

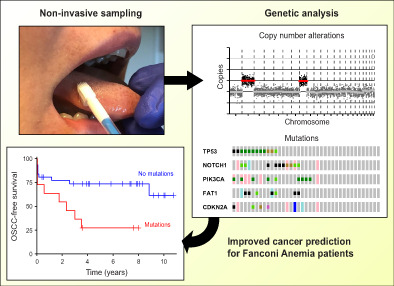
Abstract
Oral squamous cell carcinomas (OSCCs) develop in genetically altered epithelium in the mucosal lining, also coined as fields, which are mostly not visible but occasionally present as white oral leukoplakia (OL) lesions. We developed a noninvasive genetic assay using next‐generation sequencing (NGS) on brushed cells to detect the presence of genetically altered fields, including those that are not macroscopically visible. The assay demonstrated high accuracy in OL patients when brush samples were compared with biopsies as gold standard. In a cohort of Fanconi anemia patients, detection of mutations in prospectively collected oral brushes predicted oral cancer also when visible abnormalities were absent. We further provide insight in the molecular landscape of OL with frequent changes of TP53, FAT1 and NOTCH1. NGS analysis of noninvasively collected samples offers a highly accurate method to detect genetically altered fields in the oral cavity, and predicts development of OSCC in high‐risk individuals. Noninvasive genetic screening can be employed to screen high‐risk populations for cancer and precancer, map the extension of OL lesions beyond what is visible, map the oral cavity for precancerous changes even when visible abnormalities are absent, test accuracy of promising imaging modalities, monitor interventions and determine genetic progression as well as the natural history of the disease in the human patient.
Keywords: Fanconi anemia, next‐generation sequencing, noninvasive cancer screening, oral leukoplakia, oral squamous cell carcinoma
What's new?
Oral cancer can be revealed by detection of genetic alterations in epithelial cells of the mucosal lining. Here, the authors evaluated a sequencing assay to detect genetic changes in noninvasively collected cells. The noninvasive assay accurately identified genetic changes in oral lesions compared with biopsies, and the authors also demonstrated that their method could predict oral cancer in a cohort of Fanconi anemia patients who had no visible lesions. This approach could be useful as a screening tool for high‐risk populations or to monitor disease progression.
Abbreviations
- CI
confidence interval
- CNA
copy number alteration
- FA
Fanconi anemia
- FFPE
formalin‐fixed paraffin‐embedded
- HE
hematoxylin and eosin
- HR
hazard ratio
- LASA
Longitudinal Aging Study Amsterdam
- lcWGS
low‐coverage whole‐genome sequencing
- LOH
loss of heterozygosity
- NGS
next‐generation sequencing
- OL
oral leukoplakia
- OPMD
oral potentially malignant disorder
- OR
odds ratio
- OSCC
oral squamous cell carcinoma
- SNP
single nucleotide polymorphism
- VAF
variant allele frequency
1. INTRODUCTION
Oral squamous cell carcinomas (OSCC) comprise roughly a third of all malignant tumors in the upper‐aerodigestive tract. 1 This type of cancer arises in precancerous changes with dimensions of centimeters in diameter which are characterized by genetic alterations associated with the malignancy. 2 , 3 These so‐called precancerous fields may remain invisible up to the point of malignant progression as most patients present with tumors de novo. However, a precancerous field may also occasionally manifest itself as a visible lesion. Such visible clinical presentations fall under the umbrella of oral potentially malignant disorders (OPMDs). 4 In accordance with current genetic progression models and the molecular landscape of head and neck cancer, specific genetic alterations such as mutation of TP53 5 and loss of CDKN2A 6 can be detected in these potentially malignant oral lesions. Importantly, genetic alterations are strong predictors for malignant progression of OPMDs. 7 , 8 , 9
The most common OPMD is oral leukoplakia (OL). 10 The diagnosis of OL is based on the clinical presentation as a white discolored lesion and by exclusion of other known disorders or diseases. 11 The prevalence of OL is associated with age, and OL is found more frequently among smokeless tobacco users, long‐time smokers and heavy alcohol drinkers. 12 Of note, OPMDs are the visible manifestations of the precancerous mucosal fields, and form the tip of the iceberg.
Risk assessment of malignant transformation for OL is based on the absence or presence and degree of epithelial dysplasia as assessed by microscopic examination of a biopsy specimen. Moderate and severe dysplasia are considered to confer a high risk of malignant transformation, whereas mild or no dysplasia would be indicative of a low risk. 13 However, a substantial fraction of patients that develop cancer progress directly from a lesion with mild‐ or without dysplasia. 14 , 15 This could be due to sampling error, the absence of standard recognized histopathological changes that may occur in OL, or the absence of morphological changes. The recent introduction of differentiated dysplasia as a novel recognized morphological change was helpful in this respect and made epithelial dysplasia a very strong risk factor for malignant transformation of OL. 16
The current standard of care for management of oral leukoplakia is surveillance or active treatment by removal of the entire lesion by surgical excision or CO2 vaporization. 17 Large and multifocal OLs usually cannot be removed without functional sequelae and are managed by observation on a regular basis. At present the validity of active treatment is questioned, since none of the applied treatments have been proven to be more effective in preventing oral cancer than managing OL by observation only. 18 , 19
The high recurrence rate of OL and the observation that active treatment does not prevent malignant transformation may be explained by the presence of precancerous fields with genetic changes that are not visible to the naked eye and thus are currently not considered in the clinical management. Hence, appropriate clinical management to prevent malignant transformation is hampered by a lack of diagnostic tools. Therefore, a technique that enables accurate detection of precancerous cells is an urgent clinical need. Given the need of frequent surveillance of the precancerous field and monitoring treatment, a noninvasive technique would be desirable, since every biopsy is a burden for the patient. Such an assay can be applied to genetically delineate the lesion including the surrounding mucosal epithelium, monitor the result of treatment of visible lesions, identify precancerous fields that are not clinically visible, and determine progressing genetic alterations during follow‐up.
With a noninvasive test, oral screening of sub‐populations at high risk for OSCC could also become feasible. In particular Fanconi anemia (FA) patients would fall in the high‐risk category, and FA patients form therefore a unique cohort for proof of principle of noninvasive screening. FA is a rare hereditary disease that is caused by mutations in one of 22 known FA‐related genes, 20 that cause an approximately 500 to 1000‐fold increased risk of developing head and neck squamous cell carcinoma, and oral cancer in particular. 21 FA patients are particularly sensitive to cisplatin‐based chemotherapy because of their genetic defect, which presents an additional clinical need for noninvasive screening for early diagnosis and monitoring of interventions. Other high‐risk populations for oral cancers are OPMD patients, treated OSCC patients, and perhaps the aged population of heavy tobacco and alcohol users and smokeless tobacco users.
We have previously demonstrated that it is possible to detect loss of heterozygosity (LOH) using microsatellite‐PCR methods on brush samples. 7 The assay showed a specificity of 100%, but suffered from low sensitivity since it requires a relatively large proportion of cells with genetic alterations; LOH was detected in only 45% of the brushed samples for which LOH was detected in the matched biopsy. Despite the limited sensitivity, the PCR‐based assay was already able to predict malignant transformation in FA patients with noninvasive screening. 22
We hypothesized that compared with the PCR‐based assay next‐generation sequencing (NGS) approaches could improve sensitivity without impacting specificity. Here, we developed a noninvasive NGS‐based assay and tested the accuracy in a cohort of OL patients by comparing genetic alterations in brush samples vs biopsies. Next we demonstrated its clinical utility for the prediction of OSCC in FA patients.
