


Abstract
The toxic effects of compounds on environment, humans, and other organisms have been a major focus of many research areas, including drug discovery and ecological research. Identifying the potential toxicity in the early stage of compound/drug discovery is critical. The rapid development of computational methods for evaluating various toxicity categories has increased the need for comprehensive and system-level collection of toxicological data, associated attributes, and benchmarks. To contribute toward this goal, we proposed TOXRIC (https://toxric.bioinforai.tech/), a database with comprehensive toxicological data, standardized attribute data, practical benchmarks, informative visualization of molecular representations, and an intuitive function interface. The data stored in TOXRIC contains 113 372 compounds, 13 toxicity categories, 1474 toxicity endpoints covering in vivo/in vitro endpoints and 39 feature types, covering structural, target, transcriptome, metabolic data, and other descriptors. All the curated datasets of endpoints and features can be retrieved, downloaded and directly used as output or input to Machine Learning (ML)-based prediction models. In addition to serving as a data repository, TOXRIC also provides visualization of benchmarks and molecular representations for all endpoint datasets. Based on these results, researchers can better understand and select optimal feature types, molecular representations, and baseline algorithms for each endpoint prediction task. We believe that the rich information on compound toxicology, ML-ready datasets, benchmarks and molecular representation distribution can greatly facilitate toxicological investigations, interpretation of toxicological mechanisms, compound/drug discovery and the development of computational methods.
Graphical Abstract
Graphical Abstract.
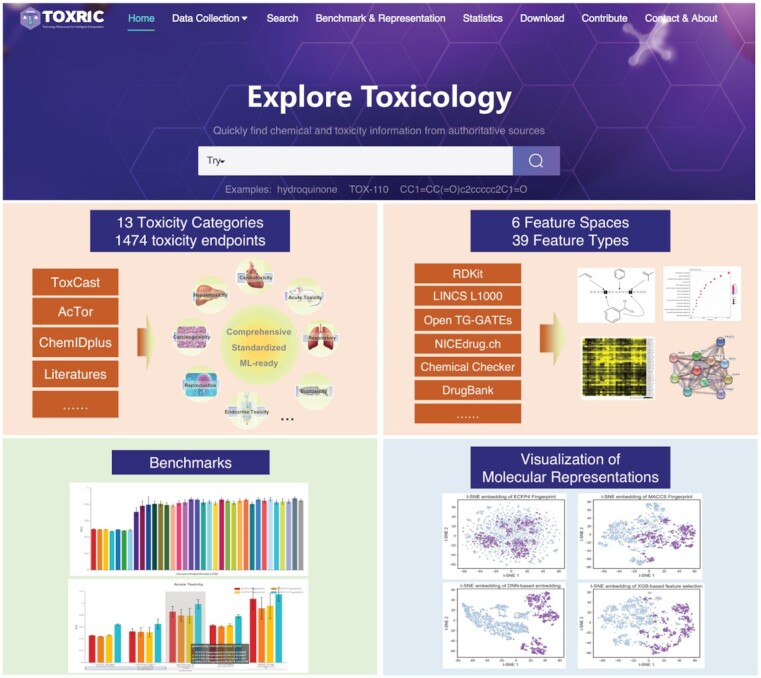
Five core functions of TOXRIC: comprehensive toxicological data, standardized feature data, practical benchmarks, informative visualization of molecular representations, and an intuitive function interface.
INTRODUCTION
Identifying the toxicity of compounds is necessary to explore their harmful effects on humans, animals, plants, and the environment (1). The toxic effects are classified into various toxicity categories, including carcinogenicity, hepatotoxicity, ecotoxicity, irritation and corrosion. The toxicity evaluation of compounds plays an important role in many areas of research. For example, during drug discovery and development, unexpected toxicities are the most significant reasons for hindering drug candidates from reaching the market (2–4). Many marketed drugs have even been withdrawn due to toxicity concerns. Potential toxicities should be extensively evaluated in the early stage of drug development (4). In ecotoxicity testing, the toxic effect of a compound is measured on both environment and organisms, such as fish, insects, microorganisms, wildlife, and plants (5,6). Elucidating the potential ecotoxicity in exploration of materials is critical.
In order to identify the toxicities of compounds, many evaluation approaches have been developed, including conventional in vivo, in vitro, and computational methods. Due to the advantages of low cost, fast speed, and high accuracy, computational methods based on a large amount of toxicological data have been widely explored, including rule-based models (7), read-across (8), QSAR (Quantitative structure-activity relationship) (9) and Machine Learning (ML)-based methods (1,10). More importantly, toxicity evaluation results can be presented before the synthesis of compounds with computational methods (4,11).
However, the major challenge in developing computational methods is the low accessibility of sufficient, reliable, and standardized toxicological and related attribute data (3,4). Despite an abundance of online databases providing access to large amounts of toxicology data (12–16), there is an increasing demand for high-quality and ML-ready toxicology, attribute datasets and benchmarks. We classified the existing online toxicological databases into four categories, toxicity category-centric, toxic feature-centric, compound-centric and ML task-centric. The toxicity category-centric and toxic feature-centric databases focus on specific toxicity categories/endpoints or toxic-related feature data (toxicogenomic data, etc.). The compound-centric databases provide multiple toxicological information, while most of them serve only to retrieve multiple toxicities and chemical information of compounds, or the downloadable toxicity datasets are not curated in ML-ready format. For toxicity evaluation, it is necessary to integrate and curate data of multiple toxicity categories and feature spaces. In addition, depending on different toxicity categories, different computational methods may apply. Methods applicable to certain types of toxicity endpoints may not work properly (or not work at all) for others (1). The benchmarks also have a critical role in facilitating progress in ML methods. According to the benchmark results, researchers can select an appropriate feature type and baseline model for different prediction task. There have been two ML task-centric databases that provide benchmarks for ML, Therapeutics Data Commons (TDC) (17) and MoleculeNet (18) databases, which are platforms to provide datasets, ML tasks, benchmarks, and other information for various fields of study, such as drug combination and biophysical-related studies. Neither is a database directly focused on toxicology, nor does it provide data retrieval. Thus, there is an urgent need for a database to retrieve comprehensive toxicology, attribute data and system-level benchmarks (19–21).
To address this issue, we propose TOXRIC (TOXicology Resources for Intelligent Computation) database (https://toxric.bioinforai.tech/), which aims to collect, curate, and disseminate comprehensive, reliable, and standardized datasets of toxicity categories and feature types. The data stored in TOXRIC contains 113,372 compounds, 13 toxicity categories, 1,474 toxicity endpoints covering in vivo/in vitro endpoints, 6 feature spaces and 39 feature types, covering structural, target, transcriptome, metabolic data, and other descriptors. The toxicity values and features of each compound can be quickly retrieved and downloaded. All the curated datasets of endpoint and feature type can be downloaded and used directly as output or input to ML models for toxicity prediction. TOXRIC also provides multiple benchmark results and spatial distribution of molecular representations for all toxicity endpoints. Based on these visualization results, researchers can better understand and select appropriate feature types, molecular representations, and baseline algorithms for each toxicity endpoint prediction task. TOXRIC offers an effective and user-friendly web interface to take full advantages of the wealthy data and available information. The detailed description of datasets, rapid search/batch search of toxicological data, download of the information on individual compound or datasets, visualization of benchmark results and molecular representations are freely available to all users through the TOXRIC web.
MATERIALS AND METHODS
Data collection and curation
TOXRIC provides toxicological data, feature, and chemical data for each compound. The chemical data include six commonly used identifier types and several physicochemical properties. Toxicological data provide toxicity values on multiple toxicity endpoints, which can be used as the label data for ML models. The feature data provide representations of compounds, which can be taken as input to ML models.
