

Abstract
Cutaneous skeletal hypophosphatemia syndrome (CSHS) is caused by somatic mosaic NRAS variants and characterized by melanocytic/sebaceous naevi, eye, and brain malformations, and FGF23‐mediated hypophosphatemic rickets. The MEK inhibitor Trametinib, acting on the RAS/MAPK pathway, is a candidate for CSHS therapy. A 4‐year‐old boy with seborrheic nevus, eye choristoma, multiple hamartomas, brain malformation, pleural lymphangioma and chylothorax developed severe hypophosphatemic rickets unresponsive to phosphate supplementation. The c.182A > G;p.(Gln61Arg) somatic NRAS variant found in DNA from nevus biopsy allowed diagnosing CSHS. We administered Trametinib for 15 months investigating the transcriptional effects at different time points by whole blood RNA‐seq. Treatment resulted in prompt normalization of phosphatemia and phosphaturia, catch‐up growth, chylothorax regression, improvement of bone mineral density, reduction of epidermal nevus and hamartomas. Global RNA sequencing on peripheral blood mononucleate cells showed transcriptional changes under MEK inhibition consisting in a strong sustained downregulation of signatures related to RAS/MAPK, PI3 kinase, WNT and YAP/TAZ pathways, reverting previously defined transcriptomic signatures. CSHS was effectively treated with a MEK inhibitor with almost complete recovery of rickets and partial regression of the phenotype. We identified “core” genes modulated by MEK inhibition potentially serving as surrogate markers of Trametinib action.
Keywords: cutaneous skeletal hypophosphatemia syndrome, MEK inhibitor, RASopathies, Schimmelpenning‐Feuerstein‐Mims syndrome, trametinib
1. INTRODUCTION
Somatic mosaicism is increasingly recognized as a fundamental etiopathogenetic mechanism contributing to human diseases as recent progresses in genomic technologies enabled characterization of several diseases deriving from somatic DNA mutation. In this setting, phenotypes associated with somatic variants of the RASopathy genes continue to emerge showing extreme clinical variability, reflecting the specific variant effects, the location of the mosaic, and timing of the DNA change during embryonic life. 3
Schimmelpenning‐Feuerstein‐Mims syndrome (SFMS, OMIM #163200), also known as linear sebaceous nevus syndrome, is a neurooculocutaneous condition caused by somatic gain‐of‐function variants in the HRAS, NRAS, or KRAS genes. SFMS is characterized by congenital linear nevus sebaceous on the face associated with brain anomalies and dysfunction, epidermal nevi, ocular malformations, and vascular abnormalities. The association of SFMS and hypophosphatemic rickets designate the cutaneous skeletal hypophosphatemia syndrome (CSHS). 4 , 5
In CSHS, mosaicism involving skeletal tissue results in increased FGF23 release by osteocytes, leading to reduced renal phosphate resorption by downregulation of the luminal expression of sodium‐phosphate cotransporters in the kidney proximal tubule. This results in hyperphosphaturic hypophosphatemia and rickets. 4 , 6 FGFR1‐RAS‐MAPK signaling pathway has an important role in human skeletal development and bone homeostasis and is a key regulator of FGF23 production. 4 , 7 CSHS has been recently described, as well as its molecular basis, with the description of a handful of patients with variants in HRAS, 4 , 5 , 8 , 9 , 10 , 11 and the pathogenic NRAS variants p.Gln61Arg, p.Gln61Leu, p.Gly12Cys, and p.Gly13Arg. 8
Recently, precision medicine offered exciting therapeutic opportunities in the RASopathies: the extensively studied role of the amplification of the signaling RAS‐MAPK cascade in cancer allowed developing targeted therapies modulating its pathogenetic oversignalling. The selective inhibition of mitogen‐activated protein/extracellular signal‐regulated kinase (MEK), the final effector signal transducer of the pathway, seems particularly promising. This strategy was effective in ameliorating both germline 12 , 13 and somatic 14 RASopathies in humans and animals. 15 Trametinib, a selective inhibitor of MEK, is approved for treatment of BRAF‐mutated metastatic melanoma. Here we report the first case of CSHS successfully treated by MEK inhibition with Trametinib.
