

Abstract
Antibacterial resistance is one of the greatest threats to human health. The development of new therapeutics against bacterial pathogens has slowed drastically since the approvals of the first antibiotics in the early and mid‐20th century. Most of the currently investigated drug leads are modifications of approved antibacterials, many of which are derived from natural products. In this review, we highlight the challenges, advancements and current standing of the clinical and preclinical antibacterial research pipeline. Additionally, we present novel strategies for rejuvenating the discovery process and advocate for renewed and enthusiastic investment in the antibacterial discovery pipeline.
Keywords: antibacterial pipeline, antibiotics, antimicrobial resistance, innovation, preclinical and clinical pipeline
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Pharmacology & Drug Discovery
Antibacterial resistance is one of the greatest threats to human health. This review highlights the challenges, advancements and current standing of the clinical and preclinical antibacterial research pipeline.

Introduction
For millennia, humans have used natural products to cure ailments and treat (superficial) infections with preparations from moulds, plants and soil (Harrison et al, 2015; Hutchings et al, 2019). During the past century, the discovery and introduction of various small‐molecule antibiotics has revolutionised health care and helped us to successfully treat life‐threatening bacterial infections. However, this progress is threatened by the rise of antimicrobial‐resistant pathogens as well as a dearth of new antibiotic classes available to clinicians. This review gives an overview of the current state of antibacterial‐compound research and development with a particular focus on antibiotics from natural sources.
A brief history of antibiotic discovery and development
Between 1890 and 1910, Emmerich and Löw, in a first modern clinical approach, used “pyocyanase,” an extract from Pseudomonas aeruginosa, to treat infectious diseases (Emmerich & Löw, 1899). The concept of small‐molecule antibiotics began with the development of the synthetic prodrug salvarsan by Paul Ehrlich (Ehrlich, 1913). Its origin, a dye that selectively stains bacterial cells, also inspired the development of prontosil by Gerhard Domagk (Otten, 1986). Prontosil was the first of the class of broad‐spectrum antibiotics called sulfonamides, which are still in use today.
The discovery of the natural product penicillin by Alexander Fleming in 1928, which was subsequently purified by Heatley and colleagues, accelerated the success story of antibiotics (Fleming, 1929; Abraham et al, 1941). The work of Selman Waksman, Albert Schatz and Elizabeth Bugie on soil‐dwelling Actinomycetales and their potential to produce antibiotic natural products was the start of the so‐called “golden age of antibiotics” from the 1940s to the 1960s (Waksman et al, 2010). Most antibiotic classes currently in use were discovered and developed during those decades.
Excluding the treatment of mycobacterial infections, 22 antibiotic classes are currently approved for systemic use by the Food and Drug Administration (FDA, USA) or the European Medicines Agency (EMA). Of these, four classes are derived from synthetic sources, 17 are from natural products, and one class, the nitroheterocycles, contains one synthetic compound and one natural product. The great impact of natural products as antibiotic scaffolds is also highlighted by the fact that more than three quarters of approved antibiotics (79%) on the market in the United States, are either natural products or derived from such compounds (Fig 1; Appendix Table S1; Werth, 2022).
Figure 1. Systemic antibiotics currently approved and marketed in the United States without antitubercular treatment.

Clock‐wise from the top: natural product‐derived antibiotics (79%): cephalosporins (24), penicillins (14), aminoglycosides (8), tetracyclins (5), glycopeptides (4), rifamycins (3), macrolides (3), carbapenems (3), polypeptides (2), amphenicol (1), daptomycin (1), fosfomycin (1)*, lincosamid (1), monobactams (1), nitroheterocycles** (1), pleuromutilins (1), streptogramins (1); antibiotics with synthetic origin (21%): sulfonamides (9), fluoroquinolones (7), oxazolidinones (2), aminopyrimidine (1), nitroheterocycles** (1). *not yet approved in the United States (Werth, 2022), **The nitroheterocyle metronidazole is derived from a natural product whereas nitroheterocycle nitrofurantoin is of synthetic origin. Source data are in Appendix Table S1.Source data are available online for this figure.
The exceptional significance of natural products as antibiotics is obvious for many reasons. To increase their fitness in their respective habitats, microbes produce antimicrobial molecules to better compete with other organisms for scarce nutrients. Owing to the co‐evolution of such organisms over millions of years, those molecules continue to be optimised in terms of structural diversity, ability to penetrate cell walls, cellular activity and selectivity for bacteria, fungi or eukaryotes (Wright, 2017; Lakemeyer et al, 2018; Hutchings et al, 2019). Obviously, this process is still ongoing, which means there is a continuous arms race in Nature between producing novel antibiotics and developing resistance mechanisms for (self)‐defence (Hegemann et al, published concurrently).
While small‐molecule antibiotics as therapeutics paved the way for modern medicine and gave mankind one of the greatest medical breakthroughs of the 20th century (Katz & Baltz, 2016; Wright, 2017; Hutchings et al, 2019), the rising impact of antimicrobial resistance (AMR) now threatens to set us back to the pre‐antibiotic era (Ventola, 2015; Lewis, 2020; Murray et al, 2022).
The impact of antimicrobial‐resistant strains
AMR is not a phenomenon of the present era. In fact, penicillin‐resistant strains were already identified 3 years before penicillin gained market approval (Fig 2A and D; Abraham & Chain, 1940). We know that resistance genes belong to the natural genetic pool of bacteria and can even be found in bacteria which were conserved in permafrost for more than 30,000 years (D'Costa et al, 2011). Considering that natural producers of antibiotics often carry resistance genes to the antibiotics they manufacture as a self‐protection mechanism and that antibiotics are produced by many ubiquitous strains, it is very likely that the development of AMR has been a continuous process ever since bacteria evolved antibiotics (Peterson & Kaur, 2018).
Figure 2. Systemically administered traditional antibiotics in clinical use and clinical trials excluding antitubercular agents.

(A) Cellular targets of traditional antibiotics in clinical use and the clinical pipeline and their resistance mechanisms; (B) Modes of action (MoA) of established and novel traditional antibiotics; (C) Modes of resistance (MoR) of traditional antibiotics; (D) Timeline of the introduction of antibiotics (blue) and examples of resistance identification (red): golden age of antibiotics (blue arrows), introduction of the WHO Pathogen Priority List in 2017 (bold blue; World Health Organization, 2017), natural products (bold), synthetic derived antibiotics (italic), agents not in clinical use anymore (grey); 1. Cell wall synthesis: β‐lactams (penicillins, cephalosporins, carbapenems, monobactams), glycopeptides, phosphonates; 2. Protein synthesis: tetracyclins, aminoglycosides, macrolides, lincosamides, streptogramins, oxazolidinones, amphenicols, pleuromutilins, fusidic acid*; 3. DNA/ RNA synthesis and replication: quinolones, nitroheterocycles (nitroimidazoles, nitrofurans), ansamycins; 4. Folic acid synthesis: sulfonamides, diaminopyrimidines; 5. Cell membrane synthesis and integrity: polymyxins, lipopeptides; 6. Cell division: benzamide; 7. Fatty acid synthesis: afabicin; *approval by European Medicines Agency (EMA).
However, the increasing and partially inappropriate use of antibiotics has increased selection pressure on bacteria and given rise to pathogenic strains that are resistant against multiple (MDR), most (XDR), or all (PDR) antibiotics available for clinical use (Cook & Wright, 2022). The global spread of these so‐called “superbugs” can trigger a pandemic as shown by the New Delhi metallo‐β‐lactamase (NDM‐l)‐producing Enterobacteriaceae. These pathogens were first isolated from a Swedish patient treated in an Indian hospital, who was colonised with XDR Klebsiella pneumoniae and Escherichia coli (Kumarasamy et al, 2010). Since then, numerous cases of infections due to NDM‐l‐producing Enterobacteriaceae have been reported all over the world (Rolain et al, 2010).
Depending on the antibiotic class, survival of the bacteria depends on different modes of resistance (MoR) to circumvent the bacteriostatic or bactericidal effect of the drug (Fig 2A and C). These mechanisms include class‐specific ones such as inactivation of antibiotics (e.g. β‐lactamases for β‐lactam antibiotics), modification or mutation of target sites (e.g. replacing the d‐Ala‐d‐Ala motif with d‐Ala‐d‐Lac or d‐Ala‐d‐Ser for glycopeptides, single‐point mutations in the target site‐encoding region of genes of ribosomal proteins for oxazolidinones), or overproduction of target enzymes (e.g. overproduction of DHPS for sulfonamides; Munita & Arias, 2016). Mechanisms for reducing the amount of antibiotic inside the bacterium, often via active export using efflux pumps or through decreasing antibiotic uptake via reduction of the cell wall permeability, exist for several antibiotic classes.
In addition to the increasing number of MoR in pathogenic bacteria, the limited number of cellular targets and modes of action of approved antibiotics play a major role in the current antibiotic crisis (Fig 2A and D).
The 22 antibiotic classes currently approved for systemic treatment of bacterial infections, excluding the treatment of tuberculosis, have five general mechanisms to kill bacteria or stop their growth: interfering with 1. cell wall synthesis, 2. protein biosynthesis, 3. DNA synthesis, 4. folic acid metabolism or 5. cell membrane synthesis and integrity (Fig 2A and B). The overall mechanisms of action seem to be related to inhibiting the cellular machineries involved in the formation of complex biological structures such as DNA, RNA, proteins and the cell wall.
In 2019, 4.95 million deaths globally were associated with bacterial AMR, 1.27 million deaths of which were directly attributed to it (Murray et al, 2022). If this trend continues without new drugs available to treat bacterial infections, it is estimated that 10 million people will die from AMR annually by 2050 (O'Neil, 2014).
In the first section of this review, we discuss the causes of the so‐called innovation gap in antibiotic discovery as well as different approaches to potentially close this gap. The second part gives an overview of the current antibacterial clinical pipeline and preclinical developments.
As the focus of this review is on antibiotics for systemic treatment, we will not cover the drug discovery pipeline for local Clostridium difficile and Helicobacter pylori infections. An overview of non‐systemic treatment options and prophylactic therapy can be found in a recent review by Cook & Wright, 2022. Furthermore, we will not discuss the treatment of infections with Mycobacteria, as they pose different challenges for antibiotic discovery and development. Several recent reviews have summarised the current antitubercular pipeline as well as strategies for overcoming the resistance crisis in the treatment of tuberculosis (He et al, 2020; Shetye et al, 2020; Black & Buchwald, 2021; Dartois & Rubin, 2022).
The need for new antibiotics
The innovation gap
The golden age of antibiotics development yielded 16 new antibiotic classes in fewer than 30 years. During the following five decades, however, only six new antibiotic classes were introduced on the market (Fig 2D). The lack of innovation is alarming, especially regarding direct‐acting antibacterials against critical Gram‐negative bacteria, given that no new class targeting those bacteria has been approved since the introduction of the monobactam aztreonam in 1986 (Hutchings et al, 2019). However, two novel classes of β‐lactamase inhibitors (BLIs) were approved during the past decade in different fixed drug combinations with β‐lactams, reinforcing treatment options regarding critical priority pathogens. The three antibiotic classes approved within the last two decades exhibit activity mostly against Gram‐positive bacteria. Furthermore, the oxazolidinones (linezolid, approved 2000) and the lipopeptide daptomycin (approved 2003) were discovered in the 1970s and 1980s, respectively (Fugitt & Luckenbaugh, 1978; Allen et al, 1987; Slee et al, 1987), highlighting a long time gap between discovery and approval. The pleuromutilins were already discovered in 1951 and even approved for topical application in humans and systemic use in animals before the introduction of lefamulin for systemic use in humans in 2019 (Kavanagh et al, 1951).
The driving force of antibiotic discovery from natural sources during the golden age was the so‐called Waksman platform, an agar‐overlay assay to screen for (mostly) antibiotic activity. Following the marketing of streptomycin by Merck in 1946, the pharmaceutical industry started screening strain collections of Actinomycetes, focusing on the promising genus Streptomyces (Katz & Baltz, 2016). This approach yielded the majority of natural product‐derived antibiotic classes. However, (over‐)mining of Actinomycetes, a phylogenetically limited phylum of antibiotic‐producing bacteria, led to an increasing number of compound rediscoveries (Lewis, 2017; Wright, 2017; Hutchings et al, 2019; Ribeiro da Cunha et al, 2019). At around this time, major progress was also made with the synthetic (fluoro)quinolones, starting with the discovery, development and authorisation of nalidixic acid in 1962 followed by four generations of (fluoro)quinolone drugs (Lesher et al, 1962; Oliphant & Green, 2002). When these were approved in the 1980s, it became clear that easy chemical access, superior tissue penetration and broad spectrum of activity of these unique drugs gave them advantages over many other marketed antibiotics (Oliphant & Green, 2002).
With decreasing rates of discovery of novel antibiotic classes, (semi‐)synthetic development of established compound classes became a main source for new antibiotics. Chemical modifications of antibiotics increased their stability, bioavailability and tolerability as well as their spectrum of target bacteria. Furthermore, newly developed derivatives overcame the resistance some bacteria had developed against older generations of antibiotics. However, as all generations of an antibiotic class share the same target and binding site, cross‐resistances quickly emerged (Ribeiro da Cunha et al, 2019; Hobson et al, 2021).
The sequencing of the full genome of Haemophilus influenzae in 1995 and the subsequent rapid expansion of bacterial genome sequencing, rekindled investments into the antibacterial pipeline by pharmaceutical companies, now using target‐based approaches. For example, GlaxoSmithKline (GSK) identified more than 160 novel essential bacterial targets (Payne et al, 2007). In 70 high‐throughput screening (HTS) campaigns, they tested more than half a million compounds from their synthetic chemical library against these targets. Other companies tried similar approaches (Tommasi et al, 2015), yet, all of them failed to produce a single candidate drug for clinical testing. The main reason for the high attrition rate of HTS was attributed to the inefficient uptake of hit compounds into the bacterial cells, especially in case of Gram‐negative pathogens, and therefore insufficient whole‐cell activity. Other compounds showed non‐specific toxicity to both bacterial and human cells, or failed to show the desired broad‐spectrum antibacterial activity (Payne et al, 2007).
The stringent requirements for a safe and potent antibiotic combined with the high attrition rate of HTS campaigns might explain the resulting loss of interest by pharmaceutical companies to pursue research in this field (Fig 3A–C). The main obstacles are as follows:
Novel antibacterials are technically challenging to discover and optimise for systemic therapy in humans. Most synthetic chemical libraries are designed around concepts of lead‐ and drug‐likeness (Lipinski et al, 2001) with physicochemical properties predicted to afford oral bioavailability and decent hit rates for human targets. However, penetrating the cell barrier of bacterial, especially Gram‐negative cells, requires different physicochemical properties. We and others therefore continue to advocate natural products as starting points for new drug leads (Miethke et al, 2021). Once promising molecules are validated hits, researchers are confronted with project requirements such as bacterial spectrum‐coverage, mechanism‐ and frequency of resistance formation in vitro and ADMET (absorption, distribution, metabolism, excretion, and toxicity) requirements for in vivo experiments. Once an in vivo proof‐of‐concept is established, typically in a rodent infection model, optimisation of pharmacokinetic properties and development of phamacokinetic and pharmacodynamic (PK/PD) indices of efficacy must be accomplished. On the positive side, mouse models of infections are generally considered predictive of human disease (Byrne et al, 2020).
Novel and resistance‐breaking antibacterials for key nosocomial infections are difficult to develop. Renal and hepatic toxicity may limit clinical dosing (Lewis, 2013). Clinical trial feasibility beyond the key Gram‐negative and Gram‐positive indications of complicated urinary tract infections (cUTI) and acute bacterial skin and skin structure infections (ABSSSIs), respectively, can be challenging. Especially the limited availability of patients infected with AMR pathogens may increase cost and limit progress. Development is difficult for certain body sites and indications‐for‐use, such as hospital‐acquired pneumonia (HAP). Specific clinical trial designs and endpoints of efficacy are laid out in guidance documents issued by regulatory bodies (Bax & Green, 2015; Theuretzbacher et al, 2020a). Recruitment of patients into clinical trials for an empirical therapy may be complicated and entirely hinge on the availability of rapid diagnostic tests (Fig 3A and B; Box 1).
Novel and resistance‐breaking antibacterials are difficult to market. They may be strictly reserved for treating patients with limited treatment options. This is good antibiotic stewardship but incompatible with a for‐profit business model relying on high volumes of sales. Current legislation therefore tries to delink antibiotic consumption from remuneration.
Figure 3. The path of a broad‐spectrum antibiotic to approval.
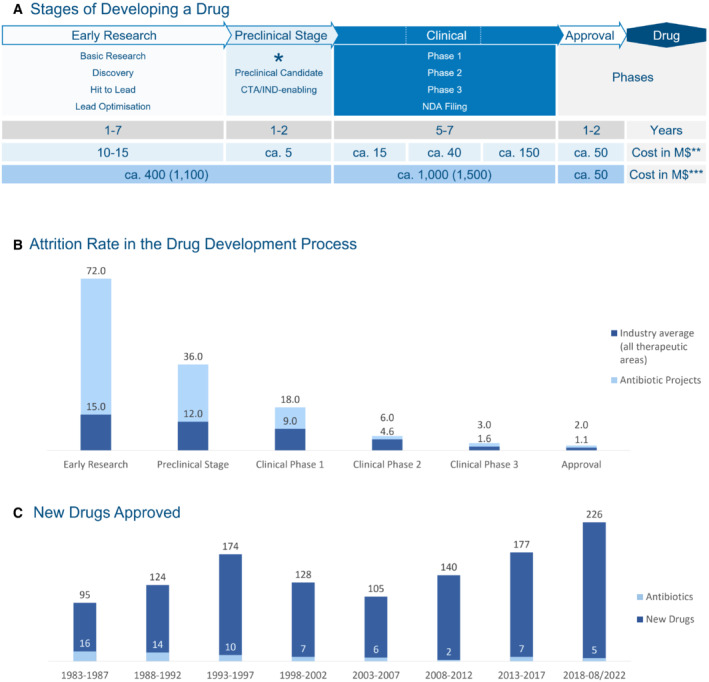
(A) Stages of developing a drug, low profitability leads to market gap, CTA/IND: clinical trial application/ investigational new drug, M$: Million US Dollar (2010), the time it takes to develop a successful drug in years—in grey, *preclinical development starts with Good Laboratory and Good Manufacturing Practises (GLP and GMP), **mean cost per successful project in 2010 (Paul et al, 2010) —in light blue—and ***mean out of pocket cost (mean capitalised cost)—in darker blue—of developing a drug in 2010 including attrition rate (Rex, 2014; DiMasi et al, 2016; Miethke et al, 2021); (B) Attrition rate of antibiotic projects vs. other drugs in the drug development process (Rex, 2014) which increases the costs shown in (A) in case of developing antibiotics; in addition to scientific factors high development costs have an impact on (C) number of antibiotics versus other new drugs approved between 1983 and August 2020 (U.S. Department of Health and Human Services, n.d.; FDA, 2015; FDA, 2021; FDA, 2022).Source data are available online for this figure.
Box 1. In need for answers.
How much are we willing to pay to prevent a pandemic of resistant bacteria? To combat AMR, we need to act fast—which depends on sufficient funding throughout the whole pipeline.
How (and how soon) can we develop rapid and highly specific diagnostics? Can they be used in a wide range of settings (clinical/communal, high‐/low‐income countries)? Diagnostics are a fundamental requirement for selectively acting antibiotics and non‐traditional antibacterials.
Can non‐traditional antibacterials pass clinical trials and advance antibacterial treatment options?
Will we be able to apply scientific accomplishments and lessons learned in a collaborative way to build a new platform?
Owing to these challenges, development and marketing of antibiotics pose great financial risks. Consequently, the bulk of the innovation in the field comes from small‐ and medium‐sized enterprises with limited financial resources; this has led to a situation where the clinical trial pipeline is considered insufficient with respect to clinical need (Fig 3C; Food and Drug Administration, 2017; Theuretzbacher et al, 2020b; Miethke et al, 2021; European Medicines Agency, 2022).
Approaches addressing the innovation gap
Regulation and funding
In publishing the global Pathogen Priority List (PPL) in 2017, the World Health Organisation (WHO) officially recognised AMR, especially among Gram‐negative pathogens, as a global health problem (Fig 4; World Health Organization, 2017). Earlier work by the Infectious Disease Society of America (IDSA) coined the term ESKAPE to highlight unmet medical needs in the therapy of infections due to Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumonia, Acinetobacter baumanni, Pseudomonas aeruginosa and Enterobacter spp. (Boucher et al, 2009). The global PPL and previous ESKAPE rankings aim to prioritise research and development, funding and incentives to fight antibiotic‐resistant bacteria.
Figure 4. WHO prioritisation of pathogens to guide research and development of new antibiotics.

(World Health Organization, 2017); ESKAPE pathogens play a critical role in nosocomial infections with critical resistance development and limited treatment options (red box; Rice, 2008; Boucher et al, 2009). The category “critical priority” includes Gram‐negative ESKAPE pathogens; the category “high priority” includes Gram‐positive ESKAPE and high community‐burden Gram‐negative pathogens, which have limited treatment options and critical resistance development; the category “medium priority” includes community‐relevant Gram‐negative and Gram‐positive pathogens with limited treatment options.
Since then, new economic models were developed to better support the different stages of product development (Miethke et al, 2021). Firstly, several national market entry rewards encourage industry to take the financial risk of development and application for authorisation of new antibiotics (e.g. UK [Mahase, 2020], US [116th Congress, 2019], and Sweden [The Public Health Agency of Sweden, 2017]).
Furthermore, competitive investment funds such as the AMR Action Fund can actively support cost‐intensive (later) phases of clinical trials that are mostly conducted by industry (IFPMA, 2020). Preclinical and translational stages can be funded by initiatives like public‐private partnerships (PPPs; e.g. IMI [Kostyanev et al, 2016], GARDP [Balasegaram & Piddock, 2020], CARB‐X [Alm & Gallant, 2020] or REPAIR [Engel, 2020]) to advance innovations to the clinical pipeline.
Regrettably, early research stages—which produce a hit, progress into hit‐to‐lead‐development, and perform in vitro and in vivo profiling as well as first animal models—are mostly conducted by academia or at small research institutes and are still chronically under‐funded (Fig 3A; Zuegg et al, 2020). New incentives and additional funding for antibacterial research in the academic sector are necessary (Box 1).
The scientific toolbox is sufficient to replenish and sustain the antibiotic pipeline, if implemented effectively and in a coordinated manner (Miethke et al, 2021). Currently, it is up to organisation and proper financial support whether or not innovation will prevail (Box 1).
Improved screening methods to find new antibiotics
After the failure of target‐based HTS campaigns to restock the antibacterial pipeline, phenotypic assays remain a mainstay to identify novel antibiotic lead structures. Moreover, phenotypic assay technologies have been refined to be of higher predictive value for later stages of preclinical and clinical development.
Direct screening of (resistant) pathogens ensures that potential hits are active against clinically relevant bacteria (Wong et al, 2012), as shown for screens against K. pneumoniae (Smith & Kirby, 2016) or Vibrio cholerae (Peach et al, 2011). Targeted whole‐cell screenings can be used to screen for antibiotics with new or unexploited modes of action (Hutter et al, 2004; Habich & von Nussbaum, 2006).
An issue of the traditional in vitro antibiotic susceptibility tests (ASTs) is that they do not take into account the in vivo environment during an infection. Changes in the availability of nutrients or oxygen, host metabolism and the presence of other microorganisms at the site of infection can greatly affect the in vivo bioactivity (Lakemeyer et al, 2018). Modified ASTs mimicking host conditions, for example, in minimal media (Zlitni et al, 2013) with physiological concentrations of NaHCO3 (Farha et al, 2018) or in artificial urine (Hennessen et al, 2020) altered minimal inhibitory concentrations (MICs), and improved prediction accuracy in mouse infection models (Ersoy et al, 2017). Actual pathogen–host interactions to find new potent antibiotics are used in high‐throughput infection screening (Clatworthy et al, 2018), employing, for example, MRSA infection models in the nematode Caenorhabditis elegans (Kim et al, 2018).
Technical advances further enable bacterial cytological profiling to find new antibiotics. Observation of various cellular characteristics with high‐resolution imaging provides information about how known antibiotics and screened compounds affect bacteria and, therefore, allows the rapid identification of their modes of action (Nonejuie et al, 2013).