Curation process of toxicological and chemical data
TOXRIC collects four toxicology groups, 13 toxicity categories and 1474 endpoint datasets across >15 species. The four groups include toxic effect, target organ toxicology, applied toxicology, and other toxicology datasets. According to the groups, relevant literatures and publicly available databases are retrieved. The main sources include ToxCast/Tox21 (22,23), LTKB (24), DILIrank (16), Livertox (25), ChemIDplus (26) database, and studies by Jain et al. (10), Wu et al. (3), Jiang et al. (27), etc. All the sources are commonly used for toxicity prediction (28–33). In addition, the 13 categories contain both in vitro and in vivo toxicity endpoints. Among them, Acute Toxicity and Ecotoxicity contain the in vivo endpoints across different species under specific exposure times and doses. Hepatotoxicity, Developmental and Reproductive Toxicity, Carcinogenicity, Respiratory Toxicity, and Clinical Toxicity are in vivo endpoints without specific exposure conditions. ToxCast&Tox21 Assay, CYP450, Cardiotoxicity and Endocrine Disruption are based on in vitro cytotoxicity assays under specific exposure conditions (34).
The curation process of toxicology data consists of three parts: compound screening, data source integration, and unit standardization. First, the compounds of all data sources are extracted, which is usually represented by different identifiers. The identifiers are matched to PubChem CID and Canonical SMILES. Duplicate compounds are removed according to Canonical SMILES. Then, salts and solvents are removed, followed by counterions, large organic compounds (Da ≥ 2000), mixtures, and inorganic compounds. This is implemented in accordance with a protocol previously developed by Jain et al. (10) and Jiang et al. (27). To distinguish the salts, compounds containing metal atoms and involved in the USAN Council's list of pharmacological salts (https://www.ama-assn.org/system/files/2019-04/radicals-and-anions-list.pdf) are removed, following the protocol developed by Bento et al. (35). Next, a unique identifier, TAID, is assigned for each compound in TOXRIC. Relevant chemical information of compounds is integrated into the database. In addition, the endpoint datasets with <10 samples are removed.
Second, in order to avoid compatibility issue, the toxicity values for all endpoints except hepatotoxicity are collected from a single reliable source. Hepatotoxicity induced by compounds is a significant problem in toxicology. To expand the amount of hepatotoxicity data, we collect toxicity values from seven sources, including LTKB database, DILIrank database, Livertox database, and four literatures (28,36–38). For compounds with inconsistent toxicity values from multiple sources, the true result can only be determined when more than 80% of the sources belong to the same toxicity value. Otherwise, the toxicity of the compound is considered ambiguous, and the compound will be removed.
Third, the datasets provided in TOXRIC include classification and regression datasets. Toxicity values in classification datasets are binary values of 0 or 1 with no units, indicating toxic or non-toxic class. Toxicity values in regression datasets (Acute Toxicity and Ecotoxicity) are quantitative results, containing LD50, LDLo, TDLo, LC50, etc. Datasets from different sources use different units, including (mg/kg), (gm/kg), (ug/kg), (ng/kg), (uL/kg), (mL/kg) and (mg/L). The units of the endpoints tested in some typical species are listed in Supplementary Table S1. We standardized the units in two steps. First, all the units are converted to (mg/kg) for Acute Toxicity or (mg/L) for Ecotoxicity to get version-1 of both datasets. The curated data of Ecotoxicity is in (mg/L) unit. Then, the formula for converting other units to (mg/kg) is as follows.
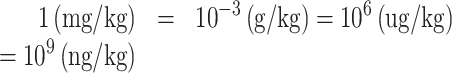 |
(1) |
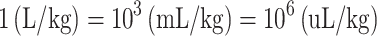 |
(2) |
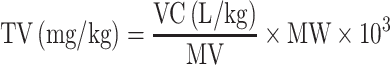 |
(3) |
The units of (gm/kg), (ug/kg) and (ng/kg) are converted to (mg/kg) through Eq. (1). The units of (uL/kg) and (mL/kg) are converted to (L/kg) through Eq. (2), and then (L/kg) is convert to (mg/kg) through Eq. (3). In Eq. (3), TV stands for toxicity value, VC stands for volume concentration, MV stands for molar volume, MW stands for molecular weight. MV and MW of compounds are obtained from PubChem using the Python PubChemPy package (https://pubchempy.readthedocs.io/en/latest/). Second, to facilitate the development of ML prediction models, the dimensionless values are introduced. The units are standardized into (−log(mol/kg)) or (−log(mg/L)) to get the dimensionless values (32). Then the version-2 of the datasets with dimensionless values can be obtained. On TOXRIC website, version-1 of regression datasets with toxicity values in (mg/kg) or (mg/L) unit is provided for retrieval, and both version-1 and version-2 are available for download.
Curation process of feature data
Multiple feature types of these compounds are computed and curated, including seven molecular fingerprints, target protein, three transcriptome profiles, metabolic reaction, two categories of drugs, and 25 descriptors collected from the Chemical Checker (CC) database (39). Seven common molecular fingerprints are computed by RDKit to represent the structure of compounds. Target proteins of compounds are collected from DrugBank (40) and BindingDB (41) database in text format. Category information are collected from DrugBank database. The 328 192 reaction equations of 9598 compounds are obtained from NICEdrug.ch (42), a database for systems-level analysis of drug metabolism. The InchIKey identifier is used to match the compounds between TOXRIC and NICEdrug.ch.
The transcriptome profiles are collected from three data sources, LINCS L1000 (43), Open TG-GATEs (44) and DrugMatrix (45) databases. Data from the LINCS L1000 database contains the expression levels of 978 landmark genes under compound perturbations (not covering the complete transcriptome). In LINCS database, the expression profiles are measured by the L1000 method, which contains only 1058 probes for 978 landmark transcripts and 80 control transcripts. The remainder of the transcriptome is inferred using the landmark transcript measurements, and 81% of the inference is accurate according to the study by Subramanian et al (43). Level 5 data is obtained from the LINCS L1000 database, including the gene expression with multiple doses and times. A unique transcriptional profile of each compound is obtained using the moderated z-score (MODZ) approach, and the z-scores are weighted and averaged according to Spearman correlations. Transcriptional profiles of compounds in primary human hepatocytes (in vitro) and rat kidney and liver organs (in vivo) are collected and curated from the Open TG-Gates database. DrugMatrix (45,46) is produced by the U.S. National Toxicology Program, containing transcriptomic profiles of more than 200 compounds tested in vivo in rat tissues such as liver and 125 compounds in the in vitro rat hepatocytes. The microarray data is obtained from GEO. In the microarray data, the gene symbols are converted to EntrezID through the annotation file, and the original probes are batch normalized by RMA using R package affy (47). The expression matrix is obtained by averaging the duplicate probe and duplicate gene. The missing values are filled using interpolation. Differential expression results are then calculated. Finally, data of different doses and exposure times are weighted averaged.
In addition, 25 features are collected from CC database (39). Different from other original features, the feature vectors provided by CC are 128-dimensional embedding processed by the node2vec algorithm. Among the CC descriptors, the 2D fingerprint is involved, which is different from the seven fingerprints provided by TOXRIC. The latter is the original feature of molecular structure, and each bit represents the presence or absence of a particular substructure. In contrast, the CC descriptors provide feature embedding, which transform features from the original space into a new low-dimensional space. Every dimension has no actual meaning. And the missing values of the original feature are filled by the similarity principle.
Benchmark construction
TOXRIC provides ML tasks and benchmarks for all the endpoint datasets. The ML tasks include classification and regression tasks. Tested on these tasks, TOXRIC provides two benchmarks on 36 feature types and four typical ML algorithms on all endpoint datasets.