2. MATERIALS AND METHODS
2.1. Case report
The boy was born from an uneventful pregnancy at 38 weeks of gestational age with normal birth parameters. At birth, a large skin malformation involving the left side of the scalp, face, neck and shoulder was evident (Figure 1A–F). The vertex showed multiple lipo‐hamartomas, an epidermal nevus was present on the left forehead and periocular skin, with a choristoma of the eye. Brain MRI showed hypotrophy of the left hemisphere with polymicrogyria. He was admitted to our hospital at 2 months of age for respiratory distress caused by a left pleural lymphangioma with chylothorax (Figure 1H). He has been empirically treated with rapamycin aiming at reducing the multiple skin lesions and chylothorax, with transient success and short periods of remissions. At 4 years and 6 months of age he presented signs of rickets (cupped enlargement of the metaphyses and bowing of the lower limbs, Figure 1C,J,M,P), and a decline in the growth percentiles. Severe hypophosphatemic rickets was diagnosed consistent with serum phosphate concentration of 0.47 mmol/L (normal for age 1.2–1.8 mmL/L), renal tubular phosphate reabsorption 72%–80% (normal >85%), and rickets score 10/10. Sequencing (NimbleGen SeqCap Target Enrichment kit, Roche, on the sequencing platform NextSeq550, Illumina) of the DNA extracted from skin biopsy of the epidermal nevus revealed the somatic mosaic pathogenic variant NRAS (NM_002524.5): c.182A > G, p.Gln61Arg with a variant allele frequency of 29% consistent with the diagnosis of CSHS.
FIGURE 1.
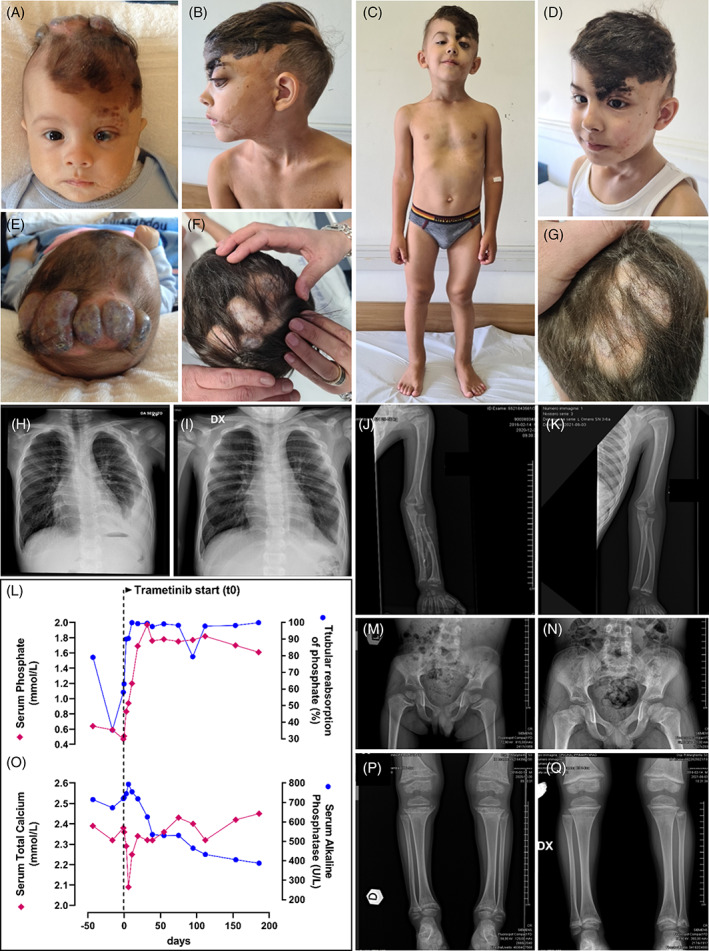
Patient at 3 months of age showed large epidermal nevus of the left side of the scalp, sebaceous nevus of the left hemi‐face, neck and shoulder (A), multiple lipo‐amarthromas at the vertex (E), and choristoma of the left eye (A) that evolved over time before treatment start (B, C, F) with clinical (C) and radiological signs (J, M, P) of rickets at 4 years and 6 months of age, consistent with recurrent chylothorax with ipsilateral pleural lymphangioma (H). After 15 months of treatment, there was an improvement in the thickness and color of the sebaceous nevus (D) and of the lipo‐amarthromatous lesion at the vertex (G), with regression of the radiological signs of rickets (K, N, Q). The left panels display the course of serum and renal tubular resorption of phosphate (L) and of serum total calcium and alkaline phosphatase (O) before and during the first 6 months of Trametinib administration
Treatment with oral phosphate (35 mg/kg/day in four aliquots) and calcitriol (0.5 μg once a day) was started. One month later, as no biochemical effect was evident, oral phosphate was titrated up to 60 mg/kg/day, but serum phosphate levels continued to decrease. Given the unresponsiveness to standard therapy, treatment with Trametinib (Mekinist®, Novartis; 0.032 mg/kg/day) for compassionate use was started after having obtained informed consent from the parents and local ad‐hoc Ethic Committee approval and under IRB approval protocol 0068301‐ID‐256‐2022, June 17, 2022, Comitato Etico Interaziendale AOU Città della Salute e della Scienza di Torino.
2.2. RNA sequencing
