

Graphical abstract
Keywords: HFpEF, iNOS, Akt S-nitrosylation, Mitochondrial function
Highlights
-
•
Combination of HDF and L-NAME induced cardiac mitochondrial dysfunction and HFpEF phenotype.
-
•
Both long-term and short-term iNOS inhibition mitigated mitochondrial dysfunction and oxidative stress in HFpEF heart.
-
•
Akt S-nitrosylation was involved into the development of HFpEF.
-
•
Both long-term and short-term iNOS inhibition reduced Akt S-nitrosylation and rescued HFpEF phenotype.
Abstract
Introduction
Despite the high morbidity and mortality of heart failure with preserved fraction (HFpEF), there are currently no effective therapies for this condition. Moreover, the pathophysiological basis of HFpEF remains poorly understood.
Objective
The aim of the present study was to investigate the role of inducible nitric oxide synthase (iNOS) and its underlying mechanism in a high-fat diet and Nω-nitro-L-arginine methyl ester-induced HFpEF mouse model.
Methods
The selective iNOS inhibitor L-NIL was used to examine the effects of short-term iNOS inhibition, whereas the long-term effects of iNOS deficiency were evaluated using iNOS-null mice. Cardiac and mitochondrial function, oxidative stress and Akt S-nitrosylation were then measured.
Results
The results demonstrated that both pharmacological inhibition and iNOS knockout mitigated mitochondrial dysfunction, oxidative stress and Akt S-nitrosylation, leading to an ameliorated HFpEF phenotype in mice. In vitro, iNOS directly induced Akt S-nitrosylation at cysteine 224 residues , leading to oxidative stress, while inhibiting insulin-mediated glucose uptake in myocytes.
Conclusion
Altogether, the present findings suggested an important role for iNOS in the pathophysiological development of HFpEF, indicating that iNOS inhibition may represent a potential therapeutic strategy for HFpEF.
Introduction
Due to its complexity and heterogeneity, heart failure is the leading cause of mortality worldwide and results in a heavy economic burden [1]. According to epidemiological studies, approximately half of the patients with heart failure experience preserved ejection fraction (EF), a condition referred to as heart failure with preserved EF (HFpEF) [2]. The incidence of HFpEF has increased as a result of obesity, diabetes and aging in the global population [3], [4]. However, there are currently no effective therapeutic strategies for HFpEF. Thus, it is critical to explore the underlying pathophysiological mechanisms of HFpEF and identify novel therapeutic targets.
Despite the heterogeneity of HFpEF clinical presentation, patients with this condition often display features of metabolic syndrome and exercise intolerance [5], [6]. In a recent study, a HFpEF mouse model was established using a high-fat diet (HFD) and Nω-nitro-L-arginine methyl ester (L-NAME), which recapitulates the systemic and cardiovascular features of the disease observed in humans, including metabolic syndrome and exercise intolerance [7].
Inducible nitric oxide (NO) synthase (iNOS) is expressed at low levels in normal heart tissue. However, upon stress, such as inflammation and hypoxia large quantities of NO are generated as a result of iNOS activation [8]. Although NO is a crucial mediator for maintaining vascular tone and preventing platelet aggregation and adhesion, imbalanced NO production induces inflammation and cytotoxic injury [9]. It has also been reported that iNOS is associated with excessive oxidative stress and insulin resistance [10], both of which are involved in the development of HFpEF. In addition, iNOS-induced nitrative stress contributes to the development of HFpEF, and the inhibition of iNOS synthesis and/or activity ameliorates the HFpEF phenotype in mouse models [7]. However, the mechanisms underlying the protective role of iNOS inhibition in HFpEF, especially short-term iNOS inhibition, remains unclear.
To maintain its pumping function, the heart needs to generate a large quantity of ATP. Approximately 70% of ATP is generated from the oxidation of fatty acids in mitochondria via oxidative phosphorylation, and the remaining 30% from glucose and lactate [11]. Usually, the utilization of metabolic substrates is highly flexible and dependent on changes in physiological and pathological conditions [12]. Impaired use of any of the aforementioned substrates can hinder mitochondrial function and inhibit ATP production, which contribute the development of HFpEF [6]. Insulin resistance is a typical feature of HFpEF that inhibits glucose metabolism in the heart, thereby impairing cardiac function [13], [14]. Protein kinase B (Akt) is important for insulin-mediated glucose utilization in the heart, and inhibition of Akt signaling has been implicated in the development of various cardiovascular diseases [15], [16]. Previous studies suggested that upregulated iNOS expression negatively modulated insulin-mediated glucose uptake by increasing Akt S-nitrosylation [9], [10]. However, the insulin-mediated glucose pathway has not been examined in HFpEF. In particular, whether iNOS promotes Akt S-nitrosylation in HFpEF is still unknown.
Therefore, the aim of the present study was to investigate the long-term and short-term effects of iNOS inhibition on Akt S-nitrosylation and Akt-mediated insulin signaling in heart tissue samples from an HFpEF mouse model using genetic knockout or pharmacological inhibition of iNOS.