Furthermore, screening for molecules that overcome resistance mechanisms (King et al, 2014) or inhibit biofilm formation to sensitise bacteria to antibiotic treatment (Peach et al, 2011) can support the fight against AMR. Moreover, machine‐learning approaches can help to identify effects of established antibiotics that go beyond their primary modes of actions and could thus point researchers to other promising targets for future antibiotics (Yang et al, 2019).
Optimising the physicochemical property space
The shortcomings of target‐based HTS demonstrated that compounds require special properties to penetrate the cell membrane and accumulate in bacteria (Payne et al, 2007). Especially the additional outer membrane of Gram‐negative bacteria, in combination with many efflux pumps proved to be a major hurdle for hit compounds to display whole‐cell activity (Nikaido & Pagès, 2012; Masi et al, 2017).
By studying the physicochemical properties of antibiotics compared with general drugs against human targets, O'Shea and Moser identified chemical property trends that differentiate between both drug categories. For example, antibiotics on average show a higher molecular mass, a higher polar surface area, and a higher number of hydrogen bond acceptors and ‐donors than general drugs. Physicochemical properties of anti‐Gram‐negative drugs also differ from those of anti‐Gram‐positive drugs (O'Shea & Moser, 2008), as can be expected given the different cell envelopes. Implementing these physicochemical property‐activity correlations has the potential to lead to new rules analogous to Lipinski's rules. This information will help to prioritise chemical libraries and influence the synthesis of pharmacophores to pass the bacterial cell envelope and avoid efflux (Masi et al, 2017).
Based on the analysis of 180 compounds that accumulated in E. coli, the Hergenrother group was able to correlate anti‐Gram‐negative compounds to the physicochemical properties that were facilitating said accumulation. They synthetically implemented a set of rules, named eNTRy rules, to convert anti‐Gram‐positive‐only compounds into broad‐spectrum antibiotics. For example, the anti‐Gram‐positive antibiotic Debio‐1452, a FabI inhibitor and derivative of afabicin (phase II), was a good model to demonstrate the application of the eNTRy rules. It already had good activity against permeability‐defective Gram‐negative strains and showed favourable globularity as well as rigidity in accordance with the eNTRy rules. Further SAR studies including X‐ray structures of the target‐compound complex were already available for this compound. Based on this information, compound accumulation in Gram‐negative bacteria was optimised without disturbing target engagement. This feat was accomplished by implementing the last missing rule, having a primary amine present, which yielded Debio‐1452‐NH3 (Muñoz & Hergenrother, 2021). Although this example is highly promising, some word of caution regarding the generalisability of eNTRy rules is necessary. It is currently impossible to estimate the applicability to other chemical programs as negative results typically do not get published; at least one recent publication describes the failure of this approach (Ropponen et al, 2021).
Unlike compounds from chemical libraries, potent natural products often already cover the physicochemical property space to achieve antibacterial activity (Grabowski & Schneider, 2007; Newman et al, 2015; Wright, 2017), making them a great starting point for identifying novel antibiotics. Although these compounds are evolutionarily optimised to achieve such potential, it should be mentioned that they frequently lack the ADME profiles required for antibiotic development and as such require improvement via synthetic modifications.
Improved approaches to find new antibacterial natural products
The increasing rediscovery rates of known antibiotics led to the assumption that it was nearly impossible to find new antibiotics from microorganisms. However, this assumption was based on three shortcomings that natural‐product discovery faced in the 20th century.
First, most screening efforts were built around Actinomycetes, largely ignoring the biosynthetic and antibiotic potential of other microorganisms. Second, the hidden or cryptic biosynthetic potential of microorganisms (including Actinomycetes) was not known. This only changed when complete genome sequences of organisms became available. For example, the genome sequence of the well‐studied bacterium Streptomyces coelicolor revealed that the number of biosynthetic gene clusters (BGCs) by far exceeded the number of natural products previously connected to the strain (Bentley et al, 2002). In routine cultivation settings, most microorganisms produce natural products that account only for fewer than 10% of their BGCs (Katz & Baltz, 2016). Third, with insufficient analytical methods and computational resources it was impossible to quickly de‐replicate extracts of newly cultivated microorganisms for known natural products. Without this knowledge, it was difficult to directly de‐prioritise extracts featuring known antibiotics or to estimate whether those extracts contained additional antibiotics.
Next to Actinobacteria, many well‐studied classes, including Myxobacteria, Cyanobacteria, Pseudomonas, Burkholderia, insect pathogenic bacteria and Firmicutes, have shown potential to produce a wide range of bioactive natural products (Wright, 2017; van Santen et al, 2022). Following the observation that chemical diversity correlates with taxonomic distance (Hoffmann et al, 2018), there is hope to find new antibiotics in understudied or even new bacterial taxa. Considering that to date 99.9% of bacterial taxa remain uncultivated (Locey & Lennon, 2016), it is reasonable to assume that at least a subset of them bears the potential to produce interesting and novel kinds of natural products (Crits‐Christoph et al, 2018). Recent comparisons between publicly available bacterial genomes and published bacterial natural products led to the estimation that about 97% of bacterial secondary metabolites have not been characterised yet (Gavriilidou et al, 2022).
In addition to isolating new bacteria from soil or marine samples, the investigation of animal and human microbiomes shows promise (Hegemann et al, 2022). The antibiotics lugdunin (Zipperer et al, 2016), lactocillin (Donia et al, 2014) and cutimycin (Claesen et al, 2020), isolated from human commensal bacteria, show activity against Gram‐positive bacteria. The antibiotics odilorhabdin (Pantel et al, 2018) and darobactin (Imai et al, 2019) are interesting because of their activity against Gram‐negative bacteria; both were isolated from nematode‐associated bacteria.
Techniques such as microfluidics (Mahler et al, 2021) or the “isolation chip” (iChip; Nichols et al, 2010) enable high‐throughput isolation of microorganisms, only limited by their ability to grow in the chosen environment. Due to the primary in situ cultivation in the environment the original sample was taken from, the iChip greatly improves the number and diversity of isolated strains, compared with conventional Petri dish‐based strain‐isolation procedures (Nichols et al, 2010). Screening of iChip isolates for antibacterial activity resulted in the discovery of the antibiotic teixobactin (Ling et al, 2015). Nevertheless, better knowledge of the microbiology of known, but understudied producers of natural products can lead to a constant increase of accessible biodiversity, as demonstrated for Myxobacteria (Garcia et al, 2009; Garcia et al, 2010). By way of example, bioactivity screening of new or little studied myxobacterial genera resulted in the discovery of the antibiotic cystobactamid (Herrmann et al, 2016). According to a recent metagenomic analysis, the majority of myxobacterial genera has not been cultivated so far (Petters et al, 2021), highlighting the potential for future discoveries from this class.
Methods to access the cryptic biosynthetic potential of microorganisms can be divided into cultivation‐based and molecular biological approaches.
Considering that their natural habitats are crowded with millions of different (micro‐) organisms, co‐cultivation of two or more microorganisms is probably the most intuitive approach. A variety of co‐cultivation settings has been successful during the past decades (Bertrand et al, 2014) and still leads to the discovery of new natural products with antibiotic properties which are not produced in pure culture (Adnani et al, 2017; Pishchany et al, 2018).
Further successful cultivation approaches to trigger the biosynthesis of cryptic natural products include changes in media composition, inducing stress in producing microorganisms by changes in temperature, pH or aeration, and addition of molecules, called elicitors, with known or unknown impact on the producing strain (Scherlach & Hertweck, 2009; Yoon & Nodwell, 2014). Within the past decade, high‐throughput elicitor screening of small‐molecule libraries has demonstrated that small molecules can trigger the biosynthesis of cryptic natural products in Burkholderia (Seyedsayamdost, 2014), Streptomyces spp. (Craney et al, 2012) and other Actinomycetes (Moon et al, 2019).
The most frequently used molecular biological approach is genome mining by searching DNA sequences for potential homologues of known biosynthetic genes to identify potential BGCs. Useful bioinformatic tools like the community‐curated repository MIBiG (Kautsar et al, 2020), prediction software like antiSMASH (Blin et al, 2021) or PRISM (Skinnider et al, 2020) that give information about the location, type and even product of a BGC, or tools to find antibiotic resistance genes like ARTS (Mungan et al, 2020) support the search for BGCs. BGCs thus identified—and potentially cryptic—are then activated in the native producer or heterologously expressed in a different host (Rutledge & Challis, 2015; Zhang & Hindra, 2019). Prioritisation of BGCs can, for example, be based on the underlying biosynthetic pathway (Hug et al, 2019) or, especially in the case of antibiotic discovery, on the presence of self‐resistance genes within or nearby the cluster (Johnston et al, 2016; Alanjary et al, 2017; Panter et al, 2018).
To improve the production of compounds from heterologously expressed BGCs and to simplify the subsequent purification, specialised host strains were created (Myronovskyi et al, 2018; Ahmed et al, 2020). One major drawback in heterologous expression is the need to find suitable and genetically tractable hosts that ideally are closely related to the native producers. Application of transposition‐based approaches (Fu et al, 2008) or the chassis‐independent recombinase‐assisted genome engineering (CRAGE) method (Wang et al, 2019) enable integration of BGCs into different heterologous hosts. The benefits of these approaches include improved production titres, depending on the integration position (Bilyk et al, 2017; Pogorevc et al, 2019) and an increased diversity of derivatives from an individual BGC, depending on different heterologous hosts (Wang et al, 2019).
Using heterologous expression even allows in some cases to screen for new natural products without the need to cultivate the original producing bacteria. Indeed, prospecting metagenomic datasets or environmental DNA (eDNA) sequences for BGCs in combination with subsequent heterologous expression of these BGCs led to the discovery of new natural products with antibiotic activities (Kallifidas et al, 2012; Katz et al, 2016; Hover et al, 2018).
Furthermore, CRISPR/Cas‐based genome engineering (Tong et al, 2019), gene or BGC synthesis (Groß et al, 2021b) and deeper understanding of biosynthetic machineries (Skinnider et al, 2020) allow the modification and even de novo design of modular biosynthetic pathways (Bozhüyük et al, 2019; Cummings et al, 2019; Hwang et al, 2020; Groß et al, 2021b).
We would like to refer the interested reader to the associated Science & Society article “Current Developments in Antibiotic Discovery—Global Microbial Diversity as Source for Evolutionary Optimized Anti‐infectives” for an in‐depth discussion of several examples (Hegemann et al, published concurrently).
Technological advances in chemoinformatics, especially in high‐resolution mass spectrometry (hrMS) combined with bioinformatic tools to analyse and compare the resulting datasets, enable early analysis of complex extracts from bacterial cultures or other natural sources (Wang et al, 2016; Schorn et al, 2021). HPLC‐hrMS or NMR‐based untargeted metabolomics provide an early overview of components in tested extracts and allow de‐replication against databases of known natural products (Zani & Carroll, 2017; van Santen et al, 2022). Furthermore, MS/MS‐based fragmentation of the compounds found in those extracts can be used to cluster the components into so‐called molecular networks, based on similar fragmentation patterns (Quinn et al, 2017). To facilitate these analyses for researchers around the world, open‐access programs were created to analyse MS/MS and subsequently annotate data (Wang et al, 2016) and predict sub‐structures (Ernst et al, 2019) of created molecular networks. Combining untargeted metabolomics data or molecular networks with the results of bioactivity screens of (fractionated) extracts allows reliable predictions of bioactivity‐driving natural products (Kurita et al, 2015; Lee et al, 2022) or molecular networks (Nothias et al, 2018).
As demonstrated by these examples, technical and methodological developments of the past decades have uncovered the vast potential of microorganisms to produce antibiotics or natural products in general. However, production, purification and structure elucidation of those new compounds remain challenging and include manual and time‐consuming method optimisation for every individual compound. Moreover, upscaling production and purification of promising candidates for further MoA, MoR‐ and pharmacokinetic studies are often a bottleneck in antibiotic development (Wilson et al, 2020).
Given the historical and current impact of bacteria in antibiotic research, this chapter and the examples therein focus on developments and techniques based on bacteria. However, this selection should not be understood as an assessment about the capacities of other sources to produce bioactive natural products.
The approved antibiotic classes of penicillins, cephalosporins, pleuromutilins and fusidic acid demonstrate that fungi also have enormous potential to produce natural products with antibacterial properties. Although working with fungi can be more complicated and time‐consuming compared with bacteria, their great taxonomic diversity and biosynthetic potential makes them interesting and promising sources of new antibiotics (van der Lee & Medema, 2016; Hyde et al, 2019).
Plants present another well‐established source for drug development. Plant‐based natural products with antibacterial activity often have the drawback of considerable toxicity to humans due to unspecific targets (Wright, 2017). On the other hand, some plant extracts and the resulting isolated and purified natural products have shown potential in potentiating or rescuing the activity of antibiotics, making them interesting as potential adjuvants in antibiotic treatment (Sadeer & Mahomoodally, 2021).
Promising approaches in medicinal chemistry
Although most of the approved antibacterial compounds are natural products or inspired by them, there are also some successful synthetic compounds and medicinal‐chemistry approaches. For instance, the FDA‐approved omadacycline is a semi‐synthetic tetracyclin derived from minocycline (Honeyman et al, 2015). It is used for the treatment of ABSSSIs and community‐acquired bacterial pneumonia (Burgos & Rodvold, 2019). Several isolated tetracyclins were approved for clinical use, but their intensive use led to the emergence of resistance and the semi‐synthetic approach only allowed limited modifications. A convergent total synthesis of tetracyclins helped to overcome this issue, opening the way for diversity‐oriented synthesis of 3,000 analogues (Charest et al, 2005). The extensive structure–activity relationships thus obtained eventually inspired eravacycline, designed to be a more potent antibiotic with optimised pharmacokinetic properties that should be less prone to resistance development (Clark et al, 2012; Xiao et al, 2012; Wright et al, 2014). In a similar fashion, the development of the well‐known macrolides made a huge step forward when a convergent synthesis was established and 300 new candidates were thus obtained (Seiple et al, 2016). This also provided easier access to already approved semi‐synthetic macrolides such as azithromycin, telithromycin, and solithromycin (Dinos, 2017). Other successful medicinal‐chemistry programs include the development of cefiderocol (Aoki et al, 2018; Sato & Yamawaki, 2019) or the optimisation of arylomycins from Gram‐positive‐specific antibacterial agents to a powerful, novel class of anti‐Gram‐negative antibiotics, thanks to a careful study of a co‐crystal structure in complex with the target (E. coli signal peptidase) and previous SAR studies (Smith et al, 2018). As elaborated in section Optimising the physicochemical property space, the permeation of the Gram‐negative bacterial cell wall represents a significant challenge. Attempts to tackle it include the design of screening libraries compliant with the properties needed to target ESKAPE pathogens (Fleeman et al, 2015).
Quinolones also have a long history of medicinal‐chemistry optimisation, starting with the synthesis of the first 6‐fluoroquinolone norfloxacin that extended the spectrum of this class to Gram‐negative pathogens (Koga et al, 1980). Several generations of fluoroquinolones followed, each one overcoming some limitations of the previous one (Zhanel et al, 1999; Zhanel et al, 2002) as some of their properties can be tuned, such as the pharmacokinetic profile (Itoh et al, 2015) or the ability to permeate membranes and overcome efflux (Gorityala et al, 2016). Finafloxacin is an example of an optimised fluoroquinolone that has the advantage of increasing potency in an acidic environment (Stubbings et al, 2011). This rare property among the class of quinolones granted it a global approval for the treatment of ear infections (McKeage, 2015).
Knowledge of the target also allows the use of alternative hit‐finding strategies such as target‐directed dynamic combinatorial synthesis that led to the identification and optimisation of an MEP pathway enzyme inhibitor in E. coli and Mycobacterium tuberculosis (Jumde et al, 2021). The use of computational tools, such as molecular‐dynamics simulations (Choudhury et al, 2015; Pavlova & Gumbart, 2015; Choudhury & Narahari Sastry, 2019) or molecular docking brought precious insights for the rational design of new antibiotic classes. For instance, molecular dynamics can be used to elucidate the reasons for microbial resistance or poor Gram‐negative permeation and how to escape them (Isabella et al, 2015). Durand‐Reville et al reported a new, highly potent class of diazabicyclooctane (DBO) that was designed in silico to inhibit multiple penicillin‐binding proteins (PBPs) and that represents the first use of structure‐porin permeation relationships to improve its permeation (Durand‐Reville et al, 2021). Chemists at Hoechst Marion Roussel had first designed DBOs as β‐lactam mimics that would not undergo β‐lactamase hydrolysis, thus restoring the antibacterial activity. However, the first compounds of this new class did not display any interesting antibacterial activities, but surprisingly inhibited β‐lactamases (Coleman, 2011). Further development of this BLI class led to the approval of avibactam and relebactam in combination with β‐lactam antibiotics (Ehmann et al, 2012; Deja, 2021). However, some DBOs also inhibit PBP2 and display an antibacterial activity in addition to their anti‐β‐lactamase activity (Lampilas et al, 2008; King et al, 2016; Levy et al, 2019). Nevertheless, that is not sufficient for a monotherapy due to a lack of in vivo efficacy and high frequency of resistance. The strategy of Durand‐Reville et al was to obtain a more broad‐spectrum derivative that would also target PBP1 and PBP3, rationally designed thanks to the use of molecular dynamics simulations (Durand‐Reville et al, 2021). They further improved the permeation of the drug by studying and optimising the porin permeation, while conserving the antibacterial activity (Cully, 2021). This careful design led to the discovery of ETX0462, that showed promising in vitro and in vivo properties, good permeation and no emergence of resistance, and thus the first DBO potentially used in a monotherapy (Cully, 2021; Durand‐Reville et al, 2021).
In a similar fashion, boronic acids have been explored as potential BLIs in a target‐based approach, as they are known to bind serine proteases (Smoum et al, 2012). The use of in silico docking led to the discovery of a cyclic boronic acid core (Hecker et al, 2015). This core became the foundation for a new class of antibacterials, among which are vaborbactam, approved in combination (Messner et al, 2022), and taniborbactam, still in clinical development (Liu et al, 2020). Finally, drug repurposing by deep‐learning analysis is another use of computational tools for the discovery of novel antibacterial scaffolds (Stokes et al, 2020; Bremner, 2021).
Another strategy is the synthesis of hybrid molecules to obtain a dual or synergistic effect (Maier, 2015; Surur & Sun, 2021), such as cadazolid, which is a quinolonyl‐oxazolidinone chimera (Baldoni et al, 2014; Locher et al, 2014a; Locher et al, 2014b). Such a dual mode of action can also lead to decreased resistance formation, as with SCH‐79797 and its more potent analogue irresistin‐16 (Martin et al, 2020). These compounds both target the dihydrofolate reductase and disrupt the bacterial membrane polarity and permeability in both Gram‐positive and Gram‐negative pathogens. More importantly, the frequency of resistance to SCH‐79797 is extremely low, compared with its constituting units separately. Intelligently designed prodrugs also achieved interesting activities, for example by taking advantage of the β‐lactamase resistance mechanism to selectively release ciprofloxacin in resistant strains (Evans et al, 2019; Jubeh et al, 2020).
The re‐exploration of targets of old natural products such as the sliding clamp of DNA polymerase, DnaN, either with new griselimycins obtained by total synthesis (Kling et al, 2015), or with small molecules discovered by iterative structure‐based synthesis (Monsarrat et al, 2021) or kinetic target‐guided synthesis (Mancini et al, 2020) also proved to be promising. The exploration of new targets such as energy‐coupling factor (ECF) transporters (Bousis et al, 2019) or dapE‐encoded N‐succinyl‐l,l‐diaminopimelic acid desuccinylase (DapE; Reidl et al, 2020), although challenging, opens the path to medicinal‐chemistry programs for the identification of novel antibiotic scaffolds with unprecedented modes of action.
Finally, medicinal chemists also explored targeting of virulence factors (“pathoblockers”) as an alternative strategy. Elastase B (LasB), a metalloprotease with an essential zinc atom in its binding site, is one of the most significant virulence factors in P. aeruginosa (Everett & Davies, 2021). As a result, numerous LasB inhibitors with a range of zinc binding moieties have been discovered (Galdino et al, 2019; Velázquez‐Libera et al, 2019; Leiris et al, 2021; Kaya et al, 2022). Other targeted virulence factors in P. aeruginosa include the lectins A and B responsible for the formation of biofilms which can be inhibited by carbohydrate derivatives (Sommer et al, 2019; Madaoui et al, 2020; Schütz et al, 2021), or the transcriptional regulator PqsR (Soheili et al, 2019). In S. aureus, the discovery of quorum‐quenching agents (Kuo et al, 2015) or adhesion inhibitors (Fernandes de Oliveira et al, 2021) gave promising hits. The various pathoblocker projects are at different stages, and the most advanced have now reached preclinical development.
Three different approaches to sustain the antibiotic pipeline
Combining the different lessons thus learned will enable us to close the antibiotic discovery gap. Here, we categorise these approaches into three categories.
The first category “quick fix, short‐term approaches” will lead to synthetically optimised compounds of known antibiotic classes. For example, applying the eNTRY rules to suitable drug candidates can enhance their activity profile (Muñoz & Hergenrother, 2021). Other synthetic or semi‐synthetic approaches can lead to decreased toxicity and fewer side effects, optimised stability, or better pharmacokinetic profiles of already established antibiotic classes (Wright et al, 2014; Grandclaudon et al, 2019). However, known scaffolds cannot be exploited indefinitely and the establishment of new modes of actions and the avoidance of daunting cross‐resistance remain challenging in this approach (Walsh & Wencewicz, 2014).
“Hard to find but long‐term approaches” will (eventually) lead to novel anti‐infective agents and new antibiotic classes. Compounds with completely new scaffolds are more likely to have a new mode of action and, in turn, have a smaller risk of showing cross‐resistance to antibiotics already on the market (Walsh & Wencewicz, 2014; Fiers et al, 2017; Kaur et al, 2021). To find and develop those innovations in a timely manner, we need to use our scientific toolbox efficiently. Implementing the lessons learned to build new HTS platforms and mining the natural product potential with new technologies will sustain the antibacterial pipeline in the long term (Hutchings et al, 2019; Ribeiro da Cunha et al, 2019; Grkovic et al, 2020). It is important to note that this development is time‐consuming and financially riskier than “short‐term” approaches. This is compounded by the limited resources at the initial discovery stages in academia and smaller institutes relative to the high‐throughput and high‐capacity resources in industry. Implementing these early‐phase discovery techniques at a more industrial scale, as was done during the golden age of antibiotics, may help resupply the pipeline.
The third, most novel, and possibly most inventive, yet most controversial category is the “alternative approaches.” These agents, such as monoclonal antibodies, enzymes, virulence inhibitors or immunomodulating agents, do not fit the definition of traditional antibiotics as direct‐acting small molecules. Since their roles in the development of antibacterials are on the rise and first candidates have already entered the clinical pipeline, the WHO included these “non‐traditional antibacterials” into their analysis of “antibacterial agents in clinical and preclinical development” for the first time in 2020 (World Health Organization, 2021). However, most non‐traditional antibacterial agents are in early stages of clinical and preclinical development. They still need to demonstrate their safety, efficacy and approvability based on carefully designed clinical trials.
Analysis of the clinical and preclinical antibacterial pipeline
This analysis covers antibacterial agents in clinical and preclinical development for the systemic treatment of the 12 bacteria listed on the WHO PPL (World Health Organization, 2017). It includes all antibacterial agents in clinical development that have not been approved for treatment in humans yet and are classified by the WHO to be active or possibly active against at least one of the priority pathogens. Peer‐reviewed data on the activity and safety of most agents in clinical trials are available, but little to no information is published in some cases, leaving the WHO or companies' homepages as the only sources of information.
In contrast to antibacterial agents registered in clinical trial databases, projects in preclinical stages are more difficult to track to estimate modes of action and activity spectra. Hence, we rely on data published by the WHO in their annual review of antibacterial agents in clinical and preclinical development (World Health Organization, 2022). We excluded agents active against M. tuberculosis or C. difficile, those with non‐systemic routes of administration, and prophylactics (World Health Organization, 2022). Information regarding some agents has been corrected with publicly available information. Projects in lead optimisation were separated from our list of preclinical agents as they have not qualified for preclinical development yet (Appendix Table S4–S9). Moreover, the reader should keep in mind that this list is not complete, as some companies or institutes did not disclose any or all information about their preclinical‐stage projects. Furthermore, claimed activities and properties cannot be verified in many cases, as no peer‐reviewed data are available.
