

Abstract
Folded molecules provide complex interaction interfaces amenable to sophisticated self‐assembly motifs. Because of their high conformational stability, aromatic foldamers constitute suitable candidates for the rational elaboration of self‐assembled architectures. Several multiturn helical aromatic oligoamides have been synthesized that possess arrays of acridine appendages pointing in one or two directions. The acridine units were shown to direct self‐assembly in the solid state via aromatic stacking leading to recurrent helix‐helix association patterns under the form of discrete dimers or extended arrays. In the presence of Pd(II), metal coordination of the acridine units overwhelms other forces and generates new metal‐mediated multihelical self‐assemblies, including macrocycles. These observations demonstrate simple access to different types of foldamer‐containing architectures, ranging from discrete objects to 1D and, by extension, 2D and 3D arrays.
Keywords: aromatic stacking, foldamer, helical conformation, metal coordination, self-assembly
Aromatic foldamers helices constitute convenient rigid scaffolds but rarely form ordered arrays upon self‐assembly or in crystals. We show that placing acridine appendages at defined position at the surface of aromatic helices guide their organization in the solid state to form discrete dimers or 1D arrays. Furthermore, metal‐coordination at the acridine units of the same compounds drives self‐assembly in solution.

Introduction
As exemplified by the large architectures formed by proteins and nucleic acids in nature, folded molecules provide complex interaction interfaces amenable to sophisticated self‐assembly motifs. Reprogramming protein and nucleic acid interaction interfaces has given access to original and useful artificial assemblies such as protein‐metal–organic frameworks [1] and DNA origamis. [2] At a smaller scale, the potential at self‐assembly in solution and in the solid state of synthetic foldamers is also emerging. [3] Occasionally, this potential can be expressed without any added features, as in the so‐called foldectures of β‐peptide helices discovered by Lee. [4] However, most cases exploit the introduction of some specific interacting functional groups. Thus, amphipathicity may promote helix bundling in water. [5] Aromatic appendages with a strong propensity to stack have been introduced as side chains of polyproline and α‐helices, [6] and of oligoamide rods. [7] Polyproline helices have also served to form peptide‐metal frameworks using terminal carboxylate functions to coordinate the metal. Similarly, metal coordination to β‐peptide helices having pyridine functions in their side chains result in the formation of discrete objects with complex topologies [8] as well as frameworks. [9] All these assemblies rely on folded scaffolds, that is, structures with potential flexibility but whose preferred conformations are nevertheless generally preserved upon assembly. They thus constitute a sort of mid‐way between, on one hand, the rigid building blocks used to produce metal–organic cages [10] and frameworks [11] and, on the other hand, more flexible multitopic ligands that have no preferred conformation but nevertheless produce well‐defined objects such as helicates [12] or knots [13] in the presence of metal ions.
Aromatic foldamers constitute a vast class of oligomers with aryl rings in their main chain that have been shown to form diverse helix and sheet structures. [14] Their conformations tend to be very stable. They thus constitute suitable candidates to elaborate complex assemblies. Yet this potential is far from being fulfilled. Stacks of helices or crescents have been observed in crystal structures [15] and in colloidal assemblies [16] and may be promoted by templates [17] or when the helices are functionalized by hydrogen bonding [18] or halogen bonding [19] sticky ends. However, more often than not, crystal structures show no defined organization patterns. Along this line, we have introduced hydroxy groups at the surface of aromatic helices to promote the assembly of multihelical structures via hydrogen bonding. [20] We have also used metal coordination to orient two helices at a specific angle. [21] Here, we show that acridine appendages may be used to generate well‐defined packing motifs of aromatic helices in the solid state, and to promote metal‐mediated self‐assembly in solution.
Results and Discussion
Design and synthesis
Oligoamides of 8‐amino‐2‐quinolinecarboxylic acid (Q in Figure 1a) adopt stable helical conformations in various solvents. [22] The well‐defined shapes of these helices with 2.5 units per turn and a pitch of 3.5 Å allow for the introduction of functional groups at specific positions on their external surface in order to promote interactions with biomolecules [23] or self‐assembly. [20] Inspired by earlier work on peptide helices,[ 6 , 8 , 9 ] we devised that large, rigidly linked, aromatic appendages at the surface of aromatic foldamer helices may give rise to new modes of assembly. The acridine group was selected for this purpose because it combines several advantages (Figure 1b, c). It is electron poor and may promote anti‐parallel aromatic stacking guided by dipolar interactions. In addition, it can coordinate metal ions. New monomer Qa bearing an acridyl‐ethynylene side chain in position 4 (Figure 1a) was thus designed and synthesized. For this, a 4‐bromoquinoline derivative was functionalized with an ethynylene linker and the acridine unit using two consecutive Sonogashira couplings (see Supporting Information for synthetic schemes and detailed experimental procedures).
Figure 1.

Foldamer sequences with acridine appendages and expected interactions. a) Chemical structures of units Qa and Q. Interactions enabled by the acridine group: π‐π and dipole‐dipole interactions (b), and metal coordination with Pd(II) (c). Sequences of foldamers with acridine groups on one side of the helix (d) and two neighboring sides of the helix (e). Schematic top and side views indicate the positions of acridine units on helices.
Sequences 1 a–1 d and 2 a–2 c were designed to display linear arrays of acridine units at their surface (Figure 1d, e) so that multiple units may simultaneously engage in similar interactions. Helix curvature is such that placing two Qa monomers at positions i and i+5 of a sequence amounts to displaying two acridine units on the same face of the helix, that is, separated by exactly two helix turns and a vertical rise of 7 Å. This orientation and spacing might allow for the interdigitation – i. e., the reciprocal intercalation – of acridine appendages belonging to different helices in a tweezer fashion. [24] However, we eventually found that interactions take place via other modes of assembly. Oligomers 1 a, 1 b and 1 c composed of 5, 10 or 15 units, respectively, possess 1, 2 or 3 acridine appendages along one line, parallel to the helix axis, on one face of the helix. These sequences bear no stereogenic centers and are expected to fold as a racemate of right‐handed (P) and left‐handed (M) helical conformers. In contrast, the N‐terminal camphanyl group of 1 d, an analogue of 1 b, completely biases helicity in favor of the M‐handedness. [25] Similarly, sequences 2 a, 2 b, and 2 c display acridine appendages along two parallel lines at the helix surface. All sequences were prepared using convergent solution phase synthesis as described for Q n oligomers. [26]
Folding and assembly of helices displaying all acridine units on the same side
The 1H NMR spectra of the (Q2QaQ2)n sequences in CDCl3 show typical features of helically folded conformations (Figures 2a–c, S1):[ 22b , 22c ] the peaks are sharp and spread over a wide range of chemical shift values despite the repetitive nature of the main chain; the CH2 protons of the isobutyl side chains are anisochronous due to their diastereotopic nature resulting from slow helix handedness inversion on the NMR time scale; and the signals of amide, aromatic and methyl ester protons shift upfield upon increasing oligomer length as a result of ring current effects associated with intramolecular aromatic stacking. [27] Some signals assigned to acridine protons based on their integration and their chemical shift value also shift upfield up to 0.3 ppm upon increasing oligomer length. These protons are not part of the stacks of the main chain aromatic helix. Upfield shifts nevertheless suggest that they may be involved in some sort of aromatic stacking.
