

Abstract
INTRODUCTION:
As many as 100 million people in the US have nonalcoholic fatty liver disease (NAFLD), characterized by increased liver lipid accumulation, which often leads to hepatocyte injury and fibrosis, characteristics of nonalcoholic steatohepatitis (NASH). NASH in turn can progress to cirrhosis and hepatocellular carcinoma. There are currently no US Food and Drug Administration–approved therapies for NAFLD or NASH. NAFLD occurs when there is disequilibrium between the processes of hepatic lipid synthesis and consumption. The nutrient sensor mechanistic target of rapamycin complex 1 (mTORC1) regulates several of these pathways. mTORC1 is thus an attractive target to modulate lipid homeostasis in the liver. However, mTORC1 also regulates numerous other cellular pathways, and blunting of mTORC1 modulation can lead to unexpected feedback loops and unwanted effects.
RATIONALE:
We hypothesized that selective modulation of hepatic mTORC1 signaling could benefit liver lipid metabolism and prevent NAFLD. In non-liver cell types, the protein folliculin (FLCN) has been shown to confer substrate specificity to mTORC1. Deletion of FLCN inhibits mTORC1-mediated phosphorylation of the transcription factor E3/B (TFE3/B) family of transcription factors, without affecting mTORC1-driven phosphorylation of its canonical substrates ribosomal protein S6 kinase beta-1 (S6K1) and eukaryotic translation initiation factor 4E–binding protein 1 (4E-BP1). Unphosphorylated TFE3 translocates to the nucleus and activates genes that promote lysosomal biogenesis, mitochondrial biogenesis, and oxidative metabolism. We reasoned that suppression of FLCN in the liver might promote fatty acid oxidation and lipid clearance without untoward effects of generalized mTORC1 inhibition.
RESULTS:
Hepatocyte-specific genetic deletion of Flcn in adult mice selectively inhibited mTORC1-mediated cytoplasmic sequestration of TFE3, with little effect on other mTORC1 targets, including S6K, 4E-BP1, and Lipin1. Hepatocyte loss of Flcn protected mice from both NAFLD and NASH and partially reversed these processes when already established. The protection against NAFLD and NASH required TFE3, which activated lipid clearance. Unleashed TFE3 additionally suppressed de novo lipogenesis. The latter was mediated in part by TFE3-mediated induction of insulin-induced gene 2 (Insig2) to inhibit proteolytic activation of sterol regulatory element–binding protein-1c (SREBP-1c), a critical lipogenic transcription factor.
CONCLUSION:
Our data establish FLCN as a critical regulator of lipid homeostasis in the liver. Flcn deletion affords selective inhibition of mTORC1, leading to nuclear translocation and activation of the transcription factor TFE3, which coordinates hepatic lipid metabolic pathways to protect against NAFLD and NASH in mice. Thus, our data reveal FLCN as a promising target for the treatment of NAFLD and NASH. The data also illuminate previously published and seemingly conflicting data, which likely reflected different effects on each arm of mTORC1 signaling. There have been numerous attempts by many to develop disease-specific treatments for NAFLD and NASH, thus far without success. A recurrent problem has been the many compensatory responses by the liver to targeting any one pathway; for example, inhibitors of acetyl–coenzyme A carboxylase led to compensatory activation of SREBP-1c and consequent hyperlipidemia. Targeting FLCN is thus particularly attractive, in that loss of FLCN simultaneously and favorably affects multiple aspects of hepatic lipid homeostasis, including promoting fatty acid oxidation and lysosomal biogenesis and inhibiting de novo lipogenesis.
Graphical Abstract
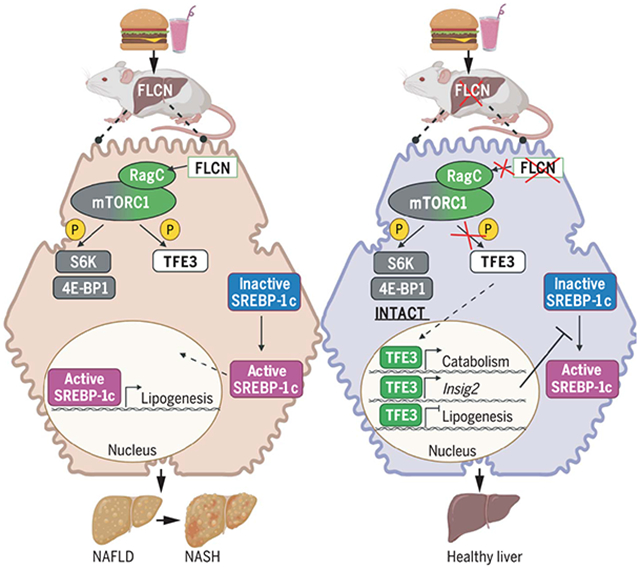
Deletion of Flcn in the liver protects mice from NAFLD and NASH through selective suppression of mTORC1. Diets high in fat, carbohydrates, and cholesterol lead to NAFLD and NASH. When Flcn is simultaneously deleted, mTORC1 is selectively inhibited, preserving phosphorylation of canonical substrates S6K and 4E-BP1 while blocking phosphorylation of the transcription factor TFE3. Unphosphorylated TFE3 is released to the nucleus, where it activates lipid catabolism genes while suppressing de novo lipogenesis genes. [Image created using Biorender]
Nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) remain without effective therapies. The mechanistic target of rapamycin complex 1 (mTORC1) pathway is a potential therapeutic target, but conflicting interpretations have been proposed for how mTORC1 controls lipid homeostasis. We show that selective inhibition of mTORC1 signaling in mice, through deletion of the RagC/D guanosine triphosphatase–activating protein folliculin (FLCN), promotes activation of transcription factor E3 (TFE3) in the liver without affecting other mTORC1 targets and protects against NAFLD and NASH. Disease protection is mediated by TFE3, which both induces lipid consumption and suppresses anabolic lipogenesis. TFE3 inhibits lipogenesis by suppressing proteolytic processing and activation of sterol regulatory element–binding protein-1c (SREBP-1c) and by interacting with SREBP-1c on chromatin. Our data reconcile previously conflicting studies and identify selective inhibition of mTORC1 as a potential approach to treat NASH and NAFLD.
As many as 100 million people in the US have nonalcoholic fatty liver disease (NAFLD), characterized by increased liver lipid accumulation (1), which often leads to hepatocyte injury and fibrosis, characteristics of nonalcoholic steatohepatitis (NASH) (2, 3). NASH can, in turn, progress to cirrhosis and hepatocellular carcinoma (1, 4). To date, there is no US Food and Drug Administration–approved therapy for NAFLD or NASH (4). NAFLD stems from a disequilibrium between hepatic lipid flux processes (5). Attempts at therapeutic modulation of any one of these processes often lead to feedback modulation of the others, limiting efficacy or causing unwanted side effects. Identifying therapeutic nodes that strike the proper balance has therefore been challenging.
The mechanistic target of rapamycin (mTOR) pathway is a critical nutrient-sensing pathway in many cell types, including hepatocytes (6). The mechanistic target of rapamycin complex 1 (mTORC1), nucleated around the adaptor protein Raptor, activates anabolic pathways, such as lipid and protein synthesis, and inhibits catabolic pathways, such as autophagy and oxidative metabolism (6, 7). For this reason, mTORC1 has been studied as an attractive target to modulate lipid homeostasis in the liver, but its role in this process remains unclear, and studies point to seemingly opposing conclusions. Deletion of Raptor in hepatocytes suppresses de novo lipogenesis (DNL) through inhibition of the lipogenic transcription factor sterol regulatory element–binding protein-1c (SREBP-1c) and protected mice from liver steatosis (8). These results were consistent with an anabolic role of mTORC1 activity (8) and with the requirement of mTORC1 for activation of SREBP-1c by the protein kinase Akt (9). However, different studies reported that hepatocyte deletion of the TSC complex subunit 1 gene (Tsc1) (10, 11), and therefore activation of mTORC1, also protected mice from liver steatosis. Other groups have observed increased steatosis (12) and liver injury (13) in mice lacking Raptor in the liver. Thus, the role of mTORC1 in liver steatosis remains poorly understood.