Based on these criteria, there are 26 antibiotics (Table 1; 20 derivatives of approved classes and 6 novel scaffolds) and 14 non‐traditional antibacterial agents (Table 2) in clinical development (as of 1 November 2021). Based on our analysis, a total of 74 antibacterial projects are in preclinical development, including 29 novel antibiotics, 20 projects based on derivatives and established antibiotics and 25 non‐traditional antibacterials (Appendix Table S4–S6).
Table 1.
Antibiotics in clinical development; adapted from WHO analysis (current to November 2021; World Health Organization, 2022).
| INN (company code) | Phase | Antibiotic class | Route of administra‐tion | Developer | Expected activity against priority pathogens | Innovation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CRAB | CRPA | CRE | OPP a | NCR | CC | T | MoA | |||||
| Solithromycin (T‐4288) | NDA b | Macrolide | i.v. and oral | Fujfilm Toyama Chemical | / | / | / | ● | ‐ | ‐ | ‐ | ‐ |
| Sulopenem; sulopenem etzadroxil / probenecid | 3 c | β‐Lactam (penem) | i.v. and oral | Iterum Therapeutics | ○ | ○ | ○ d | / | ‐ | ‐ | ‐ | ‐ |
| Durlobactam (ETX‐ 2514) + sulbactam | 3 | DBO‐BLI/PBP2 binder + β‐lactam‐BLI/ PBP1,3 binder | i.v. | Entasis Therapeutics | ● | ○ | ○ | / | ‐ | ‐ | ‐ | ‐ |
| Taniborbactam (VNRX‐5133) + cefepime | 3 | Boronate BLI + β‐lactam (cephalosporin) | i.v. | VenatoRx Pharmaceuticals / GARDP | ○ | ● | ● | / | ? | ✓ | ‐ | ‐ |
| Enmetazobactam (AAI‐101) + cefepime | 3 | BLI + β‐lactam (cephalosporin) | i.v. | Allecra Therapeutics | ○ | ○ | ○ e | / | ‐ | ‐ | ‐ | ‐ |
| Zoliflodacin | 3 | Spiropyrimidenetrione (topoisomerase inhibitor) | oral | Entasis Therapeutics / GARDP | / | / | / | ● | ✓ | ✓ | ‐ | ✓ |
| Gepotidacin | 3 | Triazaacenaphthylene (topoisomerase inhibitor) | i.v. and oral | GSK | / | / | / | ● | ? | ✓ | ‐ | ✓ |
| Nafithromycin (WCK‐4873) | 3 | Macrolide | Oral | Wockhardt | / | / | / | ● | ‐ | ‐ | ‐ | ‐ |
| Benapenem | 2/3 | β‐Lactam (carbapenem) | i.v. | Xuanzhu Biopharm f | ○ | ○ | ○ | / | ‐ | ‐ | ‐ | ‐ |
| Afabicin (Debio‐1450) | 2 | Pyrido‐enamide (FabI inhibitor) | i.v. and oral | Debiopharm | / | / | / | ● | ✓ | ✓ | ✓ | ✓ |
| Zidebactam + cefepime | 1 g | DBO‐BLI/ PBP2 binder h + cephalosporin | i.v. | Wockhardt | ● | ● | ● | / | ‐ | ‐ | ‐ | ‐ |
| OP0595 (nacubactam) + meropenem | 1 | DBO‐BLI/PBP2 binder h + β‐lactam (carbapenem) | i.v. | Meiji Seika | ○ | ○ i | ● | / | ‐ | ‐ | ‐ | ‐ |
| ETX0282 + cefpodoxime proxetil | 1 | DBO‐BLI/PBP2 binder h + β‐lactam (cephalosporin) | Oral | Entasis Therapeutics | ○ | ○ | ● | / | ‐ | ‐ | ‐ | ‐ |
| ARX‐1796 (oral avibactam prodrug) | 1 | DBO‐BLI + β‐lactam (undisclosed) | oral | Arixa Pharmaceuticals / Pfizer j | ○ | ○ | ● k | / | ‐ | ‐ | ‐ | ‐ |
| XNW4107 + imipenem + cilastatin | 1 | BLI + β‐lactam (carbapenem) / degradation inhibitor | i.v. | Sinovent | ? | ? | ? | ? | ? | ? | ? | ? |
| VNRX‐7145 + ceftibuten | 1 | Boronate‐BLI + β‐lactam (cephalo‐sporin) | Oral | VenatoRx Pharmaceuticals | ○ | ○ | ● | / | ? | ✓ | ‐ | ‐ |
| QPX7728 + QPX2014 | 1 | Boronate‐BLI + undisclosed | i.v. | Qpex Biopharma | ● | ● | ● | / | ? | ‐ | ‐ | ‐ |
| QPX7728 + QPX2015 | 1 | Boronate‐BLI + undisclosed oral β‐lactam | Oral and i.v. | Qpex Biopharma | ○ | ○ | ● | / | ? | ‐ | ‐ | ‐ |
| SPR‐206 | 1 | Polymyxin | i.v. | Spero Therapeutics | ● | ● | ● | / | ‐ | ‐ | ‐ | ‐ |
| MRX‐8 | 1 | Polymyxin | i.v. | MicuRx | ● | ● | ● | / | ‐ | ‐ | ‐ | ‐ |
| QPX9003 | 1 | Polymyxin | i.v. | Qpex Biopharma | ? | ? | ? | ? | ? | ? | ? | ? |
| KBP‐7072 | 1 | Tetracyclin | Oral | KBP BioSciences | ● | ○ | ○ | ● | ‐ | ‐ | ‐ | ‐ |
| EBL‐1003 (apramycin) | 1 l | Aminoglycoside | i.v. | Juvabis | ● | ? | ● | / | ‐ | ‐ | ‐ | ‐ |
| TXA709 | 1 | Difluorobenz‐amide (FtsZ inhibitor) | Oral and i.v. | TAXIS Pharmaceuticals | ○ | ○ | ○ | ● | ✓ | ✓ | ✓ | ✓ |
| RG6006 (Abx MCP) | 1 | Macrocyclic peptide | i.v. | Roche | ? m | ? | ? | ? | ? | ? | ? | ? |
| BWC0977 | 1 | Topo‐isomerase | i.v. | Bugworks Research | ? | ? | ? | ? | ? | ? | ? | ? |
Pathogen activity: ● active;? possibly active; ○ not or insufficiently active; / activity not assessed, as the antibiotic is focused and developed for only either Gram‐positive cocci or Gram‐negative rods. Agents not active against critical priority pathogens were assessed for activity against OPP, which includes the WHO high and medium priority pathogens.
Innovation assessment: ✓ criterion fulfilled;? inconclusive data; − criterion not fulfilled.
BLI, β‐lactamase inhibitor; CC, chemical class; CRAB, carbapenem‐resistant A. baumannii; CRE, carbapenem‐resistant Enterobacterales; CRPA, carbapenem‐resistant P. aeruginosa; ESBL, extended‐spectrum β‐lactamase; FabI, enoyl‐acyl carrier protein reductase; DBO, diazabicyclooctane; FtsZ, cell division protein named after corresponding mutant phenotype, filamenting temperature‐sensitive mutant Z; GARDP, Global Antibiotic Research and Development Partnership; i.v., intravenous; KPC, K. pneumoniae carbapenemase; MoA, new mode of action; NCR, no cross‐resistance; NDA, new drug application; OPP, other priority pathogen; PBP2, penicillin‐binding protein 2; T, new target; uUTI, uncomplicated urinary tract infection; WHO, World Health Organisation.
OPP target pathogens – solithromycin: S. pneumoniae; nafithromycin: S. aureus and S. pneumoniae; zoliflodacin: N. gonorrhoeae; gepotidacin: N. gonorrhoeae and E. coli; TNP‐2198: H. pylori; afabicin, TNP‐2092, KBP‐7072 and TXA‐109: S. aureus.
Solithromycin NDA for otorhinolaryngological infections was submitted in Japan in April 2019.
Iterum will undertake an additional Phase 3 uUTI study of sulopenem etzadroxil before any NDA resubmission.
Active against ESBL‐producing cephalosporin‐resistant Enterobacterales but not carbapenem‐resistant Enterobacterales.
Active against ESBL‐producing cephalosporin‐resistant Enterobacterales and some KPC‐producing CRE.
Xuanzhu Biopharm is a subsidiary of Sichuan Pharmaceutical Holdings but possesses fully independent intellectual property rights.
A Phase 3 trial for zidebactam + cefepime was registered in July 2021 for cUTI or acute pyelonephritis (NCT04979806).
The DBO‐BLIs zidebactam, OP0595 (nacubactam) and ETX0282 also have some antibacterial activity and have been classified as β‐lactam enhancers.
Activity against AmpC‐β‐lactamase producing and KPC‐producing CRPA.
The original developer, Arixa Pharmaceuticals, was acquired by Pfizer in October 2020.
Active against KPC but not MBL‐producing Enterobacterales.
Previously used as an antibacterial treatment in animals.
RG6006 is being developed to treat A. baumannii infections.
Table 2.
Non‐traditional antibacterial agents in clinical development; adapted from WHO analysis (current to November 2021; World Health Organization, 2022).
| Name (synonym) | Phase | Antibacterial class | Route of administration | Developer | Expected activity against priority pathogens |
|---|---|---|---|---|---|
| Reltecimod (AB103) | NDA a | Synthetic peptide antagonist of both superantigen exotoxins and the CD28 T‐cell receptor | i.v. | Atox Bio | S. aureus |
| Tosatoxumab (AR‐301) | 3 | Anti‐S. aureus IgG1 antibody | i.v. | Aridis Pharmaceuticals | S. aureus |
| Exebacase (CF‐301) | 3 | Phage endolysin | i.v. | ContraFect | S. aureus |
| AR‐320 (MEDI‐ 4893, suvratoxumab) | 2 | Anti‐S. aureus IgG mAb | i.v. | Aridis Pharmaceuticals, licenced from AstraZeneca | S. aureus |
| LSVT‐1701 (N‐Rephasin SAL200, tonabacase) | 2a/1 | Phage endolysin | i.v. | Roivant Sciences, licenced from iNtRON | S. aureus |
| Phage | 1/2 | Phage | i.v. | Adaptive Phage Therapeutics | E. coli |
| Rhu‐pGSN (rhu‐plasma gelsolin) | 1b/2a | Recombinant human plasma gelsolin protein | i.v. | BioAegis Therapeutics | Non‐specific Gram‐positive and Gram‐negative |
| Ftortiazinon (fluorothyazinone) + cefepime | 2 | Thyazinone (type III secretion system inhibitor) + cephalosporin | Oral | Gamaleya Research Institute of Epidemiology and Microbiology | P. aeruginosa |
| TRL1068 | 1 | mAb | i.v. | Trellis Bioscience | Gram‐positive and Gram‐negative biofilms |
| 9 MW1411 | 1 | mAb (α‐toxin) | i.v. | Mabwell Bioscience | S. aureus |
| LBP‐EC01 | 1b | CRISPR‐Cas3 enhanced phage | i.v. | Locus Biosciences | E. coli |
| SVT‐1C469 | 1 | Live biotherapeutic product | Oral | Servatus | H. pylori |
| CAL02 | 1 | Broad‐spectrum anti‐toxin liposomal agent and nanoparticle | i.v. | Eagle Pharmaceuticals, licenced from Combioxin | S. pneumoniae b |
| GSK3882347 | 1 | Undisclosed (FimH antagonist) | Oral | GSK | E. coli |
| ALS‐4 | 1 | Anti‐virulence (staphyloxanthin biosynthesis inhibition) | Oral | Aptorum Group | S. aureus |
FimH, type‐1 fimbrin D‐mannose‐specific adhesin; IgG1, immunoglobulin G1; mAb, monoclonal antibody; NDA, New Drug Application; WHO, world health organisation.
Submitted to the US FDA as a potential treatment for necrotizing soft tissue infections in December 2020.
While the Phase 1 trial evaluated CAL02 on patients with severe pneumonia caused by S. pneumoniae, CAL02 has broad‐spectrum effects against other bacteria, such as P. aeruginosa, A. baumannii, Enterobacterales and S. aureus.
Recently approved compounds and antibiotics discontinued from clinical development
Within the past 5 years (1 July 2017–1 November 2021), 11 new antibiotics against priority pathogens were approved, either as single agents or in combination with activity‐enhancing compounds (Appendix Table S2; World Health Organization, 2022).
Only two of these represent first‐in‐class antibiotics or enhancers. The pleuromutilin lefamulin was introduced in 2019 for oral or intravenous (i.v.) treatment of community‐acquired pneumonia (CAP), caused by Gram‐positive pathogens. Other pleuromutilin derivatives were previously approved for the topical treatment of bacterial infections in humans and the systemic treatment in animals. The second new first‐in‐class drug is vaborbactam, a boronate‐type BLI that forms a reversible covalent bond with catalytic serine residues in many serine‐β‐lactamases and carbapenemases (Plescia & Moitessier, 2020). Vaborbactam was approved in combination with the β‐lactam antibiotic meropenem for the treatment of complicated urinary‐tract and intra‐abdominal infections (cUTI and IAI) and HAP with cephalosporine and carbapenem‐resistant Enterobacteriaceae and Enterobacterales.
Among the remaining nine improved derivatives of existing antibiotic classes, cefiderocol, the first siderophore‐cephalosporine is particularly noteworthy. Its iron‐chelating siderophore‐moiety enables active uptake into a wide range of Gram‐negative pathogens, including Enterobacteriaceae and other Enterobacterales, A. baumannii and P. aeruginosa (Abdul‐Mutakabbir et al, 2020). Furthermore, cefiderocol shows good activity against pathogens harbouring a wide range of serine‐ and metallo‐β‐lactamases, but with increased MICs compared with non‐resistant strains (Abdul‐Mutakabbir et al, 2020). It was approved in 2019 for the treatment of cUTI and HAP with (multi‐) drug‐resistant aerobic Gram‐negative bacteria and further explored as combination therapy with other agents (Corcione et al, 2022). However, cefiderocol should be closely monitored for resistances to occur (Theuretzbacher et al, 2020a; Karakonstantis et al, 2022).
During the same period, the clinical development of 14 antibiotics was discontinued or suspended (Appendix Table S3). Possible reasons include toxicity issues, failure to reach anticipated endpoints as well as financial considerations and, more recently, clinical trial issues owing to the COVID‐19 pandemic (World Health Organization, 2022). While a majority of these projects was discontinued in or after phase 1 clinical development, three of these antibacterial agents were tested until phase 3 clinical trials. One of these drugs was the P. aeruginosa‐selective antibiotic murepavadin, a synthetic cyclic peptide, based on protegrin 1. By binding to the lipopolysaccharide transport protein D (LptD) and thus inhibiting its LPS transport function, murepavadin causes changes in the outer membrane that lead to cell death (Martin‐Loeches et al, 2018). The clinical development of murepavadin for the systemic treatment of HAP and ventilator‐associated pneumonia (VAP), caused by P. aeruginosa, was ended in 2019, due to an increased number of kidney injuries in patients (Polyphor AG, 2019). If not discontinued, murepavadin could have presented the first novel antibiotic class for the systemic treatment of Gram‐negative bacteria since the approval of the monobactam aztreonam in 1986.
Novel derivatives of approved antibiotic‐classes
Almost 80% (20 out of 26) of antibiotics in clinical development are improved derivatives of antibiotic classes that are already in use for systemic treatment of bacterial infections in humans (Table 1). They either target cell wall synthesis, the cell membrane or protein biosynthesis. In preclinical development, 20 out of 74 (about 27%) projects are based on derivatives of established antibiotics, however, this number may be inaccurate as not all information about some compounds is disclosed (Appendix Table S4).
Cell wall synthesis
The biggest group of antibiotics are the β‐lactams, consisting of chemically different penicillins, cephalosporins, carbapenems and monobactams, all with a β‐lactam ring as their active centre. These stop bacterial cell wall synthesis by covalently binding to PBPs and thus inhibiting the formation of peptidoglycan, a key part of bacterial cell walls.
The most important mode of resistance are β‐lactamases that cleave the β‐lactam ring; these enzymes are typically categorised into four classes A–D. Class B β‐lactamases are metalloproteases that are difficult to inhibit, and the other three are serine proteases with different target specificities (Ambler, 1980; Bush & Jacoby, 2010; Lakemeyer et al, 2018). While older BLIs are β‐lactams themselves and form covalent bonds to the active centres of β‐lactamases, recently approved DBO‐BLIs and the aforementioned boronate‐BLIs inhibit enzymes by a reversible covalent mechanism (Bush, 2015). Almost half of the antibiotics currently in clinical development belong to the β‐lactams, including 11 combinations with new BLIs and two novel β‐lactam antibiotics.
The synthetic thio‐penem sulopenem (phase 3, i.v.) and its orally available prodrug sulopenem etzdroxil/probenecid are developed for the treatment of UTI and IAI caused by cephalosporin‐resistant Enterobacterales (World Health Organization, 2022). Unfortunately, just like the novel carbapenem benapenem (phase 2/3, i.v., only developed in China; World Health Organization, 2022), sulopenem is likely to exhibit cross‐resistance to carbapenems, as no activity was reported against carbapenem‐resistant bacteria (Karlowsky et al, 2019; Ji et al, 2020; World Health Organization, 2022).
Three of the new BLI/β‐lactam combinations are currently in phase 3, all of which are administered intravenously. The DBO‐BLI durlobactam displays activity against all classes of serine β‐lactamases and intrinsic activity against Enterobacterales (Durand‐Réville et al, 2017). Furthermore, it restores and increases the activity of the β‐lactam‐BLI sulbactam against A. baumannii (Penwell et al, 2015). The combination of durlobactam and sulbactam shows a good activity against carbapenem‐resistant A. baumannii, has a low frequency of resistance and is developed for HAP and VAP caused by this pathogen (Petropoulou et al, 2021; Shapiro et al, 2021; World Health Organization, 2022).
Taniborbactam is the first boronate‐BLI with activity against all β‐lactamases (Liu et al, 2020). In combination with the fourth‐generation cephalosporin cefepime, it shows good activity against carbapenem‐resistant Enterobacterales and P. aeruginosa and is developed for the treatment of acute pyelonephritis, cUTI, HAP and VAP with these pathogens (Hamrick et al, 2020; World Health Organization, 2022). The β‐lactam‐BLI enmetazobactam has an improved penetration into bacterial cells (Papp‐Wallace et al, 2019). In combination with cefepime, it shows activity against cephalosporin‐resistant and some carbapenem‐resistant Enterobacterales and is developed for the treatment of cUTI (Papp‐Wallace et al, 2019; Tselepis et al, 2020).
The remaining eight BLI/β‐lactam antibiotic combinations are currently in phase 1 clinical development. Three DBO‐BLIs—zidebactam (Moya et al, 2017), nacubactam (Morinaka et al, 2015) and ETX0282 (Durand‐Réville et al, 2020) —are antibiotic enhancers. In addition to their characteristic serine β‐lactamase inhibition, they have intrinsic activity against some Gram‐negative bacteria due to PBP2‐binding, and enhance the activity of other β‐lactam antibiotics through synergistic effects. Combined with cefepime, zidebactam shows good in vitro activity against Enterobacterales, A. baumannii and P. aeruginosa strains, which display a wide range of β‐lactamases, including metallo β‐lactamases (Sader et al, 2017). Similar to the previous combination, nacubactam and meropenem show activity against Enterobacterales that possess β‐lactamases including metallo β‐lactamases (Mushtaq et al, 2019). The combination ETX0282 and cefpodoxime proxetil is tested for the oral treatment of Enterobacterales and is active against serine β‐lactamases, but not against metallo β‐lactamases (O'Donnell et al, 2020).
ARX‐1796 is an orally administered pro‐drug of the first‐in‐class DBO‐BLI avibactam that inhibits serine β‐lactamases (Gordon et al, 2018). It is developed to treat infections with carbapenem‐resistant Enterobacterales in combination with an undisclosed β‐lactam‐antibiotic (World Health Organization, 2022).
Similarly to Taniborbactam, the boronate‐BLI QPX7728 displays a broad activity against all classes of β‐lactamases (Lomovskaya et al, 2021). It is in clinical development with the undisclosed β‐lactam antibiotics QPX2014 and QPX2015 for the intravenous treatment of infections with multidrug‐resistant Enterobacterales, A. baumannii and P. aeruginosa in high‐risk patients and the oral treatment with cephalosporin‐ and carbapenem‐resistant Enterobacterales (Qpex Biopharma, 2022). Another orally bioavailable boronate‐BLI, VNRX‐7145, displays activity against most serine β‐lactamases (Trout et al, 2021). It is in clinical development in combination with the cephalosporin ceftibuten, the in vitro activity of which against cephalosporin‐resistant Enterobacterales could be restored by VNRX‐7145 (Mendes et al, 2022).
No substantial information can be found about BLI XNW4107, being developed in combination with the carbapenem imipenem and cilastatin, an inhibitor of the renal dehydropeptidase that increases extraction of active antibiotic into the bladder (Clissold et al, 1987).
Cell envelope
Polymyxins bind to the lipopolysaccharide layer of the outer membrane of Gram‐negative bacteria, alter its permeability, and disrupt membrane integrity (Kwa et al, 2007). Given their poor selectivity for bacterial membranes, approved polymyxins exhibit severe nephrotoxic and neurotoxic adverse effects and are therefore used only as last‐resort antibiotics (Falagas & Kasiakou, 2006).
All three new polymyxins are currently in phase 1 clinical development for the intravenous treatment of infections with multidrug‐resistant Gram‐negative priority pathogens. SPR‐206 exhibits improved in vitro activities compared with polymyxin B and is less nephrotoxic in in vivo infection models in mice (Brown et al, 2019). Another polymyxin‐derivative, MRX‐8, shows good activity against multidrug‐resistant Gram‐negative bacteria in vitro and in mouse infection models, but neither its structure nor any information about potential toxicity are available (Lepak et al, 2020). The synthetic polymyxin derivative QPX9003 displays improved in vitro activity compared with polymyxin B and is effective against polymyxin B‐resistant Gram‐negative bacteria in infection models in mice. Furthermore, it shows reduced toxicity in mouse and cynomolgus monkey infection models (Roberts et al, 2022).
Protein biosynthesis
Inhibition of protein biosynthesis is a successful MoA for antibiotics, as evidenced by seven approved antibiotic classes against different targets in the bacterial ribosome. The four antibiotics in clinical development that target protein biosynthesis belong to three of these classes.
The ketolides solithromycin (new drug application, i.v. and oral) and nafithromycin (phase 3, oral) were developed for the treatment of CAP caused by Gram‐positive cocci (Fernandes et al, 2016; World Health Organization, 2022). Both antibiotics have a similar activity in vitro as the first‐in‐class ketolide telithromycin. Nafithromycin does not exhibit cross‐resistance with older macrolides and ketolides in pneumococci, and solithromycin shows the same effect in streptococci (Farrell et al, 2015; Flamm et al, 2017). However, solithromycin has not been approved by the FDA yet, as its potential for liver toxicity was not adequately characterised (Owens, 2017). Other than most other macrolides, nafithromycin does not inhibit human CYP enzymes in vitro, thereby reducing the risk of interactions with other drugs (Chavan et al, 2016).
The aminomethylcycline KBP‐7072 (phase 1, oral) is a third‐generation tetracyclin with good in vitro activities against Gram‐positive bacteria, Enterobacteriaceae and other Enterobacterales, and A. baumannii (Huband et al, 2022). Moreover, acquired resistance genes against other tetracyclins seem to have only a minimal effect on KBP‐7072 (Pfaller et al, 2021).
Apramycin (EBL‐1003, phase 1, i.v.) is an aminoglycoside that displays activity against carbapenem‐ and cephalosporin‐resistant Enterobacteriaceae, Enterobacterales and A. baumannii and is potentially active against drug‐resistant P. aeruginosa (World Health Organization, 2022). It shows only little cross‐resistance with multi‐drug resistant Gram‐negative bacteria, including strains with resistance against many aminoglycosides (Juhas et al, 2019). Nonetheless, considering that apramycin was licenced for systematic use in animals in 1980 and that apramycin‐resistance was reported only a few years later, it seems likely that resistance will also emerge upon clinical use in humans (Wray et al, 1986; Theuretzbacher et al, 2020a).
