

Abstract
α-Synuclein (α-syn) is an intrinsically disordered protein involved in a group of diseases collectively termed synucleinopathies, characterized by the aggregation of α-syn to form insoluble, β-sheet-rich amyloid fibrils. Amyloid fibrils are thought to contribute to disease progression through cell-to-cell transmission, templating and propagating intracellular amyloid formation. Raman spectral imaging offers a direct characterization of protein secondary structure via the amide-I backbone vibration; however, specific detection of α-syn conformational changes against the background of other cellular components presents a challenge. Here, we demonstrate the ability to unambiguously identify cellularly internalized α-syn fibrils by coupling Raman spectral imaging with the use of a genetically encoded aryl alkyne, 4-ethynyl-L-phenylalanine (FCC), through amber codon suppression. The alkyne stretch (C≡C) of FCC provides a spectrally unique molecular vibration without interference from native biomolecules. Cellular uptake of FCC-α-syn fibrils formed in vitro was visualized in cultured human SH-SY5Y neuroblastoma cells by Raman spectral imaging. Fibrils appear as discrete cytosolic clusters of varying sizes, found often at the cellular periphery. Raman spectra of internalized fibrils exhibit frequency shifts and spectral narrowing relative to in vitro fibrils, highlighting the environmental sensitivity of the alkyne vibration. Interestingly, spectral analysis reveals variations in lipid and protein recruitment to these aggregates, and in some cases, secondary structural changes in the fibrils are observed. This work sets the groundwork for future Raman spectroscopic investigations using a similar approach of an evolved aminoacyl-tRNA synthetase/tRNA pair to incorporate FCC into endogenous amyloidogenic proteins to monitor their aggregation in cells.
Keywords: Parkinson’s disease, amyloid, unnatural amino acid, 4-ethynylphenylalanine, SH-SY5Y
Graphical Abstract
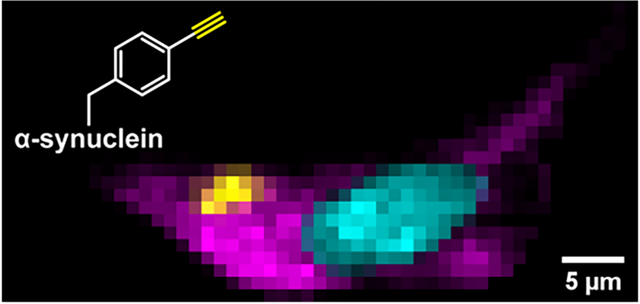
Introduction
Synucleinopathies, including Parkinson’s disease (PD), dementia with Lewy bodies, and multiple system atrophy, are a collection of protein misfolding diseases characterized by the formation of intracellular inclusions of the protein α-synuclein (α-syn) in neuronal cells [1–3]. Although α-syn is intrinsically disordered, the protein adopts a β-sheet-rich structure termed amyloid in the disease state [4]. Amyloids are formed by a number of proteins in other human diseases, including amyloid-β in Alzheimer’s disease, TAR DNA binding protein 43 (TDP-43) in amyotrophic lateral sclerosis, and islet amyloid polypeptide in type-2 diabetes mellitus [5–8]. Although atomistic structural details differ, the fundamental feature of amyloids is the formation of intermolecular β-sheets, giving rise to unbranched fibrils that may be hundreds of nanometers in length [9, 10].
Amyloid structures, including those formed by α-syn, have been shown to be highly polymorphic [11, 12], with disease-related missense mutations (E46K [13] and H50Q [14]) and post-translational modifications such as phosphorylation (pY39) altering the fibril structure [15]. Recent evidence suggests that discrete fibril polymorphs may be associated with specific synucleinopathies [16–19]. A defining property of amyloids is their ability to be propagated in vitro via a nucleation-polymerization mechanism, where the presence of a small population of fibrils serve as a template, triggering the rapid aggregation of soluble proteins to the amyloid state. The same self-templating mechanism is thought to occur in vivo, in which amyloid fibrils transmit and propagate from sick to healthy cells, leading to disease progression [20, 21].
Vibrational spectroscopies are well-suited to examine the protein structural changes associated with amyloidogenesis [22]. Infrared spectroscopy in particular has been extensively employed in studies of amyloids in vitro and demonstrated in tissues [23–25]. Raman spectral imaging has been comparatively underexplored, but is also an attractive technique for cell and tissue studies [26–28]. This experiment couples the spatial and spectral resolution of a microscope and a Raman spectrometer, respectively. Specifically, Raman spectra are acquired and analyzed at discrete points across the cell, allowing the distribution of individual chemical species to be mapped based on their vibrational modes (i.e. chemical functional groups). For example, Raman spectra of proteins are characterized by the C–H stretching (~3000 cm−1) and deformation (1450 cm−1) bands and the amide-I (~1700 cm−1) and -III bands (~1250 cm−1) [29]. Arising from backbone vibrational modes, the amide bands report on secondary structural changes, with the cross-β-sheet structure of amyloids producing a particularly sharp, intense amide-I band near 1669 cm−1 (Figure 1) [30].
Figure 1.
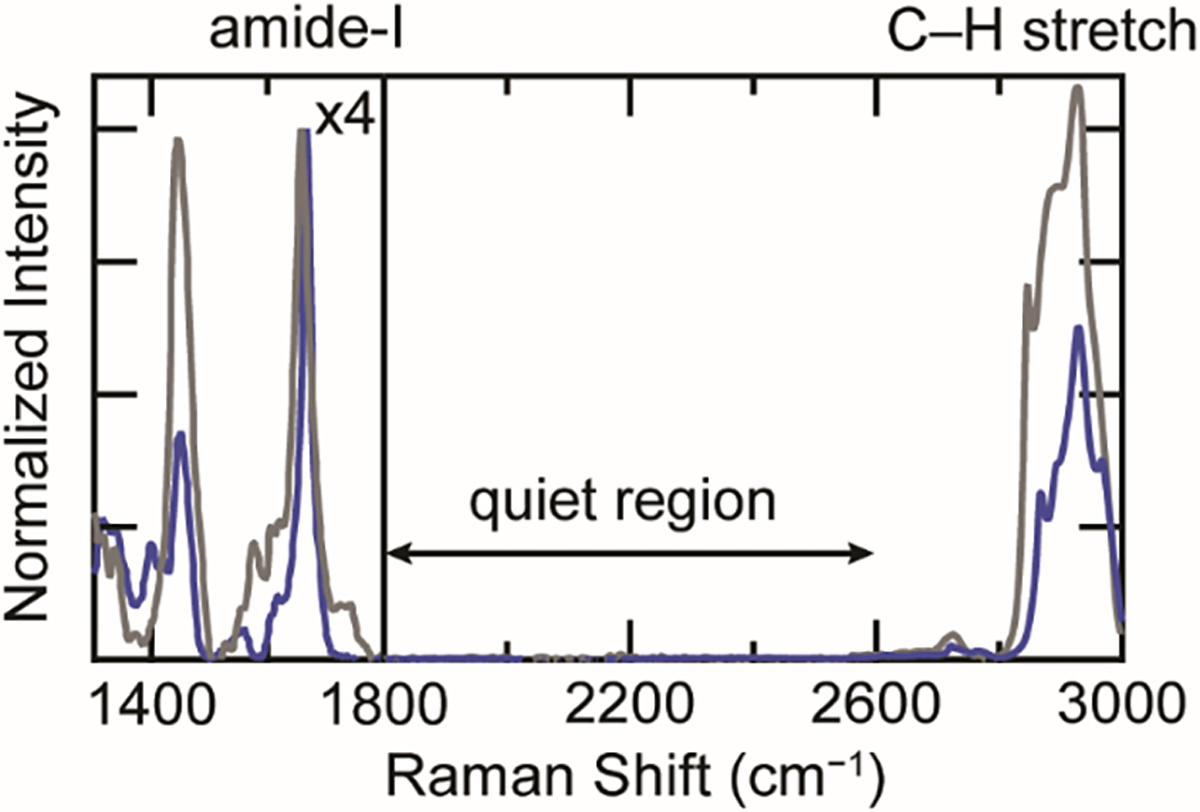
The cellular background interferes with Raman analysis of proteins. Comparison of Raman spectra of WT-α-syn fibrils (blue) and the cytosol of an SH-SY5Y cell (gray). The intensity for region 1300–1800 cm−1 is scaled ×4 for clarity. Amide-I, C–H stretching, and cellularly quiet regions are indicated.
Despite the potential of Raman spectral imaging, a major challenge of interpreting Raman spectra of proteins in a cellular environment is the interference not only from other endogenous proteins, but also other biomolecular vibrational modes, most notably the C=C and C–H stretches of lipids that overlap with the amide-I and C–H stretches of proteins, respectively. Various methods to overcome interference from these vibrational modes have been demonstrated in prior work. One such approach is incorporation of vibrational signatures that are unique against a cellular background, including isotopic labeling (13C and 2H [27]) and metabolic labeling with homopropargylglycine (HPG [26]), a methionine analogue containing a terminal alkyne. Both the C–2H stretches of isotopically-labeled protein and the alkyne stretching band (C≡C) of HPG appear in the cellularly quiet region (1800–2800 cm−1) of the Raman spectrum, where no other biological vibrational bands exist (Figure 1). Each approach has a distinct advantage: the C–2H stretching bands are extremely intense as all sidechains are labeled, which produces a stronger signal, whereas the C≡C stretch is sensitive to the local chemical environment [26], providing additional conformational information independent of amide-I analysis. The greatest benefit of these Raman probes is that they are minimally perturbative, intrinsic probes. By comparison, while amyloid stains such as thioflavin T (ThT) are the gold standard for in vitro studies, they are rather non-specific in cells (e.g. can bind to RNA and other proteins [31]). Although specific protein imaging can be accomplished by conjugation to a fluorescent protein (FP), α-syn and many other amyloidogenic proteins are much smaller than an FP, so such constructs could alter conformational dynamics and behave differently from the native protein. While having certain advantages, both isotopic and HPG-labeling suffer from a lack of site specificity, which could obscure valuable structural insights on amyloid fibrils.
