

Abstract
Pathogenic fungi have emerged as significant causes of infectious morbidity and death in patients with acquired immunodeficiency conditions such as HIV/AIDS and following receipt of chemotherapy, immunosuppressive agents or targeted biologics for neoplastic or autoimmune diseases, or transplants for end organ failure. Furthermore, in recent years, the spread of multidrug-resistant Candida auris has caused life-threatening outbreaks in health-care facilities worldwide and raised serious concerns for global public health. Rapid progress in the discovery and functional characterization of inborn errors of immunity that predispose to fungal disease and the development of clinically relevant animal models have enhanced our understanding of fungal recognition and effector pathways and adaptive immune responses. In this Review, we synthesize our current understanding of the cellular and molecular determinants of mammalian antifungal immunity, focusing on observations that show promise for informing risk stratification, prognosis, prophylaxis and therapies to combat life-threatening fungal infections in vulnerable patient populations.
Subject terms: Antimicrobial responses, Fungal host response, Fungal infection
This Review synthesizes our current mechanistic understanding of the cellular and molecular determinants of tissue-specific antifungal host defences derived from animal models of fungal disease, humans with fungal infection-manifesting inborn errors of immunity and patients treated with fungal infection-promoting, immune-targeted biologics.
Introduction
Of the approximately five million fungal species, only a few regularly infect humans. However, in recent decades, the AIDS pandemic and the increasing number of patients who have received myeloablative chemotherapy and/or solid organ or haematopoietic stem cell transplantation (HSCT) have led to a marked increase in the global burden of fungal disease in these immunocompromised individuals (Table 1 and Supplementary Table 1). Furthermore, new at-risk populations have emerged, including patients with severe respiratory virus-associated mould disease and individuals being treated with novel, precision immune-targeted biologics (monoclonal antibodies or small-molecule kinase inhibitors) for autoimmune, inflammatory and/or neoplastic disorders1–3. Emerging fungi such as multidrug-resistant Candida auris present new threats to public health4 (Box 1). Fungi exhibit distinct morphotypes (such as yeast and hyphae) and can cause superficial and life-threatening invasive infections, primarily in immunocompetent individuals and immunocompromised individuals, respectively (Table 1 and Supplementary Table 1).
Table 1.
Clinical features of the most prevalent human fungal infections
| Infection (most common fungal genera or species) | Fungal morphotype | Clinical presentation | Commonly affected patient populationa | Annual global incidence; all-cause mortality | Current principles of treatment |
|---|---|---|---|---|---|
| Mucocutaneous candidiasisb (Candida albicans, Candida dubliniensis, Candida glabrata) | Yeast (with pseudohyphae and hyphae depending on the species) | Oropharyngeal or oesophageal candidiasis | HIV/AIDS, corticosteroid use | ~3.3 million (HIV/AIDS only); NA | Topical or oral azoles; echinocandins (for azole-resistant Candida strains) |
| Vaginal candidiasis | Healthy women, antibiotic use | ~138 million; NA | |||
| Invasive candidiasis (C. albicans, C. glabrata, Candida tropicalis, Candida parapsilosis, Candida auris) | Yeast (with pseudohyphae and hyphae depending on the species) | Candidaemia | Critical illness (ICU), after COVID-19 | ~750,000; ~30–40% | Echinocandins; removal of central venous catheter |
| Intra-abdominal candidiasis | Abdominal surgery | ||||
| Disseminated candidiasis | Neutropenia, corticosteroid use, SOT, low birthweight premature neonates | ||||
| Invasive aspergillosis (Aspergillus fumigatus, Aspergillus flavus, Aspergillus terreus, Aspergillus niger) | Filamentous mould | Pulmonary or disseminated aspergillosis | Neutropenia, corticosteroid use, HSCT, SOT, after influenza or COVID-19, ibrutinib therapy | ~300,000; ~30–50% | Voriconazole (first line), isavuconazole or posaconazole; reduction of immunosuppression |
| Allergic bronchopulmonary aspergillosis | Atopic individuals | ~5 millionc; NA | Itraconazole and corticosteroids | ||
| Cryptococcosisb (Cryptococcus neoformans, C. gattii) | Yeast | Pneumonia | HIV/AIDS, corticosteroid use | ~223,000 (HIV/AIDS only); ~20–70% | Amphotericin B (+ 5-flucytosine for CNS disease) followed by fluconazole; reduction of immunosuppression |
| CNS or disseminated cryptococcosis | HIV/AIDS, HSCT, ibrutinib therapy | ||||
| Pneumocystosisb (Pneumocystis jirovecii) | Cysts and trophozoites | Pneumonia | HIV/AIDS, corticosteroid use | ~500,000; ~20–80% | Trimethoprim–sulfamethoxazole; reduction of immunosuppression |
| Disseminated infection | HIV/AIDS |
An expanded version of Table 1 is presented in Supplementary Table 1. CNS, central nervous system; HSCT, haematopoietic stem cell transplantation; ICU, intensive care unit; NA, not applicable; SOT, solid organ transplantation. aAt-risk conditions caused by inborn errors of immunity or administration of immune pathway-targeted biologics are presented separately in Tables 2 and 3. bAIDS-defining illness. cGlobal burden of infection.
Thus, interest in fungal immunology research has increased in recent years, catalysed by our growing understanding of the biology and functions of C-type lectin receptors (CLRs)5,6, which are the main class of fungal-sensing pattern recognition receptors (reviewed in ref. 5). Notably, humans with inherited deficiency of the CLR adaptor protein CARD9 are susceptible to severe mucocutaneous and invasive fungal disease, without developing other infectious or non-infectious complications; this selective susceptibility to fungal infection is a unique feature among the more than 450 known inborn errors of immunity (see later)7. In this Review, we synthesize immunological insights from the expanding number of inborn errors of immunity that predispose to fungal infection (Table 2 and Supplementary Table 2) and from fungal infection-promoting, immune-targeted biologics (Table 3 and Supplementary Table 3), together with mechanistic insights from animal models of fungal disease. We focus on observations that have translational implications for informing risk stratification, prognosis, treatment and vaccination of fungus-infected individuals. The text is structured according to the clinical disease phenotypes that are caused by various pathogenic yeasts, moulds and dimorphic endemic fungi.
Table 2.
Key human inborn errors of immunity that predispose to fungal infection
| Gene [protein name] (chromosome arm) | Predominant cellular expression | OMIM no. (clinical syndrome) | Mode of inheritance | Fungal infection susceptibility | Other clinical phenotypes | Antifungal immunological defects |
|---|---|---|---|---|---|---|
| CARD9 (9q) | Myeloid phagocytes and to a lesser extent epithelial cells | 212050 | AR (LOF) | CMC, CNS candidiasis, phaeohyphomycosis, aspergillosis, skin mucormycosis, onychomycosis, deep dermatophytosis | None | Decreased number of blood TH17 cells, impaired cytokine responses by PBMCs and microglia, impaired neutrophil recruitment to the CNS and killing of unopsonized Candida yeasts |
| STAT1 (2q) | Broadly expressed | 614162 | AD (GOF) | Invasive candidiasis, CMC, aspergillosis, mucormycosis, PJP, cryptococcosis, histoplasmosis, coccidioidomycosis | Bacterial, NTM and viral infections, autoimmunity, aneurysms, carcinomas | Decreased number of blood TH17 cells and decreased IL-17 production by PBMCs associated with increased cellular responses to IFNα/β, IFNγ and IL-27 |
| STAT3 (17q) | Broadly expressed | 147060 (AD HIES, Job syndrome) | AD (DN) | CMC, dermatophytosis, aspergillosis, cryptococcosis, histoplasmosis, gastrointestinal tract coccidioidomycosis, skin fusariosis, PJP | Skin staphylococcal infections, bacterial pneumonias, eczema, pneumatoceles, aneurysms, skeletal abnormalities, increased IgE | Decreased number of blood TH17 cells |
| IL12RB1 (19p) | Lymphoid and myeloid cells | 614891 (MSMD) | AR (LOF) | CMC, cryptococcosis, histoplasmosis, coccidioidomycosis, paracoccidioidomycosis | Salmonella and NTM infections | Impaired TH17 cell differentiation, impaired IL-12-dependent and IL-23-dependent IFNγ production |
| IL17RA (22q) | Myeloid cells and epithelial cells | 613953 | AR (LOF) | CMC | Skin staphylococcal infections, bacterial pneumonias, eczema | Abolished IL-17 cellular responses |
| IL17RC (3p) | Epithelial cells | 616445 | None | |||
| AIRE (21q) | mTECs and eTACs | 240300 (APECED) | AR (LOF) or AD (DN) | CMC | Multiorgan autoimmunity, ectodermal dystrophy, severe COVID-19 | Neutralizing serum autoantibodies to IL-17A, IL-17F and IL-22, epithelial barrier disruption caused by IFNγ produced by mucosal T cells |
| CYBB [gp91phox] (Xp) | Myeloid phagocytes | 306400 (X-linked CGD) | X-linked (LOF) | Aspergillosis, invasive candidiasis | Invasive infections with Nocardia, Staphylococcus, Serratia and Burkholderia, IBD | Impaired generation of superoxide |
| NCF2 [p67phox] (1q) | 233710 (AR CGD) | AR (LOF) | ||||
| CYBA [p22phox] (16q) | 233690 (AR CGD) | |||||
| NCF1 [p47phox] (7q) | 233700 (AR CGD) | |||||
| IFNGR1 (6q) | Broadly expressed | 209950 (AR MSMD) | AR (LOF) | Histoplasmosis, coccidioidomycosis | Salmonella and NTM infections | Impaired IFNγ cellular responses |
| 615978 (AD MSMD) | AD (DN) | |||||
| GATA2 (3q) | Neutrophils, and to a lesser extent mononuclear phagocytes and T cells | 614172 (MonoMAC syndrome, Emberger syndrome) | AD (HI) | Aspergillosis, cryptococcosis, histoplasmosis, coccidioidomycosis, blastomycosis, PJP | Viral and NTM infections, PAP, MDS, leukaemia, lymphedema | Decreased monocyte and DC counts, neutrophil granule abnormalities |
An expanded version of Table 2 is presented in Supplementary Table 2. AD, autosomal dominant; APECED, autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy; AR, autosomal recessive; CGD, chronic granulomatous disease; CMC, chronic mucocutaneous candidiasis; CNS, central nervous system; DC, dendritic cell; DN, dominant negative; eTAC, extrathymic AIRE-expressing cell; GOF, gain of function; HI, haploinsufficiency; HIES, hyper-IgE syndrome; IBD, inflammatory bowel disease; IFN, interferon; LOF, loss of function; MDS, myelodysplastic syndrome; MSMD, Mendelian susceptibility to mycobacterial disease; mTEC, medullary thymic epithelial cell; NTM, non-tuberculous mycobacteria; OMIM, Online Mendelian Inheritance in Man; PAP, pulmonary alveolar proteinosis; PBMC, peripheral blood mononuclear cell; PJP, Pneumocystis jirovecii pneumonia; TH17, T helper 17.
Table 3.
Key FDA-approved immune-targeted biologics associated with the development of human fungal infections
| Molecular target | Name (and type) of biologic | Approved indications | Fungal infection susceptibility | Risk of fungal infection (mean frequency) | Non-fungal infection susceptibility | Antifungal immunological defects (when known) |
|---|---|---|---|---|---|---|
| TNF | Infliximab (mouse–human chimeric IgG1κ mAb) | RA, AS, psoriasis, IBD | Histoplasmosis, coccidioidomycosis, blastomycosis, PJP | Moderate/high | Mycobacterial infections (including disseminated TB) | Impaired IFNγ production and granuloma formation; impaired phagocyte trafficking and function |
| Etanercept (p75 TNF soluble receptor fused to the Fc portion of IgG1) | ||||||
| Adalimumab (humanized IgG1κ mAb) | Mucosal candidiasis, invasive candidiasis, aspergillosis, phaeohyphomycosis | Low (<5%) | ||||
| Golimumab (humanized IgG1κ mAb) | ||||||
| CD52 | Alemtuzumab (humanized IgG1κ mAb) | MS, CLL | Cryptococcosis, PJP, mucosal candidiasis | Moderate/high | Herpetic infections, CMV infection, toxoplasmosis, nocardiosis, pneumonias | Prolonged and profound T lymphocytopenia |
| IL-17A | Ixekizumab (humanized IgG4 mAb) | Psoriasis, AS, IBD | Mucosal candidiasis | Low/moderate (~2–12% depending on the biologic) | None | Impaired IL-17 cellular responses |
| Secukinumab (humanized IgG1κ mAb) | ||||||
| IL-17RA | Brodalumab (humanized IgG2 mAb) | |||||
| IL-12p40 | Ustekinumab (humanized IgG1κ mAb) | |||||
| IL-23p19 | Risankizumab (humanized IgG1κ mAb) | |||||
| Guselkumab (humanized IgG1λ mAb) | ||||||
| Tildrakizumab (humanized IgG1κ mAb) | ||||||
| JAK1/2/3 | Ruxolitinib, tofacitinib, baricitinib, upadacitinib, fedratinib (kinase inhibitors) | MF, PV, GvHD, RA, IBD | Histoplasmosis, coccidioidomycosis, cryptococcosis, talaromycosis, PJP, aspergillosis | Moderate/high | Herpetic infections, CMV infection, mycobacterial infections | Impaired IFNγ and IFNλ responses, impaired lymphocyte and macrophage activation, lymphopenia |
| BTK | Ibrutinib, acalabrutinib, zanubrutinib (kinase inhibitors) | CLL, MZL, MCL, WM, GvHD | Aspergillosis (with CNS involvement), mucormycosis, cryptococcosis, blastomycosis, PJP | Moderate/high | Viral and bacterial infections | Impaired myeloid phagocyte activation and function |
| PI3K (p110δ) | Idelalisib (kinase inhibitor) | CLL, lymphomas | PJP | Moderate (~5–10%) | CMV infection, pneumonias | Impaired T cell activation |
An expanded version of Table 3 is presented in Supplementary Table 3. AS, ankylosing spondylitis; BTK, Bruton’s tyrosine kinase; CLL, chronic lymphocytic leukaemia; CMV, cytomegalovirus; CNS, central nervous system; GvHD, graft-versus-host disease; IBD, inflammatory bowel disease; IFN, interferon; mAb, monoclonal antibody; MCL, mantle-cell lymphoma; MF, myelofibrosis; MS, multiple sclerosis; MZL, marginal zone lymphoma; PJP, Pneumocystis jirovecii pneumonia; PV, polycythaemia vera; RA, rheumatoid arthritis; TB, tuberculosis; TNF, tumour necrosis factor; WM, Waldenström macroglobulinaemia.
Box 1 Candida auris.