Novel classes of antibiotics
Out of the 26 traditional antibiotics, there are six novel agents in the clinical pipeline (Table 1). In the preclinical pipeline, 29 out of 74 agents (about 39%; Appendix Table S5) are considered “innovative” and urgently needed by the WHO as they display a new chemical class, hit a new target or display a new MoA, while not showing any cross‐resistance with approved antibiotic classes, yet (Theuretzbacher, 2017).
DNA: Novel bacterial topoisomerase inhibitors
The fluoroquinolones are well‐established bacterial topoisomerase inhibitors, synthetically developed in the 1960s to 80's, and target type II topoisomerases DNA gyrase (at subunit GyrA) and topoisomerase IV (at subunit ParC), thereby stabilising double‐strand breaks of DNA with bactericidal effects (Laponogov et al, 2009). Three novel bacterial topoisomerase inhibitors (NBTIs), with new chemical scaffolds and against new targets, are in the clinical pipeline.
The orally administered NBTI zoliflodacin (phase 3) displays as new chemical scaffold a spiropyrimidenetrione. It was discovered through high‐throughput MIC screens by the Pharmacia Research Compound Collection, and developed through directed medicinal chemistry (Miller et al, 2008; Bradford et al, 2020). It is active against Neisseria gonorrhoeae and Gram‐positive cocci, and currently in phase 3 clinical trials for the treatment of uncomplicated gonorrhoea. Zoliflodacin has a distinct binding site in GyrB, which differentiates it from the binding mode displayed by fluoroquinolones; accordingly no cross‐resistance with fluoroquinolones has been reported yet.
Gepotidacin (phase 3) is an oral and i.v. bioavailable NBTI with the novel chemical scaffold triazaacenaphthalene. It was discovered through antibacterial screenings of a chemical library and optimised based on X‐ray analysis of the protein‐DNA‐NBTI complex and directed medicinal chemistry (Bax et al, 2010; Gibson et al, 2019). It is active against N. gonorrhoeae and Gram‐positive strains, and in clinical phase 3 development for the treatment of uncomplicated urogenital gonorrhoea and uncomplicated urinary tract infections (uUTI). However, due to poor gastro‐intestinal absorption, high doses are required, which leads to adverse side effects such as diarrhoea (World Health Organization, 2022). Compared with fluoroquinolones, gepotidacin has a unique binding site on GyrA and topoisomerase IV (subunit ParC). Nevertheless, early findings indicate that fluoroquinolone resistance might be a stepping stone for gepotidacin resistance as the target sites on topoisomerase IV are in close proximity to each other (preprint: Szili et al, 2018; World Health Organization, 2022).
In November 2021, a third NBTI BWC0977 (phase 1) entered clinical trials. It is developed as an i.v. and oral treatment of infections with MDR Gram‐positive and Gram‐negative bacteria. Unfortunately, no information regarding its origin, structure or MoA has been disclosed (Bugworks Research Inc., 2022).
Fatty acid synthesis: FabI Inhibitor
FabI, an enoyl acyl carrier protein reductase, is a key enzyme in the bacterial fatty acid biosynthesis. Owing to the fundamental differences of mammalian and bacterial fatty acid biosynthesis, FabI has been a potential selective antibacterial target for many years (Heath & Rock, 1995).
Currently, afabicin (phase 2) is the first and only FabI inhibitor in the clinical pipeline. It is the only compound that was identified through the genomics‐based HTS campaigns of individual targets that evolved to the clinical stage (Payne et al, 2007). The hit compound was identified as a weak inhibitor of the S. aureus FabI enzyme, which was then optimised by structure‐based design leading to a 350‐fold more potent drug candidate (Payne et al, 2002). Afabicin itself is the prodrug of Debio‐1452 and was tested in oral as well as i.v. formulations against intra‐ and extracelluar S. aureus bone and joint infections. So far, no cross‐resistance of afabicin with established antibiotics has emerged and even if resistance were to emerge, such a development may still be offset by the high target affinity of the drug.
Cell division: FtsZ inhibitor
Another agent in clinical phase 1 trials is TXA‐709, an FtsZ inhibitor with a new chemical scaffold that inhibits a novel target and therefore has a new MoA. FtsZ is a key protein of the Z‐ring, which is an essential structure formed in the middle of the bacterial cell during cell division. As the target has not yet been tapped by established antibiotics, FtsZ inhibitors have a high potential to become attractive novel antibiotics. Although several compounds have been reported to have in vitro activity against FtsZ, no agent has entered the clinical pipeline to that point.
Based on SAR studies, the benzamide scaffold was investigated and optimised through medicinal chemistry to yield an in vivo active FtsZ inhibitor (Haydon et al, 2008). A highly potent hit against S. aureus was further optimised to finally yield TXA‐709 (phase 1), a benzamide‐prodrug, which is being tested per i.v. application and orally against S. aureus infections (Kaul et al, 2015).
Undisclosed projects
Recently, an antibiotic macrocyclic peptide RG6006 (phase 1) has entered the clinical pipeline (Roche, 2022). It possesses anti‐Gram‐negative activity, will be applied i.v. and tested against A. baumannii infections. Further information regarding its structure or MoA has not been disclosed (Roche, 2022).
New scaffolds in preclinical development and promising candidates for preclinical development
We further highlight a few examples of agents with novel scaffolds in the preclinical pipeline (Appendix Table S5) and promising candidates for preclinical development (Appendix Table S8). The selection was made on the basis of available published and peer‐reviewed data.
Odilorhabdins
Nematode‐symbiotic bacteria of the genera Xenorhabdus and Photorhabdus were cultivated under various conditions and bioactivity‐guided fractionation of the supernatants led to the identification of the causal compounds, odilorhabdins, which are non‐ribosomal peptide synthetase (NRPS)‐derived ribosome‐targeting antimicrobial peptides (Pantel et al, 2018). The ability to cure infections in animal models and a validated method for chemical synthesis encouraged Nosopharm to initiate SAR studies and a medicinal‐chemistry program. They identified and optimised a lead compound among 500 analogues, evaluating in vitro activity and in vivo efficacy. The compound with best antibacterial, ADME, toxicology and pharmacology results, NOSO‐502, is currently undergoing clinical trial application and investigational new drug (CTA/IND)‐enabling studies (Racine & Gualtieri, 2019; Nosopharm, 2022).
Teixobactin
The macrocyclic depsipeptide teixobactin was discovered from the previously uncultured bacterium Eleftheria terrae using the iChip technology (Ling et al, 2015) followed by scale‐up fermentation of that strain, fractionation of its crude extract, and compound purification. This novel scaffold shows a unique MoA by blocking cell wall synthesis through binding and inhibiting the synthesis of lipid II and lipid III, which are precursors for peptidoglycan and teichoic acid biosynthesis. As no cross‐resistance to any established antibacterial has been observed so far and because of the promising in vitro toxicology and in vivo efficacy results, total synthesis efforts were initiated to enable SAR studies (Karas et al, 2020). The main limitation of teixobactin is that it is only active against Gram‐positive bacteria, a limitation which has been attempted to be overcome by synthesis of analogues and carrying out tests in combination with enhancers and anti‐Gram‐negative compounds (Chiorean et al, 2019). Teixobactin entered preclinical development recently although the resistance mechanism is still unknown and needs to be elucidated before further development can be conducted.
Darobactin
Similarly to the olidorhabdins, darobactins originate from nematode‐symbiotic bacteria of the genus Photorhabdus and were identified and isolated through bioactivity‐guided fractionation and compound purification (Imai et al, 2019). These ribosomally synthesised and post‐translationally modified peptides (RiPPs) are selectively active against Gram‐negative pathogens, including MDR K. pneumoniae, P. aeruginosa and E. coli. Interestingly, the agent is not active against gut commensals, which will possibly have a positive impact on the patients' gut health. The darobactin scaffold targets the outer membrane protein BamA (Kaur et al, 2021), which is the central component of the β‐barrel assembly machinery responsible for the folding and incorporation of outer membrane proteins and, therefore, membrane integrity (Konovalova et al, 2017; Ritzmann et al, 2022). Given the intriguing MoA, the in vitro bioactivity and in vivo efficacy, numerous derivatives were generated by both total synthesis (preprint: Lin et al, 2022; preprint: Nesic et al, 2022) and heterologous expression (Böhringer et al, 2021; Groß et al, 2021a; Seyfert et al, 2022). The lead optimisation phase is ongoing and will hopefully result in a preclinical candidate.
ULT3
Antimicrobial peptides of the ULT3 program were discovered through selective isolation of MDR bacteria from soil samples (Bjerketorp et al, 2021) which led to the identification of novel NRPS‐derived lipopeptides from Pedobacter spp. that are active against MDR Gram‐negative bacteria. As the antibacterial activity of these lipopeptides looks promising and as their cytotoxicity is acceptable, ULT3 peptides are currently in lead optimisation (Nord et al, 2020).
Corallopyronin A
The bacterial DNA‐dependent RNA polymerase (RNAP)‐inhibitor corallopyronin A with activity against S. aureus and N. gonorrhoeae was already discovered in the 1980s through screening of myxobacterial extracts for antibacterial activity (Irschik et al, 1985). Due to poor yield of the chemical synthesis, it required the elucidation of the biosynthetic pathway and development of a heterologous expression system to produce corallopyronin A in sufficient amounts for preclinical development (Erol et al, 2010; Pogorevc et al, 2019). Interestingly, and noteworthy outside the pathogen priority list, it showed highly effective eradication of the Gram‐negative intracellular Wolbachia bacteria. The main application of corallopyronin A is as an anti‐wolbachial treatment of filarial worm infections (Schiefer et al, 2020).
Owing to its novel binding site at the “switch region” on RNAP, no cross‐resistance with known RNAP inhibitors has been reported so far. Poor dissolution, stability and solubility of the compound could be addressed by modern formulation like embedment in polymeric matrices (Krome et al, 2022). Corallopyronin A is currently undergoing dose‐range finding toxicology studies in rodent and non‐rodent species (Krome et al, 2022).
Cystobactamids
Cystobactamids were discovered through screening of myxobacterial isolates for novel bioactive compounds and bioactivity‐guided fractionation and demonstrate potent activity against Gram‐negative and Gram‐positive bacteria (Baumann et al, 2014). They are biosynthesised by a NRPS machinery and form a scaffold of tailored para‐aminobenzoic acids, which enables them to inhibit gyrase and bacterial type IIa topoisomerase via a different binding site than quinolones with no cross‐resistance to quinolones reported so far (Hüttel et al, 2017; Groß et al, 2021b). After total synthesis was established (Moreno et al, 2015) with significant improvements in scalability and activity of novel derivatives (Moeller et al, 2019; Elgaher et al, 2020), cystobactamids are now in the lead optimisation phase for MDR‐Gram‐negative bacterial infections. It is worth mentioning that albicidins show a similar scaffold as cystobactamids and have completed lead optimisation for serious bacterial infections (Cociancich et al, 2015; Kretz et al, 2015; von Eckardstein et al, 2017; Behroz et al, 2019; Zborovsky et al, 2021).
Corramycin
The NRPS‐polyketide synthase (NRPS‐PKS) hybrid corramycin is produced by myxobacteria and exhibits anti‐bacterial activity against E. coli (Couturier et al, 2022) and in in vivo models of septicemia and lung infection. Lead optimisation led to derivatives with activity against Enterobacteriaceae and A. baumannii (Renard et al, 2022). The MoA has not been fully elucidated yet, but it could be shown that corramycin inhibits DNA replication and does not show cross resistance with quinolones so far (Bacqué et al, 2022; Couturier et al, 2022).
Analysis of the antibiotic pipeline
A look at the clinical pipeline of antibiotics raises hopes that, unlike after the introduction of linezolid, it will not take another 15 years until the next first‐in‐class antibiotic will be approved. It seems likely that at least one of the NBTIs zoliflodacin and gepotidacin will reach the market within the next years, given their advanced clinical development. Furthermore, it is noteworthy that the FabI inhibitor afabicin as well as the FtsZ inhibitor TXA‐709 target cellular processes outside the “typical” scope of approved antibiotics (see Fig 2B). As both antibiotics are still at an early stage of their clinical development, more time and more trials are needed to demonstrate their utility in a clinical setting.
The only representatives of new antibiotic classes against critical priority Gram‐negative bacteria that have entered the clinical pipeline within the past year are the NBTI BWC0977 and the macrocyclic peptide RG6006, respectively (World Health Organization, 2022).
While most of the novel antibiotics in clinical development target S. aureus or N. gonorrhoeae, 18 out of 20 derivatives of approved antibiotics are being developed for the treatment of critical priority MDR Gram‐negative bacteria. Within this subset, the group of BLI/β‐lactam combinations should be emphasised, as several BLIs provide unprecedented features and clinical potential. Due to their activity against all described classes of β‐lactamases, the boronate‐BLIs taniborbactam and QPX7728 could provide a valuable alternative for the treatment of infections with metallo‐β‐lactamase bearing pathogens. Moreover, it will be interesting to observe the further development of the DBO‐BLIs and the antibiotic enhancers zidebactam, nacubactam and ETX0282.
The three new polymyxin derivatives as well as the tetracyclin KBP‐7072 and the aminoglycoside apramycin are in phase 1 clinical development. Further trials will show if these three polymyxins exhibit reduced toxicity and whether both inhibitors of protein biosynthesis can overcome class‐specific resistances.
Although the global clinical pipeline of antibiotics for systemic use is rather small, it features promising aspects. Regarding late‐stage clinical development, the five antibiotics or combinations with activity against MDR Gram‐negative bacteria as well as the two first‐in‐class antibiotics will improve the treatment options in their respective indications. Moreover, several antibiotics in early clinical development represent new antibiotic classes or are promising improvements of their respective classes. However, novel antibiotic classes that do not display cross‐resistances with approved antibiotics and meet stringent performance characteristics to treat critical priority Gram‐negative pathogens remain underrepresented in the clinical pipeline (Butler et al, 2022; World Health Organization, 2022).
According to the WHO data, about 39% (29 out of 74) of antibiotics in preclinical development belong to new chemical classes. Furthermore, 48% of these antibiotics display activity against, and are being developed for treatment of, critical priority Gram‐negative pathogens. These numbers are encouraging, but given the high attrition rates in antibiotic development, it can be assumed that only one or two of these antibiotics will find their way into the market (Fig 3B).
Non‐traditional antibacterial agents in clinical and preclinical development
The rise of AMR and the challenge to find and develop new antibiotics have led to an increased interest in general antibacterial therapies that differ from the “inhibition of an essential target” approach of traditional small‐molecule antibiotics. Those non‐traditional antibacterials include any strategy to treat bacterial infections from strain‐specific inhibition of bacterial growth by bacteriophages to inhibition of virulence factors and modulation of the host immune system (Theuretzbacher & Piddock, 2019).
Following the classification by the WHO, we grouped the 14 non‐traditional antibacterials in clinical (Table 2) and 25 agents in preclinical development (Appendix Table S6) into four categories: agents targeting virulence factors, antibodies and biologics, bacteriophages or phage‐derived products, and immunomodulating agents (World Health Organization, 2022). Not included in these classes are two antibacterial agents classified as potentiators/enablers, and one agent against another cellular target (preclinical development, Appendix Table S6).
Agents targeting virulence factors (Pathoblockers)
Virulence factors are small molecules, enzymes and proteins that pathogenic bacteria rely on to infect, colonise, dominate and damage its host as well as to evade the host immune response and treatment with antibiotics. Targeting virulence factors, hence disarming bacteria instead of killing them, has attracted increasing attention within the past two decades (Clatworthy et al, 2007; Calvert et al, 2018). This approach is promising, because it reduces the damage caused by the pathogen and facilitates its clearance by the immune system, while commensal bacteria remain unaffected. Furthermore, virulence factors are non‐essential targets, which should reduce the selective pressure for resistance to evolve (Calvert et al, 2018; Lakemeyer et al, 2018; Theuretzbacher & Piddock, 2019). While screening for agents that target virulence factors can be done in biochemical assays or in in vivo infection models (Miethke et al, 2021) preclinical and clinical proof‐of‐concept are more challenging to demonstrate.
Common targets in research and preclinical development include bacterial communication by quorum sensing, direct inhibition of exotoxin function, toxin secretion via type III secretion systems (T3SS), bacterial adhesion and biofilm formation, enzymes for host invasion (e.g. LasB) or immune evasion as well as virulence‐regulating enzymes (Calvert et al, 2018; Lakemeyer et al, 2018; Theuretzbacher & Piddock, 2019). Those targets can be addressed with small or large molecules or with biologics, as reviewed in the next chapter.
Ftorziazinone (phase 2, oral) reduces the adverse effects of infections with Gram‐negative bacteria by inhibiting T3SS. While not displaying antibacterial properties in vitro, it showed activity against P. aeruginosa and other Gram‐negative pathogens in murine infection models (Sheremet et al, 2018; Zigangirova et al, 2021). Ftorziazinone is tested in combination with the cephalosporin cefepime for the treatment of cUTI caused by P. aeruginosa (World Health Organization, 2022).
CAL02 (phase 1, i.v.) was designed to reduce the adverse effects of bacterial infections. It consists of engineered liposomes to intercept bacterial toxins by mimicking the cell‐membrane lipids these toxins bind to (Da Azeredo & Shorr, 2020). The benefits of CAL02 as an addition to standard therapy of pneumonia caused by Streptococcus pneumoniae were demonstrated in murine models and first‐in‐human trials (Da Azeredo Silveira & Shorr, 2020; Laterre et al, 2019).
GSK3882347 (phase 1, oral), which is developed for treating uUTI caused by E. coli, pursues a different approach: this compound prevents the binding of the pathogen to the bladder wall by inhibiting the E. coli adhesive protein FimH (World Health Organization, 2022).
The small molecule ALS‐4 (phase 1, oral) inhibits a key enzyme in the staphyloxanthin biosynthesis in S. aureus (Aptorum Group Limited, 2022; World Health Organization, 2022). As this pigment protects S. aureus from the oxidative stress of neutrophils (Beavers & Skaar, 2016), ALS‐4 increased clearance of S. aureus in vitro and in in vivo animal infection models (Aptorum Group Limited, 2022).
Currently, only one molecule with the overall MoAs “anti‐virulence” is in preclinical development but little information is available (Appendix Table S6).
Antibodies and biologics
During the past 30 years, monoclonal antibodies have become an important modality in the treatment of cancer and autoimmune diseases. Yet, no antibodies are marketed to directly target bacteria in infections. The only three antibodies blocking toxins or other virulence factors of Bacillus anthracis (2) or C. difficile (1) were all approved within the last decade (McConnell, 2019).
Antibodies have two main modes of action. First, by binding to antigens located on the bacterial surface with subsequent opsonophagocytic killing by the immune system. Second, antibodies can bind and neutralise bacterial virulence factors like exotoxins or biofilms, reducing the negative effects on the host or helping the host to clear the infection (Lakemeyer et al, 2018; McConnell, 2019). It seems that the second approach is more promising, as all three approved antibodies target bacterial toxins, while antibodies targeting antigens on the bacterial surface have failed so far to demonstrate their value in clinical studies (Chastre et al, 2020; National Library of Medicine NCT03027609, 2022).
The four monoclonal antibodies (mAb) in clinical development therefore do not target the bacterial surface (World Health Organization, 2022). Tosatoxumab (phase 3, i.v.) is an immunoglobulin G1 (IgG1) antibody against S. aureus α‐toxin and was discovered in a screening for α‐toxin neutralisation activity in B cells from a S. aureus pneumonia patient (François et al, 2018). It is developed for the adjunctive treatment of patients with VAP, caused by S. aureus (World Health Organization, 2022). Suvratoxumab (phase 2, i.v.) is also an IgG1 antibody, but with enhanced serum half‐life and a dual mechanism of α‐toxin neutralisation to block binding to its cell receptor and inhibit the formation of the lytic heptameric trans‐membrane conformation (Oganesyan et al, 2014). It is developed to prevent S. aureus caused pneumonia in mechanically ventilated patients (François et al, 2021). 9MW1411 (phase 1, i.v.) is a monoclonal antibody to neutralise S. aureus α‐toxin by blocking its binding to the ADAM10 receptor on the cell membrane (World Health Organization, 2022).
A broader spectrum of bacteria can be targeted by TRL1068 (phase 1, i.v.), a human antibody that binds to proteins from the DNABII family, which is conserved among many Gram‐positive and ‐negative bacteria (Estellés et al, 2016). Biofilm disruption of S. aureus, P. aeruginosa and A. baumannii was achieved in vitro and TRL1068 potentiated antibiotic activities in murine and rodent infection models with S. aureus and A. baumannii (Estellés et al, 2016; Xiong et al, 2017). It is developed as an adjunctive to standard‐of‐care treatment in periprosthetic joint infections with a broad range of Gram‐positive and Gram‐negative bacteria (World Health Organization, 2022). However, the potential effect of this antibody on commensals and their biofilms requires further studies.
Three other antibodies or biologics are currently in preclinical development (Appendix Table S6).
Bacteriophages or phage‐derived products
Bacteriophages are viruses that infect bacteria and archaea (Kortright et al, 2019). After their discovery in the early 20th century, they were used to treat bacterial infections, but the use of and interest in bacteriophages decreased greatly after antibiotics became widely available (Lakemeyer et al, 2018). Phages have been extensively studied and used in Georgia, Poland and Russia (Abedon et al, 2017), but most existing data are based on anecdotal case studies or reports of compassionate use (Theuretzbacher & Piddock, 2019).
The potential benefits and opportunities of phage therapy are well known: good activity against sensitive bacteria, even in biofilms, no cross‐resistance with antibiotics, and a very high selectivity for pathogens (Iskandar et al, 2022). However, application of phage therapy in broad clinical settings faces several difficulties and concerns. This includes limited knowledge about the pharmacokinetics of the phage, the risk of immunogenic reactions of the host to the treatment, possible gene transfer to commensal bacteria, and rapid evolution of resistance against the administered bacteriophages (Lakemeyer et al, 2018; Theuretzbacher & Piddock, 2019; Vázquez et al, 2022). Furthermore, since phages are exclusive to the bacterial species or even only subsets of strains of the particular species they infect (Kortright et al, 2019), cocktails of different phages must be used (Theuretzbacher & Piddock, 2019). This requires appropriate diagnostic tools and potentially the adaptation of cocktails, depending on the infecting pathogen (Box 1; Theuretzbacher & Piddock, 2019).
Many of these problems do not apply for the use of phage‐derived products, such as phage endolysins. These are endopeptidases that degrade the peptidoglycans of bacteria and result in cell lysis (Abdelrahman et al, 2021). By analogy to bacteriophages, endolysins have a high specificity towards the targeted bacteria, and are both potent as well as bactericidal (Abdelrahman et al, 2021). Hitherto, the published data on safety, immunogenicity and resistance formation are sparse, but endolysins seem to be well tolerated by mammals, including humans, and no resistance has been reported so far (Jun et al, 2017; Abdelrahman et al, 2021).
Currently, two bacteriophage projects and two phage endolysins are in clinical development (World Health Organization, 2022). The project “phage” (Phase 1/2, i.v.) is undergoing clinical trials for the treatment of UTIs with E. coli or K. pneumoniae using personalised combinations of bacteriophages (ClinicalTrials.gov NCT04287478, 2022; World Health Organization, 2022). LBP‐EC01 (phase 1b, i.v.) is a cocktail of bacteriophages that are engineered with CRISPR to target the genome of E. coli (World Health Organization, 2022). This concoction combines the lytic activity of phages with Cas3‐activity against DNA to treat patients with lower urinary‐tract colonisation with E. coli (National Library of Medicine NCT04191148, 2022; World Health Organization, 2022).
Exebase (phase 3, i.v.) is a recombinantly produced phage endolysin against Staphylococci and Streptococci that was found in a prophage of a Streptococcus suis genome (Gilmer et al, 2013). Tonacabase (SAL200, phase 2a/1, i.v.) is another recombinantly produced endolysin with anti‐staphylococcal activity (Jun et al, 2011). Both phage endolysins are in clinical development as addition to standard treatment for blood‐stream infections with S. aureus (World Health Organization, 2022).
