

Abstract
Background:
We report the creation and evaluation of a de novo assembly of the genome of the spontaneously hypertensive rat, the most widely used model of human cardiovascular disease.
Methods:
The genome is assembled from long read sequencing (PacBio HiFi and CLR) and scaffolded with long range structural information obtained from Bionano optical maps and Hi-C proximity analysis of the genome. The genome assembly was polished with Illumina short reads. Completeness of the assembly was investigated using BUSCO analysis. The genome assembly was also evaluated with the rat reference gene set using NCBI automated protocols. We also generated orthogonal single molecule transcript sequence reads (Iso-Seq) from 8 tissues and used them to validate the coding assembly, to annotate the assembly with RNA transcripts representing unique full length transcript isoforms for each gene and to determine whether divergences between RefSeq sequences and the assembly were attributable to assembly errors or polymorphisms.
Results:
The assembly analysis indicates that this assembly is comparable in contiguity and completeness to the current rat reference assembly while the use of HiFi sequencing yields an assembly that is more correct at the single base level. Synteny analysis was performed to uncover the extent of synteny and the presence and distribution of chromosomal rearrangements between the reference and this assembly.
Conclusions:
The resulting genome assembly is reference quality and captures significant structural variation.
Keywords: De novo assembly, rat genome, long reads, optical mapping, chromosome conformation capture
Graphical Abstract
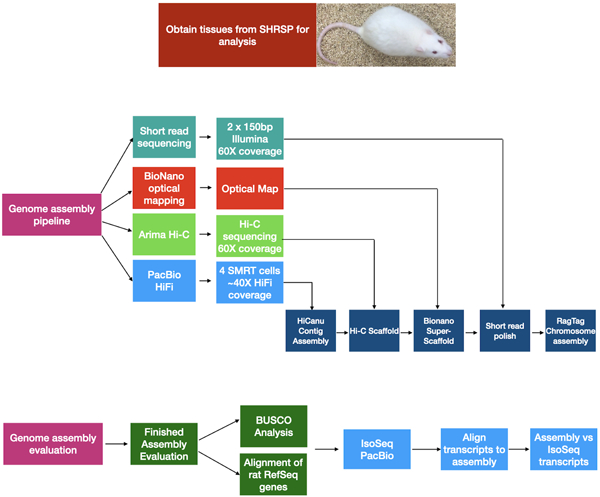
Background
During 2001–2003 the National Human Genome Research Institute of NIH awarded nearly $60M to support the creation of the first draft assembly of the genome of the brown rat (Rattus norvegicus) using DNA obtained from the inbred Brown-Norway strain (BN/SsNHsd/Mcwi substrain). After human and mouse genome assemblies, this was the third mammalian genome assembly NIH had supported and the first to be limited to draft, rather than finished quality. In a landmark 2004 Nature paper, the Rat Genome Sequencing Project Consortium reported that an assembly of ~2.75 billion bases had been produced as a result of a hybrid approach combining whole genome shotgun sequencing with bacterial artificial chromosome (BAC) clone sequencing, employing a novel assembly software to integrate the resulting “bactigs” with longer distance genome information to generate the assembly which was named Rnor3.1 [1]. This resulted in 128,000 contigs of ~38kb N50 length that were linked into 738 superbactigs (N50 = 5.4Mb) which could eventually be conjoined to form ~300 larger assembly subunits. Subsequent revision included Y chromosome sequencing from a male SHR/Akr rat to supplement the two female rats used as the DNA source for Rnor3.1. This genome assembly has been progressively improved through the introduction of both additional assembly methods and sequence data with a notable change being the inclusion of single molecule, real-time (SMRT) sequencing of continuous long read (CLR) data (Pacific Biosciences) to enhance assembly of regions with structural variation in the 2014 release of Rnor_6 release. A brief summary of the properties of these assemblies is provided in Table 1.
Table 1.
The progression of rat genome de novo assemblies available at NCBI
| Assembly name | Strain | Ploidy | Total ungapped length | NCBI Biosample | NCBI BioProject | Date | Submitter |
|---|---|---|---|---|---|---|---|
| RGSC v3.1 | BN/SsNHsdMCW | haploid | ~2,750,000,000 | none | PRJNA10629 | 3/10/04 | Rat Genome Sequencing Consortium |
| RGSC_v3.4 | BN/SsNHsdMCW | haploid | ~2,750,000,000 | none | PRJNA10629 | 12/13/04 | Rat Genome Sequencing Consortium |
| Rn_Celera | BN; Sprague-Dawley | haploid | 2,807,682,847 | SAMN03000701 | PRJNA13999 | 12/6/06 | Celera Genomics |
| Rnor4.1 | BN/SsNHsdMCW | 2,471,873,957 | none | PRJNA10629 | 2/5/10 | Rat Genome Sequencing Consortium | |
| Rnor_5.0 | BN/SsNHsdMCW | haploid | 2,573,083,111 | SAMN02808218 | PRJNA10629 | 3/16/12 | Rat Genome Sequencing Consortium |
| Rnor_6.0 | mixed BN and SHR/Akr | haploid | 2,729,985,504 | SAMN02808228 | PRJNA10629 | 7/1/14 | Rat Genome Sequencing Consortium |
| mRatBN7.2 | BN/SsNHsdMCW | diploid | 2,626,580,772 | SAMN16261960 | PRJNA662791 | 11/10/20 | Wellcome Sanger Institute |
| mRatBN7.1 alternate haplotype | BN/SsNHsdMCW | diploid | 286,931,055 | SAMN16261960 | PRJNA663241 | 11/4/20 | Wellcome Sanger Institute |
| UTH_Rnor_SHRSP_BbbUtx_1.0 | SHRSP/BbbUtx | haploid | 2,905,913,608 | SAMN24538170 | PRJNA793432 | 1/24/22 | Inbred Rat Genome Sequencing Project (UTH/UK/UofL) |
During the last 4–5 years, important technology developments have taken place that have had a major impact on whole genome sequencing and genome assembly. These have centered on methods that provide increased long-range sequence and genome structure information that provide a means to increase contiguity of the genome assembly. One such method is the use of optical genome mapping and assembly [2]. Use of high molecular weight genomic DNA allows single DNA molecules to be analyzed that often exceed 300kb in length. Optical map coverage depth of 100–200-fold is easily accomplished and the resulting molecules can provide a low resolution, but large scale sequence imaging of the genome that can be assembled across long distances. This optical map can be combined with long range scaffolding using data obtained by chromosome conformation capture and analysis via proximity ligation sequencing (Hi-C) [3]. This method extracts the long-range orientation of genomic DNA sequence that derives from the predictable and consistent spatial relationships between proximal and distant regions of a chromosome. The result is another long-range scaffold providing orthogonal information that can further assess and correct sequence-based assemblies. These two technologies can also complement one another by running them serially to improve contiguity in addition to correcting mis-joins.
