

ABSTRACT
Clostridioides difficile is the most common cause of nosocomial antibiotic-associated diarrhea, and is responsible for a spectrum of diseases characterized by high levels of recurrence, morbidity, and mortality. Treatment is complex, since antibiotics constitute both the main treatment and the major risk factor for infection. Worryingly, resistance to multiple antibiotics is becoming increasingly widespread, leading to the classification of this pathogen as an urgent threat to global health. As a consummate opportunist, C. difficile is well equipped for promoting disease, owing to its arsenal of virulence factors: transmission of this anaerobe is highly efficient due to the formation of robust endospores, and an array of adhesins promote gut colonization. C. difficile produces multiple toxins acting upon gut epithelia, resulting in manifestations typical of diarrheal disease, and severe inflammation in a subset of patients. This review focuses on such virulence factors, as well as the importance of antimicrobial resistance and genome plasticity in enabling pathogenesis and persistence of this important pathogen.
KEYWORDS: c. difficile, virulence factors, antimicrobial resistance, toxins, spores, CDI
Introduction
Clostridioides difficile is a gram-positive obligate anaerobe, capable of causing disease through the fecal-oral transmission of robust endospores. These metabolically dormant spores are able to persist in a range of environments, being resistant to oxygen, heat, and many common disinfectants – contributing to both the organism’s success as a pathogen, and the associated healthcare costs and difficulty of treating infection [1]. C. difficile is responsible for over 120,000 infections per year in the EU alone [2] and is the leading cause of hospital-associated diarrhea. As well as being an important nosocomial pathogen, a recent paradigm-shift has seen increasing reports of community-acquired C. difficile infection (CDI). Although often less severe, community-acquired CDI is responsible for an estimated 20–27% of all cases, resulting in a significant burden [3]. Clinical presentation of CDI covers a large spectrum of diseases, with diarrhea and colitis being the most common. The significant mortality associated with C. difficile typically arises from more severe manifestations, including pseudomembranous colitis, fulminant colitis, and toxic megacolon [4]. Infection recurrence, characterized by the reappearance of symptoms after treatment completion, is also common, largely due to the nature of available CDI treatments. This results in complex treatment plans and worsened prognosis [5,6].
Paradoxically, antibiotics constitute both the main treatment and a main risk factor for C. difficile infection. Administration of broad-spectrum antimicrobials, either prophylactically or to treat another infection, lead to disruption of the gut microbiota, resulting in a dysbiotic state in which C. difficile thrives [7]. As well as being associated with the broad-spectrum antimicrobials cephalosporins, clindamycin, and fluoroquinolones; antibiotics commonly used to treat C. difficile itself can also contribute to CDI, by exacerbating dysbiosis and leaving the patient acutely sensitive to reinfection or relapse [8]. This recurrence is the most common complication of CDI, arising in up to 30% of patients [9]. Collectively, this combination of factors warrants the recent classification of C. difficile as an “urgent threat” [10]. This review provides an overview of the C. difficile lifecycle, virulence factors, and antimicrobial resistance in the context of pathogenicity.
The changing epidemiology of C. difficile
The phylogenetic diversity of C. difficile has allowed for the emergence of several epidemic strains in recent years. In particular, the ribotype 027 lineage was responsible for a 2001 North American epidemic, which spread to the UK, peaking in 2004–2006 [11]. This hypervirulent lineage is associated with increased transmission and mortality, although the underlying reasons for the apparent increase in pathogenicity are far from clear. Ribotype 027 strains display increased expression of toxins, due to a deletion in tcdC (encoding a negative regulator of toxin expression) [12–14], and production of an additional binary toxin [15]. These strains also displayed high-level resistance to fluoroquinolone antibiotics, including the commonly used ciprofloxacin [16]. In the UK, improved hospital management strategies have resulted in an 80% reduction in cases from the height of the 2004–2006 outbreak, with current annual infection rates lying at 13,000 [17; 18]. C. difficile is responsible for an estimated 29,000 and 1,800 deaths per year in the USA and UK, respectively, [19; 20], with a case fatality rate of approximately 15%. However, this increases with each subsequent infection recurrence [5]. Despite strategies to reduce CDI, the costs associated with CDI have increased, with infections costing between $436 million to $3 billion per year in the USA, with total CDI-attributable costs excelling $6.3 billion [21]. In England, CDI costs an additional £5,000-£15,000 per case [21]. This burden is not solely economic – with the average UK patient stay being 37 days, CDI puts huge pressures on healthcare facilities [22]. Despite less emphasis being placed on the burden of CDI in lower-income countries, it is clear that the lack of diagnosis and prevention has led to a severe underestimation of CDI. In many African countries, due to reduced regulation of antibiotics and high HIV prevalence, CDI burden is likely to be high [23]. Similarly, in South Africa, CDI incidence was shown to be 9.2%, a third of which was community-acquired [24].
Life cycle and disease transmission
As an anaerobe, C. difficile must overcome the formidable barrier of atmospheric oxygen to spread to a new host. This is achieved through the formation of metabolically inert but incredibly resilient spores (Figure 1(a)). Mutants defective in sporulation do not transmit efficiently in an animal model that closely mimics both direct patient to patient spread and infection from contaminated surfaces in the environment [28]. In addition to providing resistance to oxygen, the spore form is also resistant to UV, desiccation, heat, many disinfectants, and antibiotics, which creates significant additional decontamination challenges in health-care facilities and also dramatically extends the maximum time interval between hosts [29; 30]. This complicates outbreak control and analysis of chains of transmission by traditional methods [31]. The efficiency of sporulation also varies widely between C. difficile strains [32] and it has been suggested that this feeds into the differences observed in transmission. Early in the ribotype 027 epidemic, it was thought that increased sporulation efficiency accounted for the enhanced transmission seen in hospitals, at least in part [33], however this has been disputed [32].
Figure 1.
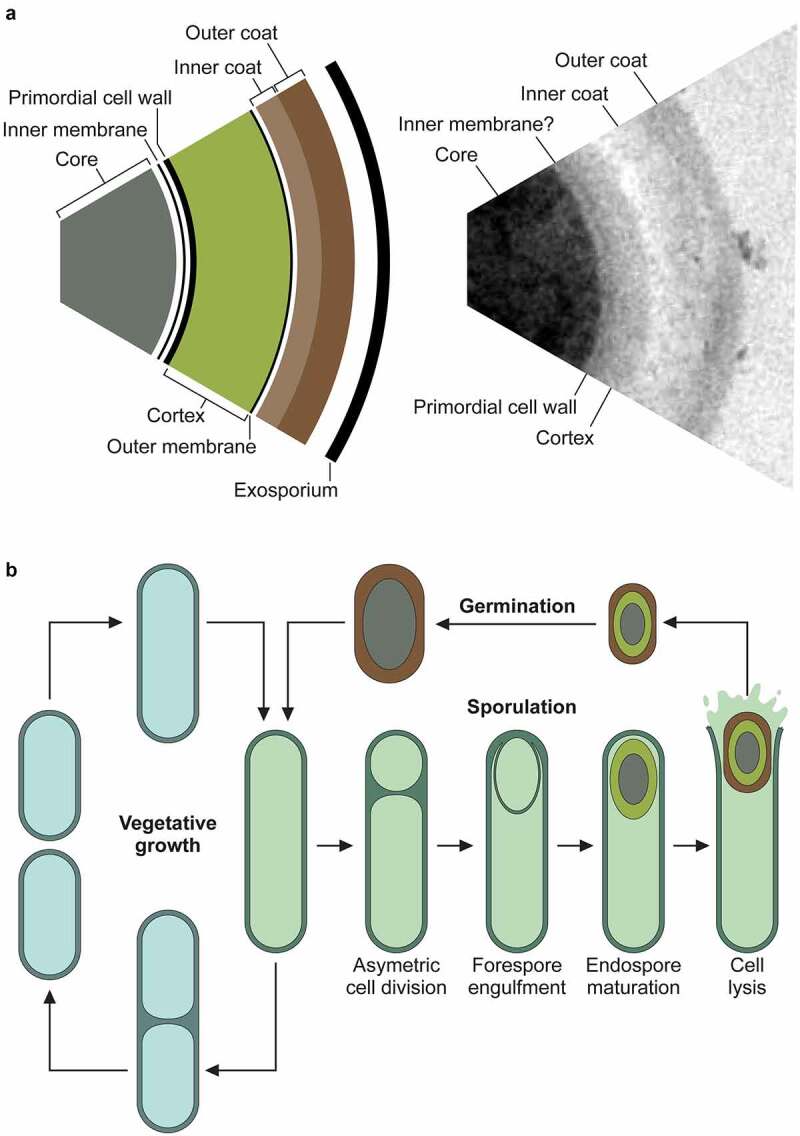
Structure of the spore and life cycle.
a. The robust properties of the spore are due to its multi-layered structure, with each layer contributing to the overall resilience [25]. The dense core is dehydrated due to the presence of up to 25% Ca-DPA, and the DNA is bound to and protected by the small acid-soluble proteins. Surrounding the core is an extremely impermeable inner membrane and the germ (primordial) cell wall. This thin layer of peptidoglycan has the same composition as in vegetative cells – and will become the nascent cell wall during germination. Around the germ cell wall is a much thicker layer of peptidoglycan, the cortex. Within the cortex peptidoglycan approximately 25% of the N-acetylmuramic acid moieties are modified to muramic-δ-lactam, and there are few crosslinks between adjacent N-acetylmuramic acid-N-acetylglucosamine polymers [26]. This results in a much more flexible peptidoglycan structure, with a distinct chemical signature that allows specific degradation during germination, without risk of compromising the germ cell wall. The cortex is surrounded by a second membrane, derived from the mother cell during engulfment, and then finally, the protein coat. The coat is a lamellar structure consisting of a large number of often highly crosslinked proteins. The outermost layers of the coat in C. difficile appear less organized, with an amorphous structure, and vary in thickness. This layer has been described as exosporium but does not appear to have the same loose-attachment and hexameric organization seen in other spore-formers [27]. Some of these structures can be seen in the transmission electron micrograph of a negative-stained, thin-sectioned spore on the right. b. When conditions are favorable, a C. difficile cell will normally be divided by binary fission. However, when environment conditions are less than ideal, most likely due to nutrient limitation, the cell can enter the sporulation pathway instead. Upon initiation of sporulation, the cell first undergoes asymmetric septation, producing the mother cell compartment and smaller forespore. A copy of the genome is transferred into the nascent spore, and the forespore is then engulfed by the mother cell - a phagocytosis-like event that results in an immature spore, bounded by two membranes, free in the cytoplasm of the mother cell. The spore then undergoes a maturation process whereby the DNA is compacted by the small acid-soluble proteins, Ca-DPA is synthesized, the core is dehydrated, and cortex and protein coats are synthesized. The final mature spore is released by lysis of the mother cell.
Sporulation
Given the pivotal role the spore plays in disease transmission, it is not surprising that this has been an area of intense research in recent years. Sporulation begins with an asymmetric septation event that separates a larger mother cell from the smaller forespore compartment (Figure 1(b)). The mother cell then engulfs the forespore, resulting in an immature double-membraned prespore contained within the mother cell cytoplasm. A thick layer of cortex peptidoglycan is then synthesized between the two spore membranes but exterior to the existing primordial cell wall, and proteinaceous coat layers are assembled on the outer surface. The core contains a high concentration of dipicolinic acid coordinated to calcium ions (Ca-DPA) which functions to dehydrate the spore and protects the DNA from heat-induced damage [34–36]. The DNA is further protected from UV damage by small acid-soluble proteins (SASPs) [37]. Upon completion of synthesis of the cortex and coat layers the now mature spore is released by lysis of the mother cell. We will not attempt to exhaustively summarize the molecular basis of sporulation here, as this is already the subject of many excellent reviews in recent years [38,39].
Sporulation in C. difficile has many parallels with the well-studied Bacilli. However, this complex cell differentiation pathway has diverged significantly since the last common ancestor, likely over 2 billion years ago [40; 41], so caution must be exercised in extrapolating protein function based on homology, for even conserved components. The master regulator Spo0A and the subsequent sigma factor cascade is largely conserved, although with some difference in temporal regulation and mechanisms of sigma factor activation [42]. The signaling events upstream of Spo0A are not conserved and, despite the identification of several regulators that feed into C. difficile Spo0A expression, it is not yet clear how its activation is controlled [43–45]. Regulation of sporulation involves integration of multiple environmental and nutritional cues. Sporulation is very sensitive to environmental pH, with production of viable spores reduced in low pH, albeit with strain–strain differences observed [46]. The nutritional status of the cell is sensed via the catabolite control protein CcpA and CodY [45,47]. CcpA regulates approximately 9% of all C. difficile genes, ensuring a broad transcriptional response to glucose availability, and directly represses the expression of both Spo0A and the first forespore-specific sigma factor SigF [47]. CodY has a similarly broad role in gene regulation in response to nutrient status, sensing the availability of branched chain amino acids and GTP [45]. Expression of Spo0A and the first mother cell-specific sigma factor SigE is repressed by CodY and this repression is relieved under nutrient limitation. However, rather than directly regulating the spo0A gene as in Bacillus spp., in C. difficile CodY appears to act indirectly via the SinRI regulatory system. In addition to these well-characterized regulatory systems with parallels in other spore-forming Firmicutes, several Clostridia-specific regulators have also been described. The RNPP family regulator RstA has been shown to have pleiotropic roles in C. difficile gene regulation including activation of sporulation [48], via effects on the sigma factor cascade downstream of Spo0A. The signal sensed by RstA is currently unknown, although this family of proteins are often regulated by quorum sensing systems and can require direct binding to the autoinducing peptide [49]. Interestingly, sporulation is also subject to epigenetic regulation via the type II 6 mA methyltransferase CamA [50; 51]. Inactivation of camA and loss of the associated 6 mA DNA modification reduced sporulation by approximately 50%. Although the exact mechanism of this sporulation defect is unclear, it appears to halt sporulation following asymmetric septation and the expression of a large number of genes with putative roles in sporulation are affected. These pleiotropic effects are not unprecedented. We have previously shown that transposon mediated disruption of 798 individual genes in strain R20291 has a significant impact on sporulation efficiency, many of which with putative roles in gene regulation, including 53 annotated as “regulator” and 21 genes encoding parts of two component systems [50]. It is clear that much of the complex process of sporulation regulation remains to be elucidated.