Material and Methods
Experimental animals
Eight-week-old male C57BL/6J mice were used for the establishment of the HFpEF model. The mice were housed in a standard environment with a 12-h light/dark cycle and free access to food and water. To induce HFpEF, a 60% HFD, combined with 0.5 g/l L-NAME (MilliporeSigma, USA) administered through the drinking water, were used. To investigate the role of short-term iNOS inhibition, L-NIL (MedChemExpress, USA) was administered intraperitoneally at a dose of 80 mg/kg of body weight for 3 days after 8 weeks of HFD and L-NAME, as previously described [7]. iNOS-null (iNOS-/-) mice were used to examine the long-term effects of iNOS inhibition on HFpEF. All experimental procedures were approved by The Chongqing Medical University Committee (Number of permit:SYXK 2018–0003) on Animal Care and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Echocardiography and Doppler imaging
The VINNO 6 VET ultrasound system for small animals was used to assess cardiac function. Anesthesia was induced with 4% isoflurane for ∼ 1–2 min, then maintained using 1–1.5% isoflurane in pure oxygen until the examination was completed. Chest fur was cleared with a chemical hair remover. M−mode measurements were performed to measure cardiac systolic function. Pulsed-wave and tissue Doppler imaging at the level of the mitral valve were used to measure diastolic function.
Exercise exhaustion test
Exercise exhaustion tests were conducted following 3 times of adaptive running on three consecutive days on a treadmill (SANA Biotechnology, China). The mice were first allowed to run uphill at 20˚C for 4 min with a speed of 5 m/min. The speed was then increased to 14 m/min for 2 min. Subsequently, the speed was increased by an additional 2 m/min every 2 min until exhaustion, as described previously [7].
Hematoxylin and eosin (HE) staining
At the end of the experiment, the animals were sacrificed, and their hearts were excised. After clean with 10-ml cold PBS, the hearts were fixed using 4% paraformaldehyde. The hearts were embedded in optimum cutting temperature compound, then cut to 7-μm-thick sections. In order to examine the global change in cardiac hypertrophy, the sections were stained with HE using an HE staining kit (Beijing Solarbio Science & Technology Co., Ltd., China) according to the manufacturer’s instructions.
Isolation of cardiac mitochondria
Cardiac mitochondria were isolated from murine heart tissue using a Tissue Mitochondria Isolation Kit (Beyotime Institute of Biotechnology, China) according to the manufacturer’s instructions. After extraction, the mitochondria were lysed in lysis buffer, and the protein samples were stored at −80 °C until further use.
Western blotting
Protein was extracted from frozen murine heart tissue or isolated cardiac myocytes using a Column Tissue and Cell Protein Extraction Kit (EpiZyme, China). BCA assay kits were used to measure the protein concentration. After boiling 10 min at 100 °C with loading buffer (EpiZyme, China), the protein samples were separated using 4–12% FuturePAGE™ gradient gels and MOPS-SDS running buffer (Nanjing ACE Biotechnology, China). The PVDF membrane (for iNOS detection, nitrocellulose membranes were used) was blocked with 5 % skimmed milk for 60–90 min, then incubated with primary antibody overnight at 4 °C. The secondary antibody was then added for 60 min at room temperature. An ECL regent (BioSharp, China) was used to visualize the protein bands using a gel imaging system (Bio-Rad Laboratories, Inc., USA). The following antibodies were used in the present study: iNOS (BD Transduction Laboratories™), Akt, nuclear factor erythroid 2–related factor 2 (Nrf2; Affinity Biosciences, China), phosphorylated (p)-Akt (Ser 473) (Wanleibio, China), NADPH oxidase 4 (NOX4), superoxide dismutase 2 (SOD2), p-Nrf2, translocase of outer mitochondrial membrane 20, heme oxygenase 1 (HO-1) (ProteinTech Group, Inc., China) and OXPHOS antibody cocktail (Abcam, UK).
Measurement of citrate synthase activity, glutathione peroxidase (GSH-Px) activity, ATP levels and malondialdehyde (MDA) levels
To assess mitochondrial function and oxidative stress, citrate synthase and GSH-Px activity, as well as ATP and MDA levels were measured in heart tissue samples using corresponding assay kits, according to the manufacturer’s instructions. All kits were purchased from Nanjing Jiancheng Bioengineering Institute (China).
Detection of nitrite and nitrate
To evaluate nitrative stress, nitrite and nitrate levels were measured in urine samples using a Nitrate/nitrite Colorimetric Assay Kit (Amyjet Scientific, China) according to the manufacturer’s instructions. The urine was diluted 1:30 before use. The absorbance was read at 540 nm.
Cardiomyocyte isolation and measurement of glucose uptake
Neonatal ventricular myocytes were isolated from 1 to 3-day-old neonatal Sprague-Dawley pups. The pups were sacrificed using anesthesia with 5% isoflurane for 3–5 min. The myocytes were cultured in M199 medium (Hyclone, Cytiva, China) supplemented with 10% FBS (PAN-Biotech GmbH, Germany). After 4 days, iNOS plasmids were transfected using the jetOPTIMUS system (Polyplus-Transfection SA, France).
Adult myocytes were isolated from the hearts of the mice using enzymatic digestion. The myocytes were allowed to adhere to culture dishes coated with laminin for 1 h in an incubator, then incubated with 5 μM 2-NBDG for 30 min. Fluorescence intensity was then detected using a microplate reader (Thermo Fisher Scientific, Inc., USA) at 485 nm.
