

Abstract
Brucellosis is one of the most common zoonoses in the Middle East. It is causing economic losses to the livestock industry and has a great public health concern. Little is known about the genetic diversity and distribution of brucellae in Iran. Therefore, forty Brucella spp. strains (B. abortus and B. melitensis) isolated from animals and humans were analyzed by whole genome sequencing (WGS) technology using single nucleotide polymorphism (SNP) analysis and core genome multilocus sequence typing (cgMLST). Brucella isolates were obtained from lymph nodes (cows and camels), milk (cows, camels and sheep), and aborted foetus samples (sheep and goats), as well as cerebrospinal fluid and blood of humans. The isolates were originating from thirteen provinces of Iran and isolated between 2015 and 2020. According to in-silico MLST, ST8 and ST2 were the most frequent sequence types in B. melitensis and B. abortus, respectively. Based on phylogeographic reconstruction using cgSNP analysis, the investigated Iranian B. melitensis strains belonged to the American and Mediterranean lineages of the B. melitensis phylogeny. Furthermore, cgSNP analysis revealed a similarity between Iranian B. abortus isolates and strains from Iraq and Egypt. Therefore, the origin of the Iranian strains can be suggested to be strains from neighboring and Middle East countries. Moreover, cgMLST analysis showed that the Iranian B. melitensis strains were closely relative to strains recovered from sheep and humans in Iraq, Afghanistan, Syria, Turkmenistan, and Pakistan. In the current panel of strains, cgMLST and cgSNP analysis provided an appropriate and accurate tool for effective traceback analyses for Brucella spp. from Iran. The results of cgSNP and cgMLST helped to understand the geographic distribution and interspecies transmission of Iranian strains and highlight the importance of specific brucellosis control measures in Iran with regard to the One-Health approach.
Keywords: Whole genome sequencing, cgSNP, cgMLST, Brucella abortus, Brucella melitensis, Genetic diversity
Highlights
-
•
Forty Brucella isolates were characterized by phenotypic typing, AMOS-PCR, Bruce-ladder and WGS.
-
•
cgMLST showed ST8 and ST2 were the most frequent sequence types in B. melitensis and B. abortus, respectively
-
•
The study provides the first data describing how Brucella isolates from Iran relate to the global population structure.
-
•
Based on cgSNP analysis, the Iranian B. melitensis strains belonged to the American and Mediterranean lineages.
-
•
Based on cgSNP analysis, a similarity between Iranian B. abortus isolates and strains from Iraq and Egypt was seen.
1. Introduction
Brucellosis is a notorious zoonotic disease affecting a wide range of animals and humans worldwide. It is considered one of the most prevalent zoonoses in the Middle East and North African (MENA) countries with public health significance and substantial economic losses in the livestock industry [1]. The genus Brucella (B.) currently comprises twelve highly genetically related species [2]. However, the inclusion of closely related Ochrobactrum species in the genus Brucella has been proposed lately [3]. Both genera must be kept separated also in the future to avoid confusion which will bring evident risks for occupational personnel who are confronted brucellosis [4]. The most virulent species for humans are B. melitensis and B. suis (except B. suis biovar 2), whereas B. abortus causes milder illness [5,6]. Bovines and small ruminants are the primary hosts for B. abortus and B. melitensis, respectively, although trans-species transmission of Brucella spp. has been reported [[7], [8], [9]]. Brucellosis is transmitted to humans through close contact with infected animals or infected materials e.g. aborted fetuses or placentas, and via consumption of unpasteurized dairy products [10]. Although the World Organization for Animal Health (WOAH) and the World Health Organization (WHO) have suggested strategies for brucellosis control, only a few countries in Europe, New Zealand, Japan, Australia, and Canada are considered free from brucellosis in livestock. The highest incidence of brucellosis is reported in the Mediterranean region, the Middle East, India, China, Sub-Saharan Africa, Mexico, and Peru [11,12]. In Iran, brucellosis is an endemic disease in animals and humans causing huge economic losses and critically impacting human health [13,14]. B. abortus biovar (bv) 3 in cattle and B. melitensis bv 1 in small ruminants and humans were the most prevalent agents causing brucellosis followed by B. abortus bv 1 and B. melitensis bv 3 [[14], [15], [16]].
The extreme genetic homogeneity of Brucella of >90% is one of the notable challenges for typing approaches of the genus Brucella [2]. The genome of Brucella is composed of two circular chromosomes of 1.2 and 2.1 Mb, which are highly conserved [17]. The 16S rRNA sequence is 100% identical between all Brucella spp. [18]. Therefore, different genetic tools such as whole genome sequencing (WGS) have to be employed to trace the geographic origin of isolates and their phylogenetic connection [[19], [20], [21], [22], [23]]. Multiple Locus Variable Number Tandem Repeat (VNTR) Analysis (MLVA) has been introduced as a valuable PCR-based molecular typing approach with high discriminatory power to differentiate Brucella isolates in epidemiological investigations [[24], [25], [26], [27]]. However, it cannot accurately reveal transmission routes and the origin of outbreak strains [25]. Therefore, genetic typing approaches have recently changed towards WGS based analysis that enables the revelation of phylogenetic correlations and a more in-depth resolution of genotypes.
There is limited knowledge about the molecular epidemiology, distribution, and diversity of Brucella spp. in Iran. The growing number of human and animal brucellosis cases led us to evaluate the geographic origin, diversity, and phylogeographic distribution of Iranian strains. Thus, WGS based analysis of B. melitensis and B. abortus isolates recovered from animals and humans over a period of five years (2015–2020) was applied to get an overview of the epidemiological situation. Additionally, isolates were compared to strains from other Middle East countries to investigate the presence of possible lineages in the region.
2. Material and methods
2.1. Ethics committee
This survey was a part of the national surveillance plan for brucellosis in the period 2015–2020 and all isolates were selected from the strain collection of Razi Vaccine and Serum Research Institute (RVSRI). This study was approved by the Ethics committee of the Iran National Science Foundation (INFS) with reference number (INFS. 99,030,922) confirming that all experiments were performed following relevant guidelines and regulations.
2.2. Brucella isolates and identification
Forty Brucella isolates recovered from animals and humans between 2015 and 2020 were used in this study (Table S1). All samples were cultured on Brucella agar (Himedia, India) for 5 days at 37 °C and 5% CO2. A panel of biotyping tests was performed for all strains as described previously [14,28]. Total genomic DNA was extracted from fresh cultures using the Exgene Cell SV kit (GeneAll, South Korea) according to the manufacturer's instructions for Gram-negative bacteria. All strains were confirmed as Brucella species by multiplex PCR using previously published approaches [14]. The AMOS PCR [29] and Bruce-ladder PCR [30] were used to confirm Brucella species differentiation.
