


Abstract
Transcriptome-wide association studies (TWASs), as a practical and prevalent approach for detecting the associations between genetically regulated genes and traits, are now leading to a better understanding of the complex mechanisms of genetic variants in regulating various diseases and traits. Despite the ever-increasing TWAS outputs, there is still a lack of databases curating massive public TWAS information and knowledge. To fill this gap, here we present TWAS Atlas (https://ngdc.cncb.ac.cn/twas/), an integrated knowledgebase of TWAS findings manually curated from extensive literature. In the current implementation, TWAS Atlas collects 401,266 high-quality human gene–trait associations from 200 publications, covering 22,247 genes and 257 traits across 135 tissue types. In particular, an interactive knowledge graph of the collected gene–trait associations is constructed together with single nucleotide polymorphism (SNP)–gene associations to build up comprehensive regulatory networks at multi-omics levels. In addition, TWAS Atlas, as a user-friendly web interface, efficiently enables users to browse, search and download all association information, relevant research metadata and annotation information of interest. Taken together, TWAS Atlas is of great value for promoting the utility and availability of TWAS results in explaining the complex genetic basis as well as providing new insights for human health and disease research.
INTRODUCTION
Transcriptome-wide association studies (TWASs), by integrating genome-wide association studies (GWASs) and data from expression quantitative trait locus (eQTL) studies, have become an effective approach for identifying trait-related genes (1). Although more and more GWASs have uncovered thousands of genomic loci associated with complex traits, the majority of trait-related variants are located in non-coding regions that are difficult to interpret (2,3). Plus, the detection of variants with small or moderate effects requires a fairly large sample size to achieve reliable statistical capability (4,5). TWAS can overcome these issues by aggregating regulatory effects of multiple eQTLs on genes and directly performing transcriptome-level testing to establish more explainable associations between genes and complex traits/diseases (6,7). A variety of public GWAS resources [i.e. GWAS Catalog (8), dbGAP (9) and GWAS ATLAS (10)] and eQTL resources [i.e. GTEx (11), BLUEPRINT (12) and eQTLGen (13)] provide a wealth of opportunities for TWAS implementation. As the first implementation of TWASs, PrediXcan identified 41 genes associated with five complex diseases. It not only recapitulated several known loci, but also identified many novel genome-wide significant genes (14). Subsequently, in recent years, hundreds of TWASs have successfully been carried out and have identified numerous important genes associated with different types of cancers, complex diseases of diverse body systems as well as physiological measurements (15–21). TWASs, undoubtedly, are leading us to better understand the complex mechanisms of genetic variants in regulating various diseases and traits.
The information and knowledge provided by TWASs has proven to be of great reference value to researchers investigating complex traits and diseases (1,5); however, a comprehensive source of TWAS publications is still out of sight. To the best of our knowledge, two available databases, TWAS-hub (http://twas-hub.org/) and webTWAS, have been set up to integrate TWAS-related datasets (22). In short, TWAS-hub is the first database for TWAS findings released in 2018, storing 75,951 gene–trait associations covering 342 disease/non-disease traits. These associations were identified by TWAS-FUSION with expression reference panels mainly from GTEx and TCGA databases (11,23). The webTWAS is a comprehensive resource of TWAS associations for human diseases, containing 235,064 gene–trait associations across 887 diseases from 1,298 curated GWAS datasets. It implemented three TWAS tools (PrediXcan/S-PrediXcan, TWAS-FUSION and UTMOST) with eQTL panels from the GTEx database (22). These two databases have provided very useful resources for TWAS by optimizing and profiling public datasets; however, they still have a few limitations. Both databases are data-oriented resources because they exclusively collect limited GWAS/expression datasets and just calculate TWAS results using specified methods. They only demonstrate the associations between genes and traits, and lack the integration of other data resources, such as gene expression and gene regulation networks. Moreover, the calculation results of gene–trait associations in both databases are only in the format of listings, instead of in intuitive visualizations of knowledge graphs. Given the fact that the number of TWASs is ever increasing, there is still a lack of cutting-edge databases for TWAS information and knowledge curation, in collecting an extensive range of current data resources, in improving new methodologies applicable to corresponding datasets and, last but not least, in integrating the association visualizations across single nucleotide polymorphisms (SNPs), genes and traits.
To address these issues, here we present TWAS Atlas, a curated resource of transcriptome-wide association studies. First, TWAS Atlas manually collected high-quality gene–trait associations from a large number of publications annotated with relevant research metadata. Second, TWAS Atlas also established an ontology mapping and classification system for traits to unify trait name, definition and category. Further, it constructed a knowledge graph of all collected gene–trait associations with significant genomic regulatory information from GTEx; thus, it can integrate and visualize the SNP–gene–trait associations at multi-omics levels. To sum up, TWAS Atlas is equipped with multiple functions, the latest updated resources and interpretation tools for gene–trait association research. Therefore, we believe that TWAS Atlas will facilitate the application and advancement of TWASs in clinical and human health studies.
