

Abstract
Heteroleptic metal‐organic cages, formed through integrative self‐assembly of ligand mixtures, are highly attractive as reduced symmetry supramolecular hosts. Ensuring high‐fidelity, non‐statistical self‐assembly, however, presents a significant challenge in molecular engineering due to the inherent difficulty in predicting thermodynamic energy landscapes. In this work, two conceptual strategies are described that circumvent this issue, using ligand design strategies to access structurally sophisticated metal‐organic hosts. Using these approaches, it was possible to realise cavity environments described by two inequivalent, unsymmetrical ligand frameworks, representing a significant step forward in the construction of highly anisotropic confined spaces.
Keywords: Cage, Heteroleptic, Low-Symmetry, Metallosupramolecular, Self-Assembly
Heteroleptic metal‐organic cages are prized for their structural complexity and multi‐functionality. Controlling integrative self‐sorting processes necessary to achieve defined self‐assembly of these supramolecular hosts is a significant design challenge. In this work, ligand‐tethering strategies are described that allow access to metal‐organic cages with highly anisotropic cavities described by two inequivalent, unsymmetrical ligand scaffolds.
Introduction
Metal‐organic self‐assembly has proven to be a powerful tool to construct functional supramolecular architectures from relatively simple components. [1] Of particular interest are systems possessing internal cavities [2] capable of binding ions or small molecules—typically referred to as metal‐organic cages (MOCs) or polyhedra (MOPs). [3] The ability to bind guest species, [4] such as drugs, [5] pollutants [6] and anions, [7] within these cavities has been exploited for catalysis, [8] stabilising reactive species [9] and modulation of photophysical properties, [10] amongst other applications.
To advance the functionality of these systems, and move towards more structurally sophisticated assemblies, researchers have recently begun to develop strategies to access MOCs of reduced symmetry in order to tailor the shape and functionality of the cavity environment. [11] Two main approaches have shown success in this endeavour. Employing unsymmetrical ligands will inherently generate MOCs of reduced symmetry (Figure 1b). [12] Alternatively, the incorporation of more than one ligand type to generate heteroleptic, [13] or mixed‐ligand, cages is also effective (Figure 1c). [14] The latter approach has the added benefit of being able to introduce different endo‐/exohedral functional moieties into a single structure. [15] For both of these strategies, ensuring high‐fidelity self‐sorting in the assembly process is essential to avoid formation of mixtures of cage structures, which makes their design challenging. It can further be imagined that combining heteroleptic and low‐symmetry strategies through the incorporation of multiple, unsymmetrical ligands into a single MOC in a defined manner (Figure 1d) would be especially difficult, which likely explains why no examples have been reported to date.
Figure 1.

a) Self‐assembly of C 2v‐symmetric ditopic ligands with square‐planar PdII ions leads to formation of Pd2L4 cages with (pseudo‐)D 4h symmetry. b) Ditopic ligands lacking bilateral symmetry (C 2 rotation axis) can assemble into four possible Pd2L4 cage diastereoisomers. c) Integrative self‐assembly of ligand mixtures leading to selective formation of heteroleptic structures. d) Integrative self‐assembly of unsymmetrical ligands into heteroleptic cages has not been previously reported.
Herein are detailed two strategies to access cavity environments within MOCs that are described by two, unsymmetrical ligand structures. The first employs a covalent‐tethering strategy to specifically constrain ligand fragments in a trans orientation within a polytopic framework. The second exploits the geometrically‐enforced arrangement of ligands with C s symmetry in the selective assembly of C 2h—symmetry palladium nanocages (Figure 1b) within a previously unreported class of double‐cavity Pd3L4 cages. With both of these approaches, the cavities generated are heteroleptic environments—i.e. they are defined by two inequivalent, unsymmetrical ligand frameworks. With the cages as a whole assembled from one type of ligand, the term pseudo‐heteroleptic is proposed to describe these structures.
The significant advantage of these pseudo‐heterolepticity approaches lies in the ability to construct sophisticated, multi‐functional internal spaces without the requirement for integrative self‐sorting of multiple components, a prerequisite of traditional heteroleptic assemblies. The delineation of these strategies opens the door to develop more sophisticated and functional confined spaces in combination with facile self‐assembly.