In this work, we employ the aryl alkyne, 4-ethynyl-L-phenylalanine (FCC), as a site-specifically incorporated Raman probe in α-syn. Although others have demonstrated incorporation of FCC via amber codon suppression both for use as a Förster resonance energy transfer partner for Trp [32] and as a reactive moiety for protein labeling [33], it has yet to be utilized as a Raman probe in a protein. Through in vitro aggregation kinetics monitored by ThT emission and fibril morphology characterization by transmission electron microscopy (TEM), we show for the first time that FCC is a minimally perturbative probe of α-syn amyloid formation. Importantly, we demonstrate the utility of FCC in cellular Raman spectral imaging experiments by examining the distribution and localization of internalized FCC fibrils in cultured human neuronal SH-SY5Y cells. The spectrally unique C≡C stretching band enables detailed analysis of internalized fibrils, revealing recruitment of endogenous lipids and proteins to these sites as well as providing evidence for secondary structural remodeling of fibrils following cellular uptake. Collectively, this work establishes the utility of FCC and Raman spectral imaging for amyloid structural studies in the cellular environment.
Materials and Methods
Reagents
Unless otherwise noted, all compounds were purchased from Millipore-Sigma.
Raman spectral imaging
Samples were imaged in 8-well #1.0 borosilicate LabTek chambers (Thermo) using a home-built Raman microscope [34]. The 514-nm line of a 200 mW argon ion laser (CVI Melles Griot, 35-MAP-431-200) was directed through a clean-up filter (Semrock, LL01-514-25) into an Olympus IX-71 inverted microscope. A dichroic mirror (Semrock, LPD01-514RU-25×36-1.1) and a PlanApo N 60×/1.42 NA oil objective (Olympus) were used to excite the sample and collect scattered light. The light was directed through a notch filter (Semrock, NF03-514E-25) and into a Horiba iHR320 spectrometer equipped with a 1200 mm−1 grating and a Symphony II liquid-nitrogen cooled back illuminated deep-depletion CCD detector (Horiba Scientific, 1024×256 px, 26.6 mm × 6.6 mm, 1 MHz repetition rate, high-gain enabled). The CCD image was binned from 118 to 134 pixels in the y-dimension to achieve confocality. Calibration was performed daily using neat cyclohexane (200 μL in an 8-well LabTek chamber). The bandwidth of the system was determined to be ≤3 cm−1. Protein spectra were recorded as the average of 32 × 4-s accumulations for fibrillar samples and 32 × 16-s accumulations for soluble samples. Spectra of the FCC (MedChem Express) and HPG (Click Chemistry Tools) amino acids were recorded as the average of 64 × 1-s accumulations. For cellular imaging, spectra were recorded as the average of 6 × 1-s accumulations collected in 1-μm steps in both the x- and y- dimensions using a SCAN IM 180×80 motorized stage (Märzhäuser Wetzlar) with a Tango controller. For z-dimension scans, spectra were recorded for 1-s in 0.2-μm steps using a MA42 focus drive (Märzhäuser Wetzlar, 27-53-406-0000). Collectively, 51 cells were collected and analyzed. Bright-field images were collected using a USB 2.0 camera (iDS, UI-1220-C).
Protein expression
Proteins were expressed using a modified version of a protocol described previously [35]. pDule2.pCNF, which codes for a mutant M. janaschii aminoacyl tRNA synthetase/tRNA pair that recognizes para-substituted Phe-derivatives was a gift from Ryan Mehl (Addgene plasmid # 85495 [32]) and pET21a(+).SNCA (with TAT for Y136 [36]) containing an Amber stop codon (TAG) at positions 4 and 94 were purchased from GenScript. Amber stop codons at positions 39 and 125 were introduced by site-directed mutagenesis. Briefly, E. coli BL21(DE3) (Invitrogen) were co-transformed with pET21a(+).SNCA (Genscript) and pDule2.pCNF under selective pressure of carbenicillin (50 μg/mL) and spectinomycin (60 μg/mL), respectively. A freshly transformed single colony was used to inoculate a 5-mL LB culture overnight at 37 °C with shaking. The starter culture was used to inoculate large cultures (1.5 L) in minimal media supplemented with glycerol, aspartate, amino acids and trace metals and grown at 37 °C with shaking. Once the OD600 reached ~1.0, FCC (MedChem Express) was added to a concentration of 20 mg/L and after 20 min, protein expression was induced with 1 mM IPTG for 3 h. Cells were harvested by centrifugation, and cell pellets were stored at −80 °C until purification.
Protein purification
Proteins were purified as previously reported [37]. Cells were resuspended in 100 mM tris(hydroxymethyl)aminomethane (Tris) buffer (pH 8) with 300 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), and 1 mM phenylmethylsulfonyl fluoride (PMSF) and lysed by sonication on ice. The lysate was heated in a boiling water bath under N2 atmosphere for 20 min and insoluble material was pelleted by centrifugation at 30,000 rpm in a Beckman Coulter 45Ti rotor for 20 min at 4 °C. The pH of the supernatant was adjusted to 3.5 by addition of HCl, and the resulting precipitated material was removed by centrifugation as before. The supernatant was dialyzed against 20 mM Tris with 0.5 mM EDTA and 0.5 mM PMSF at 4 °C twice for 8 h each time. Proteins were first purified using a HiPrep DEAE FF 16/10 column (GE Healthcare) equilibrated in 20 mM Tris, pH 8 buffer and eluted with a linear NaCl gradient, and fractions containing α-syn were concentrated by ultrafiltration using a 3-kD MWCO membrane filter and incubated in 8 M guanidinium hydrochloride (MP Biomedicals) for 16 h at RT. After buffer exchange using a HiPrep 26/10 desalting column (GE Healthcare), α-syn was applied to a MonoQ HR 16/10 column (GE Healthcare) and eluted using a linear NaCl gradient (20 mM Tris, pH 8). Proteins were concentrated to 200 μM by ultrafiltration using a 3-kD MWCO membrane filter and stored at −80 °C in 1 mL aliquots. Protein homogeneity was determined by SDS-PAGE and molecular weights were confirmed by LC-ESI-MS (NHLBI Biochemistry Core facility). F4FCC: 14484.68 amu (expected 14484.18 amu); Y39FCC: 14468.66 amu (expected 14468.18 amu); F94FCC: 14484.73 amu (expected 14484.18 amu); Y125FCC: 14468.52 amu (expected 14468.18 amu).
Fibril formation
Proteins were buffer exchanged into pH 5 buffer (20 mM sodium acetate, 50 mM NaCl) on PD10 desalting columns (GE Healthcare) and filtered through 100-kD MWCO centrifugal filter units (Millipore) to remove preformed aggregates. The protein concentration was determined by UV absorbance at 280 nm based on extinction coefficients of 6,280 M−1 cm−1 (F4FCC and F94FCC) and 4,790 M−1 cm−1 (Y39FCC and Y125FCC). α-Syn variants (300 μM) were aggregated in 1.5 mL low protein-binding tubes (Eppendorf) by incubating in a shaker (Mini-Micro 980140 shaker, VWR) at 37 °C and 600 rpm for 3 days.
Aggregation kinetics
Proteins were desalted into pH 5 buffer and filtered as described. WT and FCC-labeled α-syn (25 μM) with ThT (2.5 μM) were aggregated in 40 μL aliquots in a 384-well black polypropylene microplate (Grenier Bio-One) with a 2.0 mm glass bead in each well, sealed with an optically clear adhesive film (Thermo 4311971) and incubated at 37 °C with 6.0 mm linear shaking (330 rpm) using a Tecan Spark microplate reader. ThT fluorescence was monitored at 15 min intervals using an excitation wavelength of 415 nm and an emission wavelength of 480 nm.
TEM
Fibrils were deposited on 400 mesh formvar coated copper grids (Electron Microscopy Sciences) for 1 min, wicked with filter paper, washed briefly with a drop of water, wicked with filter paper, stained with uranyl acetate (1%) for 1 min and again wicked with filter paper. Grids were imaged on a JEOL EM-1200 EXII electron microscope (accelerating voltage 80 keV) equipped with an AMT XR-60 digital camera (NHLBI EM Core Facility).
