 2018;36(4_suppl):470. 10.1200/JCO.2018.36.4_suppl.470. [DOI] [Google Scholar]
- 15.Kamath SD, Kalyan A, Kircher S, et al. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. The Oncologist (Dayton, Ohio). 2020;25(5):e808–e815. https://onlinelibrary.wiley.com/doi/abs/10.1634/theoncologist.2019-0473. 10.1634/theoncologist.2019-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. The New England Journal of Medicine. 2012;366(26):2455–2465. 10.1056/NEJMoa1200694. 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Reilly EM, Oh D, Dhani N, et al. Durvalumab with Or without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncology. 2019;5(10):1431–1438. 10.1001/jamaoncol.2019.1588. 10.1001/jamaoncol.2019.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terrero G, Pollack T, Sussman DA, Lockhart AC, Hosein PJ. Exceptional Responses to Ipilimumab/Nivolumab (Ipi/Nivo) in Patients (Pts) with Refractory Pancreatic Ductal Adenocarcinoma (PDAC) and Germline BRCA Or RAD51 Mutations. Jco. 2020;38(4):754. https://doi.org/10.1200/JCO.2020.38.4_suppl.754. 10.1200/JCO.2020.38.4_suppl.754. [DOI] [Google Scholar]
- 19.Marabelle A, Le DT, Ascierto PA, et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. Jco. 2020;38(1):1–10. https://doi.org/10.1200/JCO.19.02105. 10.1200/JCO.19.02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Lint S, van Nuffel AM, Wilgenhof S, et al. Priming of cytotoxic T lymphocyte responses by dendritic cells: induction of potent anti-tumor immune responses. Priming of cytotoxic T lymphocyte responses by dendritic cells: induction of potent anti-tumor immune responses. Nova Science; 2013. [Google Scholar]
- 21.Katz SG, Rabinovich PM. T Cell Reprogramming Against Cancer. Methods Mol Biol. 2020;2097:3–44. https://pubmed.ncbi.nlm.nih.gov/31776916 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7063988/. 10.1007/978-1-0716-0203-4_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zamora AE, Crawford JC, Thomas PG. Hitting the Target: How T Cells Detect and Eliminate Tumors. The Journal of Immunology (1950). 2018;200(2):392–399. https://www.ncbi.nlm.nih.gov/pubmed/29311380. 10.4049/jimmunol.1701413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yarchoan M, Johnson Burles A 3, Lutz ER, Laheru DA, Jaffee EM. Targeting Neoantigens to Augment Antitumour Immunity. Nature Reviews. Cancer. 2017;17(4):209–222. https://www.ncbi.nlm.nih.gov/pubmed/28233802. 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. The New England Journal of Medicine. 2017;377(25):2500–2501. 10.1056/NEJMc1713444. 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szeto C, Gounder MM, Parulkar R, Nguyen A, Rabizadeh S, Reddy SK. High Correlation between TMB, Expressed TMB, and Neoantigen Load using Tumor: Normal Whole Exome DNA and Matched Whole Transcriptome RNA Sequencing. Jco. 2020;38(15):e15238. https://doi.org/10.1200/JCO.2020.38.15_suppl.e15238. 10.1200/JCO.2020.38.15_suppl.e15238. [DOI] [Google Scholar]
- 26.Wood MA, Weeder BR, David JK, Nellore A, Thompson RF. Burden of Tumor Mutations, Neoepitopes, and Other Variants are Weak Predictors of Cancer Immunotherapy Response and overall Survival. Genome Medicine. 2020;12(1):33. https://www.ncbi.nlm.nih.gov/pubmed/32228719. 10.1186/s13073-020-00729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balachandran VP, Luksza M, Zhao JN, et al. Identification of Unique Neoantigen Qualities in Long-Term Survivors of Pancreatic Cancer. Nature. 2017;551. https://doi.org/10.1038/nature24462. 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balachandran VP, Łuksza M, Zhao JN, et al. Identification of Unique Neoantigen Qualities in Long-Term Survivors of Pancreatic Cancer. Nature (London). 2017;551(7681):512–516. https://search.datacite.org/works/10.1038/nature24462. 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craig DJ, Nanavaty NS, Devanaboyina M, et al. The Abscopal Effect of Radiation Therapy. Future Oncology (London, England). 2021;17(13):1683–1694. 10.2217/fon-2020-0994. 10.2217/fon-2020-0994. [DOI] [PubMed] [Google Scholar]
- 30.Reits EA, Hodge JW, Herberts CA, et al. Radiation Modulates the Peptide Repertoire, Enhances MHC Class I Expression, and Induces Successful Antitumor Immunotherapy. The Journal of Experimental Medicine. 2006;203(5):1259–1271. https://www.narcis.nl/publication/RecordID/oai:pure.amc.nl:publications%2F47bd0662-ffa3-4794-9962-f1c3eb685fd9. 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using Immunotherapy to Boost the Abscopal Effect. Nature Reviews. Cancer. 2018;18(5):313–322. https://www.ncbi.nlm.nih.gov/pubmed/29449659. 10.1038/nrc.2018.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dovedi SJ, Cheadle EJ, Popple AL, et al. Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by both Resident and Infiltrating Polyclonal T-Cell Populations when Combined with PD-1 Blockade. Clinical Cancer Research. 2017;23(18):5514–5526. https://www.ncbi.nlm.nih.gov/pubmed/28533222. 10.1158/1078-0432.CCR-16-1673. [DOI] [PubMed] [Google Scholar]
- 33.Rudqvist N, Pilones KA, Lhuillier C, et al. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunology Research. 2018;6(2):139–150. https://www.ncbi.nlm.nih.gov/pubmed/29180535. 10.1158/2326-6066.CIR-17-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and Dual Checkpoint Blockade Activate Non-Redundant Immune Mechanisms in Cancer. Nature (London). 2015;520(7547):373–377. https://www.ncbi.nlm.nih.gov/pubmed/25754329. 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parikh A, Wo JY, Ryan DP, et al. A Phase II Study of Ipilimumab and Nivolumab with Radiation in Metastatic Pancreatic Adenocarcinoma. Journal of Clinical Oncology. 2019;37(4_suppl):391. 10.1200/JCO.2019.37.4_suppl.391. [DOI] [Google Scholar]
- 36.Perkhofer L, Gout J, Roger E, et al. DNA Damage Repair as a Target in Pancreatic Cancer: State-of-the-Art and Future Perspectives. Gut. 2021;70(3):606–617. 10.1136/gutjnl-2019-319984. 10.1136/gutjnl-2019-319984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong W, Raufi AG, Safyan RA, Bates SE, Manji GA. BRCA Mutations in Pancreas Cancer: Spectrum, Current Management, Challenges and Future Prospects. Cancer Management and Research. 2020;12:2731–2742. https://www.ncbi.nlm.nih.gov/pubmed/32368150. 10.2147/CMAR.S211151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. The New England Journal of Medicine. 2019;381(4):317–327. 10.1056/NEJMoa1903387. 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vikas P, Borcherding N, Chennamadhavuni A, Garje R. Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Frontiers in Oncology. 2020;10:570. https://www.ncbi.nlm.nih.gov/pubmed/32457830. 10.3389/fonc.2020.00570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiao S, Xia W, Yamaguchi H, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clinical Cancer Research. 2017;23(14):3711–3720. https://www.ncbi.nlm.nih.gov/pubmed/28167507. 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Higuchi T, Flies DB, Marjon NA, et al. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunology Research. 2015;3(11):1257–1268. https://www.ncbi.nlm.nih.gov/pubmed/26138335. 10.1158/2326-6066.CIR-15-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robillard L, Nguyen M, Loehr A, et al. Abstract 3650: Preclinical Evaluation of the PARP Inhibitor Rucaparib in Combination with PD-1 and PD-L1 Inhibition in a Syngeneic BRCA1 Mutant Ovarian Cancer Model. Cancer Research (Chicago, Ill.). 2017;77(13 Supplement):3650. 10.1158/1538-7445.AM2017-3650. [DOI] [Google Scholar]