A total of 17 bacteriophages or their products are currently in preclinical development. Twelve of these projects are intended to treat infections with Gram‐negative critical‐priority pathogens and three to treat infections with S. aureus (Appendix Table S6). However, there is no conclusive information about the exact nature of treatment or the anticipated type of application.
Immunomodulating agents
In contrast to all the other described approaches, immunomodulating agents do not target pathogens or their toxins, but modify the response of the patients' immune system to bacterial infection. Immunomodulation has demonstrated its usefulness in cancer therapy during the past decades and some of the applied approaches might be extrapolated for the treatment of infectious diseases (Naran et al, 2018; McCulloch et al, 2022).
The peptide reltecimod (new drug application, i.v.) mimics the T‐lymphocyte CD28 receptor and reduces inflammatory cytokine expression caused by exotoxins from Gram‐positive bacteria or lipopolysaccharides from Gram‐negative bacteria (Ramachandran et al, 2013; Ramachandran et al, 2015). Addition of reltecimod to standard treatment improved the resolution of organ dysfunction and hospital discharge status in patients with necrotising soft‐tissue infections compared with standard treatment alone (Bulger et al, 2020). However, no update was provided since its new drug application with the FDA in December 2020 (World Health Organization, 2022).
Recently, low levels of the human plasma protein gelsolin (pGSN) were associated with an increased risk of severe outcomes in patients with CAP. Gelsolin is an abundant protein that modulates inflammatory responses and reinforces the antimicrobial activity of macrophages in alveoli (Self et al, 2019). Rhu‐pGSN (phase 1b/2a, i.v.) is recombinantly produced pGSN that improves survival and reduces lung injuries in animal models and is well‐tolerated in patients with CAP (DiNubile et al, 2020; Tannous et al, 2020).
Two further immunomodulators are currently in preclinical development (Appendix Table S6).
Analysis of the non‐traditional antibacterial pipeline
The strategy to target and reduce bacterial virulence is an interesting and promising field with diverse approaches. While most of these are toxin‐ or pathogen‐specific, there is the possibility for a broad‐spectrum treatment, as demonstrated by CAL02. So far, only limited data is available for the utility of these approaches in clinical settings. In the case of toxin‐targeting mAbs, a proof of concept and value proposition has already been provided. Other approaches to antivirulence therapy still have to confirm their promise in further preclinical and proof‐of‐concept clinical trials. Generally, mAbs and other biologics need to demonstrate their value and their superiority to current treatment to make up for their higher cost (Lakemeyer et al, 2018).
The development of bacteriophages as antibacterial treatment is an emerging and dynamic field. Of course, further research has to be undertaken in diagnostics to quickly choose the fitting phage (−cocktail) and clinical studies to expand the knowledge about the efficacy, effects and requirements of bacteriophages as antibacterial treatment (Box 1; Suh et al, 2022). While the approval of a bacteriophage preparation for broad application seems to be unlikely in the near future, specialised hospitals and centres can already treat patients with individualised phage cocktails, based on expanded access regulations in the USA or magistral formulations in several EU countries (Suh et al, 2022; Vázquez et al, 2022). In doing so, experiences and data will become available to further facilitate the approval of phage therapy as antibacterial treatment.
Treating an infection via immune‐system modulating or supporting agents has the advantage to be pathogen‐agnostic. Given the success of therapies targeting the immune system in other indications and the advancement of reltecimod through clinical trials, it seems a promising approach. However, the pending approval of reltecimod raises the question whether further problems must be addressed.
Compared with traditional antibiotics, these approaches share the advantages of avoiding negative effects on the host commensal bacteria and resistances against antibiotics. Nonetheless, the knowledge about and experience with non‐traditional antibacterial agents is still limited, and many more problems must be overcome before their broad application can be established. For example, the high selectivity that is often required by virulence‐targeting agents, biologics and bacteriophages raises the need for simple, quick and accurate identification of the infecting pathogen (Theuretzbacher & Piddock, 2019). Furthermore, most non‐traditional antibacterial therapies are developed as add‐ons to standard treatment regimes, with the view to demonstrate superiority in confirmatory clinical trials. While some non‐traditional antibacterial therapies might not need the concomitant use of antibiotics in the future, combinations of traditional and non‐traditional antibacterial therapies seem likely. Therefore, most non‐traditional approaches will not have the potential to solve AMR on their own (Box 1; Czaplewski et al, 2016; Theuretzbacher & Piddock, 2019).
Conclusion and outlook
Considering the ever‐increasing prevalence of AMR, it is crucial to find and develop new antibiotics and non‐traditional antibacterials. As the emergence of resistance against antibiotics in use is inevitable, a constant flow of new antibacterials into and out of the clinical pipeline is important. The latter must be enabled and reinforced by vibrant early, translational and preclinical research.
These new antibiotics should address the problems and needs in treatment of bacterial infections. Currently, two major problems need particular attention.
First, very few antibiotics are available to treat nosocomial infections with MDR Gram‐negative bacteria classified as critical priority pathogens by the WHO. While the clinical pipeline harbours several improved and promising β‐lactam/BLI, it lacks new antibiotic scaffolds with direct antibacterial activity. We therefore need increased efforts to find and develop new antibiotic classes with activity against MDR Enterobacteriaceae, A. baumannii and P. aeruginosa.
Second, further treatment options, in particular with oral bioavailability for community‐acquired UTIs with MDR pathogens are required, especially considering the decreased use of fluoroquinolones due to blackbox warnings (Yarrington et al, 2019). Regarding the treatment of gonorrhoeae, the two first‐in‐class antibiotics gepotidacin and zoliflodacin will complete their clinical trials in the near future and become available to address this problem. However, these two candidates will not be sufficient to tackle these infections alone.
Two new antibiotic classes are in clinical development against MDR S. aureus—only as infections with this pathogen can be hard to treat. The difficulty is caused by production of biofilms or insufficient antibiotic concentrations at the site of infection and not due to a lack of antibiotics with appropriate activities. While it is a welcome trend that new antibiotic classes are developed, time will show if these can overcome this difficulty or if additional strategies are required.
With the exception of mAbs targeting bacterial toxins, non‐traditional antibacterials still need to show their value in a broad clinical setting. Until they have done so, we need to rely on traditional antibiotics for the treatment of bacterial infections. Nevertheless, many of these approaches show encouraging potential to reinforce our capability to fight bacterial infections. Depending on their success in clinical development, combinations of antibiotics with agents targeting virulence factors or immunomodulating agents might be promising future strategies for the treatment of bacterial infections (Box 1).
Regardless of their target, a constant supply of new agents is needed to maintain the antibacterial pipeline and thus additional resources must be channelled into respective research in natural products and medicinal chemistry.
Natural product–based drug discovery continues to be one of the most reliable sources of novel chemical agents. In part, this has been driven by comprehensive investigation of priority producer microorganisms, such as Streptomyces. Evaluation of natural products databases, such as the NPAtlas (van Santen et al, 2022) shows a very unbalanced investigation of producer organisms (Fig 5A). Members of the phylum Actinobacteria dominate the chemical space of naturally produced compounds in the database with the single genus Streptomyces accounting for more than 70% of the compounds from the entire phylum. When excluding this extensively studied genus, the top three phyla show a more equitable distribution of compounds. However, as has been mentioned in recent publications, the current state of bacterial compound discovery does not reflect the potential of natural compound producers (Fig 5B; Gavriilidou et al, 2022). Investigation of these organisms and advances in compound production, purification, identification and screening methodologies will expand the chemistry available for the antibacterial discovery pipeline.
Figure 5. Current bacterial natural products and the encoded potential for others.

(A) The number of compounds per bacterial phylum as found in the NPAtlas_v2021_08. Inset shows phyla with fewer than 200 compounds. Phyla are distributed as shown in the legend. Some phyla have since been subdivided and are not representative of the tree in part B (e.g. Myxococcota (“Myxobacteria”) is found in the NPAtlas within the phylum Proteobacteria). The total number of compounds includes analogues of the same compound family and does not necessarily represent distinct chemical scaffolds. Source data in Appendix Tables S10 and S11. (B) Representation of the biosynthetic potential of bacterial phyla based on the number of genomes per phylum and the estimated number of gene cluster families. This number estimates the number of different compounds possible from members of these phyla. Part (B) reproduced from Garvriilidou et al with permission from Springer Nature.Source data are available online for this figure.
Intriguing discoveries were made in medicinal chemistry with computational tool‐based rational design, such as the novel DBO‐BLIs with antibacterial activity. Hopefully, it will become apparent in the near future whether the new derivative in preclinical development, ETX0462, with activity against Gram‐negative pathogens can be advanced into clinical development as monotherapy (Durand‐Reville et al, 2021). This would be exciting news as it would elevate DBOs as a novel class of antibiotics with anti‐Gram‐negative activity. Moreover, the future will show whether this approach can lead to further novel classes of antibacterials.
With the experiences gained from the shortcomings of the Waksman platform and HTS campaigns and the past decade in natural product research and medicinal chemistry, we might have the tools to tackle the antibiotic crisis (Box 1). Given the high attrition rates, high research and development costs and low profit margins associated with antibacterial therapies, sufficient funding and concerted efforts must be ensured (Box 1). Furthermore, approval processes should reflect the special characteristics of traditional and non‐traditional antibacterials.
Considering the high attrition rates of antibacterials in clinical trials, a great variety of these agents in early development is necessary to ensure a constant supply to the market. As increasing rates of AMR threaten to leave us without treatment options for certain bacterial infections, we cannot afford to waste time and resources by performing redundant or futile research. Therefore, it would be immensely helpful for researchers to have more access to peer‐reviewed data of early and translational research as well as preclinical studies. As there is no valid information available for the majority of antibacterial projects in lead optimisation and in preclinical studies, it is difficult to estimate their potential for broad clinical use. Moreover, in most cases the reason for the failure of candidates, especially in early and preclinical development, is not published. To minimise repetition of mistakes and speed up the development process, it would be beneficial to have a platform where negative results can be made publicly available.
AMR is a major problem, which can only be tackled when we join forces.
Author contributions
Sebastian Walesch: Conceptualization; writing—original draft; writing—review and editing. Joy Birkelbach: Conceptualization; visualization; writing—original draft; writing—review and editing. Gwenaëlle Jézéquel: Writing—original draft; writing—review and editing. FP Jake Haeckl: Visualization; writing—original draft; writing—review and editing. Julian D Hegemann: Writing—review and editing. Thomas Hesterkamp: Writing—original draft; writing—review and editing. Anna KH Hirsch: Supervision; writing—review and editing. Peter Hammann: Conceptualization; visualization; writing—review and editing. Rolf Müller: Conceptualization; supervision; writing—review and editing.
Disclosure and competing interest statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 5
Acknowledgements
We thank Daniel Krug and Jordan Espenshade for scientific discussions, literature recommendations and proof reading. Research in R.M.'s laboratory is funded by the Helmholtz Association (HGF), the German Research Foundation (DFG), the Federal Ministry of Education and Research, Germany (BMBF) and the German Center for Infection Research (DZIF). Open Access funding enabled and organized by Projekt DEAL.
EMBO Reports (2023) 24: e56033.
See also: J D Hegemann et al (January 2023)
References
- Abdelrahman F, Easwaran M, Daramola OI, Ragab S, Lynch S, Oduselu TJ, Khan FM, Ayobami A, Adnan F, Torrents E et al (2021) Phage‐encoded endolysins. Antibiotics 10: 124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul‐Mutakabbir JC, Alosaimy S, Morrisette T, Kebriaei R, Rybak MJ (2020) Cefiderocol: a novel Siderophore cephalosporin against multidrug‐resistant gram‐negative pathogens. Pharmacotherapy 40: 1228–1247 [DOI] [PubMed] [Google Scholar]
- Abedon ST, García P, Mullany P, Aminov R (2017) Editorial: Phage therapy: past, present and future. Front Microbiol 8: 981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham EP, Chain E (1940) An enzyme from bacteria able to destroy penicillin. Nature 146: 837 [PubMed] [Google Scholar]
- Abraham EP, Chain E, Fletcher CM, Gardner AD, Heatley NG, Jennings MA, Florey HW (1941) Further observations on penicillin. Lancet 238: 177–189 [PubMed] [Google Scholar]
- Adjunctive Therapeutic Treatment With Human Monoclonal Antibody AR-105 (Aerucin®) in P. Aeruginosa Pneumonia (2022) ClinicalTrials.gov NCT03027609 (https://clinicaltrials.gov/show/NCT03027609) [DATASET]
- Adnani N, Chevrette MG, Adibhatla SN, Zhang F, Yu Q, Braun DR, Nelson J, Simpkins SW, McDonald BR, Myers CL et al (2017) Coculture of marine invertebrate‐associated bacteria and interdisciplinary technologies enable biosynthesis and discovery of a new antibiotic, Keyicin. ACS Chem Biol 12: 3093–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed Y, Rebets Y, Estévez MR, Zapp J, Myronovskyi M, Luzhetskyy A (2020) Engineering of Streptomyces lividans for heterologous expression of secondary metabolite gene clusters. Microb Cell Fact 19: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanjary M, Kronmiller B, Adamek M, Blin K, Weber T, Huson D, Philmus B, Ziemert N (2017) The antibiotic resistant target seeker (ARTS), an exploration engine for antibiotic cluster prioritization and novel drug target discovery. Nucleic Acids Res 45: W42–W48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NE, Hobbs JN, Alborn WE (1987) Inhibition of peptidoglycan biosynthesis in gram‐positive bacteria by LY146032. Antimicrob Agents Chemother 31: 1093–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm RA, Gallant K (2020) Innovation in antimicrobial resistance: The CARB‐X perspective. ACS Infect Dis 6: 1317–1322 [DOI] [PubMed] [Google Scholar]
- Ambler RP (1980) The structure of beta‐lactamases. Philos Trans R Soc Lond B Biol Sci 289: 321–331 [DOI] [PubMed] [Google Scholar]
- Aoki T, Yoshizawa H, Yamawaki K, Yokoo K, Sato J, Hisakawa S, Hasegawa Y, Kusano H, Sano M, Sugimoto H et al (2018) Cefiderocol (S‐649266), a new siderophore cephalosporin exhibiting potent activities against Pseudomonas aeruginosa and other gram‐negative pathogens including multi‐drug resistant bacteria: structure activity relationship. Eur J Med Chem 155: 847–868 [DOI] [PubMed] [Google Scholar]
- Aptorum Group Limited (2022) ALS‐4 non‐confidential overview . (http://www.aptorumgroup.com/ALS_Jun2022.pdf)
- Bacqué E, Leroi‐Geissler C, Fievet A, Dubarry N, Silve S, Cazals V, Rey A, Sordello S, Sentausa E, Vermat T et al (2022) Corramycin2, a new class potent antibacterial candidate with a novel mode of penetration into bacteria, a novel mechanism of action and compelling activities in animal models of infection . ASM Microbe 2022
- Bacteriophage therapy in patients with urinary tract infections (2022) ClinicalTrials.gov NCT04287478 (https://clinicaltrials.gov/show/NCT04287478) [DATASET]
- Balasegaram M, Piddock LJV (2020) The Global Antibiotic Research and Development Partnership (GARDP) not‐for‐profit model of antibiotic development. ACS Infect Dis 6: 1295–1298 [DOI] [PubMed] [Google Scholar]
- Baldoni D, Gutierrez M, Timmer W, Dingemanse J (2014) Cadazolid, a novel antibiotic with potent activity against Clostridium difficile: Safety, tolerability and pharmacokinetics in healthy subjects following single and multiple oral doses. J Antimicrob Chemother 69: 706–714 [DOI] [PubMed] [Google Scholar]
- Baumann S, Herrmann J, Raju R, Steinmetz H, Mohr KI, Huttel S, Harmrolfs K, Stadler M, Muller R (2014) Cystobactamids: myxobacterial topoisomerase inhibitors exhibiting potent antibacterial activity. Angew Chem Int Ed Engl 53: 14605–14609 [DOI] [PubMed] [Google Scholar]
- Bax R, Green S (2015) Antibiotics: the changing regulatory and pharmaceutical industry paradigm. J Antimicrob Chemother 70: 1281–1284 [DOI] [PubMed] [Google Scholar]
- Bax BD, Chan PF, Eggleston DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M et al (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466: 935–940 [DOI] [PubMed] [Google Scholar]
- Beavers WN, Skaar EP (2016) Neutrophil‐generated oxidative stress and protein damage in Staphylococcus aureus . Pathog Dis 74: ftw060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behroz I, Durkin P, Grätz S, Seidel M, Rostock L, Spinczyk M, Weston JB, Süssmuth RD (2019) Extensive structure‐activity relationship study of Albicidin's C‐terminal Dipeptidic p‐Aminobenzoic acid moiety. Chemistry 25: 16538–16543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley SD, Chater KF, Cerdeno‐Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D et al (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417: 141–147 [DOI] [PubMed] [Google Scholar]
- Bertrand S, Bohni N, Schnee S, Schumpp O, Gindro K, Wolfender J‐L (2014) Metabolite induction via microorganism co‐culture: a potential way to enhance chemical diversity for drug discovery. Biotechnol Adv 32: 1180–1204 [DOI] [PubMed] [Google Scholar]
- Bilyk B, Horbal L, Luzhetskyy A (2017) Chromosomal position effect influences the heterologous expression of genes and biosynthetic gene clusters in Streptomyces albus J1074. Microb Cell Fact 16: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerketorp J, Levenfors JJ, Nord C, Guss B, Öberg B, Broberg A (2021) Selective isolation of multidrug‐resistant Pedobacter spp., producers of novel antibacterial peptides. Front Microbiol 12: 642829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black TA, Buchwald UK (2021) The pipeline of new molecules and regimens against drug‐resistant tuberculosis. J Clin Tuberc Other Mycobact Dis 25: 100285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, Shaw S, Kloosterman AM, Charlop‐Powers Z, van Wezel GP, Medema MH, Weber T (2021) antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res 49: W29–W35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhringer N, Green R, Liu Y, Mettal U, Marner M, Modaresi SM, Jakob RP, Wuisan ZG, Maier T, Iinishi A et al (2021) Mutasynthetic production and antimicrobial characterization of Darobactin analogs. Microbiol Spectr 9: e0153521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48: 1–12 [DOI] [PubMed] [Google Scholar]
- Bousis S, Setyawati I, Diamanti E, Slotboom DJ, Hirsch AKH (2019) Energy‐coupling factor transporters as novel antimicrobial targets. Adv Therap 2: 1800066 [Google Scholar]
- Bozhüyük KAJ, Linck A, Tietze A, Kranz J, Wesche F, Nowak S, Fleischhacker F, Shi Y‐N, Grün P, Bode HB (2019) Modification and de novo design of non‐ribosomal peptide synthetases using specific assembly points within condensation domains. Nat Chem 11: 653–661 [DOI] [PubMed] [Google Scholar]
- Bradford PA, Miller AA, O'Donnell J, Mueller JP (2020) Zoliflodacin: an oral spiropyrimidinetrione antibiotic for the treatment of neisseria gonorrheae, including multi‐drug‐resistant isolates. ACS Infect Dis 6: 1332–1345 [DOI] [PubMed] [Google Scholar]
- Bremner J (2021) Multiple action‐based design approaches to antibacterials. Singapore City: Springer Singapore; [Google Scholar]
- Brown P, Abbott E, Abdulle O, Boakes S, Coleman S, Divall N, Duperchy E, Moss S, Rivers D, Simonovic M et al (2019) Design of Next Generation Polymyxins with lower toxicity: the discovery of SPR206. ACS Infect Dis 5: 1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugworks Research Inc. (2022) Pipeline . (https://bugworksresearch.com/pipeline/)
- Bulger EM, May AK, Robinson BRH, Evans DC, Henry S, Green JM, Toschlog E, Sperry JL, Fagenholz P, Martin ND et al (2020) A novel immune modulator for patients with necrotizing soft tissue infections (NSTI): results of a multicenter, phase 3 randomized controlled trial of Reltecimod (AB 103). Ann Surg 272: 469–478 [DOI] [PubMed] [Google Scholar]
- Burgos RM, Rodvold KA (2019) Omadacycline: a novel aminomethylcycline. Infect Drug Resist 12: 1895–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K (2015) A resurgence of β‐lactamase inhibitor combinations effective against multidrug‐resistant gram‐negative pathogens. Int J Antimicrob Agents 46: 483–493 [DOI] [PubMed] [Google Scholar]
- Bush K, Jacoby GA (2010) Updated functional classification of beta‐lactamases. Antimicrob Agents Chemother 54: 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MS, Gigante V, Sati H, Paulin S, Al‐Sulaiman L, Rex JH, Fernandes P, Arias CA, Paul M, Thwaites GE et al (2022) Analysis of the clinical pipeline of treatments for drug‐resistant bacterial infections: despite Progress, more action is needed. Antimicrob Agents Chemother 66: e0199121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JM, Waack U, Weinstein EA, Joshi A, Shurland SM, Iarikov D, Bulitta JB, Diep BA, Guina T, Hope WW et al (2020) FDA public workshop summary: advancing animal models for antibacterial drug development. Antimicrob Agents Chemother 65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert MB, Jumde VR, Titz A (2018) Pathoblockers or antivirulence drugs as a new option for the treatment of bacterial infections. Beilstein J Org Chem 14: 2607–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG (2005) A convergent enantioselective route to structurally diverse 6‐deoxytetracycline antibiotics. Science 308: 395–398 [DOI] [PubMed] [Google Scholar]