Since 2009, when it was initially identified in Japan, Candida auris has rapidly spread across six continents, causing multiple outbreaks of skin colonization and subsequent life-threatening bloodstream infections in vulnerable patients; C. auris accounts for up to 30% of cases of candidaemia in certain health care settings4,185,186. C. auris is a major threat to public health owing to its unique propensity for long-term skin colonization, antimicrobial resistance and high person-to-person transmissibility within health care facilities, leading the US Centers for Disease Control and Prevention to designate it as an urgent threat in 2019 (ref. 187). The predilection of C. auris to colonize the skin, which distinguishes it from Candida albicans and other Candida species that reside within the gastrointestinal tract, has been verified both ex vivo in human and pig skin models and in vivo in mouse skin, where it establishes long-term residence within deep skin tissue and hair follicles188,189. C. auris has structurally unique mannoproteins compared with other Candida species, and its immune recognition by human circulating mononuclear cells and their induction of cytokine production is primarily mediated by complement receptor 3 and the mannose receptor, not dectin 1 (ref. 190). In a mouse model of skin colonization, IL-17A and IL-17F, but not IL-22, produced by innate and adaptive lymphocytes were crucial for curtailing C. auris skin colonization189, and chlorhexidine application ameliorated fungal burden189, which is in keeping with a similar finding in human patients185. In invasive candidiasis in mice, C. auris is less lethal than Candida albicans189–191, yet neutrophils have been shown to be poorly recruited to sites of C. auris infection and to ineffectively produce neutrophil extracellular traps and kill C. auris relative to C. albicans, which suggests that C. auris has specific immune-evading properties that are driven by its unique mannoprotein structure191. A better understanding of the fungal and host determinants of the unique skin tropism of C. auris may help to develop improved preventive and therapeutic strategies against this pathogen.
Mucocutaneous candidiasis
Candida species (primarily Candida albicans) are commensal yeasts of the gastrointestinal and reproductive tracts in most humans. When disruptions to the immune system and/or the resident microbiota occur, Candida can cause opportunistic infections at mucocutaneous or deep-seated anatomical sites, which have different requirements for immune defence8 (Table 1). Specifically, whereas innate and adaptive lymphocytes mediate protection against mucocutaneous candidiasis, myeloid phagocytes are crucial for protection against invasive candidiasis9. Indeed, patients with HIV/AIDS, having low CD4+ T cell counts, are at risk of developing mucocutaneous candidiasis, whereas patients with neutropenia are susceptible to invasive candidiasis, but not vice versa8.
Mucocutaneous candidiasis presents clinically as oropharyngeal candidiasis (OPC), oesophageal candidiasis or vulvovaginal candidiasis (VVC). A major breakthrough in the field of fungal immunology has been the discovery that IL-17 receptor (IL-17R) signalling is a crucial defence pathway against mucocutaneous candidiasis10 (Fig. 1a). Mice and humans with complete genetic deficiencies in IL-17RA, IL-17RC or their adaptor protein ACT1 (also known as TRAF3IP2) are highly susceptible to fully penetrant chronic mucocutaneous candidiasis (CMC)10–16. Several other CMC-manifesting inborn errors of immunity exhibit varying degrees of impaired IL-17 cellular responses and/or decreased frequencies of circulating T helper 17 (TH17) cells9,17 (Table 2 and Supplementary Table 2). Moreover, administration of IL-17 pathway-targeting biologics for the treatment of psoriasis has been associated with mild, treatment-responsive OPC, with a mean frequency of ~2–12%, depending on the biologic agent18,19 (Table 3). The relatively low frequency of OPC and the lack of CMC in these patients likely reflect that mucosal IL-17 responses are not completely inhibited by these biologics and are consistent with the notion that profound and sustained inhibition of the IL-17 pathway is required for the development of mucosal fungal disease18.
Fig. 1. Host defence against Candida at the oral mucosal interface.

a, Protective responses to Candida at mucosal surfaces are mediated by IL-17 and IL-22 produced by T helper 17 (TH17) cells, CD8+ T cells, type 3 innate lymphoid cells (ILC3s) and γδ T cells. TH17 cell differentiation depends on signal transducer and activator of transcription 3 (STAT3)-mediated retinoic acid receptor-related orphan receptor-γt (RORγt) induction downstream of signalling through IL-6 receptor (IL-6R) and IL-23R, which is defective in individuals with mutations in RORC (which encodes RORγt), STAT3 or ZNF341 (which regulates STAT3 expression and function), making them highly susceptible to chronic mucocutaneous candidiasis (CMC). TH17 cells produce IL-17A, IL-17F and IL-22. IL-17A, IL-17F and IL-17A–IL-17F bind to IL-17R, consisting of IL-17RA and IL-17RC, on suprabasal epithelial cells, which leads to the activation of ACT1 and induces the production of antimicrobial peptides (AMPs; such as β-defensin 3 and S100A8/S100A9) that restrict Candida growth. CMC also develops in patients with mutations in IL17F, IL17RA, IL17RC or TRAF3IP2 (which encodes ACT1), and in patients with thymoma or AIRE mutations, both of which are associated with autoantibodies to IL-17 and/or IL-22. Furthermore, mild oral thrush can develop in a subset of patients receiving IL-17 pathway-targeted biologics such as those inhibiting IL-17A, IL-17RA, IL-12p40 (not shown) or IL-23 (Table 3). IL-22 produced by TH17 cells, CD8+ T cells, ILC3s and γδ T cells binds to the IL-22 receptor consisting of IL-22RA1 and IL-10RB on basal epithelial cells, which activates STAT3 and facilitates epithelial cell proliferation and repair, thereby enabling the replenishment and responsiveness of the IL-17R-expressing suprabasal epithelial cell layer. b, Mucosal type II interferonopathy. In the setting of autoimmunity caused by autoimmune regulator (AIRE) deficiency, mucosal CD4+ TH1 cells and CD8+ T cells locally produce increased levels of interferon-γ (IFNγ), which binds to the IFNγ receptor consisting of IFNGR1 and IFNGR2 on epithelial cells, activates STAT1 and impairs oral epithelial barrier integrity. This leads to increased susceptibility to oropharyngeal candidiasis. JAK, Janus kinase; TYK2, tyrosine kinase 2.
At the cellular level, IL-17A and IL-17F are produced by αβ T cells, γδ T cells and type 3 innate lymphoid cells during OPC14,15 (Fig. 1a). The C. albicans toxin candidalysin, Langerin-expressing dendritic cells (DCs) and microbiota-derived retinoic acid prime natural TH17 cell responses initially, followed by Candida-specific TH17 cell generation that is facilitated by antigen presentation by CCR2+ monocyte-derived DCs10,20–22. By contrast, the C. albicans lipase Lip2 was recently shown to suppress IL-17 responses by γδ T cells indirectly through inhibition of IL-23 production by tissue-resident DCs23. Notably, mucosal CD8+ T cells, γδ T cells and type 3 innate lymphoid cells can compensate for IL-17 production when TH17 cells are absent, as Cd4−/− mice are not susceptible to OPC in primary or recall models and patients with loss-of-expression CD4 mutations or idiopathic CD4 lymphocytopenia who lack CD4+ T cells are resistant to CMC18,24–26. Thus, decreased levels of circulating TH17 cells may not in isolation be a reliable immunological biomarker for risk assessment of human CMC, and the direct evaluation of global mucosal IL-17 responses may be needed to determine the mechanisms of CMC.
Following IL-17RA–IL-17RC engagement on epithelial cells by IL-17A and/or IL-17F, the generation of antimicrobial peptides restricts local Candida growth and epithelial invasion10,13,20. Furthermore, the epithelial cell pattern recognition receptor EPHA2 recognizes fungal β-glucan, activates STAT3 and epidermal growth factor receptor 2 (EGFR2) signalling, and boosts mucosal IL-17 responses likely through priming the production of IL-1α/IL-1β during mucosal infection, which facilitates natural TH17 cell proliferation20,22,27. IL-22, another type 17 cytokine produced by lymphocytes, binds to IL-22RA1–IL-10RB on epithelial cells, activates STAT3 and promotes epithelial cell proliferation and repair, thereby replenishing IL-17R-expressing epithelial cells and enabling their responsiveness to IL-17A and IL-17F28 (Fig. 1a).
Whereas IL-17 is protective during OPC, in certain settings — such as the autoimmune syndrome of AIRE deficiency, which features an incompletely penetrant association between CMC and IL-17-neutralizing autoantibodies18,29,30 — excessive interferon-γ (IFNγ) production by mucosal αβ T cells can promote OPC susceptibility by driving epithelial barrier disruption, which is ameliorated by IFNγ inhibition or inhibition of JAK–STAT signalling downstream of the IFNγ receptor31 (Fig. 1b). This finding suggests that OPC susceptibility could be classified across a spectrum that encompasses impaired type 17 mucosal defences and/or immunopathology-promoting type 1 mucosal inflammation. Indeed, the recent discovery of CMC-manifesting inborn errors of immunity with intact or enhanced IL-17 responses in the setting of autoinflammation or autoimmunity (Supplementary Table 2) may uncover additional conditions in which OPC may be driven by mucosal immunopathology rather than by impaired host resistance. Notably, in patients with STAT1 gain-of-function mutations, who universally develop CMC associated with decreased circulating TH17 cells and increased IFNγ cellular responses, pharmacological JAK–STAT inhibition ameliorates CMC, pointing to a beneficial immunotherapeutic strategy in this setting32.
VVC occurs at least once in ~75% of healthy women during their reproductive years. An estimated 6–9% of these women have more than three recurrent episodes of VVC per year, a pervasive condition termed ‘recurrent VVC’. As opposed to OPC, VVC is characterized by maladaptive, fungus-driven, neutrophil-associated inflammation with excessive local production of alarmins and pro-inflammatory cytokines33. Correspondingly, genetic polymorphisms in NLRP3, SIGLEC15, IDO1 and other genes that augment the vaginal inflammatory milieu are associated with a greater risk of recurrent VVC33,34. Interestingly, whereas patients with HIV/AIDS and patients receiving IL-17 pathway-targeted biologics are at increased risk of developing OPC, they are not at risk of developing VVC, in agreement with the finding that IL-17R deficiency does not impair fungal control during mouse VVC35. By contrast, antibiotics predispose women to VVC, but not OPC, which indicates that the vaginal microbiota has a role in restricting local growth of Candida through poorly defined mechanisms that require further investigation36. Thus, the oral and vaginal mucosal immune responses to C. albicans are distinct. Importantly, VVC has been a target disease for protection with the C. albicans agglutinin-like sequence 3 (Als3)-based vaccine, which was shown to be immunogenic and efficacious in mouse models of VVC and in women with recurrent VVC37,38. The Als3-based vaccine is the only fungal vaccine that has thus far exhibited clinical protection in humans37 (Box 2).
C. albicans is effectively controlled at the cutaneous barrier by an IL-17-enforced neuroimmune axis and rarely causes clinical skin disease in humans39. Specifically, cutaneous C. albicans infection in mice promotes the activation of TRPV1+ sensory neurons expressing the neuropeptide calcitonin gene-related peptide, which is both necessary and sufficient to drive protective IL-17 responses39,40. These neuroimmune findings paved the way for the recent demonstration that gut fungi drive IL-17-dependent neuroimmune modulation of mouse behaviour41, further expanding our knowledge of the essential roles of neuroimmune circuits in tissue homeostasis and immunity (reviewed in ref. 42).
Box 2 Fungal vaccines.
Despite the substantial global burden, cost, morbidity and mortality associated with opportunistic fungal infections, no human fungal vaccine has been licensed to date. Major challenges in developing human fungal vaccines include concerns about their safety and efficacy in the immunocompromised patients who are typically at risk of serious fungal disease and difficulties in attracting pharmaceutical industry interest. Promising results have been obtained with various vaccine formulations (for example, killed or live attenuated fungi, crude fungal extracts, recombinant fungal subunits and nucleic acids encoding fungal antigens) in animal models of candidiasis, aspergillosis, cryptococcosis, coccidioidomycosis, histoplasmosis and blastomycosis (reviewed elsewhere192,193). Among these, only three have reached human clinical trials. The first-in-human fungal vaccine was a formalin-killed spherule vaccine for coccidioidomycosis, which showed no efficacy and caused topical and systemic adverse reactions in a phase III trial in the 1980s194. The other two are recombinant Candida vaccines. The first vaccine, termed ‘PEV7’, was based on recombinant C. albicans secreted aspartic protease 2 (Sap2) formulated with influenza virosomes. It protected rats from vulvovaginal candidiasis, and a phase I clinical trial found it to be safe and immunogenic, but no further clinical development has occurred since 2012 (ref. 195). The second vaccine, termed ‘NDV-3’, is based on recombinant amino terminus of C. albicans agglutinin-like sequence 3 (Als3) formulated with alum adjuvant. NDV-3 protected mice against mucosal and invasive candidiasis caused by several Candida species, including C. auris, cross-protected against Staphylococcus aureus owing to structural homology between Als3 and an S. aureus clumping factor, and stimulated T helper 1 (TH1) cell, TH17 cell and B cell responses38,196–198. In a phase I clinical trial, NDV-3 was safe and immunogenic, eliciting antigen-specific IgA and IgG and TH1 cell- and TH17 cell-associated cytokine responses199. In a recent double-blind, placebo-controlled phase Ib/IIa trial involving 188 women with recurrent vulvovaginal candidiasis, the vaccine was safe and immunogenic, and it decreased the frequency of vulvovaginal candidiasis in women older than 40 years37. A forthcoming goal is to test the efficacy of the vaccine in preventing candidaemia in high-risk patients in intensive care. Moving forward, harnessing fungal cell wall constituents as adjuvants and exploiting conserved fungal epitopes to induce cross-protection against multiple fungi are exciting strategies towards pan-fungal vaccine development192. For example, vaccine delivery of the conserved Blastomyces-derived chaperone calnexin in glucan particles protected mice against endemic dimorphic fungi, aspergillosis, chromoblastomycosis and Pseudogymnoascus destructans, which causes the life-threatening white-nose syndrome in bats146,200,201.
Invasive candidiasis
Invasive candidiasis is a leading cause of nosocomial bloodstream infection in higher-income countries, with mortality that can exceed 40% despite treatment8. It typically occurs in patients who are critically ill in whom Candida transitions from a commensal pathogen to an opportunistic pathogen by translocating into the bloodstream and seeding deep-seated organs such as liver, spleen and kidney8. Although C. albicans is most often responsible for invasive candidiasis, Candida glabrata, Candida tropicalis and Candida parapsilosis also cause disease with increasing frequency, and the multidrug-resistant Candida auris has spread rapidly worldwide in recent years8 (Box 1).
Certain iatrogenic factors are typically associated with the development of invasive candidiasis in humans8. Broad-spectrum antibiotics induce gut dysbiosis, impair lymphocyte-mediated IL-17A-dependent and granulocyte–macrophage colony-stimulating factor (GM-CSF)-dependent intestinal immune responses and inhibit the local generation of antimicrobial peptides, thereby enhancing Candida gut colonization and invasion43,44; this antibiotic-induced susceptibility was ameliorated by IL-17A or GM-CSF immunotherapy in a mouse model of invasive candidiasis44. Central venous catheters or chemotherapy-induced mucositis and surgery, which breach the integrity of the cutaneous barrier or the intestinal barrier, respectively, can allow Candida to translocate into the bloodstream8. Iatrogenic immunosuppression (for example, chemotherapy-induced neutropenia or use of corticosteroids), which impairs protective myeloid phagocyte-dependent antifungal responses, facilitates the invasion of deep-seated organs by Candida8. Rapid neutrophil influx and activation in Candida-infected tissues are crucial for effective host defence in mice, in agreement with the profound susceptibility of humans with neutropenia to invasive candidiasis8.
