

Summary
Although systemic chemotherapy remains the standard of care for TNBC, even combination chemotherapy is often ineffective. The identification of biomarkers for differential chemotherapy response would allow for the selection of responsive patients, thus maximizing efficacy and minimizing toxicities. Here, we leverage TNBC PDXs to identify biomarkers of response. To demonstrate their ability to function as a preclinical cohort, PDXs were characterized using DNA sequencing, transcriptomics, and proteomics to show consistency with clinical samples. We then developed a network-based approach (CTD/WGCNA) to identify biomarkers of response to carboplatin (MSI1, TMSB15A, ARHGDIB, GGT1, SV2A, SEC14L2, SERPINI1, ADAMTS20, DGKQ) and docetaxel (c, MAGED4, CERS1, ST8SIA2, KIF24, PARPBP). CTD/WGCNA multigene biomarkers are predictive in PDX datasets (RNAseq and Affymetrix) for both taxane- (docetaxel or paclitaxel) and platinum-based (carboplatin or cisplatin) response, thereby demonstrating cross-expression platform and cross-drug class robustness. These biomarkers were also predictive in clinical datasets, thus demonstrating translational potential.
Subject areas: Immune response, Cancer, Omics
Graphical abstract
Highlights
-
•
Predicting response to chemotherapy is vital to reducing toxicity and improving outcomes
-
•
PDXs are biologically consistent with patients and can serve as preclinical cohorts
-
•
The CTD/WGCNA approach improves the identification of specific biomarkers
-
•
The multigene biomarkers are predictive in both PDX and patient cohorts
Immune response; Cancer; Omics
Introduction
Triple-negative breast cancer (TNBC) is an aggressive clinical subtype of breast cancer that is characterized by the absence of expression of the steroid hormone receptors (HR) estrogen receptor alpha (ESR1) and progesterone receptor (PGR), as well as the absence of overexpression and/or genomic amplification of oncogenic epidermal growth factor receptor 2 (ERBB2 or HER2).1 Relative to patients diagnosed with HR+ and ERBB2-driven breast cancers, patients with TNBC are typically diagnosed at a younger age, at a higher risk of relapse, and with lower overall survival rates. The median overall survival time for patients with metastatic TNBC remains less than 18 months.2 Unlike HR+ and ERBB2-driven tumors, which are treated with either endocrine therapy or ERBB2-targeted therapies, respectively, TNBC largely lacks biomarker-guided selection for treatment with targeted agents.3,4 Thus, treatment options for many TNBC patients remain limited to regimens using cytotoxic chemotherapies, which themselves lack predictive biomarkers for guided agent selection.5
In recent years, there have been some advances for treating TNBC more effectively. Immune checkpoint inhibitors, including atezolizumab, which targets programmed cell death ligand 1 (PD-L1), and pembrolizumab, which targets programmed cell death receptor 1 (PD-1), are approved by the Food and Drug Administration (FDA) in locally advanced, non-resectable, and metastatic patients (www.fda.gov).6,7,8,9,10,11,12,13,14,15,16 Atezolizumab is given with nab-paclitaxel,11 and pembrolizumab is given with a broader range of chemotherapeutics.10 Pembrolizumab has also been approved with chemotherapy (various) in high-risk primary TNBC in the neoadjuvant setting.17 In addition to immunotherapies, PARP inhibitors olaparib and talazoparib are also approved for HER2-negative patients with advanced disease that carry a germline BRCA1/2 mutation and who have had prior chemotherapy.18,19 Finally, the FDA recently approved sacituzumab govitecan, for locally advanced, unresectable primary tumors, as well as metastatic TNBC.20,21 Sacituzumab govitecan acts by recognizing Trop2 (trophoblast cell-surface antigen 2) with an antibody conjugated to SN38 (the active metabolite of irinotecan targeting topoisomerase I).22 Patients treated with this therapy must have received two prior systemic treatments, with at least one of these being for metastatic breast disease (i.e., chemotherapy).23 Strikingly, despite these major advances, systemic chemotherapies remain an integral part of the TNBC treatment landscape, and the therapies described above are largely still not biomarker guided despite having well-defined targets.
Reduction in tumor volume is relevant in multiple clinical settings.24,25 In the neoadjuvant setting, systemic treatment is used to shrink tumors prior to surgery.26,27,28 There are two main reasons that tumor responsiveness is important in this clinical setting. First, patients whose tumors achieve a pathologic complete response (pCR) show better overall and disease-free survival.29 Second, even in those patients not showing pCR, tumor shrinkage can enhance surgical outcomes.30 Postoperative adjuvant chemotherapy is also used to attempt to eliminate residual tumor cells after surgery that may be capable of re-growing a tumor either as a local or as a distant recurrence.31 Finally, in the metastatic setting, agents are used in an attempt to either eliminate tumors, or reduce tumor burden, to extend life.32 Thus, development of methods to determine which patients, in which clinical setting, will respond to which agents, systemic chemotherapies or otherwise, is an important clinical/translational goal.
With respect to single-agent/regimen treatment response in the neoadjuvant setting (taxane, platinum, anthracycline/cyclophosphamide), only 25%–33% of TNBC tumors achieve pCR to any given treatment.33 This mediocre response rate has led to the combination of chemotherapeutic agents, with the combinations and dose schedules determined to be most effective identified over time in human clinical trials. Combination chemotherapy is now first-line standard of care for TNBC in the neoadjuvant setting, with patients receiving up to five chemotherapeutic agents over the course of treatment.34 Even with the most recently used chemotherapy combinations, pCR rates still only reach 55%–65%.35,36,37,38 Because 35%–45% of tumors do not respond even when challenged with multiple cytotoxic chemotherapy agents, it is clear that many patients receive toxic, and ultimately ineffective, treatments for little or no clinical benefit.
If systemic combination chemotherapy is to remain at the forefront of treatment for TNBC, in any clinical setting, it is critical that molecular biomarkers of differential response to individual chemotherapy agents be identified. If successful, patients can be selected prospectively as a likely responder to one or more agents and then treated with the agent(s) most likely to be effective against their unique tumor. If successful, efficacy rates could increase dramatically. Conversely, identification of patients unlikely to respond to a given agent/regimen can be treated with alternative therapies. Thus, there should be the opportunity for treatment de-escalation, which should decrease the frequency of severe or life-threatening toxicities. Finally, these data may also provide insights into molecular mechanisms of chemotherapy resistance that may be targetable to enhance responses.
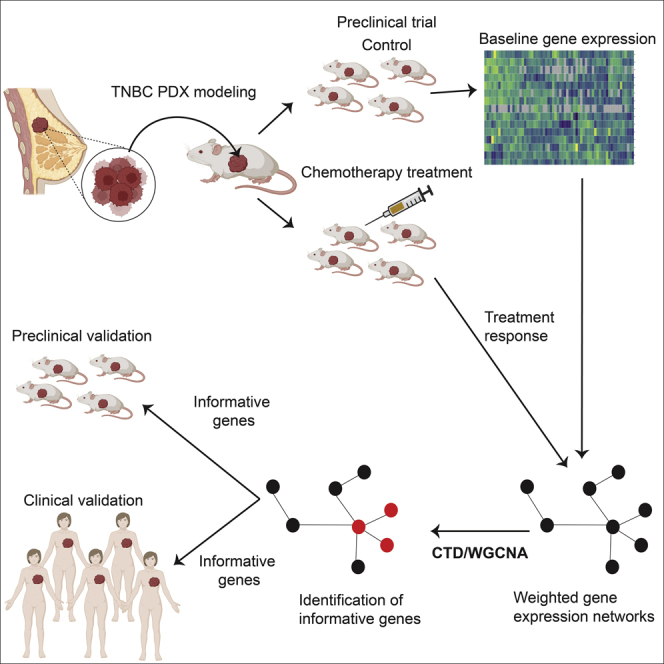
Historically, attempts to develop response predictors have been limited to either in vitro cell-line-based studies, cell line xenografts, or human clinical trials. However, to date, predictive molecular signatures of chemotherapy response are not used clinically. More recently, patient-derived xenograft (PDX) models of human breast cancer have emerged as potential surrogates for primary tumors of origin.39 We and others have shown remarkable biological consistency between patient tumors and their corresponding PDX with respect to histology, cellular heterogeneity, biomarker expression, mutations, genomic copy number alterations, variant allele frequencies, and mRNA expression patterns.40,41,42,43,44,45,46,47 Most importantly, we and others have made progress in demonstrating that treatment responses in PDX are qualitatively similar to those of the tumor of origin.40,42,43 Based on these commonalities, and the availability of a comparatively large collection of TNBC PDX, we hypothesized that a collection of PDX models can be used as a cohort comparable in size to many phase 1/2 human cohorts in clinical trials (n = 30+). If so, it should be possible to derive pre-clinically and clinically relevant molecular signatures of treatment response using a collection of PDX as the discovery platform.48
To evaluate the degree to which our PDX collection could function as a “patient cohort” in preclinical trials that might also inform tumor responses in clinical trials, we conducted a baseline proteogenomic characterization using DNA whole-exome sequencing (copy number alterations and mutations), mRNA transcriptomics (deep RNAseq ∼200M reads/sample), and mass-spectrometry-based total proteomics) and compared these data with large patient datasets to demonstrate the range of representation of the PDX models. We then aggregated single-agent responses to docetaxel or carboplatin across three recently completed PDX-based preclinical trials designed to approximate human equivalent dosing and scheduling in the mouse (to be described in full elsewhere) for analysis. Because of the sequencing depth, and the availability of both PDX-based and clinical datasets with which to validate our observations, we ultimately selected mRNA for generation of differential response signatures.
The transcriptomic data contain a wealth of information, but as with other “omics” studies, these data are high dimensional, and the number of features (genes) vastly exceeds the number of observations (PDXs). Because there are many more features than observations, feature selection is crucial in identifying informative genes, and in avoiding overfitting, to develop robust models that are generalizable across datasets. Commonly used methods to identify genes informative for prediction of response and resistance include the simple correlation of gene expression with the phenotype of interest, recursive feature elimination in machine-learning models,49 wrappers for feature selection such as Bortua,50 models using the F-test and mutual information regression in the scikit-learn python package,51 and the identification of hub genes with graph-based methods such as WGCNA (Weighted Gene Co-expression Network Analysis).52,53,54,55 Determining the correlation between genes and the phenotype of interest is perhaps the most straightforward approach for the identification of potentially informative genes. However, a problem with using gene-phenotype correlations is that a gene may be correlated with response not because of a direct role in response, but because it is correlated with other factors that are functionally relevant for response.
Another standard approach is to identify genes that add predictive power to a machine learning model of response. An example of this approach is the recursive feature elimination method. This method begins with all potentially informative features and eliminates features recursively based on the importance of individual features to the model. Unfortunately, recursive feature elimination can eliminate features that, although weak on their own, could contribute predictive power in the context of other features. Network-based methods such as WGCNA56 have also been developed to identify informative and biologically connected sets of genes. These methods are often more reliable for the identification of biomarkers than single gene comparison methods. WGCNA is a widely exploited network method in the field, and it has been used to identify informative modules and their hub genes in complex diseases including cancer and Alzheimer.52,57,58,59,60,61,62,63,64
The WGCNA approach is not without limitations itself. The standard application of WGCNA includes building a network over all samples, detecting informative modules through the correlation of the representative module eigengene profile to the phenotype of interest, and finally the identification of highly connected hub genes in informative modules.56 There are three primary issues with the standard WGCNA approach. The modules identified by WGCNA can be large and thus biologically unwieldy (hundreds to thousands of genes). Results may also be associated with inter-sample biological heterogeneity rather than to actual response. Furthermore, the use of module eigengenes to determine if a module is associated with response may miss more highly informative genes in an otherwise less informative module. This happens when a large module contains a small, more highly informative, submodule whose signal is diluted by the presence of the large number of less informative genes.
To test our hypothesis that PDX-based cohorts can yield translationally relevant predictive information, and to address one of the limitations of WGCNA, we adapted a newly developed network algorithm, CTD,65 “Connect The Dots,” in our molecular data relative to single-agent chemotherapy responses. Although this method has been used previously to identify transcriptomic differences between breast cancer subtypes, this is the first time that CTD has been used as a feature selection method to identify informative genes relative to a phenotype of interest. CTD is used in addition to WGCNA to identify highly connected sets of analyte nodes and to assign an upper-bounded p value to sets of nodes. Although other network-based algorithms also estimate p values, they use permutation testing, which is not feasible for large gene co-expression graphs with thousands of genes without prior feature selection. Our ability to assign p values without the need for permutation testing allows the identification of small sets of nodes that are significantly associated with a phenotype. This algorithmic advancement allowed for the identification of a specific set of genes that were differentially associated with docetaxel and carboplatin response/resistance in our PDX cohort. These PDX-derived informative panels were validated for their predictive power in other PDX collections, as well as in two clinical datasets. If validated in subsequent retrospective and prospective clinical trials, this approach to feature selection coupled with PDX-based discovery could accelerate development of predictive signatures that are useful clinically.
Results
PDX models of TNBC closely resemble human tumors represented in the cancer genome atlas and clinical proteomic tumor analysis consortium cohorts
At BCM, we have amassed a collection of 160 PDX models of human breast cancer that is now large enough that a subset of the collection could potentially be used as a preclinical cohort whose biological characteristics should closely resemble clinical cohorts of comparable size (typical phase 1/2 studies consist of a roughly 30 patient cohort) (publicly available models can be evaluated on https://pdxportal.research.bcm.edu/pdxportal). These models are summarized in Figure S1 (related to Figures 1, 2, and 3), which was generated with ComplexHeatmap.66
Figure 1.

