

The structure of Drosophila melanogaster frataxin (Dfh) was derived by X-ray crystallography and studied by NMR spectroscopy. The crystal and solution structures are extremely similar. NMR spectroscopy was also used to identify the iron-binding location on helix 1 and strand 1 of Dfh.
Keywords: Friedreich’s ataxia, frataxin, iron–sulfur clusters, Drosophila melanogaster, ISC pathway
Abstract
Friedreich’s ataxia (FRDA) is a hereditary cardiodegenerative and neurodegenerative disease that affects 1 in 50 000 Americans. FRDA arises from either a cellular inability to produce sufficient quantities or the production of a nonfunctional form of the protein frataxin, a key molecule associated with mitochondrial iron–sulfur cluster biosynthesis. Within the mitochondrial iron–sulfur cluster (ISC) assembly pathway, frataxin serves as an allosteric regulator for cysteine desulfurase, the enzyme that provides sulfur for [2Fe–2S] cluster assembly. Frataxin is a known iron-binding protein and is also linked to the delivery of ferrous ions to the scaffold protein, the ISC molecule responsible for the direct assembly of [2Fe–2S] clusters. The goal of this report is to provide structural details of the Drosophila melanogaster frataxin ortholog (Dfh), using both X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, in order to provide the foundational insight needed to understand the structure–function correlation of the protein. Additionally, NMR iron(II) titrations were used to provide metal contacts on the protein to better understand how it binds iron and aids its delivery to the ISC scaffold protein. Here, the structural and functional similarities of Dfh to its orthologs are also outlined. Structural data show that bacterial, yeast, human and Drosophila frataxins are structurally similar, apart from a structured C-terminus in Dfh that is likely to aid in protein stability. The iron-binding location on helix 1 and strand 1 of Dfh is also conserved across orthologs.
1. Introduction
With an incidence of 1 in 50 000 (Leone et al., 1990 ▸; López-Arlandis et al., 1995 ▸) and a carrier prevalence in some locations of up to 1 in 100 (Lodi et al., 2006 ▸), Friedreich’s ataxia (FRDA) is by far the most prevalent disease linked to defective iron–sulfur (Fe–S) cluster biosynthesis. FRDA is an autosomal recessive genetic disease caused by a GAA-trinucleotide repeat expansion in the first intron of the gene for frataxin, a protein essential for Fe–S cluster assembly (Campuzano et al., 1996 ▸). The repeat expansion leads to the underexpression of frataxin (Ohshima et al., 1998 ▸), while a unique subset of FRDA patients (5%) have a disease origin linked to frataxin point mutations (Campuzano et al., 1996 ▸). Frataxin deficiency leads to the pathophysiology observed in FRDA patients, which includes mitochondrial iron overload, disruption of Fe–S cluster biosynthesis and the increased production of reactive oxygen species (Babcock et al., 1997 ▸; Foury & Cazzalini, 1997 ▸; Stehling et al., 2004 ▸; Rötig et al., 1997 ▸). When combined, these phenotypes typically lead to cell death in metabolically active tissues that include cardiomyocytes and neurons of the dorsal root ganglia (Campuzano et al., 1996 ▸). FRDA presents early in adolescence through progressive ataxia, difficulty coordinating movement, sensory loss, weakness and dysarthria (Delatycki & Corben, 2012 ▸). FRDA patients are usually wheelchair-bound in their teens, have a significantly reduced quality of life and a shortened life expectancy. The median age of survival for FRDA patients is 35 years, with cardiac dysfunction usually being the cause of death (Tsou et al., 2011 ▸).
Frataxin activity is directly linked to the mitochondrial iron–sulfur cluster (ISC) assembly pathway. Frataxin serves as a modulator for the ISC pathway protein cysteine desulfurase, the enzyme which provides sulfur to the [2Fe–2S] cluster-assembly scaffold protein, an additional component of the pathway (Patra & Barondeau, 2019 ▸; Adinolfi et al., 2009 ▸). In humans, the cysteine desulfurase (NFS1) works with frataxin (FXN) and the accessory proteins ISD11 and ACP to form a complex that generates a persulfide as the sulfur source. The persulfide is then delivered to the scaffold (ISCU2) to complete [2Fe–2S] cluster assembly. The entire protein complex is simply referred to as NIAUF (Boniecki et al., 2017 ▸). Based on the ability of frataxin to interact with iron, additional roles in the cluster-assembly pathway have also been attributed to the protein, including serving in an iron-storage capacity (Cavadini et al., 2002 ▸) or as the iron chaperone (Rodrigues et al., 2015 ▸); however, these functions have lost favor due in part to the discovery of a frataxin-suppressing scaffold-protein mutant that maintains pathway functionality (Yoon et al., 2014 ▸). Recently, the Markley laboratory confirmed that the iron(II) utilized by the human NIAUF complex during [2Fe–2S] cluster assembly is provided to the scaffold by frataxin when in the combined presence of ferrodoxin (the reducing agent in the ISC pathway) or in the presence of a chemical reductant (Cai et al., 2018 ▸). A direct role for the metal-binding ability of frataxin as it relates to [2Fe–2S] cluster assembly is therefore worth further exploration.