Figure 2.
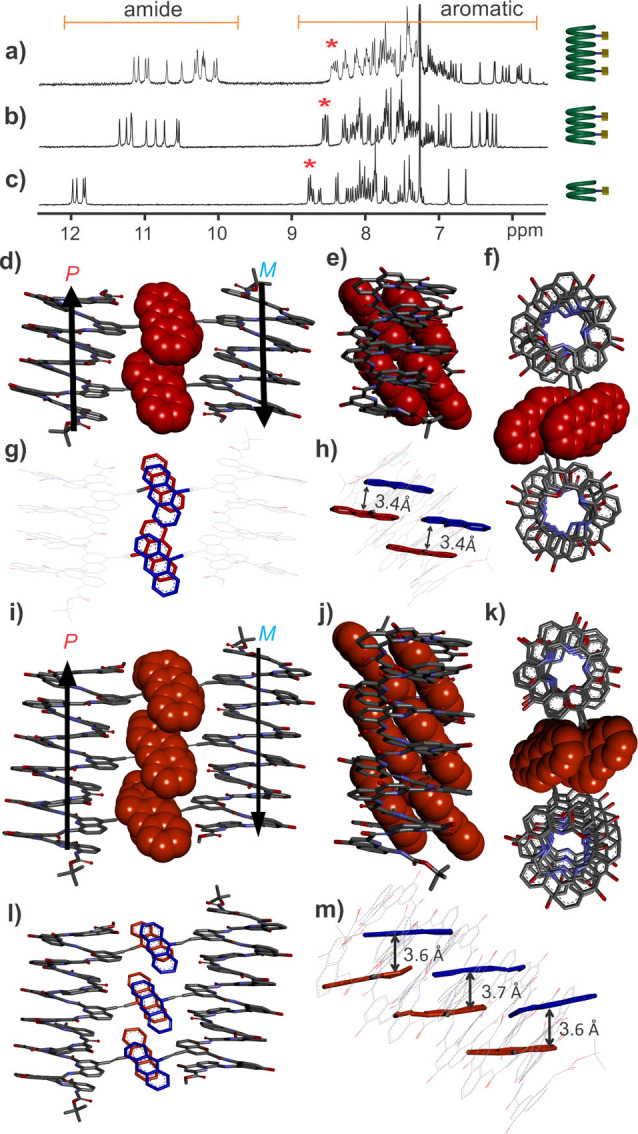
300 MHz 1H NMR spectra of 1 c (a), 1 b (b) and 1 a (c) in CDCl3. Stars indicate signals belonging to acridine moieties. Different views of the crystal structures of 1 b (d–h) and 1 c (i–m). Hydrogen atoms, isobutyl groups, and solvent molecules are omitted for clarity. Arrows indicate the N‐to‐C helix orientation. Acridine groups are shown in space filling representation in (d–f) and (i–k). Intramolecular and intermolecular average distances from the atoms of acridine groups to the aromatic planes are indicated in h) and m), respectively.
The 1H NMR spectra of 1 b and 1 c in CDCl3 show only minor changes upon increasing concentration from 0.1 mM to 10 mM (Figures S2, S3). The signals remain sharp and do not shift significantly. This trend is in principle compatible with very stable discrete aggregates that would only dissociate at much lower concentrations. However, multiple aggregates would be expected to form depending on the P or M handedness and head‐to‐head or head‐to‐tail helix‐helix orientation of the aggregated helices. Instead, we inferred that aggregation is not substantial in solution. However, the structures of 1 b and 1 c obtained by X‐ray diffraction analysis of single crystals did reveal acridine‐mediated aggregation in the solid state (Figures 2d–m, S4 and S5). Both structures were solved in space group P‐1 with one molecule in the asymmetric unit. The structures confirmed helix folding and the positioning of the isobutoxy and acridine side chains. Particularly significant is the fact that the assembly mode is similar for both compounds. Thus, in both crystal structures, two helices of opposite handedness (i. e., P and M) assemble into centrosymmetric pairs with extensive intermolecular contacts between their acridine units. Within a pair, helices are arranged head‐to‐tail with their axes parallel to each other. Within each helix, the long axis of the acridine moieties is tilted by 40° to 50° with respect to the helix axis. It follows that the benzene rings of acridine units within a helix are in close proximity, forming a sort of offset stack. [28] This arrangement naturally forbids any intermolecular interdigitation. Nevertheless, the surface available for intermolecular stacking of the acridine units remains considerable, eventually giving rise to antiparallel orientation of the dipoles of the acridine units, as generally seen in polar dye assemblies. [29] The surface involved in intermolecular aromatic stacking in 1 b and 1 c was estimated by subtracting the solvent‐accessible surface of a dimer from the solvent‐accessible surface of two monomers, yielding values of 113 and 215 Å2, respectively. The distances between aromatic surfaces range from 3.4 to 3.7 Å. A CH…O hydrogen bond was also found between the H2‐proton of an acridine and the carbonyl oxygen of an amide (d CH‐O=2.53 Å). The triple bonds linkages between acridines and quinolines show deviations from ideal bond angles of 180° (typically around 174° in 1 b, and 161°, 169°, and 166° in 1 c), as has been observed in some macrocycles. [30] Altogether, the structures of 1 b and 1 c show that the acridine appendages create well defined interaction interfaces leading to some ordering of aromatic helices in the solid state. Since these compounds display acridine on one face of the helix only, ordering is limited to the formation of dimers.
We attempted to assess the bulk phase purity by measuring powder diffraction. Unfortunately, ground crystals suspended in cold oil completely lost their diffraction power. The fragility of foldamer crystals has been frequently observed and reflects the absence of sufficiently tight contacts between molecules in the crystal lattice as well as the high solvent content. The crystals all had the same appearance which, in itself does not prove they belong to the same phase. Nevertheless, multiple single crystals were mounted and all showed the same unit cell parameters, suggesting that this may be the case.