The endpoint datasets can be used as label data for ML models, while the feature datasets can be used as input. Four typical ML algorithms widely used for toxicity prediction are evaluated (48), including eXtreme Gradient Boosting (XGB) (49), Random Forest (RF) (50), Support Vector Machine (SVM) (51) and Deep Neural Network (DNN) (10). XGB and RF are two advanced ensemble learning algorithms, which are representative of sequential ensemble and parallel ensemble, respectively. Both algorithms have performed well in small to medium datasets. SVM constructs a hyperplane or a set of hyperplanes to classify data points and is effective in high-dimensional spaces. DNNs consist of several sequential hidden layers, and have been reported to outperform most other ML methods in predicting molecular properties in large-scale datasets. In contrast, in most small or medium datasets, classical ML methods perform better than DNN with less hyperparameter tuning (52). In the benchmark construction of TOXRIC, XGB, RF and SVM are implemented in scikit-learn or XGB python module with the default configuration. DNN model is followed by Jain et al (10).
To evaluate the performance of benchmarks, Root Mean Squared Error (RMSE), R2 or F1 metrics are computed according to five-fold cross-validation. The mean and standard deviation are reported. RMSE reflects the average error between the predicted values and the ground-truth labels. Lower RMSE and higher R2 value represents higher prediction performance in regression datasets. As most of the endpoint datasets are imbalanced, F1 metric is chosen to evaluate the performance in classification tasks. The higher the F1 value, the higher the prediction performance.
TOXRIC provides two types of benchmarks. For the benchmarks on feature types, 36 features are tested using XGB algorithm. Among them, target proteins are encoded as one-hot-encoded vectors. In the training datasets of different feature types, samples with missing features are removed. And the endpoint datasets with the sample numbers <10 are removed. For the benchmarks on different algorithms, four algorithms with the same input are tested. According to the benchmark results of feature types, two fingerprints (PubChem fingerprint and RDKit2D descriptor) provided the highest average prediction performance. Taking the concatenation of these two types of fingerprints as input data, the performance results of XGB, RF, SVM and DNN algorithms are evaluated.
Molecular representation evaluation
TOXRIC visually shows the spatial distribution of multiple molecular representations on the classification tasks. Representations in multiple spaces are explored, including seven original molecular fingerprints, target protein, three transcriptome profiles and three ML-based feature embedding. The ML-based feature embedding refer to the representations after representation learning or feature selection by three ML models, i.e. XGB, RF, and DNN. The representation of XGB and RF model is based on feature selection. The top 100 important features are extracted through feature importance values provided by scikit-learn library (53). The DNN-based feature embedding is an internal representation extracted from the output of the last hidden layer of the above trained DNN. The t-SNE (t-distributed stochastic neighbor embedding) algorithm is applied to generate and visualize the two-dimensional embeddings of the representations. A scatter plot can intuitively show the clustering effect of each representation on the toxic or non-toxic classes.
Online database implementation
TOXRIC is built with a separated frontend and backend framework. The backend is implemented using the Springboot framework, and the frontend uses VUE framework. NGINX is used as a reverse proxy, enabling strong performance at scale. All the data are stored and managed using MySQL, and Elasticsearch is deployed as the search engine. Elasticsearch is a distributed, highly scalable, high real-time search and analysis engine. It can quickly search and analyze large amounts of data due to the advantage of the horizontal scalability. All the online data visualizations, including the bar charts and pie charts of statistics and benchmark results, are supported by ECharts 4.0 (54), an open-sourced JavaScript library for the rapid construction of interactive visualization. Software development tools include Python 3.8.5, PubChemPy 1.0.4, RDKit 2022.03.2, scikit-learn 0.24.2 (53), xgboost 1.4.2, and tensorflow 2.5.0. The website has been tested thoroughly to ensure functionality across multiple operating systems and web browsers.
DATABASE CONTENT AND USAGE
Data summary and analysis
The latest release of TOXRIC contains and catalogs toxicology information on 113 372 compounds, 13 toxicity categories and 1474 toxicity endpoints of more than 15 species. In addition, 39 feature types of these compounds are computed or curated. Importantly, all the datasets stored in TOXRIC are ML-ready and can be used directly as input or output to ML models.
The 13 toxicity categories contain a different number of endpoints (Figure 1A). ToxCast&Tox21 Assay contains the largest number of endpoints (1381 endpoints), followed by Acute Toxicity (59 endpoints) and Endocrine Disruption (12 endpoints), while other toxicity categories contain less than 5 endpoints. The number of compounds in each toxicity category is counted (Figure 1B). Acute Toxicity contains the largest number of compounds (79 725 compounds), accounting for 59.68% of all compounds, followed by CYP450 (16 280 compounds), accounting for 12.19%. Developmental and Reproductive Toxicity contains the least number of compounds (218 compounds), accounting for 0.16%. As shown in Figure 1C, each compound has multiple endpoint results (label data). Statistical results show that 10 668 compounds have only one endpoint, while 345 compounds have more than 180 endpoint results, which are distributed in Acute Toxicity and ToxCast&Tox21 Assay datasets.
Figure 1.

The statistical results of data stored in TOXRIC. (A) The number of endpoints for toxicity categories. (B) The number of compounds for toxicity categories. (C) The number of compounds with multiple toxicity endpoints. (D) The number of compounds for feature spaces. More statistical results are shown on Statistics page on TOXRIC web.
In the feature space, TOXRIC collects and calculates six feature spaces related to toxicity prediction, including molecular fingerprint, target protein, category of drug, transcriptome profile, metabolic reaction, and CC descriptor. For compounds, structural data is always available, while transcriptome data is scarce, covering only 2587 compounds, as shown in Figure 1D. Features of molecular fingerprints, transcriptome profiles, and CC descriptors are in a common vector format. The length of the feature vectors is between 128 (CC descriptor) and 15 406 (transcriptome profile in Open TG-GATEs) components. Among the features, RDKit2D descriptor, three transcriptome profiles and 25 CC descriptors consist of continuous values, while other features are composed of binary discrete values. More statistical results are shown on Statistics page on TOXRIC website.
Web design and interface
TOXRIC offers an effective and user-friendly web interface to take full advantage of the wealthy data, benchmarks and molecular representation distribution. Users can query the toxicological data, features, and chemical information of compounds in the search box of the Home page or on the Search page. The Data Collection page provides an overview of the toxicological and feature datasets, and the Statistics page shows the statistical results of the datasets. The Benchmark&Representation page displays visualization of benchmarks and molecular representation distribution. The Download page provides links to download data of all the endpoint and feature type datasets. Users can contribute their toxicology data on Contribute page. A detailed step-by-step tutorial of TOXRIC (Supplementary Data) and contact information is readily accessible on the Contact&About page.
Data browsing
On the Home page, the number of datasets of both toxicity categories and feature spaces is displayed in the form of two-layer concentric circles. The inner layer and outer layer represent toxicity category and feature space respectively. When clicking on the dataset field, users will be linked to the corresponding dataset description on the Data Collection page.
On the Data Collection page, three types of entries can be browsed, i.e. toxicity category, feature space and external database links. The basic information of the toxicity category dataset, toxicity endpoint, and feature type dataset is provided, including dataset description, number of compounds, sources and feature dimension. The interactive filter located on the left side of the pages allows users to explore the endpoint and feature type datasets. Users can click the details button to query the detailed information of the selected datasets. On the detail information page, all compounds contained in a dataset are listed in the form of a molecular graph (Figure 2A). Clicking on a specific compound will open the compound information page that displays affluent chemical, toxicological, and feature data of the compound.
Figure 2.

The web interface of TOXRIC. (A) The details page of a queried endpoint dataset (mouse_intraperitoneal_LD50, Acute Toxicity category). (B) Basic chemical information of a queried compound (menadione, TOX-672). (C) The list of toxicity values (Ecotoxicity category) with the queried compound. (D) The feature list (metabolic reactions) and (E) targets’ enrichment results of the queried compound. Visualization of the benchmarks for (F) feature types and (G) algorithms on the queried endpoint. (H) Visualization of two representations on CYP1A2 endpoint, i.e. ECFP6 fingerprint, DNN-based embedding.