- Chastre J, François B, Bourgeois M, Komnos A, Ferrer R, Rahav G, de Schryver N, Lepape A, Koksal I, Luyt C‐E et al (2020) 635. Efficacy, pharmacokinetics (PK), and safety profile of MEDI3902, an anti‐pseudomonas aeruginosa bispecific human monoclonal antibody in mechanically ventilated intensive care unit patients; results of the phase 2 EVADE study conducted by the public‐private COMBACTE‐MAGNET consortium in the innovative medicines initiative (IMI) program. Open Forum Infect Dis 7: S377–S378 [Google Scholar]
- Chavan R, Zope V, Yeole R, Patel M (2016) WCK 4873 (Nafithromycin): assessment of In vitro human CYP inhibitory potential of a novel lactone‐Ketolide. Open Forum Infect Dis 3: 1808 [Google Scholar]
- Chiorean S, Antwi I, Carney DW, Kotsogianni I, Giltrap AM, Alexander FM, Cochrane SA, Payne RJ, Martin NI, Henninot A et al (2019) Dissecting the binding interactions of teixobactin with the bacterial cell wall precursor lipid II. Chembiochem [DOI] [PubMed] [Google Scholar]
- Choudhury C, Narahari Sastry G (2019) Pharmacophore modelling and screening: concepts, recent developments and applications in rational drug design. In Structural bioinformatics: applications in preclinical drug discovery process, Mohan CG (ed), pp 25–53. Cham: Springer International Publishing; [Google Scholar]
- Choudhury C, Priyakumar UD, Sastry GN (2015) Dynamics based pharmacophore models for screening potential inhibitors of mycobacterial cyclopropane synthase. J Chem Inf Model 55: 848–860 [DOI] [PubMed] [Google Scholar]
- Claesen J, Spagnolo JB, Ramos SF, Kurita KL, Byrd AL, Aksenov AA, Melnik AV, Wong WR, Wang S, Hernandez RD et al (2020) A Cutibacterium acnes antibiotic modulates human skin microbiota composition in hair follicles. Sci Transl Med 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB, Hunt DK, He M, Achorn C, Chen C‐L, Deng Y, Fyfe C, Grossman TH, Hogan PC, O'Brien WJ et al (2012) Fluorocyclines. 2. Optimization of the C‐9 side‐chain for antibacterial activity and oral efficacy. J Med Chem 55: 606–622 [DOI] [PubMed] [Google Scholar]
- Clatworthy AE, Pierson E, Hung DT (2007) Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol 3: 541–548 [DOI] [PubMed] [Google Scholar]
- Clatworthy AE, Romano KP, Hung DT (2018) Whole‐organism phenotypic screening for anti‐infectives promoting host health. Nat Chem Biol 14: 331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clissold SP, Todd PA, Campoli‐Richards DM (1987) Imipenem/cilastatin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 33: 183–241 [DOI] [PubMed] [Google Scholar]
- Cociancich S, Pesic A, Petras D, Uhlmann S, Kretz J, Schubert V, Vieweg L, Duplan S, Marguerettaz M, Noëll J et al (2015) The gyrase inhibitor albicidin consists of p‐aminobenzoic acids and cyanoalanine. Nat Chem Biol 11: 195–197 [DOI] [PubMed] [Google Scholar]
- Coleman K (2011) Diazabicyclooctanes (DBOs): a potent new class of non‐β‐lactam β‐lactamase inhibitors. Curr Opin Microbiol 14: 550–555 [DOI] [PubMed] [Google Scholar]
- Cook MA, Wright GD (2022) The past, present, and future of antibiotics. Sci Transl Med 14: eabo7793 [DOI] [PubMed] [Google Scholar]
- Corcione S, de Benedetto I, Pinna SM, Vita D, Lupia T, Montrucchio G, Brazzi L, de Rosa FG (2022) Cefiderocol use in gram negative infections with limited therapeutic options: Is combination therapy the key? J Infect Public Health 15: 975–979 [DOI] [PubMed] [Google Scholar]
- Couturier C, Groß S, von Tesmar A, Hoffmann J, Deckarm S, Fievet A, Dubarry N, Taillier T, Pöverlein C, Stump H et al (2022) Structure elucidation, Total synthesis, antibacterial in vivo efficacy and biosynthesis proposal of myxobacterial corramycin. Angew Chem Int Ed Engl 10.1002/anie.202210747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craney A, Ozimok C, Pimentel‐Elardo SM, Capretta A, Nodwell JR (2012) Chemical perturbation of secondary metabolism demonstrates important links to primary metabolism. Chem Biol 19: 1020–1027 [DOI] [PubMed] [Google Scholar]
- Crits‐Christoph A, Diamond S, Butterfield CN, Thomas BC, Banfield JF (2018) Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 558: 440–444 [DOI] [PubMed] [Google Scholar]
- Cully M (2021) A novel single‐agent antibiotic. Nat Rev Microbiol 19: 743 [DOI] [PubMed] [Google Scholar]
- Cummings M, Peters AD, Whitehead GFS, Menon BRK, Micklefield J, Webb SJ, Takano E (2019) Assembling a plug‐and‐play production line for combinatorial biosynthesis of aromatic polyketides in Escherichia coli . PLoS Biol 17: e3000347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplewski L, Bax R, Clokie M, Dawson M, Fairhead H, Fischetti VA, Foster S, Gilmore BF, Hancock REW, Harper D et al (2016) Alternatives to antibiotics‐a pipeline portfolio review. Lancet Infect Dis 16: 239–251 [DOI] [PubMed] [Google Scholar]
- Da Azeredo SS, Shorr AF (2020) Critical parameters for the development of novel therapies for severe and resistant infections‐a case study on CAL02, a non‐traditional broad‐spectrum anti‐virulence drug. Antibiotics 9: 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartois VA, Rubin EJ (2022) Anti‐tuberculosis treatment strategies and drug development: challenges and priorities. Nat Rev Microbiol 20: 685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Costa VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R et al (2011) Antibiotic resistance is ancient. Nature 477: 457–461 [DOI] [PubMed] [Google Scholar]
- Deja EN (2021) Novel ß‐lactamase inhibitors: new weapons in the arms race against antimicrobial resistance. Clin Microbiol Newsl 43: 119–125 [Google Scholar]
- DiMasi JA, Grabowski HG, Hansen RW (2016) Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ 47: 20–33 [DOI] [PubMed] [Google Scholar]
- Dinos GP (2017) The macrolide antibiotic renaissance. Br J Pharmacol 174: 2967–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNubile MJ, Levinson SL, Stossel TP, Lawrenz MB, Warawa JM (2020) Recombinant human plasma gelsolin improves survival and attenuates lung injury in a murine model of multidrug‐resistant Pseudomonas aeruginosa pneumonia. Open Forum Infect Dis 7: ofaa236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA (2014) A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158: 1402–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand‐Réville TF, Guler S, Comita‐Prevoir J, Chen B, Bifulco N, Huynh H, Lahiri S, Shapiro AB, McLeod SM, Carter NM et al (2017) ETX2514 is a broad‐spectrum β‐lactamase inhibitor for the treatment of drug‐resistant gram‐negative bacteria including Acinetobacter baumannii . Nat Microbiol 2: 17104 [DOI] [PubMed] [Google Scholar]
- Durand‐Réville TF, Comita‐Prevoir J, Zhang J, Wu X, May‐Dracka TL, Romero JAC, Wu F, Chen A, Shapiro AB, Carter NM et al (2020) Discovery of an orally available Diazabicyclooctane inhibitor (ETX0282) of class A, C, and D serine β‐lactamases. J Med Chem 63: 12511–12525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand‐Reville TF, Miller AA, O'Donnell JP, Wu X, Sylvester MA, Guler S, Iyer R, Shapiro AB, Carter NM, Velez‐Vega C et al (2021) Rational design of a new antibiotic class for drug‐resistant infections. Nature 597: 698–702 [DOI] [PubMed] [Google Scholar]
- von Eckardstein L, Petras D, Dang T, Cociancich S, Sabri S, Grätz S, Kerwat D, Seidel M, Pesic A, Dorrestein PC et al (2017) Total synthesis and biological assessment of novel Albicidins discovered by mass spectrometric networking. Chemistry 23: 15316–15321 [DOI] [PubMed] [Google Scholar]
- Ehmann DE, Jahić H, Ross PL, Gu R‐F, Hu J, Kern G, Walkup GK, Fisher SL (2012) Avibactam is a covalent, reversible, non‐β‐lactam β‐lactamase inhibitor. Proc Natl Acad Sci U S A 109: 11663–11668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich P (1913) Address in pathology, ON CHEMIOTHERAPY: delivered before the seventeenth international congress of medicine. Br Med J 2: 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgaher WAM, Hamed MM, Baumann S, Herrmann J, Siebenbürger L, Krull J, Cirnski K, Kirschning A, Brönstrup M, Müller R et al (2020) Cystobactamid 507: concise synthesis, mode of action and optimization toward more potent antibiotics. Chemistry 26: 7219–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich R, Löw O (1899) Bakteriolytische Enzyme als Ursache der erworbenen Immunität und die Heilung von Infectionskrankheiten durch dieselben. Med Microbiol Immunol 31: 1–65 [Google Scholar]
- Engel A (2020) Fostering antibiotic development through impact funding. ACS Infect Dis 6: 1311–1312 [DOI] [PubMed] [Google Scholar]
- Ernst M, Kang KB, Caraballo‐Rodríguez AM, Nothias L‐F, Wandy J, Chen C, Wang M, Rogers S, Medema MH, Dorrestein PC et al (2019) MolNetEnhancer: enhanced molecular networks by integrating metabolome mining and annotation tools. Metabolites 9: 144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erol Ö, Schäberle TF, Schmitz A, Rachid S, Gurgui C, El Omari M, Lohr F, Kehraus S, Piel J, Müller R et al (2010) Biosynthesis of the myxobacterial antibiotic corallopyronin a. Chembiochem 11: 1235–1265 [DOI] [PubMed] [Google Scholar]
- Ersoy SC, Heithoff DM, Barnes L, Tripp GK, House JK, Marth JD, Smith JW, Mahan MJ (2017) Correcting a fundamental flaw in the paradigm for antimicrobial susceptibility testing. EBioMedicine 20: 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estellés A, Woischnig A‐K, Liu K, Stephenson R, Lomongsod E, Nguyen D, Zhang J, Heidecker M, Yang Y, Simon RJ et al (2016) A high‐affinity native human antibody disrupts biofilm from Staphylococcus aureus bacteria and potentiates antibiotic efficacy in a mouse implant infection model. Antimicrob Agents Chemother 60: 2292–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Medicines Agency (2022) Guideline on the evaluation of medicinal products indicated for treatment of bacterial infections . (https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐evaluation‐medicinal‐products‐indicated‐treatment‐bacterial‐infections‐revision‐3_en.pdf)
- Evans LE, Krishna A, Ma Y, Webb TE, Marshall DC, Tooke CL, Spencer J, Clarke TB, Armstrong A, Edwards AM (2019) Exploitation of antibiotic resistance as a novel drug target: Development of a β‐lactamase‐activated antibacterial prodrug. J Med Chem 62: 4411–4425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett MJ, Davies DT (2021) Pseudomonas aeruginosa elastase (LasB) as a therapeutic target. Drug Discov Today 26: 2108–2123 [DOI] [PubMed] [Google Scholar]
- Falagas ME, Kasiakou SK (2006) Toxicity of polymyxins: a systematic review of the evidence from old and recent studies. Crit Care 10: R27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farha MA, French S, Stokes JM, Brown ED (2018) Bicarbonate alters bacterial susceptibility to antibiotics by targeting the proton motive force. ACS Infect Dis 4: 382–390 [DOI] [PubMed] [Google Scholar]
- Farrell DJ, Mendes RE, Jones RN (2015) Antimicrobial activity of solithromycin against serotyped macrolide‐resistant Streptococcus pneumoniae isolates collected from U.S. medical centers in 2012. Antimicrob Agents Chemother 59: 2432–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA (2015) Novel Drugs 2015: Summary . (http://wayback.archive‐it.org/7993/20161022195939/http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugInnovation/UCM481709.pdf)
- FDA (2021) Advancing health trough innovation: new drug therapy approvals 2021 . (https://www.fda.gov/media/155227/download)
- FDA (2022) New drugs at FDA: CDER's new molecular entities and new therapeutic biological products . (https://www.fda.gov/drugs/development‐approval‐process‐drugs/new‐drugs‐fda‐cders‐new‐molecular‐entities‐and‐new‐therapeutic‐biological‐products)
- Fernandes de Oliveira LM, Steindorff M, Darisipudi MN, Mrochen DM, Trübe P, Bröker BM, Brönstrup M, Tegge W, Holtfreter S (2021) Discovery of Staphylococcus aureus adhesion inhibitors by automated imaging and their characterization in a mouse model of persistent nasal colonization. Microorganisms 9: 631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes P, Martens E, Bertrand D, Pereira D (2016) The solithromycin journey—it is all in the chemistry. Bioorg Med Chem 24: 6420–6428 [DOI] [PubMed] [Google Scholar]
- Fiers WD, Craighead M, Singh I (2017) Teixobactin and its analogues: a new hope in antibiotic discovery. ACS Infect Dis 3: 688–690 [DOI] [PubMed] [Google Scholar]
- Flamm RK, Rhomberg PR, Sader HS (2017) In vitro activity of the novel lactone ketolide nafithromycin (WCK 4873) against contemporary clinical bacteria from a global surveillance program. Antimicrob Agents Chemother 61: e01230‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleeman R, LaVoi TM, Santos RG, Morales A, Nefzi A, Welmaker GS, Medina‐Franco JL, Giulianotti MA, Houghten RA, Shaw LN (2015) Combinatorial libraries As a tool for the discovery of novel, broad‐spectrum antibacterial agents targeting the ESKAPE pathogens. J Med Chem 58: 3340–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A (1929) On the antibacterial action of cultures of a penicillium: With special reference to their use in isolation of B. influenzæ. Br J Exp Pathol 10: 266–236 [Google Scholar]
- Food and Drug Administration (2017) Antibacterial therapies for patients with an unmet medical need for the treatment of serious bacterial diseases . (https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/antibacterial‐therapies‐patients‐unmet‐medical‐need‐treatment‐serious‐bacterial‐diseases)
- François B, Mercier E, Gonzalez C, Asehnoune K, Nseir S, Fiancette M, Desachy A, Plantefève G, Meziani F, de Lame P‐A et al (2018) Safety and tolerability of a single administration of AR‐301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: first‐in‐human trial. Intensive Care Med 44: 1787–1796 [DOI] [PubMed] [Google Scholar]
- François B, Jafri HS, Chastre J, Sánchez‐García M, Eggimann P, Dequin P‐F, Huberlant V, Viña Soria L, Boulain T, Bretonnière C et al (2021) Efficacy and safety of suvratoxumab for prevention of Staphylococcus aureus ventilator‐associated pneumonia (SAATELLITE): a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, phase 2 pilot trial. Lancet Infect Dis 21: 1313–1323 [DOI] [PubMed] [Google Scholar]
- Fu J, Wenzel SC, Perlova O, Wang J, Gross F, Tang Z, Yin Y, Stewart AF, Muller R, Zhang Y (2008) Efficient transfer of two large secondary metabolite pathway gene clusters into heterologous hosts by transposition. Nucleic Acids Res 36: e113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugitt RB, Luckenbaugh RW (1978) 5‐Halomethyl‐3‐phenyl‐2‐oxazolidinones . US19780876679 A01N43/76;C07D263/20;A61K31/42;C07D263/38
- Galdino ACM, Viganor L, de Castro AA, Da Cunha EFF, Mello TP, Mattos LM, Pereira MD, Hunt MC, O'Shaughnessy M, Howe O et al (2019) Disarming Pseudomonas aeruginosa virulence by the inhibitory action of 1,10‐Phenanthroline‐5,6‐Dione‐based compounds: Elastase B (LasB) as a chemotherapeutic target. Front Microbiol 10: 1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia RO, Krug D, Muller R (2009) Chapter 3. Discovering natural products from myxobacteria with emphasis on rare producer strains in combination with improved analytical methods. Methods Enzymol 458: 59–91 [DOI] [PubMed] [Google Scholar]
- Garcia R, Gerth K, Stadler M, Dogma JR, Irineo J, Muller R (2010) Expanded phylogeny of myxobacteria and evidence for cultivation of the ‘unculturables’. Mol Phylogenet Evol 57: 878–887 [DOI] [PubMed] [Google Scholar]
- Gavriilidou A, Kautsar SA, Zaburannyi N, Krug D, Müller R, Medema MH, Ziemert N (2022) Compendium of specialized metabolite biosynthetic diversity encoded in bacterial genomes. Nat Microbiol 7: 726–735 [DOI] [PubMed] [Google Scholar]
- Gibson EG, Bax B, Chan PF, Osheroff N (2019) Mechanistic and structural basis for the actions of the antibacterial gepotidacin against Staphylococcus aureus gyrase. ACS Infect Dis 5: 570–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmer DB, Schmitz JE, Euler CW, Fischetti VA (2013) Novel bacteriophage lysin with broad lytic activity protects against mixed infection by streptococcus pyogenes and methicillin‐resistant Staphylococcus aureus . Antimicrob Agents Chemother 57: 2743–2750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon EM, Duncton MAJ, Gallop MA (2018) Orally absorbed derivatives of the β‐lactamase inhibitor avibactam. Design of novel prodrugs of sulfate containing drugs. J Med Chem 61: 10340–10344 [DOI] [PubMed] [Google Scholar]
- Gorityala BK, Guchhait G, Fernando DM, Deo S, McKenna SA, Zhanel GG, Kumar A, Schweizer F (2016) Adjuvants based on hybrid antibiotics overcome resistance in Pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew Chem Int Ed Engl 55: 555–559 [DOI] [PubMed] [Google Scholar]
- Grabowski K, Schneider G (2007) Properties and architecture of drugs and natural products revisited. Curr Chem Biol 1: 115–127 [Google Scholar]
- Grandclaudon C, Birudukota NVS, Elgaher WAM, Jumde RP, Yahiaoui S, Arisetti N, Hennessen F, Hüttel S, Stadler M, Herrmann J et al (2019) Semisynthesis and biological evaluation of amidochelocardin derivatives as broad‐spectrum antibiotics. Eur J Med Chem 188: 112005 [DOI] [PubMed] [Google Scholar]
- Grkovic T, Akee RK, Thornburg CC, Trinh SK, Britt JR, Harris MJ, Evans JR, Kang U, Ensel S, Henrich CJ et al (2020) National Cancer Institute (NCI) program for natural products discovery: rapid isolation and identification of biologically active natural products from the NCI Prefractionated library. ACS Chem Biol 15: 1104–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groß S, Panter F, Pogorevc D, Seyfert CE, Deckarm S, Bader CD, Herrmann J, Müller R (2021a) Improved broad‐spectrum antibiotics against gram‐negative pathogens via darobactin biosynthetic pathway engineering. Chem Sci 12: 11882–11893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groß S, Schnell B, Haack PA, Auerbach D, Müller R (2021b) In vivo and in vitro reconstitution of unique key steps in cystobactamid antibiotic biosynthesis. Nat Commun 12: 1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habich D, von Nussbaum F (2006) Platensimycin, a new antibiotic and “superbug challenger” from nature. ChemMedChem 1: 951–954 [DOI] [PubMed] [Google Scholar]
- Hamrick JC, Docquier J‐D, Uehara T, Myers CL, Six DA, Chatwin CL, John KJ, Vernacchio SF, Cusick SM, Trout REL et al (2020) VNRX‐5133 (Taniborbactam), a broad‐Spectrum inhibitor of serine‐ and Metallo‐β‐lactamases, restores activity of cefepime in enterobacterales and Pseudomonas aeruginosa . Antimicrob Agents Chemother 64: e01963‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison F, Roberts AEL, Gabrilska R, Rumbaugh KP, Lee C, Diggle SP (2015) A 1,000‐year‐old antimicrobial remedy with Antistaphylococcal activity. MBio 6: e01129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE et al (2008) An inhibitor of FtsZ with potent and selective anti‐staphylococcal activity. Science 321: 1673–1675 [DOI] [PubMed] [Google Scholar]
- He Y, Fan A, Han M, Zhang Y, Tong Y, Zheng G, Zhu S (2020) New perspectives on the treatment of mycobacterial infections using antibiotics. Appl Microbiol Biotechnol 104: 4197–4209 [DOI] [PubMed] [Google Scholar]
- Heath RJ, Rock CO (1995) Enoyl‐acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli . J Biol Chem 270: 26538–26542 [DOI] [PubMed] [Google Scholar]
- Hecker SJ, Reddy KR, Totrov M, Hirst GC, Lomovskaya O, Griffith DC, King P, Tsivkovski R, Sun D, Sabet M et al (2015) Discovery of a cyclic boronic acid β‐lactamase inhibitor (RPX7009) with utility vs. class A serine carbapenemases. J Med Chem 58: 3682–3692 [DOI] [PubMed] [Google Scholar]
- Hegemann JD, Birkelbach J, Walesch S, Müller R (2022) Current developments in antibiotic discovery. EMBO Rep 24: e56184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessen F, Miethke M, Zaburannyi N, Loose M, Lukežič T, Bernecker S, Hüttel S, Jansen R, Schmiedel J, Fritzenwanker M et al (2020) Amidochelocardin overcomes resistance mechanisms exerted on tetracyclines and natural chelocardin. Antibiotics 9: 619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann J, Lukezic T, Kling A, Baumann S, Huttel S, Petkovic H, Muller R (2016) Strategies for the discovery and development of new antibiotics from natural products: three case studies. Curr Top Microbiol Immunol 398: 339–363 [DOI] [PubMed] [Google Scholar]
- Hobson C, Chan AN, Wright GD (2021) The antibiotic resistome: a guide for the discovery of natural products as antimicrobial agents. Chem Rev 121: 3464–3494 [DOI] [PubMed] [Google Scholar]
- Hoffmann T, Krug D, Bozkurt N, Duddela S, Jansen R, Garcia R, Gerth K, Steinmetz H, Müller R (2018) Correlating chemical diversity with taxonomic distance for discovery of natural products in myxobacteria. Nat Commun 9: 803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeyman L, Ismail M, Nelson ML, Bhatia B, Bowser TE, Chen J, Mechiche R, Ohemeng K, Verma AK, Cannon EP et al (2015) Structure‐activity relationship of the aminomethylcyclines and the discovery of omadacycline. Antimicrob Agents Chemother 59: 7044–7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hover BM, Kim S‐H, Katz M, Charlop‐Powers Z, Owen JG, Ternei MA, Maniko J, Estrela AB, Molina H, Park S et al (2018) Culture‐independent discovery of the malacidins as calcium‐dependent antibiotics with activity against multidrug‐resistant gram‐positive pathogens. Nat Microbiol 3: 415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huband MD, Thompson JD, Gurung ND, Liu Q, Li L, Zhang J, Streit JM, Castanheira M (2022) Activity of the novel Aminomethylcycline KBP‐7072 and comparators against 1,057 geographically diverse recent clinical isolates from the SENTRY surveillance program, 2019. Antimicrob Agents Chemother 66: e0139721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug JJ, Panter F, Krug D, Müller R (2019) Genome mining reveals uncommon alkylpyrones as type III PKS products from myxobacteria. J Ind Microbiol Biotechnol 46: 319–334 [DOI] [PubMed] [Google Scholar]
- Hutchings M, Truman A, Wilkinson B (2019) Antibiotics: past, present and future. Curr Opin Microbiol 51: 72–80 [DOI] [PubMed] [Google Scholar]
- Hüttel S, Testolin G, Herrmann J, Planke T, Gille F, Moreno M, Stadler M, Brönstrup M, Kirschning A, Müller R (2017) Discovery and Total synthesis of natural cystobactamid derivatives with superior activity against gram‐negative pathogens. Angew Chem Int Ed Engl 56: 12760–12764 [DOI] [PubMed] [Google Scholar]
- Hutter B, Fischer C, Jacobi A, Schaab C, Loferer H (2004) Panel of Bacillus subtilis reporter strains indicative of various modes of action. Antimicrob Agents Chemother 48: 2588–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Lee N, Cho S, Palsson B, Cho B‐K (2020) Repurposing modular polyketide synthases and non‐ribosomal peptide synthetases for novel chemical biosynthesis. Front Mol Biosci 7: 87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde KD, Xu J, Rapior S, Jeewon R, Lumyong S, Niego AGT, Abeywickrama PD, Aluthmuhandiram JVS, Brahamanage RS, Brooks S et al (2019) The amazing potential of fungi: 50 ways we can exploit fungi industrially. Fungal Diversity 97: 1–136 [Google Scholar]
- IFPMA (2020) New AMR Action fund steps in to save collapsing antibiotic pipeline with pharmaceutical industry investment of US$1 billion: partnership aims to bring 2 to 4 new antibiotics to patients by the end of the decade and facilitate needed long‐term policy solutions. Geneva.