Assembly of helices displaying acridine units in two directions
We next examined the behavior of oligomers 2 a–c with acridine units on two faces of the helices, i. e., with potential for acridine‐acridine interactions between more than just pairs of helices. The 1H NMR spectra in CDCl3 of oligomers 2 a–c display sharp signals (Figures 3a, c, S6), like those of 1 a–c, and chemical shift values typical of helically folded aromatic amide foldamers. NMR signals shift slightly upfield upon increasing concentration from 0.1 to 10 mM (ΔΔ up to 0.17 ppm for 2 c, Figures S7–S9). These variations are larger than for 1 a–c, even when comparing compounds with the same overall number of acridine groups (2 a vs. 1 b, and 2 b vs. 1 c), suggesting some aggregation in solution. Yet no indication of long‐lived (e. g., slow exchange on the NMR time scale) discrete aggregate was found.
Figure 3.

300 MHz 1H NMR spectra of oligomers of 2 c (a), 2 b (b) and 2 a (c) in CDCl3. Stars indicate signals belonging to acridine moieties. (d–i) Different views of the crystal structure of 2 b. Hydrogen atoms, isobutyl groups and solvent molecules, are omitted for clarity. Angles between acridine moieties and the helix are indicated in d) and e). Acridine moieties are shown as space‐filling models in g–i).
Oligomer 2 b containing three acridine units crystallized by slow diffusion of n‐hexane into a chloroform solution. The crystal structure of 2 b could be solved in body‐centered monoclinic space group I2/a (Z=8) with one molecule of 2 b in the asymmetric unit. The positioning and orientation of acridine units at the surface of the helix (acr1 to acr3 numbered from the N‐ to the C‐terminus of the sequence) are highlighted in Figure 3(d–i). Differences from the arrangement of acridines found in 1 b and 1 c can be observed. The top view shows an angle of ca. 60° between the two pointing directions of the acridine units (Figure 3d, g). Acr1 and acr3, which protrude on the same side of the helix, are not stacked and parallel in this case (Figure 3 h). Unlike other acridine units, acr3 has its plane almost perpendicular to the helix axis. In contrast, acr2 and acr1 are almost parallel to each other and tilted by 55° with respect to the helix axis (Figure 3e, h).
The packing of 2 b in the solid state revealed continuous 1D arrays of helices mediated by intermolecular acridine‐acridine contacts (Figure 4a, b). All helices have their axis parallel but their P or M handedness as well as their N‐terminus to C‐terminus orientation alternate along the 1D array. In this continuous 1D arrangement, each helix is in contact with two other helices through two different interaction interfaces corresponding to the two boxes in Figure 4(a and b). Thus, both interfaces involve helices of opposite handedness and of opposite N‐terminus to C‐terminus orientation. One intermolecular interaction interface consists of the tight face‐to‐face stacking (aryl‐aryl distance of 3.4 Å) of the acr2 units of two different helices (Figure 4d, f). The two acr2 units are slightly offset and have their dipole moments antiparallel. Six acridine units are involved at the second intermolecular interaction interface. Four of them form a continuous acr2‐acr1’‐acr1‐acr2’ face‐to‐face stack (aryl‐aryl distance of 3.3 to 3.4 Å) where two pairs of acridine units from two different helices interdigitate (Figure 4c, e), leading to a stack of four acridine units that belong alternatively to one helix or the other. Offsets between stacked rings are more significant in this case. Together with the acr2‐acr2’ contacts of the first interface, a continuous face‐to‐face stack of acridine rings results. Acr3 and acr3’ come above and below the face‐to‐face stack and engage in edge‐to‐face CH‐π interactions.
Figure 4.

The assembly of 2 b in the solid state. Top view (a) and side view (b) of one dimensional continuous ladder organization of P and M helices mediated by stacking between acridine units. Acr1 and acr3 are shown in red and acr2 is shown in blue, all in space filling representation. In a), arrows indicate N‐to‐C helix orientation. In b), black circles and white circles indicate that the C‐terminus or the N‐terminus of the helix is visible, respectively. (c, e and g) show different views of helix‐helix interface B and (d, f, and g) show the same views of helix‐helix interface B. In (c–h), acr units borne by the M helix are shown in green, and acr units borne by the P helix are shown in gold. Acr units are shown either in tube representation (g, h) or in space‐filling representation (c–f). Average distances from atoms of acridine rings to the close aromatic planes are indicated in (g and h). Hydrogen atoms, isobutyl groups and solvent molecules, are omitted for clarity.
The structures of 1 b, 1 c and 2 b demonstrate that acridine‐acridine interactions determine the solid state arrangements of the foldamer helices. When all acridine units protrude on one face of the helix, discrete dimers form. When they protrude in two directions, 1D arrays of helices form. One could anticipate that 2D ordered arrays would be produced with acridine units pointing in three different directions, perhaps enabling the organization of self‐assembled monolayers on solid support. This prospect has not yet been explored. It is reasonable to expect that 2D or 3D arrays would endow the crystals with sufficient integrity to make packing better resist the loss or replacement of solvent molecules. This would allow to obtain powder diffraction and verify phase homogeneity on large samples.
Pd‐mediated assembly of helices with acridine appendages
In a second approach, we used metal ions to guide the assembly of helices through metal complexation (Figure 1c). While metal coordination is widely used to stabilize foldamer structures, [31] its use to form self‐assemblies of foldamers is less common[ 8 , 9 , 21 ] perhaps precisely because metals may interfere with folding. The endocyclic nitrogen of acridine can be used to coordinate a metal ion with good directionality, [32] and this ligand‐metal coordination was expected to prevail over aromatic stacking. Pd(II) was chosen for its well‐known ability to form stable complexes with pyridine rings, as was extensively illustrated in the field of self‐assembled metal–organic coordination cages and macrocycles.[ 10a , 10b , 33 ] Unlike Cu(II), which forms stable helicates within Qn helices, Pd(II) does not coordinate to Q n oligomers and does not interfere with helical folding. [34] Complex formation was first validated upon mixing PdCl2(CH3CN)2 with a Qa derivative in CDCl3 (Figure S10). A distinctive downfield shift of the acridine H4/H5 NMR signals above 11 ppm was observed. Upon mixing PdCl2(CH3CN)2 with two equivalents of sequence 1 a, complexation was found to be slow and could be monitored by 1H NMR spectroscopy (Figure 5b‐d). An intermediate species was observed that was assigned to a 1 : 1 complex. The final product was unambiguously assigned to a 2 : 1 Pd(1 a)2Cl2 complex based on mass spectrometric measurements (peak at m/z 3123.063 with the correct isotopic distribution, see Figure S19), DOSY (Figure S11) which clearly showed that the complex has a larger hydrodynamic radius than 1 a alone, and X‐ray crystallography (Figure 5f–h). Quite remarkably, differences in the 1H NMR spectra of the 1 : 1 intermediate and 2 : 1 final complex did not only concern protons belonging to acridine moieties. A quinoline H3 proton near 6.6 ppm presumably belonging to Qa, [35] some amide NH, and even the extremely remote methyl ester protons near 3.3 ppm had different chemical shift values depending on acridine coordination to Pd(II), suggesting long range transmission of electronic effects (Figure 5b–e).