Results and Discussion
First demonstrated by McMorran and Steel, [16] ditopic ligands are able to assemble with PdII ions to form quadruply‐stranded, Pd2L4‐type cage structures. [17] For ligands with C 2v symmetry there is only one possible configurational isomer of these cages, with (pseudo‐)D 4h symmetry (Figure 1a). Ligands with reduced C s symmetry, however, can assemble into four possible Pd2L4 diastereoisomers (Figure 1b). Unless sufficient directing effects are employed in the design of the ligand, a statistical mixture of these isomers will be generated. [18] For example, Chand and co‐workers have previously reported the self‐assembly of ditopic ligand L (Figure 2a), which is unsymmetrical by virtue of the methylene ester backbone that separates the two coordinating pyridine units. Upon self‐assembly with PdII, a statistical mixture of the four possible Pd2L4 cage isomers was observed to form both in solution and the solid state. [19]
Figure 2.
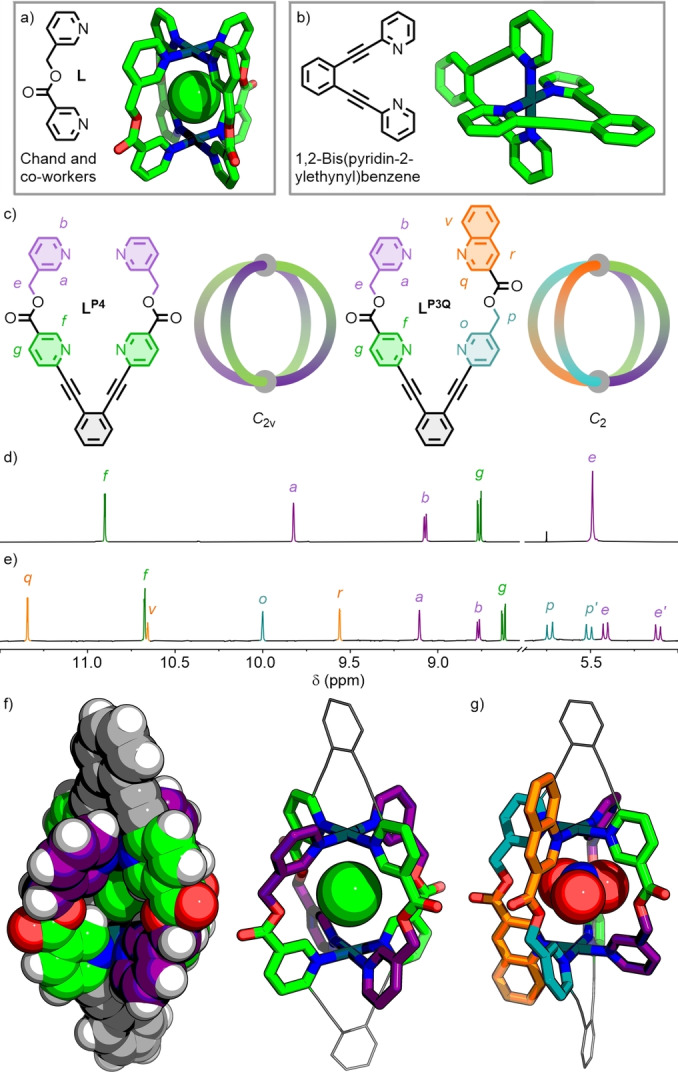
a) Chand's low‐symmetry ditopic ligand, L, and SCXRD structure of the dinuclear PdII cage structure [Pd2 L 4⊃Cl]3+ (CCDC# 1941617; only C 4v isomer shown for clarity); b) 1,2‐bis(pyridin‐2‐ylethynyl)benzene and the SCXRD structure of its mononuclear PdII complex (CCDC# 185471); c) tetratopic ligands LP4 and LP3Q with cartoon diagrams representing the relative orientations of the ligand fragments within CP4 and CP3Q ; partial 1H NMR spectra (500 MHz, d 6‐DMSO, 298 K) of d) CP4 and e) CP3Q ; SCXRD structures of f) [Pd2(LP4 )2⊃Cl]3+ and g) [Pd2(LP3Q )2⊃NO3]3+.