Peripheral blood mononuclear cells (PBMCs) were isolated form fresh blood samples within 2 h from collection and stored frozen. RNA was extracted using the Direct‐Zol RNA kit ZY‐R2052 (Euroclone). RNA concentration was quantified using the Qubit Fluorometer (Thermo Fisher Scientific). RNA quality was assessed by verifying RNA Integrity Number and percentage of RNA fragments >200 nucleotides in size (DV200) with Agilent RNAKits on a Bioanalyzer 2100 (Agilent). Total RNA was processed for RNA‐seq analysis with the TruSeq RNA Library PrepKit v2 (Illumina). Library yield was quantified using the Qubit Fluorometer (ThermoFisher Scientific). Library correct size and purity were checked on a Bioanalyzer 2100 (Agilent), using Agilent DNA High Sensitivity kit. Libraries were sequenced on a NextSeq500 system (Illumina).
2.3. Gene expression quantification
Each fastq file was aligned using Spliced Transcripts Alignment to a Reference (STAR) software (https://github.com/alexdobin/STAR/releases) to human genome version hg38. Gencode 27 was used as the transcriptome reference database and gene quantification was performed with featureCounts (http://subread.sourceforge.net). To avoid noise from low‐abundance genes, genes not reaching four counts in at least one sample were removed, and a thresholding was applied by assigning a random value between three and four counts to values below four counts. The final processing step was trimmed mean of M values normalization using the EdgeR package (available at http://bioconductor.org).
2.4. Gene set enrichment analysis
For each gene, expression values from the two samples at each day of treatment were averaged, then the log2 ratio versus Day 0 was calculated at each time point. For each time point, log2ratio values were used to rank genes for preranked GSEA, using standard parameters. 1 Log2 ratio between average expression at all treatment points versus Day 0 was also calculated for preranked GSEA.
3. RESULTS
3.1. Treatment
Trametinib administration resulted in a prompt and stable normalization of blood phosphate (1.21 mml/L, day 11 from treatment start) and increase of urinary phosphate resorption (95%–99%) (Figure 1L). After 15 days of therapy, the patient presented hungry bone syndrome (serum calcium nadir 2.1 mmol/L, Figure 1O) resolved with a short calcium therapy (1500 mg/days), and tendency to hyperphosphatemia (1.9 mmol/L) which required Trametinib reduction to 0.025 mg/kg/day: serum alkaline phosphatase decreased consistently and normalized over time. DXA total body scan revealed a significant improvement in bone mineralization, from a whole body BMD Z‐score −2.3 SDS before treatment start to +1.1 SDS at 15 months. In 2 months, catch‐up growth recovery was noticed, and continued all throughout the follow‐up. A discoloration of skin lesion and a reduction of the clinical features of rickets were evident and a stable regression of the chylothorax was observed (Figure 1i). After 5 months of therapy, reduction of the thickness of the hamartomas at the scalp and improvement in bone architecture at total body X‐ray were observed (Figure 1D, G, K, N, Q). After 6 months, RMN showed a substantial stability of the anatomical anomalies seen before treatment and cardiac function was stable. A subclinical mild increase in serum potassium concentration was constantly observed (nadir plasmatic K+ 5.4 mEq/L) but no rhythm abnormalities were evident at serial ECG nor alterations in the other ions. Serum creatinine‐phosphate‐kinase (CPK) increased from normal to 565 U/L (+37 days) and remained steadily mildly increased from there on (range 305–367) during the follow‐up observation period (currently 15 months), in spite of a reported improvement in strength and movement performance.