The CLRs dectin 1 (also known as CLEC7A), dectin 2 (also known as CLEC6A) and dectin 3 (also known as CLEC4D) recognize Candida β-glucans and α-mannans and signal through the kinase SYK and the CARD9–MALT1–BCL-10 signalling complex to activate NF-κB, collectively promoting TH17 cell development and pro-inflammatory cytokine production5,8. CARD9 deficiency is so far the only known inborn error of immunity that predisposes to both mucosal candidiasis and invasive candidiasis, with a unique tropism for the central nervous system (CNS)45 (Table 2). Mechanistically, in response to fungal candidalysin, CNS-resident microglia produce IL-1β in a CARD9-dependent manner, which mediates protective neutrophil recruitment to the CNS through CXCL1–CXCR2-dependent chemotaxis46. In line with this, pharmacological blockade of the CLR–SYK–CARD9 signalling axis with the SYK inhibitor fostamatinib has been associated with opportunistic fungal infections (Supplementary Table 3).
Invasive candidiasis also occurs in a small proportion of patients with severe congenital neutropenia syndromes or inborn errors of immunity that affect the phagocyte oxidative burst, such as complete myeloperoxidase deficiency or chronic granulomatous disease (caused by mutations in NADPH oxidase complex subunits)9 (Table 2 and Supplementary Table 2). Although invasive candidiasis is not a recognized phenotype of inherited deficiency of complement component C5 (ref. 9), acute pharmacological inhibition of C5a with eculizumab increases susceptibility to Candida infection, which suggests a crucial role of complement activation in mounting innate antifungal responses in vulnerable patients47 (Supplementary Table 3).
Within Candida-infected tissues, an intricate intercellular crosstalk orchestrates the candidacidal activity of neutrophils. Specifically, Ly6C+ inflammatory monocytes and monocyte-derived DCs produce IL-15 and IL-23, respectively, which in turn activate natural killer cells to release GM-CSF, which primes neutrophils to kill Candida48,49. Consistent with these results, a phase IV clinical trial showed that GM-CSF administration in allogeneic HSCT recipients decreased the incidence of invasive candidiasis and associated mortality50. Neutrophil reactive oxygen species (ROS)-dependent killing of Candida involves dectin 1-dependent metabolic rewiring of neutrophils and dectin 1-mediated activation of the calcineurin–NFAT and MAC1 (CD11B–CD18)–VAV–PKCδ pathways8,51. Uraemia, which is another known risk factor for human invasive candidiasis, impairs neutrophil ROS production and Candida killing by reducing GLUT1-dependent glucose uptake through hyperactivation of glycogen synthase kinase-3β (GSK3β)52. Pharmacological inhibition of GSK3β restores neutrophil candidacidal activity in mice and humans with uraemia, pointing to a potential therapeutic intervention in this setting52.
Although neutrophils are protective during invasive candidiasis, their excessive accumulation and activation may also have detrimental effects, as seen in patients with neutropenia upon neutrophil recovery. Corticosteroids ameliorate inflammation and clinical symptoms in this setting, but certain immunopathogenic factors have been delineated in mice and humans — for example, the chemokine receptor CCR1, the CLR DNGR1, the endoribonuclease MCPIP1, the secreted growth factor progranulin and IFNγ — that show promise for eventually developing more selective therapeutic approaches9,53–57. Moreover, immunopathology during renal candidiasis can be ameliorated through activation of the IL-17-dependent kallikrein–kinin system, which prevents renal tubular cell apoptosis and preserves kidney function58. Administration of bradykinin combined with antifungal therapy provided a synergistic survival benefit during mouse kidney infection with Candida58, raising the possibility that the antihypertensive class of angiotensin-converting enzyme inhibitors, which increase bradykinin levels, might have protective effects in this context.
In mouse invasive candidiasis, in addition to neutrophils, CCR2+ infiltrating monocytes and CX3CR1+ tissue-resident macrophages also contribute to protective immunity in the kidney and CNS by promoting Candida uptake and killing59,60. Importantly, beyond acute experimental candidiasis, monocytes and macrophages also protect against systemic C. albicans reinfection through a process termed ‘trained immunity’. Mechanistically, protection in this setting is conferred through activation of dectin 1, RAF1, AKT, mTOR and hypoxia-inducible factor 1α (HIF1α) signalling that drives epigenetic reprogramming and cellular metabolic rewiring of monocytes and macrophages, with a switch to aerobic glycolysis9,61. By contrast, activation of macrophage JNK1 increases mortality during invasive candidiasis by downregulating expression of the CLR CD23 (also known as FcεRII) and impairing macrophage production of nitric oxide62. Thus, whereas JNK1 deficiency predisposes to CMC in humans (Supplementary Table 2), it may protect against invasive candidiasis62,63. Interestingly, although patients with humoral defects are not considered at risk of bloodstream candidiasis, mouse gut-educated IgA+ meningeal plasma cells located near dural venous sinuses were recently shown to entrap C. albicans and restrict its spread in the CNS, uncovering a potential role for humoral immunity in protection against invasive candidiasis at this anatomical site64.
Population studies in patients who are critically ill have shown that genetic polymorphisms in genes including TLR1, CXCR1, CX3CR1, STAT1 and TAGAP, many of which have shown corroborating protective effects in mouse models of invasive candidiasis, are associated with greater risk of invasive candidiasis and/or worse outcome after infection. Notably, the combined presence of certain polymorphisms can increase susceptibility to invasive candidiasis by up to ~20-fold8,65. Collectively, these findings may provide a foundation for the eventual development of immunogenomic approaches for personalized risk stratification, prophylaxis and prognostication in high-risk patients.
Aspergillosis
Aspergillosis is an opportunistic, life-threatening fungal infection that develops following inhalation of ubiquitous Aspergillus moulds (primarily Aspergillus fumigatus) in humans with numeric or functional deficits in myeloid phagocytes, such as individuals with neutropenia, who receive corticosteroids or who are HSCT recipients, and, recently, in patients with severe influenza or COVID-19 pneumonia1,3,66 (Table 1). Humans continuously inhale Aspergillus conidia (2–3 μm in diameter in the case of A. fumigatus), and an important immunological checkpoint involves preventing the formation of tissue-invasive filamentous hyphae in the respiratory tree. Myeloid phagocytes are necessary and sufficient to maintain this checkpoint as Rag2−/−Il2rg−/− mice that lack innate and adaptive lymphocytes clear conidia without developing invasive disease67, similarly to humans with defects in lymphoid function or numbers9. Although lymphocytes seem to be redundant for protection against aspergillosis in hosts with intact myeloid cells, it is possible that adoptively transferred lymphocytes may provide benefit in hosts with an injured myeloid compartment68,69. Aspergillus also causes allergic disease and chronic cavitation in individuals with atopy and structural lung damage, respectively (reviewed in refs. 70,71).
A central theme of defence against inhaled Aspergillus is molecular and cellular redundancy in achieving sterilizing immunity and an essential role of intercellular crosstalk to regulate leukocyte infiltration and effector functions within the lung (Fig. 2). In immunocompetent individuals, conidia are engulfed by alveolar macrophages72 and conidial germination within phagosomes triggers a pro-inflammatory cascade through dectin 1-dependent recognition of β-1,3-glucan73, which is exposed on the surface of the germinating conidia74. Moreover, exposure of fungal mannans engages soluble pentraxin 2 (ref. 75) and pentraxin 3 (ref. 76) and the CLRs dectin 2 and dectin 3, sequentially activating SYK, CARD9–MALT1–BCL-10, NF-κB and the NLRP3 inflammasome, and thereby resulting in pro-inflammatory cytokine production9. CLR-dependent activation of phospholipase-γ induces Ca2+–calcineurin phosphatase activity and nuclear translocation of the transcription factor NFAT9. In addition to these immunostimulatory Aspergillus polysaccharides, conidial dihydroxynaphthalene melanin activates the CLR MelLec (also known as CLEC1A) and induces downstream signalling through an unknown pathway77. The Aspergillus thaumatin-like protein CalA binds to epithelial and endothelial cells through α5β1 integrin to promote its internalization and spread, and antibody-mediated blockade of CalA ameliorates mortality in Aspergillus-infected mice78.
Fig. 2. Cellular crosstalk regulates antifungal innate immunity in the lung.

a, Fungal recognition and epithelial cell–leukocyte crosstalk coordinate the recruitment of neutrophils and monocytes to the alveoli. Alveolar macrophages and lung-resident dendritic cells (DCs; not shown) engulf Aspergillus fumigatus conidia that reach the terminal airways, which are the site of gas exchange. Rapid release of IL-1α/β by macrophages and DCs stimulates the IL-1 receptor (IL-1R)–MYD88-dependent production of the neutrophil-recruiting chemokines CXCL1 and CXCL5 by lung epithelial cells. The release of CXCL2 by haematopoietic cells, including macrophages, through C-type lectin receptor (CLR)–CARD9 activation, and the rapid production of leukotriene B4 (LTB4) and complement component C5a also contribute to sustained neutrophil recruitment. In addition, CCR2+ circulating monocytes enter the lung parenchyma and differentiate into monocyte-derived DCs. Opsonization of A. fumigatus conidia by pentraxins enables their efficient uptake and killing by neutrophils. b, The recruited neutrophils and monocyte-derived DCs engulf conidia into phagolysosomes, which are the site of intracellular conidial killing. Both resident myeloid cells and infiltrating CCR2+ monocytes produce type I interferon early in the response and regulate ensuing type III interferon release. Fungus-engaged neutrophils and monocyte-derived DCs secrete CXCL9 and CXCL10 (through CLR–CARD9–SYK and type I interferon-dependent pathways, respectively), which recruit CXCR3+ plasmacytoid DCs (pDCs) to the lung. c, Innate crosstalk licenses the full spectrum of neutrophil antifungal activity. Conidial killing by neutrophils and monocyte-derived DCs is enhanced by the entry of CXCR3+ pDCs and by type I and type III interferons that converge on signal transducer and activator of transcription 1 (STAT1) signalling in neutrophils. Fungus-induced release of granulocyte–macrophage colony-stimulating factor (GM-CSF) also potentiates neutrophil antifungal properties by enhancing NADPH oxidase activity and the production of reactive oxygen species (ROS). In the context of blastomycosis, CCR6+ innate lymphocytes and natural T helper 17 cells contribute to GM-CSF production during acute pulmonary infection.
Among the inborn errors of immunity, aspergillosis is a signature infection of chronic granulomatous disease9, which highlights the importance of ROS generation in maintaining sterilizing lung immunity (Table 2). Aspergillus hyphae induce neutrophil production of ROS through β2 integrin and SYK79, which triggers a regulated cell death process in Aspergillus that is opposed by the fungal anti-cell-death gene Afbir1 (ref. 80). On the basis of the fact that IFNγ stimulates phagocyte ROS production, recombinant IFNγ has been approved for use as an immunotherapy to prevent invasive (including fungal) infections in patients with chronic granulomatous disease81. In addition to licensing neutrophil fungicidal activity, ROS also regulate LC3-associated phagocytosis, an adjunctive mechanism of fungal clearance82. Aspergillus conidial melanin hinders dectin 1-dependent and SYK-dependent activation of LC3-associated phagocytosis in macrophages, and phagosomal removal of melanin enhances macrophage antifungal activity by increasing calcium flux through glycolysis83.
In pulmonary aspergillosis in mice, infiltrating neutrophils, Ly6C+ inflammatory monocytes and CXCR3+ plasmacytoid DCs are essential cellular effectors67,84 (Fig. 2). Rapid neutrophil influx depends on sequential IL-1R–MYD88 signalling-dependent release of CXCL1 and CXCL5 by the lung epithelium, including club cells85,86. Thereafter, CLR–CARD9-dependent signalling in haematopoietic cells, including macrophages, sustains pulmonary neutrophil influx through CXCL2 production, which is consistent with a model of sequential and compartment-specific regulation of pulmonary neutrophil recruitment in this context. In CARD9-deficient humans, aspergillosis is typically extrapulmonary, which supports the idea that CARD9-dependent and CARD9-independent pathways of neutrophil recruitment collaborate in the lung, but that CARD9 predominantly controls neutrophil recruitment to extrapulmonary sites of Aspergillus infection87. In addition to IL-1R–MYD88-dependent and CLR–CARD9-dependent leukocyte recruitment to the Aspergillus-infected lung, signalling through the RNA sensor MDA5–MAVS88 and leukotriene B4 biosynthesis89 also contribute. Within the Aspergillus-infected lung, the lectin family member galectin 3 increases neutrophil motility and facilitates neutrophil extravasation into bronchoalveolar air spaces90.
In addition to neutrophils, mononuclear phagocytes also promote pulmonary defences against Aspergillus both directly and indirectly by priming neutrophil effector functions. CCR2+ lung-infiltrating monocytes differentiate into monocyte-derived DCs that engulf, inactivate and transport conidia to lung-draining lymph nodes67,91. CCR2+ monocyte-derived DCs also produce type I interferons, which in turn control type III interferon production within the fungus-infected lung. Type I and type III interferon receptor engagement and STAT1 signalling are essential for host defence by enhancing neutrophil ROS production and fungicidal activity, and JAK–STAT inhibitors have been reported to promote aspergillosis in humans92 (Table 3). In addition, STAT1 signalling in CCR2+ monocytes facilitates the formation of monocyte-derived DCs and enhances their fungicidal activity; this process is supported by reciprocal interferon–STAT1 crosstalk with neutrophils93. Fungus-induced release of GM-CSF also stimulates neutrophil ROS production and promotes fungal clearance94. Concordantly, humans with mutations in GM-CSF receptor signalling develop pulmonary alveolar proteinosis, a condition that is associated with opportunistic pulmonary infections, including aspergillosis95 (Supplementary Table 2).
Upon conidial engulfment, lung-infiltrating neutrophils and monocytes produce CXCR3-targeted chemokines (CXCL9 and CXCL10) to recruit circulating CXCR3+ plasmacytoid DCs into the lung, where they boost neutrophil ROS production, neutrophil extracellular trap formation and fungicidal activity84. Thus, both epithelial cell-derived chemokines and haematopoietic cell-derived chemokines coordinate early neutrophil influx to the Aspergillus-infected lung, and tissue-infiltrating mononuclear phagocytes license neutrophil functions to achieve sterilizing immunity. Nutrient limitation via metal chelators (for example, lactoferrin or calprotectin) also contributes to tissue-specific control of Aspergillus, with a role shown for calprotectin in protecting against Aspergillus keratitis but not pulmonary infection in mice96,97. Corroborating these mechanistic studies of protective immunity in mice, human population studies have identified genetic polymorphisms in APCS (encoding pentraxin 2), PTX3, CLEC7A (encoding dectin 1), CLEC1A and CXCL10 that predispose to aspergillosis following allogeneic HSCT75,77,98,99.
Moreover, pharmacological susceptibility to aspergillosis has provided additional insights into host defence pathways (Table 3 and Supplementary Table 3). Chief among these is the observation that ibrutinib-induced inhibition of Bruton’s tyrosine kinase (BTK) promotes aspergillosis in patients with lymphoma100,101, which is recapitulated in Btk−/− mice, consistent with BTK having an evolutionarily conserved role in defence against Aspergillus100. In macrophages, phagocytosis of Aspergillus conidia triggers dectin 1-dependent and Toll-like receptor 9 (TLR9)-dependent activation of BTK, which regulates calcineurin–NFAT activity and synergizes with NF-κB to promote the production of tumour necrosis factor (TNF)102. The mechanisms by which BTK mediates neutrophil anti-Aspergillus defence remain poorly defined and require further study.