These scaffolds provide frameworks that must be filled with accurate, nucleotide-resolved whole genome sequencing information, which itself is made up of correctly oriented and assembled sequence contigs. Sanger sequencing and subsequently short-read next generation sequencing had been the bedrock of genome assembly, but the inability to extend contigs through repeat elements impedes their generation and extension. This limitation has been reduced by the introduction of long-read sequencing methods. Commercial implementations of long-read genome sequencing methods have tackled the problem of high error rates associated with long reads so that now accurate, highly contiguous assemblies are possible. The most recent rat reference genome, mRatBN7.2, was produced by combining the optical and Hi-C scaffolding methods described above with long-read genome sequencing using the CLR approach [4]. Although CLR reads are noisy, the unique advantage they provide is the generation of contiguous sequence data ranging in length from 10s to 100s of kilobases, which is long enough to span many repetitive sequence elements. As the cost to generate these data continues to decrease, sufficient read coverage can be used to reduce base level errors in the final consensus sequence. Each of the scaffolding and sequencing methods depends on suitable software and computing capabilities to extract, process and utilize the information they provide. Remarkably, the cost of implementing these assembly and computation methods continues to decline so that a contemporary reference quality de novo assembly of a mammalian genome can be achieved for less than 1/1000th the cost of the original rat draft genome assembly.
This reduction in cost has posed a new dilemma in the exploitation of inbred rodent models in genetic and genomic studies: is the optimal approach to rely on common short-read WGS data (Illumina) derived from a multitude of rat strains and crosses in order to draw conclusions by comparison to a reference genome; or, is a new era emerging when phenotypes divergent in contrasting strains can be better investigated through the generation of accurate de novo genome assemblies of each individual strain? The former approach has dominated, but creates an important limitation, referred to as reference bias, that arises when sequence variation present in the subject (SHR) genome is not recognized because reads representing it do not align to the reference. In order to begin to provide a feasibility analysis of the latter approach we have generated a de novo genome assembly of the widely used inbred rat strain, the spontaneously hypertensive rat. There are nearly 25,000 PubMed citations to this model which describe a wide range of interesting heritable cardiovascular and other phenotypes. In spite of many years of active investigation, most of these phenotypes remain unresolved at the sequence level. We created our assembly using optical mapping, chromosomal conformation capture, standard short-read whole genome shotgun sequence data, long-read CLR data, and the recently introduced SMRT sequencing-derived highly accurate (“HiFi”) reads. Here we report on the methods and results of this assembly and compare this assembly to the CLR-based current rat reference assembly, mRatBN7.2.
Methods
All primary data have been made publicly available at the Ntional Center for Biotechnology Information and can be accessed via https://www.ncbi.nlm.nih.gov/bioproject?LinkName=biosample_bioproject&from_uid=24538170.
An Extended Methods section is available as a supplementary file, a brief outline of technical aspects of the assembly is provided below.
Whole genome sequencing and assembly
Pacific Biosciences (PacBio) methods were used to generate HiFi and CLR reads from genomic DNA following the manufacturer’s protocol. Short-read sequences (150 base paired end) with ~50X coverage depth (TruSeq, Illumina HiSeqX), were obtained from an animal of the same strain to polish the final assembly using Pilon software [11]. HiFi reads were assembled into contigs with HiCanu software (version 2.1.1), We used Arima Hi-C methods to generate chromosome conformation data [13]. The resulting Hi-C library was sequenced on the Illumina Novaseq 6000 platform and contigs were scaffolded using the resulting contact data and SALSA2 software [3]. Optical mapping data was obtained using the Bionano methods and materials and a de novo assembly was created that was integrated with the Hi-C scaffolds used Bionano Saphyr Solve software (v3.3) to generate hybrid scaffolds.
Chromosome builds, gap filling and polishing
We evaluated and corrected scaffolds and integrated sub-chromosomal scaffolds into full length chromosome builds using RagTag v2.1.0 software (https://github.com/malonge/RagTag) [14] using the existing rat reference assembly mRatBN7.2 (NCBI ID: GCF_015227675.2) [4] to template chromosome level scaffolding. Polishing was performed with Pilon version 1.24 [11].
Assembly Annotation and Evaluation
An annotation of the assembly was produced by NCBI using automated methods after masking of repetitive sequences in the genome [18]. The best alignment for each transcript was selected based on identity and coverage. Completeness of the assembly was assessed using BUSCO (Benchmarking Universal Single Copy Orthologs, version 5.2.2 with the MetaEuk predictor [20]. Structural variation was assessed using Sniffles2 software [21] followed by SURVIVOR software to compare structural variants across samples [22]. We also analysized synteny and structural rearrangements between the two completed assemblies using SyRI software [23,24].
Alignment of full length SHRSP gene transcripts to the assembly
Samples were collected from 8 tissues of an18 week old SHRSP male animal. RNA was sequenced using PacBio Iso-Seq protocols and analysis pipelines. To investigate specific genes, transcript reads were in silico translated and the resulting coding analyzed using NCBI Blastp. We examined the Iso-Seq reads to assess whether they differed from the genome assembly. This allowed orthogonal evaluation of coding sequence correctness.
