

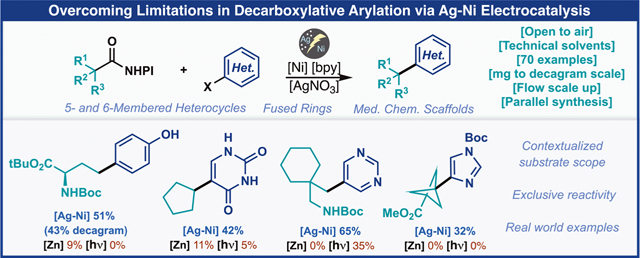
Abstract
A useful protocol for achieving decarboxylative cross-coupling (DCC) of redox-active esters (RAE, isolated or generated in situ) and halo(hetero)arenes is reported. This pragmatically focused study employs a unique Ag–Ni electrocatalytic platform to overcome numerous limitations that have plagued this strategically powerful transformation. In its optimized form, coupling partners can be combined in a surprisingly simple way: open to the air, using technical-grade solvents, an inexpensive ligand and Ni source, and substoichiometric AgNO3, proceeding at room temperature with a simple commercial potentiostat. Most importantly, all of the results are placed into context by benchmarking with state-of-the-art methods. Applications are presented that simplify synthesis and rapidly enable access to challenging chemical space. Finally, adaptation to multiple scale regimes, ranging from parallel milligram-based synthesis to decagram recirculating flow is presented.
Graphical Abstract
INTRODUCTION
The use of radicals has a rich history in the progression of chemical synthesis.1,2 In the early days, proving the very existence of radicals was an exciting area.3 Following that, their exploration in organic and organometallic chemistry led to a wellspring of useful discoveries. The classic studies of Stork,4 Curran,5 Barton,6,7 Zard,8 Minisci,9,10 and others11–13 showed how radicals could be tamed to make useful bonds. The modern manifestation of radical retrosynthesis has the potential to transform conventional logic and enable dramatically simplified routes to seemingly trivial and complex structures.14 It has now been demonstrated multiple times that such logic can obviate the need for otherwise arduous multistep synthetic sequences that are wedded to polar (2 electron) disconnections.15 Polar bond analysis, a useful rubric for synthesis planning, is often plagued by a reliance on protecting group chemistry, superfluous redox fluctuations, and functional group manipulations that lower the ideality of a synthetic design.16 In contrast, radical-based transformations offer new disconnection opportunities that are often far more convergent, enabling access to an expanded chemical space. This is in part due to the higher chemoselectivity of single electron processes, leading to a lower reliance on protecting groups. Such approaches have been highly impactful in the context of medicinal chemistry efforts, where expedient and modular access to a broad range of analogs is critical for exploring key biological hypotheses.17 For instance, the construction of potential drug candidates is often guided by the presence of C(sp2)–C(sp3) linkages, which are an essential motif in countless active pharmaceutical ingredients and are often a vector where SAR studies are performed with great diligence.18 Therefore, the development of synthetic methods that give access to a vast swath of chemical space by merging two accessible and benchtop-stable partners has attracted great attention. To this end, cross-coupling partners such as carboxylic acids and aryl halides represent an attractive choice owing to their widespread commercial availability. For this reason, numerous radical decarboxylative cross-coupling methodologies have been developed, thereby opening up a more intuitive retrosynthesis of high-value targets.19–23
As an example, trivial looking unnatural amino acids like homotyrosine (Figure 1A) have historically been accessed through linear multistep syntheses using exclusively polar transformations that require a careful reaction setup, preparation of organometallic reagents, multiple protecting group manipulations, and unstable intermediates that do not intuitively map onto the final target.24–27 Strikingly, these multistep routes, ranging from 5–10 steps, feature only one strategic bond forming step to forge a single C–C bond. Alternatively, using radical retrosynthesis, homotyrosine and other analogs could easily be traced back to glutamic acid and an appropriate aryl halide in a single step operation.
Figure 1.

(A) Prior synthetic strategies guided by polar bond analysis toward homotyrosine and the proposed radical decarboxylative arylation strategy. (B) State-of-the-art DCC methods toward the synthesis of 1 failed to generate serviceable amounts of product for a deceptively simple coupling. (C) Limitations of state-of-the-art DCC with aryl electrophiles include limited scope with complex arenes and a variety of decomposition pathways of the radical precursors.
To test the viability of this strategy, numerous decarboxylative cross-coupling (DCC) methods that represent the current state of the art were tested (Figure 1B). First, cross-electrophile coupling using zinc as a terminal reductant delivered only 9% of 1.28 Next, an electrochemical approach was pursued but no product formation with this challenging coupling partner 3 was observed.29 We next turned our attention toward photochemically enabled reactivity platforms. First, a reductive cross-electrophile coupling leveraging electron donor acceptor complexes was evaluated.30 In the event, no desired product was formed. Finally, an oxidative activation of the carboxylic acid was evaluated, using photoinduced-electron transfer (PET, Ir-based) and a key phthalimide additive.31 However, even with this method, no desired product was observed. At first glance, these results are surprising given the extensive body of literature around such transformations. However, upon closer examination of our desired cross-coupling reaction and the surrounding literature, several key challenges were limiting the success of this coupling (Figure 1C). First, the scope of aryl electrophiles for C(sp2)–C(sp3) reductive cross-coupling generally favors electron-deficient arenes and heterocycles over their electron-rich counterparts even without considering sensitive functionality. Second, the carboxylic acid or corresponding redox active ester is prone to several decomposition pathways if it is not engaged in productive catalysis. These include H-atom abstraction by transient radicals, dimerization, and in the case of redox active esters, unproductive N–O bond heterolysis resulting in recovery of the acid starting material. In extreme cases, where the reaction does not proceed, only starting materials are recovered. In this article, a practical method to overcome the limitations of decarboxylative arylation is presented. It features a wide scope amongst challenging real-world substrates, extreme operational simplicity, and a versatile scale regime from parallel screening (mg) to recirculating flow scale up (decagram). Most importantly, the results herein are presented in context with existing methods for achieving the same transformation and conducted in collaboration with a diverse array of practitioners across five different pharmaceutical partners (the typical end user of this method).
RESULTS AND DISCUSSION
Considering the aforementioned limitations of chemical, electrochemical, and photochemical methods, our recent exploration in the area of terpene synthesis was particularly inspiring.32 In that study, the unique ability of an Ag-functionalized electrode surface was exploited to enable DCC-alkenylation when chemical and electrochemical means faltered. The role of the in situ-formed electrode-bound Agnanoparticles (AgNP) was extensively studied and found to play three key roles: (1) extending catalyst lifetime, (2) preventing background decomposition of the redox active ester, and most importantly, (3) lowering the required overpotential, which in turn greatly expanded functional group compatibility. Upon closer examination of the mass balance from the prior attempts, we had observed full consumption of the redox active ester but very little product formation alongside homocoupling byproducts. These starting points were strikingly similar to the limitations encountered in the initial exploration of Ag-functionalized electrode surfaces for decarboxylative alkenylation. It was therefore hypothesized that for the same reasons, the Ag–Ni electrocatalytic manifold may provide similar advantages in DCC-arylation.