Germination
Once ingested, spores can readily survive the incredibly harsh low pH conditions of the stomach and, upon transit into the duodenum, begin to germinate back to vegetative cells that can colonize and proliferate in the colon. Germination is initiated in response to germinants, chemical signals that indicate the spore is in an environment that is conducive to vegetative cell survival and growth. In the case of C. difficile, the major germinant is the bile acid taurocholate [52]. Bile acids are surfactants that are produced in the liver from cholesterol, stored in the gallbladder and then secreted into the duodenum in response to food intake [53], where they emulsify dietary fats, aiding in their absorption and in the uptake of fat-soluble micronutrients [54]. Taurocholate is detected by the pseudoprotease CspC, which induces a signal cascade via CsbB to activate the cortex lytic hydrolase SleC that initiates germination [55]. Degradation of the cortex precedes Ca-DPA release [56], and the concomitant rehydration of the core allows metabolism to resume. Primary bile acids (those synthesized by the liver) are subject to chemical modification and degradation by members of the intestinal microbiota, generating the so-called secondary bile acids, with profound impacts on C. difficile germination and colonization (reviewed in [57]. Some secondary bile acids, including cholate, also act as C. difficile germinants while others, such as chenodeoxycholate, appear to act as direct inhibitors. Interestingly, deoxycholate can act as a C. difficile germinant but then inhibits outgrowth [52]. This cascade of bile acid metabolism likely represents one of the fundamental mechanisms of microbiota-bestowed colonization resistance [58]. Once induced, germination proceeds rapidly, with the new vegetative cells undergoing their first round of cell division within approximately 90–180 min [59]. Toxin production by these vegetative cells leads to the commonly seen symptoms of disease (see below), while subsequent rounds of sporulation generate a subpopulation of spores that ensures onward transmission to new hosts [28]. Spores also provide a reservoir of surviving viable bacteria that can lead to recurrent infection upon antibiotic cessation [60]. It was long assumed that this phenomenon was solely due to the intrinsic resistance of spores to the antibiotics that are commonly used to treat C. difficile infection. However, it has recently been reported that spores can enter intestinal epithelial cells, in an active process that involves BclA3 on the surface of the spores and host fibronectin and vitronectin and their cognate integrin receptors α5β1 and αvβ1 [61]. This surprising observation hints at a previously unexpected C. difficile reservoir in which spores are shielded from germinants, allowing later reseeding of the colon should conditions be conducive for colonization.
Virulence factors
Clinical presentation of CDI is influenced by a range of C. difficile virulence factors, including production of various toxins and surface proteins (Table 1). Primarily, pathogenesis is driven by the activity of toxins A and B, encoded within the pathogenicity locus (PaLoc). These toxins are internalized by gut epithelial cells, where they glucosylate small Rho proteins, resulting in cell death and loss of intestinal barrier function [62; 63]. Symptoms are further exacerbated by the host immune response, which involves an acute intestinal inflammatory response and neutrophil infiltration, further damaging the epithelia [64].
Table 1.
Virulence factors of C. difficile.
| Virulence Factor | Function/Evidence | References |
|---|---|---|
| Toxin A (tcdA) | Inactivate Rho GTPases. Disrupts the cytoskeleton resulting in disruption of tight junctions and loss of intestinal barrier function. | (Barth et al., 2001; Egerer et al., 2009; Gerhard et al., n.d.; Jank et al., 2007; Just et al., 1995; Madan and Petri, 2012; Oezguen et al., 2012; Papatheodorou et al., 2010; Qa’Dan et al., 2000) |
| Toxin B (tcdB) | Inactivate Rho GTPases. Disrupts the cytoskeleton resulting in disruption of tight junctions and loss of intestinal barrier function. Huge diversity of subtypes, undergoes accelerated evolution. | (Barth et al., 2001; Egerer et al., 2009; Gerhard et al., n.d.; Jank et al., 2007; Just et al., 1995; Madan and Petri, 2012; Oezguen et al., 2012; Papatheodorou et al., 2010; Qa’Dan et al., 2000; Shen et al., 2020) |
| C. difficile binary toxin (CDT) | ADP-ribosyltransferase which causes depolymerisation of the actin cytoskeleton (leading to loss of barrier function and disruption of tight junctions) and microtubule protrusions (leading to increased C. difficile adherence). | (Aktories et al., 2011; Gerding et al., 2014; Hemmasi et al., 2015; Papatheodorou et al., 2010; Schwan et al., 2009) |
| SlpA | Major S-layer constituent. S-layer null strain avirulent in hamster model, and more susceptible to lysozyme and immune effectors. Mutants making more porous S-layer display increased lysozyme sensitivity. | (Calabi et al., 2002; Kirk et al., 2017; Lanzoni-Mangutchi et al., 2022; Merrigan et al., 2013) |
| Cwp2 | Implicated in adhesion. Dominant antigen in patient sera. | (Bradshaw et al., 2017) |
| Cwp84 | Required for normal S-layer production. Dominant antigen in patient sera. However, mutants fully virulent in hamster models. | (Wright et al., n.d.; Kirby et al., 2009) |
| Cwp66 | Implicated in adhesion and stress tolerance. | (Waligora et al., 2001; Zhou et al., 2022) |
| Cwp19 | Transglycosylase involved in autolysis, resulting in toxin release. | (Wydau-Dematteis et al., 2018) |
| Cwp22 | Peptidoglycan cross-linking enzyme (L,D-transpeptidase). Supports cell wall integrity. Mutation reduced toxin production, increased cell permeability and autolysis, and reduced adherence. | (Peltier et al., 2011; Zhu et al., 2019) |
| CwpV | Large phase-variable CWP. Displays auto-aggregative properties. Putatively involved in colonisation and biofilm in vivo. Confers resistance to some bacteriophage. | (Lawley et al., 2009; Reynolds et al., 2011; Sekulovic et al., 2015) |
| CD2831 | Collagen binding protein involved in adhesion, biofilm formation and immune evasion. | (Arato et al., 2019) |
| CpbA | Involved in adherence through enhancing collagen interaction and extracellular matrix adherence. | (Tulli et al., 2013) |
| Broader virulence traits | ||
| Lysozyme resistance | Resistance to hydrolysis via lysozyme due to σV activation of PgdA and PdaV. S-layer provides barrier protection. Required for successful pathogenesis in hamster models. | (Callewaert and Michiels, 2010; Fagan et al., 2009; Ho et al., 2014; Kaus et al., 2020; Lanzoni-Mangutchi et al., 2022) |
| Biofilm | Contributes to antimicrobial resistance, resistance to oxygen stress, persistence and recurrence of CDI. | (Bordeleau et al., 2014; Ðapa et al., 2012; Dawson et al., 2012; Frost et al., 2021; Poquet et al., 2018; Semenyuk et al., 2015; Soavelomandroso et al., 2017) |
| Spore formation | Essential for transmission of C. difficile and resistance to environmental stressors, such as oxygen, heat and UV damage. Enables disease persistence. Increased sporulation efficiency possibly increases disease transmission. | (Burns et al., 2011; Donnelly et al., 2016; Fimlaid et al., 2013; Merrigan et al., 2013; Nerber and Sorg, 2021; Setlow, 2007, 2006) |
| tcdC truncation | Truncation thought to increase production of toxins A and B, associated with hypervirulence in ribotype 027 strains. | (Carter et al., 2011; Gerding et al., 2014; Warny et al., 2005) |
The PaLoc
The C. difficile PaLoc spans a 19.6 kb region, with a typically highly conserved genomic localization and organization, and encodes 5 proteins involved in toxin-mediated pathogenesis (Figure 2(a)). This locus is finely regulated on multiple levels. First by environmental factors – toxin production is suppressed in nutritional excess, and transcribed during stationary phase or nutrient limitation [66]. Regulation also occurs at the population level – a recent visualization utilizing a dual-transcriptional reporter system demonstrated expression of toxin and sporulation genes rarely overlap, and solid growth results in subpopulations expressing either toxin (virulence) or sporulation (transmission) genes [67]. The 5 genes in the PaLoc include tcdA and tcdB, encoding toxins A and B, respectively; and tcdR, tcdE and tcdC. TcdR is an alternative sigma factor and likely positive regulator of toxin production, since purified C. difficile RNA polymerase was unable to bind to the tcd promoter regions in the absence of TcdR, and interaction of TcdR with the RNA polymerase holoenzyme allowed transcriptional activation [68; 69]. TcdR also activates its own promoter, in a positive feedback loop, allowing regulation of the entire PaLoc operon. TcdC is thought to be an anti-sigma factor involved in modulating toxin expression through sequestration of TcdR [70], however this requires further classification. The exact function of TcdE has previously been controversial, however it seems likely that this holin-like protein is involved in toxin secretion – as recently demonstrated in clinical strains [69,71]. Holins are membrane proteins, commonly encoded by double-stranded DNA phage, which are required for host cell lysis. The tcdE open reading frame contains three translational start sites resulting in TcdE isoforms of three different sizes. The involvement of combinations of these isoforms in both toxin release and cell death was demonstrated in the hypervirulent strain R20291 [68].
Figure 2.
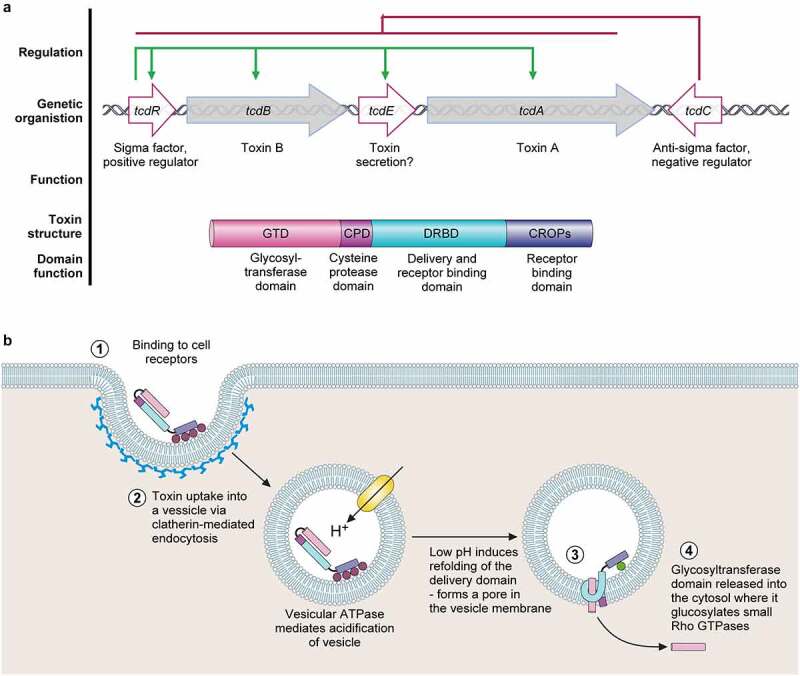
The pathogenicity locus and toxin mode of action. .
a. The pathogenicity locus (PaLoc) is composed of 5 genes: tcdA and tcdB, encoding toxins A and B respectively; tcdR, encoding an alternative sigma factor and likely positive regulator of the PaLoc (regulation shown above in green); tcdE, encoding a holin-like protein putatively involved in toxin secretion; and tcdC, an anti-sigma factor and negative regulator of the PaLoc genes (regulation shown above in red). Toxins A and B both consist of a broadly similar four-domain structure. At the N-terminal, the glucosyltransferase domain (GTD) is the active toxin moiety which inactivates members of the Rho GTPase family. A cysteine protease domain is next to the GTD, and is involved in auto-processing and release of the GTD. The next domain, often called the Delivery and Receptor Binding Domain (DRBD), contains a hydrophobic region and is thought to be involved in translocation of the GTD from the lumen of endocytic vesicles into the host cell cytoplasm. The final C-terminal receptor-binding domain (also known as C-terminal combined repetitive oligopeptides (CROPS) domain) binds to a range of cellular receptors.b. Toxin mode of action [65]. The toxins bind to various cellular receptors via the C-terminal CROPs domain, triggering clathrin-dependent endocytosis (1) followed by acidification of the resulting vesicle (2). The drop in pH triggers a conformational change in the delivery domain which inserts into, and forms a pore in, the vesicle membrane, through which the GTD transits into the host cytoplasm (3). The GTD is then released by a cleavage event mediated by the cysteine protease domain, in a process that is dependent on host inositol hexakisphosphate (4). The GTD is then able to glucosylate and inactive members of the small Rho GTPase family, including Rho, Rac, and Cdc42. Inactivation of Rho GTPases results in multi-level cellular disruption, including dysregulation of actin depolymerization, which causes disruption of tight junctions and loss of intestinal barrier function, induction of proinflammatory cytokines and activation of programmed cell death.
Toxins A (TcdA) and B (TcdB) consist of a broadly similar four-domain structure and are highly similar, with 47% amino acid identity, suggestive of an originating gene duplication event (Figure 2(a)). The N-terminal consists of a glucosyltransferase domain (GTD), next to which is a small cysteine protease domain, involved in auto-processing for the release of the GTD [72]. The next domain, often called the Delivery and Receptor Binding Domain (DRBD), contains a hydrophobic region and is thought to be involved in translocation of the GTD from the lumen of endocytic vesicles into the host cell cytoplasm. The C-terminal receptor-binding domain (also known as C-terminal combined repetitive oligopeptides (CROPS) domain) can bind to a range of carbohydrates, likely facilitating toxin binding to the cell surface [73].
Binding and internalization are essential prerequisites of C. difficile toxin-mediated pathogenesis. Despite their structural similarities, the binding repertoires of TcdA and TcdB are independent from one another. Early studies of TcdA receptors showed TcdA bound carbohydrate domains on the glucosidase enzyme sucrase-isomaltase [74], however this is not expressed in the colonic epithelium. Glycoprotein 96 was also identified to bind TcdA [75], however resides mainly in the endoplasmic reticulum, and therefore is unlikely to be the primary TcdA receptor [76]. More recently, CRISPR-Cas9 screens found that low-density lipoprotein receptors and sulfated glycosaminoglycans bound TcdA. Sulfated glycosaminoglycans were proposed as the major receptors, and were shown to be abundant in the colonic epithelium [76]. Although exploring binding interactions is essential to identify putative receptors, a recent study showed the importance of further characterization of such interactions. Low Density Lipoprotein Receptor-Related Protein-1 was proposed to serve as an endocytic receptor for TcdA, an important advancement, since TcdA must be internalized in order to act [77].
TcdB was shown to enter a variety of cell lines, indicating the existence of either multiple, or widely expressed, receptors [78]. The first recognized TcdB receptor was chondroitin sulfate proteoglycan 4 (CSPG4), a highly conserved protein, identified through shRNAmir library screening [79]. Cryo-EM structures of CSPG4-TcdB indicate binding is mediated through autoprocessing and delivery domains [80; 81]. However, CSPG4 receptors are abundantly expressed on subepithelial myofibroblasts, and not in the colonic epithelium, suggesting CSPG4 is not the dominant TcdB receptor [82]. Disruption of poliovirus receptor-like 3 (PVRL3) resulted in cells which were resistant to TcdB, leading to identification of a further TcdB binding partner [83]. PVRL3 is highly expressed in the colonic epithelia, however its role in TcdB-mediated pathogenesis has been disputed [84], and its contribution to infection remains unclear. More recently, the Frizzled receptors 1, 2, and 7 have been described as physiologically relevant binding partners of TcdB, due to their expression in the colonic epithelium [84]. Frizzled receptors interact with TcdB in a CROPS-independent manor, the crystal structure of which has recently been characterized [85]. Finally, clade 2 C. difficile, which includes hypervirulent 027 strains, express TcdB2 and TcdB4 – variants of TcdB which bind to distinct receptors. One such receptor, binding TcdB4, is tissue factor pathway inhibitor (TFPI) [86]. TFPI is expressed in the colonic crypt, and is therefore physiologically relevant.