DATA CURATION AND DATABASE DEVELOPMENT
Data curation and integration
First, we conducted a literature search in NCBI PubMed using pre-defined keywords including ‘transcriptome-wide association study’, ‘(GWAS) AND (eQTL)’ and ‘(GWAS) AND (transcriptome)’. Second, we manually reviewed the retrieved results thoroughly to remove irrelevant publications. Then we curated the study information for each qualified publication, including reported trait, applied software/method, ancestry of population, tissue type and expression data links. The computational methods in TWAS Atlas for screening these 200 publications not only included the three aforementioned procedures, but also involved many other TWAS-related approaches, such as Summary data-based Mendelian Randomization (SMR) (17), transcriptome-wide summary statistics-based Mendelian Randomization (TWMR) (24), Kernel-based TWAS (kTWAS) (25), Multi-Omic Strategies TWAS (MOSTWAS) (26) and Joint-Tissue Imputation (JTI) (27). Further, we carefully extracted information of eligible gene–trait associations (those at a significant level reported in the publication or with a P-value <1.0E-4 or an adjusted P-value <0.05), including the P-value and effect size of the association with tissue information. An overview of the data structure in TWAS Atlas is shown in Supplementary Figure S1.
To construct genome-level regulatory relationships of genes in TWASs, we integrated eQTLs of genes in 49 human tissues from GTEx version 8. We excluded regulatory loci in high linkage disequilibrium (LD) in order to provide reliable and pruned eQTLs for genes. Specifically, for each gene in each tissue, we clumped all regulatory variants of the gene based on the LD-clump strategy using Swiss (https://github.com/statgen/swiss, parameter –clump-p 1.0E-5 –clump-r2 0.1) with only the best variants by P-value kept first and the remaining variants in LD regions dropped. In addition, detailed gene information was annotated based on GENCODE version 26 (GRCh38) and information of variants was mapped to dbSNP (GRCh38) to be consistent.
Trait ontology mapping and classification
To unify trait name, definition and category, we mapped trait ontology and established a classification system. First, we integrated and normalized the collected traits by manually conducting entity alignment and combining those traits that could be expressed by the same entity. Second, we mapped and classified the normalized traits by reusing ontologies and classification hierarchies of existing and well-known biomedical resources. In brief, we mainly referred to the Experimental Factor Ontology (EFO) (28) and other resources such as MeSH (https://www.ncbi.nlm.nih.gov/mesh), NCIt (29), UMLS (30), HPO (31) and SNOMED CT (https://www.snomed.org/) to define annotation properties for every trait including trait label, ontology ID, description, synonyms and mapped term(s) ID. According to these ontological levels, traits were divided into four subcategories: disease, measurement, phenotypic abnormality and others. We also listed the third-level subcategory in Supplementary Figure S2. These categories can be added later continuously with increasingly collected features, aiding users to better locate and comprehend the traits of interest.
Knowledge graph construction
A knowledge graph was freshly constructed to enhance the visualization and interpretation of the TWAS associations. Initially, we defined three main entities (trait, gene and eQTL) by TWAS research contents,i.e. by trait entities displayed as four main categories or by gene entities divided into non-/protein-coding genes. Subsequently, we characterized semantic relationships and property relationships among these entities. There are two aspects of relationships, namely the relationship between trait entities and gene entities, and the relationship between gene entities and eQTL entities (Supplementary Figure S2). Given that tissue specificity prevails in these associations, we defined the second-order relational classes according to different tissue types. In addition, we also defined another two attributes for the relationship,i.e.P-value and effect size of the association.
Database implementation
TWAS Atlas was constructed based on MVC architecture, and standard database development technologies Thymeleaf (a Java template engine), Vue (front-end framework), HTML5, CSS, AJAX, JQuery and Bootstrap were used for rendering and interactive operations of front-end pages. Spring Boot and Django were used as the basic architecture of the back-end system. MySQL, MariaDB and Neo4J served as a container for data storage, and Mybatis as an accessor to the container. Echarts.js, D3.JS and plotly.js were adopted for building interactive graphs. Bootstrap Table and Element-ui were used to construct data tables. To be more compatible with the TWAS Atlas infrastructure, we advise using the following browsers: Google Chrome (v56.0 and up), Opera (v53.0 and up), Safari (v11.1 and up) or Firefox (v64.0 and up).