Akt expression in AC16 cells
AC16 cells were provided by Immocell Biotechnology Co.,Ltd and cultured in DMEM medium supplemented with 10% FBS. The wild-type (WT) and mutant Akt phasmids at C224, C296 and C310 S-Nitrosylation residues were generated by Tsingke Biotechnology Co., Ltd. iNOS and Akt plasmids transfection was carried out when cell were 80% confluent. And after 48 h of the initiation of transfection, cells were harvested for detection.
Detection of mitochondrial membrane potential and mitoSOX
After iNOS plasmids transfection and 10 μM L-NIL treatment, neonatal ventricular myocytes were resuspend and washed with warm HBSS buffer. Then, cells were loaded with 5 μM mitoSOX for 10 min or 15 μM TMRE for 20 min at 37 °C incubator. After washing the cells with warm HBSS for 2 times, flow cytometry was performed on a CytoFLEx platform (Beckman) and data were analyzed with CytExpert.
Detection of S-nitrosylated Akt
The S-nitrosylation of Akt was assessed using a biotin switch assay. The heart tissue was homogenized with lysis buffer (Beyotime Institute of Biotechnology). After centrifugation at 9,000 × g for 20 min, the protein concentrations of the supernatants were measured using a BCA assay, then adjusted to 1 μg/μl. The samples were then blocked with 20 mM MMTS at 50 °C for 20 min. After blocking, the protein was precipitated using cold acetone and resuspended in HENS buffer. The samples were then incubated with 4 mM HPDP-biotin (Thermo Fisher Scientific, Inc., USA) and 2.5 mM ascorbic acid for 1 h at 23 °C. After precipitating with cold acetone and resuspending in HENS buffer again, the protein was incubated with streptavidin-agarose beads at 23 °C for 1 h. After washing and incubating at 37 °C for 20 min with elution buffer, the supernatants were collected for SDS-PAGE immunoblotting with the anti-Akt antibody, as aforementioned.
Statistical analysis
The data are presented as the mean ± SEM. The differences between the groups were determined using unpaired Student’s t-test, one-way ANOVA followed by Tukey’s post hoc test or two-way ANOVA followed by Bonferroni correction where appropriate. P-value < 0.05 was considered to be statistically significant.
Results
Pharmacological inhibition and gene knockout of iNOS ameliorate HFpEF in vivo
iNOS is expressed at low levels in normal heart, however, HFD combined with L-NAME significantly increased iNOS expression in HFpEF hearts (Fig. S1A). To gain insight into the effect of iNOS on HFpEF, pharmacological and genetic iNOS inhibition were used in the HFpEF model. We first evaluated the systolic and diastolic function of the hearts using echocardiography (Fig. 1A). The heart rate did not significantly differ between the experiment groups (Fig. 1B). The results showed that the HFD + L-NAME regimen significantly increased the mitral E to A-wave (E/A) ratio and the mitral E to E’-wave (E/E’) ratio, whereas the left ventricular EF was not significantly affected (Fig. 1C-F). These features suggested that the HFpEF model was successfully established. Short-term iNOS blockade using L-NIL significantly improved the cardiac diastolic function in HFpEF mice, as evidenced by reduced E/A and E/E’ ratios (Fig. 1D–F). Moreover, iNOS knockout also resulted in improved cardiac diastolic function (Fig. 1D–F). The HFD + L-NAME regimen also led to increased body weight (Fig. 1G), systolic blood pressure (Fig. 1H), diastolic blood pressure (Fig. S1B) and cardiac hypertrophy, as evidenced by increased heart weight/tibia length ratios (Fig. S1C). Although iNOS inhibition couldn’t ameliorate increased body weight, blood pressure and cardiac hypertrophy significantly (Fig. 1G and Fig. S1B and C) of HFpEF, the increased pulmonary edema and reduced running distance were partly rescued following short-term iNOS inhibition or iNOS gene knockout (Fig. S1D and E). These results suggested that iNOS was critical for cardiac diastolic dysfunction following HFD + L-NAME administration and that iNOS inhibition could ameliorate the HFpEF phenotype in mice.
Fig. 1.

Pharmacological inhibition and genetic knockdown of iNOS ameliorate the HFpEF phenotype in mice. (A) Typical images of the left-ventricular M−mode echocardiogram of mice in different experimental groups. (B-C) The heart rate (B) and left-ventricular ejection fraction (LVEF) (C) was assessed using echocardiographic analyses. (D) Typical pulsed-wave Doppler images and tissue Doppler traces of mice in different experimental groups. (E) Mitral E to A-wave (E/A) ratios. (F) Mitral E to E’-wave (E/E’) ratios. (G-H) The change of Body weight and systolic pressure (SBP) between groups. n = 6. The data are shown as mean ± SEM and were analyzed using two-way ANOVA with Bonferroni post hoc test. *, P < 0.05. **, P < 0.01. ****, P < 0.0001. ns, no significant.