The Statistics page describes some statistical results of datasets provided by TOXRIC in pie and bar charts, including the number of compounds for the toxicity categories and feature datasets, the number of endpoints/types for toxicity categories and feature spaces, and the number of compounds with multiple labels and features.
Data retrieval
The toxicological information of compounds can be quickly searched in the search box of the Home page or on the Search page. The search box accepts both complete or partial keywords of TAID, name, IUPAC name, PubChem CID, SMILES, InChIKey and InChI identifiers. Fuzzy search is allowed. If the typed terms can match multiple compound entities in the database, a list of suitable suggestions will be provided. The query keyword is highlighted in red. In addition to searching for individual compound, TOXRIC also provides a batch search for a list of compounds on the Search page. After selecting an identifier, users can enter a compound list or upload an EXCEL or TXT file to query the information of the compounds.
The compound information page consists of three sections, i.e. chemical information, toxicity category, and feature space. The chemical information section provides seven commonly used identifier types and physicochemical properties of compounds (Figure 2B). In the toxicity category section, a list of toxicity values of 13 toxicity categories is provided in tabular format (Figure 2C). Users can view the toxicity values of various endpoints by selecting a toxicity category. Below is a list of feature spaces (Figure 2D). The targets, categories, and metabolic reactions of compounds are listed in text format to be queried, while the feature vectors of transcriptome profiles, molecular fingerprints, and CC descriptors should be downloaded to use because the length of the vectors is too long to display. Users can download the toxicity endpoints or feature types of a compound by clicking on the download button in the upper-right corner. Below the feature list, the top 10 GOBP (Gene Ontology Biological Process) and KEGG pathway enrichment results of compounds' target proteins are displayed in a bubble plot (Figure 2E).
Benchmark and representation distribution display
The Benchmark&Representation page displays visualization of two types of benchmarks and a molecular representation distribution. On the Benchmarks for Feature Types page, the bar charts intuitively show the predictive effect of 36 feature types on all toxicity endpoints (Figure 2F). The feature types include seven molecular fingerprints, a target, three transcriptome profiles, and 25 CC descriptors. The mean and standard deviation are showed when the mouse is suspended on the bar. In classification datasets, F1 metric is used to evaluate the performance of features. For regression datasets (Acute Toxicity and Ecotoxicity), users can select RMSE or R2 metric to view the results by clicking the buttons above the bar chart. Lower RMSE value or higher R2/F1 value represent higher prediction performance. It should be noted that if the value of metric is 0, it represents the number of samples with the feature type at the endpoint dataset is <10 and no benchmark experiment is performed. Click on a bar or the title of an endpoint, the corresponding feature or endpoint dataset on the Download page will open in a new tab. In addition, users can enter the keywords of required endpoint and feature in the search box at the top right corner to search.
The Benchmarks for Algorithms page shows the benchmark results of four typical algorithms on all toxicity endpoints, i.e. XGB, RF, SVM, and DNN (Figure 2G). Each picture shows 10 endpoints, slide the mouse on the bar chart and drag the scroll bar below the chart to view the results of 10 endpoints. The mean and standard deviation are shown when the mouse is suspended on the bar.
The T-SNE Embedding of Molecular Representations page shows the clustering effects of multiple representations in scatter plots on the classification endpoint datasets (Figure 2H). The representations include 11 original features and three ML-based representations. The original features are seven molecular fingerprints, a target, and three transcriptome profiles. The ML-based representations refer to vectors after representation learning or feature selection by three typical ML models, i.e. DNN, RF, and XGB.
Downloads
All toxicological and feature data can be downloaded from the website without login or registration. Datasets of different endpoints and feature types can be downloaded separately.
Contribute
In order for the TOXRIC to continue to grow and expand, we rely on the community to help us through contributions. Users can contribute toxicology data to TOXRIC by uploading data in the contribute box on Contribute page or contacting us.
CASE STUDY
Example application
This section describes how to use TOXRIC for toxicity prediction using the mouse_intraperitoneal_LD50 dataset (Acute Toxicity category) as an example (Figure 3). First, this endpoint dataset can be downloaded on the Download page as the label data (Figure 3A). By searching the benchmarks of feature types on the Benchmark&Representation page, it is found that the MACCS molecular fingerprint achieves the best performance (RMSE metric) on this endpoint (Figure 3B). Then, the MACCS fingerprint dataset can be downloaded as the input feature (Figure 3C). The input and output datasets are ready for toxicity prediction. By searching the benchmarks of algorithms, the RF algorithm achieves the best performance (RMSE metric) on this endpoint (Figure 3D). Therefore, in this dataset, RF can be considered as the baseline for the development of new ML algorithms. A step-by-step example application for toxicity prediction is provided on Supplementary data and Contact&About page of TOXRIC website. In addition, considering potential relationships between different toxicity endpoints, the TOXRIC database can be fully utilized to explore multitask learning (MTL) and transfer learning (TL) methods (3).
Figure 3.

An application examples of data and benchmarks provided in TOXRIC. (A) The Download page of toxicity dataset . The download button can be clicked to download the endpoint dataset. (B) The Benchmarks for Feature Types page. (C) The Download page of feature dataset. The download button can be clicked to download the endpoint dataset. (D) The Benchmarks for Algorithms page.
Analysis of benchmark results
For computational prediction, it is of vital importance to select an appropriate feature type and baseline model according to the benchmark results. TOXRIC provides two types of benchmarks for all endpoints, i.e. benchmarks for feature types and typical algorithms. Taking the regression dataset as an example, we compare and analyze the performance of different feature types. We average the RMSE values on all regression datasets for each feature type. Molecular fingerprints show significant performance advantages over other features. Among them, RDKit2D descriptor has the best performance (0.8596), and ECFP6 fingerprint has the worst performance (0.9104). However, in the mammal (species unspecified)_subcutaneous_LD50 dataset, ECFP6 has the best performance, which is 9.55% higher than RDKit2D. It is observed that there is no optimal feature type that performs best in all endpoints. In most cases, the RDKit2D descriptor and PubChem fingerprint can achieve better performance. Researchers should select appropriate feature types for different endpoints according to the benchmark results.
Next, the benchmark results of the four algorithms (XGB, RF, SVM, DNN) are analyzed. In terms of the average F1 and RMSE values for all datasets, XGB achieves the best average F1 value and RF achieves the best average RMSE value. SVM has the lowest average F1 value and DNN get the worst regression performance. But in some specific datasets, SVM or DNN can get the best performance. For example, in the Endocrine Disruption category, the F1 value of DNN in the NA-AR endpoint is 0.529, which is 8.78% worse than the RF (0.616). While in the SR-ARE endpoint, the performance of DNN is the best (0.514), outperforming RF by 9.60% (0.371). Thus, the optimal feature types or baseline models are different in different endpoints. Researchers should select appropriate feature type or baseline model at each endpoint according to the benchmarks provided by TOXRIC.
Benchmark results can also verify the quality of training dataset from an applied perspective. For example, we average the performance results on seven molecular fingerprints for each endpoint dataset. In classification datasets, the average F1 values under two categories (Irritation and Corrosion, Developmental and Reproductive Toxicity) exceeds 0.89, indicating the quality of training dataset. Except for the ToxCast&Tox21 Assay, 21 of 30 endpoints have average F1 values over 0.5. In ToxCast&Tox21 Assay, 1110 of 1381 endpoints have average F1 values <0.5, which may be due to the small-sized and class-imbalanced characteristics. There are 514 endpoints with an imbalance rate >9:1, and even 148 endpoints with an imbalance rate >50:1. The datasets with poor benchmark results are also provided in TOXRIC. This may motivate researchers to develop new prediction methods to solve the problem of small-sized and class-imbalanced datasets, which are significant issues in computational toxicology communities.
