

Abstract
Orthogonal joints, understood as connections with an angle of 90°, were introduced in the design of the “Geländer” model compounds 1 and 2. The banister, consisting of a conjugated carbazole dimer linked by either 1,3‐butadiyne (2) or a single thiophene (1), wraps around an axis composed of a phthalimide dimer due to the dimensional mismatch of both subunits, which are interconnected by phenylene rungs. The “Geländer” structure was assembled from a monomer comprising the 1,4‐diaminobenzene rung with one amino substituent as part of a 4‐bromo phthalimide subunit forming the orthogonal junction to the axis, and the other as part of a masked 2‐ethynyl carbazole as orthogonal joint to the banister. The macrocycle was obtained by two sequential homocoupling steps. A first dimerization by a reductive homocoupling assembled the axis, while an oxidative acetylene coupling served as ring‐closing reaction. The formed butadiyne was further derivatized to a thiophene, rendering all carbons of the model compound sp2 hybridized. Both helical structures were fully characterized and chirally resolved. Assignment of the enantiomers was achieved by simulation of chiroptical properties and enantiopure synthesis.
Keywords: atropisomer, Geländer molecules, helical chirality, heteroarenes, macrocyclization
A new design for “Geländer” architectures is presented with orthogonal joints between axis, rungs, and banister. Following this design concept, two macrocyclic model compounds are synthesized, fully characterized, and separated into room temperature stable enantiomers with the banister helically wrapped around the axis.
Introduction
The term “Geländer”‐molecule was introduced by Vögtle and co‐workers in the late nineties, pointing at the helical arrangement in their model compounds resembling the banister (in German “Geländer”) of a spiral staircase (Figure 1A). In their conceptual approach, a terphenyl backbone (axis) was decorated with elongated linkers wrapped helically around the axis (Figure 1B). [1] A comparable molecular design was reported by Rathore and co‐workers, avoiding heteroatoms in the banister subunits (Figure 1C). [2] While the concept to force helical chirality was visionary, an intrinsic shortcoming of the molecular design was the independence of both intramolecular junctions of the terphenyl backbone. As a result, besides the helical chiral (M,M) and (P,P) isomers, the achiral (M,P) meso‐form was not only present but even the statistically dominant species. To overcome this drawback, we further developed the ”Geländer” molecule concept to ladder‐type oligomers with length mismatch in both ladder stringers. In the last years, we synthesized various “Geländer” structures with oligophenylene (OP) backbones, wrapped by benzylic ethers,[ 3 , 4 , 5 ] sulfones, [6] or oligophenylene ethanes (Figure 1D). [7] Recently, we even enlarged the radius of the design by interlinking both oligomers by alkynes and wrapped para‐oligophenylene diyne around the OP backbone as a conjugated banister (Figure 1E).[ 8 , 9 ] However, the strained diynes in the banister subunits displayed enlarged reactivity towards oxygen, reducing the stability of these model compounds considerably.
Figure 1.
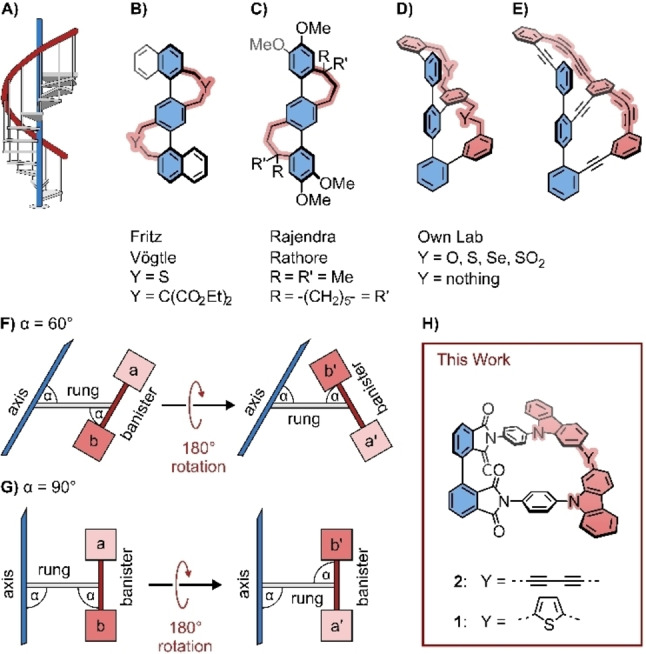
A) Sketch of a spiral staircase as inspiration of “Geländer” molecules with an axis (blue) and a helical banister (red). “Geländer” helices of B) Vögtle, C) Rathore, and D) and E) from our own lab. Geometrical consideration for rotations around the rungs for “Geländer” monomers based on F) 60° and G) 90° angles between subunits. H) New “Geländer” macrocycles 1 and 2 with rectangular arrangements between axle and rung, as well as between rung and banister
Due to the geometric boundary conditions of the benzene synthons present in both the OP backbone and the various wrapping oligophenylenes, the joints between the ladder stringers and the rungs were set at 60°. This architecture based on 60° angled parallelograms causes a variety of challenges. As sketched in Figure 1F), the rotation of the banister subunit around the rung alters the spatial arrangement of the involved building blocks considerably. Neither the distance to the axis nor the spatial arrangement of the involved building blocks are maintained. For example, a 180° rotation around the rung moves not only the initial upper building block (a) into the lower position (a’) but also places it at a further distance to the axis and mounts it at a different angle. As a consequence, each rung subunit of the oligomer has to be developed individually, and its spatial arrangement has to be considered during the formation of the targeted “Geländer” structure. Both aspects are impeding the syntheses of elongated “Geländer” oligomers considerably, and so, mainly trimeric structures with a terphenyl as OP backbone have been reported so far. In order to ease the formation of longer “Geländer” oligomers, repeat units with increased symmetry would be helpful. Particular appealing would be ladder‐type oligomers based on orthogonally (90°) arranged subunits. The conceptual idea is displayed in Figure 1G). An oligomer repeat unit with a rectangular arrangement at both joints, between the axis and rung, and between rung and banister subunit, would no longer require distinguishing between both sides of the banister subunit. A 180° rotation around the rung still relocates the initial upper building block (a) into the lower position (a’), but when the two building blocks (a) and (b) are the same, there is no difference between the two rotamers. From a synthetic point of view, in a ring closing step, both rotamers from the rectangular architecture result in the same product, avoiding “Geländer” objects with dissimilar ring sizes, which were already isolated in the case of 60° angled molecular designs.[ 5 , 6 , 7 ]
The search for molecular fragments, allowing to interlink both linear oligomers representing axle and banister with perpendicularly arranged rungs, guided our interest to five‐membered aromatic subunits comprising a nitrogen atom as the mounting point for the rigid subunit acting as rung. Guided by these design considerations, we came up with the new “Geländer” design displayed in Figure 1H), with phthalimides as axle subunits, carbazoles as banister building blocks, and para‐diaminobenzenes acting as rungs. To explore the synthetic chemistry required for their assembly and first physicochemical properties of this new molecular design, we focus here on the macrocycles 1 and 2 (Figure 1H) as the smallest members of the series of “Geländer” oligomers with orthogonal joints.
Results and Discussion
Design and Retrosynthesis
The symmetry advantage of rectangular joints in the “Geländer” architecture has already been elaborated as motivation in the introduction above. Our retrosynthetic considerations are sketched in Scheme 1. Due to our successful experience in the past,[ 8 , 9 , 10 , 11 , 12 , 13 ] an oxidative acetylene coupling was chosen as macrocyclization reaction. The formed banister structure in macrocycle 2 obtained upon ring closing consists exclusively of sp2 and sp hybridized carbon atoms, and thus, its π‐electrons are delocalized over the entire banister. However, in the past, the strained 1,3‐butadiyne motif in the banister turned out to be the origin of pronounced reactivity handicapping the “Geländer” molecule's stability.[ 8 , 9 ] To avoid such stability limitations, we profit from the reactivity of the 1,3‐butadiyne subunit in 2 as the precursor of a thiophene connecting banister building block in 1. This sulfide heterocycle is not only expected to improve the stability of the target structure 1 but also integrates perfectly into its π‐conjugated banister architecture.