2. MATERIALS AND METHODS
2.1. Patients and samples
A consecutive cohort of 68 patients with the clinical diagnosis of OL was enrolled between 2016 and 2019 at Amsterdam UMC, location VUmc. OL was defined according to the WHO definition: “a predominantly white plaque of questionable risk having excluded (other) known diseases or disorders that carry no increased risk for cancer”. 23 , 24 In all patients a biopsy was obtained to exclude other known diseases and to determine the presence or absence of oral epithelial dysplasia or invasive carcinoma. In total 41 patients, of whom an oral brush sample and a tissue biopsy were available, were included in this study. Patients underwent an incisional or an excisional biopsy, depending on the extent and location of the lesion. Before taking the biopsy, the respective area was brushed with an Orgenex brush, a derivative of the Orcellex (Rovers Medical Devices) by the practitioner and collected in 500 μl PBS. Cells were spun down and the cell pellet stored at −20°C until further processing. Biopsy samples were formalin‐fixed and paraffin‐embedded for routine diagnosis. These were subsequently retrieved from the Pathology Biobank at Amsterdam UMC, location VUmc, after approval of a research protocol.
Oral brush samples of healthy controls were collected, pelleted and stored at −20°C between October 2005 and November 2006 for the Longitudinal Aging Study Amsterdam (LASA). 25 From this cohort, 11 heavy drinkers (self‐reported excessive or very excessive) and heavy smokers (>20 pack‐years) and 12 never‐smokers and light or nondrinkers were randomly selected from all samples in the respective categories. Median age of healthy controls at time of brushing was 70 years. Control brush samples were obtained from six healthy volunteers with a median age of 30, amounting to 29 total healthy control brush samples.
Collection of brushed samples from FA patients is described elsewhere. 22 Samples were taken of standardized locations in the oral cavity: the left and right retromolar trigone, both sides of the lateral border of the mobile tongue, and the left and right side of the floor of the mouth. Locations of visible lesions outside these areas were specifically noted, and these lesions were separately brushed. Samples were collected prospectively between 2008 and 2013 and archived in Cytolyt (Hologic Benelux) at −20°C. Samples were selected retrospectively to include samples from patients who developed OSCC (n = 17). Each patient was matched with two patients who did not develop OSCC. Patients were matched by age, sex and whether or not the patient had undergone stem cell transplantation, all confounders of oral cancer risk. Samples were available for 25 of the matched samples. Clinical information was shared by the Fanconi‐Anämie Hilfe e.V. A portion of the samples of each location per patient was pooled and pelleted. Brushed samples corresponding to lesions were processed separately.
2.2. Histopathological assessment of OL
Hematoxylin‐ and eosin‐stained sections of OL‐derived biopsies were requested from the biobank of the Department of Pathology of the Amsterdam UMC, location VUmc. Tissue samples were histopathologically graded by an expert oral and head and neck pathologist (EBl). Grading adhered to WHO standards for oral epithelial dysplasia. 26 Differentiated dysplasia was scored according to criteria described for vulvar epithelium. 27
2.3. DNA extraction
Between 9 and 15 sections of 10 μm of formalin‐fixed paraffin‐embedded (FFPE) biopsy tissue were collected on microscopic glass slides. In addition, two 3 μm sections were collected as first and last and stained with hematoxylin and eosin (HE) to monitor differences in histology while cutting through the sample. The 10 μm sections were deparaffinized in xylene and rehydrated in a series of ethanol: water dilutions. Sections were briefly stained with methylene blue (0.1%) and toluidine blue (1%), rinsed and epithelial layers were manually microdissected under a stereomicroscope, followed by DNA isolation using the QIAamp FFPE DNA tissue kit (QIAGEN) according to the manufacturer's instructions.
DNA from pelleted brushed or cultured cells was isolated using the PureLink Genomic DNA Mini Kit (Life Technologies) following the manufacturer's protocol for isolation of mammalian cells and tissues, and eluted with 50 μl elution buffer. DNA concentration was measured using a Qubit dsDNA BR Assay Kit (ThermoFisher). DNA from UM‐SCC‐22A was obtained previously. 28 , 29
2.4. NGS sample preparation and sequencing
For FFPE biopsy samples up to 250 ng DNA and for frozen tissue up to 100 ng DNA was sheared using a Covaris S2 sonicator optimized to yield a peak fragment size of 200 base pairs. Sheared DNA was subjected to a 1.6× bead‐based clean‐up (Ampure Beads, Beckman Coulter) to deplete small DNA fragments (<100 base pairs) and concentrate the samples. Sheared DNA was used as input for KAPA HyperPrep NGS library preparation (KAPA Biosystems, Roche) according to the manufacturer's instructions with the following modification: only a single‐sided size selection was performed using 0.8× bead‐based clean‐up. After library amplification, products were inspected for appropriate library fragment size distribution by TapeStation (D5000 ScreenTape, Agilent Technologies). Samples were equimolarly pooled based on concentration of the PCR product and submitted for single‐end 50 bp sequencing (24 samples per lane) on an Illumina HiSeq 4000. The same pooled library was used for target enrichment sequencing. In brief, 1.5 μg of pooled library was subjected to the SeqCap EZ HyperCap Workflow (Roche, Nimblegen) using a custom designed probe set targeting TP53, CDKN2A, AJUBA, KMT2D, FBXW7, NSD1, NOTCH1, PIK3CA, PTEN, HRAS, CASP8 and FAT1 (design #0200241539, covering 72 695 bases). Due to the relatively small probe set, we performed a double capture, using a briefly amplified (five PCR cycles) product of a first round of capture for a second round of capture. The final capture product was amplified in 14 PCR cycles, measured and submitted for paired‐end 150 bp sequencing (24 samples per lane) on an Illumina HiSeq 4000.
2.5. Analysis of low‐coverage whole genome sequencing data
Single‐end 50 bp NGS reads were mapped to human genome build hg19 using BWA aln version 0.7. 30 The sequencing depth and quality statistics for each sample are summarized in Table S1. Mapped reads were converted to copy number profiles using 500 kb bin size with the R package QDNAseq, 31 version 1.24. Subsequently, FFPE‐derived data were “dewaved” using an adaptation of NOWAVES that is amenable to NGS data (available at https://github.com/tgac-vumc/QDNAseq.dev/tree/dewave). 32 Finally, QDNAseq incorporates DNAcopy 33 for segmentation. Copy number profiles, including ploidy and cellularity estimates, were obtained with the R package ACE 28 (version 1.6). For DNA of frozen samples a copy number alteration (CNA) was called if a segment significantly deviated from the mean of all corresponding segments of samples from healthy controls. The cutoff for this deviation was at least 0.0125 standardized signal, corresponding to a single copy gain or loss at 2.5% purity. Additionally, the null hypothesis that the mean of the sample segment was equal or less extreme than the 1/1000th quantile of the distribution of healthy samples needed to be rejected with multiple‐testing corrected P value <.001. The procedure was similar for FFPE samples, except that the healthy controls comprised FFPE samples derived from normal colorectal epithelium 34 and the cutoff for minimum deviation of standardized signal was 0.025. In all cases only autosomes were analyzed. All analyses were performed in R version 3.6 (www.r-project.org).
2.6. Analysis of target‐enriched deep sequencing
Paired‐end 150 bp sequencing reads were mapped to human genome build hg19 using BWA mem (version 0.7). The sequencing coverage and quality statistics for each sample are summarized in Table S2. A pileup created by samtools 35 (version 1.2) was used as input for VarScan 2 mpileup2snp and mpileup2indel. 36 Mutations, both single nucleotide variants and indels were called with at least 20 reads coverage, five variant‐supporting reads. Additionally, single nucleotide variants required a minimum variant allele frequency of 2.5% for fresh and frozen material or 5% for FFPE material, whereas indels required a minimum variant allele frequency of 5% and 10%, respectively. Variants in homopolymers, variants with low impact, and known single nucleotide polymorphisms (SNPs) with population frequencies over 0.01 (dbSNP build 142) were filtered out using SnpEff 37 and SnpSift, 38 both version 4.1b. Nonhotspot variants (www.cancerhotspots.org 39 ) were further filtered out when occurring in multiple unmatched samples. Remaining variants were manually curated for somatic status by considering variant allele frequency, aberrant cell percentage and genomic copies at the locus of the variant. Germline SNP information (variants marked as SNPs by SnpSift) was used for quality control to (1) ascertain that biopsy and brushed cells were derived from the same individual and (2) to monitor potential contamination. Samples failing quality control (>3 mismatched SNPs or >5% contaminating material) were excluded from analysis.
