

Abstract
Haploinsufficiency of the methyl-CpG-binding domain protein 5 (MBD5) gene causes a neurodevelopmental disorder that includes intellectual disability, developmental delay, speech impairment, seizures, sleep disturbances, and behavioral difficulties. Microdeletion of 2q23.1 is the most common cause of haploinsufficiency, although MBD5 haploinsufficiency may also cause this genetic disorder. We report a family harboring a heterozygous loss-of-function variant in MBD5 (NM_018328.5:c.728delC; p.Pro243Hisfs*26), which includes three affected siblings with varying phenotypic features. Both parents were phenotypically normal but deep coverage sequencing of the parents showed germline mosaicism in the mother.
Keywords: generalized tonic seizures; intellectual disability, mild; intellectual disability, moderate
INTRODUCTION
Clinical Features
Methyl-CpG-binding domain protein 5 (MBD5) haploinsufficiency, also known as MBD5-associated neurodevelopmental disorder (MAND), is characterized by intellectual disability (ID) and developmental delay, evident within the first year of life (Mullegama et al. 2022). Severe speech impairment is a frequent finding, with most individuals lacking speech entirely or having only single words, short phrases, or short sentences (Mullegama et al. 2022). In addition, 80% of affected individuals have seizures, with the average age of onset at 2 yr; 80% have sleep disturbances; and 80% have autistic-like behaviors (Mullegama et al. 2022). Self-injury and aggression have been reported in ∼60% of affected individuals (Mullegama et al. 2022). Craniofacial abnormalities may be subtle and variable depending on the underlying pathogenic variant type, as some differences in phenotype have been reported when comparing individuals with a microdeletion of 2q23.1 to those with other pathogenic variants in MBD5 (Mullegama et al. 2013). Clinical findings such as short stature, microcephaly, a wide mouth with a thick everted lower lip, and downturned corners of the mouth are more common in those with a whole gene or focal deletion of 2q23.1 (Amberger et al. 2015). Individuals with point variants or deletions in MBD5 are more likely to have seizures and repetitive behaviors, but all other associated findings are similar to individuals with deletion of 2q23.1 (Mullegama et al. 2013). Deletions/ duplications of 2q23.1 and MBD5 pathogenic sequence variants may occur as a de novo event or may be inherited from a mildly affected parent or an unaffected parent with germline mosaicism (Mullegama et al. 2022).
Molecular Pathogenesis
MBD5 is a dosage-sensitive gene located on Chromosome 2q23.1 and is a regulator of gene transcription through chromatin remodeling (Tao et al. 2014; Mullegama and Elsea 2016; Mullegama et al. 2021). Mullegama et al. demonstrated that differentially expressed genes from induced pluripotent stem cells (iPSCs) from individuals with MBD5 haploinsufficiency overlapped considerably with biological processes altered in neurodevelopmental phenotypes compared to unrelated controls (Mullegama et al. 2021). Two isoforms have been described: Isoform 1 is highly expressed in the brain and testis, whereas isoform 2 is highly expressed in oocytes. This suggests a role in cerebral function and epigenetic reprogramming after fertilization (Bonnet et al. 2013). Gene expression analysis of mouse brains by Seabra et al. (2020) found modest and context-dependent transcriptional consequences of MBD5 disruption (Seabra et al. 2020). Pathogenic variants in 2q23.1 led to haploinsufficiency of MBD5 (Mullegama et al. 2022). In total, 90% of affected individuals had a deletion of 2q23.1 that encompassed all or nearly all of the MBD5 gene, 5% had an intragenic deletion involving one or more exons from MBD5, and 5% were heterozygous for a pathogenic single-nucleotide variant (SNV) or small insertion/deletion (indel) variant in MBD5 (Mullegama et al. 2022).
RESULTS
Clinical Presentation and Family History
Individual II-1 and his unaffected twin sister II-2, are the firstborn children of healthy, nonconsanguineous parents who were both 25 yr old at the time of their birth (Fig. 1). He was born by cesarean delivery and was diagnosed with a tracheoesophageal fistula at birth, which was successfully repaired in the neonatal period. He had frequent bouts of pneumonia as a child.