3.2. Transcriptomics
We recently described that Trametinib treatment of a RAF1‐associated Noonan syndrome patient induces transcriptional changes in PBMCs. 13 These changes underpin strong and sustained downregulation of signatures related to the RAS/MAPK, PI3 kinase, WNT and YAP/TAZ pathways. Moreover, we found consistent downregulation of a PBMC signature reflecting PTPN11 germline mutation previously defined for NS patients, 2 indicating that MEK inhibition exerts a broad effect on signal transduction and that transcriptional profiling of PBMCs could be used to explore surrogate markers of Trametinib action. To further evaluate the possibility of monitoring Trametinib activity through transcriptome analysis, PBMCs were obtained from the NRAS‐mutant patient at days 0, 6, 19, 95, and 154 of treatment and processed for global RNA sequencing analysis by GeneSet Enrichment Analysis (GSEA) 1 to explore functional pathway alterations (Table S1). As shown in Figure 2, all pathways previously identified as Trametinib‐downmodulated in the RAF1 NS patient 13 are consistently downregulated also in the NRAS patient, apart from four out of eight EGFR/RAS/MEX pathway signatures (Figure 2A) and two out of three PI‐3 kinase pathway signatures (Figure 2B), displaying oscillatory behavior. Most interestingly, Trametinib completely reverted the expression of PBMC signatures previously identified in NS patients (Figure 2C). 2 In addition, GSEA provided a list of “core” genes that are the most up‐ or down‐regulated in that gene set. We therefore explored the overlap between the RAF1 and NRAS patients core genes in each of the concordantly modulated gene sets (Table S2): this analysis highlighted a striking overlap in the core genes, indicating that, for a given gene set, the most Trametinib‐modulated genes tend to be the same across NS patients.
FIGURE 2.
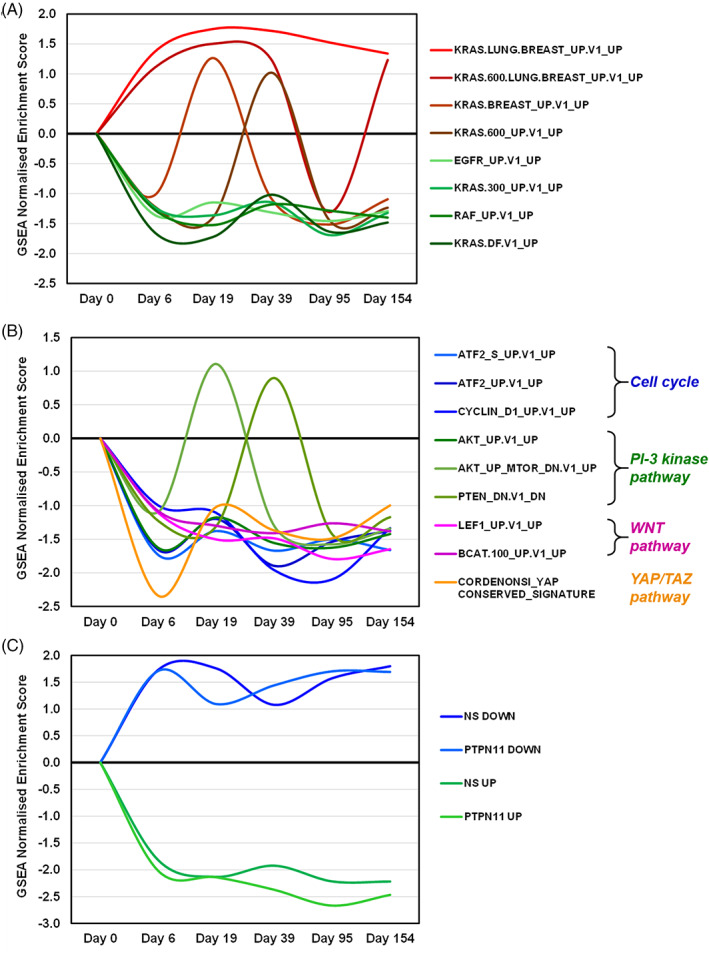
Trametinib‐induced changes in signaling pathway‐associated gene expression signatures in PBMCs of the NRAS‐associated NS patient. Signature modulation is estimated by the GSEA normalized enrichment score (y‐axis), at various times of Trametinib treatment versus pretreatment (x‐axis). (A) EGFR/RAS/MEK pathway signatures previously found downmodulated by Trametinib in PBMCs of a RAF1 NS patient. 13 (B) Cell cycle and other signaling pathway signature, as indicated, previously found downmodulated by Trametinib in PBMCs of a RAF1 NS patient. 13 (C) Reverted expression by Trametinib of previously published PBMC gene expression signatures of NS patients, 2 as indicated. PTPN11 UP/DOWN = genes up‐ or down‐regulated in PBMCs of PTPN11 NS patients versus healthy controls; NS UP/DOWN = genes up‐ or down‐regulated in PBMCs of PTPN11/SOS1/SHOC2 NS patients versus healthy controls