DATABASE CONTENTS AND USAGE
Comprehensive association knowledge for diverse traits and genes
A huge amount of effort has been devoted to the identification of trait-associated genes to understand the genetic architecture of complex diseases and traits by implementation of TWASs in the last few years (4,17). Candidate markers of multiple TWAS for the same trait/disease are complementary and verifiable with each other. In addition, comprehensive and integrated knowledge resources could add weights for TWAS research. Therefore, TWAS Atlas gathered and curated massive cutting-edge TWAS publications with remarkable gene–trait associations and relevant research metadata in a standardized manner, forming an information-intensive knowledge relationship for traits and genes. An overview of TWAS Atlas is shown in Figure 1. In the current version, we manually collected a total of 401,266 high-quality gene–trait association terms of human from 200 qualifying publications, encompassing 257 unique mapped ontological terms involving four main categories and 41 subcategories: diseases (e.g. cancer, immune system diseases and nervous system diseases); phenotypic abnormalities (e.g. abnormality of eyes, head and neck); measurements (e.g. height, body weight and metabolite measurements); and others (Figure 2A). The number of curated elements for each trait category is listed in Supplementary Table S1. Most traits collected in our knowledgebase interact with multiple genes, with a median of 54 associated genes per trait. Remarkably, Alzheimer's disease happened to be the most popular trait, being annotated with 2,140 association terms with 585 genes from 24 publications. Previous studies suggested that Alzheimer's disease was the most common cause of dementia with high genetic heritability of 79%, and many essential genetic biomarkers are associated with the pathology of Alzheimer's disease, such as APOE, CD33, MS4A4E, CLU, CR1, CD2AP, MS4A6A and EPHA1 (32–35). All these biomarkers are inclusively covered by TWAS Atlas.
Figure 1.
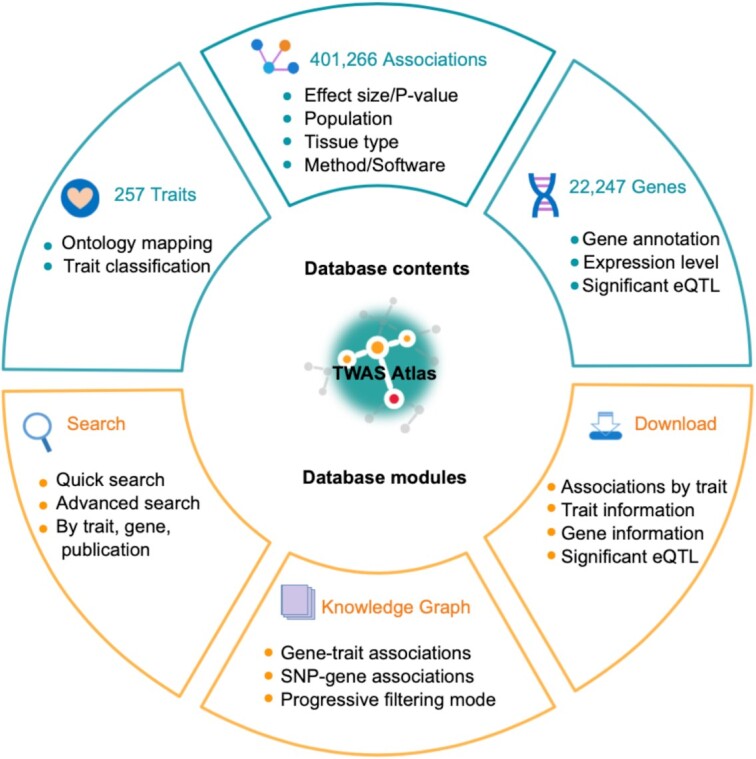
An overview of TWAS Atlas. The main data contents and statistics including associations, traits and genes embraced in TWAS Atlas (above), and three functional modules, i.e. multiple search channels, interactive knowledge graph and comprehensive information available for downloads (below).
Figure 2.

Illustration of browse and search interfaces in TWAS Atlas. (A) The distribution of mapped traits involved in the four main categories and 41 subcategories. (B) The proportional distribution of biotypes of genes collected in the atlas. (C) Screenshot of the browse table of a trait with basic information and statistics (above) and detailed trait information with ovarian epithelial tumor as a demonstration, including trait details and relevant publications and associations (below). (D) Screenshot of the browse table of a gene with basic information and statistics (above) and detailed gene information with MAPT as an example, including gene details, significant eQTLs and expression level from GTEx (below). (E, F) Three search channels for one specific trait (E) or gene (F), respectively.