Figure 1.
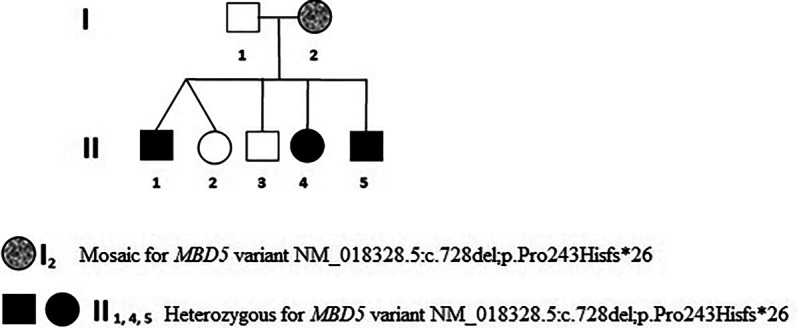
Pedigree showing individuals who are heterozygous for the variant NM_018328.5:c.728delC. Affected individuals are marked as black filled symbols and the mosaic individual is indicated with a gray filled symbol.
He had mild developmental delay, has an apparently mild ID, and has been diagnosed with autism and anxiety, although objective data to support these diagnoses are not available to us.
He had febrile seizures from the age of 9 mo until 4.5 to 5 yr of age (10 in total were recorded) and was seizure-free until nonfebrile seizures began at 14–15 yr of age. He had generalized tonic–clonic and generalized tonic seizures (focal-onset with secondary generalization), which were well controlled with anti-epileptic medication: levetiracetam (1250 mg twice daily) and valproate (1200 mg daily). When first diagnosed with epilepsy he was started on valproate; levetiracetam was added 10 yr later because of ongoing seizures. He has not been on any other medicines for epilepsy. Video-electroencephalograph (EEG) monitoring over 5 d showed rare frontal spikes that occurred more frequently on the right than left side, but no seizure episodes were recorded. A second EEG showed focal sharp and slow waves in the left frontocentral area. A cranial MRI on Individual II-1 was reported as normal. Neurological examination showed a minor postural tremor which may have been related to valproate, but no other abnormal signs were detected.
On examination at 33 yr of age his height was at the third centile and his head circumference was greater than the 97th centile. He had brachycephaly, a low anterior hairline with a widow's peak, heavy eyebrows, short palpebral fissures, high nasal bridge, large ears, small mouth, short fingers, and a sandal gap between the first and second toes (Figs. 2–4).
Figure 2.

Individual II-1: widow's peak, narrow bitemporal diameter, thick eyebrows, small mouth, and short palpebral fissures.
Figure 3.
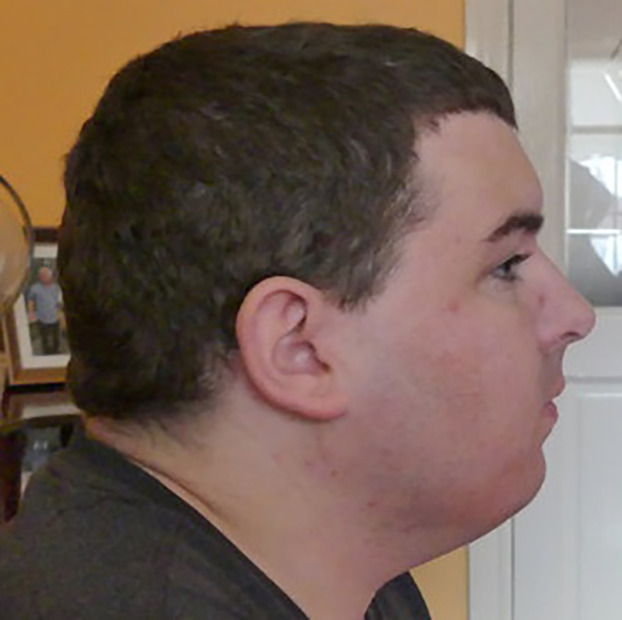
Individual II-1: brachycephaly, low anterior hairline, high nasal bridge, mild micrognathia, and large low-set ears with mild posterior rotation.