Previous reports have highlighted the ability of several frataxin orthologs to bind iron(II) within the micromolar affinity range. In the Drosophila (fly) model system, using isothermal titration calorimetry, we reported that homogeneous Dfh binds a single iron(II) atom in an enthalpically favorable manner with a K d of 6.0 ± 0.2 µM (Kondapalli et al., 2008 ▸). Under competition binding conditions performed with the iron(II) chelator Rhod-5N or Mag-Fura-2, the Dfh iron(II) binding shifts to between 7.0 ± 4.0 and 16 ± 9 µM (Koebke et al., 2020 ▸). Regardless, these affinities are within the range of free iron concentrations found within the mitochondria (∼150 µM), suggesting that Dfh is likely to be iron(II)-loaded when in the cellular mitochondrial matrix milieu (Garber Morales et al., 2010 ▸). Structural studies of iron bound to Dfh confirm that the protein coordinates iron(II) as a high-spin, six-coordinate metal utilizing only oxygen- and nitrogen-based ligands (Kondapalli et al., 2008 ▸). Given the stability of Dfh compared with its orthologs and its ability to bind iron and participate in Fe–S cluster assembly, we chose to characterize both the crystal structure and solution structure of fly frataxin to better understand how this protein functions. Our solution structure resonance assignments were used to map the iron-binding residues on Dfh through NMR titration of ferrous ions into 15N-labeled protein (Rawat et al., 2019 ▸). Although similar studies have been performed on frataxin orthologs, this report sets the foundation for characterizing the fly ISC assembly proteins in vitro and in cellulo.
2. Methods
2.1. Purification and isolation of Dfh
DNA for Dfh, acquired from the FlyBase fly genebank (FlyBase ID FBgn0030092), was transfected into a pET-101/D-TOPO vector with ampicillin resistance and placed in Escherichia coli BL21 (DE3) cells for expression (Kondapalli et al., 2008 ▸). The unlabeled protein used for crystallographic studies was overexpressed in cells grown in LB medium to an optical density (OD600) of 0.6. At this optical density, the cells were induced with 0.8 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and grown for an additional 4 h at 37°C while also being aerated by shaking at 250 rev min−1. The 133-amino-acid protein corresponding to residues 59–190 within the fly gene was overexpressed with 100 µg l−1 ampicillin added to the medium during cell growth. Cells were harvested by centrifugation at 15 970g at 4°C for 20 min. The cell pellets were stored at −20°C until lysis for isolation.
The 15N-labeled Dfh used for NMR secondary-structure characterization and the iron titrations was grown in M9 minimal medium with 15NH4Cl as the only nitrogen source, as reported previously (Rawat et al., 2019 ▸). In brief, cells were grown for 4 h in 200 ml unlabeled LB medium and the cell pellet was then used to inoculate 1 l flasks of labeled M9 medium. Each flask also contained 40% glucose solution, 5 mM 15NH4Cl, 1 mM MgSO4 and 1 mM CaCl2. Cell growth was maintained at 25°C by shaking the flask at 250 rev min−1 for 12 h. After reaching an OD600 of 0.6 the cells were induced with 1 mM IPTG and they were harvested after 6 h. Pellets were centrifuged at 4000 rev min−1 and stored at −20°C.
All steps during the protein purification were carried out at 4°C in a cold room. A lysis cocktail consisting of lysozyme (100 µg µl−1), cOmplete EDTA-free protease inhibitor (one tablet per 50 ml, Roche), DNAse (10 µg µl−1) and MgCl2 (5 mM) was transferred to the thawing cells, which were allowed to incubate for 25 min. The cells were lysed using an Emulsiflex cell-lysing apparatus and the lysate was centrifuged at 53 343g for 1 h at 4°C. The supernatant was passed through a 0.20 µm filter before being subjected to two ammonium sulfate precipitation steps: first salting at 40% ammonium sulfate to remove low-molecular-weight impurities and then salting at 65% ammonium sulfate to produce a Dfh pellet by centrifugation. Protein was recovered by solubilization in lysis buffer. This solubilized pellet was dialyzed twice against a 1:200 volume of dilution buffer A (25 mM Tris–HCl pH 8.0, 10 mM EDTA, 5 mM β-mercaptoethanol) at 4°C over a 6 h period. Following dialysis, the protein was filtered using a 0.2 µm syringe filter and loaded onto a Q-Sepharose column (Pharmacia) for anion-exchange chromatography. Before loading, the column was equilibrated with six column volumes of buffer A. Once loaded, the column was washed with one column volume of buffer A. The protein was eluted using a salt gradient produced by mixing in an increasing amount of buffer B (buffer A + 1 M NaCl). Dfh eluted at a sodium chloride concentration of 500 mM. Fractions containing Dfh were pooled and subjected to dialysis in buffer A to remove the NaCl. The dialyzed protein was then exposed to 1 M ammonium sulfate, filtered with a 0.2 µm syringe filter and loaded onto a Phenyl-Sepharose column (Pharmacia) using buffer C (buffer A + 1 M ammonium sulfate ). The column was run as a reverse salt gradient by eluting with buffer A. Dfh eluted at 800 mM ammonium sulfate. Fractions containing Dfh were pooled and concentrated to 1 ml in volume.