Cell culture and fibril treatment
SH-SY5Y cells (ATCC) were maintained in high glucose Dulbecco’s modified Eagle medium (DMEM, ATCC 30-2002) supplemented with 25 mM HEPES, 44 mM sodium bicarbonate, 10% fetal bovine serum (FBS, ATCC 30-2020) and 2% penicillin/streptomycin at 37 °C in 5% CO2 atmosphere. To prepare cells for imaging, trypsinized cells were seeded into 8-well #1.0 borosilicate LabTek chambers (Thermo) at a density of 46,000 cells/cm2 and incubated at 37 °C in 5% CO2 atmosphere for 72 h prior to fibril treatment. Preformed α-syn fibrils were made as described above in 384-well plates and diluted into phosphate buffered saline (Gibco, PBS, pH 7.4), sonicated using a probe tip for 1 s at minimum power (Branson Sonifier 450), and pelleted by centrifugation at 100,000 rpm in a Beckman Coulter TLA 100.2 rotor for 20 min at 20 °C. Half of the pelleted fibrils were disaggregated in 8 M guanidinium hydrochloride (MP Biomedicals) and α-syn concentration was determined by UV absorbance at 280 nm. The remaining fibrils were resuspended in growth media at a concentration of 0.5 μM and added to cells for 48 h at 37 °C in 5% CO2 atmosphere. After 48 h of fibril treatment, cells were washed three times with stabilization buffer (30 mM HEPES, 70 mM KCl, 5 mM MgCl2, 3 mM EGTA, pH 7.4) and fixed in 2% paraformaldehyde in stabilization buffer. Fixed cells were washed and stored in PBS at 4 °C until imaging. Multiple cell treatments (n = 22) were performed with both Y39FCC and F94FCC fibrils from at least two independent fibril preparations.
Widefield fluorescence microscopy
ThT-stained fibrils were visualized in cells using wide-field illumination with laser excitation (CrystaLaser, BCL-040-440). A dichroic mirror (Semrock, FF452-Di01-25×36) and a PlanApo N 60×/1.42 NA oil objective (Olympus) were used to excite the sample and collect emitted light. Fluorescence was directed through a bandpass filter (Semrock, FF02-485/20-25) and into an XM10 camera (Olympus).
Raman data analysis
Raman spectrum background subtraction and baseline correction were performed in LabSpec6 (Horiba). Background subtraction was performed using a buffer spectrum collected at a matching z-position. For amino acids and soluble α-syn, background spectra were collected from an aliquot of matched buffer in an adjacent LabTek well. For fibrillar α-syn and cell maps, background spectra were collected adjacent to the region of interest. Following background subtraction, spectra were fit to a two degree polynomial baseline. The baseline fitting procedure used was chosen to minimize the processing required to flatten and bring the spectrally quiet region to zero intensity. Spectra of in vitro fibrils were averaged from several aggregates. Raman maps were generated by integrating over spectral regions as indicated with a linear baseline applied over each range to reduce contributions from nearby Raman bands.
Image analysis was performed in ImageJ 1.53. A gaussian blur background (sigma 16) was subtracted from maps and an automatic threshold mask (Yen) was applied. Colocalization maps were generated by calculating a mask of colocalized pixels from individual channel masks using the Boolean AND function and applying this mask to the C≡C stretching band map.
Spectral analysis was performed in Igor Pro 7.0.8.1 (WaveMetrics). Peak positions were determined by fitting to a Lorentzian function. Difference spectra were calculated by normalizing spectra to the peak intensity of the C≡C stretching band and subtracting a spectrum of in vitro fibrils from Raman spectra collected in cells. The in vitro spectrum used was collected from the same fibril preparation used to treat the cultured cells.
Results and Discussion
FCC as an environment sensitive Raman probe
The unnatural amino acid 4-ethynyl-L-phenylalanine (FCC) was chosen for site-specific incorporation of a terminal alkyne into α-syn. FCC is not recognized by any native E. coli aminoacyl tRNA synthetases (aaRS), but has previously been incorporated into T4-lysozyme and GFP using an evolved aaRS which recognizes a variety of para-substituted Phe derivatives [32, 38]. Due to the conjugation to the aryl ring, the alkyne stretching band of FCC is red-shifted and more intense relative to HPG, with a maximum peak frequency (νmax) of 2112.3 cm−1 compared to 2123.8 cm−1 for HPG (Figure 2A), which is in agreement with prior work [39]. Similar to the reported solvent sensitivity of HPG [26], the alkyne frequency of FCC is also sensitive to the local chemical environment, which can be seen in mixed DMSO/phosphate buffer solutions (Figure S1). With decreasing H2O content, both alkynes shift to lower energies with FCC exhibiting a larger shift (8.7 cm−1) compared to HPG, which shifts by 6.1 cm−1 (Figure 2B). Due to this spectral responsiveness to its surroundings, FCC in a protein will serve as an intrinsic reporter of sidechain burial upon protein-protein interactions during fibril formation. It is anticipated that the C≡C band of FCC located within the amyloid core will red-shift upon conversion from a soluble to an aggregated state as the sidechain transfers from an water-exposed environment to a hydrophobic fibril interior. Importantly, the FCC alkyne stretching band appears in the cellularly quiet region, offering a unique Raman spectroscopic handle and enables the identification of α-syn fibrils in cells.
Figure 2.
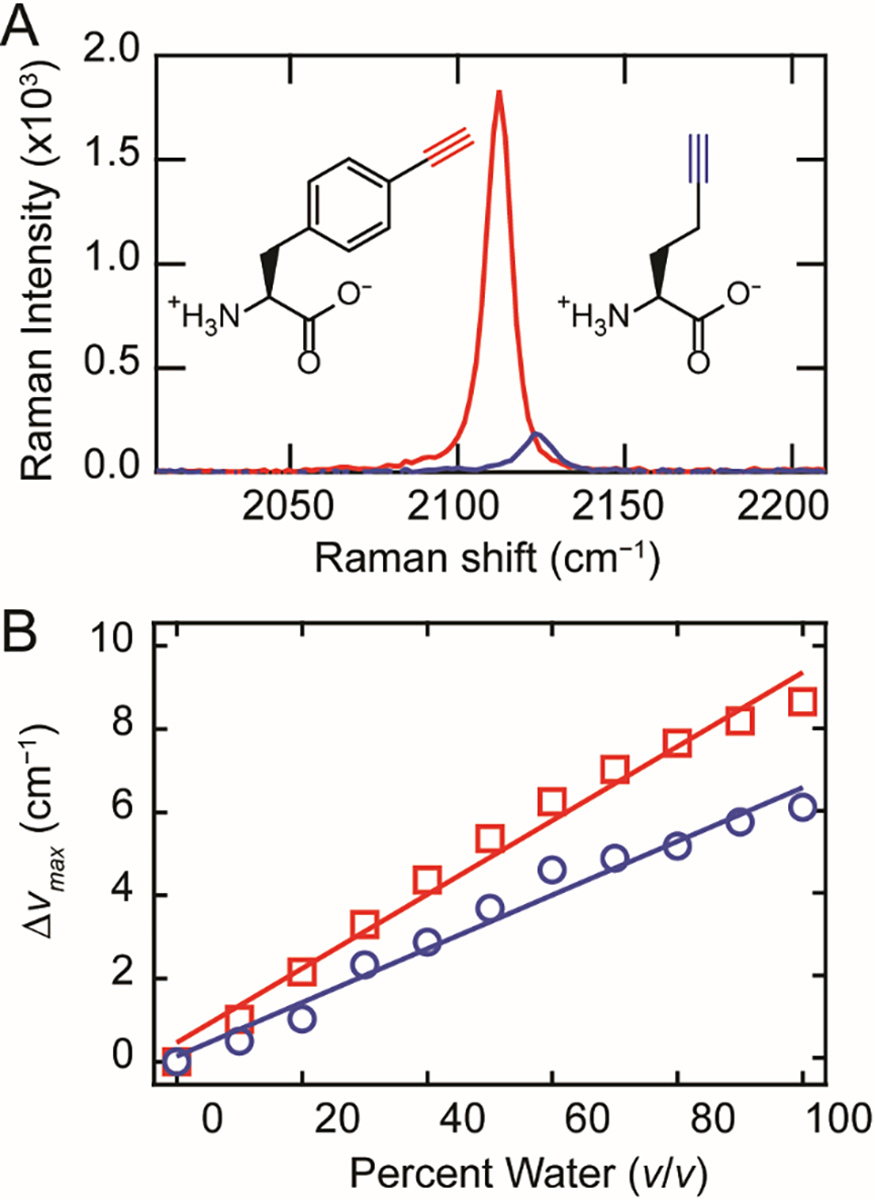
Comparison between FCC and HPG. (A) The C≡C stretching bands of FCC (red) and HPG (blue) in phosphate buffer (10 mM in 20 mM sodium phosphate, 100 mM NaCl, pH 7.4). Structures of HPG and FCC are also shown. (B) The change in the C≡C stretching peak intensity (Δνmax) from 100% DMSO to 100% phosphate buffer for FCC (red squares) and HPG (blue circles). Linear fits to the data are included as a visual guide.
FCC fibrils exhibit site-specific alkyne peak shifts
FCC was introduced at residues 4, 39, 94, and 125 in α-syn (Figure 3A, herein referred to as F4FCC, Y39FCC, F94FCC, and Y125FCC, respectively) by using an Amber stop codon (TAG) and an evolved aaRS/tRNA pair. Only aromatic sites were chosen in order to minimize structural perturbations of FCC substitution. F4FCC and Y125FCC are outside the structured amyloid core while Y39FCC and F94FCC sit within it [40]. Attempts to produce sufficient amounts of Y133FCC for study were unsuccessful. As anticipated, the conservative choice of Phe/Tyr mutations to FCC resulted in minimal effects on α-syn amyloid formation as all variants exhibited prototypical sigmoidal aggregation kinetics and fibril morphologies, similar to that of the wild-type protein as assessed by ThT fluorescence (Figure 3B) and negative stain TEM, respectively (Figure 3C–G). Thus, these variants can be used for Raman spectroscopic characterization to reveal site-specific conformational changes upon fibrillation.