Substrate specificity can be conferred to mTORC1 by the protein folliculin (FLCN) (14-16). Heterozygous loss-of-function mutations in the FLCN gene cause Birt-Hogg-Dubé syndrome, characterized by benign skin fibrofolliculomas, lung cysts, and, in a subset of patients, renal cell carcinoma caused by loss of heterozygosity (17). FLCN is a guanosine triphosphatase (GTPase)–activating protein (GAP) for the GTPases RagC and RagD, which, in their guanosine diphosphate–bound state, promote activation of mTORC1 (18). Deletion of Flcn in various cell types inhibits mTORC1-mediated phosphorylation of one set of targets, the transcription factor E3/B (TFE3/B) family of transcription factors, without affecting mTORC1-driven phosphorylation of its canonical substrates ribosomal protein S6 kinase beta-1 (S6K1) and eukaryotic translation initiation factor 4E–binding protein 1 (4E-BP1) (14-16). As a result, modulation of FLCN allows separation of these two pathways downstream of mTORC1 (Fig. 1, A and B). Unphosphorylated TFE3 translocates to the nucleus and activates genes that promote lysosomal biogenesis, mitochondrial biogenesis, and oxidative metabolism (15, 19-22). Thus, in adipose tissue, for example, FLCN deletion promotes beiging and thermogenesis (14, 23), without suppressing S6K-mediated anabolism or causing lipodystrophy (14).
Fig. 1. Liver FLCN selectively promotes mTORC1-mediated cytoplasmic sequestration of TFE3, without affecting canonical mTORC1 signaling.

(A and B) Model of selective mTORC1 regulation. P, phosphorylated; PRAS40, proline-rich AKT substrate of 40 kDa; DEPTOR, DEP domain-containing mTOR-interacting protein; mLST8, target of rapamycin complex subunit LST8. (C) Schematic of FLCN liver deletion to yield control and hepatocyte-specific Flcn-null (LiFKO) mice. (D) FLCN protein expression in livers of control and LiFKO mice fed normal chow and euthanized after overnight fast followed by 4 hours of refeeding. HSP90, heat shock protein 90. (E) TFE3 protein expression in the indicated subcellular fractions from livers of control, LiFKO, and Tfe3 knockout mice fed normal chow and euthanized ad lib. S.E., short exposure; L.E., long exposure; H3, histone H3. (F) TFE3 protein expression in the nucleus from livers of control (Con.) and Raptor liver-KO mice, euthanized after overnight fasting and 4 hours of refeeding. See fig. S1A for whole cell and cytoplasmic fractions. p:total, fraction of phosphorylated protein to total protein. (G and H) Phosphorylation of mTORC1 targets in control and LiFKO mice fed either (G) normal chow or (H) 7 or 8 days of FPC diet (TD190142) and sugar water, and euthanized after overnight fast and ~4 hours of refeeding. pS6 240/4, phospho-ribosomal protein S6 (Ser240/244); S6, ribosomal protein S6; p4E-BP1 S65, phospho-4E-BP1 (Ser65); p4E-BP1 Th37, phospho-4E-BP1 (Thr37). (I) Phosphorylation of Lipin1, assayed by immunoprecipitation (IP) with anti-Lipin1 followed by immunoblotting for phospho-Lipin1, in liver lysates of control and LiFKO mice fed 9 days of FPC diet (TD190142) and sugar water and euthanized at 10 p.m. ad lib. IgG, immunoglobulin G. (J) TFE3 protein expression in the indicated subcellular fractions from control and liver Tsc1 KO mice fed normal chow and euthanized ad lib. *P < 0.05, **P < 0.01; Student’s two-tailed t test. Data are depicted as mean ± SEM.
We reasoned that suppression of FLCN in the liver might promote fatty acid oxidation and lipid clearance without untoward effects of generalized mTORC1 inhibition. We found that deletion of Flcn in hepatocytes promoted oxidative metabolism and lysosomal biogenesis. Unexpectedly, we also found that deletion of Flcn in hepatocytes suppressed SREBP-1c activity and de novo lipogenesis through multiple mechanisms. In addition, activation of canonical mTORC1 signaling triggered a feedback loop that suppressed noncanonical FLCN-mTORC1 activity on TFE3, thereby in part explaining discordant findings reported in the literature. Ultimately, suppression of liver FLCN led to protection against, and reversal of, NAFLD and NASH.
Liver FLCN selectively promotes mTORC1-mediated cytoplasmic sequestration of TFE3, without affecting canonical mTORC1 signaling
Mice with liver-specific deletion of Flcn (LiFKO mice) were generated by infecting Flcnlox/lox mice with hepatotropic adeno-associated virus serotype 8 (AAV8) expressing Cre recombinase driven by the hepatocyte-specific thyroxine-binding globulin (TBG) promoter (Fig. 1C). FLCN content in LiFKO livers was decreased by more than 90% (Fig. 1D). TFE3 protein was enriched in the nuclei of cells from livers lacking Flcn (Fig. 1E), as was a slower migrating form of TFE3, as previously observed with Flcn deletion in different cell types (15, 22). Similar experiments in mice lacking Raptor in hepatocytes (12) demonstrated comparable accumulation of nuclear TFE3 (Fig. 1F and fig. S1A). Thus, hepatic FLCN appears to activate mTORC1 and promote retention of TFE3 in the cytoplasm.
Phosphorylation of canonical mTORC1 targets, including S6 and 4E-BP1, was mostly unaffected in livers of LiFKO mice, whether fed normal chow (Fig. 1G) or a NAFLD-inducing diet [the fructose, palmitate, and cholesterol (FPC) diet] (Fig. 1H). Phosphorylation of S6 was unchanged in mice fasted overnight and then refed normal chow (Fig. 1G), and only mildly decreased in mice fed an FPC diet (Fig. 1H). Phosphorylation of 4E-BP1 was unchanged (Fig. 1H). Phosphorylation of Lipin1, an mTORC1 substrate reported to inhibit SREBP-1c in the liver (8), was also unaffected in LiFKO livers (Fig. 1I). Together, these data indicate that hepatic FLCN activates mTORC1 selectively to regulate TFE3 localization, which is known to be regulated by mTORC1-mediated phosphorylation, with little impact on canonical mTORC1 substrates (Fig. 1, A and B).
FLCN binds to and inhibits or activates adenosine monophosphate–activated protein kinase (AMPK) in various cell types (24-26). We observed a small increase in phospho-AMPK (active AMPK) in livers from chow-fed LiFKO mice (fig. S1B) but no change in mice fed a NAFLD-inducing diet (fig. S1C). Phosphorylation of AMPK substrate acetyl–coenzyme A (acetyl-CoA) carboxylase (ACC) was unchanged in animals fed normal chow but increased in mice fed an FPC diet, whereas phosphorylation of unc-51–like autophagy kinase 1 (ULK1) was largely unchanged in both conditions (fig. S1, B and C). Loss of FLCN thus appears to have variable but mild effects on AMPK signaling in the liver.
We next tested the impact of Tsc1 deletion on TFE3 nuclear localization. The TSC complex suppresses canonical mTORC1 activity (27). TFE3 was localized to the nucleus in livers depleted of Tsc1 (Fig. 1J), despite activation of canonical mTORC1 signaling (fig. S1D). We also observed selective stabilization of the slower migrating TFE3 band, a signature of FLCN suppression (Fig. 1J). Canonical mTORC1 signaling (e.g., through S6K) therefore appears to exert negative feedback on the FLCN:TFE3 axis of mTORC1 signaling, such that chronic activation of the canonical arm, achieved by Tsc1 deletion, might suppress FLCN activity (Fig. 1, A and B). FLCN protein abundance was unchanged in livers lacking Tsc1 (Fig. 1J), indicating that inhibition of FLCN occurs after translation. Thus, activation of TFE3 in the liver can be achieved, seemingly paradoxically, by either inhibition of (noncanonical) mTORC1 or activation of (canonical) mTORC1. The presence of this feedback might explain the observations that both activation and inhibition of mTORC1 protect against steatosis (8, 11). In this model (Fig. 1, A and B), loss of Flcn should protect against NAFLD, and that protection should be dependent on TFE3 activity.