Copy number variation is quantitatively and qualitatively similar in TNBC PDX models and TNBC clinical samples
(A) Copy number variation comparison between the PDXs and the TNBC clinical samples in TCGA with a heatmap.
(B) Hierarchical clustering of PDXs among the TCGA TNBC samples with copy number variations to illustrate the distribution of PDX models within human clinical samples. PDX samples are in red, and TCGA samples are in black.
Figure 2.

Mutational load is similar in TNBC PDXs and TNBC clinical samples
(A) Mutational load plot of PDX models and TNBC TCGA samples show similar mutational profiles in highly mutated genes.
(B)Waterfall plot showing the distribution of the types of mutations found in the PDX models.
Figure 3.

Deep RNAseq and proteomics reveal similarities between PDX models and TCGA clinical breast cancer samples
(A) Hierarchical clustering of TCGA and PDX models over the PAM50 gene signature.
(B) Hierarchical clustering of TCGA and PDX models over the top 1,000 most variable proteins.
For this study, we make use of a subset of 45 PDX models of TNBC, all of which were treated with single-agent docetaxel or carboplatin at human equivalent doses and in a preclinical platform that closely mimics a multi-cycle clinical trial in human patients.67 This PDX collection shows a wide range of responses to docetaxel and carboplatin, with some PDX being cross-resistant or cross-responsive, but others showing differential responses to these two agents, suggesting that molecular signatures of differential treatment response could be generated that might also have predictive power in clinical datasets.40
To evaluate the quality of our TNBC PDX multi-omic data and to determine if our collection of TNBC PDX reflected a significant cross section of TNBC clinical samples represented in TCGA (genome and transcriptome) and CPTAC (proteome) to be considered a representative preclinical cohort, we compared DNA, RNA, and protein-based assays.
At the DNA level, we first compared genomic copy number alteration patterns in our cohort relative to TCGA samples using whole-exome sequencing (WES) data. Figure 1A shows that the overall pattern of gains and loses across our PDX collection and the TCGA is qualitatively similar upon visual inspection. Similar results were obtained for ER+ and HER2+ PDX, but our sample size was not large enough for full analysis (Figures S2A and S2B which are related to Figure 1). To demonstrate similarities in a more quantitative manner, we clustered the gain and loss patterns of both the human and PDX cohorts using Pearson’s correlation coefficient. Figure 1B shows these hierarchical clustering results. Not only could PDX gain/loss patterns be compared directly with TCGA samples, but the results also show that our PDX collection represents a wide range of TNBC at the copy number alteration level.
Next, we evaluated the spectrum of mutations in our TNBC PDX cohort relative to TNBC in TCGA to determine whether the distribution of mutations was indeed similar. Figure 2A demonstrates that the spectrum of genes most highly mutated in TNBC TCGA samples (n = 115) were also mutated at largely similar frequencies in TNBC PDXs (n = 65). Similar results were observed for ER+ and HER2+ PDX models, (Figures S3A and S3B, respectively which are related to Figure 2). Although our sample sizes for both the ER+ and HER2+ subtypes were not large enough to be representative, these results suggested that the mutation spectrum observed in our PDX collection is similar to the mutation spectrum observed in human patients, particularly TNBC patients. To characterize the mutational profiles of the BCM PDX models further, we then characterized the mutational type of the most frequently mutated genes in PDXs collection (Figure 2B). The results of this analysis show that the mutational profiles of the PDX models recapitulate those of primary TNBC tumors.
With respect to gene expression analysis at the transcriptomic level, deep RNAseq (∼200M reads per sample) GSE183187 data from untreated PDX models (n = 93) were hierarchically clustered with patient tumors represented in TCGA that have been classified with PAM50 (n = 849) using the PAM50 signature (Figure 3A).68 Although the PAM50 gene set does not perform well for subtyping PDX relative to its patient-matched tumor of origin, it is useful for demonstrating that the PDX models do not cluster together as a single group but are interspersed among basal-like TCGA samples (88%) and luminal TCGA samples (12%), suggesting transcriptomic heterogeneity in PDX TNBC cohort that models primary human tumors.
With respect to gene expression analysis at the proteomic level, we detected 8,346 human proteins in at least half of the PDX models by mass spectrometry (MSV000088233, Table S4 which is related to Figure 3). As with the RNAseq analysis, PDX models were again distributed across the samples represented in the CPTAC proteomic dataset using the top 1,000 most variable proteins (Figure 3B). The clustering pattern was remarkably like that derived from mRNA data, again with two main groups clustering with luminal tumors (16%), and the majority clustering in the basal-like group (84%), with PDX models distributed across the full spectrum of basal-like tumors. For PDXs that had both proteomics and expression data available, we found that 89% of the PDXs were assigned to the same group by clustering with proteomics and expression data from primary TCGA tumors.
Responses of PDX models to human-equivalent doses of docetaxel are largely consistent with clinical responses of the tumor-of-origin when challenged with a taxane
To evaluate concordance of treatment responses across a range of chemotherapies, we compared the “best clinical response” of PDX using a modified Response Evaluation Criteria in Solid Tumors (RECIST version 1.1) classification69 to clinical imaging (typically magnetic resonance imaging [MRI], sometimes positron emission tomography (PET) or ultrasound, and rarely clinical exam alone). In addition, for most patients in the neoadjuvant setting, results of pathologic responses in their tumor of origin when challenged with the same (or same class of) agent were available. Best clinical responses, corroborated by pathologic evaluation when possible, are shown in Table 1, Table 2, Table 3, 2, and 3.
Table 1.
Patient tumor/PDX docetaxel (or paclitaxel) response concordance analysis
| Patient Number (Arbitrary) | PDX | Clinical Setting | Patient Treatment | Clinical Response | PDX D20 Response | D20 Patient Concordance? | PDX D30 Response | D30 Patient Concordance? |
|---|---|---|---|---|---|---|---|---|
| 1 | BCM-0002 | Neo | Docetaxel (w/ Doxorubicin) | PR | SD | Yesa | SD | Yesa |
| 2 | BCM-0046 | Neo | Docetaxel | SD | PD | Yesb | ND | NA |
| 3 | BCM-0104 | Neo | Paclitaxel | PR | PR | Yes | PR | Yes |
| 4 | BCM-0132 | Neo | Paclitaxel (w/ carboplatin) | PR | PR | Yes | PR | Yes |
| 5 | BCM-2665 | Neo | Docetaxel | PD | SD | Yesb | PR | No |
| 6 | BCM-3107 | Neo | Docetaxel | PR | PR | Yes | ND | NA |
| 7 | BCM-3469 | Neo | Docetaxel (w/ cyclophosphamide) | PR | PR | Yes | ND | NA |
| 8 | BCM-3611 | Neo | Docetaxel | PD | PR | No | PR | No |
| 9 | BCM-4013 | Neo | Docetaxel | PD | PD | Yes | ND | NA |
| 10 | BCM-4195 | Neo | Docetaxel | SD | PR | Yesa | ND | NA |
| 11 | BCM-4272/4849 | Neo | Docetaxel (w/ experimental drug) | PR | PR | Yes | ND | NA |
| 12 | BCM-4664 | Neo | Docetaxel (w/ experimental drug) | PR | PR | Yes | PR | Yes |
| 13 | BCM-4913/5438 | Neo | Docetaxel | PD | PR | No | CR | No |
| 14 | BCM-5471 | Neo | Docetaxel | PD | PD | Yes | CR | No |
| 15 | BCM-6257 | Neo | Docetaxel | PR | PR | Yes | ND | NA |
| 16 | BCM-7482 | Neo | Docetaxel | PR | PD | No | PR | Yes |
| 17 | BCM-7649 | Neo | Docetaxel | PD | SD | Yesb | ND | NA |
| 18 | BCM-7821 | Neo | Docetaxel | PR | SD | Yesa | SD | Yesa |
| 19 | BCM-15029 | Neo | Docetaxel (w/ carboplatin) | PR | PR | Yes | ND | NA |
| 20 | BCM-15006/15046 | Neo | Paclitaxel (w/ carboplatin) | PD | PD | Yes | ND | NA |
| 21 | HCI-001 | Neo | Paclitaxel | PD | PD | Yes | SD | Yesb |
| 22 | HCI-015 | Neo | Paclitaxel (w/ AC) | PD | PR | No | ND | NA |
| 23 | WHIM2 | Neo | AC→Paclitaxel | PR | PR | Yes | PR | Yes |
| 24 | WHIM14 | Neo | EC (w/ 5FU)→Docetaxel | PR | PR | Yes | ND | NA |
| 25 | WHIM30 | Neo | Paclitaxel | CR | PR | Yesc | CR | Yes |
| 26 | WHIM68 | Neo | Docetaxel (w/ carboplatin) | PR | PR | Yes | PR | Yes |
| 26 | WHIM74 | Neo | Docetaxel (w/ carboplatin) | PR | PR | Yes | ND | NA |
- PR and SD are adjacent RECIST 1.1 classifications.
- SD and PD are adjacent RECIST 1.1 classifications.
- PR and CR are adjacent RECIST 1.1 classifications.
Table 2.
Patient tumor/PDX carboplatin (or cisplatin) response concordance analysis
| Patient Number (Arbitrary) | PDX | Clinical Setting | Patient Treatment | Clinical Response | PDX C50 Response | C50 Patient Concordance? |
|---|---|---|---|---|---|---|
| 28 | BCM-15006/15046 | Neo | Carboplatin (with paclitaxel) | PD | PR | No |
| 28 | BCM-15006/15046 | Neo | Cisplatin (with radiation) | CR | PR | Yesa |
| 29 | BCM-15029 | Neo | Carboplatin (with docetaxel) | PR | PR | Yes |
| 30 | BCM-0132 | Neo | Carboplatin (with paclitaxel) | CR | PD | Nob |
| 26 | WHIM68 | Neo | Docetaxel (w/ carboplatin) | PR | PR | Yes |
| 26 | WHIM74 | Neo | Docetaxel (w/ carboplatin) | PR | PR | Yes |
- PR and CR are adjacent RECIST 1.1 classifications. Patient response likely affected by concurrent radiation treatment.
- Carboplatin response possibly confounded by simultaneous treatment with paclitaxel, as BCM-0132 was responsive to docetaxel.
Table 3.
Patient tumor/PDX doxorubicin/cyclophosphamide (AC) or epirubicin/cyclophosphamide (EC) response concordance analysis
| Patient Number (Arbitrary) | PDX | Clinical Setting | Patient Treatment | Clinical Response | PDX AC Response | AC/EC Patient Concordance? |
|---|---|---|---|---|---|---|
| 32 | BCM-0046 | Neo | EC | PR | PD | No |
| 33 | BCM-2665 | Neo | AC | PR | PD | No |
| 34 | BCM-3469 | Neo | AC | PR | PD | No |
| 35 | BCM-3936 | Neo | AC | PR | PD | No |
| 36 | BCM-15006/15046 | Neo | AC | PR | PR | Yes |
| 23 | WHIM2 | Neo | AC→Paclitaxel | PR | PR | Yes |
| 24 | WHIM14 | Neo | EC (w/ 5FU)→Docetaxel | PR | PR | Yes |
| 38 | WHIM6 | Neo | EC (with 5FU) | PR | PR | Yes |
| 40 | WHIM30 | Neo | AC (with paclitaxel)→ddAC | CR | PR | Yesa |
- PR and CR are adjacent RECIST 1.1 classifications.
In the patient-matched PDX models, treated with 20 mg/kg docetaxel (Table 1), 16/27 (59%) showed unqualified concordance with the tumor of origin. An additional seven (26%) showed “qualified” concordance, in which the PDX response fell into an adjacent RECIST response category in the patient. Given that the preclinical regimen we used may not yet be fully equivalent to that used in a human clinical trial, we reasoned that the use of “qualified concordance” was justified. If this argument is accepted, a remarkable 23/27 (85%) of observations were considered concordant. Only 4/27 (15%) PDXs received an unqualified discordant response from the original tumor at this dose (No), possibly due to differences in drug metabolism or the difference between the mouse and human microenvironments. More likely, the low human-equivalent dose of docetaxel may be insufficient to treat some tumors.
For the four unqualified non-concordant PDX models at D20 (BCM-3611, BCM-4913, BCM-7482, and HCI-015), three tumors of origin showed progressive disease (PD), whereas the corresponding PDX (BCM-3611, BCM-4913, and HCI-015) showed a partial response (PR). The reason for the enhanced response relative to the tumor of origin is not known, but it is possibly due to the absence of an intact immune system in host mice. Conversely, PDX BCM-7482 showed PD, whereas the tumor of origin showed a PR. For this line specifically, we hypothesized that the difference in response may be because we evaluated docetaxel at the low human equivalent dose (20 mg/kg), rather than the high human equivalent dose (30 mg/kg), to which the tumor of origin would likely have been exposed. If so, BCM-7482 may show response to the higher dose and thus then show concordance. Similarly, some of the qualified concordances may convert to unqualified. To test this possibility, we challenged BCM-7482 with four cycles of docetaxel at the higher 30 mg/kg dose. Several other lines were also evaluated. As hypothesized, BCM-7482 became concordant. Of the qualified concordance lines tested at the higher dose, only WHIM30 converted from a qualified to an unqualified concordance. Thus, overall, only 10% (3/27) of the PDX evaluated showed unqualified discordance with patient tumor response.