To begin this investigation, the electrochemical coupling of redox active ester 2 with 4-iodophenol (3) under recently disclosed Ag–Ni electrocatalytic vinylation conditions was pursued (Figure 2). In the event, these coupling partners were combined with NiCl2·6H2O (10 mol %), 2–2′-bipyridine (10 mol %) and the key AgNO3 additive (0.3 equiv) in dimethylformamide (DMF) under constant current electrolysis with a reticulated vitreous carbon (RVC) cathode and sacrificial magnesium anode (Figure 2). To our delight, the desired homotyrosine derivative was generated in 42% yield (entry 1). Importantly, the absence of AgNO3 resulted in a low yield of the desired product along with substantial amounts of decomposed RAE, thus highlighting one of the critical roles of the Ag-functionalized electrode in mitigating background consumption of the limiting reagent (entry 2). Omission of ligand or the nickel catalyst resulted in substantially lower yields, poor reaction performance, and quantitative recovery of the aryl halide, thus ruling out the mechanistic possibility of direct cathodic reduction of the C(sp2) electrophile (entries 3–4). Additionally, the role of electricity was crucial as simply allowing the reaction mixture to stir in the presence of the magnesium electrode, which in principle could act as a terminal reductant, only gave a trace yield of product (entry 5). Increasing the nickel and ligand loading to 20 mol % and slightly increasing the amount of silver provided a significant boost in yield to 59% (entries 6–7). Additionally, the reaction could be performed equally well in NMP and to our surprise could even be run open to air in the absence of any Schlenck techniques, delivering the desired compound in 51% isolated yield (entries 8–9). Despite the slightly lower yields observed for this less rigorous setup, it was selected as the preferred general conditions based on the practical advantages as it enables easy parallel synthesis set-ups and reduces preparation time for single reactions. Even technical-grade NMP could be used in this cross-coupling reaction without affecting the yield, thus highlighting the robust nature of this cross-coupling (entry 10). Unfortunately, the corresponding aryl bromide did not couple as efficiently, likely due to sluggish oxidative addition to the highly electron-rich arene (entry 11). An important point of differentiation to the developed method is the stoichiometry of the starting materials. All the previously mentioned protocols use an excess of the redox active ester while this approach uses a slight excess of the aryl halide coupling partner in the optimized conditions. However, this reaction can be run using excess RAE with minimal losses in yield if the aryl halide is more precious (see the SI).
Figure 2.

Development of the Ag–Ni electrocatalytic DCC reaction. aYield determined by 1H-NMR with 1,3,5-trimethoxybenzene as an internal standard. Permitted use of logo is credit to IKA.
In the final optimized manifestation of this protocol, the reaction was carried out as follows. First, all the reagents except AgNO3 were added to a 5 mL ElectraSyn 2.0 vial directly. It was observed that extended stirring of the solution with AgNO3 led to detrimental outcomes, likely due to decomposition of the in situ-generated AgCl salt.32 Then, NMP or DMF was added, resulting in a homogeneous solution. Finally, silver nitrate was added directly as a solid, and the contents were allowed to undergo constant current electrolysis at 12 mA for 2.5 F/mol (around 2.5 h). No precautions were taken to exclude water or air, and all reagents were used as delivered from a commercial supplier. The catalyst (NiCl2·6H2O) is amongst the most inexpensive sources of Ni available (ca. $57/mole). The ligand (2,2′-bpy) can be obtained for approximately $1.5/g. Finally, AgNO3 is a benchtop stable reagent that used to be widely used in the photographic industry (ca. $10/g). As previously reported, recycled Ag-electrodes can be employed.
EXPLORATION OF SCOPE
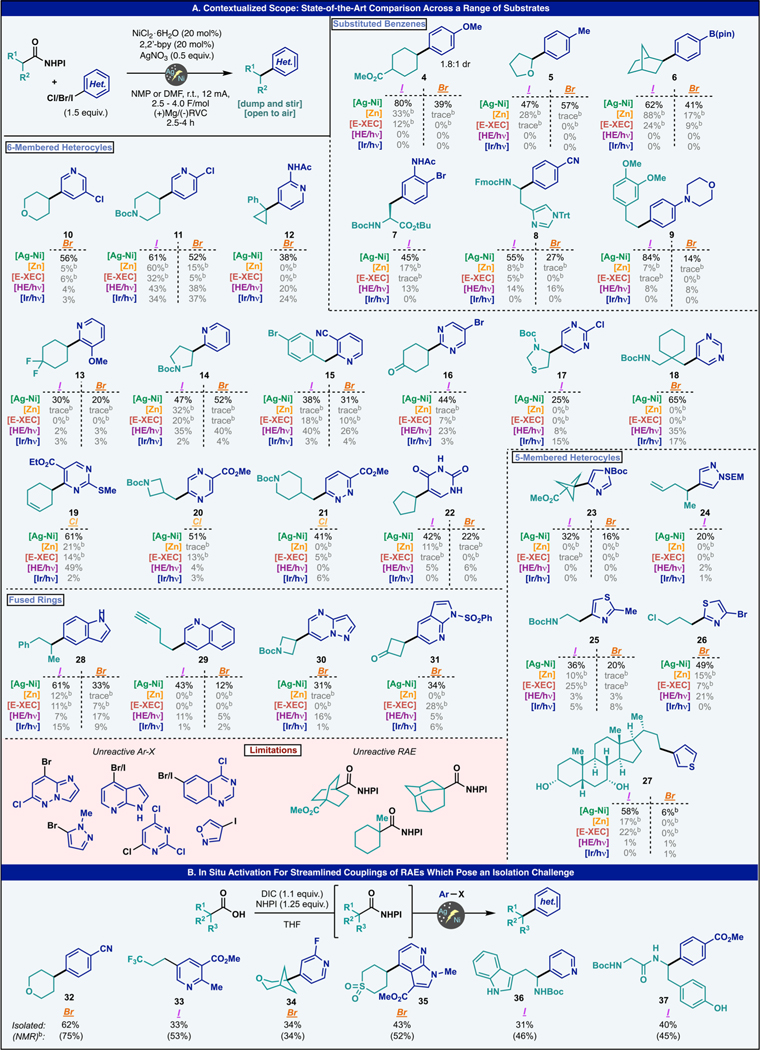
The generalization of the Ag-enabled DCC-arylation is outlined in Table 1. In consultation with our pharma collaborators, a vast range of functionality relevant to medicinal chemistry campaigns were curated and benchmarked against contemporaneous decarboxylative arylation methods. Selection criteria for substrates were biased toward historically challenging coupling partners and pharmacophores of high interest. A diverse array of both arene and RAE subunits were evaluated, and the arenes can be segregated into four categories: (1) substituted benzenes, (2) six-membered heterocycles, (3) five-membered heterocycles, and (4) fused heterocycles. Across these four categories, a variety of alkyl components were evaluated and include primary, secondary, strained tertiary, and alpha-heteroatom redox active esters.