Figure 5.

A model to show the interconversion between PP/PM/MM helices connected with a single metal coordination (a). Formation of complex Pd(1 a)2Cl2 followed by NMR spectroscopy (300 MHz, CDCl3) at different time intervals after mixing PdCl2(CH3CN)2 with two equiv. of 1 a (b–e). Different views of the crystal structure of Pd(1 a)2Cl2 (f–h). In (f), arrows indicate N‐to‐C helix orientation. In (g and h), the side views of two inequivalent complexes found in the asymmetric unit are shown and the distinct helix‐helix orientation indicated. Hydrogen atoms, isobutyl groups and solvent molecules, are omitted for clarity.
The solid state structure of Pd(1 a)2Cl2 was solved in space group C2/c with two independent complexes in the asymmetric unit. Both complexes were meso conformers with one P helix and one M helix (Figure 5f). The two helices are kept 1.7 nm apart by the acridine‐Pd complex. It is thus unlikely that any contact between them could mediate helix‐helix handedness communication. [36] One may thus infer that the chiral PP/MM conformer also exist in solution (Figure 5a). The fact that the 1H NMR spectrum shows only one set of signals (Figure 5b) indicates that the PM and PP/MM conformations have perfectly overlapping signals. Indeed, fast exchange on the NMR time scale that would average the signals of the diastereomers is excluded for both metal‐ligand exchange and helix handedness inversion. The presence of meso conformers in the solid state thus reflects preferential crystallization of this compound. The two complexes of the asymmetric unit differ in that they have different helix‐helix relative orientations. One complex shows almost parallel helix axes with opposite orientation of the N‐ and C‐termini (Figure 5f, g) whereas in the other complex, the helix axes are tilted by an angle of 40° (Figure 5 h). These different orientations just represent two out of numerous possible helix‐helix orientations and reflect the free rotation about aryl‐alkyne bonds. Pd ions are in the expected square planar coordination geometry. The two acridine rings, in trans, are parallel in one complex of the asymmetric unit and slightly tilted in the other (tilt angle of 16°). The alkyne spacers substantially deviate from linearity which gives a wavy shape to the linker between the helices. This distortion may come from crystal packing and illustrates the flexibility of the linkages, as seen above in the structure of 1 c. Molecular packing in the crystal lattice revealed very reduced acridine‐acridine contacts (limited to one contact between the two complexes of the asymmetric unit), confirming that metal coordination overwhelms the attractive interactions that prevail in the structures of 1 b, 1 c and 2 b.
We next investigated Pd coordination to longer oligomers containing more than one acridine unit. With 1 b, which displays two acridine units on the same face of the helix, a 2 : 2 helix/metal complex may be expected that would include a 96 atom macrocycle. Thus, 1 b was mixed with 1 equiv. of PdCl2(CH3CN)2 in CDCl3 and heated to 60 °C. After one night, the 1H NMR spectrum of the reaction showed multiple peaks indicating a complex mixture (Figure S12). This complicated NMR spectrum was assigned to the numerous possible 1 : 1, 2 : 1, and 1 : 2 intermediates towards the final expected macrocycle (Figure S13). Upon prolonged heating (seven days) the spectrum simplified and four methyl ester peaks of similar intensity could be distinguished near 3.1 ppm (Figure 6b). A high resolution mass spectrum confirmed the formation of a 2 : 2 complex Pd2(1 b)2Cl4 with the correct isotopic distribution (m/z 2990.987, z=2, Figure S20) and DOSY NMR further validated that the complex has a larger hydrodynamic radius than 1 b alone (Figure S14). The presence of four species in the 1H NMR spectrum is consistent with the fact that acridines are presented on the same face of a helix regardless of helix handedness and helix N‐to‐C orientation. Thus, all parallel‐PP/MM, parallel‐PM, antiparallel‐PP/MM, and antiparallel‐PM 2 : 2 Pd2(1 b)2Cl4 complexes may form with equal probability. In order to simplify NMR spectra and validate our analysis, enantiopure helical oligomer 1 d was produced. Its N‐terminal chiral camphanyl group is known to completely bias helix handedness. [25] Thus, 1 d only exists as an M‐helix due to the S configuration of its camphanyl group. Upon mixing 1 d and Pd(II) (1 equiv.), Pd2(1 d)2Cl4 was produced and its NMR spectrum showed only two methyl ester peaks (Figure S15) which were assigned to parallel‐MM and antiparallel‐MM complexes.
Figure 6.

The 1H NMR (300 MHz) spectra of 1 b (c) and its complex with palladium Pd2(1 b)2Cl4 containing four isomers (b) as well as one of the isomers purified (a) in CDCl3, (d–h) the crystal structure of the complex Pd2(1 b)2Cl4 showing different views, d) front view, e) top view through the helices; f) side view showing the angle of acridine to the axis of helix; g) side view with the angle of palladium atoms to the axis of helices; h) views of the palladium coordination. One dimensional solvent channel formed by the stacking of the complex Pd2(1 b)2Cl4, on the view from (i) b‐axis and (j) c‐axis. All the hydrogen atoms (d–j) and side chains (d–h) in the crystal structures are removed for clarity.
Single crystals of Pd2(1 b)2Cl4 were obtained by slow diffusion of acetonitrile into a chloroform solution of Pd2(1 b)2Cl4. The structure was solved in the P‐1 space group with one complex in the asymmetric unit and proved to be the antiparallel‐PM conformer (Figure 6d). However, upon redissolving the crystals and immediately measuring an NMR spectrum, the four species were still observed. This suggested that all four species may have been present in the initially isolated crystals, that is, in separate single crystals. [37] Indeed, the interconversion between the different species require complex dissociation and helix handedness inversion and is therefore not expected to be a fast process. Upon recrystallizing the mixture several times, it was eventually enriched into one of the four species only which confirmed its kinetic stability at room temperature in CDCl3 (Figures 6a, S16).