Mucormycosis
Mucormycosis is a life-threatening infection caused by ubiquitous inhaled moulds of the order Mucorales (primarily Rhizopus species)103. As is the case for aspergillosis, mucormycosis typically occurs in patients with quantitative and/or qualitative phagocyte defects, such as individuals with neutropenia, who receive corticosteroids or who are HSCT recipients103. However, Mucorales members are unique among the human mould pathogens in terms of their predilection to cause disease in the settings of diabetic ketoacidosis or iron overload103 (Supplementary Table 1). Moreover, mucormycosis has emerged as a devastating complication of COVID-19, particularly in India (where it had a prevalence of 0.27% among hospitalized patients with COVID-19), associated with corticosteroid use and uncontrolled hyperglycaemia1. Strikingly, mucormycosis has distinct infection patterns in patients with different risk factors. Specifically, diabetic ketoacidosis typically predisposes to the rhino-orbital–cerebral form of the disease, whereas patients with neutropenia or who are HSCT recipients typically develop sinopulmonary mucormycosis103.
During infection in mice, Mucorales members produce mucoricin, a plant ricin-like toxin that promotes cellular necrosis, vascular permeability and death104. Recent work has uncovered tissue-specific mechanisms by which hyperglycaemia and iron overload increase susceptibility to mucormycosis. Specifically, increased levels of glucose, iron and ketone bodies, all of which occur during diabetic ketoacidosis, attenuate neutrophil-mediated damage to Rhizopus and induce expression of the glucose-regulated receptor GRP78 on the surface of endothelial cells and nasal epithelial cells and of the Mucorales invasin CotH3 (refs. 105–108). The CotH3–GRP78 interaction enables fungal invasion and damage of the nasal epithelium, and can be targeted with therapeutic benefit in Rhizopus-infected mice with diabetic ketoacidosis105–107. Because Aspergillus does not encode CotH3, this functional attribute of Mucorales may underlie the selective predisposition of patients with diabetes to mucormycosis. By contrast, in the setting of pulmonary mucormycosis, Rhizopus CotH7 interacts with β1 integrin, not GRP78, on the surface of alveolar epithelial cells to promote fungal invasion, EGFR activation and cellular damage108,109. Pharmacological inhibition of β1 integrin or EGFR with the FDA-approved inhibitors cetuximab or gefitinib blocks fungal invasion of alveolar epithelial cells and prolongs mouse survival during pulmonary mucormycosis108,109. Thus, Rhizopus interacts with different tissue-specific mammalian receptors depending on the underlying predisposing factor to instigate opportunistic disease. Furthermore, during pulmonary mucormycosis in mice, alveolar macrophages contribute to fungal clearance and host survival by stimulating an intracellular iron restriction programme that curbs Rhizopus growth within phagosomes, underscoring the importance of protective nutritional immunity in this setting110.
Chromoblastomycosis and phaeohyphomycosis
Chromoblastomycosis is a disfiguring, tumour-like, progressive and often treatment-unresponsive infection in tropical areas, which has been designated as a neglected tropical disease by the World Health Organization. It is primarily caused by the pigmented fungus Fonsecaea pedrosoi, which typically enters the skin via traumatic inoculation (Supplementary Table 1). In F. pedrosoi-infected mice, CD4+ T cells are essential for fungal clearance, primarily involving TH1 cell and TH17 cell responses111,112. Unlike Candida and Aspergillus species, F. pedrosoi stimulates modest CLR responses through the dectin 2-dependent and Mincle-dependent SYK–CARD9 axis and fails to effectively activate TLRs, resulting in inadequate local inflammatory responses in infected mice and humans111,113. Topical administration of the TLR7 agonist imiquimod combined with antifungal therapy markedly improved clinical outcomes in patients with refractory chromoblastomycosis114, demonstrating a key adjunctive role for this immunotherapeutic intervention.
Phaeohyphomycosis is another mycosis caused by melanin-bearing, pigmented fungi that typically enter the skin via traumatic inoculation. Although infection is self-limited in most healthy individuals, it can cause severe subcutaneous or disseminated disease in patients who are HSCT recipients or who have CARD9 deficiency7,115 (Supplementary Table 1). Exserohilum rostratum, one of the agents of phaeohyphomycosis, caused a meningitis outbreak following epidural injections of contaminated methylprednisolone in the USA in 2012 (ref. 116). CARD9-dependent defence against phaeohyphomycosis in mice was recently shown to rely on macrophages, whereas neutrophil recruitment to the infected skin was CARD9 independent117. Specifically, CARD9 primes macrophages to produce IL-1β and TNF, which promote macrophage killing of fungi in an interdependent manner117. Consistent with this finding, phaeohyphomycosis has been reported following treatment with TNF-targeted biologics such as infliximab118 (Table 3). Upstream of CARD9, dectin 1 is also crucial for macrophage-mediated defence against phaeohyphomycosis in mice, and deleterious mutations in the gene encoding dectin 1 (CLEC7A) that impair fungal sensing and TNF production were found in ~70% of putatively immunocompetent patients with severe forms of phaeohyphomycosis117. These findings suggest that human dectin 1 deficiency may contribute to susceptibility to severe phaeohyphomycosis in the setting of traumatic inoculation.
Cryptococcosis
Cryptococcosis is caused by the encapsulated yeasts Cryptococcus neoformans and Cryptococcus gattii, which have distinct infection patterns in humans119. C. neoformans is more common, and typically causes meningoencephalitis in immunocompromised patients with CD4+ T cell depletion and/or dysfunction (Table 1). Cryptococcal meningoencephalitis is an AIDS-defining illness and remains a significant cause of death in resource-limited areas (for example, some parts of Africa) that are associated with limited availability of antiretroviral therapy and first-line antifungal drugs. The incidence of cryptococcal meningoencephalitis is increasing in non-HIV-infected individuals in the Western hemisphere. By contrast, C. gattii primarily causes pulmonary disease in putatively immunocompetent individuals and rarely affects individuals with HIV/AIDS.
Human exposure to inhaled Cryptococcus is widespread by early childhood. The crucial immunological checkpoint for preventing local proliferation of yeast in the lung and extrapulmonary dissemination is the containment and killing of inhaled Cryptococcus by lung-resident macrophages, which is enabled by their crosstalk with IFNγ-producing CD4+ T cells119 (Fig. 3). Notably, Cryptococcus yeasts secrete a capsule containing glucuronoxylomannan and galactoxylomannan that shields its pathogen-associated molecular patterns from immune recognition119,120. Unsurprisingly, therefore, defective TLR-mediated and CLR-mediated responses in the setting of human MYD88 deficiency and CARD9 deficiency, respectively, do not predispose to cryptococcosis9. By contrast, a few inborn errors of immunity primarily affecting numbers of lymphocytes and/or monocytes and macrophages, and/or their activation, underlie human cryptococcosis (Table 2 and Supplementary Table 2), which supports the crucial role of effective crosstalk between lymphocytes and mononuclear phagocytes in cryptococcal clearance9,119.
Fig. 3. Crosstalk between macrophages and T cells during infection with intracellular fungi.

Type 1 immune responses, characterized by interferon-γ (IFNγ)-producing T cells and macrophages, are crucial for protection against intramacrophagic fungi (such as Cryptococcus and Histoplasma). Infected macrophages produce IL-12, which drives T helper 1 (TH1) cell polarization of CD4+ T cells through Janus kinase 2 (JAK2)–signal transducer and activator of transcription 4 (STAT4) signalling. TH1 cells produce IFNγ, which binds to the IFNγ receptor, comprising IFNGR1 and IFNGR2, on macrophages and activates JAK1/JAK2–STAT1-dependent signalling to induce expression of inducible nitric oxide synthase (iNOS). In turn, iNOS mediates fungal killing together with the production of reactive oxygen species (ROS), which is partly controlled by sphingosine 1-phosphate receptor 3 (S1PR3) in the setting of cryptococcal infection. Tumour necrosis factor (TNF) activates macrophages for fungal killing and promotes the formation of granulomas and IFNγ production by lymphocytes. Granulocyte–macrophage colony-stimulating factor (GM-CSF) also primes macrophages to effectively kill fungi intracellularly. The transcription factor GATA2 regulates fungal uptake by macrophages184. Disruption of these protective antifungal immune pathways by biologics, inborn errors of immunity or anti-cytokine autoantibodies enhances susceptibility to infections with intracellular fungi in humans.
Loss-of-function mutations in IL12RB1 are associated with cryptococcosis, which is explained by impaired IL-12-driven IFNγ production by T cells and the resulting inability of macrophages to promote intracellular killing of fungi9 (Fig. 3). Consistent with this notion, cryptococcosis develops in adult patients with neutralizing autoantibodies to IFNγ121, which may be amenable to anti-CD20 or anti-CD38 targeted biologics that deplete B cells and plasma cells, respectively122,123. The higher prevalence of autoantibodies to IFNγ in individuals of Asian ancestry who were born in Asia but not elsewhere suggests that both environmental and genetic factors are involved121. Moreover, impaired T cell responses promote cryptococcosis in the setting of CD40L deficiency124. GATA2 deficiency predisposes to infection with various intramacrophagic pathogens, including mycobacteria and Cryptococcus, associated with profound monocytopenia125. GM-CSF is a key factor that promotes anticryptococcal immunity by activating macrophages to a functional state that is associated with efficient fungal killing126. Inherited deficiency of GM-CSF receptor signalling or neutralizing autoantibodies to GM-CSF can underlie cryptococcosis, with an enrichment for C. gattii infections in particular121,127. A genome-wide association study in individuals with HIV/AIDS of African descent uncovered a correlation between CSF1 genetic variation and cryptococcosis128. CSF1 encodes macrophage colony-stimulating factor (M-CSF), another growth factor that is crucial for macrophage activation and survival, particularly for CNS-resident microglia129.
Pharmacological susceptibility to cryptococcosis has provided additional evidence for the importance of crosstalk between TH1 cells and macrophages (Table 3 and Supplementary Table 3). For example, JAK–STAT inhibitors (which block IFNγ receptor signalling), TNF inhibitors (which impair IFNγ production and granuloma formation) and BTK inhibitors (which impair macrophage activation) all increase the risk of cryptococcosis9. Alemtuzumab (which targets CD52 on lymphocytes) and natalizumab (which targets α4β1 integrin on lymphocytes) promote cryptococcosis through profound and prolonged T cell depletion and through impaired integrin-mediated T cell trafficking to Cryptococcus-infected tissues, respectively9. The sphingosine 1-phosphate receptor (S1PR) inhibitor fingolimod was recently reported to promote cryptococcosis. In a mouse model of cryptococcosis, fingolimod enhanced susceptibility by impairing S1PR3-dependent macrophage phagocytosis and granuloma formation and by driving a macrophage phenotype with poor oxidative killing capacity, not by affecting S1PR1-mediated T cell trafficking130.
Defective IFNγ signalling in macrophages impairs the activity of inducible nitric oxide synthase (iNOS) and hence macrophage oxidative killing pathways, which enables Cryptococcus to survive within phagosomes, where it can be shielded from immune recognition, as has been shown in infected brain tissue of patients with HIV/AIDS131. Intracellular growth in macrophages is promoted by fungal-derived urease and modification of the phagosome microenvironment132,133. In patients with HIV/AIDS, a lack of CD4+ T cells and paucity of pro-inflammatory cytokines (IFNγ and TNF) in the cerebrospinal fluid were the strongest predictors of cryptococcosis severity in the CNS, and T cell responses producing IFNγ and TNF were associated with increased survival134–136. Consistent with the idea that IFNγ may boost macrophage antifungal activity in patients with HIV/AIDS in the absence of T cell help, a clinical trial showed that adjunctive IFNγ immunotherapy rapidly reduced fungal burden in the cerebrospinal fluid, the level of which is a known surrogate of patient survival137.
Although they are crucial for fungal control, TH1 cells may also promote pathogenic effects in the settings of non-HIV cryptococcosis and HIV-associated immune reconstitution inflammatory syndrome (IRIS), a condition characterized by the development of life-threatening immunopathology upon initiation of antiretroviral therapy. In these patients, corticosteroids ameliorate inflammatory sequelae and neurological symptoms138. In patients without HIV infection with cryptococcosis, significant infiltration of CXCR3+ TH1 cells into the CNS is observed, associated with high levels of CXCL10 in cerebrospinal fluid, poor colocalization of macrophages and fungus, and neuronal damage139. Correspondingly, in a mouse model of non-HIV cryptococcosis, CXCR3 inhibition abrogated the recruitment of TH1 cells into the CNS by glial-derived CXCL10 and increased survival140. Furthermore, patients with AIDS without IRIS have increased numbers of TH1 cells and increased serum levels of IFNγ relative to patients without IRIS141. In a mouse model of cryptococcus-associated IRIS, IFNγ-producing CD4+ T cells were sufficient to mediate CNS damage associated with upregulation of aquaporin 4, which modulates water influx and swelling within the brain142. Better understanding of the molecular underpinnings of pathogenic inflammation during cryptococcus-associated IRIS may help to identify targeted, corticosteroid-sparing immunomodulatory strategies for affected patients.
Other endemic fungal infections
Histoplasma, Coccidioides, Paracoccidioides, Talaromyces and Blastomyces species are geographically restricted dimorphic fungi that typically cause self-limited pneumonia in healthy individuals but that can lead to progressive pulmonary and/or extrapulmonary disease in immunocompromised patients with HIV/AIDS, corticosteroid use or transplantation (Table 1 and Supplementary Table 1). Race, ethnicity and hormonal factors can influence the risk and severity of these infections, through mechanisms that remain poorly understood. For example, African American, Native American and Asian ancestries are associated with increased risk of blastomycosis and disseminated coccidioidomycosis, pregnancy increases the risk of severe coccidioidomycosis, and chronic paracoccidioidomycosis more commonly affects males9.
As for cryptococcosis, defence against histoplasmosis, coccidioidomycosis, paracoccidioidomycosis, talaromycosis and blastomycosis depends on the coordinated crosstalk between IFNγ-producing T cells and fungicidal macrophages (Fig. 3). In line with this crucial role of granulomatous inflammation, inborn errors of immunity affecting type 1 immune responses caused by mutations in the genes encoding IL-12RB1, IFNγR1, STAT1, STAT3, CD40L or GATA2 (Table 2 and Supplementary Table 2) predispose to severe, disseminated infections by these fungi, as do neutralizing autoantibodies to IFNγ121 and receipt of TNF inhibitors, JAK–STAT inhibitors or the IFNγ-targeted biologic emapalumab9 (Table 3 and Supplementary Table 3). In Coccidioides-infected patients, genetic variants in β-glucan sensing and response genes (for example, CLEC7A and PLCG2) that impair TNF production were recently shown to underlie disseminated disease in ~50% of evaluated patients143. By contrast, type 2 immune responses are detrimental for pathogen control by dampening type 1 responses and skewing macrophages towards a phenotype that is permissive for intracellular fungal persistence144. Macrophage-mediated nutritional immunity to Histoplasma is also mediated by TH1 cell responses, particularly GM-CSF, which promotes metallothionein-mediated zinc sequestration that starves the pathogen. By contrast, the TH2 cell cytokine IL-4 promotes zinc acquisition by yeast, which enables its intracellular survival145. The Blastomyces adhesin BAD1 inhibits CD4+ T cell-derived IFNγ release, and Blastomyces uses additional immune-evasion strategies, including cleavage and inactivation of GM-CSF and of monocyte-recruiting CC chemokines146.