2.7. Statistical analysis
All statistical analyses were performed in R version 3.6. Associations in 2×2 contingencies were tested for significance using two‐sided Fisher' exact test. OSCC‐free survival was analyzed by the Kaplan‐Meier method and hazard ratio determined by Cox's proportional hazard and significance calculated by log‐rank test.
3. RESULTS
3.1. Validation of NGS approach
We developed an NGS‐based workflow that can detect both genome‐wide CNAs and mutations using the same sequencing library. Part of the library (~20%) was subjected to low‐coverage whole genome sequencing (lcWGS), a powerful NGS method to detect CNAs. 40 , 41 Subsequently, the remaining library was enriched for sequences of 12 genes frequently mutated in OSCC 42 using bead‐based hybridization capture. Sequencing of these fragments yielded high‐coverage results (median coverage of 1073, Table S2) that allowed confident calling of single nucleotide variants and small (< 50 bases) insertions and deletions.
To assess the analytical lower detection limit of the assay, DNA from squamous carcinoma cell line UM‐SCC‐22A diluted with healthy control DNA was sequenced (Figure S1A). A dilution with 30% cell line DNA, which is the limit of the previously used PCR‐based assay, and a dilution with 5% cell line DNA was tested. Both at 30% and 5% cell line DNA CNAs were called. Additionally, all expected driver mutations known in UM‐SCC‐22A (two TP53 mutations and a NOTCH1 mutation) were detected, without detection of any nonspecific mutations at higher allele frequencies (Figure S1B). Based on these observations and the noise levels in the data we set the detection threshold at a variant allele frequency of 2.5%. In summary, NGS can reliably detect CNAs and mutations in samples containing DNA of as little as 5% aberrant cells.
To assess the specificity of the assay with the defined cutoffs, we analyzed DNA collected from brush samples of healthy individuals: 23 participants of LASA 25 and six young adults. In these 29 samples, no CNAs were found, expectantly as somatic CNAs are uncommon in the normal population (specificity = 1; 95% CI: 0.88‐1). Similarly, we did not detect any somatic mutations (specificity = 1; 95% CI: 0.88‐1). This indicates that the detection of somatic genetic variants in brush samples is a specific biomarker.
3.2. Clinical validation of genetic alterations in brush and biopsy samples of OL
We next investigated whether DNA of brushed cells from OL reflects the presence of mutations and CNAs identified in biopsy material of the corresponding lesion. Brushed samples and matched biopsies of a cohort of 41 OL patients were examined and copy number data were reported for all samples and mutation data for all but two (Table 1 and genetic profiles of all OL samples in Figure S2). Concordance between matched brushes and biopsies for detection of copy number alterations and mutations is shown in Figure 1A and B, respectively, and for detection of any genetic alteration in Figure 1C. In 26 patients genetic alterations were found both in brushed and biopsy material, indicating that these are common in oral leukoplakia, and can generally be detected in the brushed samples. However, in five patients we were unable to detect genetic alterations in the brushed cells that were clearly present in biopsy material, yielding a sensitivity of 84% (Figure 1C). In nine patients, genetic alterations were neither detected in the brush nor in the biopsy sample. Surprisingly, in one patient we found genetic alterations in the brush sample, but not in the biopsy. This formally indicates an imperfect specificity of analyzing brush samples, but given the superior DNA quality of brushed samples and the very high confidence of the observed CNAs (Figure S2, LP67), this more likely points to a false negative biopsy result. We note that the exact combination of CNAs and mutations was generally not completely concordant between biopsy and brush sample. This was either due to higher detection sensitivity (especially for mutations) in the frozen brush samples as compared with the FFPE biopsies, or because of subclonal differences between the biopsy and brush sample (Figure S2, notably LP12 and LP28 for respective examples). The accuracy to detect any genetic alteration in a brush sample when one or more are detected in the corresponding biopsy was 85% including the likely false negative biopsy finding (Figure 1).
TABLE 1.
Summary of results from genetic analysis of all OL brush samples with matched biopsy sample
| ID | Dysplasia | Diff. dysp. | CNAs | Mutations | Genetic event | |||
|---|---|---|---|---|---|---|---|---|
| Biopsy | Brush | Biopsy | Brush | Biopsy | Brush | |||
| LP09 | None | Yes | 12 | 2 | 5 | 2 | Yes | Yes |
| LP10 | None | Yes | 2 | 1 | 0 | 0 | Yes | Yes |
| LP11 | None | No | 0 | 0 | 2 | 0 | Yes | No |
| LP12 | None | No | 2 | 4 | 1 | 2 | Yes | Yes |
| LP13 | Mild | Yes | 4 | 7 | 1 | 1 | Yes | Yes |
| LP14 | Mild | No | 0 | 0 | 0 | 0 | No | No |
| LP15 | None | No | 0 | 0 | 0 | 0 | No | No |
| LP17 | None | Yes | 0 | 0 | 0 | 0 | No | No |
| LP19 | Mild | No | 2 | 3 | 1 | 2 | Yes | Yes |
| LP20 | None | No | 1 | 1 | 0 | 0 | Yes | Yes |
| LP21 | None | No | 1 | 1 | 0 | 2 | Yes | Yes |
| LP22 | None | No | 0 | 0 | 0 | 0 | No | No |
| LP25 | Moderate | No | 6 | 5 | 5 | 5 | Yes | Yes |
| LP26 | None | No | 2 | 3 | 5 | 5 | Yes | Yes |
| LP27 | Mild | No | 18 | 0 | 2 | 1 | Yes | Yes |
| LP28 | Mild | No | 2 | 9 | 0 | 4 | Yes | Yes |
| LP29 | ‐ | ‐ | 5 | 8 | 2 | 1 | Yes | Yes |
| LP31 | None | No | 2 | 2 | 3 | 4 | Yes | Yes |
| LP32 | ‐ | ‐ | 1 | 0 | 4 | 0 | Yes | No |
| LP33 | Moderate | No | 0 | 1 | 5 | 1 | Yes | Yes |
| LP35 | None | Yes | 0 | 0 | 0 | 0 | No | No |
| LP39 | Severe | Yes | 22 | 3 | 3 | 1 | Yes | Yes |
| LP40 | None | No | 0 | 1 | 5 | 8 | Yes | Yes |
| LP41 | None | No | 0 | 0 | 0 | 0 | No | No |
| LP42 | None | No | 0 | 0 | 0 | 0 | No | No |
| LP43 | Moderate | No | 2 | 5 | 1 | 1 | Yes | Yes |
| LP45 | Mild | No | 1 | 0 | 0 | 0 | Yes | No |
| LP46 | None | No | 10 | 8 | 0 | 0 | Yes | Yes |
| LP47 | None | Yes | 14 | 4 | 0 | 2 | Yes | Yes |
| LP48 | None | No | 2 | 0 | 1 | 0 | Yes | No |
| LP49 | None | No | 0 | 0 | 0 | 0 | No | No |
| LP51 | None | Yes | 0 | 0 | 0 | 0 | No | No |
| LP52 | None | Yes | 3 | 4 | 0 | 0 | Yes | Yes |
| LP53 | None | Yes | 2 | 2 | 2 | 3 | Yes | Yes |
| LP56 | None | Yes | 0 | 0 | 1 | 1 | Yes | Yes |
| LP57 | Mild | No | 3 | 3 | 0 | 0 | Yes | Yes |
| LP58 | None | No | 1 | 0 | 0 | 0 | Yes | No |
| LP59 | None | No | 14 | 16 | 3 | 2 | Yes | Yes |
| LP65 | Moderate | No | 11 | 22 | ‐ | ‐ | Yes | Yes |
| LP67 | None | Yes | 0 | 18 | 0 | ‐ | No | Yes |
| LP68 | None | Yes | 0 | 0 | 1 | 1 | Yes | Yes |
Note: Biopsy material and brushed cells from matching OL lesions were analyzed for presence of mutations and CNAs. Number of CNAs is subject to the segmentation algorithm. This table also reports the grade of dysplasia and the presence of differentiated dysplasia observed in the biopsy. Presence of either mutations or CNAs is summarized as the detection of any genetic event in the last two columns.
