

Abstract
The discovery of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway arose from investigations of how cells respond to interferons (IFNs), revealing a paradigm in cell signaling conserved from slime molds to mammals. These discoveries revealed mechanisms underlying rapid gene expression mediated by a wide variety of extracellular polypeptides including cytokines, interleukins and related factors. This knowledge has provided numerous insights into human disease, from immune deficiencies to cancer, and was rapidly translated to new drugs for autoimmune, allergic, and infectious disease, including Covid-19. Despite these advances, major challenges and opportunities remain.
Introduction
The molecular components of the JAK-STAT pathway were identified 30 years ago. We celebrate this birthday by providing a brief history (Figure 1) of how this discovery allowed us to understand mechanisms through which extracellular polypeptides drive physiological and pathological responses, including cellular and organismal development, proliferation, metabolism, infection, inflammation, and cancer. We also consider how this information has been leveraged to develop novel therapeutic modulators of the pathway. A wealth of new evidence has been obtained concerning how this pathway impacts the genome to induce and repress gene expression. In recognition of the many publications and reviews published since the birth of the JAK-STAT pathway, we now strive to synthesize the salient findings and concepts, hoping to summarize the current state of knowledge, aware that each major sub-topic is itself worthy of a detailed review. Despite the extraordinary advances achieved so far, no shortage of profound questions remains. Indeed, some of the questions posed 30 years ago and revisited at year 20 (Stark and Darnell, 2012) are still being addressed, tremendously aided by contemporary technologies and perspectives.
Figure 1. JAK-STAT Timeline.
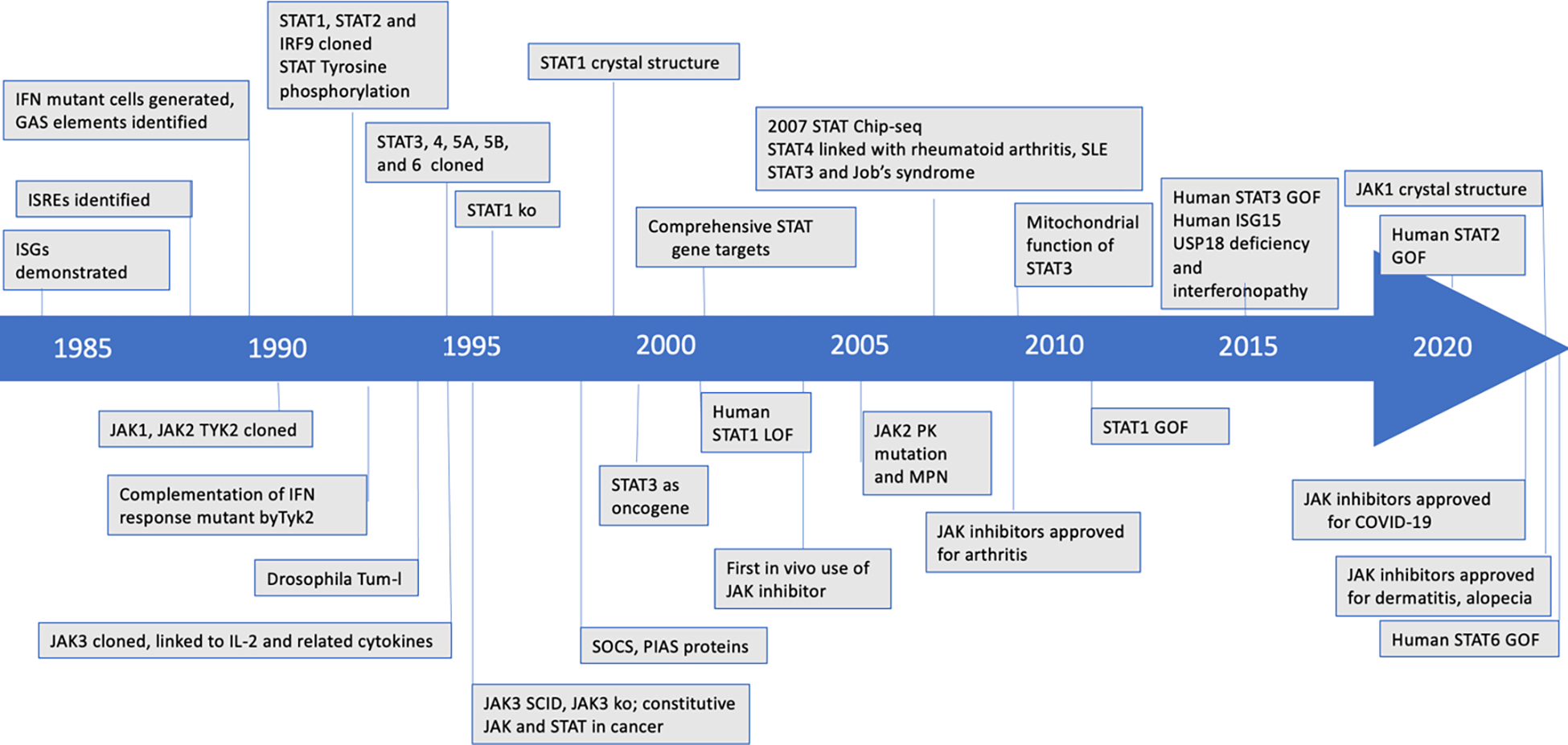
This timeline highlights various milestones in understanding the JAK-STAT pathway since 1984. GAS, gamma activated sequence; IFN, interferon; ISGs, IFN stimulated genes, ISRE, interferon-sensitive response element; SCID, severe combined immune deficiency; KO, knockout; SLE, systemic lupus erythematosus; PK, pseudokinase; MPN, myeloproliferative neoplasms; GOF, gain-of-function; SOCS, suppressor of cytokine signaling; PIAS, protein inhibitor of activated STAT, Tum-l, tumorous lethal.
In the beginning: a new paradigm in signal transduction
Discovered in 1957, interferon (IFN) was quickly recognized to rapidly induce a vital antiviral program. The discovery of the JAK-STAT pathway arose from a very simple but profound question: How do cells respond to IFN? More broadly, the challenge faced by investigators in pre-genomic era of the late 1980s and early 1990s was to elucidate mechanisms underlying how gene expression is mediated in response to a variety of exogenous signals. Receptor-mediated signal transduction via G protein coupled receptors and receptor tyrosine kinases was being probed using the tools of biochemistry and of molecular and cell biology, simultaneously with the identification of specific DNA binding proteins and an array of transcription factors. How the steps of signal transduction and transcriptional activation came together to provide a pathway from membrane-bound receptors to gene transcription remained a mystery at the time.
The discovery of reversible protein phosphorylation by Krebs and Fischer, coupled with the discovery of oncogenic serine/threonine and tyrosine kinases, sparked a race to discover new tyrosine kinases. Using the relatively new method of degenerate polymerase chain reaction to generate protein kinase libraries, a new family of tyrosine kinases was identified that included tyrosine kinase 2, (TYK2) (Firmbach-Kraft et al., 1990; Krolewski et al., 1990), Janus kinase 1 (JAK1), and JAK2 (Harpur et al., 1992; Wilks et al., 1991). The name JAK arose from the two-faced Roman god Janus, since this family of kinases has a characteristic two-faced structure with an amino-terminal kinase domain preceded by a regulatory pseudo-kinase domain. However, since the function of the JAK family of kinases was still elusive, the alternative definition of JAK as “Just Another Kinase” was also appropriate.
To celebrate the 20th anniversary of the discovery of the JAK-STAT pathway, Stark and Darnell (Stark and Darnell, 2012) wrote individual detailed accounts of the history of the work in their laboratories and in the laboratory of the co-discoverer Ian Kerr, allowing us to provide only a brief summary here. Through the pioneering efforts of many others, IFN was readily available as a reagent. The ability to produce cDNA libraries from mRNAs induced by IFN treatment allowed several IFN-induced genes to be found and thus the promoters and regulatory elements that governed their expression were identified (Levy et al., 1986). The Stark and Kerr labs used the promoter of an IFN-induced gene to drive the expression of a lethal marker that killed all cells that responded to IFN. Following extensive mutagenesis, a resistant cell clone was isolated, which through genetic complementation, yielded TYK2 as the essential gene inactivated by the mutagen (Velazquez et al., 1992). The Darnell, Stark, and Kerr labs showed that the signal transducing agent was present in the cytoplasm and rapidly moved to the nucleus when cells were stimulated with IFN, enabling the Darnell lab to purify the first STATs (STAT1 and STAT2, along with IRF9), determine their partial amino acid sequences and, from this information, deduce enough of the coding sequence to enable cloning of the genes. Jim Darnell christened the factors that his lab discovered “Signal Transducers and Activators of Transcription” (STATs) to signify their dual (in other words, two-headed?) functions and to emphasize how rapidly their functions were carried out (namely, stat in medical terminology) (Stark and Darnell, 2012).
With the wisdom of hindsight, the discovery of the JAK-STAT pathway was facilitated greatly by the choice of IFN as the initial target of investigation. Other factors that use this pathway had been discovered long before IFN, with growth hormone and prolactin being discovered 20–30 years earlier. However, focusing on IFNs was fortuitous since the gene expression they induce is very rapid and the pattern of antiviral genes is very distinct compared to the programs of gene expression regulating cell growth and differentiation.
Filling out the pathway
Using a set of cell lines deficient in each of the proteins required for both type I and type II IFN signaling, along with the power of genetic complementation, the Stark, Kerr, and Darnell labs and collaborators rapidly defined the broad outlines of JAK-STAT signaling. This information fueled an explosive effort by many labs that converged on the realization that JAK-STAT pathways are used by many cytokines, growth factors, and other ligands to regulate gene expression in target cells. We now know that nearly 60 cytokines including many interleukins, colony stimulating factors, hormone-like cytokines and growth factors use the JAK-STAT pathway as their predominant, requisite mode of initiating transcription.
With the completion of the human and other genomes, it became clear that there are only 4 members of the JAK family based on their distinctive structure. The final member of the JAK family identified was JAK3, which has more selective expression, unlike other family members (Figure 2). Consistent with its expression, JAK3 activation is restricted to a subset of cytokines that employ the common gamma chain (γc) (Johnston et al., 1994).
Figure 2. Overall scheme of JAK STAT signaling.
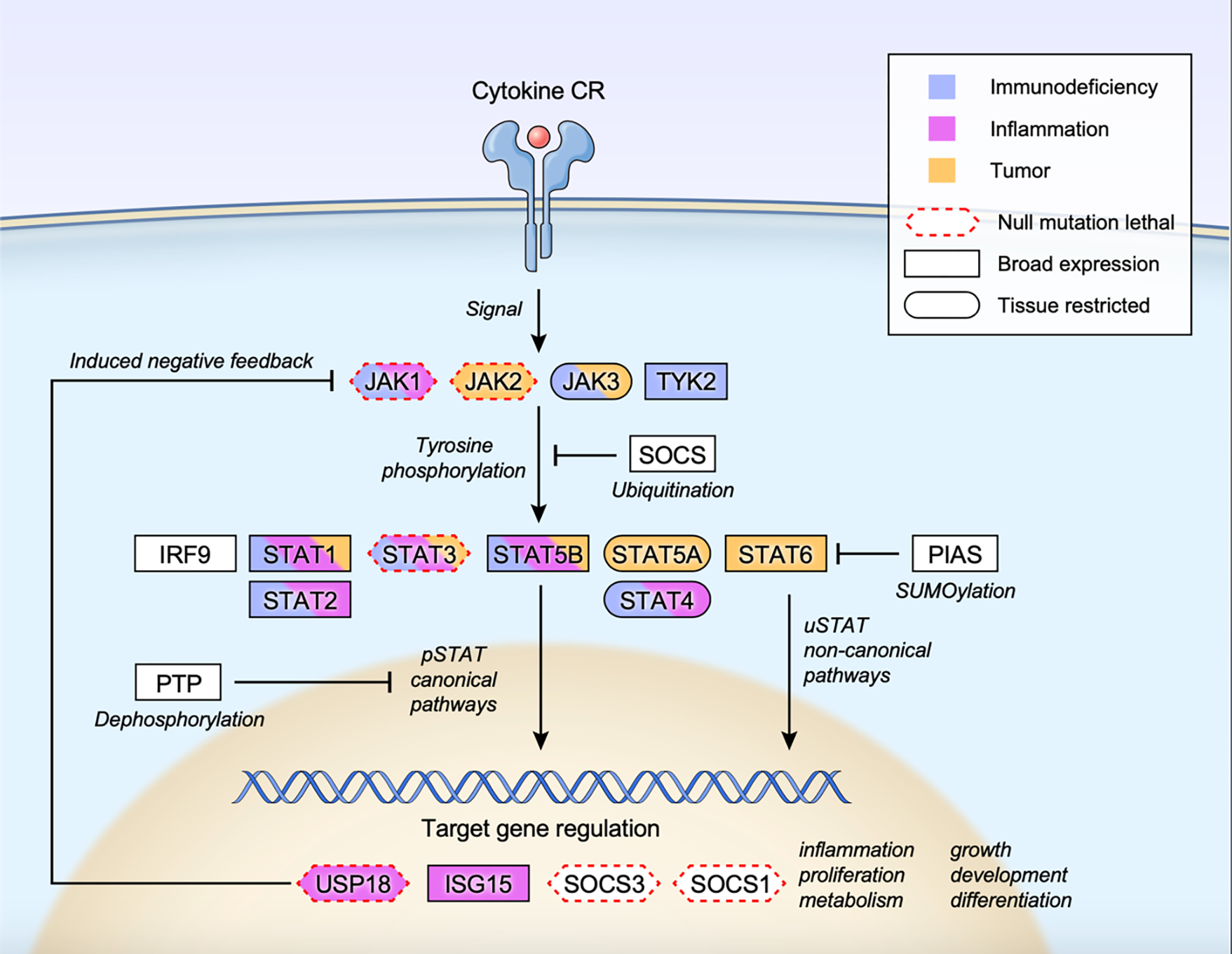
Cytokine-receptor engagement transmits signal to tyrosine phosphorylate and activate receptor associated JAKs, which subsequently phosphorylate and activate STATs. Tyrosine phosphorylated STATs (pSTATs) form dimers, translocate to the nucleus, bind to target DNA sequences, and regulate gene expression (pSTAT canonical pathways). Unphosphorylated STATs also form dimers, enter the nucleus and regulate transcription (uSTAT non-canonical pathways). Multiple inducible negative feedback systems are in place to constrain the signaling circuit that include SOCS (suppressor of cytokine signaling) family proteins, PIAS (protein inhibitor of activated STAT) family proteins, PTP (protein tyrosine phosphatase), USP18 and ISG15. Human monogenic mutations that lead to gain-of-function and/or loss-of-function phenotypes are color coded as immunodeficiency (blue), inflammation (pink) or tumor (orange) for each constituent gene. Genes whose null mutation in mice leads to lethality is marked with red dotted border (Mouse Genome Informatics; http://www.informatics.jax.org). Tissue expression pattern of each protein is also marked as broad expression (square border) or tissue restricted (oval border) (The human protein atlas; https://www.proteinatlas.org/search).