Released Assembly
The finished assembly and underlying data are available from NCBI at https://www.ncbi.nlm.nih.gov/assembly/GCA_021556685.1 and https://www.ncbi.nlm.nih.gov/bioproject/793432. Iso-Seq data can be accessed via the bioproject link.
Results
Contig assembly
Genomic DNA was sequenced with ~36X HiFi read coverage. These HiFi reads were assembled using HiCanu software to generate contigs. Output statistics for HiCanu contig formation are provided in Table S1.
Assembly completeness, accuracy and annotation.
Comparison of the assembly structure to that of the current rat reference assembly was made using assembly-stats software [25] and is presented in Figure 1. BUSCO analysis is summarized in Table 2 and compares the SHRSP genome assembly scores with those of the current rat reference genome, mRatBN7.2. Analysis was performed using conserved ortholog lineage databases for the class mammalia and the clade glires.
Figure 1.
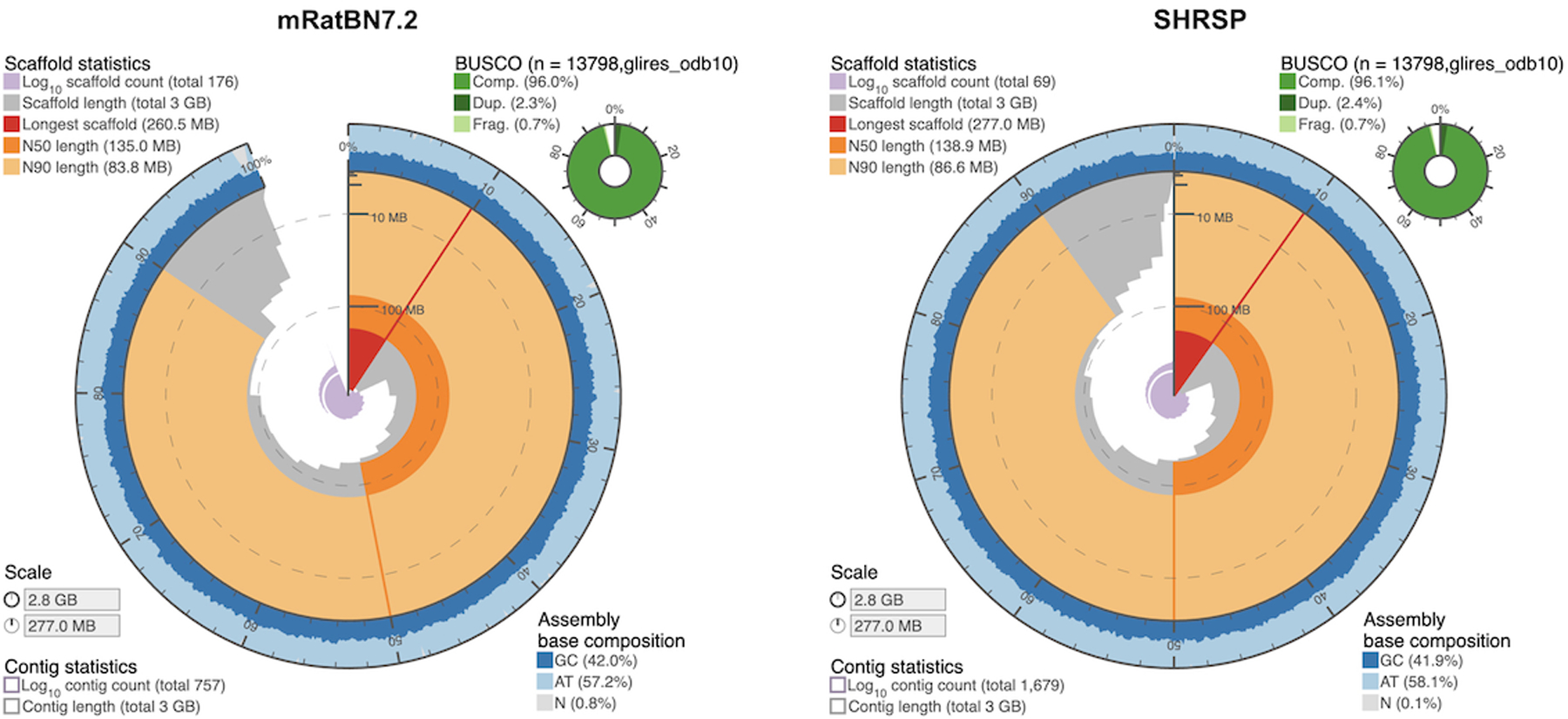
Visual representation of mRatBN7.2 and SHRSP genome assemblies made using assembly-stats software.
Table 2.
Use of Benchmarking Universal Single Copy Ortholog (BUSCO) analysis of recently assembled rat genomes to establish a measure of completeness using the ortholog databases for mammalia and glires.
| Strain | %Complete* | %Fragmented | %Missing | OrthoDB | Groups searched |
|---|---|---|---|---|---|
| mRatBN7.2 | 95.98 | 0.74 | 3.28 | glires.odb10 2021–01-19 | 13798 |
| SHR | 96.10 | 0.71 | 3.19 | glires.odb10 2021–01-19 | 13798 |
| mRatBN7.2 | 96.25 | 0.91 | 2.84 | mammalia.odb10 2021–01-19 | 9226 |
| SHR | 96.22 | 0.91 | 2.87 | mammalia.odb10 2021–01-19 | 9226 |
complete and single copy and complete and duplicated
We used short-read sequencing of the SHRSP genome to estimate accuracy of the HiFi-derived assembly. Variant call rate between these reads and the assembly was 1 variant per 20,833 assembly bases. This call rate combines errors in both the HiFi and short reads but still represents a base call concordance greater than 99.99%, representing a Q score of 43.