The protein was incubated on ice for 20 min with 5 mM EDTA to remove any bound metal, followed by a final size-exclusion chromatography purification using a Sephacryl 75 column (GE) equilibrated in buffer D (20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM β-mercaptoethanol). Dfh eluted at a volume corresponding to a protein monomer. Isolated Dfh was concentrated to 1 ml and dialyzed in two volumes of 2 l anaerobic buffer D over a 6 h period. Individual samples were aliquoted anaerobically in 100 µl samples tubes within a Coy wet anaerobic chamber, flash-cooled in liquid nitrogen and stored at −80°C. The yield was 23 mg per litre of growth medium. Inductively coupled plasma mass spectrometry was used to verify that no trace amounts of divalent metal were bound to the isolated protein.
2.2. X-ray crystallography
Crystal trays of purified 2 mM apo Dfh in buffer D were utilized during the protein crystallization process. Crystals of native Dfh were obtained using sitting-drop vapor diffusion by mixing 7.2 mg ml−1 protein with 0.1 M citric acid pH 5.0, 1.6 M ammonium sulfate in a 1:1 ratio and grown at 20°C. Crystals were harvested, soaked in mother liquor with 25% glycerol and flash-cooled in liquid nitrogen. Data were collected on the 21-ID-G beamline (LS-CAT Sector 21) at the Advanced Photon Source (Argonne National Laboratory, Argonne, Illinois, USA). A 1.405 Å resolution data set was processed and scaled using autoPROC (Vonrhein et al., 2011 ▸). The Dfh crystal structure was solved by molecular replacement with Phenix AutoMR (McCoy et al., 2007 ▸) using the atomic coordinates of PDB entry 1ekg (Dhe-Paganon et al., 2000 ▸) and was further built and refined manually using Coot (Emsley et al., 2010 ▸) and phenix.refine with TLS refinement (Adams et al., 2010 ▸), respectively. The final model was validated in MolProbity (Davis et al., 2007 ▸).
2.3. NMR backbone assignments of apo Dfh
Previously, 15N- and 15N,13C-labeled Dfh in NMR buffer (20 mM HEPES, 150 mM NaCl pH 7.5, 90% H2O/7% D2O) were used to complete backbone resonance assignments (Rawat et al., 2019 ▸). NMR data were collected on both a Varian 720 MHz (a previous National High Magnetic Field Laboratory instrument) and our in-house 600 MHz Varian Inova spectrometer, both equipped with triple-resonance probes. The chemical shifts obtained from data analysis of the full complement of triple-resonance experiment sets have been deposited in the BioMagRes Data Bank under accession code BMRB 17135. Dfh chemical shifts were utilized with the Chemical Shift Index analysis program to predict the secondary structure for each amino acid in the protein solution (Wishart & Sykes, 1994 ▸) and these were compared with the secondary structures of the residues in the crystal structure.
2.4. Identifying iron-binding residues in Dfh by NMR
NMR amide chemical shift perturbation experiments were used to identify amino acids impacted by the presence of ferrous ions as an initial step towards identifying the iron-binding site in Dfh. Anaerobic 15N-labeled apo Dfh, at ∼0.5 mM concentration, was prepared in NMR buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 2 mM DTT, 93% H2O/7% D2O). The protein was anaerobically transferred into an NMR tube within a Coy anaerobic wet chamber and capped using an airtight septum. A ferrous ammonium sulfate solution at 1.5 mM concentration was also prepared anaerobically in NMR buffer within the anaerobic chamber. During data collection for both the apo and holo Dfh samples, separate 15N heteronuclear single quantum coherence (HSQC) spectra were collected from three independent sample sets to ensure spectral reproducibility. Iron was added to the protein while in the anaerobic chamber at metal:protein ratios of 0.5:1, 1:1 and 2:1; following the addition of the metal, the samples were recapped with rubber septa. NMR spectra for each sample at each iron concentration were collected on a Varian INOVA 600 MHz spectrometer, replicating the collection conditions used during the backbone assignment experiments. Once inserted into the magnet, samples were allowed to equilibrate to 298 K prior to data collection. Full 1H/15N-HSQC spectra were collected at each titration point using a 1H sweep width of 7804 Hz (2048 points and 64 transients) and a 15N sweep width of 2500 Hz (512 increments). Spectra were transformed, as reported previously, using NMRPipe and peak center positions were determined using SPARKY (Delaglio et al., 1995 ▸; Goddard & Kneller, 2001 ▸). Complementary apoprotein spectra were collected on a 720 MHz Varian Inova Spectrometer (National High Magnetic Field Laboratory, Tallahassee, Florida, USA) and on a Bruker Advance III 900 MHz spectrometer (National Magnetic Resonance Facility at Madison, University of Madison, Wisconsin, USA) to measure field effects. Amide chemical shifts were measured using XEASY and normalized chemical shift changes (δ) resulting from the addition of iron(II) to 15N Dfh were determined using the equation δ = 25[(δHN)2 + (δN/5)2]0.5 (Bartels et al., 1995 ▸).