JAKs contain a carboxy terminal catalytic or kinase domain preceded by a pseudokinase (PK) or kinase-like domain, a feature that prompted the name of the family. JAKs are constitutively associated with the intracellular Box1 and Box2 domains of cytokine receptors via the amino-terminal band four point one ezrin radixin, moesin (FERM) domain and Src Homology 2 (SH2) domains (Figure 3). A detailed structure of a full-length JAK was remarkably challenging to obtain, and only recently has the structure of full-length JAK1 complexed with the intracellular domain of a cytokine receptor been obtained (Figure 3) (Glassman et al., 2022). This work revealed that JAK1 domains form an extended structural unit that facilitates the dimerization of the cytokine receptor/JAK complex via packing of the PK domains from the monomeric receptor/kinase complex. The regulatory function of the PK domain and its mechanistic biological impact will be discussed below.
Figure 3. Structure of Janus kinases.
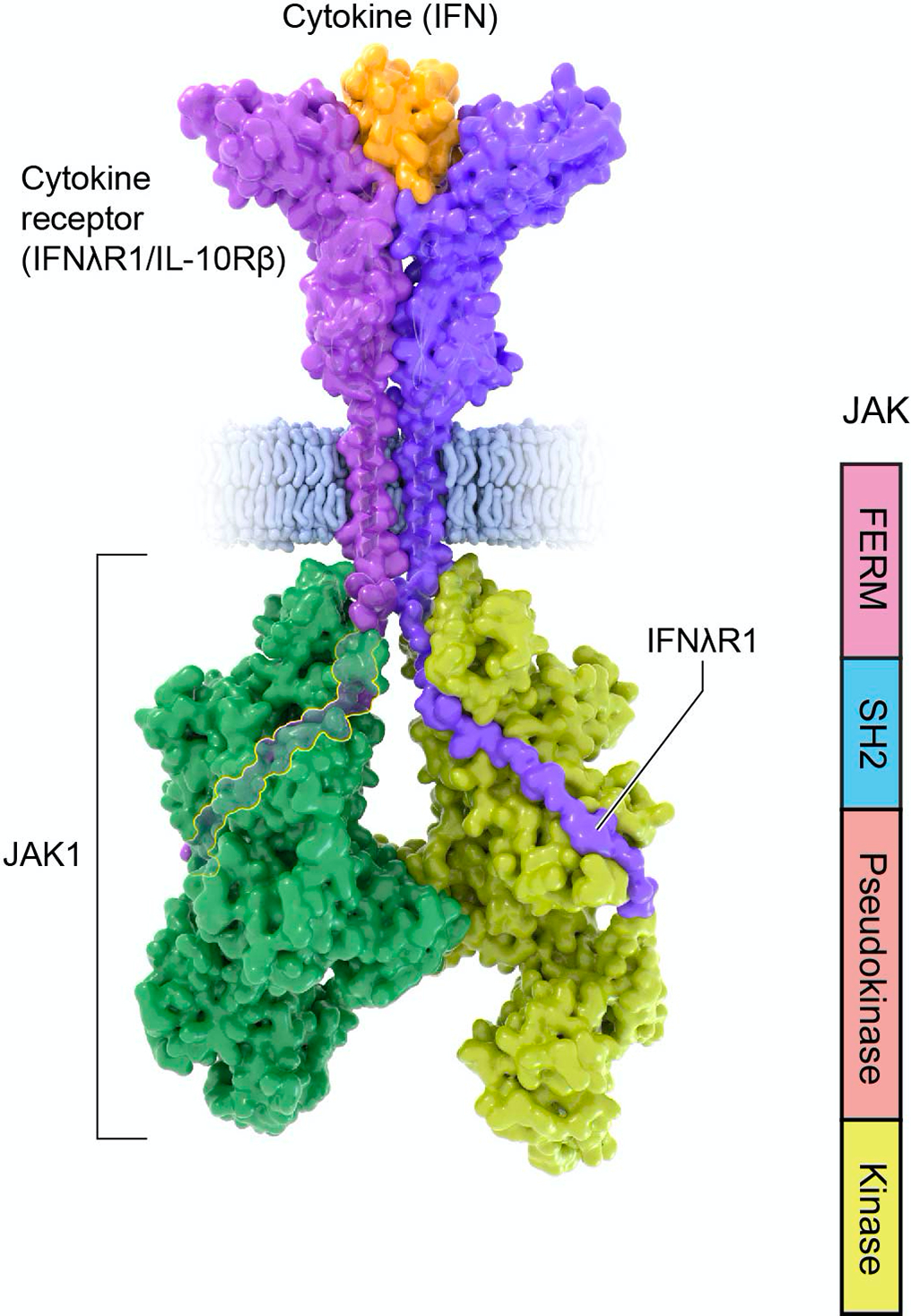
Structure of activated Janus Kinase dimer (green; PDB 7T6F) complexed with the intracellular domain of matured IFNlR1/IL-10Rb (purple) bound to IFN-l (orange) (PDB 5T5W). Of note, the intracellular portion of the receptor binds JAK1 FERM and SH2 domains through N-terminal Box1 and C-terminal Box2 motifs. The JAK kinase-like or pseudokinase domain promotes dimerization of the cytokine receptor/JAK complex.
Following the discovery of STAT1 and STAT2, additional members of the STAT family were rapidly discovered in library or functional screens. STAT3 was identified in response to IL-6 as a DNA-binding “acute phase response factor” that behaved like IFN-activated STAT1 and was found as both a homodimer and heterodimer with STAT1 (Akira et al., 1994; Zhong et al., 1994b). Of note, epidermal growth factor and other growth factors were also recognized as STAT3 activators. Correspondingly, while “JAK-STAT pathway” is a useful shorthand, JAKs are not required for STAT activation by receptor tyrosine kinases. Next, STAT4 was identified based on homology to STAT1 and STAT2 (Zhong et al., 1994a) and was found to be predominantly expressed in lymphoid tissues and activated by IL-12, IL-23, and type I IFNs. A “mammary gland specific nuclear factor” activated by prolactin was identified and designated STAT5 (Gouilleux et al., 1994; Liu et al., 1995; Wakao et al., 1994), and further clarified as two STAT5 genes, STAT5A and STAT5B, with a very high degree of homology that can be activated by factors such as IL-2, IL-3, GM-CSF, and IL-5. Lastly, STAT6 was identified and cloned as an IL-4-inducible factor (Hou et al., 1994).
These discoveries led to a coherent view of the JAK-STAT pathway (Figure 2). Cytokine-receptor ligation activates JAKs that are constitutively associated with cytokine receptors, leading to JAK auto/transphosphorylation, activation, and phosphorylation of tyrosine residues in the receptor tails. Receptor phosphorylation creates docking sites for latent, cytoplasmic STATs, which are recruited to the receptor complex via tyrosine-phosphate-binding SH2 domains and become phosphorylated themselves (Greenlund et al., 1994). The activated phosphorylated STATs homo- or heterodimerize via intermolecular SH2-phosphotyrosine interactions and acquire the ability to translocate to the nucleus and bind DNA regulatory elements to alter chromatin accessibility and induce gene transcription. While a preferential/dominant STAT can often be identified for a given cytokine, many cytokines have been reported to activate nearly all STATs in some circumstance or tissue, and all STATs have been shown to be activated by the major cytokines that have been extensively studied. This is not a trivial finding; given the large number of cytokines, the relative paucity of JAKs and STATs is inadequate to explain the assorted actions of so many diverse factors while still achieving specificity in signaling.
The JAK-STAT pathway is widely conserved amongst metazoans, from Caenorhabditis elegans and Drosophila melanogaster to vertebrates. Drosophila has three related cytokine-like ligands – Unpaired (Upd), Upd2 and Upd3, one cytokine-like receptor, Domeless (Dome) that leads to the activation of one JAK, termed Hopscotch (Hop) and one STAT transcription factor, Stat92E. Dictyostelium discoideum has four STAT proteins, but only in Bilateria is convergence of the entire pathway evident.
Even though the JAK-STAT pathway is a major component of cytokine signaling, other SH2 signaling molecules are also recruited, and pathways beyond STATs are engaged by cytokine receptors. These include Ras, MAP kinase, AKT, PI3K, and mTOR pathways (Silvennoinen et al., 1993; Yu et al., 1995). Based on proteomic analysis of IL-2-stimulated cells, 90% of the phospho-proteome of IL-2 activated T cells are JAK-dependent, with the remainder being mediated by SRC family kinase signaling, encompassing the production of phosphatidylinositol (3,4,5)-trisphosphate and AKT activity (Ross et al., 2016).
Negative Regulation of Cytokine Signaling
Physiological negative regulation.
As with many biological settings, negative feedback systems are induced by a positive-acting stimuli; this is certainly the case for cytokines and interferons, and the suppressor of cytokine (SOCS family) of STAT-target genes is an outstanding example (Figure 2) (Linossi and Nicholson, 2015). Eight SOCS proteins are encoded in the human genome, including SOCS1-7 and CIS, which contain a central SH2 domain and a carboxy-terminal SOCS box. SOCS proteins interfere with cytokine signaling by binding to cytokine receptors, competing with STATs, and inducing their ubiquitination and degradation via elongin BC and the E3 ligase Cullin5. SOCS1 and SOCS3 contain a short motif called the Kinase Inhibitory Region (KIR) that interacts with JAKs directly (JAK2, JAK1, and TYK2). The SOCS1 KIR inhibits JAK2 by obstructing the access of both ATP and substrate (Giordanetto and Kroemer, 2003) whereas SOCS3 interacts with JAK2 associated with gp130 (Kershaw et al., 2013). SOCS3 is preferentially induced by IL-6, whereas SOCS1 predominantly inhibits IFNs but also IL-12, IL-4/13, and IL-2 family of cytokines.
Other key negative feedback regulators are ubiquitin-specific peptidase 18 (USP18) and interferon stimulated gene 15 (ISG15) (Figure 2). ISG15 is a ubiquitin-like protein that is covalently conjugated to target proteins (ISGylation) to modulate the function of multiple host and viral proteins, and USP18 is a protease that deconjugates ISG15 from target proteins. Both are induced by interferons and are key negative regulators of type I IFN signaling (Figure 2). USP18 competes with JAK1 for binding to IFNAR2, and STAT2 is an essential adaptor that recruits USP18 to IFNAR2. Additionally, ISG15 non-covalently binds to USP18 to prevent its ubiquitination/degradation.
Negative regulators also target downstream of the JAK-STAT pathway (Figure 2). The protein inhibitor of activated STAT (PIAS) family comprises multiple members that act on STATs and other factors including NF-κB, and interfere with signaling via multiple mechanisms, such as blocking the DNA-binding activity of transcription factors, recruiting transcriptional co-repressors and promoting protein sumoylation (Shuai, 2006). Multiple phosphatases are reported to dephosphorylate STATs, including CD45, PTP1B, TC-PTP, PHLPP1, PTPRT, PRPRK, PTPN9, and PTPN2. Notably, SHP-1 and SHP-2 have both positive and negative effects on signaling.
Negative regulators are also conserved, as Drosophila have a single SOCS protein designated Socs36E and a single PIAS protein desginated dPIAS.
Negative regulation adapted by viruses.
The importance of STAT-mediated IFN-signaling as a fundamental component of antiviral defense is highlighted by the repeated evolution of viral anti-immune strategies that target STATs directly or indirectly. Mechanisms of STAT-directed IFN antagonism include targeted STAT degradation, sequestration, and cytoplasmic retention. Variations on these basic themes of STAT inhibition have been reinvented by many viruses to escape IFN antiviral responses. Paramyxovirus nonstructural proteins (V, C, and W) engage STATs directly and in combination with other factors. Parainfluenza viruses PIV5 and PIV2, and mumps virus use their V proteins to assemble STAT1 or STAT2-targeting ubiquitin ligase complexes to induce proteasomal degradation (Ulane and Horvath, 2002). Flaviviruses, including Dengue virus and Zika virus, target STAT2 for proteasome-dependent degradation (Ashour et al., 2009; Grant et al., 2016), and a porcine deltacoronavirus nsp5 protease can cleave STAT2 (Zhu et al., 2017).
The Henipaviruses, Nipah virus and Hendra virus, and measles virus bind STAT1 and STAT2 with their V proteins, preventing oligomerization, sequestration in cytosolic aggregates, nuclear translocation, and import. Dimeric STAT1 nuclear import uses a unique binding site on importin 5, and the Ebola VP24 protein specifically blocks this interaction (Xu et al., 2014). Disruption of STAT nuclear import by karyopherin is also found in enteroviruses and rotaviruses (Wang et al., 2017a). The SARS-CoV-2 ORF6 protein interferes STAT import via the Nup98-Rae1 nuclear pore sub-complex (Miorin et al., 2020).
Other viruses can modulate STATs by sequestration in cytosolic aggregates or promote nuclear export. SARS-CoV-2 N protein can mediate STAT cytosolic retention (Mu et al., 2020) and both Henipavirus V and Chikungunya virus can actively export STATs via a CRM-1 export signal (Goertz et al., 2018).
STAT structure, complex formation, and impact on function
All the STATs share similar N-terminal, coiled-coil, DNA-binding, linker, SH2, and transactivation domains (Figure 4). In general, activated JAKs phosphorylate the conserved tyrosine residue of one STAT monomer, which then binds to the SH2 domain of another monomer, driving a conformational change that enables STATs to translocate into the nucleus to initiate the transcription of target genes. In this review, we use P-STAT to designate ‘canonical’ tyrosine phosphorylation (i.e., STAT1-Y701, STAT2-Y690, STAT3-Y705, STAT4-Y693, STAT5-Y694, SYAY6-Y641) and U-STAT to designate a STAT without tyrosine phosphorylation. Other residues may or may not be phosphorylated.
Figure 4. STAT domains and structure.