- 43.Prevo R, Fokas E, Reaper PM, et al. The Novel ATR Inhibitor VE-821 Increases Sensitivity of Pancreatic Cancer Cells to Radiation and Chemotherapy. Cancer Biology & Therapy. 2012;13(11):1072–1081. http://www.tandfonline.com/doi/abs/10.4161/cbt.21093. 10.4161/cbt.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheng H, Huang Y, Xiao Y, et al. ATR Inhibitor AZD6738 Enhances the Antitumor Activity of Radiotherapy and Immune Checkpoint Inhibitors by Potentiating the Tumor Immune Microenvironment in Hepatocellular Carcinoma. Journal for Immunotherapy of Cancer. 2020;8(1):e000340. 10.1136/jitc-2019-000340. 10.1136/jitc-2019-000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wengner AM, Siemeister G, Luecking U, et al. Abstract 321: Synergistic Activity of the ATR Inhibitor BAY 1895344 in Combination with DNA Damage Inducing and DNA Repair Compromising Therapies in Preclinical Tumor Models. Cancer Research (Chicago, Ill.). 2018;78(13 Supplement):321. 10.1158/1538-7445.AM2018-321. [DOI] [Google Scholar]
- 46.Zhang Q, Green MD, Lang X, et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Research (Chicago, Ill.). 2019;79(15):3940–3951. https://www.ncbi.nlm.nih.gov/pubmed/31101760. 10.1158/0008-5472.CAN-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo W, Yang G, Luo W, et al. Novel Therapeutic Strategies and Perspectives for Metastatic Pancreatic Cancer: Vaccine Therapy is More than just a Theory. Cancer Cell International. 2020;20(1):66. https://www.ncbi.nlm.nih.gov/pubmed/32158356. 10.1186/s12935-020-1147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.LUTZ E, YEO CJ, ONNERS B, et al. A Lethally Irradiated Allogeneic Granulocyte-Macrophage Colony Stimulating Factor-Secreting Tumor Vaccine for Pancreatic Adenocarcinoma. A Phase II Trial of Safety, Efficacy, and Immune Activation. Annals of Surgery. 2011;253(2):328–335. https://www.ncbi.nlm.nih.gov/pubmed/21217520. 10.1097/SLA.0b013e3181fd271c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LE DT, BROCKSTEDT DG, GIEDLIN M, et al. A Live-Attenuated Listeria Vaccine (ANZ-100) and a Live-Attenuated Listeria Vaccine Expressing Mesothelin (CRS-207) for Advanced Cancers: Phase I Studies of Safety and Immune Induction. Clinical Cancer Research. 2012;18(3):858–868. https://www.ncbi.nlm.nih.gov/pubmed/22147941. 10.1158/1078-0432.CCR-11-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.BERNHARDT SL, GJERTSEN MK, TRACHSEL S, et al. Telomerase Peptide Vaccination of Patients with Non-Resectable Pancreatic Cancer: A Dose Escalating Phase I II Study. British Journal of Cancer. 2006;95(11):1474–1482. 10.1038/sj.bjc.6603437. 10.1038/sj.bjc.6603437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu A, Jaffee E, Lee V. Current Status of Immunotherapies for Treating Pancreatic Cancer. Curr Oncol Rep. 2019;21(7):1–11. https://www.ncbi.nlm.nih.gov/pubmed/31101991. 10.1007/s11912-019-0811-5. [DOI] [PubMed] [Google Scholar]
- 52.Hassan R, Thomas A, Alewine C, Le DT, Jaffee EM, Pastan I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? Journal of Clinical Oncology. 2016;34(34):4171–4179. https://www.ncbi.nlm.nih.gov/pubmed/27863199. 10.1200/JCO.2016.68.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimizu A, Hirono S, Tani M, et al. Coexpression of MUC16 and Mesothelin is Related to the Invasion Process in Pancreatic Ductal Adenocarcinoma. Cancer Science. 2012;103(4):739–746. https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1349-7006.2012.02214.x. 10.1111/j.1349-7006.2012.02214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Le DT, Wang-Gillam A, Picozzi V, et al. Safety and Survival with GVAX Pancreas Prime and Listeria Monocytogenes-Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. Journal of Clinical Oncology. 2015;33(12):1325–1333. https://www.ncbi.nlm.nih.gov/pubmed/25584002. 10.1200/JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le DT, Picozzi VJ, Ko AH, et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clinical Cancer Research. 2019;25(18):5493–5502. https://www.ncbi.nlm.nih.gov/pubmed/31126960. 10.1158/1078-0432.CCR-18-2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy Converts Nonimmunogenic Pancreatic Tumors into Immunogenic Foci of Immune Regulation. Cancer Immunology Research. 2014;2(7):616–631. https://search.datacite.org/works/10.1158/2326-6066.cir-14-0027. 10.1158/2326-6066.cir-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le DT, Lutz E, Uram JN, et al. Evaluation of Ipilimumab in Combination with Allogeneic Pancreatic Tumor Cells Transfected with a GM-CSF Gene in Previously Treated Pancreatic Cancer. Journal of Immunotherapy (1997). 2013;36(7):382–389. https://www.ncbi.nlm.nih.gov/pubmed/23924790. 10.1097/CJI.0b013e31829fb7a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsujikawa T, Crocenzi T, Durham JN, et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with Or without Nivolumab in Patients with Pancreatic Cancer. Clinical Cancer Research. 2020;26(14):3578–3588. https://www.ncbi.nlm.nih.gov/pubmed/32273276. 10.1158/1078-0432.CCR-19-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng L, Judkins C, Hoare J, et al. 812 Urelumab (Anti-CD137 Agonist) in Combination with Vaccine and Nivolumab Treatments is Safe and Associated with Pathologic Response as Neoadjuvant and Adjuvant Therapy for Resectable Pancreatic Cancer. Journal for Immunotherapy of Cancer. 2020;8(Suppl 3):A862. 10.1136/jitc-2020-SITC2020.0812. 10.1136/jitc-2020-SITC2020.0812. [DOI] [Google Scholar]
- 60.Lepisto AJ, Moser AJ, Zeh H, et al. A Phase I/II Study of a MUC1 Peptide Pulsed Autologous Dendritic Cell Vaccine as Adjuvant Therapy in Patients with Resected Pancreatic and Biliary Tumors. Cancer Therapy. 2008;6(B):955–964. https://www.ncbi.nlm.nih.gov/pubmed/19129927. [PMC free article] [PubMed] [Google Scholar]
- 61.Gatti-Mays ME, Strauss J, Donahue RN, et al. A Phase I Dose-Escalation Trial of BN-CV301, a Recombinant Poxviral Vaccine Targeting MUC1 and CEA with Costimulatory Molecules. Clinical Cancer Research. 2019;25(16):4933–4944. https://www.ncbi.nlm.nih.gov/pubmed/31110074. 10.1158/1078-0432.CCR-19-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suzuki N, Hazama S, Iguchi H, et al. Phase II Clinical Trial of Peptide Cocktail Therapy for Patients with Advanced Pancreatic Cancer: VENUS‐ PC Study. Cancer Science. 2017;108(1):73–80. https://onlinelibrary.wiley.com/doi/abs/10.1111/cas.13113. 10.1111/cas.13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Middleton G, Prof, Silcocks P, BMBCh, Cox T, PhD, et al. Gemcitabine and Capecitabine with Or without Telomerase Peptide Vaccine GV1001 in Patients with Locally Advanced Or Metastatic Pancreatic Cancer (TeloVac): An Open-Label, Randomised, Phase 3 Trial. The Lancet Oncology. 2014;15(8):829–840. https://www.clinicalkey.es/playcontent/1-s2.0-S1470204514702360. 10.1016/S1470-2045(14)70236-0. [DOI] [PubMed] [Google Scholar]
- 64.Mirandola L, Chiriva-Internati M, Bresalier R, Marincola FM, Figueroa JA, Dahlbeck S. Preliminary Report of a Novel Formulation of Clinical-Grade, Fully Matured, Tumor-Associated Peptide-Loaded Dendritic Cells for Cancer Immunotherapy. Translational Medicine Communications. 2019;4(1):1–10. https://explore.openaire.eu/search/publication?articleId=dedup_wf_001∷cd3fa7e5e8d0a2fdac3aae255cd6d281. 10.1186/s41231-019-0049-0. [DOI] [Google Scholar]