Meanwhile, there were 22,247 genes related to at least one trait in the atlas. These genes consist of 67% protein-coding genes and 33% non-coding genes (Figure 2B). Statistically, on average, each gene is associated with 5.0 different traits. Take gene ATF6B for instance; it is a transcription factor in the unfolded protein response pathway during endoplasmic reticulum (ER) stress (36) and it has connections with 64 distinguished traits from 20 publications. It is worth noting that TWASs focus on associations between genetically regulated gene expression and traits, and it is necessary to integrate eQTL data for complete regulatory relationships among SNPs, genes and traits. In order to provide reliable regulatory relationships, we excluded regulatory loci in high LD, and consequently ∼65% of genes in the atlas are associated with at least one SNP. Strikingly, ATF6B is closely related to hundreds of flanking variants located in chromosome 6.
User-friendly browse, search and download modules
TWAS Atlas can navigate through the ‘Browse’ page where the indexed publications, traits and genes are listed in three browsable and interactive tables. Traits are the core objects described in the atlas with basic information (e.g. trait name, mapped ontology and trait type) and brief summarized statistics (e.g. number of publications and associations), as listed in the trait browse table (Figure 2C, upper panel). All detailed information for one trait can be viewed on each specified trait's page, which records the trait information and all association terms about this trait (Figure 2C, bottom panel with an example of ovarian epithelial tumor). Genes are another key object reported in TWAS Atlas. Basic gene information (e.g. gene symbol, Ensembl ID, gene location and gene type) and brief summarized statistics (e.g. associated traits and the most associated traits) are listed in the gene browse table (Figure 2D, upper panel). Each gene's page exhibits not only all association terms, but also the external expression level and eQTL information across different tissues (Figure 2D, bottom panel with an example of MAPT). Additionally, TWAS Atlas provides users with hyperlinks to the external databases dbSNP (37), Ensembl (38), EMBL-EBI Expression Atlas (39), OMIM (40) and GeneCards (41) for extra information requisition.
To efficiently query contents of interest, TWAS Atlas is equipped with several search channels: (i) a quick search box on the home page is provided for a real-time querying service by trait label, trait ontology ID, gene label or Ensembl ID; (ii) an advanced search function on the ‘Search’ page is available for directly accessing TWAS Atlas by terms of interest, including certain trait label or ontology ID and detailed gene information (e.g. gene symbol, Ensembl ID or genomic location); and (iii) a search mode for an intuitive graph by trait label and gene symbol is available on the ‘Knowledge Graph’ page (Figure 2E, F). Moreover, an auto-suggestion function is supported in TWAS Atlas, providing candidate query terms for users even based on short inputs. To facilitate the global usability of TWAS findings, all data stored in TWAS Atlas are publicly accessible. All query results displayed on the webpage can be downloaded. Meanwhile, a summarized list of curated publications, traits and genes, and significant eQTL information in the atlas are accessible on the ‘Download’ page as well.
Highly integrated knowledge graph with interactive visualization
To better integrate and visualize knowledge embraced in the atlas, we systematically combined gene–trait associations with SNP–gene associations to form comprehensive and interactive knowledge graphs of regulatory relationships. Meanwhile, we were also making it possible to find indirect links between traits, genes and variants. Overall, three types of entities are defined in the knowledge graph,i.e. 257 mapped traits, 22,247 genes and 153,623 eQTLs. The knowledge graph mainly contains two aspects: (i) 366,936 gene–trait associations noted with tissue type; and (ii) 238,147 SNP–gene associations from 49 tissues (the same associations in different tissues are considered as different associations). In addition to browsing information in tables, users can directly search for the gene and trait of interest on the ‘Knowledge Graph’ page to get an overview of a specifically centralized knowledge graph. By default, only the top 20 associations are displayed, sorted in ascending order by adjustable P-value. To help users accurately and quickly capture the content of interest, nodes on the graph can be extended by double-clicking, and networks can be filtered by gene type, effect direction and tissue type. In particular, all nodes and lines in the graph are editable, allowing the user to adjust the display, extend networks and export in high resolution.
Case study: ovarian epithelial tumor as an example
The knowledge graph in TWAS Atlas not only provides direct and comprehensive references for individual traits and genes, but can also be used to explore indirect and extensional relationships among them. Here we take ovarian epithelial tumor as an example to demonstrate how to use the knowledge graph to explore traits or genes of interest.