Immunomodulatory restoration of the TH1 cell–TH2 cell balance during endemic mycoses can be exploited with therapeutic benefit. Specifically, a child with no known inborn error of immunity and refractory disseminated coccidioidomycosis who had increased levels of IL-4 and decreased levels of IFNγ achieved rapid symptom resolution and infection remission after adjunctive administration of recombinant IFNγ and the IL-4/IL-13 receptor inhibitor dupilumab147.
Pneumocystis jirovecii pneumonia
Pneumocystis jirovecii pneumonia (PJP) is an AIDS-defining illness that develops with declining CD4+ T cell counts and carries a high global disease burden and mortality. In HIV-negative individuals, PJP is most often seen following corticosteroid administration (Table 1). Epidemiological data indicate that inhalational exposure to P. jirovecii is universal during infancy without causing severe clinical disease, which is indicative of potent, protective host responses to Pneumocystis148. In mice infected with Pneumocystis murina, the mouse-specific Pneumocystis species, CD4+ T cells are crucial for defence as CD4-deficient animals are highly susceptible, which is consistent with the increased risk of PJP in individuals with HIV/AIDS or patients treated with the T cell-depleting biologic alemtuzumab (Table 3). Although TH1 cells, TH2 cells and TH17 cells are all induced during mouse infection, none of them has been shown individually to promote disease resolution using corresponding gene-deficient mouse strains149. Recently, CD4+ T cell-intrinsic signalling through IL-21R and STAT3 was found to be crucial for defence in Pneumocystis-infected mice, by priming T cell effector functions and protective class-switched antibody responses150. These findings recapitulate the susceptibility to PJP that is seen in patients with STAT3 and IL21R mutations (Table 2 and Supplementary Table 2). In addition to T cells, B cells are also crucial for anti-Pneumocystis defence through antigen presentation to and priming of CD4+ T cell responses in the lung, by facilitating opsonic phagocytosis by alveolar macrophages, and through secreted IgM151–153. In agreement with this role for B cells, recipients of CD20-targeted B cell-depleting antibodies are at risk of developing PJP (Supplementary Table 3). Among myeloid phagocytes, alveolar macrophages and eosinophils, but not neutrophils, contribute to Pneumocystis clearance during mouse infection154,155. Other PJP-manifesting inborn errors of immunity that involve impaired T cell and/or B cell responses include severe combined immunodeficiency, anhidrotic ectodermal dysplasia with immune deficiency due to IKBA deficiency, and mutations in CD40L, IKZF1 or IKZF3 (ref. 149) (Supplementary Table 2). Biologics that predispose humans to PJP include TNF, PI3K and JAK–STAT inhibitors, which impair the responses of T cells and macrophages to Pneumocystis149 (Table 3).
Endogenous fungi: protection and pathology
Fungi are core constituents of endogenous microbial communities at several human barrier sites (Box 3). Recent studies have advanced the concept that tissue-resident fungi shape immune homeostasis and development, function as reservoirs for invasive fungal infections and contribute to the pathogenesis of autoimmune, inflammatory and neoplastic diseases (Fig. 4). Here we focus primarily on human studies of the mycobiota, with mouse work having been reviewed elsewhere156,157.
Fig. 4. Endogenous fungal communities and their relationship to invasive fungal disease.
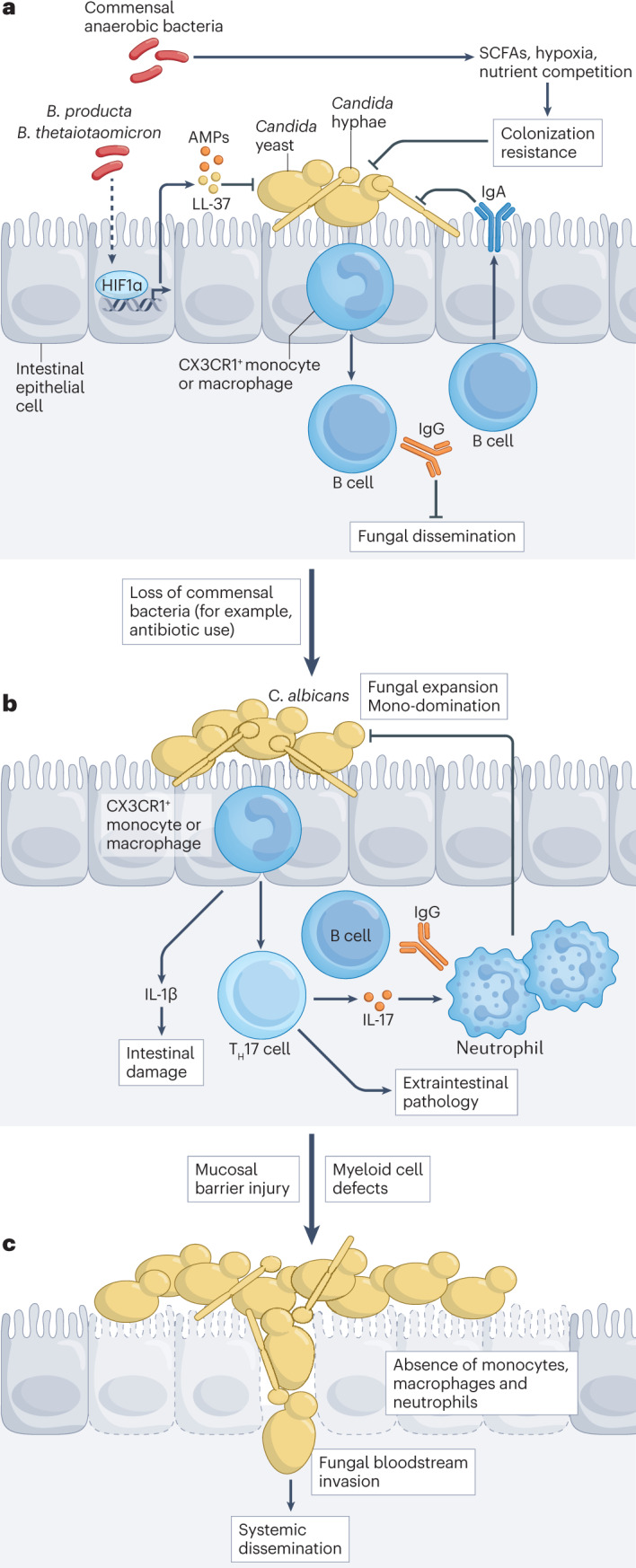
a, Mucosal and intestinal bacterial communities outnumber and compete with site-specific endogenous fungi. In the intestine, commensal anaerobic bacteria, exemplified by Blautia producta and Bacteroides thetaiotaomicron, provide colonization resistance against Candida species by activating the transcription factor hypoxia-inducible factor 1α (HIF1α) in intestinal epithelial cells to regulate the release of LL-37, an antimicrobial peptide (AMP) that restricts Candida albicans growth. Bacterial products of fermentation, specifically short-chain fatty acids (SCFAs), and metabolites likely also contribute to fungal colonization resistance and inhibition of Candida filamentation. Nutrient competition and regulation of tissue oxygen levels contribute to the balance between endogenous bacteria and competing fungal species in the gastrointestinal and reproductive tracts. CX3CR1+ mononuclear phagocytes recognize intestinal fungi through C-type lectin receptor (CLR)–CARD9 signalling and regulate the production of IgA and IgG antibodies to Candida by B cells. IgA antibodies recognize and target C. albicans adhesins (such as agglutinin-like sequence 3 (Als3)) that are primarily expressed by pseudohyphae and thus promote maintenance of the commensal yeast cell morphology. Candida-targeted IgG antibodies can protect against bloodstream invasion and disseminated candidiasis. b, In the intestinal and reproductive tracts, antibiotic-induced loss of predominantly anaerobic bacterial taxa reduces Candida colonization resistance and facilitates domination by individual fungal taxa. C. albicans strains from patients with inflammatory bowel disease with fungal dysbiosis can aggravate tissue damage, correlating with fungal strain-specific induction of IL-1β production by immune cells. The induction of T helper 17 (TH17) cells and IL-17 production can promote neutrophil recruitment to inhibit fungal tissue invasion and dissemination. However, Candida-specific TH17 cells can also promote extraintestinal inflammation. For example, mould aeroallergens can stimulate C. albicans-primed TH17 cells and aggravate fungus-associated asthma. c, Systemic bloodstream infections can arise from intestinal fungal dysbiosis with concomitant epithelial cell damage (for example, following chemotherapy) and with defects in the number or function of circulating myeloid phagocytes.
In early human life, fungal–bacterial interactions shape the orderly assembly of the microbiota158. For example, ecological modelling in preterm neonates in an intensive care unit predicted that high abundance of intestinal Candida inhibited the expansion of Enterobacteriaceae, which facilitate the ensuing transition to commensal bacterial anaerobes159. This prediction was validated in interkingdom co-culture experiments with infant-derived Candida strains and in sequential intestinal colonization studies in mice, which showed that gut-resident Candida decreased the load of sequentially introduced Enterobacteriaceae. A recent review provides an excellent summary of numerous studies on the functional interactions between commensal, probiotic and potentially pathogenic bacteria and fungi160.
In addition to the role of fungi in microbiota assembly, the density of intestinal fungi and, in particular, the abundance of Pichia kudriavzevii (the sexual state of Candida krusei) have been linked to subsequent atopy in independent birth cohorts161. Transient P. kudriavzevii colonization exacerbated house dust mite-induced allergic airway disease in neonatal specific pathogen-free mice161. In human adults, circulating fungus-reactive memory TH17 cells preferentially target C. albicans-derived antigens, consistent with the idea that intestinal colonization with C. albicans induces TH17 cell differentiation162. Circulating C. albicans-induced TH17 cells cross-react with other fungi and can exacerbate the pathology of A. fumigatus-triggered allergic lung disease.
Malassezia, the major fungal commensal of human skin163, can cause pityriasis versicolor, characterized by hyperpigmented or hypopigmented flaky skin lesions. In a mouse epicutaneous infection model, Malassezia induced TH17 cell differentiation and elicited IL-23-dependent IL-17 release by γδ T cells and type 3 innate lymphoid cells164. These sources of IL-17 restrict fungal growth in intact skin but can exacerbate Malassezia-driven inflammation in injured skin. Local IL-17 recall responses to Malassezia and Candida also aggravated psoriasiform skin inflammation in mice, which recapitulates transcriptional features of human psoriatic skin lesions165.
In the gut, the humoral immune response can shape mutualism between intestinal Candida and its human host by preferentially targeting adhesins expressed by C. albicans hyphae but not the yeast form. This IgA-dependent selection favours the growth of yeast rather than hyphal morphotypes in the intestine and enhances the competitive fitness of yeast in antibiotic-treated mice166,167. Furthermore, the human repertoire of antibodies to C. albicans includes IgG antibodies, which depend on CARD9+ intestinal macrophages to promote their production and are protective in mice infected systemically with C. albicans and C. auris168.
Commensal anaerobic bacteria can regulate intestinal fungal density, partly by inducing the production of antimicrobial peptides, by producing short-chain fatty acids through fermenting dietary fibre and by maintaining a low-oxygen environment. Bacteroides and Blautia bacteria activate intestinal epithelial cell signalling through HIF1α, which regulates enterocyte release of the antimicrobial peptide LL-37 and C. albicans colonization resistance43 (Fig. 4a). Antibiotics that promote the loss of anaerobic bacterial taxa drive intestinal fungal expansion. For example, antibiotic-induced injury to the bacterial microbiota facilitates intestinal expansion and domination by pathogenic Candida (Fig. 4b), which in the context of chemotherapy-induced mucosal injury can translocate across the intestinal barrier to cause life-threatening bloodstream infections in HSCT recipients169 (Fig. 4c). Intestinal fungal dysbiosis during the peri-HSCT period is also linked to reduced long-term survival and increased non-relapse mortality170,171.
In human cohorts, fungal gut dysbiosis characterized by Malassezia, Debaromyces or Candida expansion is associated with inflammatory bowel disease (IBD) phenotypes. In mice, during intestinal wound healing and dextran sodium sulfate (DSS)-induced colitis, the introduction of representative clinical strains from individual fungal genera (Malassezia, Debaromyces or Candida) caused exuberant tissue pathology, whereas laboratory-passaged strains did not172–175. Genetic variation in CLEC7A (Y238*) and CARD9 (S12N) is associated with increased risk and severity of IBD157,176,177. Responsiveness to faecal microbiota transplantation in patients with IBD is associated with high levels of Candida before faecal microbiota transplantation, and disease amelioration is associated with low levels of Candida after faecal microbiota transplantation, which is consistent with the idea that faecal microbiota transplantation may reduce the contribution of fungus-associated inflammation to IBD severity178.
A recent study explored how fungal strain-specific attributes affect functional interactions with host cells to affect IBD severity. A collection of C. albicans strains cultured from colonic lavages of patients with IBD and non-disease controls was classified on the basis of filamentation properties and ability to induce IL-1β production by macrophages as a measure of strain-dependent aggravation of intestinal inflammation179 (Fig. 4b). High damage-inducing strains induced IL-1β-dependent tissue damage and instructed TH17 cell differentiation through recognition of candidalysin. These strain attributes were linked to IBD severity in humans and were recapitulated in a mouse DSS-induced colitis model, in which IL-1 blockade ameliorated inflammation.
In the pancreas, tissue-resident fungi (particularly Malassezia) are markedly enriched in pancreatic ductal adenocarcinoma compared with non-cancerous tissue180. In a mouse model of pancreatic ductal adenocarcinoma, antifungal drugs ameliorated tumour progression and the reintroduction of Malassezia accelerated tumour growth. Mechanistically, mannose-binding lectin bound to Malassezia glycans and activated the C3 complement cascade, which enhanced the growth of tumour cells expressing C3a receptor180. Depletion of commensal fungi was recently shown to enhance responsiveness to radiotherapy in a mouse model of breast cancer, and intratumoural dectin 1 expression negatively correlated with survival of patients with breast cancer181. The recent comprehensive characterization of the composition of the tumour-associated mycobiota across multiple human cancers has revealed the low-level, yet ubiquitous, presence of fungal DNA and cells in a tumour type-specific manner, often in close spatial association with macrophages and malignant cells182,183. Clinically, certain plasma or tumour-associated mycobiota subtypes were associated with differential response to immunotherapy, disease severity and/or survival, which indicates that cell-free or tumour-associated fungal DNA may be a potentially promising diagnostic and prognostic biomarker in patients with cancer182,183.
Box 3 The human mycobiota: localization, species and biomass.
Fungi are core constituents of microbial communities in human tissues, including the skin, mouth and respiratory, intestinal and genitourinary tracts160, although they typically account for less than 1% of total microbial amplicon sequences, with 50–100 fungal genera being reliably identified202. A core fungal signature at different tissue sites has been challenging to define, as humans inhale and ingest fungi daily, blurring the distinction between passenger and colonizer species. Longitudinally collected faecal samples show high intra-individual and interindividual variability of fungal taxa, and low day-to-day stability of intestinal fungi, which is in contrast to the relative intra-individual stability of bacterial communities170,203. Frequent tooth brushing reduced Candida levels in dental plaque, saliva and stool, which is consistent with the oral reservoir contributing to intestinal flux204. Saccharomyces stool levels dropped precipitously when human volunteers consumed Saccharomyces-free diets, only to increase again when the volunteers ate a single bite of bread204. Notwithstanding this variability, certain genera predominate at distinct sites: Saccharomyces, Candida, Debaromyces and Pichia are commonly found in the digestive and genital tracts, Malassezia is commonly found in the skin, and Aspergillus, Fusarium, Alternaria and Cladosporium moulds are commonly found in the airways, albeit at lower densities. The figure depicts commensal fungal communities that are adapted to specific tissue niches of humans. Common taxa found under homeostatic conditions are indicated, as are common diseases associated with some of these taxa, such as invasive candidiasis, vulvovaginal candidiasis (VVC) and pityriasis versicolor.