FIGURE 1.
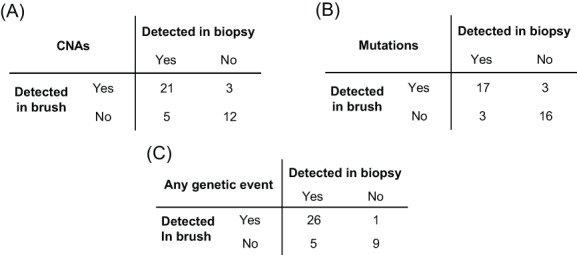
Accuracy of detection of genetic changes in brush samples compared with biopsy samples. Each OL was categorized based on detection of genetic alterations in biopsy and brush samples. Mutations were detected with an accuracy of 85%, sensitivity of 85% and specificity of 86%. CNAs were detected with an accuracy of 80%, sensitivity of 81% and specificity of 80%. Categorizing samples on the basis of detecting any genetic alteration led to a sensitivity of 84%, a specificity of 90% and an accuracy of 85%
While OSCC genomes are characterized by an abundance of chromosomal gains and losses, 29 , 42 only few OL samples displayed many copy number changes (for instance LP39 and LP59, Figure S2). Instead, most lesions only harbored one or a few CNAs, and mostly chromosomal gains, as exemplified by the copy number profile in Figure 2A. Occasionally copy number profiles of OL indicate the presence of multiple subclones in the lesion, which become apparent when CNA deflections do not align with integer copy numbers. Discordance of a CNA from an integer implies that at least two subclones exist with a differential copy number. This is evident in the copy number profile of LP25 (Figure 2B). This lesion harbors a homozygous focal deletion of the complete CDKN2A locus at chromosome 9p. The deflections of copy number aberrations at chromosomes 6, 7 and 15 indicate that there are more than two, but fewer than three copies of these chromosomes, suggesting that the gain of these chromosomes was not present in all cells with the CDKN2A deletion, and was therefore subclonal.
FIGURE 2.
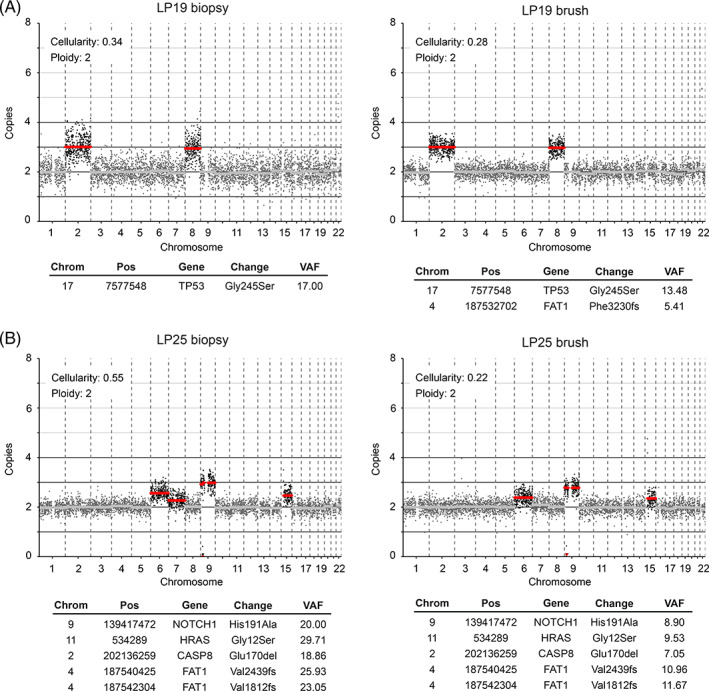
Genetic analysis of two OL lesions. Lesions were analyzed for mutations and CNAs using biopsy material (left) and brushed cells (right). LP19 (A) represents a classic OL copy number profile with single copy gains of chromosome 2 and 8, and a mutation in a single copy of TP53, all of which appear clonal in both biopsy and brush. Brush data exhibits less noise than the corresponding biopsy, indicating superior DNA quality of the freshly frozen brush cells compared with FFPE tissue in case of the biopsy, which allowed us to detect the subclonal FAT1 frameshift variant. (B) LP25 reflects a burgeoning precancerous lesion. Mutations and CNAs are identical in biopsy and brush material of LP25. While mutations and CNAs on chromosome 9 appear clonal, CNAs on chromosomes 6, 7 and 15 are subclonal
3.3. The genomic landscape of OL
Although the primary aim of the study in OL was to test the accuracy of the brush analysis, it also encompassed an extensive body of descriptive data on the genomic landscape of OL. In total we obtained lcWGS for 41 OLs, and for 39 of these we also obtained target‐selected deep sequencing data. In this consecutive cohort, we discovered at least one genetic event in 32 out of 41 patients (78%, Table 1 and Figure 3). As observed in OSCC, 43 the most frequently mutated gene in OL is TP53, followed by NOTCH1, PIK3CA, FAT1 and CDKN2A (Figure 3A).
FIGURE 3.
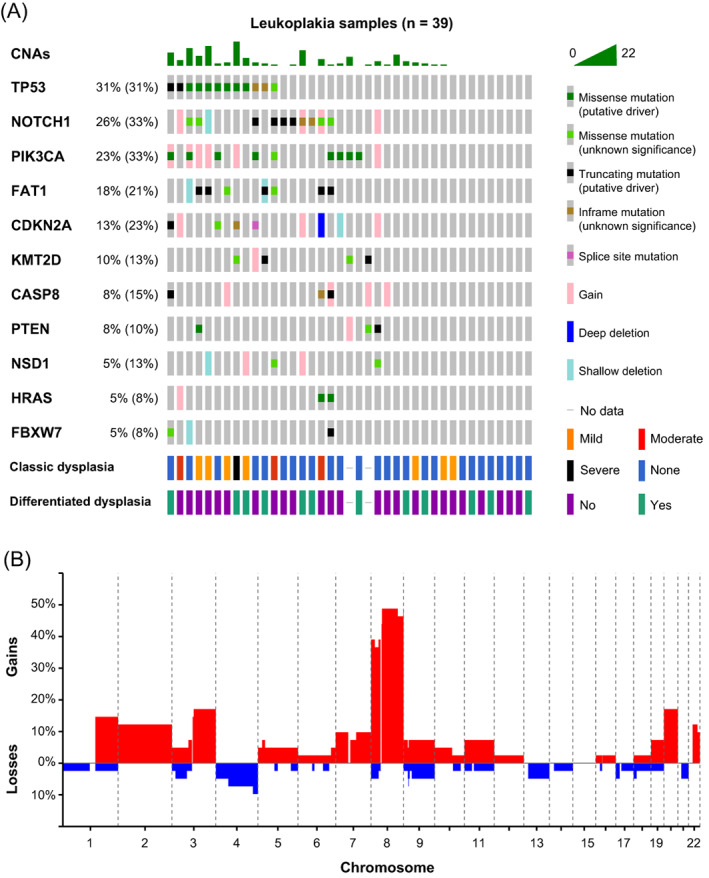
Genetic characterization of OL samples. (A) Genetic data for 39 OL lesions in an oncoprint graph. Percentages of samples with alterations excluding single copy deletions and gains are given for the respective genes, with in parentheses percentages including deletions and gains. Gains but no amplifications were detected. Additionally, histopathological grading of dysplasia and differentiated dysplasia is shown. (B) Genome‐wide frequencies of copy number gains (red) and losses (blue) in 41 OL lesions. Genetic data were pooled from both brush and biopsy samples
Despite the frequent co‐occurrence of mutations and CNAs, three samples only harbored mutations whereas seven samples only harbored CNAs. Although we cannot exclude the presence of mutations in rare driver genes that are not in our capture panel, this suggests that a relevant proportion of OL initially accumulate CNAs without any mutations in canonical driver genes such as TP53 and CDKN2A. Interestingly, most of the CNAs are single copy gains. OLs generally contained 0 to 10 CNAs, with relatively few losses or amplifications (five or more genomic copies). Only a few OL samples, one of which displayed severe dysplasia, were characterized by extensive copy number changes causing aneuploidy. This is also reflected in the low frequencies of gains and losses across the genome, with the notable exception of chromosome 8 (Figure 3B). This is in sharp contrast with OSCCs that are generally aneuploid by many CNAs. Only a third of OSCCs exhibit a near‐diploid genome. 43
We also studied the relationship between dysplasia and genetic alterations. Oral epithelial dysplasia as judged by WHO classification standards was observed in approximately one third of OL (12 out of 39, histopathological grading could not be reviewed for two biopsies). Strikingly, TP53 mutations were significantly enriched in dysplastic lesions; seven out of 11 dysplastic lesions were TP53 mutant, while 21 out of 26 nondysplastic lesions (with deep sequencing data available) were TP53 wild‐type (OR: 6.9, P = .018, Fisher's exact test). Samples were also interrogated for the presence of differentiated dysplasia, 27 recently shown to contribute to prediction of malignant progression. 16 , 44 There were no significant associations with differentiated dysplasia.