4. DISCUSSION
We report the first case of severe CSHS treated with a precision medicine approach with the MEK inhibitor Trametinib. In CSHS, successful treatment with oral phosphate supplements and calcitriol was reported. 5 , 9 Although an age‐related spontaneous regression of hypophosphatemia and improvement of the skeletal phenotype has been described, 5 progression of the phenotype to extensive bone dysplasia in adulthood is common. 16 In our case, rickets became acutely prominent at 4 years of age with a rapidly evolving skeletal demineralization, growth arrest and life‐threatening hypophospatemia. No response to oral phosphate supplementation was observed, despite the administration of sustained high doses aimed at compensating for the large renal loss. With failure of the conventional treatment, we took into consideration two strategies: the recently introduced anti‐FGF23 antibody burosumab, approved for treating X‐linked hypophosphatemia 17 and downregulation the RAS/MAPK cascade by MEK‐inhibition. Despite that burosumab is usually proposed in children to treat X‐linked hypophosphatemia, we chose the MEK‐inhibition strategy in order to modulate the molecular pathogenetic mechanism in all the tissues involved in the genetic NRAS mosaic affecting several apparatuses. This strategy was successful in normalizing phosphate metabolism, inducing a regression of recurrent chylothorax and mitigating skin manifestations. Shortly after the treatment began, we observed a prompt normalization of the phosphaturia and phosphatemia. Phosphate plasmatic levels and urinary excretion remained constantly normal throughout the treatment and allowed a rapid remineralization of bone evaluated by improvement in the classical rickets signs, radiological rickets score, and bone densitometry. Treatment response was also accompanied by catch‐up growth and resolution of muscle weakness, bone pain, chronic fatigue, and appetite loss. As often observed in rickets, treatment resulted in the “hungry bone syndrome” phenomenon, and hypocalcemia linked to the massive calcium incorporation into the bone, effectively managed with calcium supplements. The therapy was well tolerated, showing no clinically relevant side effects. From the biochemical point of view, a transient moderate increase in muscle CPK and serum potassium were noted: common side effects previously observed under Trametinib therapy.