Concordance of response with the tumor of origin: PDX responses to carboplatin or AC were equivocal
With respect to platinum-based agents, we identified only six PDX-matched patients who had been treated with a platinum-containing regimen, none of which were single agent. Of these, there were three unqualified concordances (50%) and one qualified concordance (most likely due to the addition of radiation with cisplatin) for an overall concordance of 66%. Countering this observation, there were two unqualified discordances (33%) (Table 2). In the two discordant cases, the patients were treated with combination carboplatin/taxane. For BCM-0132, the tumor of origin showed a CR to combination treatment, whereas the PDX showed PD to single-agent carboplatin, a result consistent with BCM-0132 being responsive to docetaxel. Conversely, although BCM-15006 showed PR to single-agent carboplatin, the tumor of origin unexpectedly showed PD with combination treatment, suggesting the intriguing possibility that carboplatin and paclitaxel may have been antagonistic to one another in this patient, a phenomenon long known to occur in some cell lines in vitro. 70,71,72,73,74,75,76,77 Although overall these results look promising, it remains to be determined whether PDX responses to platinum-containing regimens truly recapitulate patient tumor-of-origin responses.
Finally, with respect to multi-cycle AC (anthracycline and cyclophosphamide), nine patient-matched PDXs could be evaluated for concordance. In contrast to docetaxel-treated PDX, AC-treated PDX showed only four unqualified concordances and two qualified concordances (Table 3). Four of nine (44%) showed unqualified concordance compared with the tumor of origin, with an additional one of nine (11%) showing qualified concordance. This yielded an overall concordance of 55%. In the four of nine cases of discordance (44%), the clinical response of the tumor of origin was uniformly PR, whereas the corresponding PDX was uniformly PD. This is most likely due to the fact that we could not achieve human equivalent doses for either the doxorubicin or cyclophosphamide, or there may be an effect of the immune system on response.78 Thus, PDX responses to AC are unlikely to be fully reflective of the responses in patients, at least under the conditions achievable in SCID/Bg mice.
Identification of multigene biomarker panels predictive of differential chemotherapy response
Given the high degree of concordance in the treatment response analysis (at least for taxanes), we hypothesized that the PDX could be used to develop differential taxane/platinum response predictors. Here, we used the RNAseq transcriptomic data, as this was the most robust “omic” type. Widely used tools such as Xenome79 allow for the separation of murine and human genes. In this study we performed deep RNA-seq (∼200M paired end reads per sample), which allowed us to identify far more unique genes (25,384 human genes and 21,552 murine) than proteins (8,346 human proteins) that were identified by our proteomics analysis. Finally, transcriptomic datasets are more widely available for the validation of candidate biomarkers. In fact, there are no relevant proteomics datasets in existence by which we could evaluate a protein-derived signature.
For each PDX, epithelial gene expression and stromal gene expression were evaluated by separating the RNAseq reads by species using Xenome79 This separation allowed us to evaluate both epithelial and stromal genes that may be associated with response for each approach.
For both the WGCNA and combined CTD/WGCNA approaches, we first built a pair of weighted gene-gene co-expression networks for each cell type (stromal or epithelial)/response combination using WCGNA only (biweight midcorrelation). For each condition, one of these graphs was built over all the PDX models, whereas the other was built only over the responsive PDXs (CR and PR). We first removed all weak edges (edge weight <0.2). Because gene-gene correlation could be caused by heterogeneity in the samples that is not related to the responsiveness of the samples to the agents in question, we then pruned or removed edges with the CTD graph naive pruning function,80 that were found in the graph built over just the responsive PDXs to ensure that we eliminated noise not related to response. By removing these edges, we both eliminated gene-gene variance that may not be associated with response, and made the networks more sparse, and thus easier to evaluate. This approach produced four pruned networks that were specific to both a tissue compartment and chemotherapy treatment (i.e., human epithelial carboplatin, human epithelial docetaxel, murine stromal carboplatin, murine stromal docetaxel).
Pruned networks were then broken into modules with WGCNA using the standard approach (described in detail at https://horvath.genetics.ucla.edu/htl/CoexpressionNetwork/Rpackages/WGCNA/Tutorials). The method identified between 3 and 99 gene modules for each of the graphs. These modules were large (30–200 genes each), and not biologically tractable, as they contained too many genes representing several pathways. Thus, WGCNA alone did not allow us to determine which specific genes were most closely related to response in a straightforward manner. To determine how informative the WGCNA modules were for response, we first calculated each module’s eigengenes using WGCNA’s module Eigengenes57 function. We then identified modules whose eigengenes had at least a 35% correlation with response as informative. For carboplatin, the WGCNA approach identified 97 genes in informative modules for carboplatin. To determine if these genes were informative for chemotherapy response, we applied a random forest LOOCV approach and found that the models built using these genes had low predictive power (AUC 0.55–0.56). Likewise, the WGCNA approach identified 322 genes in informative modules for docetaxel. Here we also found that LOOCV random forest models built from these genes had low predictive power (AUC 0.6–0.79).
As previously mentioned, the WGCNA approach may overlook modules with informative genes whose signal is overwhelmed by less-informative genes, because it utilizes module eigengenes to determine if a specific module is related to the phenotype of interest (in this case response to chemotherapy). Once module eigengenes are used to identify informative module, the standard WGCNA workflow then identifies hub genes that may be particularly informative. Following this approach (see STAR Methods), we identified 14 hub genes for carboplatin (AUC with a GLM LOOCV 0.62–0.77) and 27 hub genes for docetaxel (AUC with a GLM LOOCV 0.58–0.68). Although these genes are representative of each module’s eigengenes, they are not necessarily the genes that carry the predictive power. To address these issues, we applied a novel adaptation of CTD to determine which sets of genes in these large WGCNA modules were significantly connected in the pruned networks.
CTD is an information-theoretic-based network method that identifies patterns of connectedness between analytes and assigns p values to highly connected subsets of analytes within large modules without computationally costly permutation testing (Figure 4A).65 With respect to gene expression data, CTD is used to “connect the dots” and identify these highly connected gene sets by finding subsets that are more connected in a weighted graph than by chance. Because response has not been added as a node in the expression graph, we hypothesize that significantly connected sets within the large modules may be connected due to their latent connection to response (Figure 4B). For each of the pruned networks, we identified small CTD submodules, which then allowed for the generation of generalized linear models (GLMs), and the identification of specific genes that are predictive for differential response to the two agent classes chosen. The p value calculated by CTD for these modules, as well as the significance of their linear regression with response, and whether the larger module was identified as informative by WGCNA can be found in Table S1 related to Figure 4. In Table S2 (related to Figure 4), the genes used for the multigene biomarkers and their modules are identified.
Figure 4.

CTD connects the dots to identify multigene biomarker panels of response to carboplatin and docetaxel
(A) CTD is a network method that can be used to “connect the dots” and identify highly connected sets of genes. CTD takes a weighted graph and a set of nodes of interest (gray) and outputs a set of highly connected genes (purple) with a p value of connectedness.
(B) Highly connected nodes may be connected through their latent connection to response.
(C) CTD carboplatin epithelial submodule 5, which contains 3 informative genes highlighted with a star.
(D) CTD docetaxel epithelial submodule 23, which contains one informative gene highlighted with a star.
(E) Table of all the informative genes for carboplatin identified with the CTD/WGCNA approach.
(F) Table of all the informative genes for docetaxel identified with the CTD/WGCNA approach.
An example of an informative submodule for the carboplatin epithelial network is shown in Figure 4C, whereas an example of an informative docetaxel epithelial submodule is shown in Figure 4D. For carboplatin, five genes were identified as informative for response in the epithelial network (MSI1, TMSB15A, ARHGDIB, GGT1, SV2A) and four genes were identified as informative in the stromal network (SEC14L2, SERPINI1, ADAMTS20, DGKQ) (Figure 4E). For docetaxel, four genes were identified as informative in the epithelial network (ITGA7, MAGED4, CERS1, ST8SIA2) and two genes were identified as informative in the stromal network (KIF24, PARPBP) (Figure 4F).
We then combined the stromal and epithelial RNAseq expression profiles for each of the PDX models to generate a pseudo-bulk gene expression profile for each PDX. To show the directionality of these informative genes relative to response, we plotted the median log2 normalized expression of our informative panel in the pseudo-bulk expression profiles for the carboplatin genes (Figure 5A) and the docetaxel genes (Figure 5B) relative to quantitative drug responses. In both cases, an expression gradient can be observed across PDX with respect to response.
Figure 5.

CTD outperforms other feature selection methods when selecting informative genes and is predictive in our PDX cohort
(A) Heatmap of informative genes for carboplatin across all PDXs from most responsive (left) to most resistant (right). CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease.
(B) Heatmap of informative genes for docetaxel across all PDXs from most responsive (left) to most resistant (right). CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease.
(C) Overlap between genes predicted to be informative for carboplatin by commonly used feature selection methods visualized with an UpSet plot.
(D) Overlap between genes predicted to be informative for docetaxel by commonly used feature selection methods visualized with an UpSet plot.
(E) MAE comparison of 4 different methods (red = CTD/WGCNA approach, lime green = correlation, turquoise = recursive feature extraction, orange WGCNA hub genes, purple = Boruta, pink = F-test, green = mutual information regression).
(F) RMSE comparison of 4 different methods (red = CTD/WGCNA approach, lime green = correlation, turquoise = recursive feature extraction, orange WGCNA hub genes, purple = Boruta, pink = F-test, green = mutual information regression).
Multigene biomarker panel genes in the literature
To determine more rigorously if our multigene biomarkers were associated with cancer progression or response to therapy previously, we conducted a literature search. For the carboplatin multigene biomarker panel, we found that increased MSI1 expression was associated with lung cancer malignancy81 and stem cells in breast cancer.82 In addition, TMSB15A is predictive of response to combination chemotherapy in two cohorts (one cohort was treated with epirubicin-/cyclophosphamide-based chemotherapy, whereas the other was treated with docetaxel, doxorubicin, and cyclophosphamide).83 ARHGDIB is associated with breast cancer progression and is associated with drug resistance to etoposide and doxorubicin.84,85 High levels of pretreatment GGT1 are linked to breast cancer progression and prognosis.86 SERPINI1 may be associated with the survival of cancer cells,87 and ADAMTS20 is associated with the grade of breast cancer, and its expression varies in different breast cancer subtypes.88
For the docetaxel multigene biomarkers, we found that ITGA7 is associated with worse prognosis and increased migration in breast cancer89,90 and is a stem cell marker in squamous cell carcinoma,91 breast cancer patients with elevated expression of MAGED4 had worse survival in a large cohort of breast cancer patients,92 CERS1 is associated with cancer cell death in head and neck squamous cell carcinomas,93 and PARPBP was identified previously in breast cancer as a marker of resistance to epirubicin.94 In addition, SV2A95 and KIF24 96 were found to be overexpressed in breast cancer cell line (MCF7, ZR57-1, MDA-MB-231, MDA-MB-468, Hs578Tm MCF10A) data when compared with an immortalized, but not transformed, human mammary epithelial cell line (HMEC).
Comparison of the CTD/WGCNA approach with other feature selection methods
We then investigated how the combined CTD/WGCNA approach was compared with six widely used feature selection methods (WGCNA hub genes,52 correlation,49 recursive feature elimination with a random forest model,49 Boruta,50 F-test,51 and Mutual Information Regression).51 To identify the WGCNA hub genes, we identified modules whose eigengenes were correlated with response and then determined which genes had the highest significance. For the correlation and recursive feature selection approaches, we identified the top 10 genes (as our CTD/WGCNA approach identified 9 genes for carboplatin and 6 genes for docetaxel) that were related to response in both the stromal and epithelial compartments. Boruta50 is a random forest wrapper method that was applied using the Boruta function in R to identify informative genes. We also used the SelectKBest function from the python scikit-learn package to perform the F-test and mutual information regression analysis. The UpSetR package was then used to visualize the number of unique genes identified by each method as well as the number of genes multiple methods identified (Figures 5C and 5D).
Although a few genes were identified with multiple methods, most potentially informative genes were unique to the feature selection method used. Genes that were identified as potentially informative by at least three feature selection methods for carboplatin included SV2A (5 methods), MAGED4 (4 methods), MAGED4B (4 methods), TRHDE (4 methods), and MED12L (3 methods). Genes that were identified by at least three feature selection methods for docetaxel included only ASGR1 (4 methods) and CRMP1 (3 methods).
CTD/WGCNA outperforms other methods of feature selection
To evaluate CTD as an informative feature selection method, we tested each of these feature selection methods for their performance predicting treatment response using “pseudo-bulk” RNAseq data reconstructed from the PDX epithelial and stromal RNAseq gene expression datasets. We built these pseudo-bulk mRNA expression profiles by summing the epithelial and stromal mRNA expression values for common genes to approximate human clinical bulk RNAseq. To evaluate the performance of each feature selection method with a generalized linear model within our dataset, we compared the Mean Absolute Error (MAE) (Figure 5E) and root-mean-square error (RMSE) (Figure 5F) from the quantitative response predictions using a 70/30 training/test set splitting approach of the source data, which were reshuffled 10 times. Although each feature selection method identified a largely unique set of genes, we found that the CTD/WGCNA approach outperformed all the others, including WGCNA alone, when predicting quantitative response of PDX models to both carboplatin and docetaxel (Figures 5E and 5F, respectively).