In the solid state structure, the two helices of the complex are separated by ∼1.8 nm. The helix axes are perfectly parallel. The conformation of each helix is similar to its conformation in absence of Pd(II) (Figure 2d) with the acridine planes almost parallel to each other and tilted with respect to the helix axis. The metal complexes show similar features as those mentioned for Pd(1 a)2Cl2, including some distortions. The Pd−Pd distance is only 6.7 Å. This proximity results in CH…Cl contacts between a chloride of each complex and the acridine units of the other complex. The four acridines are thus sterically intricated in a way reminiscent of some molecular gears used in molecular machinery. [38] If the acridine units are allowed some motions, for example change in the tilt angles, these motions must be coupled and synchronous for the four acridines. The crystal packing of Pd2(1 b)2Cl4 revealed large rectangular pores, with dimensions around 30×5 Å (Figure 6i, j). This possibly reflects the rigid dumbbell shape of the complex which may make it difficult to achieve tight packing.
Complexation of Pd(II) to longer oligomer 1 c containing three acridine units on the same face of the helix showed a behavior similar to that of 1 b (Figure S17). The 1H NMR spectrum showed four methyl ester signals, which may be assigned, as before, to four isomeric macrocycles comprised of two helices. The mass expected for a Pd3(1 c)2Cl6 complex with three Pd(II) ions bridging two helices was detected (Figure S21). However, in the absence of a crystal structure, the analysis was not pushed further. Complexation of Pd(II) to oligomer 2 a containing two acridine units pointing in different directions was also investigated. The orientation of the acridine units may not allow for the formation of a 2+2 macrocycle, as with 1 b. After more than five days at 60 °C, the NMR spectrum showed multiple ester peaks (Figure S18). HRMS analysis indicated the presence of a 3 : 3 (Pd3(2 a)3Cl6+3H)3+ complex (Figure S22), overlapping with (Pd(2 a)Cl2+H)+, perhaps a fragment of the latter. A triangular shaped macrocycle with three helices at each summit is a plausible object, but again, investigations were not pushed further due to the number of possible isomers with various combinations of helix handedness and N‐to‐C orientation.
Conclusion
In conclusion, we have demonstrated that acridine units appended at precise positions of an aromatic foldamer helix efficiently guide helix‐helix interactions in the solid state, giving rise to the formation of discrete dimers and 1D ordered arrays. Such control of molecular packing where one structural feature prevails allows to establish a hierarchy between interactions that may be useful in crystal engineering and to direct the organization of self‐assembled monolayers on surfaces. Ultimately, the goal is to convert structural control in specific materials properties. For instance, ferromagnetic exchange between paramagnetic Cu(II) centers has been measured on crystals of Cu‐containing aromatic helical foldamers, and electron transport properties have been evidenced in monolayers of the same compounds. [34]
Furthermore, the ability of acridine units to form complexes with Pd(II) was efficiently used to form stable cyclic supramolecular assemblies of helices in solution. Self‐assembly was somewhat complicated by the presence of isomers associated with helix handedness and helix C‐to‐N orientation. The system may be simplified by controlling helix handedness independently, using chiral monomers or end‐groups. Also, helices could in principle lack C‐to‐N polarity and be symmetrical upon using a central diamine or diacid unit. The inter‐helix associations we have evidenced in the solid state and in solution operate orthogonally to the interactions responsible for folding and do not alter helix shape. Thus, these results further establish aromatic foldamer helices as versatile programmable scaffolds for exomolecular recognition and molecular assembly.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The China Scholarship Council is gratefully acknowledged for a predoctoral fellowship to J. Wang. The work benefited from the facilities and expertise of the Biophysical and Structural Chemistry platform at IECB, CNRS UAR3033, INSERM US001, Université de Bordeaux. We thank B. Kauffmann for assistance with crystallographic measurements and F. Rosu for assistance with MS measurements. Open Access funding enabled and organized by Projekt DEAL.
J. Wang, B. Wicher, V. Maurizot, I. Huc, Chem. Eur. J. 2022, 28, e202201345.
Contributor Information
Dr. Victor Maurizot, Email: victor.maurizot@u-bordeaux.fr
Prof. Ivan Huc, Email: ivan.huc@cup.lmu.de.
Data Availability Statement
The data that support the findings of this study are openly available free of charge from the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe at https://ccdc.cam.ac.uk, reference numbers 2166701, 2166702, 2166703, 2166704, 2166705.
References
- 1.
- 1a. Zhu J., Avakyan N., Kakkis A., Hoffnagle A. M., Han K., Li Y., Zhang Z., Choi T. S., Na Y., Yu C.-J., Tezcan F. A., Chem. Rev. 2021, 121, 13701–13796; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Kakkis A., Gagnon D., Esselborn J., Britt R. D., Tezcan F. A., Angew. Chem. Int. Ed. 2020, 59, 21940–21944, [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22124–22128; [Google Scholar]
- 1c. Sontz P. A., Bailey J. B., Ahn S., Tezcan F. A., J. Am. Chem. Soc. 2015, 137, 11598–11601; [DOI] [PubMed] [Google Scholar]
- 1d. Bailey J. B., Tezcan F. A., J. Am. Chem. Soc. 2020, 142, 17265–17270; [DOI] [PubMed] [Google Scholar]