The proposed mode of action, known as the ABCD model (activity (A), binding (B), cutting (C), delivery (D)), is similar between both TcdA and TcdB (Figure 2(b)) [65]. Here, TcdA and TcdB bind to cellular receptors as described above. Once bound, the toxins undergo endocytosis, through a clathrin-dynamin-dependent pathway [87]. Subsequent acidification of the endosome results in a conformational change of the toxin, allowing membrane insertion and channel formation [88; 89]. In the cytosol, the toxins undergo a further change, induced by the host virulence cofactor inositol hexakisphosphate. This allows activation of the toxin cysteine protease domain, and results in toxin autocleavage at a position between the cysteine protease and glucosyltransferase domains, releasing the glucosyltransferase domain into the cytosol [90;91]. Despite TcdB being able to induce cellular toxicity independent of the GTD, recent evidence suggests glucosyltransferase activity is still key for disease pathogenesis [92]. In the cytosol, the GTD is able to inactivate members of the Rho GTPase family, including Rho, Rac and Cdc42, via transfer of glucose from UDP-glucose to these proteins at a conserved threonine residue [93; 94]. Since Rho GTPases control pleiotropic signal transduction pathways, disruption to the host cell is widespread. Most notably, dysregulation of actin depolymerisation leads to disruption of the cytoskeleton, resulting in cell rounding, apoptosis, disruption of tight junctions and loss of intestinal barrier function [95; 96]. In fact, TcdA and TcdB are capable of causing both type I (apoptosis) and type III (necrosis) programmed cell death [97]. Changes in Rho GTPase function also evoke changes in proinflammatory signalling pathways, resulting in the production of proinflammatory cytokines IL-1β, TNF-α, IL-8. This, coupled with the subsequent influx of neutrophils, leads to further host tissue damage characteristic of CDI [98].
The relative contributions of TcdA and TcdB to virulence have commonly been disputed – historically, TcdA was accepted as the major virulence factor in C. difficile, owing to the significant immune response to TcdA, but not TcdB, observed in animal infection models [99]. However, the emergence of C. difficile clinical isolates producing only TcdB led to a re-evaluation of the roles of each toxin in disease [100]. Isogenic ribotype 027 toxin mutants demonstrated that both toxins contribute to fulminant disease in hamster models independently [101]. However, contending studies using a variety of animal models showed attenuated virulence in TcdA-producing isogenic strains, with full virulence in TcdB-producing strains [102]. Despite such discrepancies, TcdB is now widely accepted as the major C. difficile virulence factor, due to its involvement in invoking both local and systemic host damage, and activation of the host inflammatory response [102; 103]. Interestingly, a variety of toxin variants of TcdB, but not TcdA, have been reported. Owing to this, a recent global comparison of available TcdB sequences found huge diversity in this protein, allowing an 8-subtype classification. TcdB undergoes accelerated evolution, maximizing diversity and impacting pathogenicity and disease progression [104].
The location of tcdC at the end of the PaLoc, its divergent transcription and its inverse expression profile (high transcription during exponential, low transcription during stationary phase) compared with the rest of the PaLoc genes has led to its common association with PaLoc repression [70,105]. The role of TcdC as a modulator of toxin expression is abundantly apparent in vivo – mutations truncating TcdC are widespread in hypervirulent clinical isolates, and are commonly acknowledged to contribute to the high mortality of ribotype 027 strains [15]. However, the diverse genomic backgrounds of such clinical strains have made confirming this relationship difficult. In an attempt to combat this, analysis of isogenic C. difficile ribotype 027 strains revealed that mutation of tcdC led to hypervirulence, and complementation reduced virulence in hamster models [106]. Despite the wealth of functional evidence for TcdC as a negative regulator of toxin expression, mechanistic detail is still lacking. TcdC has been proposed to act as an anti-sigma factor, through interfering with the binding of RNAP to the tcdA promoter. The mechanism of this inhibition is unclear – TcdC may inhibit interaction of the TcdR sigma factor with RNAP, or prevent recognition of the tcdA promoter [70]. More recently, a detailed topological analysis of C-terminally tagged TcdC suggested an extracellular localization. This localization is discordant with previous work, suggesting TcdC may not act as an anti-sigma factor, and highlighting that the functional characterization of TcdC is far from complete [107].
Binary toxin
Characterization of the hypervirulent ribotype 027 epidemic strain, first reported at the start of the millennium, showed a combination of factors putatively involved in increased virulence: high-level fluoroquinolone resistance, tcdC mutation leading to increased PaLoc expression, and possession of a further toxin – C. difficile binary toxin (CDT) [108]. CDT is an ADP-ribosylating toxin, composed of 2 proteins, the crystal structures of which have been reported recently [109; 110]. CDTa is an ADP-ribosyltransferase, the enzymatic component involved in modifying host cell actin; while CDTb is involved in binding to host cells and translocating CDTa to the host cytosol. CDT first binds to host cells via the lipolysis-stimulated lipoprotein receptor, present in host cell liver, kidney, small intestine and colon [111]. This binding is followed by accumulation of lipid rafts, oligomerization and induction of endocytosis [87,112]. In the resulting endosome, acidification triggers membrane insertion and pore formation by CDTb, allowing translocation of CDTa into the cytosol. Refolding of CDTa after translocation is mediated by host chaperones, including Hsp90 and Cyp40 [113]. CDTa then ADP-ribosylates cellular actin at Arg177, producing ADP-ribose and nicotinamide as biproducts. The modified actin is prevented from further polymerization due to the ADP-ribose moiety. Eventually, this leads to complete depolymerization of the actin cytoskeleton, resulting in phenotypes typical for toxins affecting the cytoskeleton, including loss of barrier function and disruption of tight junctions [114; 115]. However, CDT displays a multifaceted approach to host cell toxicity, since actin polymerization results in redistribution of the microtubule network. Microtubules are involved in a range of cellular processes, including intracellular transport, cell division and cilia formation [116]. CDT hijacks this network, resulting in formation of long cellular protrusions which increase C. difficile adherence to host cells, both in vitro and in mouse models [115,117]. Details of the mechanism of CDT are reviewed in detail elsewhere [15,114].
The regulation of CDT is distinct from, but entwined with, the PaLoc; since cdtA and cdtB are located on a separate 6.2 kb chromosomal region of the genome, known as CdtLoc. CdtLoc contains the two genes encoding CDT, and cdtR – a LytTR family orphan response regulator. CdtR is a positive regulator of both CDT and the PaLoc in hypervirulent strains [118]. Non-CDT producing C. difficile strains contain either a truncated version of the CdtLoc, or a 68 bp insertion sequence at this site [119].
The substantial contribution of CDT to C. difficile pathogenesis has become increasingly apparent through both clinical and experimental works. Several cases of patients with C. difficile infections, with the unusual toxinotype of TcdA and TcdB negative/CDT positive have been reported [120]. Although rare, these cases show CDT alone is capable of causing symptomatic infection. These observations have been confirmed experimentally – an isogenic TcdA/TcdB negative ribotype 027 C. difficile strain, expressing only CDT, caused disease phenotypes in hamster models [101]. However, such phenotypes were different to typical CDI, with symptoms reminiscent of small intestine involvement. Further analysis of the role of CDT in conjunction with the PaLoc showed CDT contributes to increased virulence and disease severity, with mouse models exhibiting increased weight loss and higher mortality compared to a CDT-negative strain [121]. This also highlighted the role of CDT in activating the inflammatory response, with elevated IL-6 cytokine levels observed in mouse models, compared to CDT-negative C. difficile. CDT also induces inflammation via the Toll-like receptor 2-dependent pathway, and suppresses the protective host eosinophil response [121]. Recent evidence has also implicated CDT in the activation of cytotoxic responses in human mucosal-associated invariant T-cells, leading to further aggravation of the pro-inflammatory response [122]. Collectively, these studies suggest CDT is an important virulence factor in C. difficile pathogenesis, particularly in hypervirulent strains.
Surface proteins: S-layer
C.difficile surface proteins are a group of important virulence factors which support C. difficile colonization through adherence to the gut epithelium, activation of the host immune response, and other aspects of pathogenesis. The S-layer is an evolutionary conserved paracrystalline array of protein which envelops the cell, and is ubiquitous among C. difficile strains [123; 124]. It is one of the most metabolically expensive components of the cell, consuming a large percentage of the total cellular protein production [124]. Primarily, this layer is composed of two subunits – the low-molecular weight and high-molecular weight S-layer proteins, which are derived from the post-translational cleavage of a single precursor (SlpA) [125]. These subunits form a heterodimer, which self-assembles to form the S-layer [126; 127]. Accompanying this core structure are 28 cell wall proteins (CWP), which comprise 5–20% of the S-layer and provide a range of additional functions [124].
The essentiality of SlpA, and thus the lack of available isogenic slpA mutants, has impeded the complete functional characterization of the S-layer. However, isolation and analysis of a spontaneous S-layer null strain has implicated the S-layer in sporulation, and resistance to innate immune effectors including lysozyme and LL-37 [128]. Collectively, these findings suggest the S-layer plays an important multifunctional role in successful pathogenesis. Further, the spontaneous S-layer null strain was avirulent in the acute hamster model of CDI, despite persistent colonization in the cecum and colon – suggesting the S-layer is a crucial virulence factor. Both in vivo and in vitro, toxin release of the S-layer null strain was markedly reduced compared to wildtype, associating the S-layer with toxin production, albeit via an unknown mechanism [128]. Strong and specific binding of S-layer proteins to human gastrointestinal tissue specimens have previously been reported, with the strongest binding on the surface epithelium lining the lumen [129]. Binding of the S-layer to HEp-2 cell lines has also been shown, and addition of anti-SlpA antisera led to a 50% reduction in binding to monolayers of multiple C. difficile strains – collectively suggesting an important role in colonization and disease establishment [130]. The apparent contradiction between these observations and the lack of a colonization defect seen with the S-layer null strain in hamsters suggests that there are aspects of C. difficile ecology in the gut that we do not fully understand.
Surface proteins: CWPs
The 28 members of the CWP family are defined by the presence of three tandem copies of the cell wall-binding 2 domain (PF04122), with many also having additional individual domains conferring function [124]. Twelve members of the family are encoded within the CWP gene cluster, a 36.6 kb region including slpA, and related genes, and adjacent to a cluster of genes thought to be involved in the synthesis of the cell wall polysaccharide PS-II [131]. The remaining CWP genes are distributed throughout the genome [132].
Many CWPs are associated with pathogenesis, and are often highly immunogenic. Antibodies to a range of CWPs, most prominently Cwp2 and Cwp84, were found in convalescent patient sera [133]. Such CWPs are therefore likely to be expressed and surface accessible during colonization and/or pathogenesis. Since Cwp2 is the most highly expressed constitutive CWP, and is highly immunogenic, it is perhaps surprising that a cwp2 knockout displayed no defects in growth, sporulation, or virulence in hamster models [134]. However, a significant reduction in adherence to Caco-2 cells suggests this protein functions mainly as an adhesin. Similarly, the immunogenicity of Cwp84 is not concordant with virulence – despite being highly conserved, and responsible for SlpA cleavage, cwp84 mutants were fully virulent in hamster models [135]. Cwp66 is a 66 kDa protein, containing the typical cell wall binding domains, and an additional domain of unknown function. This protein was the first C. difficile classified adhesin, since antibodies to Cwp66 reduced cellular adherence [136]. Recently, molecular characterisation of a Δcwp66 mutant implicated this surface protein in adhesion, motility and stress tolerance [137]. Further, transcriptomic analysis of Δcwp66 suggested a wider cellular involvement of the protein in antimicrobial resistance and metabolism – signifying a multifactorial role in pathogenesis [137]. Cwp22 is an L,D-transpeptidase, a peptidoglycan cross-linking enzyme, that contributes to multiple aspects of pathogenesis [138; 139]. Mutation of cwp22 led to reduced toxin production, and increased cell permeability and autolysis, as well as reduced cellular adherence. Further, cwp22 mutants displayed reduced fitness compared to WT in mice, collectively suggesting that this protein plays important roles in cell envelope integrity and pathogenesis [139]. A further CWP linked to the C. difficile toxins, Cwp19, has been identified as a transglycosylase, contributing to C. difficile pathogenesis through autolysis, resulting in toxin release in specific environmental conditions [140]. CwpV is the largest protein in the CWP family, and exhibits phase-variable expression [141]. As with SlpA, CwpV is subject to post-translational cleavage followed by stable interaction between the resulting cleavage products [142]. The CwpV-specific C-terminal domain consists of a series of repeats that are highly variable between C. difficile strains, with five distinct repeat types identified to date [143]. Functional characterization has demonstrated that CwpV can contribute to auto-aggregative cell–cell interactions and as such, is postulated to be involved in colonization and the biofilm-like growth that is observed in vivo [60].
Surface proteins: Collagen-binding proteins
As with all pathogenic bacteria, multiple complex mechanisms allow fine-tuned host interactions and immune evasion. One such mechanism, binding to the host extracellular matrix, has recently been described in C. difficile. CD2831 is a collagen binding protein, which further promotes adhesion and biofilm formation [144]. CD2831 also enhances immune evasion, through binding the collagen-like domain of C1q of the complement pathway, modulating the classical immune response [144]. Similarly, C. difficile produces an additional collagen-binding surface protein, CbpA. Despite the cpbA knockout being indistinguishable from its respective WT during immobilized collagen V binding assays, this protein enhances collagen interaction and extracellular matrix adherence, demonstrating the large redundancy involved in host interaction and pathogenesis [145].
Lysozyme resistance
Lysozyme is a ubiquitous and highly conserved antimicrobial protein involved in the innate immune response. This antimicrobial targets the bacterial cell wall, cleaving the β(1–4) bond between N-acetylglucosamine and N-acetylmuramic acid of peptidoglycan [146]. C. difficile is highly resistant to cell wall hydrolysis via lysozyme, due to a combination of important virulence factors. The C. difficile S-layer clearly provides some protection against lysozyme, since an S-layer null mutant becomes sensitive to physiological concentrations of the enzyme [128]. Deletion of domain 2 of the low-molecular weight S-layer protein [126] increased the apparent permeability of the assembled S-layer and rendered C. difficile susceptible to lysozyme, suggesting that a steric barrier function contributes to S-layer-mediated resistance to innate immune effectors [147]. In addition to this barrier protection, C. difficile also has a further inducible resistance system that is controlled by the extracytoplasmic sigma factor σV [148]. σV is activated upon lysozyme detection, resulting in expression of peptidoglycan deacetylases PgdA and PdaV [148]. Classical microbiology has shown the synergistic effects of these two proteins in their contribution to lysozyme resistance – deletion of a single protein resulted in small reductions in resistance, whereas deletion of both proteins simultaneously resulted in 1000× reduction in resistance, through almost complete loss of peptidoglycan deacetylation [149]. Deacetylation is an effective method of lysozyme resistance, since interaction between the activate site of lysozyme, and acetyl groups on peptidoglycan enables efficient hydrolysis [150]. Moreover, σV is required for successful pathogenesis in hamster models, demonstrating the importance of lysozyme resistance mechanisms as virulence factors [148].
Biofilm
The clear contribution of biofilms to both virulence and antimicrobial resistance, along with improved tools and novel study methods, has led to the emergence of biofilms as a “hot topic” in microbiology over the last decade. Despite this, little is known about the formation, regulation and maintenance of C. difficile biofilms. C. difficile forms part of the healthy, multi-species biofilm during asymptomatic carriage [151]. However, it has been hypothesized that biofilms may, in fact, also play a role in the persistence and recurrence of CDI [152]. Despite the picture being far from complete, multiple factors have been associated with biofilm formation and regulation in C. difficile. Of note, the sporulation master-regulator Spo0A is associated with regulation of biofilm formation, with mutants exhibiting significantly reduced biofilm [153]. The second messenger c-di-GMP, known to regulate the switch from motile single-cellular to multicellular formations in gram-negative organisms has also been implicated in C. difficile biofilm formation [154]. Increased c-di-GMP reduced flagellar motility and upregulated type 4 pili – increasing cell aggregation [154; 155]. Multiple other genes have also been implicated in biofilm formation – cwp84 mutants displayed a severe defect in biofilm formation, as did mutants lacking the quorum sensing regulator LuxS [153]. The biofilm lifestyle is thought to be dampened through expression of DnaK, a stress response protein, since alterations to dnaK result in stronger biofilms [156]. Likewise, LexA, the global transcriptional repressor and inducer of the SOS response, also reduces biofilm formation, with ΔlexA mutants showing reduced sporulation, motility, and increased biofilm formation [157]. The complex regulation of biofilm formation is therefore clearly mediated in part by both stress and quorum sensing. A detailed review of C. difficile biofilm regulation can be found here [158].