Preclinical validation of differential chemotherapy response predictors
For the purposes of validation in other PDX datasets, and in clinical trial response assessments, we converted our quantitative response assessments to a qualitative modified RECIST 1.1 classification for each PDX. We then determined how predictive our models were for both complete response (CR) versus all else (progressive disease [PD], stable disease (SD), a partial response [PR]) and CR/PR versus SD/PD using the full 45 PDX dataset, and how they were compared with models built using the informative module and hub genes identified with the standard WGCNA approach.
ROC curves were constructed using LOOCV models for each of the treatments for two response cutoffs: complete response (CR) versus all else and complete response plus partial response (CR/PR) versus all else. For both the CTD biomarkers and the hub genes identified by WGCNA, GLM LOOCV models were used to predict the qualitative response. However, due to the large number of genes identified as informative in the WGCA models, Random Forest (RF) models with default caret49 parameters were used instead of GLMS to predict qualitative response to therapy. For carboplatin CR versus all else, the AUCs of these models are as follows: CTD = 0.85, hub genes = 0.77, and WGCNA modules = 0.56 (Figure 6A). Likewise, the AUCs for carboplatin CR/PR versus all else are CTD = 0.80, hub genes = 0.62, and WGCNA modules = 0.55 Figure 6B). All these methods had low predictive power for docetaxel CR (Figure 6C), which we hypothesize may be associated with the unbalanced nature of responses in this collection (9 CR, 36 nCR). For docetaxel CR/PR the AUCS were CTD = 0.86, hub genes = 0.69, and WGCNA modules = 0.79 (Figure 6D). Overall, we found that the CTD/WGCNA method was more predictive than the standard WGCNA approach and that both the carboplatin CTD/WGCNA multigene biomarker panel and the docetaxel biomarker panel were informative for both quantitative and qualitative response.
Figure 6.

CTD multigene biomarkers are predictive of qualitative response and are also predictive of the best therapy for each PDX
(A) ROC for carboplatin complete response (CR) versus all else. The ROC for the CTD genes is in red, the ROC for WGCNA hub genes is in green, and the ROC for the WGCNA modules is in blue.
(B) ROC for carboplatin complete and partial response (CR/PR) versus all else. The ROC for the CTD genes is in red, the ROC for WGCNA hub genes is in green, and the ROC for the WGCNA modules is in blue.
(C) ROC for docetaxel complete response (CR) versus all else. The ROC for the CTD genes is in red, the ROC for WGCNA hub genes is in green, and the ROC for the WGCNA modules is in blue.
(D) ROC for docetaxel complete and partial response (CR/PR) versus all else. The ROC for the CTD genes is in red, the ROC for WGCNA hub genes is in green, and the ROC for the WGCNA modules is in blue.
(E) The best response predictions for all PDXs. If both the actual best therapy and the predicted best therapy is carboplatin, the dot is blue. If both the actual best therapy and the predicted best therapy is docetaxel, the dot is red. If the best therapy and the predicted best therapy do not match, the dot is black.
(F) The best response predictions for PDXs that are predicted to have a log2 fold-change difference of at least 2 between their response to carboplatin and their response to platinum. If both the actual best therapy and the predicted best therapy is carboplatin, the dot is blue. If both the actual best therapy and the predicted best therapy is docetaxel, the dot is red. If the best therapy and the predicted best therapy do not match, the dot is black.
To be maximally useful, biomarker panels predictive of differential drug response should be informative for choosing which drug might be more effective in which tumors. To evaluate our ability to choose which chemotherapy agent, docetaxel, or carboplatin, would have the best response in each PDX, we used a leave one out cross-validation (LOOCV) approach with the CTD multigene biomarkers to predict the quantitative response of each PDX to both carboplatin and docetaxel.
Across all the PDX models, the accuracy in predicting the best response was 73% (Figure 6E). However, we are especially interested in predicting the differential response in PDXs with large differences in their carboplatin and docetaxel response. Although some PDXs show similar responses to treatment by carboplatin and docetaxel, it is particularly important to correctly predict the best therapy for PDXs that have fundamental differences in response to these two chemotherapy agents.
By limiting our scope to PDXs whose predicted responses to both agents differed by a log2 fold-change of more than two (n = 27, 60%), the accuracy of our predictions rose to 85% (Figure 6F). Thus, using this approach, we can (1) identify the subset of models for which an accurate prediction of differential response can be made and (2) predict the best therapy for these models, both with good precision.
Preclinical validation: Multigene biomarker panels are predictive for chemotherapy response in other PDX datasets
To test the predictive power of our gene panels, we tested them using on other PDX datasets (Table 1) with either associated RNAseq (within platform) or Affymetrix array (across platform) gene expression data. With respect to within platform validation, the carboplatin and docetaxel were tested in a PDX dataset with RNAseq and qualitative drug response from the Rosalind & Morris Goodman Cancer Research Center (GSE142767).42 Of the PDXs in this dataset (n = 37), 30 had mRECIST-like single-agent response data for cisplatin and paclitaxel. An LOOCV approach was used with the XGBoost tree model over the nine informative genes for carboplatin to predict cisplatin response, and a similar LOOCV approach was used over the six docetaxel genes to predict response to paclitaxel. Despite the differences in the PDX composition and the specific therapies used (cisplatin versus carboplatin and paclitaxel versus docetaxel), we found that our multigene biomarker panels were predictive in this dataset. The AUCs for cisplatin response in this dataset were 0.72 for CR versus all else and 0.54 for CR/PR versus all else (Figures 7A and 7B). The AUCs for paclitaxel response were 0.72 for CR versus all else and 0.73 for CR/PR versus all else (Figures 7A and 7B). Thus, our biomarker panels had predictive power for therapies that were in the same class (taxane versus platinum-based).
Figure 7.

Multigene biomarkers are predictive of qualitative response in other PDX datasets and in a clinical human dataset with response to cisplatin
(A) ROC of response predictions in the RMGCRC PDX cohort for complete and partial response (CR/PR) versus all else. The ROC for predictions for cisplatin is in blue, and the ROC for predictions to paclitaxel is red.
(B) ROC of response predictions in the RMGCRC PDX cohort for complete response (CR) versus all else. The ROC for predictions for cisplatin is in blue, and the ROC for predictions to paclitaxel is red.
(C) ROC of response prediction for cisplatin response in the Curie cohort.
(D) ROC of response prediction for cisplatin response in a human clinical cohort (DFCI).
(E) ROC of response prediction for paclitaxel prediction in the BrighTNess cohort. Note that the response was measured after treatment with AC as well.
To test the cross-platform performance of our biomarker panel, the predictive power of our carboplatin biomarker was tested in a gene expression Affymetrix array dataset from the Curie Institute (n = 37) evaluating qualitative responses following treatment with cisplatin. To determine the predictive power of the biomarkers, an XGBoost tree model was built with LOOCV over the 9 informative genes for carboplatin. The AUC of this model was 0.66 (Figure 7C). Thus, by leveraging other publicly available PDX datasets, we have shown that the multigene panels are predictive across preclinical datasets and across gene expression platforms.
Clinical validation: Predictive power of the platinum multigene biomarker panel
To investigate the predictive power of our PDX-based platinum response signature in a clinical dataset, we tested our carboplatin gene biomarker on an array dataset with 24 TNBC patient samples (GSE18864).97 These patients were treated with a cisplatin monotherapy and had response to therapy measured qualitatively with the RECIST 1.1 classification system.69 For this analysis, we also used an XGBoost tree model (LOOCV), this time using the nine informative genes for carboplatin response. The AUC for this model was 0.73 (Figure 7D), demonstrating that we have predictive power in a clinical dataset for platinum response, again across expression platforms.
To determine if the expression of stromal genes indeed contributed to the predictive power of the clinical model, we determined the variable importance rankings for this model using the varImp function from caret. The stromal genes were ranked second (DGKQ), third (ADAMTS20), fifth (SEC14L2), and ninth (SERPINI1) out of the nine genes for importance. This suggests that both stromal genes and epithelial genes contribute predictive power to the multigene biomarker panel.
Clinical validation: Predictive power of the taxane multigene biomarker panel
Unfortunately, a single-agent taxane dataset could not be identified. However, we also tested the predictive power of our taxane biomarker panel for “benefit from taxane” by evaluating RNAseq data from a subset of TNBC patient samples from the BrighTNess clinical trial who were treated with paclitaxel followed by doxorubicin and cyclophosphamide using a LOOCV XGBoost approach. In this trial, response was evaluated only after the AC treatment was completed. Although full RECIST 1.1 69 calls were not available, patients were evaluated for pCR versus not (GSE164458) (Loibl et al., 2018). Although a different taxane agent was used (paclitaxel) and response was not evaluated immediately after the paclitaxel treatment, we still had some predictive power in this clinical dataset (AUC = 0.59) (Figure 7E). The contribution of the stromal genes to the benefit from taxane prediction in the BrighTNess trial was also evaluated using the variable importance rankings function from caret. We found that here too the stromal genes contributed to the model and were ranked first (PARPBP) and fourth (KIF24) out of the six genes for informativeness.
Discussion
PDX models can be used to identify biomarkers of response to therapy
PDX models have emerged as useful preclinical tools to model cancer biology. The TNBC PDXs characterized in this study is highly representative of two widely used cohorts’ human tumors (TCGA and CPTAC) with respect to copy number alteration patterns, mutation frequency, and gene expression profiles at both the transcriptomic and proteomic levels. Further, we demonstrate that, at least for taxanes, the responses of PDX to docetaxel are qualitatively similar to the responses of the tumors of origin in the patient when treated with docetaxel or paclitaxel. This high degree of concordance allowed us to treat the TNBC as a preclinical cohort to develop multigene biomarker panels predictive of differential response to two commonly used first-line agents: platinum agents (carboplatin and cisplatin) and taxane agents (docetaxel and paclitaxel).
CTD allows for the identification of biologically tractable multigene biomarker
To identify our gene biomarker panels, we first had to address a methodological issue. The initial gene modules identified by WGCNA were large, and it was difficult to determine which specific genes were contributing to the modules being associated with response. Through the application of CTD, we identified highly connected sets of genes and a specific set of genes that are predictive of response to therapy. The biomarker panels identified by this approach outperformed several methods that are commonly used for feature selection including correlation analysis, a recursive feature extraction approach, and the hub genes identified with the standard WGCNA approach at predicting quantitative response. We also compared the performance of CTD and the standard WGCNA approach of identifying informative modules and hubs genes for predicting qualitative response to therapy. Here, we again observe that CTD outperforms other feature selection methods. Moreover, a literature search confirmed that many of the biomarker genes have been shown to play a role in cancer progression or resistance to therapy.
Within the carboplatin and docetaxel response prediction biomarkers, we identified both stromal and epithelial genes whose differential expression is predictive of differential response to both classes of chemotherapy agents. Although murine stromal genes were identified as part of the original biomarker panels, their human homologs were also shown to be predictive in the clinical cohort. The identification of predictive stromal genes in this analysis is also supported by studies that have previously associated breast cancer stromal cells with both progression and resistance.98,99,100,101,102
Multigene biomarkers are predictive across expression platforms and within chemotherapy classes
We then used our multigene biomarkers to predict response to therapy in other data collections. Because of biological differences between PDX models and patients, differences in data type (array vs RNAseq), and differences in chemotherapy agents (e.g., carboplatin vs cisplatin), we built new models over our multigene biomarkers for each dataset.
Several publications have demonstrated phenotypic and gene expression stability over multiple transplant generations, and in some cases, our response data were derived from PDX models at multiple passages (to be described in full elsewhere). Thus, a given PDX generally represents the tumor of origin with high fidelity.40,46,47,103,104 However, in patients, several biological factors can lead to heterogeneity in breast cancer tumors and affect a tumor’s response to therapy including tumor stage, location, grade/stage, and race/ethnicity (Mermut et al., 2021). Treatment of tumors also affects a tumor’s expression pattern105,106 and its subsequent response to therapy. Therefore, the patient’s prior treatment regimen may impact a PDX’s response to therapy. Because these biological covariates may affect each of our datasets differently, we fix our multigene biomarkers, thereby allowing the models to be flexible, which, in turn, allows us to predict chemotherapy response in diverse datasets.
By testing our multigene biomarkers across datasets from different cohorts, analyzed with different gene expression platforms, treated with different agents, we determined that (1) our multigene panels are predictive in both RNAseq and microarray datasets and (2) the multigene panels developed for carboplatin and docetaxel are predictive for other chemotherapy agents of the same class (platinums and taxanes, respectively).
Although this study focuses on identifying mRNA-based biomarkers of response, CTD is a flexible algorithm that can be applied to other data types.65 As cancer research shifts to multi- “omics” approaches of tumor characterization, CTD could be utilized to integrate these “omics” layers. By applying the CTD/WGCNA approach to many “omics” types (metabolomics, genomics, proteomics, and so forth), we could develop multi-omic biomarkers of response. This approach could also be used to address other biological questions and identify signatures of progression, metastasis, clonal evolution, and other phenotypes of interest.