Figure 3.
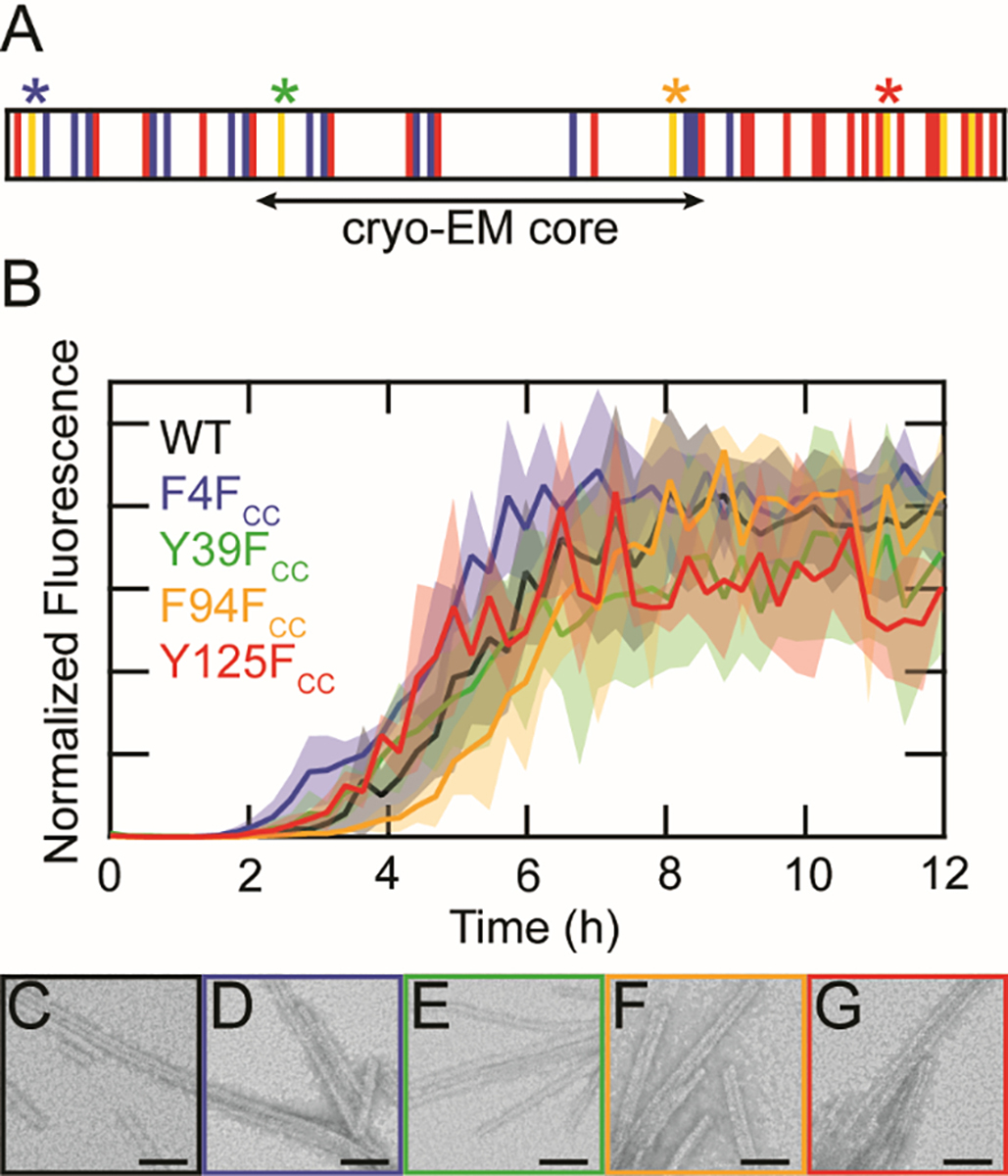
Substitution of FCC at native aromatic sites is minimally perturbative to α-syn fibril formation. (A) Schematic representation of the α-syn primary sequence. Basic, acidic and aromatic residues are shown in blue, red, and yellow, respectively. The β-sheet structured core determined by cryo-EM is indicated. Sites of FCC incorporation (4, 39, 94 and 125) are marked with asterisks. (B) ThT-monitored aggregation kinetics of WT (black), F4FCC (blue), Y39FCC (green), F94FCC (orange), and Y125FCC (red). Solid lines represent averages with shaded areas indicating the standard deviation (n = 3). Proteins were aggregated at 25 μM in 20 mM sodium acetate, 50 mM NaCl, pH 5.0 at 37 °C with shaking. Data from individual wells were normalized to the maximum fluorescence value before averaging. TEM images of (C) WT, (D) F4FCC (E) Y39FCC (F) F94FCC, and (G) Y125FCC taken 40 h post-aggregation. Scale bars are 50 nm.
Raman spectra of the soluble proteins exhibited nearly identical peak center positions (2109.1 ± 0.3 cm−1) for the four variants, indicating that the FCC sidechains experience similar aqueous surroundings, as expected for a disordered state (Figure 4, top panel and Table S1). In contrast, different peak positions were observed upon aggregation (Figure 4, bottom panel and Table S1). The two sites within the amyloid core, 39 and 94, showed red-shifts as the sidechains become sequestered from water in a fibrillar state with the change for F94FCC (Δνmax = 3 cm−1) greater than Y39FCC (Δνmax = 1.5 cm−1). While both positions 4 and 125 are located outside the amyloid core, Y125FCC had a measurable shift (Δνmax = 1 cm−1), whereas F4FCC remains unchanged, indicating that this site in the C-terminal domain is more protected than the N-terminus. The amide bands in for all four mutants were very similar, indicative of a common fibrillar structure (Figure S2). Taken together, these results demonstrate the versatility of FCC as a site-specific, environmentally sensitive, minimally perturbative Raman probe of protein structure, and to our knowledge, this is the first demonstration of its kind using FCC to characterize amyloid fibrils.
Figure 4.
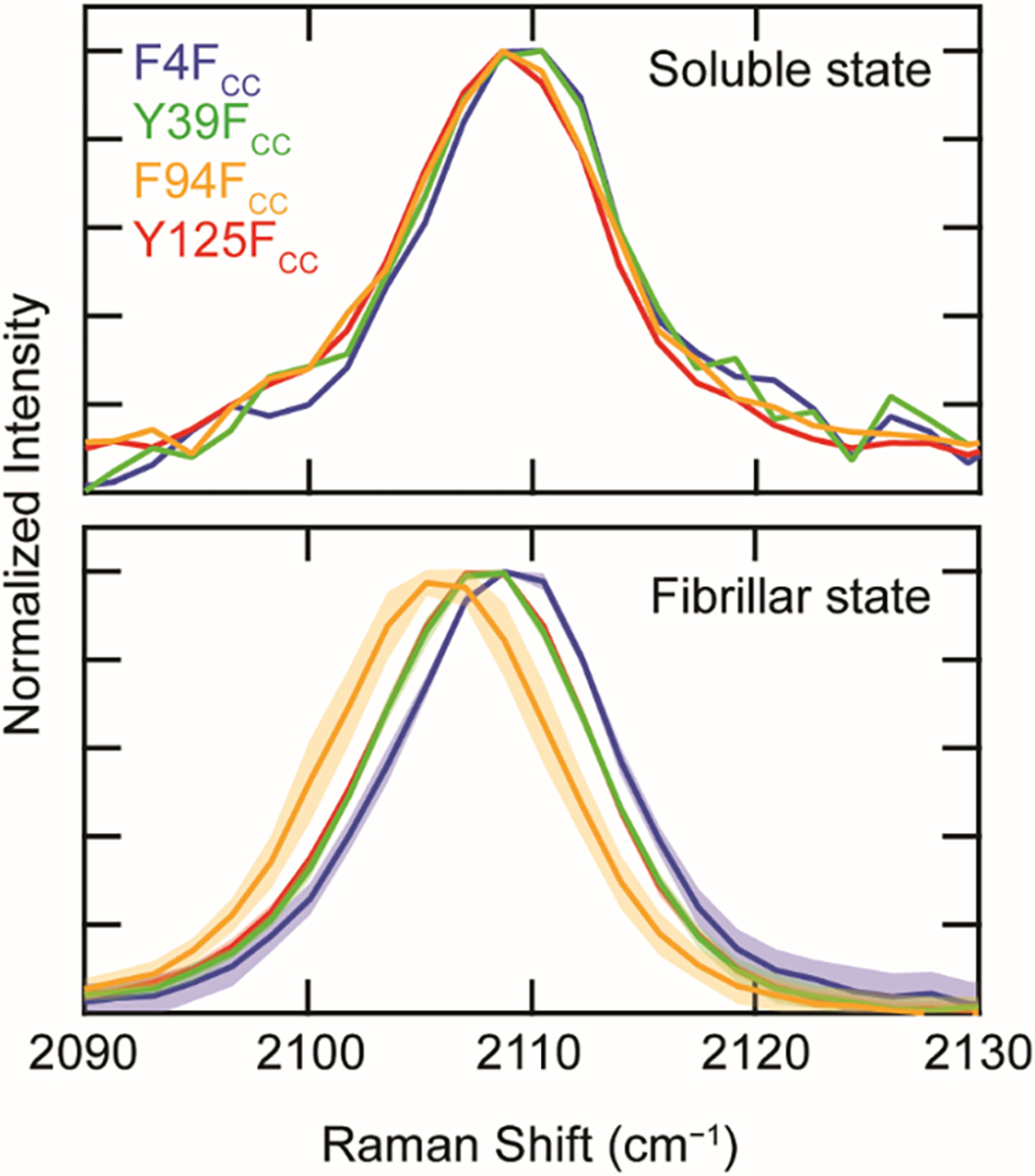
The C≡C stretching band of FCC-α-syn mutants in soluble vs. fibrillar state. Spectra of proteins (F4FCC (blue), Y39FCC (green), F94FCC (orange), and Y125FCC (red)) collected prior to (top panel) and following aggregation (bottom panel) ([α-syn] = 300 μM in 20 mM sodium acetate, 50 mM NaCl, pH 5.0). Note that the fibrillar spectra of Y125FCC and Y39FCC overlay. For fibrillar spectra, solid lines represent averages across multiple aggregates with shaded areas indicating the standard deviation (n = 8). All spectra are normalized to the peak intensity of the C≡C stretching band.