Characteristics of molecular representations
In addition to the two types of benchmarks, TOXRIC also displays the spatial distribution of multiple molecular representations on the Benchmark&Representation page. The features of structural, target, transcriptome profile and ML-based representation are projected to two dimensions in scatter plots by the t-SNE algorithm. Compared to the representations trained by ML models, the original features can hardly show the clustering effect on all the classification tasks. There is a lot of overlap between clusters, indicating that the features cannot well distinguish the classes. However, for the ML-based feature spaces especially DNN-based embedding, there is a clear separation of the two classes in all tasks. After mapping the original features to the new feature embedding space, the DNN can better learn the rules in the input data. In addition, the features of target and transcriptome profiles show completely different distributions from the molecular fingerprints, which may provide new insights into the representations of compounds for related studies.
Application scenarios
Application scenarios of TOXRIC includes:
Individual compound searches and downloads are available for toxicological investigations, interpretation of toxicological mechanisms, and compound/drug discovery. The toxicity values, chemical information, transcriptional profiles, metabolic reaction equations, targets and enrichment results of each compound can be queried through the TOXRIC website. All the related toxicological and feature data for an individual compound can be downloaded on the compound page.
To help researches better understand the representation of compounds in different feature spaces, TOXRIC provides visualization of molecular representation distribution of multiple feature types, including original structure, target, transcriptome feature and embedding after representation learning of ML models. The clustering effects of molecular representations can help researchers intuitively understand and interpret the mechanisms of toxicities in different spaces.
For development of computational methods in toxicity prediction, TOXRIC provides comprehensive ML-ready datasets and benchmarks. The ML-ready endpoint and feature type datasets can be downloaded separately on the Download page and can be used directly as output or input to ML models for toxicity prediction. The two types of benchmarks can help researchers select appropriate feature types and baseline algorithms for each toxicity endpoint prediction task.
COMPARISON WITH EXISTING DATABASES
There is an abundance of online databases providing access to large amounts of toxicology data. For example, researchers within U.S. Environmental Protection Agency (EPA)’s National Center for Computational Toxicology (NCCT) have developed several research programs, databases and web-based interfaces (dashboards), including the ToxCast program and the affiliated Tox21 program (22,23), Aggregated Computational Toxicology Online Resource (ACToR) (55), DSSTox database (56) and CompTox Chemistry Dashboard (Dashboard) (57,58), which provides comprehensive in vitro bioassay data and other chemical information of compounds. We classified the existing online toxicological databases into four categories, toxicity category-centric, toxic feature-centric, compound-centric, and ML task-centric. Some statistics and comparisons of the databases are listed in Table 1.
Table 1.
Statistics and comparison between TOXRIC and existing toxicological databases
Note: The list of existing databases is referenced from the study by Lin et al. (71), Jeong et al. (48), Vo et al. (72) and the resources provided by American Society of Cellular and Computational Toxicology (https://www.ascctox.org/resources).
Toxicity category-centric databases
Databases in this category target specific toxicity categories or testing programs of compounds. For instance, EPA’s ToxCast and Tox21 program are engaged in the generation and analysis of in vitro bioassay data for thousands of chemicals evaluated in high-throughput (HTS) assays. CEBS (13) collects animal public environmental health data primarily from National Toxicology Program (NTP) testing program. EnviroTox (59) aims at ecotoxicity. DILIrank database (16) focuses on data of drug-induced liver toxicity. The above databases focus on a single toxicity category, and the data available in most of these databases are integrated into TOXRIC.
In addition, The Integrated Chemical Environment (ICE) database (60) has collected multiple toxicity categories from multiple sources, including ToxCast data, acute toxicity, endocrine, developmental and reproductive toxicity. The functions of data retrieval and dataset download are provided. Whereas the endpoints of the collected datasets are not further processed and curated into ML-ready format. TOXRIC not only curated more endpoints into a standardized format, but also provides multiple curated feature types and benchmark results of each endpoint dataset.
Toxic feature-centric databases
The toxic feature-centric databases provide toxic-related omics or other feature data, such as ToxicoDB (67), DrugMatrix (45), and Open TG-GATEs databases (61). All of them are toxicogenomic database that stores gene expression profiles derived from in vivo, in vitro exposure to compounds. DrugMatrix contains gene expression data of >200 compounds tested in the in vivo rat tissues (e.g. liver), and 125 compounds tested in the in vitro rat hepatocytes. Open TG-GATEs stores gene expression profiles derived from in vivo (rat) and in vitro (primary rat hepatocytes, primary human hepatocytes) exposure to 170 compounds at multiple dosages and time points. Data in ToxicoDB is collected from the above two databases, which are also collected into the feature space of TOXRIC. Besides the transcriptome profiles, TOXRIC provides 39 feature types covering structural, target, metabolic data and other descriptors.
Compound-centric databases
The compound-centric databases provide comprehensive chemical and toxicological information on compounds. EPA’s DSSTox database (launched in 2004) and Dashboard (launched in 2016) are used as integration platforms that combined the ToxCast/Tox21 programs with other research efforts. The EPA’s ACToR database contains knowledge extracted from multiple collections and has been retired now. The above EPA’s databases focus primarily on data from in vitro bioassays. The most widely used database, Dashboard, provides chemical/toxicology data retrieval, and compound/assay dataset download. However, except for ToxCast/Tox21 assay, data on other toxicity categories is not organized into datasets for download. For other compound-centric databases, T3DB database (15) focuses on information of toxic exposome, including detailed chemical and target information of toxins. The eChemPortal database provides free access to chemicals and associated properties, allowing searches by compounds and Global Hazard Summary (GHS) classification. These databases serve only to retrieve multiple toxicities and chemical information of compounds. There is no curated endpoint dataset. Compared to these databases, TOXRIC not only provides data retrieval of compounds, but also provides the curated endpoint and feature type datasets that can be used directly in computational methods.
In addition, there are some large chemically-indexed databases, such as PubChem (62), ChEMBL (63), and CTD (64), which are not focus on toxicology, but provide toxicology-related information. However, the toxicological data provided by them is not organized into dataset. Most of the information provided for retrieval is the text content of literatures, and no qualitative or quantitative toxicity value is provided.
ML task-centric databases
There are two ML task-centric databases, TDC (17) and MoleculeNet (18), which are platforms to provide datasets, ML tasks, benchmarks, and other information for various fields of study, such as ADMET, drug combination, quantum mechanical- and biophysical-related studies. Neither is a database directly focused on toxicology, nor does it provide data retrieval. In TDC, the datasets need to be obtained by calling the functions from Python code, and the benchmarks need to be obtained after the training process. In MoleculeNet, the benchmarks of four toxicology datasets and eight algorithms are provided in bar charts. Neither is a database provide benchmark for features and representation distribution. And the toxicology datasets are not categorized by toxicity categories. Compared to both databases, TOXRIC focuses on toxicity studies, covering chemical information, more toxicity categories, and feature types of compounds, which can be quickly retrieved on the web. The benchmark results displayed on TOXRIC web are intuitive and contain more feature types on >1000 endpoint datasets.
Taken together, TOXRIC covers a wider range of toxicity categories and feature spaces. The toxicity values, chemical information, transcriptional profiles, metabolic reaction equations, targets, and enrichment results of each compound can be retrieved through the TOXRIC website. The endpoint and feature type datasets have been organized into ML-ready format to be downloaded. The benchmarks for feature types and algorithms can facilitate the development of AI-based computational toxicology. The visualization of molecular representation distributions can help researchers better understand and interpret the mechanisms of toxicities in different spaces.
CONCLUSION AND DISCUSSION
TOXRIC has five core functions, comprehensive data, ML-ready downloadable datasets, practical benchmarks, informative visualization of molecular representations, and an intuitive function interface. To accelerate the development of toxicity evaluation and compound discovery, TOXRIC provides comprehensive and standardized toxicological, feature, and chemical data for toxicity retrieval. To better utilize and understand the data, TOXRIC performs a system-level analysis and displays informative visualization of benchmarks and molecular representations on all the toxicity endpoints. TOXRIC offers an effective and user-friendly web interface to take full advantage of the wealthy data, ML-ready datasets, benchmarks, and molecular representation distribution. Users can retrieve the toxicological information of compounds, browse the detailed information of datasets, download the curated datasets, view the results of benchmarks and molecular representation distribution on TOXRIC web.