2.3. Whole genome sequencing (WGS) and bioinformatic processing
WGS was carried out by paired-end sequencing (2 × 300 bp) on a MiSeq machine (Illumina, San Diego, CA, USA) as previously described [31]. Briefly, genomic library was prepared using the Nextera XT library preparation kit (Illumina Inc., San Diego, CA, USA). FastQC (v 0.11.8, Babraham Bioinformatics, Babraham Institute, Cambridge, UK) was applied to evaluate the quality metrics of Illumina sequence data. For contamination assessment and classification of reads at the genus and species level, Kraken2 (v 2.0.7_beta) [32] was applied. De novo genome assembly was conducted with SPAdes within Shovill (v. 1.0.4) (https://github.com/tseemann/shovill). Quality control of assembled contigs was performed using QUAST (v 5.0.2) [33] and potential coding genome regions were predicted by Prokka (v 1.14.5) [34].
To place the sequenced Iranian strains into an international context, the NCBI Sequence Read Archive (SRA) was searched for paired-end Illumina sequencing data of B. melitensis and B. abortus (accessed on 27th February 2022) (Table S2). Those foreign data were processed as described above.
2.4. Core genome SNP (cgSNP) analysis
Core genome SNP calling was conducted with snippy (v 4.3.6) (https://github.com/tseemann/snippy) using B. abortus strain 2308 (ASM54005.1) and B. melitensis strain 16 M (NC_003317 and NC_003318) as references. Core genome SNP alignments served as the basis for a maximum likelihood analysis performed by RAxML (v 8.2.12) [35]. The SNP distance between each pair of genomes was calculated from this alignment using SNP-dists (v 0.6.3) [36].
2.5. Core genome multilocus sequence typing (cgMLST) and in silico MLST
Ridom SeqSphere+ v7.7 (Ridom GmbH, Münster, Germany) was applied for the cgMLST of Iranian B. melitensis isolates using the corresponding typing scheme [37] and default target quality control parameters. The allelic profiles served as the basis for minimum spanning tree construction with pairwise ignoring missing values. In silico MLST was conducted by scanning the genome assemblies against the 9 loci typing scheme [38] with the tool mlst (https://github.com/tseemann/mlst).
3. Results
3.1. Brucella identification and differentiation
Forty bacterial isolates were obtained from eight different geographical areas (Table 1, Fig. 1) and identified as Brucella spp. by classical typing tools. AMOS-PCR and Bruce-ladder PCR confirmed three B. abortus isolates (two from cattle and one from a human) and thirty-seven B. melitensis isolates (21 from human blood, one from human cerebrospinal fluid, four from bovine milk, three from bovine lymph nodes, one from camel milk, one from camel lymph node, four from an ovine aborted fetus, one from ovine milk and one from an aborted fetus of a goat (Supplementary Table S1). The classical AMOS PCR failed to identify B. abortus biovar 3 that the failure of classical AMOS PCR to identify B. abortus biovar 3 was seen in this study. Biovars 1, 2, and 3 in B. abortus isolates and biovars 1, 2, and 3 in B. melitensis were confirmed by applying classical biotyping methods. The predominant brucellae were B. melitensis bv 1, which was found in 30 strains isolated from all investigated host species and numerous provinces of Iran (Fig. 1, Supplementary Table S1).
Table 1.
Numbers and geographical origin of Brucella strains recovered from humans and animals in Iran collected over 5 years (2015–2020).
| Geographic Area | Province | City | B. melitensis | B. abortus |
|---|---|---|---|---|
| North | Mazandaran, Semnan | Amol, Semnan | 2 | – |
| Northwest | Alborz, Tehran, Zanjan | Karaj, Tehran, Gharchak, Shahr Ray, Zanjan | 16 | 1 |
| Northeast | Kermanshah | Kermanshah | 6 | – |
| South | Hormozgan | Jask | 1 | – |
| Southeast | Kerman | Kerman | 1 | |
| Southwest | Fars | Shiraz, Fasa, Eghlid | 4 | 1 |
| West | Hamedan | Hamedan | 3 | – |
| Central | Qom, Yazd, Isfahan | Qom, Yazd, Isfahan | 4 | 1 |
| Total | 37 | 3 | ||
Fig. 1.
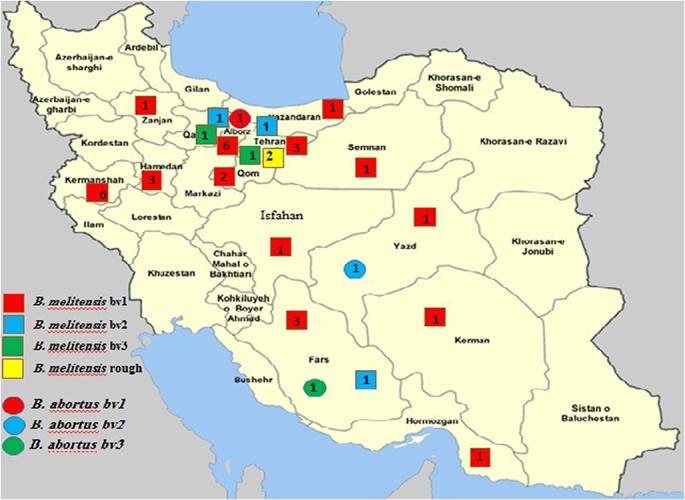
The geographic distribution of Brucella species/biovars from animals and humans in Iran. The numbers inside the boxes indicate the frequencies of Brucella biovars.
3.2. Genome sequencing and assembly
The average number of reads was 1,645,251 (min 1,217,718, max 2,835,032) for each isolate, which yields an average genome coverage of 105.9-fold (min 99, max 202) (Supplementary Table S3). Genome assemblies comprised between 17 and 26 contigs covering 98.5% to 99.4% of the respective reference genome (B. melitensis 16 M and B. abortus 2308). The GC content was within the expected range (57.24–57.28%). Between 3090 and 3143 coding regions were predicted.
3.3. Core genome SNP analysis of B. abortus and B. melitensis
The cgSNP analysis for B. abortus is based on an alignment of 1751 core genome SNPs. Three sequence types could be differentiated for the Iranian B. abortus strains (Fig. 2). Among these, the isolates 20Y0156 and 20Y0153, which were both isolated from cattle in 2019 and 2018, respectively, showed the highest identity (252 SNPs difference). However, this difference is comparably high, as the Egyptian strains included in this analysis merely differed in maximally 8 SNPs. B. abortus bv 1 strain 20Y0140 that was isolated from a human sample in 2015 differed in 1171 SNPs and 1179 SNPs from the other two Iranian B. abortus isolates, which were assigned to bv 2 (20Y0153) and bv 3 (20Y0156). When compared to global strains, the highest identity to Iranian isolates was observed for Egyptian B. abortus strains isolated from cattle, which differed in 80–82 SNPs from the Iranian isolate of human origin (20Y0140). The more recent Iranian isolates from 2018 and 2019 exhibited a higher similarity to a strain isolated from cattle (67/93) in 1967 in Iraq (608 and 614 SNPs difference).