Raman spectral imaging of internalized FCC fibrils in SH-SY5Y cells
After establishing the in vitro reference Raman spectra for FCC fibrils, we next turned towards their utility in cellular studies by performing a widely used cellular treatment method in which preformed fibrils are fed exogenously and internalized by cultured cells [41, 42]. This experiment models the latter stages of cell-to-cell transmission, in which the fibrils are uptaken by a cell. While in our prior work rat dopaminergic N27 and human SK-MEL28 melanoma cells were used [43], here we use human SH-SY5Y neuroblastoma cells, a broadly accepted cell model for neurodegenerative disease research, including synucleinopathies [44].
SH-SY5Y cells were treated with preformed FCC fibrils (0.5 μM) directly added to the growth media for 48 h and fixed with paraformaldehyde prior to imaging. Treatments of F4FCC, Y39FCC, and F94FCC fibrils were conducted (yield of Y125FCC was too low for cell treatments), but the alkyne stretching band could only be consistently detected for treatments with Y39FCC and F94FCC. A representative bright-field (BF) image of a cell treated with F94FCC fibrils is shown in Figure 5A. Cells with internalized fibrils were initially identified by ThT fluorescence using widefield fluorescence imaging (Figure S3). Raman spectra were collected at 1 μm steps across the cell and were background subtracted using a spectrum collected outside the cell. Spectral quality is exemplified by the selected Raman spectra at indicated numbered locations in Figure 5B, where the alkyne peak is clearly identifiable in the cellularly quiet region in spectrum 1. Similarly, in spectrum 2 and 3, the peaks attributable to the nucleotides in the nucleus and lipids in the cytosol are evident in the highlighted regions.
Figure 5.
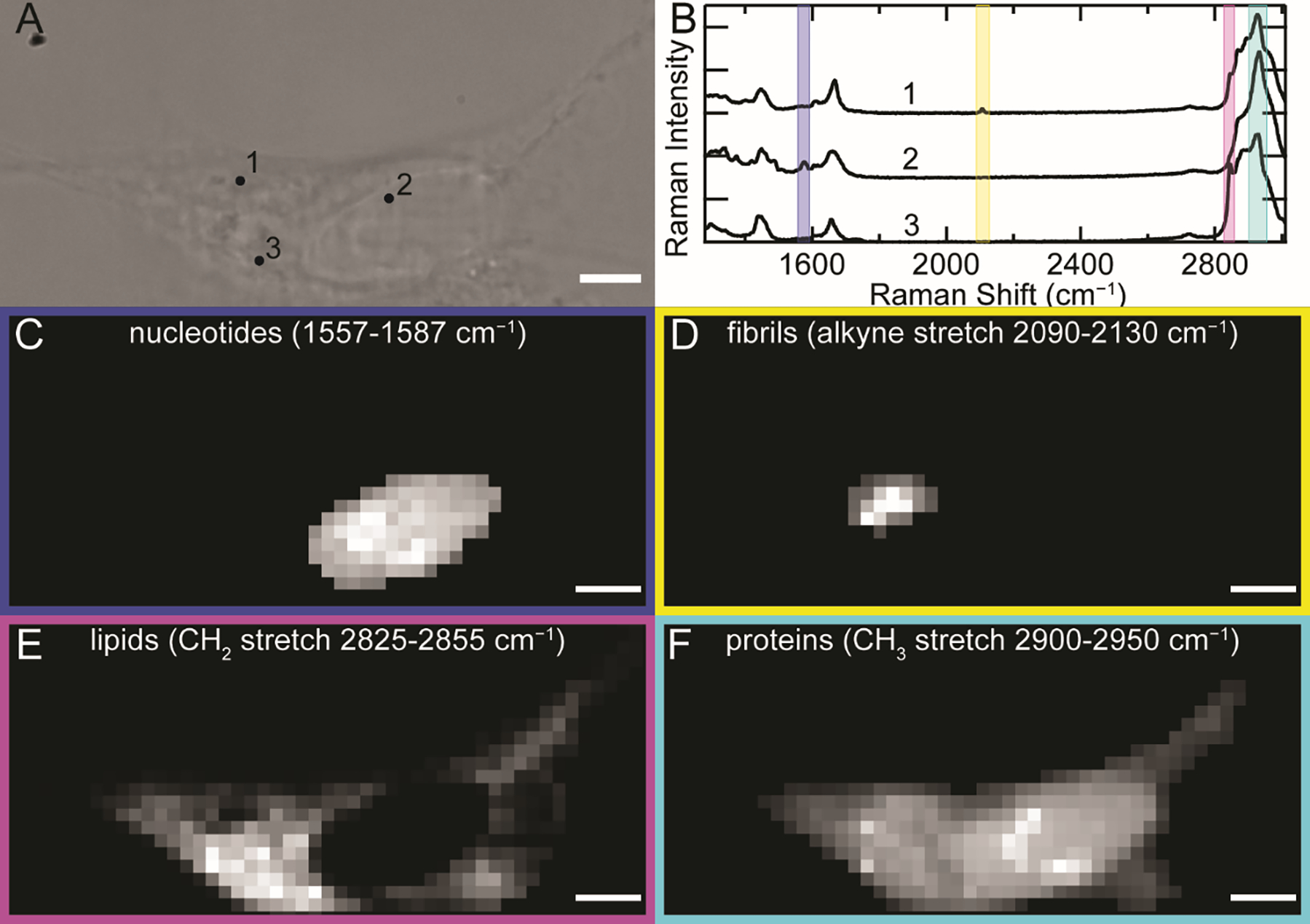
The FCC C≡C stretch unambiguously reports on the localization of F94FCC fibrils uptaken by SH-SY5Y cells. (A) BF image of an SH-SY5Y cell treated with 0.5 μM F94FCC fibrils for 48 h. (B) Raman spectra collected (6 × 1 s accumulation time) at the corresponding numbers in (A). Colored boxes represent spectral regions of nucleotide ring breathing (blue, 1557–1587 cm−1), FCC C≡C stretching (yellow, 2090–2130 cm−1), methylene (magenta, 2825–2855 cm−1) and methyl (cyan, 2900–2950 cm−1) C–H stretching modes. Corresponding Raman maps generated by integration over the different spectral regions representing (C) nucleotides, (D) FCC-containing fibrils, (E) lipids, and (F) proteins as indicated. Scale bars are 5 μm.
By integrating over spectral regions of interest, Raman maps were generated for the subcellular distribution of endogenous biomolecules [45–47]: 1557–1587 cm−1 for nucleotide ring breathing modes (Figure 5C), 2090–2130 cm−1 for the alkyne stretching mode that unambiguously identifies α-syn (Figure 5D), 2825–2855 cm−1 for the methylene C–H stretch that reports predominantly on lipids (Figure 5E), and 2900–2950 cm−1 for the methyl C–H stretch which arises from proteins with some contribution from lipids (Figure 5F). Maps generated using these regions clearly distinguish the nucleus, which is rich in nucleotides, from the cytoplasm which contains both strong methylene and methyl stretching modes throughout, indicating the presence of membranous organelles and endogenous proteins, respectively. Notably, lipids are absent in the nucleus. Maps of the alkyne stretching mode reveal discrete clusters of FCC fibrils within the cell with varying intensities. Scans in the z-direction show that these clusters are localized within the interior of the cell (Figure S4). For all the maps, there are clear variations in the chemical species at different spatial locations, reinforcing the power of this approach in examining the endogenous populations of biomolecules without the need for extrinsic probes.
Raman spectral analysis comparing individual FCC clusters within a cell
To realize the utility of a site-specific, environmentally sensitive Raman probe, we asked whether the alkyne stretch could delineate differences amongst fibril-containing regions found in an individual cell. In Figure 6A, three F94Fcc clusters of varying sizes were discernible within the cytosol of the cell. Raman spectra averaged across these fibril clusters in the specific regions of interest (ROI, Figure 6B) reveal that the alkyne band is centered at 2107.5 cm−1, which is very similar to the νmax of 2107.2 cm−1 measured for in vitro fibrils (Figures 6D, 6G, and 6K, red vs. black curves), indicating that the FCC sidechain environment in the fibrillar structure is preserved upon internalization. Notably, the internalized fibrils exhibit a narrower peak than that of the in vitro spectrum, which indicates a more homogeneous environment around the FCC sidechain. This may reflect the packaging of fibrils into a well-defined subcellular compartment, or it could potentially be related to remodeling of the fibrils (i.e. limited proteolysis [48]), leading to a more homogeneous structure.