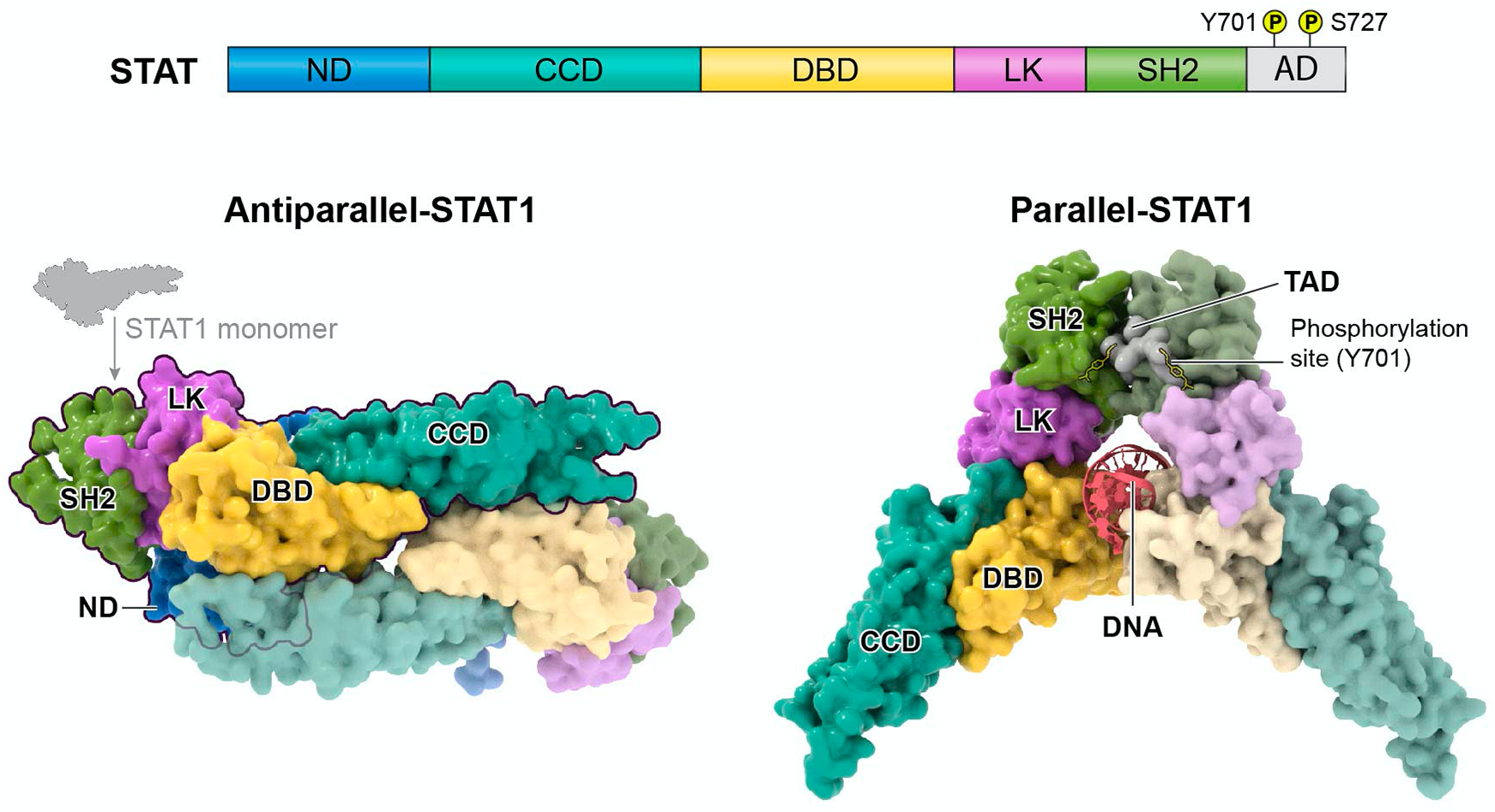
STAT proteins contain the following domains: N-terminal domain (ND), coiled-coil domain (CCD), DNA-binding domain (DBD), linker domain (LK), Src Homology 2 domain (SH2), and transactivation domain (AD) (upper panel). Key phosphorylated tyrosine and serine amino acids reside within the AD for STAT dimerization and functional modulation. Below are the structures of STAT1 dimers, specifically, in its unphosphorylated anti-parallel conformation (PDB 1YVL) and phosphorylated parallel conformation bound to DNA (PDB 1BF5). Notably, the antiparallel confirmation prevents the DBD from interacting with DNA through reciprocal interaction with the opposing dimer’s CCD. Like STAT1, parallel structures of homodimers bound to DNA have been shown for STAT3 (PDB 1BG1) and STAT6 (PBD 4Y5W) and anti-parallel confirmation of STAT dimers have been described for STAT3 (PDB 6TLC) and STAT5a (PDB 1Y1U).
STATs can form a variety of hetero- or homodimers. STAT1, STAT3, and STAT5a have been shown to form homodimers in either a parallel or antiparallel conformation (Figure 4; STAT1 shown as an example). The DNA-binding domains (DBDs) of two P-STATs bind to DNA as a C-shaped clamp in the parallel conformation (Chen et al., 1998). However, in the antiparallel homo-dimer, the DBDs reciprocally interact through their coiled-coil domains (CCDs), preventing access to DNA (Zhong et al., 2005). Tyrosine phosphorylation regulates the equilibrium between these two conformations. For example, P-STAT1 is primarily parallel and U-STAT1 is primarily antiparallel, but low levels of the alternative conformations are also present. Notably, U-STAT3, U-STAT5 and U-STAT6 also have functional roles in the nucleus in regulating gene expression or heterochromatin formation. U-STAT3 competes with IκB and binds to NF-κB, which then with the help of importin-α3, brings the U-STAT3/p65/p50 complex to the nucleus to bind to a κB site for RANTES gene expression (Yang et al., 2007). Rather than initiating transcription, U-STAT5 functions as a tumor suppressor capable of repressing multiple oncogenes via heterochromatin formation (Hu et al., 2013b). U-STAT6, associated with p300, binds directly to the COX-2 GAS element to drive its expression in NSCLC cells (Cui et al., 2007).
The N-terminal domain is critical for regulating STAT homodimer formation and function. When P-STAT1 disengages from DNA, the N-terminal domain dimerizes and helps to expose the phosphorylated tyrosine as a target of the TC45 phosphatase (Chen et al., 2003; Wenta et al., 2008). Additionally, STAT tetramers form by the cooperative recruitment of pairs of homodimers via interactions between N-terminal domains. (Begitt et al., 2014; Lin et al., 2012; Vinkemeier et al., 1998). Such tetramers bind to adjacent DNA loci that combine high-affinity and low-affinity binding sites. (Mandal et al., 2011). Consequently, tetramers form more stable DNA complexes and have more contact with DNA than dimers. Tetramer formation is essential for the complete transcriptional response of STAT1 to type II IFN (Begitt et al., 2014) and of STAT3 to IL-6 (Zhang and Darnell, 2001). For STAT5, the tetramers not only directly induce gene expression but also indirectly regulate gene repression, presumably through the positive regulation of an intermediate repressor (Lin et al., 2012; Mandal et al., 2011).
Unlike the other STATs, which form homodimers, gene expression in response to IFNs I and III is driven by STAT1:STAT2 heterodimers that, together with IRF9, comprise ISGF3 (Figure 5). In P-ISGF3, the parallel conformation of the P-STAT1:P-STAT2 dimer is stabilized by reciprocal phosphotyrosine-SH2 interactions. In this conformation, the DBDs of the two STATs bind to DNA simultaneously, together with the DBD of IRF9 (Rengachari et al., 2018). In U-ISGF3, the two DBDs can still bind to DNA coordinately. Modular associations between STATs and IRFs can contribute to non-canonical complexes, including IRF9 in complex with variations of unphosphorylated STAT1 or STAT2 (Majoros et al., 2017). Recent evidence from electron microscopic and biochemical analyses reveals that U-STAT1 and U-STAT2 heterodimers adopt a very stable antiparallel conformation (Wang et al., 2021). It is clear though that U-STAT1:U-STAT2 and U-STAT2:IRF9 dimers are mutually exclusive; that is, IRF9 cannot bind to the U-STAT1:U-STAT2 dimer and U-STAT1 cannot bind to U-STAT2:IRF9. From this new information, the functional complex previously called “U-ISGF3” (Cheon et al., 2013) is now primarily the U-STAT2:IRF9 dimer and that U-STAT1 plays a more peripheral role. The U-STAT2:IRF9 complex drives the expression of the majority of ISGs in unstimulated cells and switches to an ISGF3 complex containing STAT1, STAT2 and IRF9 in response to IFN-I or IFN-γ stimulation (Platanitis et al., 2019).
Figure 5. Conformational changes of STAT1 and STAT2 complexes.
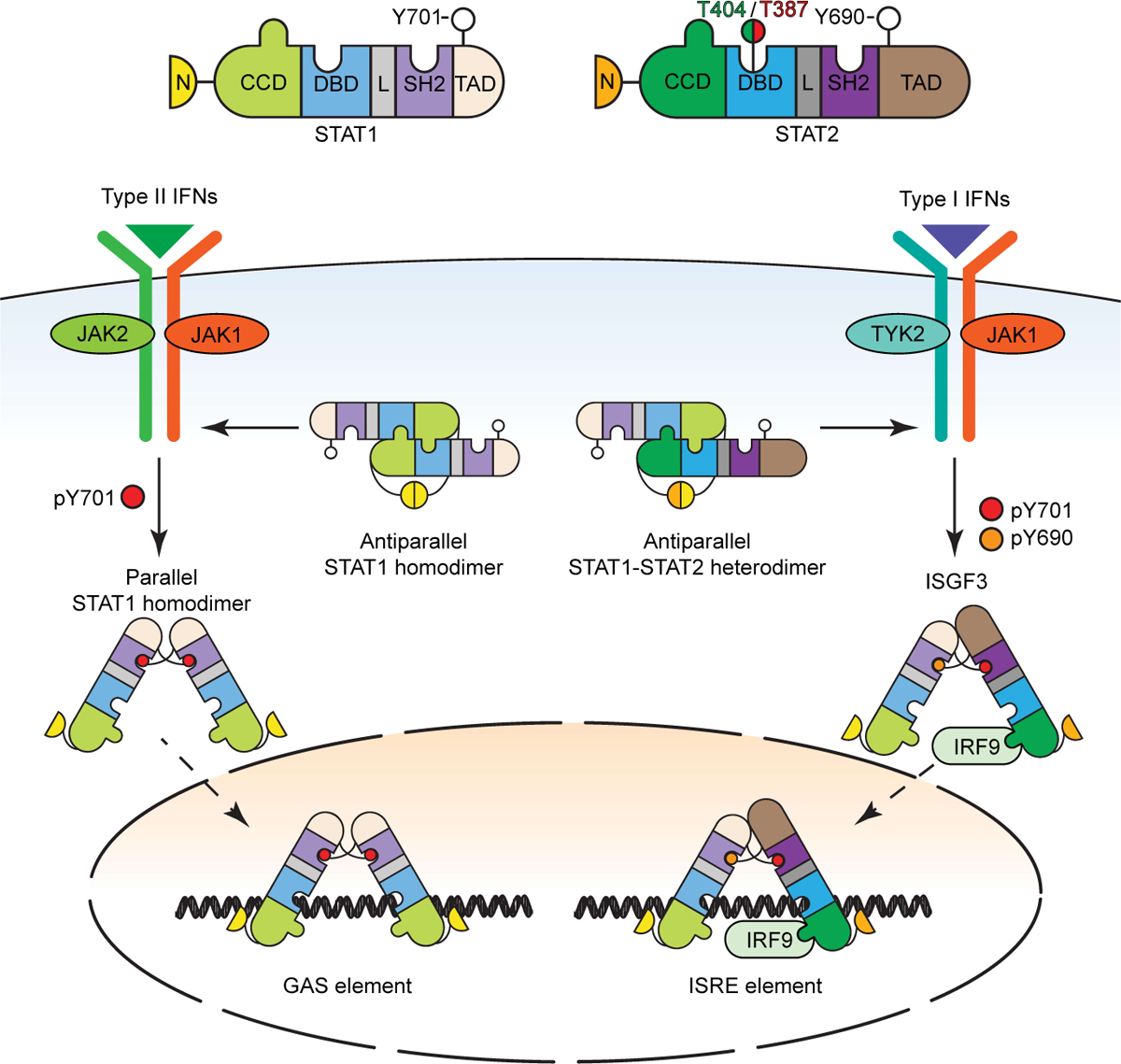
See Figure 4 for definitions of the STAT structural domains. Structural insights indicate that the STAT1 homodimer and the STAT1:STAT2 heterodimer each adapt two conformations, termed parallel and antiparallel. The parallel dimers are stabilized by interactions between the SH2 domains and the phosphorylated tyrosine residues, whereas the antiparallel dimers are stabilized by ND interactions. In addition to the tyrosine phosphorylations, two newly discovered phosphorylations of T387 and T404 of STAT2 regulate U-STAT1:U-STAT2 heterodimer stability. See the text for details.
Furthermore, functional Interferon-Stimulated Regulatory Elements (ISREs) that bind to U-STAT2:IRF9 are also present and functional in regions upstream of regulatory elements of a subset of NF-κB-driven genes, for example, IL6 (Nan et al., 2018). It is important to stress that high levels of U-STAT2 and IRF9 are needed to drive substantial gene expression, as seen in cancer (discussed later in Complex roles of JAKs and STATs in cancer). The U-STAT1:U-STAT2 dimer also acts pervasively in an IFN-I-independent manner to inhibit STAT1 function in multiple signaling pathways, such as IFN-γ and IL-27 (Ho et al., 2016; Wang et al., 2021). In this U-dimer, STAT2 binds to STAT1 through exceptionally strong interactions between the two N-termini. Since U-STAT2 is mainly located in the cytoplasm in unstimulated cells (Martinez-Moczygemba et al., 1997), it inhibits the phosphorylation and nuclear translocation of STAT1 in multiple signaling pathways (Wang et al., 2021).
The U-STAT1:U-STAT2 dimer not only suppresses multiple P-STAT1 homodimer functions but also inhibits ISGF3 formation. Although we know that robust stimulation with IFN-I drives the formation of P-STAT2, key molecular events that regulate U-STAT2 function are just now being revealed, including the recent discovery of two important regulatory phosphorylations of STAT2 on T387 and T404 (Figure 5). The phosphorylation of T387 favors the formation of U-STAT1:U-STAT2 heterodimers at the expense of tyrosine-phosphorylated heterodimers and ISGF3 (Wang et al., 2017b). On the other hand, T404 is phosphorylated by IKKi or TBK1, disrupting the very stable U-STAT1:U-STAT2 dimer, facilitating the response to IFN-I and dramatically increasing the affinity of ISGF3 affinity for ISREs. Phosphorylation of T404 also facilitates the binding of U-STAT2 to STING, inhibiting the ability of STING to drive the production of IFN-I (Wang et al, submitted). Further research will expand our knowledge of the mechanisms underlying these novel non-canonical functions of U-STAT2 and of the range of roles that U-STAT2 plays in biology.
Complex formation also affects nucleocytoplasmic shuttling of STATs (Reich and Liu, 2006). This occurs through nuclear pore complexes and is regulated by interactions with transporter proteins of the karyopherin-β family (i.e. importins and exportins). STAT nuclear trafficking is not always dependent on tyrosine phosphorylation, and the regulation of nuclear trafficking is diverse among different STATs. The dynamic redistribution of STAT1 and STAT2 between the cytoplasm and the nucleus is coordinate with their gain of ability to bind DNA. Nuclear trafficking of STAT1 homodimers is dependent on tyrosine phosphorylation where importin-α5 recognizes a conditional nuclear localization signal (NLS) in the P-STAT1 dimer. The constitutive NLS of IRF9 facilitates nuclear localization of the U-STAT2:IRF9 complex. Additionally, nuclear export of STAT2 is independent of its phosphorylation status and is mediated by a nuclear export signal (NES) in its carboxyl terminus. The STAT3 NLS is in its coiled-coil domain and nuclear localization of STAT3 is independent of tyrosine phosphorylation but dependent on recognition by importin-α3. The regulation of nuclear trafficking of STATs is complex and much remains to explore.