3. Results
3.1. Comparison of frataxin orthologs
Comparison of amino-acid sequences between bacterial, yeast, human and D. melanogaster frataxins (CyaY, Yfh1, FXN and Dfh, respectively) suggests that these eukaryotic orthologs are highly similar. Sequence-identity correlations measured by Clustal Omega indicate that Dfh and FXN are 54% identical, while Dfh and Yfh1 are 38% identical, with the sequences represented in Fig. 1 ▸; in addition, a comparison with the prokaryotic ortholog shows that Dfh and CyaY are 22% identical (Sievers et al., 2011 ▸). Interestingly, the high amino-acid conservation between eukaryotes does not translate into similar protein stability. While homogeneous Dfh is extremely stable against aggregation and precipitation, with a melting temperature (T m) of 59°C (Kondapalli et al., 2008 ▸), Yfh1 has a T m of 35.8°C and becomes insoluble unless stored anaerobically at 4°C (Adinolfi et al., 2002 ▸). In the case of FXN, the T m is 69.4°C (Adinolfi et al., 2002 ▸), but this ortholog is prone to N-terminal autodegradation. Under in vitro conditions, Dfh can substitute for Yfh1 deletion within the yeast NIAUFX complex to carry out Fe–S cluster assembly (Kondapalli et al., 2008 ▸). Interestingly, CyaY has a reduced sequence-identity overlap with Dfh (22% identical), with a T m of 50°C (Adinolfi et al., 2002 ▸). In bacteria, CyaY inhibits cysteine desulfurase activity during [2Fe–2S] cluster assembly; however, substitution of FXN in bacteria has the reverse effect (Bridwell-Rabb et al., 2012 ▸).
Figure 1.

Clustal Omega sequence alignment between frataxin orthologs from D. melanogaster (Dfh), H. sapiens (FXN), S. cerevisiae (Yfh1) and E. coli (CyaY). Fully conserved residues are indicated by an asterisk (*) under the residue. Iron-binding residues are highlighted in gray. The C-terminal regions are indicated with a black box.
3.2. X-ray crystal structure of Dfh
Crystallographic results indicate that the Dfh structure is well folded at 1.4 Å resolution (Table 1 ▸). The Dfh fold is a α–β sandwich structural motif comprised of two flanking α-helices that form one plane of the protein that borders a succession of five β-strands which form the second plane of the molecule, with a sixth β-strand intersecting the two planes and a structured 310-helical feature on the C-terminal tail (Fig. 2 ▸). Electrostatic potential plots show that acidic residues on helix 1 and strands 1 and 2 are surface-exposed, projecting away from the protein core, and these generate a charged patch on both planes of the molecule. A comparison of the Dfh structure with frataxin orthologs (Fig. 3 ▸) shows a high degree of structural reproducibility despite the number of β-strands of the molecules being slightly variable. Yeast frataxin and the bacterial homolog have a total of six β-strands, while a seventh is observed in the structure of the human ortholog which projects from the N-terminus of helix 2 (Dhe-Paganon et al., 2000 ▸). R.m.s.d. values between the Dfh structure and the human and yeast frataxin orthologs in regions of secondary structure are 0.49 and 1.17 Å, as measured using the ‘superposition’ tool in the Protein Data Bank (PDB).
Table 1. Crystallographic data-collection and refinement statistics for Dfh (PDB entry 7n9i).
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength (Å) | 0.979 |
| Space group | C2 |
| a, b, c (Å) | 78.90, 38.51, 46.30 |
| α, β, γ (°) | 90, 123.45, 90 |
| Resolution (Å) | 37–1.405 (1.410–1.405) |
| Observed reflections | 74124 |
| Unique reflections | 21840 |
| R merge † (%) | 3.7 (32.8) |
| Multiplicity | 3.4 (2.1) |
| Completeness (%) | 95.9 (75.6) |
| Refinement statistics | |
| Resolution range (Å) | 37–1.405 |
| R factor‡ (%) | 15.24 |
| R free § (%) | 17.39 |
| No. of protein atoms | 1986 |
| No. of nonprotein atoms | 10 |
| No. of water molecules | 140 |
| MolProbity score | 3.92 |
| Ramachandran plot¶ (%) | 97.0/3.0/0.0 |
| 〈I/σ(I)〉 | 1.65 |
| Average B factor (Å2) | |
| Protein atoms | 20.9 |
| Nonprotein atoms | 46.2 |
| Waters | 31.4 |
| R.m.s.d. | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.191 |
R
merge =
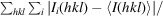
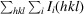 , where I(hkl) is the intensity of reflection hkl and 〈I(hkl)〉 is the average intensity over all equivalent reflections.
, where I(hkl) is the intensity of reflection hkl and 〈I(hkl)〉 is the average intensity over all equivalent reflections.