Scheme 1.

Retrosynthetic analysis: Assembly strategy for the macrocycles 1 and 2 from accessible precursors.
As the precursor of macrocycle 2, the dimer D is required, which should be accessible by reductive homocoupling of the monomer M. This monomer combines with the bromine substituent of the phthalic anhydride part and with the masked ethynyl group decorating the carbazole subunit all the functional groups required to subsequently assemble the axes subunit of the “Geländer” macrocycle 2. The monomer M should be assembled by a few classical chemical reactions from either commercially available or already reported precursors like monoprotected ethyne, 2‐bromo‐9H‐carbazole, 4‐fluoronitrobenzene, and 3‐bromophthalic anhydride.
Synthesis
As shown in Scheme 2, monomer 6 was obtained in four steps from commercially available compounds. Nucleophilic aromatic substitution of the fluorine substituent in 4‐fluoronitro‐benzene with the nitrogen of 2‐bromo‐9H‐carbazole provided the N‐substituted carbazole 3. In the first attempts, 3 was isolated in about 70 % yield using dimethylformamide (DMF) as solvent and potassium carbonate as base. The isolated yield was improved to 92 % with dry dimethyl sulfoxide (DMSO) as solvent in the presence of caesium fluoride. [14] In either case, the product was precipitated from the reaction mixture with deionized water as mustard yellow powder. The only observed impurities originated from 4‐fluoronitrobenzene and were removed by precipitating 3 from a saturated dichloromethane (CH2Cl2) solution.
Scheme 2.

Synthesis of the monomer 6. Reagents and conditions: a) CsF, 4‐fluoronitrobenzene, DMSO, 150 °C, 20 h, 92 %; b) HCCSi(CH(CH3)2)3, Pd(PPh3)4, CuI, DIPA, THF, 90 °C, 20 h, 94 %; c) SnCl2, THF, EtOH, 50 °C, 20 h; d) 3‐bromophthalic anhydride, AcOH, 70 °C, 48 h, 84 % isolated yield over both steps c) and d).
In the next step, the bromine substituent of 3 was replaced by a triisopropylsilane masked ethynyl group in a Sonagashira‐Hagihara cross‐coupling reaction. Bromide 3 and the masked ethynyl were heated to 90 °C in a mixture of tertrahydrofuran (THF) and diisopropylamine (volume 1 : 3) in the presence of Pd (PPh3)4 and CuI as catalysts. After purification by column chromatography (CC), carbazole 4 was obtained as a yellow solid in 94 % isolated yield. The nitro group of 4 was reduced to the amino group in 5 using SnCl2 in a mixture of THF and ethanol. The course of the transformation could be monitored by bare eyes as the reaction mixture turned from a turbid yellow mixture into an orange clear solution. After aqueous work‐up, 5 was obtained quantitatively as an orange viscous oil. The reaction between crude 5 and 3‐bromophtalic anhydride in acetic acid yielded 6 in 84 % after filtration as an off‐white solid.
With the adequately functionalized monomeric building block 6 in hands, the assembly of the macrocycles was pursued, as shown in Scheme 3. Reductive nickel mediated homocoupling was performed under an argon protection gas atmosphere. The reaction flask was charged with zinc as a reducing agent and nickel bromide together with triphenylphosphine forming the active catalyst in N,N‐dimethylacetamide (DMAc) at 80 °C, before the monomer 6 was added. After keeping the reaction mixture at 80 °C overnight, aqueous workup and CC provided the desired dimer 7 in 80 % isolated yield as a faint yellow powder. Apart from the desired dimer 7, traces of the debrominated monomer 8 were isolated by CC as a side product. The formation of the catalyst before the addition of the monomer [15] had an important impact on the yield of the dimer. In initial attempts, the isolated yield of the dimer did not exceed 50 %. Increasing the catalyst load from 2.5 mol % over 10 up to 25 mol % and lowering the ligand to catalyst ratio from 7 : 1 to 2 : 1 improved the yield slightly above 60 %. With dry NiBr2 and activated zinc dust, the yield was further improved to 68 %. Interestingly, Ni(PPh3)2Cl2 without additional ligand provided 7 in poorer yields. Replacing zinc as reducing agent by indium or samarium was not successful, as the desired bisphthalimide was not detected in the reaction mixture. With the linear dimer 7 accessible in good yields and reasonable quantities, the macrocyclization to 2 was investigated. In first attempts a stepwise alkyne deprotection by tetrabutylammonium fluoride (TBAF) in wet tetrahydrofuran (THF) followed by an oxidative acetylene coupling using typical Glaser‐Hay conditions[ 10 , 16 ] was considered. Even so complete consumption of deprotected 7 was indicated by matrix‐assisted laser desorption‐ionization mass spectrometry (MALDI‐MS), neither the desired macrocycle nor a conclusive side product could be isolated by CC from the reaction mixture. As alternative Eglinton‐Breslow conditions were explored, which were previously successfully used in our laboratories to form strained butadiynes.[ 8 , 9 ] Again the desired macrocycle was observed by both MALDI‐MS and high‐performance liquid chromatography coupled electron‐spray ionization mass spectrometry (HPLC‐ESI‐MS). It is noteworthy that we were not able to detect the dimer 7 by ESI‐MS, while the macrocyclic product 2 was observed as radical anion. [17] But still, all our attempts to isolate the macrocycle 2 failed. During the struggles optimizing the reaction conditions and isolating 2, we observed that the macrocycle is formed in DMSO with a copper(II) source present after deprotection of the alkynes, even in degassed solvents and under an argon atmosphere. Thus the deprotection of the acetylenes and subsequent oxidative coupling by the same reagent was considered. And indeed, using CuF2 in DMSO under air‐free conditions without any additional base or ligand provided the macrocycle 2, which was precipitated form the reaction mixture by water and isolated by filtration in 50 % yield. To the best of our knowledge, such a one‐pot deprotection and oxidative alkyne homocoupling has not been reported so far. With the desired macrocycle 2 in hands, also the mystery of its challenging isolation was uncovered. The compound displayed poor stability on silica and thus, avoiding conventional CC was crucial for its isolation. Highest yields were observed between 1 mM and 2 mM of 7, lowering the concentration led to increased reaction times and depleted yields. Performing the reaction under aerated conditions, the isolated yield even increased to 94 %. An analytical sample of the macrocycle 2 was purified by gel permeation chromatography (GPC). For our further synthetic endeavors however, precipitation with petroleum ether from a CH2Cl2 solution provided 2 as faint yellow powder in sufficient purity (see Supporting Information for NMR after precipitation and GPC, respectively). To our delight, the isolated macrocycle 2 turned out to be stable at ambient conditions for several months. To circumvent the reactivity of the bent buta‐1,3‐diyn and to provide a conjugated banister consisting exclusively of sp2 hybridized carbon atoms, the transformation of the butadiene motif of 2 into the bridging thiophene of 1 was investigated. To our surprise, this derivatization turned out to be quite challenging. The transformation is usually performed in alkaline solutions of disodium sulfide at elevated temperatures with water acting as proton source.[ 16 , 18 , 19 ] Applying such conditions did neither provide the macrocycle 1, nor any conclusive product emerging from a side reaction. To increase the reaction temperature above the boiling point of water, 2‐methoxy ethanol is suggested as proton source combined with high boiling solvents such as toluene or DMSO.[ 10 , 11 , 20 ] Although under all investigated conditions, consumption of the starting material was observed, only traces (<1 % yield) of the macrocycle 1 could be isolated. It thus seems that the phthalimide backbone lacks the stability required for the alkaline environment at the elevated temperature. [21] As milder method for the transformation, Zhang et al. reported the use of trisulfur radical anions. [22] The radical anion is formed in situ from elemental sulfur and a base in DMF. A more recent variation of the procedure used sodium hydrogen sulfide as base. [23] Applying these conditions at room temperature for three hours provided the desired macrocycle 1 in moderate isolated yields of about 50 %. Monitoring the course of the reaction by TLC showed that the conversion was completed within the first 10 minutes. Quenching the reaction after this period with saturated ammonium chloride solution provided the macrocycle 1 in excellent isolated yields of 96 % after precipitation from CH2Cl2 with petroleum ether as pale yellow powder.