This group, [20] and others,[ 21 , 22 ] have previously used geometric design principles within reduced symmetry, ditopic ligands to successfully bias the self‐assembly process with PdII ions towards cis‐Pd2L4 cages with C 2h symmetry, due to the favourable antiparallel arrangement of trans‐oriented ligands (Figure 1b). Accessing Pd2L4 cage isomers of alternative symmetries represents a significant design challenge. The use of geometric parameters to prepare C 2v trans‐Pd2L4 cages, for example, would require enforcement of a parallel relationship between trans‐oriented ligands, and antiparallel configuration of cis‐oriented ligands. Recently, an M2L4 system with C 4v symmetry was able to be realised by Crowley and co‐workers, using a sub‐component self‐assembly strategy in the synthesis of a heteronuclear PdPtL4 cage. [23]
The 1,2‐bis(pyridin‐2‐ylethynyl)benzene motif has previously been used as a trans‐chelating unit for square‐planar PdII ions (Figure 2b), [24] and has been investigated for its potential as a trans analogue of Fujita's cis‐protection strategy [25] in metal‐organic assembly. [26] It was envisaged that covalently tethering ligand fragments [27] via this unit could be used to constrain the relative orientations of ligands held trans to each other. In this manner it might be possible to obtain cages of C 2v symmetry that have previously proved difficult to access.
trans‐Pd2L2 C2v Cage
LP 4 was readily synthesised from 1,2‐diethynylbenzene and commercially available reagents in 2 steps in 73 % overall yield. It was anticipated that two LP4 ligands would assemble in an anti‐parallel arrangement around two PdII ions. Combining LP4 with Pd(NO3)2 ⋅ 2H2O in d 6‐DMSO at room temperature gradually led to quantitative formation of a single species (CP4 ), as observed by 1H NMR (Figure 2d). Formation of an assembly with the anticipated [Pd2(LP4 )2]4+ formula was confirmed by electrospray ionisation mass spectrometry (ESI‐MS; Fig S19 and S20). Ultimately, despite the poor quality of the crystals, unambiguous confirmation of the structure was achieved via single crystal X‐ray diffraction (SCXRD) studies of the host–guest complex with an encapsulated chloride anion, [Pd2(LP4 )2⊃Cl]3+ (Figure 2f). [28]
Due to the ligand structure and the steric bulk of the diethynylbenzene linker, CP4 exclusively assembles as a Pd2L2 cage with C 2v symmetry. Consequently, the cavity of the cage can be described as being surrounded by four iterations of the ligand L, reported by Chand, in a (pseudo‐)trans‐Pd2L4 configuration (Figure 1b). As with L, efficient formation of CP4 required the presence of a suitable anion template such as NO3 − or Cl−, with BF4 − alone resulting in a mixture of products (Figure S22). Cl− appeared to be a better guest than NO3 −, evidenced by an increased downfield shift of the endohedral protons H a and H f (Δδ=0.43 and 0.41 ppm, respectively).
trans‐Pd2L2 Pseudo‐heteroleptic C2 Cage
Having successfully demonstrated the viability of this tethering strategy to constrain two dipyridyl ligand moieties in a trans arrangement, the thought occurred that the two ditopic units need not be identical. Consequently, LP3Q (Figure 2c) was prepared incorporating one of the previously described dipyridyl fragments, and a second ditopic ligand with one pyridine and one quinoline coordinating unit. Self‐assembly of a stoichiometric mixture of LP3Q with Pd(NO3)2 ⋅ 2H2O in d 6‐DMSO at 50 °C for 2 h again yielded a single species, determined to be [Pd2(LP3Q )2](NO3)4 (CP3Q ) by NMR and ESI‐MS. Diastereotopic splitting of the signals assigned to the methylene units was observed (Figure 2e), giving two pairs of doublets (5.74/5.51 ppm and 5.42/5.12 ppm), indicative of inequivalent chemical environments on either face of the ditopic ligand fragments. Vapour diffusion of Et2O into a solution of the assembly in 1 : 1 DMSO/CH3CN yielded X‐ray quality crystals, with SCXRD revealing the anticipated [Pd2(LP4 )2⊃NO3]3+ structure (Figure 2g). [28] By virtue of the inequivalence of the two ditopic ligand fragments within LP3Q , the structural framework of CP3Q is analogous to that of a heteroleptic Pd2L2L′2 cage [29] assembled from two unsymmetrical ligands (L and L′). Within the assembly, pairs of identical ligands are held cis to each other in an anti‐parallel arrangement, giving a structure with C 2 symmetry.
Of note, due to the (bis‐)trans‐heterobidentate nature of LP3Q , each of the PdII centres in CP3Q possesses axial chirality. [30] From the SCXRD structure it could be shown that within CP3Q both ions are of the same stereochemistry, and the system crystallises as a racemic mixture of both (S,S)‐ and (R,R)‐ CP3Q enantiomers (see Supporting Information section 6).