3.4. Oral cancer screening in Fanconi anemia patients
The developed genetic assay, validated in OL and noncancer controls, was designed to identify precancerous changes that might cause an increased cancer risk, regardless of whether or not a macroscopic lesion is visible. As the oral cancer risk in the general population is low and precancer screening would demand a very large study, we performed a proof of concept clinical validation study in FA patients who are at high risk for development of oral cancer. In analogy with our previous efforts to demonstrate the added value of oral precancer screening in FA patients, 22 NGS analysis for mutations and CNAs of brushed oral mucosa samples were performed. Brushed samples, derived from locations in the oral cavity without visible abnormalities, were analyzed for 17 FA patients who developed OSCC and matched patients who did not develop OSCC (Table 2 and Figure S3). While we aimed for two matched controls per case, we found only one matched control for nine cases, yielding a total of 25 controls. Age and stem cell transplantation, all risk factors for oral cancer in FA patients, were not significantly different between groups (Table S3). Alcohol and tobacco use were not sufficiently reported to account for. CNAs and mutations were more common in patients who proceeded to develop OSCC in the observational period (CNAs observed in 12 and mutations in eight out of 17 progressors) compared with nonprogressors (11 and 3 out of 25, respectively). When considering time to malignant transformation, FA patients are at statistically significant 3.4‐fold greater hazard (95% CI 1.3‐9.2, P = .009, log‐rank test) of developing OSCC if mutations were detected in their brushed cells (Figure 4A). Presence of CNAs trended towards increased hazard ratio (HR = 2.6, 95% CI 0.91‐7.4, P = .066, log‐rank test), but was not statistically significant (Figure 4B). In several cases, cancer was reported shortly (<3 months) after brushing, which suggests that in these patients cancer might have been present at the moment of sampling. Exclusion of these samples did not alter the outcome and conclusion of this analysis (Figure S4A,B), but in fact only increased the hazard ratio between patients with and without mutations detected in the oral cavity. Presence of mutations in brushed cells remained a significant predictor of OSCC when the potential confounding factors age, sex and stem cell transplantation were included in the Cox proportional hazards model (Table S4). Combining copy number and mutation data did not improve stratification of FA patients compared with using only mutation data (not shown).
TABLE 2.
Clinical and sample characteristics of FA patient cohort
| Patient | Age when brushed | SCT | Sex | CNAs | Mutations | Months follow‐up | Location tumor |
|---|---|---|---|---|---|---|---|
| DE009 | 20 | Yes | M | 0 | 0 | 106 | Gingiva |
| DE019 | 16 | Yes | F | 1 | 1 | 42 | Tongue |
| DE050 | 28 | Yes | M | 2 | 0 | 31 | Lower lip |
| DE074 | 17 | Yes | M | 0 | 0 | 14 | Gingiva |
| DE076 | 41 | No | M | 2 | 0 | <1 a | Tongue |
| FR017 | 26 | Yes | M | 1 | 4 | <1 a | Tongue |
| FR037 | 33 | Yes | F | 5 | 4 | 36 | Tongue |
| UK015 | 23 | Yes | F | 2 | 0 | 1 a | Gingiva |
| US001 | 55 | No | F | 3 | 4 | <1 a | Gingiva |
| US003 | 43 | No | F | 0 | 0 | <1 a | Gingiva |
| US009 | 37 | No | M | 0 | 0 | 2 a | Gingiva |
| US035 | 16 | Yes | F | 26 | 1 | <1 a | Tongue |
| US076 | 49 | No | F | 1 | 0 | 1 | Gingiva |
| US082 | 24 | Yes | F | 1 | 4 | 21 | Tongue |
| US139 | 31 | No | M | 5 | 2 | 7 | Gingiva |
| US162 | 30 | Yes | M | 1 | 1 | 28 | Tongue |
| US175 | 35 | Yes | M | 0 | 0 | 1 a | Tongue |
| AU001 | 18 | Yes | F | 1 | 1 | 91 | n/a |
| BR029 | 37 | No | M | 0 | 2 | 96 | n/a |
| BR127 | 31 | Yes | M | 3 | 1 | 43 | n/a |
| BR163 | 33 | Yes | F | 0 | 0 | 43 | n/a |
| DE005 | 23 | Yes | F | 1 | 0 | 88 | n/a |
| DE007 | 24 | Yes | F | 3 | 0 | 59 | n/a |
| DE008 | 26 | Yes | M | 1 | 0 | 95 | n/a |
| DE016 | 16 | Yes | M | 0 | 0 | 47 | n/a |
| DE017 | 25 | No | F | 2 | 0 | 115 | n/a |
| DE037 | 44 | No | F | 0 | 0 | 100 | n/a |
| DE038 | 40 | Yes | F | 0 | 0 | 35 | n/a |
| DE066 | 16 | Yes | M | 1 | 0 | 129 | n/a |
| DE072 | 19 | Yes | F | 0 | 0 | 120 | n/a |
| DE075 | 21 | Yes | F | 0 | 0 | 121 | n/a |
| ES026 | 29 | Yes | M | 0 | 0 | 110 | n/a |
| FR041 | 34 | Yes | M | 0 | 0 | 89 | n/a |
| NL021 | 27 | Yes | F | 0 | 0 | 95 | n/a |
| UK006 | 48 | No | F | 0 | 0 | 81 | n/a |
| US002 | 25 | No | F | 0 | 0 | 50 | n/a |
| US045 | 17 | Yes | M | 2 | 0 | 6 | n/a |
| US063 | 18 | Yes | M | 0 | 0 | 49 | n/a |
| US094 | 26 | Yes | M | 1 | 0 | 5 | n/a |
| US118 | 35 | Yes | M | 1 | 0 | 97 | n/a |
| US140 | 47 | No | F | 0 | 0 | 94 | n/a |
| US182 | 51 | No | F | 1 | 0 | 70 | n/a |
Note: Age at time of brushing, whether or not the patient received a stem cell transplantation (SCT), detection of CNAs and mutations in the brushed sample, the follow‐up time and the location of the tumor, if applicable, were recorded. Follow‐up from the date of sample collection until the occurrence of OSCC, death, or last moment of follow‐up.
