

Abstract
Triple-negative breast cancer (TNBC) is an aggressive disease that disproportionately affects African American (AA) women. Limited targeted therapeutic options exist for TNBC patients. Here, we employ spatial transcriptomics to interrogate tissue from a racially diverse TNBC cohort to comprehensively annotate the transcriptional states of spatially resolved cellular populations. A total of 38,706 spatial features from a cohort of 28 sections from 14 patients were analyzed. Intratumoral analysis of spatial features from individual sections revealed heterogeneous transcriptional substructures. However, integrated analysis of all samples resulted in nine transcriptionally distinct clusters that mapped across all individual sections. Furthermore, novel use of join count analysis demonstrated non-random directional spatial dependencies of the transcriptionally defined shared clusters, supporting a conserved spatio-transcriptional architecture in TNBC. These findings were substantiated in an independent validation cohort comprising 17,861 spatial features representing 15 samples from 8 patients. Stratification of samples by race revealed race-associated differences in hypoxic tumor content and regions of immune-rich infiltrate. Overall, this study combined spatial and functional molecular analyses to define the tumor architecture of TNBC, with potential implications in understanding TNBC disparities.
Introduction
Breast cancer – the most commonly diagnosed cancer in women – is a disease spectrum that can be classified into several histological and molecular subtypes (1). Triple-negative breast cancer (TNBC) is a form of breast cancer that does not express estrogen receptor, progesterone receptor, or amplified HER2, and comprises approximately 15% of invasive breast cancers (2). TNBC remains clinically challenging, as it is associated with more aggressive disease, earlier metastasis, higher rates of relapse, and poorer outcomes than other breast cancer subtypes (3). TNBC is known to be a genetically heterogenous disease, with many driver events giving rise to several intrinsic subtypes with varied clinical outcomes (4, 5). This, in addition to the lack of expression of actionable targets, has made development of effective therapies for TNBC difficult (6).
TNBC disproportionately affects African American (AA) women and women of sub-Saharan African ancestry, who have higher incidence, greater TNBC-related mortality, and poorer 5-year survival rates compared to Caucasian women (7–9). The association of TNBC risk with African ancestry suggests a genetic component to the disparity (10), and genomic studies have identified unique characteristics of AA, African, and Caucasian breast cancers (11, 12). However, the resulting effects on tumor biology are poorly understood. Thus, descriptive and clinical studies of TNBC must prioritize diverse representation in study cohorts, which has historically been inadequate (13).
Advanced molecular profiling provides an opportunity to further our understanding of disease biology and therapeutic vulnerabilities through comprehensive characterization of tumor specimens. Transcriptomic profiling technologies have evolved to allow the measurement of comprehensive gene expression from single cells, which can resolve the composition of cell types within the tumor microenvironment to reveal the extent of tumor heterogeneity (14). Further advancement now allows for spatially resolved whole-transcriptome measurements, where tissue architecture is maintained and provides critical context for higher-dimensional data interpretation (15, 16). Spatial transcriptomics (ST) technologies are particularly suitable for the study of solid tumors, which develop in a complex environment where communication between malignant cells, immune cells, fibroblasts, and vasculature can influence disease progression (17). For instance, cellular interactions in breast tumors can take the form of metabolic crosstalk (18), immune crosstalk (19), and subclonal communication (20–22). Preservation of spatial context provides a critical framework in which to dissect these relationships.
In this study, we applied ST to comprehensively describe the transcriptional substructure and heterogeneity defining the cellular architecture of TNBC. We performed manual and expression-informed molecular annotation of 28 TNBC tissue sections from 14 patients, of whom seven self-identified as AA and seven as Caucasian. We then utilized clustering analysis to summarize and assess transcriptionally defined intratumoral heterogeneity. We further integrated expression data from all samples to generate a reference from which we defined nine shared clusters represented across the entire dataset. Molecular annotation tools were used to understand the biological context of cells represented by transcriptionally distinct clusters. Moreover, to analyze spatial relationships between clusters, we made novel use of join count analysis, which revealed consistent patterns of spatial exclusion and dependency in TNBC. We validated our identified reference clusters and their spatial relationships in an independent cohort of 15 additional samples from eight AA patients. Finally, functional annotation of shared clusters revealed racial differences in the expression of clinically significant immune- and hypoxia-related genes, which may shed light on racial disparities in TNBC clinical outcomes.
Materials & Methods
Sample selection
De-identified human breast tissues were procured from the Virginia Commonwealth University (VCU) Massey Cancer Center Tissue and Data Acquisition and Analysis Core under a protocol approved by the VCU Institutional Review Board (VCU IRB HM2471). All patients provided written informed consent. Specimens were obtained during standard-of care procedures and banked when judged to be in excess of that required for patient diagnosis and treatment. Fresh samples procured during routine gross examination in surgical pathology were deposited in 2ml cryovials, snap-frozen in liquid nitrogen, then stored at −80°C. Tissue samples were embedded in Scigen Tissue Plus O.C.T. Compound (Fisher Scientific) prior to cryosectioning.
All samples selected for study were primary triple-negative breast cancer (TNBC) tumors collected between the years of 2002 and 2019. Samples were divided into two cohorts: a reference cohort of 28 samples, and a validation cohort of 15 samples. Reference cohort samples were obtained from 14 TNBC patients, 7 of whom identified as Black or African American (AA) and 7 who identified as Caucasian or Non-Hispanic White. Every effort was made to stage-match and age-match the samples, with age < 50 and stage II tumors preferred. 11 of 14 tumors represented in the reference cohort did not receive neoadjuvant treatment; these patients may have declined therapy or were not candidates for neoadjuvant treatment at the time of collection. Validation cohort samples were obtained from 8 patients, all of whom were AA, and varied from stage II to stage III. 6 of 8 tumors represented in the validation cohort received neoadjuvant treatment.
Two frozen blocks representing non-adjacent tumor sections were available for all patients, with the exception of three cases with limited tissue available, for which adjacent sections were provided, and one case for which a single section was provided. Sections were randomly sampled from frozen blocks to reduce selection bias. Distances between paired non-adjacent sections varied based on the size of the tumor and its handling at the time of banking; most sections sampled the original tumor centrally. Full specimen information is available in Supplemental File 1. In total, we applied spatial transcriptomics (ST) to 43 tissue sections, representing 22 tumor specimens.
Visium Spatial Gene Expression assay and sequencing
Fresh frozen, OCT-embedded tissues were cryosectioned at a thickness of 10μm and mounted on Visium spatial gene expression slides (10x Genomics, #1000184), which contain four 6.5mm x 6.5mm capture areas comprising 5000 barcoded spatial features each. The samples were processed as described in the manufacturer’s protocols. Briefly, tissues were stained with hematoxylin and eosin (H&E) and microscopic images were captured with a Zeiss Axioscan2 microscope using a 10x objective. Tissues were then permeabilized, allowing RNA to diffuse and bind to the slide surface via polyA capture. Permeabilization was performed for 9 minutes, which was determined using the Spatial Tissue Optimization procedure (10x Genomics, #1000193). Following permeabilization, cDNA was synthesized on-slide from the immobilized RNA. cDNA was then collected and used to generate sequencing libraries containing unique sample indices, as well as P5 and P7 primers for Illumina-compatible sequencing. Paired-end sequencing (2×100) was performed on an Illumina NovaSeq 6000 instrument to produce a minimum of ~250 million read pairs per sample (Supp. Fig. 1).
Data pre-processing and normalization
Sequencing data was pre-processed using the Space Ranger pipelines (version 1.1.0) (10x Genomics). BCL data was demultiplexed and converted to FASTQ format using the spaceranger mkfastq pipeline. The spaceranger count pipeline was then used for read alignment to human reference genome GRCh38, Unique Molecular Identifier (UMI) counting, and generation of feature-spot matrices corresponding to the microscopic tissue image. The pipeline also performs automatic tissue detection and fiducial alignment from the image. The raw UMI counts and spatial barcodes were imported to the R program Seurat (RRID:SCR_016341, version 4.0.4) (23) with the Load10x_Spatial command. Gene expression count data was then normalized using the SCtransform method from the R package sctransform (RRID:SCR_022146, version 0.3.2) (24). We chose this method due to its ability to correct for technical read depth variation while preserving biological variation, which is often present in ST data due to the varying cellularity across a tissue section (24).
Histopathological annotation
Frozen tissues from each block selected for study were stained with H&E and evaluated for tumor adequacy. The images captured from Visium slides were reviewed and annotated by a pathologist, using Adobe Photoshop software (RRID:SCR_014199). For each slide, areas of carcinoma were delineated from the intervening and surrounding desmoplastic stromal tissue. The degree of tumoral and stromal inflammatory cell infiltration was also assessed and reported quantitatively. Where applicable, areas of necrosis, dense immune infiltrates, or background stromal fibrosis or desmoplasia were indicated.
ESTIMATE analysis
ESTIMATE analysis was performed with the R package estimate (version 1.0.13) (25). ESTIMATE utilizes gene signatures derived from immune and stromal cells to calculate two corresponding enrichment scores: Immune Score and Stromal Score. Tumor Purity is then computed from these scores as described by Yoshihara et al (25). ESTIMATE was applied to normalized expression data from each spatial feature. The analysis returned a Stromal Score, Immune Score, and Tumor Purity for each feature, allowing visualization on a spatial map or as a violin plot.
Dimensionality reduction and clustering analysis
Normalized counts were subjected to principal component analysis (PCA) through 30 principal components. A shared nearest neighbor graph was generated, and clustering was performed with the Louvain modularity optimization algorithm at a resolution of 0.4. This resolution produced a stable set of clusters without over- or under-clustering the data (Supp. Fig 2A–B). Resolution determination was aided by the R package clustree (RRID:SCR_016293, version 0.5.0)(26), and by evaluation of ESTIMATE scores, UMAP projections, and spatial profiles of clusters. Dimensionality reduction was performed by UMAP (Uniform Manifold Approximation and Projection). Seurat was used for all clustering and dimensionality reduction analyses.
TNBC subtype determination
Subtype determination at the cluster level was performed with the TNBCtype webtool, which classifies samples based on centroid correlation to 6 TNBC subtypes (27). Normalized gene expression was averaged across all features in each cluster and provided as input. All reported results had a p-value < 0.05.
Gene set enrichment analysis (GSEA)
GSEA was applied at both the feature level and the cluster level using the Broad Institute’s GenePattern software (RRID:SCR_003199) (28). At the feature level, single sample GSEA (ssGSEA) (29) was applied to expression data from each spatial feature, using gene sets from the Hallmarks collection (30) and infiltrating immune cell signatures defined by Yu et al (31) to yield feature enrichment scores. To apply GSEA at the cluster level, differentially expressed cluster markers were first determined by Wilcoxon rank sum testing, using Seurat’s FindAllMarkers feature. Gene lists pertaining to each cluster were analyzed by Pre-ranked GSEA against the Hallmark gene sets.