The automated NCBI annotation QC pipeline was used to evaluate the SHRSP assembly and to compare the results with the same analysis of the mRatBN7.2 reference assembly. The results of this analysis are summarized in Table 3. The reference mRatBN7.2 assembly has 200 frame-shifted gene coding sequences, while the SHRSP assembly has less than half this number. This may reflect an impact of the improved base level accuracy obtained by HiFi sequencing methods used for SHRSP compared to the CLR sequencing used in the mRatBN7.2 assembly [26]. Among the 91 frameshifted genes in the SHRSP assembly, 8 were also frame-shifted in the mRatBN7.2 assembly (possibly indicating errors in the curated RefSeq transcript sequence) and 43 were in genes encoding the highly duplicated odorant receptor (olfactory receptor and vomeronasal organ receptor) gene families.
Table 3.
Assembly metrics and RefSeq automated analysis of recent rat genome assemblies.
| Assembly Name | mRatBN7.2 | SHR |
|---|---|---|
| NCBI Accession # | GCA_015227675.2 | GCA_021556685.1 |
| Total Transcripts evaluated | 20479 | 20479 |
| Total Genes evaluated | 18260 | 18260 |
| Unaligned Transcripts | 18 | 27 |
| Unaligned Genes | 18 | 27 |
| Split Transcripts | 0 | 2 |
| Split Genes | 0 | 2 |
| Low Coverage CDS | 16 | 35 |
| Low Coverage CDS Genes | 16 | 31 |
| Frameshifted CDS | 205 | 100 |
| Frameshifted CDS Genes | 180 | 95 |
| Transcripts with start codon changes | 4 | 7 |
| Genes with start codon changes | 1 | 7 |
| Transcripts with stop codon changes | 15 | 47 |
| Genes with stop codon changes | 15 | 46 |
| TOTAL Genes | 230 | 208 |
Notes: Unaligned transcripts and genes are transcripts and protein coding sequences for which no alignment was found. Split alignment transcripts and genes are those for which the best alignment is split across more than one locus. Low coding sequences (CDS) coverage transcripts and genes have alignments covering less than 95% of the CDS. Dropped transcripts and genes are excluded from the final annotation because they overlap or conflict with other gene(s) with higher precedence. Frameshifts are transcripts with CDS that project imperfectly on the assembly. Start and stop changes are transcripts and genes with a mismatch versus the genome in the start codon or the stop codon causing it to become coding, or in an internal codon causing it to become a stop codon.
We have investigated these problem genes to assess whether they reflect assembly errors using the orthogonal Iso-Seq data obtained from 8 unique tissues from an SHRSP animal. This SHRSP Iso-Seq data allows expressed full-length transcripts to be aligned directly to the SHRSP genome. The presence of one or more transcripts that correctly align across any flagged problem in a given gene corroborates that the flagged region is correctly assembled. In SHRSP a total of 272 unique RefSeq genes were flagged during annotation as containing a possible problem. The same analysis flagged 287 genes in mRatBN7.2. “Problem” genes fall into several categories: 1) missing from assembly, 2) split alignments, 3) low coverage genes, 4) frameshifted genes, 5) start-stop changes, and 6) dropped genes. Table S2 provides a definition of these changes, identifies the affected genes in both strains and summarizes the results of Iso-Seq evaluation. There were 80 genes that were flagged in both SHRSP and mRatBN7.2 assemblies (Table S2), suggesting possible problems associated with the RefSeq transcript sequence. Odorant receptor (OR) genes represent a highly duplicated and diverse family of genes and the majority of the RefSeq OR genes were identified from the BN-derived rat reference genome by computational analysis. Thus, high divergence between these genes can be expected. Likewise, BN and SHRSP belong to different major histocompatibility (MHC) groups so divergence in rat RT-1 genes is expected [27]. In SHRSP, 80 of the flagged genes were odorant receptor or MHC genes. This leaves 112 genes flagged uniquely in SHRSP that were not odorant receptor or MHC genes and not shared in common with mRatBN7.2. Although Iso-Seq data from only 8 tissues provides limited coverage of the complete set of rat RefSeq genes, some of the remaining 112 flagged genes could be investigated by analysis of aligned Iso-Seq transcripts. We identified 51 of 112 genes with SHRSP full length transcripts that aligned to the SHRSP genome assembly. Among the 14 problem genes with transcripts and identified as having divergent stop or start codons, transcript alignment verified 13 of these are accurately described in our assembly. Among 37 other problem genes (principally frameshifts) with Iso-Seq transcripts, 9 were found to have transcripts fully aligned to the SHRSP assembly that encoded full length proteins identical to the NCBI reference protein. We found no evidence that genes with Iso-Seq transcripts had assembled genomic sequences that were incongruent with the Iso-Seq transcript. Thus, all 51 of the flagged genes with full length transcripts were correctly represented in our assembly.
Structural variation and synteny analysis
Structural variation was detecting using Sniffles2 software [21] by direct analysis of SHRSP PacBio genomic HiFi reads with the mRatBN7.2 assembly and generating a variant call file (File S1). The structural variations between the assemblies were compared using SURVIVOR software (https://github.com/fritzsedlazeck/SURVIVOR) and a summary of variant types and affected genome lengths was generated (Table S3). Synteny analysis of the two assemblies was performed with SyRI software [23,24] and indicated that 96.0% of the mRatBN7.2 was syntenous to SHRSP, while 91.7% of the SHRSP was syntenous to the mRatBN7.2 assembly (Table 4). This difference in estimated synteny across the bi-directional comparisons reflects the fact that the SHRSP assembly is ~0.2Gb larger than the mRatBN7.2 primary assembly, thus a smaller fraction of the SHRSP assembly is syntenous to the mRatBN7.2 assembly because SHRSP contains regions likely not incorporated into the mRatBN7.2 assembly. The mRatBN7.2 assembly had much less sequence that was duplicated in the SHRSP assembly (~6Mb in 359 duplications) compared to ~57Mb of SHRSP, which duplicated in 4247 duplications in mRatBN7.2. This difference may arise from the assembly pipeline used for mRatBN7.2 which anticipated distinct maternal and paternal haplotypes and in which some sequence duplications were computationally purged from the primary assembly [4]. There were nearly 5 million single nucleotide polymorphisms (SNPs) across the two assemblies, indicating a genome-wide base level divergence of 0.18%. In addition SyRI identified ~1.66 million indels. Files S1–11 provide complete information regarding variation between SHRSP and the mRatBN7.2 genome including a variant call file and tables identifying single nucleotide polymorphisms, structural variations, inversions, duplications, inverted duplications, translocations, inverted translocations, cross-chromosomal events and syntenic/non-syntenic (unaligned) regions [23]. Figure 2 provides a visualization of synteny between SHRSP’s genome and the mRatBN7.2 reference generated from the synteny and rearrangement analysis performed using SyRI software [23] and visualized with plotsr software [28].