- 1e. Churchfield L. A., Tezcan F. A., Acc. Chem. Res. 2019, 52, 345–355; [DOI] [PubMed] [Google Scholar]
- 1f. Rittle J., Field M. J., Green M. T., Tezcan F. A., Nat. Chem. 2019, 11, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Li Q., Zhao J., Liu L., Jonchhe S., Rizzuto F. J., Mandal S., He H., Wei S., Sleiman H. F., Mao H., Mao C., Nat. Mater. 2020, 19, 1012–1018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Platnich C. M., Rizzuto F. J., Cosa G., Sleiman H. F., Chem. Soc. Rev. 2020, 49, 4220–4233; [DOI] [PubMed] [Google Scholar]
- 2c. Trinh T., Liao C., Toader V., Barłóg M., Bazzi H. S., Li J., Sleiman H. F., Nat. Chem. 2018, 10, 184–192; [DOI] [PubMed] [Google Scholar]
- 2d. Li J., Pei H., Zhu B., Liang L., Wei M., He Y., Chen N., Li D., Huang Q., Fan C., ACS Nano 2011, 5, 8783–8789; [DOI] [PubMed] [Google Scholar]
- 2e. Hu Q., Li H., Wang L., Gu H., Fan C., Chem. Rev. 2019, 119, 6459–6506; [DOI] [PubMed] [Google Scholar]
- 2f. Rothemund P. W. K., Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- 3. Rinaldi S., Molecules 2020, 25, 3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Kwon S., Kim B. J., Lim H.-K., Kang K., Yoo S. H., Gong J., Yoon E., Lee J., Choi I. S., Kim H., Lee H.-S., Nat. Commun. 2015, 6, 8747; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Yoo S. H., Eom T., Kwon S., Gong J., Kim J., Cho S. J., Driver R. W., Lee Y., Kim H., Lee H.-S., J. Am. Chem. Soc. 2015, 137, 2159–2162; [DOI] [PubMed] [Google Scholar]
- 4c. Kwon S., Shin H. S., Gong J., Eom J.-H., Jeon A., Yoo S. H., Chung I. S., Cho S. J., Lee H.-S., J. Am. Chem. Soc. 2011, 133, 17618–17621; [DOI] [PubMed] [Google Scholar]
- 4d. Kwon S., Jeon A., Yoo S. H., Chung I. S., Lee H.-S., Angew. Chem. Int. Ed. 2010, 49, 8232–8236, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8408–8412; [Google Scholar]
- 4e. Eom J.-H., Gong J., Kwon S., Jeon A., Jeong R., Driver R. W., Lee H.-S., Angew. Chem. Int. Ed. 2015, 54, 13204–13207, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13402–13405; [Google Scholar]
- 4f. Yoon E., Gong J., Jung Y., Lee W., Driver R. W., Lee H.-S., Chem. Commun. 2016, 52, 5250–5253. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Daniels D. S., Petersson E. J., Qiu J. X., Schepartz A., J. Am. Chem. Soc. 2007, 129, 1532–1533; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Pomerantz W. C., Yuwono V. M., Pizzey C. L., Hartgerink J. D., Abbott N. L., Gellman S. H., Angew. Chem. Int. Ed. 2008, 47, 1241–1244, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1261–1264; [Google Scholar]
- 5c. Collie G. W., Pulka-Ziach K., Lombardo C. M., Fremaux J., Rosu F., Decossas M., Mauran L., Lambert O., Gabelica V., Mackereth C. D., Guichard G., Nat. Chem. 2015, 7, 871–878; [DOI] [PubMed] [Google Scholar]
- 5d. Lombardo C. M., Collie G. W., Pulka-Ziach K., Rosu F., Gabelica V., Mackereth C. D., Guichard G., J. Am. Chem. Soc. 2016, 138, 10522–10530; [DOI] [PubMed] [Google Scholar]
- 5e. Collie G. W., Bailly R., Pulka-Ziach K., Lombardo C. M., Mauran L., Taib-Maamar N., Dessolin J., Mackereth C. D., Guichard G., J. Am. Chem. Soc. 2017, 139, 6128–6137. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Lewandowska U., Zajaczkowski W., Corra S., Tanabe J., Borrmann R., Benetti E. M., Stappert S., Watanabe K., Ochs N. A. K., Schaeublin R., Li C., Yashima E., Pisula W., Müllen K., Wennemers H., Nat. Chem. 2017, 9, 1068–1072; [DOI] [PubMed] [Google Scholar]
- 6b. Foletti C., Trapp N., Loosli S., Lewandowski B., Wennemers H., Helv. Chim. Acta 2019, 102, e1900052; [Google Scholar]
- 6c. Ochs N. A. K., Lewandowska U., Zajaczkowski W., Corra S., Reger S., Herdlitschka A., Schmid S., Pisula W., Müllen K., Bäuerle P., Wennemers H., Chem. Sci. 2019, 10, 5391–5396; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Heinz-Kunert S. L., Pandya A., Dang V. T., Tran P. N., Ghosh S., McElheny D., Santarsiero B. D., Ren Z., Nguyen A. I., J. Am. Chem. Soc. 2022, 144, 7001–7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maity D., Hamilton A. D., Chem. Commun. 2021, 57, 9192–9195. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Sawada T., Fujita M., Chem 2020, 6, 1861–1876; [Google Scholar]
- 8b. Sawada T., Inomata Y., Shimokawa K., Fujita M., Nat. Commun. 2019, 10, 5687; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Sawada T., Saito A., Tamiya K., Shimokawa K., Hisada Y., Fujita M., Nat. Commun. 2019, 10, 921; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Inomata Y., Sawada T., Fujita M., Chem 2020, 6, 294–303; [Google Scholar]
- 8e. Sawada T., Yamagami M., Akinaga S., Miyaji T., Fujita M., Chem. Asian J. 2017, 12, 1715–1718; [DOI] [PubMed] [Google Scholar]
- 8f. Yamagami M., Sawada T., Fujita M., J. Am. Chem. Soc. 2018, 140, 8644–8647. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Jeong S., Zhang L., Kim J., Gong J., Choi J., Ok K. M., Lee Y., Kwon S., Lee H.-S., Angew. Chem. Int. Ed. 2022, 61, e202108364, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202108364; [DOI] [PubMed] [Google Scholar]
- 9b. Misra R., Saseendran A., Dey S., Gopi H. N., Angew. Chem. Int. Ed. 2019, 58, 2251–2255, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 2273–2277; [Google Scholar]
- 9c. Schnitzer T., Paenurk E., Trapp N., Gershoni-Poranne R., Wennemers H., J. Am. Chem. Soc. 2021, 143, 644–648; [DOI] [PubMed] [Google Scholar]
- 9d. Sawada T., Matsumoto A., Fujita M., Angew. Chem. Int. Ed. 2014, 53, 7228–7232, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7356–7360; [Google Scholar]
- 9e. Dey S., Misra R., Saseendran A., Pahan S., Gopi H. N., Angew. Chem. Int. Ed. 2021, 60, 9863–9868; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 9951–9956. [Google Scholar]
- 10.