Figure 4.
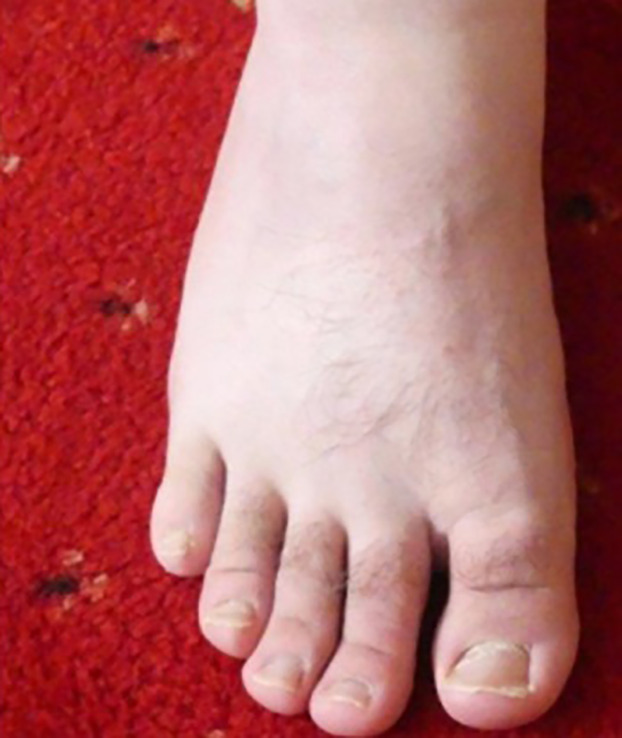
Individual II-1: sandal gap between first and second toes.
Individual II-1 attended mainstream school where he had resource teaching in some subjects and graduated from high school, passing the regular final examinations. He is good at languages and has always been sociable. In recent years, however, his mother has noted that his intellectual skills are slowly regressing, and his behavior is becoming more aggressive.
Individual II-4 is the younger sister of Individual II-1 (Fig. 1). She walked at 18 mo of age, had delayed speech development, and, as an adult, her speech is indistinct. She has an apparently mild ID, severely challenging behavior, a complex mood disorder, and bipolar disorder, although objective data to support these diagnoses are not available to us. She has never had seizures. On examination at 27 yr of age her height was at the 25th centile, her head circumference was at the 97th centile, and she had brachycephaly, narrow bifrontal diameter, low anterior hairline, thick hair, heavy eyebrows, short philtrum, small mouth, low-set ears with mild posterior rotation, a high palate, short fingers, stiff wrist joints, and a sandal gap between her first and second toes.
Individual II-5 is the youngest of the three affected siblings. He never developed speech and uses sign language to indicate his needs. He was a “bottom shuffler” until learning to walk with help at the age of 5 yr. He has never walked independently, however, as he has spastic diplegia of his legs, which makes it difficult for him to stand alone. Currently, he uses a wheelchair or moves around on his knees, which results in frequent dislocation of his patellae. At the age of 18, he had hamstring lengthening surgery on both his legs because of contractures. He has poor social skills and demonstrates hand flapping, although formal cognitive testing results were not available to us to confirm this diagnosis. He is computer-literate and loves computer games.
Individual II-5 experienced numerous febrile seizures, which began at 2 mo of age and ended at 5 yr. He was then seizure-free until he developed nonfebrile seizures at the age of 17 yr. His seizures were generalized tonic–clonic, and his aura was bending down to scratch his leg. In adulthood, his seizures consisted of daily myoclonus, atypical absences, and convulsive seizures without warning. In adulthood he was initially prescribed levetiracetam to control his seizures, but it was poorly tolerated because of mood changes. Lacosamide was then tried but was ineffective. As of this report, his seizures were well-controlled with sodium valproate (500 mg twice daily). A cranial magnetic resonance imaging (MRI) was reported as normal (see Supplemental Materials).