Fig. 2.
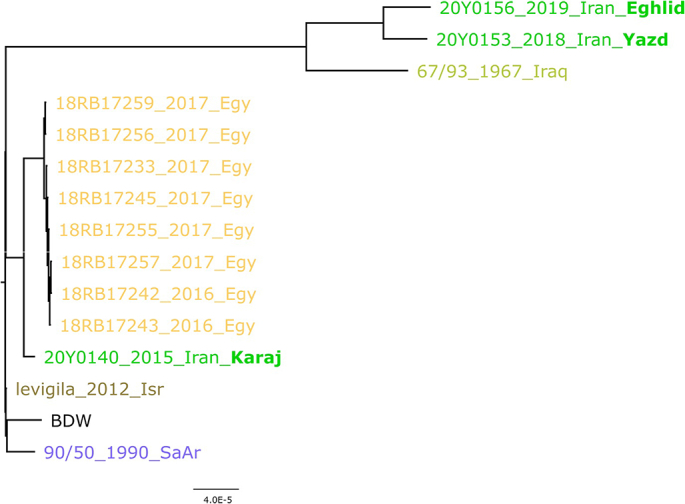
Maximum likelihood tree based on cgSNP alignment of Iranian B. abortus strains and strains of similar geographic origin to the reference strain BDW. Besides the strain names, the year and country of isolation are also given (Egy – Egypt; Isr – Israel; SaAr – Saudi Arabia). For the Iranian strains, the isolation location is printed in bold letters (Eghl-Eghlid; Yazd; Karaj). The scale bar indicates the number of nucleotide changes per site.
Brucella melitensis isolates were assigned to two cgSNP lineages: American (genotype Va) and East Mediterranean (genotypes IIb and IIf) (Fig. 3). Most of them represented unique SNP sequence types differing in more than five SNPs.
Fig. 3.
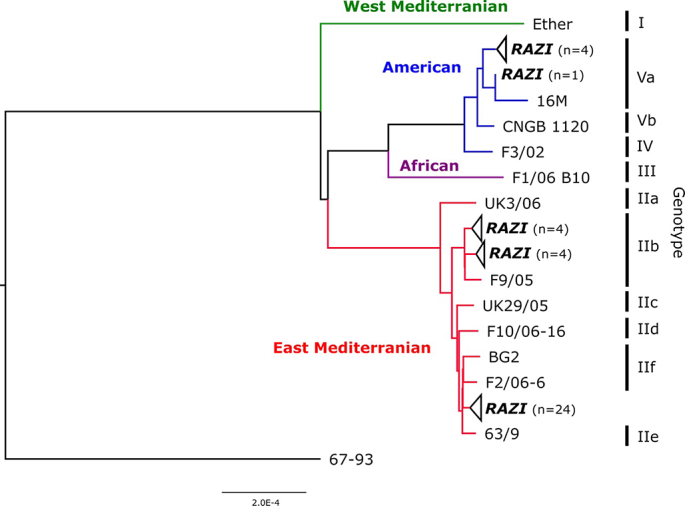
Maximum likelihood tree based on cgSNP alignment of Iranian B. melitensis strains and representatives of B. melitensis lineages to the reference strain 16 M. For better visualization, branches containing exclusively Iranian strains are collapsed and named RAZI, with numbers of strains given in brackets. For a complete version of the tree, see Supplementary Fig. S1. B. abortus 67/93 was used for rooting the tree. Designation of lineages and genotypes are according to [39,40], respectively. The scale bar indicates the number of nucleotide changes per site.
Four Iranian B. melitensis bv 1 isolates that were obtained from cows (Qom in 2015 and Fars in 2020), sheep (Mazandaran in 2018), and camel (Hormozgan in 2020) belonged to the same genotype (Va) within the American lineage and showed only two cgSNP differences (cluster No. 1, Table 2, Fig. 4). However, another human isolate from Alborz province isolated in 2015 was assigned to the same genotype but differed in 187 cgSNPs from the other four Va genotype isolates. The difference between these isolates and the reference strain 16 M, a member of the Va genotype, was found to be 404 and 219 cgSNPs, respectively.
Table 2.
Clusters of Iranian B. melitensis strains that differed in ≤2 SNPs.
| No. | Strain | Province | Year | Host | Source | Biovar |
|---|---|---|---|---|---|---|
| 1 | 20Y0151 | Qom | 2016 | Cow | Milk | 1 |
| 1 | 20Y0155 | Mazandaran | 2018 | Sheep | Milk | 1 |
| 1 | 20Y0173 | Fars | 2020 | Cow | Lymph node | 1 |
| 1 | 20Y0179 | Hormozgan | 2020 | Camel | Lymph node | 1 |
| 2 | 20Y0177 | Alborz | 2019 | Goat | Aborted fetus | 2 |
| 2 | 20Y0178 | Fars | 2020 | Sheep | Aborted fetus | 2 |
| 3 | 20Y0171 | Fars | 2020 | Sheep | Aborted fetus | 1 |
| 3 | 20Y0172 | Yazd | 2020 | Sheep | Aborted fetus | 1 |
| 4 | 20Y0150 | Tehran | 2017 | Cow | Milk | 1 |
| 4 | 20Y0154 | Fars | 2019 | Cow | Milk | 1 |
| 5 | 20Y0166 | Kermanshah | 2019 | Human | Blood | 1 |
| 5 | 20Y0167 | Alborz | 2019 | Human | Blood | 1 |
| 5 | 20Y0168 | Alborz | 2019 | Human | Blood | 1 |
| 5 | 20Y0169 | Kermanshah | 2019 | Human | Blood | 1 |
| 5 | 20Y0170 | Tehran | 2019 | Human | Blood | 1 |
| 6 | 20Y0162 | Tehran | 2019 | Human | Blood | 1 |
| 6 | 20Y0165 | Hamedan | 2019 | Human | Blood | 1 |
| 7 | 20Y0174 | Semnan | 2019 | Cow | Lymph node | 1 |
| 7 | 20Y0175 | Isfahan | 2019 | Cow | Lymph node | 1 |
| 8 | 20Y0159 | Alborz | 2019 | Human | Blood | 1 |
| 8 | 20Y0163 | Hamedan | 2019 | Human | Blood | 1 |
| 8 | 20Y0164 | Hamedan | 2019 | Human | Blood | 1 |
Fig. 4.