Table 4.
Assessment of structural variations identified by computational analysis of two recent rat genome assemblies.
| Structural annotations mRatBN7.2 vs SHRSP | |||
|---|---|---|---|
| Variation_type | Count | Length_mRatBN7.2 | Length_SHR |
| Syntenic regions | 702 | 2,551,196,998 | 2,558,120,777 |
| Inversions | 159 | 13,116,928 | 33,862,589 |
| Translocations | 323 | 28,291,472 | 28,840,750 |
| Duplications (reference) | 359 | 6,163,791 | - |
| Duplications (query) | 4247 | - | 56,897,605 |
| Not aligned (reference) | 723 | 50,927,996 | - |
| Not aligned (query) | 4451 | - | 115,219,184 |
| Sequence annotations mRatBN7.2 vs SHRSP | |||
| Variation_type | Count | Length_mRatBN7.2 | Length_SHR |
| SNPs | 4915618 | 4,915,618 | 4,915,618 |
| Insertions | 814647 | - | 32,299,090 |
| Deletions | 841660 | 32,076,370 | - |
| Copy gains | 244 | - | 9,571,191 |
| Copy losses | 227 | 969,154 | - |
| Highly diverged | 11426 | 126,628,517 | 146,308,512 |
| Tandem repeats | 60 | 46,456 | 45,078 |
| Synteny between mRatBN7.2 vs SHRSP | |||
| Strain | Genome sizes | Amount syntenous | |
| mRatBN7.2 | 2,658,743,687 | 96.0 | |
| SHR | 2,788,855,122 | 91.7 | |
Figure 2.
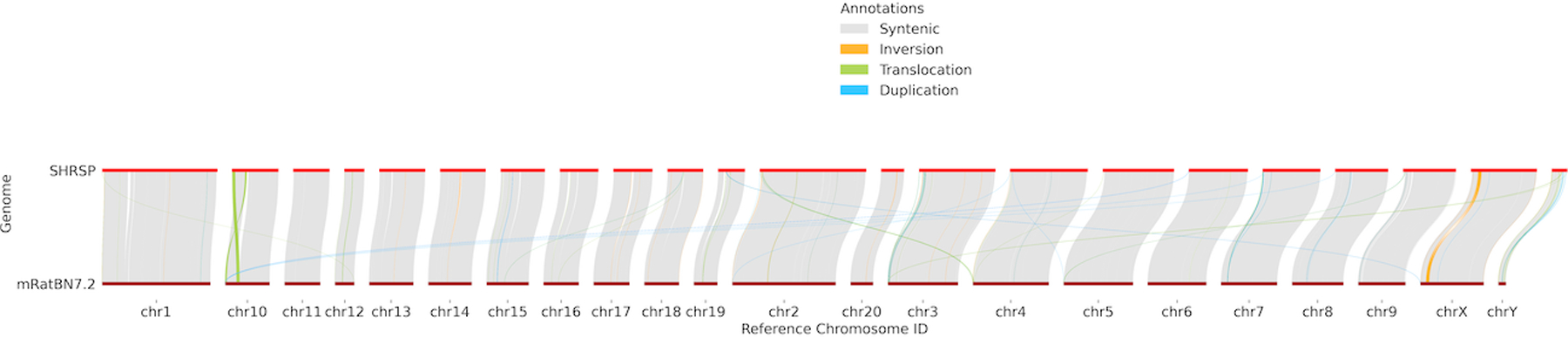
Synteny and Rearrangement plot of the current rat reference (mRatBN7.2) compared with the SHRSP genome assembly. This plot reveals both within and across chromosome re-arrangements.
Discussion.
Rattus norvegicus, the brown rat, is a relatively recent invasive species that evolved in east Asia and, during the last two millennia, entered and rapidly spread throughout the western hemisphere. While multiple routes of colonization to the western hemisphere occurred, the degree of genetic diversity in the rat suggests some impact of genetic founder effects, which are especially apparent in the rat strains that have been developed for use as research model organisms [29]. The current rat reference genome was derived from wild-caught (Northeastern US) rats that were inbred in the laboratory setting and thus may be considered to be a representative of the species that was free from human selection forces arising from domestication and breeding of rats for sporting activities, for generating fancy breeds maintained as pets, and for laboratory research. However, genome-wide analysis of genetic markers indicate that BN and the numerous inbred laboratory rat strains do in fact represent a cluster of genomes more closely related to each other than to globally dispersed representatives of the species. Within this cluster, the Brown Norway reference strain is somewhat less closely related to the other studied strains which show greater similarity to each other [29]. Thus, the development of a de novo genome assembly for the Wistar-derived spontaneously hypertensive rat (SHRSP) provides an additional resource that, by virtue of its proximity to other research strains and its value as a highly utilized research model of cardiovascular disease, adds to the capacity to define the genetic basis of phenotypes that diverge across inbred strains.