On examination at 23 yr of age, his height was at the third centile for age and he had brachycephaly, a low anterior hairline, widow's peak, thick arched eyebrows, short palpebral fissures, bulbous nasal tip, small mouth, low-set ears, short fingers, and a sandal gap between the first and second toes (Fig. 5).
Figure 5.

Individual II-5: Low anterior hairline, widow's peak, thick arched eyebrows, short palpebral fissures, bulbous nasal tip, small mouth, and low-set ears.
The phenotypes of Individuals II-1, II-4, and II-5 are shown in Table 1 and are compared with those of individuals with deletions of 2q23.1, and with MBD5-specific disruptions. Table 1 shows that Individuals II-1, II-4, and II-5 more closely resemble individuals with MBD5-specific gene deletion than those with deletion 2q23.1. They also have some previously unreported minor craniofacial abnormalities.
Table 1.
Phenotypic comparison of individuals with 2q23.1 deletions and MBD5-specific disruptions and Individuals II-1, II-4, and II-5 of this report
| Common features | 2q23.1 deletions: % frequency | MBD5-specific disruption: % frequency | Current report | Frequency | ||
|---|---|---|---|---|---|---|
| Individual II-1 | Individual II-4 | Individual II-5 | ||||
| Neurological and/or behavioral | ||||||
| Developmental delay | 100 | 100 | Mild | + | + | 3/3 |
| Motor delay | 100 | 100 | Moderate | + | + | 3/3 |
| Language impairment | 100 | 71 | Mild | + | Severe | 3/3 |
| Intellectual disability | NK | NK | Apparent | + | Apparent | 3/3 |
| Ataxia | 71 | 0 | − | − | − | 0/3 |
| Infantile hypotonia | 93 | 75 | − | − | Spastic diplegia of lower limbs | 1/3 |
| Infantile feeding difficulties | 100 | 67 | − | − | Reflux until 4 yr old | 1/3 |
| Seizures | 82 | 86 | + | − | + | 2/3 |
| Behavioral problems | 100 | 100 | Since teenage years | Severe | Mild | 3/3 |
| Sleep disturbances | 73 | 50 | − | − | − | 0/3 |
| Short attention span | 100 | 100 | + | + | + | 3/3 |
| Self-injurious behavior | 61 | 50 | − | + | + | 2/3 |
| Stereotypic repetitive behavior | 88 | 60 | + | + | + | 3/3 |
| Aggression | 100 | 50 | + | + | − | 2/3 |
| Hyperphagia | 78 | 0 | − | − | − | 0/3 |
| Autistic-like symptoms | 100 | 100 | + | − | + | 2/3 |
| Anti-epileptic drugs (AEDs) | ||||||
| Current therapy | NK | NK | Levetiracetam (1250 mg twice daily), valproate (1200 mg daily) | NA | Sodium valproate (500 mg twice daily) | 2/3 |
| Previous therapies | NK | NK | − | NA | Levetiracetam, lacosamide | 1/3 |
| Growth abnormalities | ||||||
| Postnatal growth retardation | 91 | 40 | − | − | − | 0/3 |
| Short stature | 86 | 40 | third centile | 25th centile | third centile | 2/3 |
| Craniofacial abnormalities | ||||||
| Craniofacial manifestations | 100 | 67 | + | + | + | 3/3 |
| Tracheo-esophagealfistula | + | − | − | 1/3 | ||
| Microcephaly | 90 | 0 | − | − | − | 0/3 |
| Macrocephaly | − | − | + | + | + | 3/3 |
| Brachycephaly | 36 | 0 | + | + | + | 3/3 |
| Broad forehead | 88 | 40 | − | − | − | 0/3 |
| Narrow bitemporal diameter | NK | NK | + | + | + | 3/3 |
| Synophrys | 47 | 20 | − | − | − | 0/3 |
| Thick/arched eyebrows | 75 | 100 | + | + | + | 3/3 |
| Eye abnormalities | 73 | 60 | + | + | + | 3/3 |
| Closely spaced eyes | 33 | 0 | − | − | − | 0/3 |