This covalent tethering strategy was clearly highly effective at constraining the relative orientations of ligand fragments, and even for the incorporation of more than one framework into an assembly. The bent, or V‐shaped, nature of LP3Q gave rise to the C 2 symmetry of CP3Q . To widen the scope of accessible Pd2L2L′2 cavity symmetries, there was motivation to investigate alternative modes of tethering that might lead to C s symmetry structures (Figure 1d). These would be formed from pairs of unsymmetrical ligands with identical ligands arranged cis to each other, but now in a parallel orientation. Tethering of ligand fragments in a linear fashion appeared to present a solution to accessing these structures.
cis‐Pd2L4 Cages from Unsymmetrical Ditopic Ligands
Based on principles of geometric complementarity delineated in previous work, [20] two low‐symmetry, ditopic ligands were synthesised with isostructural core frameworks (Figure 3a). The ligands differed only in the identity of the flanking aromatic units—1,4‐phenyl (L1Ph ) and 2,5‐m‐xylyl (L1Xy ). In each case, self‐assembly with PdII as the tetrafluoroborate salt in a 2 : 1 ligand/metal ratio resulted in formation of the anticipated cis‐[Pd2L4](BF4)4 architectures (C1Ph and C1Xy ), as determined by NMR (Figure 3b and c), ESI‐MS (Figure S75–77 and S100–102) and, for C1Xy , SCXRD (see below).
Figure 3.
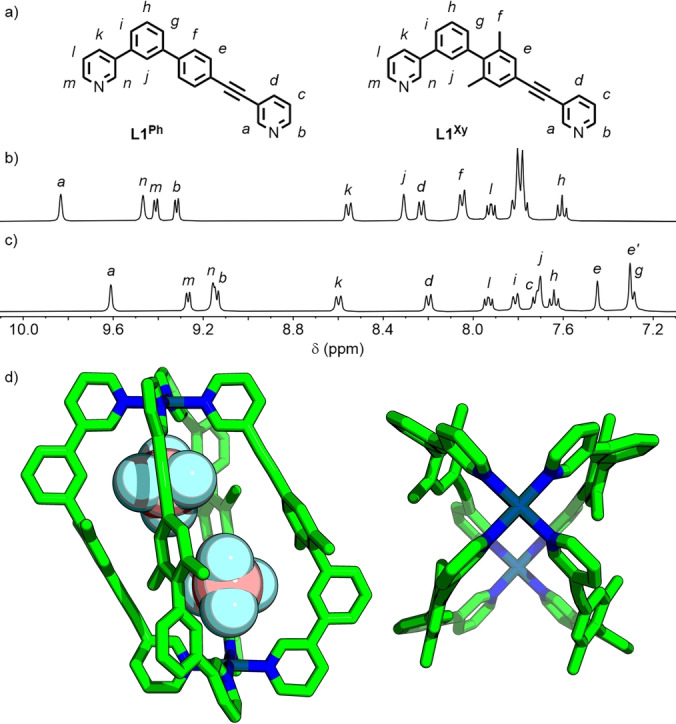
a) Ligands L1Ph and L1Xy ; partial 1H NMR spectra (400 MHz, d 6‐DMSO, 298 K) of b) C1Ph and c) C1Xy ; d) SCXRD structure of [Pd2(L1Xy )4⊃2BF4]2+.
The impact of exchanging the flanking phenyl group in C1Ph for a xylyl unit in C1Xy resulted in some spectroscopic differences, noticeably a shift in the resonance of the signal assigned to endohedral proton H j from 8.31 ppm to 7.71 ppm. The symmetry of the 1H NMR spectra of C1Ph and C1Xy , through‐space interactions between ortho‐pyridyl protons observed by NOESY (H a ⋅⋅⋅H n , H b ⋅⋅⋅H m ; Figure S73 and S98) and splitting of the H e (7.46 and 7.31 ppm, Figure 3c) and H f (2.13 and 1.99 ppm) resonances within C1Xy into two distinct sets of signals, were all congruent with the C 2h symmetry associated with selective formation of the cis assembly (Figure 1b). This was ultimately further confirmed by the solid‐state SCXRD structure of C1Xy (Figure 3d). [28]
cis‐Pd3L4 Double‐Cavity Cages
There has been some interest in recent years in the synthesis of linearly‐elongated, polytopic ligand scaffolds, and their self‐assembly to form multi‐cavity cage architectures. [31] In this manner, larger assemblies can be formed that possess several small cavities [32] instead of a single large one. [33] The formation of such a species from low‐symmetry ligands has, to the best of my knowledge, never been reported. [34]
To probe whether the geometric complementarity approach would remain effective for the assembly of low‐symmetry, multi‐cavity systems, tritopic ligand L2Ph (Figure 4a) was synthesised, incorporating two linearly‐fused iterations of the L1Ph scaffold. Pleasingly, self‐assembly with [Pd(CH3CN)4](BF4)2 in a 3 : 4 metal/ligand ratio in 3 : 1 d 6‐DMSO/CDCl3 successfully yielded the desired cis‐Pd3L4 double‐cavity cage (C2Ph ), determined by NMR (Figure 4b; see below) and ESI‐MS (Figure S151–153).