R factor =
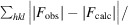
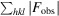 , where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively.
, where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively.
R free is the same as the R factor but for a 5% subset of all reflections that were not used in the refinement.
Core/allowed/disallowed.
Figure 2.

The crystal structure of the Drosophila frataxin homolog Dfh. (a) Ribbon structure and (b) surface electrostatic potential plots for Dfh at three different orientations around the vertical axis. (a) Labels for the different secondary-structural elements are marked on the corresponding helix or strand (PDB entry 7n9i). (b) To the left is a surface electrostatic potential plot for Dfh in an orientation that corresponds to the ribbon structure. Electrostatic surface plots were calculated using PDB2PQR and are labeled with the APBS plugin for PyMOL using a solvent radius of 1.4 Å and a contour value of 10 kT e−1 for both charged isoforms. Secondary-structural landmarks on the rotated structures are labeled for clarity.
Figure 3.

Comparison of different orthologs of the frataxin protein. From left to right, E. coli (PDB entry 1soy), S. cerevisiae (PDB entry 2ga5), D. melanogaster (PDB entry 7n9i) and H. sapiens (PDB entry 1ekg).
3.3. Comparison of crystal and solution-state structures of Dfh
Chemical Shift Index (CSI) analysis was used to generate predicted secondary-structural characteristics for the Dfh solution structure, which we compare with the crystal structure of Dfh in Fig. 4 ▸. CSI analysis compared our published NMR chemical shift values for each residue with the values listed for structures in the BioMagRes Data Bank. This methodology predicts the probability of an amino acid existing in an unstructured or structured (helix or strand) environment. Comparing our CSI-based predicted solution structure with the crystal structure shows a high degree of similarity between the predicted solution and the crystal structure. Slight differences exist between the crystal and the predicted solution structure in the following regions: the N-terminal α-helix predicted in the solution structure extends slightly in both the N-terminal and C-terminal residues compared with the crystal structure, the length of the strand and the origin/terminal residues in each of the β-strands vary slightly between the two structurally characterized species and, finally, the 310-helix observed in the crystal structure is not apparent in the chemical shift prediction of the solution structure. These differences may reflect the dynamic nature of the protein in solution or possibly structural aspects influenced by crystal packing in the crystal structure. However, the general orientation of secondary structures is conserved between ortholog structures, suggesting that the overall tertiary structure is also maintained between the solution and crystal structures.
Figure 4.

The predicted NMR and solved crystal structure-secondary element specifics of Dfh. The predicted solution equivalents (orange) were generated using the CSI algorithm and the published chemical shifts for Dfh from BMRB 17135. The crystal structure equivalents (cyan) were generated using AutoMR, Coot and Phenix.
3.4. Identification of iron-binding residues on Dfh
NMR spectroscopy aided in the identification of potential residues involved in iron binding in Dfh and their locations on the structured protein. A series of 1H/15N-HSQC spectra were collected by the anaerobic titration of buffered (NH4)2Fe(SO4)2·6H2O into an apo Dfh solution. Both the Dfh and the iron(II) solutions were prepared under strict anaerobic conditions to stabilize the reduced oxidation state of iron. There are minimal differences between apo Dfh HSQC spectra collected at 600 MHz compared with the published spectrum at 900 MHz (Rawat et al., 2019 ▸); chemical shift perturbations observed due to the presence of iron were however observed more dramatically at 600 MHz. Backbone assignments for apo Dfh at 600 MHz were used as the control for signal perturbation in the iron-titration experiments outlined in Supplementary Fig. S1. Based on an overlay of backbone amide chemical shifts in the apo and holo forms (at 1:1 Fe:Dfh), spectral differences indicated that eight Dfh residues underwent substantial chemical shift changes (δ) in the range 1–4 (Fig. 5 ▸). The average δ for residues marked as unshifted was 0.3. Residues that dramatically shifted include Ala26, Leu27, Glu36, Asn37, Asp45, Val55, Asn56 and Thr70. In addition, there were five amide backbone signals that were line-broadened beyond detection due to iron(II) addition. The locations of perturbed and broadened residues in the helix 1 and strands 1/2 region, as displayed in Fig. 6 ▸, are similar in position to the iron-binding residues observed in the bacterial, yeast and human ortholog iron titrations (see shaded residues in Fig. 1 ▸). In each ortholog, the iron-binding regions are composed predominantly of acidic residues identified on the same helical turn, most of which are conserved. Finally, an electron-density map of the iron-binding regions of the Dfh protein is presented in Fig. 6 ▸.
Figure 5.

Histogram of the 2D 1H/15N-HSQC spectra of Dfh. Normalized amide chemical shift changes in the presence of 1.5 mM ferrous ammonium sulfate. Residues with shift changes greater than 1 are identified. Residues marked with an asterisk (*) on the x axis are identified as prolines. The horizontal black marker at 1 on the y axis designates the chemical shift cutoff, which is 2× the data resolution. Secondary-structure information for Dfh is depicted below the histogram.
Figure 6.