More broadly, the contribution of biofilm to C. difficile-host interactions has been explored through confocal laser scanning microscopy in mouse models. In a mono-associated mouse model, which simplifies analysis of pathogen–host interactions without competition from the microbiota, C. difficile was found to produce a 3D biofilm associated with the mucus layer [159]. Cells were entrapped in a glycan matrix, composed largely of the bacterial polysaccharide PS-II. To attain a more realistic view of the role of commensal C. difficile biofilm in relation to the host gut, 16S rRNA analysis identified C. difficile as a minor part of the complex multispecies host biofilm, composed of Bacteroidetes and Firmicutes [160]. C. difficile biofilms may be important for virulence, since they enhance survival through improved resistance to antibiotics and oxygen stress [161]. However, more in-depth studies of the dynamics of such in vivo biofilms are needed to fully understand the contribution of this lifestyle to pathogenesis.
The contribution of antibiotic resistance to pathogenesis
One of the most important factors in C. difficile pathogenesis is antibiotic resistance (Figure 3). Prior exposure to antibiotics has long since been accepted as the primary risk factor for CDI, since increased abundance of C. difficile in the colon correlates with dysbiosis, most commonly caused through antibiotic exposure [60]. Being intrinsically highly resistant to a multitude of antibiotics further increases virulence, and significantly reduces treatment options. The major complication of CDI, recurrence, is also attributed to exacerbation of gut dysbiosis due to antibiotic treatment. Thus, antibiotic resistance allows colonization, avoidance of clearance, persistence, and recurrence – impacting all aspects of infection.
Figure 3.
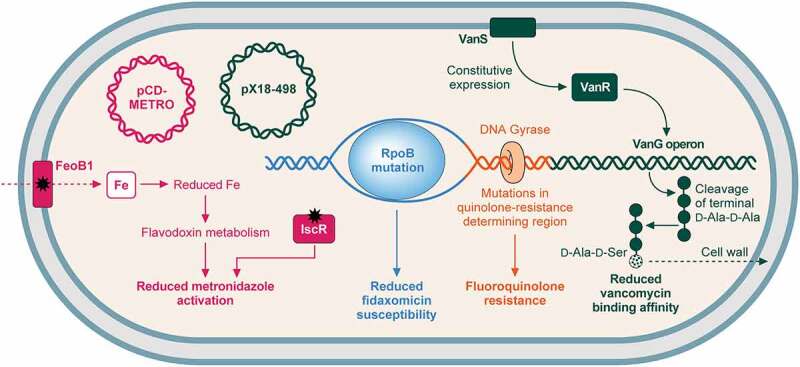
Mechanisms of resistance to commonly used antibiotics. .
Mechanisms of C. difficile resistance to antibiotics commonly used to treat CDI (vancomycin, fidaxomicin, and metronidazole) and fluoroquinolones. (i) metronidazole (pink): resistance can be gained via the plasmid pCD-METRO [162]. Metronidazole resistance may also be gained through mutation of either FeoB1, which reduces intracellular iron, reducing flavodoxin metabolism and metronidazole activation; or IscR, which also reduces metronidazole activation [163]. (ii) fidaxomicin (blue): mutations in RpoB reduce fidaxomicin susceptibility [164]. (iii) vancomycin (green): Mutations in vanSR two-component system allow constitutive expression of the vanG-like operon, which aids vancomycin resistance through replacement of the terminal d-alanine in peptidoglycan pentapeptide sidechains with d-serine, reducing vancomycin binding affinity [104,165]. Plasmid pX18-498 has also recently been associated with resistance, although the mechanism is not understood [166]. (iv) fluoroquinolones (orange): mutations in the genes encoding DNA gyrase, particularly gyrA results in fluoroquinolone resistance [167].
A multitude of evidence supports antibiotics as the major risk factors for CDI. The mechanism of microbiome-associated colonization resistance is far from clear, but is likely a multi-faceted phenomenon involving competition for nutrients, immune modulation, and production of harmful metabolites [168]. The best understood factor is the impact on bile acid metabolism described above, and in particular, the conversion of deconjugated primary bile acids to deoxycholate and lithocholate by members of the microbiome with 7α-dehydroxylase activity [169]. Treatment with antibiotics, either prophylactically or for another infection, causes severe and unpredictable disruption to the resident microbiome. Changes in diversity and relative abundance of species within the microbiome reduces colonisation resistance in the colon, allowing C. difficile to colonize and flourish [60]. Supporting this, a wealth of clinical evidence links antibiotic exposure to CDI: retrospective cohort studies have implicated dose, number of antibiotics used, and days of antibiotic exposure with CDI, with risk increasing in a dose-dependent manner [170]. Further, a striking recent study suggested that odds of infection increased by 12.8% with every day of antibiotic therapy – however this was dependent on both the antibiotic used, and route of administration [171]. Although many antimicrobials are associated with CDI, risk is most highly associated with broad-spectrum antibiotics, including cephalosporins, carbapenems, fluoroquinolones, and clindamycin [171]. In mouse models, clindamycin reduced microbiome diversity by 90% for 28 days. This increased CDI mortality and led to colonic inflammation even in recovering mice – suggesting antibiotic exposure increases not only the risk but severity of CDI [172]. Of course, recurrence – either through relapse or reinfection – is also highly associated with antibiotic use [173]. In pediatric recurrent CDI patients, antibiotic exposure and recent surgery were significant recurrence risk factors [174]. Other clinical studies report similar outcomes, with antibiotics, and previous use of fluoroquinolones, being independent risk factors for recurrence [9]. Therefore, antibiotic use undoubtedly has a large impact on the ability of C. difficile to act as an opportunistic pathogen.
The success of C. difficile as a pathogen is inherently linked to its ability to resist antibiotics. The 4.29 Mb genome of C. difficile has demonstrated an extraordinary ability to gain resistance to a multitude of antibiotics, including aminoglycosides, tetracyclines, erythromycin, clindamycin, beta-lactams, and cephalosporins [175; 176]. This multidrug resistance was the driving force of the CDI epidemic at the start of the millennium, in addition to emergence of novel epidemic lineages, highlighting the importance of such factors in pathogenesis. Resistance to the macrolide-lincosamide-streptograminB (MLSB) family of antibiotics, encompassing erythromycin and clindamycin, is achieved through ribosomal methylation, and is gained via acquisition of transposons, such as Tn5398, containing erm genes [175,177]. erm encodes a 23S rRNA methylase, which modifies the 23S rRNA of the 50S ribosomal subunit, reducing drug binding affinity [178]. However, several C. difficile erythromycin-resistant strains have been identified which lack erm genes – suggesting the presence of yet uncharacterized alternative resistance mechanisms [179]. Tetracycline resistance is less widespread in C. difficile, however conjugative transposons have allowed transfer of tetM to certain strains, providing a mechanism of ribosome protection against tetracycline [180]. Perhaps, the most intriguing capability is that of fluoroquinolone resistance. Not unusually, resistance occurs via alterations to the DNA gyrase subunits, typically GyrA (Figure 3) [181]. However, the emergence of ribotype 027 was associated with widespread fluoroquinolone use, and the epidemic strains possessed recently acquired high-level fluoroquinolone resistance. Since antibiotics target essential cellular processes, resistance is often associated with large fitness burdens. However, competition analysis using mutations seen in C. difficile 027 clinical isolates found fluoroquinolone resistance did not lead fitness costs in vitro, suggesting that this property will persist in the species even in the context of improved fluoroquinolone stewardship [167].
Resistance to antibiotics used to treat C. difficile
Of course, being resistant to a wealth of antibiotics poses two challenges: (i) the extensive resistance displayed greatly reduces treatment options for CDI, warranting the status of C. difficile as an urgent threat; and (ii) such treatment options are likely to be further limited through the high degree of adaptation and flexibility in the C. difficile genome. Until recently, three antibiotics were commonplace for the treatment of CDI. Metronidazole was typically the antibiotic of choice for mild-to-moderate CDI in first instance of infection, while vancomycin was reserved for severe and severe-complicated disease. Fidaxomicin – a narrow-spectrum antibiotic, effective against gram-positive anaerobes – was often overlooked due to higher cost, being significantly more expensive than metronidazole [182; 183]. In 2021, vancomycin became the NICE-recommended front-line antibiotic for CDI, replacing metronidazole as the first-instance treatment [184]. This move reflects both high metronidazole-related recurrence rates, and increasing reports of metronidazole resistance, but poses risks of its own in terms of increasing vancomycin selection pressures.
Metronidazole
Metronidazole is a nitroimidazole antibiotic, effective against anaerobes via formation of unstable nitroimidazole anions, which, when converted into reactive intermediates, react with cellular components to form harmful adducts [163]. Alongside the recent emergence of various epidemic lineages, there has been an increase in metronidazole treatment failure [185]. Resistance in C. difficile was previously thought to be transient, however a recent explosion in research focussed on characterizing metronidazole resistance has led to the discovery of multiple heritable pathways to reduced susceptibility (Figure 3). One such mechanism involved a 7-kb plasmid, dubbed pCD-METRO, that increased resistance 25-fold, and conferred stable resistance to metronidazole [162]. Worryingly, pCD-METRO is thought to be horizontally transferrable, and is already internationally disseminated. However, the lack of universality of this mechanism implies the existence of multiple pathways to metronidazole resistance. Further clinical isolate studies found multiple SNPs in genes affecting iron utilization and electron transport – hinting at the molecular mechanism of resistance [186]. This mechanism was later uncovered using an evolutionary approach, which demonstrated the involvement of redox and iron homoeostasis genes, in a deterministic route to resistance [163]. The existence of multiple routes of resistance to what was once the first-instance treatment for CDI demonstrates how even antibiotics used to treat C. difficile can further contribute to pathogenesis through treatment failure and recurrence.
Fidaxomicin
The current second-line antibiotic, fidaxomicin, acts to inhibit RNA-polymerase at a site distinct from rifamycin through binding to the DNA-template-RNA-polymerase complex prior to transcription initiation. This traps the complex in an “open clamp” position, preventing interaction with the −35 and −10 sequence [187; 188]. Despite the use of fidaxomicin being curtailed due to cost, it displays clear benefits to CDI treatment – its narrower-spectrum of activity results in reduced rates of recurrence compared to alternative treatments [62]. It is worrying, therefore, that resistance has recently been described (Figure 3). Clinical isolate Goe-91 was found to have mutations in rpoB, seen previously in laboratory studies [164]. However, this clinical isolate displayed no apparent fitness burden in terms of growth and sporulation [189]. Since fidaxomicin is already rarely used, emerging resistance casts doubts over the longevity of this CDI treatment.
Vancomycin
Vancomycin is a glycopeptide antibiotic used for treatment of Gram-positive pathogens. Vancomycin impacts the cell wall biogenesis at multiple levels – binding the terminal D-ala-D-ala to prevent crosslinking of peptide chains by transpeptidases, whilst also binding and inhibiting the glycosyltransferase enzyme involved in polymerization of the NAM-NAG sugar backbone. Such activities have bactericidal action on cells through osmotic stress [190]. Despite being well-characterized in other species, and now predominantly used for CDI, vancomycin resistance in C. difficile has been poorly defined. That said, vancomycin resistance rates have increased substantially since 2012, correlating with an increased usage worldwide [191]. The whole-genome sequence of C. difficile published in 2006 revealed a vanG-like cluster, proposed to confer resistance through changing the terminal D-Ala-D-Ala residues to D-Ala-D-Ser, reducing drug binding affinity [165; 192]. Using an evolutionary approach, the two-component system vanSR, responsible for regulating the vanG operon, was shown to be constitutively expressed in isolates with reduced vancomycin susceptibility (Figure 3) [104]. Recent detection of vancomycin-resistant clinical isolates did not, however, identify mutations in this cluster, again suggesting multiple mechanisms of resistance, and demonstrating the frightening plasticity of the C. difficile genome [193]. 2021 also marked the first documentation of plasmid-mediated vancomycin resistance in C. difficile, through a broad-host-range and highly transferable plasmid. Plasmid p × 18–498 was associated with reduced vancomycin susceptibility in vitro, and more severe CDI in vivo in mouse models, highlighting the role of both resistance and plasmid carriage in C. difficile pathogenesis [166].
Therapeutics: Current and future
The increasing threat of antimicrobial resistance, coupled with the diminishing number of available treatments has driven interest in both novel antimicrobials and alternative therapeutics for the treatment of CDI. Since the root of the problem lies with broad-spectrum antibiotics, new approaches aim to shift the archetypal C. difficile treatment to be more targeted and narrower spectrum, reducing further exacerbations of dysbiosis and risk of recurrence. Such treatments include faecal microbial transplantation (FMT), phage therapy, and narrow-spectrum antimicrobials.
FMT
FMT involves administration of faeces from a healthy individual (heterologous), or from one’s own previously healthy microbiome (autologous) to restore the natural gut flora. This has gained popularity as a treatment for CDI over the last decade, however the procedure has yet to be standardized, and there have been reports of adverse events post-transplantation [194]. Typically, faeces can be delivered via colonoscopy, enema, nasogastric tube or oral capsules [195]. The virtues of FMT are well established, as both a standalone and combination therapy: one trial suggested clinical resolution following FMT was 92% [196], while another found FMT with vancomycin provided an 81% clinical resolution of CDI, compared to just 31% for vancomycin alone [197]. Moreover, the potential of FMT to treat the major complication, recurrent CDI, should not be forgotten – a recent study found a 68% success rate across complex patients with recurrent CDI alongside multiple co-morbidities and extended antibiotic use [198]. Despite intense effort in recent years, the underlying mechanism of FMT-mediated restoration of colonization resistance is still disputed but likely involves a combination of competition for resources, immune modulation and production of inhibitory metabolites. Intriguingly however, a very small trial of only 5 patients demonstrated a high rate of CDI resolution using a sterile fecal filtrate, hinting that resident bacteriophage could also be a contributory factor in the effectiveness of FMT [199].
Despite the clear effectiveness of FMT, its unconventional nature has limited public acceptance, and the lack of process standardization poses a worry to clinicians. Further, upon progression to pseudomembranous colitis, FMT has reduced efficacy and often requires repeat treatment [200]. There is also a question mark over manipulation of the microbiome – despite huge advancement in metagenomics, a complete understanding of the gut microbiome, and essential constituents, is lacking [201]. Most importantly, larger, randomized-controlled clinical trials are required to fully understand the efficacy and safety of FMT, since the nature of the therapy holds the risk of transferring pathogens to already-vulnerable patients [194]. Taken together, FMT provides a feasible alternative therapy for CDI, however there are many challenges to overcome before it becomes mainstream. A more refined approach to FMT is clearly desirable, and this is reflected in the array of new microbiome-based therapeutics in clinical development or already undergoing clinical trials for the treatment of CDI. Among these are those derived from donor feces, such as SER-109 from Seres Therapeutics, consisting of spores of mixed Firmicute species purified following ethanol treatment [202], and suspensions of defined bacterial communities grown in pure culture such as the 8-species VE303 from Vedanta Biosciences [203]. Both approaches show great promise and highlight the progression toward more targeted manipulation of the microbiome for therapeutic purposes.