Figure 6.
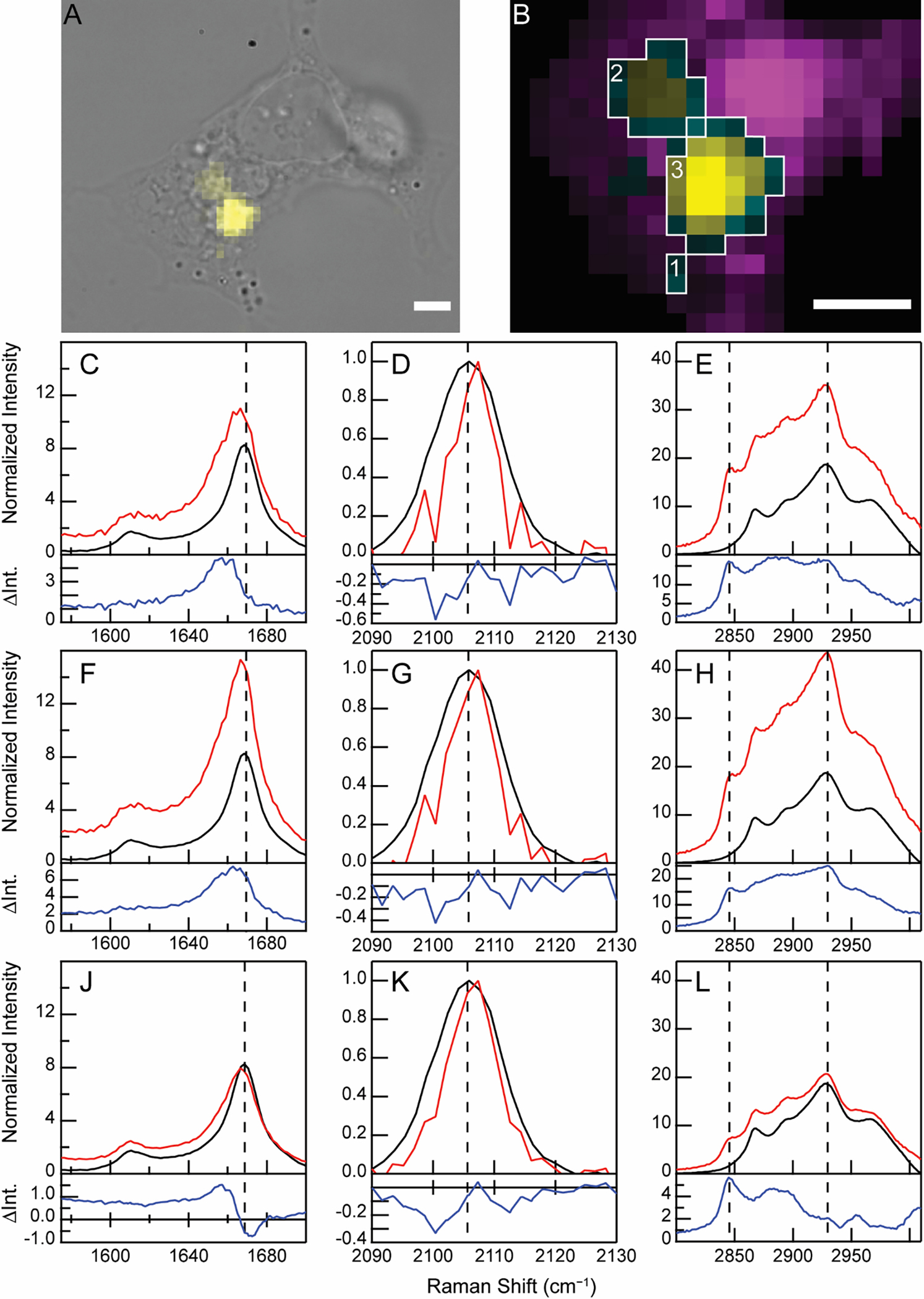
Discrete clusters of cellularly internalized FCC fibrils exhibit distinct spectral features. (A) BF image of an SH-SY5Y cell treated with F94FCC fibrils (0.5 μM) overlaid with a Raman map of the C≡C stretching band (yellow). (B) Expanded view of the fibril-containing regions from (A) shown as Raman maps of lipids (methylene stretch, magenta) and fibrils (C≡C stretch, yellow). Co-localization of lipids and fibrils is shown in cyan. The intensity of the cyan channel is determined from the alkyne signal. Scale bars are 5 μm. Boxed regions indicate pixels averaged to generate spectra in (1: C–E), (2: F–H), and (3: J–L). Amide-I (C, F, J), C≡C stretching (D, G, K), and C–H stretching (E, H, L) regions from the Raman spectra of the ROIs (red) as indicated in (B) are shown. Raman spectra collected for in vitro fibrils (black) are also shown for comparison with the difference spectra (cell−in vitro fibril, blue) shown below in each panel. Data are normalized to the intensity of the C≡C stretching band. The dashed lines serve as guides for comparison.
To further interrogate the local environment of internalized fibrils, we took advantage of the absence of interfering signals in the C≡C stretching region to generate difference spectra (Figure 6, blue curves). Spectra from three ROIs (red curves) and in vitro spectra of the same fibril preparation (black curves) were normalized to the C≡C stretching band intensity and subtracted. The appearance of a higher background in the cellular data is indicative of the presence of other biomolecules. The resulting spectra reveal environmental differences between fibril clusters within a single cell. Changes in the amide-I region relative to the alkyne stretching band are challenging to interpret in isolation due to contributions from both protein amide-I and lipid C=C stretching modes, but can be better understood in combination with inspection of the C–H stretching bands. In the smallest cluster, a red-shift is observed in the amide-I band (Figure 6C), accompanied by increases in intensity in both the methylene (2845 cm−1) and methyl (2930 cm−1) C–H stretching bands (Figure 6E), suggestive that both lipids and proteins are enriched. The second fibril cluster is broadly similar (Figure 6F); however, the intensity of the methyl stretching band is higher relative to the methylene band, indicating additional recruitment of endogenous proteins (Figure 6H). Interestingly, the difference spectrum of the last ROI reveals an amide-I shift (Figure 6J) that is explained not only from increased Raman scattering at lower wavenumbers, but also a reduction in amyloid β-sheet character relative to the alkyne stretching band intensity. This observation lends further support to the interpretation of C≡C stretching band narrowing as indicative of fibril remodeling. Consistent with the other fibril-containing regions, lipids appeared enriched. The observed molecular differences are reminiscent of the cryoelectron micrographs of intracellular deposits of α-syn, known as Lewy bodies, which contain membranous materials [49].
Raman spectral analysis comparing individual FCC-containing regions from different cells
To provide additional support for the redistribution of endogenous lipids and proteins and to evaluate cell-to-cell variations, other cells were analyzed (Figure 7) including those treated with Y39FCC fibrils (Figure S6). In addition to the range of subcellular locations, the size of internalized fibril clusters also differs dramatically across cells, in some cases spanning 10 μm (Figure 7D). It is highly improbable that such large fibril clumps could be internalized as a whole. Thus, we hypothesize that these clusters represent fibrils that are trafficked and accumulated in specific compartments within the cell. This is reasonable as in dealing with misfolded and aggregated proteinaceous materials, the cell collects and sequesters them in inclusion bodies such as the juxtanuclear quality control compartment (JUNQ) and insoluble protein deposits (IPOD) [50].
Figure 7.