Nucleic acid-based methods underestimate fungal biomass given the size difference of bacteria (~0.5–2-μm diameter) and fungi (~3–5-μm diameter for yeasts and ~10 to >100 μm and greater diameter for hyphae) and metagenomic sequencing has not been widely applied to analyse fungal communities, owing to large and complex fungal genomes (typically more than 20 Mb in size, with both haploid and diploid genomes), low fungal reads compared with bacteria and incomplete, poorly annotated reference libraries202. Thus, the true size and composition of the mycobiome in humans remain incompletely understood.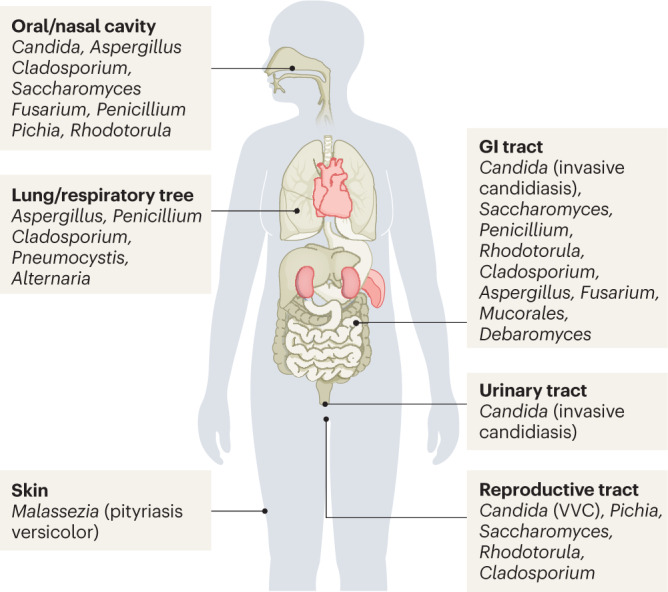
Conclusions and future perspectives
Opportunistic fungal infections cause a substantial disease burden globally and are a major threat to the well-being of vulnerable patients. The past decade of research on fungal immunology has moved at a remarkable pace and has yielded exciting advances in our basic understanding of the cellular and molecular determinants of antifungal defences. In this Review, we have delineated promising immunogenomic factors that may enable personalized risk assessment and prognosis of patients at risk of fungal disease, and adjunctive immunotherapeutic strategies that may protect fungus-infected patients by modulating antifungal responses. We have highlighted the expanding universe of inherited and iatrogenic immunodeficiency conditions that predispose to opportunistic fungal disease, as well as the conceptual advances in our understanding of protective and pathogenic antifungal immune pathways that have been derived from studying these conditions. We have also discussed how the endogenous mycobiota shape immune homeostasis and regulate responses to neoplasia, infections, autoimmunity and other exogenous insults. Moving forward, further increasing our understanding of tissue-specific mechanisms of antifungal defence should help to improve the management of life-threatening fungal infections and curtail their detrimental impact on human health.
Supplementary information
Acknowledgements
The authors apologize to their many colleagues whose valuable contributions they were not able to cite because of space constraints. This work was supported by the Division of Intramural Research of the NIAID, NIH (ZIA AI001175 to M.S.L.). R.A.D. was supported by the UK Medical Research Council (awards MR/S024611 and MR/T029137) and Academy of Medical Sciences (award SBF004_1008). T.M.H. was supported by NIH grants R01 AI093808, R01 AI139632, R21 AI156157 and P30 CA008748 (to Memorial Sloan Kettering Cancer Center).
Glossary
- Agglutinin-like sequence 3
(Als3). A Candida albicans hyphal protein that facilitates adhesion and invasion in host epithelial and endothelial cells through binding to E-cadherin and N-cadherin, respectively, and that has formed the basis for developing the investigational vaccine NDV-3.
- Candidalysin
A pore-forming, cytolytic peptide toxin produced by Candida albicans hyphae that promotes damage to oral epithelial cells and is required for virulence during mucosal candidiasis.
- Chronic mucocutaneous candidiasis
(CMC). A clinical condition characterized by severe, recurrent and treatment-refractory episodes of candidiasis of mucosal surfaces, skin and/or nails.
- Diabetic ketoacidosis
A life-threatening complication of uncontrolled diabetes that is characterized by insulin deficiency driving excessive accumulation of ketone bodies in the blood, which leads to acidosis.
- Hyphae
Long filamentous branching structures of certain fungi (for example, Aspergillus) that grow by apical extension.
- Inborn errors of immunity
Conditions caused by deleterious germ line mutations in single immune-related genes that manifest themselves with various infectious complications with or without other accompanying clinical manifestations, such as autoimmunity, autoinflammation, atopy and/or malignancy.
- Kallikrein–kinin system
A family of serine proteases (kallikreins) that cleave kininogens to generate the kinin peptides bradykinin and kallidin, which as a collective system have important roles in renal homeostasis and blood pressure regulation, with additional roles now emerging during inflammation, immunity and tissue repair.
- LC3-associated phagocytosis
A non-canonical autophagy pathway activated downstream of pattern recognition receptors which promotes phagosome maturation and intracellular killing of pathogens within phagocytes and regulates other mononuclear phagocyte functions, such as antigen presentation, efferocytosis and anti-inflammatory cytokine responses.
- Natural TH17 cell
A type 17 cell subpopulation that develops in the thymus, expresses CD4, TCRαβ, RORγt, CCR6 and IL-23R and, in contrast to inducible TH17 cells, is characterized by an intrinsic capacity for rapid activation in response to T cell receptor-independent stimulation in naive hosts.
- Trained immunity
The ability of the innate immune system, through epigenetic and metabolic reprogramming, to develop a form of immunological memory that can provide protection against a subsequent challenge with homologous or even heterologous pathogens.
- Uraemia
A clinical syndrome characterized by increased concentrations of urea in the blood and electrolyte, volume and metabolic abnormalities that are collectively caused by deteriorating kidney function.
- Yeast
A eukaryotic, unicellular fungal organism that reproduces by budding (for example, Candida).
Author contributions
The authors contributed equally to all aspects of the article.
Peer review
Peer review information
Nature Reviews Immunology thanks A. Ibrahim, S. Levitz and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Competing interests
T.M.H. participated in a scientific advisory board for Boehringer Ingelheim in 2020. The other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41577-022-00826-w.
References
- 1.Hoenigl M, et al. COVID-19-associated fungal infections. Nat. Microbiol. 2022;7:1127–1140. doi: 10.1038/s41564-022-01172-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lionakis MS, Hohl TM. Call to action: how to tackle emerging nosocomial fungal infections. Cell Host Microbe. 2020;27:859–862. doi: 10.1016/j.chom.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salazar F, Bignell E, Brown GD, Cook PC, Warris A. Pathogenesis of respiratory viral and fungal coinfections. Clin. Microbiol. Rev. 2022;35:e0009421. doi: 10.1128/CMR.00094-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow NA, et al. Multiple introductions and subsequent transmission of multidrug-resistant Candida auris in the USA: a molecular epidemiological survey. Lancet Infect. Dis. 2018;18:1377–1384. doi: 10.1016/S1473-3099(18)30597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown GD, Willment JA, Whitehead L. C-type lectins in immunity and homeostasis. Nat. Rev. Immunol. 2018;18:374–389. doi: 10.1038/s41577-018-0004-8. [DOI] [PubMed] [Google Scholar]
- 6.Borriello F, et al. An adjuvant strategy enabled by modulation of the physical properties of microbial ligands expands antigen immunogenicity. Cell. 2022;185:614–629 e621. doi: 10.1016/j.cell.2022.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corvilain E, Casanova JL, Puel A. Inherited CARD9 deficiency: invasive disease caused by ascomycete fungi in previously healthy children and adults. J. Clin. Immunol. 2018;38:656–693. doi: 10.1007/s10875-018-0539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pappas PG, Lionakis MS, Arendrup MC, Ostrosky-Zeichner L, Kullberg BJ. Invasive candidiasis. Nat. Rev. Dis. Prim. 2018;4:18026. doi: 10.1038/nrdp.2018.26. [DOI] [PubMed] [Google Scholar]
- 9.Lionakis MS, Levitz SM. Host control of fungal infections: lessons from basic studies and human cohorts. Annu. Rev. Immunol. 2018;36:157–191. doi: 10.1146/annurev-immunol-042617-053318. [DOI] [PubMed] [Google Scholar]
- 10.Gaffen SL, Moutsopoulos NM. Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci. Immunol. 2020;5:eaau4594. doi: 10.1126/sciimmunol.aau4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puel A, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boisson B, et al. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 2013;39:676–686. doi: 10.1016/j.immuni.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conti HR, et al. IL-17 receptor signaling in oral epithelial cells is critical for protection against oropharyngeal candidiasis. Cell Host Microbe. 2016;20:606–617. doi: 10.1016/j.chom.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conti HR, et al. Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J. Exp. Med. 2014;211:2075–2084. doi: 10.1084/jem.20130877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conti HR, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy R, et al. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc. Natl Acad. Sci. Usa. 2016;113:E8277–E8285. doi: 10.1073/pnas.1618300114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puel A, et al. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr. Opin. Allergy Clin. Immunol. 2012;12:616–622. doi: 10.1097/ACI.0b013e328358cc0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oikonomou V, Break TJ, Gaffen SL, Moutsopoulos NM, Lionakis MS. Infections in the monogenic autoimmune syndrome APECED. Curr. Opin. Immunol. 2021;72:286–297. doi: 10.1016/j.coi.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davidson L, et al. Risk of candidiasis associated with interleukin-17 inhibitors: a real-world observational study of multiple independent sources. Lancet Reg. Health Eur. 2022;13:100266. doi: 10.1016/j.lanepe.2021.100266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swidergall M, LeibundGut-Landmann S. Immunosurveillance of Candida albicans commensalism by the adaptive immune system. Mucosal Immunol. 2022;15:829–836. doi: 10.1038/s41385-022-00536-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aggor FE, et al. A gut-oral microbiome-driven axis controls oropharyngeal candidiasis through retinoic acid. JCI Insight. 2022;7:e160348. doi: 10.1172/jci.insight.160348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verma AH, et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci. Immunol. 2017;2:eaam8834. doi: 10.1126/sciimmunol.aam8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basso P, et al. Deep tissue infection by an invasive human fungal pathogen requires lipid-based suppression of the IL-17 response. Cell Host Microbe. 2022;30:1589–1601 e1585. doi: 10.1016/j.chom.2022.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lisco A, et al. Lost in translation: lack of CD4 expression due to a novel genetic defect. J. Infect. Dis. 2021;223:645–654. doi: 10.1093/infdis/jiab025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cudrici CD, et al. Characterization of autoantibodies, immunophenotype and autoimmune disease in a prospective cohort of patients with idiopathic CD4 lymphocytopenia. Clin. Immunol. 2021;224:108664. doi: 10.1016/j.clim.2021.108664. [DOI] [PubMed] [Google Scholar]
- 26.Hernandez-Santos N, et al. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. 2013;6:900–910. doi: 10.1038/mi.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swidergall M, Solis NV, Lionakis MS, Filler SG. EphA2 is an epithelial cell pattern recognition receptor for fungal beta-glucans. Nat. Microbiol. 2018;3:53–61. doi: 10.1038/s41564-017-0059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aggor FEY, et al. Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis. Sci. Immunol. 2020;5:eaba0570. doi: 10.1126/sciimmunol.aba0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puel A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010;207:291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kisand K, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 2010;207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Break TJ, et al. Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science. 2021;371:eaay5731. doi: 10.1126/science.aay5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toubiana J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127:3154–3164. doi: 10.1182/blood-2015-11-679902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yano J, et al. Current patient perspectives of vulvovaginal candidiasis: incidence, symptoms, management and post-treatment outcomes. BMC Women’s Health. 2019;19:48. doi: 10.1186/s12905-019-0748-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopes JP, Lionakis MS. Pathogenesis and virulence of Candida albicans. Virulence. 2022;13:89–121. doi: 10.1080/21505594.2021.2019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peters BM, et al. The interleukin (IL) 17R/IL-22R signaling axis is dispensable for Vulvovaginal candidiasis regardless of estrogen status. J. Infect. Dis. 2020;221:1554–1563. doi: 10.1093/infdis/jiz649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacAlpine J, et al. A small molecule produced by Lactobacillus species blocks Candida albicans filamentation by inhibiting a DYRK1-family kinase. Nat. Commun. 2021;12:6151. doi: 10.1038/s41467-021-26390-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edwards JE, Jr., et al. A fungal immunotherapeutic vaccine (NDV-3A) for treatment of recurrent vulvovaginal candidiasis-a phase 2 randomized, double-blind, placebo-controlled trial. Clin. Infect. Dis. 2018;66:1928–1936. doi: 10.1093/cid/ciy185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ibrahim AS, et al. NDV-3 protects mice from vulvovaginal candidiasis through T- and B-cell immune response. Vaccine. 2013;31:5549–5556. doi: 10.1016/j.vaccine.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kashem SW, et al. Nociceptive sensory fibers drive interleukin-23 production from CD301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity. 2015;43:515–526. doi: 10.1016/j.immuni.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen JA, et al. Cutaneous TRPV1+ neurons trigger protective innate type 17 anticipatory immunity. Cell. 2019;178:919–932 e914. doi: 10.1016/j.cell.2019.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leonardi I, et al. Mucosal fungi promote gut barrier function and social behavior via type 17 immunity. Cell. 2022;185:831–846 e814. doi: 10.1016/j.cell.2022.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huh JR, Veiga-Fernandes H. Neuroimmune circuits in inter-organ communication. Nat. Rev. Immunol. 2020;20:217–228. doi: 10.1038/s41577-019-0247-z. [DOI] [PubMed] [Google Scholar]
- 43.Fan D, et al. Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 2015;21:808–814. doi: 10.1038/nm.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drummond RA, et al. Long-term antibiotic exposure promotes mortality after systemic fungal infection by driving lymphocyte dysfunction and systemic escape of commensal bacteria. Cell Host Microbe. 2022;30:1020–1033.e6. doi: 10.1016/j.chom.2022.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glocker EO, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 2009;361:1727–1735. doi: 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drummond RA, et al. CARD9+ microglia promote antifungal immunity via IL-1β - and CXCL1-mediated neutrophil recruitment. Nat. Immunol. 2019;20:559–570. doi: 10.1038/s41590-019-0377-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jodele S, et al. Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood. 2020;135:1049–1057. doi: 10.1182/blood.2019004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bar E, Whitney PG, Moor K, Reis e Sousa C, LeibundGut-Landmann S. IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity. 2014;40:117–127. doi: 10.1016/j.immuni.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 49.Dominguez-Andres J, et al. Inflammatory Ly6Chigh monocytes protect against candidiasis through IL-15-driven NK cell/neutrophil activation. Immunity. 2017;46:1059–1072 e1054. doi: 10.1016/j.immuni.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 50.Wan L, et al. Effect of granulocyte-macrophage colony-stimulating factor on prevention and treatment of invasive fungal disease in recipients of allogeneic stem-cell transplantation: a prospective multicenter randomized phase IV trial. J. Clin. Oncol. 2015;33:3999–4006. doi: 10.1200/JCO.2014.60.5121. [DOI] [PubMed] [Google Scholar]