CIBERSORTx analysis
Deconvolution of immune content in clusters was performed with CIBERSORTx (RRID:SCR_016955) (32). Normalized gene expression was summed across all features in each cluster to create a pseudo-bulk profile. This was provided as a mixture file and analyzed against the LM22 immune cell signatures (33). The analysis was run in absolute mode with B-mode batch correction at 500 permutations.
Data integration of reference cohort
Integration of transcriptional data from all 28 reference cohort samples was performed with Seurat using the method described by Stuart et al (34) with the goal of identifying shared cell populations. Briefly, the method identifies transcriptionally similar anchor cells in all samples, which are used to correct technical batch effect and integrate the datasets. After individual SCTransform normalization and PCA of samples, 3000 integration features (genes) were determined for use in anchor identification. Because of the large size of our dataset, use of a canonical correlation analysis – which determines anchors between all possible sample pairings – for anchor determination proved to be computationally prohibitive. We instead used reciprocal PCA, with sample 092A as the reference. The anchors identified were then used for integration. PCA was performed on the integrated data and integrated clusters (ICs) were defined at a clustering resolution of 0.4.
Label transfer to validation cohort
Integrated cluster assignments were transferred to the validation cohort utilizing Seurat’s reference mapping feature. Following normalization, each sample in the validation cohort was individually queried against the integrated reference to find transcriptionally aligned anchors, using canonical correlation analysis. Identified anchors were then used to project IC labels from the reference onto the query sample.
Join count statistics
Join count analysis (JCA) is a statistical method for quantifying spatial correlation of contiguous nominal data. For application to our ST data, spatial maps were converted to a raster format, in which barcodes were represented by pixels coded by cluster assignment, as follows.
Spatial coordinates for each barcode were retrieved and annotated with the corresponding Integrated Cluster (IC) assignment. Using the R package raster (version 3.5-2), a rectangular raster was created with an extent encompassing the area of the tissue, and a resolution that captured one barcode per pixel with no empty space between. The resolution varied between samples, likely due to slight variations in the printing of the capture features on the slides. Each IC was then subset and rasterized individually using the raster parameters previously determined. Rasterizing ICs individually allowed each to be given a unique identifier coded in the resulting pixels. After each rasterization, a check was performed to ensure the number of pixels matched the number of barcodes in the subset. All rasterized IC subsets were then merged to recreate the full tissue section as a raster.
The rasterized sample was then compatible with JCA. Using the R package spdep (RRID:SCR_019294, version 1.1-11) (35), a neighbors list was created in the “queen” style, which identifies adjacent and diagonal neighbors for each feature. The resulting neighbors list was then subset to remove empty background space, as well as single features with no neighbors (e.g. features removed from the main tissue). The neighbors list was then supplemented with binary spatial weights, and multi-categorical join count analysis was performed with the command joincount.multi, which tabulates the joins of all possible combinations of cluster pairings within the sample. The test returns the observed join counts, the expected count under conditions of spatial randomness, and the variance of observed to expected calculated under non-free sampling. The z-score is then calculated as the difference between observed and expected counts, divided by the square root of the variance. Since this analysis does not correct for multiple testing, we selected a stringent z-score cutoff of +/− 3 (equivalent to a p-value of 0.00135, or 99.99% confidence) to assess significance.
We developed an R Bioconductor package, stJoincount (https://github.com/Nina-Song/stJoincount), to facilitate JCA of ST data produced by the 10x Genomics Visium platform. The program finds optimal parameters for sample rasterization, and performs JCA followed by result visualization.
Survival analysis
Relapse-free survival in our reference cohort was defined as days to first recurrence or days to death, if no recurrence first occurred. If no death event was recorded, patients were considered censored at the date of their last clinical encounter. Survival data can be found in Supplemental File 1. NDRG1 and IGKC expression were averaged across all features in each sample to produce a summary expression value. Cutoff values to differentiate low from high expression were determined with the webtool CutoffFinder (36) to optimize the significance of correlation with survival. Survival analysis was then performed with R package survival (RRID:SCR_021137, version 3.3-1) (37). Significance was determined by Cox-Mantel logrank test.
Survival analysis of publicly available breast cancer microarray-based expression datasets was performed using the web-based Kaplan-Meier Plotter tool (RRID:SCR_018753) (38). Datasets queried are listed in Supplemental File 2. NDRG1 expression was examined with Probe ID 200632_s_at, and IGKC with Probe ID 211643_x_at. Analysis was performed with NDRG1 and IGKC individually, and with the ratio of NDRG1 expression (as the numerator) to IGKC expression (as the denominator). Optimal cutoff values to define high and low expression were determined based on an iterative approach as described in Lanczky et al (38). Biased arrays and redundant samples were removed from the analysis, resulting in a total cohort size of 4929 patients. The cohort was analyzed as a whole, and was also subset into PAM50 molecular subtypes: basal (n = 953), luminal A (n = 1809), luminal B (n = 1353), and HER2+ (n = 695). Significance was determined by Cox-Mantel logrank test.
Data availability
Expression data and tissue images for all samples are available in the Gene Expression Omnibus (GEO) under record GSE210616. Datasets analyzed for survival analysis are all publicly available and are listed in Supplemental File 2.
Results
Sample selection and spatial transcriptomics
We selected 14 primary TNBC tumors for study from 14 patients, including seven that self-identified as AA and seven that identified as Caucasian. Most tumors in this reference cohort were histopathologically classified as stage II and 11 of 14 were chemo-naïve (Supp File 1). Of the samples that received neoadjuvant chemotherapy, two were AA and one was Caucasian. From each tumor, we obtained two non-adjacent tissue sections, except for one case in which we used serial sections due to limited material. Thus, this reference cohort was comprised of 28 TNBC tissue sections, to which we applied spatial transcriptomics (ST).
The ST workflow is described in Methods and summarized in Figure 1. Following sequencing, all data proceeded through a pipeline for demultiplexing, genome alignment, and spatial alignment. Data were quality assessed and high-quality data was obtained from all samples (Supp Fig.1). On average, each sample produced expression data from 1,382 individual spatial features, and 22,855 genes were detected on average per section. Expression data for each sample was normalized using SCTransform to best correct for technical artifacts while preserving biological variation (24). All subsequent analysis was conducted with the resulting normalized expression values.
Figure 1. Overview of spatial transcriptomics workflow and study design.
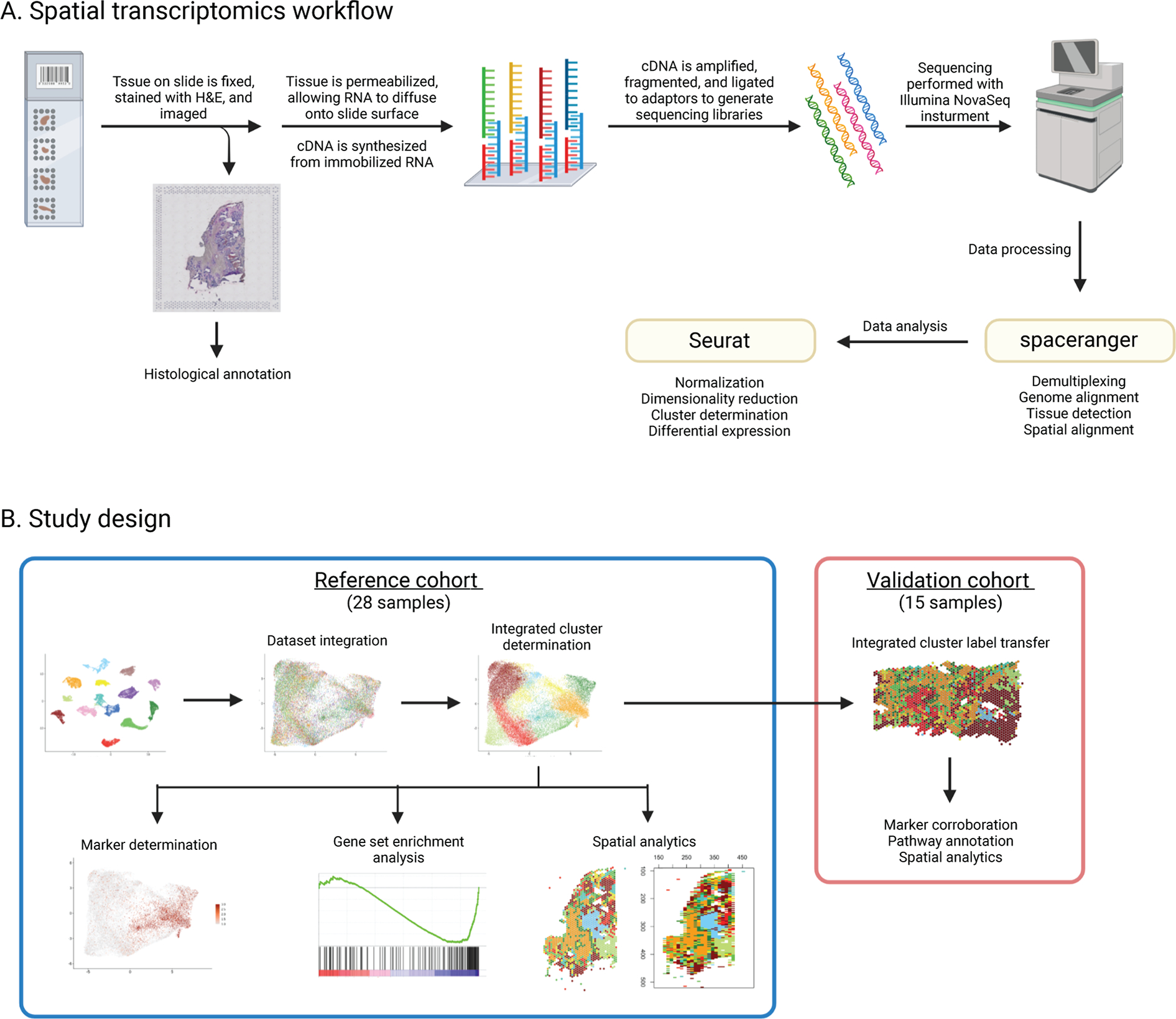
(A) An overview of the 10x Genomics Visium spatial transcriptomics workflow, beginning with affixation of tissue to Visium spatial gene expression slides and ending with sequencing data processing and analysis. (B) An overview of the study design, including analysis strategies for a reference cohort of 28 samples, and a validation cohort of 15 samples. Image created with BioRender.com.