Maximum likelihood tree based on cgSNP alignment of Iranian B. melitensis strains and strains of similar geographic origin to the reference strain 16 M. Besides the strain names, also the year and country of isolation are given (Afgh - Afghanistan; Cyp - Cyprus; Egy - Egypt; Isr - Israel; Jor - Jordan; Kuw - Kuwait; Pak - Pakistan; SaAr - Saudi Arabia; Syr - Syria; Turk - Turkey; Turkm - Turkmenistan). For the Iranian strains, the location of isolation is printed in bold letters (Alb - Alborz; Fars; Ham - Hamedan; Hor - Hormozgan; Ila - Ilam; Isf - Isfahan; Kersh - Kermanshah; Qom; Sem - Semnan; Teh - Tehran; Yaz - Yazd; Zan - Zanjan). Grey lines and numbers indicate cgSNP groups as given in Table 3. The scale bar indicates the number of nucleotide changes per site.
Eight Iranian B. melitensis isolates that were isolated from animals (cows in Semnan and Isfahan provinces in 2019) and humans (Kerman, Tehran, Hamedan, Alborz, and Ilam provinces) were assigned to the IIb genotype of the East Mediterranean lineage. Among these, two clusters (No. 7 and 8; Table 2, Fig. 4) were highly similar strains. These two isolates were isolated from human (cluster No. 8) and cow samples (cluster No. 7) in 2019 but from different places in Iran. Cluster 8 displayed the lowest SNP differences of all analyzed Iranian isolates to foreign strains (38–46 SNPs), i.e., strains from Syria, Iran, Iraq, and Jordan. The other Iranian strains were separated by at least 56 SNPs from foreign strains.
For the majority of Iranian B. melitensis isolates (24 strains), the exact genotype could not be determined, as the strains clustered between genotypes IIe and IIf of the East Mediterranean lineage (Fig. 3, Fig. 4, Supplementary Fig. S1). Also, the similarity to foreign strains was comparably low: the highest homology was observed in a strain isolated in Afghanistan in 2015 (NIPH-Bru43). However, within this group of strains, five clusters of highly identical strains were identified that differed in at most two SNPs (clusters No. 2–6, Table 2, Fig. 4). Remarkably, no cluster was isolated exclusively from one place. Strains within the clusters mostly originated from places hundreds of kilometers apart, e.g. Fars and Alborz (cluster 2). However, isolates of the same SNP cluster were always either exclusively of animal or human origin.
3.4. Allele based typing methods
A total of four MLST genotypes were identified in all B. melitensis isolates using an in silico MLST analysis approach (Supplementary Table S1). Four B. melitensis isolates represented a novel sequence type (ST) that could not be assigned to existing groups. There was no apparent connection between sequence type and source or place of isolation. Of the 22 isolates of human origin, 19 (86.4%) were assigned to ST8, one was assigned to ST7 (4.5%), and two were assigned to the novel ST (9%) (Table 3). Among the seven isolates of cow origin, three (42.8%) belonged to ST8, two to ST7 (28.6%), and two were assigned to the novel ST (28.6%). Further, three out of five isolates of sheep origin (60%) belonged to ST8, while the others were each assigned to ST7 (20%) and ST71 (20%). One strain of camel origin belonged to ST7 and the other was assigned to ST8. The strain isolated from a goat was assigned to ST71. MLST ST1 and ST2 were only identified in the Iranian B. abortus strains, but not B. melitensis.
Table 3.
Affiliation of isolates to MLST sequence types (ST) according to the isolation source and Brucella spp.
| ST | Human | Sheep | Goat | Cow | Camel | Brucella spp. |
|---|---|---|---|---|---|---|
| 1 | 1 | – | – | – | – | B. abortus |
| 2 | – | – | – | 2 | – | B. abortus |
| 7 | 1 | 1 | – | 2 | 1 | B. melitensis |
| 8 | 19 | 3 | – | 3 | 1 | B. melitensis |
| 71 | – | 1 | 1 | – | – | B. melitensis |
| Novel | 2 | – | – | 2 | – | B. melitensis |
To obtain a higher level of differentiation and to compare the Iranian B. melitensis isolates with strains from neighboring and Middle East countries, a cgMLST analysis was conducted (Fig. 5). The resulting clusters of the Iranian strains were in accordance with the results of the SNP typing. The Iranian isolates that were assigned to the American SNP lineage before showed a high distance to the strains from Asia and were clearly distinct from the other Iranian isolates. In contrast, the cgMLST profiles of the 24 isolates that clustered in the IIe/IIf SNP group, exhibited high similarities to strains from Afghanistan, Syria, Kuwait, and Turkey. Likewise, the three strains of SNP cluster No. 8 (Table 2) clustered with strains from Syria, Iraq, and Turkey. However, in accordance with the SNP analysis, there was no apparent connection between the cgMLST sequence type and the place or time of strain isolation.
Fig. 5.

Minimum spanning tree based on cgMLST distances between Iranian B. melitensis strains and strains of similar geographic origin. The circles are colored according to the country of isolation and the size corresponds to the number of isolates. Numbers at the branches indicate allelic differences between the strains. For the Iranian isolates, abbreviations in the circles indicate the location of isolation (Alb -Alborz; Fars; Ham - Hamedan; Hor - Hormozgan; Ila - Ilam; Isf - Isfahan; Kersh - Kermanshah; Qom; Sem - Semnan; Teh - Tehran; Yaz - Yazd; Zan - Zanjan). For better visibility, not all numbers of allelic differences are displayed.
4. Discussion
Brucellosis is a neglected zoonotic disease in most developing countries despite its significant effects on livestock industries and public health [41,42]. Humans and animals can be infected by different Brucella species. However, B. melitensis is the most frequently observed causative agent and the most virulent species of brucellae in Iran [14,15]. Therefore, identifying the circulating Brucella species, genotypes, and biovars is crucial for monitoring transmission routes and tracing infection sources [40] to help control the disease. However, the PCR-based identification of species is only sufficient for detecting human/animal brucellosis or food contamination but not for tracing infection events [43]. Here, we characterized 37 B. melitensis and three B. abortus strains isolated from humans and various animal hosts over a period of five years in different locations in Iran using whole genome sequencing.
Our results confirm that both B. abortus and B. melitensis are causative agents of brucellosis in humans as reported for other countries as well [31,[44], [45], [46]]. Furthermore, isolation of B. melitensis from cattle and camels highlights the growing incidence of Brucella infection of species in non-preferred hosts, especially where host species mix freely with each other [47]. The two B. melitensis strains isolated from domestic camels indicate the importance of B. melitensis in camelid brucellosis in Iran. In other countries with camel husbandry, B. abortus and B. suis have also been reported in camels [48]. Previous investigations on circulating brucellae in Iran have identified B. melitensis biovars 1, 2, and 3 (predominantly 1) in sheep, cattle, goats, camels, dogs and humans, as well as B. abortus biovars 1, 2, 3 (predominantly 3) in cattle, humans and sheep [14,49,50]. Considering the predominance of B. melitensis biovar 1 in the present study, it can be assumed that this biovar is indeed now the dominating biovar of B. melitensis in Iran.