The use of multiple orthogonal contemporary genome assembly methods has resulted in a genome assembly that is at least equal to the current rat reference in measures such as contiguity, completeness, and correctness. Indeed, the mBN7.2 reference assembly included somewhat less accurate CLR sequencing and utilized an assembler that anticipated an outbred genome with divergent paternal and maternal haplotypes [4]. Because BN is, in fact, fully inbred [9,30], this may have resulted in the mis-assembly of gene duplications, as such assemblers anticipate that a divergent sequence region represents parental alleles at the same locus and removes one of the haplotypes from the primary assembly into an alternative haplotype. When such divergent sequences are the result of authentic gene duplication events with subsequent evolution of the duplicated regions, these “purged” primary assemblies can be reduced in their representation of authentic duplications. The occurrence of segmental duplication in gene coding regions is an important mechanism of evolution and a systematic removal of evidence of such events from the primary assembly may introduce biologically important inaccuracy. This type of assembly artifact may be reflected in the divergence in the number of duplicated segments detected in the BN assembly compared to the SHR assembly (Table 1).
The smaller size of the mRatBN7.2 genome assembly (2.65Gb vs 2.86Gb) may reflect, in part, the placement of authentic haploid genome sequences from the primary haplotype into the alternative haplotype. Use of HiFi reads in the assembly of the SHRSP genome may also facilitate contiguity of complex repetitive genomic regions. Thus, telomeres, centromeres and the Y chromosome may have fuller, though incomplete, representation in the SHRSP assembly than in the reference assembly. It is notable that prior assemblies of the BN reference that correctly treated the genome as fully haploid were generally closer in overall size to the SHR assembly than the current reference. Nonetheless, the recent human genome telomere-to-telomere (T2T) project has demonstrated that mammalian reference genome assemblies may lack representation of substantially sized, highly repetitive or duplicated genome segments that cannot be incorporated by assembly from long reads, including large-scale scaffolding methods such as we have used here [12,31]. The T2T project has effectively increased the extent of the human genome by incorporating over 200Mb of sequence that could not previously be assembled. Undoubtedly the SHRSP genome assembly, which includes unincorporated contigs, may be further improved by the emerging technical approaches unique to the T2T project. Trio binning has been proposed as a means to improve genome assembly of outbred diploid species [32,33]. However, because of the impossibility of introduction of wild animals to research animal housing facilities, the fully inbred state of SHRSP would require forming a trio by crossing to another laboratory strain. Such trios can improve assembly if there is sufficient genome-wide divergence in the parental genomes. Since BN and SHRSP are among the most distant of laboratory strains they might be considered in such an effort [29]. However, the overall frequency of SNP’s between these strains is only 0.18% and consequently trio binning is unlikely to be a useful tool to improve genome assembly.
Although the Iso-Seq data set we have generated is somewhat limited in tissue representation, the utility of these reads to authenticate the coding regions of the genome assembly and resolve ambiguities in annotation is established by our application of this approach. Furthermore, the method provides other opportunities. For example, the expression of RNA splice variation may diverge across tissues within an inbred strain. This may or may not reflect splicing patterns in tissues from a different strain, which may be relevant to biological traits. In addition, alternative isoform usage may be associated with underlying sequence variation in the genome (allele specific splicing) and the Iso-Seq data can provide insight into such events across strains. Our analysis of genes indicated by annotation as non-congruent with RefSeq sequences shows examples in which there is concurrence to RefSeq splice variants (XP_) of the corresponding gene (Table S2, see “SHRSP with transcript” and “Start stop evaluation” tabs). Finally, genetic variation can affect the level of gene expression. While Iso-Seq is semi-quantitative [34], it is still able to provide some useful quantitative information [34]. For example, inbred rat strains are known to vary in the expression of the important AP-1 transcription factor, JunD. This allelic effect occurs in cis and has been attributed to promoter polymorphism [35]. The SHRSP strain used in the current genome assembly contains the promoter variation previously associated with low JunD expression. Allelic JunD gene expression has been reported in macrophages, renal glomeruli, myocardium and prostate [35–37]. Our Iso-Seq data failed to uncover JunD transcripts in any of the 8 SHRSP tissues studied. Thus, multi-tissue Iso-Seq data may have the capacity to discern whether promoter variation that drives allelic expression shows a global effect on expression across tissues or is related to tissue-specific transcriptional control. As this long-read RNA sequencing method continues to develop, it may advance as a quantitative tool for studies of strain-specific gene expression patterns [38] and faciliate strain and phenotype-specific expression and alternative splicing.
The present study demonstrates that the construction of a contiguous, complete and correct genome assembly of an inbred rat strain is readily accomplished and can support improved utilization of genetic and genomic studies of rat models. This assembly reveals divergence between the existing reference and the subject genome that cannot emerge in alignment of short reads to the reference, exposing the potential for reference bias [5,39]. Linkage mapping, the primary tool of phenotype localization within the genomes of inbred animal models, may be affected by such differences because markers near a trait-associated locus may not display Hardy-Weinberg equilibrium when such markers lie within genomic regions that are duplicated in only one haplotype. Similarly, translocation of chromosomal regions within or between chromosomes may be specific to the reference or subject genomes, interfering with the utility of markers located within such syntenous, but translocated regions. Thus, the creation of high quality genome assemblies can advance the investigation of the many complex traits for which inbred rat strains serve as model organisms. The comparison between mRatBN7.2 and SHRSP genomes has delineated the potential for reference bias to affect genetic studies in this model of hypertension and related cardiovascular disease. Further de novo assemblies of strains used in crosses to perform genetic studies of SHR now seem warranted and will provide outstanding resources to advance a field that has struggled to make progress during more than 25 years of effort [40].
Perspectives
When knowledge of the genomic structure of an inbred gentic model of cardiovascular disease is limited to what can be observed by aligning short genome sequence reads to the genome assembly of a different inbred (reference) strain substantial limitations emerge. These include inaccuracies in the reference strain assembly and the presence of genomic sequence that is present in the genetic model, but absent in the reference and so cannot be aligned. Sequences from transposable elements comprise a significant fraction of the genome. These sequences can be highly conserved and can contribute to trait variation. However, they cannot be investigated without a genome assembly that correctly incorporates them. The production of a structurally accurate and complete genome assembly for the SHRSP strain will accelerate insights that this valuable model strain can provide into the genetic basis of hypertension and end organ damage.
Supplementary Material
Table S1 HiCanu assembly statistics for the SHRSP genome.