Limitations of the study
Although this study has shown that PDXs can serve as preclinical cohorts, there are elements of the human immune system that are not replicated in immunodeficient PDX models. Creating humanized PDX mice via immune system reconstitution may allow for further insight into how the immune system contributes to resistance to chemotherapy in TNBC. The use of humanized PDX models may also improve the concordance between patient and PDXs response to therapy. In addition, in the clinical paclitaxel dataset (Loibl et al., 2018), clinical response was not evaluated immediately after treatment with paclitaxel. Testing our taxane biomarker on a clinical dataset where response was measured after immediately following taxane treatment would allow us to determine better the predictive power of this biomarker for patient response. Finally, although the CTD/WGCNA approach was applied to TNBC in this study, its application to other subtypes of breast cancer would potentially allow for the identification of chemotherapy biomarkers that cut across breast cancer subtypes.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| BCM/HCI PDX models | https://pdxportal.research.bcm.edu/ | 45 models Table S3 |
| Rosalind & Morris Goodman Cancer Research Centre RNAseq PDX models | GSE142767 | All models |
| Curie PDX models | Curie institute | All models |
| Dana-Farber PDX models | GSE18864 | All models |
| BrighTNess TNBC patient data | GSE164458 | All models |
| Deposited data | ||
| BCM/HCI PDX RNAseq | GSE183187 | All models |
| Software and algorithms | ||
| Code to recreate analysis | https://github.com/vpetrosyan/biomarkers_chemotherapy | All models (all input includes) |
Resource availability
Lead contact
Further question as requests for PDX resources should be addressed to Michael T. Lewis at mtlewis@bcm.edu.
Material availability
Information related to publicly available PDX models from Baylor College of Medicine (BCM) and the Huntsman Cancer Institute (HCI) can be found on the BCM PDX portal (https://pdxportal.research.bcm.edu/).
Experimental model and subject details
The BCM institutional review board has approved all experiments and all the experiments described here conform to all relevant standards. Female SCID/bg mice between 4 and 5 weeks of age were used for PDX generation in this study.
Patient derived xenograft model selection and chemotherapy response
Single agent docetaxel, carboplatin and untreated/vehicle control data were derived from three separate preclinical trials (to be described in full elsewhere). In total, 50 TNBC PDX models were treated across these three preclinical trials. To define a homogeneous set of tumors that did not over-represent any particular group of patient samples, we eliminated PDX models that did not match an immunosuppressed basal profile or were derived from the same patient as other models in this collection. Ultimately, 45 PDX were used for the generation of predictive models this study (Table S3).
Each of the three preclinical trials used the same treatment strategy. Briefly, PDX-bearing mice were treated with four weekly cycles of docetaxel (20 mg/kg, IP) (low human equivalent dose), docetaxel (30 mg/kg, IP) (high human equivalent dose), carboplatin (50 mg/kg, IP) (human equivalent dose AUC6), or left untreated for four weeks. A subset of models also received docetaxel at 30 mg/kg (high human-equivalent dose). For PDXs with both docetaxel (20 mg/kg, IP) and docetaxel (30 mg/kg, IP) response data, the best response to docetaxel was used. In addition, a subset of PDX models for whom we had clinical responses of the tumor-of-origin were also treated with doxorubicin (3 mg/kg)/cyclophosphamide (100 mg/kg) (AC), but human-equivalent doses could not be achieved under the conditions used.
Tumor volume was assessed twice weekly using calipers. Treatment responses were evaluated quantitatively by the log2 fold-change in tumor volume relative to their volume prior to treatment (∼200mm3). A qualitative “best clinical response” was assigned using a modified RECIST 1.1 69 criteria allowing for more direct comparison with tumor response in patients. Complete response (CR) was defined as non-palpable. Partial response (PR) was defined as those PDX showing >30% reduction in tumor volume, but not reaching the non-palpable state. PDX lines showing stable disease (SD) showed a <30% decrease, and no more than a 20% increase over starting volume. PDX lines showing progressive disease (PD) increased >20% over starting volume vs. vehicle control at the end of the study. Quantitative response is summarized in Table S3 (related to Figure 6).
Methods details
Genomic sequencing
DNA sequencing was performed by core facilities at Cornell University and BCM. At Cornell Exome Sequencing Libraries were prepared using the Agilent SureSelect XT v6.0 Human kit. Sequencing was performed on an Illumina HiSeq 4000 machine (PE, 2 × 100 cycles, ∼133X estimated coverage per sample). At BCM, sequencing was performed on Illumina NovaSeq machine (PE, 2 × 100, ∼200M read pairs per sample). Separation of human and mouse reads, alignment to the reference genome, variant calling, and annotation were performed using the PDXNet 105Tumor-Only Variant Calling pipeline developed by the Jackson Laboratory and hosted on the Cancer Genomics Cloud.
Baseline RNAseq transcriptomics
For RNA extraction from untreated PDX tissue, snap frozen tissue from an early transplant generation (e.g., TG1-TG5) xenograft-derived tumors were harvested and stored at −80 °C prior to use. For library preparation, the NuGEN Ovation RNAseq v2 Kit (protocol p/n 7102, reagent kit p/n 7102-08) along with ThermoFisher’s ERCC RNA Spike-In Control Mixes Protocol (publication number 4455352, rev. D) and either the Illumina TruSeq DNA-Seq (protocol p/n 15,005,180 Rev. C, June 2011, reagent kit p/n FC-121-2001) or the Rubicon ThruPlex DNA-Seq (protocol: QAM-108-002, kit p/n R400428) protocols were used.
Baseline mass spectrometry-based proteomics
The mass spectrometry proteomic iBAQ values were obtained for the same set of PDX models. To identify proteins that are specifically expressed in the human cancer cells, gpGrouper was used to deconvolute murine vs human proteins. The iBAQ values were then used to identify differentially expressed proteins.
Bioinformatics analyses and comparisons with TCGA and CPTAC samples
DNA sequencing data processing
All raw FASTQ files were subjected to QC verification by FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and were trimmed for adapter sequences with TrimGalore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Whole Exome FASTQ files were processed using the Tumor-Only Variant Calling pipeline46 developed by the Jackson Laboratory for PDXNet hosted on the Cancer Genomics Cloud. This pipeline uses Xenome79 to separate human epithelial reads and mouse stromal reads, then aligns human reads against the GRCh38 human genome using bwa.107 It then uses GATK MuTect2108,109,110 to call variants and SnpEff/SnpSift111,112 to annotate variants, generating per-sample VCF and tabular outputs for downstream analyses. Further annotation with gnomAD113 and CLINVAR114 was performed which added information by matching locus fields (Chromosome, Position, Reference, and Alternate alleles) to standard annotation VCF files. Multi-allelic sites were decomposed to ensure Alternate alleles matched exactly with annotation sources.
Copy number analysis
A set of 6 normal breast tissue samples were subjected to Whole Exome Sequencing both at Cornell and Baylor to serve as platform-matched normal samples. These platform-matched normal samples were run through the Variant Calling pipeline to obtain platform-matched normal BAMs. CopywriteR115 was used on each tumor BAM file obtained from the Variant Calling pipeline, and a random platform-matched normal BAM was used as control. CopywriteR v 2.28.0 uses depth information from off-target reads to calculate segment-level copy number data. These segment level data were then processed using GISTIC2.0116 to obtain raw and threshold gene-level and focal-level copy number information.
Comparison with TCGA samples
Mutation, Copy Number and Clinical Biomarker data for TCGA-BRCA samples was obtained using the TCGABiolinks package. TCGA-BRCA samples and PDX models were grouped based on their ER/PR and HER2 IHC status, and each TCGA group was compared with its equivalent group of PDX models.
Mutation frequency was computed per gene for all TCGA and PDX groups and compared among TNBC, ER+ and HER2+ groups. Similarly, Copy Number segment data were plotted per group to visualize common patterns in each group. To examine similarities between TNBC TCGA samples and TNBC PDX models, the segment level data for all TNBC samples across the groups were analyzed with GISTIC2.0 and the focal level copy number changes were used to generate a distance matrix and dendrogram using the R packages ggplot2 v 2.3.6117, dendextend v1.16118 and circlize v 0.4.15.119 A similar analysis was also done for ER+ and HER2+ groups.
Gene expression quantification
RNAseq FASTQ files were processed using Xenome to separate murine stromal reads and human epithelial reads, which were both then quantified with rsem-calculate-expression. A reference index was created for RSEM (v1.3.0)120 using rsem-prepare-reference on the hg38 and mm10 genome assembly (FASTA) and transcript feature (GTF) files. Expected counts from RSEM were collated to create a genes-by-samples matrix. This matrix was Upper-Quantile normalized per-sample to account for differences in sequencing depth.
Clustering of PDXs with primary tumors
For the RNA-seq clustering of PDX with TCGA primary tumors, the log2(upper quantile normalized) RSEM counts that were classified as cancer reads in the PDXs by Xenome were clustered with log2(upper quantile normalized) RSEM counts from TCGA BRCA tumors. Clustering was done over the PAM50 genes with the hclust function from the stats v 4.2.1 package with Pearson’s correlation coefficient.
For the protein clustering, the z-scores of proteins were computed (TNBC vs non TNBC) for both the primary CPTAC tumors and the PDX tumors. These z-scores were then used to cluster the PDXs with the primary tumors with hclust over the 1,000 most variable proteins with Pearson’s correlation coefficient using the stats v 4.2.1 package.
Network building
To identify genes that could be predictive of response to chemotherapy, we used the network approach described below. For both the human and mouse reads, a network was built over all the PDXs and the responsive PDXs for both carboplatin and docetaxel independently. The gene expression data was obtained from deep RNAseq (∼200M reads/sample) of the PDXs. Preprocessing included removing genes that were expressed in less than 20% of the samples or had a low expression across all the samples. The five thousand most variable genes were then selected for the human and mouse reads using the mad function from the stats v 4.2.1 package. These genes were used to build the species-specific networks using the WGNCA package v 1.71. The following graphs were built: an epithelial graph with all the PDXs, an epithelial graph with the samples that are responsive to carboplatin, an epithelial graph with the samples that are responsive to docetaxel, a stromal graph from all the samples, a stromal graph with the samples that are responsive to carboplatin, and a stromal graph with the samples that are responsive to docetaxel. The graphs were then pruned to remove weak edges (defined as having an edge weight of <0.2) using the delete.edges function from igraph v 1.3.4. The graphs built from all the samples were then pruned to remove the edges that were also found in the responsive graphs using CTD’s v 1.1.0 graph.naivePruning function. This was done for both carboplatin and docetaxel in both the epithelial and stromal compartments to identify signatures linked to resistance and potentially resistance mechanisms. As well as removing signal, which was not associated with response, this pruning step also reduced the number of edges making these graphs sparser.
WGCNA module discovery and the application of CTD
Coarse modules were defined in the pruned graphs by applying WGCNA v 1.71.’s cutreeDynamic function. After the initial module detection with the cutreeDynamic function, related modules were merged with WGCNA v 1.71’s mergeCloseModules function. The module WGCNA v 1.71.’s moduleEigengenes function was then used to calculate the module eigengenes for each of these modules.
To identify significantly connected submodules within these modules with CTD v 1.1.0 (https://github.com/BRL-BCM/CTD),65 we used the single-node diffusion walker (singleNode.getNodeRanksN function) CTD v 1.1.0 to get the ranks of each node. The mle.getPtBSbyK CTD v 1.10 function was then used to encode each set of connected nodes, and the mle.getEncodingLength CTD v 1.1.0 function was used to get the encoding length of these node sets. We then calculated the probability that the nodes were more connected than by chance by using the d-score calculated by the mle.getEncodingLength CTD v 1.1.0 function. This d-score represents how concisely the gene set can be encoded, and the most significantly connected set will be the one that is the most concisely encoded. The p value for each set of genes is then calculated using the following function.
A linear regression was then used to determine which genes within these submodules were informative for response. 9 informative genes were identified for carboplatin (5 epithelial and 4 stromal) and 6 informative genes were identified for docetaxel (4 epithelial and 2 stromal) (Thistlethwaite et al., 2021).
To empower other users to utilize CTD, we have deployed CTD as a tool on the Seven Bridge’s Cancer Genomic Cloud as a public application:
(https://cgc.sbgenomics.com/public/apps/pedjaoetf/ctd-commit-project/ctd)
Users will only need to upload their graphs as an adjacency matrix, as well as a list of genes in each module.
CTD’s code has been deposited in public GitHub repository at https://github.com/BRL-BCM/CTD where users can either download and use the package locally in R or run it inside Docker Desktop.
Predicting response to chemotherapy
The genes identified as informative were used to predict the response to carboplatin and docetaxel and the combination treatment. To model bulk patient data better, a pseudo-bulk profile was first made for each of the PDXs in our collection by summing the human epithelial reads and the murine stromal reads. Ten balanced 30% test and 70% training sets were defined using caret v. 6.0–93’s (Kuhn, 2008) createDataPartition function for each treatment. For each of these training/test permutations, the training set was used to create a quantitative GLM, whose performance was then tested on the test set. The mean absolute error and root-mean-square error were calculated for each training/test permutation and the observed vs. predicted log2 (fold change) was plotted. An LOOCV approach was used to determine the predictive power of our model in determining resistance vs. response as defined by the modified RECIST 1.1 69 criteria. The quantitative response for each sample was predicted, then the AUC was calculated to determine based on the qualitative response.