Ovarian epithelial tumor is a heterogeneous disease with a major heritable component, and GWAS has identified ∼40 loci associated with ovarian epithelial tumor in recent years (42,43). To explore the mediators of mutations affecting ovarian epithelial tumors by TWAS Atlas, initially, we screened two TWASs on European populations, and found 38 and 42 association terms for ovarian epithelial tumor by implementing different methods (Figure 2C). Then, we used the knowledge graph of ovarian epithelial tumor to visualize and interpret the results. We found that both protein-coding genes and non-coding genes regulated by the genomic variation are related to ovarian epithelial tumor, and mostly were detected in ovary tissue and other hormone-related tissues (e.g. breast and prostate tissues) (Figure 3A). For instance, the expression level of MAPT, significantly regulated by the genetic variants rs2425557 and rs2532395, is positively related to the risk of ovarian epithelial tumor (15,44). Furthermore, the overview of MAPT in the knowledge graph indicated that it was associated with various measurements and specifically associated with Parkinson's disease in various brain tissues (Figure 3B). Intriguingly, the expanded graph revealed that another five genes, namely WNT3, LRRC37A, CRHR1, LRRC37A2 and KANSL1-AS1, were also involved in both ovarian epithelial tumor and Parkinson's disease (Figure 3C). Interestingly, all of these genes are located at chromosome 17q21.31 with a common inversion. The structural variation was first known to be strongly associated with neurodegenerative diseases, including progressive supranuclear palsy, corticobasal degeneration, Parkinson's disease and Alzheimer's disease (45,46). It has also been reported as a risk region for ovarian cancer in recent years (15,44,47). Neurodegenerative disorders and cancer may appear to be unrelated illnesses, however, there is emerging evidence for an implication of the neurodegeneration-associated protein TAU/MAPT in cancer (48,49).
Figure 3.

An application case for using the knowledge graph in TWAS Atlas. (A) Knowledge graph centered on the specific trait, ovarian epithelial tumor, with filtering selections for associations including eQTL, gene type, effect size and tissue category. (B) Knowledge graph centered on the specific gene, MAPT, a gene associated with ovarian epithelial tumor. (C) Shared regulatory genes between ovarian epithelial tumor and Parkinson's disease indicated potential connections.
Taken together, despite the limited TWAS publications reporting associations between genes and traits, with high integrity and connectivity, the knowledge graph in TWAS Atlas implies an inestimable amount of information and will add application usefulness to provide reliable references and insights for researchers on life and health.
DISCUSSION AND FUTURE DEVELOPMENTS
As an effective research mode based on large-scale and multi-omics data, TWASs are increasingly being applied to understand genetic mechanisms of diverse phenotypes and complex diseases. Unfortunately, a large number of TWAS-related results emerge without a platform to accommodate them. Up to now, TWAS Atlas is the first available knowledge resource that collects, curates and integrates the published TWAS findings and presents them in the form of a visualized and interactive knowledge graph. Compared with the exiting TWAS databases, TWAS-hub and webTWAS, TWAS Atlas features the following: (i) manual curation of high-quality gene–trait associations from extensive publications annotated with relevant research metadata, involving a wider range of data resources and improved TWAS methodologies for corresponding datasets (not limited to fixed data and methods); (ii) construction of a knowledge graph, integrating and visualizing SNP–gene–trait associations at multi-omics levels for users to browse, adjust, download and perform other subsequent usages, and a trait ontology classification system adopted to unify trait name, definition and category, effectively improving the readability and applicability of TWAS results; and (iii) integration of various external data and hyperlinks, such as SNP–gene regulatory information, gene expression information and variant information, serving as a more centralized and comprehensive resource in the perspective of multiple omics.
By combining TWAS findings from studies worldwide, TWAS Atlas forms an information-intensive and highly connected knowledge graph for diverse traits, genes and variants. As one of most important database resources in the National Genomics Data Center (NGDC, https://ngdc.cncb.ac.cn), TWAS Atlas will be continuously maintained and updated by collecting and integrating the latest transcriptome-wide association findings. As TWASs are further extended and more widely used, our knowledgebase, beyond humans, will expand to more species. Moreover, growing single-cell RNA sequencing (scRNA-seq) data and analysis methods have provided a huge opportunity for single-cell eQTL mapping, allowing the exploration of transcriptome heterogeneity across cell types at a refined resolution (50,51). At the same time, several large-scale projects of mapping single-cell eQTLs across different cell types have emerged recently (50,52–54), thus making it possible to perform TWASs at the single-cell level in the near future, which will be considered for integrating in our updated version. Additionally, regulatory information between genes, such as protein–protein interaction and mRNA–lncRNA interaction from known databases, such as STRING (55) and starBase (56), will be integrated into the knowledge graph to establish multi-level regulatory networks. We believe that TWAS Atlas will be a valuable resource for facilitating TWAS in gene expression regulation and providing comprehensive up to date knowledge for human health and disease studies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank a number of users for reporting bugs and providing suggestions. The expression level and eQTL information of genes collected in TWAS Atlas are based on the GTEx project: https://gtexportal.org/.