- Imai Y, Meyer KJ, Iinishi A, Favre‐Godal Q, Green R, Manuse S, Caboni M, Mori M, Niles S, Ghiglieri M et al (2019) A new antibiotic selectively kills gram‐negative pathogens. Nature 576: 459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irschik H, Jansen R, Höfle G, Gerth K, Reichenbach H (1985) The corallopyronins, new inhibitors of bacterial RNA synthesis from Myxobacteria. J Antibiot 38: 145–152 [DOI] [PubMed] [Google Scholar]
- Isabella VM, Campbell AJ, Manchester J, Sylvester M, Nayar AS, Ferguson KE, Tommasi R, Miller AA (2015) Toward the rational design of carbapenem uptake in Pseudomonas aeruginosa . Chem Biol 22: 535–547 [DOI] [PubMed] [Google Scholar]
- Iskandar K, Murugaiyan J, Hammoudi Halat D, Hage SE, Chibabhai V, Adukkadukkam S, Roques C, Molinier L, Salameh P, van Dongen M (2022) Antibiotic discovery and resistance: the chase and the race. Antibiotics 11: 182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Kuramoto Y, Amano H, Kazamori D, Yazaki A (2015) Discovery of WQ‐3810: design, synthesis, and evaluation of 7‐(3‐alkylaminoazetidin‐1‐yl)fluoro‐quinolones as orally active antibacterial agents. Eur J Med Chem 103: 354–360 [DOI] [PubMed] [Google Scholar]
- Ji X‐W, Xue F, Kang Z‐S, Zhong W, Kuan IH‐S, Yang X‐P, Zhu X, Li Y, Lv Y (2020) Model‐informed drug development, pharmacokinetic/pharmacodynamic cutoff value determination, and antibacterial efficacy of benapenem against Enterobacteriaceae . Antimicrob Agents Chemother 64: e01751‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston CW, Skinnider MA, Dejong CA, Rees PN, Chen GM, Walker CG, French S, Brown ED, Bérdy J, Liu DY et al (2016) Assembly and clustering of natural antibiotics guides target identification. Nat Chem Biol 12: 233–239 [DOI] [PubMed] [Google Scholar]
- Jubeh B, Breijyeh Z, Karaman R (2020) Antibacterial prodrugs to overcome bacterial resistance. Molecules 25: 1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhas M, Widlake E, Teo J, Huseby DL, Tyrrell JM, Polikanov YS, Ercan O, Petersson A, Cao S, Aboklaish AF et al (2019) In vitro activity of apramycin against multidrug‐, carbapenem‐ and aminoglycoside‐resistant Enterobacteriaceae and Acinetobacter baumannii . J Antimicrob Chemother 74: 944–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumde RP, Guardigni M, Gierse RM, Alhayek A, Zhu D, Hamid Z, Johannsen S, Elgaher WAM, Neusens PJ, Nehls C et al (2021) Hit‐optimization using target‐directed dynamic combinatorial chemistry: development of inhibitors of the anti‐infective target 1‐deoxy‐d‐xylulose‐5‐phosphate synthase. Chem Sci 12: 7775–7785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun SY, Jung GM, Son J‐S, Yoon SJ, Choi Y‐J, Kang SH (2011) Comparison of the antibacterial properties of phage endolysins SAL‐1 and LysK. Antimicrob Agents Chemother 55: 1764–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun SY, Jang IJ, Yoon S, Jang K, Yu K‐S, Cho JY, Seong M‐W, Jung GM, Yoon SJ, Kang SH (2017) Pharmacokinetics and tolerance of the phage endolysin‐based candidate drug SAL200 after a single intravenous administration among healthy volunteers. Antimicrob Agents Chemother 61: e02629‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallifidas D, Kang H‐S, Brady SF (2012) Tetarimycin a, an MRSA‐active antibiotic identified through induced expression of environmental DNA gene clusters. J Am Chem Soc 134: 19552–19555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakonstantis S, Rousaki M, Kritsotakis EI (2022) Cefiderocol: systematic review of mechanisms of resistance, heteroresistance and in vivo emergence of resistance. Antibiotics 11: 723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas JA, Chen F, Schneider‐Futschik EK, Kang Z, Hussein M, Swarbrick J, Hoyer D, Giltrap AM, Payne RJ, Li J et al (2020) Synthesis and structure‐activity relationships of teixobactin. Ann N Y Acad Sci 1459: 86–105 [DOI] [PubMed] [Google Scholar]
- Karlowsky JA, Adam HJ, Baxter MR, Denisuik AJ, Lagacé‐Wiens PRS, Walkty AJ, Puttagunta S, Dunne MW, Zhanel GG (2019) In vitro activity of sulopenem, an oral penem, against urinary isolates of Escherichia coli . Antimicrob Agents Chemother 63: e01832‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L, Baltz RH (2016) Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol 43: 155–176 [DOI] [PubMed] [Google Scholar]
- Katz M, Hover BM, Brady SF (2016) Culture‐independent discovery of natural products from soil metagenomes. J Ind Microbiol Biotechnol 43: 129–141 [DOI] [PubMed] [Google Scholar]
- Kaul M, Mark L, Zhang Y, Parhi AK, Lyu YL, Pawlak J, Saravolatz S, Saravolatz LD, Weinstein MP, LaVoie EJ et al (2015) TXA709, an FtsZ‐targeting Benzamide prodrug with improved pharmacokinetics and enhanced In vivo efficacy against methicillin‐resistant Staphylococcus aureus. Antimicrob Agents Chemother 59: 4845–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H, Jakob RP, Marzinek JK, Green R, Imai Y, Bolla JR, Agustoni E, Robinson CV, Bond PJ, Lewis K et al (2021) The antibiotic darobactin mimics a β‐strand to inhibit outer membrane insertase. Nature 593: 125–129 [DOI] [PubMed] [Google Scholar]
- Kautsar SA, Blin K, Shaw S, Navarro‐Muñoz JC, Terlouw BR, van der Hooft JJJ, van Santen JA, Tracanna V, Suarez Duran HG, Pascal Andreu V et al (2020) MIBiG 2.0: a repository for biosynthetic gene clusters of known function. Nucleic Acids Res 48: D454–D458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh F, Hervey A, Robbins WJ (1951) Antibiotic substances from basidiomycetes: VIII. Pleurotus Multilus (Fr.) Sacc. And Pleurotus Passeckerianus Pilat. Proc Natl Acad Sci U S A 37: 570–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya C, Walter I, Yahiaoui S, Sikandar A, Alhayek A, Konstantinović J, Kany AM, Haupenthal J, Köhnke J, Hartmann RW et al (2022) Substrate‐inspired fragment merging and growing affords efficacious LasB inhibitors. Angew Chem Int Ed Engl 61: e202112295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Zhu W, Hendricks GL, van Tyne D, Steele AD, Keohane CE, Fricke N, Conery AL, Shen S, Pan W et al (2018) A new class of synthetic retinoid antibiotics effective against bacterial persisters. Nature 556: 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AM, Reid‐Yu SA, Wang W, King DT, de Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD (2014) Aspergillomarasmine A overcomes metallo‐β‐lactamase antibiotic resistance. Nature 510: 503–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AM, King DT, French S, Brouillette E, Asli A, Alexander JAN, Vuckovic M, Maiti SN, Parr TR, Brown ED et al (2016) Structural and kinetic characterization of Diazabicyclooctanes as dual inhibitors of both serine‐β‐lactamases and penicillin‐binding proteins. ACS Chem Biol 11: 864–868 [DOI] [PubMed] [Google Scholar]
- Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, Zaburannyi N, Herrmann J, Wenzel SC, König C et al (2015) Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 348: 1106–1112 [DOI] [PubMed] [Google Scholar]
- Koga H, Itoh A, Murayama S, Suzue S, Irikura T (1980) Structure‐activity relationships of antibacterial 6,7‐ and 7,8‐disubstituted 1‐alkyl‐1,4‐dihydro‐4‐oxoquinoline‐3‐carboxylic acids. J Med Chem 23: 1358–1363 [DOI] [PubMed] [Google Scholar]
- Konovalova A, Kahne DE, Silhavy TJ (2017) Outer membrane biogenesis. Annu Rev Microbiol 71: 539–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortright KE, Chan BK, Koff JL, Turner PE (2019) Phage therapy: a renewed approach to combat antibiotic‐resistant bacteria. Cell Host Microbe 25: 219–232 [DOI] [PubMed] [Google Scholar]
- Kostyanev T, Bonten MJM, O'Brien S, Steel H, Ross S, François B, Tacconelli E, Winterhalter M, Stavenger RA, Karlén A et al (2016) The innovative medicines initiative's new drugs for bad bugs programme: European public–private partnerships for the development of new strategies to tackle antibiotic resistance. J Antimicrob Chemother 71: 290–295 [DOI] [PubMed] [Google Scholar]
- Kretz J, Kerwat D, Schubert V, Grätz S, Pesic A, Semsary S, Cociancich S, Royer M, Süssmuth RD (2015) Total synthesis of albicidin: a lead structure from Xanthomonas albilineans for potent antibacterial gyrase inhibitors. Angew Chem Int Ed Engl 54: 1969–1973 [DOI] [PubMed] [Google Scholar]
- Krome AK, Becker T, Kehraus S, Schiefer A, Gütschow M, Chaverra‐Muñoz L, Hüttel S, Jansen R, Stadler M, Ehrens A et al (2022) Corallopyronin a: Antimicrobial discovery to preclinical development. Nat Prod Rep 39: 1705–1720 [DOI] [PubMed] [Google Scholar]
- Kumarasamy KK, Toleman MA, Walsh TR, Bagaria J, Butt F, Balakrishnan R, Chaudhary U, Doumith M, Giske CG, Irfan S et al (2010) Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis 10: 597–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo D, Yu G, Hoch W, Gabay D, Long L, Ghannoum M, Nagy N, Harding CV, Viswanathan R, Shoham M (2015) Novel quorum‐quenching agents promote methicillin‐resistant Staphylococcus aureus (MRSA) wound healing and sensitize MRSA to β‐lactam antibiotics. Antimicrob Agents Chemother 59: 1512–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita KL, Glassey E, Linington RG (2015) Integration of high‐content screening and untargeted metabolomics for comprehensive functional annotation of natural product libraries. Proc Natl Acad Sci U S A 112: 11999–12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwa A, Kasiakou SK, Tam VH, Falagas ME (2007) Polymyxin B: similarities to and differences from colistin (polymyxin E). Expert Rev Anti Infect Ther 5: 811–821 [DOI] [PubMed] [Google Scholar]
- Lakemeyer M, Zhao W, Mandl FA, Hammann P, Sieber SA (2018) Thinking outside the box‐novel antibacterials to tackle the resistance crisis. Angew Chem Int Ed Engl 57: 14440–14475 [DOI] [PubMed] [Google Scholar]
- Lampilas M, Rowlands DA, Kebsi A, Ledoussal B, Pierres C, Lampilas M, Rowlands DA, Kebsi A, Ledoussal B, Pierres C (2008) Nitrogenous heterocyclic compounds, preparation thereof and use thereof AS antibacterial medicaments . FR20070002663 A61K31/529;A61P31/00;A61P31/04;C07D211/56;C07D241/00;C07D471/08;C07D471/18
- Laponogov I, Sohi MK, Veselkov DA, Pan X‐S, Sawhney R, Thompson AW, McAuley KE, Fisher LM, Sanderson MR (2009) Structural insight into the quinolone‐DNA cleavage complex of type IIA topoisomerases. Nat Struct Mol Biol 16: 667–669 [DOI] [PubMed] [Google Scholar]
- Laterre P‐F, Colin G, Dequin P‐F, Dugernier T, Boulain T, Da Azeredo Silveira S, Lajaunias F, Perez A, François B (2019) CAL02, a novel antitoxin liposomal agent, in severe pneumococcal pneumonia: a first‐in‐human, double‐blind, placebo‐controlled, randomised trial. Lancet Infect Dis 19: 620–630 [DOI] [PubMed] [Google Scholar]
- van der Lee TAJ, Medema MH (2016) Computational strategies for genome‐based natural product discovery and engineering in fungi. Fungal Genet Biol 89: 29–36 [DOI] [PubMed] [Google Scholar]
- Lee S, van Santen JA, Farzaneh N, Liu DY, Pye CR, Baumeister TUH, Wong WR, Linington RG (2022) NP analyst: an open online platform for compound activity mapping. ACS Cent Sci 8: 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiris S, Davies DT, Sprynski N, Castandet J, Beyria L, Bodnarchuk MS, Sutton JM, Mullins TMG, Jones MW, Forrest AK et al (2021) Virtual screening approach to identifying a novel and tractable series of Pseudomonas aeruginosa elastase inhibitors. ACS Med Chem Lett 12: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepak AJ, Wang W, Andes DR (2020) Pharmacodynamic evaluation of MRX‐8, a novel polymyxin, in the neutropenic mouse thigh and lung infection models against gram‐negative pathogens. Antimicrob Agents Chemother 64: e01517‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesher GY, Froelich EJ, Gruett MD, Bailey JH, Brundage RP (1962) 1,8‐Naphthyridine derivatives. A new class of chemotherapeutic agents. J Med Chem 5: 1063–1065 [DOI] [PubMed] [Google Scholar]
- Levy N, Bruneau J‐M, Le Rouzic E, Bonnard D, Le Strat F, Caravano A, Chevreuil F, Barbion J, Chasset S, Ledoussal B et al (2019) Structural basis for E. coli penicillin binding protein (PBP) 2 inhibition, a platform for drug design. J Med Chem 62: 4742–4754 [DOI] [PubMed] [Google Scholar]
- Lewis K (2013) Platforms for antibiotic discovery. Nat Rev Drug Discov 12: 371–387 [DOI] [PubMed] [Google Scholar]
- Lewis K (2017) New approaches to antimicrobial discovery. Biochem Pharmacol 134: 87–98 [DOI] [PubMed] [Google Scholar]
- Lewis K (2020) The science of antibiotic discovery. Cell 181: 29–45 [DOI] [PubMed] [Google Scholar]
- Lin Y‐C, Schneider F, Eberle K, Chiodi D, Nakamura H, Reisberg S, Chen J, saito m, Baran PS (2022) Atroposelective total synthesis of darobactin A. ChemRxiv 10.26434/chemrxiv-2022-28tbx [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schäberle TF, Hughes DE, Epstein S et al (2015) A new antibiotic kills pathogens without detectable resistance. Nature 517: 455–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46: 3–26 [DOI] [PubMed] [Google Scholar]
- Liu B, Trout REL, Chu G‐H, McGarry D, Jackson RW, Hamrick JC, Daigle DM, Cusick SM, Pozzi C, de Luca F et al (2020) Discovery of Taniborbactam (VNRX‐5133): a broad‐spectrum serine‐ and metallo‐β‐lactamase inhibitor for carbapenem‐resistant bacterial infections. J Med Chem 63: 2789–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locey KJ, Lennon JT (2016) Scaling laws predict global microbial diversity. Proc Natl Acad Sci U S A 113: 5970–5975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher HH, Caspers P, Bruyère T, Schroeder S, Pfaff P, Knezevic A, Keck W, Ritz D (2014a) Investigations of the mode of action and resistance development of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob Agents Chemother 58: 901–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher HH, Seiler P, Chen X, Schroeder S, Pfaff P, Enderlin M, Klenk A, Fournier E, Hubschwerlen C, Ritz D et al (2014b) In vitro and in vivo antibacterial evaluation of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob Agents Chemother 58: 892–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomovskaya O, Tsivkovski R, Sun D, Reddy R, Totrov M, Hecker S, Griffith D, Loutit J, Dudley M (2021) QPX7728, an ultra‐broad‐spectrum B‐lactamase inhibitor for intravenous and oral therapy: overview of biochemical and microbiological characteristics. Front Microbiol 12: 697180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madaoui M, Vidal O, Meyer A, Noël M, Lacroix J‐M, Vasseur J‐J, Marra A, Morvan F (2020) Modified Galacto‐ or Fuco‐clusters exploiting the siderophore pathway to inhibit the LecA‐ or LecB‐associated virulence of Pseudomonas aeruginosa . Chembiochem 21: 3433–3448 [DOI] [PubMed] [Google Scholar]
- Mahase E (2020) UK launches subscription style model for antibiotics to encourage new development. BMJ 369: m2468 [DOI] [PubMed] [Google Scholar]
- Mahler L, Niehs SP, Martin K, Weber T, Scherlach K, Hertweck C, Roth M, Rosenbaum MA (2021) Highly parallelized droplet cultivation and prioritization of antibiotic producers from natural microbial communities. Elife 10: e64774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier ME (2015) Design and synthesis of analogues of natural products. Org Biomol Chem 13: 5302–5343 [DOI] [PubMed] [Google Scholar]
- Mancini F, Unver MY, Elgaher WAM, Jumde VR, Alhayek A, Lukat P, Herrmann J, Witte MD, Köck M, Blankenfeldt W et al (2020) Protein‐templated hit identification via an Ugi four‐component reaction. Chemistry 26: 14585–14593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JK, Sheehan JP, Bratton BP, Moore GM, Mateus A, Li SH‐J, Kim H, Rabinowitz JD, Typas A, Savitski MM et al (2020) A dual‐mechanism antibiotic kills gram‐negative bacteria and avoids drug resistance. Cell 181: 1518–1532.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Loeches I, Dale GE, Torres A (2018) Murepavadin: a new antibiotic class in the pipeline. Expert Rev Anti Infect Ther 16: 259–268 [DOI] [PubMed] [Google Scholar]
- Masi M, Réfregiers M, Pos KM, Pagès J‐M (2017) Mechanisms of envelope permeability and antibiotic influx and efflux in gram‐negative bacteria. Nat Microbiol 2: 17001 [DOI] [PubMed] [Google Scholar]
- McConnell MJ (2019) Where are we with monoclonal antibodies for multidrug‐resistant infections? Drug Discov Today 24: 1132–1138 [DOI] [PubMed] [Google Scholar]
- McCulloch TR, Wells TJ, Souza‐Fonseca‐Guimaraes F (2022) Towards efficient immunotherapy for bacterial infection. Trends Microbiol 30: 158–169 [DOI] [PubMed] [Google Scholar]
- McKeage K (2015) Finafloxacin: first global approval. Drugs 75: 687–693 [DOI] [PubMed] [Google Scholar]
- Mendes RE, Rhomberg PR, Watters AA, Castanheira M (2022) In vitro activity of the orally bioavailable ceftibuten/VNRX‐7145 (VNRX‐5236 etzadroxil) combination against a challenge set of Enterobacterales pathogens carrying molecularly characterized β‐lactamase genes. J Antimicrob Chemother 77: 689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner K, Vuong B, Tranmer GK (2022) The boron advantage: the evolution and diversification of Boron's applications in medicinal chemistry. Pharmaceuticals 15: 264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethke M, Pieroni M, Weber T, Brönstrup M, Hammann P, Halby L, Arimondo PB, Glaser P, Aigle B, Bode HB et al (2021) Towards the sustainable discovery and development of new antibiotics. Nat Rev Chem 5: 726–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AA, Bundy GL, Mott JE, Skepner JE, Boyle TP, Harris DW, Hromockyj AE, Marotti KR, Zurenko GE, Munzner JB et al (2008) Discovery and characterization of QPT‐1, the progenitor of a new class of bacterial topoisomerase inhibitors. Antimicrob Agents Chemother 52: 2806–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller M, Norris MD, Planke T, Cirnski K, Herrmann J, Müller R, Kirschning A (2019) Scalable syntheses of methoxyaspartate and preparation of the antibiotic Cystobactamid 861‐2 and highly potent derivatives. Org Lett 21: 8369–8372 [DOI] [PubMed] [Google Scholar]
- Monsarrat C, Compain G, André C, Engilberge S, Martiel I, Oliéric V, Wolff P, Brillet K, Landolfo M, Da Silva VC et al (2021) Iterative structure‐based optimization of short peptides targeting the bacterial sliding clamp. J Med Chem 64: 17063–17078 [DOI] [PubMed] [Google Scholar]
- Moon K, Xu F, Zhang C, Seyedsayamdost MR (2019) Bioactivity‐HiTES unveils cryptic antibiotics encoded in Actinomycete bacteria. ACS Chem Biol 14: 767–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno M, Elgaher W, Herrmann J, Schläger N, Hamed M, Baumann S, Müller R, Hartmann R, Kirschning A (2015) Synthesis and biological evaluation of cystobactamid 507: a bacterial topoisomerase inhibitor from Cystobacter sp. Synlett 26: 1175–1178 [Google Scholar]
- Morinaka A, Tsutsumi Y, Yamada M, Suzuki K, Watanabe T, Abe T, Furuuchi T, Inamura S, Sakamaki Y, Mitsuhashi N et al (2015) OP0595, a new diazabicyclooctane: mode of action as a serine β‐lactamase inhibitor, antibiotic and β‐lactam ‘enhancer’. J Antimicrob Chemother 70: 2779–2786 [DOI] [PubMed] [Google Scholar]
- Moya B, Barcelo IM, Bhagwat S, Patel M, Bou G, Papp‐Wallace KM, Bonomo RA, Oliver A (2017) WCK 5107 (Zidebactam) and WCK 5153 are novel inhibitors of PBP2 showing potent “β‐lactam enhancer” activity against Pseudomonas aeruginosa, including multidrug‐resistant metallo‐β‐lactamase‐producing high‐risk clones. Antimicrob Agents Chemother 61: e02529‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungan MD, Alanjary M, Blin K, Weber T, Medema MH, Ziemert N (2020) ARTS 2.0: Feature updates and expansion of the antibiotic resistant target seeker for comparative genome mining. Nucleic Acids Res 48: W546–W552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munita JM, Arias CA (2016) Mechanisms of antibiotic resistance. Microbiol Spectr 10.1128/microbiolspec.VMBF-0016-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz KA, Hergenrother PJ (2021) Facilitating compound entry as a means to discover antibiotics for gram‐negative bacteria. Acc Chem Res 54: 1322–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, Han C, Bisignano C, Rao P, Wool E et al (2022) Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399: 629–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushtaq S, Vickers A, Woodford N, Haldimann A, Livermore DM (2019) Activity of nacubactam (RG6080/OP0595) combinations against MBL‐producing Enterobacteriaceae . J Antimicrob Chemother 74: 953–960 [DOI] [PubMed] [Google Scholar]
- Myronovskyi M, Rosenkranzer B, Nadmid S, Pujic P, Normand P, Luzhetskyy A (2018) Generation of a cluster‐free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab Eng 49: 316–324 [DOI] [PubMed] [Google Scholar]
- Naran K, Nundalall T, Chetty S, Barth S (2018) Principles of immunotherapy: Implications for treatment strategies in cancer and infectious diseases. Front Microbiol 9: 3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesic M, Ryffel DB, Maturano J, Shevlin M, Pollack SR, Gauthier DR Jr, Trigo‐Mouriño P, Zhang L‐K, Schultz D, McCabe Dunn JM et al (2022) Total synthesis of Darobactin A. ChemRxiv 10.26434/chemrxiv-2022-c4mz6 [PREPRINT] [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM, Kingston DGI (2015) Natural products as pharmaceuticals and sources for lead structures. In The practice of medicinal chemistry, Wermuth CG, Aldous D, Rognan D (eds), pp 101–139. San Diego, CA: Academic Press; [Google Scholar]
- Nichols D, Cahoon N, Trakhtenberg EM, Pham L, Mehta A, Belanger A, Kanigan T, Lewis K, Epstein SS (2010) Use of Ichip for high‐throughput In situ cultivation of “uncultivable” microbial species. Appl Environ Microbiol 76: 2445–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H, Pagès J‐M (2012) Broad‐specificity efflux pumps and their role in multidrug resistance of gram‐negative bacteria. FEMS Microbiol Rev 36: 340–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonejuie P, Burkart M, Pogliano K, Pogliano J (2013) Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc Natl Acad Sci U S A 110: 16169–16174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord C, Bjerketorp J, Levenfors JJ, Cao S, Strömstedt AA, Guss B, Larsson R, Hughes D, Öberg B, Broberg A (2020) Isopedopeptins A‐H: cationic cyclic lipodepsipeptides from Pedobacter cryoconitis UP508 targeting WHO top‐priority carbapenem‐resistant bacteria. ACS Chem Biol 15: 2937–2944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosopharm (2022) Pipeline . (https://www.nosopharm.com/news/press‐release/nosopharm‐and‐gna‐now‐announce‐positive‐results‐for‐the‐late‐preclinical‐development‐of‐the‐first‐in‐class‐antibiotic‐noso‐502/)
- Nothias L‐F, Nothias‐Esposito M, da Silva R, Wang M, Protsyuk I, Zhang Z, Sarvepalli A, Leyssen P, Touboul D, Costa J et al (2018) Bioactivity‐based molecular networking for the discovery of drug leads in natural product bioassay‐guided fractionation. J Nat Prod 81: 758–767 [DOI] [PubMed] [Google Scholar]
- O'Donnell J, Tanudra A, Chen A, Hines D, Tommasi R, Mueller J (2020) Pharmacokinetic/Pharmacodynamic determination and preclinical pharmacokinetics of the β‐lactamase inhibitor ETX1317 and its orally available prodrug ETX0282. ACS Infect Dis 6: 1378–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oganesyan V, Peng L, Damschroder MM, Cheng L, Sadowska A, Tkaczyk C, Sellman BR, Wu H, Dall'Acqua WF (2014) Mechanisms of neutralization of a human anti‐α‐toxin antibody. J Biol Chem 289: 29874–29880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant CM, Green G (2002) Quinolones: a comprehensive review. Am Fam Physician 65: 455 [PubMed] [Google Scholar]
- O'Neil J (2014) Antimicrobial resistance: tackling a crisis for the health and wealth of nations .