- 10a. Fujita M., Oguro D., Miyazawa M., Oka H., Yamaguchi K., Ogura K., Nature 1995, 378, 469–471; [Google Scholar]
- 10b. Takeda N., Umemoto K., Yamaguchi K., Fujita M., Nature 1999, 398, 794–796; [Google Scholar]
- 10c. Percástegui E. G., Ronson T. K., Nitschke J. R., Chem. Rev. 2020, 120, 13480–13544; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10d. Leininger S., Fan J., Schmitz M., Stang P. J., Proc. Natl. Acad. Sci. USA 2000, 97, 1380–1384; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e. Mal P., Schultz D., Beyeh K., Rissanen K., Nitschke J. R., Angew. Chem. Int. Ed. 2008, 47, 8297–8301, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8421–8425; [Google Scholar]
- 10f. Zhang D., Ronson T. K., Nitschke J. R., Acc. Chem. Res. 2018, 51, 2423–2436; [DOI] [PubMed] [Google Scholar]
- 10g. Byrne K., Zubair M., Zhu N., Zhou X.-P., Fox D. S., Zhang H., Twamley B., Lennox M. J., Düren T., Schmitt W., Nat. Commun. 2017, 8, 15268; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10h. Fujita D., Ueda Y., Sato S., Mizuno N., Kumasaka T., Fujita M., Nature 2016, 540, 563–566; [DOI] [PubMed] [Google Scholar]
- 10i. Sun Q.-F., Iwasa J., Ogawa D., Ishido Y., Sato S., Ozeki T., Sei Y., Yamaguchi K., Fujita M., Science 2010, 328, 1144–1147; [DOI] [PubMed] [Google Scholar]
- 10j. Mal P., Breiner B., Rissanen K., Nitschke J. R., Science 2009, 324, 1697–1699; [DOI] [PubMed] [Google Scholar]
- 10k. Han M., Engelhard D. M., Clever G. H., Chem. Soc. Rev. 2014, 43, 1848–1860; [DOI] [PubMed] [Google Scholar]
- 10l. Freye S., Hey J., Torras-Galán A., Stalke D., Herbst-Irmer R., John M., Clever G. H., Angew. Chem. Int. Ed. 2012, 51, 2191–2194, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2233–2237; [Google Scholar]
- 10m. Tessarolo J., Lee H., Sakuda E., Umakoshi K., Clever G. H., J. Am. Chem. Soc. 2021, 143, 6339–6344; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10n. Yamashina M., Tanaka Y., Lavendomme R., Ronson T. K., Pittelkow M., Nitschke J. R., Nature 2019, 574, 511–515; [DOI] [PubMed] [Google Scholar]
- 10o. Chen B., Horiuchi S., Holstein J. J., Tessarolo J., Clever G. H., Chem. Eur. J. 2019, 25, 14921–14927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Stock N., Biswas S., Chem. Rev. 2012, 112, 933–969; [DOI] [PubMed] [Google Scholar]
- 11b. Zhou H.-C., Long J. R., Yaghi O. M., Chem. Rev. 2012, 112, 673–674; [DOI] [PubMed] [Google Scholar]
- 11c. Wang Q., Astruc D., Chem. Rev. 2020, 120, 1438–1511. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Lehn J. M., Rigault A., Siegel J., Harrowfield J., Chevrier B., Moras D., Proc. Natl. Acad. Sci. USA 1987, 84, 2565–2569; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Kramer R., Lehn J. M., Marquis-Rigault A., Proc. Natl. Acad. Sci. USA 1993, 90, 5394–5398; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Wood C. S., Ronson T. K., McConnell A. J., Roberts D. A., Nitschke J. R., Chem. Sci. 2016, 7, 1702–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Carpenter J. P., McTernan C. T., Greenfield J. L., Lavendomme R., Ronson T. K., Nitschke J. R., Chem 2021, 7, 1534–1543; [Google Scholar]
- 13b. Leigh D. A., Danon J. J., Fielden S. D. P., Lemonnier J.-F., Whitehead G. F. S., Woltering S. L., Nat. Chem. 2021, 13, 117–122; [DOI] [PubMed] [Google Scholar]
- 13c. Leigh D. A., Schaufelberger F., Pirvu L., Stenlid J. H., August D. P., Segard J., Nature 2020, 584, 562–568; [DOI] [PubMed] [Google Scholar]
- 13d. Zhong J., Zhang L., August D. P., Whitehead G. F. S., Leigh D. A., J. Am. Chem. Soc. 2019, 141, 14249–14256; [DOI] [PubMed] [Google Scholar]
- 13e. Katsonis N., Lancia F., Leigh D. A., Pirvu L., Ryabchun A., Schaufelberger F., Nat. Chem. 2020, 12, 939–944. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Huc I., Eur. J. Org. Chem. 2004, 2004, 17–29; [Google Scholar]
- 14b. Zhang D.-W., Zhao X., Hou J.-L., Li Z.-T., Chem. Rev. 2012, 112, 5271–5316. [DOI] [PubMed] [Google Scholar]
- 15. Haldar D., Jiang H., Léger J.-M., Huc I., Tetrahedron 2007, 63, 6322–6330. [Google Scholar]
- 16.
- 16a. Cai W., Wang G.-T., Xu Y.-X., Jiang X.-K., Li Z.-T., J. Am. Chem. Soc. 2008, 130, 6936–6937; [DOI] [PubMed] [Google Scholar]
- 16b. You L.-Y., Wang G.-T., Jiang X.-K., Li Z.-T., Tetrahedron 2009, 65, 9494–9504; [Google Scholar]
- 16c. Cuccia L. A., Lehn J.-M., Homo J.-C., Schmutz M., Angew. Chem. Int. Ed. 2000, 39, 233–237, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 239–243. [Google Scholar]
- 17.
- 17a. Petitjean A., Cuccia L. A., Schmutz M., Lehn J.-M., J. Org. Chem. 2008, 73, 2481–2495; [DOI] [PubMed] [Google Scholar]
- 17b. Gan Q., Wang X., Kauffmann B., Rosu F., Ferrand Y., Huc I., Nat. Nanotechnol. 2017, 12, 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Huo Y., Zeng H., Acc. Chem. Res. 2016, 49, 922–930; [DOI] [PubMed] [Google Scholar]
- 18b. Zhao H., Sheng S., Hong Y., Zeng H., J. Am. Chem. Soc. 2014, 136, 14270–14276; [DOI] [PubMed] [Google Scholar]
- 18c. Zhao H., Ong W. Q., Zhou F., Fang X., Chen X., Li S. F. Y., Su H., Cho N.-J., Zeng H., Chem. Sci. 2012, 3, 2042–2046; [Google Scholar]
- 18d. Bindl D., Mandal P. K., Allmendinger L., Huc I., Angew. Chem. Int. Ed. 2022, 61, e202116509, [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202116509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu C.-Z., Koppireddi S., Wang H., Zhang D.-W., Li Z.-T., Angew. Chem. Int. Ed. 2019, 58, 226–230, [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 232–236. [Google Scholar]
- 20.