Notes
Present address: Qianwen Gao, Beijing Novogene Bioinformatics Technology Co., Ltd, Beijing 100000, China.
Contributor Information
Mingming Lu, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Yadong Zhang, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China.
Fengchun Yang, Institute of Medical Information, Chinese Academy of Medical Sciences/Peking Union Medical College, Beijing 100020, China.
Jialin Mai, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Qianwen Gao, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Xiaowei Xu, Institute of Medical Information, Chinese Academy of Medical Sciences/Peking Union Medical College, Beijing 100020, China.
Hongyu Kang, Institute of Medical Information, Chinese Academy of Medical Sciences/Peking Union Medical College, Beijing 100020, China.
Li Hou, Institute of Medical Information, Chinese Academy of Medical Sciences/Peking Union Medical College, Beijing 100020, China.
Yunfei Shang, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Qiheng Qain, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Jie Liu, North China University of Science and Technology Affiliated Hospital, Tangshan 063000, China.
Meiye Jiang, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Hao Zhang, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Congfan Bu, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China.
Jinyue Wang, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Zhewen Zhang, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China.
Zaichao Zhang, Department of Biology, The University of Western Ontario, London, Ontario, N6A 5B7, Canada.
Jingyao Zeng, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China.
Jiao Li, Institute of Medical Information, Chinese Academy of Medical Sciences/Peking Union Medical College, Beijing 100020, China.
Jingfa Xiao, National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
DATA AVAILABILITY
TWAS Atlas is a curated knowledge database of transcriptome-wide association studies at https://ngdc.cncb.ac.cn/twas/.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by grants from the Strategic Priority Research Program of the Chinese Academy of Sciences [XDB38030400]; the CAMS Innovation Fund for Medical Sciences (CIFMS) [2021-I2M-1-056]; National Natural Science Foundation of China [31970634 and 32170669]; National Key Research Program of China [2020YFA0907001, 2016YFC0901901 and 2016YFB0201702]; Specialized Research Assistant Program of the Chinese Academy of Sciences [202044]; The Youth Innovation Promotion Association of Chinese Academy of Science [2022098]; and China Postdoctoral Science Foundation [2021M693109]. Funding for open access charge: The Strategic Priority Research Program of the Chinese Academy of Sciences [XDB38030400].
Conflict of interest statement. None declared.
REFERENCES
- 1. Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W.J.H., Jansen R., de Geus E.J.C., Boomsma D.I., Wright F.A.et al.. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016; 48:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J.. 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017; 101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gallagher M.D., Chen-Plotkin A.S.. The post-GWAS era: from association to function. Am. J. Hum. Genet. 2018; 102:717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cano-Gamez E., Trynka G.. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet. 2020; 11:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li B., Ritchie M.D.. From GWAS to gene: transcriptome-wide association studies and other methods to functionally understand GWAS discoveries. Front. Genet. 2021; 12:713230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao C., Ding B., Li Q., Kwok D., Wu J., Long Q.. Power analysis of transcriptome-wide association study: implications for practical protocol choice. PLos Genet. 2021; 17:e1009405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wainberg M., Sinnott-Armstrong N., Mancuso N., Barbeira A.N., Knowles D.A., Golan D., Ermel R., Ruusalepp A., Quertermous T., Hao K.et al.. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 2019; 51:592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buniello A., Macarthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E.et al.. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019; 47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tryka K.A., Hao L., Sturcke A., Jin Y., Wang Z.Y., Ziyabari L., Lee M., Popova N., Sharopova N., Kimura M.et al.. NCBI’s database of genotypes and phenotypes: dbGaP. Nucleic Acids Res. 2014; 42:D975–D979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Watanabe K., Stringer S., Frei O., Umićević Mirkov M., de Leeuw C., Polderman T.J.C., van der Sluis S., Andreassen O.A., Neale B.M., Posthuma D.. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019; 51:1339–1348. [DOI] [PubMed] [Google Scholar]