STATs and transcriptional regulation
Once STAT complexes arrive in the nucleus, they accomplish all the tasks associated with transcriptional regulation: engagement of chromatinized target genes, binding response elements, and recruitment of transcriptional co-activators that can recruit and activate RNA Pol II (Au-Yeung and Horvath, 2018).
STAT regulation of ISGs.
Induction of ISG transcription is typically fast, considering the necessity of a quick response to viral infection. This corresponds with an increased transcription rate, fast mRNA processing and degradation rates, and little or no evidence for post-transcriptional control (i.e. microRNA, translation) (Jovanovic et al., 2015). RNA polymerase II (RNA Pol II) recruitment to ISGs de novo is stimulated by IFN, but pause-release mechanisms are also used in ISG regulation (Patel et al., 2013), sometimes in association with a TBP-free TAFII130/GCN5 transcription complex (Paulson et al., 2002). Here, phosphorylation of its C-terminal domain by pTEFb via BRD4 results in the release of the RNA Pol II-associated repressors NELF and DSIF, enabling ISG mRNA elongation (Patel et al., 2013). Coordinately with RNA Pol II recruitment, STAT2 transactivation domain engages Mediator via MED14 and MED17 subunits (Lau et al., 2003) and enables STAT2 to activate RNA Pol II.
Additionally, after IFN treatment, there is an increase in chromatin accessibility at promoters and gene bodies at or near ISGF3 binding sites (Mostafavi et al., 2016) as well as recruitment of SWI/SNF nucleosome remodeling complex subunits. BRG1, an ATPase subunit of the SWI/SNF nucleosome remodeling complex (or mammalian BAF/pBAF complex), interacts with STAT2 to promote transcription. Both RVB1 and RVB2, components of SRCAP, TIP60, URI and INO80 chromatin remodeling complexes also associate with STAT2 (Gnatovskiy et al., 2013). Interference with RVB1 prevents Pol II recruitment to ISG promoters, but the exact role is unclear. Additionally, chromatin remodelers can influence histone acetylation, as knockdown of BAF47 leads to decreased histone H4 acetylation at ISG promoters (Cui et al., 2004). Likewise, acetylated chromatin can enhance bromodomain recruitment, bringing BRG1 to ISGF3-proximal histones.
H2A.Z nucleosomes play a regulatory role in ISG transcription, as shRNA interference with H2A.Z results in greater ISGF3 occupancy, increased ISG mRNA production, and created a more potent antiviral state leading to more robust inhibition of virus replication. Correspondingly, ISGF3 arrival induces H2A.Z loss from the promoter, and inhibition of either the HAT GCN5 or the bromodomain protein BRD2 disrupts both H2A.Z removal and ISG transcription. It is important to note that well known coactivators including CBP, HDACs, and RVB1 are dispensable for H2A.Z removal but strictly needed for ISG transcription. Along with H2A.Z remodeling, histone H3.3 is deposited in the gene body, and genes with this mark are transcriptionally sensitized due to prior activation.
Both histone acetylation and deacetylation activities are required for ISG transcription activation. Multiple control points contribute to acetylation-based regulation of interferon/cytokine signaling, as acetylation and deacetylation target the chromatin and transcriptional apparatus and many proteins beyond STATs themselves (Wieczorek et al., 2012). Multiple STATs associate with histone acetyltransferases (HAT), as STAT2 associates with CBP/p300 and GCN5 and p300 associates with STAT1, STAT3, STAT5 and STAT6 (Bhattacharya et al., 1996; Paulson et al., 1999). Interference with either GCN5 or CBP inhibits ISG transcription, but H2A.Z eviction from ISG promoters depends only on GCN5, not CBP, demonstrating specificity rather than redundancy in HAT function and reflecting the potential for multiple regulatory outcomes (Au-Yeung and Horvath, 2018). Likewise, ISG transcription is blocked by both interference with HDAC proteins and HDAC inhibitors (Chang et al., 2004; Nusinzon and Horvath, 2003), as RNA Pol II recruitment to ISG promoters is impaired by HDAC inhibitors. In addition, HDAC inhibition may create an acetylation sink for BRD4, restricting its availability for STAT co-activation (Marie et al., 2018). The importance of both BRD4 and HDACs to ISG regulation may enable cross-regulation due to reversible sequestration of BRD4 on hyperacetylated versus hypoacetylated chromatin.
STATs and other transcription factors.
Under physiological conditions, cells receive multiple concurrent signals that activate multiple signaling pathways. A classic example is toll-like receptor (TLR) plus a cytokine signal (such as IFN-γ) that activate the NF-κB and JAK-STAT pathway, respectively. Multiple observations have been made in the literature describing the interaction between STATs and other transcription factors, including regulation of transcription factor synthesis, regulation of transcription factor activity, and direct or indirect interactions within the genome that affect transcription (Farlik et al., 2010; Platanitis and Decker, 2018). Most notably, some key genes in an immune response require synergy between multiple pathways, namely NK cells require synergistic IL-18 (NF-κB) and IL-12 (STAT4) for maximal IFN-γ expression (Takeda et al., 1998).
Cooperativity and antagonism between STATs.
Whether it is from two different signals, or the same, different STATs can synergize or antagonize each other to affect transcriptional output. These cross-talks can occur by physical interaction, serine phosphorylation of the transactivation domain, cooperative DNA binding, and synthesis of specific signaling inhibitors. STAT1 and STAT3 represent clear examples whereby crosstalk can occur in either a synergistic or antagonistic manner. Synergy mediated by IL-2 and IL-12 leads to greater IFN-γ expression due to serine phosphorylation of STAT1 and STAT3 (Gollob et al., 1999). Antagonism between these STATs are commonly described between interferons and STAT3-activating cytokines, which have biological consequences as seen in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) or STAT1 gain-of-function (GOF) patients where an exaggerated STAT1 response leads to chronic mucocutaneous candidiasis (CMC), an immune response dependent on STAT3 (Break et al., 2021; Liu et al., 2011). In many cases, STAT1 and STAT3 are activated by many of the same cytokines (i.e. IL-6, IL-21, and IL-27). Therefore, the mechanisms underlying antagonizing effects of these transcription factors is more elusive. Efforts have been made to try to address this conundrum and in the case of IL-6 and IL-27, STAT3 is responsible for transcriptional output, whereas STAT1 is the driver of specificity (Hirahara et al., 2015). With IL-21 signaling, STAT1 and STAT3 have partially opposing roles in CD4+ T cells, even though both are critical for IL-21-mediated gene transcription (Wan et al., 2015).
STAT1 and STAT4 can also antagonize each other. For example, IL-12 and IL-18 antagonizes IFN-α signaling by reducing STAT1 transcript levels as well as type I IFN transcriptional output (Wiedemann et al., 2021). However, in response to type I IFNs both STAT1 and STAT4 are activated in NK and CD8 T cells (Nguyen et al., 2002); yet STAT1 antagonizes STAT4, specifically by impairing IFN-γ production and proliferation in NK cells and CD8 T cells, respectively. This effect is modified by alteration of levels of each STAT during viral infection. STAT4 levels are initially high in comparison to STAT1, but as STAT1 protein levels increase during infection, STAT1 antagonistic functions begin to take hold presumably to limit potential damaging responses.
Furthermore, crosstalk occurs between ISGF3 and GAF after IFN-I induction. When IFN-I signaling is impaired with STAT2 or IRF9 deficiency, induction of negative regulators such as USP18 is lost, resulting in a prolonged GAF transcriptional response (Gothe et al., 2022). In principle, the antagonistic actions of STATs can help explain specificity of cytokine signaling; however, this is an area that clearly needs further investigation to truly understand the logic and mechanisms.
Genomic views of cytokine action
Technological advances facilitated new discoveries through the investigation of cytokine and interferon action. The ability to comprehensively define the entire transcriptome induced by cytokines and interferons began with microarray technology (Aberger et al., 2001; Chatterjee-Kishore et al., 2000; Ramana et al., 2002) but was quickly supplanted with massively parallel sequencing platforms. These efforts revealed that cytokines and interferons induce thousands of genes, including the expected genes participating in host defense responses, but also genes encoding proteins that regulate cell cycle and metabolism. During this time, it became possible to comprehensively define STAT binding sites throughout the genome by using chromatin immunoprecipitation sequencing (ChIP-seq) (Robertson et al., 2007) archived in relevant databases (http://cistrome.org/db/#/). This work revealed tens of thousands of cytokine-induced genomic binding sites. At the time, the significance of this binding was unclear. That is, while much work had been devoted to enumerating conventional, protein-coding genes, we now know that much of the genome is transcribed in cell- and activation-specific manners, and that microRNAs and long noncoding RNAs (lncRNAs) exceed the number of conventional genes. Unsurprisingly, STATs have major effects on all these transcripts, including Ifngas-1 and others (Hu et al., 2013a; Witte and Muljo, 2014).
STAT-Regulated Enhancers.
Enhancers can be operationally defined as regions of DNA that in their genomic, cellular, and organismal context interact with proteins that augment the likelihood of transcription of one or more genes through cis-regulatory mechanisms. Along with STAT ChIP-seq, antibodies directed against chromatin modifications or the histone acetyltransferase p300 were used in concert with STAT-deficient activated cells to decipher the genomic impact of STATs. Newer methodologies for measuring genomic accessibility, including assessment of small numbers of cells or even single cells, enable us to map cells activated in vivo without extensive manipulation and expansion in vitro. This has further accelerated understanding the genomic impact of STATs.
It has become clear that the binding of STATs to the genome has multiple consequences. Diseases associated with a transcriptomic “interferon signature” (i.e. Interferonopathies) are associated with an interferon epigenomic signature that primes cells for subsequent responses (Barrat et al., 2019). Cytokines like IFN-γ and IL-4 can mutually inhibit epigenomic and transcriptomic alterations induced by each cytokine alone. Thus, STATs can mediate induction and repressive epigenomic and transcriptomic effects (Piccolo et al., 2017).
STATs can have broad impacts on active enhancers (Figure 6). They can influence proximal histone modifications and are not replaced by the action of lineage-defining transcription factors (LDTFs) (Vahedi et al., 2012). STATs can also participate in the formation of latent enhancers, regions that acquire accessibility via LDTFs but become active after cellular stimulation (Ostuni et al., 2013). On the other hand, STATs can drive accessibility in regions that lack LDTF-binding motifs. Though, LDTFs can be enriched in these regions by presumed non-canonical interactions and/or association with STATs (Sciume et al., 2020). That is, for unmarked (latent) enhancers, the canonical roles of LDTFs and STATs vis-à-vis accessibility can be inverted. This calls into question a “chicken and egg” scenario for the temporal primacy of LDTFs as “pioneer factors” over STATs, as is the case for STATs and T-bet in NK cells (Sciume et al., 2020). Consistent with the primacy of STATs over LDTFs in establishing enhancer recognition, JAK inhibition prevents histone marking and PU.1 recruitment (Ostuni et al., 2013). Thus, interdependence of LDTFs and STATs in both directions provides yet another layer of combinatorial variability contributing to specificity of gene programs.
Figure 6. STAT and chromatin organization.
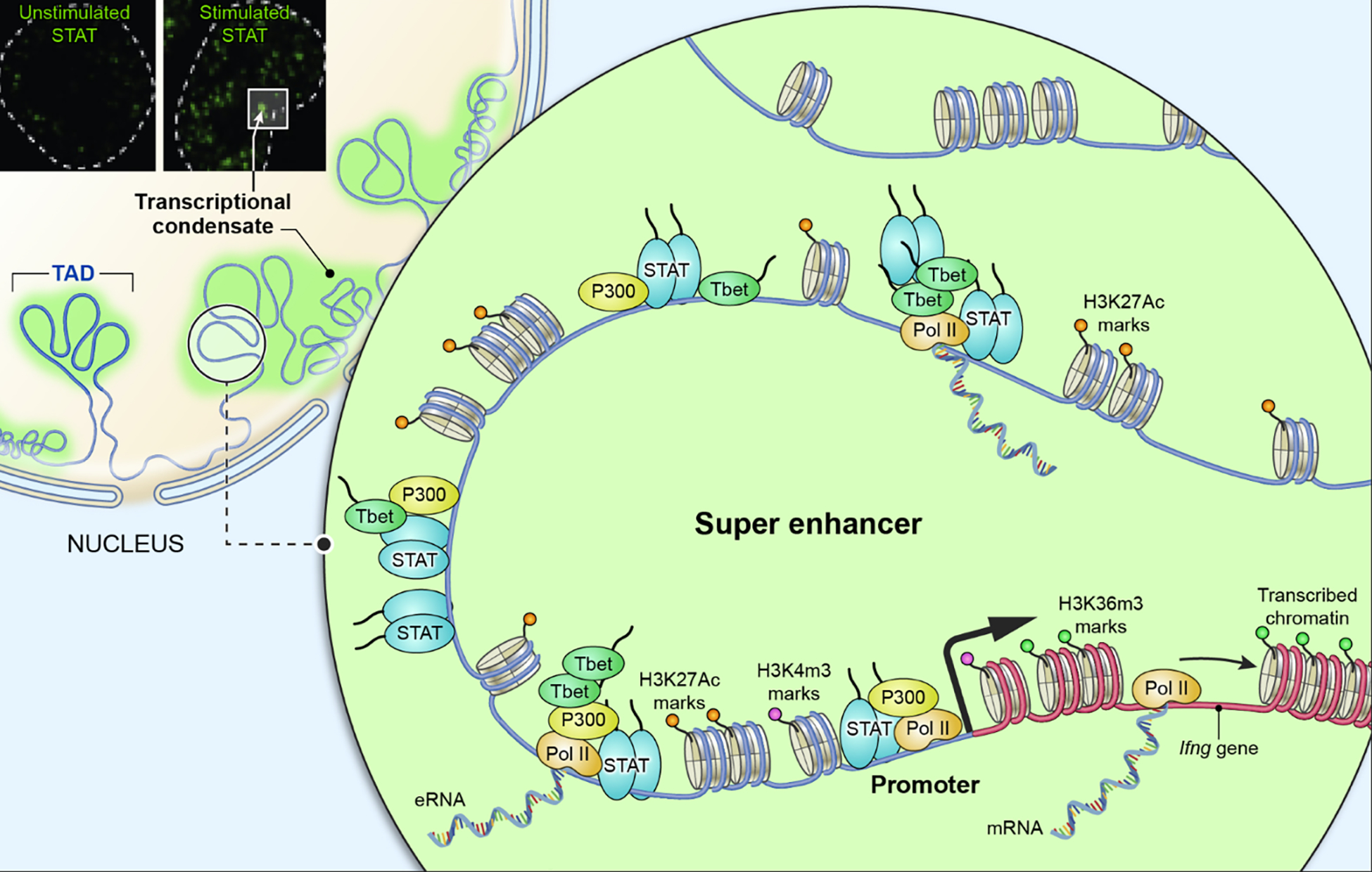
STATs bind to DNA at epigenetically marked promoters and enhancers of target genes to regulate transcription. Distinct histone epigenetic marks are observed at promoters (H3K4 trimethylation), enhancers (H3K27 acetylation) and pol II transcribed region (H3K36 trimethylation). As an example, the Ifng locus is depicted, which is regulated by a super enhancer composed by multiple tandem enhancer elements occupied by and enriched for STAT, T-bet, as well as p300, in response to cytokine stimulation. Confocal microscopy (upper left corner) documents signal dependent aggregation of STAT transcriptional nuclear condensates (Zamudio et al., 2019). Super enhancers, transcriptional condensates, and topologically associating domains (TADs) may all converge into a functional unit establishing a physical platform to regulate dynamic transcription of target genes.