iNOS inhibition reduced nitrative stress and Akt S-nitrosylation in HFpEF heart
According to the STRING database version 11.5 (https://string-db.org), the physical and functional interaction of iNOS with Akt was significant (Fig. 2A–B). Previous studies have also suggested that iNOS mediates Akt S-nitrosylation and reduces Akt activity [17], [18]. We then investigated the effects of iNOS inhibition on Akt S-nitrosylation. Despite no obvious effects of iNOS inhibition were observed in the heart tissue from normal mice (Fig. S2), both short-term iNOS inhibition and iNOS knockout reduced the urine nitrite and nitrate levels, as well as Akt S-nitrosylation in HFpEF mice (Fig. 2C–E). These findings suggested that short-term iNOS inhibition was as efficient at reducing nitrative stress and Akt S-nitrosylation as iNOS knockout in HFpEF mice. To confirm the effect of iNOS on Akt S-nitrosylation in the myocardium, iNOS-containing plasmids were transfected into isolated neonatal rat cardiac myocytes (Fig. 2F and G). It was observed that iNOS overexpression could upregulate Akt S-nitrosylation in cardiomyocytes (Fig. 2F and H). Moreover, iNOS-induced Akt S-Nitrosylation was significantly blunted when the C224 cysteine residues was mutated, but not C296 and C310 (Fig. 2I and J), indicating that C224 is the main S- Nitrosylation residues of cardiomyocytes upon iNOS expression.
Fig. 2.

iNOS inhibition reduced nitrative stress and Akt S-nitrosylation in HFpEF heart. (A-B) The interactions of iNOS with other proteins were identified using the STRING database. iNOS (Nos2) directly interacted with Akt (A). The confidence score of the interaction of iNOS and other proteins is shown in B. (C) Urinary nitrite/nitrate concentration in mice from different experimental groups. n = 4 per group. (D-E) Representative immunostaining and semi-quantification of S-nitrosylated Akt (SNO-Akt) levels in heart tissue samples. n = 6 per group. (F-H) Representative immunostaining and semi-quantification of S-nitrosylated Akt and iNOS expression levels in neonatal rat cardiomyocytes transfected with plasmids. n = 6 per group for the detection of S-nitrosylated Akt. n = 3 per group for the detection of iNOS expression. (I-J) Representative immunostaining and semi-quantification of S-nitrosylated Akt in AC16 human cardiomyocyte cells transfected with mutated Akt plasmids. The data are shown as mean ± SEM and were analyzed using one-way ANOVA followed by Tukey’s post hoc test (C-E), or Student’s t-tests (F-H) ,or two-way ANOVA with Bonferroni post hoc test (I-J). *, P < 0.05. **, P < 0.01. ***, P < 0.0005. ****, P < 0.0001. ns, no significant.
iNOS inhibition improved Akt-mediated insulin signal
Akt S-nitrosylation impairs its activity and increases insulin resistance, leading to reduced glucose utilization [19], [20]. Akt phosphorylation at the Ser473 active site was significantly reduced in the heart tissue samples from HFpEF model mice; Although no significantly effects on Akt phosphorylation in normal heart was observed (Fig. S3A–B), iNOS inhibition restored the Akt phosphorylation levels (Fig. 3A and B). Moreover, although glucose uptake in ex vivo cardiomyocytes was markedly reduced in HFpEF model mice, this was notably upregulated following both pharmacological inhibition and knockout of iNOS, as evidenced by increased 2-NBDG uptake (Fig. 3C). In isolated neonatal rat cardiac myocytes, iNOS did not affect basal Akt phosphorylation and 2-NBDG uptake; However, insulin-mediated Akt phosphorylation and glucose uptake were significantly inhibited following iNOS overexpression (Fig. 3D-F). Moreover, those effects of iNOS on Akt phosphorylation and glucose uptake were significantly rescued by mutation of C224 S- nitrosylation residues (Fig. 3D–F) This suggested that iNOS impaired the Akt-mediated insulin pathway via S-nitrosylation and that iNOS inhibition alleviated Akt S-nitrosylation and increased Akt-mediated insulin signal, consistent with reduced Akt S-nitrosylation in the heart of HFpEF mice.
Fig. 3.

iNOS inhibition improved Akt-mediated insulin signaling. (A-B) Representative immunostaining and semi-quantification of phosphorylated Akt (Ser473 site) in heart tissue samples. n = 6 per group. (C) Glucose uptake in isolated cardiac myocytes from adult mice. n = 6 per group. (D-E) Representative immunostaining and semi-quantification of phosphorylated Akt at Ser473 site in AC16 human cardiomyocyte cells transfected with iNOS or mutated Akt plasmids. (F) Glucose uptake in AC16 human cardiomyocyte cells transfected with iNOS or mutated Akt-containing plasmids. n = 4 per group. The data are shown as mean ± SEM and were analyzed using one-way ANOVA followed by Tukey’s post hoc test (A-C), or two-way ANOVA with Bonferroni post hoc test (D-F). *, P < 0.05. **, P < 0.01. ***, P < 0.0005. ****, P < 0.0001. ns, no significant.
iNOS inhibition improved cardiac mitochondrial function in HFpEF mice
Intact mitochondrial function and sufficient ATP supply are important for normal cardiac function [21]. In addition, Akt-mediated insulin signaling has been shown to be critical for energy metabolism. To determine whether iNOS impairs mitochondrial function and ATP supply, we next examined the expression of components of the mitochondrial electron transport chain complex (COX proteins). In HFpEF model mice, a marked reduction in COX II, -III and -IV expression was observed, although COX I and -V expression levels were not significantly affected (Fig. 4A–F). Both pharmacological inhibition and gene knockout of iNOS upregulated COX II and -III expression (Fig. 4C and E), although only the knockout increased the COX IV expression in the HFpEF model mice (Fig. 4D). Moreover, iNOS knockout, but not short-term pharmacological iNOS inhibition, increased citrate synthase activity (Fig. 4G). However, both approaches improved the ATP supply in HFpEF (Fig. 4H). However, as the low expression of iNOS expression, iNOS inhibition had no obvious effects on ATP content and citrate synthase activity in normal heart (Fig. S3C-D). These results indicated that iNOS inhibition improved cardiac mitochondrial function and increased ATP supply in the hearts of HFpEF model mice, which may account for the attenuated HFpEF phenotype.
