

Summary
Molecular machines, such as polymerases, ribosomes or proteasomes, fulfil complex tasks requiring the thermal energy of their environment. They achieve this by restricting random motion along a path of possible conformational changes. These changes are often directed through engagement with different cofactors, which can best be compared to a Brownian ratchet. Many molecular machines undergo three major steps throughout their functional cycles, including initialisation, repetitive processing, and termination. Several of these major states have been elucidated by cryogenic electron microscopy (cryoEM). However, the individual steps for these machines are unique and multi-step processes, and their coordination in time is still elusive. To measure these short-lived intermediate events by cryoEM, the total reaction time needs to be shortened to enrich for respective pre-equilibrium states. This approach is termed time-resolved cryoEM (trEM). In this review, we sum up the methodological development of trEM and its application to a range of biological questions.
Graphical Abstract
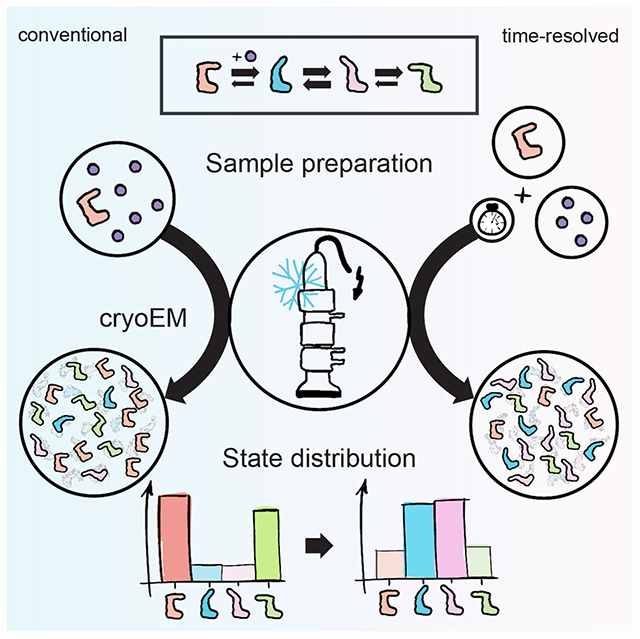
eTOC blurb
In this Review article, Amann et al. discuss the state of the art in time-resolved cryoEM and how it advanced during the last three decades. They focus on the molecular mechanisms of processive molecular machines and how time-resolved cryoEM can be applied for the investigation of their conformational complexity.
Introduction
Most cellular processes are driven by macromolecular assemblies made of proteins and nucleic acids often termed molecular machines. These proteins evolved in a way that random noise energy of the environment powers specific conformational changes along a restricted path, often directed by chemical energy in the form of nucleotides, to achieve various complex tasks. These tasks are for instance nucleic acid or protein synthesis as well as protein degradation, the assembly or disassembly of larger cellular structures or the transport of larger assemblies through the cell 1–4. In an abstract view one can distinguish three major steps that are consecutive in terms of the canonical work cycles of many processive molecular machines. From this perspective the machine is initialised by recognizing the target. Secondly, a mostly repetitive productive cycle achieving the main task follows. Finally, the reaction stops in some sort of termination. While this simplistic view does not apply to every molecular machine, it is true for many involved in important cellular processes such as the ribosome, the RNA polymerase, the spliceosome, the proteasome, motor proteins such as dynein, large enzyme complexes like the Anaphase-Promoting Complex or the fatty acid synthase and even chaperones like GroEL 1–3,5–8. The individual steps in the processes carried out by these machines are themselves complex multi-step processes and are unique for any of these machines. Nevertheless, almost every step will involve either the dissociation or binding of a new partner, or a conformational change. In most cases, while we often have a higher-level understanding of the necessary steps and their order as well as the components involved, the precise interactions and how these are coordinated in time is still elusive. This is especially evident when we try to assign functions to individual domains or subunits of molecular machines. Few subunits/domains within the proteome can be clearly assigned to a specific catalytic activity, most domains therefore get attributed to serving a as so-called “scaffolds”. In reality, it is probably more likely that at least some of these subunits have some secondary role in acting as hinges or transducers of long-range conformational changes. We have not yet been able to either systematically describe or predict these more subtle roles.
A reason for our lack in understanding is of course the very different modes through which molecular and macroscopic machines act. While all macroscopic machines behave in a deterministic manner, molecular machines behave stochastically. This means that many of the movements seen in molecular machines may not be productive and do not contribute to the overall productive cycle. However, despite their stochastic nature they are intrinsically biased towards a productive outcome, meaning that while they constantly step forward and backward the net direction is forward. This is especially evident at the different scales of sensitivity attainable through various biophysical measuring techniques. While almost all molecular machines look like very productive directed machines at the second time scale, they look very stochastic at the micro and millisecond time scales 9.
To understand the basic physical mechanisms underlying molecular machines, we need to access the short time scales in which stochastic movements occur. So far, these time scales have only been accessible for fast kinetic measurements that usually give limited structural information 10–17. For the complete understanding of a molecular machine, we require a complete understanding of the structural changes within the complex. In an ideal scenario, we would be able to visualize the conformational movement of any given molecular machine in full structural detail during its productive cycle. In this review, we discuss recent advances in cryoEM that allow for a biochemical and structural depiction of molecular machines through a single set of experiments.
Many processive molecular machines follow a repetitive cycle
The best studied molecular machines are probably those that are involved in the life cycle of proteins, i.e., the machines that carry out the central dogma of molecular biology. Still, despite this process being intensively characterized structurally and functionally, we are yet to pinpoint a consecutive work mechanism involving its many parts. To this end, structural insights of these machines in action are especially necessary. In this section, we will concentrate our discussion on our current understanding of four processive molecular machines and their timing of processing substrate, as trEM might prove especially useful in the investigation of these complexes.
As stated above, the actions of many processive molecular machines can be distinguished into an initialization, processivity and termination phase. While the processivity phase takes the longest time, it is also the most repetitive pattern and thus experimentally harder to grasp. Protein synthesis starts at transcription with the RNA polymerase II (Pol II) being responsible for the transcription of pre-mRNA in eukaryotic cells (Figure 1A). Prior to transcription, Pol II needs to be initialized on the DNA by multiple factors. For the canonical pathway, TFIID recognizes the TATA box on the DNA leading to the loading of TFIIB and TFIIF bound Pol II. Furthermore, TFIIE and TFIIH are incorporated to form the pre-initiation complex 18. This will happen on a time scale of seconds. After a licensing event, a processive elongation cycle starts at an average rate of 60-70 nt/s 19. This means that transcription of a gene may be ongoing for many minutes to a few hours. Ultimately transcription will terminate through one of several different pathways. This again most likely happens on a per-second time scale, but exact times are not yet conclusively measured.
Figure 1: Four cartoons depicting a selection of molecular machines where time-resolved cryoEM may prove particularly useful in illuminating their function.
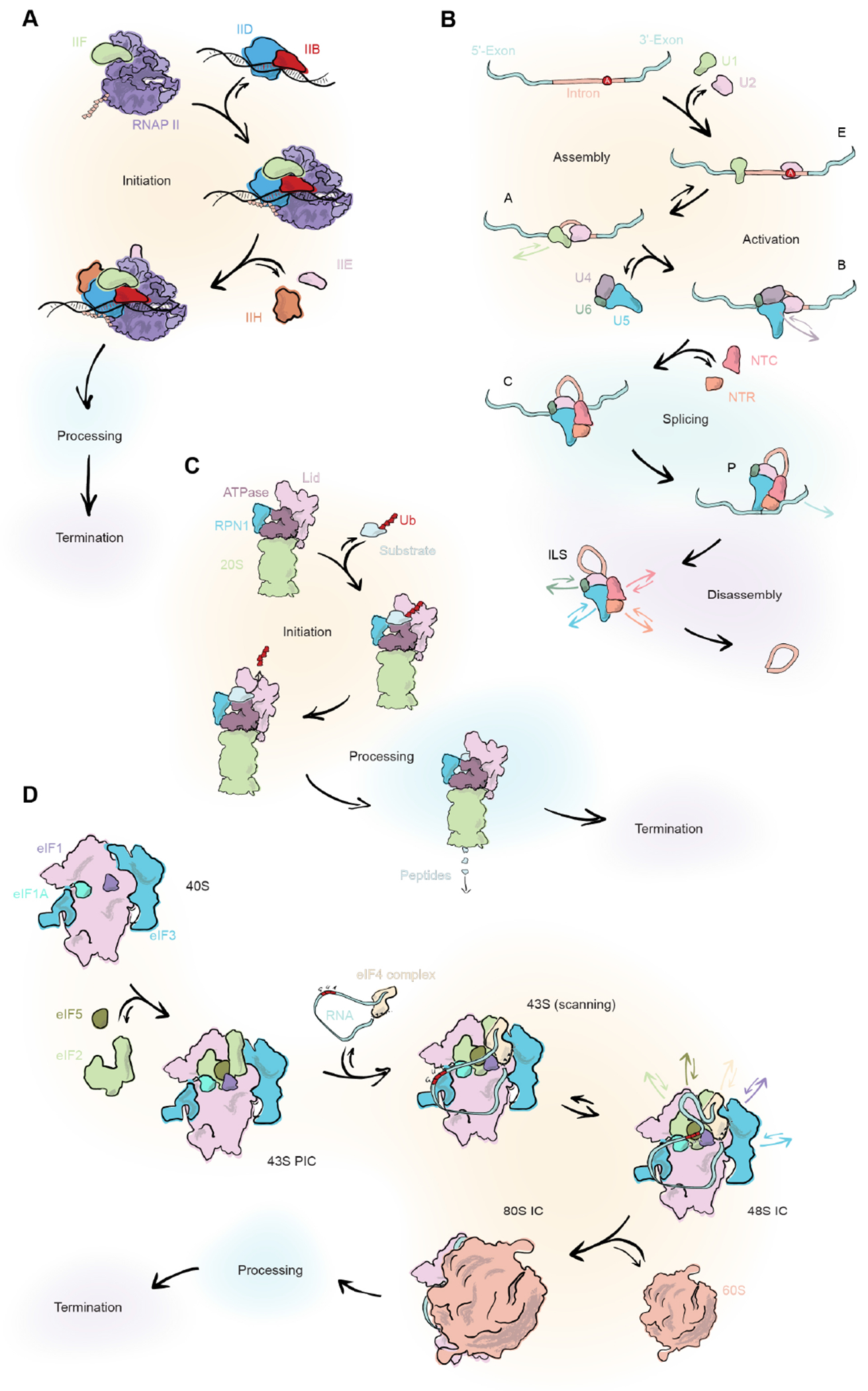
A: Translation by the RNA polymerase II
B: RNA splicing by the spliceosome
C: 26S Proteasomal degradation
D: Eukaryotic ribosome translation
After, or already during the transcription, splicing of the pre-mRNA is carried out by the eukaryotic spliceosome to remove introns (Figure 1B). snRNPs U1 and U2 interact with an intron and incorporate the tri-snRNP U4/U6.U5. The 5’ splice site is transferred from U1 to U6 leading to the unwinding of the U6 snRNA by the U4 snRNA. The U6 snRNA then folds with the U2 snRNA into a complex that stabilizes the hydrolysis of the 5’ splice site by a lariat intron formation between the 5’ and a downstream adenine. The 3’ splice site is then positioned in a way that it is accessible by the free 3’ hydroxy group of the 5’ splice site for a nucleophilic attack, resulting in the excision of the lariat intron and a mature mRNA 7. The timing of splicing is not clear as measurements between 15 s and 15 mins exist 20. However, as transcription itself is in most cases slower, splicing is not rate limiting for gene expression.