- O'Shea R, Moser HE (2008) Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem 51: 2871–2878 [DOI] [PubMed] [Google Scholar]
- Otten H (1986) Domagk and the development of the sulphonamides. J Antimicrob Chemother 17: 689–696 [DOI] [PubMed] [Google Scholar]
- Owens B (2017) Solithromycin rejection chills antibiotic sector. Nat Biotechnol 35: 187–188 [DOI] [PubMed] [Google Scholar]
- Pantel L, Florin T, Dobosz‐Bartoszek M, Racine E, Sarciaux M, Serri M, Houard J, Campagne J‐M, de Figueiredo RM, Midrier C et al (2018) Odilorhabdins, antibacterial agents that cause miscoding by binding at a new ribosomal site. Mol Cell 70: 83–94.e7 [DOI] [PubMed] [Google Scholar]
- Panter F, Krug D, Baumann S, Müller R (2018) Self‐resistance guided genome mining uncovers new topoisomerase inhibitors from myxobacteria. Chem Sci 9: 4898–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp‐Wallace KM, Bethel CR, Caillon J, Barnes MD, Potel G, Bajaksouzian S, Rutter JD, Reghal A, Shapiro S, Taracila MA et al (2019) Beyond piperacillin‐tazobactam: cefepime and AAI101 as a potent β‐lactam‐β‐lactamase inhibitor combination. Antimicrob Agents Chemother 63: e00105‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL (2010) How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov 9: 203–214 [DOI] [PubMed] [Google Scholar]
- Pavlova A, Gumbart JC (2015) Parametrization of macrolide antibiotics using the force field toolkit. J Comput Chem 36: 2052–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DJ, Miller WH, Berry V, Brosky J, Burgess WJ, Chen E, De Wolf Jr WE, Fosberry AP, Greenwood R, Head MS et al (2002) Discovery of a novel and potent class of FabI‐directed antibacterial agents. Antimicrob Agents Chemother 46: 3118–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6: 29–40 [DOI] [PubMed] [Google Scholar]
- Peach KC, Bray WM, Shikuma NJ, Gassner NC, Lokey RS, Yildiz FH, Linington RG (2011) An image‐based 384‐well high‐throughput screening method for the discovery of biofilm inhibitors in vibrio cholerae. Mol Biosyst 7: 1176–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penwell WF, Shapiro AB, Giacobbe RA, Gu R‐F, Gao N, Thresher J, McLaughlin RE, Huband MD, DeJonge BLM, Ehmann DE et al (2015) Molecular mechanisms of sulbactam antibacterial activity and resistance determinants in Acinetobacter baumannii . Antimicrob Agents Chemother 59: 1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson E, Kaur P (2018) Antibiotic resistance mechanisms in bacteria: relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front Microbiol 9: 2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulou D, Siopi M, Vourli S, Pournaras S (2021) Activity of sulbactam‐durlobactam and comparators against a national collection of carbapenem‐resistant acinetobacter baumannii isolates from greece. Front Cell Infect Microbiol 11: 814530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petters S, Groß V, Söllinger A, Pichler M, Reinhard A, Bengtsson MM, Urich T (2021) The soil microbial food web revisited: predatory myxobacteria as keystone taxa? ISME J 15: 2665–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, Li L, Liu Q, Zhang J, Huband MD, Lindley JM, Mendes RE (2021) In vitro activity of a novel aminomethylcycline antibacterial (KBP‐7072), a third‐generation tetracycline, against clinical isolates with molecularly characterized tetracycline resistance mechanisms. JAC Antimicrob Resist 3: dlab177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishchany G, Mevers E, Ndousse‐Fetter S, Horvath DJ, Paludo CR, Silva‐Junior EA, Koren S, Skaar EP, Clardy J, Kolter R (2018) Amycomicin is a potent and specific antibiotic discovered with a targeted interaction screen. Proc Natl Acad Sci U S A 115: 10124–10129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plescia J, Moitessier N (2020) Design and discovery of boronic acid drugs. Eur J Med Chem 195: 112270 [DOI] [PubMed] [Google Scholar]
- Pogorevc D, Panter F, Schillinger C, Jansen R, Wenzel SC, Müller R (2019) Production optimization and biosynthesis revision of corallopyronin a, a potent anti‐filarial antibiotic. Metab Eng 55: 201–211 [DOI] [PubMed] [Google Scholar]
- Polyphor AG (2019) Polyphor closes the Phase III PRISM studies of murepavadin intravenous formulation and evaluates further product improvement options .
- Qpex Biopharma (2022) Pipeline . (https://www.qpexbio.com/pipeline)
- Quinn RA, Nothias L‐F, Vining O, Meehan M, Esquenazi E, Dorrestein PC (2017) Molecular networking as a drug discovery, drug metabolism, and precision medicine strategy. Trends Pharmacol Sci 38: 143–154 [DOI] [PubMed] [Google Scholar]
- Racine E, Gualtieri M (2019) From Worms to drug candidate: the story of Odilorhabdins, a New class of antimicrobial agents. Front Microbiol 10: 2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran G, Tulapurkar ME, Harris KM, Arad G, Shirvan A, Shemesh R, Detolla LJ, Benazzi C, Opal SM, Kaempfer R et al (2013) A peptide antagonist of CD28 signaling attenuates toxic shock and necrotizing soft‐tissue infection induced by streptococcus pyogenes. J Infect Dis 207: 1869–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran G, Kaempfer R, Chung C‐S, Shirvan A, Chahin AB, Palardy JE, Parejo NA, Chen Y, Whitford M, Arad G et al (2015) CD28 homodimer interface mimetic peptide acts as a preventive and therapeutic agent in models of severe bacterial sepsis and gram‐negative bacterial peritonitis. J Infect Dis 211: 995–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidl CT, Heath TK, Darwish I, Torrez RM, Moore M, Gild E, Nocek BP, Starus A, Holz RC, Becker DP (2020) Indoline‐6‐sulfonamide inhibitors of the bacterial enzyme DapE. Antibiotics 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard S, Versluys S, Taillier T, Dubarry N, Leroi‐Geissler C, Rey A, Carry J‐C, Angouillant O, Monget S, Gouyon T et al (eds) (2022) Corramycin: enlarging the antibacterial spectrum and optimizing the developability profile of corramycin, a novel class natural product antibacterial: ASM Microbe 2022 .
- Rex J (2014) Enabling drug discovery and development to address the crisis of antimicrobial resistance: new tools, new pathways, and remaining challenges .
- Ribeiro da Cunha B, Fonseca LP, Calado CRC (2019) Antibiotic discovery: where have we come from, where do we go? Antibiotics 8: 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice LB (2008) Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 197: 1079–1081 [DOI] [PubMed] [Google Scholar]
- Ritzmann N, Manioglu S, Hiller S, Müller DJ (2022) Monitoring the antibiotic darobactin modulating the β‐barrel assembly factor BamA. Structure 30: 350–359.e3 [DOI] [PubMed] [Google Scholar]
- Roberts KD, Zhu Y, Azad MAK, Han M‐L, Wang J, Wang L, Yu HH, Horne AS, Pinson J‐A, Rudd D et al (2022) A synthetic lipopeptide targeting top‐priority multidrug‐resistant gram‐negative pathogens. Nat Commun 13: 1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche (2022) Roche Group development pipeline . (https://www.roche.com/solutions/pipeline/)
- Rolain JM, Parola P, Cornaglia G (2010) New Delhi metallo‐beta‐lactamase (NDM‐1): towards a new pandemia? Clin Microbiol Infect 16: 1699–1701 [DOI] [PubMed] [Google Scholar]
- Ropponen H‐K, Diamanti E, Siemens A, Illarionov B, Haupenthal J, Fischer M, Rottmann M, Witschel M, Hirsch AKH (2021) Assessment of the rules related to gaining activity against gram‐negative bacteria. RSC Med Chem 12: 593–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge PJ, Challis GL (2015) Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol 13: 509–523 [DOI] [PubMed] [Google Scholar]
- Sadeer NB, Mahomoodally MF (2021) Antibiotic potentiation of natural products: a promising target to fight pathogenic bacteria. Curr Drug Targets 22: 555–572 [DOI] [PubMed] [Google Scholar]
- Sader HS, Rhomberg PR, Flamm RK, Jones RN, Castanheira M (2017) WCK 5222 (cefepime/zidebactam) antimicrobial activity tested against gram‐negative organisms producing clinically relevant β‐lactamases. J Antimicrob Chemother 72: 1696–1703 [DOI] [PubMed] [Google Scholar]
- Safety, Tolerability, and PK of LBP-EC01 in Patients With Lower Urinary Tract Colonization Caused by E. Coli (2022) ClinicalTrials.gov NCT04191148 (https://clinicaltrials.gov/show/NCT04191148) [DATASET]
- van Santen JA, Poynton EF, Iskakova D, McMann E, Alsup TA, Clark TN, Fergusson CH, Fewer DP, Hughes AH, McCadden CA et al (2022) The natural products atlas 2.0: A database of microbially‐derived natural products. Nucleic Acids Res 50: D1317–D1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Yamawaki K (2019) Cefiderocol: discovery, chemistry, and in vivo profiles of a novel Siderophore cephalosporin. Clin Infect Dis 69: S538–S543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherlach K, Hertweck C (2009) Triggering cryptic natural product biosynthesis in microorganisms. Org Biomol Chem 7: 1753–1760 [DOI] [PubMed] [Google Scholar]
- Schiefer A, Hübner MP, Krome A, Lämmer C, Ehrens A, Aden T, Koschel M, Neufeld H, Chaverra‐Muñoz L, Jansen R et al (2020) Corallopyronin a for short‐course anti‐wolbachial, macrofilaricidal treatment of filarial infections. PLoS Negl Trop Dis 14: e0008930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorn MA, Verhoeven S, Ridder L, Huber F, Acharya DD, Aksenov AA, Aleti G, Moghaddam JA, Aron AT, Aziz S et al (2021) A community resource for paired genomic and metabolomic data mining. Nat Chem Biol 17: 363–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz C, Ho D‐K, Hamed MM, Abdelsamie AS, Röhrig T, Herr C, Kany AM, Rox K, Schmelz S, Siebenbürger L et al (2021) A new PqsR inverse agonist potentiates tobramycin efficacy to eradicate Pseudomonas aeruginosa biofilms. Adv Sci 8: 2004369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiple IB, Zhang Z, Jakubec P, Langlois‐Mercier A, Wright PM, Hog DT, Yabu K, Allu SR, Fukuzaki T, Carlsen PN et al (2016) A platform for the discovery of new macrolide antibiotics. Nature 533: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self WH, Wunderink RG, DiNubile MJ, Stossel TP, Levinson SL, Williams DJ, Anderson EJ, Bramley AM, Jain S, Edwards KM et al (2019) Low admission plasma gelsolin concentrations identify community‐acquired pneumonia patients at high risk for severe outcomes. Clin Infect Dis 69: 1218–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost MR (2014) High‐throughput platform for the discovery of elicitors of silent bacterial gene clusters. Proc Natl Acad Sci U S A 111: 7266–7271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfert CE, Porten C, Yuan B, Deckarm S, Panter F, Bader C, Coetzee J, Deschner F, Tehrani K, Higgins PG et al (2022) Darobactins exhibiting superior antibiotic activity by Cryo‐EM structure guided biosynthetic engineering. Angew Chem Int Ed Engl 10.1002/ange.202214094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro AB, Moussa SH, McLeod SM, Durand‐Réville T, Miller AA (2021) Durlobactam, a new diazabicyclooctane β‐lactamase inhibitor for the treatment of Acinetobacter infections in combination with sulbactam. Front Microbiol 12: 709974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheremet AB, Zigangirova NA, Zayakin ES, Luyksaar SI, Kapotina LN, Nesterenko LN, Kobets NV, Gintsburg AL (2018) Small molecule inhibitor of type three secretion system belonging to a class 2,4‐disubstituted‐4H‐1,3,4‐thiadiazine‐5‐ones improves survival and decreases bacterial loads in an airway Pseudomonas aeruginosa infection in mice. Biomed Res Int 2018: 5810767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetye GS, Franzblau SG, Cho S (2020) New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl Res 220: 68–97 [DOI] [PubMed] [Google Scholar]
- Skinnider MA, Johnston CW, Gunabalasingam M, Merwin NJ, Kieliszek AM, MacLellan RJ, Li H, Ranieri MRM, Webster ALH, Cao MPT et al (2020) Comprehensive prediction of secondary metabolite structure and biological activity from microbial genome sequences. Nat Commun 11: 6058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee AM, Wuonola MA, McRipley RJ, Zajac I, Zawada MJ, Bartholomew PT, Gregory WA, Forbes M (1987) Oxazolidinones, a new class of synthetic antibacterial agents: in vitro and in vivo activities of DuP 105 and DuP 721. Antimicrob Agents Chemother 31: 1791–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KP, Kirby JE (2016) Validation of a high‐throughput screening assay for identification of adjunctive and directly acting antimicrobials targeting carbapenem‐resistant Enterobacteriaceae . Assay Drug Dev Technol 14: 194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PA, Koehler MFT, Girgis HS, Yan D, Chen Y, Chen Y, Crawford JJ, Durk MR, Higuchi RI, Kang J et al (2018) Optimized arylomycins are a new class of gram‐negative antibiotics. Nature 561: 189–194 [DOI] [PubMed] [Google Scholar]
- Smoum R, Rubinstein A, Dembitsky VM, Srebnik M (2012) Boron containing compounds as protease inhibitors. Chem Rev 112: 4156–4220 [DOI] [PubMed] [Google Scholar]
- Soheili V, Tajani AS, Ghodsi R, Bazzaz BSF (2019) Anti‐PqsR compounds as next‐generation antibacterial agents against Pseudomonas aeruginosa: a review. Eur J Med Chem 172: 26–35 [DOI] [PubMed] [Google Scholar]
- Sommer R, Rox K, Wagner S, Hauck D, Henrikus SS, Newsad S, Arnold T, Ryckmans T, Brönstrup M, Imberty A et al (2019) Anti‐biofilm agents against Pseudomonas aeruginosa: a structure‐activity relationship study of C‐Glycosidic LecB inhibitors. J Med Chem 62: 9201–9216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes JM, Yang K, Swanson K, Jin W, Cubillos‐Ruiz A, Donghia NM, MacNair CR, French S, Carfrae LA, Bloom‐Ackermann Z et al (2020) A deep learning approach to antibiotic discovery. Cell 180: 688–702.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbings W, Leow P, Yong GC, Goh F, Körber‐Irrgang B, Kresken M, Endermann R, Labischinski H (2011) In vitro spectrum of activity of finafloxacin, a novel, pH‐activated fluoroquinolone, under standard and acidic conditions. Antimicrob Agents Chemother 55: 4394–4397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh GA, Lodise TP, Tamma PD, Knisely JM, Alexander J, Aslam S, Barton KD, Bizzell E, Totten KMC, Campbell JL et al (2022) Considerations for the use of phage therapy in clinical practice. Antimicrob Agents Chemother 66: e0207121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surur AS, Sun D (2021) Macrocycle‐antibiotic hybrids: a path to clinical candidates. Front Chem 9: 659845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szili P, Draskovits G, Revesz T, Bogar F, Balogh D, Martinek T, Daruka L, Spohn R, Vasarhelyi BM, Czikkely M et al (2018) Antibiotic usage promotes the evolution of resistance against gepotidacin, a novel multi‐targeting drug. bioRxiv 10.1101/495630 [PREPRINT] [DOI] [Google Scholar]
- Tannous A, Levinson SL, Bolognese J, Opal SM, DiNubile MJ (2020) Safety and pharmacokinetics of recombinant human plasma gelsolin in patients hospitalized for nonsevere community‐acquired pneumonia. Antimicrob Agents Chemother 64: e00579‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Public Health Agency of Sweden (2017) Availability of antibiotics: reporting of Government commission .
- Theuretzbacher U (2017) Antibiotic innovation for future public health needs. Clin Microbiol Infect 23: 713–717 [DOI] [PubMed] [Google Scholar]
- Theuretzbacher U, Piddock LJV (2019) Non‐traditional antibacterial therapeutic options and challenges. Cell Host Microbe 26: 61–72 [DOI] [PubMed] [Google Scholar]
- Theuretzbacher U, Bush K, Harbarth S, Paul M, Rex JH, Tacconelli E, Thwaites GE (2020a) Critical analysis of antibacterial agents in clinical development. Nat Rev Microbiol 18: 286–298 [DOI] [PubMed] [Google Scholar]
- 116th Congress (2019) To amend title XVIII of the Social Security Act to encourage the development and use of DISARM antimicrobial drugs, and for other purposes .
- Theuretzbacher U, Outterson K, Engel A, Karlén A (2020b) The global preclinical antibacterial pipeline. Nat Rev Microbiol 18: 275–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA (2015) ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov 14: 529–542 [DOI] [PubMed] [Google Scholar]
- Tong Y, Weber T, Lee SY (2019) CRISPR/Cas‐based genome engineering in natural product discovery. Nat Prod Rep 36: 1262–1280 [DOI] [PubMed] [Google Scholar]
- Trout RE, Zulli A, Mesaros E, Jackson RW, Boyd S, Liu B, Hamrick J, Daigle D, Chatwin CL, John K et al (2021) Discovery of VNRX‐7145 (VNRX‐5236 Etzadroxil): An orally bioavailable β‐lactamase inhibitor for Enterobacterales expressing Ambler class A, C, and D enzymes. J Med Chem 64: 10155–10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tselepis L, Langley GW, Aboklaish AF, Widlake E, Jackson DE, Walsh TR, Schofield CJ, Brem J, Tyrrell JM (2020) In vitro efficacy of imipenem‐relebactam and cefepime‐AAI101 against a global collection of ESBL‐positive and carbapenemase‐producing Enterobacteriaceae . Int J Antimicrob Agents 56: 105925 [DOI] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services New drugs at FDA: CDER's new molecular entities and new therapeutic biological products . (http://wayback.archive‐it.org/7993/20161022052126/http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/default.htm)
- Vázquez R, Díez‐Martínez R, Domingo‐Calap P, García P, Gutiérrez D, Muniesa M, Ruiz‐Ruigómez M, Sanjuán R, Tomás M, Tormo‐Mas MÁ et al (2022) Essential topics for the regulatory consideration of phages as clinically valuable therapeutic agents: a perspective from Spain. Microorganisms 10: 717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velázquez‐Libera JL, Murillo‐López JA, de la Torre SF, Caballero J (2019) Structural requirements of N‐alpha‐Mercaptoacetyl dipeptide (NAMdP) inhibitors of pseudomonas aeruginosa virulence factor LasB: 3D‐QSAR, molecular docking, and interaction fingerprint studies. Int J Mol Sci 20: 6133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventola CL (2015) The antibiotic resistance crisis: Part 1: causes and threats. P T 40: 277–283 [PMC free article] [PubMed] [Google Scholar]
- Waksman SA, Schatz A, Reynolds DM (2010) Production of antibiotic substances by actinomycetes. Ann N Y Acad Sci 1213: 112–124 [DOI] [PubMed] [Google Scholar]
- Walsh CT, Wencewicz TA (2014) Prospects for new antibiotics: a molecule‐centered perspective. J Antibiot 67: 7–22 [DOI] [PubMed] [Google Scholar]
- Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto‐Knaan T et al (2016) Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat Biotechnol 34: 828–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhao Z, Ke J, Engel Y, Shi Y‐M, Robinson D, Bingol K, Zhang Z, Bowen B, Louie K et al (2019) CRAGE enables rapid activation of biosynthetic gene clusters in undomesticated bacteria. Nat Microbiol 4: 2498–2510 [DOI] [PubMed] [Google Scholar]
- Werth BJ (2022) Overview of antibacterial drugs . (https://www.merckmanuals.com/professional/infectious‐diseases/bacteria‐and‐antibacterial‐drugs/overview‐of‐antibacterial‐drugs)
- Wilson BAP, Thornburg CC, Henrich CJ, Grkovic T, O'Keefe BR (2020) Creating and screening natural product libraries. Nat Prod Rep 37: 893–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WR, Oliver AG, Linington RG (2012) Development of antibiotic activity profile screening for the classification and discovery of natural product antibiotics. Chem Biol 19: 1483–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2017) Global priority list of antibiotic‐resistant bacteria to guide research, discovery, and development of new antibiotics – 2017. Geneva, Switzerland: WHO; [Google Scholar]
- World Health Organization (2021) 2020 antibacterial agents in clinical and preclinical development: an overview and analysis. Geneva, Switzerland: World Health Organization; [Google Scholar]
- World Health Organization (2022) 2021 antibacterial agents in clinical and preclinical development: an overview and analysis. Geneva, Switzerland: World Health Organization; [Google Scholar]
- Wray C, Hedges RW, Shannon KP, Bradley DE (1986) Apramycin and gentamicin resistance in Escherichia coli and salmonellas isolated from farm animals. J Hyg 97: 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GD (2017) Opportunities for natural products in 21st century antibiotic discovery. Nat Prod Rep 34: 694–701 [DOI] [PubMed] [Google Scholar]
- Wright PM, Seiple IB, Myers AG (2014) The evolving role of chemical synthesis in antibacterial drug discovery. Angew Chem Int Ed Engl 53: 8840–8869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X‐Y, Hunt DK, Zhou J, Clark RB, Dunwoody N, Fyfe C, Grossman TH, O'Brien WJ, Plamondon L, Rönn M et al (2012) Fluorocyclines. 1. 7‐fluoro‐9‐pyrrolidinoacetamido‐6‐demethyl‐6‐deoxytetracycline: a potent, broad spectrum antibacterial agent. J Med Chem 55: 597–605 [DOI] [PubMed] [Google Scholar]
- Xiong YQ, Estellés A, Li L, Abdelhady W, Gonzales R, Bayer AS, Tenorio E, Leighton A, Ryser S, Kauvar LM (2017) A human biofilm‐disrupting monoclonal antibody potentiates antibiotic efficacy in rodent models of both Staphylococcus aureus and Acinetobacter baumannii infections. Antimicrob Agents Chemother 61: e00904‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Wright SN, Hamblin M, McCloskey D, Alcantar MA, Schrübbers L, Lopatkin AJ, Satish S, Nili A, Palsson BO et al (2019) A white‐box machine learning approach for revealing antibiotic mechanisms of action. Cell 177: 1649–1661.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarrington ME, Anderson DJ, Dodds Ashley E, Jones T, Davis A, Johnson M, Lokhnygina Y, Sexton DJ, Moehring RW (2019) Impact of FDA black box warning on fluoroquinolone and alternative antibiotic use in southeastern US hospitals. Infect Control Hosp Epidemiol 40: 1297–1300 [DOI] [PubMed] [Google Scholar]
- Yoon V, Nodwell JR (2014) Activating secondary metabolism with stress and chemicals. J Ind Microbiol Biotechnol 41: 415–424 [DOI] [PubMed] [Google Scholar]
- Zani CL, Carroll AR (2017) Database for rapid dereplication of known natural products using Data from MS and fast NMR experiments. J Nat Prod 80: 1758–1766 [DOI] [PubMed] [Google Scholar]
- Zborovsky L, Kleebauer L, Seidel M, Kostenko A, von Eckardstein L, Gombert FO, Weston J, Süssmuth RD (2021) Improvement of the antimicrobial potency, pharmacokinetic and pharmacodynamic properties of albicidin by incorporation of nitrogen atoms. Chem Sci 12: 14606–14617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel GG, Walkty A, Vercaigne L, Karlowsky JA, Embil J, Gin AS, Hoban DJ (1999) The new fluoroquinolones: a critical review. Can J Infect Dis 10: 207–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel GG, Ennis K, Vercaigne L, Walkty A, Gin AS, Embil J, Smith H, Hoban DJ (2002) A critical review of the fluoroquinolones: focus on respiratory infections. Drugs 62: 13–59 [DOI] [PubMed] [Google Scholar]
- Zhang X, Hindra EMA (2019) Unlocking the trove of metabolic treasures: activating silent biosynthetic gene clusters in bacteria and fungi. Curr Opin Microbiol 51: 9–15 [DOI] [PubMed] [Google Scholar]
- Zigangirova NA, Nesterenko LN, Sheremet AB, Soloveva AV, Luyksaar SI, Zayakin ES, Balunets DV, Gintsburg AL (2021) Fluorothiazinon, a small‐molecular inhibitor of T3SS, suppresses salmonella oral infection in mice. J Antibiot (Tokyo) 74: 244–254 [DOI] [PubMed] [Google Scholar]
- Zipperer A, Konnerth MC, Laux C, Berscheid A, Janek D, Weidenmaier C, Burian M, Schilling NA, Slavetinsky C, Marschal M et al (2016) Human commensals producing a novel antibiotic impair pathogen colonization. Nature 535: 511–516 [DOI] [PubMed] [Google Scholar]
- Zlitni S, Ferruccio LF, Brown ED (2013) Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat Chem Biol 9: 796–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuegg J, Hansford KA, Elliott AG, Cooper MA, Blaskovich MAT (2020) How to stimulate and facilitate early stage antibiotic discovery. ACS Infect Dis 6: 1302–1304 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 5