Fig. 4.

iNOS inhibition improved cardiac mitochondrial function in HFpEF mice. (A) The levels of mitochondrial respiratory chain complexes in heart mitochondria were evaluated using immunostaining with an OXPHOS antibody cocktail. (B-F) Semi-quantification of COX I-V immunostaining. (G) Citrate synthase activity in the heart tissue samples from mice in different experimental groups. (H) Myocardial ATP content in mice. n = 6. The data are shown as mean ± SEM and were analyzed using one-way ANOVA followed by Tukey’s post hoc test. *, P < 0.05. **, P < 0.01. ***, P < 0.0005. ****, P < 0.0001. ns, no significant.
iNOS induces oxidative stress and mitochondrial injury in isolated caridomyotes
Mitochondria are the main source of reactive oxygen species (ROS) [22]. iNOS dysregulation in mitochondria causes excessive ROS production, leading to oxidative stress and mitochondrial dysfunction [23], [24]. It was observed that iNOS overexpression significantly increased Nox4 expression (Fig. 5A–B) and superoxide level according to increased mitoSOX (Fig. 5C–D). Moreover, iNOS reduced mitochondrial membrane potential according to decreased TMRE in cardiomyocytes (Fig. 5E–F). Those results suggested that iNOS expression induced oxidative stress injury and mitochondrial dysfunction. However, inhibiting iNOS with L-NIL blunted the adverse effect of iNOS expression in cardiomyocytes (Fig. 5).
Fig. 5.

iNOS induces oxidative stress and mitochondrial injury in cardiomyocyte. (A-B) Representative immunostaining and semi-quantification of NOX4 expression in neonatal rat cardiomyocytes transfected with plasmids encoding iNOS. n = 6. (C-D) The mitoSOX was used as an mitochondrial ROS indicator and assessed by flow cytometry. (E-F) The TMRE was used as an mitochondrial membrane potential indicator and assessed by flow cytometry. n = 4. The data are shown as mean ± SEM and were analyzed using one-way ANOVA followed by Tukey’s post hoc test. *, P < 0.05. **, P < 0.01. ***, P < 0.0005. ****, P < 0.0001. ns, no significant.
iNOS inhibition alleviates oxidative stress in the heart of HFpEF mice
We next sought to determine the effects of iNOS inhibition on oxidative stress in the hearts of HFpEF model mice. Although there was no difference in SOD2 expression, Nox4 expression was significantly inhibited following iNOS pharmacological inhibition and iNOS gene knockout (Fig. 6A-C). In addition, myocardial MDA levels (Fig. 6D) significantly increased, while GSH-Px activity decreased (Fig. 6E) in the HFpEF hearts. By contrast, for both approaches, iNOS mitigated excessive oxidative stress in the heart (Fig. 6D and E), as evidenced by reduced MDA content and increased GSH-Px activity. Under excessive oxidative stress, Nrf2 pathway is activated and leads to antioxidant reactions. Interestingly, we did not observe increased Nrf2 phosphorylation in the hearts of HFpEF model mice, however, both iNOS pharmacological inhibition and iNOS gene knockout upregulated Nrf2 phosphorylation and HO-1 expression in HFpEF heart (Fig. 6F–H). In normal heart, no significant effects of iNOS inhibition on Nox4 and SOD2 as well as Nrf2 pathway were observed (Fig. S4). Those results suggested that the Nrf2 pathway may mediate the protective effects of iNOS inhibition on HFpEF.
Fig. 6.

iNOS inhibition alleviated oxidative stress in the heart of HFpEF mice. (A-C) Representative immunostaining and semi-quantification of NOX4 and SOD2 in heart tissue samples. (D) Myocardial malondialdehyde levels. (E) GSH-Px activity. (F-H) Representative immunostaining and semi-quantification of p-Nrf2/Nrf2 as well as HO-1 in the heart tissue samples from mice in different experimental groups. n = 6. The data are shown as mean ± SEM and were analyzed using one-way ANOVA followed by Tukey’s post hoc test. *, P < 0.05. **, P < 0.01. ***, P < 0.0005. ****, P < 0.0001. ns, no significant.
Discussion
Although HFpEF is a main form of heart failure with high morbidity, evidenced-based efficacy pharmacological therapy for HFpEF is still lacking, owing to a lack of preclinical models and insufficient understanding of the pathophysiological process of HFpEF [25]. Recently, a ‘two-hit’ mouse model of HFpEF induced by HFD and L-NAME that recapitulates the numerous systemic and cardiovascular features of HFpEF has been developed [7]. Using this model, it was demonstrated that inflammation, metabolic disorder and nitrative stress were involved in the development of HFpEF. In another study, a ‘three-hit’ mouse model was generated by targeting systemic metabolism [26], which also mimicked the key hemodynamic features of HFpEF. Similar to the ‘two-hit’ model, this model also confirmed that mitochondrial dysfunction, oxidative stress and inflammation are critical for the development of HFpEF.