Short time to progression (<3 months).
FIGURE 4.
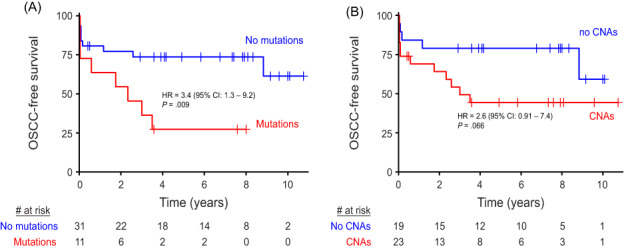
OSCC‐free survival of FA patients. Time to event from the moment the brush sample was taken until end of follow‐up (censored) or detection of OSCC (event) stratified by whether mutations (A) or CNAs (B) were detected in the brush sample of the respective patients
While the analysis above was performed on brushed cells of visually normal epithelium, we also obtained brush samples taken from lesions of two FA patients. In both cases, additional genetic alterations were detected in the lesions (Figure S3, DE019 and US082). Given the finding that both lesions and the visually normal epithelium contained roughly the same percentage of aberrant cells, it is plausible that large parts of the oral mucosa in these patients consist of epithelial cells carrying a subset of mutations and CNAs, whereas visible lesions developed by clonal expansion of cells that acquired additional mutations.
4. DISCUSSION
We show that noninvasive detection of genetic alterations, using DNA extracted from brushed cells, is feasible. The observed sensitivity of 84% is a significant improvement compared with the 45% previously reported for the PCR‐based LOH assay. 7 We envision that the sensitivity may yet be further improved by multiple sampling, by enriching for epithelial cells (for instance using surface markers such as EpCam or EGFR), or by depleting leukocytes. The current study was entirely based on oral brushes. Arguably, oral rinse or saliva samples may be more comprehensive to assess the genetic status of the oral cavity. Future studies will need to determine whether the sensitivity attained by these samples remains sufficient for the detection of genetic alterations of OL lesions and other precancerous changes.
While a noninvasive genetic detection method is somewhat less important in the diagnostic setting of OL, which is a visible lesion, it could very well be applied to map the extension of the lesion beyond what is visible as well as monitor whether the OL is completely excised or vaporized. OL commonly recurs after clinically complete excision, 45 and the benefit of excision to prevent cancer has never been formally demonstrated. 18 We speculate that an OL lesion is larger than what is clinically visible, and the normal‐appearing but genetically altered surrounding mucosa may cause recurrence after excision of the OL. The presence of genetic alterations in the mucosa outside the visible lesions has previously been reported, supporting this hypothesis. 3 , 46 Therefore, the described noninvasive NGS assay can be used to examine a patient after excision of OL to determine whether all aberrant cells have been excised. Should OL recur after excision, then the assay can be applied to establish clonality (or lack thereof) with the index lesion. In addition, the oral cavity of OL patients can be screened at other sites, to detect genetically altered fields that appear clinically normal. Particularly when patients with low risk OLs are referred to the first line for surveillance, invisible high risk changes elsewhere in the oral cavity need to be excluded. Whether the absence of aberrant cells as indicated by negative noninvasive genetic test results is sufficient evidence to reduce clinical monitoring of OL needs to be addressed in further studies with longitudinal sampling.
Besides the proof of concept that noninvasive genetic cytology works well to detect precancerous changes in the oral cavity, this study also greatly expands the knowledge of the genomic landscape of OL. A previous study that performed whole exome sequencing only included 13 OL patients. 47 More recently a genetic study of a much larger cohort of 188 precancer cases was reported, 48 but these cases were not restricted to OL, and the study analyzed CNAs only in specific genomic loci and did not include mutation sequencing. We show that the majority of OL harbor CNAs and/or mutations in known OSCC driver genes. Strikingly, TP53 and CDKN2A were mutated or lost at a much lower frequency than in OSCC, while the percentages for the other most frequent driver genes, NOTCH1, FAT1 and PIK3CA, were not significantly different. Mutated TP53 has been commonly listed as being associated with an increased risk of progression. 49 , 50 Interestingly, it was also significantly associated with the presence of dysplasia in our cohort.
The other genetic discrepancy between OL and OSCC is the prevalence of CNAs and whole genome duplications. Genome instability leading to aneuploidy and whole‐genome duplication have been found to constitute early events in the lifetime of several cancer types including HNSCC, of which OSCC is the major subsite, 51 but our data indicate they are uncommon in the precancerous stage. Future studies with OL samples and matched oral carcinoma may shed more light on molecular events that constitute the key steps in malignant transformation. Particularly longitudinal noninvasive sampling of precancerous changes that remain in situ, either when these are not visible or too large for resection, will reveal detailed knowledge on carcinogenesis and malignant transformation. While our study expands our knowledge of genetic alterations in OL, larger studies with extended patient follow‐up are required to unravel which, if any, specific genetic alterations impart the greatest risk for malignant transformation.
We demonstrated the added value of genetic alterations in brushed cells from normal‐appearing mucosa in FA patients, who are at higher risk for developing oral cancer than the general population. Genetic analyses of brushed samples of FA patients is complicated since FA patients frequently receive stem cell transplants, and donor DNA is often present in the brushed samples. 22 This hampered the microsatellite PCR assay, but SNP filtering proved sufficient to exclude germline variants from donor leukocytes.
FA patients with mutations in brushed material were at significantly increased risk of developing oral cancer than patients without mutations, providing the proof of principle that the noninvasive NGS assay presented here provides a screening tool to stratify patients at different risks of oral cancer. Noteworthy is that many FA patient develop gingival tumors, a site not brushed in the presented cohort. We were unable to study whether the detected genetic alterations were also present in the consecutive tumor. Nonetheless, sampling the gingiva might be important particularly in FA patients.
In this study, we demonstrate the ability to detect genetic alterations from noninvasive samples. This opens the door to prospective studies to determine whether implementing this information in the clinic will lead to earlier detection of OSCC and better patient outcome.
AUTHOR CONTRIBUTIONS
Ruud H. Brakenhoff and Bauke Ylstra conceived the study. Jan G. de Visscher, Elisabeth R. Brouns, Ilkay Evren, Ralf Dietrich, Christine Krieg and Eunike Velleuer collected patient samples and maintained clinical databases. Elisabeth Bloemena performed histopathological assessment. Jos B. Poell and Arjen Brink developed methodology. Jos B. Poell and Leon J. Wils performed experiments. Jos B. Poell analyzed data. Jos B. Poell, Ruud H. Brakenhoff and Bauke Ylstra wrote the manuscript. All authors reviewed the manuscript. The work reported in the paper has been performed by the authors, unless clearly specified in the text.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
The studies reported were approved by the Institutional Review Board of Amsterdam UMC. Studies in oral leukoplakia were registered under number 2014.139 including amendments. Studies in healthy controls of the LASA cohort and FA patients were registered under number 2005.156 including amendments. All participants in all studies have provided written informed consent.