Loss of FLCN in the liver protects against NAFLD
LiFKO mice fed standard rodent chow appeared grossly normal, gained body weight at similar rates to control mice (Fig. 2A), and exhibited normal liver histology, as shown by hematoxylin and eosin (H&E) staining (Fig. 2B, top, and fig. S2A). When fed an Amylin liver NASH (AMLN) diet—a NAFLD-inducing diet high in trans fat, fructose, and cholesterol (28)—control mice gained weight (Fig. 2C) and developed severe liver steatosis, as shown by H&E staining (Fig. 2B, bottom), liver triglyceride quantification (Fig. 2D), and blinded histological evaluation of H&E slides (Fig. 2E). In contrast, LiFKO mice were mostly protected from both body weight gain and liver steatosis (Fig. 2, B to E). LiFKO mice also revealed lower levels of plasma triglycerides (fig. S2B) and nonesterified fatty acids (fig. S2C) and were protected from diet-induced elevations in total cholesterol, both HDL and non-HDL (fig. S2, D to F). Similar protection from weight gain and steatosis was observed in mice fed a Gubra AMLN (GAN) diet, which is a modified AMLN diet without trans fat that has replaced the AMLN diet since the ban on trans fats (29) (fig. S2G). Loss of Flcn is thus protective against NAFLD in these models.
Fig. 2. Loss of FLCN in the liver potently protects against NAFLD, and the protection requires TFE3.

(A to E) Control, LiFKO, and DKO mice were fed a normal chow (n = 7) or AMLN diet (n = 3 to 9) for 17 to 18.5 weeks and euthanized after a 4- to 6-hour fast. (A) Body weights on normal chow. (B) Representative images of liver H&E staining. Scale bars, 500 μm. (C) Body weights on AMLN diet. (D) Quantification of liver triglycerides. (E) Blinded histological evaluation of liver H&E slides. (F to J) Control, LiFKO, Tfe3 KO, and DKO mice were fed normal chow (n = 10 to 13) or FPC diet regimen (TD160785 with sugar water; n = 5 to 11) for 16 weeks and euthanized after removing their food for 4 to 6 hours. (F) Body weights on normal chow. (G) Representative images of liver H&E staining. Scale bars, 200 μm. (H) Body weights on FPC diet. (I) Quantification of hepatic liver triglycerides. (J) Blinded histological evaluation of liver H&E slides. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant. Statistical values are indicated for the final body weight measurement. Statistical analysis was done with two-way repeated measures or mixed effects analysis of variance (ANOVA) with multiple comparisons test in (A), (C), (F), and (H). Student’s two-tailed t test was used for normal chow controls for the AMLN diet experiment. One-way ANOVA with Tukey’s multiple comparisons test was used otherwise. Data are depicted as mean ± SEM.
One possible explanation for the observed protection against steatosis in LiFKO mice was the protection against weight gain and concomitant reductions in plasma insulin and homeostatic model assessment for insulin resistance (HOMA-IR) (fig. S2, H to J). The mechanism for the protection against weight gain in LiFKO mice on an AMLN diet is not clear, as comprehensive lab animal monitoring system (CLAMS) studies with control and LiFKO mice fed a GAN diet revealed no significant differences in food consumption, water consumption, ambulatory or locomotor activity, or energy expenditure (fig. S3). We thus sought a diet where body weight gains were equivalent between genotypes. Control and LiFKO mice were subjected to an FPC diet regimen, consisting of high fat, sucrose, and cholesterol with reduced vitamin E and choline, in conjunction with fructose and glucose in the drinking water—often called the “Big Mac and Coke” diet (30). With this FPC regimen, in which diminished choline intake can suppress weight gain (30, 31), there were no significant differences in body weight between the genotypes (Fig. 2H and fig. S4A), despite an apparent mild reduction in energy expenditure (fig. S4B). Plasma triglycerides, nonesterified fatty acids, insulin, glucose, and HOMA-IR were also unaffected (fig. S4, C to G). However, as with the AMLN diet, LiFKO mice were almost entirely protected against NAFLD induced by the FPC regimen (Fig. 2, G to J). The beneficial effects of Flcn deletion on the liver thus appear to be independent of body weight.
NAFLD protection through loss of FLCN requires TFE3
Loss of Flcn induces TFE3 translocation to the nucleus (Fig. 1E). Notably, codeletion of Flcn and Tfe3 [double knockout (DKO) mice] completely prevented the protection against NAFLD seen with Flcn deletion alone, in both the AMLN and FPC diets (Fig. 2, B, D, E, G, I, and J). Tfe3 deletion alone had little impact compared with control mice (Fig. 2, I to J). In the AMLN diet cohort, deletion of Tfe3 also reversed the reductions in body weight seen with Flcn deletion (Fig. 2C), whereas no impact on body weight was seen in the FPC diet cohort (Fig. 2H). The protection against NAFLD in Flcn liver-null mice therefore requires TFE3. Together, these data demonstrate that loss of liver Flcn strongly protects against the development of NAFLD, in at least two NAFLD-inducing dietary conditions; that the protection is independent of effects on body weight; and that the protection is mediated by TFE3, most likely through TFE3 dephosphorylation, nuclear translocation, and transcriptional activation of an antisteatotic program.
Loss of FLCN in the liver activates pathways of lipid catabolism
In many cell types, TFE3 activates the expression of genes that drive lysosomal biogenesis (15, 32), an important step in the breakdown of lipids through lipophagy (33), and the expression of PGC-1α and PGC-1β (14), drivers of mitochondrial biogenesis, fatty acid oxidation, electron transport chain, and tricarboxylic acid (TCA) cycle genes. RNA sequencing (RNA-seq) analysis (Fig. 3, A to C) and protein immunoblotting (Fig. 3D) showed that both of these TFE3-mediated gene programs were activated in livers from LiFKO mice, whether fed normal chow or a NAFLD-inducing diet. Moreover, the induction of both programs was abrogated in DKO livers (Fig. 3, B and D), demonstrating their dependence on TFE3. PGC-1α mRNA (Ppargc1a) abundance was increased in LiFKO livers in a TFE3-dependent manner (Fig. 3E), and TFE3 occupancy on the chromatin at Ppargc1a, as well as lysosomal genes [e.g., cathepsin z (Ctsz)], was increased in LiFKO livers, coinciding with PolII occupancy and epigenetic markers of transcriptionally active chromatin (Fig. 3F). In line with these data, fatty acid oxidation, measured by conversion of radiolabeled palmitate to water, was increased in hepatocytes isolated from LiFKO mice (Fig. 3G). Gluconeogenic targets of PGC-1α (34) did not show increased expression in Flcn KO livers (fig. S5A), indicating that deletion of Flcn activates PGC-1α–driven oxidation programs specifically, without adversely affecting gluconeogenesis. Consistent with this observation, glucose sensitivity was unchanged in mice fed a long-term AMLN diet (fig. S5B) or a short-term GAN diet (fig. S5C) and, if anything, improved in LiFKO mice fed normal chow (fig. S5B). Together, these data indicate that Flcn deletion, by releasing TFE3 to the nucleus, allows TFE3 to activate a coordinated program of lipid catabolism, thereby potentially limiting liver steatosis. However, the overall increase in systemic fatty acid oxidation seemed limited, as measured by β-hydroxybutyrate levels in plasma (fig. S6A) and whole-body respiratory exchange ratio values (fig. S6B). In light of the strong protection against NAFLD observed in LiFKO mice, these observations suggested that loss of Flcn might promote other antisteatotic programs.
Fig. 3. Loss of FLCN in the liver activates pathways of lipid catabolism.