- 65.Peng M, Mo Y, Wang Y, et al. Neoantigen Vaccine: An Emerging Tumor Immunotherapy. Molecular Cancer. 2019;18(1):128. https://search.datacite.org/works/10.1186/s12943-019-1055-6. 10.1186/s12943-019-1055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Research. 2020;48(W1):W449–W454. https://www.ncbi.nlm.nih.gov/pubmed/32406916. 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ott PA, Hu Z, Keskin DB, et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature (London). 2017;547(7662):217–221. https://search.datacite.org/works/10.1038/nature22991. 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hilf N, Kuttruff-Coqui S, Frenzel K, et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature (London). 2018;565(7738):240–245. https://search.datacite.org/works/10.1038/s41586-018-0810-y. 10.1038/s41586-018-0810-y. [DOI] [PubMed] [Google Scholar]
- 69.Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature (London). 2018;565(7738):234–239. https://search.datacite.org/works/10.1038/s41586-018-0792-9. 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fang Y, Mo F, Shou J, et al. A Pan-Cancer Clinical Study of Personalized Neoantigen Vaccine Monotherapy in Treating Patients with various Types of Advanced Solid Tumors. Clinical Cancer Research. 2020:clincanres.2881.2019. https://www.ncbi.nlm.nih.gov/pubmed/32439700. 10.1158/1078-0432.CCR-19-2881. [DOI] [PubMed] [Google Scholar]
- 71.Cohen RB, Twardowski P, Johnson ML, et al. GEN-009, a Neoantigen Vaccine Containing ATLAS Selected Neoantigens, to Generate Broad Sustained Immunity Against Immunogenic Tumor Mutations and Avoid Inhibitory Peptides. Journal of Clinical Oncology. 2020;38(15_suppl):3107. 10.1200/JCO.2020.38.15_suppl.3107.32687451 [DOI] [Google Scholar]
- 72.Mueller S, Taitt JM, Villanueva-Meyer JE, et al. Mass Cytometry Detects H3.3K27M-Specific Vaccine Responses in Diffuse Midline Glioma. The Journal of Clinical Investigation. 2020;130(12):6325–6337. https://www.ncbi.nlm.nih.gov/pubmed/32817593. 10.1172/JCI140378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ott PA, Hu-Lieskovan S, Chmielowski B, et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-Small Cell Lung Cancer, Or Bladder Cancer. Cell (Cambridge). 2020;183(2):347–362.e24. 10.1016/j.cell.2020.08.053. 10.1016/j.cell.2020.08.053. [DOI] [PubMed] [Google Scholar]
- 74.Kinkead HL, Hopkins A, Lutz E, et al. Combining STING-Based Neoantigen-Targeted Vaccine with Checkpoint Modulators Enhances Antitumor Immunity in Murine Pancreatic Cancer. JCI Insight. 2018;3(20). https://search.datacite.org/works/10.1172/jci.insight.122857. 10.1172/jci.insight.122857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ryschich Eduard, Nötzel Tanja, Hinz Ulf, et al. Control of T-Cell–Mediated Immune Response by HLA Class I in Human Pancreatic Carcinoma. Clinical Cancer Research. 2005;11(2):498–504. http://clincancerres.aacrjournals.org/content/11/2/498.abstract. [PubMed] [Google Scholar]
- 76.Yamamoto K, Venida A, Yano J, et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature (London). 2020;581(7806):100–105. https://www.ncbi.nlm.nih.gov/pubmed/32376951. 10.1038/s41586-020-2229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mukhopadhyay S, Biancur DE, Parker SJ, et al. Autophagy is Required for Proper Cysteine Homeostasis in Pancreatic Cancer through Regulation of SLC7A11. Proceedings of the National Academy of Sciences - PNAS. 2021;118(6):e2021475118. https://www.ncbi.nlm.nih.gov/pubmed/33531365. 10.1073/pnas.2021475118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wabitsch S, McVey JC, Ma C, et al. Hydroxychloroquine can Impair Tumor Response to Anti-PD1 in Subcutaneous Mouse Models. iScience. 2021;24(1):101990. 10.1016/j.isci.2020.101990. 10.1016/j.isci.2020.101990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolpin BM, Rubinson DA, Wang X, et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition using Hydroxychloroquine in Patients with Metastatic Pancreatic Adenocarcinoma. The Oncologist (Dayton, Ohio). 2014;19(6):637–638. https://onlinelibrary.wiley.com/doi/abs/10.1634/theoncologist.2014-0086. 10.1634/theoncologist.2014-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karasic TB, O’Hara MH, Loaiza-Bonilla A, et al. Effect of Gemcitabine and Nab-Paclitaxel with Or without Hydroxychloroquine on Patients with Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncology. 2019;5(7):993–998. 10.1001/jamaoncol.2019.0684. 10.1001/jamaoncol.2019.0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Samaras P, Tusup M, Nguyen-Kim T, et al. Phase I Study of a Chloroquine–gemcitabine Combination in Patients with Metastatic Or Unresectable Pancreatic Cancer. Cancer Chemother Pharmacol. 2017;80(5):1005–1012. https://www.ncbi.nlm.nih.gov/pubmed/28980060. 10.1007/s00280-017-3446-y. [DOI] [PubMed] [Google Scholar]
- 82.Yang A, Herter-Sprie G, Zhang H, et al. Autophagy sustains pancreatic cancer growth through both cell autonomous and non-autonomous mechanisms. Cancer discovery. 2018;8(3):276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Piffoux M, Eriau E, Cassier PA. Autophagy as a Therapeutic Target in Pancreatic Cancer. British Journal of Cancer. 2021;124(2):333–344. https://www.ncbi.nlm.nih.gov/pubmed/32929194. 10.1038/s41416-020-01039-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wabitsch S, McVey JC, Ma C, et al. Hydroxychloroquine can Impair Tumor Response to Anti-PD1 in Subcutaneous Mouse Models. iScience. 2021;24(1):101990. 10.1016/j.isci.2020.101990. 10.1016/j.isci.2020.101990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and Autophagy Inhibition as a Treatment Approach for Pancreatic Cancer. Nature Medicine. 2019;25(4):628–640. https://www.ncbi.nlm.nih.gov/pubmed/30833752. 10.1038/s41591-019-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seton-Rogers S Eliminating Protective Autophagy in KRAS-Mutant Cancers. Nature Reviews. Cancer. 2019;19(5):247. https://www.ncbi.nlm.nih.gov/pubmed/30936466. 10.1038/s41568-019-0137-5. [DOI] [PubMed] [Google Scholar]
- 87.Raufi A, Wong W, Lee SM, Manji GA. MEKiAUTO: A Phase I/II Open-Label Study of Combination Therapy with the MEK Inhibitor Cobimetinib, Immune-Checkpoint Blockade with Atezolizumab, and the AUTOphagy Inhibitor Hydroxychloroquine in KRAS-Mutated Advanced Malignancies. Jco. 2021;39(3):TPS450. 10.1200/JCO.2021.39.3_suppl.TPS450. 10.1200/JCO.2021.39.3_suppl.TPS450. [DOI] [Google Scholar]
- 88.Hong DS, Fakih MG, Strickler JH, et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. The New England Journal of Medicine. 2020;383(13):1207. https://www.ncbi.nlm.nih.gov/pubmed/32955176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li B, Skoulidis F, Falchook G, et al. PS01.07 Registrational Phase 2 Trial of Sotorasib in KRAS P.G12C Mutant NSCLC: First Disclosure of the Codebreak 100 Primary Analysis. Journal of Thoracic Oncology. 2021;16(3):S61. 10.1016/j.jtho.2021.01.321. 10.1016/j.jtho.2021.01.321. [DOI] [Google Scholar]
- 90.Fakih M, Durm GA, Govindan R, et al. Trial in Progress: A Phase Ib Study of AMG 510, a Specific and Irreversible KRASG12C Inhibitor, in Combination with Other Anticancer Therapies in Patients with Advanced Solid Tumors Harboring KRAS P.G12C Mutation (CodeBreak 101). Jco. 2020;38(15):TPS3661. 10.1200/JCO.2020.38.15_suppl.TPS3661. 10.1200/JCO.2020.38.15_suppl.TPS3661. [DOI] [Google Scholar]