Whole genome sequencing-based typing methods provide the discriminatory power to differentiate closely related strains [23,51]. Using in silico analysis, three MLST types of B. melitensis were identified with the most common being ST8 (n = 26) isolated from a wide variety of host species. This substantiates the previous findings of molecular typing for Brucella spp. isolated from livestock and humans in Iran by MLST-9 analysis, which also showed that ST8 is one of the most prevalent sequence types for B. melitensis in Iran besides ST7 and ST102 [52]. Likewise, B. abortus ST1 and ST2 were identified in the present and other previous studies in isolates from humans and cattle [46,53,54] supposing that both are apparently the most prevalent STs of B. abortus.
Interestingly, B. melitensis strains that were assigned to different biovars (1 and 3) were clustered in the same MLST sequence type (ST8). That was also the case for B. abortus ST2 isolates, which were identified as biovar 2 and 3. This agrees with other findings that biovars especially those of B. melitensis do not correlate well with defined genetic entities [55] highlighting the importance of molecular analysis for rapid and accurate characterization of Brucella isolates [23,51]. WGS analysis provides also detailed genomic data on microbial taxonomy and enables in-depth comparative analysis especially genome wide SNP-based analysis can be used as an accurate genotyping approach for molecular epidemiological investigations [56]. The present study is the first extensive genetic diversity analysis of a larger panel of Brucella strains isolated in Iran based on WGS technology. It could be shown that all investigated Iranian B. melitensis strains belonged to the American (13.5%) and East Mediterranean (86.5%) lineages. In contrast, no members of the West Mediterranean and African lineages have been defined. It has been reported before that most of the Asian B. melitensis strains belong to genotype II (Mediterranean lineage) [57,58], while genotypes III (African lineage), IV (European lineage), and V (American lineage) have limited geographical distribution.
In the current study, five strains were assigned to the American B. melitensis lineage which is rarely reported in Asia except in Azerbaijan and China [59,60]. Here, these strains were isolated from cows, sheep, camels, and humans in different locations of Iran. In accordance with other studies, our findings also showed that the Mediterranean strains occupied the basal node of the phylogenetic tree indicating the possible origin of B. melitensis from Mediterranean countries [55]. Furthermore, SNP analysis of the B. abortus strains from Eghlid and Yazd indicated that these isolates were more similar to isolates from Iraq, while the B. abortus strain from Karaj showed higher similarity to isolates from Egypt which indicates the different origin of the strains.
Previous molecular study in Iran reported three MLST-9 genotypes for B. melitensis e.g. ST8, ST7, and ST102 and two for. B. abortus e.g. ST1 and ST2. [61]. In another study in Iran, three isolates of B. abortus and 51 isolates of B. melitensis were analyzed through the MLVA16 (multiple-locus variable number tandem repeat analysis based on 16 markers) and represented genotypes 46 and genotypes 22 for 80% of isolates [62].
The results of the present study did not show direct transmission of strains between animals and humans, as the most identical strains were exclusively always either of human or animal origin, i.e. none of the highly identical strains were present in both. Animal brucellosis cases from neighboring provinces, e.g. Fars and Yazd, might be connected to the usage of common pastures. As most of the human patients were farmers, the source of brucellosis can be assumed to be infected animals or even contaminated feed. Stray dogs and cats are proven to play a role in spreading brucellosis in dairy farms [63], how far pet animals and wildlife species plays a role in spreading brucellosis in Iran remains elusive. The discriminatory power of cgMLST makes it the most widely used pathogen sub-typing method [64]. Brucellosis is an endemic disease in Iran and several cases were reported every year in humans and animals, therefore, the relatively restricted number of isolates from human and animals available in the current study is the only limitation as it does not cover the full geographical areas of Iran. Moreover, Brucella isolates were collected via opportunistic sampling, but rather not were obtained via a prospective epidemiological study. Therefore, firm conclusions regarding the prevalence of zoonotic Brucella spp. in specific hosts, including different animal hosts and humans, cannot be drawn. However, this study despite its limitation provided a valuable update of epidemiological information on this endemic area. To our knowledge, this is the first study in Iran that used the NGS technology to trace back the genetic diversity of Brucella species in Iran.
5. Conclusions
Our results showed the close epidemiologically connection of Iranian B. melitensis strains with strains from Pakistan, Afghanistan, Turkey, and Syria based on cgMLST profile similarity. This finding reflects the frequent and uncontrolled livestock exchange between these countries in the past and still nowadays the transboundary movement of nomadic animals. Thus, the outbreak of animal and human brucellosis in Iran may be attributed to a lack of controlled movement of infected animals and livestock at Iran's border. This highlights the importance of improving the inspection and control approaches for livestock exported from endemic countries, including import only from herds guaranteed to be free or from free countries. Moreover, collaboration between animal and human health services, as well as wildlife authorities could improve the surveillance of disease and control brucellosis outbreaks through the one health approaches. This can be done by the effective collaboration of multiple disciplines and sectors working locally, nationally and globally to attain optimal health for people, animals and the environment. Furthermore, information and education works that should be undertaken to prevent the transmission of B. melitensis in the interface between human and livestock animals can be categorized by the five aspects such as building healthy public policy; creating supportive environments for developing a strong national and local professional veterinary service; strengthening community action for better collaboration between farmers and government; developing personal skills as well as reorienting health services. All in all, cgSNP analysis is a powerful tool for brucellosis tracking and intraspecies differentiation of closely related strains in Iran. Further investigation using a larger number of isolates might help to better understand the diversity of brucellae in Iran and the global distribution of lineages.
Funding
This work is based upon research funded by Iran National Science Foundation (INFS) under project No: 99030922.
CRediT authorship contribution statement
Maryam Dadar: Conceptualization, Methodology, Software. Hanka Brangsch: Conceptualization, Methodology, Software. Saeed Alamian: Visualization, Investigation. Heinrich Neubauer: Software, Validation, Writing – review & editing. Gamal Wareth: Data curation, Writing – original draft, Supervision.
Declaration of Competing Interest
None declared.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.onehlt.2023.100483.
Contributor Information
Maryam Dadar, Email: dadar.m77@gmail.com.
Gamal Wareth, Email: gamal.wareth@fli.de.