Phage therapy
Phage therapy involves the use of naturally-occurring bacteriophages to infect and lyse pathogenic bacteria [204]. This has been long-established as a potential therapeutic approach, especially at the coal face of the antimicrobial resistance crisis. The major benefit of using phage, as opposed to static antimicrobial agents, is the ability of phage to evolve. Much like with antibiotics, bacteria can, and have, evolved mechanisms of resistance to phage invasion [205; 206] – however unlike antibiotics, phage have evolved numerous ways to overcome such defences [207]. This continual evolutionary arms-race means phage therapies will not become obsolete [208]. Multiple phages infecting C. difficile have been identified, and increasing evidence suggests at least some of these are viable therapeutic agents [209], for example, ФCD27 was shown to reduce C. difficile growth and toxin levels in a batch fermentation model of CDI [210]. To date, no strictly lytic C. difficile phage have been identified, and all of the characterized temperate phage are double-strand DNA viruses, either contractile myoviruses or non-contractile siphoviruses [211]. Additionally, few C. difficile cell surface receptors have been identified, however the S-layer seems to be a common target [128; 212; 213; 214; 215]. Given the high degree of sequence variability seen in the S-layer [123], it is likely that a cocktail of phage would be required in an effective CDI therapeutic. One such combination of phages has been shown to cause complete C. difficile lysis in vitro, and to reduce disease symptoms and bacterial colonization in hamster models, suggesting targeted phage cocktails may be feasible treatment options [216]. More recently, this particular cocktail was further optimized to a combination of 4 phage, which showed complete C. difficile eradication in fermentation vessels [217].
R-type bacteriocins, phage tail-like particles that are structurally similar to the contractile myoviruses, have also been explored as potential therapeutic agents [128,218]. These have the benefit of bypassing many natural phage resistance systems, as they have no genome, but have the same host-range limitations as phage and present additional production challenges as they are not self-perpetuating. Genetic engineering could allow us to overcome many of the limitations of naturally occurring phage and phage tail-like particles. In recent years engineered phage have had some high-profile clinical successes, for example in the treatment of a recalcitrant Mycobacterium abscessus infection [219]. Similar approaches have been adopted for CDI, including altering the target spectrum of phage tail-like particles by swapping receptor binding proteins [128] and enhancement of killing, both in vitro and in vivo, by engineered reduction of lysogeny and redirection of the endogenous type 1-B CRISPR Cas system to target the host’s own genome [220]. This latter approach was the first demonstration that a C. difficile phage could be engineered to be lytic, albeit not completely, and showed that cargo DNA could be added to the phage genome with no apparent impact on the efficiency of infection or formation of progeny phage. As our understanding of both phage infection and host resistance improves it seems likely that engineered optimized phage will play an important role in the treatment of bacterial infections, particularly those such as CDI, where species-specificity is paramount.
As with all novel therapeutics, phage therapy does not come without limitations. While phage therapy is generally regarded as safe, and small human trials have not reported adverse effects, the possibility of harm to the patient cannot be ruled out [221]. Host inflammatory responses to phage have been reported in in vivo models, suggesting the possibility of adverse reactions which may worsen disease [222]. One possible therapeutic approach to avoid such downsides is to use engineered phage-derived biomolecules rather than whole phage [223], with phage endolysins emerging as a clear favorite in recent years [224–226]. Overall, the specificity, coupled with the ability to counter bacterial resistance, suggests great promise for phage treatment as a C. difficile therapeutic.
Antibody therapies
Few novel C. difficile specific therapeutics have come to market in recent years. Fidaxomicin was approved by the FDA in 2011 and the human monoclonal antibody bezlotoxumab followed in 2016. Bezlotoxumab binds to two highly similar sites within the TcdB CROPs domain, thereby blocking binding of the toxin to carbohydrate receptors [227]. Interaction between the antibody and TcdB prevents intoxication and, in combination with an anti-TcdA monoclonal actoxumab, was found to highly protective in animal models of infection, including in hamsters, a species that is acutely sensitive to the C. difficile toxins [228]. Given the specificity of these antibodies it is not surprising that they have minimal adverse impact on the microbiota [229]. In two large-scale human phase III trials, bezlotoxumab alone was shown to dramatically reduce the rate of recurrence [230], a finding that has been confirmed in later studies [231]. Interestingly, it has recently been shown that bezlotoxumab also blocks extraintestinal organ damage that occurs due to systemic dissemination of the toxins following damage to the intestinal mucosa in a mouse model of CDI [232]. This highlights a potential important application of antibody therapy in ameliorating the worst effects of CDI in severe infections. Bezlotoxumab is a clear success story for monoclonal antibody therapy but this approach is not without limitations, not least challenging production and resulting high cost, estimated to be in excess of $6,000 per patient [233]. One possible avenue to avoid these drawbacks is in the development of therapeutic nanobodies instead. Nanobodies are cheaper to mass produce and can have superior pharmacokinetic properties to traditional monoclonal antibodies [234], leading to significant interest in their potential as therapeutic agents for many pathogenic bacteria, including C. difficile (reviewed [235]. Nanobodies that effectively target TcdA or TcdB receptor binding [236] and glucosyltransferase domains [237], the binary toxin [238] and the S-layer [239] have been described but, despite their promise, none of these have progressed to human trials as of yet.
Novel small molecule antimicrobials
The limited arsenal of C. difficile antibiotics, and the additional problems of collateral damage to the microbiome posed by broad-spectrum antimicrobials, has led to a more concentrated search for species-specific antimicrobials. Such narrow spectrum antimicrobials could have several possible advantages over traditional agents, including reduced impact on the microbiome, reduced recurrence rates, superiority over conventional treatment, and improved pharmacokinetic profiles [240]. Despite the intense interest and investment in this space, it has proven challenging to develop antimicrobials of sufficient specificity that are also superior to the current gold standards vancomycin and fidaxomicin. As a result, several promising agents have ceased development after disappointing trial results. One such agent, lacticin 3147, is a two-component lantibiotic produced by Lactococcus lactis that targets the cell wall precursor lipid II to inhibit peptidoglycan biosynthesis, as well as forming pores in the cell membrane to achieve cell death [241]. The potent cell killing activities of lacticin 3147 to a range of C. difficile isolates was demonstrated in a faecal fermentation model, achieving complete elimination of C. difficile in 30 min [242]. However, whilst leaving non-spore-forming anaerobes and total Gram-negative anaerobes intact, this antimicrobial had negative impacts on lactobacilli and bifidobacteria – a likely reason why this treatment has not been taken forward over the last decade [243]. Another promising small molecule antibiotic, cadazolid, has also been abandoned after disappointing phase III results [244]. Cadazolid is a structural hybrid of the oxazolidinone and quinolone classes which had previously demonstrated impressive activity against C. difficile in vitro [245] and had performed well in phase II but was inferior to vancomycin in two phase III trials. Surotomycin, a membrane-targeting cyclic lipopeptide with excellent activity against C. difficile in vitro [246] was similarly abandoned by Merck following a phase III trial which failed to demonstrate superiority over vancomycin [247]. Despite these recent disappointments, several interesting candidate drugs remain at various stages in the development pipeline. Ibezapolstat, a potent DNA polymerase IIIC inhibitor [248], is currently in phase IIb after a successful initial phase II trial [249]. Ibezapolstat has a favorable pharmacokinetic profile, promoting high concentrations at the site of infection in the colon [249], and appears to induce less harmful changes in microbiota composition and diversity than vancomycin [250].
Ridinilazole, a novel small-molecule antimicrobial with highly specific activities against C. difficile [251], is currently the only anti-C. difficile antibiotic in phase III (reviewed in [252]). Whilst the mechanism of action of ridinilazole is yet to be fully characterised, phase II trials demonstrated superiority compared to vancomycin. Ridinilazole sustained a 66.7% clinical response rate, compared to 42.4% for vancomycin, and a higher clinical cure rate; whilst showing markedly reduced recurrence rates. Ridinilazole is also poorly absorbed, thus achieving high concentrations in the colon [253]. Recently, C. difficile strains from Asia, which were broadly resistant to several antimicrobials, were all shown to be highly susceptible to ridinilazole [252]. Together, these studies suggest great promise for ridinilazole as a novel C. difficile-specific antimicrobial agent.
In addition to the novel compounds that are currently in clinical development, there has also been significant interest in repurposing existing licensed drugs. Among these are the antirheumatic agent auranofin [254] and antibiotics such as fusidic acid [255], rifampin [256], and tigecycline [257]. These and further alternative therapeutics are reviewed in depth here [240].
Conclusions
The increasing disease incidence, coupled with growing reports of community-acquired CDI and the threat of antimicrobial resistance has focussed efforts on characterization of C. difficile as an opportunistic pathogen. Over the last decade, work on C. difficile has exploded, owing greatly to the ever-expanding array of genetic tools available. Despite such victories, there are still many research gaps to address. Further understanding of virulence factors, resistance mechanisms and host interactions will no doubt aid development of novel therapeutics, and exploring alternative therapeutic avenues may also prove fruitful. It should not be forgotten, however, that the success of C. difficile as a pathogen is owed largely to its remarkable genome plasticity – allowing the acquisition of virulence factors and an array of resistance mechanisms. With this in mind, it is clear that the road to combatting this pathogen is far from complete [56,258,259,260,261].
Acknowledgments
Work in the RPF laboratory is currently supported by grants from the BBSRC (BB/P02002X/1), MRC (MR/S009272/1), and Wellcome Trust (204877/Z/16/Z). JEB is supported by a studentship from the MRC Discovery Medicine North (DiMeN) Doctoral Training Partnership (MR/R015902/1).
Funding Statement
The work was supported by the BBSRC [BB/P02002X/1]; Medical Research Council (MRC) [MR/R015902/1]; Medical Research Council (MRC) [MR/S009272/1]; Wellcome Trust [204877/Z/16/Z].
Disclosure statement
RPF has an active collaboration with Summit Therapeutics and they are CASE partner for JEB’s PhD studentship.
References
- [1].Adams CM, Eckenroth BE, Putnam EE, et al. Structural and functional analysis of the CspB protease required for Clostridium spore germination. PLoS Pathog. 2013;9(2):e1003165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].ECDC , n.d. Clostridium difficile infections - facts and surveillance [WWW Document]. European Centre for Disease Prevention and Control [cited 2022 Mar 22]. Available from https://www.ecdc.europa.eu/en/Clostridium-difficile-infections/facts
- [3].Liao F, Li W, Gu W, et al. A retrospective study of community-acquired Clostridium difficile infection in southwest China. Sci Rep. 2018;8(1):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rupnik M, Wilcox MH, Gerding DN.. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nature rev Microbiol. 2009;7(7):526–536. [DOI] [PubMed] [Google Scholar]
- [5].Cole SA, Stahl TJ. Persistent and recurrent Clostridium difficile colitis. Clin Colon Rectal Surg. 2015;28(02):65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Napolitano LM, Edmiston CE. Clostridium difficile disease: diagnosis, pathogenesis, and treatment update. Surgery. 2017;162(2):325–348. [DOI] [PubMed] [Google Scholar]
- [7].Lawley TD, Clare S, Walker AW, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012;8(10):e1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Owens RC, Donskey CJ, Gaynes RP, et al. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin Infect Dis. 2008;46(s1):S19–31. [DOI] [PubMed] [Google Scholar]
- [9].Song JH, Kim YS. Recurrent Clostridium difficile infection: risk factors, treatment, and prevention. Gut Liver. 2019;13(1):16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].CDC , 2019. Antibiotic-resistant Germs: new threats [WWW Document].Centers for Disease Control and Prevention [cited 2020 Oct 29]. Available from https://www.cdc.gov/drugresistance/biggest-threats.html
- [11].He M, Miyajima F, Roberts P, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. 2013;45(1):109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Curry SR, Marsh JW, Muto CA, et al. tcdC genotypes associated with severe TcdC truncation in an epidemic clone and other strains of Clostridium difficile. J Clin Microbiol. 2007;45(1):215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Spigaglia P, Mastrantonio P. Molecular analysis of the pathogenicity locus and polymorphism in the putative negative regulator of toxin production (TcdC) among Clostridium difficile clinical isolates. J Clin Microbiol. 2002;40(9):3470–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Warny M, Pepin J, Fang A, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366(9491):1079–1084. [DOI] [PubMed] [Google Scholar]
- [15].Gerding DN, Johnson S, Rupnik M, et al. Clostridium difficile binary toxin CDT. Gut Microbes. 2014;5(1):15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Razavi B, Apisarnthanarak A, Mundy LM. Clostridium difficile: emergence of hypervirulence and fluoroquinolone resistance. Infection. 2007;35(5):300. [DOI] [PubMed] [Google Scholar]
- [17].Mansfield MJ, Tremblay BJ-M, Zeng J, et al. Phylogenomics of 8,839 Clostridioides difficile genomes reveals recombination-driven evolution and diversification of toxin a and B. PLoS Pathog. 2020;16(12):e1009181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].U PHE. 2018. Clostridium difficile infection: mandatory surveillance 2017/18 summary of the mandatory surveillance annual epidemiological commentary 2017/18.