Manual and gene signature-informed sample annotation
We first analyzed whether gene expression data could be used for discernment of carcinoma from surrounding tissue. For comparison, H&E-stained images of each sample were manually annotated by a board certified anatomic pathologist (Fig 2A), which revealed that sections were dominated by regions of tumor and fibrosis. Less common were regions of necrosis, adipose tissue, and immune infiltrate (Supp Fig. 3A). We then applied ESTIMATE to normalized expression data to concordantly define the transcriptional substructure of each sample. ESTIMATE was developed and validated on bulk transcriptional data to infer tumor, stromal, and immune content using gene expression signatures (25). Application of ESTIMATE at the feature level closely mirrored manual histological annotation. In representative sample 094D (Fig. 2A), ESTIMATE’s stromal score highlighted extensive regions of fibrosis (Fig. 2B, Supp Fig. 3B) and tumor purity score accurately identified regions of tumor (Fig. 2B–C).
Figure 2. Transcriptional estimation of tumor purity approximates expert pathological annotation.
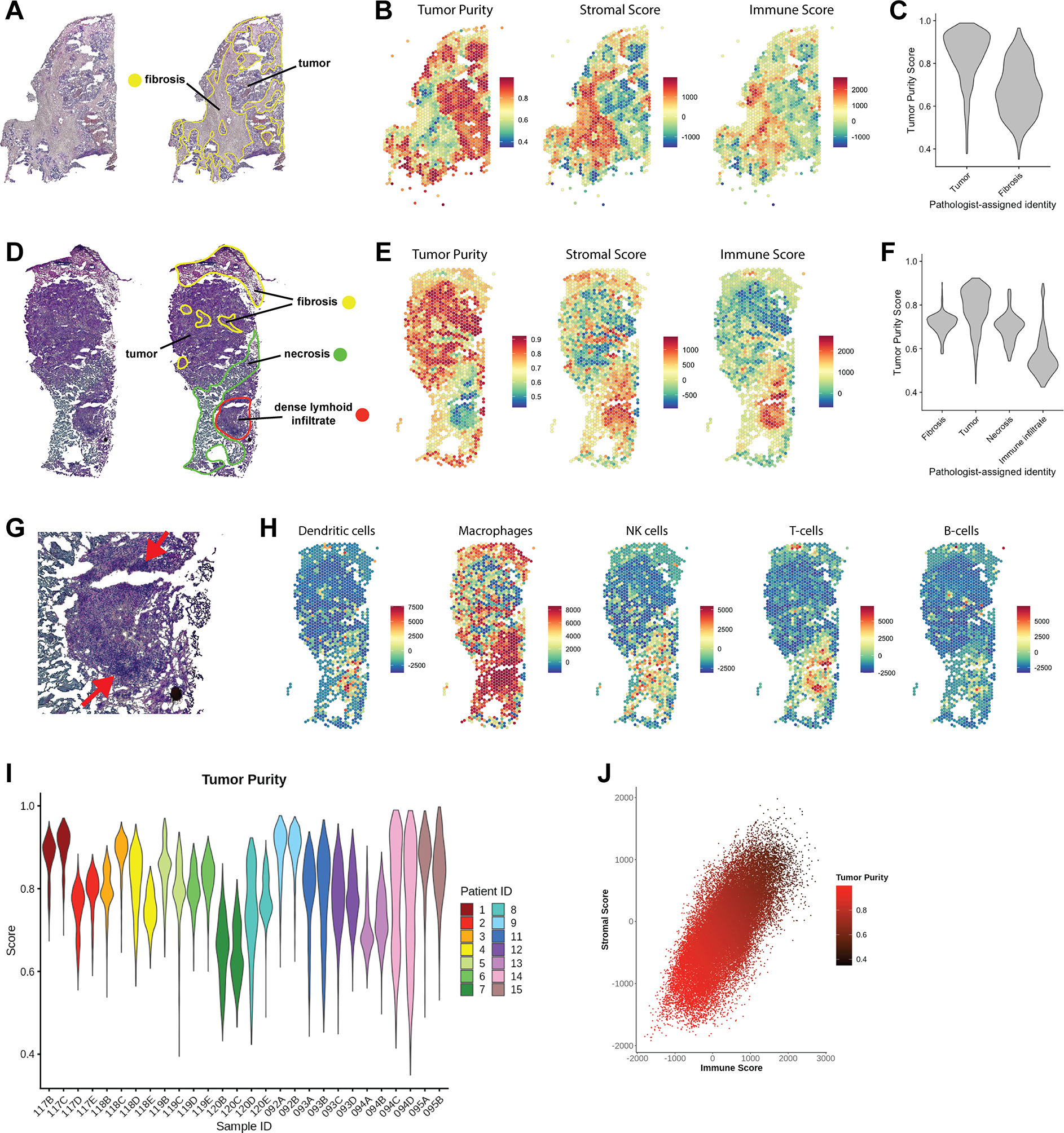
(A) H&E stained images of sample 094D. Pathologist’s annotation highlights extensive regions of fibrosis. (B) Expression data per spatial feature was analyzed using ESTIMATE to generate a Tumor Purity score, Stromal Score, and Immune Score. (C) Violin plot of tumor purity scores of features in the regions identified as tumor and fibrosis by manual annotation of sample 094D. (D) Pathologist’s annotation of sample 120D, which contained a region of dense lymphoid infiltrate in addition to tumor, fibrotic, and necrotic regions. (E) Per-feature ESTIMATE analysis of sample 120D. (F) Comparison of pathologist-assigned identity to Tumor Purity score for sample 120D. (G) Magnified region of immune infiltrate in sample 120D, with an arrow indicating dense outer edge. (H) Per-feature enrichment scores for several tumor-associated immune cell signatures (31) as determined by ssGSEA. (I) Violin plot of tumor purity profiles of all 28 samples. Samples are colored by patient ID; two samples were available for each patient. (J) Scatter plot of immune and stromal scores per feature in all samples, with a correlation coefficient of r = 0.7. Data points are colored by Tumor Purity, which is inversely related to immune and stromal scores.
In independent representative case 120D, manual annotation identified fibrotic and necrotic regions, in addition to a region of dense lymphoid infiltrate (Fig. 2D). Once again, ESTIMATE scores corresponded with the histopathological assessment (Fig. 2E–F, Supp Fig 3C). To further characterize the identity of infiltrating immune cells, we applied a set of cell-type specific expression signatures derived from tumor-associated immune cells (31). The spatial pattern of T-cell enrichment matches a dense ring of lymphoid cells, while the remainder of the region strongly expresses a macrophage signature (Fig. 2G–H). We can thus apply ESTIMATE and cell-type specific gene signatures to ST data to quantify tumor and immune content, and augment manual annotation.
Samples in the reference cohort varied in their tumor content, with samples originating from the same patient displaying similar tumor purity profiles (Fig. 2I) and ESTIMATE scores (Supp Fig. 3D–E). UMAP embeddings also reflect the general transcriptional similarity of paired samples (Supp. Fig. 3F). Furthermore, the immune score and stromal score were closely correlated in all samples, with both enriched in histologically defined regions of fibrosis (Fig. 2J).
Clustering reveals intra-tumor heterogeneity
TNBC is genetically heterogeneous (1), which is illustrated when ST data is subject to unsupervised clustering within single specimens. Clustering of spatial features at low resolution broadly allows discernment of tumor and stromal regions in agreement with histological and informatics-based annotation (Supp. Fig. 2A–B). Increasing the resolution produces more clusters, revealing transcriptionally distinct subregions of tumor and stroma (Supp Fig. 2A–B). We determined a resolution of 0.4 to capture maximum sample heterogeneity without over-clustering the data (Supp. Fig. 2A). At this resolution, samples contained an average of four tumor clusters and one non-tumor cluster (Supp Fig. 2C).
Clustering analysis of sample 094D based on expression data produced six clusters (Fig. 3A–B). ESTIMATE analysis revealed three clusters (2, 5, and 6) with high tumor purity, while cluster 4 demonstrated low tumor purity and represents fibrosis (Fig. 3C–D). Determination of TNBC subtypes defined by Lehmann et al (39) for each cluster revealed subtype mixing within the sample (Fig. 3E). Basal like-1 (BL1) and mesenchymal (M) subtypes were represented in tumor regions, while cluster 4 was classified as mesenchymal stem like (MSL), a subtype that represents tumor associated stromal cells (40). Cluster 1, which separates regions of tumor from regions of fibrosis was classified as immunomodulatory (IM), which reflects the presence of tumor-infiltrating lymphocytes (40).
Figure 3. Clustering reveals transcriptionally distinct regions of tumor and stroma.
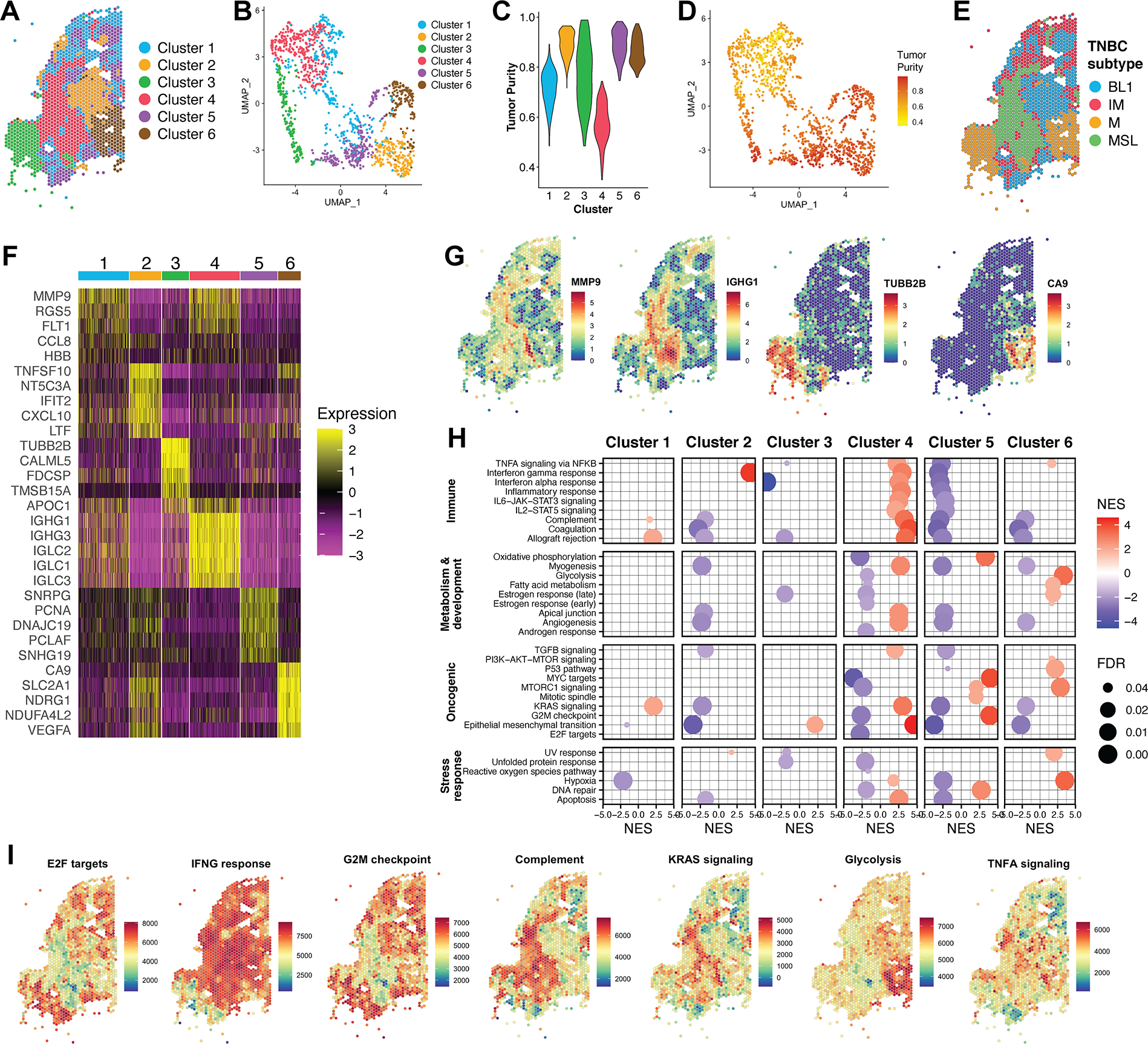
(A) Graph-based clustering of sample 094D resulted in 6 transcriptionally distinct clusters. (B) UMAP projection of sample 094D, colored by cluster assignment. (C) Tumor purity profiles of clusters identified in sample 094D. (D) UMAP projection of sample 094D, colored by Tumor Purity score. (E) TNBC subtypes assigned to each cluster. (F) Heatmap of top differentially expressed genes per cluster for sample 094D. Colors represent the Pearson residuals of normalized and variance-stabilized expression data (24). Each column represents a single spatial feature, and features are grouped by their cluster assignment. (G) Spatial mapping of representative marker genes for clusters in sample 094D. Color scale represents log-transformed normalized gene expression. (H) GSEA for clusters of sample 094D against Hallmark collection gene sets. Only results with a false discovery rate (FDR) < 0.05 are displayed. NES = Normalized enrichment score. (I) Spatially mapped feature-level ssGSEA scores for selected Hallmark gene sets. Color scale represents enrichment scores.