NO is an important signaling molecule regulating vascular tone [27]. There are three different NOS enzymes responsible for NO production, namely, endothelial NOS, neuronal NOS and iNOS [28]. Among them, iNOS generates excessive NO leading to nitrative stress and tissue injury [29]. Thus, iNOS expression levels are very low under physiological conditions [30]. Aberrant iNOS upregulation has been implicated in several cardiovascular diseases, including myocardial infarction, atherosclerosis, and heart failure [31], [32]. A previous study suggested that iNOS expression was upregulated in heart tissue during HFpEF and that inhibiting iNOS attenuates the HFpEF phenotype [7], which was also confirmed in the present study. While iNOS deficiency protects the heart from HFpEF by restoring X-box binding protein 1 (XBP1s) levels in the myocardium, short-term iNOS inhibition can improve cardiac function in HFpEF model mice independently of XBP1s [7]. Thus, there may be additional mechanisms mediating the protective effects of iNOS inhibition.
In the present study, both the pharmacological inhibition and the knockout of iNOS alleviated mitochondrial dysfunction and oxidative stress, resulting in increased ATP supply in the hearts of HFpEF model mice. These findings suggest the important role of iNOS in regulating mitochondrial function in the process of HFpEF and iNOS inhibition may protect cardiac function by mitigating mitochondrial dysfunction and oxidative stress.
Akt is a serine/threonine protein kinase that is essential for the insulin signaling pathway, glucose homeostasis and oxidative stress [33]. Impaired Akt activity is associated with increased insulin resistance and dysregulated energy metabolism. The activity of Akt is regulated by post-transcriptional modification [15]. Indeed, Akt is activated by phosphorylation at the Ser473 residue, but iNOS leads to Akt S-nitrosylation and suppresses its activity [29], [34]. However, whether Akt S-nitrosylation is involved into the progress of HFpEF and associated with the protective effects of iNOS inhibition have not been investigated. In the present study, elevated S-nitrosylation of Akt was observed in the heart of HFpEF model mice. However, iNOS inhibition reduced the S-nitrosylation and increased the phosphorylation of Akt (Ser473). Consistent with this observation, Akt-mediated glucose uptake in cardiomyocytes was also partly rescued by iNOS inhibition. In cells, we furtherly confirmed that iNOS induced the S-nitrosylation at C224 residues. Moreover, mutation of Akt S-nitrosylation site blunted the adverse effects of iNOS on insulin signal. Those data suggested the important roles of Akt S-nitrosylation in HFpEF and inhibiting iNOS may function via reducing Akt S-nitrosylation.
Previous studies have indicated that Akt contributed to Nrf2 phosphorylation, thus exerting antioxidant effects. In the present study, increased Nrf2 phosphorylation and reduced oxidative stress were also observed in the iNOS-/- HFpEF mice, which may be attributed to the reduced Akt S-nitrosylation levels. These results further indicated that iNOS inhibition may alleviate the HFpEF phenotype by reducing Akt S-nitrosylation and enhancing Akt activity.
However, the present study has its limitations. The role of iNOS was only examined in global iNOS-/- mice. Thus, the effects of iNOS deficiency on other cell and tissue types could not be excluded. Additionally, the C57BL/6J background was used in this study, although previous studies have used C57BL/6N to induce HFpEF model. Thus, our results should be confirmed in other strains to confirm the role of iNOS on Akt S-nitrosylation.
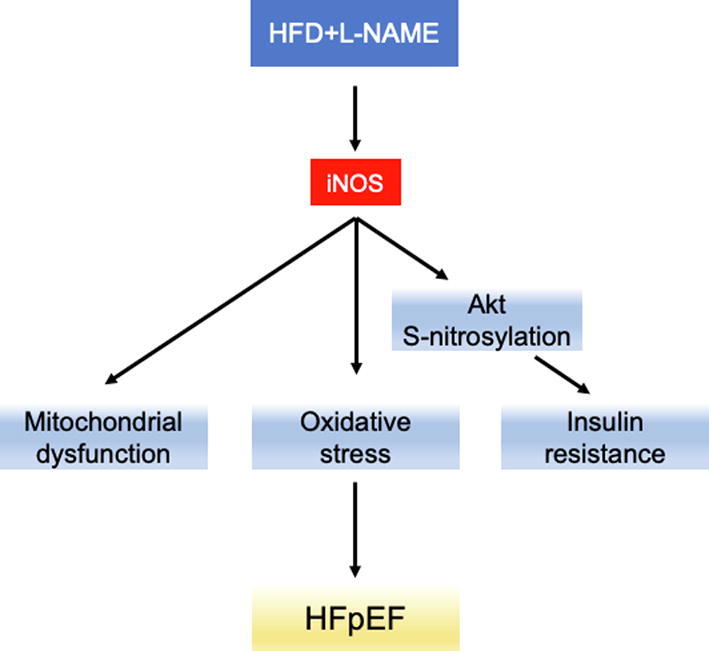
In conclusion, the present study confirmed the critical role of iNOS as a pathophysiological driver of HFpEF (Fig. 7). Both short-term iNOS inhibition or its knockout improved mitochondrial function and reduced Akt S-nitrosylation, leading to an attenuated HFpEF phenotype. As selective iNOS inhibitors are both safe and accessible, iNOS may represent an immediate, potential therapeutic target for HFpEF.
Fig. 7.