- 11. Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N.et al.. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen L., Ge B., Casale F.P., Vasquez L., Kwan T., Garrido-Martín D., Watt S., Yan Y., Kundu K., Ecker S.et al.. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell. 2016; 167:1398–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Võsa U., Claringbould A., Westra H.-J., Jan Bonder M., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Yazar S.et al.. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021; 53:1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gamazon E.R., Wheeler H.E., Shah K.P., Mozaffari S.v., Aquino-Michaels K., Carroll R.J., Eyler A.E., Denny J.C., Nicolae D.L., Cox N.J.et al.. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015; 47:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gusev A., Lawrenson K., Lin X., Lyra P.C., Kar S., Vavra K.C., Segato F., Fonseca M.A.S., Lee J.M., Pejovic T.et al.. A transcriptome-wide association study of high-grade serous epithelial ovarian cancer identifies new susceptibility genes and splice variants. Nat. Genet. 2019; 51:815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gandal M., Zhang P., Hadjimichael E., Walker RL., Chen C., Liu S., Won H., van B.H., Varghese M., Wang Y.et al.. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018; 362:eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M.et al.. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016; 48:481–487. [DOI] [PubMed] [Google Scholar]
- 18. Liao C., Laporte A.D., Spiegelman D., Akçimen F., Joober R., Dion P.A., Rouleau G.A.. Transcriptome-wide association study of attention deficit hyperactivity disorder identifies associated genes and phenotypes. Nat. Commun. 2019; 10:4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang C.P., Li X., Wu Y., Shen Q., Zeng Y., Xiong Q., Wei M., Chen C., Liu J., Huo Y.et al.. Comprehensive integrative analyses identify GLT8D1 and CSNK2B as schizophrenia risk genes. Nat. Commun. 2018; 9:838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gillies C.E., Putler R., Menon R., Otto E., Yasutake K., Nair V., Hoover P., Lieb D., Li S., Eddy S.et al.. An eQTL landscape of kidney tissue in human nephrotic syndrome. Am. J. Hum. Genet. 2018; 103:232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mancuso N., Shi H., Goddard P., Kichaev G., Gusev A., Pasaniuc B.. Integrating gene expression with summary association statistics to identify genes associated with 30 complex traits. Am. J. Hum. Genet. 2017; 100:473–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao C., Wang J., Kwok D., Cui F., Zhang Z., Zhao D., Li M.J., Zou Q.. webTWAS: a resource for disease candidate susceptibility genes identified by transcriptome-wide association study. Nucleic Acids Res. 2022; 50:D1123–D1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weinstein J.N., Collisson E.A., Mills G.B., Mills Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Porcu E., Rüeger S., Lepik K., Santoni F.A., Reymond A., Kutalik Z.. Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. Nat. Commun. 2019; 10:3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cao C., Kwok D., Edie S., Li Q., Ding B., Kossinna P., Campbell S., Wu J., Greenberg M., Long Q.. kTWAS: integrating kernel machine with transcriptome-wide association studies improves statistical power and reveals novel genes. Brief. Bioinform. 2021; 22:bbaa270. [DOI] [PubMed] [Google Scholar]
- 26. Bhattacharya A., Li Y., Love M.I.. MOSTWAS: multi-omic strategies for transcriptome-wide association studies. PLoS Genet. 2021; 17:e1009398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou D., Jiang Y., Zhong X., Cox N.J., Liu C., Gamazon E.R.. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat. Genet. 2020; 52:1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malone J., Holloway E., Adamusiak T., Kapushesky M., Zheng J., Kolesnikov N., Zhukova A., Brazma A., Parkinson H.. Modeling sample variables with an experimental factor ontology. Bioinformatics. 2010; 26:1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hartel F.W., de Coronado S., Dionne R., Fragoso G., Golbeck J.. Modeling a description logic vocabulary for cancer research. J. Biomed. Inform. 2005; 38:114–129. [DOI] [PubMed] [Google Scholar]
- 30. Bodenreider O. The unified medical language system (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004; 32:D267–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Köhler S., Vasilevsky N.A., Engelstad M., Foster E., McMurry J., Aymé S., Baynam G., Bello S.M., Boerkoel C.F., Boycott K.M.et al.. The human phenotype ontology in 2017. Nucleic Acids Res. 2017; 45:D865–D876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hao S., Wang R., Zhang Y., Zhan H.. Prediction of Alzheimer's disease-associated genes by integration of GWAS summary data and expression data. Front. Genet. 2019; 9:653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang S., Wang Y.J., Guo J.. Biofluid biomarkers of Alzheimer's disease: progress, problems, and perspectives. Neurosci. Bull. 2022; 38:677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raghavan N., Tosto G.. Genetics of Alzheimer's disease: the importance of polygenic and epistatic components. Curr. Neurol. Neurosci. Rep. 2017; 17:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adams P.M., Albert M.S., Albin R.L., Apostolova L.G., Arnold S.E., Asthana S., Atwood C.S., Baldwin C.T., Barber R.C., Barmada M.M.et al.. Assessment of the genetic variance of late-onset alzheimer's disease. Neurobiol. Aging. 