We determined expression markers for each cluster by differential expression, and spatial visualization of these genes confirms their discrete localization (Fig. 3F–G). To better understand the biological context of cells represented by each cluster, we applied gene set enrichment analysis (GSEA) to identify molecular pathways enriched among each cluster (Fig. 3H). In agreement with ESTIMATE analysis, cluster 4 was enriched in immune-related pathways, while tumor clusters were enriched in various oncogenic and metabolic pathways. For example, GSEA implies the activation of MYC and cell cycle signaling driving cancer cells within cluster 5, while cluster 6 likely includes tumor cells that are hypoxic and glycolysis dependent. Furthermore, the results of cluster-level GSEA are consistent with feature-level enrichment analysis when plotted spatially (Fig. 3I).
Dataset integration and identification of shared cell populations
We next sought to perform clustering of all features across all samples to investigate shared TNBC spatio-transcriptional characteristics. To do this, we integrated expression data from all features to consolidate the natural deviation across tumors from multiple individuals with varying genetic backgrounds (Supp Fig. 4). Integration allows us to use diverse individual datasets to create a comprehensive reference from which common transcriptionally defined TNBC cell types can be annotated (34). The resulting integrated dataset comprised 38,706 spatial features, with nearly equal representation of AA and Caucasian features (19,401 and 19,305, respectively).
The integrated data was subject to clustering analysis to produce nine integrated clusters (ICs) (Fig. 4A). Cluster assignments could then be mapped back to features in individual samples to assess the substructure of clusters across cases and sections (Fig. 4B). ESTIMATE analysis indicated that IC3, IC6, and IC7 represent likely stromal-immune cell types, while the remaining clusters are defined by tumor cell types, with varying degrees of tumor purity (Fig. 4C). Importantly, all ICs were represented in all individual samples in varying proportions (Fig. 4D).
Figure 4. Clustering of an integrated dataset reveals shared cell populations.
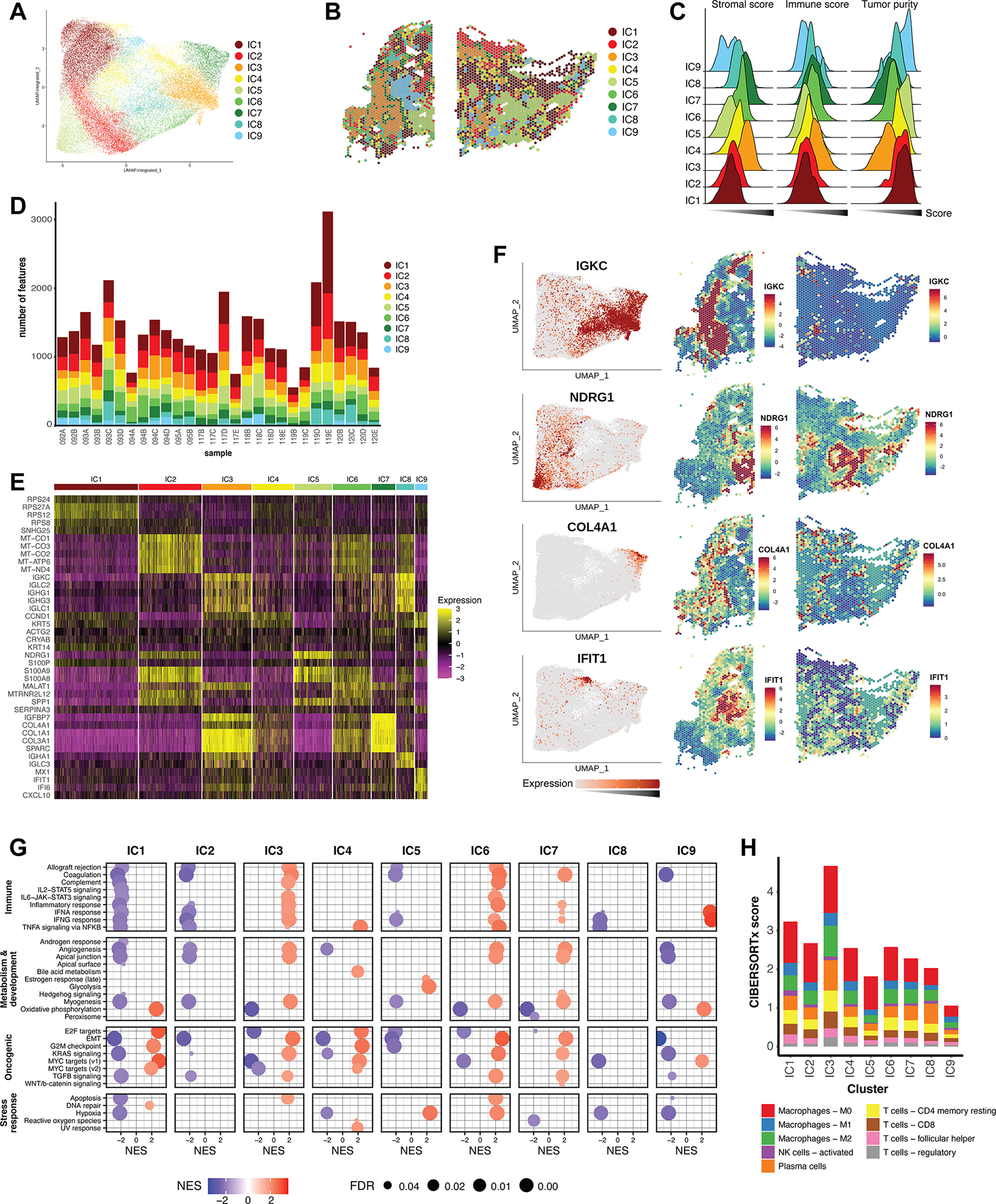
A) Graph-based clustering analysis of the integrated reference dataset resulted in nine integrated clusters (ICs), depicted here on the integrated UMAP embeddings. (B) ICs mapped back to individual representative samples. (C) Ridgeline plot of ESTIMATE scores for all ICs, calculated for all corresponding features in the reference dataset. The x-axis represents score, and increases from left to right. (D) Bar graph representing the distribution of the nine ICs across the individual samples of the dataset. (E) Heatmap of top differentially expressed marker genes of the ICs across the integrated dataset. Color scale represents the Pearson residuals of normalized and variance-stabilized expression data (24). (F) Expression of selected marker genes represented on the integrated UMAP embedding, as well as spatially in individual samples. Color scales represent Pearson residuals. (G) GSEA of ICs using the Hallmark gene sets. Only results with a false discovery rate (FDR) < 0.05 are displayed. NES = Normalized enrichment score. (H) CIBERSORTx was used to deconvolute immune mixtures in each ICs. Absolute CIBERSORTx scores are represented on the y-axis, and immune identities of selected cell types are differentiated by color.
Assessment of marker genes revealed further characteristics of ICs (Fig. 4E). The most widely represented tumor clusters, IC1 and IC2, are enriched in ribosomal and mitochondrial genes, respectively. We interpret these as archetypal characteristics of highly metabolic tumors. The remaining tumor ICs have more distinctive expression patterns. For example, IC5 expressed high levels of NDRG1, a hypoxia-inducible gene (41) that has been associated with breast cancer progression and brain metastasis (42). IC9 distinctly expressed the antiviral gene IFIT1, which has been studied in breast cancer as a predictive marker for chemotherapy response (43). The non-tumor clusters also expressed distinguishing markers: IC3 was enriched in immunoglobulin genes, and IC7 in collagens. Gene expression mapping on individual samples confirms the co-localization of marker genes with their respective ICs (Fig 4F).
GSEA of IC expression data revealed enrichment of a hypoxic signature in IC5, while IC9 was distinctly enriched for genes involved in interferon alpha and gamma response pathways (Fig 4G). IC1 and IC4 were enriched in several cell cycle-promoting pathways and likely represent actively dividing tumor cells. On the other hand, IC5 may reflect dormant cells with inactive E2F signaling and G2M checkpoint signaling, consistent with a hypoxic environment. In agreement with ESTIMATE analysis, IC3, IC6, and IC7 were enriched in immune signaling pathways, while tumor clusters IC1, IC2, and IC5 were deficient in these (Fig. 4G).
To further explore the immune effector cell composition of ICs, we applied CIBERSORTx [29] to IC expression data. Consistent with ESTIMATE and GSEA, IC3 had the highest overall CIBERSORTx score, suggesting that it contains the highest immune content (Fig. 4H). Notably, tumor cluster IC5 has a high proportion of tumor associated macrophages but is relatively deficient in CD4 and CD8 T cells (Fig. 4H), which is supported by enrichment analysis of lymphocyte signatures (Supp Fig. 5A).