Table 1.
Contextualized Scope of the Ag–Ni Electrocatalytic DCCa
|
Yields of isolated products are indicated in each case unless otherwise specified.
Yield determined by 1H-NMR with 1,3,5-trimethoxybenzene as an internal standard.
Substituted benzenes represent the largest class of reported substrates for DCC-type couplings. However, numerous limitations remain, some of which are not widely advertised. For instance, electron neutral or rich arenes are often problematic. Substrates 4 and 5 are emblematic of this issue. Whereas photochemical-based DCC reactions of simple carboxylate precursors to these compounds are well documented, they fail when employing anisole or toluene-based haloarenes. Although chemical activation provides serviceable yields of 4 and 5 (when using Ar-I), the current protocol proved superior. In these cases, even the corresponding aryl bromides also work, whereas none of the other methods evaluated provided any product. From a strategic standpoint, the value of accessing 4 in one step is undeniable as prior routes required laborious multistep processes based on 2-electron logic.33 The boron-containing substrate 6 has been synthesized before using an exotic organosilicate coupling partner.34 While the current method enabled its synthesis in reasonable yields from the aryl iodide, it outshone other methods when using the corresponding bromide, which may be the only option a medicinal chemist has when advancing a complex chemical series. Aryl iodides that bear the acetylated ortho bromoaniline motif were efficiently while preferentially reacting at the aryl iodide to afford unnatural amino acid derivative 7 in 45% yield. This product represents a useful starting material for a Larock/Castro indole synthesis.33–37 Histidine, an amino acid bearing an oxidatively-labile imidazole motif, was derivatized effectively, affording 8 in 55% yield with 4-iodobenzonitrile and in 27% yield with the corresponding bromide. Interestingly, existing literature methods failed to deliver more than 20% of the desired product with iodide and only gave trace product with aryl bromide. Morpholines are an important motif in medicinal chemistry owing to their favorable physiochemical, biological, and metabolic properties.38 To probe the robustness of the reaction to this valuable functional group, compound 9 was accessed efficiently from the corresponding aryl iodide yet faltered with the aryl bromide. Comparatively, existing methods failed to deliver more than 10% yield with either halide.
Six-membered heterocycles such as pyridine, pyrimidine, pyrazine, and pyridazine are ubiquitous in marketed therapeutics.39 Despite their prevalence in this sector, their behavior in cross-coupling reactions is often hard to predict owing to the vast range of electronic properties they can adopt as well as the presence of Lewis-basic nitrogen atoms that can complicate transition-metal-mediated processes.40,41 Chloropyridines 10 and 11 are emblematic of this problem. Whereas 10 was barely accessible with existing literature methods, 11 was produced far more efficiently across the board when the chlorine atom was moved from the C-3 to C-2 position. In contrast, the current DCC protocol coupled both in a highly efficient manner. Functionalized pyridines such as 12 could be accessed efficiently with strained and stabilized cyclopropyl radicals. C2-substituted pyridines are a challenging class of compounds to access through cross-coupling methods.42 To this end, compounds 13–15 were selected to encompass a wide range of electronic and steric properties. Electron-rich, neutral, and -deficient C2-halo pyridines were coupled in serviceable yields using the developed protocol. Importantly, existing methods, while sporadically working with the 2-iodopyridine derivatives, generally failed with the corresponding bromides, highlighting the challenges associated with this specific pyridine substitution pattern. Pyrrolidine adduct 14 has previously been prepared either through Suzuki/hydrogenation of the corresponding pyrrolidinone or via de novo pyrrolidine synthesis (dipolar cycloaddition), both of which are nonideal in a medicinal chemistry setting. Substituted pyrimidines coupled efficiently as well.43,44 Notably, excellent halo selectivity was observed when bromo-iodopyrimidines were employed delivering, for example, ketone bearing 16 in 44% yield. Notably, cysteine in its protected thiazoline form could be coupled with 5-iodo-2-chloropyrimidine to afford 17 in 25% yield. This coupling is noteworthy in the context of the existing methods as it bears a sulfide functional group which can poison catalytic processes and easily engage in exogenous redox events. Unsubstituted pyrimidines perform efficiently as well as the redox active ester derived from gabapentin, which was coupled with 5-bromopyrimidine to afford 18 in 65% yield. A chlorine atom adjacent to a nitrogen atom in a six-membered heterocycle is a versatile intermediate toward further derivatization. Additionally, such motifs are easily accessed in highly complex ring systems owing to the numerous deoxychlorination methods that map onto the products of heterocycle ring synthesis products.45 Therefore, countless methods have emerged to engage them in cross-coupling reactions to further extend their utility.46–48 Ester-bearing chloropyrimidine was coupled efficiently with an alkene bearing redox active ester to afford 19 in 61% yield. It is instructive to point out that pyrimidines did not perform well in zinc or electrochemically mediated reductive cross-coupling reactions unless they were activated by a neighboring electron withdrawing group (EWG). To further probe the reactivity of heteroaryl chlorides, pyrazine-containing 20 was generated in 51% yield, and pyridazine-containing 21 was accessed in 41% yield from the respective heteroaryl chlorides. In a final test of functional group compatibility for six-membered heterocycles, uracil derivative 22 (ca. $1000/g) was targeted as there are limited protecting group options for this heterocycle, rendering uracil ring synthesis as the most logical current option.49 Despite these challenges, coupling with iodo- and bromouracil afforded 22 in 42% yield and 22% yield, respectively.
Five-membered heterocycles represent an important challenge in the context of radical cross-coupling methodology.50–52 Such molecules are often underrepresented in the current C(sp2)–C(sp3) radical cross-coupling literature. Given the functional group compatibility demonstrated above, several five-membered heterocycles were targeted. Imidazole-containing propellane 23 was generated from the corresponding iodo- and bromo-imidazole coupling partners in 32% yield and 16% yield, respectively. SEM-protected iodo-pyrazole was coupled with a secondary acyclic homoallylic redox active ester giving 24 in 20% yield. It is important to note that all the existing methods tested for DCC transformations failed to give more than trace amounts of product for these challenging cross-coupling partners. Thiazoles were able to perform well in this cross-coupling platform. Beta-glycine derivative 25 was generated from the corresponding bromo- and iodo-methylthiazole. Activation of the thiazole with an electron withdrawing bromine atom significantly improved the results of the cross-coupling as evidenced by 26, which formed in 49% from the corresponding bromide. Notably, excellent chemoselectivity was observed between the second bromine atom on the thiazole as well as the pendant alkyl chloride on the redox active ester fragment. Finally, the RAE derived from unprotected chenodeoxycholic acid was coupled efficiently with 3-iodothiophene to provide 27 in 58% yield. Such arylated cholic acid derivatives are usually synthesized through Minisci transformations limited to six-membered heterocycles.53,54
Higher order heterocycles that bear fused aromatic rings are important subclasses of arenes as well.55 Unprotected indole motifs were well tolerated in this reaction as 5-iodoindole was coupled efficiently to afford 28 in 61% yield while giving a modest 33% yield with the corresponding bromide. Quinoline, another important bicycle which is usually functionalized via Minisci approaches,56 was coupled to the free alkyne bearing RAE to give 29 in 42% yield from the iodo-quinoline and 12% from the bromide. Pyrazolopyrimidine derivate 30 was successfully accessed in 31% yield from the corresponding bromide. Prior published routes to 30 involved the coupling of an alkyl bromide in a reductive cross-electrophile coupling mediated by zinc in 15% yield.57 Finally, sulphonamide-protected azaindole-containing cyclobutanone 31 was synthesized in 34% yield with this cross-coupling protocol, while only the previously reported electrochemical approach gave comparable yields.