For data collection, TOXRIC has always been committed to implementing the FAIR principles (73) to enhance the findability, accessibility, interoperability, and reusability of its data. To be findable, all compounds in TOXRIC come with a unique and persistent identifier, TAID, which is used to link the toxicology and feature data of each compound. TOXRIC is freely accessible for academics from a stable, permanent web address (https://toxric.bioinforai.tech/), and does not require account creation or logon. TOXRIC can also be dynamically queried through HTML direct links. To be interoperable, TOXRIC incorporates identifiers and physicochemical properties of compounds from PubChem, and provides external links to PubChem on compound page. Users can click on the PubChem CID to link to the corresponding compound page of PubChem website. In addition, TOXRIC web provide links to all the related databases on the External Database Links page. To be reusable, TOXRIC’s curation paradigms are fully described in the manuscript. All datasets collected in the database are described in detail on Data Collection page and can be reused in downloadable files with machine-readable formats (.csv) on Download page of the website.
The 1474 endpoint datasets stored in TOXRIC contain in vivo and in vitro datasets. For the in vivo endpoints, the ADME process of various pharmacokinetic effects can be measured. However, live animal experiments are flawed with ethical, economic, and scientific limitations. Compared with in vivo endpoints, in vitro experiment has advantages in terms of economy, condition control and safety. Computational methods can be used to relate in vitro to in vivo endpoints, i.e. in vitro to in vivo extrapolation (IVIVE) method (74). The IVIVE is often performed with a toxicokinetic (TK) or a physiologically pharmacokinetic (PBPK) modeling-based reverse dosimetry, to estimate the in vivo dose required to achieve an in vitro bioactive concentration in the blood or target tissue (74–76). The bioactive concentration includes point of departures (PODs), activity concentration causing 50% maximum activity (AC50), or lowest effective concentration. In TOXRIC database, there are a large amount of in vitro data stored in ToxCast&Tox21 Assay, which uses HTS techniques to study the perturbations provoked by chemicals to biochemical and biological pathways in isolated systems in vitro. Some IVIVE approaches have been applied to translate in vitro bioactivity concentrations from the ToxCast&Tox21 data to the daily human oral doses (i.e. human equivalent doses, HEDs) (75–78).
Several limitations in the TOXRIC should be elaborated. First, TOXRIC simply provide both in vitro and in vivo endpoints, without further distinguishing and linking the in vitro and in vivo data. In vitro results do not necessary, neither always, correlate to in vivo. This is an issue that needs to be taken care of whether using in vitro data for model building, or using IVIVE methods to extrapolate in vitro data into in vivo. Second, the data provided by TOXRIC is still insufficient. There are still many resources involved toxicology data, such as PubChem, ChEMBL and related literatures. The NOAEL or LOAEL data should also be integrated into TOXRIC. For feature spaces, multi-omics integration will be a core content of TOXRC in the future. Currently, there is only transcriptome data integrated in TOXRIC, other omics data should be added. Third, some datasets show poor performance in benchmarks may be due to the characteristics such as small-sized samples and class imbalance. Thus, more data should be collected in these datasets. At the same time, these characteristics of datasets should be further analyzed, which will motivate ML scientists to develop new algorithms for this problem.
In the future, we plan to integrate more data from other resources and literatures into TOXRIC, including more toxicity categories, toxicity values, compounds, multi-omics and multi-views feature types. The data quality will be controlled manually and the data management will follow the FAIR principles. In addition, the detailed analysis of characteristics of endpoint datasets will be added according to benchmarks and representation distributions. More visualization tools will be developed to display the characteristics of datasets. Then researchers can develop better methods to solve the problems of datasets.
TOXRIC will be continuously updated with new entries, datasets, and benchmarks to facilitate the development of compound discovery and toxicity prediction efforts of ML scientists. We hope that TOXRIC can serve as a comprehensive resource for the toxicologists and a benchmark platform for the computational toxicology community to drive algorithmic and scientific advances.
DATA AVAILABILITY
The data is provided via TOXRIC website (https://toxric.bioinforai.tech/).
Supplementary Material
Contributor Information
Lianlian Wu, Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing 100850, China; Academy of Medical Engineering and Translational Medicine, Tianjin University, Tianjin 300072, China.
Bowei Yan, Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing 100850, China; State Key Laboratory of Genetic Engineering and Collaborative Innovation Center for Genetics and Development, School of Life Sciences, Institute of Biomedical Sciences, Human Phenome Institute, Fudan University, Shanghai 200433, China; State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences, Beijing 102206, China.
Junshan Han, Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing 100850, China.
Ruijiang Li, Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing 100850, China.
Jian Xiao, Department of Pharmacy, Xiangya Hospital, Central South University, Changsha 410008, Hunan, China; Institute for Rational and Safe Medication Practices, National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha 410008, Hunan, China.
Song He, Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing 100850, China.
Xiaochen Bo, Department of Bioinformatics, Institute of Health Service and Transfusion Medicine, Beijing 100850, China; Academy of Medical Engineering and Translational Medicine, Tianjin University, Tianjin 300072, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [62103436]. Funding for open access charge: National Natural Science Foundation of China [62103436].
Conflict of interest statement. None declared.
REFERENCES
- 1. Raies A.B., Bajic V.B.. In silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev. Comput. Mol. Sci. 2016; 6:147–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giri S., Bader A.. A low-cost, high-quality new drug discovery process using patient-derived induced pluripotent stem cells. Drug Discov. Today. 2015; 20:37–49. [DOI] [PubMed] [Google Scholar]
- 3. Wu Z., Jiang D., Wang J., Hsieh C.Y., Cao D., Hou T.. Mining toxicity information from large amounts of toxicity data. J. Med. Chem. 2021; 64:6924–6936. [DOI] [PubMed] [Google Scholar]
- 4. Yang H., Sun L., Li W., Liu G., Tang Y.. Silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front. Chem. 2018; 6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roy K., Kar S.. Silico models for ecotoxicity of pharmaceuticals. Methods Mol. Biol. 2016; 1425:237–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kapanen A., Itavaara M.. Ecotoxicity tests for compost applications. Ecotoxicol. Environ. Saf. 2001; 49:1–16. [DOI] [PubMed] [Google Scholar]
- 7. Venkatapathy R., Wang N.C.. Developmental toxicity prediction. Methods Mol. Biol. 2013; 930:305–340. [DOI] [PubMed] [Google Scholar]
- 8. Modi S., Hughes M., Garrow A., White A.. The value of in silico chemistry in the safety assessment of chemicals in the consumer goods and pharmaceutical industries. Drug Discov. Today. 2012; 17:135–142. [DOI] [PubMed] [Google Scholar]
- 9. Deeb O., Goodarzi M.. In silico quantitative structure toxicity relationship of chemical compounds: some case studies. Curr. Drug Saf. 2012; 7:289–297. [DOI] [PubMed] [Google Scholar]