The final step of protein expression is translation of mature mRNA carried out by the 80S ribosome (Figure 1D). For the initiation of translation, Met-tRNA in complex with GTP-eIF2 is brought to the 40S ribosomal subunit with eIF5, −3, −1 and −1A forming the 43S preinitiation complex (PIC). Mature mRNA is unwound and brought into a closed loop by the eIF4 complex interacting with both the 5’ cap and the 3’ tail which is then recruited to the 43S PIC. eIF1 and −1A are bound to the A and P site that allows mRNA binding in the 40S cleft and the scanning which requires ATP hydrolysis for 43S PIC movement along the mRNA until the AUG start codon is at the P-site interacting with the Met-tRNA anticodon. Upon that, eIF1 relocates and stabilizes GTP-eIF2 in a way that favours GTP hydrolysis, resulting in the release of GDP-eIF2 and eIF5 and the recruitment of GTP-eIF5B that favours the joining with the 60S ribosomal subunit. Hydrolysed eI5B bound to GDP is released together with eIF1A and leads to the formation of the 80S ribosome that can begin with the translation 21. This process is indicated to take between 4 and 240 s 22 and is followed by a processive elongation cycle that add about 6 amino acids per s 23.
At the end of the life cycle of a protein, the 26S proteasome is responsible for the degradation of ubiquitinated proteins with a loosely folded domain in eukaryotes (Figure 1C). Four different main conformational states have been identified during substrate processing using cryoEM that encompass a rotation and a tilting of the regulatory particle, both thought to be a prerequisite for substrate deubiquitination and degradation 24–27. FRET and fluorescence anisotropy analyses showed that the loosely folded domain insertion into the central pore, lid rotation, and deubiquitylation takes around 1.6 seconds, 0.6 seconds, and 4.6 seconds, respectively. The remaining unfolding and degradation progresses at a rate of around 10 amino acids per second 12.
In all these cases, the three main steps (initiation, processing, termination) are happening on a second time scale which makes them accessible to many modern techniques. However, substeps such as individual elongation steps or conformational changes during initialisation will happen on shorter time scales in the range between 100s of microseconds and 10s of milliseconds. To measure these events by any biophysical method, special apparatuses such as stopped-flow devices or microfluidic chips need to be used to reduce the reaction time to a minimum.
Solutions of biochemical macromolecules are shock-frozen in cryoEM
Transmission electron microscopy (TEM) is a powerful method used to analyse structures of soft matter. CryoEM in particular can resolve structures of biological macromolecules such as proteins in near-native states to high resolution 28. Though strongly dependent on the protein, it is possible to resolve structures from several angstroms up to atomic resolution, which two recent cryoEM studies achieved with the highly symmetric apoferritin 29,30. Cryo-grid preparation relies on the fixation of macromolecules in a thin film of frozen buffer, while preserving a near-native structure. Therefore, pure samples need to be frozen rapidly on grids that usually are made of a copper mesh or other metals, coated with holey carbon 31. To retain the structural integrity of the particles, the surrounding water in the solvent must not crystalize during freezing but instead form a thin vitreous layer. This requires a quick cooling step to ensure that the solvent remains in an amorphous and non-crystalline state 32. Conventionally, this is achieved by blotting a grid that is covered with sample using a filter paper to remove an excess of solvent. This is followed by rapidly plunging the grid into a container of liquid ethane at around 90 K in a bath of liquid nitrogen 33.
Energetically stable conformational states dominate in conventional cryoEM
To achieve high-resolution structures in X-ray crystallography, the proteins need to be grown in rigid, highly ordered, three-dimensional crystals, whereas for single particle cryoEM, the proteins are vitrified in a near native state 32–34. This difference in sample preparation not only circumvents the tedious work of growing protein crystals but also allows the observation of proteins carrying out biochemical reactions when they are mixed with their substrates and necessary cofactors. In TEM, the structural information of single molecules is encoded in their 2D projections 35. Through computational analysis 3D maps that represent an average of the entire dataset can be retrieved from these 2D projections. The population of single particle 2D projections is usually not homogeneous and consists of subsets of particles in different conformational or compositional states during a biochemical reaction. These subsets can be identified by state-of-the-art sorting techniques, which themselves can be resolved to consensus 3D maps that represent the different conformational states 36–40.
Many biochemical reactions progress in a multistep manner, for example substrate recognition, processing, and release 41–48. In a cryoEM dataset of a multiple turnover reaction in equilibrium, particle subsets of the most energetically stable conformational states are predominant, as reaction progression is slowed down by higher energy barriers. As a result, particle subsets in transient states that are often very short-lived are heavily under-represented which renders a 3D reconstruction of such states nearly impossible.
Energy landscapes reconstructed from equilibrium lack procedural information
The preparation and imaging of cryoEM grids yields 2D projections of particles usually in the range of 105-107 images that can be used for further processing. Generally, the sample will be in a thermodynamic and kinetic equilibrium state at room temperature and thus the population of particle images will represent the distribution of conformational states under these conditions. However, the representativeness is limited by interactions induced by sample preparation. Notoriously, protein absorption at the air-water interface has the potential to skew the distribution of conformations in comparison to conditions in vivo 49–52. Keeping these limitations in mind, cryoEM still provides a powerful tool for the analysis of conformational dynamics, especially when complemented with biochemical or biophysical experiments 43,53–57. Through modern computational approaches, one can disentangle the conformational states of the individual particles and thus approximate reaction coordinates of the conformational space from particle images and use these to construct so-called energy landscapes 25,36,38,43,54,58,59. Such landscapes map conformational states to their potential energy resulting from their thermodynamic distribution and can be interpreted to infer possible pathways and intermediate states a macromolecule goes through 60. Notwithstanding, energy landscapes are purely a representation of probabilities of a molecule being in a particular state, not of their probability of changing from one state into another 43. Thus, they lack any kind of directional or procedural information. This means that any interpretation is based on assumptions of the behaviour of the complex and needs to be validated by additional time-resolved experiments. In energy landscapes, the transient states of a conformational mechanism might not be visible at all or appear as energy hills due to their low probability in equilibrium, independent from their importance in understanding the mechanism. These limitations call for the need to resolve discrete time steps in cryoEM, which gives us the probability of a molecule being in certain conformational state at a specific timepoint of a reaction. This allows us to make informed interpretations of possible pathways.
Time-resolved cryoEM allows for assessment of the directionality of energy landscape trajectories
In X-ray crystallography molecules are trapped in a specific conformational state by crystallisation. Adding compounds such as substrates, cofactors or drugs, can lead to the enrichment of other conformations. However, these compounds stabilise a specific conformation, rendering a majority of states inaccessible with this method. Time-resolved cryoEM aims at shifting the conformational distribution towards intermediate states of a biochemical reaction by capturing a pre-equilibrium time point precisely after the reaction was initiated. This is achieved by sufficiently reducing the reaction time in a controlled manner before vitrification. This increases the probability of a particle being in the state of question, ideally allowing for 3D reconstruction. In conventional cryoEM sample preparation, the minimal reaction time that can be achieved is limited by the blotting and plunging steps of the grid which add up to several seconds 41,43,48,59,61. However, many biochemical reactions have interesting but structurally unresolved initial or early intermediate states or interactions with lifetimes in the micro- to millisecond range upon mixing of the reactants 62–67. Therefore, novel grid preparation methods that minimize the total reaction time of biochemical reactants need to be developed.
Time-resolved cryoEM
Since the mid 1990s, several methods have emerged that aim to increase the temporal resolution of trapping biochemical reactions on cryoEM grids to fractions of a second (Figure 2). Most of these methods follow an approach that uses sprays to deposit the sample on a grid that is plunged into liquid ethane for vitrification. The reactants are either mixed and sprayed on a grid or one reactant is sprayed on a grid that is already covered with a solution of the other reactant 50,68–73. Since these methods omit the blotting step, which removes excess solvent, the volume of sprayed sample needs to be sufficiently low and should spread evenly so that the ice thickness is thin enough for TEM imaging 74. Other approaches make use of chemically caged cofactors that can be activated upon laser flash photolysis 75,76, or involve laser-induced melting of the vitrified sample followed by immediate re-vitrification 77,78. Tables 1 and 2 give an overview of the different techniques that have been proposed.
Figure 2: An illustrated overview of the different time-resolved cryoEM approaches reviewed in this article.