Spatial distribution of integrated clusters
Following IC mapping across individual samples, visual inspection suggested that non-random spatial patterns exist. To quantitively evaluate spatial correlation between ICs, we employed join count analysis (JCA) (Fig. 5A), which is appropriate for determination of the structures of spatially contiguous nominal data of two or more categories (44). In the simplest form of data, in which only two categories are present, JCA determines the degree of dispersion or interspersion of the observations. When applied to multi-categorical data, as we have, JCA determines the strength and directionality of the spatial correlation between all category pairings (45).
Figure 5. Spatial analytics of integrated clusters reveals a consistent molecular topography.
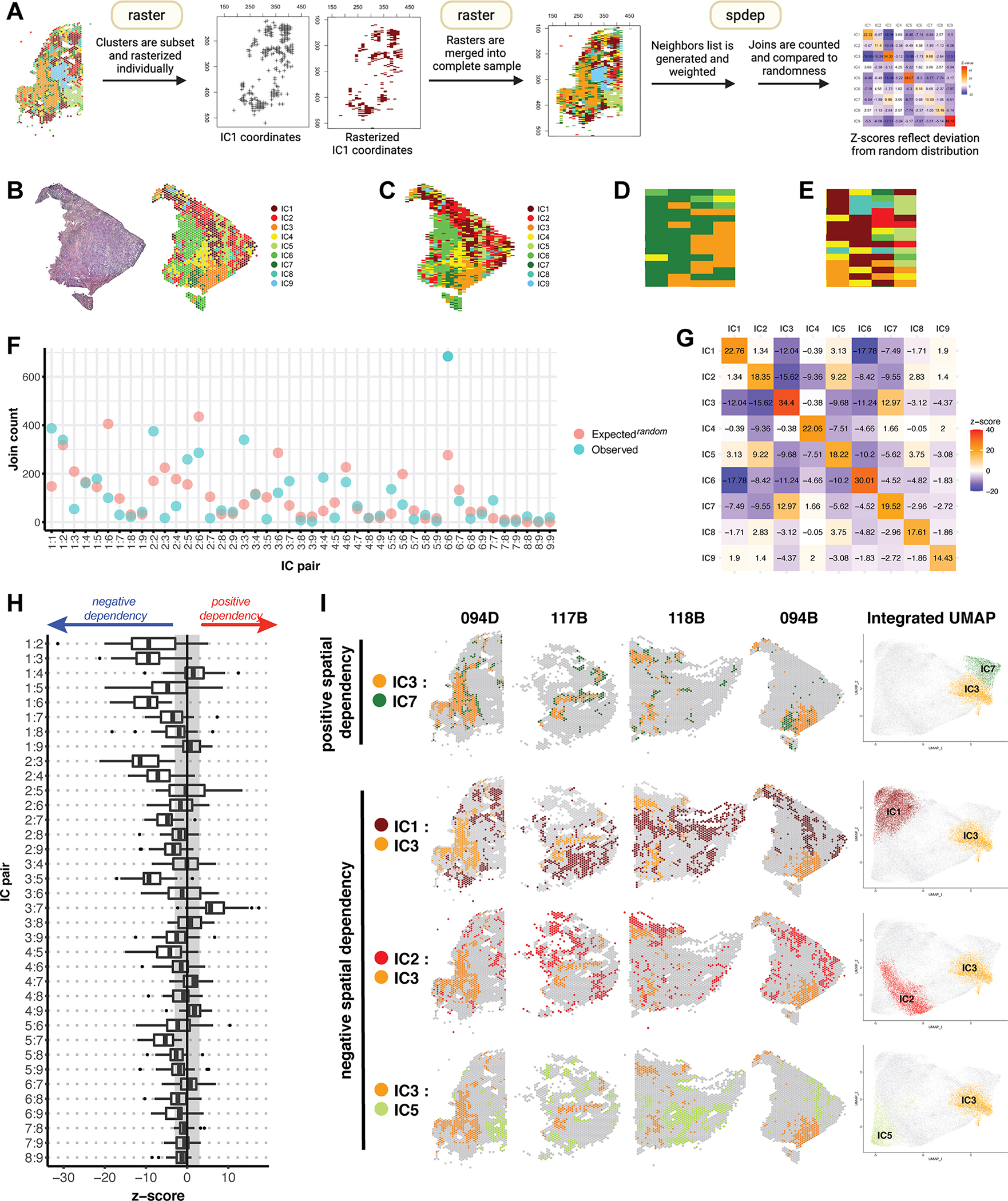
(A) An overview of join count analysis (JCA). Each sample is subset by integrated cluster (IC), and corresponding coordinates are rasterized individually using R package raster. Subsetting allows each IC to be coded uniquely during rasterization. Individual rasters for all 9 ICs are then merged to recreate the complete sample. R package spdep is then used to generate a neighbors list with spatial weights for each observation. Join counts for all cluster pairs are then tabulated and compared to a spatially random distribution. (B) Mapping of ICs on sample 094B. (C) Raster of sample 094B, in which each spatial feature is represented by a pixel coded by IC assignment. (D) A region of sample 094B representative of clustering, in which any given pixel is likely to have a similar group of neighbors. (E) A region representative of dispersion, in which most pixels have dissimilar neighbors. (F) JCA results for sample 094B. For each IC pair, the observed join count is indicated, along with what would be expected under conditions of spatial randomness. (G) Z-values corresponding to the analysis in (F), reflecting the deviation of the observed join count from theoretical randomness. A positive z-value indicates spatial clustering, a negative value indicates spatial dispersion, and a value close to 0 indicates no spatial dependency, i.e. a near random distribution. (H) A summary of z-values resulting from JCA of all 28 samples represented in a box plot. The box represents the interquartile range, with the median marked by a vertical line. Outliers are represented by points outside the range of the whiskers. The shaded region represents our selected z-value cutoff of +/−3 (as described in Methods) to assess significance of results. All autocorrelations were strongly positive (z ≥ 10) and are not represented here. (I) Spatial mapping and UMAP embeddings of selected IC pairs found to trend towards positive and negative spatial dependency.
The workflow for JCA of ST data is summarized in Fig. 5A and described in Methods. To apply JCA to our samples, we first rasterize our spatial data while maintaining categorical information (Fig. 5B–C). In the resulting raster, each spatial barcode is represented by a pixel coded by IC assignment. Neighbors for each observation are then identified, and JCA is performed to quantify “joins”, paired categories of neighboring observations. Spatially proximate observations will have many similar neighbors and joins, which would be greater than expected in a random arrangement of features (Fig. 5D). By the same rationale, spatially dispersed observations will manifest as dissimilarity of neighbors and fewer joins than in a random arrangement (Fig. 5E). The observed join count for each pair of observations can then be compared to what would be expected under spatially random conditions (Fig. 5F). The statistical significance of the spatial correlation is represented by the z-score, which reflects the degree and directionality of deviation from randomness (Fig. 5G). A positive z-score indicates clustering, while a negative z-score indicates dispersion. Each IC is typically characterized by strong positive autocorrelation, while IC pairings vary on a spectrum of spatial dependencies (Fig. 5G).
We applied JCA to all 28 samples, and Fig. 5H summarizes the resulting z-scores. We observed consistent patterns in the spatial relationships of several cluster pairs. IC3 and IC7 were positively correlated in nearly all samples. We had previously characterized these as stromal-immune clusters distinguished by unique gene expression programs (Fig. 4E, G). Conversely, IC3 was consistently dispersed from IC1, IC2, and IC5, which represent tumor clusters. Thus, JCA results demonstrate that some TNBC tumor cells are non-randomly distanced from immunologically rich regions within the tumor microenvironment. The results are reinforced when presented spatially in representative samples and are further reflected in the UMAP embeddings of the integrated dataset (Fig. 5I).
Annotation of an independent validation cohort
We sought to validate the ICs defined from our reference cohort in a validation cohort of 15 additional, independent TNBC samples from 8 AA patients. Samples in the validation cohort varied from stage II to III and in tumor purity scores (Fig. 6A). Following data normalization, we annotated the validation cohort via reference mapping to the integrated reference dataset. Each validation sample was individually queried against the reference, and IC labels were transferred for each feature, analogous to reference mapping of single-cell RNA-seq data (34).
Figure 6. Annotation of a validation cohort with IC labels.