| Narrow palpebral fissures | NK | NK | + | + | + | 3/3 |
| Midface retrusion (hypoplasia) | 50 | 17 | Mild | − | Mild | 2/3 |
| Nasal abnormalities | 100 | 100 | − | − | + | 1/3 |
| Outer ear abnormalities | 88 | 20 | + | + | + | 3/3 |
| Wide mouth | 80 | 0 | Small mouth | Small mouth | Small mouth | 0/3 |
| Open mouth | 90 | 33 | Mild | − | Mild | 2/3 |
| Thin upper lip | 7 | 100 | + | + | + | 3/3 |
| Tented upper lip | 65 | 20 | − | − | − | 0/3 |
| Thick or everted lower lip | 67 | 33 | − | − | − | 0/3 |
| Downturned corners of mouth | 72 | 0 | − | − | − | 0/3 |
| Dental abnormalities | 52 | 50 | − | − | − | 0/3 |
| Widely spaced teeth | 47 | 50 | − | − | − | 0/3 |
| Large tongue, macroglossia | 19 | 0 | − | − | − | 0/3 |
| Micrognathia/retrognathia | 60 | 60 | + | + | − | 2/3 |
| Skeletal abnormalities | ||||||
| Hand/foot anomalies | 96 | 67 | + | + | + | 3/3 |
| Small hands and feet | 75 | 20 | − | − | − | 0/3 |
| Clinodactyly, fifth finger | 68 | 20 | + | + | − | 2/3 |
| Brachydactyly | 41 | 0 | + | + | + | 3/3 |
| Short fifth digit of hands/feet | 41 | 0 | + | − | − | 1/3 |
| Sandal gap | 25 | 50 | + | + | + | 3/3 |
| Other abnormalities | ||||||
| Cardiovascular abnormalities | 11 | 0 | − | − | − | 0/3 |
| Urogenital abnormalities | 29 | 0 | − | − | − | 0/3 |
| Constipation | 90 | 100 | − | − | + | 1/3 |
| Localized hirsutism | 22 | 0 | Generalized hirsutism | − | Generalized hirsutism | 0/3 |
| Bipolar disorder | + | + | − | + | − | 1/3 |
| Menarche | NK | NK | NA | 15 yr | NA | |
| Developmental regression | + | + | + | − | + | 2/3 |
| Behavioral regression | + | + | + | − | + | 2/3 |
Adapted from Table 1, Talkowski et al. (2011).
(NK) Not known, (NA) not applicable, (+) present, (−) not present.
Genomic Analysis
Individuals II-1, II-4, and II-5 and their parents underwent exome sequencing (ES). All three affected siblings in this family harbored the same frameshift deletion in MBD5 (NM_018328.5:c.728delC:p.Pro243Hisfs*26) (Table 2), which was detected using ES (Benson et al. 2020). The variant was classified using American College of Medical Genetics and Genomics (ACMG) guidelines as pathogenic (criteria PVS1, PM2). This variant, however, was not detected in either parent by Sanger sequencing or next-generation sequencing (NGS) using blood-derived DNA. The variant was not detected in Individual II-1's unaffected twin sister by Sanger sequencing and the unaffected brother declined testing. Frameshift deletions in MBD5 have been reported as pathogenic in only 30 individuals to date (according to ClinVar records, correct as of 6th September 2021). Although it is possible for a pathogenic variant to arise de novo in an affected individual, it is extremely unlikely for the same pathogenic variant to arise de novo in three siblings. Thus, targeted high-coverage panel sequencing was performed on the mother's sample. This deep sequencing (conducted using the same blood-derived blood sample used for ES) identified the same variant as was present in the three affected children in her peripheral blood sample with a variant allele fraction of 0.09 (128/1458) of the mother's sequencing reads, indicating probable gonadosomatic mosaicism (Benson et al. 2020).