- 10. Jain S., Siramshetty V.B., Alves V.M., Muratov E.N., Kleinstreuer N., Tropsha A., Nicklaus M.C., Simeonov A., Zakharov A.V.. Large-Scale modeling of multispecies acute toxicity end points using consensus of multitask deep learning methods. J. Chem. Inf. Model. 2021; 61:653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Segall M.D., Barber C.. Addressing toxicity risk when designing and selecting compounds in early drug discovery. Drug Discov. Today. 2014; 19:688–693. [DOI] [PubMed] [Google Scholar]
- 12. Dix D.J., Houck K.A., Martin M.T., Richard A.M., Setzer R.W., Kavlock R.J.. The toxcast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 2007; 95:5–12. [DOI] [PubMed] [Google Scholar]
- 13. Lea I.A., Gong H., Paleja A., Rashid A., Fostel J.. CEBS: a comprehensive annotated database of toxicological data. Nucleic Acids Res. 2017; 45:D964–D971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmidt U., Struck S., Gruening B., Hossbach J., Jaeger I.S., Parol R., Lindequist U., Teuscher E., Preissner R.. SuperToxic: a comprehensive database of toxic compounds. Nucleic Acids Res. 2009; 37:D295–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wishart D., Arndt D., Pon A., Sajed T., Guo A.C., Djoumbou Y., Knox C., Wilson M., Liang Y., Grant J.et al.. T3DB: the toxic exposome database. Nucleic Acids Res. 2015; 43:D928–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen M., Suzuki A., Thakkar S., Yu K., Hu C., Tong W.. DILIrank: the largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov Today. 2016; 21:648–653. [DOI] [PubMed] [Google Scholar]
- 17. Huang K., Fu T., Gao W., Zhao Y., Roohani Y., Leskovec J., Coley C.W., Xiao C., Sun J., Zitnik M.. Artificial intelligence foundation for therapeutic science. Nat. Chem. Biol. 2022; 18:1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu Z.Q., Ramsundar B., Feinberg E.N., Gomes J., Geniesse C., Pappu A.S., Leswing K., Pande V.. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 2018; 9:513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ai H., Chen W., Zhang L., Huang L., Yin Z., Hu H., Zhao Q., Zhao J., Liu H.. Predicting drug-induced liver injury using ensemble learning methods and molecular fingerprints. Toxicol. Sci. 2018; 165:100–107. [DOI] [PubMed] [Google Scholar]
- 20. Sosnin S., Karlov D., Tetko I.V., Fedorov M.V.. Comparative study of multitask toxicity modeling on a broad chemical space. J. Chem. Inf. Model. 2018; 59:1062–1072. [DOI] [PubMed] [Google Scholar]
- 21. Aguirre-Plans J., Piñero J., Souza T., Callegaro G., Kunnen S.J., Sanz F., Fernandez-Fuentes N., Furlong L.I., Guney E., Oliva B.. An ensemble learning approach for modeling the systems biology of drug-induced injury. Biol. Direct. 2021; 16:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kavlock R, Chandler K, Houck K, Hunter S, Judson R, Kleinstreuer N, Knudsen T, Martin M, Padilla S, Reif Det al.. Update on EPA’s toxcast program: providing high through-put decision support tools for chemical risk management. Chem. Res. Toxicol. 2012; 25:1287–1302. [DOI] [PubMed] [Google Scholar]
- 23. Thomas R.S., Paules R.S., Simeonov A., Fitzpatrick S.C., Crofton K.M., Casey W.M., Mendrick D.L.. The US federal tox21 program: a strategic and operational plan for continued leadership. ALTEX. 2018; 35:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen M., Zhang J., Wang Y., Liu Z., Kelly R., Zhou G., Fang H., Borlak J., Tong W.. The liver toxicity knowledge base: a systems approach to a complex end point. Clin. Pharmacol. Ther. 2013; 93:409–412. [DOI] [PubMed] [Google Scholar]
- 25. Hoofnagle J.H., Serrano J., Knoben J.E., Navarro V.J.. LiverTox: a website on drug-induced liver injury. Hepatology. 2013; 57:873–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tomasulo P. ChemIDplus-super source for chemical and drug information. Med. Ref. Serv. Q. 2002; 21:53–59. [DOI] [PubMed] [Google Scholar]
- 27. Jiang D., Wu Z., Hsieh C.Y., Chen G., Liao B., Wang Z., Shen C., Cao D., Wu J., Hou T.. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminform. 2021; 13:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. He S., Ye T., Wang R., Zhang C., Zhang X., Sun G., Sun X.. An in silico model for predicting drug-induced hepatotoxicity. Int. J. Mol. Sci. 2019; 20:1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Banerjee P., Eckert A.O., Schrey A.K., Preissner R.. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018; 46:W257–W263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garcia de Lomana M., Morger A., Norinder U., Buesen R., Landsiedel R., Volkamer A., Kirchmair J., Mathea M.. ChemBioSim: enhancing conformal prediction of in vivo toxicity by use of predicted bioactivities. J. Chem. Inf. Model. 2021; 61:3255–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang C., Cheng F., Li W., Liu G., Lee P.W., Tang Y.. In silico prediction of drug induced liver toxicity using substructure pattern recognition method. Mol. Inform. 2016; 35:136–144. [DOI] [PubMed] [Google Scholar]
- 32. Jain S., Siramshetty V.B., Alves V.M., Muratov E.N., Kleinstreuer N., Tropsha A., Nicklaus M.C., Simeonov A., Zakharov A.V.. Large-Scale modeling of multispecies acute toxicity end points using consensus of multitask deep learning methods. J. Chem. Inf. Model. 2021; 61:653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sosnin S., Karlov D., Tetko I.V., Fedorov M.V.. Comparative study of multitask toxicity modeling on a broad chemical space. J. Chem. Inf. Model. 2019; 59:1062–1072. [DOI] [PubMed] [Google Scholar]
- 34. Liu Z.C., Huang R.L., Roberts R., Tong W.D.. Toxicogenomics: a 2020 vision. Trends Pharmacol. Sci. 2019; 40:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bento A.P., Hersey A., Felix E., Landrum G., Gaulton A., Atkinson F., Bellis L.J., De Veij M., Leach A.R.. An open source chemical structure curation pipeline using RDKit. J Cheminform. 2020; 12:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Greene N., Fisk L., Naven R.T., Note R.R., Patel M.L., Pelletier D.J.. Developing structure-activity relationships for the prediction of hepatotoxicity. Chem. Res. Toxicol. 2010; 23:1215–1222. [DOI] [PubMed] [Google Scholar]
- 37. Xu Y., Dai Z., Chen F., Gao S., Pei J., Lai L.. Deep learning for drug-induced liver injury. J. Chem. Inf. Model. 2015; 55:2085–2093. [DOI] [PubMed] [Google Scholar]
- 38. Chen M.J., Vijay V., Shi Q., Liu Z.C., Fang H., Tong W.D.. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov Today. 2011; 16:697–703. [DOI] [PubMed] [Google Scholar]
- 39. Duran-Frigola M., Pauls E., Guitart-Pla O., Bertoni M., Alcalde V., Amat D., Juan-Blanco T., Aloy P.. Extending the small-molecule similarity principle to all levels of biology with the chemical checker. Nat. Biotechnol. 2020; 38:1087–1096. [DOI] [PubMed] [Google Scholar]
- 40. Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z.. DrugBank 5.0: a major update to the drugbank database for 2018. Nucleic Acids Res. 2018; 46:D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gilson M.K., Liu T., Baitaluk M., Nicola G., Hwang L., Chong J.. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016; 44:D1045–D1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. MohammadiPeyhani H., Chiappino-Pepe A., Haddadi K., Hafner J., Hadadi N., Hatzimanikatis V.. NICEdrug.ch, a workflow for rational drug design and systems-level analysis of drug metabolism. Elife. 2021; 10:e65543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K.et al.. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017; 171:1437–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Igarashi Y., Nakatsu N., Yamashita T., Ono A., Ohno Y., Urushidani T., Yamada H.. Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Res. 2015; 43:D921–D927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ring C., Sipes N.S., Hsieh J.H., Carberry C., Koval L.E., Klaren W.D., Harris M.A., Auerbach S.S., Rager J.E.. Predictive modeling of biological responses in the rat liver using in vitro tox21 bioactivity: benefits from high-throughput toxicokinetics. Comput. Toxicol. 2021; 18:100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ganter B., Snyder R.D., Halbert D.N., Lee M.D.. Toxicogenomics in drug discovery and development: mechanistic analysis of compound/class-dependent effects using the drugmatrix database. Pharmacogenomics. 2006; 7:1025–1044. [DOI] [PubMed] [Google Scholar]