Table S2 RefSeq genes identified by annotation as being incongruently represented in the SHRSP assembly. Each type of alignment imperfection is represented in a single tab in the file. Iso-Seq analysis of these genes is provided in additional tabs.
Table S3 Structural variations between the assemblies identified by Sniffles software and compared using SURVIVOR software (https://github.com/fritzsedlazeck/SURVIVOR). A summary of variant types and affected genome lengths is provided
Files S1–11
Files S1–3 are large files and are available from the Zenodo persistent public repository website at the links provided in the files.
File S1 vcf file generated by Sniffles2/SURVIVOR analysis.
File S2 vcf file generated by SyRI analysis.
File S3 list of SNPs between the SHRSP and mRatBN7.2 genome assemblies
File S4 list of structural variants
File S5 list of syntenous regions
File S6 list of duplicated genome regions
File S7 list of interchromosomal translocations
File S8 list of unaligned regions
File S9 list of duplicated regions
File S10 list of translocated (within chromosome) regions)
File S11 list of inverted genome regions
File S12 list of both inverted and translocated regions
Pathophysiological Novelty and Relevance:
What is new?
A de novo assembly of the SHRSP genome made using current methods that capture larger structural genomic features while providing highly accurate and contiguous assembly at reference quality level.
What is Relevant?
The SHR and SHRSP strains provide a model of genetic hypertension. Progress in defining the genetic variation contributing to hypertension is scant. Limitations that result from aligning short genomic sequence reads to a rat reference genome from a different strain impede progress. This new assembly provides a reference quality genome sequence that will permit strain specific variation to be fully identified.
Clinical/Pathophysiological Implications?
SHR remains a valuable model og genetic hypertension and exists in strains susceptible (SHRSP) and resistant (SHR) to hypertensive end organ injury. This new assembly will aid in identifying the clinical mechanisms by which the naturally occuring genetic variation present in these strains can lead to cardiovascular disease.
Sources of Funding
This work was supported in part by the National Center for Biotechnology Information of the National Library of Medicine (NLM) at the National Institutes of Health. This work was also supported by National Institutes of Health Award Numbers: NIH R01HG011252 to PAD/MLS/TSK and R01DK081866 to PAD.
Abbreviations
- SMRT
single molecule, real-time sequencing
- CLR
continuous long read data
- Hi-C
proximity ligation sequencing
- BUSCO
Benchmarking Universal Single Copy Orthologs
References
- 1.Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 2004;428: 493–521. doi: 10.1038/nature02426 [DOI] [PubMed] [Google Scholar]
- 2.Jeffet J, Margalit S, Michaeli Y, Ebenstein Y. Single-molecule optical genome mapping in nanochannels: multidisciplinarity at the nanoscale. Essays Biochem 2021;65: 51–66. doi: 10.1042/EBC20200021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghurye J, Rhie A, Walenz BP, Schmitt A, Selvaraj S, Pop M, et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol 2019;15: e1007273. doi: 10.1371/journal.pcbi.1007273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howe K, Dwinell M, Shimoyama M, Corton C, Betteridge E, Dove A, et al. The genome sequence of the Norway rat, Rattus norvegicus Berkenhout 1769. Wellcome Open Res 2021;6: 118. doi: 10.12688/wellcomeopenres.16854.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martiniano R, Garrison E, Jones ER, Manica A, Durbin R. Removing reference bias and improving indel calling in ancient DNA data analysis by mapping to a sequence variation graph. Genome Biol 2020;21: 250. doi: 10.1186/s13059-020-02160-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paten B, Novak AM, Eizenga JM, Garrison E. Genome graphs and the evolution of genome inference. Genome Res 2017;27: 665–676. doi: 10.1101/gr.214155.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Y, Zhang Z, Bao Z, Li H, Lyu Y, Zan Y, et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 2022;606: 527–534. doi: 10.1038/s41586-022-04808-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okamoto K, Yamori Y, Nagaoka A. Establishment of the Stroke-prone Spontaneously Hypertensive Rat (SHR). Circ Res 1974;14: I143–I153. [Google Scholar]
- 9.Hermsen R, de Ligt J, Spee W, Blokzijl F, Schäfer S, Adami E, et al. Genomic landscape of rat strain and substrain variation. BMC Genomics 2015;16: 357. doi: 10.1186/s12864-015-1594-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vollger MR, Logsdon GA, Audano PA, Sulovari A, Porubsky D, Peluso P, et al. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann Hum Genet 2020;84: 125–140. doi: 10.1111/ahg.12364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014;9: e112963. doi: 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res 2020;30: 1291–1305. doi: 10.1101/gr.263566.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shultzaberger RK, Abrams RE, Sullivan CJ, Schmitt AD, Thompson TWJ, Dresios J. Agnostic detection of genomic alterations by holistic DNA structural interrogation. PLoS One 2018;13: e0208054. doi: 10.1371/journal.pone.0208054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alonge M, Soyk S, Ramakrishnan S, Wang X, Goodwin S, Sedlazeck FJ, et al. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol 2019;20: 224. doi: 10.1186/s13059-019-1829-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience 2021;10: giab008. doi: 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 2017;27: 722–736. doi: 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34: 3094–3100. doi: 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thibaud-Nissen F, Souvorov A, Murphy T, DiCuccio M, Kitts P. Eukaryotic Genome Annotation Pipeline. The NCBI Handbook [Internet] 2nd edition. National Center for Biotechnology Information (US); 2013. Available: https://www.ncbi.nlm.nih.gov/books/NBK169439/ [Google Scholar]