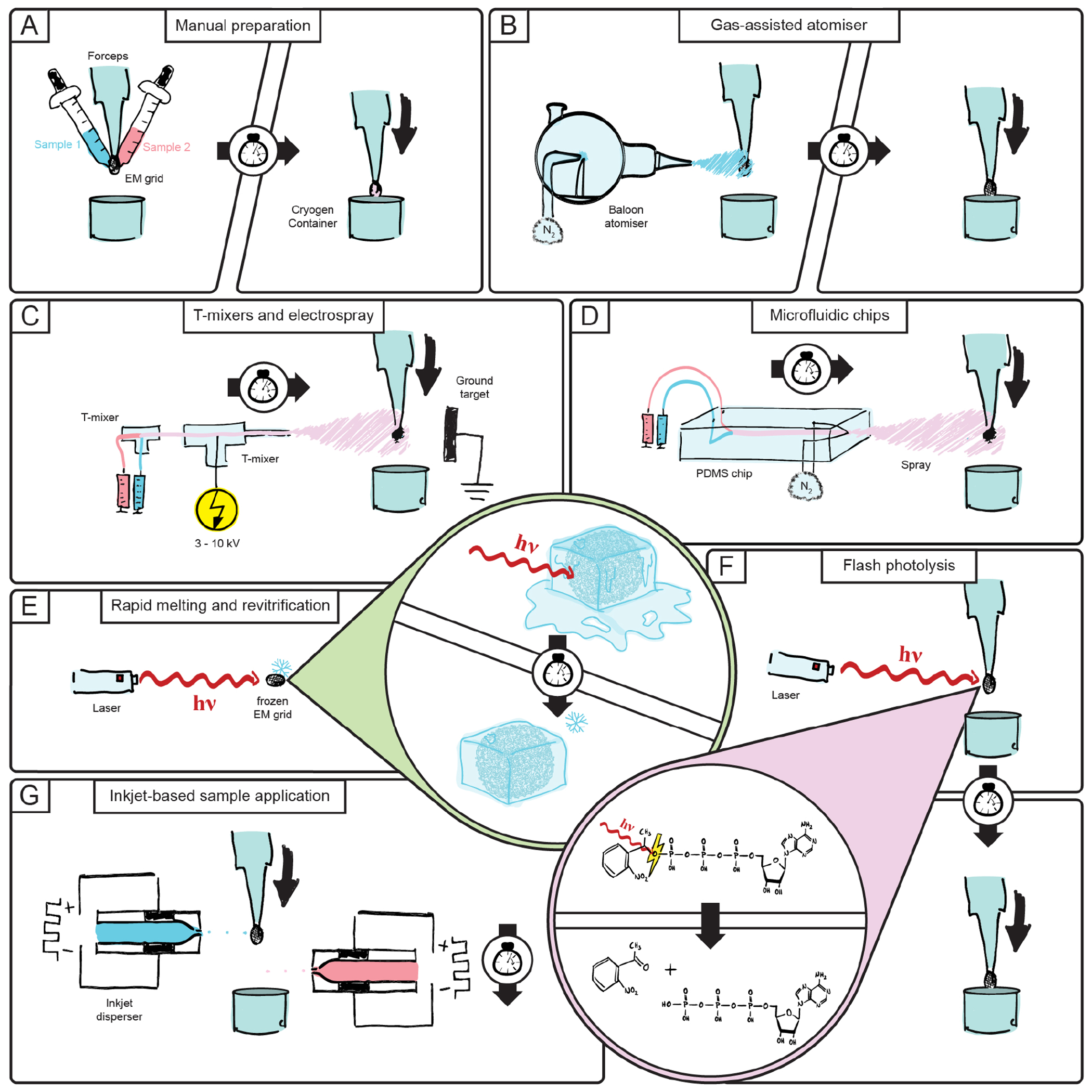
A: Manually timed sample mixing 43. B: Gas-assisted sample atomization 68. C: Voltage-assisted sample spray 72. D: Microfluidic mixing in silicon chips with gas-assisted spray 50. E: Laser-induced rapid in situ melting and revitrification 78. F: Laser-induced photolysis of caged reactants 75. G: Inkjet-based sample droplet application 83.
Table 1: A comparison of the different approaches for time-resolved cryoEM by their quantifiable properties.
Publications that propose several methods are split into separate rows. N / S (not stated) means that the publication does not specify a value. N / A (not applicable) means that the value is not applicable to the method.
| Studied system | trEM approach | Time resolution | Time frame recorded | Minimum reaction time | Required sample amount | Quant. mixing efficiency |
|---|---|---|---|---|---|---|
| Dissociation of actomyosin75 | Flash photolysis of caged components before freezing | N / S | 20 - 80 ms | 20 ms | N / S | N / S |
| NaCl induced changes of Erythrocyte68 | Gas-assisted spray of one reactant on blotted grid | N / S | 5 ms | 5 ms | N / S | N / S |
| Dissociation of actomyosin73 | Voltage-assisted spray of one reactant on blotted grid | ~ ms | 5 - 50 ms, >50 ms | N / S | 100 μL | N / S |
| Voltage-assisted spray of reactants mixed in Berger ball | (as above) | (as above) | N / S | (as above) | N / S | |
| Mixed particle colocalisation50 | Voltage-assisted spray | N / S | N / S | N / S | 1 - 2 μL | N / S |
| Microfluidic chip mixing channel82 | Voltage-assisted spray of one reactant on blotted grid | 1 s | > 1 s | N / S | 5 - 10 μL | N / S |
| Microfluidic chip sprayer nozzle47 | Gas-assisted spray of reactants mixed in microfluidic silicon chip (butterfly mixers) | N / S | 9 ms | 9 ms | 80μL (for 4 grids) | N / S |
| 70S Ribosome tRNA retro-translocation43 | characterisation experiments of device from above | N / S | 0.49 ms (only mixing) | (as above) | (as above) | 96 - 98% |
| Sprayed apoferritin81 | Gas-assisted spray of reactants mixed in microfluidic silicon chip, improved spray | N / S | N / S | (as above) | (as above) | (as above) |
| Mixed particle colocalisation50 | manual trEM | 1 min | 1, 2, 5, 20 | 1 min | 2-4 μL | N / S |
| Microfluidic chip mixing channel82 | Gas-assisted spray from microfluidic silicon chip | N / S | N / A | N / S | 9 μL | N / A |
| Mixed particle colocalization and dissociation of actomyosin70 | Voltage-assisted spray of reactants mixed in T-mixers | 10 - 15 ms | > 15 ms | 15 ms | 33 μL | N / S |
| Voltage-assisted spray of one reactant on blotted grid | 10 ms | > 11 ms | 11 ms | (33 + 3) μL | N / S | |
| 70S ribosome association, MthK Ca2+ binding, RNA pol DNA association, GTP hydrolysis dependent rearrangement of dynamin lipid tubes69 | Inkjet application of one reactant onto the other | N / S | 90 ms | 90 ms | 2.5 - 16 μL | N / S |
| RecA filament growth71 | Gas-assisted spray of reactants mixed in microfluidic silicon chip | 10 ms | 40 - 1360 ms | 30 ms | 30 μL | max. 92% |
| Actomyosin dissociation113 | Gas-assisted spray of one reactant on blotted grid | ~ 6 ms | 7 - 640 ms | 7 ms | > 1.25 μL | N / S |
| Gas-assisted spray of reactants mixed in a T-mixer | (as above) | 400 - 700 ms | 400 ms | > 0.35 - 2 μL | N / S | |
| GroEL disassembly upon electron-beam damage78 | Rapid melting and revitrification inside the electron microscope | 5 μs | 10-50μs | N / S | 3 μL | N / A |
| Apoferritin revitrification114 | (as above) | (as above) | 20 μs | N / S | 3 - 3.5 μL | N / A |
Table 2: A comparison of the different approaches for time-resolved cryoEM according by their technical properties.
Publications that propose several methods are split into separate rows. N / S (not stated) means that the publication does not specify a value.
| Studied system | trEM approach | Time resolution calibration | Time adjustment | Needed equipment / Availability |
|---|---|---|---|---|
| Dissociation of actomyosin75 | Flash photolysis of caged components before freezing | Actomyosin-S1 dissociation | Falling height between flash lamp and liquid ethane container | Flash lamp and photo-electric trigger |
| NaCl induced changes of Erythrocytes68 | Gas-assisted spray of one reactant on blotted grid | N / S | varying plunging time (velocity or distance) | widely available components, legacy atomizer |
| Dissociation of actomyosin73 | Voltage-assisted spray of one reactant on blotted grid | N / S | Plunge speed (via plunging pressure), plunge time (via plunging distance), intermediate stop | Syringe pump, high voltage DC converter and custom-built machine with widely available components |
| Voltage-assisted spray of reactants mixed in Berger ball | N / S | (as above) | (as above) | |
| Mixed particle colocalisation50 | Voltage-assisted spray | N / S | N / S | (as above) |
| Microfluidic chip mixing channel82 | Voltage-assisted spray of one reactant on blotted grid | N / S | N / S | (as above) |
| Microfluidic chip sprayer nozzle47 | Gas-assisted spray of reactants mixed in microfluidic silicon chip (butterfly mixers) | N / S | Different chips with varying delay length | Microfabrication equipment |
| 70S Ribosome tRNA retro-translocation43 | characterisation experiments of device above | measurement and CFD simulation of mixing index and residence time distribution | (as above) | (as above) |
| Sprayed apoferritin81 | Gas-assisted spray of reactants mixed in microfluidic silicon chip, improved spray | (as above) | (as above) | 3D MEMS fabrication equipment |
| Mixed particle colocalisation50 | manual trEM | stopwatch | longer incubation in the minute range | controlled environment vitrification system/ standard EM equipment |
| Microfluidic chip mixing channel82 | Gas-assisted spray from microfluidic silicon chip | N / S | N / S | Microfabrication equipment |
| Mixed particle colocalization and dissociation of actomyosin70 | Voltage-assisted spray of reactants mixed in T-mixers | N / S | Residence time and plunge speed (via plunging pressure) | widely available components |
| Voltage-assisted spray of one reactant on blotted grid | N / S | Plunge speed (via plunging pressure) | (as above) | |
| 70S ribosome association, MthK Ca2+ binding, RNA pol DNA association, GTP hydrolysis dependent rearrangement of dynamin69 | Inkjet application of one reactant onto the other | 70S ribosome assembly was monitored | varying plunging time (velocity or distance) (90-500ms tested so far | proprietary device |
| RecA filament growth71 | Gas-assisted spray of reactants mixed in microfluidic silicon chip | Time dependent RecA + ssDNA/ATPyS filament growth | Different chips with varying delay length | Microfabrication equipment |
| Actomyosin dissociation113 | Gas-assisted spray of one reactant on blotted grid | Actomyosin dissociation, Plunge speed measurement + liquid flow rate + droplet | plunge stop | Microfabrication equipment |
| Gas-assisted spray of reactants mixed in a T-mixer | (as above) | varying delay line inner diameter | (as above) | |
| GroEL disassembly upon electron-beam damage78 | Rapid melting and revitrification inside the electron microscope | time series of refraction at ice and gold film in EM | Varying laser pulse times | Customization of the TEM with lasers for electron beam pulsing and heating |
| Apoferritin revitrification114 | (as above) | (as above) | (as above) | (as above) |
Manual Time-resolved cryoEM in the seconds to minutes range
The simplest way to achieve time-resolved cryoEM is the usage of a conventional blotting and plunge freezer system and a timer to achieve a specific incubation time (Figure 2A). However, this limits the utilisation of this method only to very slow biochemical processes that do not reach equilibrium before one to ten seconds 43,59,61. To mitigate this, the method is often combined with reaction speed decelerating measures such as a lowered incubation temperature or specific inhibitors that can block the biomolecules at a specific state 41,43,48.