Orientation of iron-impacted residues and electron-density mapping on the crystal structure of Dfh as determined by 2D 1H/15N-HSQC NMR spectroscopy and modeled by PDB entry 7n9i. Left: orientation 1 showing the α-helical plane of the protein. Right: orientation 2 showing the β-sheet plane of the molecule. Residues perturbed upon iron addition with δ > 1.0 (colored green, labeled, ball-and-stick structure) include Ala26, Leu27, Glu36 and Asn37 on helix 1, Asp45 on strand 1, Val55 and Asn56 on strand 2 and Thr70 on strand 3. Residues that disappeared (have lines that are broadened beyond recognition) upon iron addition (colored red) include Cys28, Asp29 and Thr35 located on helix 1, Ala47 on strand 2 and Asp50 on the strand 1/2 loop. Electron-density mapping on helix 1 and strand 1 show the interaction of the iron-binding residues with iron (red spheres).
4. Discussion
The Dfh structure determined by X-ray crystallography is similar to those obtained for frataxin orthologs. This structural similarity is expected, given the high sequence conservation, and it suggests that eukaryotic frataxin orthologs have a common function within cells during mitochondrial Fe–S cluster bioassembly. It is noteworthy to mention that although the eukaryotic frataxins are very similar in structure, key differences arise in their biophysical characteristics regarding protein stability and the potential for aggregation and autodegradation.
Regarding acidic (Asp and Glu) residues in the orthologs, Dfh, FXN, Yfh1 and CyaY contain 12.8%, 16.7%, 18.7% and 20.4% in total, respectively, with approximately equivalent percentages of basic residues (Lys and Arg) at 6.8%, 10%, 6.5% and 7.4% for Dfh, FXN, Yfh1 and CyaY, respectively. As shown by the Pastore laboratory (Adinolfi et al., 2004 ▸), the length of the frataxin C-terminus directly impacts protein thermal stability, while the N-terminus does not contribute. Yfh1, with the lowest melting temperature and a propensity for precipitation, has a truncated four-residue C-terminus, while CyaY and FXN have longer C-termini (Adinolfi et al., 2004 ▸) that fold back on the protein between helices 1 and 2, all of which lack structure. The C-termini of Dfh and FXN are the largest of the orthologs and both show some degree of structural similarity in the region where the 310-helix is observed in Dfh. These additional C-terminal structured residues may allow Dfh and FXN to form direct interhelical contacts between both helices 1 and 2 that lead to the higher thermal stability for these two orthologs in particular. This is specifically true in Dfh for residues Phe126 and Arg128 on the C-terminus, the latter which interacts with Tyr30 on helix 1 and the former of which interacts with His108 and Glu109 on helix 2. Aromatic stacking, additional hydrophobic residues and a lengthened, more structured C-terminus in the Dfh protein may attribute to the increased stability of the fly ortholog.
Our comparison of the crystal structure and predicted solution structure of Dfh provides additional insight into protein regions that might also be functionally significant. Although a full NMR structural characterization is needed for a direct comparison between crystal structure and solution molecules, the CSI prediction results do allow a quick comparison. The extension of structured regions in the N-terminal helix in the predicted solution structure may arise from the sequence extension of residues in the N-terminus of the peptide and its influence on structural stability throughout this secondary-structural element. In the opposite observation, unstructured residues in the N-terminal region of strand 1, containing Asp45, may also suggest enhanced flexibility for the protein in solution to better accommodate metal binding in the strand 1/2 region. Additionally, structural stability differences in the β-sheet plane between the crystal structure and the predicted solution structure may indicate an additional level of molecular flexibility present in this region that may assist Dfh when sampling intermolecular binding surfaces during multiprotein association. These dynamic regions could also provide flexible surfaces that help accommodate interactions with substrates during Fe–S cluster assembly, either directly or indirectly.
The exposed acidic residues on α-helix 1 and β-strands 1 and 2 of Dfh provide a negatively charged region that is amenable to connect with partner molecules with a positive charge. A conserved acidic residue patch in this region is common in frataxin orthologs (see Fig. 1 ▸), so this is likely to also be of functional significance (Cook et al., 2006 ▸). In the structure of the entire human ISC multiprotein complex identified as NIAUF, FXN interacts with the cysteine desulfurase utilizing residues on the helical plane (Fox et al., 2019 ▸). The importance of these acidic residues in relation to their known iron-binding ability is not likely to be functionally significant, since FXN interacts with NFS1 in an iron-independent manner utilizing this protein region. In contrast, acidic residues on the FXN β-sheet surface are known to form an intermolecular interface with the yeast scaffold protein in an iron-dependent manner, while acidic residues in strands 1 and 2 are also implicated in iron binding. Given the orientation of FXN in the NIAUF complex, where the β-sheet surface is positioned to interact with ISCU2, and acidic residues in strands 1 and 2 in this region bind iron, it is worth further exploring the role of the Dfh β-strands with iron(II)-binding residues in promoting scaffold iron loading. The dynamic mobility of Dfh β-sheet residues seen in the solution structure analysis may help to direct the targeted association of FXN–ISCU in such a way as to assist metal delivery and [2Fe–2S] cluster assembly.