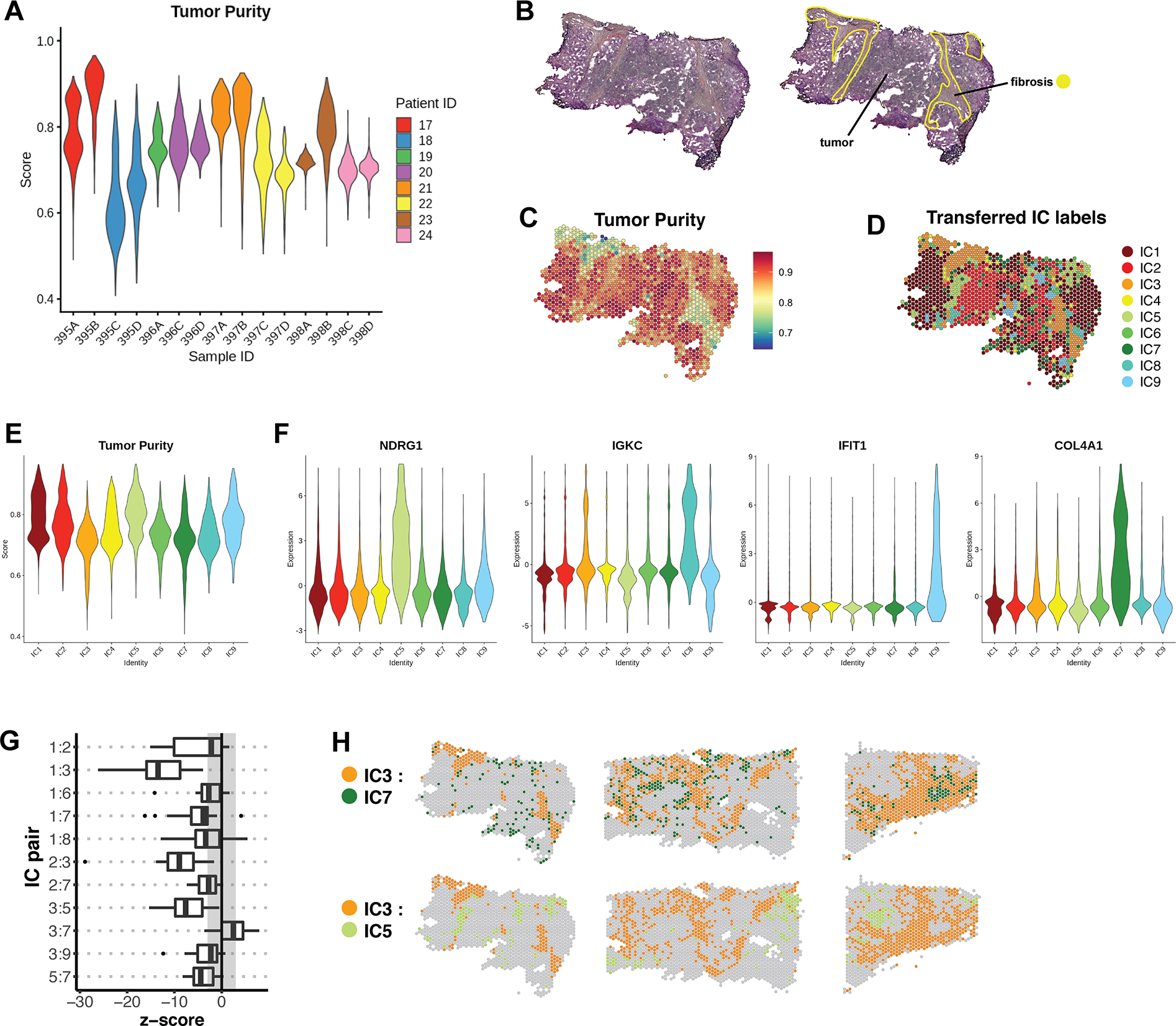
(A) Tumor purity profiles of 15 samples belonging to the validation cohort. (B) Representative sample 395B from the validation cohort, with pathologist’s manual annotation. (C) Per-feature Tumor Purity scores for sample 395B. (D) IC assignments for sample 395B, which were transferred from the integrated dataset by reference mapping. (E) Cumulative Tumor Purity scores for all samples in the validation cohort after each was individually queried for reference mapping. The x-axis represents transferred IC labels. (F) The expression of selected marker genes in the labeled validation cohort. Expression is represented by Pearson residuals. (G) Join count analysis was performed on labeled samples of the validation cohort. The box plot represents a summary of the resulting z-scores for a subset of cluster pairs that were found to be significant in the reference cohort. (H) Spatial validation of cluster pairs with positive and negative spatial dependencies in representative samples of the validation cohort.
On an individual basis, transferred IC labels were compared to manual annotations and tumor purity scores for each sample (Fig. 6B–D). In general, features assigned to the stromal-immune clusters (IC3, IC6, and IC7) had low tumor purity, and matched regions annotated as fibrosis. When tumor purity scores were cumulatively examined in all 15 samples, the pattern matched that seen in the reference cohort (Fig. 6E, 4C). We further validated that IC marker genes identified in the reference dataset corresponded with the label-transferred identities in the validation set (Fig. 6F). Additionally, JCA revealed that all strong spatial dependencies observed in the reference cohort were replicated in the validation samples (Fig. 6G–H). Thus, we observe a common spatio-transcriptional architecture in TNBC.
Racial differences revealed by TNBC spatial transcriptomics
Because our reference dataset contained a near-equal number of features from AA and Caucasian samples, we examined whether IC representation varied by race. We found a higher proportion of features representing IC3, IC6, and IC8 in Caucasian samples, while features representing IC5, IC7 and IC9 were predominantly contributed by AA samples (Fig. 7A). Furthermore, Caucasian samples contained significantly more IC3 by area, while AA samples contained more IC5 by area (Fig. 7B).
Figure 7. Annotation of ICs in the context of racial disparities.
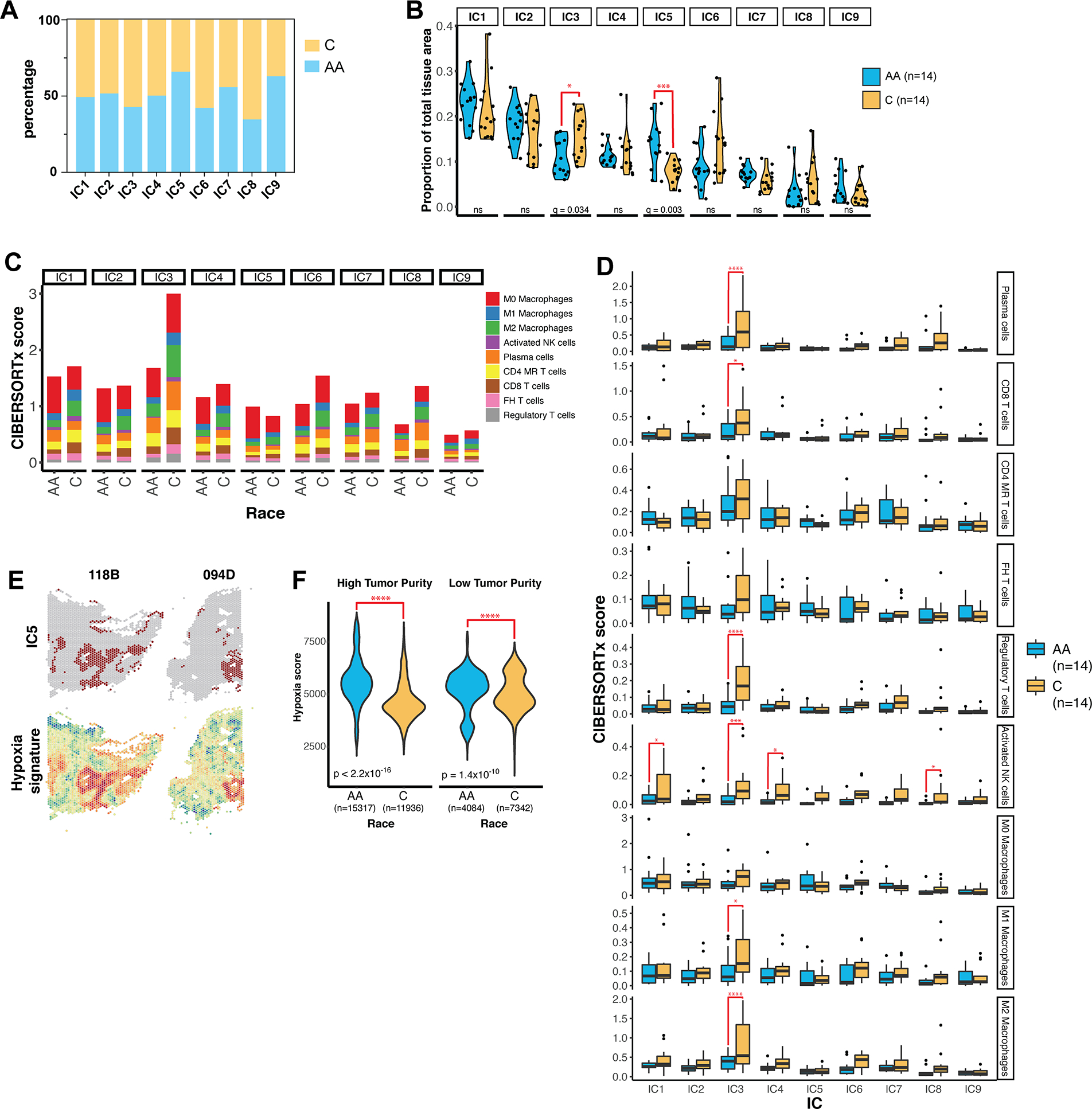
(A) The percentage of features in each IC contributed by Caucasian (C) and African American (AA) samples in the cohort. (B) The proportion of each IC by total tissue area in each sample was calculated as the number of spatial features belonging to the IC divided by the total number of features in the tissue. IC proportions were compared between AA and C samples using multiple t-test corrected by false discovery rate (FDR). (C) Immune content of ICs in AA and Caucasian samples as determined by CIBERSORTx. Absolute CIBERSORTx scores are represented on the y-axis. MR = memory resting, FH = follicular helper. (D) Boxplots summarizing the immune content determined by IC in individual samples. q-values were determined by multiple t-testing corrected by FDR. * = q < 0.05; *** = q < 0.001; **** = q < 0.0001. (E) Spatial maps of features assigned to IC5 and hypoxia signature scores in representative samples. (F) Violin plots of hypoxia enrichment score by race in features with high tumor purity (score > 0.75) and low tumor purity (score < 0.75). n indicates the number of spatial features in each group. P-value was determined by Welch’s two-sample t-test.
Given the disparity in clinical outcomes for AA and Caucasian TNBC patients, we sought to further investigate both IC3 and IC5. Annotation of IC3 previously revealed a strong immune identity (Fig. 4). Further CIBERSORTx analysis revealed that lymphocyte and macrophage content within IC3 was significantly greater in Caucasian samples, while AA samples contained lower immune content overall (Fig. 7C–D). Interestingly, M0 macrophage content was disproportionately high in AA samples, particularly in IC5 (Supp. Fig. 5B). GSEA revealed IC5 to be distinctly hypoxic and coordinately glycolytic (Fig. 4G), characteristics which are associated with cell growth, metastasis, and resistance to radiotherapy in breast cancer (46–48). We verified that IC5 corresponded to features with high hypoxia scores in both AA and Caucasian samples (Fig. 7E). However, features from AA samples had greater cumulative hypoxia scores than features from Caucasian samples (Supp Fig. 5C), and the average hypoxia score per section was higher in AA samples (Supp Fig. 5D). Furthermore, this difference was more pronounced in tumor regions, defined as features with high tumor purity scores (Fig. 7F).
JCA revealed a negative spatial association between IC5 and IC3 (Fig. 5H), which agrees with hypoxia having several established roles in immune suppression (49). The relationship of hypoxia to immune content in our cohort can be summarized by the expression ratio of NDRG1 to IGKC, the top marker genes for IC5 and IC3, respectively (Fig. 4E). We verify that the NDRG1 to IGKC expression ratio was significantly higher in AA features in our cohort, particularly in IC5 (Fig. 8A). Furthermore, we determined that high NDRG1 expression was associated with poor survival in our cohort, while high IGKC expression was protective (Fig. 8B–C).
Figure 8. NDRG1 and IGKC expression correlate with survival in TNBC and basal breast cancer.