In general, many of the known methods for achieving DCC-arylation feature complementary scope. Empirical screening is often needed to deconvolute which particular method is suitable for a particular substrate class, akin to ligand-enabled Pd-based cross-couplings.58 Some methods fare well with electron-deficient arenes but not with electron-rich arenes. Similarly, certain couplings do not tolerate sensitive functional groups (e.g., free phenols, indoles, histidines, acidic heterocyclic N–H) or Lewis-basic heteroatoms. The present method appears to be a particularly general protocol for achieving such couplings in consistent and meaningful yields by leveraging the unique reactivity benefits of Ag-functionalized electrodes in Nielectrocatalysis32 (vida infra); however, it is not without its limitations. Throughout the course of this study, heterocycles were encountered that were unreactive, such as N-alkyl pyrazoles, tricholoropyrimidines, quinazolines, and other fused heterocycles. Additionally, isoxazole motifs were found to undergo reductive ring opening during the reaction and led to no desired cross-coupling products. With respect to the alkyl coupling partner, [2.2.2] redox active esters failed to couple as well as adamantyl and simple tertiary radical precursors. In such couplings, only the reduced redox active ester was observed.
In real-world scenarios, it may be an inconvenience to isolate and purify RAE coupling partners. In other cases, the RAE may be too hydrolytically labile to be easily isolated. Thus, an in situ protocol for RAE generation was developed as depicted in Table 1B. The stable RAE precursor to 32 was used as a test case for this purpose. The only major procedural difference here involved conducting activation and cross-coupling steps under a balloon of Ar. This was necessary due to the empirical observation that the presence of the urea byproduct (from RAE formation) rendered the reaction slightly more air-sensitive (~10% yield depreciation relative to using a pre-made RAE). This is likely due to the urea’s ability to complex the nickel catalyst and interfere with productive cross-coupling under air. Using this modified in situ protocol, 32 was obtained in 62% isolated yield (75% NMR yield). Substrates 33–37, unlike 32, utilized RAEs that were either unstable or difficult to purify. Because of the polar nature of products 33–37, NMR yields are also given as some material was lost during purification. RAE’s bearing trifluoromethyl motifs were surprisingly hydrolytically labile. Fortunately, utilizing the in situ protocol, trisubstituted pyridine 33 was accessed in 33% isolated yield (53% NMR yield). Next, a 3-oxabicyclo[3.1.1]-heptane bearing RAE was coupled with 2-fluoro-4-bromo pyridine to afford 34 in 34% isolated yield. Sulfone bearing redox active esters were highly challenging to purify because of degradation on silica as well as coelution with coupling reagent byproducts. However, 35 was accessed efficiently in 43% isolated yield (52% NMR yield) after coupling with a densely functionalized bromoazaindole. RAEs generated from amino acid derivatives and dipeptides were particularly challenging to purify when reactive residues were used but were coupled efficiently using this protocol. Tryptophan analog 36, bearing an unprotected indole, was accessed in 31% isolated yield (46% NMR yield). Finally, a tyrosine bearing dipeptide was engaged in successful cross-coupling, delivering 37 in 40% yield (45% NMR yield) in the presence of the unprotected phenol.
APPLICATIONS TO SIMPLIFY SYNTHESIS
The broadened scope and enhanced chemoselectivity of this electrocatalytic DCC enable distinct access to structures of medicinal importance. To demonstrate this point, five case studies were chosen, four of which are derived from the literature and one of which was encountered internally by our pharma collaborators (Figure 3). Each of these provide unique examples of how the current protocol proved superior to both conventional and newly emerging methods. Pyridine-containing amino acid 38 has previously been accessed through a Negishi-coupling strategy from glutamic acid, necessitating several FG-interconversions to access the requisite organozinc derivative.59 In contrast, the same starting materials can be employed directly to arrive at 38 in 63 or 45% from either the pyridyl iodide or bromide, respectively. Gram quantities of both are easily accessible with the same potentiostat by simply increasing the reaction vial size and current delivered with a similar yield. Pyrazole-cyclohexanone derivative 40 was previously accessed through a very common Suzuki-hydrogenation approach.60 This route, which is emblematic of hundreds if not thousands of examples found in the patent literature, necessitates formation of a vinyl boronate (41) via a vinyl triflate and subsequent Suzuki cross-coupling followed by hydrogenation. In this case, two protecting group steps are needed because of the presence of another ketone, and three different Pd catalysts are required across three steps, adding drastically to the cost of the route. In contrast, 40 was accessed directly from a cyclohexanone bearing redox active ester (42) and an appropriate iodopyrazole (43) in 31% yield. Morpholine 44 was previously accessed through a lengthy synthetic sequence that was centered around stepwise construction of the morpholine (via 46) onto the acid bearing pyrimidine starting material 45.61 Instead, 44 could be accessed from coupling of a morpholine-derived redox active ester 47 (derived from the commercial acid) and 2-iodo-5-bromo pyrimidine in 28% yield, thus bypassing the need for a tedious ring synthesis strategy for such compounds. Indoline 48 is an intermediate toward (R)-silodosin (Rapaflo), an FDA-approved therapeutic for the treatment of benign prostatic hyperplasia. In collaboration with Minakem, our laboratory previously pursued an approach toward 48 that utilized a reductive cross-electrophile coupling between 49 and 50.62 In that initial approach, 50 must first be converted to the more reactive iodide (via Cu-catalyzed exchange)63 prior to a Zn-mediated reductive DCC, as utilizing the aryl bromide directly failed. In contrast, the newly developed protocol described herein was used to couple the bromide directly in 37% yield without the need for the aromatic halogen exchange step. Finally, intermediate 51, a useful scaffold for a current medicinal chemistry program, was specifically targeted as its access proved elusive under a variety of known DCC conditions. Thus, DCC of RAE 52 with pyrimidine 53 (Y = ZnX) under Negishi-like conditions failed as well as chemical and photochemical cross-electrophile strategies (Y = Br, 54).28,30,64 Oxidative PET-based DCC using acid 55 and pyrimidine 56 (a C2-Cl reagent was used rather than the photochemically labile SMe group) was also unsuccessful.65 In contrast, the current protocol delivered 41% isolated yield of the target from 52 and 54, thereby opening the door to exploring SAR on this valuable series.