- 20a. De S., Chi B., Granier T., Qi T., Maurizot V., Huc I., Nat. Chem. 2018, 10, 51–57; [DOI] [PubMed] [Google Scholar]
- 20b. Mazzier D., De S., Wicher B., Maurizot V., Huc I., Chem. Sci. 2019, 10, 6984–6991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20c. Mazzier D., De S., Wicher B., Maurizot V., Huc I., Angew. Chem. Int. Ed. 2020, 59, 1606–1610, [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1623–1627. [Google Scholar]
- 21. Delsuc N., Hutin M., Campbell V. E., Kauffmann B., Nitschke J. R., Huc I., Chem. Eur. J. 2008, 14, 7140–7143. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Qi T., Maurizot V., Noguchi H., Charoenraks T., Kauffmann B., Takafuji M., Ihara H., Huc I., Chem. Commun. 2012, 48, 6337–6339; [DOI] [PubMed] [Google Scholar]
- 22b. Jiang H., Léger J.-M., Huc I., J. Am. Chem. Soc. 2003, 125, 3448–3449; [DOI] [PubMed] [Google Scholar]
- 22c. Jiang H., Léger J.-M., Dolain C., Guionneau P., Huc I., Tetrahedron 2003, 59, 8365–8374. [Google Scholar]
- 23.
- 23a. Hu X., Dawson S. J., Mandal P. K., de Hatten X., Baptiste B., Huc I., Chem. Sci. 2017, 8, 3741–3749; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Zwillinger M., Reddy P. S., Wicher B., Mandal P. K., Csékei M., Fischer L., Kotschy A., Huc I., Chem. Eur. J. 2020, 26, 17366–17370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jang H. J., Lee S., An B. J., Song G., Jeon H.-G., Jeong K.-S., Chem. Commun. 2022, 58, 1410–1413. [DOI] [PubMed] [Google Scholar]
- 25. Kendhale A. M., Poniman L., Dong Z., Laxmi-Reddy K., Kauffmann B., Ferrand Y., Huc I., J. Org. Chem. 2011, 76, 195–200. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Li X., Qi T., Srinivas K., Massip S., Maurizot V., Huc I., Org. Lett. 2016, 18, 1044–1047; [DOI] [PubMed] [Google Scholar]
- 26b. Qi T., Deschrijver T., Huc I., Nat. Protoc. 2013, 8, 693–708. [DOI] [PubMed] [Google Scholar]
- 27. Delsuc N., Kawanami T., Lefeuvre J., Shundo A., Ihara H., Takafuji M., Huc I., ChemPhysChem 2008, 9, 1882–1890. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Desiraju G. R., Gavezzotti A., J. Chem. Soc. Chem. Commun. 1989, 10, 621–623; [Google Scholar]
- 28b. Martinez C. R., Iverson B. L., Chem. Sci. 2012, 3, 2191–2201; [Google Scholar]
- 28c. Hunter C. A., Lawson K. R., Perkins J., Urch C. J., J. Chem. Soc. Perkin Trans. 2 2001, 5, 651–669. [Google Scholar]
- 29.
- 29a. Würthner F., Acc. Chem. Res. 2016, 49, 868–876; [DOI] [PubMed] [Google Scholar]
- 29b. Würthner F., Yao S., Debaerdemaeker T., Wortmann R., J. Am. Chem. Soc. 2002, 124, 9431–9447; [DOI] [PubMed] [Google Scholar]
- 29c. Kirchner E., Bialas D., Fennel F., Grüne M., Würthner F., J. Am. Chem. Soc. 2019, 141, 7428–7438; [DOI] [PubMed] [Google Scholar]
- 29d. Zitzler-Kunkel A., Kirchner E., Bialas D., Simon C., Würthner F., Chem. Eur. J. 2015, 21, 14851–14861. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Kaiser K., Scriven L. M., Schulz F., Gawel P., Gross L., Anderson H. L., Science 2019, 365, 1299–1301; [DOI] [PubMed] [Google Scholar]
- 30b. O'Sullivan M. C., Sprafke J. K., Kondratuk D. V., Rinfray C., Claridge T. D. W., Saywell A., Blunt M. O., O'Shea J. N., Beton P. H., Malfois M., Anderson H. L., Nature 2011, 469, 72–75; [DOI] [PubMed] [Google Scholar]
- 30c. Rickhaus M., Vargas Jentzsch A., Tejerina L., Grübner I., Jirasek M., Claridge T. D. W., Anderson H. L., J. Am. Chem. Soc. 2017, 139, 16502–16505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Metallofoldamers, Wiley-VCH, Weinheim, 2013.
- 32. Ha K., Acta Crystallogr. 2012, E68, m54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.
- 33a. Fujita M., Fujita N., Ogura K., Yamaguchi K., Nature 1999, 400, 52–55; [Google Scholar]
- 33b. Leininger S., Olenyuk B., Stang P. J., Chem. Rev. 2000, 100, 853–908; [DOI] [PubMed] [Google Scholar]
- 33c. Chakrabarty R., Mukherjee P. S., Stang P. J., Chem. Rev. 2011, 111, 6810–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang J., Wicher B., Méndez-Ardoy A., Li X., Pecastaings G., Buffeteau T., Bassani D. M., Maurizot V., Huc I., Angew. Chem. Int. Ed. 2021, 60, 18461–18466, [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 18609–18614. [Google Scholar]
- 35.The most upfield shifted aromatic proton likely belongs to the central unit of the helix.
- 36. Dolain C., Léger J.-M., Delsuc N., Gornitzka H., Huc I., Proc. Natl. Acad. Sci. USA 2005, 102, 16146–16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crystals were too small to allow for the NMR analysis of a single freshly disolved crystal.
- 38.
- 38a. Ube H., Yasuda Y., Sato H., Shionoya M., Nat. Commun. 2017, 8, 14296; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38b. Sanada K., Ube H., Shionoya M., J. Am. Chem. Soc. 2016, 138, 2945–2948; [DOI] [PubMed] [Google Scholar]
- 38c. Setaka W., Nirengi T., Kabuto C., Kira M., J. Am. Chem. Soc. 2008, 130, 15762–15763; [DOI] [PubMed] [Google Scholar]
- 38d. Jiang X., Duan H.-B., Khan S. I., Garcia-Garibay M. A., ACS Cent. Sci. 2016, 2, 608–613; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38e. Jellen M. J., Liepuoniute I., Jin M., Jones C. G., Yang S., Jiang X., Nelson H. M., Houk K. N., Garcia-Garibay M. A., J. Am. Chem. Soc. 2021, 143, 7740–7747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are openly available free of charge from the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe at https://ccdc.cam.ac.uk, reference numbers 2166701, 2166702, 2166703, 2166704, 2166705.