Scheme 3.

Synthesis of the macrocycles 1 and 2. Reagents and conditions: a) Zn, NiBr2, PPh3, DMAc, 80 °C, 20 h, 80 %; b) CuF2, aerated DMSO, 100 °C, 20 h, 94 %; c) S8, NaHS, DMF, 25 °C, 10 min, 96 %.
All reported compounds have been characterized by 1H‐ and 13C NMR spectroscopy and high‐resolution mass spectrometry. Furthermore, the solid‐state structure of macrocycle 2 corroborated its identity.
Structural analysis
The macrocycles 1 and 2 have both been synthesized as racemates. Due to the substantial length mismatch between the phthalimide backbone and the carbazole banister, the latter is expected to wrap helically around the backbone, providing both macrocycles as pairs of enantiomers. Thus, the structural features of these “Geländer” model compounds were of particular interest, such as the integrity and dynamic of their spatial arrangement.
Already the 1H NMR of the racemate of both macrocycles displayed interesting dynamic features. In Figure 2, the 1H NMR spectra of rac‐1 and rac‐2 are displayed with the assignment of all protons. While reasonably sharp resonances were recorded for most protons, the ones of the para‐phenylene rungs interlinking banister and backbone were broadened. The effect was particular pronounced for the phenyl protons in the proximity of the phthalimides’ carbonyl groups and points at the rotation of the para‐phenylene rung along the interlinking axis on the NMR time scale. And indeed, variable temperature NMR experiments (see Supporting Information) displayed a resolved spin system below 220 K for both macrocycles rac‐1 and rac‐2, while fast exchange regimes were reached above 320 K. The analysis of temperature dependence of the spin system provided the coalescence temperature as well as the chemical shift differences in the slow exchange regime, which allowed to determine the rotation barrier. The obtained values are 51.9 kJmol−1 for the para‐phenylene rungs in rac‐1 and 53.7 kJmol−1 for the ones in rac‐2 very comparable. The slightly lower rotation barrier of rac‐1 compared with rac‐2 is already visible in their NMR spectra at 298 K (Figure. 2) as the spin pairs of rac‐1 (dd’ and ee’) are less broadened than the ones of rac‐2 (qq’ and rr’). In summary, the only dynamic motion observed by 1H NMR spectroscopy with samples of rac‐1 and rac‐2 is the rotation of the para‐phenylene rungs.
Figure 2.

1H NMR of the macrocycles rac‐1 and rac‐2 at 298 K measured at 600 MHz in THF‐d8 and the assignment of the signals. Substantial broadening of the peaks from the para‐phenylene rungs labeled by green circles is displayed for both macrocycles pointing at rotation in the time scale of the experiment. VT NMR experiments determining the rotation barrier are provided in the Supporting Information.
First insights concerning the spatial arrangement of these new “Geländer” architectures were obtained by DFT (density functional theory) calculations. The optimized geometries of (P)‐ 1 and (P)‐ 2 are shown in Figure 3 A) and B), respectively, and display the intended helical wrapping of the banister subunit. The conformational analysis based on a combination of an automated sampling method using the tight‐binding model [24] and refinement by DFT showed that the bridging para‐phenylenes avoid the steric bulk in the center of the macrocycle. Geometries with the hydrogens of the bridging para‐phenylene avoiding the center of cavity are found with the lowest energy (see Supporting Information for the calculated structures). In the case of (P)‐ 1, the thiophene unit interlinking both carbazoles in the banister is slightly tilted out of the plane both carbazoles span, thus only partially optimizing the stabilization through increased conjugation. The models further suggest that the helical turn becomes tighter upon the structural change from the 1,3‐butadiyne linker in 2 to the thiophene linker in 1 (Table 1). To corroborate the calculated geometries, the solid‐state structures of the macrocycles were of particular interest. After numerous unsuccessful attempts with both macrocycles, single crystals suitable for X‐ray analysis were obtained from vapor diffusion of n‐hexane into a chloroform solution at room temperature for rac‐ 2. The macrocycle crystalizes in the point group P21/n with an asymmetric unit cell comprising both enantiomers (see Figure S10 in the Supporting Information for the unit cell) and has a herringbone‐type packing (Figure S11). For comparison with the calculated structure, the solid‐state structure of (P)‐ 2 is displayed in Figure 3 C). In the focus of the structural analysis were the angles α and β between backbone and rung of the “Geländer” architecture and between rung and banister, respectively. As shown in Figure 3 A) two dummy atoms (green spheres) were introduced to (P)‐1 to determine these angles. The first one allowed to obtain the angle α between the biphenyl backbone provided by the linked phthalimides and the para‐phenylene rungs. It was placed in the center of the benzene ring of a phthalimide backbone subunit. From this dummy atom, the angle α was defined as the one opened by the rays through the connected carbon atoms of the neighboring subunits, i. e., the carbon atom of the neighboring phthalimide engaged in the backbone single bond and the carbon atom of the para‐phenylene rung connected to the phthalimide nitrogen. To determine the angle β between the rung and the banister, a dummy atom was placed in the center of the line connecting the C2 and C7 of the carbazole banister subunit. Form this dummy atom, the angle between the C2−C7 connecting line and the ray through the carbon atom of the para‐phenylene rung connected to the carbazole nitrogen was measured as β.
Figure 3.

Optimized geometries (B3LYP/6‐311G**) of (P)‐1 in A) and of (P)‐2 in B). C) The arrangement of the (P)‐2 enantiomer extracted from the solid‐state structure of rac‐2. The dummy atoms and the subsidiary lines to obtain the angles α and β between the subunits of the “Geländer” structure are displayed in A). The various dihedral angles φ analyzed to compare the structures are sketched in B). Note that for dihedral angles φ1‐4 only one of both angles is highlighted to improve visibility.
Table 1.
Angles of the orthogonal joints (α and β), the helical turn (φa), the dihedral angles between the axis and rung (φ1 and φ2), and the dihedral angles between the banister and rung (φ3 and φ4) in the optimized structures of (P)‐1, (P)‐2, and in the solid‐state structure of rac‐2. [i] averaged value
|
|
Α |
β |
φa |
φ1 [i] |
φ2 [i] |
φ3 [i] |
φ4 [i] |
|---|---|---|---|---|---|---|---|
|
(P)‐2 X‐ray |
+91° |
+90° |
+63° |
+50° |
+47° |
+63° |
+53° |
|
(P)‐2 model |
+92° |
+90° |
+68° |
+45° |
+46° |
+66° |
+66° |
|
(P)‐1 model |
+91° |
+90° |
+59° |
+47° |
+47° |
+67° |
+64° |
As listed in Table 1, the angles α and β are not only very comparable for the calculated model structures and the solid‐state structure; they are in all cases equal to or very close to 90°. This orthogonal arrangement of the subunits was the major intention of the revised “Geländer” design. Indeed, the obtained angles corroborate the spatial symmetry of the motive and thus its suitability even for elongated model compounds. A variety of dihedral angles φ were analyzed to compare the structures further. The different dihedral angles are displayed in Figure 3B). The most interesting one is φa, which is the angle between the planes defined by the phthalimide subunits of the backbone and is a measure for the helical turn achieved by the particular “Geländer” architecture. The values of φa are listed in Table 1 and allow an in‐depth comparison of the simulated and recorded structure of (P)‐2 with 68° and 63°, respectively. The difference of 5° points at a more spacious arrangement of the macrocycle in the calculated structure compared to its solid‐state packing in the racemate. The difference might reflect the different boundary conditions of the simulation in open space (vacuum) and the x‐ray diffraction obtained from the close‐packed form in a single crystal (see Figure S11 for the packing of rac‐2). The even smaller angle of 59° for (P)‐1 compared with the values observed in (P)‐2 was expected, due to the more compact arrangement of the four bridging carbon atoms in the heterocyclic thiophene linker in the banister of (P)‐1, compared to the elongated arrangement in the 1,3‐butadiyne‐1,4‐diyl linker in the banister of (P)‐2. The dihedral angles between the phthalimide subunits of the backbone and the para‐phenylene rungs were divided into the outer ones (φ1) and inner ones (φ2). The same applies to the dihedral angles between the carbazole subunits of the banister and the para‐phenylene rungs resulting in (φ3) and (φ4), respectively. Note that each of these angles φ1‐φ4 was measured twice, even though only one of the two is highlighted in Figure 3B) to improve the visibility. While the measured values for each individual angle are provided in the Supporting Information, the values listed in Table 1 are the average of both measurements. The values between 45° and 67° are in the expected range considering the fact that the para‐phenylene rungs are interlinking the backbone and the banister, which are, in first approximation, perpendicularly (90°) arranged. To prevent steric repulsion between the hydrogen atoms of the para‐phenylene rungs and the carbonyl oxygen atoms of the phthalimide subunits on the backbone side and the hydrogen atoms at C1 and C8 of the carbazole subunits on the banister side angles around 90°/2=45° were expected. However, already the NMR experiments discussed above point at the rotation of the para‐phenylene rungs at room temperature such that the minima of the dihedral angles φ1‐φ4 cannot be very deep.