Figure 3.

Strategic applications of the Ag–Ni electrocatalytic DCC to medicinally relevant motifs across a wide range of literature sources and active collaborations.
SCALE REGIMES: PARALLEL SYNTHESIS TO FLOW-BASED SCALE UP
While the aforementioned attributes of Ag–Ni based electrocatalytic DCC are attractive from the standpoint of enabling access to new chemical space, the ability to generate libraries and scale up promising structures is critical. In 2019, commercial vendors began selling standardized equipment geared toward synthetic chemists for parallel electrochemical synthesis. More recently, the Lin group in collaboration with Merck revealed another interesting approach to reaction miniaturization.66 For this study, we elected to use the commercial “E-hive” module sold by IKA that conveniently attaches to ElectraSyn 2.0 (Figure 4A). This device is capable of running 24 reactions in parallel at constant potential. It features a unique design wherein the reaction vessel is itself an electrode (stainless steel) and a small graphite rod (which is part of the cap) is the other electrode. Either one can serve as a cathode or anode depending on the desired setting. To demonstrate the utility of such a setup for library synthesis, a set of 12 arenes (screened against 55) and 12 RAEs (screened against 56) was selected. To replicate an actual discovery setting, we deliberately chose substrates that had not been tested before. Furthermore, the electrode materials, concentration, and current density were completely different from the standardized procedure, rendering e-hive screening a real-world robustness test for this reaction.
Figure 4.

(A) Parallel synthesis on IKA e-hive enables rapid small-scale analog screening. (B) Preparative scale parallel synthesis enables access to serviceable amounts of derivatives and serves to validate selected entries from the e-hive entries. (C) Recirculating-flow scale up enables access to decagram quantities of 1 and 38 leveraging the developed Ag–Ni electrocatalytic protocol. Permitted use of logo is credit to IKA.
In practice, the above concerns proved unnecessary as the experiment was both simple and intuitive to setup and worked well, largely owing to the reaction’s innate insensitivity to air, current, and excess electricity. Thus, of the 24 substrates run in parallel on this device, 20 gave detectable amounts of product by HPLC/MS analysis. The functionality present across the successful entries of this screen covers a wide range of chemical space and allowed us to test various functional groups that were not explored in the prior scope study. With respect to the arene, several entries are worth noting. Thiazoles and benzothiazole (57, 66), an anisole derivative (58), sulfide bearing pyrimidines (59), a variety of C2-substituted pyridines (60, 61, 67), the nucleobase found in remdesivir (62), as well as a Boc-protected aniline (63) and fluorobenzene (64) all gave detectable amounts of products. Unfortunately, aldehyde bearing azaindole (65) and a complex pyrazole (68) failed to give any detectable amounts of product. The chemical space of the RAE screen led to some unique discoveries of functional group compatibility. [2.2.1] Bicycle (69), homoproline (70), cyclopropyl (71), protected hydroxyproline (72), monofluoromethyl (74), alpha-amide (75), pyranyl (76), nitrile bearing [1.1.1] propellane (77), neopentyl (78), and diols derived from tartaric acid (80) all gave the substituted azaindole product in this screen. However, the hindered sesquiterpene fragment derived from gibberellic acid (73) and the tertiary substructure (79) derived from clofibric acid failed to give product likely due to steric issues.
With a panel of successful couplings demonstrated on the 15–20 mg scale, validation under optimized reaction conditions was pursued in parallel using the IKA carousel (Figure 4B). Electron-poor 2-bromo-3-fluoro-6-chloropyridine coupled efficiently with 55 to afford 81 in 44% isolated yield (51% NMR yield). Inverting the electronics of the pyridine and coupling with 2-bromo-5-methoxypyridine gave pyran derivative 82 in 32% yield (34% NMR yield). Adding an ortho substituent was still tolerated and 83 was accessed in 28% yield (28% NMR yield) after coupling with 2-iodo-3-methylpyridine. On the other three positions of the carousel, a series of azaindole derivatives were accessed simultaneously. Pyrrolidine bearing 84 was synthesized in 46% isolated yield (57% NMR yield). Neopentyl-substituted 85 was similarly accessed in 37% isolated yield (47% NMR yield). Finally, cyclopropyl substrate 86 represented an interesting opportunity to couple strained radicals using this chemistry. While this compound was detected in the e-hive screen, it was low yielding in the preparative reaction, giving 86 in only 5% isolated yield (11% NMR yield) and was found to readily decompose upon standing. Consistent with the above miniaturization in the e-hive, these couplings gave serviceable yields of products in this parallel preparative procedure.
Finally, to probe the scalability of this reaction, a 100 mmol scale synthesis of unnatural amino acids 1 and 38 was pursued utilizing recirculating flow (Figure 4C).67 Additionally, given the inconvenience of synthesizing and isolating 100 mmol of the necessary RAE, an in situ protocol was targeted to avoid an intermediate purification step en route to multigram quantities of 1 and 38. In practice, 100 mmol (ca. 34 g) of glutamic acid derived 87/88 was activated in THF with DIC and NHPI for 3 h. Given the empirical observation that the urea byproducts from in situ activation can hinder the performance of the reaction, a means by which to remove this byproduct was needed in order to carry out this large-scale reaction open to air. Fortunately, the diisopropylurea is not completely soluble in THF and can simply be filtered off using filter paper as the RAE is being transferred to the round-bottom flask containing the nickel catalyst and the iodoarene coupling partner together with lithium chloride as a supporting electrolyte. Once the addition was complete, the key silver nitrate additive was added directly to the flask as a solid. A peristaltic pump was enlisted to recirculate the reaction mixture through the reactor bearing a sacrificial magnesium plate anode and carbon felt cathode (see the SI for full details and graphical guide). In the initial hour of the reaction, the reaction mixture progressed from a heterogenous suspension of silver halide salts to a homogenous green solution as the silver is plated within the electrode chamber on the carbon felt cathode. After electrolysis at 1 A for 12 h, the reaction mixture was worked up following the standard protocol, and 1 and 38 were accessed in 15 g (43%) and 18 g (49%), respectively.
OUTLOOK AND CONCLUSIONS
Owing to the ubiquitous nature of carboxylic acids in nature and within the catalogs of commercial suppliers, the utilization of decarboxylative cross-coupling methods is seeing increasing use. In essence, carboxylic acids (or their RAE congeners) can be viewed as easily accessible and stable surrogates for alkyl halides (of which relatively few are available) and thus represent ideal radical synthons. The current study capitalizes on a unique observation made during the course of a terpene total synthesis campaign involving in situ-deposited Agnanoparticles on an electrode surface.32 Such functionalized electrodes were shown to dramatically improve the yield of DCC-based alkenylation in a variety of contexts. The mechanistic aspects of this phenomenon were explored in great detail and laid a groundwork for the current study. Specifically, the issues encountered in that study (low yields and chemoselectivity) were quite reminiscent of the challenges facing DCC-based arylation.