- 91.Peng Y, Zhang J, Liang W, et al. Elevation of MMP-9 and IDO Induced by Pancreatic Cancer Cells Mediates Natural Killer Cell Dysfunction. BMC Cancer. 2014;14(1):738. https://www.ncbi.nlm.nih.gov/pubmed/25274283. 10.1186/1471-2407-14-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jun E, Song AY, Choi J, et al. Progressive Impairment of NK Cell Cytotoxic Degranulation is Associated with TGF-B1 Deregulation and Disease Progression in Pancreatic Cancer. Frontiers in Immunology. 2019;10:1354. https://www.ncbi.nlm.nih.gov/pubmed/31281312. 10.3389/fimmu.2019.01354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Peng Y, Xi C, Zhu Y, et al. Altered Expression of CD226 and CD96 on Natural Killer Cells in Patients with Pancreatic Cancer. Oncotarget. 2016;7(41):66586–66594. https://www.ncbi.nlm.nih.gov/pubmed/27626490. 10.18632/oncotarget.11953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lim SA, Kim J, Jeon S, et al. Defective Localization with Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Frontiers in Immunology. 2019;10:496. https://www.ncbi.nlm.nih.gov/pubmed/31024520. 10.3389/fimmu.2019.00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peng Y, Zhu Y, Zhang J, et al. Comprehensive Analysis of the Percentage of Surface Receptors and Cytotoxic Granules Positive Natural Killer Cells in Patients with Pancreatic Cancer, Gastric Cancer, and Colorectal Cancer. Journal of Translational Medicine. 2013;11(1):262. https://www.ncbi.nlm.nih.gov/pubmed/24138752. 10.1186/1479-5876-11-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Freeman AJ, Vervoort SJ, Ramsbottom KM, et al. Natural Killer Cells Suppress T Cell-Associated Tumor Immune Evasion. Cell Reports (Cambridge). 2019;28(11):2784–2794.e5. 10.1016/j.celrep.2019.08.017. 10.1016/j.celrep.2019.08.017. [DOI] [PubMed] [Google Scholar]
- 97.Lee J, Kang TH, Yoo W, et al. An Antibody Designed to Improve Adoptive NK-Cell Therapy Inhibits Pancreatic Cancer Progression in a Murine Model. Cancer Immunology Research. 2019;7(2):219–229. https://www.ncbi.nlm.nih.gov/pubmed/30514792. 10.1158/2326-6066.CIR-18-0317. [DOI] [PubMed] [Google Scholar]
- 98.Segal NH, Infante JR, Sanborn RE, et al. Safety of the Natural Killer (NK) Cell-Targeted Anti-KIR Antibody, Lirilumab (Liri), in Combination with Nivolumab (Nivo) Or Ipilimumab (Ipi) in Two Phase 1 Studies in Advanced Refractory Solid Tumors. Annals of Oncology. 2016;27:vi372. 10.1093/annonc/mdw378.40. 10.1093/annonc/mdw378.40. [DOI] [Google Scholar]
- 99.Wainberg ZA, Diamond JR, Curigliano G, et al. First-Line Durvalumab + Monalizumab, mFOLFOX6, and Bevacizumab Or Cetuximab for Metastatic Microsatellite-Stable Colorectal Cancer (MSS-CRC). Jco. 2020;38(4):128. https://doi.org/10.1200/JCO.2020.38.4_suppl.128. 10.1200/JCO.2020.38.4_suppl.128. [DOI] [Google Scholar]
- 100.Seery TE, Kistler M, Nangia CS, et al. Immunotherapy Combining NK and T Cell Activation with IL-15 Super Agonist (N-803), Off-the-Shelf High-Affinity CD16 NK (haNK) Or PDL1 Targeted haNK and Checkpoint Inhibitor in Relapsed/Refractory Advanced Pancreatic Cancer. Journal of Clinical Oncology. 2020;38(15_suppl):e15015. 10.1200/JCO.2020.38.15_suppl.e15015. [DOI] [Google Scholar]
- 101.Seery TE, Nangia CS, Sender LS, Reddy SK, Soon-Shiong P. Trial in Progress: Open-Label, Randomized, Comparative Phase 2/3 Study of Combination Immunotherapy Plus Standard-of-Care Chemotherapy and SBRT Versus Standard-of-Care Chemotherapy for the Treatment of Locally Advanced Or Metastatic Pancreatic Cancer. Jco. 2021;39(15):TPS4174. https://doi.org/10.1200/JCO.2021.39.15_suppl.TPS4174. 10.1200/JCO.2021.39.15_suppl.TPS4174. [DOI] [Google Scholar]
- 102.Hegde S, Krisnawan VE, Herzog BH, et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell. 2020;37(3):289–307.e9. 10.1016/j.ccell.2020.02.008. 10.1016/j.ccell.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.DeVito NC, Plebanek MP, Theivanthiran B, Hanks BA. Role of Tumor-Mediated Dendritic Cell Tolerization in Immune Evasion. Frontiers in Immunology. 2019;10:2876. https://www.ncbi.nlm.nih.gov/pubmed/31921140. 10.3389/fimmu.2019.02876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.BEATTY GL, CHIOREAN EG, TORIGIAN DA, et al. CD40 Agonists Alter Tumor Stroma and show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science (American Association for the Advancement of Science). 2011;331(6024):1612–1616. https://www.jstor.org/stable/29783936. 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ma HS, Poudel B, Torres ER, et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell–Mediated Anticancer Activity. Cancer Immunology Research. 2019;7(3):428–442. https://www.ncbi.nlm.nih.gov/pubmed/30642833. 10.1158/2326-6066.CIR-18-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH. Sufficiency of CD40 Activation and Immune Checkpoint Blockade for T Cell Priming and Tumor Immunity. Proceedings of the National Academy of Sciences - PNAS. 2020;117(14):8022–8031. https://www.ncbi.nlm.nih.gov/pubmed/32213589. 10.1073/pnas.1918971117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Soong R, Song L, Trieu J, et al. Direct T Cell Activation Via CD40 Ligand Generates High Avidity CD8+ T Cells Capable of Breaking Immunological Tolerance for the Control of Tumors. PloS One. 2014;9(3):e93162. https://www.ncbi.nlm.nih.gov/pubmed/24664420. 10.1371/journal.pone.0093162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Von Hoff DD, Ramanathan RK, Borad MJ, et al. Gemcitabine Plus Nab-Paclitaxel is an Active Regimen in Patients with Advanced Pancreatic Cancer: A Phase I/II Trial. Journal of Clinical Oncology. 2011;29(34):4548–4554. http://jco.ascopubs.org/content/29/34/4548.abstract. 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Hara MH, O’Reilly EM, Varadhachary G, et al. CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab) and Chemotherapy, with Or without Nivolumab, for the Treatment of Metastatic Pancreatic Adenocarcinoma: An Open-Label, Multicentre, Phase 1b Study. The Lancet Oncology. 2021;22(1):118–131. 10.1016/S1470-2045(20)30532-5. 10.1016/S1470-2045(20)30532-5. [DOI] [PubMed] [Google Scholar]
- 110.O’Hara MH, O’Reilly EM, …, Vonderheide RH. 4019: Gemcitabine (Gem) and Nab-Paclitaxel (NP) ± Nivolumab (Nivo) ± CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab), in Patients (Pts) with Untreated Metastatic Pancreatic Adenocarcinoma (mPDAC): Phase (Ph) 2 Final Results. J Clin Oncol. 2021;39. https://meetinglibrary.asco.org/record/196811/abstract. [Google Scholar]
- 111.Thomas LJ, He L, Gergel LE, et al. Abstract 3217: Preclinical Evaluation of the Recombinant Dendritic Cell Growth Factor CDX-301 (Flt3L), and AST-008, a TLR9 Agonist SNA. Cancer Res. 2019;79(13):3217. http://cancerres.aacrjournals.org/content/79/13_Supplement/3217.abstract. 10.1158/15387445.AM2019-3217. [DOI] [Google Scholar]
- 112.Yu Q, Kovacs C, Yue FY, Ostrowski MA. The Role of the p38 Mitogen-Activated Protein Kinase, Extracellular Signal-Regulated Kinase, and Phosphoinositide-3-OH Kinase Signal Transduction Pathways in CD40 Ligand-Induced Dendritic Cell Activation and Expansion of Virus-Specific CD8+ T Cell Memory Responses. The Journal of Immunology (1950). 2004;172(10):6047–6056. http://www.jimmunol.org/cgi/content/abstract/172/10/6047. 10.4049/jimmunol.172.10.6047. [DOI] [PubMed] [Google Scholar]
- 113.Sanborn RE, Gabrail NY, Bhardwaj N, et al. Abstract LB-194: First-in-Human Phase I Study of the CD40 Agonist mAb CDX-1140 and in Combination with CDX-301 (rhFLT3L) in Patients with Advanced Cancers: Interim Results. Cancer Res. 2019;79(13 Supplement):LB-194. http://cancerres.aacrjournals.org/content/79/13_Supplement/LB-194.abstract. 10.1158/1538-7445.AM2019-LB-194. [DOI] [Google Scholar]