Frataxin orthologs consistently bind ferrous ions with intermediate to weaker binding affinities (1 < K d < 50 µM; Cook et al., 2006 ▸; Lewis et al., 2019 ▸; Bou-Abdallah et al., 2004 ▸; Kondapalli et al., 2008 ▸). X-ray absorption spectroscopic analysis of iron(II) bound to frataxin orthologs indicate that iron is coordinated to the protein as high-spin iron(II) and is held in a six-coordinate ligand environment constructed only by low-Z (O and N) atoms (Bencze et al., 2007 ▸; Cook et al., 2006 ▸; Lewis et al., 2019 ▸; Kondapalli et al., 2008 ▸). Acidic residues that have deprotonated carboxylate side chains would be suitable ligands to bind a positively charged metal, although oxygen ligands tend to favor metals with higher valence states. Metal binding by the acidic residues in β-strands 1 and 2 of frataxin, with support by adjacent polar residues, would be consistent with the structural binding data from all frataxin orthologs. In the electron-density representation of the structure of Dfh, iron-binding residues in the helix 1 and β-strand 1 and 2 regions do form a close organization point for interaction with the identified binding acidic residues, indicating that both could serve as metal-binding sites. Functionally, the β-sheet position could help to accommodate the direct transfer of iron(II) by FXN to ISCU2, as noted by the Markley group (Cai et al., 2018 ▸).
Supplementary Material
PDB reference: Drosophila melanogaster frataxin, 7n9i
Supplementary Figure S1. DOI: 10.1107/S2059798322011639/ni5025sup1.pdf
Acknowledgments
Crystallographic data were collected on the LS-CAT beamline at the Advanced Photon Source as part of an effort funded by the Office for the Vice President for Research at Wayne State University. The authors report no declarations of conflict of interest.
Funding Statement
AVR was supported by the American Heart Association and the Friedreich’s Ataxia Research Alliance collaborative grant (12PRE11720005). This work was supported by National Institutes for Health grant DK068139 to TLS. TVH is supported by a Wayne State University Dean’s Diversity grant.
References
- Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Zwart, P. H. (2010). Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed]
- Adinolfi, S., Iannuzzi, C., Prischi, F., Pastore, C., Iametti, S., Martin, S. R., Bonomi, F. & Pastore, A. (2009). Nat. Struct. Mol. Biol. 16, 390–396. [DOI] [PubMed]
- Adinolfi, S., Nair, M., Politou, A., Bayer, E., Martin, S., Temussi, P. & Pastore, A. (2004). Biochemistry, 43, 6511–6518. [DOI] [PubMed]
- Adinolfi, S., Trifuoggi, M., Politou, A. S., Martin, S. & Pastore, A. (2002). Hum. Mol. Genet. 11, 1865–1877. [DOI] [PubMed]
- Babcock, M., de Silva, D., Oaks, R., Davis-Kaplan, S., Jiralerspong, S., Montermini, L., Pandolfo, M. & Kaplan, J. (1997). Science, 276, 1709–1712. [DOI] [PubMed]
- Bartels, C., Xia, T., Billeter, M., Güntert, P. & Wüthrich, K. (1995). J. Biomol. NMR, 6, 1–10. [DOI] [PubMed]
- Bencze, K. Z., Yoon, T., Millán-Pacheco, C., Bradley, P. B., Pastor, N., Cowan, J. A. & Stemmler, T. L. (2007). Chem. Commun., pp. 1798–1800. [DOI] [PMC free article] [PubMed]
- Boniecki, M. T., Freibert, S. A., Mühlenhoff, U., Lill, R. & Cygler, M. (2017). Nat. Commun. 8, 1287. [DOI] [PMC free article] [PubMed]
- Bou-Abdallah, F., Adinolfi, S., Pastore, A., Laue, T. M. & Chasteen, N. D. (2004). J. Mol. Biol. 341, 605–615. [DOI] [PubMed]
- Bridwell-Rabb, J., Iannuzzi, C., Pastore, A. & Barondeau, D. P. (2012). Biochemistry, 51, 2506–2514. [DOI] [PMC free article] [PubMed]
- Cai, K., Frederick, R. O., Tonelli, M. & Markley, J. L. (2018). J. Inorg. Biochem. 183, 107–116. [DOI] [PMC free article] [PubMed]
- Campuzano, V., Montermini, L., Moltò, M. D., Pianese, L., Cossée, M., Cavalcanti, F., Monros, E., Rodius, F., Duclos, F., Monticelli, A., Zara, F., Cañizares, J., Koutnikova, H., Bidichandani, S. I., Gellera, C., Brice, A., Trouillas, P., De Michele, G., Filla, A., De Frutos, R., Palau, F., Patel, P. I., Di Donato, S., Mandel, J. L., Cocozza, S., Koenig, M. & Pandolfo, M. (1996). Science, 271, 1423–1427. [DOI] [PubMed]