Gas-assisted atomizer
The first generation of specialised trEM sample preparation devices aimed to control and minimize the total reaction time of two compounds by using a gas-assisted atomizer that sprays droplets containing one component onto a grid containing the other component in a thin aqueous film (Figure 2B). The droplets that impinge on the liquid film spread rapidly over the surface as a result of surface tension. This approach limits the minimal reaction time to the duration of of the plunge-freezing step with achievable time resolutions of 1-100 ms proposed. To demonstrate the mixing capabilities of this approach, crystallized acetylcholine receptor tubes and Ferritin were colocalized and actomyosin disassembly at 10 ms after the addition of ATP was observed which was shown to be in line with stopped-flow light scattering measurements 68,73. Furthermore, this method was used to study transient states such as the channel-opening and gating movement of the Acetylcholine receptor upon the addition of Acetylcholine 79,80.
Voltage-assisted sprays
The second generation of trEM sample preparation devices followed the same principle as the first generation but focussed on increasing the quality of the produced grids in order to increase the amount of collectible area for single particle analysis. The gas-assisted atomizer was replaced by a 7 kV electrospray that reduced the sprayed droplet size to less than 1 μm which led to a 10-fold increase in the number of droplets that impinge on the pre-applied liquid on the grid (Figure 2C). To show the mixing capabilities of this generation of devices, BSA-conjugated 10 nm gold fiducials were sprayed on top of pre-applied F-actin and an ATP-dependent actomyosin disassembly was once again observed. To prove that folded proteins retain structural integrity by the electro-spray, Apoferritin, E. coli ribosome, and Porcine cardiac thin filaments were 3D refined to 3.6, 4.3, and 5.6 Å resolution, respectively 70,72.
Microfluidic mixing
The first two generations of time-resolved grid preparation devices were mainly based on spraying one reactant onto a second to initiate a biochemical reaction. This approach limits the mixing capability to the diffusion rates of each sample. The third generation of trEM mixing devices (Figure 2D) relies on microfluidic silicon chips that incorporate mixing units and a gas-assisted sprayer outlet to achieve mixing times in the range of a few milliseconds 47,71,81. The mixing capabilities of these chips have been shown by the colocalization 70S ribosomes and ferritin, as well as fluorescence microscopy of nanoparticles and CFD simulations 50,82. Lu et al. suggested the integration of delay lines between the mixing and spraying of samples to vary the total reaction times which was implemented in practice by Mäeots et al. Using chips with different retention times after mixing, they were able to show the time-dependent filament growth of RecA when mixed with single-stranded DNA and ATPγS 71. To show that proteins survive this rapid mixing, the 70S ribosome and apoferritin were 3D refined to 18.9 and 2.8 Å, respectively 50,71. Despite possibly having the best mixing capabilities, the microfluidic chips proposed so far require high amounts of sample, which limits their usage to proteins that can be purified in large amounts. The method also poses challenges for single molecule processing as the collectible area on grids is fairly limited, resulting in low particle counts 47,50,71,81.
Inkjet-based sample application
All aforementioned trEM mixing devices consume fairly high amounts of sprayed sample. To address this, the Carragher lab is developing the fourth generation of trEM mixing devices using piezo assisted jets (Figure 2G). These work in similar fashion to inkjet printers to deposit samples in the nanolitre range onto an EM grid 83. To further improve the ice quality of EM grids, the inkjet technology was combined with so-called self-wicking nanowire grids. These grids contain nanotubes on the grating around the grid squares which wick away excess solvent, leaving only a thin layer of liquid before freezing 74,84. By stacking two jets on top of each other and spraying a different sample from each onto a plunging grid, samples can be mixed and vitrified within 90 ms. Using this device, the assembly of 70S ribosomes was monitored upon mixing 30S and 50S ribosomal subunits it was possible to refine the assembled 70S ribosome to 4.75 Å spatial resolution. The calcium-gated potassium channel MthK was mixed with Ca2+ and the Ca2+-bound conformational state of MthK was observed. In another experiment, RNA polymerase was mixed with DNA and their interaction could be seen after 2D classification of the particle images. After mixing of GTP with dynamin decorated lipid tubes, the constriction of the latter could be observed 69.
Flash-photolysis of caged reactants
While sample application via different forms of sprays brings several advantages, it also comes with negative side effects. Multiple hydrodynamic and physicochemical parameters must be controlled in order to monitor droplet size and mixing and, depending on the method used, some of these parameters might elude control. A first attempt to have a fully mixed solution of reactants while maintaining control over the reaction time was proposed by Ménétret et al. by using the actin-myosin S1 complex in conjunction with biologically inactive but photoactivatable “caged” ATP 75. The method consists of holding an electron microscopy grid in a guillotine-type setup commonly used in rapid freezing, where the reactant mixture is applied on the grid and blotted. During the plunging step, the grid falls through an area illuminated by a flash lamp which causes the caged compound to become bioactive via photolysis. The reaction time is controlled by varying the distance that the grid travels after photolysis and before the grid is immersed in the cryogen (Figure 2F). With this setup, Ménétret et al. report a minimum reaction time of 20 ms and claim to resolve timesteps with 10 ms precision, although a detailed description of how the reaction time was adjusted and monitored is lacking. A similar device was successfully used to trap transient states of bacteriorhodopsin in cryo electron diffraction crystallography experiments by Subramaniam et al. 85,86. However, the protein in question is intrinsically sensitive to photons and caged cofactors were not used. Building on the works of Ménétret et al. and White et al., Shaikh et al. have implemented a time-resolved rapid freezing apparatus using flash photolysis and sample application by either quench-flow or electrospray 72,76. In their characterization studies, they have shown that the flashing step heats bare copper grids by more than 40 K, making it necessary to use custom-made grids to mitigate structural damage to protein complexes induced by thermal energy while maintaining optimal imaging conditions for single particle analysis. Shaikh et al. have investigated two further case studies in addition to the caged ATP used by Ménétret et al., namely caged GTP and caged calcium, which play a role in several biochemical processes in their uncaged states 76. However, no conformational change of a macromolecule was investigated in the work in question. While flash photolysis represents an elegant answer to many problems in trEM, it can only be applied to processes where cofactors can be obtained in a caged form 72,76. Furthermore, the method poses another challenge by inducing temperature rises in the order of 8 K even when using the adapted grids 76. Recently, Yoder et al. have demonstrated an accessible device for flashing and plunging cryoEM grids which might serve as a base for future time-resolved experiments 87.
Rapid melting and revitrification
Another unconventional approach to overcome the shortcomings of spray-based trEM has been proposed by Voss et al. (Figure 2E), who suggest the rapid in situ melting of the vitrified grid using a laser, followed by a sufficiently fast revitrification within the environment of the cryo electron microscope itself 78. Controlling the laser power and pulse duration, a localized portion of the grids’ gold film is heated, inducing the melting of the adjacent ice to room temperature which remobilizes the molecules in question for the duration of the laser pulse. The implementation of this method requires the modification of a cryo-capable TEM by adding lasers and control hard- and software for heating of the sample and pulsing of the electron beam 88. In their study, Voss et al. show that the heating and cooling dynamics of vitrified water on a gold cryoEM grid are sufficiently fast to melt and revitrify within 1.2 μs and 3.6 μs respectively, allowing for a theoretical temporal resolution of 5 μs. The method has been controlled by inducing irradiation damage onto GroEL and monitoring its consecutive disassembly. While the approach suggested by Voss et al. is significantly faster than other trEM methods reviewed in this article so far, no native molecular dynamics have been examined with it and the method must be combined with some form of controlled mixing, flash photolysis of caged components or another external trigger for the mechanism of interest. The requirement to modify or custom-build a cryo-capable TEM to use the method might prove prohibitive for many groups.