- 114.Sun Z, Fourcade J, Pagliano O, et al. IL10 and PD-1 Cooperate to Limit the Activity of Tumor-Specific CD8+ T Cells. Cancer Research (Chicago, Ill.). 2015;75(8):1635–1644. https://www.ncbi.nlm.nih.gov/pubmed/25720800. 10.1158/0008-5472.CAN-14-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Naing A, Papadopoulos KP, Autio KA, et al. Safety, Antitumor Activity, and Immune Activation of Pegylated Recombinant Human Interleukin-10 (AM0010) in Patients with Advanced Solid Tumors. Journal of Clinical Oncology. 2016;34(29):3562–3569. https://www.ncbi.nlm.nih.gov/pubmed/27528724. 10.1200/JCO.2016.68.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hecht JR, Lonardi S, Bendell J, et al. Randomized Phase III Study of FOLFOX Alone Or with Pegilodecakin as Second-Line Therapy in Patients with Metastatic Pancreatic Cancer that Progressed After Gemcitabine (SEQUOIA). Journal of Clinical Oncology. 2021;39(10):1108–1118. https://www.ncbi.nlm.nih.gov/pubmed/33555926. 10.1200/JCO.20.02232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Spigel D, Jotte R, Nemunaitis J, et al. Randomized Phase 2 Studies of Checkpoint Inhibitors Alone Or in Combination with Pegilodecakin in Patients with Metastatic NSCLC (CYPRESS 1 and CYPRESS 2). Journal of Thoracic Oncology. 2021;16(2):327–333. 10.1016/j.jtho.2020.10.001. 10.1016/j.jtho.2020.10.001. [DOI] [PubMed] [Google Scholar]
- 118.Phan U, Ueda R, Mangadu R, et al. Development of the Anti-IL-10 mAb MK-1966 in Combination with in Situ Vaccination of a TLR9 Agonist SD-101 for Cancer Immunotherapy. Eur J Cancer. 2016;69:S91–S92. 10.1016/S0959-8049(16)32870-2. [DOI] [Google Scholar]
- 119.Ni G, Zhang L, Yang X, et al. Targeting Interleukin-10 Signalling for Cancer Immunotherapy, a Promising and Complicated Task. Human Vaccines & Immunotherapeutics. 2020;16(10):2328–2332. http://www.tandfonline.com/doi/abs/10.1080/21645515.2020.1717185. 10.1080/21645515.2020.1717185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Slaney CY, Kershaw MH, Darcy PK. Trafficking of T Cells into Tumors. Cancer Research (Chicago, Ill.). 2014;74(24):7168–7174. https://www.ncbi.nlm.nih.gov/pubmed/25477332. 10.1158/0008-5472.CAN-14-2458. [DOI] [PubMed] [Google Scholar]
- 121.Hilmi M, Nicolle R, Bousquet C, Neuzillet C. Cancer-Associated Fibroblasts: Accomplices in the Tumor Immune Evasion. Cancers. 2020;12(10):1–23. https://search.proquest.com/docview/2451852116. 10.3390/cancers12102969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grauel AL, Nguyen B, Ruddy D, et al. TGFβ-Blockade Uncovers Stromal Plasticity in Tumors by Revealing the Existence of a Subset of Interferon-Licensed Fibroblasts. Nature Communications. 2020;11(1):6315. https://www.ncbi.nlm.nih.gov/pubmed/33298926. 10.1038/s41467-020-19920-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Moore MJ, Hamm J, Dancey J, et al. Comparison of Gemcitabine Versus the Matrix Metalloproteinase Inhibitor BAY 12–9566 in Patients with Advanced Or Metastatic Adenocarcinoma of the Pancreas: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. Journal of Clinical Oncology. 2003;21(17):3296–3302. http://jco.ascopubs.org/content/21/17/3296.abstract. 10.1200/JCO.2003.02.098. [DOI] [PubMed] [Google Scholar]
- 124.BRAMHALL SR, SCHULZ J, NEMUNAITIS J, BROWN PD, BAILLET M, BUCKETS JAC. A Double-Blind Placebo-Controlled, Randomised Study Comparing Gemcitabine and Marimastat with Gemcitabine and Placebo as First Line Therapy in Patients with Advanced Pancreatic Cancer. British Journal of Cancer. 2002;87(2):161–167. 10.1038/sj.bjc.6600446. 10.1038/sj.bjc.6600446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Whatcott CJ, Diep CH, Jiang P, et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clinical Cancer Research. 2015;21(15):3561–3568. https://www.ncbi.nlm.nih.gov/pubmed/25695692. 10.1158/1078-0432.CCR-14-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut. 2013;62(1):112–120. 10.1136/gutjnl-2012-302529. 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ramanathan RK, McDonough SL, Philip PA, et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. Journal of Clinical Oncology. 2019;37(13):1062–1069. https://www.ncbi.nlm.nih.gov/pubmed/30817250. 10.1200/JCO.18.01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hingorani SR, Zheng L, Bullock AJ, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients with Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. Journal of Clinical Oncology. 2018;36(4):359–366. https://www.ncbi.nlm.nih.gov/pubmed/29232172. 10.1200/JCO.2017.74.9564. [DOI] [PubMed] [Google Scholar]
- 129.Van Cutsem E, Tempero MA, Sigal D, et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. Journal of Clinical Oncology. 2020;38(27):3185–3194. https://www.ncbi.nlm.nih.gov/pubmed/32706635. 10.1200/JCO.20.00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Blair AB, Kim VM, Muth ST, et al. Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clinical Cancer Research. 2019;25(17):5351–5363. https://www.ncbi.nlm.nih.gov/pubmed/31186314. 10.1158/1078-0432.CCR-18-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yao W, Maitra A, Ying H. Recent Insights into the Biology of Pancreatic Cancer. EBioMedicine. 2020;53:102655. 10.1016/j.ebiom.2020.102655. 10.1016/j.ebiom.2020.102655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lo A, Li C, Buza EL, et al. Fibroblast Activation Protein Augments Progression and Metastasis of Pancreatic Ductal Adenocarcinoma. JCI Insight. 2017;2(19). https://www.ncbi.nlm.nih.gov/pubmed/28978805. 10.1172/jci.insight.92232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ostermann Elinborg, Garin-Chesa Pilar, Heider Karl Heinz, et al. Effective Immunoconjugate Therapy in Cancer Models Targeting a Serine Protease of Tumor Fibroblasts. Clinical Cancer Research. 2008;14(14):4584–4592. http://clincancerres.aacrjournals.org/content/14/14/4584.abstract. 10.1158/1078-0432.CCR-07-5211. [DOI] [PubMed] [Google Scholar]
- 134.WANG LS, LO A, SOLOMIDES CC, et al. Targeting Fibroblast Activation Protein in Tumor Stroma with Chimeric Antigen Receptor T Cells can Inhibit Tumor Growth and Augment Host Immunity without Severe Toxicity. Cancer Immunology Research. 2014;2(2):154–166. https://www.ncbi.nlm.nih.gov/pubmed/24778279. 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nugent FW, Cunningham C, Barve MA, et al. Phase 2 Study of Talabostat/Gemcitabine in Stage IV Pancreatic Cancer. Journal of Clinical Oncology. 2007;25(18_suppl):4616. 10.1200/jco.2007.25.18_suppl.4616.17925557 [DOI] [Google Scholar]
- 136.Hofheinz R-, al-Batran S-, Hartmann F, et al. Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients with Metastatic Colorectal Cancer . Oncology Research and Treatment. 2003;26(1):44–48. https://www.karger.com/Article/Abstract/69863. 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 137.Boyd LNC, Andini KD, Peters GJ, Kazemier G, Giovannetti E. Heterogeneity and Plasticity of Cancer-Associated Fibroblasts in the Pancreatic Tumor Microenvironment. Seminars in Cancer Biology. 2021. 10.1016/j.semcancer.2021.03.006. 10.1016/j.semcancer.2021.03.006. [DOI] [PubMed] [Google Scholar]
- 138.Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell. 2015;28(6):831–833. 10.1016/j.ccell.2015.11.002. 10.1016/j.ccell.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 139.Rhim A, Oberstein P, Thomas D, et al. Stromal Elements Act to Restrain, rather than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2014;25(6):735–747. 10.1016/j.ccr.2014.04.021. 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ene–Obong A, Clear AJ, Watt J, et al. Activated Pancreatic Stellate Cells Sequester CD8+ T Cells to Reduce their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology (New York, N.Y. 1943). 2013;145(5):1121–1132. https://www.clinicalkey.es/playcontent/1-s2.0-S0016508513010767. 10.1053/j.gastro.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proceedings of the National Academy of Sciences of the United States of America. 2013. https://explore.openaire.eu/search/publication?articleId=dedup_wf_001∷a4554aee871787ca179594681f5fbc79. 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Seo YD, Jiang X, Sullivan KM, et al. Mobilization of CD8 + T Cells Via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clinical Cancer Research. 2019;25(13):3934–3945. https://www.ncbi.nlm.nih.gov/pubmed/30940657. 10.1158/1078-0432.CCR-19-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bockorny B, Semenisty V, Macarulla T, et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nature Medicine. 2020;26(6):878–885. https://www.ncbi.nlm.nih.gov/pubmed/32451495. 10.1038/s41591-020-0880-x. [DOI] [PubMed] [Google Scholar]