Another particularly interesting aspect of these new “Geländer” structures is their chirality. The helical wrapping of the banister was expected to yield in an (M) and (P) enantiomer for both model compounds 1 and 2. Based on the experience with our earlier “Geländer” designs,[ 3 , 5 , 6 , 7 ] the stability of the individual enantiomers was the focus of interest. Both racemic macrocycles rac‐1 and rac‐2 were separated into their enantiomers by high‐performance liquid chromatography (HPLC) using a chiral stationary phase, as displayed in Figure 4.
Figure 4.
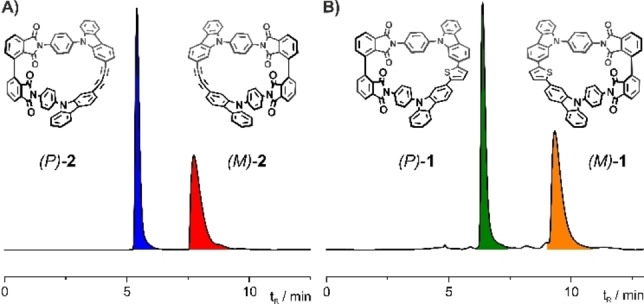
Chromatogram of A) rac‐2 and B) rac‐1 using a chiral stationary phase (Chiralpak IG, 30 mL min−1, 7 : 3=CH2Cl2 :n‐heptane, 20 °C).
Reinjecting the separated enantiomers (M)‐2 and (P)‐2 on an analytical column after three days stored at room temperature in solution in the solvent mixture of CH2Cl2 and n‐heptane exclusively displayed the pure enantiomer without any signs of racemization. Also, exposing the enantiopure samples to elevated temperatures like 120 °C in xylene did not result in racemization, documenting the pronounced thermal stability of the enantiomers (Figure S1 in the Supporting Information shows the chromatogram after 20 h at 120 °C). The high enantiostability of these “Geländer” macrocycles arises most likely from the steric clash of both phthalimide subunits of the backbone, which are mechanically interlocked due to their overhanging carbonyl substituents. The enantiostability of both enantiomers (M)‐2 and (P)‐2 also allowed the synthesis of (M)‐1 and (P)‐1 from enantiopure precursors and thus, draw not only the relationship between all signals of the chiral HPLC analyses but also the complete assignment of all enantiomers (see Figure S2 in the Supporting Information). Even so, initial attempts to grow single crystals from enantiopure samples of 1 and 2 failed, the simulation of the CD spectra of enantiopure (P)‐1 (Figure 7A and C) allowed the assignment of the peaks. As discussed below in more detail, the first eluted isomer during the chiral resolution of rac‐1 was assigned as (P)‐1, based on the good agreement of the calculated spectrum with the experimental CD spectrum (Figure 7C). Optical rotation further corroborates our assignment, (P)‐1 and (P)‐2 are both dextrorotary (positive optical rotation), while (M)‐1 and (M)‐2 are both levorotary (negative optical rotation).
Figure 7.
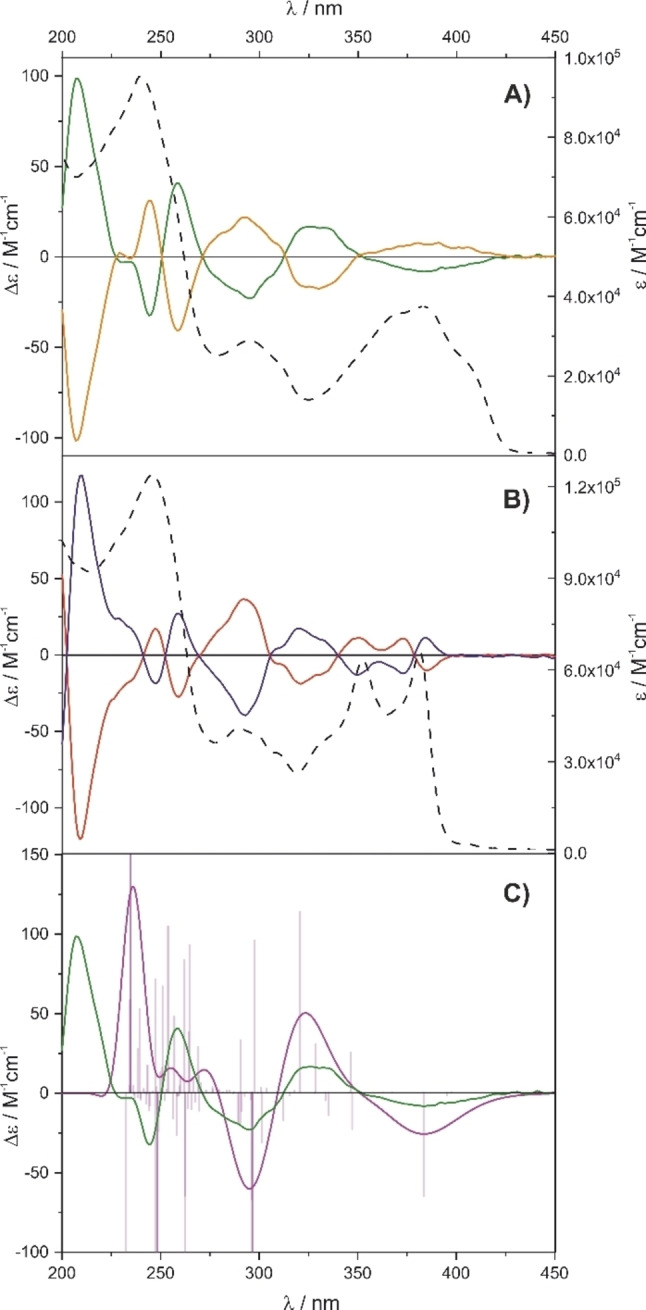
Circular dichroism (solid lines) and absorption spectra (dashed lines) of the enantiomers of 1 in A) and of 2 in B). (M)‐1: orange, (P)‐1: green, (M)‐2: red, and (P)‐2: blue). Spectra were recorded at 25 °C at c=10−6 mol⋅dm−3 concentration in CH3CN. C) Calculated CD spectra at TD−B3LYP/6‐311G(d,p) level of theory of the lowest energy conformer. Calculated (pink, fitted with σ=0.2 eV, shifted by 0.1 eV) and experimental (green) spectrum of (P)‐1. The computed transitions are indicated as bars (purple).
Optical properties
With the macrocycles 1 and 2 and the dimeric precursor 7 available, their optical properties were investigated. In Figure 5, the UV/Vis absorption spectra and the fluorescence spectra of the target structures 1 and 2 and their precursor 7 are displayed. Carbazoles typically show a weak absorption around 350 nm with a distinct double peak shape due to their vibronic resolution and strong absorption at 250 nm. [25] N‐Phenylphtalimide has its absorption maxima at 294 nm and 244 nm in methanol. The combination of these features is visible in the absorption spectra of 7. The weak signals at 343 and 357 nm fit in wavelength and shape to the absorption signature expected from the carbazole moiety, as well as the strong absorption at 254 nm. The absorption at 315 nm is found red shifted for the bisphthalimide 7 compared to N‐phenylphthalimide.