Graphical mechanistic model. The model illustrates how iNOS causes mitochondrial dysfunction and Akt S-nitrosylation contributing to HFpEF.
Ethics statement
All experiments involving animals were conducted according to the ethical policies and procedures approved by The Chongqing Medical University Committee on Animal Care, China (permit of animal housing no. SYXK (YU) 2018–0003).
Study approval
All experimental procedures were approved by The Chongqing Medical University Committee on Animal Care and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Funding sources
This work was supported by the National Natural Science foundation of China grants (81270210, 82070238), Cultivating fund of The First Affiliated Hospital of Chongqing Medical university (PYJJ2021-05) and High-quality Development Fund of West China Guang’an Hospital of Sichuan University (21FZ009).
CRediT Author Statement
Conceptualization:YZ Guo, SX Luo and XW Wang. Data curation: YZ Guo, JJ Wen, SX Luo and XW Wang. Formal analysis: YZ Guo, JJ Wen, A He, and C Qu. Funding acquisition: SX Luo, YZ Guo and JJ Wen. Investigation:YZ Guo, YC Peng and A He. Methodology:YZ Guo, JJ Wen, YC Peng and C Qu. Project administration: XW Wang and SX Luo. Resources: XW Wang and SX Luo. Software: None. Supervision: SX Luo and XW Wang. Validation: SX Luo and XW Wang. Visualization: YZ Guo and XW Wang. Writing-original draft: YZ Guo. Writing-review&editing: XW Wang and Suxin Luo.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
None.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2022.03.003.
Contributor Information
Suxin Luo, Email: luosuxin@hospital.cqmu.edu.cn.
Xiaowen Wang, Email: xiaowenwang@cqmu.edu.cn.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., et al. Heart disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139(10):e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Mesquita T., Lin Y.-N., Ibrahim A. Chronic low-grade inflammation in heart failure with preserved ejection fraction. Aging Cell. 2021;20(9) doi: 10.1111/acel.v20.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sani C.M., Pogue E., Hrabia J.B., Zayachkowski A.G., Zawadka M.M., Poniatowski A.G., et al. Association between low-grade chronic inflammation and depressed left atrial compliance in heart failure with preserved ejection fraction: A retrospective analysis. Folia Med Cracov. 2018;58(2):45–55. doi: 10.24425/fmc.2018.124657. [DOI] [PubMed] [Google Scholar]
- 4.Hong G.-R., Vannan M.A., Bossone E. Heart Failure with Preserved Ejection Fraction: Current Opinion and Future Perspectives. Heart Fail Clin. 2021;17(3):xiii–xiv. doi: 10.1016/j.hfc.2021.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Kumar A.A., Kelly D.P., Chirinos J.A. Mitochondrial Dysfunction in Heart Failure With Preserved Ejection Fraction. Circulation. 2019;139(11):1435–1450. doi: 10.1161/CIRCULATIONAHA.118.036259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miranda‐Silva D., Wüst R.C.I., Conceição G., Gonçalves‐Rodrigues P., Gonçalves N., Gonçalves A., et al. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol (Oxf) 2020;228(3) doi: 10.1111/apha.v228.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schiattarella G.G., Altamirano F., Tong D., French K.M., Villalobos E., Kim S.Y., et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568(7752):351–356. doi: 10.1038/s41586-019-1100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pérez-Torres I., Manzano-Pech L., Rubio-Ruíz M.E., Soto M.E., Guarner-Lans V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules. 2020;25(11):2555. doi: 10.3390/molecules25112555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasukawa T., Tokunaga E., Ota H., Sugita H., Martyn J.A.J., Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280(9):7511–7518. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 10.Ozbayer C., Kebapci M.N., Kurt H., Colak E., Gunes H.V., Degirmenci I. Potential associations between variants of genes encoding regulators of inflammation, and mediators of inflammation in type 2 diabetes and insulin resistance. J Clin Pharm Ther. 2021;46(5):1395–1403. doi: 10.1111/jcpt.13471. [DOI] [PubMed] [Google Scholar]
- 11.Taegtmeyer H., Wilson C.R., Razeghi P., Sharma S. Metabolic energetics and genetics in the heart. Ann N Y Acad Sci. 2005;1047:208–218. doi: 10.1196/annals.1341.019. [DOI] [PubMed] [Google Scholar]
- 12.Crewe C., Kinter M., Szweda L.I. Rapid inhibition of pyruvate dehydrogenase: an initiating event in high dietary fat-induced loss of metabolic flexibility in the heart. Plos One. 2013;8(10) doi: 10.1371/journal.pone.0077280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boudina S., Bugger H., Sena S., O'Neill B.T., Zaha V.G., Ilkun O., et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119(9):1272–1283. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scherbakov N., Bauer M., Sandek A., Szabó T., Töpper A., Jankowska E.A., et al. Insulin resistance in heart failure: differences between patients with reduced and preserved left ventricular ejection fraction. Eur J Heart Fail. 2015;17(10):1015–1021. doi: 10.1002/ejhf.317. [DOI] [PubMed] [Google Scholar]