Key genes controlling cell type-specific function tend to be regulated by clusters of enhancers densely occupied by transcription factors, coactivators, RNA polymerase II, and elongation factors (Vahedi et al., 2015; Whyte et al., 2013), a structure designated super- or stretch-enhancers (SE) (Figure 6). In B cells, the balance between STAT5 and other transcription factor network at SE is key and act as a molecular switch to govern proliferation, survival, and differentiation of progenitors (Katerndahl et al., 2017).
In summary, STATs can deliver their effects via multiple context dependent mechanisms—direct transcriptional induction/repression, epigenetic conditioning, or both. On a genome-wide scale, activated STATs contribute significantly to the modulation of epigenetic processes.
Mitochondrial localization of STATs
In addition to their prominent role in the cytoplasm and nucleus, a small fraction of STAT1, STAT3, and STAT5 (5–10% of the total) is found in the mitochondria where it functions independent of cytoplasmic signaling and nuclear gene activation. Mitochondrial STAT3 is studied the best, as it directly contributes to Ras-induced transformation by regulating respiration, redox homeostasis, and mitochondrial transcription (Carbognin et al., 2016; Garama et al., 2015). In Ras-transformed cells, ATP levels are decreased, and the activities of Complexes II and V of the electron transport chain are diminished in the absence of STAT3 (Gough et al., 2009). Tyrosine 705 phosphorylation, which is crucial for canonical nuclear functions of STAT3, is not needed to drive its mitochondrial functions but phosphorylation of STAT3 on S727 seems to be needed instead (Peron et al., 2021). Access of STATs to the mitochondria, as well as to other cellular organelles, including the endoplasmic reticulum (Avalle et al., 2019), and their potential organellar functions in cellular physiology remains an open and active research area.
In vivo functions of JAKs and STATs
As expected, gene targeting of mice provided great insights into actions of JAKs, STATs and related molecules; however, mutations and variants very quickly revealed relevance to human disease (Figure 2) Consequences of germline mutations in the JAK-STAT pathway vary widely in severity (Table I), but the salient clinical findings are summarized below.
Table I.
JAK STAT pathway genetics and in vivo functions in mice and humans
| Knockout phenotype | Mendelian Disorders | Somatic GOF mutations | GWAS links | |
|---|---|---|---|---|
| JAK1 | Perinatal lethality Impaired responses to multiple cytokines including type I and II IFNs, IL-2, IL-4, IL-10, IL-27 |
LOF: Mycobacterial disease, warts parasitic and fungal infection Progressive T lymphopenia, increased IgG and IgA levels, GOF: atopy, autoimmunity |
Acute lymphoblastic leukemia Acute myeloid leukemia |
JIA, MS, autoimmune thyroid disease, CRP levels, acute myeloid leukemia, lymphocyte and eosinophil counts, body mass and height |
| JAK2 | Embryonic lethal, absence of definitive hematopoiesis | Myeloproliferative neoplasms: Polycythemia vera, Essential thrombocythaemia Acute lymphoblastic leukemia Acute myeloid leukemia Chronic myelogenous leukemia |
SLE, allergy, eczema, AS, RA, T1DM, Psoriasis, IBD, cholesterol level, eosinophil basophil platelet erythrocyte count, body height | |
| JAK3 | Defective T and B cell development, Impaired responses to IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 immunodeficiency | LOF: T-B+NK- SCID | Myeloproliferative neoplasms Acute lymphoblastic leukemia Myelomonocytic leukaemia NK Lymphoma/Leukemia T cell Lymphoma/Leukemia |
|
| TYK2 | Bacterial and viral infection Impaired responses to type I interferon, IL-12, IL-23 and IL-10 | LOF: Mycobacterial disease and infections with other bacteria and viruses | SLE, RA, JIA, psoriasis, AS, IBD, MS, T1DM, T2DM, COVID-19, mycobacterial susceptibility, platelet and lymphocyte count, | |
| STAT1 | Bacterial and viral infection | LOF: Mycobacterial disease, viral infections GOF: Autoimmunity Mucocutaneous candidiasis, Invasive fungal infections, viral susceptibility |
T cell Lymphoma-Leukemia Other hematopoietic and solid organ cancers |
SLE, JIA, reticulocyte count, eosinophil and neutrophil count, brain measurement, body height |
| STAT2 | Impaired type I IFN responses Viral susceptibility |
LOF: Severe viral infections mitochondrial abnormalities, | Hematopoietic and solid organ cancers | Psoriasis, psoriatic arthritis, diabetic retinopathy, lymphocyte count, height |
| STAT3 | Impaired placental development, defective signaling by multiple cytokines | LOF: Hyperimmunoglobulin E recurrent infection syndrome GOF: Multisystem autoimmune disease |
NK Cell Lymphoma-Leukemia T cell Lymphoma -Leukemia B cell lymphoma Myelodysplastic syndrome Solid organ tumors (Head and neck, breast, skin, gastrointestinal, neural cancers) | eczema, IBD, MS, SLE, T2DM, Mean corpuscular volume, CRP, blood pressure, mosquito bite reaction, |
| STAT4 | Impaired IL-12 and IL-23 signaling, and type 1 immune responses | LOF: fungal disease | Hematopoietic and solid organ cancers | SLE, RA, Sjogren's syndrome |
| STAT5A | Defective mammary gland development and impaired GM-CSF signaling | Leukemia Lymphoma Solid organ tumors (Head and neck, breast, skin, gastrointestinal, neural, prostate cancers) |
Asthma, cholesterol, bone density, obesity | |
| STAT5B | Impaired growth, liver gene expression, innate lymphocyte homeostasis | Growth hormone insensitivity, immunodeficiency, autoimmunity atopy | Acute and chronic lymphocytic leukemia Lymphoma Large granular lymphocytic leukemia Solid organ tumors (Head and neck, breast, skin, gastrointestinal, neural, prostate cancers) Atopy, autoimmunity, eosinophilia |
Asthma, allergy, cholesterol level, neutrophil, eosinophil, and lymphocyte count, bone density, blood pressure, obesity |
| STAT6 | Impaired IL-4 and IL-13 signaling, and type 2 immune response | GOF: treatment resistant allergic disease | B Cell Lymphoma Follicular lymphoma Other hematopoietic and solid organ cancers |
Asthma, allergy, eczema,eosinophil count, IgE levels, mosquito bite size, |
AS- ankylosing spondyloarthritis; CRP-C-reactive protein, GOF – gain of function; IBD – inflammatory bowel disease, JIA – juvenile idiopathic arthritis; LOF – loss of function; MS – multiple sclerosis; RA- rheumatoid arthritis; SCID – severe combined immunodeficiency; SLE-systemic lupus erythematosus, T1DM – type 1 diabetes mellitus; T2DM-type 2 diabetes mellitus; (https://www.ebi.ac.uk/gwas)
JAK mutations.
The critical role of JAKs was first shown in humans with severe combined immunodeficiency (SCID). Mutations of IL2RG, which encodes gc, underlies X-linked SCID. Because JAK3 associates with γc, JAK3 autosomal recessive loss-of-functions (LOF) mutations were also sought and found to be a cause of SCID (Macchi et al., 1995; Russell et al., 1995) and confirmed in mouse models (Park et al., 1995; Thomis et al., 1995). Notably, these patients and mice are otherwise normal, indicative of JAK3’s very specific function compared to other JAKs.
TYK2-deficient mice are viable and have selective impairment of cytokine responses that include loss of IFN and IL-12/23 family cytokine responses and thus increases susceptibility to infections. TYK2 deficiency in humans is associated with bacterial and viral infections, as well as allergic disease (Minegishi et al., 2006). A LOF variant of TYK2 in humans (P1104A) is associated with reduced incidence of autoimmune disease and increased risk of tuberculosis.
JAK2 has essential roles for many cytokines including hematopoietic growth factors. JAK2-deficient mice die in utero with bone marrow failure (Neubauer et al., 1998; Parganas et al., 1998). In a classic series of papers, JAK2 GOF mutations were identified in patients with myeloproliferative neoplasms (MPN), including polycythemia vera, primary myelofibrosis and essential thrombocytopenia (Kralovics et al., 2005; Zhang et al., 2012). The most common mutation is V617F located in the JAK2 PK domain, which results in constitutive activity and cytokine-independent proliferation (Hammaren et al., 2015); other mutations also associated with MPN include R683G, R683S, and R683T. The first evidence of JAK GOF mutations were revealed by the Drosophila Tumorous-lethal (Tum-l) mutation in the kinase-like domain of the hopscotch locus, which resulted in malignant neoplasia of blood cells (Hanratty and Dearolf, 1993; Harrison et al., 1995; Luo et al., 1995). The structure of a full-length JAK now provides detailed insights into the mechanism by which PK mutations lead to cytokine-independent signaling, with GOF mutations residing within the core of the PK interdimer interface, enhancing packing complementarity to facilitate ligand-independent activation (Glassman et al., 2022).
In keeping with the many cytokines that rely on this kinase, JAK1-deficient mice die perinatally with impaired organogenesis and dwarfism (Rodig et al., 1998). In humans, a JAK1 LOF mutation was associated with recurrent mycobacterial infection, developmental delay, and early-onset metastatic bladder carcinoma (Eletto et al., 2016). JAK1 GOF mutations lead to an autoinflammatory-related disease and hyperimmunoglobulin E syndrome (HIES) (Del Bel et al., 2017; Gruber et al., 2020). Genome-wide association studies (GWAS) have revealed polymorphisms of JAK1 in juvenile idiopathic arthritis, multiple sclerosis, autoimmune thyroid disease, C-reactive protein levels, lymphocyte and eosinophil counts.
STAT mutations.
Although, STAT1 is activated by many cytokines, the predominant phenotype of STAT1 knockout mice and mutations in humans is susceptibility to intracellular pathogens such as bacteria and viruses. But in addition, STAT1 is important for hematopoietic precursor function (Li et al., 2022). STAT1-GOF patients have autoimmunity and increased susceptibility to fungal and viral infection; however, the molecular mechanisms underlying STAT-GOF mutations is very incomplete. GOF mutations may favor STAT1’s parallel phosphorylated state, however, these observations have yet to be confirmed by detailed structural analysis. Notably, these “accidents of nature” provide substantial opportunities to link structure and function.
Germline STAT2 LOF mutations in mice and humans confer susceptibility to viral infections. However, some STAT2 mutations do not affect phosphorylation or activation; rather, they prevent STAT2 from recruiting USP18 to the IFN receptor subunit, IFNAR2. These STAT2 mutations can be associated with severe interferonopathy due to defective negative regulation of type I interferon signaling (Gothe et al., 2022).
Germline deletion of STAT3 is lethal in mice because of its importance in placental function. Conditional knockouts have revealed broad roles of STAT3 in multiple tissues (Levy and Lee, 2002), with important roles in innate and adaptive immunity, stem cells, wound healing, tissue repair, and cancer. Dominant negative LOF mutations of STAT3 underlie hyperimmunoglobulin E (Job’s) syndrome, a primary immunodeficiency syndrome also associated with craniofacial, skeletal and vascular abnormalities (Holland et al., 2007; Minegishi et al., 2007). STAT3 GWAS variants are linked with inflammatory bowel disease, multiple sclerosis, type 2 diabetes mellitus, and myocardial infarction and patients with germline STAT3 GOF mutations exhibit lymphoproliferation and poly-autoimmunity (Milner et al., 2015).
STAT4 knockout mice have impaired IFN-γ production and increased susceptibility to T. gondii and other pathogens that rely on a type 1/IFN-γ response. STAT4 LOF mutations in humans are associated with susceptibility to fungal infections and viral infections. STAT4 variants are associated with lupus, rheumatoid arthritis (RA), and Sjogren’s syndrome with the presence of the risk allele being associated with more severe disease (Hasni et al., 2021; Remmers et al., 2007). Moreover, a STAT4 coding variant (Glu128Val) doubles the risk of RA (Saevarsdottir et al., 2022).
Adjacent in the genome, STAT5A and STAT5B are >90% identical. Stat5a knockout mice have loss of prolactin-mediated mammary gland development, whereas Stat5b-deficient mice have impaired sexually dimorphic growth. Stat5a/b double-deficient mice die within a few weeks of birth, are infertile and have defective mammary gland development, as well as hypocellular bone marrow. Homozygous STAT5B LOF mutations in humans result in growth failure, facial dysmorphism, and autoimmunity. Activating somatic mutations in hematopoietic progenitors can also cause nonclonal hypereosinophilia, eczema, urticaria, and diarrhea. STAT5A variants are linked with obesity, whereas STAT5B GWAS reveal links to lymphocyte and neutrophil counts, asthma and allergy, and bone density.
Consistent with the role of STAT6 in IL-4 and IL-13 signaling, STAT6-deficient mice fail to mount type 2 immune responses and impaired parasite elimination and reduced allergic disease. Germline STAT6 GOF mutations are associated with severe, treatment resistant allergic disease with eosinophilia (Sharma et al., 2022). GWAS links with STAT6 include allergy, asthma, and eosinophilia.
Mutations in negative feedback regulators.
Homozygous deficiency of SOCS1 in mice is lethal with hypersensitivity to IFN-γ, and SOCS1-deficient humans exhibit early onset autoimmune and allergic disease (Hadjadj et al., 2020). A major consequence of USP18 and ISG15 deficiency in humans is severe, systemic interferonopathy (Alsohime et al., 2020; Martin-Fernandez et al., 2020; Martin-Fernandez et al., 2022; Meuwissen et al., 2016; Speer et al., 2016; Zhang et al., 2015).
Complex roles of JAKs and STATs in cancer
Constitutive activation of JAKs and STATs in cancer was quickly recognized shortly after the discovery of the pathway, particularly in the setting of viral or oncogene-mediated transformation (Meydan et al., 1996; Migone et al., 1995). Subsequently, there are many examples of overexpression and hyperactivation of JAKs and STATs associated with cancer. However, cancers evade immune cell killing, abrogating IFN-γ signaling by inactivating JAK1 mutations (Zaretsky et al., 2016) and altering immune cell function in the tumor microenvironment. Thus, the roles of cytokines and the JAK-STAT pathway in cancer and the tumor microenvironment is anything but simple – and a particular challenge to review in just a few paragraphs.