This comprehensive study, conducted in collaboration with multiple companies, addresses many of the less-publicized limitations with this powerful transformation. Foremost in our view were the scope limitations apparent on both the arene and carboxylic acid-based fragments. To be sure, electron-rich arenes, diverse heterocycles, and highly functionalized carboxylic acids are all highly coveted coupling partners. To vividly demonstrate this challenge, the present work benchmarks state-of-the-art conditions across the entire scope explored. Overall, the present Ag-Ni electrocatalytic protocol appears to have superior generality. In addition, it features high simplicity, robustness, and short reaction times (2.5 h). Several examples are highlighted, wherein the success of these uniquely enabled reactions can have a simplifying effect on synthesis. This reaction has been extensively field-tested amongst our collaborators and works on a wide range of additional proprietary structures not depicted herein. Finally, for this reaction to have maximal impact, it must be useful in real-world scale regimes. Thus, a demonstration of parallel synthesis to rapidly generate analog libraries, subsequent scale up for more meaningful quantities, and a simple recirculating-flow setup for decagram scale and beyond are reported.
From a strategic standpoint, this study adds another chapter to the growing body of literature, showing that in many contexts, a one-electron based retrosynthetic plan should be considered alongside more conventional polar bond analysis. It is anticipated that this work will therefore find utility in a variety of settings and contribute to the more mainstream adoption of electrochemical tactics and radical retrosynthetic strategies.
Supplementary Material
ACKNOWLEDGMENTS
We thank D.-H. Huang. and L. Pasternack. for assistance with NMR spectroscopy; J. Chen, B Sanchez, and E. Sturgell (Scripps Automated Synthesis Facility) for assistance with HPLC, HRMS, and LCMS; B. P. Smith, R. A. Bauer, S. Gnaim, Y. Kawamata, A. Pollatos, and A. Garrido-Castro for helpful discussions; D. Chiodi for donation of starting materials; M.D.P. thanks Richard and Nicola Lerner for the Endowed Fellowship and BMS for the Graduate Research Fellowship.
Funding
Financial support for this work was provided by the NIH (MIRA GM-118176), NSF Center for Synthetic Organic Electrochemistry (CHE-2002158). M.D.P. thanks Richard and Nicola Lerner for the Endowed Fellowship and BMS for the Graduate Research Fellowship. G.L. thanks the George E. Hewitt Foundation for the postdoctoral fellowship. J.C. thanks Vivozon, Inc. for the Young Scientist Grant and Postdoctoral Fellowship.
ABBREVIATIONS
- AgNP
silver nanoparticle
- DCC
Decarboxylative cross-coupling
- RAE
Redox active esters
- PET
Photoinduced electron transfer
- DMF
N,N′-dimethylformamide
- NMP
N-methyl-2-pyrrolidone
- EWG
Electron withdrawing group
- SAR
Structure–activity relationship
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c08006.
Additional experimental details, materials, methods, and characterization data for all new compounds (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c08006
Contributor Information
Maximilian D. Palkowitz, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States.
Gabriele Laudadio, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States.
Simon Kolb, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States.
Jin Choi, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States.
Martins S. Oderinde, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States
Tamara El-Hayek Ewing, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States.
Philippe N. Bolduc, Biogen Inc., Cambridge, Massachusetts 02142, United States
TeYu Chen, Biogen Inc., Cambridge, Massachusetts 02142, United States.
Hao Zhang, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States.
Peter T. W. Cheng, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States
Benxiang Zhang, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States.
Michael D. Mandler, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States
Vanna D. Blasczak, Biogen Inc., Cambridge, Massachusetts 02142, United States
Jeremy M. Richter, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States
Michael R. Collins, Oncology Medicinal Chemistry Department, Pfizer Pharmaceuticals, San Diego, California 92121, United States
Ryan L. Schioldager, Oncology Medicinal Chemistry Department, Pfizer Pharmaceuticals, San Diego, California 92121, United States
Martin Bravo, Oncology Medicinal Chemistry Department, Pfizer Pharmaceuticals, San Diego, California 92121, United States.
T. G. Murali Dhar, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States.
Benjamin Vokits, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States.
Yeheng Zhu, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States.
Pierre-Georges Echeverria, Minakem Recherche, Beuvry-La-Forêt 59310, France.
Michael A. Poss, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States
Scott A. Shaw, Bristol Myers Squibb Research and Development, Princeton, New Jersey 08534, United States
Sebastian Clementson, Research and Early Development, LEO Pharma A/S, 2750 Ballerup, Denmark.
Nadia Nasser Petersen, Research and Early Development, LEO Pharma A/S, 2750 Ballerup, Denmark.
Pavel K. Mykhailiuk, Enamine Ltd., 02094 Kyiv, Ukraine
Phil S. Baran, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States
REFERENCES
- (1).Romero KJ; Galliher MS; Pratt DA; Stephenson CRJ Radicals in natural product synthesis. Chem. Soc. Rev 2018, 47, 7851–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Jasperse CP; Curran DP; Fevig TL Radical reactions in natural product synthesis. Chem. Rev 1991, 91, 1237–1286. [Google Scholar]
- (3).Gomberg M.An instance of trivalent carbon: triphenylmethyl. J. Am. Chem. Soc 1900, 22, 757–771. [Google Scholar]
- (4).Stork G; Mook R; Biller SA; Rychnovsky SD Free-radical cyclization of bromo acetals. Use in the construction of bicyclic acetals and lactones. J. Am. Chem. Soc 1983, 105, 3741–3742. [Google Scholar]
- (5).Curran DP; Rakiewicz DM Tandem radical approach to linear condensed cyclopentanoids. Total synthesis of (±)-hirsutene. J. Am. Chem. Soc 1985, 107, 1448–1449. [Google Scholar]