Whole blood RNA‐seq was performed before and during treatment to define the transcriptional effects of MEK inhibition. As recently described in a RAF1 NS patient treated with Trametinib, 13 we performed PBMC transcriptome profiling to monitor gene expression changes and explore functional readouts of Trametinib efficacy. At odds with the exploratory type of analysis performed in our previous work, we could here focus on Trametinib‐modulated pathways and signatures already observed in our RAF1 patient and found remarkable concordance. Although some of the signatures displayed oscillatory behavior, concordant modulation of all pathways was confirmed, reflecting Trametinib‐driven downregulation of EGFR/RAS/MEK, cell cycle, PI‐3 kinase, WNT and YAP/TAZ pathways. Moreover, reversion of the previously identified NS‐associated PBMC signatures 2 was even more evident than in our RAF1 patient, confirming that MEK inhibition exerts a broad effect on signal transduction. Among the newly identified functional changes induced by Trametinib in the NRAS patient, we observed that the most heavily upregulated gene set (“RELA_DN.V1_UP”) reflects downregulation by Trametinib of the NFKB pathway, confirming a previously identified cross‐talk between the two pathways. 18
In conclusion, a biological mechanism‐guided treatment in CSHS by MEK‐inhibition seems promising and contributes further to a proof of principle supporting future studies and trials on MEK‐inhibition strategies for mosaic RASopathies.
FUNDING INFORMATION
This research received no external funding.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
ETHICS STATEMENT
The study was approved by the Hospital Ethics Committee (Comitato Etico Interaziendale dell' A.O.U. Città della Salute e della Scienza di Torino, dell'A.O. Ordine Mauriziano di Torino e dell'A.S.L. Città di Torino; IRB approval protocol 0068301‐ ID‐256‐2022, June 17, 2022).
Supporting information
Table S1. Results of GeneSet Enrichment Analysis (GSEA) 1 on C6 “Oncogenic signatures” and Noonan signature, 2 comparing different days of treatment versus pretreatment control. Previously identified NS signatures in peripheral blood mononuclear cells (PBMC) are highlighted in yellow. Previously identified Trametinib‐modulated gene sets are highlighted as follows: EGFR/RAS/MEK pathway gene sets: bold, red text; Cell cycle gene sets: bold, blue text; PI3Kinase pathway gene sets: bold, green text; WNT pathway gene sets: bold, purple text; YAP/TAZ gene set: bold, orange text.
Table S2. Hypergeometric distribution of cores intersections.
ACKNOWLEDGEMENT
Open Access Funding provided by Universita degli Studi di Torino within the CRUI‐CARE Agreement.
Carli D, Cardaropoli S, Tessaris D, et al. Successful treatment with MEK‐inhibitor in a patient with NRAS ‐related cutaneous skeletal hypophosphatemia syndrome. Genes Chromosomes Cancer. 2022;61(12):740‐746. doi: 10.1002/gcc.23092
Giovanni Battista Ferrero is Co‐corresponding author.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545‐15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ferrero GB, Picco G, Baldassarre G, et al. Transcriptional hallmarks of Noonan syndrome and Noonan‐like syndrome with loose anagen hair. Hum Mutat. 2012;33(4):703‐709. doi: 10.1002/humu.22026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mussa A, Carli D, Cardaropoli S, Ferrero GB, Resta N. Lateralized and segmental overgrowth in children. Cancers (Basel). 2021;13(24):6166. doi: 10.3390/cancers13246166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lim YH, Ovejero D, Sugarman JS, et al. Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Hum Mol Genet. 2014;23(2):397‐407. doi: 10.1093/hmg/ddt429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ovejero D, Lim YH, Boyce AM, et al. Cutaneous skeletal hypophosphatemia syndrome: clinical spectrum, natural history, and treatment. Osteoporos Int. 2016;27(12):3615‐3626. doi: 10.1007/s00198-016-3702-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Castro LF, Ovejero D, Boyce AM. Diagnosis of endocrine disease: mosaic disorders of FGF23 excess: fibrous dysplasia/McCune‐Albright syndrome and cutaneous skeletal hypophosphatemia syndrome. Eur J Endocrinol. 