- 15.Tsuzuki T., Shinozaki S., Nakamoto H., Kaneki M., Goto S., Shimokado K., et al. Voluntary Exercise Can Ameliorate Insulin Resistance by Reducing iNOS-Mediated S-Nitrosylation of Akt in the Liver in Obese Rats. Plos One. 2015;10(7) doi: 10.1371/journal.pone.0132029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao J., Yamashita H., Qiao L., Friedman J.E. Decreased Akt kinase activity and insulin resistance in C57BL/KsJ-Leprdb/db mice. J Endocrinol. 2000;167(1):107–115. doi: 10.1677/joe.0.1670107. [DOI] [PubMed] [Google Scholar]
- 17.Moldogazieva N.T., Mokhosoev I.M., Feldman N.B., Lutsenko S.V. ROS and RNS signalling: adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic Res. 2018;52(5):507–543. doi: 10.1080/10715762.2018.1457217. [DOI] [PubMed] [Google Scholar]
- 18.Charbonneau A., Marette A. Inducible nitric oxide synthase induction underlies lipid-induced hepatic insulin resistance in mice: potential role of tyrosine nitration of insulin signaling proteins. Diabetes. 2010;59(4):861–871. doi: 10.2337/db09-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneki M., Shimizu N., Yamada D., Chang K. Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal. 2007;9(3):319–329. doi: 10.1089/ars.2006.1464. [DOI] [PubMed] [Google Scholar]
- 20.Musso G., Gambino R., De Michieli F., Biroli G., Premoli A., Pagano G., et al. Nitrosative stress predicts the presence and severity of nonalcoholic fatty liver at different stages of the development of insulin resistance and metabolic syndrome: possible role of vitamin A intake. Am J Clin Nutr. 2007;86(3):661–671. doi: 10.1093/ajcn/86.3.661. [DOI] [PubMed] [Google Scholar]
- 21.Kiyuna L.A., Albuquerque R., Chen C.H., Mochly-Rosen D., Ferreira J. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic Biol Med. 2018;129:155–168. doi: 10.1016/j.freeradbiomed.2018.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou B., Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128(9):3716–3726. doi: 10.1172/JCI120849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pall M.L. The NO/ONOO-cycle as the central cause of heart failure. Int J Mol Sci. 2013;14(11):22274–22330. doi: 10.3390/ijms141122274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cyr A., Chambers L., Waltz P.K., Whelan S.P., Kohut L., Carchman E., et al. Endotoxin Engages Mitochondrial Quality Control via an iNOS-Reactive Oxygen Species Signaling Pathway in Hepatocytes. Oxid Med Cell Longev. 2019;2019:1–9. doi: 10.1155/2019/4745067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitakata H., Endo J., Hashimoto S., Mizuno E., Moriyama H., Shirakawa K., et al. Imeglimin prevents heart failure with preserved ejection fraction by recovering the impaired unfolded protein response in mice subjected to cardiometabolic stress. Biochem Biophys Res Commun. 2021;572:185–190. doi: 10.1016/j.bbrc.2021.07.090. [DOI] [PubMed] [Google Scholar]
- 26.Deng Y., Xie M., Li Q., Xu X., Ou W., Zhang Y., et al. Targeting Mitochondria-Inflammation Circuit by beta-Hydroxybutyrate Mitigates HFpEF. Circ Res. 2021;128(2):232–245. doi: 10.1161/CIRCRESAHA.120.317933. [DOI] [PubMed] [Google Scholar]
- 27.Gkaliagkousi E., Ferro A. Nitric oxide signalling in the regulation of cardiovascular and platelet function. Front Biosci (Landmark Ed) 2011;16:1873–1897. doi: 10.2741/3828. [DOI] [PubMed] [Google Scholar]
- 28.Kone B.C., Kuncewicz T., Zhang W., Yu Z.-Y. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am J Physiol Renal Physiol. 2003;285(2):F178–F190. doi: 10.1152/ajprenal.00048.2003. [DOI] [PubMed] [Google Scholar]
- 29.Carvalho-Filho M.A., Ropelle E.R., Pauli R.J., Cintra D.E., Tsukumo D.M., Silveira L.R., et al. Aspirin attenuates insulin resistance in muscle of diet-induced obese rats by inhibiting inducible nitric oxide synthase production and S-nitrosylation of IRbeta/IRS-1 and Akt. Diabetologia. 2009;52(11):2425–2434. doi: 10.1007/s00125-009-1498-1. [DOI] [PubMed] [Google Scholar]
- 30.Akaike T., Maeda H. Nitric oxide and virus infection. Immunology. 2000;101(3):300–308. doi: 10.1046/j.1365-2567.2000.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nathan C., Calingasan N., Nezezon J., Ding A., Lucia M.S., La Perle K., et al. Protection from Alzheimer's-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202(9):1163–1169. doi: 10.1084/jem.20051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang P., Xu X., Hu X., van Deel E.D., Zhu G., Chen Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ Res. 2007;100(7):1089–1098. doi: 10.1161/01.RES.0000264081.78659.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Q., Gao F., Ma X.L. Insulin says NO to cardiovascular disease. Cardiovasc Res. 2011;89(3):516–524. doi: 10.1093/cvr/cvq349. [DOI] [PubMed] [Google Scholar]
- 34.Liang X.-X., Wang R.-y., Guo Y.-Z., Cheng Z., Lv D.-y., Luo M.-H., et al. Phosphorylation of Akt at Thr308 regulates p-eNOS Ser1177 during physiological conditions. Febs Open Bio. 2021;11(7):1953–1964. doi: 10.1002/2211-5463.13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.