- [19].GOV.UK , n.d. Clostridioides difficile: guidance, data and analysis [WWW Document]. [cited 2022 Mar 3]. Available from https://www.gov.uk/government/collections/Clostridium-difficile-guidance-data-and-analysis
- [20].Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Heimann SM, Cruz Aguilar MR, Mellinghof S, et al. Economic burden and cost-effective management of Clostridium difficile infections. Médecine et maladies infectieuses. 2018;48(1):23–29. [DOI] [PubMed] [Google Scholar]
- [22].Reigadas Ramírez E, Bouza ES. Economic burden of Clostridium difficile infection in European Countries. Adv Exp Med Biol. 2018;1050:1–12. [DOI] [PubMed] [Google Scholar]
- [23].Roldan GA, Cui AX, Pollock NR. Assessing the burden of Clostridium difficile infection in low- and middle-income countries. J Clin Microbiol. 2018;56(3). DOI: 10.1128/JCM.01747-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rajabally YA, Afzal S. Clinical and economic comparison of an individualised immunoglobulin protocol vs. standard dosing for chronic inflammatory demyelinating polyneuropathy. J Neurol. 2019;266(2):461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Paredes-Sabja D, Shen A, Sorg JA. Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol. 2014;22(7):406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Coullon H, Candela T. Clostridioides difficile peptidoglycan modifications. Curr Opin Microbiol. 2022;65:156–161. [DOI] [PubMed] [Google Scholar]
- [27].Janganan TK, Mullin N, Dafis-Sagarmendi A, et al. Architecture and self-assembly of Clostridium sporogenes and Clostridium botulinum spore surfaces illustrate a general protective strategy across spore formers. mSphere. 2020;5(4). DOI: 10.1128/mSphere.00424-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Deakin LJ, Clare S, Fagan RP, et al. The Clostridium difficile spo0a gene is a persistence and transmission factor. Infect Immun. 2012;80(8):2704–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].MacLeod-Glover N, Sadowski C. Efficacy of cleaning products for C. difficile: environmental strategies to reduce the spread of Clostridium difficile-associated diarrhea in geriatric rehabilitation. Can Fam Physician. 2010. 56(5):417–423. [PMC free article] [PubMed] [Google Scholar]
- [30].Rodriguez-Palacios A, LeJeune JT. Moist-heat resistance, spore aging, and superdormancy in Clostridium difficile. Appl Environ Microbiol. 2011;77(9):3085–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Murray SG, Yim JWL, Croci R, et al. Using spatial and temporal mapping to identify nosocomial disease transmission of Clostridium difficile. JAMA Intern Med. 2017;177(12):1863–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Burns DA, Heeg D, Cartman ST, et al. Reconsidering the sporulation characteristics of hypervirulent Clostridium difficile BI/NAP1/027. PLoS One. 2011;6(9):e24894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Merrigan M, Venugopal A, Mallozzi M, et al. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol. 2010;192(19):4904–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Donnelly ML, Fimlaid KA, Shen A. Characterization of Clostridium difficile spores lacking either SpoVAC or dipicolinic acid synthetase. J Bacteriol. 2016;198(11):1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Setlow P. I will survive: DNA protection in bacterial spores. Trends Microbiol. 2007;15(4):172–180. [DOI] [PubMed] [Google Scholar]
- [36].Setlow P. Spores of Bacillus subtilis: their resistance to and killing by radiation, heat and chemicals. J Appl Microbiol. 2006;101(3):514–525. [DOI] [PubMed] [Google Scholar]
- [37].Nerber HN, Sorg JA, McClane BA. The small acid-soluble proteins of Clostridioides difficile are important for UV resistance and serve as a check point for sporulation. PLoS Pathog. 2021;17(9):e1009516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee CD, Rizvi A, Edwards AN, et al. Genetic mechanisms governing sporulation initiation in Clostridioides difficile. Curr Opin Microbiol. 2022;66:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shen A. Clostridioides difficile spore formation and germination: new insights and opportunities for intervention. Annu Rev Microbiol. 2020;74(1):545–566. [DOI] [PubMed] [Google Scholar]
- [40].Battistuzzi FU, Feijao A, Hedges SB. A genomic timescale of prokaryote evolution: insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol Biol. 2004;4(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ramos-Silva P, Serrano M, Henriques AO. From root to tips: sporulation evolution and specialization in Bacillus subtilis and the intestinal pathogen Clostridioides difficile. Mol Biol Evol. 2019;36(12):2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fimlaid KA, Bond JP, Schutz KC, et al. Global analysis of the sporulation pathway of Clostridium difficile. PLoS Gene. 2013;9(8):e1003660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Edwards AN, Wetzel D, DiCandia MA, et al. Three orphan Histidine kinases inhibit Clostridioides difficile sporulation. J Bacteriol. 2022;204(5). DOI: 10.1128/jb.00106-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Martins D, DiCandia MA, Mendes AL, et al. CD25890, a conserved protein that modulates sporulation initiation in Clostridioides difficile. Sci Rep. 2021;11(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nawrocki KL, Edwards AN, Daou N, et al. CodY-dependent regulation of sporulation in Clostridium difficile. J Bacteriol. 2016;198(15):2113–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wetzel D, McBride SM. The impact of pH on Clostridioides difficile sporulation and physiology. Appl Environ Microbiol. 2019. DOI: 10.1128/AEM.02706-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Antunes A, Camiade E, Monot M, et al. Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Res. 2012;40(21):10701–10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Edwards AN, Tamayo R, McBride SM. A novel regulator controls Clostridium difficile sporulation, motility and toxin production. Mol Microbiol. 2016;100(6):954–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rocha-Estrada J, Aceves-Diez AE, Guarneros G, et al. The RNPP family of quorum-sensing proteins in Gram-positive bacteria. Appl Microbiol Biotechnol. 2010;87(3):913–923. [DOI] [PubMed] [Google Scholar]
- [50].Dembek M, Barquist L, Boinett CJ, et al. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. MBio. 2015;6(2). DOI: 10.1128/mBio.02383-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Oliveira PH, Ribis JW, Garrett EM, et al. Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis. Nat Microbiol. 2020;5(1):166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190(7):2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gerskowitch VP, Russell RI. The physiology of bile acids in duodenum and jejunum. Scott Med J. 1973;18(5):138–141. [DOI] [PubMed] [Google Scholar]
- [54].de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17(5):657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kevorkian Y, Shen A, Schneewind O. Revisiting the role of Csp family proteins in regulating Clostridium difficile spore germination. J Bacteriol. 2017;199(22). DOI: 10.1128/JB.00266-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Francis MB, Sorg JA, Limbago BM. Dipicolinic acid release by germinating Clostridium difficile spores occurs through a mechanosensing mechanism. mSphere. 2016;1(6). DOI: 10.1128/mSphere.00306-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Britton RA, Young VB. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol. 2012;20(7):313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pike CM, Theriot CM. Mechanisms of colonization resistance against Clostridioides difficile. J Infect Dis. 2020;223(Supplement_3):S194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Dembek M, Stabler RA, Witney AA, et al. Transcriptional analysis of temporal gene expression in germinating Clostridium difficile 630 endospores. PLoS One. 2013;8(5):e64011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lawley TD, Clare S, Walker AW, et al. Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect Immun. 2009;77(9):3661–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Castro-Córdova P, Mora-Uribe P, Reyes-Ramírez R, et al. Entry of spores into intestinal epithelial cells contributes to recurrence of Clostridioides difficile infection. Nat Commun. 2021;12(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Burke KE, Lamont JT. Clostridium difficile infection: a worldwide disease. Gut Liver. 2014;8(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. [DOI] [PubMed] [Google Scholar]
- [64].Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol. 2011;60(8):1070–1079. [DOI] [PubMed] [Google Scholar]
- [65].Jank T, Aktories K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 2008;16(5):222–229. [DOI] [PubMed] [Google Scholar]
- [66].Bouillaut L, Dubois T, Sonenshein AL, et al. Integration of metabolism and virulence in Clostridium difficile. Res Microbiol. 2015;166(4):375–383. DOI: 10.1016/j.resmic.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Donnelly ML, Shrestha S, Ribis JW, et al. Development of a dual-fluorescent-reporter system in Clostridioides difficile Reveals a Division of Labor between Virulence and Transmission Gene Expression. mSphere. 2022;7(3). DOI: 10.1128/msphere.00132-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Govind R, Fitzwater L, Nichols R. Observations on the role of TcdE isoforms in Clostridium difficile toxin secretion. J Bacteriol. 2015;197(15):2600–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Mani N, Dupuy B. Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc Natl Acad Sci USA. 2001;98(10):5844–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Matamouros S, England P, Dupuy B. Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol Microbiol. 2007;64(5):1274–1288. [DOI] [PubMed] [Google Scholar]
- [71].Majumdar A, Govind R. Regulation of Clostridioides difficile toxin production. Curr Opin Microbiol. 2022;65:95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Pruitt RN, Chambers MG, Ng KK-S, et al., 2010. Structural organization of the functional domains of Clostridium difficile toxins a and B. Proceedings of the National Academy of Sciences 107, 13467–13472. 10.1073/pnas.1002199107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hartley-Tassell LE, Awad MM, Seib KL, et al. Lectin activity of the TcdA and TcdB toxins of Clostridium difficile. Infect Immun. 2018;87(3). DOI: 10.1128/IAI.00676-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Pothoulakis C, Gilbert RJ, Cladaras C, et al. Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin a. J Clin Invest. 1996;98(3):641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Na X, Kim H, Moyer M, et al. gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infection and immunity. 2008;76(7): 2862–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tao L, Tian S, Zhang J, et al. Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin a entry into cells. Nat Microbiol. 2019;4(10):1760–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Schöttelndreier D, Langejürgen A, Lindner R, et al. Low density Lipoprotein Receptor-related Protein-1 (LRP1) is involved in the uptake of Clostridioides difficile toxin a and serves as an internalizing receptor. Front Cell Infect Microbiol. 2020;10. DOI: 10.3389/fcimb.2020.565465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Pan Z, Zhang Y, Luo J, et al. Functional analyses of epidemic Clostridioides difficile toxin B variants reveal their divergence in utilizing receptors and inducing pathology. PLoS Pathog. 2021;17(1):e1009197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Yuan P, Zhang H, Cai C, et al. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res. 2015;25(2):157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chen P, Zeng J, Liu Z, et al. Structural basis for CSPG4 as a receptor for TcdB and a therapeutic target in Clostridioides difficile infection. Nat Commun. 2021;12(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Jiang M, Shin J, Simeon R, et al. Structural dynamics of receptor recognition and pH-induced dissociation of full-length Clostridioides difficile Toxin B. PLoS Biol. 2022;20(3):e3001589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Terada N, Ohno N, Murata S, et al. Immunohistochem-ical study of NG2 chondroitin sulfate proteoglycan expression in the small and large intestines. Histochem Cell Biol. 2006;126(4):483–490. [DOI] [PubMed] [Google Scholar]
- [83].LaFrance ME, Farrow MA, Chandrasekaran R, et al. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proceedings of the National Academy of Sciences. 2015;112(22):7073–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Tao L, Zhang J, Meraner P, et al. Frizzled are colonic epithelial receptors for Clostridium difficile toxin B. Nature. 2016;538(7625):350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chen P, Tao L, Wang T, et al. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science. 2018;360(6389):664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Luo J, Yang Q, Zhang X, et al. TFPI is a colonic crypt receptor for TcdB from hypervirulent clade 2 C. difficile. Cell. 2022;185(6):980–994.e15. [DOI] [PubMed] [Google Scholar]
- [87].Papatheodorou P, Zamboglou C, Genisyuerek S, et al. Clostridial glucosylating toxins enter cells via Clathrin-mediated endocytosis. PLoS One. 2010;5.(5):e10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Barth H, Pfeifer G, Hofmann F, et al. Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J Biol Chem. 2001;276(14):10670–10676. [DOI] [PubMed] [Google Scholar]
- [89].Qa’Dan M, Spyres LM, Ballard JD. pH-induced conformational changes in Clostridium difficile toxin B. Infect Immun. 2000;68(5):2470–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Egerer M, Giesemann T, Herrmann C, et al. Autocatalytic Processing of Clostridium difficile toxin B: binding of inositol hexakisphosphate*. J Biol Chem. 2009;284:3389–3395. [DOI] [PubMed] [Google Scholar]
- [91].Oezguen N, Power TD, Urvil P, et al. Clostridial toxins. Gut Microbes. 2012;3(1):35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Bilverstone TW, Garland M, Cave RJ, et al. The glucosyltransferase activity of C. difficile toxin B is required for disease pathogenesis. PLoS Pathog. 2020;16(9):e1008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Jank T, Giesemann T, Aktories K. Rho-glucosylating Clostridium difficile toxins a and B: new insights into structure and function. Glycobiology. 2007;17(4):15R–22R. [DOI] [PubMed] [Google Scholar]
- [94].Just I, Selzer J, Wilm M, et al. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375(6531):500–503. [DOI] [PubMed] [Google Scholar]
- [95].Gerhard R, Nottrott S, Schoentaube J, et al. Glucosylation of Rho GTPases by Clostridium difficile toxin a triggers apoptosis in intestinal epithelial cells. J Med Microbiol. 2008;57(6):765–770. [DOI] [PubMed] [Google Scholar]
- [96].Janoir C, 2016. 37 13–24 . Virulence factors of Clostridium difficile and their role during infection. Anaerobe []. [DOI] [PubMed] [Google Scholar]
- [97].Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18(2):247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Madan R, Petri WA. Immune responses to Clostridium difficile infection. Trends Mol Med. 2012;18(11):658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lyerly DM, Lockwood DE, Richardson SH, et al. Biological activities of toxins A and B of Clostridium difficile. Infect Immun. 1982;35(3):1147–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Drudy D, Fanning S, Kyne L. Toxin A-negative, toxin B-positive Clostridium difficile. Inter J Infect Dis. 2007;11(1):5–10. [DOI] [PubMed] [Google Scholar]
- [101].Kuehne SA, Collery MM, Kelly ML, et al. Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J Infect Dis. 2014;209(1):83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Carter GP, Chakravorty A, Nguyen TAP, et al. Defining the Roles of TcdA and TcdB in localized gastrointestinal disease, systemic organ damage, and the host response during Clostridium difficile infections. MBio. 2015;6(3). DOI: 10.1128/mBio.00551-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Li Z, Lee K, Rajyaguru U, et al. Ribotype classification of Clostridioides difficile isolates is not predictive of the amino acid sequence diversity of the toxin virulence factors TcdA and TcdB. Front Microbiol. 2020;11. DOI: 10.3389/fmicb.2020.01310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Shen W-J, Deshpande A, Hevener KE, et al. Constitutive expression of the cryptic vanGcd operon promotes vancomycin resistance in Clostridioides difficile clinical isolates. J Antimicrob Chemother. 2020;75(4):859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Govind R, Vediyappan G, Rolfe RD, et al. Evidence that Clostridium difficile TcdC is a membrane-associated protein. J Bacteriol. 2006;188(10):3716–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Carter GP, Douce GR, Govind R, et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog. 2011;7(10):e1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Paiva AMO, Jong LD, Friggen AH, et al. The C-terminal domain of Clostridioides difficile TcdC is exposed on the bacterial cell surface. J Bacteriol. 2020. DOI: 10.1128/JB.00771-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Loo VG, Poirier L, Miller MA, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile–associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353(23):2442–2449. [DOI] [PubMed] [Google Scholar]