Figure 4.
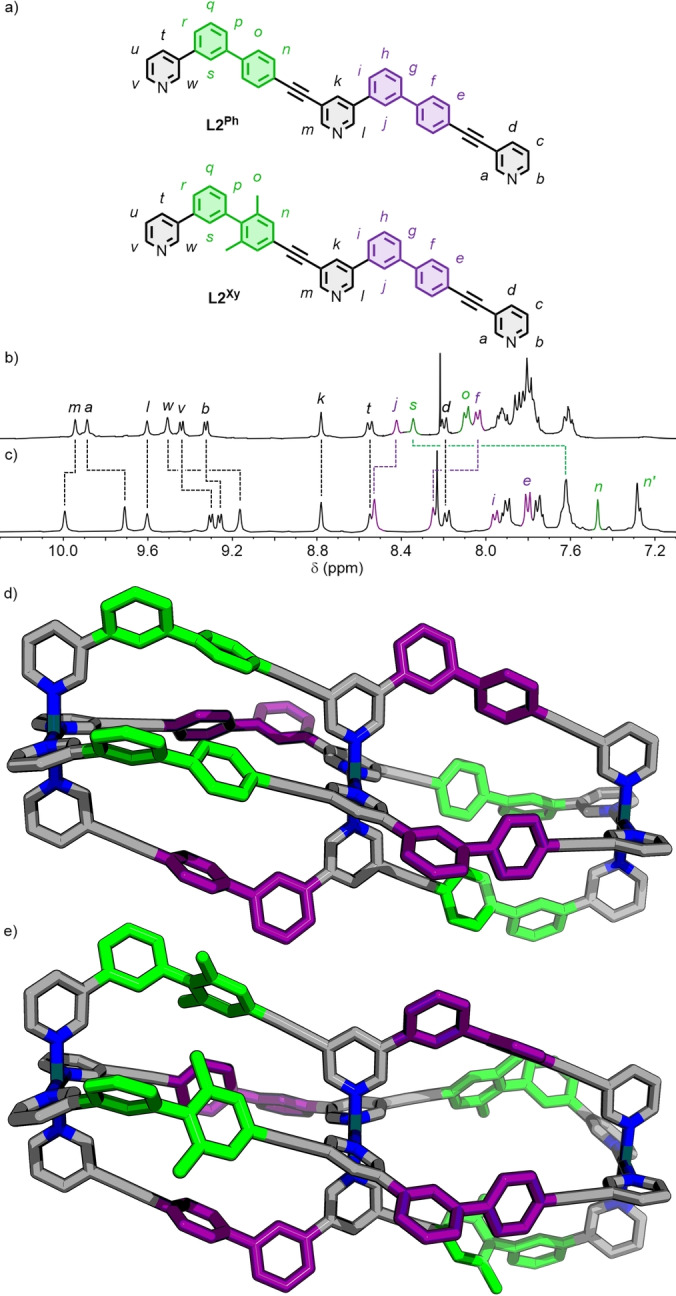
a) Tritopic ligands L2Ph and L2Xy ; partial 1H NMR spectra (400 MHz, 3 : 1 d 6‐DMSO/CDCl3, 298 K) of b) C2Ph and c) C2Xy ; molecular models (PM6) of d) C2Ph and e) C2Xy with some chemically inequivalent core ligand fragments shown in green and purple, highlighting the pseudo‐heteroleptic nature of the cavities.