- Cavadini, P., O’Neill, H. A., Benada, O. & Isaya, G. (2002). Hum. Mol. Genet. 11, 217–227. [DOI] [PubMed]
- Cook, J. D., Bencze, K. Z., Jankovic, A. D., Crater, A. K., Busch, C. N., Bradley, P. B., Stemmler, A. J., Spaller, M. R. & Stemmler, T. L. (2006). Biochemistry, 45, 7767–7777. [DOI] [PMC free article] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., Snoeyink, J., Richardson, J. S. & Richardson, J. S. (2007). Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed]
- Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995). J. Biomol. NMR, 6, 277–293. [DOI] [PubMed]
- Delatycki, M. B. & Corben, L. A. (2012). J. Child Neurol. 27, 1133–1137. [DOI] [PMC free article] [PubMed]
- Dhe-Paganon, S., Shigeta, R., Chi, Y. I., Ristow, M. & Shoelson, S. E. (2000). J. Biol. Chem. 275, 30753–30756. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Foury, F. & Cazzalini, O. (1997). FEBS Lett. 411, 373–377. [DOI] [PubMed]
- Fox, N. G., Yu, X., Feng, X., Bailey, H. J., Martelli, A., Nabhan, J. F., Strain-Damerell, C., Bulawa, C., Yue, W. W. & Han, S. (2019). Nat. Commun. 10, 2210. [DOI] [PMC free article] [PubMed]
- Garber Morales, J., Holmes-Hampton, G. P., Miao, R., Guo, Y., Münck, E. & Lindahl, P. A. (2010). Biochemistry, 49, 5436–5444. [DOI] [PMC free article] [PubMed]
- Goddard, T. D. & Kneller, D. G. (2001). SPARKY 3. University of California, San Francisco, California, USA.
- Koebke, K. J., Batelu, S., Kandegedara, A., Smith, S. R. & Stemmler, T. L. (2020). J. Inorg. Biochem. 203, 110882. [DOI] [PubMed]
- Kondapalli, K. C., Kok, N. M., Dancis, A. & Stemmler, T. L. (2008). Biochemistry, 47, 6917–6927. [DOI] [PMC free article] [PubMed]
- Leone, M., Brignolio, F., Rosso, M. G., Curtoni, E. S., Moroni, A., Tribolo, A. & Schiffer, D. (1990). Clin. Genet. 38, 161–169. [DOI] [PubMed]
- Lewis, B. E., Mason, Z., Rodrigues, A. V., Nuth, M., Dizin, E., Cowan, J. A. & Stemmler, T. L. (2019). Metallomics, 11, 1820–1835. [DOI] [PMC free article] [PubMed]
- Lodi, R., Tonon, C., Calabrese, V. & Schapira, A. H. (2006). Antioxid. Redox Signal. 8, 438–443. [DOI] [PubMed]
- López-Arlandis, J. M., Vílchez, J. J., Palau, F. & Sevilla, T. (1995). Neuroepidemiology, 14, 14–19. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Ohshima, K., Montermini, L., Wells, R. D. & Pandolfo, M. (1998). J. Biol. Chem. 273, 14588–14595. [DOI] [PubMed]
- Patra, S. & Barondeau, D. P. (2019). Proc. Natl Acad. Sci. USA, 116, 19421–19430. [DOI] [PMC free article] [PubMed]
- Rawat, S., Kondapalli, K. C., Rodrigues, A. V. & Stemmler, T. L. (2019). Biomol. NMR Assign. 13, 377–381. [DOI] [PMC free article] [PubMed]
- Rodrigues, A. V., Kandegedara, A., Rotondo, J. A., Dancis, A. & Stemmler, T. L. (2015). Biometals, 28, 567–576. [DOI] [PMC free article] [PubMed]
- Rötig, A., de Lonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., Munnich, A. & Rustin, P. (1997). Nat. Genet. 17, 215–217. [DOI] [PubMed]
- Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J., Thompson, J. D. & Higgins, D. G. (2011). Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed]
- Stehling, O., Elsässer, H. P., Brückel, B., Mühlenhoff, U. & Lill, R. (2004). Hum. Mol. Genet. 13, 3007–3015. [DOI] [PubMed]
- Tsou, A. Y., Paulsen, E. K., Lagedrost, S. J., Perlman, S. L., Mathews, K. D., Wilmot, G. R., Ravina, B., Koeppen, A. H. & Lynch, D. R. (2011). J. Neurol. Sci. 307, 46–49. [DOI] [PubMed]
- Vonrhein, C., Flensburg, C., Keller, P., Sharff, A., Smart, O., Paciorek, W., Womack, T. & Bricogne, G. (2011). Acta Cryst. D67, 293–302. [DOI] [PMC free article] [PubMed]
- Wishart, D. S. & Sykes, B. D. (1994). J. Biomol. NMR, 4, 171–180. [DOI] [PubMed]
- Yoon, H., Knight, S. B., Pandey, A., Pain, J., Zhang, Y., Pain, D. & Dancis, A. (2014). Biochem. J. 459, 71–81. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Drosophila melanogaster frataxin, 7n9i
Supplementary Figure S1. DOI: 10.1107/S2059798322011639/ni5025sup1.pdf