Supporting information
TABLE S3 Distribution of confounding variables in Fanconi Anemia cases and controls. Age, sex and stem cell transplantation are reported separately for the cases (FA patients who developed OSCC) and the controls (FA patients who did not develop OSCC). Age was compared between cases and controls by unpaired t‐test assuming unequal variance. Sex and stem cell transplantation were compared using Fisher's exact test (FET)
TABLE S4. Cox proportional hazards model for development of OSCC. A Cox model for time‐to‐event (development of OSCC) was fitted for the variables mutations, age, sex and stem cell transplantation. The coefficient of the variables, which is the natural logarithm of the proportional hazard associated with the variable, is reported, along with its SD, and the hazard with 95% confidence interval and P value. Hazard applies to the status of the variable as indicated in parentheses for binary variables
FIGURE S1. Detection of CNAs and mutations in cell line DNA diluted with DNA from healthy control cells. DNA of tumor cell line UM‐SCC‐22A were analyzed separately and in dilutions of 30% UM‐SCC‐22A DNA and 5% UM‐SCC‐22A DNA. The respective pure and diluted DNA samples were subjected to low‐coverage WGS to determine CNAs (A) and targeted deep sequencing to detect mutations in known HNSCC driver genes (B). Variant allele frequencies are in accordance with four out of four mutant NOTCH1 copies in UM‐SCC‐22A and both copies of TP53 containing a separate mutation
FIGURE S4. OSCC‐free survival of FA patients with minimum follow‐up more than 3 months. Time to event from the moment the brush sample was taken until end of follow‐up (censored) or detection of OSCC (event) stratified by whether mutations (A) or CNAs (B) were detected in the brush sample of the respective patients. Patients with diagnosis of OSCC within 3 months after brushing were excluded in this analysis
FIGURE S2 Copy number profiles and somatic mutations of all OL brush samples and matching biopsies. Copy number profiles scaled to optimally fit integer copy numbers and somatic mutations with variant allele frequency over 2.5% for brush samples and over 5% for biopsy samples are depicted for all OL samples. Genetic alterations found in matched brushes and biopsies are largely concordant, although the exact composition is often not identical. This figure is provided in a separate PDF‐file
FIGURE S3 Copy number profiles and somatic mutations of all brush samples of FA patients. Copy number profiles scaled to optimally fit integer copy numbers and somatic mutations with variant allele frequency over 2.5% are depicted for all brush samples obtained from FA patients. Of all patients, brush samples taken from visually normal‐appearing mucosa were analyzed. In addition, visible lesions were analyzed for DE019 and US082. This figure is provided in a separate PDF‐file
TABLE S1 Coverage statistics for low‐coverage whole genome sequencing of all samples. Sequencing summary statistics for each individual sample, including total reads sequenced, uniquely mapped nonduplicate reads and used reads for copy number profiling. For coverage the minimum, mean and maximum sequencing depth, as well as the fraction of bases with coverage ≥10× is given. Reads were mapped to build hg19 of the human genome. The table is provided in a separate file
TABLE S2 Coverage statistics for target‐enriched sequencing of all samples. Sequencing summary statistics for each individual sample, including total reads sequenced, uniquely mapped nonduplicate reads and total targeted bases covered. For coverage the minimum, mean and maximum sequencing depth, as well as the fraction of bases with coverage ≥20×, 50×, 100× and 200× is given. Reads were mapped to build hg19 of the human genome. The table is provided in a separate file
ACKNOWLEDGEMENT
The authors thank all participating individuals. This study was funded by a Cancer Center Amsterdam Institutional Grant and the Hanarth Foundation.
Poell JB, Wils LJ, Brink A, et al. Oral cancer prediction by noninvasive genetic screening. Int J Cancer. 2023;152(2):227‐238. doi: 10.1002/ijc.34277
Funding information Hanarth Foundation; Cancer Center Amsterdam Institutional Grant
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Califano J, van der Riet P, Westra W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488‐2492. [PubMed] [Google Scholar]
- 3. Tabor MP, Brakenhoff RH, van Houten VM, et al. Persistence of genetically altered fields in head and neck cancer patients: biological and clinical implications. Clin Cancer Res. 2001;7:1523‐1532. [PubMed] [Google Scholar]
- 4. Sarode SC, Sarode GS, Tupkari JV. Oral potentially malignant disorders: precising the definition. Oral Oncol. 2012;48:759‐760. [DOI] [PubMed] [Google Scholar]
- 5. Ries JC, Schreiner D, Steininger H, Girod SC. p53 mutation and detection of p53 protein expression in oral leukoplakia and oral squamous cell carcinoma. Anticancer Res. 1998;18:2031‐2036. [PubMed] [Google Scholar]
- 6. Papadimitrakopoulou V, Izzo J, Lippman SM, et al. Frequent inactivation of p16INK4a in oral premalignant lesions. Oncogene. 1997;14:1799‐1803. [DOI] [PubMed] [Google Scholar]
- 7. Graveland AP, Bremmer JF, de Maaker M, et al. Molecular screening of oral precancer. Oral Oncol. 2013;49:1129‐1135. [DOI] [PubMed] [Google Scholar]
- 8. Zhang L, Poh CF, Williams M, et al. Loss of heterozygosity (LOH) profiles: validated risk predictors for progression to oral cancer. Cancer Prev Res (Phila). 2012;5:1081‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mao L, Lee JS, Fan YH, et al. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat Med. 1996;2:682‐685. [DOI] [PubMed] [Google Scholar]
- 10. Dionne KR, Warnakulasuriya S, Zain RB, Cheong SC. Potentially malignant disorders of the oral cavity: current practice and future directions in the clinic and laboratory. Int J Cancer. 2015;136:503‐515. [DOI] [PubMed] [Google Scholar]
- 11. van der Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol. 2009;45:317‐323. [DOI] [PubMed] [Google Scholar]
- 12. Mello FW, Miguel AFP, Dutra KL, et al. Prevalence of oral potentially malignant disorders: a systematic review and meta‐analysis. J Oral Pathol Med. 2018;47:633‐640. [DOI] [PubMed] [Google Scholar]
- 13. Speight PM, Khurram SA, Kujan O. Oral potentially malignant disorders: risk of progression to malignancy. Oral Surg Oral Med Oral Pathol Oral Radiol. 2018;125:612‐627. [DOI] [PubMed] [Google Scholar]
- 14. Dost F, le Cao K, Ford PJ, Ades C, Farah CS. Malignant transformation of oral epithelial dysplasia: a real‐world evaluation of histopathologic grading. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;117:343‐352. [DOI] [PubMed] [Google Scholar]
- 15. Chaturvedi AK, Udaltsova N, Engels EA, et al. Oral leukoplakia and risk of progression to oral cancer: a population‐based cohort study. J Natl Cancer Inst. 2020;112:1047‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wils LJ, Poell JB, Evren I, et al. Incorporation of differentiated dysplasia improves prediction of oral leukoplakia at increased risk of malignant progression. Mod Pathol. 2020;33:1033‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kumar A, Cascarini L, McCaul JA, et al. How should we manage oral leukoplakia? Br J Oral Maxillofac Surg. 2013;51:377‐383. [DOI] [PubMed] [Google Scholar]
- 18. Lodi G, Franchini R, Warnakulasuriya S, et al. Interventions for treating oral leukoplakia to prevent oral cancer. Cochrane Database Syst Rev. 2016;7:CD001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brouns E, Baart J, Karagozoglu K, Aartman I, Bloemena E, van der Waal I. Malignant transformation of oral leukoplakia in a well‐defined cohort of 144 patients. Oral Dis. 2014;20:e19‐e24. [DOI] [PubMed] [Google Scholar]
- 20. Nalepa G, Clapp DW. Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer. 2018;18:168‐185. [DOI] [PubMed] [Google Scholar]
- 21. Kutler DI, Auerbach AD, Satagopan J, et al. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg. 2003;129:106‐112. [DOI] [PubMed] [Google Scholar]
- 22. Smetsers SE, Velleuer E, Dietrich R, et al. Noninvasive molecular screening for oral precancer in Fanconi anemia patients. Cancer Prev Res (Phila). 2015;8:1102‐1111. [DOI] [PubMed] [Google Scholar]
- 23. Warnakulasuriya S, Johnson NW, van der Waal I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J Oral Pathol Med. 2007;36:575‐580. [DOI] [PubMed] [Google Scholar]
- 24. Warnakulasuriya S. Oral potentially malignant disorders: a comprehensive review on clinical aspects and management. Oral Oncol. 2020;102:104550. [DOI] [PubMed] [Google Scholar]