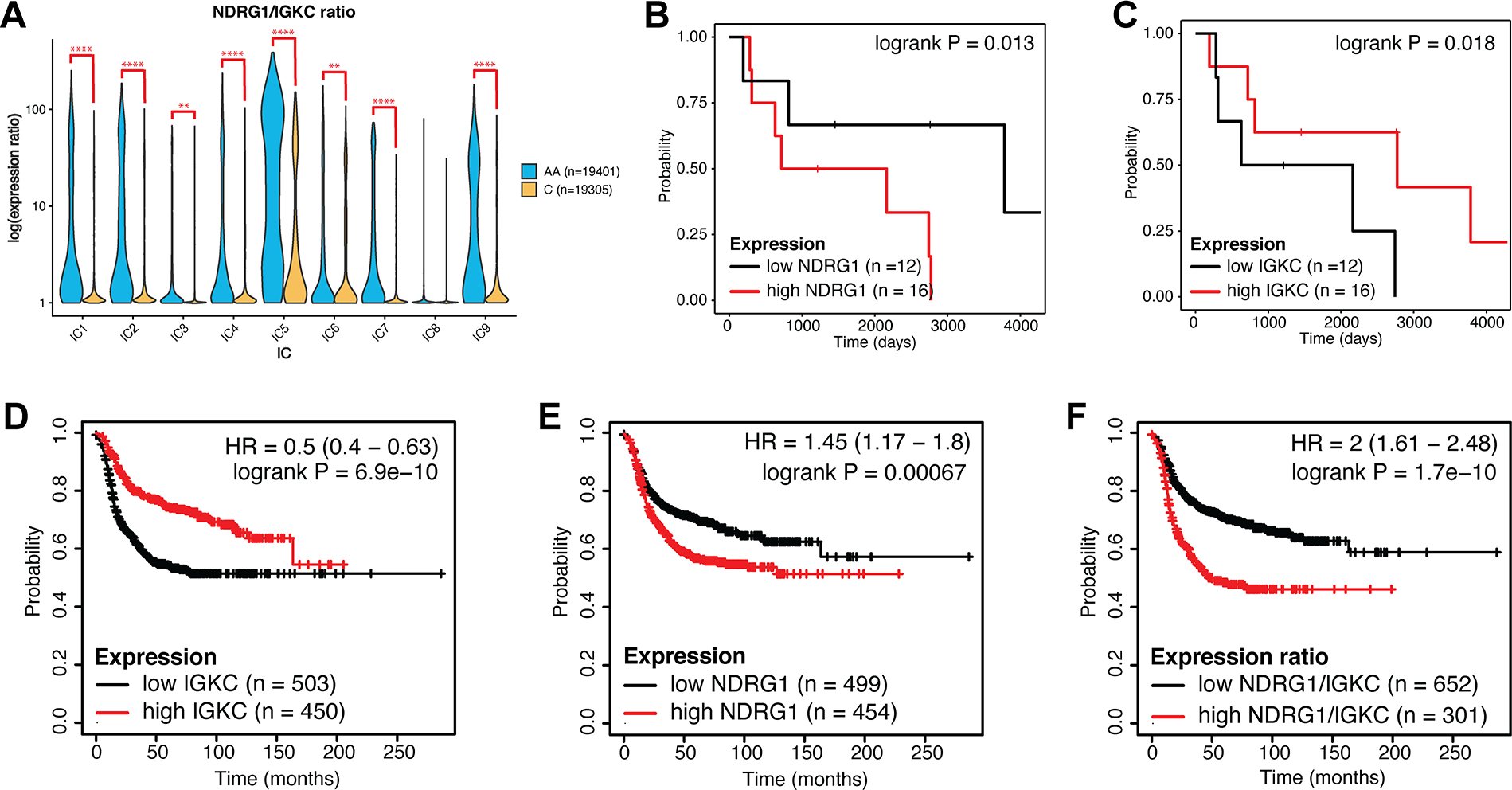
(A) Violin plot of the ratio of expression of NDRG1 to IGKC by race in each IC. Log-transformed expression ratio is represented on the y-axis. q-value was determined by multiple t-test corrected by false discovery rate. ** = q < 0.01; **** = q < 0.0001. (B-C) Kaplan-Meier plots of relapse-free survival in our reference cohort, when samples were stratified by NDRG1 (B) or IGKC (C) expression as detailed in Methods. (D-F) Kaplan-Meier plots of relapse-free survival stratified by IGKC expression (D), NDRG1 expression (E), and NDRG1/IGKC ratio (F) in molecularly subtyped basal breast cancer. Analysis is based on publicly available microarray expression datasets as detailed in Methods. Hazard ratio (HR) and p-value are indicated.
We verified these findings in large, publicly available breast cancer expression datasets. In basal-like breast cancer, the molecular subtype most similar to histologically defined TNBC (50), high IGKC expression was significantly protective (Fig. 8D), more so than in Luminal A, Luminal B, and HER2+ subtypes (Supp. Fig 6). This is consistent with previous findings that IGKC is prognostic in breast and other cancers (51). Conversely, high NDRG1 expression was associated with worse outcomes (Fig. 8E), and a high ratio of NDRG1 to IGKC expression was most strongly indicative of poor survival (Fig. 8F), especially in basal-like breast cancer (Supp. Fig 6).
Discussion
In this study, we utilize spatial transcriptomics (ST) technology to perform comprehensive spatial and transcriptional characterization of a cohort of triple-negative breast cancer (TNBC) tumors from African American (AA) and Caucasian patients.
We describe several levels of data annotation. The first is at the level of the spatial feature, which encompasses approximately 5 to 10 cells in a diameter of 55 μm. By treating each feature individually, we can apply single sample gene set enrichment (ssGSEA) (29) and ssGSEA-based analysis, such as ESTIMATE (25), to accurately define regions of tumor, stroma, and immune infiltrate. Spatially-mapped ESTIMATE signatures were comparable to a pathologist’s manual annotations in identification of tumor regions. Additionally, signatures pertaining to cell identity markers or signaling pathways provide further insight that cannot be deduced histologically. To summarize gene expression profiles within a sample, we move to cluster-level annotation. Clustering analysis revealed several transcriptionally distinct tumor and stromal cell populations within each sample, representing multiple TNBC subtypes and oncogenic processes. This is consistent with several single cell RNA-sequencing (scRNA-seq) studies of TNBC that have revealed the high degree of subclonal heterogeneity in tumors (52–54).
Finally, to obtain the most comprehensive view of ST profiles in TNBC, we perform an integrative analysis to define and annotate nine shared integrated clusters (ICs) represented in all samples. While previous studies have generated breast cancer cell atlases from scRNA-seq data (55), the same has not been done with ST data alone, as most ST technologies do not allow single-cell resolution. With this constraint in mind, we designed a study that could lay the groundwork for developing such ST tumor atlases. Our reference dataset comprising 38,706 spatial features is the largest integrated ST dataset to be described, to the best of our knowledge. Furthermore, we validate the applicability of 9 ICs in an additional 17,861 features, representing 15 independent samples. Therefore, despite the resolution limitations of ST, our study utilizes a moleculary-rich TNBC cohort, and provides the unique opportunity to interrogate the TNBC tumor and immune microenvironment in an unbiased manner through informatics-based molecular annotation coupled with spatial analysis.
When considering spatial statistics, it is essential to apply the appropriate analysis for the data type and structure. When ICs were mapped spatially on individual samples, our data took the form of nominal labels in a latticed arrangement. To describe spatial relationships within this scheme, we make novel use of join count analysis (JCA), which, to our knowledge, has not previously been applied to ST data. We demonstrate an approach to make ST data derived from the 10x Genomics Visium platform compatible with JCA through rasterization. Subsequent analysis revealed the direction and degree of spatial dependency between ICs in all samples. Interestingly, several tumor cluster pairs were consistently non-randomly dispersed. This, in addition to the strong spatial autocorrelation of each IC, may reflect compartmentalized clonal evolution and cellular substructure (56). We also observed dispersion between IC3, a stromal-immune cluster, and several tumor clusters, indicating that immune content is largely sequestered in the fibrotic compartment in TNBC.
The spatial incompatibility between hypoxic cells (IC5) and the primary immune cluster (IC3) is particularly noteworthy. A growing body of evidence supports both active and passive roles for tumor hypoxia in immune evasion (49, 57, 58). Indeed, hypoxic regions in our samples exhibited greater lymphocyte deficiency than other tumor regions. Hypoxia itself is associated with metastasis and radiotherapy resistance (47, 59), and exerts a selective pressure for more aggressive tumor clones (60). Hypoxia is therefore mechanistically significant to disease progression, and its association with immunosuppression has further implications for the use of immunotherapy in TNBC treatment.
Our finding that IC5 was overrepresented in AA samples, while IC3 was overrepresented in Caucasian samples, supports the notion that the tumor microenvironment differs in AA and Caucasian breast cancer (61). It has been reported that AA breast cancers have greater densities of microvessels and tumor-associated macrophages (TAMs) (62, 63), which have recently been linked to aggressive disease and reduced survival in breast cancer (64). This is consistent with our annotation of IC5, which we found to have a higher proportion of TAMs to lymphocytes. Furthermore, TAMs in AA breast cancers favor the pro-tumorigenic M2 phenotype and have greater proliferative capacity (63). Interestingly, these findings resulted from studies utilizing laser-capture microdissection and manual histological annotation of tumor tissue. In contrast, a study utilizing bulk RNA-sequencing data found no significant difference in the general immunological profiles of AA and Caucasian TNBC (65). Because the relevant cell populations are small, the use of bulk RNA-sequencing data may obscure results. The proportion of IC3 in our study samples varied between 6–23%. Thus, detection of subtle differences in these cell populations may require additional studies with larger specimens.
Our results suggest that NDRG1 expression is representative of the hypoxic expression profile in AA tumors, while IGKC expression represents the immune-supportive environment in Caucasian tumors. By calculating the ratio of expression between the two, we approximate the balance between hypoxia and immune infiltrate in our study cohort. We illustrate that a high ratio of NDRG1 to IGKC is associated with race and with significantly reduced survival in our cohort. Our survival analysis was validated in publicly available datasets of basal-like breast cancer. While basal-like is a molecular subtype of breast cancer that is not interchangeable with histologically-defined TNBC (66, 67), there is large overlap between the two (3, 50), and both disproportionately affect AA women (7, 68). Thus, these results suggest a link between differences in the tumor microenvironment and disparities in TNBC clinical outcomes.
It is important to acknowledge several variables present in our study. First, clinical disparities in TNBC outcome are multifactorial, influenced by both biological factors and broader disparities (8). While we control for patient age and tumor stage in our cohort, we do not consider factors such as socioeconomic or environmental variables. Although ST technology is robust, its relatively low throughput does not lend itself well to controlling for these multiple factors. Additionally, a minority of samples in our reference cohort received neoadjuvant therapy, which can alter tumor biology. However, because we find that all ICs are represented in all samples, we may be capturing heterogeneity at a level independent of the effects of neoadjuvant therapy. Furthermore, most validation cohort samples had received neoadjuvant treatment; we were also able to map and validate ICs in that cohort. Nevertheless, these are variables to be controlled for in larger studies.