- (6).Barton DHR; Beaton JM; Geller LE; Pechet MM A new photochemical reaction. J. Am. Chem. Soc 1960, 82, 2640–2641. [Google Scholar]
- (7).Barton DHR; Crich D; Motherwell WB The invention of new radical chain reactions. Part VIII. Radical chemistry of thiohydroxamic esters; A new method for the generation of carbon radicals from carboxylic acids. Tetrahedron 1985, 41, 3901–3924. [Google Scholar]
- (8).Delduc P; Tailhan C; Zard SZ A convenient source of alkyl and acyl radicals. J. Chem. Soc., Chem. Commun 1988, 4, 308–310. [Google Scholar]
- (9).Minisci F; Bernardi R; Bertini F; Galli R; Perchinummo M.Nucleophilic character of alkyl radicals—VI: A new convenient selective alkylation of heteroaromatic bases. Tetrahedron 1971, 27, 3575–3579. [Google Scholar]
- (10).Duncton MAJ Minisci reactions: Versatile CH-functionalizations for medicinal chemists. MedChemComm 2011, 2, 1135–1161. [Google Scholar]
- (11).Kharasch MS; Jensen EV; Urry WH Reactions of Atoms and Free Radicals in Solution. X. The Addition of Polyhalomethanes to Olefins. J. Am. Chem. Soc 1947, 69, 1100–1105. [Google Scholar]
- (12).Kochi JK; Rust FF Oxidation of Free Radicals from Unsaturated Compounds by Cupric Salts. J. Am. Chem. Soc 1962, 84, 3946–3953. [Google Scholar]
- (13).Kochi JK; Tamura M.Alkylcopper(I) in the coupling of Grignard reagents with alkyl halides. J. Am. Chem. Soc 1971, 93, 1485–1487. [Google Scholar]
- (14).Smith JM; Harwood SJ; Baran PS Radical Retrosynthesis. Acc. Chem. Res 2018, 51, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Peters DS; Pitts CR; McClymont KS; Stratton TP; Bi C; Baran PS Ideality in Context: Motivations for Total Synthesis. Acc. Chem. Res 2021, 54, 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Smith JM; Dixon JA; deGruyter JN; Baran PS Alkyl Sulfinates: Radical Precursors Enabling Drug Discovery. J. Med. Chem 2019, 62, 2256–2264. [DOI] [PubMed] [Google Scholar]
- (18).Dombrowski AW; Gesmundo NJ; Aguirre AL; Sarris KA; Young JM; Bogdan AR; Martin MC; Gedeon S; Wang Y.Expanding the Medicinal Chemist Toolbox: Comparing Seven C(sp2)–C(sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett 2020, 11, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Parida SK; Mandal T; Das S; Hota SK; De Sarkar S; Murarka S.Single Electron Transfer-Induced Redox Processes Involving N-(Acyloxy)phthalimides. ACS Catal. 2021, 11, 1640–1683. [Google Scholar]
- (20).Patra T; Maiti D.Decarboxylation as the Key Step in C–C Bond-Forming Reactions. Chem. – Eur. J 2017, 23, 7382–7401. [DOI] [PubMed] [Google Scholar]
- (21).Oderinde MS; Varela-Alvarez A; Aquila B; Robbins DW; Johannes JW Effects of Molecular Oxygen, Solvent, and Light on Iridium-Photoredox/Nickel Dual-Catalyzed Cross-Coupling Reactions. J. Org. Chem 2015, 80, 7642–7651. [DOI] [PubMed] [Google Scholar]
- (22).Chen X; Luo X; Peng X; Guo J; Zai J; Wang P.Catalyst-Free Decarboxylation of Carboxylic Acids and Deoxygenation of Alcohols by Electro-Induced Radical Formation. Chem. – Eur. J 2020, 26, 3226–3230. [DOI] [PubMed] [Google Scholar]
- (23).Murarka S.N-(Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal 2018, 360, 1735–1753. [Google Scholar]
- (24).Sabat M; Johnson CR Synthesis of Unnatural Amino Acids via Suzuki Cross-Coupling of Enantiopure Vinyloxazolidine Derivatives. Org. Lett 2000, 2, 1089–1092. [DOI] [PubMed] [Google Scholar]
- (25).Wallberg H; Xu M; Lin Q; Lei S; PSun P; Parkes K; Johnson T; Samuelson B.(2,5-dioxoimidazolidin-1-yl)-N-hydroxyacetamides a s metalloproteinase inhibitors. WO2007068474, 2007. [Google Scholar]
- (26).Do Q; Nguyen GT; Phillips RS Inhibition of tyrosine phenol-lyase by tyrosine homologues. Amino Acids 2016, 48, 2243–2251. [DOI] [PubMed] [Google Scholar]
- (27).Yoon YH Method for the preparation of unnatural amino acid. KR2016021569, 2014. [Google Scholar]
- (28).Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc 2016, 138, 5016–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Koyanagi T; Herath A; Chong A; Ratnikov M; Valiere A; Chang J; Molteni V; Loren J.One-Pot Electrochemical Nickel-Catalyzed Decarboxylative Sp2–Sp3 Cross-Coupling. Org. Lett 2019, 21, 816–820. [DOI] [PubMed] [Google Scholar]
- (30).Kammer LM; Badir SO; Hu R-M; Molander GA Photoactive electron donor–acceptor complex platform for Ni-mediated C(sp3)–C(sp2) bond formation. Chem. Sci 2021, 12, 5450–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cesar PK; Kautzky Jacob A; Krska Shane W; Nowak T; Dreher SD; MacMillan DWC Accelerating reaction generality and mechanistic insight through additive mapping. Science 2022, 376, 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Harwood SJ; Palkowitz MD; Gannett CN; Perez P; Yao Z; Sun L; Abruña HD; Anderson SL; Baran PS Modular terpene synthesis enabled by mild electrochemical couplings. Science 2022, 375, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lu Z; Chen Y-H; Smith C; Li H; Thompson C; Sweis R; Sinclair P; Kallashi F; Hunt J; Adamson S; Dong G; Ondeyka D; Qian X; Sun W; Vachal P; Zhao K.Cyclic amine-substituted oxazolidinones as CETP inhibitor and their preparation. WO2012058187, 2012. [Google Scholar]
- (34).Vara BA; Jouffroy M; Molander GA C(sp3)–C(sp2) cross-coupling of alkylsilicates with borylated aryl bromides – an iterative platform to alkylated aryl- and heteroaryl boronates. Chem. Sci 2017, 8, 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Larock RC; Yum EK Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J. Am. Chem. Soc 1991, 113, 6689–6690. [Google Scholar]
- (36).Castro CE; Havlin R; Honwad VK; Malte AM; Moje SW Copper(I) substitutions. Scope and mechanism of cuprous acetylide substitutions. J. Am. Chem. Soc 1969, 91, 6464–6470. [Google Scholar]
- (37).Shen M; Li G; Lu BZ; Hossain A; Roschangar F; Farina V; Senanayake CH The First Regioselective Palladium-Catalyzed Indolization of 2-Bromo- or 2-Chloroanilines with Internal Alkynes: A New Approach to 2,3-Disubstituted Indoles. Org. Lett 2004, 6, 4129–4132. [DOI] [PubMed] [Google Scholar]
- (38).Kourounakis AP; Xanthopoulos D; Tzara A.Morpholine as a privileged structure: A review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med. Res. Rev 2020, 40, 709–752. [DOI] [PubMed] [Google Scholar]