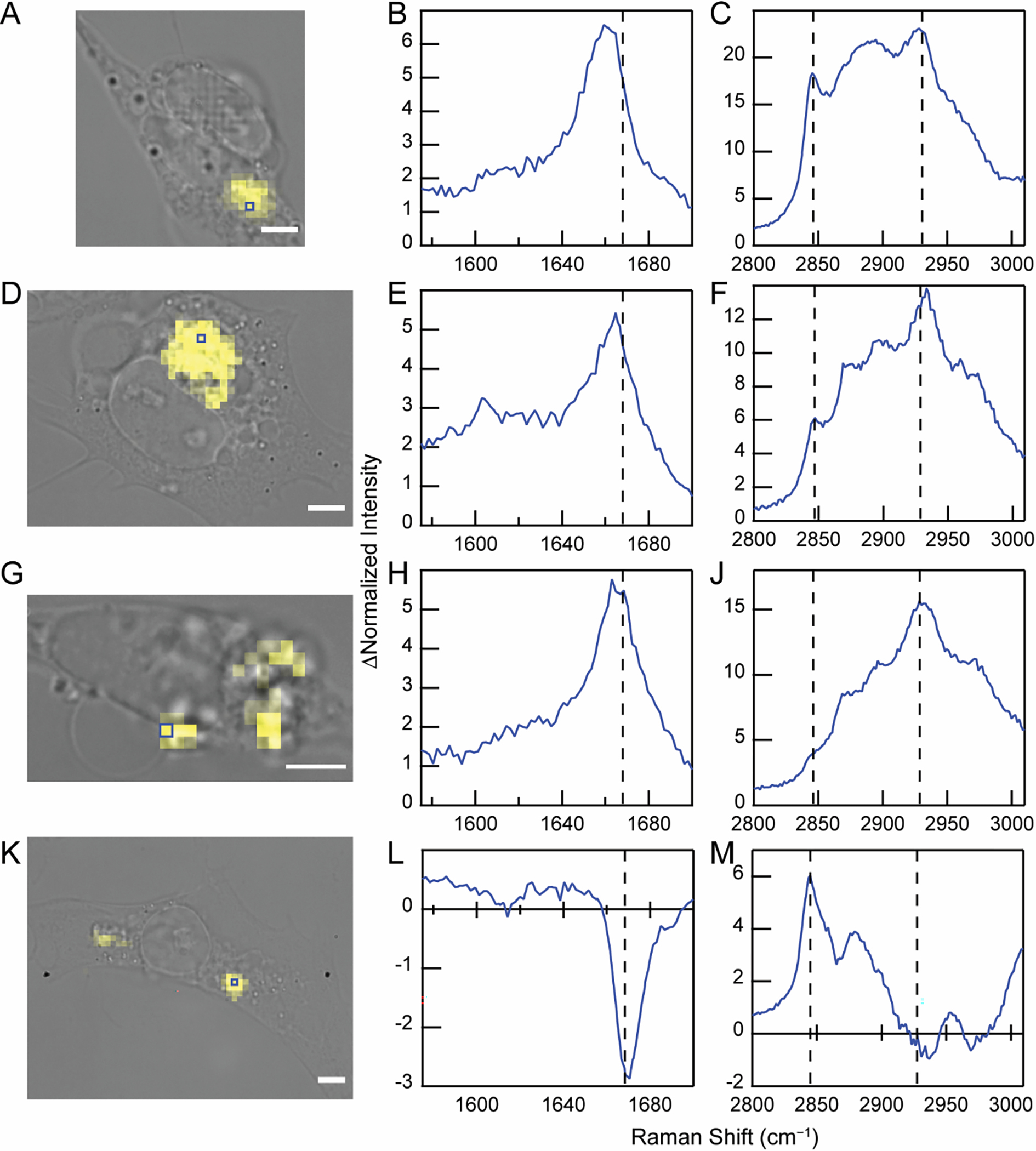
Internalized FCC fibrils from different cells exhibit distinct spectral features. BF image of SH-SY5Y cells treated with F94FCC fibrils (0.5 μM) overlaid with the Raman map generated from C≡C stretching band in yellow (A, D, G, and K). Scale bars are 5 μm. Blue boxes indicate the location of individual pixels where difference spectra were obtained. The amide-I (B, E, H, and L) and C–H stretching (C, F, J, and M) regions are shown. The dashed lines serve as guides for comparison. Corresponding spectra can be found in Figure S5. Similar data analysis for internalized Y39FCC fibrils is shown in Figure S6.
As observed above in Figure 6, difference spectra analysis reveals a range of environments within the clusters of cellularly internalized fibrils. In the case of the cell analyzed in Figures 7A–C, the intense methylene stretching band at 2845 cm−1 indicates that the amide-I spectral changes arise from lipid accumulation within the fibril cluster. By comparison, the cell shown in Figures 7D–F provides an example where both lipids and proteins are recruited, as indicated by the presence both of the methylene peak as well as the relatively high intensity methyl stretching band at 2930 cm−1 in the difference spectrum. Similar fibril distributions and spectral features were also observed for cells treated with Y39FCC fibrils, i.e. the differential recruitment of endogenous lipids and proteins to fibril-containing locations (Figure S6).
Significant lipid accumulation was apparent across fibril clusters in most cells studied; however, Figures 7G–J shows a cell in which minimal lipid co-localization is observed. Here, the difference spectrum shows large increases in both amide-I and methyl C–H peaks, representative of protein enrichment, but with minimal contribution from the methylene stretching band derived from lipids. We interpret the intensity and similarity of the amide-I shift to the distinctive νmax (1669 cm−1) of in vitro amyloids to signify that some of the recruited protein has adopted a β-sheet structure upon templating by the exogenously added fibrils. Although this experiment cannot determine the identity of the recruited protein, one possible candidate is endogenous α-syn, which is expressed in SH-SY5Y cells [51].
The most striking difference spectra are those in which the fibril clusters display negative spectral features (Figures 7K–M). As documented above in another F94FCC example (Figure 6J) and seen for Y39FCC (Figures S6F and S6K), the shift in amide-I can arise not only from additional scattering at lower wavenumbers, but also from reduced intensity of the β-sheet amide-I peak relative to the FCC alkyne stretching band. Combined with the differences in the C–H stretching region that indicate the presence of lipids but not additional proteins, these features suggest changes in the β-sheet content within the fibrils. This may be interpreted as changes in the structure due to disassembly of the fibril core, which would result in decreased β-sheet content in the structurally sensitive amide-I region without a decrease in scattering by the structurally insensitive methyl stretching band. Alternatively, these spectral features could be interpreted as partial proteolysis of fibrils by recruitment of proteases. In this case, partial degradation of α-syn will cause a loss of β-sheet structure, but not in the methyl stretching band due to recruitment of endogenous proteins which offsets the loss of intensity resulting from α-syn degradation. Indeed, we speculate that the difficulty in detecting internalized F4FCC fibrils may result from removal of the unstructured N-terminus (and consequently the FCC residue) upon internalization. Although the mechanistic explanation remains speculative at this point, the data clearly demonstrate that in some cases, packaging of internalized α-syn fibrils into subcellular deposits is accompanied by partial loss of β-sheet structure of the amyloid fibrils.
Conclusions
Collectively, these data demonstrate that FCC is an excellent site-specific Raman probe of α-syn amyloid formation. The C≡C stretching band of FCC is shown to be strong and environmentally sensitive, enabling detection of internalized fibrils in cultured cells. The heterogeneity in the fate of cellularly internalized α-syn fibrils is apparent from the range of spatial distributions and spectral characteristics seen in these experiments. The observation of lipid and protein recruitment to these internalized fibrils, combined with structural analysis of their β-sheet content demonstrate the strength of Raman spectral imaging. This detailed analysis was enabled by the intensity of the FCC C≡C stretching band and the lack of spectral interference in the cellularly quiet region of the Raman spectrum. Beyond the favorable spectral qualities of this amino acid, FCC also represents a minimally perturbative substitution for aromatic residues. This is particularly important in the context of amyloid formation which is well known to be highly sensitive to single point mutations.
The incorporation of FCC at selective residues via genetic code expansion opens a tremendous opportunity to probe the fate of different regions of an amyloidogenic protein and is in principle broadly applicable to other protein systems, in particular proteins rich in aromatic residues such as TDP-43. The characterized structural changes in α-syn fibrils upon internalization in cells is compelling, especially in the case where recruitment of endogenous proteins is accompanied by the apparent enhancement in β-sheet content. Moving forward, it would be of utmost importance to probe the process from the perspective of endogenous proteins to gain a full molecular understanding. Indeed, incorporation of para-substituted phenylalanine derivatives in mammalian cells has been reported [52–54], and thus, labeling of endogenously expressed α-syn is a viable and promising future application of FCC.
Supplementary Material
Detection of amyloids in cells requires unique spectral signatures
The aryl alkyne FCC is a site-specific conformational probe of α-syn aggregation
Cellular Raman mapping uniquely identifies internalized FCC-labeled α-syn fibrils
Spectral analysis reveals differences in lipid and protein recruitment to fibrils
Raman spectral imaging is a powerful approach to study amyloid structure in cells
Acknowledgment
This work was supported by the Intramural Research Program at the NIH, NHLBI. LC-ESI-MS and TEM were performed on instruments maintained by the NHLBI Biochemistry and EM Core, respectively.