- 47. Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P.. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–264. [DOI] [PubMed] [Google Scholar]
- 48. Jeong J., Choi J.. Artificial intelligence-based toxicity prediction of environmental chemicals: future directions for chemical management applications. Environ. Sci. Technol. 2022; 56:7532–7543. [DOI] [PubMed] [Google Scholar]
- 49. Chen T.Q., Guestrin C.. XGBoost: a scalable tree boosting system. Kdd'16: Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining. 2016; 785–794. [Google Scholar]
- 50. Breiman L. Random forests. Mach. Learn. 2001; 45:5–32. [Google Scholar]
- 51. Noble W.S. What is a support vector machine?. Nat. Biotechn. 2006; 24:1565–1567. [DOI] [PubMed] [Google Scholar]
- 52. Wu L., Wen Y., Leng D., Zhang Q., Dai C., Wang Z., Liu Z., Yan B., Zhang Y., Wang J.et al.. Machine learning methods, databases and tools for drug combination prediction. Brief. Bioinform. 2022; 23:bbab355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V.. Scikit-learn: machine learning in python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 54. Li D., Mei H., Shen Y., Su S., Zhang W., Wang J., Zu M., Chen W.. ECharts: a declarative framework for rapid construction of web-based visualization. Visual Informatics. 2018; 2:136–146. [Google Scholar]
- 55. Judson R.S., Martin M.T., Egeghy P., Gangwal S., Reif D.M., Kothiya P., Wolf M., Cathey T., Transue T., Smith D.et al.. Aggregating data for computational toxicology applications: the U.S. environmental protection agency (EPA) aggregated computational toxicology resource (ACToR) system. Int. J. Mol. Sci. 2012; 13:1805–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grulke C.M., Williams A.J., Thillanadarajah I., Richard A.M.. EPA’s DSSTox database: history of development of a curated chemistry resource supporting computational toxicology research. Comput Toxicol. 2019; 12:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Williams A.J., Lambert J.C., Thayer K., Dorne J.C.M.. Sourcing data on chemical properties and hazard data from the US-EPA comptox chemicals dashboard: a practical guide for human risk assessment. Environ. Int. 2021; 154:106566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Williams A.J., Grulke C.M., Edwards J., McEachran A.D., Mansouri K., Baker N.C., Patlewicz G., Shah I., Wambaugh J.F., Judson R.S.et al.. The comptox chemistry dashboard: a community data resource for environmental chemistry. J. Cheminform. 2017; 9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Connors K.A., Beasley A., Barron M.G., Belanger S.E., Bonnell M., Brill J.L., de Zwart D., Kienzler A., Krailler J., Otter R.et al.. Creation of a curated aquatic toxicology database: envirotox. Environ. Toxicol. Chem. 2019; 38:1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bell S.M., Phillips J., Sedykh A., Tandon A., Sprankle C., Morefield S.Q., Shapiro A., Allen D., Shah R., Maull E.A.et al.. An integrated chemical environment to support 21st-Century toxicology. Environ. Health Perspect. 2017; 125:054501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Igarashi Y., Nakatsu N., Yamashita T., Ono A., Ohno Y., Urushidani T., Yamada H.. Open TG-GATEs: a large-scale toxicogenomics database. Nucleic Acids Res. 2015; 43:D921–D927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B.et al.. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021; 49:D1388–D1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mendez D., Gaulton A., Bento A.P., Chambers J., De Veij M., Felix E., Magarinos M.P., Mosquera J.F., Mutowo P., Nowotka M.et al.. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019; 47:D930–D940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davis A.P., Grondin C.J., Johnson R.J., Sciaky D., Wiegers J., Wiegers T.C., Mattingly C.J.. Comparative toxicogenomics database (CTD): update 2021. Nucleic Acids Res. 2021; 49:D1138–D1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Richman T., Arnold E., Williams A.J.. Curation of a list of chemicals in biosolids from EPA national sewage sludge surveys & biennial review reports. Sci. Data. 2022; 9:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bitsch A., Jacobi S., Melber C., Wahnschaffe U., Simetska N., Mangelsdorf I.. REPDOSE: a database on repeated dose toxicity studies of commercial chemicals–A multifunctional tool. Regul. Toxicol. Pharmacol. 2006; 46:202–210. [DOI] [PubMed] [Google Scholar]
- 67. Nair S.K., Eeles C., Ho C., Beri G., Yoo E., Tkachuk D., Tang A., Nijrabi P., Smirnov P., Seo H.et al.. ToxicoDB: an integrated database to mine and visualize large-scale toxicogenomic datasets. Nucleic Acids Res. 2020; 48:W455–W462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Judson R., Richard A., Dix D., Houck K., Elloumi F., Martin M., Cathey T., Transue T.R., Spencer R., Wolf M.. ACToR — aggregated computational toxicology resource. Toxicol. Appl. Pharmacol. 2008; 233:7–13. [DOI] [PubMed] [Google Scholar]
- 69. Jeliazkova N., Chomenidis C., Doganis P., Fadeel B., Grafstrom R., Hardy B., Hastings J., Hegi M., Jeliazkov V., Kochev N.et al.. The eNanoMapper database for nanomaterial safety information. Beilstein J. Nanotechnol. 2015; 6:1609–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vinken M., Pauwels M., Ates G., Vivier M., Vanhaecke T., Rogiers V.. Screening of repeated dose toxicity data present in SCC(NF)P. /SCCS safety evaluations of cosmetic ingredients. Arch. Toxicol. 2012; 86:405–412. [DOI] [PubMed] [Google Scholar]
- 71. Lin Z., Chou W.-C.. Machine learning and artificial intelligence in toxicological sciences. Toxicol. Sci. 2022; 189:7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vo A.H., Van Vleet T.R., Gupta R.R., Liguori M.J., Rao M.S.. An overview of machine learning and big data for drug toxicity evaluation. Chem. Res. Toxicol. 2020; 33:20–37. [DOI] [PubMed] [Google Scholar]
- 73. Wilkinson M.D., Dumontier M., Aalbersberg I.J., Appleton G., Axton M., Baak A., Blomberg N., Boiten J.W., Santos L.B.D., Bourne P.E.et al.. The FAIR guiding principles for scientific data management and stewardship. Sci Data. 2019; 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bell S.M., Chang X., Wambaugh J.F., Allen D.G., Bartels M., Brouwer K.L.R., Casey W.M., Choksi N., Ferguson S.S., Fraczkiewicz G.et al.. In vitro to in vivo extrapolation for high throughput prioritization and decision making. Toxicol. In Vitro. 2018; 47:213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wetmore B.A. Quantitative in vitro-to-in vivo extrapolation in a high-throughput environment. Toxicology. 2015; 332:94–101. [DOI] [PubMed] [Google Scholar]
- 76. Wetmore B.A., Wambaugh J.F., Ferguson S.S., Sochaski M.A., Rotroff D.M., Freeman K., Clewell H.J. III, Dix D.J., Andersen M.E., Houck K.Aet al.. Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol. Sci. 2012; 125:157–174. [DOI] [PubMed] [Google Scholar]
- 77. Rotroff D.M., Wetmore B.A., Dix D.J., Ferguson S.S., Clewell H.J., Houck K.A., LeCluyse E.L., Andersen M.E., Judson R.S., Smith C.M.et al.. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol. Sci. 2010; 117:348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lin Y.J., Lin Z.. In vitro-in silico-based probabilistic risk assessment of combined exposure to bisphenol a and its analogues by integrating toxcast high-throughput in vitro assays with in vitro to in vivo extrapolation (IVIVE) via physiologically based pharmacokinetic (PBPK) modeling. J. Hazard. Mater. 2020; 399:122856. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data is provided via TOXRIC website (https://toxric.bioinforai.tech/).