Table 2.
Variant table
| Gene | Coordinates (hg38) | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP ID | Genotype |
|---|---|---|---|---|---|---|---|
| MBD5 | 2: 148468669 | NM_018328.5:c.728delC | NP_060798:p.Pro243Hisfs*26 | Frameshift deletion | Protein frameshift | rs1680676671 | Het |
DISCUSSION
Comparison of Phenotypic Findings
Table 1 shows the clinical findings in reported individuals with an abnormality of MBD5. Those with deletion 2q23.1 have a more severe phenotype than those with MBD5-specific disruption, most likely caused by the variable size of the deletion (Talkowski et al. 2011; Myers et al. 2021). It has been suggested that large deletions or duplications encompassing three genes (ORC4, KIF5C, and EPC2) that are close to the MBD5 gene could be responsible for this broader range of 2q23.1 deletion comorbidities compared to features in individuals with MDB5-specific deletions (Mullegama and Elsea, 2016). Fewer individuals have been reported with MBD5-specific disruption than with deletion 2q23.1, but from the small sample available it may be noted that although all individuals with an abnormality of MBD5 have global developmental delay and intellectual disability, those with MBD5-specific disruption have a slightly higher incidence of seizures and have thick arched eyebrows, thin upper lip, widely spaced teeth, and a sandal gap between the first and second toes (Talkowski et al. 2011; Myers et al. 2021).
In our report of three siblings with a frameshift deletion within MBD5, the typical clinical findings in individuals with large deletions/duplications of 2q23.1 were not observed, further supporting the hypothesis that these comorbidities are not a direct result of MBD5 haploinsufficiency specifically (Table 1; Amberger et al. 2015).
Seizures
Two of the siblings in this report have epilepsy, which is a common finding in individuals with MBD5-disruption disorder. The seizure types observed in these siblings are consistent with those in a cohort of 23 individuals with MBD5 haploinsufficiency reported by Myers et al. (2021) Generalized tonic–clonic seizures (Individual II-5) were the most common seizure type, whereas tonic and drop seizures (Individual II-1) and febrile seizures (Individual II-1 and Individual II-5) were frequently observed (Myers et al. 2021). Febrile seizure onset in Individual II-1 was at ∼9 mo of age. In Individual II-5 febrile seizures began at 2 mo of age. This age of onset is earlier than the median age of 2.9 yr reported by Myers et al. (2021) but consistent with individuals with SNVs and indels from that cohort (6–9 mo).
Both inter- and intrafamilial variability of phenotype and response to anti-epileptic drugs (AEDs) have been reported in individuals with MBD5 haploinsufficiency (Myers et al. 2018, 2021; Verhoeven et al. 2019). In this family, from the onset of nonfebrile seizures at 14 yr of age, Individual II-1 responded well to sodium valproate, although, recently, levetiracetam has been added because of an increase in symptoms. Individual II-5 had an initial trial of levetiracetam, but this was poorly tolerated because of a mood change in the individual. Lacosamide was ineffective but the individual's seizures ended when sodium valproate was prescribed and he was seizure-free on this medication at the time of this report.
Psychopathological Component of MBD5
There is an emerging body of evidence that microdeletion of 2q23.1 has a psychopathological component. MBD5 is expressed in adults as well as in early development, suggesting that it may have a role in adult neuropsychiatry (Tadros et al. 2017). Of note, Individual II-4 has a diagnosis of bipolar disorder (Table 1). Hodge et al. (2014) also reported bipolar disorder (BPD) in two individuals with pathogenic MBD5 variants, so although this could be coincidence, a diagnosis of BPD is worth considering in any individual with symptoms suggestive of MBD5 haploinsufficiency (Hodge et al. 2014). Regression in behavior and/or intellect, as reported in Individual II-1, has also been documented by Hodge et al. (2014) in three individuals with MBD5 haploinsufficiency, and by Verhoeven et al (2019) in one individual, suggesting a potential association (Hodge et al. 2014; Verhoeven et al. 2019).