- 19.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 2003;31: 3784–3788. doi: 10.1093/nar/gkg563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 2021;38: 4647–4654. doi: 10.1093/molbev/msab199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smolka M, Paulin LF, Grochowski CM, Mahmoud M, Behera S, Gandhi M, et al. Comprehensive Structural Variant Detection: From Mosaic to Population-Level. bioRxiv; 2022. p. 2022.04.04.487055. doi: 10.1101/2022.04.04.487055 [DOI]
- 22.Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun 2017;8: 14061. doi: 10.1038/ncomms14061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goel M, Sun H, Jiao W-B, Schneeberger K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol 2019;20: 277. doi: 10.1186/s13059-019-1911-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiao W-B, Schneeberger K Chromosome-level assemblies of multiple Arabidopsis genomes reveal hotspots of rearrangements with altered evolutionary dynamics. Nat Commun 2020;11: 989. doi: 10.1038/s41467-020-14779-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Challis R, Richards E, Rajan J, Cochrane G, Blaxter M. BlobToolKit - Interactive Quality Assessment of Genome Assemblies. G3 (Bethesda) 2020;10: 1361–1374. doi: 10.1534/g3.119.400908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Jong TV, Chen H, Brashear WA, Kochan KJ, Hillhouse AE, Zhu Y, et al. mRatBN7.2: familiar and unfamiliar features of a new rat genome reference assembly. Physiol Genomics 2022;54: 251–260. doi: 10.1152/physiolgenomics.00017.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Günther E, Walter L. The major histocompatibility complex of the rat (Rattus norvegicus). Immunogenetics 2001;53: 520–542. doi: 10.1007/s002510100361 [DOI] [PubMed] [Google Scholar]
- 28.Goel M, Schneeberger K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 2022;38: 2922–2926. doi: 10.1093/bioinformatics/btac196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puckett EE, Micci-Smith O, Munshi-South J. Genomic analyses identify multiple Asian origins and deeply diverged mitochondrial clades in inbred brown rats (Rattus norvegicus). Evol Appl 2018;11: 718–726. doi: 10.1111/eva.12572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cowley AW, Stoll M, Greene AS, Kaldunski ML, Roman RJ, Tonellato PJ, et al. Genetically defined risk of salt sensitivity in an intercross of Brown Norway and Dahl S rats. Physiol Genomics 2000;2: 107–115. doi: 10.1152/physiolgenomics.2000.2.3.107 [DOI] [PubMed] [Google Scholar]
- 31.Vollger MR, Guitart X, Dishuck PC, Mercuri L, Harvey WT, Gershman A, et al. Segmental duplications and their variation in a complete human genome 2021. May p. 2021.05.26.445678. doi: 10.1101/2021.05.26.445678 [DOI] [PMC free article] [PubMed]
- 32.Rice ES, Koren S, Rhie A, Heaton MP, Kalbfleisch TS, Hardy T, et al. Chromosome-length haplotigs for yak and cattle from trio binning assembly of an F1 hybrid. bioRxiv 2019; 737171. doi: 10.1101/737171 [DOI]
- 33.Koren S, Rhie A, Walenz BP, Dilthey AT, Bickhart DM, Kingan SB, et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat Biotechnol 2018. doi: 10.1038/nbt.4277 [DOI] [PMC free article] [PubMed]
- 34.Tseng E, Underwood JG, Hutzenbiler BDE, Trojahn S, Kingham B, Shevchenko O, et al. Long-read isoform sequencing reveals tissue-specific isoform expression between active and hibernating brown bears (Ursus arctos) 2021. Jul p. 2021.07.13.452179. doi: 10.1101/2021.07.13.452179 [DOI] [PMC free article] [PubMed]
- 35.Behmoaras J, Bhangal G, Smith J, McDonald K, Mutch B, Lai PC, et al. Jund is a determinant of macrophage activation and is associated with glomerulonephritis susceptibility. Nat Genet 2008;40: 553–559. doi: 10.1038/ng.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamashita S, Wakazono K, Nomoto T, Tsujino Y, Kuramoto T, Ushijima T. Expression quantitative trait loci analysis of 13 genes in the rat prostate. Genetics 2005;171: 1231–1238. doi: 10.1534/genetics.104.038174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kriegel AJ, Didier DN, Li P, Lazar J, Greene AS. Mechanisms of cardioprotection resulting from Brown Norway chromosome 16 substitution in the salt-sensitive Dahl rat. Physiol Genomics 2012;44: 819–827. doi: 10.1152/physiolgenomics.00175.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al’Khafaji AM, Smith JT, Garimella KV, Babadi M, Sade-Feldman M, Gatzen M, et al. High-throughput RNA isoform sequencing using programmable cDNA concatenation. bioRxiv; 2021. p. 2021.10.01.462818. doi: 10.1101/2021.10.01.462818 [DOI] [PubMed]
- 39.Eizenga JM, Novak AM, Sibbesen JA, Heumos S, Ghaffaari A, Hickey G, et al. Pangenome Graphs. Annu Rev Genomics Hum Genet 2020;21: 139–162. doi: 10.1146/annurev-genom-120219-080406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doris PA. Genetics of hypertension: an assessment of progress in the spontaneously hypertensive rat. Physiol Genomics 2017;49: 601–617. doi: 10.1152/physiolgenomics.00065.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 HiCanu assembly statistics for the SHRSP genome.
Table S2 RefSeq genes identified by annotation as being incongruently represented in the SHRSP assembly. Each type of alignment imperfection is represented in a single tab in the file. Iso-Seq analysis of these genes is provided in additional tabs.
Table S3 Structural variations between the assemblies identified by Sniffles software and compared using SURVIVOR software (https://github.com/fritzsedlazeck/SURVIVOR). A summary of variant types and affected genome lengths is provided
Files S1–11
Files S1–3 are large files and are available from the Zenodo persistent public repository website at the links provided in the files.
File S1 vcf file generated by Sniffles2/SURVIVOR analysis.
File S2 vcf file generated by SyRI analysis.
File S3 list of SNPs between the SHRSP and mRatBN7.2 genome assemblies
File S4 list of structural variants
File S5 list of syntenous regions
File S6 list of duplicated genome regions
File S7 list of interchromosomal translocations
File S8 list of unaligned regions
File S9 list of duplicated regions
File S10 list of translocated (within chromosome) regions)
File S11 list of inverted genome regions
File S12 list of both inverted and translocated regions