The promise of ST lies in its ability to provide unprecedented insight into the compositions and interactions of cells within the tumor. We have combined functional molecular annotation with spatial analysis to profile the tumor architecture of TNBC. Our findings suggest a conserved spatio-transcriptional architecture in TNBC, despite intratumoral and stromal heterogeneity within individual samples. Furthermore, our results indicate that hypoxia may considerably influence the tumor environment in AA TNBC. Pending validation in larger cohorts, this raises the possibility of evaluating the utility of hypoxia-targeting strategies, such as hypoxia-activated prodrugs (69) or hypoxia-targeted nanoparticles (70). We also found that hypoxic regions in AA samples were associated with disproportionately high M0 macrophage content, which may support the evaluation of macrophage reprogramming-based therapeutic strategies, some of which are undergoing clinical trials (71). Thus, our study provides novel insights into the relationship between tumor biology and racial disparities in TNBC, and could ultimately lead to improved outcomes for this clinically challenging disease.
Supplementary Material
Statement of Significance.
Spatial transcriptomics profiling of a diverse cohort of triple-negative breast cancers and innovative informatics approaches reveal a conserved cellular architecture across cancers and identify proportional differences in tumor cell composition by race.
Acknowledgements
Funding for the research project was provided by NIH-NCI Cancer Center Support Grant P30 CA014089 (D. Craig, J. Carpten), NIH-NCI CARE2 PACHE U54 CA233465 (D. Craig, J. Carpten), Tower Cancer Research Foundation (R. Bassiouni, J. Carpten), and AACR/Genentech Cancer Disparities Fellowship 20-40-18-BASS (R. Bassiouni).
Services in support of the research project were provided by the VCU Massey Cancer Center Tissue and Data Acquisition and Analysis Core, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Sequencing for the project was performed by the Keck Genomics Platform and the Molecular Genomics Core at the University of Southern California, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA014089.
Microscopy for the project was partially performed by the Optical Imaging Facility at the Eli and Edythe Broad CIRM Center at the University of Southern California. We particularly thank Dr. Seth Ruffins for his assistance in image acquisition.
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
References
- 1.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foulkes WD, Smith IE, Reis JS. Triple-Negative Breast Cancer. New Engl J Med. 2010;363(20):1938–48. [DOI] [PubMed] [Google Scholar]
- 3.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–34. [DOI] [PubMed] [Google Scholar]
- 4.Bareche Y, Venet D, Ignatiadis M, Aftimos P, Piccart M, Rothe F, et al. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann Oncol. 2018;29(4):895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–502. [DOI] [PubMed] [Google Scholar]
- 8.Dietze EC, Sistrunk C, Miranda-Carboni G, O’Regan R, Seewaldt VL. Triple-negative breast cancer in African-American women: disparities versus biology. Nat Rev Cancer. 2015;15(4):248–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amirikia KC, Mills P, Bush J, Newman LA. Higher population-based incidence rates of triple-negative breast cancer among young African-American women : Implications for breast cancer screening recommendations. Cancer. 2011;117(12):2747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman LA, Kaljee LM. Health Disparities and Triple-Negative Breast Cancer in African American Women: A Review. JAMA Surg. 2017;152(5):485–93. [DOI] [PubMed] [Google Scholar]
- 11.Keenan T, Moy B, Mroz EA, Ross K, Niemierko A, Rocco JW, et al. Comparison of the Genomic Landscape Between Primary Breast Cancer in African American Versus White Women and the Association of Racial Differences With Tumor Recurrence. J Clin Oncol. 2015;33(31):3621–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitt JJ, Riester M, Zheng Y, Yoshimatsu TF, Sanni A, Oluwasola O, et al. Characterization of Nigerian breast cancer reveals prevalent homologous recombination deficiency and aggressive molecular features. Nat Commun. 2018;9(1):4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loree JM, Anand S, Dasari A, Unger JM, Gothwal A, Ellis LM, et al. Disparity of Race Reporting and Representation in Clinical Trials Leading to Cancer Drug Approvals From 2008 to 2018. JAMA Oncol. 2019:e191870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez Castro LN, Tirosh I, Suva ML. Decoding Cancer Biology One Cell at a Time. Cancer Discov. 2021;11(4):960–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353(6294):78–82. [DOI] [PubMed] [Google Scholar]
- 16.Bassiouni R, Gibbs LD, Craig DW, Carpten JD, McEachron TA. Applicability of spatial transcriptional profiling to cancer research. Mol Cell. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dias AS, Almeida CR, Helguero LA, Duarte IF. Metabolic crosstalk in the breast cancer microenvironment. Eur J Cancer. 2019;121:154–71. [DOI] [PubMed] [Google Scholar]
- 19.Salemme V, Centonze G, Cavallo F, Defilippi P, Conti L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front Oncol. 2021;11:610303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin-Pardillos A, Valls Chiva A, Bande Vargas G, Hurtado Blanco P, Pineiro Cid R, Guijarro PJ, et al. The role of clonal communication and heterogeneity in breast cancer. BMC Cancer. 2019;19(1):666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508(7494):113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naffar-Abu Amara S, Kuiken HJ, Selfors LM, Butler T, Leung ML, Leung CT, et al. Transient commensal clonal interactions can drive tumor metastasis. Nat Commun. 2020;11(1):5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–87 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20(1):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zappia L, Oshlack A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience. 2018;7(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, et al. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Inform. 2012;11:147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–U22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, Chen YA, Conejo-Garcia JR, Chung CH, Wang X. Estimation of immune cell content in tumor using single-cell RNA-seq reference data. BMC Cancer. 2019;19(1):715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–902 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bivand RSP,E; Gomez-Rubio V Applied spatial data analysis with R, Second edition: Springer, NY; 2013. [Google Scholar]
- 36.Budczies J, Klauschen F, Sinn BV, Gyorffy B, Schmitt WD, Darb-Esfahani S, et al. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One. 2012;7(12):e51862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Therneau T A Package for Survival Analysis in R. 2022. p. R package, https://CRAN.R-project.org/package=survival.
- 38.Lanczky A, Gyorffy B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J Med Internet Res. 2021;23(7):e27633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One. 2016;11(6):e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cangul H Hypoxia upregulates the expression of the NDRG1 gene leading to its overexpression in various human cancers. BMC Genet. 2004;5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlee Villodre E, Hu X, Eckhardt BL, Larson R, Huo L, Yoon EC, et al. NDRG1 in Aggressive Breast Cancer Progression and Brain Metastasis. J Natl Cancer Inst. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105(47):18490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cliff AD, Ord JK. Spatial Processes: Models & Applications: Pion; 1981. [Google Scholar]
- 45.Upton G, Fingleton B Spatial Data Analysis by Example, Volume 1: Wiley; 1985. [Google Scholar]
- 46.Tang K, Zhu L, Chen J, Wang D, Zeng L, Chen C, et al. Hypoxia Promotes Breast Cancer Cell Growth by Activating a Glycogen Metabolic Program. Cancer Res. 2021;81(19):4949–63. [DOI] [PubMed] [Google Scholar]
- 47.Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352(6282):175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mast JM, Kuppusamy P. Hyperoxygenation as a Therapeutic Supplement for Treatment of Triple Negative Breast Cancer. Front Oncol. 2018;8:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barsoum IB, Koti M, Siemens DR, Graham CH. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014;74(24):7185–90. [DOI] [PubMed] [Google Scholar]
- 50.Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol. 2011;24(2):157–67. [DOI] [PubMed] [Google Scholar]
- 51.Schmidt M, Hellwig B, Hammad S, Othman A, Lohr M, Chen Z, et al. A comprehensive analysis of human gene expression profiles identifies stromal immunoglobulin kappa C as a compatible prognostic marker in human solid tumors. Clin Cancer Res. 2012;18(9):2695–703. [DOI] [PubMed] [Google Scholar]
- 52.Karaayvaz M, Cristea S, Gillespie SM, Patel AP, Mylvaganam R, Luo CC, et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat Commun. 2018;9(1):3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pal B, Chen Y, Vaillant F, Capaldo BD, Joyce R, Song X, et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021;40(11):e107333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou S, Huang YE, Liu H, Zhou X, Yuan M, Hou F, et al. Single-cell RNA-seq dissects the intratumoral heterogeneity of triple-negative breast cancer based on gene regulatory networks. Mol Ther Nucleic Acids. 2021;23:682–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. 2021;53(9):1334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pietrobon V, Marincola FM. Hypoxia and the phenomenon of immune exclusion. J Transl Med. 2021;19(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vito A, El-Sayes N, Mossman K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells-Basel. 2020;9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sorensen BS, Horsman MR. Tumor Hypoxia: Impact on Radiation Therapy and Molecular Pathways. Front Oncol. 2020;10:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379(6560):88–91. [DOI] [PubMed] [Google Scholar]
- 61.Kim G, Pastoriza JM, Condeelis JS, Sparano JA, Filippou PS, Karagiannis GS, et al. The Contribution of Race to Breast Tumor Microenvironment Composition and Disease Progression. Front Oncol. 2020;10:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martin DN, Boersma BJ, Yi M, Reimers M, Howe TM, Yfantis HG, et al. Differences in the tumor microenvironment between African-American and European-American breast cancer patients. PLoS One. 2009;4(2):e4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koru-Sengul T, Santander AM, Miao F, Sanchez LG, Jorda M, Gluck S, et al. Breast cancers from black women exhibit higher numbers of immunosuppressive macrophages with proliferative activity and of crown-like structures associated with lower survival compared to non-black Latinas and Caucasians. Breast Cancer Res Treat. 2016;158(1):113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell. 2019;35(4):588–602 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Meara T, Safonov A, Casadevall D, Qing T, Silber A, Killelea B, et al. Immune microenvironment of triple-negative breast cancer in African-American and Caucasian women. Breast Cancer Res Treat. 2019;175(1):247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. [DOI] [PubMed] [Google Scholar]
- 67.Rakha EA, Tan DSP, Foulkes WD, Ellis IO, Tutt A, Nielsen TO, et al. Are triple-negative tumours and basal-like breast cancer synonymous? Breast Cancer Research. 2007;9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109(9):1721–8. [DOI] [PubMed] [Google Scholar]
- 69.Li Y, Zhao L, Li XF. Targeting Hypoxia: Hypoxia-Activated Prodrugs in Cancer Therapy. Front Oncol. 2021;11:700407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Jeon J, Park JH. Hypoxia-responsive nanoparticles for tumor-targeted drug delivery. Cancer Lett. 2020;490:31–43. [DOI] [PubMed] [Google Scholar]
- 71.Tan Y, Wang M, Zhang Y, Ge S, Zhong F, Xia G, et al. Tumor-Associated Macrophages: A Potential Target for Cancer Therapy. Front Oncol. 2021;11:693517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Expression data and tissue images for all samples are available in the Gene Expression Omnibus (GEO) under record GSE210616. Datasets analyzed for survival analysis are all publicly available and are listed in Supplemental File 2.