- (39).Baumann M; Baxendale IR An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem 2013, 9, 2265–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kutchukian PS; Dropinski JF; Dykstra KD; Li B; DiRocco DA; Streckfuss EC; Campeau L-C; Cernak T; Vachal P; Davies IW; Krska SW; Dreher SD Chemistry informer libraries: a chemoinformatics enabled approach to evaluate and advance synthetic methods. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Uehling MR; King RP; Krska SW; Cernak T; Buchwald SL Pharmaceutical diversification via palladium oxidative addition complexes. Science 2019, 363, 405–408. [DOI] [PubMed] [Google Scholar]
- (42).Cook XAF; de Gombert A; McKnight J; Pantaine LRE; Willis MC The 2-Pyridyl Problem: Challenging Nucleophiles in Cross-Coupling Arylations. Angew. Chem., Int. Ed 2021, 60, 11068–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jones A; Kemp M; Stockley M; Gibson K; Whitlock G.1-Cyanopyrrolidine compounds as USP30 inhibitors and their preparation. WO2016156816, 2016. [Google Scholar]
- (44).Lai KW; Liang J; Zhang B; Labadie S; Ortwine D; Dragovich P; Kiefer J; Gehling VS; Harmange J-C N-Pyrrolidinyl amide compounds as histone demethylase inhibitors and their preparation. WO2016057924, 2016. [Google Scholar]
- (45).Ishihara Y; Montero A; Baran PS The Portable Chemist’s Consultant: A Survival Guide for Discovery, Process, and Radiolabelling; Macintosh Pusblishing, 2013. https://books.apple.com/us/book/the-portable-chemists-consultant/id618463142 (accessed May 13th, 2022). [Google Scholar]
- (46).Nimmagadda SK; Korapati S; Dasgupta D; Malik NA; Vinodini A; Gangu AS; Kalidindi S; Maity P; Bondigela SS; Venu A; Gallagher WP; Aytar S; González-Bobes F; Vaidyanathan R.Development and Execution of an Ni(II)-Catalyzed Reductive Cross-Coupling of Substituted 2-Chloropyridine and Ethyl 3-Chloropropanoate. Org. Proc. Res. Dev 2020, 24, 1141–1148. [Google Scholar]
- (47).Everson DA; Buonomo JA; Weix DJ Nickel-Catalyzed Cross-Electrophile Coupling of 2-Chloropyridines with Alkyl Bromide. Synlett 2014, 25, 233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fürstner A; Leitner A; Méndez M; Krause H.Iron-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc 2002, 124, 13856–13863. [DOI] [PubMed] [Google Scholar]
- (49).Basnak I; Balkan A; Coe PL; Walker RT The Synthesis of Some 5-Substituted and 5,6-Disubstituted 2′-Deoxyuridines. Nucleosides Nucleotides 1994, 13, 177–196. [Google Scholar]
- (50).Everson DA; Shrestha R; Weix DJ Nickel-Catalyzed Reductive Cross-Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2010, 132, 920–921. [DOI] [PubMed] [Google Scholar]
- (51).Hansen EC; Li C; Yang S; Pedro D; Weix DJ Coupling of Challenging Heteroaryl Halides with Alkyl Halides via Nickel-Catalyzed Cross-Electrophile Coupling. J. Org. Chem 2017, 82, 7085–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Tan J; Chen Y; Li H; Yasuda N.Suzuki-Miyaura Cross-Coupling Reactions of Unprotected Haloimidazoles. J. Org. Chem 2014, 79, 8871–8876. [DOI] [PubMed] [Google Scholar]
- (53).Zhang Z; He Q; Zhang X; Yang C.Photoredox-catalysed regioselective synthesis of C-4-alkylated pyridines with N-(acyloxy)-phthalimides. Org. Biomol. Chem 2022, 20, 1969–1973. [DOI] [PubMed] [Google Scholar]
- (54).Leverrier A; Bero J; Frédérich M; Quetin-Leclercq J; Palermo J.Antiparasitic hybrids of Cinchona alkaloids and bile acids. Eur. J. Med. Chem 2013, 66, 355–363. [DOI] [PubMed] [Google Scholar]
- (55).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (56).Proctor RSJ; Phipps RJ Recent Advances in Minisci-Type Reactions. Angew. Chem., Int. Ed 2019, 58, 13666–13699. [DOI] [PubMed] [Google Scholar]
- (57).Aguirre AL; Loud NL; Johnson KA; Weix DJ; Wang Y.ChemBead Enabled High-Throughput Cross-Electrophile Coupling Reveals a New Complementary Ligand. Chem. – Eur. J 2021, 27, 12981–12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Buitrago SA; Regalado EL; Pereira T; Shevlin M; Bateman K; Campeau L-C; Schneeweis J; Berritt S; Shi Z-C; Nantermet P; Liu Y; Helmy R; Welch CJ; Vachal P; Davies IW; Cernak T; Dreher Spencer D.Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science 2015, 347, 49–53. [DOI] [PubMed] [Google Scholar]
- (59).Usuki T; Yanuma H; Hayashi T; Yamada H; Suzuki N; Masuyama Y.Improved Negishi Cross-Coupling Reactions of an Organozinc Reagent Derived from L-Aspartic Acid with Monohalopyridines. J. Heterocycl. Chem 2014, 51, 269–273. [Google Scholar]
- (60).Burch JD; Lau K; Barker JJ; Brookfield F; Chen Y; Chen Y; Eigenbrot C; Ellebrandt C; Ismaili MHA; Johnson A; Kordt D; MacKinnon CH; McEwan PA; Ortwine DF; Stein DB; Wang X; Winkler D; Yuen P-W; Zhang Y; Zarrin AA; Pei Z.Property- and Structure-Guided Discovery of a Tetrahydroindazole Series of Interleukin-2 Inducible T-Cell Kinase Inhibitors. J. Med. Chem 2014, 57, 5714–5727. [DOI] [PubMed] [Google Scholar]
- (61).Galley G; Pflieger P; Norcross R; Cecere G; Shen H; Hu Y.Preparation of morpholinylpyridine derivatives for use as TAAR modulators. WO2015165835, 2015. [Google Scholar]
- (62).Chen T-G; Mele L; Jentzer O; Delbrayelle D; Echeverria P-G; Vantourout JC; Baran PS Convergent synthesis of (R)-silodosin via decarboxylative cross-coupling. Tetrahedron Lett. 2021, 79, No. 153290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Klapars A; Buchwald SL Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction. J. Am. Chem. Soc 2002, 124, 14844–14845. [DOI] [PubMed] [Google Scholar]
- (64).Cornella J; Edwards JT; Qin T; Kawamura S; Wang J; Pan C-M; Gianatassio R; Schmidt M; Eastgate MD; Baran PS Practical Ni-Catalyzed Aryl–Alkyl Cross-Coupling of Secondary Redox-Active Esters. J. Am. Chem. Soc 2016, 138, 2174–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Pinchman JR; Hopkins CD; Bunker KD; Huang PQ Methods for Cross Coupling. WO2018039232, 2018. [Google Scholar]
- (66).Rein J; Annand JR; Wismer MK; Fu J; Siu JC; Klapars A; Strotman NA; Kalyani D; Lehnherr D; Lin S.Unlocking the Potential of High-Throughput Experimentation for Electrochemistry with a Standardized Microscale Reactor. ACS Cent. Sci 2021, 7, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Noël T; Cao Y; Laudadio G.The Fundamentals Behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res 2019, 52, 2858–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.