Inheritance in Individuals with MBD5 Haploinsufficiency
The vast majority of individuals with MBD5 haploinsufficiency, regardless of variant type, do not inherit the pathogenic variant, but rather it occurs as a de novo event. A parent with germline mosaicism, occasionally limited to gonadal tissue, may transmit the pathogenic variant to an affected child or children but may not exhibit any features of the disorder themselves. Inheritance of MBD5 haploinsufficiency from an unaffected parent as a result of germline mosaicism has been described in two individuals by Tadros et al. (2017), although both had microdeletions involving MBD5 as opposed to the point variant seen in this family (Tadros et al. 2017). Individuals with a pathogenic variant in MBD5 inherited from a parent with mosaicism were observed by Myers et al. (2021) in three families (Myers et al. 2021). Two of the parents were unaffected, whereas one had mild to moderate intellectual disability without seizures (Myers et al. 2021). The frequency of parental mosaicism in families with developmental and epileptic encephalopathies and an apparent de novo inheritance pattern is 8.3% according to Myers et al. (2018). In a family with one affected child, the risk of recurrence of epileptic and developmental encephalopathies in subsequent children ranges from 1% in the case of a de novo event to as high as 50% as a result of parental mosaicism (Myers et al. 2018).
To conclude, we report on a family with three siblings with variable phenotypic features and a pathogenic frameshift deletion in MBD5 inherited from an unaffected mother with probable gonadosomatic mosaicism. Although the same pathogenic MBD5 variant was detected in both probands with epilepsy in this report, their AED requirements differed despite having a similar seizure presentation. To date, no publication has reported a precision medicine for epilepsy in individuals with MBD5 haploinsufficiency.
METHODS
In this family, ES on the Illumina platform was conducted initially on both Individuals II-1, II-4, and II-5 and on their parents (Supplemental Table 1). Segregation testing using ES was performed later on a third affected sibling who did not have epilepsy. High coverage targeted gene panel sequencing on all affected family members and their parents, as described, was also carried out (Benson et al. 2020). Pathogenic variants were confirmed using Sanger sequencing via an accredited service provider (CeGaT GmbH).
ADDITIONAL INFORMATION
Data Deposition and Access
The variant discussed in this manuscript has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession number SCV001160783. Requests for access to raw sequence data can be made via direct contact with the corresponding author.
Ethics Statement
All individuals, recruited as part of a prior study, had written informed consent provided by themselves or by a parent or legal guardian following approval of the study by a local ethics committee (Benson et al. 2020).
Acknowledgments
We thank the affected individuals and their parents and family for making this research possible. Several support groups exist for individuals and their family members affected by MBD5 haploinsufficiency.
Author Contributions
M.B., M.T.G., and K.A.B. contributed to the conception and design of the study and drafted the manuscript and related tables. G.L.C., N.D., M.T.G., and K.A.B. coordinated the sequencing efforts and interpretation of the genetic variants. M.W., B.J.S., D.J.C., and M.T.G. contributed to the acquisition of the phenotype information and recruitment of these individuals. All authors contributed to copy editing and approval of the final draft.