Comparison of our CTD/WGCNA approach to other feature selection methods
To compare the MAE and RMSE of the CTD/WGCNA approach to other commonly used feature selection methods, we also choose potentially informative genes with three other commonly used feature selection methods (correlation, recursive feature extraction, hub genes from informative WGCNA modules). Correlation - for each treatment the top 10 most correlated epithelial and stromal genes were determined (base R v 4.2.1). Recursive feature extraction – for each treatment the top 10 most informative epithelial and stromal genes were determined (caret v 6.0–93). Hub genes – to identify the hub genes for each treatment, WGCNA modules whose eigengene was correlation with response >0.35 were identified (WGCNA v 1.71). Then genes with a >0.75 correlation with the module eigengene and a >0.2 correlation with response were identified as the hub genes. For carboplatin there were 14 epithelial WGCNA hub genes and no stromal WGCNA hub genes. For docetaxel there were 26 epithelial WGCNA genes and 1 stromal WGCNA gene. Boruta – the Boruta R function was used to identify genes that were tentative and confirmed for importance in the response models (Boruta v 7.0.0). For carboplatin Boruta identified 22 genes, and for docetaxel Boruta identified 28 genes. Reticulate v 1.26 121 was used to implement the python scikit-learn package in R. The scikit-learn v 1.1 SelectKBest function was used to perform the F-test and mutual information regression feature selection methods with k = 10 (stromal and epithelial). The F-test approach identified 18 genes that are informative for carboplatin and 15 genes that are informative for docetaxel. The mutual information regression approach identified 16 genes that are informative for carboplatin and 16 genes that are informative for docetaxel.
Rosalind & Morris Goodman Cancer Research Center RNAseq PDX dataset prediction
An RNAseq dataset and qualitative response were obtained from GEO (GSE142767) (n = 37). The majority of these PDX models were Basal-like and triple negative, however there were some PDX models that were classified as HER2 (7) or positive for ER (5) or HER2 enrichment (3). A LOOCV XGBoost v 1.6.0 tree model was built over both the carboplatin genes (to predict response to cisplatin) and the docetaxel genes (to predict response to paclitaxel). A ROC curve was then calculated to determine the predictive power of predicting if the PDX has CR vs. all else or CR and PR vs. all else.
Curie dataset prediction
The Curie dataset was obtained from the Curie institute. This dataset consists of the Affymetrix array expression of 37 TNBC PDXs and the human equivalent response to cisplatin. Due to the inconsistencies between the array and RNAseq platforms, an XGBoost v 1.6.0 tree model (LOOCV) was built over the carboplatin multigene biomarker with the xgbTree option in caret for this dataset to determine the AUC and predictive nature of these genes.
Dana-Farber cancer institute TNBC patient array dataset prediction
The Dana-Farber (GSE18864) dataset was obtained from GEO.97 This dataset included array data from 24 patient biopsies as well as qualitative response of these patients to neoadjuvant cisplatin. These patients were part of a cohort of 28 women with TNBC that were treated with neoadjuvant cisplatin every 21 days. An XGBoost v 1.6.0 tree model (LOOCV) was built over the carboplatin multigene biomarker with the xgbTree option in caret for this dataset to determine the AUC and predictive nature of these genes.
BrighTNess TNBC patient RNAseq dataset prediction
RNAseq and patient response to paclitaxel followed by AC was obtained for the BrighTNess (GSE164458) dataset from GEO 38Data from arm C, single agent paclitaxel followed by AC in 123 patients were used for this study. A LOOCV XGBoost v 1.2.0.1 approach was used over the informative genes for to predict the response (pCR or not) to paclitaxel.
Quantification and statistical analysis
The quantification of the response of the BCM/HCI PDX models to therapy was assessed biweekly with calipers and reported as the log2 fold change relative to the volume before treatment (full details in the methods section). gpGrouper122 was used to determine the expression of both human and murine proteins and iBAQ values were used to compare samples. Xenome was used to separate and quantify the murine and human expression in each PDX model, and the expression values were upper quantile normalized to account for varying sequencing depth. CopywriteR115 and GISTIC2.0 116 were used to determine copy number information.
To determine the predictive power of each of the feature selection methods (full details in the methods sections) in the PDX dataset, we trained each model on 70% of the data and determined its accuracy (mean absolute error and root mean squared error) on the 30% test set. To ensure that selection bias did not play a role in this experiment, the same split was used for each feature selection method. To determine the predictive power of our multigene biomarker on validation sets, a LOOCV approach was used and an ROC curve was constructed for each validation dataset.
Acknowledgments
This work was supported by NIH/NCI grants U54 CA224076 (to A.L.W, B.E.W, and M.T.L.), U24 CA226110, (to M.T.L.), P50 CA186784 (to C.K.O and M.T.L.), Dan L. Duncan Cancer Center (P30 Cancer Center Support Grant NCI-CA125123) (to C.K.O.), a grant from the Common Fund of the National Institutes of Health (NIH) (5U54 DA036134) (to A.M) and the Henry and Emma Meyer Chair in Molecular Genetics (to A.M), and by grant T32 CA203690 from the Translational Breast Cancer Research Training Program (to J.T.L.). This work was also supported by a Core Facility grant from the Cancer Prevention and Research Institute of Texas (CPRIT Core Facilities Support Grant RP170691), a grant from the V Foundation, and a generous gift from the Korell family for the study of triple-negative breast cancer. B.Z. is a CPRIT Scholar in Cancer Research, and this work was supported by CPRIT award RR160027. This work was further supported by the Stand Up to Cancer Dream Team Translational Research Grant (SU2C-AACR-DT0209 (GMW)), DF/HCC Specialized Program of Research Excellence (SPORE) in Breast Cancer, NIH Grant P50 CA168504 (GMW) and the Breast Cancer Research Foundation. Work performed by BCM Mass Spectrometry Proteomics Core was supported by the Dan L. Duncan Comprehensive Cancer Center Award (P30 CA125123) and CPRIT Core Facility Awards (RP170005 and RP210227).
Author contributions
Conceptualization V.P., A.M., and M.T.L.; Methodology, V.P., L.T., V.K., P.O., R.R.S., J.T.L., L.E.D., A.M., and M.T.L.; Formal Analysis, V.P., A.M., and M.T. L.; Resources L.E.D., A.N.L., C.S., M.J.E., C.K.O., M.F.R., A.P., M.N.S., H.D., A.J., A.B.S., A.M., E.M., A.L.W., B.E.W., S.L., G.M.W., O.M., C.H., S.V., S.G.H., B.Z., A.M., and M.T.L.; Data Curation, V.P.; Writing V.P., A.M., and M.T.L.; Visualization, V.P., J.T.L., and A.M.; Supervision, A.M. and M.T.L.; Project Administration, A.M. and M.T.L.; Funding Acquisition, A.M. and M.T.L.
Declaration of interests
M.T.L is a Founder of, and an uncompensated Limited Partner in, StemMed Ltd., and an uncompensated Manager in StemMed Holdings L.L.C., its General Partner. MTL is also a Founder of, and equity stake holder in, Tvardi Therapeutics Inc. L.E.D. is a compensated employee of StemMed Ltd. Selected BCM PDX models described herein are exclusively licensed to StemMed Ltd., resulting in tangible property royalties to M.T.L. and L.E.D. University of Utah may license the HCI PDX models described herein to for-profit companies, which may result in tangible property royalties to A.L.W. and B.E.W. Washington University has licensed selected PDX to Envigo, which results in tangible property royalties to S.L. He also received research funding from Pfizer, Takeda Oncology, and Zenopharm, independent of this project. S.L. has received license fees from Envigo. He received research funding from Pfizer, Takeda Oncology, and Zenopharm, outside of this project. O.S, is a compensated employee of, and equity stake holder in, Bluebird Bio. M.J.E. received consulting fees from Abbvie, Sermonix, Pfizer, AstraZeneca, Celgene, NanoString, Puma, Veracyte, Eli Lilly, and Novartis, and he is an equity stockholder and Board of Directors member of BioClassifier. M.J.E. is an inventor on a patent for the Breast Cancer PAM50-based assay, Prosigna, which is marketed by Veracyte. M.J.E. also receives royalties from Washington University in St Louis when the WHIM PDX lines are licensed to for-profit companies.
Published: January 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105799.
Supplemental information
Data and code availability
-
•
All the code and inputs necessary to recreate the analyses in this manuscript have been deposited at GitHub: https://github.com/vpetrosyan/biomarkers_chemotherapy. DOI: https://doi.org/10.5281/zenodo.7140873 and are publicly available as of the date of publication.
-
•
All data required to recreate the BCM/HCI analysis has been deposited at GEO: GSE183187. The PDX Portal: https://pdxportal.research.bcm.edu/ also allows the user to easily query both patient and PDX data.
-
•
All data required to recreate the validation analysis is available from: GEO: GSE142767, GSE18864, GSE164458, or the Curie Institute upon reasonable request.
-
•
Any additional information is available from the lead contact upon request.
References
- 1.Mehanna J., Haddad F.G., Eid R., Lambertini M., Kourie H.R. Triple-negative breast cancer: current perspective on the evolving therapeutic landscape. Int. J. Womens Health. 2019;11:431–437. doi: 10.2147/ijwh.s178349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garrido-Castro A.C., Lin N.U., Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov. 2019;9:176–198. doi: 10.1158/2159-8290.cd-18-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehmann B.D., Pietenpol J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes: biomarker strategies for triple-negative breast cancer. J. Pathol. 2014;232:142–150. doi: 10.1002/path.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.da Silva J.L., Cardoso Nunes N.C., Izetti P., de Mesquita G.G., de Melo A.C. Triple negative breast cancer: a thorough review of biomarkers. Crit. Rev. Oncol. Hematol. 2020;145:102855. doi: 10.1016/j.critrevonc.2019.102855. [DOI] [PubMed] [Google Scholar]
- 5.Cocco S., Piezzo M., Calabrese A., Cianniello D., Caputo R., Lauro V.D., Fusco G., Gioia G.d., De Laurentiis M., Licenziato M., Laurentiis M. de. Biomarkers in triple-negative breast cancer: state-of-the-art and future perspectives. Int. J. Mol. Sci. 2020;21:4579. doi: 10.3390/ijms21134579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali M.A., Aiman W., Shah S.S., Hussain M., Kashyap R. Efficacy and safety of Pembrolizumab based therapies in triple-negative breast cancer: a systematic review of clinical trials. Crit. Rev. Oncol. Hematol. 2021;157:103197. doi: 10.1016/j.critrevonc.2020.103197. [DOI] [PubMed] [Google Scholar]
- 7.Kwapisz D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021;70:607–617. doi: 10.1007/s00262-020-02736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobson A. Pembrolizumab improves outcomes in early-stage and locally advanced or metastatic triple-negative breast cancer. Oncol. 2022;27:S17–S18. doi: 10.1093/oncolo/oyac014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganguly S., Gogia A. Pembrolizumab monotherapy in advanced triple-negative breast cancer. Lancet Oncol. 2021;22:e224. doi: 10.1016/s1470-2045(21)00249-7. [DOI] [PubMed] [Google Scholar]
- 10.Cortes J., Rugo H.S., Cescon D.W., Im S.-A., Yusof M.M., Gallardo C., Lipatov O., Barrios C.H., Perez-Garcia J., Iwata H., et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N. Engl. J. Med. 2022;387:217–226. doi: 10.1056/nejmoa2202809. [DOI] [PubMed] [Google Scholar]
- 11.Kang C., Syed Y.Y. Atezolizumab (in combination with nab-paclitaxel): a review in advanced triple-negative breast cancer. Drugs. 2020;80:601–607. doi: 10.1007/s40265-020-01295-y. [DOI] [PubMed] [Google Scholar]
- 12.Schmid P., Adams S., Rugo H.S., Schneeweiss A., Barrios C.H., Iwata H., Diéras V., Hegg R., Im S.-A., Shaw Wright G., et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018;379:2108–2121. doi: 10.1056/nejmoa1809615. [DOI] [PubMed] [Google Scholar]
- 13.Emens L.A., Molinero L., Loi S., Rugo H.S., Schneeweiss A., Diéras V., Iwata H., Barrios C.H., Nechaeva M., Nguyen-Duc A., et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer: biomarker evaluation of the IMpassion130 study. J. Natl. Cancer Inst. 2021;113:1005–1016. doi: 10.1093/jnci/djab004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddy S.M., Carroll E., Nanda R. Atezolizumab for the treatment of breast cancer. Expert Rev. Anticancer Ther. 2020;20:151–158. doi: 10.1080/14737140.2020.1732211. [DOI] [PubMed] [Google Scholar]
- 15.Kolbin A.S., Vilyum I.A., Proskurin M.A., Balikina Y.E., Pavlysh A.V. Pharmacoeconomic analysis of atezolizumab plus nab-paclitaxel in the treatment of the advanced or metastatic triple-negative breast cancer. Kachestvennaya Klinicheskaya Praktika. 2020:4–21. doi: 10.37489/2588-0519-2020-1-4-21. [DOI] [Google Scholar]