Applications of time-resolved cryoEM in Biology
Manual time-resolved cryoEM sample preparation in the seconds to minutes range
Among the multiple methods for trEM, only two have so far been used to answer biological questions. Unsurprisingly, the manual preparation in the seconds to minutes range is one of these methods since it only requires a common blot plunge freezing system for cryoEM sample preparation 33. The prokaryotic 70S ribosome can easily be purified in high yields by gradient centrifugation and can be refined to high resolution in cryoEM without much effort, making it an often-used system for trEM 89–91. During translation elongation, two tRNAs, the peptidyl-tRNA and the deacetylated tRNA move from the A- and P-sites to the P- and E-sites of the 70S ribosome by a ratchet-like movement of the 30S subunit 92–94. In absence of the elongation factor EF-G, this tRNA translocation can also occur in the reverse direction (retro-translocation), which is traceable within minutes 95. Fischer et al. initiated retro-translocation on a 70S post-translocation complex containing a peptidyl-tRNA in the P-site by adding a deacetylated tRNA and collected a time series of this reaction in the minute range using cryoEM. This allowed them to trace the tRNAs moving through the ribosome. Five pre-translocation states of which three states contain tRNAs in hybrid positions and three post-translocation states following the ratchet-like movement of the 30S subunit were identified. The change in distribution of the pre- and post-translocation states over time allowed the authors to determine the overall retro-translocation rate of 0.8 ± 0.1 min−1 43.
The forward-translocation step of the tRNAs and mRNA in prokaryotic 70S ribosomes is favoured by the GTPase elongation factor EF-G 96. Petrychenko et al. as well as Carbone et al. demonstrated how EF-G catalyses this translocation, using trEM in the seconds range in combination with measures that slow the reaction down, as the system would otherwise reach equilibrium within milliseconds 41,48.
Petrychenko et al. combined trEM sample preparation within 10 s from mixing to plunging, a reaction temperature of 4°C, polyamines, and apramycin, an antibiotic that inhibits the tRNA movement from the A- to the P-site, all with the aim of enriching for species in the early steps of translocation. The timepoint where GTP-hydrolysis has occurred but Pi-release is ongoing was chosen based on stopped-flow data under the same conditions. This allowed for the visualization of EF-G in its GDP–Pi- and GDP-bound states on translocating ribosomes. In the GDP-Pi form, EF-G stabilizes the rotated conformation of ribosomal subunits, while EF-G is repositioned upon Pi release such that it favours the rotation of the ribosomal subunits and a movement of the 30S head resulting in tRNA translocation 48.
Carbone et al. recorded a time series after incubation on ice for 0, 25 and 3600 s prior blotting and plunge freezing, but without the addition of any chemical inhibitors. Eight structures representing different states during ribosomal tRNA translocation were classified. The population of pre-translocating particles decreased over time, whereas the population of post-translocating particles increased. EF-G bound intermediates were only identified at the 25 s reaction timepoint, featuring three conformations. Throughout these conformations, the tRNAs translocate from the A/P, P/E to hybrid sites, accompanied by a shift of EF-G along the 30S subunit 41.
Manual trEM sample preparation was not only used for investigating the 70S ribosome translation. Another system examined is the loading of the MCM helicase double hexamer onto DNA by the origin recognition complex (ORC), a prerequisite for the bidirectional DNA replication 97,98. By analysing a reaction time series of 2, 6 and 20 minutes, novel intermediate steps during the formation of the double hexamer were identified by 2D classification. ORC recruits the first MCM hexamer by interacting with the MCM C-terminus which allows the interaction of a second ORC at the MCM N-terminus. The latter is now oriented in the direction where it can recruit a second MCM hexamer in an inverted orientation 61.
Using manual trEM in the tens of seconds to minutes range, Zhang et al. studied substrate processing of the 26S proteasome in presence of USP14 99. USP14 is a deubiquitinase that transiently associates with the 26S proteasome. It catalyses the removal of ubiquitin chains on substrates until a single chain remains 100. Zhang et al. could not identify USP14-associated 26S proteasome subsets in supposed conformational states where Rpn11 hydrolyses ubiquitin chains. However, USP14 was associated to the 26S proteasome during conformational states representing substrate unfolding, indicating that USP14 might lead to an alternative pathway of substrate deubiquitination during degradation 99.
Time-resolved cryoEM sample preparation in the millisecond range
Microfluidic mixing chips are the second method that has been used to study biological questions with trEM to this point. These chips provide excellent mixing of two compounds and allow to investigate reactions in the range of tens of milliseconds 71. As they require high sample volumes in ranging from tens to hundreds of μL per grid and samples need to be at higher concentrations than for conventional cryoEM grid preparation, the only system studied so far using microfluidic chips is the E. coli 70S ribosome.
The initial step of translation, the association of the 30S and 50S ribosomal subunits to form the 70S ribosome was the subject of the first two studies that tried to answer biological questions using trEM in the millisecond range. The 70S is held together by 12 to 14 inter-subunit bridges which can break and reform for certain ribosomal functions 94,101.
Shaikh et al. used microfluidic chips to acquire cryoEM datasets of mixed 30S and 50S subunits with incubation times of either 9.4 or 43 ms and in equilibrium. For both pre-equilibrium datasets, 3D reconstructions only show 8 out of 12 inter-subunit bridges, although all 12 were formed in the reassociated 70S of the experiment at equilibrium. This suggests that the missing bridges are formed after 43 ms 102.
Unfortunately, the resulting 3D maps of this study have a spatial resolution in the range of 23-33Å, limiting structural interpretation of these interactions. Chen et al. repeated this experiment with a focus on improved cryoEM grid quality. The implementation of an environmental chamber and the improvement of the gas pressure to produce ideal sprayed droplet sizes were done to this purpose. Based on ribosome association kinetics derived from light-scattering studies, reaction times of 60 ms, 140 ms, and the time required to reach equilibrium were chosen 103. The observed proportion of the associated 70S particles in the set of all particles containing 50S increased over time, even though slower than expected from the kinetic simulation. Three different associated ribosome conformations were identified, a non-rotated 70S, a non-rotated 70S with a swivelled 30S head and a rotated 70S ribosome. The observed proportions of these conformations were the same after 60 and 140 ms, indicating that associated ribosomes equilibrate before 60 ms after association. The three conformations were 3D refined to 9-12Å spatial resolution and the inter-subunit bridges that were missing in Shaikh et al. were found in all three of the timepoints 42,102.
The 70S initiation complex (70S IC) is formed upon the joining of the 50S ribosomal subunit to a 30S subunit containing an mRNA and initiator tRNA. This is promoted by initiation factors that must dissociate from the 30S subunit before the formation of the 70S elongation-competent complex (70S EC)104. Kaledhonkar et al. used trEM in the range of tens to hundreds of milliseconds to investigate 70S ribosome association, the dissociation of the initiation factors IF2 and the positioning of the initiator tRNA during the formation of the 70S EC complex 46. The reaction times of 20, 80, 200 and 600 ms were chosen based on a kinetic model with rate constants from literature where the 70S IC with GTP- or GDP-Pi-bound IF2 is most populated after 150 ms and the formation of the 70S EC is complete by approximately two thirds after 600 ms 105. The resulting population distributions of the 50S, 70S IC and 70S EC followed the predictions of the kinetic model, where the 50S population decreased and the 70S EC increased over time. The 70S IC population has reached its maximum after 80 ms and IF2 has dissociated from the major 70S IC population after 200 ms throughout 600 ms. Following dissociation of IF2, the 70S IC is not stabilized anymore in a semi-rotated state and the 30S subunit rotates reversely into the 70S EC state, becoming ready for translational elongation 46.
At the end of translation by the 70S ribosome, the mRNA signals termination via the stop codons. The stop codons in the A site leads to the recruitment of release factors (RF) that hydrolyse the growing peptide strand from the peptidyl-tRNA in the P site that is 70Å away from the A site. Unbound RF2 is compact and only 20Å in size, but for hydrolysation they extend to perform hydrolysis 106–109. To investigate the transient compact forms of RF1 and RF2 recruited by the 70S ribosome prior to translation termination, Fu et al. analysed trEM timepoints of the 70S ribosomes incubated with release factors for 24 and 60 ms and in equilibrium. These timepoints are based on a kinetic model with rate constants derived from quench-flow experiments, where RFs were predominantly in the compact form at 24 ms and heavily reduced after 60 ms. The kinetic model was somewhat in agreement with the trEM data, although the trEM data suggested slightly faster rate constants. Further analysis of the trEM datasets revealed that after 24 ms, the RF was mainly bound in the compact form and the peptidyl-tRNA was in the P-site. After 60 ms, the RF was mainly bound in the extended form and the hydrolysed peptide in the exit tunnel. After 45 s, the RF was still bound in the extended form and peptides were no longer found in the exit tunnel 110.
Discussion and Outlook
While trEM promises to become an important tool for the analysis of many molecular machines in the future, there are a few caveats and challenges ahead as can be seen from the lack of studies employing the method.
The main reason for the lack of studies is certainly the absence of a widespread user-friendly device for the preparation of samples. At the moment, a plethora of different approaches exist in parallel and only one is commercially available so far. Additionally, all of the described devices have the overarching disadvantage in that they require significant investments, with purchasing and assembly being only the most obvious ones. Importantly, there is still a lack in precise time calibration capabilities and orthogonal data analyses such as smFRET that are readily feasible are also missing. Along these lines, the reproducibility of the method, meaning whether the measurement of several samples from the same time point would provide the same result has not been systematically studied. Currently, that is an issue that has not been sufficiently addressed in steady state measurements. Another part of the problem is that the analysis tools we are using do not deliver high confidence results for the classification of individual particles due to the enormous levels of noise in the images. Here, characterisation through orthogonal methods, for instance a cryoCLEM approach, may also be necessary. Thus, with our current capabilities, trEM can be seen as more of a qualitative method providing structural insights into possible molecular trajectories rather than as tool for the extraction of kinetic data.
For trEM to be a generally accepted and utilised method, a well-characterized device that can be easily set up by a research institute’s workshop will be necessary such that many labs can further develop its applicability in parallel. Rubinstein et al. propose the use of self-wicking grids in combination with piezo atomizers that can be salvaged from very easily available air humidifiers for trEM studies 111. Besides the low cost and effort involved to acquire these piezo atomizers, they are capable of depositing as little as 1-4 μL of sample in the form of a spray onto a plunging grid. The required sample volume is not as low as for the aforementioned inkjets but is still in a comparable range to the typical amounts used for conventional cryoEM grid preparation 74. The produced cryoEM grids are of sufficient quality for data collection, but experiments that quantify the efficacy of mixing with this approach have yet to be performed. To examine how well the approach preserves the structural integrity of proteins, Apoferritin was sprayed on a grid and 3D-refined to 2.77Å resolution in single particle processing, indicating that the protein remains undamaged after sample deposition 111. Tan and Rubinstein further developed this device by replacing the very elaborative protocol required to produce self-wicking grids with a backside blotting system where they use commonly available glass fibre filters. Compared to commonly used filter papers these glass fibre filters can more efficiently remove excess of solvent from the sprayed grids leaving a thin film and resulting in approximately a third of collectible area on the entire grid which is sufficient for acquiring a 2 Å resolution structures of Apoferritin. In addition, it was demonstrated that this device can increase the number of rare views when the time of absorption to the air water interface is reduced. The percentage of Influenza A hemagglutinin trimer sideviews was increased from 2 % to 34 % when the absorption time was reduced from several seconds to 90-135 ms 112.