- [109].Anderson DM, Sheedlo MJ, Jensen JL, et al. Structural insights into the transition of Clostridioides difficile binary toxin from prepore to pore. Nat Microbiol. 2020;5(1):102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Xu X, Godoy-Ruiz R, Adipietro KA, et al., 2020. Structure of the cell-binding component of the Clostridium difficile binary toxin reveals a di-heptamer macromolecular assembly. Proceedings of the National Academy of Sciences 117, 1049–1058. 10.1073/pnas.1919490117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Papatheodorou P, Carette JE, Bell GW, et al. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc Natl Acad Sci U S A. 2011;108(39):16422–16427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Hemmasi S, Czulkies BA, Schorch B, et al. Interaction of the Clostridium difficile binary toxin CDT and its host cell receptor, Lipolysis-stimulated Lipoprotein Receptor (LSR). J Biol Chem. 2015;290(22):14031–14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ernst K, Langer S, Kaiser E, et al. Cyclophilin-facilitated membrane translocation as pharmacological target to prevent intoxication of mammalian cells by binary clostridial actin ADP-ribosylated toxins. J Mol Biol Elucidation Protein Translocation Pathways (Part II). 2015;427(6):1224–1238. [DOI] [PubMed] [Google Scholar]
- [114].Aktories K, Lang AE, Schwan C, et al. Actin as target for modification by bacterial protein toxins. FEBS J. 2011;278(23):4526–4543. [DOI] [PubMed] [Google Scholar]
- [115].Schwan C, Stecher B, Tzivelekidis T, et al. Clostridium difficile toxin CDT induces formation of microtubule-based protrusions and increases adherence of bacteria. PLoS Pathog. 2009;5(10):e1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol. 2015;16(12):711–726. [DOI] [PubMed] [Google Scholar]
- [117].Schwan C, Kruppke AS, Nölke T, et al. Clostridium difficile toxin CDT hijacks microtubule organization and reroutes vesicle traffic to increase pathogen adherence. Proc Natl Acad Sci U S A. 2014;111(6):2313–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lyon SA, Hutton ML, Rood JI, et al. CdtR regulates TcdA and TcdB production in Clostridium difficile. PLoS Pathog. 2016;12(7):e1005758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Carter GP, Lyras D, Allen DL, et al. Binary toxin production in Clostridium difficile is regulated by CdtR, a LytTR family response regulator. J Bacteriol. 2007;189(20):7290–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Eckert C, Emirian A, Le Monnier A, et al. Prevalence and pathogenicity of binary toxin–positive Clostridium difficile strains that do not produce toxins a and B. New Microbes New Infect. 2015;3:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Cowardin CA, Buonomo EL, Saleh MM, et al. The binary toxin CDT enhances Clostridium difficile virulence by suppressing protective colonic eosinophilia. Nat Microbiol. 2016;1(8):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Marquardt I, Jakob J, Scheibel J, et al. Clostridioides difficile toxin CDT induces cytotoxic responses in human mucosal-associated invariant T (MAIT) cells. Front Microbiol. 2021;12. DOI: 10.3389/fmicb.2021.752549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Dingle KE, Didelot X, Ansari MA, et al. Recombinational switching of the Clostridium difficile S-layer and a novel glycosylation gene cluster revealed by large-scale whole-genome sequencing. J Infect Dis. 2013;207:675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Fagan RP, Fairweather NF. Biogenesis and functions of bacterial S-layers. Nature rev Microbiol. 2014;12(3):211–222. [DOI] [PubMed] [Google Scholar]
- [125].Calabi E, Ward S, Wren B, et al. Molecular characterization of the surface layer proteins from Clostridium difficile. Mol Microbiol. 2001;40(5):1187–1199. [DOI] [PubMed] [Google Scholar]
- [126].Fagan RP, Albesa-Jové D, Qazi O, et al. Structural insights into the molecular organization of the S-layer from Clostridium difficile. Mol Microbiol. 2009;71(5):1308–1322. [DOI] [PubMed] [Google Scholar]
- [127].Mori N, Takahashi T. Characteristics and immunological roles of surface layer proteins in Clostridium difficile. Ann Lab Med. 2018;38(3):189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kirk JA, Gebhart D, Buckley AM, et al. New Class of precision antimicrobials redefines role of Clostridium difficile S-layer in virulence and viability. Sci Transl Med. 2017;9(406). DOI: 10.1126/scitranslmed.aah6813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Calabi E, Calabi F, Phillips AD, et al. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect Immun. 2002;70(10):5770–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Merrigan MM, Venugopal A, Roxas JL, et al. Surface-layer protein a (SlpA) is a major contributor to host-cell adherence of Clostridium difficile. PLoS One. 2013;8(11):e78404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Willing SE, Candela T, Shaw HA, et al. Clostridium difficile surface proteins are anchored to the cell wall using CWB2 motifs that recognise the anionic polymer PSII. Mol Microbiol. 2015;96(3):596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Knight DR, Elliott B, Chang BJ, et al. Diversity and evolution in the genome of Clostridium difficile. Clin Microbiol Rev. 2015;28(3):721–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Wright A, Drudy D, Kyne L, et al. Immunoreactive cell wall proteins of Clostridium difficile identified by human sera. J Med Microbiol. 2008;57(6):750–756. n.d. DOI: 10.1099/jmm.0.47532-0. [DOI] [PubMed] [Google Scholar]
- [134].Bradshaw WJ, Kirby JM, Roberts AK, et al. Cwp2 from Clostridium difficile exhibits an extended three domain fold and cell adhesion in vitro. FEBS J. 2017;284(17):2886–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kirby JM, Ahern H, Roberts AK, et al. Cwp84, a surface-associated cysteine protease, plays a role in the maturation of the surface layer of Clostridium difficile. J Biol Chem. 2009;284(50):34666–34673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Waligora A-J, Hennequin C, Mullany P, et al. Characterization of a cell surface protein of Clostridium difficile with adhesive properties. Infect Immun. 2001;69(4):2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Zhou Q, Rao F, Chen Z, et al. The cwp66 gene affects cell adhesion, stress tolerance, and antibiotic resistance in Clostridioides difficile. Microbiol Spectr. 2022;10(2). DOI: 10.1128/spectrum.02704-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Peltier J, Courtin P, El Meouche I, et al. Clostridium difficile has an original peptidoglycan structure with a high level of N-Acetylglucosamine deacetylation and mainly 3-3 cross-links. J Biol Chem. 2011;286(33):29053–29062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Zhu D, Bullock J, He Y, et al. Cwp22, a novel peptidoglycan cross-linking enzyme, plays pleiotropic roles in Clostridioides difficile. Environ Microbiol. 2019;21(8):3076–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Wydau-Dematteis S, Meouche IE, Courtin P, et al. Cwp19 is a novel lytic transglycosylase involved in stationary-phase autolysis resulting in toxin release in Clostridium difficile. MBio. 2018;9(3). DOI: 10.1128/mBio.00648-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Emerson JE, Reynolds CB, Fagan RP, et al. A novel genetic switch controls phase variable expression of CwpV, a Clostridium difficile cell wall protein. Mol Microbiol. 2009;74(3):541–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Dembek M, Reynolds CB, Fairweather NF. Clostridium difficile cell wall protein CwpV undergoes enzyme-independent intramolecular autoproteolysis. J Biol Chem. 2012;287(2):1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Reynolds CB, Emerson JE, de la Riva L, et al. The Clostridium difficile cell wall protein CwpV is antigenically variable between strains, but exhibits conserved aggregation-promoting function. PLoS Pathog. 2011;7(4):e1002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Arato V, Gasperini G, Giusti F, et al. Dual role of the colonization factor CD2831 in Clostridium difficile pathogenesis. Sci Rep. 2019;9(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tulli L, Marchi S, Petracca R, et al. CbpA: a novel surface exposed adhesin of Clostridium difficile targeting human collagen. Cell Microbiol. 2013;15:1674–1687. [DOI] [PubMed] [Google Scholar]
- [146].Ragland SA, Criss AK, Bliska JB. From bacterial killing to immune modulation: recent insights into the functions of lysozyme. PLoS Pathog. 2017;13(9):e1006512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Lanzoni-Mangutchi P, Banerji O, Wilson J, et al. Structure and assembly of the S-layer in C. difficile. Nat Commun. 2022;13(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Ho TD, Williams KB, Chen Y, et al. Clostridium difficile extracytoplasmic function σ factor σV regulates lysozyme resistance and is necessary for pathogenesis in the hamster model of infection. Infect Immun. 2014;82(6):2345–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Kaus GM, Snyder LF, Müh U, et al. Lysozyme resistance in C. difficile is dependent on two peptidoglycan deacetylases. bioRxiv. 2020. DOI: 10.1101/2020.07.17.209676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Callewaert L, Michiels CW, 2010. 35 127–160 . Lysozymes in the animal kingdom. J Biosci.]. [DOI] [PubMed] [Google Scholar]
- [151].Chilton CH, Pickering DS, Freeman J. Microbiologic factors affecting Clostridium difficile recurrence. Clin Microbiol Infect. 2018;24(5):476–482. [DOI] [PubMed] [Google Scholar]
- [152].Frost LR, Cheng JKJ, Unnikrishnan M. Clostridioides difficile biofilms: a mechanism of persistence in the gut? PLoS Pathog. 2021;17(3):e1009348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Ðapa T, Ng R, Leuzzi YK, et al. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J Bacteriol. 2012;195(3):545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Bordeleau E, Purcell EB, Lafontaine DA, et al. Cyclic Di-GMP Riboswitch-regulated Type IV pili contribute to aggregation of Clostridium difficile. J Bacteriol. 2014;197(5):819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Poquet I, Saujet L, Canette A, et al. Clostridium difficile biofilm: remodeling metabolism and cell surface to build a sparse and heterogeneously aggregated architecture. Front Microbiol. 2018;9. DOI: 10.3389/fmicb.2018.02084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Jain S, Smyth D, O’Hagan BMG, et al. Inactivation of the dnaK gene in Clostridium difficile 630 δerm yields a temperature-sensitive phenotype and increases biofilm-forming ability. Sci Rep. 2017;7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Walter BM, Cartman ST, Minton NP, et al. The SOS response master regulator LexA is associated with sporulation, motility and biofilm formation in Clostridium difficile. PLoS One. 2015;10(12):e0144763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Taggart MG, Snelling WJ, Naughton PJ, et al. Biofilm regulation in Clostridioides difficile: novel systems linked to hypervirulence. PLoS Pathog. 2021;17(9):e1009817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Soavelomandroso AP, Gaudin F, Hoys S, et al. Biofilm structures in a mono-associated mouse model of Clostridium difficile infection. Front Microbiol. 2017;8. DOI: 10.3389/fmicb.2017.02086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Semenyuk EG, Poroyko VA, Johnston PF, et al. Analysis of bacterial communities during Clostridium difficile infection in the mouse. Infect Immun. 2015;83(11):4383–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Dawson LF, Valiente E, Faulds-Pain A, et al. Characterisation of Clostridium difficile biofilm formation, a role for Spo0A. PLoS One. 2012;7(12):e50527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Boekhoud IM, Hornung BVH, Sevilla E, et al. Plasmid-mediated metronidazole resistance in Clostridioides difficile. Nat Commun. 2020;11(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Deshpande A, Wu X, Huo W, et al. Chromosomal resistance to metronidazole in Clostridioides difficile can be mediated by epistasis between iron homeostasis and oxidoreductases. Antimicrob Agents Chemother. 2020;64(8). bioRxiv 2020.03.04.977868. DOI: 10.1101/2020.03.04.977868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Leeds JA, Sachdeva M, Mullin S, et al. In vitro selection, via serial passage, of Clostridium difficile mutants with reduced susceptibility to fidaxomicin or vancomycin. J Antimicrob Chemother. 2014;69(1):41–44. [DOI] [PubMed] [Google Scholar]
- [165].Sassi M, Guérin F, Lesec L, et al. Genetic characterization of a VanG-type vancomycin-resistant Enterococcus faecium clinical isolate. J Antimicrob Chemother. 2018;73(4):852–855. [DOI] [PubMed] [Google Scholar]
- [166].Pu M, Cho JM, Cunningham SA, et al. Plasmid acquisition alters vancomycin susceptibility in Clostridioides difficile. Gastroenterology. 2021;160(3):941–945.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Wasels F, Kuehne SA, Cartman ST, et al. Fluoroquinolone resistance does not impose a cost on the fitness of Clostridium difficile in vitro. Antimicrob Agents Chemother. 2015;59(3):1794–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Ducarmon QR, Zwittink RD, Hornung BVH, et al. Gut microbiota and colonization resistance against bacterial enteric infection. Microbiol Mol Biol Rev. 2019;83(3). DOI: 10.1128/MMBR.00007-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Buffie CG, Bucci V, Stein RR, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517(7533):205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Stevens V, Dumyati G, Fine LS, et al. Cumulative antibiotic exposures over time and the risk of Clostridium difficile infection. Clin Infect Dis. 2011;53(1):42–48. [DOI] [PubMed] [Google Scholar]
- [171].Webb BJ, Subramanian A, Lopansri B, et al. Antibiotic exposure and risk for hospital-associated Clostridioides difficile infection. Antimicrob Agents Chemother. 2020;64(4). DOI: 10.1128/AAC.02169-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Buffie CG, Jarchum I, Equinda M, et al. Profound alterations of intestinal microbiota following a single dose of Clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun. 2011;80(1):62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Gómez S, Chaves F, Orellana MA. Clinical, epidemiological and microbiological characteristics of relapse and re-infection in Clostridium difficile infection. Anaerobe. 2017;48:147–151. [DOI] [PubMed] [Google Scholar]
- [174].Nicholson MR, Thomsen IP, Slaughter JC, et al. Novel risk factors for recurrent Clostridium difficile infection in children. J Pediatr Gastroenterol Nutr. 2015;60(1):18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Peng Z, Jin D, Kim HB, et al. Update on antimicrobial resistance in Clostridium difficile: resistance mechanisms and antimicrobial susceptibility testing. J Clin Microbiol. 2017;55(7):1998–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Spigaglia P. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther Advances Infect. 2016;3(1):23–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Farrow KA, Lyras D, Rood JI. The macrolide-lincosamide-streptogramin B resistance determinant from Clostridium difficile 630 contains two erm(b) genes. Antimicrob Agents Chemother. 2000;44(2):411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Dzyubak E, Yap M-NF. The expression of antibiotic resistance methyltransferase correlates with mRNA stability independently of ribosome stalling. Antimicrob Agents Chemother. 2016;60(12):7178–7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Spigaglia P, Barbanti F, Mastrantonio P, et al. Multidrug resistance in European Clostridium difficile clinical isolates. J Antimicrob Chemother. 2011;66(10):2227–2234. DOI: 10.1093/jac/dkr292. [DOI] [PubMed] [Google Scholar]
- [180].Dong D, Chen X, Jiang C, et al. Genetic analysis of Tn916-like elements conferring tetracycline resistance in clinical isolates of Clostridium difficile. Int J Antimicrob Agents. 2014;43(1):73–77. [DOI] [PubMed] [Google Scholar]
- [181].Dridi L, Tankovic J, Burghoffer B, et al. gyrA and gyrB mutations are implicated in cross-resistance to Ciprofloxacin and moxifloxacin in Clostridium difficile. Antimicrob Agents Chemother. 2002;46(11):3418–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Cruz MP Fidaxomicin (Dificid), a novel oral macrocyclic antibacterial agent for the treatment of Clostridium difficile-associated diarrhea in adults. P T. 2012. 37(5):278–281. [PMC free article] [PubMed] [Google Scholar]
- [183].Nelson RL, Suda KJ, Evans CT. Antibiotic treatment for Clostridium difficile -associated diarrhoea in adults. Cochrane Database Syst Rev. 2017;2017(3). DOI: 10.1002/14651858.CD004610.pub5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].NICE , 2021. Recommendations | Clostridioides difficile infection: antimicrobial prescribing | Guidance | NICE [WWW Document]. [cited 2022 Apr 13] Available from https://www.nice.org.uk/guidance/ng199/chapter/Recommendations