(A to C) RNA-seq was performed on the livers of control, LiFKO, and DKO mice (n = 3) on normal chow or AMLN diet (as described in Fig. 2). (A) −Log10(adjusted p-value) versus log2(LiFKO/control fold change) volcano plot. Dotted line: P = 0.05. (B) Heatmap of normalized expression of mitochondrial and lysosomal gene sets. (C) Top 10 differentially expressed gene sets between LiFKO and control livers. Red indicates pathways up-regulated in LiFKO, blue indicates pathways down-regulated in LiFKO. (D) Protein expression of components of mitochondrial electron transport chain complexes (C.I, C.II, C.III, and C.V), LC3B-I and LCSB-II, p62, and HSP90, in livers of control, LiFKO, and DKO mice fed an FPC diet regimen (TD160785 with sugar water) for 16 weeks. (E) mRNA expression of Ppargc1a and Ppargc1b in livers of control, LiFKO, and DKO mice fed a normal chow (n = 7) or AMLN diet (n = 3 to 9) for 17 to 18.5 weeks. (F) Genome browser tracks of the Ctsz and Ppargc1a promoters. TFE3 ChIP-seq was performed on livers of mice fed normal chow (n = 2 to 4). Depicted are tracks from one representative sample per genotype. Green indicates publicly available ENCODE liver ChIP-seq datasets. (G) Hepatocytes were isolated from three control and LiFKO mice and pooled. Fatty acid oxidation was measured from three or four wells of each genotype by incubating them with 3H-palmitate for 120 min and either vehicle or 100 μM etomoxir (an inhibitor of fatty acid oxidation). Conversion of 3H-palmitate to 3H2O was measured by scintillation counting and normalized to cell count. dpm, disintegrations per minute. One-way ANOVA with Tukey’s multiple comparisons test (for three groups) or Student’s two-tailed t test (for two groups) was used. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are depicted as mean ± SEM.
Loss of FLCN in the liver suppresses de novo lipogenesis
We had expected that Flcn deletion would promote lysosomal and mitochondrial biogenesis. However, RNA-seq and subsequent quantitative polymerase chain reaction (qPCR) and Western blotting revealed that it also caused suppression in LiFKO livers, in both normal chow and NAFLD-inducing diet conditions, of a broad program of de novo lipogenesis gene and protein expression, including adenosine triphosphate citrate lyase (Acly), fatty acid synthase (Fasn), acyl-CoA synthetase short chain family member 2 (Acss2), stearoyl-CoA desaturase-1 (Scd1), sterol regulatory element–binding transcription factor 1 (Srebf1), and specifically the Srebp1c isoform of Srebf1, which encodes for SREBP-1c, the predominant driver of de novo lipogenesis in the liver (Fig. 4, A to D, and fig. S7A). Most of these observed gene and protein expression changes were reversed in DKO livers, and Tfe3 deletion alone had little impact compared with control mice (Fig. 4, A to D, and fig. S7A). We conclude that loss of Flcn in the liver broadly suppresses the de novo lipogenesis gene program and appears to do so in a manner dependent on TFE3.
Fig. 4. Loss of FLCN in the liver suppresses de novo lipogenesis.

(A) Heatmap of normalized expression values of de novo lipogenesis genes from RNA-seq described in Fig. 3. (B and C) mRNA expression of de novo lipogenesis genes in livers of control, LiFKO, Tfe3 KO, and DKO mice fed (B) an AMLN diet (n = 3 to 9) for 17 to 18.5 weeks or (C) an FPC diet regimen (TD160785 with sugar water; n = 5 to 11) for 16 weeks. (D) Protein expression of SREBP1, ACLY, ACSS2, and FASN in livers from control, LiFKO, and DKO mice fed an FPC diet regimen (TD160785 with sugar water) for 16 weeks. HSP90 from Fig. 3D was used as loading control. Mice were euthanized after a 4- to 6-hour fast, and thus only the precursor form of SREBP1 was detected. (E) Control, LiFKO, and DKO mice (n = 6 or 7) were fed an AMLN diet for 7 to 11 days, fasted from 9 a.m. to 7 p.m., refed for 2 hours, and force-fed a bolus of 13C-fructose and 12C-glucose. The mice were fed overnight and killed the next morning. LC-MS was performed to examine the amount of 13C label incorporation into hepatic fatty acids. (F) Control, LiFKO, and DKO mice (n = 9 to 11) were fed an FPC diet regimen (TD190142 with sugar water) for 9 days and then injected intraperitoneally (i.p.) with deuterium oxide (2H2O) at ~7 p.m. Five hours later, the mice were killed, and their livers harvested. LC-MS was performed to examine the amount of deuterium label incorporation into hepatic fatty acids. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; one-way ANOVA with Tukey’s multiple comparisons test. Data are depicted as mean ± SEM.
To measure DNL directly, we force-fed mice 13C-fructose, a rich source of carbons for de novo lipogenesis in the liver, and quantified 13C incorporation into liver fatty acids. De novo lipogenesis was decreased by about one-half in LiFKO mice fed a short-term AMLN diet, in the absence of changes in body weight (Fig. 4E). DKO animals had de novo lipogenesis slightly greater than that of LiFKO alone, but the effect was not statistically significant (Fig. 4E). Similar results were observed in mice injected with deuterium oxide, followed by measuring incorporation of 2H into liver fatty acids, an alternate method of measuring de novo lipogenesis (Fig. 4F). Mild suppression of DNL was also evident in LiFKO mice fed normal chow (fig. S7B). Together, these data demonstrate that the FLCN:mTORC1:TFE3 axis in the liver both induces a catabolic (lysosomal and oxidative) gene program and simultaneously suppresses anabolic de novo lipogenesis.
TFE3 acts downstream of LXR to suppress de novo lipogenesis
Transcription of de novo lipogenesis genes is largely mediated by the transcription factor SREBP-1c (35). SREBP-1c activity is regulated at both the protein and mRNA levels. Thus, TFE3 might suppress de novo lipogenesis genes by inhibiting the SREBP-1c pathway at one or more points of regulation. The transcription factor liver X receptor (LXR) promotes transcription of SREBP-1c (35). We therefore tested whether TFE3 suppressed LXR. RNA-seq data revealed that some LXR target genes were suppressed in LiFKO livers but others were not (fig. S8A). The expression of liver X receptor alpha (LXR alpha; Lxra) and LXR beta (Lxrb) was unchanged in LiFKO livers from mice under various dietary conditions (fig. S8B). We next treated LiFKO mice with the LXR agonist T0901317 (T09) in conjunction with long-term FPC diet (fig. S8, C and D). T09 significantly exacerbated liver steatosis in control mice (36) (fig. S8E). Deletion of liver Flcn, however, suppressed liver steatosis, reversing the increase in liver triglyceride content promoted by T09 (fig. S8E). Analyses of liver de novo lipogenesis genes and other LXR target gene expression revealed similar patterns, whereby de novo lipogenesis genes were uniformly suppressed in LiFKO mice even in the presence of LXR agonism (fig. S8F). These data demonstrate that loss of Flcn and activation of TFE3 act downstream of LXR to suppress DNL.
TFE3 suppresses SREBP-1c proteolytic processing and activation
We tested whether Flcn deletion inhibited SREBP-1c posttranslational regulation. SREBP-1c exists as an inactive precursor (pSREBP-1c) in the endoplasmic reticulum (ER) and is proteolytically cleaved to liberate its basic helix–loop–helix leucine zipper (bHLH-Zip) domain (nSREBP-1c) to translocate to the nucleus and transcriptionally activate target genes, including de novo lipogenesis genes (37). The proteins insulin-induced gene 1 (INSIG1) and 2 (INSIG2) suppress processing of pSREBP-1c (38). To assay SREBP-1c processing, we killed control and LiFKO mice at night during feeding, when SREBP-1c proteolytic processing is active (39, 40). After 9 days of FPC diet, chosen to model an early steatotic state with no differences in body weight (Fig. 5A), we observed an increased abundance of the liver-specific Insig2a isoform (11, 41) of Insig2, as well as INSIG2 protein, in the livers of LiFKO mice (Fig. 5, B and C). The accumulation of Insig2 mRNA and of INSIG2 protein required TFE3, as it was absent in the DKO mice (Fig. 5, B and C). Chromatin immunoprecipitation sequencing (ChIP-seq) experiments showed chromatin occupancy by TFE3 in the Insig2 regulatory region, which was further increased in livers from LiFKO mice, consistent with direct regulation of Insig2 mRNA expression by TFE3 (Fig. 5D). In line with the established role of INSIG2 in suppressing SREBP-1c processing, abundance of the fully processed nSREBP-1c form (designated “N” in Fig. 5C) was decreased in LiFKO livers, reflecting the large amounts of INSIG2 protein (Fig. 5C). The suppression of nSREBP-1c was less in DKO livers (Fig. 5C). Thus, FLCN appears to suppress expression of de novo lipogenesis genes in large part by suppressing proteolytic processing and activation of SREBP-1c (Fig. 5E).
Fig. 5. TFE3 suppresses SREBP-1c proteolytic processing and activation.