Appendix A. Supplementary data
Supplementary material
Data availability
Raw sequencing reads were registered in the ENA database under project No. PRJEB50179.
References
- 1.Wareth G., Dadar M., Ali H., Hamdy M.E.R., Al-Talhy A.M., Elkharsawi A.R., Tawab A., Neubauer H. The perspective of antibiotic therapeutic challenges of brucellosis in the Middle East and north African countries: current situation and therapeutic management. Transbound. Emerg. Dis. 2022;69(5):e1253–e1268. doi: 10.1111/tbed.14502. [DOI] [PubMed] [Google Scholar]
- 2.Leclercq S.O., Cloeckaert A., Zygmunt M.S. Taxonomic organization of the family Brucellaceae based on a phylogenomic approach. Front. Microbiol. 2020;10:3083. doi: 10.3389/fmicb.2019.03083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hördt A., López M.G., Meier-Kolthoff J.P., Schleuning M., Weinhold L.-M., Tindall B.J., Gronow S., Kyrpides N.C., Woyke T., Göker M. Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front. Microbiol. 2020;11:468. doi: 10.3389/fmicb.2020.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreno E., Blasco J.M., Letesson J.J., Gorvel J.P., Moriyón I. Pathogenicity and its implications in taxonomy: the Brucella and Ochrobactrum case. Pathogens. 2022;11(3) doi: 10.3390/pathogens11030377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dadar M., Shahali Y., Wareth G. Microbial Technology for the Welfare of Society. Springer; 2019. Molecular diagnosis of acute and chronic brucellosis in humans; pp. 223–245. [Google Scholar]
- 6.O’callaghan D. Human brucellosis: recent advances and future challenges. Infect. Dis. Poverty. 2020;9(1):1–2. doi: 10.1186/s40249-020-00715-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Preena P. Vol. 6. 2020. Cross-Species Transmission of Brucella abortus in an Aborted Sow, Mankind; p. 22. [Google Scholar]
- 8.Hegazy Y.M., Abdel-Hamid N.H., Eldehiey M., Oreiby A.F., Algabbary M.H., Hamdy M.E., Beleta E.I., Martínez I., Shahein M.A., García N. Trans-species transmission of Brucellae among ruminants hampering brucellosis control efforts in Egypt. J. Appl. Microbiol.132. 2021;1:90–100. doi: 10.1111/jam.15173. [DOI] [PubMed] [Google Scholar]
- 9.Wareth G., Melzer F., Tomaso H., Roesler U., Neubauer H. Detection of Brucella abortus DNA in aborted goats and sheep in Egypt by real-time PCR. BMC Res. Notes. 2015;8(1):1–5. doi: 10.1186/s13104-015-1173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dadar M., Shahali Y., Whatmore A.M. Human brucellosis caused by raw dairy products: a review on the occurrence, major risk factors and prevention. Int. J. Food Microbiol. 2019;292:39–47. doi: 10.1016/j.ijfoodmicro.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Corbel M.J. CRC Press; 2020. Microbiology of the Genus Brucella, Brucellosis: Clinical and Laboratory Aspects; pp. 53–72. [Google Scholar]
- 12.Wareth G., Abdeen A., Ali H., Bardenstein S., Blasco J.M., Cardoso R., Cvetnić C.D.S.M.Ž., de Massis F., El-Diasty M., Ekateriniadou L., Gürbilek S.E., Ferreira A.C., Garin-Bastuji B., Garofolo G., Hamdy M.E.R., Hellal J., Katsiolis J.A.A., Koleci X., Kornspan D., Krt B., Laušević D., Melzer F., Moustafa S., Njeru J., Ocepek M.S., Flavio S., Sakhria S., Šerić-Haračić S., Špičić S., Sprague L.D., Tittarelli M., Villari S., Neubauer H. 2019. Brucellosis in the Mediterranean Countries: History, Prevalence, Distribution, Current Situation and Attempts at Surveillance and Control. [Google Scholar]
- 13.Shirzadi M.R., Mohammadi P., Moradi G., Goodarzi E., Khazaei S., Moayed L., Khazaei Z. The incidence and geographical distribution of brucellosis in Iran using geographic information system and prediction of its incidence in 2021. J. Prev. Med. Hyg. 2021;62(3):E635–e634. doi: 10.15167/2421-4248/jpmh2021.62.3.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dadar M., Alamian S., Behrozikhah A.M., Yazdani F., Kalantari A., Etemadi A., Whatmore A.M. Veterinary Research Forum, Faculty of Veterinary Medicine. Urmia University; Urmia, Iran: 2019. Molecular identification of Brucella species and biovars associated with animal and human infection in Iran; p. 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zowghi E., Ebadi A., Yarahmadi M. Isolation and identification of Brucella organisms in Iran. Iran. J. Clin. Infect. Dis. 2008;3(4):185–188. [Google Scholar]
- 16.Dadar M., Wareth G., Neubauer H. Brucellosis in Iranian buffalo: prevalence and diagnostic methods. Ger. J. Vet. Res. 2021;1(2):13–16. [Google Scholar]
- 17.Foster J.T., Beckstrom-Sternberg S.M., Pearson T., Beckstrom-Sternberg J.S., Chain P.S., Roberto F.F., Hnath J., Brettin T., Keim P. Whole-genome-based phylogeny and divergence of the genus Brucella. J. Bacteriol. 2009;191(8):2864–2870. doi: 10.1128/JB.01581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gee J.E., De B.K., Levett P.N., Whitney A.M., Novak R.T., Popovic T. Use of 16S rRNA gene sequencing for rapid confirmatory identification of Brucella isolates. J. Clin. Microbiol. 2004;42(8):3649–3654. doi: 10.1128/JCM.42.8.3649-3654.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Georgi E., Walter M.C., Pfalzgraf M.-T., Northoff B.H., Holdt L.M., Scholz H.C., Zoeller L., Zange S., Antwerpen M.H. Whole genome sequencing of Brucella melitensis isolated from 57 patients in Germany reveals high diversity in strains from Middle East. PLoS One. 2017;12(4) doi: 10.1371/journal.pone.0175425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansen T.B., Scheffer L., Jensen V.K., Bohlin J., Feruglio S.L. Whole-genome sequencing and antimicrobial resistance in Brucella melitensis from a Norwegian perspective. Sci. Rep. 2018;8(1):1–9. doi: 10.1038/s41598-018-26906-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z.G., Cao X.A., Wang M., Piao D.R., Zhao H.Y., Cui B.Y., Jiang H., Li Z.J. Whole-genome sequencing of a Brucella melitensis strain (BMWS93) isolated from a bank clerk and exhibiting complete resistance to rifampin. Microbiol. Resour. Announc. 