A second reason for the low number of biological studies is the lack of the “right” samples. The biochemical processes to be followed need to be synchronisable and need to stay synchronised over multiple time scales, which is a major challenge given the stochastic nature of biological molecular machines. This excludes a number of important molecular machines that exhibit a fast-processing cycle and lack characteristic initiation and termination steps, such as transporter proteins and ion channels. Given the stochasticity of biological molecular machines, trEM will only provide an enrichment of certain states but never deliver pure populations that could be assigned to a specific step in a cycle. Additionally, the time scales to be visualized need to be accessible to the method. Given the current devices, processes that occur on the high millisecond or low second time scale would be ideally suited. Additionally, the actual timing of the process should be known as precisely as possible through external methods such as single molecule or fluorescence measurements as only a few time points can be prepared and analysed due to the high demand in measurement and analysis time. This limits the possible analysis to systems that are already well characterized like ribosomes and proteasomes. However, in turn a well characterized sample may not even require an additional device. If the process of interest is on the second time scale, manual mixing will be sufficient and can provide many fascinating insights. Despite the narrow range of biological questions studied so far, we expect trEM to be applicable over a wide range of processes like signalling (E3 ligases and kinases), transport (cytoskeleton, motor proteins), macromolecular processing (Cas9, RNAi) and membrane fusion (snares). In summary, trEM is a very promising method as it is the only approach that may provide molecular movies of a molecular machine directly with full structural detail. However, it is still in its infancy and many characterizations and pioneering studies will need to continue to be done in the next decade.
Acknowledgements
S.J.A. and D.H. are supported by WWTF LS19-029, N.G.B. and T.B. are supported by NIH R35GM128855. D.K. is funded by Boehringer Ingelheim. Additionally, the Haselbach Lab and the IMP are supported by Boehringer Ingelheim.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- 1.Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, and Martin A (2018). Structure and Function of the 26S Proteasome. Annu. Rev. Biochem 87, 697–724. 10.1146/annurev-biochem-062917-011931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank J. (2017). The translation elongation cycle—capturing multiple states by cryo-electron microscopy. Philos. Trans. R. Soc. B Biol. Sci 372, 20160180. 10.1098/rstb.2016.0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osman S, and Cramer P (2020). Structural Biology of RNA Polymerase II Transcription: 20 Years On. Annu. Rev. Cell Dev. Biol 36, 1–34. 10.1146/annurev-cellbio-042020-021954. [DOI] [PubMed] [Google Scholar]
- 4.Wu W-J, Yang W, and Tsai M-D (2017). How DNA polymerases catalyse replication and repair with contrasting fidelity. Nat. Rev. Chem 1, 0068. 10.1038/s41570-017-0068. [DOI] [Google Scholar]
- 5.Beld J, Lee DJ, and Burkart MD (2015). Fatty acid biosynthesis revisited: structure elucidation and metabolic engineering. Mol. Biosyst 11, 38–59. 10.1039/C4MB00443D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt H, and Carter AP (2016). Review: Structure and mechanism of the dynein motor ATPase. Biopolymers 105, 557–567. 10.1002/bip.22856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilkinson ME, Charenton C, and Nagai K (2020). RNA Splicing by the Spliceosome. Annu. Rev. Biochem 89, 359–388. 10.1146/annurev-biochem-091719-064225. [DOI] [PubMed] [Google Scholar]
- 8.Yamano H. (2019). APC/C: current understanding and future perspectives. F1000Research 8, 725. 10.12688/flOOOresearch.18582.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Astumian RD (2015). Irrelevance of the Power Stroke for the Directionality, Stopping Force, and Optimal Efficiency of Chemically Driven Molecular Machines. Biophys. J 108, 291–303. 10.1016/j.bpj.2014.11.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acker MG, Shin B-S, Nanda JS, Saini AK, Dever TE, and Lorsch JR (2009). Kinetic Analysis of Late Steps of Eukaryotic Translation Initiation. JMB 385, 491–506. 10.1016/j.jmb.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Appling FD, Lucius AL, and Schneider DA (2015). Transient-State Kinetic Analysis of the RNA Polymerase I Nucleotide Incorporation Mechanism. Biophys. J 109, 2382–2393. 10.1016/j.bpj.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bard JAM, Bashore C, Dong KC, and Martin A (2019). The 26S Proteasome Utilizes a Kinetic Gateway to Prioritize Substrate Degradation. Cell 177, 286–298.e15. 10.1016/j.cell.2019.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goerisch H, Goss DJ, and Parkhurst LJ (1976). Kinetics of ribosome dissociation and subunit association studied in a light-scattering stopped-flow apparatus. Biochemistry 15, 5743–5753. 10.1021/bi00671a010. [DOI] [PubMed] [Google Scholar]
- 14.Henderson KL, Evensen CE, Molzahn CM, Felth LC, Dyke S, Liao G, Shkel IA, and Record MT (2019). RNA Polymerase: Step-by-Step Kinetics and Mechanism of Transcription Initiation. Biochemistry 58, 2339–2352. 10.1021/acs.biochem.9b00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoskins AA, Gelles J, and Moore MJ (2011). New insights into the spliceosome by single molecule fluorescence microscopy. Curr. Opin. Chem. Biol 15, 864–870. 10.1016/j.cbpa.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raper AT, Reed AJ, and Suo Z (2018). Kinetic Mechanism of DNA Polymerases: Contributions of Conformational Dynamics and a Third Divalent Metal Ion. Chem Rev 118, 6000–6025. 10.1021/acs.chemrev.7b00685. [DOI] [PubMed] [Google Scholar]
- 17.Sharma AK, Sormanni P, Ahmed N, Ciryam P, Friedrich UA, Kramer G, and O’Brien EP (2019). A chemical kinetic basis for measuring translation initiation and elongation rates from ribosome profiling data. PLOS Comput. Biol 15, e1007070. 10.1371/journal.pcbi.1007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sainsbury S, Bernecky C, and Cramer P (2015). Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol 16,129–143. 10.1038/nrm3952. [DOI] [PubMed] [Google Scholar]
- 19.Singh J, and Padgett RA (2009). Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol 16, 1128–1133. 10.1038/nsmb.l666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alpert T, Herzel L, and Neugebauer KM (2017). Perfect timing: splicing and transcription rates in living cells: Splicing and transcription rates in living cells. Wiley Interdiscip. Rev. RNA 8, e1401. 10.1002/wrna.l401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merrick WC, and Pavitt GD (2018). Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb. Perspect. Biol 10, a033092. 10.1101/cshperspect.a033092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan X, Hoek TA, Vale RD, and Tanenbaum ME (2016). Dynamics of Translation of Single mRNA Molecules In Vivo. Cell 165, 976–989. 10.1016/j.cell.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ingolia NT, Lareau LF, and Weissman JS (2011). Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity and Dynamics of Mammalian Proteomes. Cell 147, 789–802. 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, Wu J, Lu Y, Ma YB, Lee BH, Yu Z, Ouyang Q, Finley DJ, Kirschner MW, and Mao Y (2016). Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. U. S. A 113, 12991–12996. 10.1073/pnas.1614614113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haselbach D, Schrader J, Lambrecht F, Henneberg F, Chari A, and Stark H (2017). Long-range allosteric regulation of the human 26S proteasome by 20S proteasome-targeting cancer drugs. Nat. Commun 8, 15578. 10/f977p6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unverdorben P, Beck F, Sledz P, Schweitzer A, Pfeifer G, Plitzko JM, Baumeister W, and Förster F (2014). Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc. Natl. Acad. Sci. U. S. A 111, 5544–5549. 10.1073/pnas.l403409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wehmer M, Rudack T, Beck F, Aufderheide A, Pfeifer G, Plitzko JM, Förster F, Schulten K, Baumeister W, and Sakata E (2017). Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc. Natl. Acad. Sci. U. S. A 114, 1305–1310. 10.1073/pnas.1621129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franken LE, Grünewald K, Boekema EJ, and Stuart MCA (2020). A Technical Introduction to Transmission Electron Microscopy for Soft-Matter: Imaging, Possibilities, Choices, and Technical Developments. Small 16, 1906198. 10.1002/smll.201906198. [DOI] [PubMed] [Google Scholar]
- 29.Nakane T, Kotecha A, Sente A, McMullan G, Masiulis S, Brown PMGE, Grigoras IT, Malinauskaite L, Malinauskas T, Miehling J, et al. (2020). Single-particle cryo-EM at atomic resolution. Nature 587, 152–156. 10/gjwcfp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yip KM, Fischer N, Paknia E, Chari A, and Stark H (2020). Atomic-resolution protein structure determination by cryo-EM. Nature 587, 157–161. 10/ghtfcv. [DOI] [PubMed] [Google Scholar]
- 31.Grassucci RA, Taylor DJ, and Frank J (2007). Preparation of macromolecular complexes for cryo-electron microscopy. Nat. Protoc 2, 3239–3246. 10/fdq94g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor KA, and Glaeser RM (1976). Electron Microscopy of Frozen Hydrated Biological Specimens. J. Ultrastruct. Res 55, 448–456. 10/ccrz96. [DOI] [PubMed] [Google Scholar]