- 144.Wang-Gillam A, MD, Li C, Prof, Bodoky G, Prof, et al. Nanoliposomal Irinotecan with Fluorouracil and Folinic Acid in Metastatic Pancreatic Cancer After Previous Gemcitabine-Based Therapy (NAPOLI-1): A Global, Randomised, Open-Label, Phase 3 Trial. The Lancet (British Edition). 2016;387(10018):545–557. https://www.clinicalkey.es/playcontent/1-s2.0-S0140673615009861. 10.1016/S0140-6736(15)00986-1. [DOI] [PubMed] [Google Scholar]
- 145.Derynck R, Turley SJ, Akhurst RJ. TGFβ Biology in Cancer Progression and Immunotherapy. Nature Reviews. Clinical Oncology. 2021;18(1):9–34. https://www.ncbi.nlm.nih.gov/pubmed/32710082. 10.1038/s41571-020-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bartholin L, Cyprian FS, Vincent D, et al. Generation of Mice with Conditionally Activated Transforming Growth Factor Beta Signaling through the TbetaRI/ALK5 Receptor. Genesis (New York, N.Y. : 2000). 2008;46(12):724–731. https://www.ncbi.nlm.nih.gov/pubmed/18821589. 10.1002/dvg.20425. [DOI] [PubMed] [Google Scholar]
- 147.Park H, Bang J, Nam A, et al. The Prognostic Role of Soluble TGF‐ beta and its Dynamics in Unresectable Pancreatic Cancer Treated with Chemotherapy. Cancer Medicine (Malden, MA). 2020;9(1):43–51. https://onlinelibrary.wiley.com/doi/abs/10.1002/cam4.2677. 10.1002/cam4.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Soares KC, Rucki AA, Kim V, et al. TGF-B Blockade Depletes T Regulatory Cells from Metastatic Pancreatic Tumors in a Vaccine Dependent Manner. Oncotarget. 2015;6(40):43005–43015. https://www.ncbi.nlm.nih.gov/pubmed/26515728. 10.18632/oncotarget.5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature (London). 2018;554(7693):544–548. https://www.ncbi.nlm.nih.gov/pubmed/29443960. 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Principe DR, Park A, Dorman MJ, et al. TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Molecular Cancer Therapeutics. 2019;18(3):613–620. https://www.ncbi.nlm.nih.gov/pubmed/30587556. 10.1158/1535-7163.MCT-18-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Melisi D, Garcia-Carbonero R, Macarulla T, et al. Galunisertib Plus Gemcitabine Vs. Gemcitabine for First-Line Treatment of Patients with Unresectable Pancreatic Cancer. British Journal of Cancer. 2018;119(10):1208–1214. https://www.ncbi.nlm.nih.gov/pubmed/30318515. 10.1038/s41416-018-0246-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Melisi D, Oh D, Hollebecque A, et al. Safety and Activity of the TGFβ Receptor I Kinase Inhibitor Galunisertib Plus the Anti-PD-L1 Antibody Durvalumab in Metastatic Pancreatic Cancer. Journal for Immunotherapy of Cancer. 2021;9(3):e002068. 10.1136/jitc-2020-002068. 10.1136/jitc-2020-002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Strauss J, Heery CR, Schlom J, et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clinical Cancer Research. 2018;24(6):1287–1295. https://www.ncbi.nlm.nih.gov/pubmed/29298798. 10.1158/1078-0432.CCR-17-2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Press Release: Merck KGaA, Darmstadt, Germany, Reports Topline Data for Bintrafusp Alfa as Second-Line Monotherapy Treatment in Biliary Tract Cancer. Dow Jones Institutional NewsMar 16,, 2021.. https://global.factiva.com/en/du/article.asp?accessionno=DJDN000020210316eh3g000hi. [Google Scholar]
- 155.Zhang D, Li L, Jiang H, et al. Tumor-Stroma IL1β-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018;78(7):1700–1712. 10.1158/0008-5472.CAN-17-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Research (Chicago, Ill.). 2020;80(5):1088–1101. https://www.ncbi.nlm.nih.gov/pubmed/31915130. 10.1158/0008-5472.CAN-19-2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Johnson BE, Kim TM, Hiltermann TJ, et al. Abstract CT214: CANOPY-1: Safety Run-in Results from Phase (Ph) 3 Study of Canakinumab (CAN) Or Placebo (PBO) in Combination (Comb) with Pembrolizumab (PEM) Plus Platinum-Based Doublet Chemotherapy (Ctx) as 1St Line Therapy in Patients (Pts) with Advanced Or Metastatic NSCLC. Cancer Res. 2020;80(16):CT214. http://cancerres.aacrjournals.org/content/80/16_Supplement/CT214.abstract. 10.1158/1538-7445.AM2020-CT214. [DOI] [Google Scholar]
- 158.Osipov A, Blair AB, Liberto J, et al. Inhibition of Focal Adhesion Kinase Enhances Antitumor Response of Radiation Therapy in Pancreatic Cancer through CD8+ T Cells. Cancer Biology & Medicine. 2021;18(1):206–214. https://www.ncbi.nlm.nih.gov/pubmed/33628595. 10.20892/j.issn.2095-3941.2020.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Stokes JB, Adair SJ, Slack-Davis JK, et al. Inhibition of Focal Adhesion Kinase by PF-562,271 Inhibits the Growth and Metastasis of Pancreatic Cancer Concomitant with Altering the Tumor Microenvironment. Molecular Cancer Therapeutics. 2011;10(11):2135–2145. https://www.ncbi.nlm.nih.gov/pubmed/21903606. 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang-Gillam A, McWilliams R, Lockhart AC, et al. Abstract CT118: Phase I Study of Defactinib Combined with Pembrolizumab and Gemcitabine in Patients with Advanced Cancer: Experiences of Pancreatic Ductal Adenocarcinoma (PDAC) Patients. Cancer Res. 2020;80(16 Supplement):CT118. http://cancerres.aacrjournals.org/content/80/16_Supplement/CT118.abstract. 10.1158/1538-7445.AM2020-CT118. [DOI] [Google Scholar]
- 161.Jiang H, Liu X, Knolhoff BL, et al. Development of Resistance to FAK Inhibition in Pancreatic Cancer is Linked to Stromal Depletion. Gut. 2020;69(1):122–132. 10.1136/gutjnl-2018-317424. 10.1136/gutjnl-2018-317424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen H, Bian A, Yang L, et al. Targeting STAT3 by a Small Molecule Suppresses Pancreatic Cancer Progression. Oncogene. 2021;40(8):1440–1457. https://www.ncbi.nlm.nih.gov/pubmed/33420372. 10.1038/s41388-020-01626-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ho WJ, Jaffee EM, Zheng L. The Tumour Microenvironment in Pancreatic Cancer - Clinical Challenges and Opportunities. Nature Reviews. Clinical Oncology. 2020;17(9):527–540. https://www.ncbi.nlm.nih.gov/pubmed/32398706. 10.1038/s41571-020-0363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liou G, Döppler H, Necela B, et al. Mutant KRAS-Induced Expression of ICAM-1 in Pancreatic Acinar Cells Causes Attraction of Macrophages to Expedite the Formation of Precancerous Lesions. Cancer Discovery. 2015;5(1):52–63. https://www.ncbi.nlm.nih.gov/pubmed/25361845. 10.1158/2159-8290.CD-14-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schmiechen ZC, Stromnes IM. Mechanisms Governing Immunotherapy Resistance in Pancreatic Ductal Adenocarcinoma. Frontiers in Immunology. 2020;11:613815. https://www.ncbi.nlm.nih.gov/pubmed/33584701. 10.3389/fimmu.2020.613815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yang S, Liu Q, Liao Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Frontiers in Cell and Developmental Biology. 2020;8:607209. https://www.ncbi.nlm.nih.gov/pubmed/33505964. 