(A to D) Control, LiFKO, and DKO mice were fed an FPC diet regimen (TD190142 with sugar water) for 9 days and euthanized at 10 p.m. ad lib (n = 7 to 9). One-way ANOVA with Tukey’s multiple comparisons test was used. (A) Body weights. (B) mRNA expression of DNL genes. (C) Liver protein expression of SREBP-1, INSIG2, and beta actin. AAV-1c, positive control from liver injected with AAV8 expressing constitutively nuclear SREBP-1c; P, precursor form of SREBP-1; N, nuclear (processed) form of SREBP-1. (D) Genome browser tracks of the Insig2 promoter. Depicted are TFE3 ChIP-seq tracks (described in Fig. 3F) from one representative sample per genotype. (E) Schematic of FLCN:TFE3 regulation of SREBP-1c proteolytic processing. (F to J) Flcnlox/lox mice (n = 5 or 6) were injected with either “control virus” (AAV8-GFP or AAV8-Cre; to generate control or LiFKO mice) or AAV8-ApoE/AAT-HA-nSREBP-1c (“1c”) and then maintained for 9 days on an FPC diet (TD190142 with sugar water). Student’s two-tailed t test was used for analysis. (F) Experimental outline. (G) Body weights. (H) Liver protein expression of exogenous HA-tagged nuclear SREBP-1c (HA), total SREBP-1, INSIG2, and 14-3-3. (I) Hepatic triglyceride quantification. (J) Liver mRNA expression of indicated DNL genes. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data are depicted as mean ± SEM.
To test this notion directly, we introduced, using AAV8 delivery, hemagglutinin (HA)–tagged constitutively processed nSREBP-1c to control and LiFKO mice (Fig. 5F). After 9 days, mice displayed similar body weights (Fig. 5G) and equal amounts of HA-nSREBP-1c protein in the liver (Fig. 5H). AAV8-nSREBP-1c nearly quadrupled triglyceride content in control mice (Fig. 5I) and increased transcription of de novo lipogenesis genes (Fig. 5J) (42). Hepatic triglycerides were elevated to a nearly equal extent in LiFKO and control mice infected with AAV8-nSREBP-1c (Fig. 5I), indicating that loss of FLCN did not suppress de novo lipogenesis in the presence of constitutive nuclear nSREBP-1c. Taken together, these data are consistent with loss of liver FLCN leading to TFE3 activation, increased expression of INSIG2, and suppression of SREBP-1c processing and activation, ultimately leading to suppression of de novo lipogenesis and hepatic steatosis.
TFE3 synergistically occupies chromatin in close proximity to SREBP-1c genome-wide
Although loss of Flcn did not suppress nSREBP-1c–induced accumulation of hepatic triglycerides (Fig. 5I), it did still mildly suppress expression of nSREBP-1c–induced DNL genes (Fig. 5J). This suggested that, in addition to inhibition of SREBP-1c processing through expression of INSIG2, TFE3 may also have a direct effect on processed, nuclear SREBP-1c. We therefore used ChIP-seq experiments to test whether TFE3 and SREBP-1c occupied similar, or the same, sites on chromatin. TFE3-bound chromatin regions were enriched for the TFE family motif, and pathway analyses revealed enrichment of “vesicle-mediated transport” and “membrane trafficking” (fig. S9), consistent with the known roles of the TFE family (32, 43). The pathways of “metabolism” and “metabolism of lipids and lipoproteins” were consistently the two most enriched pathways among the TFE3 cistromes (fig. S9). Sites known to be occupied by SREBP-1c (42) overlapped with TFE3 binding, which was increased in the absence of FLCN, in mice fed normal chow or an FPC diet (fig. S10A). We conclude that TFE3 binds genome-wide at genes of lipid homeostasis, that binding is increased by loss of FLCN, and that these TFE3 binding sites overlap with SREBP-1c binding sites.
TFE3, like SREBP-1c, is a bHLH-Zip transcription factor that binds to DNA containing E-box motifs (44, 45). This raised the possibility that TFE3 and SREBP-1c may compete for access to chromatin. However, binding by HA-nSREBP-1c to its cognate chromatin sites was no different between control and LiFKO mice (fig. S10B), indicating that there is no competitive removal of SREBP-1c from chromatin by TFE3. In the converse experiment, genome-wide TFE3 binding in these same livers increased in the presence of exogenous nSREBP-1c (fig. S10C). Unbiased motif analyses of TFE3 binding sites that increased in the presence of AAV8-HA-nSREBP-1c identified the SREBP-responsive element as enriched (fig. S10D). Thus, expression of nSREBP-1c promotes binding of TFE3 at, or near, SREBP-1c binding sites. Closer evaluation of TFE3 occupancy specifically at sites near de novo lipogenesis gene transcription start sites revealed a pattern, seen only in the absence of FLCN, whereby TFE3 bound chromatin closely downstream of SREBP-1c binding (fig. S10, E and F).
These data indicate that loss of FLCN induces changes in both TFE3 binding pattern and binding intensity and that TFE3 binding localizes near SREBP-1c binding genome-wide. However, these factors do not compete with each other for binding to chromatin; instead, the presence of SREBP-1c synergistically promotes binding of TFE3.
Loss of FLCN in mouse liver prevents and reverses NASH
NAFLD can progress to NASH, a predictor of poor clinical outcomes in humans (46). The choline-deficient amino acid–defined and high fat (CDAA-HF) diet is a rapid NASH-inducing diet that recapitulates some aspects of human physiology (47, 48). Unlike mice on other NASH-inducing diets, mice on the CDAA-HF diet do not lose weight (Fig. 6A). Control mice fed the CDAA-HF diet showed liver steatosis and NASH, as demonstrated by tripling of Sirius Red staining for fibrosis (Fig. 6, B and C); greater than 30-fold increase in collagen type I alpha 1 (Col1a1) gene expression (Fig. 6D), a marker for NASH (49); and increased expression of a range of genes involved in fibrosis and inflammation (Fig. 6E). In contrast, LiFKO mice were largely protected from NASH, as seen in H&E and Sirius Red staining (Fig. 6, B and C), suppression of many genetic markers of fibrosis (Fig. 6, D and E), and suppressed steatosis and fibrosis measured through blinded histological evaluation (fig. S11). Deletion of TFE3 in DKO mice largely abrogated the protection afforded by FLCN deletion in LiFKO mice, demonstrating the critical role of TFE3 in protection against NASH, as with NAFLD (Fig. 6, D and E). Fibrosis levels remained low in histological evaluations even in the absence of TFE3 (Fig. 6C and fig. S11), indicating that FLCN also regulates other pathways to protect against NASH. Similar reductions of NASH markers were observed in LiFKO mice fed either the AMLN diet (fig. S12, A and C) or FPC diet (fig. S12, B and D), although the induction of NASH in control mice fed these diets was milder than that seen with the CDAA-HF diet. Thus, loss of liver FLCN protects against the development of NASH and NAFLD in multiple mouse models.
Fig. 6. Loss of FLCN in the liver prevents and reverses NASH.

(A to E) Prevention of NASH. Control, LiFKO, and DKO mice were maintained on normal chow (n = 2 to 4) or CDAA-HF diet (n = 5 to 8) for 6 weeks and then euthanized after a 4- to 6-hour fast. (A) Weekly body weights. (B) Representative images of liver H&E and Sirius Red staining of mice fed CDAA-HF diet. (C) Quantification of Sirius Red positive staining. [(D) and (E)] Liver mRNA expression of the indicated fibrosis and inflammation markers. (F to J) Reversal of NASH. (F) Experimental outline. Flcnlox/lox mice were fed a CDAA-HF diet for 29 days to induce NASH, at which time “non-injected” mice were euthanized (n = 8). The remaining mice were injected with AAV8-TBG-GFP or AAV8-TBG-Cre to yield control and LiFKO mice (n = 8 in each group), and subsequently maintained for another 4 weeks on a CDAA-HF diet. Representative liver H&E and Sirius Red images depicted for each group. (G) Weekly body weights. [(H) and (I)] Liver mRNA expression of the indicated fibrosis and inflammation markers. (J) Quantification of hepatic liver triglycerides and Sirius Red positive staining. Two-way repeated measures ANOVA with multiple comparisons test were used in (A) and (G). One-way ANOVA with Tukey’s multiple comparisons test was used to assess differences between CDAA-HF–fed control, LiFKO, and DKO mice [(C) to (E)] or noninjected, control, and LiFKO mice [(H) to (J)]. Student’s two-tailed t test was used to compare control and LiFKO mice on normal chow. *P < 0.05, ** P < 0.01, *** P < 0.001, ****P < 0.0001. Scale bars, 200μm. Data are depicted as mean ± SEM.