Figure 5.
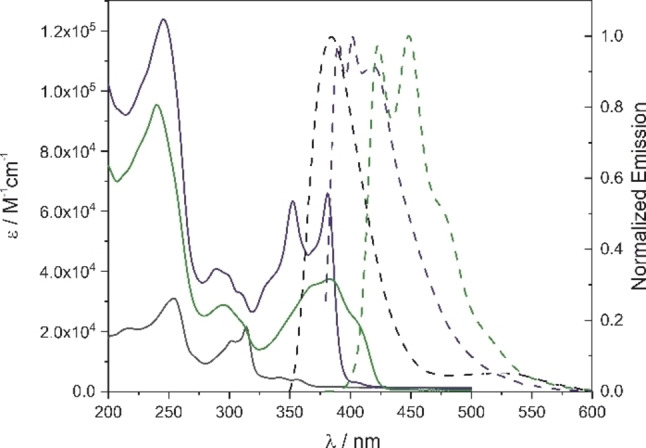
Absorption (solid lines) and normalized emission (dashed lines, excitation at 360 nm for rac‐1 and rac‐2, and at 300 nm for 7) of macrocycles 1 (green), 2 (blue), and dimer 7 (black) in acetonitrile. Spectra were recorded at 25 °C at c=10−6 mol ⋅ dm−3 concentration.
Upon macrocyclization to 2, the peaks of 7 at 343 and 357 nm shift to the vibronically resolved peaks at 353 nm, and 382 nm with a pronounced shoulder at 334 nm, transformation to 1 leads to a further red shift of these signals to 369 and 384 nm with a pronounced shoulder at 405 nm. These redshifts arise from the increased conjugation of the thiophene linked carbazole banister compared to the 1,3‐butadiyne bridged one in the precursor.[ 26 , 27 , 28 ] Furthermore, the absorption of the banister is more intense than the one recorded for the backbone subunits, indicating increased conjugation as well. The signature of the phthalimide in 2 is found blue shifted to 290 nm and in 1 to 295 nm. Transformation of the 1,3‐butadiyne to thiophene broadens the absorptions spectra leading to a less vibronically resolved spectrum than 2, probably due to the increased degree of freedom found in the thiophene linked banister in 1 compared to the butadiyne linked banister in 2.
The emission spectrum of 7 shows an intense signal at 383 nm and a broad weak one at 525 nm. The emissions of 1 and 2 are both red shifted compared to the open dimer 7, but the one of 1 to a larger extent than 2. The emission of 1 has two resolved peaks at 422 nm and 450 nm, with a shoulder at 480 nm corresponding to the shape found in the absorption. The emission of 2 displays three resolved peaks at 390, 403, and 420 nm.
The shape and the observed shifts of both absorption and emission spectra resemble the ones reported by Kato and co‐workers for conjugated carbazole oligomers interlinked at different positions by either thiophenes or 1,3‐butadiynes.[ 26 , 27 , 28 ] The main differences recorded for these oligomers as banister subunits of these “Geländer” architectures were the lower quantum yields compared to these reported linear oligomers. In the case of the thiophene interlinked carbazoles of 1, the quantum yield decreased by a factor of 20, while in the case of the 1,3‐butadiene interlinked banister of 2, the quantum yield was even 30 times less intense than the linear analogue reported by Kato et al. (recorded quantum yield for 1: Φf=2.5 ⋅ 10−2, and for 2: Φf=6.6 ⋅ 10−3, in CH3CN, absolute measurement using an integrating sphere). In analogy to the linear model carbazole oligomers, the vibronic resolution of the emission of 1 is clearly improved compared to its absorption. They hypothesize that this might point to a more rigid geometry in the excited state, arising from a quinodal electron distribution in the interlinking thiophene of the chromophore.
The very comparable electronic transitions of the banister structure of our “Geländer” macrocycles and the reported linear model compounds can be rationalized by the fact that the carbazole nitrogen is almost completely decoupled from the conjugated system of the 2,7‐linked carbazoles. This can be visualized by the highest occupied molecular orbital (HOMO), which has hardly any density on the carbazole nitrogen in our macrocyclic model compounds (Figure 6), as well as in the linear oligomers of Kato et al. Another very interesting aspect is the complete separation of the frontier orbitals in these “Geländer” architectures. While the HOMO is localized on the banister, the lowest unoccupied molecular orbital (LUMO) is exclusively localized on the backbone structure (Figure 6). As described above, both macrocycles were synthesized as racemic mixtures rac‐1 and rac‐2. They were subsequently separated into pure enantiomers (P)‐1 and (M)‐1, and (P)‐2 and (M)‐2, respectively, by semi‐preparative HPLC with a chiral stationary phase (Figure 4). To record the circular dichroism (CD) spectra of the pure enantiomers, the separated samples of the pure enantiomers were evaporated to dryness in vacuo at 25 °C and subsequently dissolved in acetonitrile (CH3CN) and taken into an acetonitrile solution to measure circular dichroism (CD). As expected and displayed in Figure 7, the separated enantiomers of both macrocycles display mirrored Cotton bands. The CD spectra of the enantiomers of the macrocycle 2 show the most pronounced Cotton effect at 210 nm, 260 nm, 292 nm, and 320 nm (Figure 7B), while for the enantiomers of macrocycle 1 the signals with significant Cotton effects were observed at 208 nm, 245 nm, 260 nm, 295 nm, and 330 nm, complemented by a shallow band at 387 nm (Figure 7A). As already discussed above, the enantiopure samples of both macrocycles did not show any tendency to racemize and were stable in solution as well as in the solid state for several weeks. The comparison between recorded and simulated CD spectra of (P)‐1 enabled the assignment of the enantiomers. The good agreement between both spectra is displayed in Figure 7C. For these calculations, the first 100 electronic transitions of the lowest energy conformer (see Supporting Information) were computed at TD−B3LYP/6‐311G(d,p) level of theory.
Figure 6.
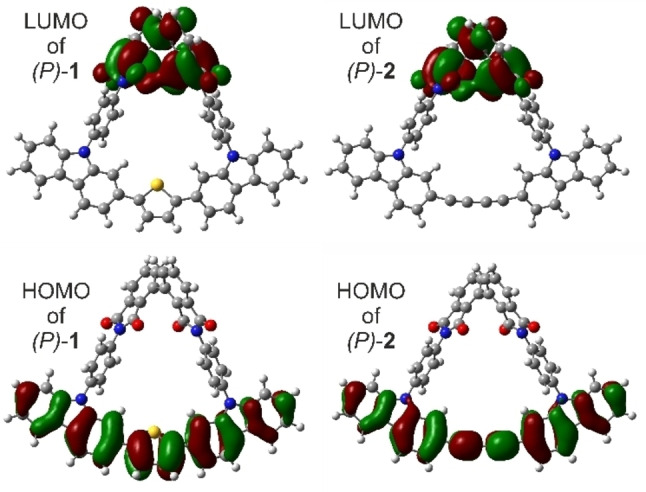
Computed frontier orbitals of the “Geländer” macrocycles (P)‐1 and (P)‐2 at B3LYP/6‐311G** level of theory.
Conclusion
The new type of “Geländer” macrocycles 1 and 2 with orthogonal joints between the subunits axis (backbone), rungs, and banister has been designed and synthesized. The thiophene linked target structure 1 was prepared in a 7 step linear synthesis with an overall yield of 52 %. The macrocyclization was achieved in excellent yields by an oxidative acetylene coupling of the TIPS masked acetylenes of the bisphthalimide 7, which underwent an in situ deprotection and subsequent homocoupling procedure using copper(II) fluoride. The “Geländer”‐type macrocycles were synthesized as racemates and subsequently separated into pure enantiomers by HPLC with a chiral stationary phase. The enantiomers of both macrocycles 1 and 2 are thermally stable and display no tendency to racemize. Spectroscopic investigations display the banister dominated optical activity with moderate circular dichroism. Simulation of the CD spectra allows the assignment of the enantiomers.
While the herein reported macrocycles are our first “Geländer” structures combining a conjugated banister with enantiomeric stability, the developed building blocks with improved symmetry enable us to transfer the design to longer oligomers, which are currently under investigation.