The simplest scenarios relate to GOF mutations. A Tel-JAK2 fusion protein associated with leukemia was identified early on (Lacronique et al., 1997), followed by the identification of other JAK2 fusion partners (e.g. PCM1, SEC1A, PAX5). Additionally, generation of a constitutively active version of STAT3 with two cysteine substitutions in the SH2 domain that causes spontaneous dimerization, binding to DNA, and transcriptional activation results in cellular transformation (Bromberg et al., 1999). This remarkable insight anticipated the common mutations in the SH2 domains of STAT3 and STAT5 often seen in hematopoietic malignancies (Tate et al., 2019); (https://cancer.sanger.ac.uk/cosmic). One common mutation, STAT5 N642H, is sufficient to drive leukemogenesis in mice (Kollmann et al., 2021).
With improvements in sequencing technology, a more comprehensive understanding of JAK/STAT germline and somatic GOF mutations or amplifications has been achieved (Tate et al., 2019) (https://cancer.sanger.ac.uk/cosmic). In fact, mutations of all JAKs and STATs are common but are seen predominantly in hematopoietic/lymphoid malignancies. For example, STAT1 is a tumor promoter in leukemia development (Kovacic et al., 2006). In B cell acute lymphocytic leukemia (ALL), STAT5- and ERK-activating mutations are commonly found but, interestingly, the two pathways are antagonistic (Chan et al., 2020; Katerndahl et al., 2017). STAT5 antagonizes NF-κB and IKAROS by opposing regulation of shared target genes; that is, super-enhancer enrichment for STAT5 binding is associated with an opposing network of transcription factors, including PAX5, EBF1, PU.1, IRF4 and IKAROS. Additionally, STAT5B-N642H is a frequently occurring driver mutation that promotes aggressive T cell leukemia/lymphoma, by which this mutation causes a structural conformation yielding a STAT5B that is resistant to dephosphorylation (de Araujo et al., 2019). Moreover, patients with a high ratio of active STAT5 to NF-κB or IKAROS had more-aggressive disease, but deletion of divergent pathway components also accelerates leukemogenesis – a more nuanced view of transformation.
While mutations of JAKs and STATs occur in nonhematopoietic tumors, including tumors affecting breast, lung, and large intestine, this is relatively uncommon; nonetheless, this does not mean that the pathway is irrelevant for solid tumors. Beyond mutations, activation of STAT3 and STAT5 is very frequent in many human cancers, both solid and liquid, and overexpression of STATs are associated with poor prognosis. STATs, especially STAT3 and STAT5, can directly regulate genes that promote proliferation (cell cycle genes like MYC) and oppose cell death (BCL2, BCLXL, MCL1, BIRC5). Additionally, STAT3 and STAT5 can promote stem cell-like characteristics, metastatic potential, and alter metabolism by canonical and non-canonical mechanisms. This includes lipid metabolism, which promotes tumor growth, chemoresistance and alter immune cell function (Zhang et al., 2020). Beyond activation of tyrosine phosphorylation, the action of STAT3 can also be mediated by serine phosphorylation. In a lung adenocarcinoma model, Ras dependent STAT3 serine phosphorylation is an important driver (Alhayyani et al., 2022).
In addition to cell intrinsic actions (i.e. role of JAKs and STATs in tumor cells themselves), STAT3 in particular has a role in inflammation, wound repair, and fibrosis. The role of non-malignant cells in the tumor microenvironment and stromal cancer-associated fibroblasts can drive development and progression of tumors by alterations in extracellular matrix production, angiogenesis, and metabolic milieu.
Importantly, IFNs have complex roles in cancer. A recent review summarizes evidence on how cancer cells control their own synthesis of IFN-I and how these cells control responses to the IFN-I that is present in the tumor microenvironment (Cheon et al, under review). In addition, the importance of IFN-I as a regulator of the immune system in cancer has been well reviewed by others recently (Snell et al., 2017; Yu et al., 2022). Namely, cancer cells minimize the pro-apoptotic and anti-growth activities of high concentrations of IFN-I and maximize the pro-tumor effects of low concentrations of this important cytokine. In many tumors, STAT1 and STAT2 lacking tyrosine phosphorylation (U-STATs) and IRF9 are expressed constitutively at high levels, forming U-ISGF3, which drives the expression of most ISGs, some of which encode important pro-tumor proteins. The levels of U-ISGF3 are low in normal cells but are increased by IFN-I (Cheon et al., 2021; Cheon et al., 2013). In many cancers, high levels of U-ISGF3 correlate with resistance to DNA damage by inducing the expression of “Interferon-Related DNA Damage resistance gene Signature”, or IRDS (Weichselbaum et al., 2008). In lung cancer cells, high levels of U-STAT2 promote an aggressive phenotype and confer resistance to cisplatin-induced cell death (Nan et al., 2018).
PD-L1, often expressed on the surfaces of tumor cells and well recognized for its importance in inactivating cytotoxic T cells, also has important tumor cell-intrinsic activities, including dampening acute responses to cytotoxic high levels of IFN-I and sustaining the expression of the low levels of IFN-I that benefit tumors (Cheon et al., 2021).
Fortunately, many strategies are being employed to optimize immuno-oncologic therapies, but in this short discussion, it should be very clear that the roles of cytokines and the JAK/STAT pathway in cancer are anything but simple.
Therapeutic targeting of the JAK/STAT pathway
JAK inhibitors.
Unsurprisingly, the discovery of JAKs and strong genetic evidence demonstrating their indispensable role provoked consideration of JAKs as therapeutic targets (Russell et al., 1995). Ten jakinibs are now approved to treat a wide, and still expanding, variety of disorders driven by both innate and adaptive immune mechanisms (Table II).
Table II.
Approved JAK inhibitors
| Indication | Drug |
|---|---|
| Rheumatoid arthritis | Tofacitinib, Baricitinib, Upadacitinib, Filgotinib, Peficitinib |
| Psoriatic arthritis | Tofacitinib, Upadacitinib |
| Juvenile arthritis (> 2 yrs) | Tofacitinib |
| Ankylosing spondyloarthritis | Tofacitinib, Upadacitinib |
| Ulcerative Colitis | Tofacitinib, Filgotinib, |
| Atopic Dermatitis | Baricitinib, Upadacitinib, Abrocitinib, Delgocitinib (topical), Ruxolitinib (topical), Oclacitinib (dogs) |
| Alopecia Areata | Baricitinib |
| Graft Versus Host Disease | Ruxolitinib |
| Myeloproliferative neoplasms | Ruxolitinib, Fedratinib, Pacritinib |
| Covid-19 | Baricitinib |
| Vitiligo | Ruxolitinib (topical) |
| Plaque Psoriasis | Deucravacitinib |
Tofacitinib was the first jakinib used in vivo using a model of allograft rejection (Changelian et al., 2003). However, ruxolitinib, a JAK1/JAK2 inhibitor, was the first jakinib to receive approval for treatment of MPN based on the discovery of JAK2 GOF mutations. Fedratinib and pacritinib are also approved for MPN. Ruxolitinib is also approved for acute and chronic graft-versus-host disease and topical ruxolitinib is approved for atopic dermatitis and vitiligo. Tofacitinib was the first jakinib approved for immune-mediated disease, including multiple forms of arthritis and inflammatory bowel disease. Baricitinib is approved for the treatment of RA, alopecia areata and COVID-19. Several other jakinibs have now been approved for other forms of immune-mediated arthritis, allergic disorders, and inflammatory bowel disease. Oclacitinib is approved to treat allergic dermatitis in dogs. Currently, numerous clinical trials are ongoing for systemic lupus erythematosus, Sjogren syndrome, sarcoidosis, psoriasis, hidradenitis suppurativa, asthma and malignancies.
Most of the adverse events observed so far can be explained by the known mechanism of action of specific cytokines with infections, anemia, and cancer, and the safety profile is largely comparable to biologics. Additional adverse interactions include increased risk of thromboembolic events, alteration in lipids, and cardiovascular complications.
Selective JAK inhibitors were developed with the motivation to interfere with the function of fewer cytokines, increase specificity, and ideally fewer adverse effects. These include JAK1 inhibitors (upadacitinib, filgotinib, and abrocitinib), TYK2 inhibitors (ropsacitinib/PF-06826647 and NDI-031407 (Gracey et al., 2020)) and deucravacitinib, an allosteric JAK2 inhibitor that targets the PK domain (Burke et al., 2019). Deucravacitinib is approved for that treatment of plaque psoriasis. An allosteric JAK1 inhibitor that targets the PK domain has also been generated (https://doi.org/10.1038/s41589-022-01098-0). In addition, topical and inhaled jakinibs are being generated to reduce side effects associated with systemic administration. Targeted protein degradation is also now an option for generation of kinase inhibitors including JAK1 (Kavanagh et al., 2022).
STAT inhibitors.
Various strategies have been employed to develop STAT inhibitors, including interfering with SH2 mediated dimerization, DNA binding, or transactivation and inducing STAT degradation. Multiple inhibitors are in clinical trials, but no agents have been approved at this time.
SOCS mimetics.
SOCS peptidomimetics have shown efficacy in immune and tumor preclinical models (Ahmed et al., 2015; La Manna et al., 2018).
Challenges and opportunities ahead
We hope that our summary has convinced the reader that the discovery of the JAK-STAT pathway 30 years ago brought a wealth of information that directly impacts human health – including treatment of COVID-19. Yet, it would be misleading to not consider the challenges ahead – particularly since the JAK-STAT pathway is a tractable tool that provides many opportunities for future investigation. In the brief section below, we reflect on a few topics, recognizing that we are just touching the tip of the iceberg – the reader may well come to different conclusions regarding priorities going forward.
Structural and cell biology JAKs, STATs and cytokine signaling.
The structural discoveries pertaining to JAK1 and STATs are illuminating but equally striking is the paucity of structural information regarding other JAKs complexed with cytokine receptors and other STATs. With the advances in imaging modalities, we clearly have a great deal to learn. Using more sophisticated tools to analyze early events in cytokine signaling, following STAT activation from membrane to the nucleus and regulation of transcription is likely to be revealing, as the canonical model of STAT phosphorylation inducing dimerization that activates nuclear translocation does not take into consideration the elaborate dynamic movements of STATs through nuclear pores in both the cytokine-activated and steady states. STAT protein associations with diverse organelles is yet to be fully appreciated functionally and physically. Capitalizing upon the many mutations in JAKs and STATs identified in human immune and malignant disorders and deciphering their mechanisms of action is likely to a useful strategy.
Towards a more complete understanding of STAT-mediated chromatin modifications and transcriptional regulation.
Dynamic modifications of complex enhancer structure, enhancer transcription, lncRNAs, histone modifications and chromatin looping and interactions in topologically associating domains (TADs) afford many opportunities for precise tissue-specific, temporal and dynamic regulation of gene expression (Figure 6). The impact of cytokines and STATs on these events is becoming clearer (Kanno et al., 2012; Platanitis et al., 2022). Furthermore, while STATs are prime examples of signal-dependent transcription factors, the documented actions of nonphosphorylated STAT1 illustrate its constitutive actions beyond a role as a signal-dependent transcription factor. How U-STAT1 modifies chromatin and the three-dimensional genome will be of interest and the extent to which other STATs have this capacity needs to be explored.
An alternative model that explains the function of complex enhancers beyond chromatin looping and the proximity of enhancers and promoters is the phenomenon termed liquid-liquid phase separation (LLPS) (Banani et al., 2017; Hnisz et al., 2017; Hyman et al., 2014). Occupancy of transcription factors at defined regulatory regions with concomitant recruitment of coactivators and transcriptional machinery complexes with intrinsically disordered regions may trigger demixing and condensate formation. STATs appear to participate in the process (Figure 6) and are endowed with characteristics conducive to demixing: high valency (ability to heterodimerize; phosphorylation) and nucleation perhaps initiated by high STATs density at enhancers (Zamudio et al., 2019). Detailed understanding of the structural and functional contributions of STATs to biophysical aspects of phase separation will be important, again taking advantage of germline and somatic GOF STAT mutations. Parenthetically, an area that has not been pursued in detail is the role of nuclear JAK2 and its ability to phosphorylate histones (Dawson et al., 2009; Dawson et al., 2011).
Achieving cell, tissue, and stimulus-specific programs.
Perhaps the greatest challenge is to understand how signals generated by a vast array of cytokines are interpreted in cells that use a limited cassette of JAKs and STATs. There is no simple response to this challenge at this time, but we will offer some brief reflections that may be considered. Deciphering how cell signaling provides specific instructions is not a problem unique to cytokines; rather, given its simplicity, the JAK-STAT pathway may have many attributes that can resolve this fundamental biological conundrum.
A first consideration is specificity and regulation of expression for signaling components. JAKs and STATs are broadly expressed, but some have more selectivity, e.g., JAK3, TYK2, and STAT4 are preferentially in immune cells. STATs also directly and dynamically regulate their own expression, STAT1 being a particularly good example. Thus, in the course of infection, STAT4 initially mediates type I IFN signaling but later STAT1 predominates (Nguyen et al., 2002). Along with cytokine receptors, the dynamic expression of JAKs, STATs, SOCS proteins and other downstream elements need to be considered in analysis of output. In this respect, both the strength and quality of the signaling can evolve during development or acutely during infections.
Second, STAT activation dynamics have been studied, but most cytokines activate more than one STAT family member with different amplitudes of activation, rates of phosphorylation and dephosphorylation, nuclear translocation and DNA binding. These measures are often not comprehensively and rigorously determined over time and improved techniques for quantitatively assessing these changes are needed. Notably, engineered cytokines and cytokine antagonists have been generated that alter these variables and are likely to be useful tools (Ren et al., 2022; Spangler et al., 2015; Yen et al., 2022). Formation of STAT heterodimers has been suggested as another way to explain specificity but is often overlooked (Delgoffe and Vignali, 2013). Notably, while genomic analysis of motifs bound by STATs always reveals the canonical palindromic motifs, a substantial proportion of STAT-bound regions depart from the classical motif. How STATs are recruited in the absence of a cognate DNA binding motif remains to be elucidated. What underlies binding to noncanonical sites?
Third, as mentioned, many stimulus-specific STAT post-translationally modified states can occur that influence intracellular trafficking, DNA binding and transcriptional potential; however, these modifications are typically not comprehensively and dynamically measured (Myers and Gottschalk, 2022). Almost no information has been provided regarding post-translational modifications of JAKs in response to different stimuli over time. This is an important topic that needs further investigation.
Finally, the diversity of cytokine receptors allows considerable variability in their engagement of pathways as discussed above (e.g., MAPKs, PI3K, etc.). During developmental, inflammatory, or other events, cells are not impacted by a single cytokine; multiple exogenous, endogenous and inducible signals participate in the outcome. Multiple SDTFs, LDTFs, and inducible TFs beyond STATs are also deployed. In this respect, STATs coordinate with other factors cooperating and antagonist pathways. Added to this is the complexity of enhancers and opportunities for dense binding of multiple factors.
Although complex, context-specific regulation is needed to achieve specificity, an obvious solution to achieving specific interpretations of signals relates to combinatorial logic. While this may seem like a daunting proposal, the generation of multiomic tools now facilitates comprehensive measurement of changes over time, with minimal manipulation of single cells responding to physiologic and relevant pathologic challenges in vivo. In short, there’s lots to do – but the tools are so much better now. Based on precedent, more sophisticated knowledge of JAKs and STATs will unquestionably offer many opportunities for new drugs with greater specificity. If you’re not already working on the JAK-STAT pathway presently, you should be thinking about doing so.