2019;8(33) doi: 10.1128/MRA.01645-18. e01645–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaeffer J., Revilla-Fernández S., Hofer E., Posch R., Stoeger A., Leth C., Schmoll F., Djordjevic V., Lakicevic B., Matovic K. Tracking the origin of Austrian human brucellosis cases using whole genome sequencing. Front. Med. 2021;8:187. doi: 10.3389/fmed.2021.635547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holzer K., El-Diasty M., Wareth G., Abdel-Hamid N.H., Hamdy M.E.R., Moustafa S.A., Linde J., Bartusch F., Sayour A.E., Elbauomy E.M., Elhadidy M., Melzer F., Beyer W. Tracking the distribution of Brucella abortus in Egypt based on Core genome SNP analysis and in silico MLVA-16. Microorganisms. 2021;9(9) doi: 10.3390/microorganisms9091942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al Dahouk S., Le Flèche P., Nöckler K., Jacques I., Grayon M., Scholz H.C., Tomaso H., Vergnaud G., Neubauer H. Evaluation of Brucella MLVA typing for human brucellosis. J. Microbiol. Methods. 2007;69(1):137–145. doi: 10.1016/j.mimet.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 25.Garofolo G., Di Giannatale E., De Massis F., Zilli K., Ancora M., Cammà C., Calistri P., Foster J.T. Investigating genetic diversity of Brucella abortus and Brucella melitensis in Italy with MLVA-16. Infect. Genet. Evol. 2013;19:59–70. doi: 10.1016/j.meegid.2013.06.021. [DOI] [PubMed] [Google Scholar]
- 26.Xiao P., Yang H., Di D., Piao D., Zhang Q., Hao R., Yao S., Zhao R., Zhang F., Tian G. Genotyping of human Brucella melitensis biovar 3 isolated from Shanxi Province in China by MLVA16 and HOOF. PLoS One. 2015;10(1) doi: 10.1371/journal.pone.0115932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wareth G., El-Diasty M., Melzer F., Schmoock G., Moustafa S.A., El-Beskawy M., Khater D.F., Hamdy M.E.R., Zaki H.M., Ferreira A.C., Ekateriniadou L.V., Boukouvala E., Abdel-Glil M.Y., Menshawy A.M.S., Sancho M.P., Sakhria S., Pletz M.W., Neubauer H. MLVA-16 genotyping of Brucella abortus and Brucella melitensis isolates from different animal species in Egypt: geographical relatedness and the Mediterranean lineage. Pathogens. 2020;9(6) doi: 10.3390/pathogens9060498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alton G., Jones L., Angus R., Verger J. Institute National de la Recherdie Agrononique; Paris: 1988. Techniques for the Brucellosis Laboratory. [Google Scholar]
- 29.Bricker B.J., Halling S.M. Differentiation of Brucella abortus bv. 1, 2, and 4, Brucella melitensis, Brucella ovis, and Brucella suis bv. 1 by PCR. J. Clin. Microbiol. 1994;32(11):2660–2666. doi: 10.1128/jcm.32.11.2660-2666.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Goñi I., Garcia-Yoldi D., Marin C., De Miguel M., Munoz P., Blasco J., Jacques I., Grayon M., Cloeckaert A., Ferreira A. Evaluation of a multiplex PCR assay (Bruce-ladder) for molecular typing of all Brucella species, including the vaccine strains. J. Clin. Microbiol. 2008;46(10):3484–3487. doi: 10.1128/JCM.00837-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wareth G., El-Diasty M., Abdel-Hamid N.H., Holzer K., Hamdy M.E.R., Moustafa S., Shahein M.A., Melzer F., Beyer W., Pletz M.W., Neubauer H. Molecular characterization and antimicrobial susceptibility testing of clinical and non-clinical Brucella melitensis and Brucella abortus isolates from Egypt. One Health. 2021;13 doi: 10.1016/j.onehlt.2021.100255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with kraken 2. Genome Biol. 2019;20(1):1–13. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurevich A., Saveliev V., Vyahhi N., Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 35.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seemann T. 2018. snp-dists; Convert a FASTA Alignment to SNP Distance Matrix. [Google Scholar]
- 37.Janowicz A., De Massis F., Ancora M., Cammà C., Patavino C., Battisti A., Prior K., Harmsen D., Scholz H., Zilli K. Core genome multilocus sequence typing and single nucleotide polymorphism analysis in the epidemiology of Brucella melitensis infections. J. Clin. Microbiol. 2018;56(9):e00517–e00518. doi: 10.1128/JCM.00517-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whatmore A.M., Shankster S.J., Perrett L.L., Murphy T.J., Brew S.D., Thirlwall R.E., Cutler S.J., MacMillan A.P. Identification and characterization of variable-number tandem-repeat markers for typing of Brucella spp. J. Clin. Microbiol. 2006;44(6):1982–1993. doi: 10.1128/JCM.02039-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karthik K., Anbazhagan S., Thomas P., Chitra M.A., Senthilkumar T.M.A., Sridhar R., Raj G.D. Genome sequencing and comparative genomics of Indian isolates of Brucella melitensis. Front. Microbiol. 2021;12 doi: 10.3389/fmicb.2021.698069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pisarenko S.V., Kovalev D.A., Volynkina A.S., Ponomarenko D.G., Rusanova D.V., Zharinova N.V., Khachaturova A.A., Tokareva L.E., Khvoynova I.G., Kulichenko A.N. Global evolution and phylogeography of Brucella melitensis strains. BMC Genomics. 2018;19(1):1–10. doi: 10.1186/s12864-018-4762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franc K., Krecek R., Häsler B., Arenas-Gamboa A. Brucellosis remains a neglected disease in the developing world: a call for interdisciplinary action. BMC Public Health. 2018;18(1):1–9. doi: 10.1186/s12889-017-5016-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaltungo B., Saidu S., Musa I., Baba A. Brucellosis: a neglected zoonosis. Microbiol. Res. J. Int. 2014:1551–1574. [Google Scholar]
- 43.De Santis R., Ciammaruconi A., Faggioni G., Fillo S., Gentile B., Di Giannatale E., Ancora M., Lista F. High throughput MLVA-16 typing for Brucella based on the microfluidics technology. BMC Microbiol. 2011;11(1):1–9. doi: 10.1186/1471-2180-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang H., Wang H., Xu L., Hu G., Ma J., Xiao P., Fan W., Di D., Tian G., Fan M. MLVA genotyping of Brucella melitensis and Brucella abortus isolates from different animal species and humans and identification of Brucella suis vaccine strain S2 from cattle in China. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0076332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan M.Z., Usman T., Sadique U., Qureshi M.S., Hassan M.F., Shahid M., Khan A. Molecular characterization of Brucella abortus and Brucella melitensis in cattle and humans at the north west of Pakistan. Pak. Vet. J. 2017;37:360–363. [Google Scholar]