Experimental Section
General remarks
All chemicals were directly used without further purification if nothing else is stated. CsF was dried overnight at 120 °C in vacuo and stored in a desiccator. NiBr2 was dried at 300 °C under a stream of nitrogen for three days and stored under an argon atmosphere in a desiccator. Zinc powder was activated by treatment with 2 M HCl followed by washings with methanol and Et2O, the activated and washed zinc was dried in vacuo and stored under an argon atmosphere. Dry solvents were used as crown capped and purchased from Acros and Sigma‐Aldrich. NMR solvents were obtained from CIL Cambridge Isotope Laboratories, Inc. (Andover, MA, USA), Acros, Sigma‐Aldrich, or Apollo Scientific. All NMR experiments were performed on Bruker Avance III or III HD, two or four‐channel NMR spectrometer operating at 400.13, 500.13, or 600.13 MHz proton frequency. The instruments were equipped with a direct‐observe 5 mm BBFO smart probe (400 and 500 MHz) or an indirect‐detection 5 mm BBI probe (600 MHz). All probes were equipped with actively shielded z‐gradients (10 A). The experiments were performed at 298 K if not differently stated. All chemical shifts (δ) are reported in ppm relative to TMS, and coupling constants (J) are given in Hertz (Hz). The measurements are performed at room temperature. The multiplicities are written as: s=singlet, d=doublet, t=triplet, q=quartet, quint=quintet, dd=doublet of doublet, m=multiplet. For column chromatography, SilicaFlashR P60 from Silicycle was used with a particle size of 40–63 μm (230–400 mesh) and performed on a Biotage Isolera. Recycling size‐exclusion chromatography (SEC or GPC) was performed with a Shimadzu Prominence System equipped with SDV preparative columns from Polymer Standards Service (two Shodex columns in series, 20×600 mm each, exclusion limit: 30 000 g mol−1) with chloroform as solvent. For analytical HPLC, a Shimadzu LC‐20AT HPLC was used, equipped with a diode‐array UV/Vis detector (SPD‐M20 A VP from Shimadzu, λ=200–600 nm) and a column oven Shimadzu CTO‐20AC. For preparative HPLC, a Shimadzu LC‐20AP HPLC was used equipped with a diode‐array UV/Vis detector (SPD‐M20 A VP from Shimadzu, λ=200–600 nm). The used column for analytical separation on chiral stationary phase was a Chiralpak IG, 5 μm, 4.6×250 mm, Daicel Chemical Industries Ltd and for preparative separation, Chiralpak IG, 5 μm, 30×250 mm, Daicel Chemical Industries Ltd. UV/Vis absorption spectra were recorded on a Jasco V‐770 Spectrophotometer. Fluorescence spectra and quantum yield (using nitrogen purged integrating sphere) measurements were performed on a Jasco FP‐8600. CD measurements were performed on a JASCO J‐1500 CD Spectrophotometer. The UV/Vis, CD, and fluorescence spectra were measured in a 1 cm quartz glass cuvettes, quantum yields in 0.5 cm quartz glass cuvettes. Optical rotations were measured at 295 K on a JASCO P‐2000 polarimeter with a 10.0 cm cell (1.00 mL). Infrared spectra were recorded neat with an ATR equipped Schimadzu IRTacer‐100. High‐resolution mass spectra (HR‐MS) were measured with a Bruker Maxis 4G ESI‐TOF instrument or a Bruker solariX spectrometer with a MALDI source.
Synthesis
2‐bromo‐9‐(4‐nitrophenyl)‐9H‐carbazole (3): In an oven‐dried and argon purged 25 mL Schlenk tube equipped with a septum and magnetic stirring bar 2‐bromo‐9H‐carbazole (5.25 g, 20.9 mmol) and dry CsF (6.35 g, 41.8 mmol) were suspended in 5 mL dry DMSO. 1‐Fluoro‐4‐nitrobenzene (4.42 g, 31.3 mmol) was added and the tube sealed. The reaction was placed in an oil bath heated to 150 °C and allowed to stir over night. Reaction control by LC–MS indicated complete conversion the next day and the reaction mixture was poured into deionized water. The precipitated solids were collected and dissolved in a small amount of CH2Cl2 and precipitated with petroleum ether. The solids were collected and dried at 45 °C in vacuo yielding the titular compound as a yellow powder (6.75 g, 18.4 mmol) in 92 % yield.
1H NMR (500 MHz, CDCl3): δ=8.55–8.48 (m, 2H), 8.12 (dt, J=7.8, 1.0 Hz, 1H), 8.02–7.97 (dd, J=8.3, 0.4 Hz, 1H), 7.82–7.69 (m, 2H), 7.63–7.57 (dd, J=1.8, 0.5 Hz, 1H), 7.52–7.41 (m, 3H), 7.40–7.33 (m, 1H); 13C{1H} NMR (126 MHz, CDCl3): δ 146.4, 143.3, 140.9, 140.3, 127.1, 127.1, 125.9, 124.5, 123.7, 123.2, 121.9, 121.8, 120.8, 120.2, 112.9, 109.9; IR (neat): =1589 (s), 1500 (s) cm−1 (NO2); HR‐MS (ESI‐ToF, +): m/z=[M+Na]+ Calcd. for C18H11BrN2NaO2: 388.9896; Found 388.9890.
9‐(4‐nitrophenyl)‐2‐((triisopropylsilyl)ethynyl)‐carbazole (4): To a 500 mL Schlenk tube equipped with a magnetic stirring bar and septum 3 (4.66 g, 12.7 mmol) was added and dissolved in a mixture of 50 mL THF and 150 mL diisopropylamin. The mixture was sparged 30 min with argon, followed by the addition of Pd(PPh3)4 (734 mg, 635 μmol), CuI (145 mg, 762 μmol), and (triisopropylsilyl)acetylene (6.95 g, 38.1 mmol). The tube was sealed, placed in a pre‐heated oil bath at 90 °C, and allowed to stir over night. Reaction control by LC–MS indicated complete conversion, thus, the reaction mixture was diluted with CH2Cl2 and filtered over a silica plug. Volatiles were removed and the crude subjected to fcc (SiO2: 19 : 1‐4 : 1=cyclohexane:toluene). The titular compound was isolated as yellow solid (5.61 g, 12.0 mmol) in 94 % yield.
1H NMR (500 MHz, CDCl3): δ=8.55–8.48 (m, 2H), 8.12 (dt, J=7.8, 1.0 Hz, 1H), 8.05 (dd, J=8.1, 0.7 Hz, 1H), 7.83–7.76 (m, 2H), 7.55–7.53 (m, 1H), 7.50–7.42 (m, 3H), 7.39 −7.32 (m, 1H), 1.25–1.05 (m, 21H); 13C NMR (126 MHz, CDCl3) δ=146.3, 143.6, 140.7, 139.6, 127.1, 127.1, 125.8, 125.7, 124.2, 123.9, 121.6, 121.5, 121.0, 120.5, 113.1, 109.9, 107.8, 91.1, 18.9, 11.5; IR (neat): =2941 (w), 2862 (w) cm−1 (CH) 2157 (w) cm−1 (C≡C) 1593 (s) 1515 (s) cm−1 (NO2); HR‐MS (ESI‐ToF, +): m/z=[M+Na]+ Calcd. for C29H32N2NaO2Si: 491.2125; Found 491.2117.
4‐(2‐((triisopropylsilyl)ethynyl)‐carbazol‐9‐yl)aniline (5): In a 100 mL round bottomed flask equipped with a magnetic stirring bar 4 (3.55 g, 7.57 mmol) and SnCl2 dihydrate (5.23 g, 22.7 mmol) were dissolved in a mixture of 40 mL of THF and 20 mL ethanol. The reaction mixture was set under an argon atmosphere, placed in a pre‐heated oil bath at 50 °C, and allowed to stir over night. LC–MS indicated complete conversion and saturated aqueous NH4Cl was added. Volatiles were removed under reduced pressure and the aqueous layer extracted thrice with ethyl acetate. The combined organic layers where sequentially washed with deionized water, saturated aqueous NaHCO3, deionized water and brine. After evaporation an orange oil (3.32 g) was isolated. NMR indicated sufficient purity and the product was used without further characterization and purification.