Funding
This publication has emanated from research supported by a research grant from Science Foundation Ireland (SFI) under Grant Number 16/RC/3948 and cofunded under the Mullegama SV.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. 2015. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an Online catalog of human genes and genetic disorders. Nucl Acids Res 43: D789–D798. 10.1093/nar/gku1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson KA, White M, Allen NM, Byrne S, Carton R, Comerford E, Costello D, Doherty C, Dunleavey B, El-Naggar H, et al. 2020. A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. Eur J Hum Genet 28: 1066–1077. 10.1038/s41431-020-0610-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet C, Ali Khan A, Bresso E, Vigouroux C, Béri M, Lejczak S, Deemer B, Andrieux J, Philippe C, Moncla A, et al. 2013. Extended spectrum of MBD5 mutations in neurodevelopmental disorders. Eur J Hum Genet 21: 1457–1461. 10.1038/ejhg.2013.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge JC, Mitchell E, Pillalamarri V, Toler TL, Bartel F, Kearney HM, Zou YS, Tan WH, Hanscom C, Kirmani S, et al. 2014. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry 19: 368–379. 10.1038/mp.2013.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama SV, Elsea SH. 2016. Clinical and molecular aspects of MBD5-associated neurodevelopmental disorder (MAND). Eur J Hum Genet 24: 1235. 10.1038/ejhg.2016.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama SV, Rosenfeld JA, Orellana C, van Bon BWM, Halbach S, Repnikova EA, Brick L, Li C, Dupuis L, Rosello M, et al. 2014. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet 22: 57–63. 10.1038/ejhg.2013.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama SV, Klein SD, Williams SR, Innis JW, Probst FJ, Haldeman-Englert C, Martinez-Agosto JA, Yang Y, Tian Y, Elsea SH, et al. 2021. Transcriptome analysis of MBD5-associated neurodevelopmental disorder (MAND) neural progenitor cells reveals dysregulation of autism-associated genes. Sci Rep 11: 11295. 10.1038/s41598-021-90798-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullegama SV, Mendoza-Londono R, Elsea SH, 2022. MBD5 haploinsufficiency. GeneReviews® (ed. Adam MP, et al.). University of Washington, Seattle. [PubMed] [Google Scholar]
- Myers CT, Hollingsworth G, Muir AM, Schneider AL, Thuesmunn Z, Knupp A, King C, Lacroix A, Mehaffey MG, Berkovic SF, et al. 2018. Parental mosaicism in “de novo” epileptic encephalopathies. N Engl J Med 378: 1646–1648. 10.1056/NEJMc1714579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KA, Marini C, Carvill GL, McTague A, Panetta J, Stutterd C, Stanley T, Marin S, Nguyen J, Barba C, et al. 2021. Phenotypic spectrum of seizure disorders in MBD5-associated neurodevelopmental disorder. Neurol Genet 7: e579. 10.1212/NXG.0000000000000579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabra CM, Aneichyk T, Erdin S, Tai DJC, De Esch CEF, Razaz P, An Y, Manavalan P, Ragavendran A, Stortchevoi A, et al. 2020. Transcriptional consequences of MBD5 disruption in mouse brain and CRISPR-derived neurons. Mol Autism 11: 45. 10.1186/s13229-020-00354-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros S, Wang R, Waters JJ, Waterman C, Collins AL, Collinson MN, Ahn JW, Josifova D, Chetan R, Kumar A. 2017. Inherited 2q23.1 microdeletions involving the MBD5 locus. Mol Genet Genomic Med 5: 608–613. 10.1002/mgg3.316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Mullegama SV, Rosenfeld JA, Van Bon BWM, Shen Y, Repnikova EA, Gastier-Foster J, Thrush DL, Kathiresan S, Ruderfer DM, et al. 2011. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet 89: 551–563. 10.1016/j.ajhg.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Wu Q, Guo X, Zhang Z, Shen Y, Wang F. 2014. MBD5 regulates iron metabolism via methylation-independent genomic targeting of Fth1 through KAT2A in mice. Br J Haematol 166: 279–291. 10.1111/bjh.12863 [DOI] [PubMed] [Google Scholar]
- Verhoeven W, Egger J, Kipp J, Verheul- aan de Wiel J, Ockeloen C, Kleefstra T, Pfundt R. 2019. A novel MBD5 mutation in an intellectually disabled adult female patient with epilepsy: suggestive of early onset dementia? Mol Genet Genomic Med 7: e849. 10.1002/mgg3.849 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The variant discussed in this manuscript has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession number SCV001160783. Requests for access to raw sequence data can be made via direct contact with the corresponding author.