Abbreviations:
- (α-syn)
α-synuclein
- (FCC)
4-ethynyl-L-phenylalanine
- (C≡C)
alkyne
- (PD)
Parkinson’s disease
- (TDP-43)
TAR DNA binding protein 43
- (HPG)
homopropargylglycine
- (ThT)
thioflavin T
- (FP)
fluorescent protein
- (TEM)
transmission electron microscopy
- (aaRS)
aminoacyl tRNA synthetase
- (νmax)
peak frequency
- (JUNQ)
juxtanuclear quality control compartment
- (IPOD)
insoluble protein deposit
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary information
Supplementary information to this article can be found online at https://doi.org/10.1016/j.jmb.XXX
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA. 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998). Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 251, 205–208. [DOI] [PubMed] [Google Scholar]
- [3].Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H (1998). α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 249, 180–182. [DOI] [PubMed] [Google Scholar]
- [4].Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, Eliezer D, Moore DJ, Schneider B, Aebischer P, El-Agnaf OM, Masliah E, Lashuel HA (2012). α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985). Amyloid plaque core protein in Alzheimer disease and down syndrome. Proc. Natl. Acad. Sci. USA. 82, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB (1987). Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. USA. 84, 8628–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314, 130–133. [DOI] [PubMed] [Google Scholar]
- [8].Westermark P, Andersson A, Westermark GT (2011). Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 91, 795–826. [DOI] [PubMed] [Google Scholar]
- [9].Jahn TR, Makin OS, Morris KL, Marshall KE, Tian P, Sikorski P, Serpell LC (2010). The common architecture of cross-β amyloid. J. Mol. Biol. 395, 717–727. [DOI] [PubMed] [Google Scholar]
- [10].Eisenberg DS, Sawaya MR (2017). Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 86, 69–95. [DOI] [PubMed] [Google Scholar]
- [11].Tycko R (2015). Amyloid polymorphism: Structural basis and neurobiological relevance. Neuron. 86, 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gallardo R, Ranson NA, Radford SE (2020). Amyloid structures: Much more than just a cross-β fold. Curr. Opin. Struct. Biol. 60, 7–16. [DOI] [PubMed] [Google Scholar]
- [13].Boyer DR, Li B, Sun C, Fan W, Zhou K, Hughes MP, Sawaya MR, Jiang L, Eisenberg DS (2020). The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc. Natl. Acad. Sci. USA. 117, 3592–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boyer DR, Li B, Sun C, Fan W, Sawaya MR, Jiang L, Eisenberg DS (2019). Structures of fibrils formed by α-synuclein hereditary disease mutant H50Q reveal new polymorphs. Nat. Struct. Mol. Biol. 26, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhao K, Lim YJ, Liu Z, Long H, Sun Y, Hu JJ, Zhao C, Tao Y, Zhang X, Li D, Li YM, Liu C (2020). Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proc. Natl. Acad. Sci. USA. 117, 20305–20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Peng C, Gathagan RJ, Lee VM (2018). Distinct α-synuclein strains and implications for heterogeneity among α-synucleinopathies. Neurobiol. Dis. 109, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Strohäker T, Jung BC, Liou S-H, Fernandez CO, Riedel D, Becker S, Halliday GM, Bennati M, Kim WS, Lee S-J, Zweckstetter M (2019). Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat. Commun. 10, 5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T, Hasegawa K, Murayama S, Yoshida M, Hasegawa M, Scheres SHW, Goedert M (2020). Structures of α-synuclein filaments from multiple system atrophy. Nature. 585, 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mehra S, Gadhe L, Bera R, Sawner AS, Maji SK (2021). Structural and functional insights into α-synuclein fibril polymorphism. Biomolecules. 11, 1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Goedert M, Falcon B, Clavaguera F, Tolnay M (2014). Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies. Curr. Neurol. Neurosci. Rep. 14, 495. [DOI] [PubMed] [Google Scholar]
- [21].Volpicelli-Daley LA, Luk KC, Lee VM (2014). Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 9, 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li H, Lantz R, Du D (2019). Vibrational approach to the dynamics and structure of protein amyloids. Molecules. 24, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moran SD, Zanni MT (2014). How to get insight into amyloid structure and formation from infrared spectroscopy. J. Phys. Chem. Lett 5, 1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Alperstein AM, Ostrander JS, Zhang TO, Zanni MT (2019). Amyloid found in human cataracts with two-dimensional infrared spectroscopy. Proc. Natl. Acad. Sci. USA. 116, 6602–6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fields CR, Dicke SS, Petti MK, Zanni MT, Lomont JP (2020). A different hIAPP polymorph is observed in human serum than in aqueous buffer: Demonstration of a new method for studying amyloid fibril structure using infrared spectroscopy. J. Phys. Chem. Lett 11, 6382–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Flynn JD, Gimmen MY, Dean DN, Lacy SM, Lee JC (2020). Terminal alkynes as Raman probes of α-synuclein in solution and in cells. ChemBioChem. 21, 1582–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Watson MD, Flynn JD, Lee JC (2021). Raman spectral imaging of 13C2H15N-labeled α-synuclein amyloid fibrils in cells. Biophys. Chem. 269, 106528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sevgi F, Brauchle EM, Carvajal Berrio DA, Schenke-Layland K, Casadei N, Salker MS, Riess O, Singh Y (2021). Imaging of α-synuclein aggregates in a rat model of Parkinson’s disease using Raman microspectroscopy. Front. Cell Dev. Biol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rygula A, Majzner K, Marzec KM, Kaczor A, Pilarczyk M, Baranska M (2013). Raman spectroscopy of proteins: A review. J. Raman Spectrosc. 44, 1061–1076. [Google Scholar]
- [30].Flynn JD, Lee JC (2018). Raman fingerprints of amyloid structures. Chem. Commun. 54, 6983–6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cooper JH (1969). An evaluation of current methods for the diagnostic histochemistry of amyloid. J. Clin. Pathol. 22, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Miyake-Stoner SJ, Miller AM, Hammill JT, Peeler JC, Hess KR, Mehl RA, Brewer SH (2009). Probing protein folding using site-specifically encoded unnatural amino acids as FRET donors with tryptophan. Biochemistry. 48, 5953–5962. [DOI] [PubMed] [Google Scholar]
- [33].Grammel M, Dossa PD, Taylor-Salmon E, Hang HC (2012). Cell-selective labeling of bacterial proteomes with an orthogonal phenylalanine amino acid reporter. Chem. Commun. 48, 1473–1474. [DOI] [PubMed] [Google Scholar]
- [34].Flynn JD, McGlinchey RP, Walker RL 3rd, Lee JC (2018). Structural features of α-synuclein amyloid fibrils revealed by Raman spectroscopy. J. Biol. Chem. 293, 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hammill JT, Miyake-Stoner S, Hazen JL, Jackson JC, Mehl RA (2007). Preparation of site-specifically labeled fluorinated proteins for 19F-NMR structural characterization. Nat. Protoc. 2, 2601–2607. [DOI] [PubMed] [Google Scholar]
- [36].Masuda M, Dohmae N, Nonaka T, Oikawa T, Hisanaga S, Goedert M, Hasegawa M (2006). Cysteine misincorporation in bacterially expressed human α-synuclein. FEBS Lett. 580, 1775–1779. [DOI] [PubMed] [Google Scholar]
- [37].Watson MD, Lee JC (2019). N-terminal acetylation affects α-synuclein fibril polymorphism. Biochemistry. 58, 3630–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dippel AB, Olenginski GM, Maurici N, Liskov MT, Brewer SH, Phillips-Piro CM (2016). Probing the effectiveness of spectroscopic reporter unnatural amino acids: A structural study. Acta Crystallogr. Sect. D. Biol. Crystallogr. 72, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yamakoshi H, Dodo K, Palonpon A, Ando J, Fujita K, Kawata S, Sodeoka M (2012). Alkyne-tag Raman imaging for visualization of mobile small molecules in live cells. J. Am. Chem. Soc. 134, 20681–20689. [DOI] [PubMed] [Google Scholar]
- [40].Ni X, McGlinchey RP, Jiang J, Lee JC (2019). Structural insights into α-synuclein fibril polymorphism: Effects of Parkinson’s disease-related C-terminal truncations. J. Mol. Biol. 431, 3913–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA. 106, 13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Domert J, Sackmann C, Severinsson E, Agholme L, Bergstrom J, Ingelsson M, Hallbeck M (2016). Aggregated α-synuclein transfer efficiently between cultured human neuron-like cells and localize to lysosomes. PLoS One. 11, e0168700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Watson MD, Lee JC (2021). Coupling chemical biology and vibrational spectroscopy for studies of amyloids in vitro and in cells. Curr. Opin. Chem. Biol. 64, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xicoy H, Wieringa B, Martens GJ (2017). The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 12, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Smith R, Wright KL, Ashton L (2016). Raman spectroscopy: An evolving technique for live cell studies. Analyst. 141, 3590–3600. [DOI] [PubMed] [Google Scholar]
- [46].Schie IW, Kiselev R, Krafft C, Popp J (2016). Rapid acquisition of mean Raman spectra of eukaryotic cells for a robust single cell classification. Analyst. 141, 6387–6395. [DOI] [PubMed] [Google Scholar]
- [47].Pezzotti G (2021). Raman spectroscopy in cell biology and microbiology. J. Raman Spectrosc. 52, 2348–2443. [Google Scholar]
- [48].McGlinchey RP, Lacy SM, Huffer KE, Tayebi N, Sidransky E, Lee JC (2019). C-terminal α-synuclein truncations are linked to cysteine cathepsin activity in Parkinson’s disease. J. Biol. Chem. 294, 9973–9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castano-Diez D, Schweighauser G, Graff-Meyer A, Goldie KN, Sutterlin R, Huisman E, Ingrassia A, Gier Y, Rozemuller AJM, Wang J, Paepe A, Erny J, Staempfli A, Hoernschemeyer J, Grosseruschkamp F, Niedieker D, El-Mashtoly SF, Quadri M, Van IWFJ, Bonifati V, Gerwert K, Bohrmann B, Frank S, Britschgi M, Stahlberg H, Van de Berg WDJ, Lauer ME (2019). Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22, 1099–1109. [DOI] [PubMed] [Google Scholar]
- [50].Raiss CC, Braun TS, Konings IBM, Grabmayr H, Hassink GC, Sidhu A, le Feber J, Bausch AR, Jansen C, Subramaniam V, Claessens MMAE (2016). Functionally different α-synuclein inclusions yield insight into Parkinson’s disease pathology. Sci Rep. 6, 23116–23116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee BR, Kamitani T (2011). Improved immunodetection of endogenous α-synuclein. PLoS One. 6, e23939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu W, Brock A, Chen S, Chen S, Schultz PG (2007). Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nat. Methods. 4, 239–244. [DOI] [PubMed] [Google Scholar]
- [53].Hino N, Okazaki Y, Kobayashi T, Hayashi A, Sakamoto K, Yokoyama S (2005). Protein photo-cross-linking in mammalian cells by site-specific incorporation of a photoreactive amino acid. Nat. Methods. 2, 201–206. [DOI] [PubMed] [Google Scholar]
- [54].Wang W, Takimoto JK, Louie GV, Baiga TJ, Noel JP, Lee KF, Slesinger PA, Wang L (2007). Genetically encoding unnatural amino acids for cellular and neuronal studies. Nat. Neurosci. 10, 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.