- 51.Li DD, et al. Fungal sensing enhances neutrophil metabolic fitness by regulating antifungal Glut1 activity. Cell Host Microbe. 2022;30:530–544 e536. doi: 10.1016/j.chom.2022.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jawale CV, et al. Restoring glucose uptake rescues neutrophil dysfunction and protects against systemic fungal infection in mouse models of kidney disease. Sci. Transl. Med. 2020;12:eaay5691. doi: 10.1126/scitranslmed.aay5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Candon S, et al. Chronic disseminated candidiasis during hematological malignancies: an immune reconstitution inflammatory syndrome with expansion of pathogen-specific T helper type 1 cells. J. Infect. Dis. 2020;221:1907–1916. doi: 10.1093/infdis/jiz688. [DOI] [PubMed] [Google Scholar]
- 54.Garg AV, et al. MCPIP1 endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity. 2015;43:475–487. doi: 10.1016/j.immuni.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lionakis MS, et al. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog. 2012;8:e1002865. doi: 10.1371/journal.ppat.1002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Del Fresno C, et al. DNGR-1 in dendritic cells limits tissue damage by dampening neutrophil recruitment. Science. 2018;362:351–356. doi: 10.1126/science.aan8423. [DOI] [PubMed] [Google Scholar]
- 57.Liu J, et al. Progranulin aggravates lethal Candida albicans sepsis by regulating inflammatory response and antifungal immunity. PLoS Pathog. 2022;18:e1010873. doi: 10.1371/journal.ppat.1010873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramani K, et al. The kallikrein-kinin system: a novel mediator of IL-17-driven anti-Candida immunity in the kidney. PLoS Pathog. 2016;12:e1005952. doi: 10.1371/journal.ppat.1005952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lionakis MS, et al. CX3CR1-dependent renal macrophage survival promotes Candida control and host survival. J. Clin. Invest. 2013;123:5035–5051. doi: 10.1172/JCI71307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ngo LY, et al. Inflammatory monocytes mediate early and organ-specific innate defense during systemic candidiasis. J. Infect. Dis. 2014;209:109–119. doi: 10.1093/infdis/jit413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng SC, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao X, et al. JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat. Med. 2017;23:337–346. doi: 10.1038/nm.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, et al. Chronic mucocutaneous candidiasis and connective tissue disorder in humans with impaired JNK1-dependent responses to IL-17A/F and TGF-beta. Sci. Immunol. 2019;4:eaax7965. doi: 10.1126/sciimmunol.aax7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fitzpatrick Z, et al. Gut-educated IgA plasma cells defend the meningeal venous sinuses. Nature. 2020;587:472–476. doi: 10.1038/s41586-020-2886-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumar V, et al. Immunochip SNP array identifies novel genetic variants conferring susceptibility to candidaemia. Nat. Commun. 2014;5:4675. doi: 10.1038/ncomms5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Latge JP, Chamilos G. Aspergillus fumigatus and aspergillosis in 2019. Clin. Microbiol. Rev. 2019;33:e00140-18. doi: 10.1128/CMR.00140-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Espinosa V, et al. Inflammatory monocytes orchestrate innate antifungal immunity in the lung. PLoS Pathog. 2014;10:e1003940. doi: 10.1371/journal.ppat.1003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt S, et al. Human natural killer cells exhibit direct activity against Aspergillus fumigatus hyphae, but not against resting conidia. J. Infect. Dis. 2011;203:430–435. doi: 10.1093/infdis/jiq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seif M, et al. CAR T cells targeting Aspergillus fumigatus are effective at treating invasive pulmonary aspergillosis in preclinical models. Sci. Transl. Med. 2022;14:eabh1209. doi: 10.1126/scitranslmed.abh1209. [DOI] [PubMed] [Google Scholar]
- 70.Cadena J, Thompson GR, III, Patterson TF. Aspergillosis: epidemiology, diagnosis, and treatment. Infect. Dis. Clin. North. Am. 2021;35:415–434. doi: 10.1016/j.idc.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 71.Agarwal R, et al. Allergic bronchopulmonary aspergillosis. Clin. Chest Med. 2022;43:99–125. doi: 10.1016/j.ccm.2021.12.002. [DOI] [PubMed] [Google Scholar]
- 72.Philippe B, et al. Killing of Aspergillus fumigatus by alveolar macrophages is mediated by reactive oxidant intermediates. Infect. Immun. 2003;71:3034–3042. doi: 10.1128/IAI.71.6.3034-3042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hohl TM, et al. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog. 2005;1:e30. doi: 10.1371/journal.ppat.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aimanianda V, et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature. 2009;460:1117–1121. doi: 10.1038/nature08264. [DOI] [PubMed] [Google Scholar]
- 75.Doni A, et al. Serum amyloid P component is an essential element of resistance against Aspergillus fumigatus. Nat. Commun. 2021;12:3739. doi: 10.1038/s41467-021-24021-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garlanda C, et al. Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature. 2002;420:182–186. doi: 10.1038/nature01195. [DOI] [PubMed] [Google Scholar]
- 77.Stappers MHT, et al. Recognition of DHN-melanin by a C-type lectin receptor is required for immunity to Aspergillus. Nature. 2018;555:382–386. doi: 10.1038/nature25974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu H, et al. Aspergillus fumigatus CalA binds to integrin alpha5beta1 and mediates host cell invasion. Nat. Microbiol. 2016;2:16211. doi: 10.1038/nmicrobiol.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leal SM, Jr., et al. Fungal antioxidant pathways promote survival against neutrophils during infection. J. Clin. Invest. 2012;122:2482–2498. doi: 10.1172/JCI63239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shlezinger N, et al. Sterilizing immunity in the lung relies on targeting fungal apoptosis-like programmed cell death. Science. 2017;357:1037–1041. doi: 10.1126/science.aan0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.International Chronic Granulomatous Disease Cooperative Study Group. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N. Engl. J. Med. 1991;324:509–516. doi: 10.1056/NEJM199102213240801. [DOI] [PubMed] [Google Scholar]
- 82.Akoumianaki T, et al. Aspergillus cell wall melanin blocks LC3-associated phagocytosis to promote pathogenicity. Cell Host Microbe. 2016;19:79–90. doi: 10.1016/j.chom.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Goncalves SM, et al. Phagosomal removal of fungal melanin reprograms macrophage metabolism to promote antifungal immunity. Nat. Commun. 2020;11:2282. doi: 10.1038/s41467-020-16120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo Y, et al. During aspergillus infection, monocyte-derived DCs, neutrophils, and plasmacytoid DCs enhance innate immune defense through CXCR3-dependent crosstalk. Cell Host Microbe. 2020;28:104–116 e104. doi: 10.1016/j.chom.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jhingran A, et al. Compartment-specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog. 2015;11:e1004589. doi: 10.1371/journal.ppat.1004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Caffrey AK, et al. IL-1alpha signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog. 2015;11:e1004625. doi: 10.1371/journal.ppat.1004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rieber N, et al. Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight. 2016;1:e89890. doi: 10.1172/jci.insight.89890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang X, et al. MDA5 is an essential sensor of a pathogen-associated molecular pattern associated with vitality that is necessary for host resistance against Aspergillus fumigatus. J. Immunol. 2020;205:3058–3070. doi: 10.4049/jimmunol.2000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Caffrey-Carr AK, et al. Host-derived leukotriene B4 is critical for resistance against invasive pulmonary aspergillosis. Front. Immunol. 2017;8:1984. doi: 10.3389/fimmu.2017.01984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Snarr BD, et al. Galectin-3 enhances neutrophil motility and extravasation into the airways during Aspergillus fumigatus infection. PLoS Pathog. 2020;16:e1008741. doi: 10.1371/journal.ppat.1008741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hohl TM, et al. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. 2009;6:470–481. doi: 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Espinosa V, et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol. 2017;2:eaan5357. doi: 10.1126/sciimmunol.aan5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Espinosa V, et al. Cutting edge: neutrophils license the maturation of monocytes into effective antifungal effectors. J. Immunol. 2022;209:1827–1831. doi: 10.4049/jimmunol.2200430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kasahara S, et al. Role of granulocyte-macrophage colony-stimulating factor signaling in regulating neutrophil antifungal activity and the oxidative burst during respiratory fungal challenge. J. Infect. Dis. 2016;213:1289–1298. doi: 10.1093/infdis/jiw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic infections in patients with pulmonary alveolar proteinosis. J. Infect. 2012;65:173–179. doi: 10.1016/j.jinf.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 96.Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI. Human polymorphonuclear leukocytes inhibit Aspergillus fumigatus conidial growth by lactoferrin-mediated iron depletion. J. Immunol. 2007;178:6367–6373. doi: 10.4049/jimmunol.178.10.6367. [DOI] [PubMed] [Google Scholar]
- 97.Clark HL, et al. Zinc and manganese chelation by neutrophil S100A8/A9 (calprotectin) limits extracellular aspergillus fumigatus hyphal growth and corneal infection. J. Immunol. 2016;196:336–344. doi: 10.4049/jimmunol.1502037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cunha C, et al. Genetic PTX3 deficiency and aspergillosis in stem-cell transplantation. N. Engl. J. Med. 2014;370:421–432. doi: 10.1056/NEJMoa1211161. [DOI] [PubMed] [Google Scholar]
- 99.Fisher CE, et al. Validation of single nucleotide polymorphisms in invasive aspergillosis following hematopoietic cell transplantation. Blood. 2017;129:2693–2701. doi: 10.1182/blood-2016-10-743294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lionakis MS, et al. Inhibition of B cell receptor signaling by ibrutinib in primary CNS lymphoma. Cancer Cell. 2017;31:833–843 e835. doi: 10.1016/j.ccell.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ghez D, et al. Early-onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood. 2018;131:1955–1959. doi: 10.1182/blood-2017-11-818286. [DOI] [PubMed] [Google Scholar]
- 102.Herbst S, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol. Med. 2015;7:240–258. doi: 10.15252/emmm.201404556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Roden MM, et al. Epidemiology and outcome of zygomycosis: a review of 929 reported cases. Clin. Infect. Dis. 2005;41:634–653. doi: 10.1086/432579. [DOI] [PubMed] [Google Scholar]
- 104.Soliman SSM, et al. Mucoricin is a ricin-like toxin that is critical for the pathogenesis of mucormycosis. Nat. Microbiol. 2021;6:313–326. doi: 10.1038/s41564-020-00837-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu M, et al. The endothelial cell receptor GRP78 is required for mucormycosis pathogenesis in diabetic mice. J. Clin. Invest. 2010;120:1914–1924. doi: 10.1172/JCI42164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gebremariam T, et al. CotH3 mediates fungal invasion of host cells during mucormycosis. J. Clin. Invest. 2014;124:237–250. doi: 10.1172/JCI71349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gebremariam T, et al. Bicarbonate correction of ketoacidosis alters host-pathogen interactions and alleviates mucormycosis. J. Clin. Invest. 2016;126:2280–2294. doi: 10.1172/JCI82744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alqarihi A, et al. GRP78 and integrins play different roles in host cell invasion during mucormycosis. mBio. 2020;11:e01087-20. doi: 10.1128/mBio.01087-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Watkins TN, et al. Inhibition of EGFR signaling protects from mucormycosis. mBio. 2018;9:e01384-18. doi: 10.1128/mBio.01384-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Andrianaki AM, et al. Iron restriction inside macrophages regulates pulmonary host defense against Rhizopus species. Nat. Commun. 2018;9:3333. doi: 10.1038/s41467-018-05820-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Siqueira IM, et al. Early immune response against Fonsecaea pedrosoi requires dectin-2-mediated Th17 activity, whereas Th1 response, aided by Treg cells, is crucial for fungal clearance in later stage of experimental chromoblastomycosis. PLoS Negl. Trop. Dis. 2020;14:e0008386. doi: 10.1371/journal.pntd.0008386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Teixeira de Sousa Mda G, Ghosn EE, Almeida SR. Absence of CD4+ T cells impairs host defence of mice infected with Fonsecaea pedrosoi. Scand. J. Immunol. 2006;64:595–600. doi: 10.1111/j.1365-3083.2006.01846.x. [DOI] [PubMed] [Google Scholar]
- 113.Sousa Mda G, et al. Restoration of pattern recognition receptor costimulation to treat chromoblastomycosis, a chronic fungal infection of the skin. Cell Host Microbe. 2011;9:436–443. doi: 10.1016/j.chom.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.de Sousa Mda G, et al. Topical application of imiquimod as a treatment for chromoblastomycosis. Clin. Infect. Dis. 2014;58:1734–1737. doi: 10.1093/cid/ciu168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Revankar SG, Patterson JE, Sutton DA, Pullen R, Rinaldi MG. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin. Infect. Dis. 2002;34:467–476. doi: 10.1086/338636. [DOI] [PubMed] [Google Scholar]
- 116.Kainer MA, et al. Fungal infections associated with contaminated methylprednisolone in Tennessee. N. Engl. J. Med. 2012;367:2194–2203. doi: 10.1056/NEJMoa1212972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Drummond RA, et al. Human dectin-1 deficiency impairs macrophage-mediated defense against phaeohyphomycosis. J. Clin. Invest. 2022;132:e159348. doi: 10.1172/JCI159348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mazzurco JD, Ramirez J, Fivenson DP. Phaeohyphomycosis caused by Phaeoacremonium species in a patient taking infliximab. J. Am. Acad. Dermatol. 2012;66:333–335. doi: 10.1016/j.jaad.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 119.Casadevall A. Immunity to invasive fungal diseases. Annu. Rev. Immunol. 2022;40:121–141. doi: 10.1146/annurev-immunol-101220-034306. [DOI] [PubMed] [Google Scholar]
- 120.Erwig LP, Gow NA. Interactions of fungal pathogens with phagocytes. Nat. Rev. Microbiol. 2016;14:163–176. doi: 10.1038/nrmicro.2015.21. [DOI] [PubMed] [Google Scholar]
- 121.Browne SK, et al. Adult-onset immunodeficiency in Thailand and Taiwan. N. Engl. J. Med. 2012;367:725–734. doi: 10.1056/NEJMoa1111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Browne SK, et al. Anti-CD20 (rituximab) therapy for anti-IFN-gamma autoantibody-associated nontuberculous mycobacterial infection. Blood. 2012;119:3933–3939. doi: 10.1182/blood-2011-12-395707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ochoa S, et al. Daratumumab (anti-CD38) for treatment of disseminated nontuberculous mycobacteria in a patient with anti-interferon-gamma autoantibodies. Clin. Infect. Dis. 2021;72:2206–2208. doi: 10.1093/cid/ciaa1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Francoise U, Lafont E, Suarez F, Lanternier F, Lortholary O. Disseminated cryptococcosis in a patient with CD40 ligand deficiency. J. Clin. Immunol. 2022;42:1622–1625. doi: 10.1007/s10875-022-01329-y. [DOI] [PubMed] [Google Scholar]
- 125.Vinh DC, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115:1519–1529. doi: 10.1182/blood-2009-03-208629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Na YR, et al. GM-CSF induces inflammatory macrophages by regulating glycolysis and lipid metabolism. J. Immunol. 2016;197:4101. doi: 10.4049/jimmunol.1600745. [DOI] [PubMed] [Google Scholar]