- 25. Huisman M, Poppelaars J, van der Horst M, et al. Cohort profile: the longitudinal aging study Amsterdam. Int J Epidemiol. 2011;40:868‐876. [DOI] [PubMed] [Google Scholar]
- 26. El‐Naggar AK, Chan JK, Grandis JR, Takata T, Slootweg PJ. WHO Classification of Head and Neck Tumours. Vol 9. 4th ed. IARC; 2017; Lyon, France. [Google Scholar]
- 27. Arsenic R, Kurrer MO. Differentiated dysplasia is a frequent precursor or associated lesion in invasive squamous cell carcinoma of the oral cavity and pharynx. Virchows Arch. 2013;462:609‐617. [DOI] [PubMed] [Google Scholar]
- 28. Poell JB, Mendeville M, Sie D, Brink A, Brakenhoff RH, Ylstra B. ACE: absolute copy number estimation from low‐coverage whole‐genome sequencing data. Bioinformatics. 2019;35:2847‐2849. [DOI] [PubMed] [Google Scholar]
- 29. van Harten AM, Poell JB, Buijze M, et al. Characterization of a head and neck cancer‐derived cell line panel confirms the distinct TP53‐proficient copy number‐silent subclass. Oral Oncol. 2019;98:53‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scheinin I, Sie D, Bengtsson H, et al. DNA copy number analysis of fresh and formalin‐fixed specimens by shallow whole‐genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 2014;24:2022‐2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van de Wiel MA, Brosens R, Eilers PH, et al. Smoothing waves in array CGH tumor profiles. Bioinformatics. 2009;25:1099‐1104. [DOI] [PubMed] [Google Scholar]
- 33. Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657‐663. [DOI] [PubMed] [Google Scholar]
- 34. van Dijk E, Biesma HD, Cordes M, et al. Loss of chromosome 18q11.2‐q12.1 is predictive for survival in patients with metastatic colorectal cancer treated with bevacizumab. J Clin Oncol. 2018;36:2052‐2060. [DOI] [PubMed] [Google Scholar]
- 35. Li H, Handsaker B, Wysoker A, et al. Genome project data processing S. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koboldt DC, Larson DE, Wilson RK. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr Protoc Bioinformatics. 2013;44:15 4 1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin). 2012;6:80‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cingolani P, Patel VM, Coon M, et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front Genet. 2012;3:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chang MT, Bhattarai TS, Schram AM, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 2018;8:174‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci U S A. 2008;105:16266‐16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kader T, Goode DL, Wong SQ, et al. Copy number analysis by low coverage whole genome sequencing using ultra low‐input DNA from formalin‐fixed paraffin embedded tumor tissue. Genome Med. 2016;8:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cancer Genome Atlas N . Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pickering CR, Zhang J, Yoo SY, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013;3:770‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Evren I, Brouns ER, Wils LJ, et al. Annual malignant transformation rate of oral leukoplakia remains consistent: a long‐term follow‐up study. Oral Oncol. 2020;110:105014. [DOI] [PubMed] [Google Scholar]
- 45. Sundberg J, Korytowska M, Holmberg E, et al. Recurrence rates after surgical removal of oral leukoplakia: a prospective longitudinal multi‐centre study. PLoS One. 2019;14:e0225682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tabor MP, Brakenhoff RH, Ruijter‐Schippers HJ, Kummer JA, Leemans CR, Braakhuis BJ. Genetically altered fields as origin of locally recurrent head and neck cancer: a retrospective study. Clin Cancer Res. 2004;10:3607‐3613. [DOI] [PubMed] [Google Scholar]
- 47. Farah CS, Jessri M, Bennett NC, Dalley AJ, Shearston KD, Fox SA. Exome sequencing of oral leukoplakia and oral squamous cell carcinoma implicates DNA damage repair gene defects in malignant transformation. Oral Oncol. 2019;96:42‐50. [DOI] [PubMed] [Google Scholar]
- 48. William WN Jr, Zhao X, Bianchi JJ, et al. Immune evasion in HPV(‐) head and neck precancer‐cancer transition is driven by an aneuploid switch involving chromosome 9p loss. Proc Natl Acad Sci U S A. 2021;118:e2022655118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shah NG, Trivedi TI, Tankshali RA, et al. Molecular alterations in oral carcinogenesis: significant risk predictors in malignant transformation and tumor progression. Int J Biol Markers. 2007;22:132‐143. [DOI] [PubMed] [Google Scholar]
- 50. Murti PR, Warnakulasuriya KA, Johnson NW, et al. p53 expression in oral precancer as a marker for malignant potential. J Oral Pathol Med. 1998;27:191‐196. [DOI] [PubMed] [Google Scholar]
- 51. Bielski CM, Zehir A, Penson AV, et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet. 2018;50:1189‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S3 Distribution of confounding variables in Fanconi Anemia cases and controls. Age, sex and stem cell transplantation are reported separately for the cases (FA patients who developed OSCC) and the controls (FA patients who did not develop OSCC). Age was compared between cases and controls by unpaired t‐test assuming unequal variance. Sex and stem cell transplantation were compared using Fisher's exact test (FET)
TABLE S4. Cox proportional hazards model for development of OSCC. A Cox model for time‐to‐event (development of OSCC) was fitted for the variables mutations, age, sex and stem cell transplantation. The coefficient of the variables, which is the natural logarithm of the proportional hazard associated with the variable, is reported, along with its SD, and the hazard with 95% confidence interval and P value. Hazard applies to the status of the variable as indicated in parentheses for binary variables
FIGURE S1. Detection of CNAs and mutations in cell line DNA diluted with DNA from healthy control cells. DNA of tumor cell line UM‐SCC‐22A were analyzed separately and in dilutions of 30% UM‐SCC‐22A DNA and 5% UM‐SCC‐22A DNA. The respective pure and diluted DNA samples were subjected to low‐coverage WGS to determine CNAs (A) and targeted deep sequencing to detect mutations in known HNSCC driver genes (B). Variant allele frequencies are in accordance with four out of four mutant NOTCH1 copies in UM‐SCC‐22A and both copies of TP53 containing a separate mutation
FIGURE S4. OSCC‐free survival of FA patients with minimum follow‐up more than 3 months. Time to event from the moment the brush sample was taken until end of follow‐up (censored) or detection of OSCC (event) stratified by whether mutations (A) or CNAs (B) were detected in the brush sample of the respective patients. Patients with diagnosis of OSCC within 3 months after brushing were excluded in this analysis
FIGURE S2 Copy number profiles and somatic mutations of all OL brush samples and matching biopsies. Copy number profiles scaled to optimally fit integer copy numbers and somatic mutations with variant allele frequency over 2.5% for brush samples and over 5% for biopsy samples are depicted for all OL samples. Genetic alterations found in matched brushes and biopsies are largely concordant, although the exact composition is often not identical. This figure is provided in a separate PDF‐file
FIGURE S3 Copy number profiles and somatic mutations of all brush samples of FA patients. Copy number profiles scaled to optimally fit integer copy numbers and somatic mutations with variant allele frequency over 2.5% are depicted for all brush samples obtained from FA patients. Of all patients, brush samples taken from visually normal‐appearing mucosa were analyzed. In addition, visible lesions were analyzed for DE019 and US082. This figure is provided in a separate PDF‐file
TABLE S1 Coverage statistics for low‐coverage whole genome sequencing of all samples. Sequencing summary statistics for each individual sample, including total reads sequenced, uniquely mapped nonduplicate reads and used reads for copy number profiling. For coverage the minimum, mean and maximum sequencing depth, as well as the fraction of bases with coverage ≥10× is given. Reads were mapped to build hg19 of the human genome. The table is provided in a separate file
TABLE S2 Coverage statistics for target‐enriched sequencing of all samples. Sequencing summary statistics for each individual sample, including total reads sequenced, uniquely mapped nonduplicate reads and total targeted bases covered. For coverage the minimum, mean and maximum sequencing depth, as well as the fraction of bases with coverage ≥20×, 50×, 100× and 200× is given. Reads were mapped to build hg19 of the human genome. The table is provided in a separate file
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.