A therapy intended to treat NASH and NAFLD would benefit from modifying established disease, in addition to preventing it. We therefore provided Flcnlox/lox mice with the CDAA-HF diet for 4 weeks to induce NASH (Fig. 6F) and then infected the mice with AAV8-GFP (green fluorescent protein) or AAV8-Cre to yield control and LiFKO mice, respectively, followed by another 4 weeks of CDAA-HF diet (Fig. 6F). There was no difference in body weights at either the 4- or 8-week time points (Fig. 6G). After 8 weeks, deletion of Flcn led to significant reversal of NASH and NAFLD when compared with both the 4-week pre-deletion mice and the 8-week control mice, as evidenced by reduced steatosis on H&E (Fig. 6F); reduced Col1a1 expression (Fig. 6H); reduced expression of other measured genes of inflammation and fibrosis (Fig. 6I); reduced hepatic triglyceride accumulation (Fig. 6J); and reduced NAFLD activity scores, steatosis levels, and inflammation levels evaluated through blinded histological evaluation (fig. S11). Sirius Red staining and fibrosis scores were not reversed by Flcn deletion, perhaps reflecting the slower process of reversing collagen deposition (Fig. 6J and fig. S11). Taken together, these data demonstrate that suppression of liver FLCN prevents NASH as efficiently as it prevents NAFLD, that the suppression is largely dependent on TFE3, and that loss of FLCN can also reverse most characteristics of NAFLD and NASH.
Discussion
Suppression of FLCN in the liver protected mice from both NAFLD and NASH and helped reverse these processes if already established. The data thus reveal FLCN as a possible target for the treatment of NAFLD and NASH. Loss-of-heterozygosity or “second hit” somatic mutations in carriers of FLCN mutations can lead to renal cell cancer (50), raising a theoretical concern for possible development of cancer with any therapy that suppresses FLCN. However, FLCN suppression can be targeted specifically to the liver through, for example, liver-targeted nanoparticles or N-acetylgalactosamine (GalNAc)–modified small interfering RNAs (siRNAs) (51-53), and hepatocellular carcinoma has not been observed in carriers of FLCN mutations. Moreover, FLCN deletion is thought to induce renal cell cancer through induction of mTORC1 signaling (54, 55), and we did not observe higher canonical mTORC1 activity in livers of LiFKO mice (Fig. 1, G and H).
Our data provide insight into the mechanisms by which mTORC1 regulates and coordinates lipid homeostasis in the liver. The mTORC1 pathway is sometimes depicted as receiving multiple inputs and integrating that information into a single on/off switch that then phosphorylates all of its targets. However, specificity of mTORC1 signal transduction in fact exists (14-16), and suppression of FLCN selectively suppresses mTORC1-mediated phosphorylation of TFE3, with minimal effect on other targets. This FLCN:mTORC1:TFE3 arm appears to be necessary for mTORC1-mediated lipid anabolism and steatosis in response to various diets. Moreover, cytoplasmic sequestration and suppression of TFE3 is the critical mediator of this anabolic signal.
This work provides at least a partial explanation for the seemingly contradictory data on the role of mTORC1 in the development of steatosis. We propose that the FLCN:mTORC1:TFE3 arm is dominant in the anabolic regulation of SREBP-1c and DNL. Consistent with this notion, deletion of Flcn itself is protective against NAFLD but has little impact on the canonical mTORC1:S6K arm. Overactivation of the mTORC1:S6K arm, achieved by deletion of Tsc1, may improve NAFLD (10-12) by activating a feedback loop that leads to suppression of the FLCN:mTORC1:TFE3 arm and to TFE3 nuclear translocation. Loss of Tsc1 may thus promote protein anabolism through activation of the mTORC1:S6K arm but suppress lipid anabolism through feedback inhibition of the FLCN:mTORC1:TFE3 arm. These results are also consistent with the notion that the mTORC2 complex, which is known to promote DNL, does so directly rather than by suppression of TSC, adding another level of complexity to this system (56-58).
Our data provide mechanistic insight into the regulation of SREBP-1c and de novo lipogenesis, a critical component of NAFLD in humans (59, 60). Releasing TFE3 to the nucleus induces INSIG2 to inhibit proteolytic processing of SREBP-1c and promotes TFE3 binding to chromatin near SREBP-1c to further suppress lipogenesis genes. This pathway of SREBP-1c regulation is independent of Lipin1 phosphorylation and of LXR signaling, indicating that Lipin1 is primarily targeted by the canonical arm of mTORC1, and that, under these conditions, Lipin1 is not necessary for efficient suppression of SREBP-1c and protection against NAFLD and NASH.
A potentially beneficial aspect of FLCN as a therapeutic target for NAFLD and NASH is its coordinated regulation of multiple programs involved in the progression to steatosis, that is, simultaneous induction of lipid consumption programs and suppression of lipid generation (de novo lipogenesis). This coordinated and synergistic effect likely drives the observed protection against NAFLD and NASH. Moreover, the coordinated effect on multiple arms of lipid handling may prevent the activation of feedback loops that may neutralize the benefits of targeting a single arm. For example, inhibitors against ACC have been tried in the clinic but were discontinued because of compensatory elevation in SREBP-1c activity and consequent hypertriglyceridemia (61). In light of these maladaptive responses to single-pathway inhibition, a strong argument has been made to identify upstream targets that coordinate control of many aspects of lipid metabolism in order to treat NASH successfully (62). We propose that FLCN is one such target.
This work has several limitations. Our studies were limited to genetic deletion of Flcn, but potential therapeutic approaches, such as liver-targeted GalNAc-modified siRNAs, are more likely to rely on incomplete suppression of FLCN. We also did not evaluate the impact of TFEB, a transcription factor with homology to TFE3 (32), the presence of which may in part explain why deletion of Tfe3 does not reverse the consequence of Flcn deletion in some of our experiments. For example, differences between TFE3 and TFEB kinetics may explain why mice lacking both Flcn and Tfe3 in the liver did not show significantly higher functional de novo lipogenesis relative to mice lacking only Flcn on a short-term NAFLD diet (Fig. 4, E and F) but did show higher de novo lipogenesis gene and protein expression on a long-term NAFLD diet (Fig. 4, B to D). For instance, it may be that the presence of TFEB compensates for TFE3 loss in the short term but not in the long term. It is also important to note that none of the diets used in our studies (AMLN, GAN, FPC, and CDAA-HF diets) perfectly recapitulate the characteristics of human NAFLD or NASH, and each diet was chosen to reflect some, but not all, characteristics (28, 30, 47, 48). Finally, it is also important to note that, in the context of at least some of these diets (AMLN and GAN diets, but not FPC and CDAA-HF diets), LiFKO mice displayed reduced weight gain compared with control animals, indicating that deletion of Flcn in the liver may have additional systemic salutary effects.
Materials and methods summary
Most studies involved either adult homozygous Flcnlox/lox mice (14, 54) or Flcnlox/lox mice also containing a loss-of-function whole-body Tfe3 mutation (14, 45), which were subsequently injected with ~1.5 × 1011 genome copies per mouse of AAV8-TBG-GFP or AAV8-TBG-Cre (Penn Vector Core, AV-8-PV0146 and AV-8-PV1091) to yield control mice (Flcnlox/lox, Tfe3+/Y with GFP), LiFKO mice (Flcnlox/lox, Tfe3+/Y with Cre), Tfe3 KO mice (Flcnlox/lox, Tfe3−/Y with GFP), and DKO mice (Flcnlox/lox, Tfe3−/Y with Cre). Mice were then fed various diets (normal chow, FPC diet regimen, AMLN diet, or GAN diet) and euthanized at various time points, and livers were harvested. For the AAV-nSREBP-1c rescue experiments, mice were additionally injected with 1.0 × 1011 genome copies per mouse of AAV8-ApoE/AAT-nSREBP-1c. We also used adult Raptorlox/lox mice injected with 1.0 × 1011 genome copies per mouse of either AAV8-TBG-GFP (control) or AAV8-TBG-Cre (“Rap KO” or “Raptor KO”) and euthanized 2 weeks later after an overnight fast followed by 4 hours of refeeding, as in (12) (Fig. 1F and fig. S1A). For the control versus Tsc1 KO subcellular fractionation experiment, mouse liver samples from the laboratory of S. Biddinger were used. Eight- to nine-week-old female Tsc1lox/lox (control) mice or Albumin-Cre Tsc1lox/lox mice (Tsc1 KO) were euthanized ad lib (63).