- 46.Ma J.-Y., Wang H., Zhang X.-F., Xu L.-Q., Hu G.-Y., Jiang H., Zhao F., Zhao H.-Y., Piao D.-R., Qin Y.-M. MLVA and MLST typing of Brucella from Qinghai, China. Infect. Dis. Poverty. 2016;5(1):1–8. doi: 10.1186/s40249-016-0123-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hegazy Y.M., Abdel-Hamid N.H., Eldehiey M., Oreiby A.F., Algabbary M.H., Hamdy M.E., Beleta E.I., Martínez I., Shahein M.A., García N. Trans-species transmission of Brucellae among ruminants hampering brucellosis control efforts in Egypt. J. Appl. Microbiol. 2022;132(1):90–100. doi: 10.1111/jam.15173. [DOI] [PubMed] [Google Scholar]
- 48.Wernery U. Camelid brucellosis: a review. Rev. Sci. Tech. 2014;33(3):839–857. doi: 10.20506/rst.33.3.2322. [DOI] [PubMed] [Google Scholar]
- 49.Alamian S., Etemadi A., Samiee M.R., Dadar M. Isolation of Brucella abortus biovar 1 from human lumbar disc bulging: a case report of brucellar discitis. BMC Infect. Dis. 2021;21(1):1–5. doi: 10.1186/s12879-021-06538-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dadar M., Alamian S. 76(1) Archives of Razi Institute; 2021. Identification of Main Brucella Species Implicated in Ovine and Caprine Abortion Cases by Molecular and Classical Methods; p. 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whatmore A.M., Foster J.T. Emerging diversity and ongoing expansion of the genus Brucella. Infect. Genet. Evol. 2021;92 doi: 10.1016/j.meegid.2021.104865. [DOI] [PubMed] [Google Scholar]
- 52.Dadar M., Alamian S., Tadayon K., Ashford R.T., Whatmore A.M. Molecular characterization of zoonotic Brucella species isolated from animal and human samples in Iran. Acta Trop. 2022;229:106363. doi: 10.1016/j.actatropica.2022.106363. [DOI] [PubMed] [Google Scholar]
- 53.Khan A.U., Melzer F., Sayour A.E., Shell W.S., Linde J., Abdel-Glil M., El-Soally S.A., Elschner M.C., Sayour H.E., Ramadan E.S. Whole-genome sequencing for tracing the genetic diversity of Brucella abortus and Brucella melitensis isolated from livestock in Egypt. Pathogens. 2021;10(6):759. doi: 10.3390/pathogens10060759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sankarasubramanian J., Vishnu U.S., Gunasekaran P., Rajendhran J. Development and evaluation of a core genome multilocus sequence typing (cgMLST) scheme for Brucella spp. Infect. Genet. Evol. 2019;67:38–43. doi: 10.1016/j.meegid.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 55.Tan K.-K., Tan Y.-C., Chang L.-Y., Lee K.W., Nore S.S., Yee W.-Y., Mat Isa M.N., Jafar F.L., Hoh C.-C., AbuBakar S. Full genome SNP-based phylogenetic analysis reveals the origin and global spread of Brucella melitensis. BMC Genomics. 2015;16(1):1–11. doi: 10.1186/s12864-015-1294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holzer K., Wareth G., El-Diasty M., Abdel-Hamid N.H., Hamdy M.E., Moustafa S.A., Linde J., Bartusch F., Abdel-Glil M.Y., Sayour A.E. Transbound. Emerg; Dis: 2022. Tracking the Distribution, Genetic Diversity, and Lineage of Brucella melitensis Recovered from Humans and Animals in Egypt Based on Core-Genome SNP Analysis and In Silico MLVA-16. [DOI] [PubMed] [Google Scholar]
- 57.Zhu X., Zhao Z., Ma S., Guo Z., Wang M., Li Z., Liu Z. Brucella melitensis, a latent “travel bacterium,” continual spread and expansion from Northern to Southern China and its relationship to worldwide lineages. Emerg. Microbes Infect. 2020;9(1):1618–1627. doi: 10.1080/22221751.2020.1788995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daugaliyeva А., Sultanov A., Usserbayev B., Baramova S., Modesto P., Adambayeva A., Acutis P.L., Peletto S. Genotyping of Brucella melitensis and Brucella abortus strains in Kazakhstan using MLVA-15. Infect. Genet. Evol. 2018;58:135–144. doi: 10.1016/j.meegid.2017.12.022. [DOI] [PubMed] [Google Scholar]
- 59.Aliyev J., Alakbarova M., Garayusifova A., Omarov A., Aliyeva S., Fretin D., Godfroid J. Identification and molecular characterization of Brucella abortus and Brucella melitensis isolated from milk in cattle in Azerbaijan. BMC Vet. Res. 2022;18(1):1–9. doi: 10.1186/s12917-022-03155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao X., Li S., Li Z., Liu Z., Ma J., Lou Z., Zhou J., Liu Y., Jing Z., Fu B. Enzootic situation and molecular epidemiology of Brucella in livestock from 2011 to 2015 in Qingyang, China. Emerg. Microbes Infect. 2018;7(1):1–8. doi: 10.1038/s41426-018-0060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dadar M., Alamian S., Brangsch H., Elbadawy M., Elkharsawi A.R., Neubauer H., Wareth G. Determination of virulence-associated genes and antimicrobial resistance profiles in Brucella isolates recovered from humans and animals in Iran Using NGS technology. Pathogens. 2023;12(2023):82. doi: 10.3390/pathogens12010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirkalantari S., Masjedian F., Fateme A. Determination of investigation of the link between human and animal Brucella isolates in Iran using multiple-locus variable number tandem repeat method comprising 16 loci (MLVA-16) Braz. J. Infect. Dis. 2021;25 doi: 10.1016/j.bjid.2020.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wareth G., Melzer F., El-Diasty M., Schmoock G., Elbauomy E., Abdel-Hamid N., Sayour A., Neubauer H. Isolation of Brucella abortus from a dog and a cat confirms their biological role in re-emergence and dissemination of bovine brucellosis on dairy farms. Transbound. Emerg. Dis. 2017;64(5):e27–e30. doi: 10.1111/tbed.12535. [DOI] [PubMed] [Google Scholar]
- 64.Revez J., Espinosa L., Albiger B., Leitmeyer K.C., Struelens M.J., Points E.N.M.F., E. Group Survey on the use of whole-genome sequencing for infectious diseases surveillance: rapid expansion of European national capacities, 2015–2016. Front. Public Health. 2017;5:347. doi: 10.3389/fpubh.2017.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Data Availability Statement
Raw sequencing reads were registered in the ENA database under project No. PRJEB50179.