- [185].Banawas SS, 2018. 8414257 . Clostridium difficile infections: a Global overview of drug sensitivity and resistance mechanisms [WWW Document]. BioMed Research International [] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Lynch T, Chong P, Zhang J, et al. Characterization of a stable, metronidazole-resistant Clostridium difficile clinical isolate. PLoS One. 2013;8(1):e53757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lin W, Das K, Degen D, et al. Structural basis of transcription inhibition by fidaxomicin (lipiarmycin a3). Mol Cell. 2018;70(1):60–71.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Venugopal AA, Johnson S. Fidaxomicin: a novel macrocyclic antibiotic approved for treatment of Clostridium difficile infection. Clin Infect Dis. 2012;54(4):568–574. [DOI] [PubMed] [Google Scholar]
- [189].Schwanbeck J, Riedel T, Laukien F, et al. Characterization of a clinical Clostridioides difficile isolate with markedly reduced fidaxomicin susceptibility and a V1143D mutation in rpoB. J Antimicrob Chemother. 2019;74(1):6–10. [DOI] [PubMed] [Google Scholar]
- [190].Patel S, Preuss CV, Bernice F. Vancomycin in: StatPearls. StatPearls Publishing, Treasure Island (FL); 2020. [PubMed] [Google Scholar]
- [191].Saha S, Kapoor S, Tariq R, et al. Increasing antibiotic resistance in Clostridioides difficile: a systematic review and meta-analysis. Anaerobe. 2019;58:35–46. ASA2018: DOI: 10.1016/j.anaerobe.2019.102072. [DOI] [PubMed] [Google Scholar]
- [192].Sebaihia M, Wren BW, Mullany P, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38(7):779–786. [DOI] [PubMed] [Google Scholar]
- [193].Darkoh C, Keita K, Odo C, et al. Emergence of Clinical Clostridioides difficile isolates with decreased susceptibility to vancomycin. Clin Infect Dis. 2022;74(1):120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Lee WJ, Lattimer LDN, Stephen S, et al. Fecal microbiota transplantation: a review of emerging indications beyond relapsing Clostridium difficile toxin colitis. Gastroenterol Hepatol (N Y). 2015. 11(1):24–32. [PMC free article] [PubMed] [Google Scholar]
- [195].Basson AR, Zhou Y, Seo B, et al. Autologous fecal microbiota transplantation for the treatment of inflammatory bowel disease. Transl Res. 2020;226:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Quraishi MN, Widlak M, Bhala N, et al. Systematic review with meta-analysis: the efficacy of faecal microbiota transplantation for the treatment of recurrent and refractory Clostridium difficile infection. Aliment Pharmacol Ther. 2017;46(5):479–493. [DOI] [PubMed] [Google Scholar]
- [197].van Nood E, Vrieze A, Nieuwdorp M. et al, et al., 2013. 368 5 407–415 . Duodenal infusion of donor feces for recurrent Clostridium difficile []. []. [DOI] [PubMed] [Google Scholar]
- [198].Nowak A, Hedenstierna M, Ursing J, et al. Efficacy of routine fecal microbiota transplantation for treatment of recurrent Clostridium difficile infection: a retrospective cohort study. Int J Microbiol. 2019;2019:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Ott SJ, Waetzig GH, Rehman A, et al. Efficacy of sterile fecal filtrate transfer for treating patients with Clostridium difficile infection. Gastroenterology. 2017;152(4):799–811.e7. [DOI] [PubMed] [Google Scholar]
- [200].Sbahi H, Palma JAD. Faecal microbiota transplantation: applications and limitations in treating gastrointestinal disorders. BMJ Open Gastroenterol. 2016;3(1):e000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Gupta S, Allen-Vercoe E, Petrof EO. Fecal microbiota transplantation: in perspective. Therap Adv Gastroenterol. 2016;9(2):229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].McGovern BH, Ford CB, Henn MR, et al. SER-109, an investigational microbiome drug to reduce recurrence after Clostridioides difficile Infection: lessons learned from a Phase 2 trial. Clin Infect Dis. 2021;72(12):2132–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Dsouza M, Menon R, Crossette E, et al. Colonization of the live biotherapeutic product VE303 and modulation of the microbiota and metabolites in healthy volunteers. Cell Host Microbe. 2022;30(4):583–598.e8. [DOI] [PubMed] [Google Scholar]
- [204].Salmond GPC, Fineran PC. A century of the phage: past, present and future. Nature Rev Microbiol. 2015;13(12):777–786. [DOI] [PubMed] [Google Scholar]
- [205].Doron S, Melamed S, Ofir G, et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science. 2018;359(6379). DOI: 10.1126/science.aar4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Tal N, Sorek R. SnapShot: bacterial immunity. Cell. 2022;185(3):578. [DOI] [PubMed] [Google Scholar]
- [207].Koonin EV, Krupovic M. Phages build anti-defence barriers. Nat Microbiol. 2020;5(1):8–9. [DOI] [PubMed] [Google Scholar]
- [208].Hampton HG, Watson BNJ, Fineran PC. The arms race between bacteria and their phage foes. Nature. 2020;577(7790):327–336. [DOI] [PubMed] [Google Scholar]
- [209].Fortier L-C, Moineau S. Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl Environ Microbiol. 2007;73(22):7358–7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Meader E, Mayer MJ, Gasson MJ, et al. Bacteriophage treatment significantly reduces viable Clostridium difficile and prevents toxin production in an in vitro model system. Anaerobe. 2010;16(6):549–554. [DOI] [PubMed] [Google Scholar]
- [211].Hargreaves KR, Clokie MRJ. Clostridium difficile phages: still difficult? Front Microbiol. 2014;5. DOI: 10.3389/fmicb.2014.00184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Dowah ASA, Xia G, Ali AAK, et al. The structurome of a Clostridium difficile phage and the remarkable accurate prediction of its novel phage receptor-binding protein. bioRxiv. 2021. DOI: 10.1101/2021.07.05.451159 [DOI] [Google Scholar]
- [213].Phothichaisri W, Ounjai P, Phetruen T, et al. Characterization of bacteriophages infecting clinical isolates of Clostridium difficile. Front Microbiol. 2018;9. DOI: 10.3389/fmicb.2018.01701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Royer ALM, Umansky AA, Allen M-M, et al. The Clostridioides difficile S-layer protein a (SlpA) serves as a general phage receptor. bioRxiv. 2022. DOI: 10.1101/2022.09.19.508581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Whittle MJ, Bilverstone TW, Esveld RJ van, et al. A novel bacteriophage with broad host range against Clostridioides difficile ribotype 078 supports SlpA as the likely phage receptor. Microbiol Spectr. 2022;10(1). DOI: 10.1128/spectrum.02295-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Nale JY, Spencer J, Hargreaves KR, et al. Bacteriophage combinations significantly reduce Clostridium difficile growth in vitro and proliferation in vivo. Antimicrob Agents Chemother. 2015. DOI: 10.1128/AAC.01774-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Nale JY, Redgwell TA, Millard A, et al. Efficacy of an optimised bacteriophage cocktail to clear Clostridium difficile in a batch fermentation model. Antibiotics. 2018;7(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Gebhart D, Lok S, Clare S, et al. A modified R-type bacteriocin specifically targeting Clostridium difficile prevents colonization of mice without affecting gut microbiota diversity. MBio. 2015;6(2). DOI: 10.1128/mBio.02368-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Dedrick RM, Guerrero-Bustamante CA, Garlena RA, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25(5):730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Selle K, Fletcher JR, Tuson H, et al. In Vivo targeting of Clostridioides difficile using phage-delivered CRISPR-Cas3 antimicrobials. MBio. 2020;11(2). DOI: 10.1128/mBio.00019-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].McCallin S, Alam Sarker S, Barretto C, et al. Safety analysis of a Russian phage cocktail: from MetaGenomic analysis to oral application in healthy human subjects. Virology. 2013;443(2):187–196. [DOI] [PubMed] [Google Scholar]
- [222].Park K, Cha KE, Myung H. Observation of inflammatory responses in mice orally fed with bacteriophage T7. J Appl Microbiol. 2014;117(3):627–633. [DOI] [PubMed] [Google Scholar]
- [223].Fischetti VA. Bacteriophage endolysins: a novel anti-infective to control Gram-positive pathogens. Int J Med Microbiol. 2010;300(6):357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Mayer MJ, Garefalaki V, Spoerl R, et al. Structure-based modification of a Clostridium difficile-targeting endolysin affects activity and host range. J Bacteriol. 2011;193(19):5477–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Phothichaisri W, Chankhamhaengdecha S, Janvilisri T, et al. Potential role of the host-derived cell-wall binding domain of endolysin CD16/50L as a molecular anchor in preservation of uninfected Clostridioides difficile for new rounds of phage infection. Microbiol Spectr. 2022;10(2). DOI: 10.1128/spectrum.02361-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Wang Q, Euler CW, Delaune A, et al. Using a novel lysin to help control Clostridium difficile infections. Antimicrob Agents Chemother. 2015;59(12):7447–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Orth P, Xiao L, Hernandez LD, et al. Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J Biol Chem. 2014;289(26):18008–18021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Warn P, Thommes P, Sattar A, et al. Disease progression and resolution in rodent models of Clostridium difficile infection and impact of antitoxin antibodies and vancomycin. Antimicrob Agents Chemother. 2016;60(11):6471–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Džunková M, D’Auria G, Xu H, et al. The monoclonal antitoxin antibodies (Actoxumab–bezlotoxumab) treatment facilitates normalization of the gut microbiota of mice with Clostridium difficile infection. Front Cell Infect Microbiol. 2016;6. DOI: 10.3389/fcimb.2016.00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Wilcox MH, Gerding DN, Poxton IR, et al. Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N Engl J Med. 2017;376(4):305–317. [DOI] [PubMed] [Google Scholar]
- [231].Thandavaram A, Channar A, Purohit A. et al, et al., 2022. 14 8 e27979 . The efficacy of bezlotoxumab in the prevention of recurrent Clostridium difficile: a systematic review. Cureus []. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Mileto SJ, Hutton ML, Walton SL, et al. Bezlotoxumab prevents extraintestinal organ damage induced by Clostridioides difficile infection. Gut Microbes. 2022;14(1):2117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Prabhu VS, Dubberke ER, Dorr MB, et al. Cost-effectiveness of bezlotoxumab compared with placebo for the prevention of recurrent Clostridium difficile infection. Clin Infect Dis. 2018;66(3):355–362. [DOI] [PubMed] [Google Scholar]
- [234].Jovčevska I, Muyldermans S. The therapeutic potential of nanobodies. BioDrugs. 2020;34(1):11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Péchiné S, Janoir C, Collignon A. Emerging monoclonal antibodies against Clostridium difficile infection. Expert Opin Biol Ther. 2017;17(4):415–427. [DOI] [PubMed] [Google Scholar]
- [236].Murase T, Eugenio L, Schorr M, et al. Structural basis for antibody recognition in the receptor-binding domains of toxins A and B from Clostridium difficile. J Biol Chem. 2014;289(4):2331–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Yang Z, Schmidt D, Liu W, et al. A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. Infection Mice J Infect Dis. 2014;210(6):964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Unger M, Eichhoff AM, Schumacher L, et al. Selection of nanobodies that block the enzymatic and cytotoxic activities of the binary Clostridium difficile toxin CDT. Sci Rep. 2015;5(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Kandalaft H, Hussack G, Aubry A, et al. Targeting surface-layer proteins with single-domain antibodies: a potential therapeutic approach against Clostridium difficile-associated disease. Appl Microbiol Biotechnol. 2015;99(20):8549–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Petrosillo N, Granata G, Cataldo MA. Novel antimicrobials for the treatment of Clostridium difficile infection. Front Med. 2018;5. DOI: 10.3389/fmed.2018.00096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Wiedemann I, Böttiger T, Bonelli RR, et al. The mode of action of the lantibiotic lacticin 3147 - a complex mechanism involving specific interaction of two peptides and the cell wall precursor lipid II. Mol Microbiol. 2006;61(2):285–296. [DOI] [PubMed] [Google Scholar]
- [242].Rea M, Clayton E, O’Connor P. et al, et al., 2007. 56 Pt7 940–946 . Antimicrobial activity of lacticin 3,147 against clinical Clostridium difficile strains. J Medical Microbiol. [cited 2022]. [DOI] [PubMed] [Google Scholar]
- [243].Rea MC, Dobson A, O’Sullivan O, et al. Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc Natl Acad Sci U S A. 2011;108():4639–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Gerding DN, Cornely OA, Grill S, et al. Cadazolid for the treatment of Clostridium difficile infection: results of two double-blind, placebo-controlled, non-inferiority, randomised phase 3 trials. Lancet Infect Dis. 2019;19(3):265–274. [DOI] [PubMed] [Google Scholar]
- [245].Rashid M-U, Lozano HM, Weintraub A, et al. In vitro activity of cadazolid against Clostridium difficile strains isolated from primary and recurrent infections in Stockholm, Sweden. Anaerobe. 2013;20:32–35. [DOI] [PubMed] [Google Scholar]
- [246].Snydman DR, Jacobus NV, McDermott LA. Activity of a novel cyclic lipopeptide, CB-183,315, against resistant Clostridium difficile and other gram-positive aerobic and anaerobic intestinal pathogens. Antimicrob Agents Chemother. 2012;56(6):3448–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Daley P, Louie T, Lutz JE, et al. Surotomycin versus vancomycin in adults with Clostridium difficile infection: primary clinical outcomes from the second pivotal, randomized, double-blind, Phase 3 trial. J Antimicrob Chemother. 2017;72(12):3462–3470. [DOI] [PubMed] [Google Scholar]
- [248].Xu W-C, Silverman MH, Yu XY, et al. Discovery and development of DNA polymerase IIIC inhibitors to treat Gram-positive infections. Bioorg Med Chem. 2019;27(15):3209–3217. [DOI] [PubMed] [Google Scholar]
- [249].Garey KW, McPherson J, Dinh AQ, et al. Efficacy, safety, pharmacokinetics, and microbiome changes of ibezapolstat in adults with Clostridioides difficile infection: a Phase 2a multicenter clinical trial. Clin Infect Dis. 2022;75(7):1164–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].McPherson J, Hu C, Begum K, et al. Functional and metagenomic evaluation of ibezapolstat for early evaluation of anti-recurrence effects in Clostridioides difficile infection. Antimicrob Agents Chemother. 2022;66(8). DOI: 10.1128/aac.02244-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Vickers RJ, Tillotson GS, Nathan R, et al. Efficacy and safety of ridinilazole compared with vancomycin for the treatment of Clostridium difficile infection: a phase 2, randomised, double-blind, active-controlled, non-inferiority study. Lancet Infect Dis. 2017;17(7):735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Collins DA, Wu Y, Tateda K, et al. Evaluation of the antimicrobial activity of ridinilazole and six comparators against Chinese, Japanese and South Korean strains of Clostridioides difficile. J Antimicrob Chemother. 2021;76(4):967–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Vickers R, Robinson N, Best E, et al. A randomised phase 1 study to investigate safety, pharmacokinetics and impact on gut microbiota following single and multiple oral doses in healthy male subjects of SMT19969, a novel agent for Clostridium difficile infections. BMC Infect Dis. 2015;15(1):91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Hutton ML, Pehlivanoglu H, Vidor CJ, et al. Repurposing auranofin as a Clostridioides difficile therapeutic. J Antimicrob Chemother. 2020;75:409–417. [DOI] [PubMed] [Google Scholar]
- [255].Wullt M, Odenholt I. A double-blind randomized controlled trial of fusidic acid and metronidazole for treatment of an initial episode of Clostridium difficile-associated diarrhoea. J Antimicrob Chemother. 2004;54(1):211–216. [DOI] [PubMed] [Google Scholar]
- [256].Chen LF, Kaye D. Current use for old antibacterial agents: polymyxins, rifamycins, and aminoglycosides. Med Clin North Am Antibacterial Ther Newer Agents. 2011;95(4):819–842. [DOI] [PubMed] [Google Scholar]
- [257].Jump RLP, Kraft D, Hurless K, et al. Impact of Tigecycline versus other antibiotics on the fecal metabolome and on colonization resistance to Clostridium difficile in mice. PAI. 2017;2(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Carlson PE, Kaiser AM, McColm SA, et al. Variation in germination of Clostridium difficile clinical isolates correlates to disease severity. Anaerobe. 2015;33:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Francis MB, Allen CA, Sorg JA. Muricholic acids inhibit Clostridium difficile spore germination and growth. PLoS One. 2013;8(9):e73653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Shen E, Zhu K, Li D, et al. Subtyping analysis reveals new variants and accelerated evolution of Clostridioides difficile toxin B. Commun Biol. 2020;3(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Sekulovic O, Bedoya MO, Fivian-Hughes AS, et al. The Clostridium difficile cell wall protein CwpV confers phase-variable phage resistance. Mol Microbiol. 2015;98(2):329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]