1H NMR (250 MHz, CD2Cl2) δ=8.11 (d, J=7.8 Hz, 1H), 8.06 (d, J=8.1 Hz, 1H), 7.52–7.33 (m, 4H), 7.33–7.16 (m, 4H), 7.00–6.81 (m, 2H), 1.20–1.10 (m, 21H); HR‐MS (ESI‐ToF, +): m/z=[M+Na]+ Calcd. For Calcd. for C29H34N2Si: 439.2564; Found 439. 2561.
Monomer (6): In an 50 mL round bottom flask equipped with a magnetic stirring bar 5 (3.32 g, 7.57 mmol) was dissolved in 20 mL glacial acetic acid. 3‐Bromophthalic anhydride (1.89 g, 8.33 mmol) was added, the mixture set under an argon atmosphere, and the reaction was placed in a preheated oil bath at 70 °C. After 1 h the product starts to precipitate and the mixture was allowed to stir for 48 h until LC–MS indicated complete conversion. The reaction mixture was allowed to cool to room temperature and diluted with deionized water. After filtration the solid was washed with deionized water, methanol, and Et2O. The powder was dried at 45 °C The titular compound 6 was isolated as beige powder (4.10 g, 6.33 mmol) in 84 % yield over two steps.
1H NMR (500 MHz, [D8]THF) δ=8.16 (dt, J=7.8, 1.0 Hz, 1H), 8.13 (dd, J=8.1, 0.7 Hz, 1H), 8.00 (dd, J=8.0, 0.9 Hz, 1H), 7.98 (dd, J=7.3, 0.8 Hz, 1H), 7.85–7.80 (m, 2H), 7.80–7.76 (m, 2H), 7.76–7.72 (m, 1H), 7.58–7.56 (m, 1H), 7.47 (dt, J=8.3, 0.9 Hz, 1H), 7.44–7.38 (m, 2H), 7.30–7.25 (m, 1H), 1.16 (m, 21H); 13C NMR (126 MHz, [D8]THF) δ=166.0, 165.6, 142.8, 141.4, 140.1, 137.6, 136.4, 135.7, 132.8, 130.8, 129.2, 128.4, 127.6, 125.5, 124.9, 123.5, 121.7, 121.5, 121.1, 119.3, 113.9, 111.0, 110.1, 97.1, 90.2, 19.9, 12.5; IR (neat): =2941 (w), 2863 (w) cm−1 (CH) 2151 (w) cm−1 (C≡C) 1720 (s) cm−1 (C=O); HR‐MS (ESI‐ToF, +): m/z=[M+Na]+ Calcd. for C37H35N2NaO2Si: 669.1543; Found 669.1530.
Dimer (7): In an oven dried 10 mL Schlenk tube equipped with an magnetic stirring bar zinc (75.5 mg, 1.16 mmol), NiBr2 (8.41 mg, 0.04 mmol), and PPh3 (77.5 mg, 0.29 mmol) were combined and the tube cycled three times between vacuum and argon. 1 mL of DMAc was added and the active catalyst was allowed to form for half an hour (until a red solution was observed) at 80 °C. In an oven dried roundbottom flask 6 (500 mg, 0.77 mmol) was dissolved in 2 mL DMAc by gentle heating with a heat gun under an argon atmosphere. The solution of 6 was added to the active catalyst, the tube sealed, and allowed to stir over night. The mixture was diluted with N,N‐dimethyacetamide and poured into ice water. The precipitate was collected over celite and washed with methanol. Afterwards, the solid was washed down with CH2Cl2 and concentrated on silica. The crude mixture was purified by automated fcc (SiO2: 1 : 9‐6 : 1=dichloromethane:cyclohexane) yielding 7 (348 mg, 0.39 mmol) in 80 % yield.
1H NMR (500 MHz, [D8]THF) δ=8.11–7.99 (m, 4H), 8.01‐7.98 (dd, J=8.1, 0.7 Hz, 2H) 7.87 (t, J=7.6 Hz, 2H), 7.79 (dd, J=7.7, 1.0 Hz, 2H), 7.73–7.65 (m, 4H), 7.65–7.58 (m, 4H), 7.43–7.39 (m, 2H), 7.36–7.29 (dt, J=8.19, 0.8 Hz, 2H), 7.29–7.22 (m, 4H), 7.16–7.10 (m, 2H), 1.09–0.83 (m, 42H); 13C NMR (126 MHz, [D8]THF) δ=166.3, 166.0, 141.6, 140.2, 136.2, 136.0, 134.8, 133.9, 132.2, 131.7, 129.4, 127.9, 127.1, 126.4, 124.3, 123.7, 123.3, 123.0, 120.5, 120.3, 119.9, 112.7, 109.8, 108.6, 89.0, 18.1, 11.3; UV/Vis (acetonitrile): λmax (ϵ)=314 (24353), 255 nm(34615 mol−1dm3cm−1); fluorescence (acetonitrile): λex=300 nm; λem=384 nm; HR‐MS (MALDI‐ToF, +): m/z=[M]+ Calcd. for C74H70N4O4Si2: 1134.4930; Found 1134.4941.
Butadiyne macrocycle (rac‐ 2): In a 500 mL round bottom flask equipped with a large magnetic stirrer 7 (890 mg, 0.78 mmol) and CuF2 ⋅ 2H2O (648 mg, 4.71 mmol) were combined and suspended. The mixture was placed in an oil bath heated to 100 °C and vigorously stirred at open ambient atmosphere overnight. The solution was poured into 400 mL deionized water and the precipitate was filtered off and washed with cold water and methanol. The obtained off‐white powder was dried in a vacuum oven at 45 °C yielding 605 mg (94 %) of macrocycle 2. Further separation of the corresponding enantiomers was achieved by preparative HPLC on a chiral stationary phase (Chiral Pak IG, eluent: 30 % n‐heptane in dichloromethane, flow rate: 30 mL min−1, T=20 °C).
[α]D22=+107.6±9.2 (c=0.23 in CHCl3, (P)‐2), [α]D22=−94.9±11.9 (c=0.098 in CHCl3, (M)‐2); 1H NMR (500 MHz, [D8]THF) δ=8.14 (dt, J=7.8, 0.8 Hz, 2H), 8.12–8.09 (m, 4H), 8.00 (td, J=7.6, 0.95 Hz, 2H) 7.81 (dt, J=7.8, 1.0 Hz, 2H), 7.76 (m, 4H), 7.70–7.64 (m, 4H), 7.63 (m, 2H), 7.50–7.34 (m, 4H), 7.28 (dd, J=8.1, 1.4 Hz, 2H), 7.27–7.22 (m, 1H); 13C NMR (126 MHz, [D8]THF) δ=167.2, 167.0, 142.8, 142.0, 137.1, 136.6, 136.2, 135.4, 133.0, 133.0, 130.9, 129.0, 128.6, 127.9, 125.2, 124.5, 124.1, 122.6, 121.7, 121.4, 121.2, 120.0, 117.5, 110.8, 85.7, 75.5; IR (neat): =2942 (br w) cm−1 (C≡C−C≡C) 1715 (s) cm−1 (C=O); UV/Vis (acetonitrile): λmax (ϵ)=381 (65972), 353 (63201), 290 (40791), 245 nm (123798 mol−1dm3cm−1); fluorescence (acetonitrile): λex=360 nm; λem=391, 403, 420 nm; HR‐MS (MALDI‐ToF, +): m/z=[M]+ Calcd. for C56H28N4O4: 820.2105; Found 820.2106.
Thiophene macrocycle (rac‐1): In a 10 mL Schlenk tube equipped with magnetic stirring bar 2 (103 mg, 0.13 mmol), NaHS⋅xH2O (56 mg, below 40 % crystal water according to Sigma‐Aldrich), and elemental sulfur (12 mg, 0.38 mmol) were combined, the tube was closed with a rubber septum and cycled three times between vacuum and Argon. In a separate flask 1.5 mL of DMF was sparged with Argon for 5 min. The solvent was added to the reaction mixture which instantaneously turned royal blue. After 10 minutes of stirring TLC (SiO2, dichloromethane) indicated full conversion and the reaction was quenched with saturated aqueous NH4Cl. The precipitate was collected and washed with cold water followed by methanol, the collected solids were dissolved in an aliquot of dichloromethane and precipitated with petroleum ether. The obtained faint yellow solid was dried in a vacuum oven at 45 °C yielding 103 mg (96 %) macrocycle 1. Further separation of the corresponding enantiomers was achieved by preparative HPLC on a chiral stationary phase (Chiralpak IG, eluent: 30 % n‐heptane in dichloromethane, flow rate: 30 mL min−1, T=20 °C).