- 127.Saijo T, et al. Anti-granulocyte-macrophage colony-stimulating factor autoantibodies are a risk factor for central nervous system infection by Cryptococcus gattii in otherwise immunocompetent patients. mBio. 2014;5:e00912-14. doi: 10.1128/mBio.00912-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kannambath S, et al. Genome-wide association study identifies novel colony stimulating factor 1 locus conferring susceptibility to cryptococcosis in human immunodeficiency virus-infected South Africans. Open Forum Infect. Dis. 2020;7:ofaa489. doi: 10.1093/ofid/ofaa489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Elmore MRP, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bryan AM, et al. FTY720 reactivates cryptococcal granulomas in mice through S1P receptor 3 on macrophages. J. Clin. Invest. 2020;130:4546–4560. doi: 10.1172/JCI136068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee SC, Dickson DW, Casadevall A. Pathology of cryptococcal meningoencephalitis: analysis of 27 patients with pathogenetic implications. Hum. Pathol. 1996;27:839–847. doi: 10.1016/S0046-8177(96)90459-1. [DOI] [PubMed] [Google Scholar]
- 132.Johnston SA, May RC. The human fungal pathogen Cryptococcus neoformans escapes macrophages by a phagosome emptying mechanism that is inhibited by Arp2/3 complex-mediated actin polymerisation. PLoS Pathog. 2010;6:e1001041. doi: 10.1371/journal.ppat.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fu MS, et al. Cryptococcus neoformans urease affects the outcome of intracellular pathogenesis by modulating phagolysosomal pH. PLoS Pathog. 2018;14:e1007144. doi: 10.1371/journal.ppat.1007144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Scriven JE, et al. The CSF immune response in HIV-1-associated cryptococcal meningitis: macrophage activation, correlates of disease severity, and effect of antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2017;75:299–307. doi: 10.1097/QAI.0000000000001382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jarvis JN, et al. Cerebrospinal fluid cytokine profiles predict risk of early mortality and immune reconstitution inflammatory syndrome in HIV-associated cryptococcal meningitis. PLoS Pathog. 2015;11:e1004754. doi: 10.1371/journal.ppat.1004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jarvis JN, et al. The phenotype of the Cryptococcus-specific CD4+ memory T-cell response is associated with disease severity and outcome in HIV-associated cryptococcal meningitis. J. Infect. Dis. 2013;207:1817–1828. doi: 10.1093/infdis/jit099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jarvis JN, et al. Adjunctive interferon-γ immunotherapy for the treatment of HIV-associated cryptococcal meningitis: a randomized controlled trial. AIDS. 2012;26:1105–1113. doi: 10.1097/QAD.0b013e3283536a93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Anjum S, et al. Outcomes in previously healthy cryptococcal meningoencephalitis patients treated with pulse taper corticosteroids for post-infectious inflammatory syndrome. Clin. Infect. Dis. 2021;73:e2789–e2798. doi: 10.1093/cid/ciaa1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Panackal AA, et al. Paradoxical immune responses in non-HIV cryptococcal meningitis. PLoS Pathog. 2015;11:e1004884. doi: 10.1371/journal.ppat.1004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu J, et al. Chemokine receptor CXCR3 is required for lethal brain pathology but not pathogen clearance during cryptococcal meningoencephalitis. Sci. Adv. 2020;6:eaba2502. doi: 10.1126/sciadv.aba2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Antonelli LR, et al. Elevated frequencies of highly activated CD4+ T cells in HIV+ patients developing immune reconstitution inflammatory syndrome. Blood. 2010;116:3818–3827. doi: 10.1182/blood-2010-05-285080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Khaw YM, et al. Th1-dependent cryptococcus-associated immune reconstitution inflammatory syndrome model with brain damage. Front. Immunol. 2020;11:529219. doi: 10.3389/fimmu.2020.529219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hsu AP, et al. Immunogenetics associated with severe coccidioidomycosis. JCI Insight. 2022 doi: 10.1172/jci.insight.159491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wuthrich M, Deepe GS, Jr, Klein B. Adaptive immunity to fungi. Annu. Rev. Immunol. 2012;30:115–148. doi: 10.1146/annurev-immunol-020711-074958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS., Jr. Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity. 2013;39:697–710. doi: 10.1016/j.immuni.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wuthrich M, Ersland K, Sullivan T, Galles K, Klein BS. Fungi subvert vaccine T cell priming at the respiratory mucosa by preventing chemokine-induced influx of inflammatory monocytes. Immunity. 2012;36:680–692. doi: 10.1016/j.immuni.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tsai M, et al. Disseminated coccidioidomycosis treated with interferon-gamma and dupilumab. N. Engl. J. Med. 2020;382:2337–2343. doi: 10.1056/NEJMoa2000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vargas SL, et al. Search for primary infection by Pneumocystis carinii in a cohort of normal, healthy infants. Clin. Infect. Dis. 2001;32:855–861. doi: 10.1086/319340. [DOI] [PubMed] [Google Scholar]
- 149.Hoving JC, Kolls JK. New advances in understanding the host immune response to Pneumocystis. Curr. Opin. Microbiol. 2017;40:65–71. doi: 10.1016/j.mib.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Elsegeiny W, et al. Murine models of Pneumocystis infection recapitulate human primary immune disorders. JCI Insight. 2018;3:e91894. doi: 10.1172/jci.insight.91894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Opata MM, et al. B lymphocytes are required during the early priming of CD4+ T cells for clearance of pneumocystis infection in mice. J. Immunol. 2015;195:611–620. doi: 10.4049/jimmunol.1500112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rapaka RR, et al. Conserved natural IgM antibodies mediate innate and adaptive immunity against the opportunistic fungus Pneumocystis murina. J. Exp. Med. 2010;207:2907–2919. doi: 10.1084/jem.20100034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Steele C, et al. Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the dectin-1 beta-glucan receptor. J. Exp. Med. 2003;198:1677–1688. doi: 10.1084/jem.20030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Eddens T, et al. Eosinophils contribute to early clearance of Pneumocystis murina infection. J. Immunol. 2015;195:185–193. doi: 10.4049/jimmunol.1403162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Limper AH, Hoyte JS, Standing JE. The role of alveolar macrophages in Pneumocystis carinii degradation and clearance from the lung. J. Clin. Invest. 1997;99:2110–2117. doi: 10.1172/JCI119384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li XV, Leonardi I, Iliev ID. Gut mycobiota in immunity and inflammatory disease. Immunity. 2019;50:1365–1379. doi: 10.1016/j.immuni.2019.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Underhill DM, Braun J. Fungal microbiome in inflammatory bowel disease: a critical assessment. J. Clin. Invest. 2022;132:e155786. doi: 10.1172/JCI155786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.van Tilburg Bernardes E, et al. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat. Commun. 2020;11:2577. doi: 10.1038/s41467-020-16431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rao C, et al. Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature. 2021;591:633–638. doi: 10.1038/s41586-021-03241-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Santus W, Devlin JR, Behnsen J. Crossing kingdoms: how the mycobiota and fungal-bacterial interactions impact host health and disease. Infect. Immun. 2021;89:e00648-20. doi: 10.1128/IAI.00648-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Boutin RC, et al. Bacterial-fungal interactions in the neonatal gut influence asthma outcomes later in life. Elife. 2021;10:e67740. doi: 10.7554/eLife.67740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bacher P, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell. 2019;176:1340–1355 e1315. doi: 10.1016/j.cell.2019.01.041. [DOI] [PubMed] [Google Scholar]
- 163.Findley K, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498:367–370. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sparber F, et al. The skin commensal yeast malassezia triggers a type 17 response that coordinates anti-fungal immunity and exacerbates skin inflammation. Cell Host Microbe. 2019;25:389–403 e386. doi: 10.1016/j.chom.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 165.Hurabielle C, et al. Immunity to commensal skin fungi promotes psoriasiform skin inflammation. Proc. Natl Acad. Sci. USA. 2020;117:16465–16474. doi: 10.1073/pnas.2003022117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ost KS, et al. Adaptive immunity induces mutualism between commensal eukaryotes. Nature. 2021;596:114–118. doi: 10.1038/s41586-021-03722-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Doron I, et al. Mycobiota-induced IgA antibodies regulate fungal commensalism in the gut and are dysregulated in Crohn’s disease. Nat. Microbiol. 2021;6:1493–1504. doi: 10.1038/s41564-021-00983-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Doron I, et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell. 2021;184:1017–1031 e1014. doi: 10.1016/j.cell.2021.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhai B, et al. High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat. Med. 2020;26:59–64. doi: 10.1038/s41591-019-0709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rolling T, et al. Haematopoietic cell transplantation outcomes are linked to intestinal mycobiota dynamics and an expansion of Candida parapsilosis complex species. Nat. Microbiol. 2021;6:1505–1515. doi: 10.1038/s41564-021-00989-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Malard F, et al. Impact of gut fungal and bacterial communities on the outcome of allogeneic hematopoietic cell transplantation. Mucosal Immunol. 2021;14:1127–1132. doi: 10.1038/s41385-021-00429-z. [DOI] [PubMed] [Google Scholar]
- 172.Sokol H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039–1048. doi: 10.1136/gutjnl-2015-310746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Leonardi I, et al. CX3CR1+ mononuclear phagocytes control immunity to intestinal fungi. Science. 2018;359:232–236. doi: 10.1126/science.aao1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Limon JJ, et al. Malassezia is associated with Crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe. 2019;25:377–388 e376. doi: 10.1016/j.chom.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jain U, et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science. 2021;371:1154–1159. doi: 10.1126/science.abd0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Iliev ID, et al. Interactions between commensal fungi and the C-type lectin receptor dectin-1 influence colitis. Science. 2012;336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.McGovern DP, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 2010;42:332–337. doi: 10.1038/ng.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Leonardi I, et al. Fungal trans-kingdom dynamics linked to responsiveness to fecal microbiota transplantation (FMT) therapy in ulcerative colitis. Cell Host Microbe. 2020;27:823–829 e823. doi: 10.1016/j.chom.2020.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li XV, et al. Immune regulation by fungal strain diversity in inflammatory bowel disease. Nature. 2022;603:672–678. doi: 10.1038/s41586-022-04502-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Aykut B, et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature. 2019;574:264–267. doi: 10.1038/s41586-019-1608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shiao SL, et al. Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell. 2021;39:1202–1213 e1206. doi: 10.1016/j.ccell.2021.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dohlman AB, et al. A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell. 2022;185:3807–3822 e3812. doi: 10.1016/j.cell.2022.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Narunsky-Haziza L, et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell. 2022;185:3789–3806 e3717. doi: 10.1016/j.cell.2022.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lasbury ME, Tang X, Durant PJ, Lee CH. Effect of transcription factor GATA-2 on phagocytic activity of alveolar macrophages from Pneumocystis carinii-infected hosts. Infect. Immun. 2003;71:4943–4952. doi: 10.1128/IAI.71.9.4943-4952.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Proctor DM, et al. Integrated genomic, epidemiologic investigation of Candida auris skin colonization in a skilled nursing facility. Nat. Med. 2021;27:1401–1409. doi: 10.1038/s41591-021-01383-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mathur P, et al. Five-year profile of candidaemia at an Indian trauma centre: high rates of Candida auris blood stream infections. Mycoses. 2018;61:674–680. doi: 10.1111/myc.12790. [DOI] [PubMed] [Google Scholar]
- 187.CDC. Antibiotic resistance threats in the United States, 2019. https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (2019).
- 188.Eix EF, et al. Ex vivo human and porcine skin effectively model Candida auris colonization, differentiating robust and poor fungal colonizers. J. Infect. Dis. 2022;225:1791–1795. doi: 10.1093/infdis/jiac094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Huang X, et al. Murine model of colonization with fungal pathogen Candida auris to explore skin tropism, host risk factors and therapeutic strategies. Cell Host Microbe. 2020;29:210–221.e6. doi: 10.1016/j.chom.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bruno M, et al. Transcriptional and functional insights into the host immune response against the emerging fungal pathogen Candida auris. Nat. Microbiol. 2020;5:1516–1531. doi: 10.1038/s41564-020-0780-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Johnson CJ, Davis JM, Huttenlocher A, Kernien JF, Nett JE. Emerging fungal pathogen Candida auris evades neutrophil attack. mBio. 2018;9:e01403-18. doi: 10.1128/mBio.01403-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Oliveira LVN, Wang R, Specht CA, Levitz SM. Vaccines for human fungal diseases: close but still a long way to go. NPJ Vaccines. 2021;6:33. doi: 10.1038/s41541-021-00294-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rivera A, Lodge J, Xue C. Harnessing the immune response to fungal pathogens for vaccine development. Annu. Rev. Microbiol. 2022;76:703–726. doi: 10.1146/annurev-micro-041020-111511. [DOI] [PubMed] [Google Scholar]
- 194.Pappagianis D, Valley Fever Vaccine Study Group. Evaluation of the protective efficacy of the killed Coccidioides immitis spherule vaccine in humans. Am. Rev. Respir. Dis. 1993;148:656–660. doi: 10.1164/ajrccm/148.3.656. [DOI] [PubMed] [Google Scholar]
- 195.De Bernardis F, Graziani S, Tirelli F, Antonopoulou S. Candida vaginitis: virulence, host response and vaccine prospects. Med. Mycol. 2018;56:26–31. doi: 10.1093/mmy/myx139. [DOI] [PubMed] [Google Scholar]
- 196.Lin L, et al. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 2009;5:e1000703. doi: 10.1371/journal.ppat.1000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Singh S, et al. The NDV-3A vaccine protects mice from multidrug resistant Candida auris infection. PLoS Pathog. 2019;15:e1007460. doi: 10.1371/journal.ppat.1007460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Spellberg BJ, et al. Efficacy of the anti-Candida rAls3p-N or rAls1p-N vaccines against disseminated and mucosal candidiasis. J. Infect. Dis. 2006;194:256–260. doi: 10.1086/504691. [DOI] [PubMed] [Google Scholar]
- 199.Schmidt CS, et al. NDV-3, a recombinant alum-adjuvanted vaccine for Candida and Staphylococcus aureus, is safe and immunogenic in healthy adults. Vaccine. 2012;30:7594–7600. doi: 10.1016/j.vaccine.2012.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wuthrich M, et al. Calnexin induces expansion of antigen-specific CD4+ T cells that confer immunity to fungal ascomycetes via conserved epitopes. Cell Host Microbe. 2015;17:452–465. doi: 10.1016/j.chom.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Fisher MC, et al. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nilsson RH, et al. Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019;17:95–109. doi: 10.1038/s41579-018-0116-y. [DOI] [PubMed] [Google Scholar]
- 203.Nash AK, et al. The gut mycobiome of the human microbiome project healthy cohort. Microbiome. 2017;5:153. doi: 10.1186/s40168-017-0373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Auchtung TA, et al. Investigating colonization of the healthy adult gastrointestinal tract by fungi. mSphere. 2018;3:e00092-18. doi: 10.1128/mSphere.00092-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.