10.3389/fcell.2020.607209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Groth C, Hu X, Weber R, et al. Immunosuppression Mediated by Myeloid-Derived Suppressor Cells (MDSCs) during Tumour Progression. British Journal of Cancer. 2019;120(1):16–25. https://www.ncbi.nlm.nih.gov/pubmed/30413826. 10.1038/s41416-018-0333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chao T, Furth EE, Vonderheide RH. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-Cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunology Research. 2016;4(11):968–982. https://www.ncbi.nlm.nih.gov/pubmed/27737879. 10.1158/2326-6066.CIR-16-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shen M, Hu P, Donskov F, Wang G, Liu Q, Du J. Tumor-Associated Neutrophils as a New Prognostic Factor in Cancer: A Systematic Review and Meta-Analysis. PloS One. 2014;9(6):e98259. https://www.ncbi.nlm.nih.gov/pubmed/24906014. 10.1371/journal.pone.0098259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lianyuan T, Gang L, Ming T, et al. Tumor Associated Neutrophils Promote the Metastasis of Pancreatic Ductal Adenocarcinoma. Cancer Biology & Therapy. 2020;21(10):937–945. http://www.tandfonline.com/doi/abs/10.1080/15384047.2020.1807250. 10.1080/15384047.2020.1807250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Beatty Gregory L., Chiorean Elena G., Fishman Matthew P., et al. CD40 Agonists Alter Tumor Stroma and show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science (American Association for the Advancement of Science). 2011;331(6024):1612–1616. https://www.jstor.org/stable/29783936. 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Leinwand J, Miller G. Regulation and Modulation of Antitumor Immunity in Pancreatic Cancer. Nature Immunology. 2020;21(10):1152–1159. https://www.ncbi.nlm.nih.gov/pubmed/32807942. 10.1038/s41590-020-0761-y. [DOI] [PubMed] [Google Scholar]
- 173.Dickson I CD11b Agonism Overcomes PDAC Immunotherapy Resistance. Nature Reviews. Gastroenterology & Hepatology. 2019;16(9):514. https://www.ncbi.nlm.nih.gov/pubmed/31332304. 10.1038/s41575-019-0191-1. [DOI] [PubMed] [Google Scholar]
- 174.Panni RZ, Herndon JM, Zuo C, et al. Agonism of CD11b Reprograms Innate Immunity to Sensitize Pancreatic Cancer to Immunotherapies. Science Translational Medicine. 2019;11(499):eaau9240. https://www.ncbi.nlm.nih.gov/pubmed/31270275. 10.1126/scitranslmed.aau9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bendell J, Messersmith W, Rasco D, et al. 388 Preliminary Results from KEYNOTE-A36, a Study of GB1275, a First-in-Class Oral CD11b Modulator, Alone and with Pembrolizumab Or Chemotherapy in Specified Advanced Solid Tumors. Journal for Immunotherapy of Cancer. 2020;8(Suppl 3):A413. 10.1136/jitc-2020-SITC2020.0388. 10.1136/jitc-2020-SITC2020.0388. [DOI] [Google Scholar]
- 176.Gunderson AJ, Kaneda MM, Tsujikawa T, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discovery. 2016;6(3):270–285. https://www.ncbi.nlm.nih.gov/pubmed/26715645. 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Das S, Bar-Sagi D. BTK Signaling Drives CD1d Hi CD5 + Regulatory B-Cell Differentiation to Promote Pancreatic Carcinogenesis. Oncogene. 2019;38(17):3316. https://www.ncbi.nlm.nih.gov/pubmed/30635655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Overman M, Javle M, Davis RE, et al. Randomized Phase II Study of the Bruton Tyrosine Kinase Inhibitor Acalabrutinib, Alone Or with Pembrolizumab in Patients with Advanced Pancreatic Cancer. Journal for Immunotherapy of Cancer. 2020;8(1):e000587. 10.1136/jitc-2020-000587. 10.1136/jitc-2020-000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li M, Li M, Yang Y, et al. Remodeling Tumor Immune Microenvironment Via Targeted Blockade of PI3K-Γ and CSF-1/CSF-1R Pathways in Tumor Associated Macrophages for Pancreatic Cancer Therapy. Journal of Controlled Release. 2020;321:23–35. 10.1016/j.jconrel.2020.02.011. 10.1016/j.jconrel.2020.02.011. [DOI] [PubMed] [Google Scholar]
- 180.Candido JB, Morton JP, Bailey P, et al. CSF1R+ Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Reports (Cambridge). 2018;23(5):1448–1460. 10.1016/j.celrep.2018.03.131. 10.1016/j.celrep.2018.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Saung MT, Muth S, Ding D, et al. Targeting Myeloid-Inflamed Tumor with Anti-CSF-1R Antibody Expands CD137+ Effector T-Cells in the Murine Model of Pancreatic Cancer. Journal for Immunotherapy of Cancer. 2018;6(1):118. https://www.ncbi.nlm.nih.gov/pubmed/30424804. 10.1186/s40425-018-0435-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-Cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Research (Chicago, Ill.). 2014;74(18):5057–5069. https://www.ncbi.nlm.nih.gov/pubmed/25082815. 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang-Gillam A, O’Reilly EM, Bendell JC, et al. A Randomized Phase II Study of Cabiralizumab (Cabira) + Nivolumab (Nivo) ± Chemotherapy (Chemo) in Advanced Pancreatic Ductal Adenocarcinoma (PDAC). Journal of Clinical Oncology. 2019;37(4_suppl):TPS465. 10.1200/JCO.2019.37.4_suppl.TPS465. [DOI] [Google Scholar]
- 184.Cassier PA, Garin G, Eberst L, et al. MEDIPLEX: A Phase 1 Study of Durvalumab (D) Combined with Pexidartinib (P) in Patients (Pts) with Advanced Pancreatic Ductal Adenocarcinoma (PDAC) and Colorectal Cancer (CRC). Journal of Clinical Oncology. 2019;37(15_suppl):2579. 10.1200/JCO.2019.37.15_suppl.2579. [DOI] [Google Scholar]
- 185.MITCHEM JB, BRENNAN DJ, HEWITT S, et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Research (Chicago, Ill.). 2013;73(3):1128–1141. https://www.ncbi.nlm.nih.gov/pubmed/23221383. 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tu MM, Abdel-Hafiz HA, Jones RT, et al. Inhibition of the CCL2 Receptor, CCR2, Enhances Tumor Response to Immune Checkpoint Therapy. Communications Biology. 2020;3(1):720. https://www.ncbi.nlm.nih.gov/pubmed/33247183. 10.1038/s42003-020-01441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Noel M, O’Reilly EM, Wolpin BM, et al. Phase 1b Study of a Small Molecule Antagonist of Human Chemokine (C-C Motif) Receptor 2 (PF-04136309) in Combination with Nab-Paclitaxel/Gemcitabine in First-Line Treatment of Metastatic Pancreatic Ductal Adenocarcinoma. Investigational New Drugs. 2020;38(3):800–811. https://www.ncbi.nlm.nih.gov/pubmed/31297636. 10.1007/s10637-019-00830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nywening TM, Belt BA, Cullinan DR, et al. Targeting both Tumour-Associated CXCR2+ Neutrophils and CCR2+ Macrophages Disrupts Myeloid Recruitment and Improves Chemotherapeutic Responses in Pancreatic Ductal Adenocarcinoma. Gut. 2018;67(6):1112–1123. 10.1136/gutjnl-2017-313738. 10.1136/gutjnl-2017-313738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Steele CW, Karim SA, Leach JDG, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016;29(6):832–845. 10.1016/j.ccell.2016.04.014. 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cohen EEW, Harrington KJ, Hong DS, et al. 1044OA Phase Ib/II Study (SCORES) of Durvalumab (D) Plus Danvatirsen (DAN; AZD9150) Or AZD5069 (CX2i) in Advanced Solid Malignancies and Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (RM-HNSCC): Updated Results. Annals of Oncology. 2018;29(suppl_8). https://explore.openaire.eu/search/publication?articleId=sygma_______∷af74c48379df5d8242647725d5958545. 10.1093/annonc/mdy287. [DOI] [Google Scholar]
- 191.AstraZeneca. Phase Ib/II Study of MEDI4736 Evaluated in Different Combinations in Metastatic Pancreatic Ductal Carcinoma (ClinicalTrials.gov Identifier: NCT02583477). https://ClinicalTrials.gov/show/NCT02583477. Updated 2019. Accessed July 17, 2021.