- 33.Dubochet J, Lepault J, Freeman R, Berriman JA, and Homo J-C (1982). Electron microscopy of frozen water and aqueous solutions. J. Microsc 128, 219–237. 10/frtwbj. [Google Scholar]
- 34.Jelsch C, Teeter MM, Lamzin V, Pichon-Pesme V, Blessing RH, and Lecomte C (2000). Accurate protein crystallography at ultra-high resolution: Valence electron distribution in crambin. Proc. Natl. Acad. Sci 97, 3171–3176. 10/bxmfbp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henderson R, and Unwin PNT (1975). Three-dimensional model of purple membrane obtained by electron microscopy. Nature 257, 28–32. 10/cvmx57. [DOI] [PubMed] [Google Scholar]
- 36.Frank J, and Ourmazd A (2016). Continuous changes in structure mapped by manifold embedding of single-particle data in cryo-EM. Methods 100, 61–67. 10/f8n36q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kimanius D, Dong L, Sharov G, Nakane T, and Scheres SHW (2021). New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J 478, 4169–4185. 10/gp5ncf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maji S, Liao H, Dashti A, Mashayekhi G, Ourmazd A, and Frank J (2020). Propagation of Conformational Coordinates Across Angular Space in Mapping the Continuum of States from Cryo-EM Data by Manifold Embedding. J. Chem. Inf. Model. 60, 2484–2491. 10/gp5ncd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong ED, Bepler T, Berger B, and Davis JH (2021). CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 18, 176–185. 10/gjv8x8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166.10/gfmxdp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carbone CE, Loveland AB, Gamper HB, Hou Y-M, Demo G, and Korostelev AA (2021). Time-resolved cryo-EM visualizes ribosomal translocation with EF-G and GTP. Nat. Commun 12, 7236. 10/gp5nch. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen B, Kaledhonkar S, Sun M, Shen B, Lu Z, Barnard D, Lu T-M, Gonzalez RL, and Frank J (2015). Structural Dynamics of Ribosome Subunit Association Studied by Mixing-Spraying Time-Resolved Cryogenic Electron Microscopy. Structure 23, 1097–1105. 10.1016/j.str.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, and Stark H (2010). Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature 466, 329–333. 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- 44.Fu Z, Kaledhonkar S, Borg A, Sun M, Chen B, Grassucci RA, Ehrenberg M, and Frank J (2016). Key Intermediates in Ribosome Recycling Visualized by Time-Resolved Cryoelectron Microscopy. Structure 24, 2092–2101. 10.1016/j.str.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greene ER, Dong KC, and Martin A (2020). Understanding the 26S proteasome molecular machine from a structural and conformational dynamics perspective. Curr. Opin. Struct. Biol 61, 33–41.10/gj6snh. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaledhonkar S, Fu Z, Caban K, Li W, Chen B, Sun M, Gonzalez RL, and Frank J (2019). Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature 570, 400–404. 10/ghfkm7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Z, Barnard D, Shaikh TR, Meng X, Mannella CA, Yassin AS, Agrawal RK, Wagenknecht T, and Lu T-M (2014). Gas-assisted annular microsprayer for sample preparation for time-resolved cryo-electron microscopy. J. Micromechanics Microengineering 24, 115001. 10.1088/0960-1317/24/11/115001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrychenko V, Peng B-Z, de A. P. Schwarzer AC, Peske F, Rodnina MV, and Fischer N (2021). Structural mechanism of GTPase-powered ribosome-tRNA movement. Nat. Commun 12, 5933.10/gp5ncg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D’Imprima E, Floris D, Joppe M, Sánchez R, Grininger M, and Kühlbrandt W (2019). Protein denaturation at the air-water interface and how to prevent it. eLife 8, e42747.10/ggktds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu Z, Shaikh TR, Barnard D, Meng X, Mohamed H, Yassin A, Mannella CA, Agrawal RK, Lu T-M, and Wagenknecht T (2009). Monolithic microfluidic mixing-spraying devices for time-resolved cryo-electron microscopy. J. Struct. Biol 168, 388–395. 10.1016/j.jsb.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noble AJ, Dandey VP, Wei H, Brasch J, Chase J, Acharya P, Tan YZ, Zhang Z, Kim LY, Scapin G, et al. (2018). Routine single particle CryoEM sample and grid characterization by tomography. eLife 7, e34257. 10/gm6nth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yano YF, Arakawa E, Voegeli W, Kamezawa C, and Matsushita T (2018). Initial Conformation of Adsorbed Proteins at an Air–Water Interface. J. Phys. Chem. B. 10/gdd8tf. [DOI] [PubMed] [Google Scholar]
- 53.de la Peña AH, Goodall EA, Gates SN, Lander GC, and Martin A (2018). Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 362, eaav0725. 10/gfggbv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haselbach D, Komarov I, Agafonov DE, Hartmuth K, Graf B, Dybkov O, Urlaub H, Kastner B, Lührmann R, and Stark H (2018). Structure and Conformational Dynamics of the Human Spliceosomal Bact Complex. Cell 172, 454–464.e11. 10/gcxqnq. [DOI] [PubMed] [Google Scholar]
- 55.Hofmann S, Januliene D, Mehdipour AR, Thomas C, Stefan E, Brüchert S, Kuhn BT, Geertsma ER, Hummer G, Tampé R, et al. (2019). Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571, 580–583. 10/gm64cg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinz-Thompson CD, Sharma AK, Frank J, Gonzalez RL, and Chowdhury D (2015). Quantitative Connection between Ensemble Thermodynamics and Single-Molecule Kinetics: A Case Study Using Cryogenic Electron Microscopy and Single-Molecule Fluorescence Resonance Energy Transfer Investigations of the Ribosome. J. Phys. Chem. B 119, 10888–10901. 10/f7pwvk. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plaschka C, Lin P-C, and Nagai K (2017). Structure of a pre-catalytic spliceosome. Nature 546, 617–621. 10/f9762d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Z, Chen E, Zhang S, Wang WL, Ma Y, Dong Y, Liu C, Yin C-C, and Mao Y (2022). Hidden dynamics of proteasome autoregulation discovered by cryo-EM data-driven deep learning. 2020.12.22.423932. 10.1101/2020.12.22.423932. [DOI] [Google Scholar]
- 59.Zhang S, Zou S, Yin D, Finley D, Wu Z, and Mao Y (2021). Mechanistic insights into dynamic mutual regulation of USP14 and proteasome (Biochemistry) 10.1101/2021.09.15.460436. [DOI] [Google Scholar]
- 60.Seitz E, and Frank J (2020). POLARIS: Path of Least Action Analysis on Energy Landscapes. J. Chem. Inf. Model. 60, 2581–2590. 10/gp5ncc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller TCR, Locke J, Greiwe JF, Diffley JFX, and Costa A (2019). Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature 575, 704–710. 10/gnr724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Basner JE, and Schwartz SD (2005). How Enzyme Dynamics Helps Catalyze a Reaction in Atomic Detail: A Transition Path Sampling Study. J. Am. Chem. Soc 127,13822–13831. 10/d44w4z. [DOI] [PubMed] [Google Scholar]
- 63.Boehr DD, Dyson HJ, and Wright PE (2006). An NMR Perspective on Enzyme Dynamics. Chem. Rev 106, 3055–3079. 10/dnmr62. [DOI] [PubMed] [Google Scholar]
- 64.Henzler-Wildman K, and Kern D (2007). Dynamic personalities of proteins. Nature 450, 964–972. 10/dhfmnk. [DOI] [PubMed] [Google Scholar]
- 65.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M, and Kern D (2007). A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 450, 913–916. 10/cpp85j. [DOI] [PubMed] [Google Scholar]
- 66.Singh V, Lee JE, Núñez S, Howell PL, and Schramm VL (2005). Transition State Structure of 5’-Methylthioadenosine/ S -Adenosylhomocysteine Nucleosidase from Escherichia coli and Its Similarity to Transition State Analogues. Biochemistry 44, 11647–11659. 10/bh48h5. [DOI] [PubMed] [Google Scholar]
- 67.Wolfenden R (1969). Transition State Analogues for Enzyme Catalysis. Nature 223, 704–705. 10/dck3bk. [DOI] [PubMed] [Google Scholar]
- 68.Berriman J, and Unwin N (1994). Analysis of transient structures by cryo-microscopy combined with rapid mixing of spray droplets. Ultramicroscopy 56, 241–252.10/ctwp5j. [DOI] [PubMed] [Google Scholar]
- 69.Dandey VP, Budell WC, Wei H, Bobe D, Maruthi K, Kopylov M, Eng ET, Kahn PA, Hinshaw JE, Kundu N, et al. (2020). Time-resolved cryoEM using Spotiton (Biophysics) 10.1101/2020.04.20.050518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kontziampasis D, Klebl DP, Iadanza MG, Scarff CA, Kopf F, Sobott F, Monteiro DCF, Trebbin M, Muench SP, and White HD (2019). A cryo-EM grid preparation device for time-resolved structural studies. IUCrJ 6, 1024–1031. 10/gm6ntv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mäeots M-E, Lee B, Nans A, Jeong S-G, Esfahani MMN, Ding S, Smith DJ, Lee C-S, Lee SS, Peter M, et al. (2020). Modular microfluidics enables kinetic insight from time-resolved cryo-EM. Nat. Commun 11, 3465. 10/gm6nts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.White HD, Thirumurugan K, Walker ML, and Trinick J (2003). A second generation apparatus for time-resolved electron cryo-microscopy using stepper motors and electrospray. J. Struct. Biol 144, 246–252. 10/fcmj2c. [DOI] [PubMed] [Google Scholar]
- 73.White HD, Walker ML, and Trinick J (1998). A Computer-Controlled Spraying-Freezing Apparatus for Millisecond Time-Resolution Electron Cryomicroscopy. J. Struct. Biol 121, 306–313. 10/b2p7ft. [DOI] [PubMed] [Google Scholar]