- 16.Marra A., Viale G., Curigliano G. Recent advances in triple negative breast cancer: the immunotherapy era. BMC Med. 2019;17:90. doi: 10.1186/s12916-019-1326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid P., Cortes J., Pusztai L., McArthur H., Kümmel S., Bergh J., Denkert C., Park Y.H., Hui R., Harbeck N., et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 2020;382:810–821. doi: 10.1056/nejmoa1910549. [DOI] [PubMed] [Google Scholar]
- 18.Tutt A.N.J., Garber J.E., Kaufman B., Viale G., Fumagalli D., Rastogi P., Gelber R.D., de Azambuja E., Fielding A., Balmaña J., et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N. Engl. J. Med. 2021;384:2394–2405. doi: 10.1056/nejmoa2105215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blum J.L., Laird A.D., Litton J.K., Rugo H.S., Ettl J., Hurvitz S.A., Martín M., Roché H.H., Lee K.-H., Goodwin A., et al. Determinants of response to talazoparib in patients with HER2-negative, germline BRCA1/2-mutated breast cancer. Clin. Cancer Res. 2022;28:1383–1390. doi: 10.1158/1078-0432.ccr-21-2080. clincanres. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spring L.M., Nakajima E., Hutchinson J., Viscosi E., Blouin G., Weekes C., Rugo H., Moy B., Bardia A. Sacituzumab govitecan for metastatic triple-negative breast cancer: clinical overview and management of potential toxicities. Oncol. 2021;26:827–834. doi: 10.1002/onco.13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiss J., Glode A., Messersmith W.A., Diamond J. Sacituzumab govitecan: breakthrough targeted therapy for triple-negative breast cancer. Expert Rev. Anticancer Ther. 2019;19:673–679. doi: 10.1080/14737140.2019.1654378. [DOI] [PubMed] [Google Scholar]
- 22.Bardia A., Messersmith W.A., Kio E.A., Berlin J.D., Vahdat L., Masters G.A., Moroose R., Santin A.D., Kalinsky K., Picozzi V., et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann. Oncol. 2021;32:746–756. doi: 10.1016/j.annonc.2021.03.005. [DOI] [PubMed] [Google Scholar]
- 23.Bardia A., Mayer I.A., Vahdat L.T., Tolaney S.M., Isakoff S.J., Diamond J.R., O’Shaughnessy J., Moroose R.L., Santin A.D., Abramson V.G., et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N. Engl. J. Med. 2019;380:741–751. doi: 10.1056/nejmoa1814213. [DOI] [PubMed] [Google Scholar]
- 24.Bottini A., Berruti A., Bersiga A., Brizzi M.P., Bruzzi P., Aguggini S., Brunelli A., Bolsi G., Allevi G., Generali D., et al. Relationship between tumour shrinkage and reduction in Ki67 expression after primary chemotherapy in human breast cancer. Br. J. Cancer. 2001;85:1106–1112. doi: 10.1054/bjoc.2001.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y., Chen W., Zhang X., He S., Shao N., Shi H., Lin Z., Wu X., Li T., Lin H., Lin Y. Prediction of tumor shrinkage pattern to neoadjuvant chemotherapy using a multiparametric MRI-based machine learning model in patients with breast cancer. Front. Bioeng. Biotechnol. 2021;9:662749. doi: 10.3389/fbioe.2021.662749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toppmeyer D.L. 2002. Breast Cancer, A Guide to Detection and Multidisciplinary Therapy; pp. 213–226. [DOI] [Google Scholar]
- 27.Huober J., von Minckwitz G. Neoadjuvant therapy – what have we achieved in the last 20 years. Breast Care. 2011;6:419–426. doi: 10.1159/000335347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mieog J.S.D., van der Hage J.A., van de Velde C.J.H., Charehbili A. Preoperative chemotherapy for women with operable breast cancer. Cochrane Database Syst. Rev. 2007;2007:CD005002. doi: 10.1002/14651858.cd005002.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spring L.M., Fell G., Arfe A., Sharma C., Greenup R., Reynolds K.L., Smith B.L., Alexander B., Moy B., Isakoff S.J., et al. Pathologic complete response after neoadjuvant chemotherapy and impact on breast cancer recurrence and survival: a comprehensive meta-analysis. Clin. Cancer Res. 2020;26:2838–2848. doi: 10.1158/1078-0432.ccr-19-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spanheimer P.M., Carr J.C., Thomas A., Sugg S.L., Scott-Conner C.E.H., Liao J., Weigel R.J. The response to neoadjuvant chemotherapy predicts clinical outcome and increases breast conservation in advanced breast cancer. Am. J. Surg. 2013;206:2–7. doi: 10.1016/j.amjsurg.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anampa J., Makower D., Sparano J.A. Progress in adjuvant chemotherapy for breast cancer: an overview. BMC Med. 2015;13:195. doi: 10.1186/s12916-015-0439-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. Oncol. 2005;10:20–29. doi: 10.1634/theoncologist.10-90003-20. [DOI] [PubMed] [Google Scholar]
- 33.Caparica R., Lambertini M., Pondé N., Fumagalli D., de Azambuja E., Piccart M. Post-neoadjuvant treatment and the management of residual disease in breast cancer: state of the art and perspectives. Ther. Adv. Med. Oncol. 2019;11 doi: 10.1177/1758835919827714. 1758835919827714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wahba H.A., El-Hadaad H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015;12:106–116. doi: 10.7497/j.issn.2095-3941.2015.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma P., López-Tarruella S., García-Saenz J.A., Ward C., Connor C.S., Gómez H.L., Prat A., Moreno F., Jerez-Gilarranz Y., Barnadas A., et al. Efficacy of neoadjuvant carboplatin plus docetaxel in triple-negative breast cancer: combined analysis of two cohorts. Clin. Cancer Res. 2017;23:649–657. doi: 10.1158/1078-0432.ccr-16-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sikov W.M., Berry D.A., Perou C.M., Singh B., Cirrincione C.T., Tolaney S.M., Kuzma C.S., Pluard T.J., Somlo G., Port E.R., et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (alliance) J. Clin. Oncol. 2015;33:13–21. doi: 10.1200/jco.2014.57.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Minckwitz G., Schneeweiss A., Loibl S., Salat C., Denkert C., Rezai M., Blohmer J.U., Jackisch C., Paepke S., Gerber B., et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol. 2014;15:747–756. doi: 10.1016/s1470-2045(14)70160-3. [DOI] [PubMed] [Google Scholar]
- 38.Loibl S., O’Shaughnessy J., Untch M., Sikov W.M., Rugo H.S., McKee M.D., Huober J., Golshan M., von Minckwitz G., Maag D., et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol. 2018;19:497–509. doi: 10.1016/s1470-2045(18)30111-6. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020;13:4. doi: 10.1186/s13045-019-0829-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X., Claerhout S., Prat A., Dobrolecki L.E., Petrovic I., Lai Q., Landis M.D., Wiechmann L., Schiff R., Giuliano M., et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–4897. doi: 10.1158/0008-5472.can-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobrolecki L.E., Airhart S.D., Alferez D.G., Aparicio S., Behbod F., Bentires-Alj M., Brisken C., Bult C.J., Cai S., Clarke R.B., et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016;35:547–573. doi: 10.1007/s10555-016-9653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Savage P., Pacis A., Kuasne H., Liu L., Lai D., Wan A., Dankner M., Martinez C., Muñoz-Ramos V., Pilon V., et al. Chemogenomic profiling of breast cancer patient-derived xenografts reveals targetable vulnerabilities for difficult-to-treat tumors. Commun. Biol. 2020;3:310. doi: 10.1038/s42003-020-1042-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whittle J.R., Lewis M.T., Lindeman G.J., Visvader J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015;17:17. doi: 10.1186/s13058-015-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Echeverria G.V., Powell E., Seth S., Ge Z., Carugo A., Bristow C., Peoples M., Robinson F., Qiu H., Shao J., et al. High-resolution clonal mapping of multi-organ metastasis in triple negative breast cancer. Nat. Commun. 2018;9:5079. doi: 10.1038/s41467-018-07406-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powell R.T., Redwood A., Liu X., Guo L., Cai S., Zhou X., Tu Y., Zhang X., Qi Y., Jiang Y., et al. Pharmacologic profiling of patient-derived xenograft models of primary treatment-naïve triple-negative breast cancer. Sci. Rep. 2020;10:17899. doi: 10.1038/s41598-020-74882-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evrard Y.A., Srivastava A., Randjelovic J., Arunachalam S., Bult C.J., Chen H., Chen L., Davies M., Davies S., Davis-Dusenbery B., et al. Systematic establishment of robustness and standards in patient-derived xenograft experiments and analysis. bioRxiv. 2019:790246. doi: 10.1101/790246. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woo X.Y., Giordano J., Srivastava A., Zhao Z.-M., Lloyd M.W., de Bruijn R., Suh Y.-S., Patidar R., Chen L., Scherer S., et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. bioRxiv. 2019:861393. doi: 10.1101/861393. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koga Y., Ochiai A. Systematic review of patient-derived xenograft models for preclinical studies of anti-cancer drugs in solid tumors. Cells. 2019;8:418. doi: 10.3390/cells8050418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuhn M. Vol. 1. Wiley Interdiscip Rev Comput Statistics; 2008. Building Predictive Models in R Using the Caret Package; pp. 128–129. [Google Scholar]
- 50.Kursa M.B., Rudnicki W.R. Feature selection with the Boruta package. J. Stat. Soft. 2010;36 doi: 10.18637/jss.v036.i11. [DOI] [Google Scholar]
- 51.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Müller A., Nothman J., Louppe G., et al. Scikit-learn: Machine Learning in Python. Arxiv. 2012 Preprint at. [Google Scholar]
- 52.Zhao W., Langfelder P., Fuller T., Dong J., Li A., Hovarth S. Weighted gene coexpression network analysis: state of the art. J. Biopharm. Stat. 2010;20:281–300. doi: 10.1080/10543400903572753. [DOI] [PubMed] [Google Scholar]
- 53.Tadist K., Najah S., Nikolov N.S., Mrabti F., Zahi A. Feature selection methods and genomic big data: a systematic review. J. Big Data. 2019;6:79. doi: 10.1186/s40537-019-0241-0. [DOI] [Google Scholar]
- 54.Tang J., Kong D., Cui Q., Wang K., Zhang D., Gong Y., Wu G. Prognostic genes of breast cancer identified by gene Co-expression network analysis. Front. Oncol. 2018;8:374. doi: 10.3389/fonc.2018.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jia R., Zhao H., Jia M. Identification of co-expression modules and potential biomarkers of breast cancer by WGCNA. Gene. 2020;750:144757. doi: 10.1016/j.gene.2020.144757. [DOI] [PubMed] [Google Scholar]
- 56.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu W., Li L., Ye H., Tu W. [Weighted gene co-expression network analysis in biomedicine research] Sheng Wu Gong Cheng Xue Bao Chin J Biotechnology. 2017;33:1791–1801. doi: 10.13345/j.cjb.170006. [DOI] [PubMed] [Google Scholar]
- 58.Di Y., Chen D., Yu W., Yan L. Bladder cancer stage-associated hub genes revealed by WGCNA co-expression network analysis. Hereditas. 2019;156:7. doi: 10.1186/s41065-019-0083-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Giulietti M., Occhipinti G., Principato G., Piva F. Identification of candidate miRNA biomarkers for pancreatic ductal adenocarcinoma by weighted gene co-expression network analysis. Cell. Oncol. 2017;40:181–192. doi: 10.1007/s13402-017-0315-y. [DOI] [PubMed] [Google Scholar]
- 60.Huang Y., Liu H., Zuo L., Tao A. Key genes and co-expression modules involved in asthma pathogenesis. PeerJ. 2020;8:e8456. doi: 10.7717/peerj.8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang L., Kang W., Lu X., Ma S., Dong L., Zou B. Weighted gene co-expression network analysis and connectivity map identifies lovastatin as a treatment option of gastric cancer by inhibiting HDAC2. Gene. 2019;681:15–25. doi: 10.1016/j.gene.2018.09.040. [DOI] [PubMed] [Google Scholar]
- 62.Liao Y., Wang Y., Cheng M., Huang C., Fan X. Weighted gene coexpression network analysis of features that control cancer stem cells reveals prognostic biomarkers in lung adenocarcinoma. Front. Genet. 2020;11:311. doi: 10.3389/fgene.2020.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Du B.S., Yuan L., Sun L.G., Zhang Z.Y. WGCNA screening of prognostic markers in medulloblastoma. Zhonghua Yixue Zazhi. 2020;100:460–464. doi: 10.3760/cma.j.issn.0376-2491.2020.06.013. [DOI] [PubMed] [Google Scholar]
- 64.Qiu J., Du Z., Wang Y., Zhou Y., Zhang Y., Xie Y., Lv Q. Weighted gene co-expression network analysis reveals modules and hub genes associated with the development of breast cancer. Medicine. 2019;98:e14345. doi: 10.1097/md.0000000000014345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thistlethwaite L.R., Petrosyan V., Li X., Miller M.J., Elsea S.H., Milosavljevic A. CTD: an information-theoretic algorithm to interpret sets of metabolomic and transcriptomic perturbations in the context of graphical models. PLoS Comput. Biol. 2021;17:e1008550. doi: 10.1371/journal.pcbi.1008550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 67.Izumchenko E., Paz K., Ciznadija D., Sloma I., Katz A., Vasquez-Dunddel D., Ben-Zvi I., Stebbing J., McGuire W., Harris W., et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017;28:2595–2605. doi: 10.1093/annonc/mdx416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cancer Genome Atlas Network. Fulton R.S., McLellan M.D., Schmidt H., Kalicki-Veizer J., McMichael J.F., Fulton L.L., Dooling D.J., Ding L., Mardis E.R., et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eisenhauer E.A., Therasse P., Bogaerts J., Schwartz L.H., Sargent D., Ford R., Dancey J., Arbuck S., Gwyther S., Mooney M., et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 70.Judson P.L., Watson J.M., Gehrig P.A., Fowler W.C., Haskill J.S. Cisplatin inhibits paclitaxel-induced apoptosis in cisplatin-resistant ovarian cancer cell lines: possible explanation for failure of combination therapy. Cancer Res. 1999;59:2425–2432. [PubMed] [Google Scholar]