- 192.Ciernikova S, Earl J, García Bermejo ML, Stevurkova V, Carrato A, Smolkova B. Epigenetic Landscape in Pancreatic Ductal Adenocarcinoma: On the Way to Overcoming Drug Resistance? International Journal of Molecular Sciences. 2020;21(11):4091. https://www.ncbi.nlm.nih.gov/pubmed/32521716. 10.3390/ijms21114091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The Emerging Role of Epigenetic Therapeutics in Immuno-Oncology. Nature Reviews. Clinical Oncology. 2020;17(2):75–90. https://www.ncbi.nlm.nih.gov/pubmed/31548600. 10.1038/s41571-019-0266-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ghoneim HE, Fan Y, Moustaki A, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell (Cambridge). 2017;170(1):142–157.e19. 10.1016/j.cell.2017.06.007. 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ritter C, Fan K, Paschen A, et al. Epigenetic Priming Restores the HLA Class-I Antigen Processing Machinery Expression in Merkel Cell Carcinoma. Scientific Reports. 2017;7(1):2290–11. https://www.ncbi.nlm.nih.gov/pubmed/28536458. 10.1038/s41598-017-02608-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Christmas BJ, Rafie CI, Hopkins AC, et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunology Research. 2018;6(12):1561–1577. https://www.ncbi.nlm.nih.gov/pubmed/30341213. 10.1158/2326-6066.CIR-18-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Xiao Q, Zhou D, Rucki AA, et al. Cancer-Associated Fibroblasts in Pancreatic Cancer are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Research (Chicago, Ill.). 2016;76(18):5395–5404. https://www.ncbi.nlm.nih.gov/pubmed/27496707. 10.1158/0008-5472.CAN-15-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Maibach F, Sadozai H, Seyed Jafari SM, Hunger RE, Schenk M. Tumor-Infiltrating Lymphocytes and their Prognostic Value in Cutaneous Melanoma. Frontiers in Immunology. 2020;11:2105. https://search.proquest.com/docview/2448640845. 10.3389/fimmu.2020.02105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zamarin D, Hamid O, Nayak-Kapoor A, et al. Mogamulizumab in Combination with Durvalumab Or Tremelimumab in Patients with Advanced Solid Tumors: A Phase I Study. Clinical Cancer Research. 2020;26(17):4531–4541. https://search.proquest.com/docview/2418118344. 10.1158/1078-0432.CCR-20-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ebert PJR, Cheung J, Yang Y, et al. MAP Kinase Inhibition Promotes T Cell and Anti-Tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity (Cambridge, Mass.). 2016;44(3):609–621. https://search.datacite.org/works/10.1016/j.immuni.2016.01.024. 10.1016/j.immuni.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 201.Liu L, Mayes PA, Eastman S, et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21(7):1639–1651. http://clincancerres.aacrjournals.org/content/21/7/1639.abstract. 10.1158/1078-0432.CCR-14-2339. [DOI] [PubMed] [Google Scholar]
- 202.Loi S, Dushyanthen S, Beavis PA, et al. RAS/MAPK Activation is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clinical Cancer Research. 2016;22(6):1499–1509. https://search.datacite.org/works/10.1158/1078-0432.ccr-15-1125. 10.1158/1078-0432.ccr-15-1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Baumann D, Hägele T, Mochayedi J, et al. Proimmunogenic Impact of MEK Inhibition Synergizes with Agonist Anti-CD40 Immunostimulatory Antibodies in Tumor Therapy. Nature Communications. 2020;11(1):2176. https://www.ncbi.nlm.nih.gov/pubmed/32358491. 10.1038/s41467-020-15979-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Poon E, Mullins S, Watkins A, et al. The MEK Inhibitor Selumetinib Complements CTLA-4 Blockade by Reprogramming the Tumor Immune Microenvironment. Journal for Immunotherapy of Cancer. 2017;5(1):63. https://www.ncbi.nlm.nih.gov/pubmed/28807001. 10.1186/s40425-017-0268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Steinberg SM, Shabaneh TB, Zhang P, et al. Myeloid cells that impair immunotherapy are restored in melanomas which acquire resistance to BRAF inhibitors. Cancer research (Chicago, Ill.). 2017;77(7):1599–1610. https://www.ncbi.nlm.nih.gov/pubmed/28202513. 10.1158/0008-5472.CAN-16-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Allegrezza MJ, Rutkowski MR, Stephen TL, et al. Trametinib Drives T-cell–Dependent Control of KRAS-Mutated Tumors by Inhibiting Pathological Myelopoiesis. Cancer Research (Chicago, Ill.). 2016;76(21):6253–6265. https://www.ncbi.nlm.nih.gov/pubmed/27803104. 10.1158/0008-5472.CAN-16-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yarchoan M, Mohan AA, Dennison L, et al. MEK Inhibition Suppresses B Regulatory Cells and Augments Anti-Tumor Immunity. Plos One. 2019;14(10):e0224600. 10.1371/journal.pone.0224600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bendell J, Ciardiello F, Tabernero J, et al. Efficacy and Safety Results from IMblaze370, a Randomised Phase III Study Comparing Atezolizumab+cobimetinib and Atezolizumab Monotherapy Vs Regorafenib in Chemotherapy-Refractory Metastatic Colorectal Cancer. Annals of Oncology. 2018;29:v123. 10.1093/annonc/mdy208.003. 10.1093/annonc/mdy208.003. [DOI] [Google Scholar]
- 209.Yarchoan M, Cope L, Anders R, Noonan A, Goff L, Azad N. CT043 - A Multicenter Randomized Phase 2 Trial of Atezolizumab as Monotherapy Or in Combination with Cobimetinib in Biliary Tract Cancers (BTCs): A NCI Experimental Therapeutics Clinical Trials Network (ETCTN) Study. 2020. https://www.abstractsonline.com/pp8/#!/9045/presentation/10752.
- 210.Dushyanthen S, Teo ZL, Caramia F, et al. Agonist Immunotherapy Restores T Cell Function Following MEK Inhibition Improving Efficacy in Breast Cancer. Nature Communications. 2017;8(1):606–18. https://search.datacite.org/works/10.1038/s41467-017-00728-9. 10.1038/s41467-017-00728-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Dennison L, Yarchoan M. Context-Dependent Immunomodulatory Effects of MEK Inhibition are Enhanced with T cell Agonist Therapy. Unpublished. [DOI] [PMC free article] [PubMed]
- 212.Largeot A, Pagano G, Gonder S, Moussay E, Paggetti J. The B-Side of Cancer Immunity: The Underrated Tune. Cells (Basel, Switzerland). 2019;8(5):449. https://www.ncbi.nlm.nih.gov/pubmed/31086070. 10.3390/cells8050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab Versus Interferon Beta-1a in Relapsing Multiple Sclerosis. The New England Journal of Medicine. 2017;376(3):221–234. 10.1056/NEJMoa1601277. 10.1056/NEJMoa1601277. [DOI] [PubMed] [Google Scholar]
- 214.Edwards JCW, Szczepanski L, Szechinski J, et al. Efficacy of B-Cell-Targeted Therapy with Rituximab in Patients with Rheumatoid Arthritis. The New England Journal of Medicine. 2004;350(25):2572–2581. http://content.nejm.org/cgi/content/abstract/350/25/2572. 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 215.DiLillo DJ, Yanaba K, Tedder TF. B Cells are Required for Optimal CD4+ and CD8+ T Cell Tumor Immunity: Therapeutic B Cell Depletion Enhances B16 Melanoma Growth in Mice. The Journal of Immunology (1950). 2010;184(7):4006–4016. http://www.jimmunol.org/cgi/content/abstract/184/7/4006. 10.4049/jimmunol.0903009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Aklilu M, Stadler WM, Markiewicz M, et al. Depletion of Normal B Cells with Rituximab as an Adjunct to IL-2 Therapy for Renal Cell Carcinoma and Melanoma. Annals of Oncology. 2004;15(7):1109–1114. https://api.istex.fr/ark:/67375/HXZ-X0MRZ9L3-X/fulltext.pdf. 10.1093/annonc/mdh280. [DOI] [PubMed] [Google Scholar]
- 217.Wejksza K, Lee-Chang C, Bodogai M, et al. Cancer-Produced Metabolites of 5-Lipoxygenase Induce Tumor-Evoked Regulatory B Cells Via Peroxisome Proliferator-Activated Receptor A. The Journal of Immunology (1950). 2013;190(6):2575–2584. https://www.ncbi.nlm.nih.gov/pubmed/23408836. 10.4049/jimmunol.1201920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Shimabukuro-Vornhagen A, Draube A, Liebig TM, Rothe A, Kochanek M, von Bergwelt-Baildon MS. The Immunosuppressive Factors IL-10, TGF-B, and VEGF do Not Affect the Antigen-Presenting Function of CD40-Activated B Cells. Journal of Experimental & Clinical Cancer Research. 2012;31(1):47. https://www.ncbi.nlm.nih.gov/pubmed/22592077. 10.1186/1756-9966-31-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Gonzalez NK, Wennhold K, Balkow S, et al. In Vitro and in Vivo Imaging of Initial B-T-Cell Interactions in the Setting of B-Cell Based Cancer Immunotherapy. Oncoimmunology. 2015;4(9):e1038684. http://www.tandfonline.com/doi/abs/10.1080/2162402X.2015.1038684. 10.1080/2162402X.2015.1038684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yarchoan M, Ho WJ, Mohan A, et al. Effects of B Cell-Activating Factor on Tumor Immunity. JCI Insight. 2020;5(10). https://www.ncbi.nlm.nih.gov/pubmed/32434989. 10.1172/jci.insight.136417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Shurin MR, Ma Y, Keskinov AA, et al. BAFF and APRIL from Activin A-Treated Dendritic Cells Upregulate the Antitumor Efficacy of Dendritic Cells in Vivo. Cancer Research (Chicago, Ill.). 2016;76(17):4959–4969. https://www.ncbi.nlm.nih.gov/pubmed/27364554. 10.1158/0008-5472.CAN-15-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy Converts Nonimmunogenic Pancreatic Tumors into Immunogenic Foci of Immune Regulation. Cancer Immunology Research. 2014;2(7):616–631. https://www.ncbi.nlm.nih.gov/pubmed/24942756. 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]