Radioimmunoprecipitation assay buffer lysis was used for protein isolation from whole liver tissues, followed by immunoblotting using standard wet-transfer methods; both commercially available and custom-made antibodies were used. To assay the cellular localization of TFE3, subcellular fractionation was done using whole cell lysis on mouse liver samples followed by centrifugation to yield nuclear and cytoplasmic fractions; immunoblotting was done using a commercially available anti-TFE3 antibody. For phospho-Lipin1 analysis, total Lipin1 was immunoprecipitated using the custom-made anti-total Lipin1 antibody (8) and subsequently subject to immunoblotting using a custom-made anti-pLipin1 S106 antibody (8). Histology analysis was performed on liver tissue using standard paraformaldehyde fixation followed by ethanol dehydration and paraffin embedding; commercially available stains were then used to evaluate for triglyceride accumulation (H&E, Oil Red O) and fibrosis (Sirius Red). Liver triglycerides were quantified using a commercially available colorimetric reagent. Plasma metabolites (triglycerides, nonesterified fatty acids, cholesterol, insulin, glucose, and β-hydroxybutyrate) were all quantified using commercially available colorimetric reagents or an Axcel autoanalyzer, as described in the supplementary materials. Blood glucose was measured with a glucometer. For fatty acid oxidation measurements, primary hepatocytes were isolated, cultured overnight, and subjected to radioactive palmitate tracing as referenced (42). The cells were incubated with 125 μM 3H-palmitate conjugated on bovine serum albumin and 1 mM carnitine for an additional 2 hours with or without 100 μM etomoxir. The media was delipidated, and 3H2O was measured by scintillation counting. Oral glucose tolerance tests was done on fasted mice gavaged with 2 g/kg D-glucose; blood glucose was then measured at serial time points. Whole-body metabolism and body composition were measured using the CLAMS and EchoMRI systems, respectively. RNA isolation and cDNA synthesis was performed with standard commercial extraction and synthesis methods. RNA-seq was done using GENEWIZ; data was analyzed with standard bioinformatic tools.
To measure de novo lipogenesis, mice were gavaged with a 1:1 mixture of 12C D-glucose:13C fructose (2 g/kg each) and euthanized; livers were subjected to liquid chromatography–mass spectrometry (LC-MS) to measure 13C label incorporation into hepatic fatty acids. Alternatively, mice were injected intraperitoneally with 30 μl per gram of body weight of 99.9% D2O; after euthanasia, livers were subjected to LC-MS to measure deuterium label incorporation into hepatic fatty acids. For the LXR agonist rescue experiment, mice were injected intraperitoneally with a commercially available LXR agonist (T0901317) or vehicle twice weekly, as described (36), while being fed an FPC diet regimen. ChIP-seq was performed by subjecting livers to sequential steps of nuclear isolation, fixation, sonication of chromatin, and immunoprecipitation with either anti-TFE3 or anti-HA antibodies; crosslinks were then reversed, protein was degraded, and the remaining immunoprecipitated DNA was isolated. The ChIP input and immunoprecipitated DNA was then sequenced with commercially available library preparation and sequencing methods; data was analyzed with standard bioinformatic tools. Quantification and statistical analysis were done using GraphPad Prism, R, Python, and other standard methods. A full description of methods is provided in the supplementary materials.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Harris for the Lipin1 antibodies; F. Foufelle for the INSIG2 antibody; L. Schmidt for the Flcnlox/lox mouse; D. Fisher for the Tfe3-null mouse; M. Giacca and L. Zentilin for the AAV-ApoE/AAT plasmid backbone; D. Cromley for Axcel analysis; L. Cheng and the University of Pennsylvania Cardiovascular Institute Histology Core for processing and staining of histology samples; the University of Pennsylvania Diabetes Research Center (DRC) for the use of the viral vector and the metabolomics cores (P30-DK19525); J. Li, I. Soaita, M. Blair, J. Axsom, M. Noji, and C. Bowman for technical assistance; Y. Kim, H. C. B. Nguyen, and M. Adlanmerini for expert advice on ChIP-seq; G. Liang for expert advice on nuclear SREBP-1 immunoblotting; M. Lazar for the AAV-nSREBP-1c plasmid; D. Salisbury for advice on LXR primers; P. Tontonoz and S. Lee for expert advice on T0901317 experiments; Penn Vector Core for production of AAVs; the Penn Rodent Metabolic Phenotyping Core for CLAMS studies; and the Penn Vet Comparative Pathology Core for blind scoring of mouse liver slides.
Funding:
This work was supported by an F30 NRSA fellowship from the NIDDK (F30 DK120096) and a Blavatnik Family Foundation Fellowship Award to B.S.G.; the DRC Regional Metabolomics Core (P30 DK19525); and NIH support for Z.A. (R01 DK107667).
Footnotes
Competing interests:
The authors declare no competing interests.
Data and materials availability: All data are available in the manuscript or the supplementary materials. Next-generation sequencing data are available through GEO accession number GSE160292.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Rinella ME, Nonalcoholic fatty liver disease: A systematic review. JAMA 313, 2263–2273 (2015). doi: 10.1001/jama.2015.5370 [DOI] [PubMed] [Google Scholar]
- 2.Schuster S, Cabrera D, Arrese M, Feldstein AE, Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol 15, 349–364 (2018). doi: 10.1038/s41575-018-0009-6 [DOI] [PubMed] [Google Scholar]
- 3.Schwabe RF, Tabas I, Pajvani UB, Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158, 1913–1928 (2020). doi: 10.1053/j.gastro.2019.11.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheka AC et al. , Nonalcoholic steatohepatitis: A review. JAMA 323, 1175–1183 (2020). doi: 10.1001/jama.2020.2298 [DOI] [PubMed] [Google Scholar]
- 5.Kawano Y, Cohen DE, Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol 48, 434–441 (2013). doi: 10.1007/s00535-013-0758-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ricoult SJH, Manning BD, The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 14, 242–251 (2013). doi: 10.1038/embor.2013.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dibble CC, Manning BD, Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol 15, 555–564 (2013). doi: 10.1038/ncb2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peterson TR et al. , mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 (2011). doi: 10.1016/j.cell.2011.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porstmann T et al. , SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236 (2008). doi: 10.1016/j.cmet.2008.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kenerson HL, Yeh MM, Yeung RS, Tuberous sclerosis complex-1 deficiency attenuates diet-induced hepatic lipid accumulation. PLOS ONE 6, e18075 (2011). doi: 10.1371/journal.pone.0018075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yecies JL et al. , Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 14, 21–32 (2011). doi: 10.1016/j.cmet.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinn WJ 3rd et al. , mTORC1 stimulates phosphatidylcholine synthesis to promote triglyceride secretion. J. Clin. Invest 127, 4207–4215 (2017). doi: 10.1172/JCI96036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Umemura A et al. , Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 20, 133–144 (2014). doi: 10.1016/j.cmet.2014.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wada S et al. , The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 30, 2551–2564 (2016). doi: 10.1101/gad.287953.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J et al. , Myeloid folliculin balances mTOR activation to maintain innate immunity homeostasis. JCI Insight 5, 126939 (2019). doi: 10.1172/jci.insight.126939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Napolitano G et al. , A substrate-specific mTORC1 pathway underlies Birt–Hogg–Dubé syndrome. Nature 585, 597–602 (2020). doi: 10.1038/s41586-020-2444-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt LS, Birt–Hogg–Dubé syndrome: from gene discovery to molecularly targeted therapies. Fam. Cancer 12, 357–364 (2013). doi: 10.1007/s10689-012-9574-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsun Z-Y et al. , The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 52, 495–505 (2013). doi: 10.1016/j.molcel.2013.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Betschinger J et al. , Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 153, 335–347 (2013). doi: 10.1016/j.cell.2013.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kennedy JC et al. , Loss of FLCN inhibits canonical WNT signaling via TFE3. Hum. Mol. Genet 28, 3270–3281 (2019). doi: 10.1093/hmg/ddz158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kennedy JC, Khabibullin D, Boku Y, Shi W, Henske EP, New developments in the pathogenesis of pulmonary cysts in Birt–Hogg–Dubé Syndrome. Semin. Respir. Crit. Care Med 41, 247–255 (2020). doi: 10.1055/s-0040-1708500 [DOI] [PubMed] [Google Scholar]