Acknowledgements
Interferons were first described over seventy years ago, leading to an extraordinary body of literature. Unfortunately, space limitations preclude including many outstanding citations; our apologies to all whose work could not be cited. This work was supported by the Intramural Research Programs of NIAMS. R.L.P. was supported by a Postdoctoral Research Associate (PRAT) fellowship from the National Institute of General Medical Sciences (NIGMS), award number 1Fi2GM137942-01. Additionally, Y.W. was supported by National Heart, Lung, and Blood Institute (NHLBI), award number R56 HL160639 and G.R.S was supported by National Institute of Allergy and Infectious Diseases (NIAID), award number R01 AI153085 and National Cancer Institute (NCI), award number P01CA062220.
Footnotes
Declaration of Interests
NIH holds a US patent related to JAK inhibitors and Dr. O’Shea receives royalty income.
References
- Aberger F, Costa-Pereira AP, Schlaak JF, Williams TM, O’Shaughnessy RF, Hollaus G, Kerr IM, and Frischauf AM (2001). Analysis of gene expression using high-density and IFN-gamma-specific low-density cDNA arrays. Genomics 77, 50–57. [DOI] [PubMed] [Google Scholar]
- Ahmed CM, Larkin J 3rd, and Johnson HM (2015). SOCS1 Mimetics and Antagonists: A Complementary Approach to Positive and Negative Regulation of Immune Function. Front Immunol 6, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, and Kishimoto T (1994). Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71. [DOI] [PubMed] [Google Scholar]
- Alhayyani S, McLeod L, West AC, Balic JJ, Hodges C, Yu L, Smith JA, Prodanovic Z, Bozinovski S, Kumar B, et al. (2022). Oncogenic dependency on STAT3 serine phosphorylation in KRAS mutant lung cancer. Oncogene 41, 809–823. [DOI] [PubMed] [Google Scholar]
- Alsohime F, Martin-Fernandez M, Temsah MH, Alabdulhafid M, Le Voyer T, Alghamdi M, Qiu X, Alotaibi N, Alkahtani A, Buta S, et al. (2020). JAK Inhibitor Therapy in a Child with Inherited USP18 Deficiency. N Engl J Med 382, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashour J, Laurent-Rolle M, Shi PY, and Garcia-Sastre A (2009). NS5 of dengue virus mediates STAT2 binding and degradation. J Virol 83, 5408–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au-Yeung N, and Horvath CM (2018). Transcriptional and chromatin regulation in interferon and innate antiviral gene expression. Cytokine Growth Factor Rev 44, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalle L, Camporeale A, Morciano G, Caroccia N, Ghetti E, Orecchia V, Viavattene D, Giorgi C, Pinton P, and Poli V (2019). STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca(2+) fluxes and apoptotic responses. Cell Death Differ 26, 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, Crow MK, and Ivashkiv LB (2019). Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol 20, 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begitt A, Droescher M, Meyer T, Schmid CD, Baker M, Antunes F, Knobeloch KP, Owen MR, Naumann R, Decker T, and Vinkemeier U (2014). STAT1-cooperative DNA binding distinguishes type 1 from type 2 interferon signaling. Nat Immunol 15, 168–176. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, and Livingston DM (1996). Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature 383, 344–347. [DOI] [PubMed] [Google Scholar]
- Break TJ, Oikonomou V, Dutzan N, Desai JV, Swidergall M, Freiwald T, Chauss D, Harrison OJ, Alejo J, Williams DW, et al. (2021). Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, and Darnell JE Jr. (1999). Stat3 as an oncogene. Cell 98, 295–303. [DOI] [PubMed] [Google Scholar]
- Burke JR, Cheng L, Gillooly KM, Strnad J, Zupa-Fernandez A, Catlett IM, Zhang Y, Heimrich EM, McIntyre KW, Cunningham MD, et al. (2019). Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med 11. [DOI] [PubMed] [Google Scholar]
- Carbognin E, Betto RM, Soriano ME, Smith AG, and Martello G (2016). Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. EMBO J 35, 618–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan LN, Murakami MA, Robinson ME, Caeser R, Sadras T, Lee J, Cosgun KN, Kume K, Khairnar V, Xiao G, et al. (2020). Signalling input from divergent pathways subverts B cell transformation. Nature 583, 845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marie I, and Levy DE (2004). Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc Natl Acad Sci U S A 101, 9578–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, Rizzuti BJ, Sawyer PS, Perry BD, Brissette WH, et al. (2003). Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 302, 875–878. [DOI] [PubMed] [Google Scholar]
- Chatterjee-Kishore M, Wright KL, Ting JP, and Stark GR (2000). How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J 19, 4111–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bhandari R, Vinkemeier U, Van Den Akker F, Darnell JE Jr., and Kuriyan J (2003). A reinterpretation of the dimerization interface of the N-terminal domains of STATs. Protein Sci 12, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE Jr., and Kuriyan J (1998). Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell 93, 827–839. [DOI] [PubMed] [Google Scholar]
- Cheon H, Holvey-Bates EG, McGrail DJ, and Stark GR (2021). PD-L1 sustains chronic, cancer cell-intrinsic responses to type I interferon, enhancing resistance to DNA damage. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, Rice CM, Jackson MW, Junk DJ, and Stark GR (2013). IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J 32, 2751–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui K, Tailor P, Liu H, Chen X, Ozato K, and Zhao K (2004). The chromatin-remodeling BAF complex mediates cellular antiviral activities by promoter priming. Mol Cell Biol 24, 4476–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Zhang L, Luo J, Rajasekaran A, Hazra S, Cacalano N, and Dubinett SM (2007). Unphosphorylated STAT6 contributes to constitutive cyclooxygenase-2 expression in human non-small cell lung cancer. Oncogene 26, 4253–4260. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, and Kouzarides T (2009). JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461, 819–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Bannister AJ, Saunders L, Wahab OA, Liu F, Nimer SD, Levine RL, Gottgens B, Kouzarides T, and Green AR (2011). Nuclear JAK2. Blood 118, 6987–6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo ED, Erdogan F, Neubauer HA, Meneksedag-Erol D, Manaswiyoungkul P, Eram MS, Seo HS, Qadree AK, Israelian J, Orlova A, et al. (2019). Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat Commun 10, 2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bel KL, Ragotte RJ, Saferali A, Lee S, Vercauteren SM, Mostafavi SA, Schreiber RA, Prendiville JS, Phang MS, Halparin J, et al. (2017). JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol 139, 2016–2020 e2015. [DOI] [PubMed] [Google Scholar]
- Delgoffe GM, and Vignali DA (2013). STAT heterodimers in immunity: A mixed message or a unique signal? JAKSTAT 2, e23060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eletto D, Burns SO, Angulo I, Plagnol V, Gilmour KC, Henriquez F, Curtis J, Gaspar M, Nowak K, Daza-Cajigal V, et al. (2016). Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun 7, 13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M, Reutterer B, Schindler C, Greten F, Vogl C, Muller M, and Decker T (2010). Nonconventional initiation complex assembly by STAT and NF-kappaB transcription factors regulates nitric oxide synthase expression. Immunity 33, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firmbach-Kraft I, Byers M, Shows T, Dalla-Favera R, and Krolewski JJ (1990). tyk2, prototype of a novel class of non-receptor tyrosine kinase genes. Oncogene 5, 1329–1336. [PubMed] [Google Scholar]
- Garama DJ, Harris TJ, White CL, Rossello FJ, Abdul-Hay M, Gough DJ, and Levy DE (2015). A Synthetic Lethal Interaction between Glutathione Synthesis and Mitochondrial Reactive Oxygen Species Provides a Tumor-Specific Vulnerability Dependent on STAT3. Mol Cell Biol 35, 3646–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordanetto F, and Kroemer RT (2003). A three-dimensional model of Suppressor Of Cytokine Signalling 1 (SOCS-1). Protein Eng 16, 115–124. [DOI] [PubMed] [Google Scholar]
- Glassman CR, Tsutsumi N, Saxton RA, Lupardus PJ, Jude KM, and Garcia KC (2022). Structure of a Janus kinase cytokine receptor complex reveals the basis for dimeric activation. Science 376, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnatovskiy L, Mita P, and Levy DE (2013). The human RVB complex is required for efficient transcription of type I interferon-stimulated genes. Mol Cell Biol 33, 3817–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goertz GP, McNally KL, Robertson SJ, Best SM, Pijlman GP, and Fros JJ (2018). The Methyltransferase-Like Domain of Chikungunya Virus nsP2 Inhibits the Interferon Response by Promoting the Nuclear Export of STAT1. J Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollob JA, Schnipper CP, Murphy EA, Ritz J, and Frank DA (1999). The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J Immunol 162, 4472–4481. [PubMed] [Google Scholar]
- Gothe F, Stremenova Spegarova J, Hatton CF, Griffin H, Sargent T, Cowley SA, James W, Roppelt A, Shcherbina A, Hauck F, et al. (2022). Aberrant inflammatory responses to type I interferon in STAT2 or IRF9 deficiency. J Allergy Clin Immunol. [DOI] [PubMed] [Google Scholar]
- Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, and Levy DE (2009). Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324, 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouilleux F, Wakao H, Mundt M, and Groner B (1994). Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J 13, 4361–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracey E, Hromadova D, Lim M, Qaiyum Z, Zeng M, Yao Y, Srinath A, Baglaenko Y, Yeremenko N, Westlin W, et al. (2020). TYK2 inhibition reduces type 3 immunity and modifies disease progression in murine spondyloarthritis. J Clin Invest 130, 1863–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, and Garcia-Sastre A (2016). Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 19, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlund AC, Farrar MA, Viviano BL, and Schreiber RD (1994). Ligand-induced IFN gamma receptor tyrosine phosphorylation couples the receptor to its signal transduction system (p91). EMBO J 13, 1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber CN, Calis JJA, Buta S, Evrony G, Martin JC, Uhl SA, Caron R, Jarchin L, Dunkin D, Phelps R, et al. (2020). Complex Autoinflammatory Syndrome Unveils Fundamental Principles of JAK1 Kinase Transcriptional and Biochemical Function. Immunity 53, 672–684 e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, Armstrong M, Ashrafian H, Cutcutache I, Ebetsberger-Dachs G, et al. (2020). Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun 11, 5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammaren HM, Ungureanu D, Grisouard J, Skoda RC, Hubbard SR, and Silvennoinen O (2015). ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc Natl Acad Sci U S A 112, 4642–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanratty WP, and Dearolf CR (1993). The Drosophila Tumorous-lethal hematopoietic oncogene is a dominant mutation in the hopscotch locus. Mol Gen Genet 238, 33–37. [DOI] [PubMed] [Google Scholar]
- Harpur AG, Andres AC, Ziemiecki A, Aston RR, and Wilks AF (1992). JAK2, a third member of the JAK family of protein tyrosine kinases. Oncogene 7, 1347–1353. [PubMed] [Google Scholar]
- Harrison DA, Binari R, Nahreini TS, Gilman M, and Perrimon N (1995). Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J 14, 2857–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasni SA, Gupta S, Davis M, Poncio E, Temesgen-Oyelakin Y, Carlucci PM, Wang X, Naqi M, Playford MP, Goel RR, et al. (2021). Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun 12, 3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahara K, Onodera A, Villarino AV, Bonelli M, Sciume G, Laurence A, Sun HW, Brooks SR, Vahedi G, Shih HY, et al. (2015). Asymmetric Action of STAT Transcription Factors Drives Transcriptional Outputs and Cytokine Specificity. Immunity 42, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, Pelzel C, Begitt A, Mee M, Elsheikha HM, Scott DJ, and Vinkemeier U (2016). STAT2 Is a Pervasive Cytokine Regulator due to Its Inhibition of STAT1 in Multiple Signaling Pathways. PLoS Biol 14, e2000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, et al. (2007). STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357, 1608–1619. [DOI] [PubMed] [Google Scholar]
- Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, and McKnight SL (1994). An interleukin-4-induced transcription factor: IL-4 Stat. Science 265, 1701–1706. [DOI] [PubMed] [Google Scholar]
- Hu G, Tang Q, Sharma S, Yu F, Escobar TM, Muljo SA, Zhu J, and Zhao K (2013a). Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat Immunol 14, 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Dutta P, Tsurumi A, Li J, Wang J, Land H, and Li WX (2013b). Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc Natl Acad Sci U S A 110, 10213–10218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, and Julicher F (2014). Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol 30, 39–58. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Kawamura M, Kirken RA, Chen YQ, Blake TB, Shibuya K, Ortaldo JR, McVicar DW, and O’Shea JJ (1994). Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2. Nature 370, 151–153. [DOI] [PubMed] [Google Scholar]
- Jovanovic M, Rooney MS, Mertins P, Przybylski D, Chevrier N, Satija R, Rodriguez EH, Fields AP, Schwartz S, Raychowdhury R, et al. (2015). Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 347, 1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno Y, Vahedi G, Hirahara K, Singleton K, and O’Shea JJ (2012). Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol 30, 707–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katerndahl CDS, Heltemes-Harris LM, Willette MJL, Henzler CM, Frietze S, Yang R, Schjerven H, Silverstein KAT, Ramsey LB, Hubbard G, et al. (2017). Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat Immunol 18, 694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh ME, Horning BD, Khattri R, Roy N, Lu JP, Whitby LR, Ye E, Brannon JC, Parker A, Chick JM, et al. (2022). Selective inhibitors of JAK1 targeting an isoform-restricted allosteric cysteine. Nature Chemical Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA, and Babon JJ (2013). SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol 20, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmann S, Grausenburger R, Klampfl T, Prchal-Murphy M, Bastl K, Pisa H, Knab VM, Brandstoetter T, Doma E, Sperr WR, et al. (2021). A STAT5B-CD9 axis determines self-renewal in hematopoietic and leukemic stem cells. Blood 138, 2347–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic B, Stoiber D, Moriggl R, Weisz E, Ott RG, Kreibich R, Levy DE, Beug H, Freissmuth M, and Sexl V (2006). STAT1 acts as a tumor promoter for leukemia development. Cancer Cell 10, 77–87. [DOI] [PubMed] [Google Scholar]
- Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, and Skoda RC (2005). A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352, 1779–1790. [DOI] [PubMed] [Google Scholar]
- Krolewski JJ, Lee R, Eddy R, Shows TB, and Dalla-Favera R (1990). Identification and chromosomal mapping of new human tyrosine kinase genes. Oncogene 5, 277–282. [PubMed] [Google Scholar]
- La Manna S, Lee E, Ouzounova M, Di Natale C, Novellino E, Merlino A, Korkaya H, and Marasco D (2018). Mimetics of suppressor of cytokine signaling 3: Novel potential therapeutics in triple breast cancer. Int J Cancer 143, 2177–2186. [DOI] [PubMed] [Google Scholar]
- Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, and Bernard OA (1997). A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 278, 1309–1312. [DOI] [PubMed] [Google Scholar]
- Lau JF, Nusinzon I, Burakov D, Freedman LP, and Horvath CM (2003). Role of metazoan mediator proteins in interferon-responsive transcription. Mol Cell Biol 23, 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Larner A, Chaudhuri A, Babiss LE, and Darnell JE Jr. (1986). Interferon-stimulated transcription: isolation of an inducible gene and identification of its regulatory region. Proc Natl Acad Sci U S A 83, 8929–8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, and Lee CK (2002). What does Stat3 do? J Clin Invest 109, 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Williams MJ, Park HJ, Bastos HP, Wang X, Prins DP, Wilson NK, Johnson C, Sham K, Wantoch M, et al. (2022). STAT1 is essential for HSC function and maintains MHCIIhi stem cells that resists myeloablation and neoplastic expansion. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JX, Li P, Liu D, Jin HT, He J, Ata Ur Rasheed M, Rochman Y, Wang L, Cui K, Liu C, et al. (2012). Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity 36, 586–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linossi EM, and Nicholson SE (2015). Kinase inhibition, competitive binding and proteasomal degradation: resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol Rev 266, 123–133. [DOI] [PubMed] [Google Scholar]
- Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, et al. (2011). Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208, 1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Robinson GW, Gouilleux F, Groner B, and Hennighausen L (1995). Cloning and expression of Stat5 and an additional homologue (Stat5b) involved in prolactin signal transduction in mouse mammary tissue. Proc Natl Acad Sci U S A 92, 8831–8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Hanratty WP, and Dearolf CR (1995). An amino acid substitution in the Drosophila hopTum-l Jak kinase causes leukemia-like hematopoietic defects. EMBO J 14, 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O’Shea JJ, and et al. (1995). Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature 377, 65–68. [DOI] [PubMed] [Google Scholar]
- Majoros A, Platanitis E, Kernbauer-Holzl E, Rosebrock F, Muller M, and Decker T (2017). Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front Immunol 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, Kee BL, Dinner AR, and Clark MR (2011). Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol 12, 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie IJ, Chang HM, and Levy DE (2018). HDAC stimulates gene expression through BRD4 availability in response to IFN and in interferonopathies. J Exp Med 215, 3194–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fernandez M, Bravo Garcia-Morato M, Gruber C, Murias Loza S, Malik MNH, Alsohime F, Alakeel A, Valdez R, Buta S, Buda G, et al. (2020). Systemic Type I IFN Inflammation in Human ISG15 Deficiency Leads to Necrotizing Skin Lesions. Cell Rep 31, 107633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fernandez M, Buta S, Le Voyer T, Li Z, Dynesen LT, Vuillier F, Franklin L, Ailal F, Muglia Amancio A, Malle L, et al. (2022). A partial form of inherited human USP18 deficiency underlies infection and inflammation. J Exp Med 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Moczygemba M, Gutch MJ, French DL, and Reich NC (1997). Distinct STAT structure promotes interaction of STAT2 with the p48 subunit of the interferon-alpha-stimulated transcription factor ISGF3. J Biol Chem 272, 20070–20076. [DOI] [PubMed] [Google Scholar]
- Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, van Unen L, Heijsman D, Goldmann T, et al. (2016). Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med 213, 1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, et al. (1996). Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 379, 645–648. [DOI] [PubMed] [Google Scholar]
- Migone TS, Lin JX, Cereseto A, Mulloy JC, O’Shea JJ, Franchini G, and Leonard WJ (1995). Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 269, 79–81. [DOI] [PubMed] [Google Scholar]
- Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, Lyons JJ, Engelhardt KR, Zhang Y, Topcagic N, et al. (2015). Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 125, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, Takada H, Hara T, Kawamura N, Ariga T, et al. (2006). Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 25, 745–755. [DOI] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, et al. (2007). Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448, 1058–1062. [DOI] [PubMed] [Google Scholar]
- Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, Cupic A, Makio T, Mei M, Moreno E, et al. (2020). SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci U S A 117, 28344–28354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Yoshida H, Moodley D, LeBoite H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang SY, Feng T, et al. (2016). Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 164, 564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SA, and Gottschalk RA (2022). Mechanisms encoding STAT functional diversity for context-specific inflammatory responses. Curr Opin Immunol 74, 150–155. [DOI] [PubMed] [Google Scholar]
- Nan J, Wang Y, Yang J, and Stark GR (2018). IRF9 and unphosphorylated STAT2 cooperate with NF-kappaB to drive IL6 expression. Proc Natl Acad Sci U S A 115, 3906–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, and Pfeffer K (1998). Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 93, 397–409. [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O’Shea JJ, and Biron CA (2002). Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 297, 2063–2066. [DOI] [PubMed] [Google Scholar]
- Nusinzon I, and Horvath CM (2003). Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc Natl Acad Sci U S A 100, 14742–14747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, and Natoli G (2013). Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171. [DOI] [PubMed] [Google Scholar]
- Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, et al. (1998). Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93, 385–395. [DOI] [PubMed] [Google Scholar]
- Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, Miyake K, Nakauchi H, Shirasawa T, and Saito T (1995). Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity 3, 771–782. [DOI] [PubMed] [Google Scholar]
- Patel MC, Debrosse M, Smith M, Dey A, Huynh W, Sarai N, Heightman TD, Tamura T, and Ozato K (2013). BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol Cell Biol 33, 2497–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson M, Pisharody S, Pan L, Guadagno S, Mui AL, and Levy DE (1999). Stat protein transactivation domains recruit p300/CBP through widely divergent sequences. J Biol Chem 274, 25343–25349. [DOI] [PubMed] [Google Scholar]
- Paulson M, Press C, Smith E, Tanese N, and Levy DE (2002). IFN-Stimulated transcription through a TBP-free acetyltransferase complex escapes viral shutoff. Nat Cell Biol 4, 140–147. [DOI] [PubMed] [Google Scholar]
- Peron M, Dinarello A, Meneghetti G, Martorano L, Betto RM, Facchinello N, Tesoriere A, Tiso N, Martello G, and Argenton F (2021). Y705 and S727 are required for the mitochondrial import and transcriptional activities of STAT3, and for regulation of stem cell proliferation. Development 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo V, Curina A, Genua M, Ghisletti S, Simonatto M, Sabo A, Amati B, Ostuni R, and Natoli G (2017). Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nat Immunol 18, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanitis E, and Decker T (2018). Regulatory Networks Involving STATs, IRFs, and NFkappaB in Inflammation. Front Immunol 9, 2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanitis E, Demiroz D, Schneller A, Fischer K, Capelle C, Hartl M, Gossenreiter T, Muller M, Novatchkova M, and Decker T (2019). A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nat Commun 10, 2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanitis E, Gruener S, Ravi Sundar Jose Geetha A, Boccuni L, Vogt A, Novatchkova M, Sommer A, Barozzi I, Muller M, and Decker T (2022). Interferons reshape the 3D conformation and accessibility of macrophage chromatin. iScience 25, 103840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana CV, Gil MP, Schreiber RD, and Stark GR (2002). Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol 23, 96–101. [DOI] [PubMed] [Google Scholar]
- Reich NC, and Liu L (2006). Tracking STAT nuclear traffic. Nat Rev Immunol 6, 602–612. [DOI] [PubMed] [Google Scholar]
- Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, et al. (2007). STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357, 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Chu AE, Jude KM, Picton LK, Kare AJ, Su L, Montano Romero A, Huang PS, and Garcia KC (2022). Interleukin-2 superkines by computational design. Proc Natl Acad Sci U S A 119, e2117401119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengachari S, Groiss S, Devos JM, Caron E, Grandvaux N, and Panne D (2018). Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function. Proc Natl Acad Sci U S A 115, E601–E609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. (2007). Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4, 651–657. [DOI] [PubMed] [Google Scholar]
- Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, et al. (1998). Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 93, 373–383. [DOI] [PubMed] [Google Scholar]
- Ross SH, Rollings C, Anderson KE, Hawkins PT, Stephens LR, and Cantrell DA (2016). Phosphoproteomic Analyses of Interleukin 2 Signaling Reveal Integrated JAK Kinase-Dependent and -Independent Networks in CD8(+) T Cells. Immunity 45, 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, Migone TS, Noguchi M, Markert ML, Buckley RH, et al. (1995). Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science 270, 797–800. [DOI] [PubMed] [Google Scholar]
- Saevarsdottir S, Stefansdottir L, Sulem P, Thorleifsson G, Ferkingstad E, Rutsdottir G, Glintborg B, Westerlind H, Grondal G, Loft IC, et al. (2022). Multiomics analysis of rheumatoid arthritis yields sequence variants that have large effects on risk of the seropositive subset. Ann Rheum Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciume G, Mikami Y, Jankovic D, Nagashima H, Villarino AV, Morrison T, Yao C, Signorella S, Sun HW, Brooks SR, et al. (2020). Rapid Enhancer Remodeling and Transcription Factor Repurposing Enable High Magnitude Gene Induction upon Acute Activation of NK Cells. Immunity 53, 745–758 e744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Lu HY, Vaseghi-Shanjani M, Del Bel KL, Fornes O, van der Lee R, Richmond PA, Lin S, Dalmann J, Lee JJ, et al. (2022). Human Germline Heterozygous Gain-of-Function STAT6 Variants Cause Severe Allergic Disease. medRxiv, 2022.2004.2025.22274265. [Google Scholar]
- Shuai K (2006). Regulation of cytokine signaling pathways by PIAS proteins. Cell Res 16, 196–202. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O, Schindler C, Schlessinger J, and Levy DE (1993). Ras-independent growth factor signaling by transcription factor tyrosine phosphorylation. Science 261, 1736–1739. [DOI] [PubMed] [Google Scholar]
- Snell LM, McGaha TL, and Brooks DG (2017). Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol 38, 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler JB, Moraga I, Mendoza JL, and Garcia KC (2015). Insights into cytokine-receptor interactions from cytokine engineering. Annu Rev Immunol 33, 139–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, Rubino E, Gardner TJ, Wedeking T, Hermann M, et al. (2016). ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun 7, 11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, and Darnell JE Jr. (2012). The JAK-STAT pathway at twenty. Immunity 36, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, and Akira S (1998). Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8, 383–390. [DOI] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomis DC, Gurniak CB, Tivol E, Sharpe AH, and Berg LJ (1995). Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science 270, 794–797. [DOI] [PubMed] [Google Scholar]
- Ulane CM, and Horvath CM (2002). Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 304, 160–166. [DOI] [PubMed] [Google Scholar]
- Vahedi G, Kanno Y, Furumoto Y, Jiang K, Parker SC, Erdos MR, Davis SR, Roychoudhuri R, Restifo NP, Gadina M, et al. (2015). Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 520, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, and O’Shea JJ (2012). STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez L, Fellous M, Stark GR, and Pellegrini S (1992). A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 70, 313–322. [DOI] [PubMed] [Google Scholar]
- Vinkemeier U, Moarefi I, Darnell JE Jr., and Kuriyan J (1998). Structure of the amino-terminal protein interaction domain of STAT-4. Science 279, 1048–1052. [DOI] [PubMed] [Google Scholar]
- Wakao H, Gouilleux F, and Groner B (1994). Mammary gland factor (MGF) is a novel member of the cytokine regulated transcription factor gene family and confers the prolactin response. EMBO J 13, 2182–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan CK, Andraski AB, Spolski R, Li P, Kazemian M, Oh J, Samsel L, Swanson PA 2nd, McGavern DB, Sampaio EP, et al. (2015). Opposing roles of STAT1 and STAT3 in IL-21 function in CD4+ T cells. Proc Natl Acad Sci U S A 112, 9394–9399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Sun M, Yuan X, Ji L, Jin Y, Cardona CJ, and Xing Z (2017a). Enterovirus 71 suppresses interferon responses by blocking Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through inducing karyopherin-alpha1 degradation. J Biol Chem 292, 10262–10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nan J, Willard B, Wang X, Yang J, and Stark GR (2017b). Negative regulation of type I IFN signaling by phosphorylation of STAT2 on T387. EMBO J 36, 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Song Q, Huang W, Lin Y, Wang X, Wang C, Willard B, Zhao C, Nan J, Holvey-Bates E, et al. (2021). A virus-induced conformational switch of STAT1-STAT2 dimers boosts antiviral defenses. Cell Res 31, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, et al. (2008). An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A 105, 18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenta N, Strauss H, Meyer S, and Vinkemeier U (2008). Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc Natl Acad Sci U S A 105, 9238–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek M, Ginter T, Brand P, Heinzel T, and Kramer OH (2012). Acetylation modulates the STAT signaling code. Cytokine Growth Factor Rev 23, 293–305. [DOI] [PubMed] [Google Scholar]
- Wiedemann GM, Santosa EK, Grassmann S, Sheppard S, Le Luduec JB, Adams NM, Dang C, Hsu KC, Sun JC, and Lau CM (2021). Deconvoluting global cytokine signaling networks in natural killer cells. Nat Immunol 22, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zurcher G, and Ziemiecki A (1991). Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol 11, 2057–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte S, and Muljo SA (2014). Integrating non-coding RNAs in JAK-STAT regulatory networks. JAKSTAT 3, e28055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, Berry KN, Yen B, Hamilton J, Brett TJ, et al. (2014). Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe 16, 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, and Stark GR (2007). Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev 21, 1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen M, Ren J, Liu Q, Glassman CR, Sheahan TP, Picton LK, Moreira FR, Rustagi A, Jude KM, Zhao X, et al. (2022). Facile discovery of surrogate cytokine agonists. Cell 185, 1414–1430 e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, and Jove R (1995). Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269, 81–83. [DOI] [PubMed] [Google Scholar]
- Yu R, Zhu B, and Chen D (2022). Type I interferon-mediated tumor immunity and its role in immunotherapy. Cell Mol Life Sci 79, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio AV, Dall’Agnese A, Henninger JE, Manteiga JC, Afeyan LK, Hannett NM, Coffey EL, Li CH, Oksuz O, Sabari BR, et al. (2019). Mediator Condensates Localize Signaling Factors to Key Cell Identity Genes. Mol Cell 76, 753–766 e756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, Zhang Z, Li W, Lee H, Aftabizadeh M, et al. (2020). STAT3 Activation-Induced Fatty Acid Oxidation in CD8(+) T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab 31, 148–161 e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. (2012). The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, et al. (2015). Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature 517, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Darnell JE Jr. (2001). Functional importance of Stat3 tetramerization in activation of the alpha 2-macroglobulin gene. J Biol Chem 276, 33576–33581. [DOI] [PubMed] [Google Scholar]
- Zhong M, Henriksen MA, Takeuchi K, Schaefer O, Liu B, ten Hoeve J, Ren Z, Mao X, Chen X, Shuai K, and Darnell JE Jr. (2005). Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc Natl Acad Sci U S A 102, 3966–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, and Darnell JE Jr. (1994a). Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc Natl Acad Sci U S A 91, 4806–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Wen Z, and Darnell JE Jr. (1994b). Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264, 95–98. [DOI] [PubMed] [Google Scholar]
- Zhu X, Wang D, Zhou J, Pan T, Chen J, Yang Y, Lv M, Ye X, Peng G, Fang L, and Xiao S (2017). Porcine Deltacoronavirus nsp5 Antagonizes Type I Interferon Signaling by Cleaving STAT2. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]