2020;182(5):R83‐R99. doi: 10.1530/EJE-19-0969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fowlkes JL, Thrailkill KM, Bunn RC. RASopathies: the musculoskeletal consequences and their etiology and pathogenesis. Bone. 2021;152:116060. doi: 10.1016/j.bone.2021.116060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Avitan‐Hersh E, Tatur S, Indelman M, et al. Postzygotic HRAS mutation causing both keratinocytic epidermal nevus and thymoma and associated with bone dysplasia and hypophosphatemia due to elevated FGF23. J Clin Endocrinol Metab. 2014;99(1):E132‐E136. doi: 10.1210/jc.2013-2813 [DOI] [PubMed] [Google Scholar]
- 9. Lim YH, Ovejero D, Derrick KM, Collins MT, Choate KA. Genomics YCfM. Cutaneous skeletal hypophosphatemia syndrome (CSHS) is a multilineage somatic mosaic RASopathy. J Am Acad Dermatol. 2016;75(2):420‐427. doi: 10.1016/j.jaad.2015.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramesh R, Shaw N, Miles EK, Richard B, Colmenero I, Moss C. Mosaic NRAS Q61R mutation in a child with giant congenital melanocytic naevus, epidermal naevus syndrome and hypophosphataemic rickets. Clin Exp Dermatol. 2017;42(1):75‐79. doi: 10.1111/ced.12969 [DOI] [PubMed] [Google Scholar]
- 11. Park PG, Park E, Hyun HS, et al. Cutaneous skeletal hypophosphatemia syndrome in association with a mosaic. Ann Clin Lab Sci. 2018;48(5):665‐669. [PubMed] [Google Scholar]
- 12. Andelfinger G, Marquis C, Raboisson MJ, et al. Hypertrophic cardiomyopathy in Noonan syndrome treated by MEK‐inhibition. J Am Coll Cardiol. 2019;73(17):2237‐2239. doi: 10.1016/j.jacc.2019.01.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mussa A, Carli D, Giorgio E, et al. MEK inhibition in a newborn with RAF1‐associated Noonan syndrome ameliorates hypertrophic cardiomyopathy but is insufficient to revert pulmonary vascular disease. Genes (Basel). 2021;13(1):6. doi: 10.3390/genes13010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Al‐Olabi L, Polubothu S, Dowsett K, et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest. 2018;128(4):1496‐1508. doi: 10.1172/JCI98589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu X, Simpson J, Hong JH, et al. MEK‐ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest. 2011;121(3):1009‐1025. doi: 10.1172/JCI44929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maia TF, Freire BL, Rodrigues AS, et al. MON‐333 cutaneous skeletal Hypophosphatemic syndrome (Cshs) caused by somatic HRAS p.G13R mutation: long follow‐up of two Brazilian women. J Endocr Soc. 2020;4(Supplement_1):A700. doi: 10.1210/jendso/bvaa046.1383 [DOI] [Google Scholar]
- 17. Smith P, Bayliss S, Shinawi M, et al. SAT‐LB085 first report of Burosumab (anti‐FGF23 monoclonal antibody) for rickets complicating HRAS‐associated cutaneous skeletal hypophosphatemia syndrome. J Endocr Soc. 2019;3(Supplement_1):SATLB085. doi: 10.1210/js.2019-SAT-LB085 [DOI] [Google Scholar]
- 18. Lim KS, Yong ZWE, Wang H, et al. Inflammatory and mitogenic signals drive interleukin 23 subunit alpha (IL23A) secretion independent of IL12B in intestinal epithelial cells. J Biol Chem. 2020;295(19):6387‐6400. doi: 10.1074/jbc.RA120.012943 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Results of GeneSet Enrichment Analysis (GSEA) 1 on C6 “Oncogenic signatures” and Noonan signature, 2 comparing different days of treatment versus pretreatment control. Previously identified NS signatures in peripheral blood mononuclear cells (PBMC) are highlighted in yellow. Previously identified Trametinib‐modulated gene sets are highlighted as follows: EGFR/RAS/MEK pathway gene sets: bold, red text; Cell cycle gene sets: bold, blue text; PI3Kinase pathway gene sets: bold, green text; WNT pathway gene sets: bold, purple text; YAP/TAZ gene set: bold, orange text.
Table S2. Hypergeometric distribution of cores intersections.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.