- 22.Hong S-B et al. , Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLOS ONE 5, e15793 (2010). doi: 10.1371/journal.pone.0015793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan M et al. , Chronic AMPK activation via loss of FLCN induces functional beige adipose tissue through PGC-1α/ERRα. Genes Dev 30, 1034–1046 (2016). doi: 10.1101/gad.281410.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baba M et al. , Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. U.S.A 103, 15552–15557 (2006). doi: 10.1073/pnas.0603781103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khabibullin D et al. , Folliculin regulates cell–cell adhesion, AMPK, and mTORC1 in a cell-type-specific manner in lung-derived cells. Physiol. Rep 2, e12107 (2014). doi: 10.14814/phy2.12107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan M et al. , The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J. Clin. Invest 124, 2640–2650 (2014). doi: 10.1172/JCI71749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J, Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol 13, 1259–1268 (2003). doi: 10.1016/S0960-9822(03)00506-2 [DOI] [PubMed] [Google Scholar]
- 28.Clapper JR et al. , Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol 305, G483–G495 (2013). doi: 10.1152/ajpgi.00079.2013 [DOI] [PubMed] [Google Scholar]
- 29.Boland ML et al. , Towards a standard diet-induced and biopsy-confirmed mouse model of non-alcoholic steatohepatitis: Impact of dietary fat source. World J. Gastroenterol 25, 4904–4920 (2019). doi: 10.3748/wjg.v25.i33.4904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X et al. , Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 24, 848–862 (2016). doi: 10.1016/j.cmet.2016.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobs RL et al. , Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem 285, 22403–22413 (2010). doi: 10.1074/jbc.M110.108514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martina JA et al. , The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal 7, ra9 (2014). doi: 10.1126/scisignal.2004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong J et al. , TFE3 alleviates hepatic steatosis through autophagy-induced lipophagy and PGC1α-mediated fatty acid β-oxidation. Int. J. Mol. Sci 17, 387 (2016). doi: 10.3390/ijms17030387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon JC et al. , Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138 (2001). doi: 10.1038/35093050 [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Viscarra J, Kim S-J, Sul HS, Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol 16, 678–689 (2015). doi: 10.1038/nrm4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao M, Liu D, The liver X receptor agonist T0901317 protects mice from high fat diet-induced obesity and insulin resistance. AAPS J. 15, 258–266 (2013). doi: 10.1208/s12248-012-9429-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horton JD, Goldstein JL, Brown MS, SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest 109, 1125–1131 (2002). doi: 10.1172/JCI0215593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yabe D, Brown MS, Goldstein JL, Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. U.S.A 99, 12753–12758 (2002). doi: 10.1073/pnas.162488899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Martelot G et al. , REV-ERBα participates in circadian SREBP signaling and bile acid homeostasis. PLOS Biol. 7, e1000181 (2009). doi: 10.1371/journal.pbio.1000181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guan D et al. , Diet-induced circadian enhancer remodeling synchronizes opposing hepatic lipid metabolic processes. Cell 174, 831–842.e12 (2018). doi: 10.1016/j.cell.2018.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yellaturu CR, Deng X, Park EA, Raghow R, Elam MB, Insulin enhances the biogenesis of nuclear sterol regulatory element-binding protein (SREBP)-1c by posttranscriptional down-regulation of Insig-2A and its dissociation from SREBP cleavage-activating protein (SCAP)·SREBP-1c complex. J. Biol. Chem 284, 31726–31734 (2009). doi: 10.1074/jbc.M109.050914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papazyan R et al. , Physiological suppression of lipotoxic liver damage by complementary actions of HDAC3 and SCAP/SREBP. Cell Metab. 24, 863–874 (2016). doi: 10.1016/j.cmet.2016.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Settembre C et al. , TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011). doi: 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horton JD, Bashmakov Y, Shimomura I, Shimano H, Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Natl. Acad. Sci. U.S.A 95, 5987–5992 (1998). doi: 10.1073/pnas.95.11.5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steingrímsson E et al. , Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc. Natl. Acad. Sci. U.S.A 99, 4477–4482 (2002). doi: 10.1073/pnas.072071099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angulo P et al. , Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149, 389–397.e10 (2015). doi: 10.1053/j.gastro.2015.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsumoto M, An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol 94, 93–103 (2013). doi: 10.1111/iep.12008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei G et al. , Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am. J. Physiol. Gastrointest. Liver Physiol 318, G174–G188 (2020). doi: 10.1152/ajpgi.00041.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lytle KA, Jump DB, Is Western diet-induced nonalcoholic steatohepatitis in Ldlr−/− mice reversible? PLOS ONE 11, e0146942 (2016). doi: 10.1371/journal.pone.0146942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vocke CD et al. , High frequency of somatic frameshift BHD gene mutations in Birt–Hogg–Dubé-associated renal tumors. J. Natl. Cancer Inst 97, 931–935 (2005). doi: 10.1093/jnci/dji154 [DOI] [PubMed] [Google Scholar]
- 51.Barba AA, Bochicchio S, Dalmoro A, Lamberti G, Lipid delivery systems for nucleic-acid-based-drugs: From production to clinical applications. Pharmaceutics 11, 360 (2019). doi: 10.3390/pharmaceutics11080360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Debacker AJ, Voutila J, Catley M, Blakey D, Habib N, Delivery of oligonucleotides to the liver with GalNAc: From research to registered therapeutic drug. Mol. Ther 28, 1759–1771 (2020). doi: 10.1016/j.ymthe.2020.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rüger J, Ioannou S, Castanotto D, Stein CA, Oligonucleotides to the (gene) rescue: FDA approvals 2017–2019. Trends Pharmacol. Sci 41, 27–41 (2020). doi: 10.1016/j.tips.2019.10.009 [DOI] [PubMed] [Google Scholar]
- 54.Baba M et al. , Kidney-targeted Birt-Hogg-Dubé gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl. Cancer Inst 100, 140–154 (2008). doi: 10.1093/jnci/djm288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hudon V et al. , Renal tumour suppressor function of the Birt–Hogg–Dubé syndrome gene product folliculin. J. Med. Genet 47, 182–189 (2010). doi: 10.1136/jmg.2009.072009 [DOI] [PubMed] [Google Scholar]
- 56.Hagiwara A et al. , Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 15, 725–738 (2012). doi: 10.1016/j.cmet.2012.03.015 [DOI] [PubMed] [Google Scholar]
- 57.Yuan M, Pino E, Wu L, Kacergis M, Soukas AA, Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J. Biol. Chem 287, 29579–29588 (2012). doi: 10.1074/jbc.M112.386854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhat N et al. , Dyrk1b promotes hepatic lipogenesis by bypassing canonical insulin signaling and directly activating mTORC2 in mice. J. Clin. Invest 132, 153724 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Donnelly KL et al. , Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest 115, 1343–1351 (2005). doi: 10.1172/JCI23621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ, Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146, 726–735 (2014). doi: 10.1053/j.gastro.2013.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim C-W et al. , Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: A bedside to bench investigation. Cell Metab. 26, 394–406.e6 (2017). doi: 10.1016/j.cmet.2017.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ, Mechanisms of NAFLD development and therapeutic strategies. Nat. Med 24, 908–922 (2018). doi: 10.1038/s41591-018-0104-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu Y et al. , Fructose and glucose can regulate mammalian target of rapamycin complex 1 and lipogenic gene expression via distinct pathways. J. Biol. Chem 293, 2006–2014 (2018). doi: 10.1074/jbc.M117.782557 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.