- 71.Levasseur L.M., Greco W.R., Rustum Y.M., Slocum H.K. Combined action of paclitaxel and cisplatin against wildtype and resistant human ovarian carcinoma cells. Cancer Chemother. Pharmacol. 1997;40:495–505. doi: 10.1007/s002800050693. [DOI] [PubMed] [Google Scholar]
- 72.Guminski A.D., Harnett P.R., deFazio A. Carboplatin and paclitaxel interact antagonistically in a megakaryoblast cell line – a potential mechanism for paclitaxel-mediated sparing of carboplatin-induced thrombocytopenia. Cancer Chemother. Pharmacol. 2001;48:229–234. doi: 10.1007/s002800100279. [DOI] [PubMed] [Google Scholar]
- 73.Tanaka R., Ariyama H., Qin B., Takii Y., Baba E., Mitsugi K., Harada M., Nakano S. In vitro schedule-dependent interaction between paclitaxel and oxaliplatin in human cancer cell lines. Cancer Chemother. Pharmacol. 2005;55:595–601. doi: 10.1007/s00280-004-0966-z. [DOI] [PubMed] [Google Scholar]
- 74.Stamelos V.A., Robinson E., Redman C.W., Richardson A. Navitoclax augments the activity of carboplatin and paclitaxel combinations in ovarian cancer cells. Gynecol. Oncol. 2013;128:377–382. doi: 10.1016/j.ygyno.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 75.Yunos N.M., Beale P., Yu J.Q., Strain D., Huq F. Studies on combinations of platinum with paclitaxel and colchicine in ovarian cancer cell lines. Anticancer Res. 2010;30:4025–4037. [PubMed] [Google Scholar]
- 76.Tanaka R., Takii Y., Shibata Y., Ariyama H., Qin B., Baba E., Kusaba H., Mitsugi K., Harada M., Nakano S. In vitro sequence-dependent interaction between nedaplatin and paclitaxel in human cancer cell lines. Cancer Chemother. Pharmacol. 2005;56:279–285. doi: 10.1007/s00280-004-0991-y. [DOI] [PubMed] [Google Scholar]
- 77.Makiyama A., Qin B., Uchino K., Shibata Y., Arita S., Isobe T., Hirano G., Kusaba H., Baba E., Akashi K., Nakano S. Schedule-dependent synergistic interaction between gemcitabine and oxaliplatin in human gallbladder adenocarcinoma cell lines. Anti Cancer Drugs. 2009;20:123–130. doi: 10.1097/cad.0b013e3283218080. [DOI] [PubMed] [Google Scholar]
- 78.Fu C., Liu Y., Han X., Pan Y., Wang H.Q., Wang H., Dai H., Yang W. An immune-associated genomic signature effectively predicts pathologic complete response to neoadjuvant paclitaxel and anthracycline-based chemotherapy in breast cancer. Front. Immunol. 2021;12:704655. doi: 10.3389/fimmu.2021.704655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Conway T., Wazny J., Bromage A., Tymms M., Sooraj D., Williams E.D., Beresford-Smith B. Xenome—a tool for classifying reads from xenograft samples. Bioinformatics. 2012;28:i172–i178. doi: 10.1093/bioinformatics/bts236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Csardi G., Nepusz T. InterJournal; 2006. The Igraph Software Package for Complex Network Research; p. 1695. [Google Scholar]
- 81.Lang Y., Kong X., He C., Wang F., Liu B., Zhang S., Ning J., Zhu K., Xu S. Musashi1 promotes non-small cell lung carcinoma malignancy and chemoresistance via activating the akt signaling pathway. Cell. Physiol. Biochem. 2017;44:455–466. doi: 10.1159/000485012. [DOI] [PubMed] [Google Scholar]
- 82.Lagadec C., Vlashi E., Frohnen P., Alhiyari Y., Chan M., Pajonk F. The RNA-binding protein Musashi-1 regulates proteasome subunit expression in breast cancer- and glioma-initiating cells. Stem Cell. 2014;32:135–144. doi: 10.1002/stem.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Darb-Esfahani S., Kronenwett R., von Minckwitz G., Denkert C., Gehrmann M., Rody A., Budczies J., Brase J.C., Mehta M.K., Bojar H., et al. Thymosin beta 15A (TMSB15A) is a predictor of chemotherapy response in triple-negative breast cancer. Br. J. Cancer. 2012;107:1892–1900. doi: 10.1038/bjc.2012.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang B., Zhang Y., Dagher M.-C., Shacter E. Rho GDP dissociation inhibitor protects cancer cells against drug-induced apoptosis. Cancer Res. 2005;65:6054–6062. doi: 10.1158/0008-5472.can-05-0175. [DOI] [PubMed] [Google Scholar]
- 85.Bozza W.P., Zhang Y., Hallett K., Rivera Rosado L.A., Zhang B. RhoGDI deficiency induces constitutive activation of Rho GTPases and COX-2 pathways in association with breast cancer progression. Oncotarget. 2015;6:32723–32736. doi: 10.18632/oncotarget.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Staudigl C., Concin N., Grimm C., Pfeiler G., Nehoda R., Singer C.F., Polterauer S. Prognostic relevance of pretherapeutic gamma-glutamyltransferase in patients with primary metastatic breast cancer. PLoS One. 2015;10:e0125317. doi: 10.1371/journal.pone.0125317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Valiente M., Obenauf A.C., Jin X., Chen Q., Zhang X.H.-F., Lee D.J., Chaft J.E., Kris M.G., Huse J.T., Brogi E., Massagué J. Serpins promote cancer cell survival and vascular Co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo X., Li J., Zhang H., Liu H., Liu Z., Wei X. Relationship between ADAMTS8, ADAMTS18, and ADAMTS20 (A disintegrin and metalloproteinase with thrombospondin motifs) expressions and tumor molecular classification, clinical pathological parameters, and prognosis in breast invasive ductal carcinoma. Med. Sci. Monit. 2018;24:3726–3735. doi: 10.12659/msm.907310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bai X., Gao C., Zhang L., Yang S. Integrin α7 high expression correlates with deteriorative tumor features and worse overall survival, and its knockdown inhibits cell proliferation and invasion but increases apoptosis in breast cancer. J. Clin. Lab. Anal. 2019;33:e22979. doi: 10.1002/jcla.22979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bhandari A., Xia E., Zhou Y., Guan Y., Xiang J., Kong L., Wang Y., Yang F., Wang O., Zhang X. ITGA7 functions as a tumor suppressor and regulates migration and invasion in breast cancer. Cancer Manag. Res. 2018;10:969–976. doi: 10.2147/cmar.s160379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ming X.-Y., Fu L., Zhang L.-Y., Qin Y.-R., Cao T.-T., Chan K.W., Ma S., Xie D., Guan X.-Y. Integrin α7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nat. Commun. 2016;7:13568. doi: 10.1038/ncomms13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jia B., Zhao X., Wang Y., Wang J., Wang Y., Yang Y. Prognostic roles of MAGE family members in breast cancer based on KM-Plotter Data. Oncol. Lett. 2019;18:3501–3516. doi: 10.3892/ol.2019.10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dany M., Ogretmen B. Ceramide induced mitophagy and tumor suppression. Biochim. Biophys. Acta. 2015;1853:2834–2845. doi: 10.1016/j.bbamcr.2014.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen B., Lai J., Dai D., Chen R., Liao N., Tang H. 2020. PARPBP Is a Prognostic Marker and Confers Chemotherapeutic Resistance to Breast Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bandala C., Cortés-Algara A.L., Mejía-Barradas C.M., Ilizaliturri-Flores I., Dominguez-Rubio R., Bazán-Méndez C.I., Floriano-Sánchez E., Luna-Arias J.P., Anaya-Ruiz M., Lara-Padilla E. Botulinum neurotoxin type A inhibits synaptic vesicle 2 expression in breast cancer cell lines. Int. J. Clin. Exp. Pathol. 2015;8:8411–8418. [PMC free article] [PubMed] [Google Scholar]
- 96.Kim S., Lee K., Choi J.-H., Ringstad N., Dynlacht B.D. Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nat. Commun. 2015;6:8087. doi: 10.1038/ncomms9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Silver D.P., Richardson A.L., Eklund A.C., Wang Z.C., Szallasi Z., Li Q., Juul N., Leong C.-O., Calogrias D., Buraimoh A., et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010;28:1145–1153. doi: 10.1200/jco.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Farmer P., Bonnefoi H., Anderle P., Cameron D., Becette V., Wirapati P., André S., Piccart M., Campone M., Brain E., et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009;15:68–74. doi: 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- 99.Plava J., Cihova M., Burikova M., Matuskova M., Kucerova L., Miklikova S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol. Cancer. 2019;18:67. doi: 10.1186/s12943-019-0960-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dittmer J., Leyh B. The impact of tumor stroma on drug response in breast cancer. Semin. Cancer Biol. 2015;31:3–15. doi: 10.1016/j.semcancer.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 101.Criscitiello C., Esposito A., Curigliano G. Tumor–stroma crosstalk: targeting stroma in breast cancer. Curr. Opin. Oncol. 2014;26:551–555. doi: 10.1097/cco.0000000000000122. [DOI] [PubMed] [Google Scholar]
- 102.Conklin M.W., Keely P.J. Why the stroma matters in breast cancer: insights into breast cancer patient outcomes through the examination of stromal biomarkers. Cell Adh. Migr. 2012;6:249–260. doi: 10.4161/cam.20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guillen K.P., Fujita M., Butterfield A.J., Scherer S.D., Bailey M.H., Chu Z., DeRose Y.S., Zhao L., Cortes-Sanchez E., Yang C.-H., et al. A breast cancer patient-derived xenograft and organoid platform for drug discovery and precision oncology. bioRxiv. 2021 doi: 10.1101/2021.02.28.433268. Preprint at. 2021.02.28.433268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sun H., Cao S., Mashl R.J., Mo C.-K., Zaccaria S., Wendl M.C., Davies S.R., Bailey M.H., Primeau T.M., Hoog J., et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidates for targeted treatment. Nat. Commun. 2021;12:5086. doi: 10.1038/s41467-021-25177-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bownes R.J., Turnbull A.K., Martinez-Perez C., Cameron D.A., Sims A.H., Oikonomidou O. On-treatment biomarkers can improve prediction of response to neoadjuvant chemotherapy in breast cancer. Breast Cancer Res. 2019;21:73. doi: 10.1186/s13058-019-1159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hannemann J., Oosterkamp H.M., Bosch C.A.J., Velds A., Wessels L.F.A., Loo C., Rutgers E.J., Rodenhuis S., van de Vijver M.J. Changes in gene expression associated with response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2005;23:3331–3342. doi: 10.1200/jco.2005.09.077. [DOI] [PubMed] [Google Scholar]
- 107.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Arxiv. 2013 doi: 10.48550/arXiv.1303.3997. Preprint at. [DOI] [Google Scholar]
- 108.Marx V. Genomics in the clouds. Nat. Methods. 2013;10:941–945. doi: 10.1038/nmeth.2654. [DOI] [PubMed] [Google Scholar]
- 109.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cingolani P., Patel V.M., Coon M., Nguyen T., Land S.J., Ruden D.M., Lu X. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kuilman T., Velds A., Kemper K., Ranzani M., Bombardelli L., Hoogstraat M., Nevedomskaya E., Xu G., de Ruiter J., Lolkema M.P., et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015;16:49. doi: 10.1186/s13059-015-0617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wickham H. ggplot2, Elegant Graphics for Data Analysis. Radiokhimiya. 2016:147–168. doi: 10.1007/978-3-319-24277-4_7. [DOI] [Google Scholar]
- 118.Galili T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015;31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gu Z., Gu L., Eils R., Schlesner M., Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 120.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tang K.U., J.A., Y. 2022. Reticulate: Interface to ‘Python’.https://rstudio.github.io/reticulate/authors.html#citation [Google Scholar]
- 122.Saltzman A.B., Leng M., Bhatt B., Singh P., Chan D.W., Dobrolecki L., Chandrasekaran H., Choi J.M., Jain A., Jung S.Y., et al. gpGrouper: a peptide grouping algorithm for gene-centric inference and quantitation of bottom-up proteomics data. Mol. Cell. Proteomics. 2018;17:2270–2283. doi: 10.1074/mcp.tir118.000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All the code and inputs necessary to recreate the analyses in this manuscript have been deposited at GitHub: https://github.com/vpetrosyan/biomarkers_chemotherapy. DOI: https://doi.org/10.5281/zenodo.7140873 and are publicly available as of the date of publication.
-
•
All data required to recreate the BCM/HCI analysis has been deposited at GEO: GSE183187. The PDX Portal: https://pdxportal.research.bcm.edu/ also allows the user to easily query both patient and PDX data.
-
•
All data required to recreate the validation analysis is available from: GEO: GSE142767, GSE18864, GSE164458, or the Curie Institute upon reasonable request.
-
•
Any additional information is available from the lead contact upon request.