[α]D22=+104.8±2.4 (c=0.19 in CHCl3, (P)‐1), [α]D22=‐101.4±8.1 (c=0.22 in CHCl3, (M)‐1); 1H NMR (500 MHz, [D8]THF) δ=8.20‐8.14 (d, J=8.3 Hz, 2H) 8.14–8.11 (m, 2H), 8.11–8.08 (d, J=1 Hz, 2H), 8.01 (t, J=7.6 Hz, 2H), 7.89 (dd, J=7.8, 1.1 Hz, 2H), 7.79 (m, 4H), 7.74 (dd, J=8.3, 1.6 Hz, 2H), 7.73–7.69 (m, 4H), 7.60 (d, J=1.5 Hz, 2H), 7.52 (s, 2H), 7.44–7.40 (m, 2H), 7.38 ‐ 7.30 (m, 2H), 7.27–7.20 (m, 2H); 13 C NMR (126 MHz, [D8]THF) δ=167.4, 167.1, 144.8, 142.5, 142.4, 137.5, 136.4, 135.7, 135.5, 133.5, 132.9, 132.6, 131.2, 129.1, 128.2, 127.0, 125.2, 124.5, 124.4, 123.9, 121.5, 121.3, 121.1, 118.2, 110.7, 107.5; IR (neat): =2981 (w), 2912 (w) cm−1 (CH) 1715 (s) cm−1 (C=O); UV/Vis (acetonitrile): λmax (ϵ)=381 (37279), 293 (28691), 241 nm (95145 mol−1dm3cm−1); fluorescence (acetonitrile): λex=360 nm; λem=422, 450 nm; HR‐MS (ESI‐ToF, +): m/z=[M+Na]+ Calcd. for C56H30N4NaO4SiS: 877.1880; Found 877.1879.
Deposition Number 2175851 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors acknowledge generous support by the Swiss National Science Foundation (SNF Grant no. 200020‐207744). M.M. acknowledges support from the 111 project (Grant No. 90002‐18011002). Open Access funding provided by Universität Basel.
A. D'Addio, J. Malinčik, O. Fuhr, D. Fenske, D. Häussinger, M. Mayor, Chem. Eur. J. 2022, 28, e202201678.
Data Availability Statement
The data that support the findings of this study are openly available in zenodo at https://doi.org/10.5281/zenodo.6614541, reference number 6614541.
References
- 1. Kiupel B., Niederalt C., Nieger M., Grimme S., Vögtle F., Angew. Chem. Int. Ed. 1998, 37, 3031–3034; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 3206–3209. [Google Scholar]
- 2. Modjewski M., Lindeman S. V., Rathore R., Org. Lett. 2009, 11, 4656–4659. [DOI] [PubMed] [Google Scholar]
- 3. Rickhaus M., Bannwart L. M., Neuburger M., Gsellinger H., Zimmermann K., Häussinger D., Mayor M., Angew. Chem. Int. Ed. 2014, 53, 14587–14591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14816–14820. [Google Scholar]
- 4. Rickhaus M., Bannwart L. M., Unke O., Gsellinger H., Häussinger D., Mayor M., Eur. J. Org. Chem. 2015, 2015, 786–801. [Google Scholar]
- 5. Rickhaus M., Unke O. T., Mannancherry R., Bannwart L. M., Neuburger M., Häussinger D., Mayor M., Chem. Eur. J. 2015, 21, 18156–18167. [DOI] [PubMed] [Google Scholar]
- 6. Mannancherry R., Šolomek T., Cavalli D., Malinčík J., Häussinger D., Prescimone A., Mayor M., J. Org. Chem. 2021, 86, 5431–5442. [DOI] [PubMed] [Google Scholar]
- 7. Mannancherry R., Rickhaus M., Häussinger D., Prescimone A., Mayor M., Chem. Sci. 2018, 9, 5758–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bannwart L. M., Jundt L., Müntener T., Neuburger M., Häussinger D., Mayor M., Eur. J. Org. Chem. 2018, 2018, 3391–3402. [Google Scholar]
- 9. Bannwart L. M., Müntener T., Rickhaus M., Jundt L., Häussinger D., Mayor M., Chem. Eur. J. 2021, 27, 6295–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weiland K. J., Brandl T., Atz K., Prescimone A., Häussinger D., Šolomek T., Mayor M., J. Am. Chem. Soc. 2019, 141, 2104–2110. [DOI] [PubMed] [Google Scholar]
- 11. Weiland K. J., Münch N., Gschwind W., Häussinger D., Mayor M., Helv. Chim. Acta 2019, 102, e1800205. [Google Scholar]
- 12. Brandl T., Hoffmann V., Pannwitz A., Häussinger D., Neuburger M., Fuhr O., Bernhard S., Wenger O. S., Mayor M., Chem. Sci. 2018, 9, 3837–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mayor M., Lehn J.-M., J. Am. Chem. Soc. 1999, 121, 11231–11232. [Google Scholar]
- 14. Liou G.-S., Hsiao S.-H., Huang N.-K., Yang Y.-L., Macromolecules 2006, 39, 5337–5346. [Google Scholar]
- 15. Percec V., Bae J.-Y., Zhao M., Hill D. H., J. Org. Chem. 1995, 60, 176–185. [Google Scholar]
- 16. Schaack C., Sidler E., Trapp N., Diederich F., Chem. Eur. J. 2017, 23, 14153–14157. [DOI] [PubMed] [Google Scholar]
- 17. Schäfer M., Drayß M., Springer A., Zacharias P., Meerholz K., Eur. J. Org. Chem. 2007, 2007, 5162–5174. [Google Scholar]
- 18. Schaack C., Arrico L., Sidler E., Górecki M., Di Bari L., Diederich F., Chem. Eur. J. 2019, 25, 8003–8007. [DOI] [PubMed] [Google Scholar]
- 19. Schulte K. E., Reisch J., Hörner L., Chem. Ber. 1962, 95, 1943–1954. [Google Scholar]
- 20. Masuda M., Maeda C., Chem. Eur. J. 2013, 19, 2971–2975. [DOI] [PubMed] [Google Scholar]
- 21. Gibson M. S., Bradshaw R. W., Angew. Chem. Int. Ed. Engl. 1968, 7, 919–930. [Google Scholar]
- 22. Zhang G., Yi H., Chen H., Bian C., Liu C., Lei A., Org. Lett. 2014, 16, 6156–6159. [DOI] [PubMed] [Google Scholar]
- 23. Feng L., Hu T., Zhang S., Xiong H.-Y., Zhang G., Org. Lett. 2019, 21, 9487–9492. [DOI] [PubMed] [Google Scholar]
- 24. Pracht P., Bohle F., Grimme S., Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [DOI] [PubMed] [Google Scholar]
- 25. Kawaguchi K., Nakano K., Nozaki K., J. Org. Chem. 2007, 72, 5119–5128. [DOI] [PubMed] [Google Scholar]
- 26. Kato S., Shimizu S., Kobayashi A., Yoshihara T., Tobita S., Nakamura Y., J. Org. Chem. 2014, 79, 618–629. [DOI] [PubMed] [Google Scholar]
- 27. Kato S., Noguchi H., Kobayashi A., Yoshihara T., Tobita S., Nakamura Y., J. Org. Chem. 2012, 77, 9120–9133. [DOI] [PubMed] [Google Scholar]
- 28. Kato S., Shimizu S., Taguchi H., Kobayashi A., Tobita S., Nakamura Y., J. Org. Chem. 2012, 77, 3222–3232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are openly available in zenodo at https://doi.org/10.5281/zenodo.6614541, reference number 6614541.