- 74.Dandey VP, Wei H, Zhang Z, Tan YZ, Acharya P, Eng ET, Rice WJ, Kahn PA, Potter CS, and Carragher B (2018). Spotiton: New features and applications. J. Struct. Biol 202, 161–169. 10/gffmjm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ménétret JF, Hofmann W, Schröder RR, Rapp G, and Goody RS (1991). Time-resolved cryo-electron microscopic study of the dissociation of actomyosin induced by photolysis of photolabile nucleotides. J. Mol. Biol 219, 139–144. 10/bv74gp. [DOI] [PubMed] [Google Scholar]
- 76.Shaikh TR, Barnard D, Meng X, and Wagenknecht T (2009). Implementation of a flash-photolysis system for time-resolved cryo-electron microscopy. J. Struct. Biol 165, 184–189. 10.1016/j.jsb.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harder OF, Voss JM, Olshin PK, Drabbels M, and Lorenz UJ (2022). Microsecond Melting and Revitrification of Cryo Samples – Protein Structure and Beam-Induced Motion (Biophysics) 10.1101/2022.02.14.480378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Voss JM, Harder OF, Olshin PK, Drabbels M, and Lorenz UJ (2021). Rapid melting and revitrification as an approach to microsecond time-resolved cryo-electron microscopy. Chem. Phys. Lett 778, undefined-undefined. 10/gpjt7d. [Google Scholar]
- 79.Unwin N (1995). Acetylcholine receptor channel imaged in the open state. Nature 373, 37–43. 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 80.Unwin N, and Fujiyoshi Y (2012). Gating Movement of Acetylcholine Receptor Caught by Plunge-Freezing. J. Mol. Biol 422, 617–634. 10/gkzpth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feng X, Fu Z, Kaledhonkar S, Jia Y, Shah B, Jin A, Liu Z, Sun M, Chen B, Grassucci RA, et al. (2017). A Fast and Effective Microfluidic Spraying-Plunging Method for High-Resolution Single-Particle Cryo-EM. Structure 25, 663–670.e3. 10.1016/j.str.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lu Z, Mcmahon J, Mohamed H, Barnard D, Shaikh TR, Mannella CA, Wagenknecht T, and Lu TM (2010). Passive Microfluidic device for Sub Millisecond Mixing. Sens Actuators B Chem 144, 301–309. 10/bzm2n9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jain T, Sheehan P, Crum J, Carragher B, and Potter CS (2012). Spotiton: A prototype for an integrated inkjet dispense and vitrification system for cryo-TEM. J. Struct. Biol 179, 68–75. 10/f32wkm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noble AJ, Wei H, Dandey VP, Zhang Z, Tan YZ, Potter CS, and Carragher B (2018). Reducing effects of particle adsorption to the air–water interface in cryo-EM. Nat. Methods 15, 793–795. 10/gd85f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Subramaniam S, Gerstein M, Oesterhelt D, and Henderson R (1993). Electron diffraction analysis of structural changes in the photocycle of bacteriorhodopsin. EMBO J. 12, 1–8. 10.1002/j.1460-2075.1993.tb05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Subramaniam S, and Henderson R (2000). Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature 406, 653–657. 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 87.Yoder N, Jalali-Yazdi F, Noreng S, Houser A, Baconguis I, and Gouaux E (2020). Light-coupled cryo-plunger for time-resolved cryo-EM. J. Struct. Biol 212, 107624. 10/gp5nbh. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Olshin PK, Drabbels M, and Lorenz UJ (2020). Characterization of a time-resolved electron microscope with a Schottky field emission gun. Struct. Dyn 7, 054304. 10/gm4zrf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Halfon Y, Matzov D, Eyal Z, Bashan A, Zimmerman E, Kjeldgaard J, Ingmer H, and Yonath A (2019). Exit tunnel modulation as resistance mechanism of S. aureus erythromycin resistant mutant. Sci. Rep 9, 11460. 10/gp5nb7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rundlet EJ, Holm M, Schacherl M, Natchiar SK, Altman RB, Spahn CMT, Myasnikov AG, and Blanchard SC (2021). Structural basis of early translocation events on the ribosome. Nature 595, 741–745. 10/gp5nb6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watson ZL, Ward FR, Méheust R, Ad O, Schepartz A, Banfield JF, and Cate JH (2020). Structure of the bacterial ribosome at 2 Å resolution. eLife 9, e60482. 10/gn9tsx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cornish PV, Ermolenko DN, Noller HF, and Ha T (2008). Spontaneous Intersubunit Rotation in Single Ribosomes. Mol. Cell 30, 578–588. 10/fp5ckw. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frank J, and Agrawal RK (2000). A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature 406, 318–322. 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- 94.Valle M, Zavialov A, Sengupta J, Rawat U, and Frank J (2003). Locking and Unlocking of Ribosomal Motions. 12. 10/ddvkv9. [DOI] [PubMed] [Google Scholar]
- 95.Konevega AL, Fischer N, Semenkov YP, Stark H, Wintermeyer W, and Rodnina MV (2007). Spontaneous reverse movement of mRNA-bound tRNA through the ribosome. Nat. Struct. Mol. Biol 14, 318–324. 10/dw3wb8. [DOI] [PubMed] [Google Scholar]
- 96.Rodnina MV, Savelsbergh A, Katunin VI, and Wintermeyer W (1997). Hydrolysis of GTP by elongation factor G drives tRNA movement on the ribosome. Nature 385, 37–41. 10/cdmw73. [DOI] [PubMed] [Google Scholar]
- 97.Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, and Speck C (2009). A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci 106, 20240–20245. 10/ck9wq7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, and Diffley JFX (2009). Concerted Loading of Mcm2–7 Double Hexamers around DNA during DNA Replication Origin Licensing. Cell 139, 719–730. 10/dp9k56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang S, Zou S, Yin D, Zhao L, Finley D, Wu Z, and Mao Y (2022). USP14-regulated allostery of the human proteasome by time-resolved cryo-EM. Nature, 1–8. 10/gp5nb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee B-H, Lu Y, Prado MA, Shi Y, Tian G, Sun S, Elsasser S, Gygi SP, King RW, and Finley D (2016). USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 532, 398–401. 10/f8jdvm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Frank J, Zhu J, Penczek P, Li Y, Srivastava S, Verschoor A, Radermacher M, Grassucci R, Lata RK, and Agrawal RK (1995). A model of protein synthesis based on cryo-electron microscopy of the E. coli ribosome. Nature 376, 441–444. [DOI] [PubMed] [Google Scholar]
- 102.Shaikh TR, Yassin AS, Lu Z, Barnard D, Meng X, Lu T-M, Wagenknecht T, and Agrawal RK (2014). Initial bridges between two ribosomal subunits are formed within 9.4 milliseconds, as studied by time-resolved cryo-EM. Proc. Natl. Acad. Sci 111, 9822–9827. 10.1073/pnas.1406744111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hennelly SP, Antoun A, Ehrenberg M, Gualerzi CO, Knight W, Lodmell JS, and Hill WE (2005). A Time-resolved Investigation of Ribosomal Subunit Association. J. Mol. Biol 346, 1243–1258. 10/c868tb. [DOI] [PubMed] [Google Scholar]
- 104.Antoun A (2003). The roles of initiation factor 2 and guanosine triphosphate in initiation of protein synthesis. EMBO J. 22, 5593–5601. 10/cn7vtz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goyal A, Belardinelli R, Maracci C, Milón P, and Rodnina MV (2015). Directional transition from initiation to elongation in bacterial translation. Nucleic Acids Res. 43, 10700–10712. 10/f8crhv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Klaholz BP, Pape T, Zavialov AV, Myasnikov AG, Orlova EV, Vestergaard B, Ehrenberg M, and van Heel M (2003). Structure of the Escherichia coli ribosomal termination complex with release factor 2. Nature 421, 90–94. 10/djjdxn. [DOI] [PubMed] [Google Scholar]
- 107.Shin DH, Brandsen J, Jancarik J, Yokota H, Kim R, and Kim S-H (2004). Structural Analyses of Peptide Release Factor 1 from Thermotoga maritima Reveal Domain Flexibility Required for Its Interaction with the Ribosome. J. Mol. Biol 341, 227–239. 10/bght95. [DOI] [PubMed] [Google Scholar]
- 108.Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, and Barford D (2000). The Crystal Structure of Human Eukaryotic Release Factor eRF1—Mechanism of Stop Codon Recognition and Peptidyl-tRNA Hydrolysis. 11. 10/bv4q9p. [DOI] [PubMed] [Google Scholar]
- 109.Vestergaard B, Van LB, Andersen GR, Nyborg J, Buckingham RH, and Kjeldgaard M (2001). Bacterial Polypeptide Release Factor RF2 Is Structurally Distinct from Eukaryotic eRFl. Mol. Cell 8, 1375–1382. 10/bkg5x5. [DOI] [PubMed] [Google Scholar]
- 110.Fu Z, Indrisiunaite G, Kaledhonkar S, Shah B, Sun M, Chen B, Grassucci RA, Ehrenberg M, and Frank J (2019). The structural basis for release-factor activation during translation termination revealed by time-resolved cryogenic electron microscopy. Nat. Commun 10, 2579. 10/gjfc2s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rubinstein JL, Guo H, Ripstein ZA, Haydaroglu A, Au A, Yip CM, Di Trani JM, Benlekbir S, and Kwok T (2019). Shake-it-off: a simple ultrasonic cryo-EM specimen-preparation device. Acta Crystallogr. Sect. Struct. Biol 75, 1063–1070. 10/gm6ntt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tan YZ, and Rubinstein JL (2020). Through-grid wicking enables high-speed cryoEM specimen preparation. Acta Crystallogr. Sect. Struct. Biol 76, 1092–1103. 10.1107/S2059798320012474. [DOI] [PubMed] [Google Scholar]
- 113.Klebl DP, White HD, Sobott F, and Muench SP (2021). On-grid and in-flow mixing for time-resolved cryo-EM. Acta Crystallogr. Sect. Struct. Biol 77, 1233–1240. 10/gp5nbg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harder OF, Voss JM, Olshin PK, Drabbels M, and Lorenz UJ (2022). Microsecond Melting and Revitrification of Cryo Samples – Protein Structure and Beam-Induced Motion (Biophysics) 10.1101/2022.02.14.480378. [DOI] [PMC free article] [PubMed] [Google Scholar]