2016; 41:200.e13–200.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haze K., Okada T., Yoshida H., Yanagi H., Yura T., Negishi M., Mori K.. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 2001; 355:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sherry S.T., Ward M.-H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cunningham F., Allen J.E., Allen J., Alvarez-Jarreta J., Ridwan Amode M., Armean I.M., Austine-Orimoloye O., Azov A.G., Barnes I., Bennett R.et al.. Ensembl 2022. Nucleic Acids Res. 2022; 50:989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moreno P., Fexova S., George N., Manning J.R., Miao Z., Mohammed S., Muñoz-Pomer A., Fullgrabe A., Bi Y., Bush N.et al.. Expression atlas update: gene and protein expression in multiple species. Nucleic Acids Res. 2022; 50:D129–D140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amberger J.S., Bocchini C.A., Ois Schiettecatte F., Scott A.F., Hamosh A.. OMIM.org: online Mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2014; 43:D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Safran M., Rosen N., Twik M., BarShir R., Stein T.I., Dahary D., Fishilevich S., Lancet D.. The genecards suite. Practical Guide to Life Science Databases. 2021; Singapore: Springer; 27–56. [Google Scholar]
- 42. Flaum N., Crosbie E.J., Edmondson R.J., Smith M.J., Evans D.G.. Epithelial ovarian cancer risk: a review of the current genetic landscape. Clin. Genet. 2020; 97:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Phelan C.M., Kuchenbaecker K.B., Tyrer J.P., Kar S.P., Lawrenson K., Winham S.J., Dennis J., Pirie A., Riggan M.J., Chornokur G.et al.. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017; 49:680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu Y., Beeghly-Fadiel A., Wu L., Guo X., Li B., Schildkraut J.M., Im H.K., Chen Y.A., Permuth J.B., Reid B.M.et al.. A transcriptome-wide association study among 97,898 women to identify candidate susceptibility genes for epithelial ovarian cancer risk. Cancer Res. 2018; 78:5419–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Jong S., Chepelev I., Janson E., Strengman E., van den Berg L.H., Veldink J.H., Ophoff R.A.. Common inversion polymorphism at 17q21.31 affects expression of multiple genes in tissue-specific manner. BMC Genomics. 2012; 13:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo P., Gong W., Li Y., Liu L., Yan R., Wang Y., Zhang Y., Yuan Z.. Pinpointing novel risk loci for Lewy body dementia and the shared genetic etiology with Alzheimer's disease and Parkinson's disease: a large-scale multi-trait association analysis. BMC Medicine. 2022; 20:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Permuth-Wey J., Lawrenson K., Shen H.C., Velkova A., Tyrer J.P., Chen Z., Lin H.Y., Ann Chen Y., Tsai Y.Y., Qu X.et al.. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nat. Commun. 2013; 4:1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Papin S., Paganetti P.. Emerging evidences for an implication of the neurodegeneration-associated protein tau in cancer. Brain Sci. 2020; 10:862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gargini R., Segura-Collar B., Sánchez-Gómez P.. Novel functions of the neurodegenerative-related gene tau in cancer. Front. Aging Neurosci. 2019; 11:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van der Wijst M.G.P., Brugge H., de Vries D.H., Deelen P., Swertz M.A., Franke L.. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 2018; 50:493–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Snijder B., Pelkmans L.. Origins of regulated cell-to-cell variability. Nat. Rev. Mol. Cell Biol. 2011; 12:119–125. [DOI] [PubMed] [Google Scholar]
- 52. Kim-Hellmuth S., Aguet F., Oliva M., Muñoz-Aguirre M., Kasela S., Wucher V., Castel S.E., Hamel A.R., Viñuela A., Roberts A.L.et al.. Cell type specific genetic regulation of gene expression across human tissues. Science. 2020; 11:eaaz8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Randolph H.E., Fiege J.K., Thielen B.K., Mickelson C.K., Shiratori M., Barroso-Batista J., Langlois R.A., Barreiro L.B.. Genetic ancestry effects on the response to viral infection are pervasive but cell type specific. Science. 2021; 374:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jerber J., Seaton D.D., Cuomo A.S.E., Kumasaka N., Haldane J., Steer J., Patel M., Pearce D., Andersson M., Bonder M.J.et al.. Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. Nat. Genet. 2021; 53:304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Szklarczyk D., Gable A.L., Nastou K.C., Lyon D., Kirsch R., Pyysalo S., Doncheva N.T., Legeay M., Fang T., Bork P.et al.. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021; 49:D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H.. StarBase v2.0: decoding miRNA–ceRNA, miRNA–ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TWAS Atlas is a curated knowledge database of transcriptome-wide association studies at https://ngdc.cncb.ac.cn/twas/.