As with previous work on the self‐assembly of unsymmetrical ditopic ligands, [20] the symmetry of the 1H NMR spectrum and through‐space interactions observed by NOESY between opposite ends of the ligand framework (H a ⋅⋅⋅H w , H l ⋅⋅⋅H m ; Figure S149) within the trinuclear architecture were consistent with cages of either C 2v (trans) or C 2h (cis) symmetry. Disappointingly, the growth of X‐ray quality crystals proved elusive. Based, however, on previous work, the dinuclear cages C1Ph and C1Xy , and molecular modelling of the cis‐Pd3L4 structure (Figure 4d), it was concluded that C2Ph must be the cis assembly. As such, the efficacy of using geometric design parameters to enforce ligands trans to each other to arrange in an anti‐parallel fashion is retained by the elongated, tritopic ligand.
Although the two cavities of the double‐decker assembly C2Ph are chemically equivalent, separate sets of signals in the 1H NMR spectrum were observed for the protons associated with each half of the ligand framework (Figure 4b), confirming the difference in their chemical environments. This can readily be seen by the different chemical shifts in the 1H NMR spectrum of the homologous ortho‐pyridyl protons H a and H m (δ=9.89 and 9.94 ppm, respectively) and H l and H w (δ=9.60 and 9.51 ppm, respectively), and endohedral protons H j and H s (δ=8.42 and 8.34 ppm, respectively).
The consequence of the combination of chemically inequivalent fragments of L2Ph and the symmetry of C2Ph is that each of the identical cage cavities are encompassed by two chemically distinct, unsymmetrical ligand fragments (Figure 4d). As a result, the cavities can be visualised as (pseudo‐)heteroleptic Pd2L2L′2 environments of Cs symmetry.
This concept was further demonstrated through the self‐assembly of ligand L2Xy (Figure 4a) with [Pd(CH3CN)4](BF4)2, under the same conditions as L2Ph , to give C2Xy , in which one half of the ligand scaffold possesses a phenyl ring as the flanking aromatic unit, the other a xylyl moiety. As with the dinuclear C1 cages and C2Ph , confirmation of successfully forming the targeted cis‐Pd3L4 isomer came from ESI‐MS (Figure S168–170), the high symmetry of the NMR spectrum (Figure 4c), NOE interactions only feasible between ligands in an anti‐parallel arrangement (H a ⋅⋅⋅H w , H l ⋅⋅⋅H m ; Figure S165) and, consistent with C1Xy , splitting of the aromatic (H n ; δ=7.47 and 7.28 ppm) and methyl (H o ; δ=2.03 and 1.92 ppm) protons of the xylyl unit in C2Xy into two distinct signals.
Within the trinuclear C2Xy , two pairs of ligands surround each individual cage cavity (Figure 4e)—one with phenyl units as the flanking aromatic moieties (as per C1Ph ), the other with m‐xylyl (as per C1Xy ), the close proximity of which was demonstrated by NOE interactions observed between the two (H e ⋅⋅⋅H o ; Figure S165 and S166). Again, the difference in the ligand environments was readily shown by 1H NMR (Figure 4c) in the chemical shifts of homologous endohedral protons (e.g. H j and H s —8.53 and 7.62 ppm, respectively).
The geometric parameters that drive selective formation of the cis‐[Pd3(L2)4]6+ cage structures mean that ligands positioned trans from each other across the PdII ions are in an anti‐parallel relative orientation. The result is that each cavity is described by two chemically distinct, unsymmetrical ligand environments (Figure 4d and e). Within a single, dinuclear cage structure, such an assembly would be termed heteroleptic. As the cis‐[Pd3(L2)4]6+ assemblies are homoleptic as a whole, and it is the individual cavities that possess an induced heteroleptic environment, the term pseudo‐heteroleptic is suggested to describe the cavity environments that result from the self‐assembly of the low‐symmetry ligands. This term would also seem appropriate for the CP3Q architecture described above, in which two distinct, unsymmetrical ligand fragments were incorporated into a single cage structure in a configurationally defined manner. As such, these pseudo‐heterolepticity approaches offer the potential for incorporating multiple functionalities into the cavity environment of the cage that could not otherwise be achieved via integrative self‐sorting of multiple ligands, and under facile self‐assembly conditions afforded by the formally homoleptic architectures.
Conclusion
With general principles of metal‐organic self‐assembly well evolved, and growing interest in the various applications of metal‐organic cages, the development of strategies to access more structurally sophisticated systems would greatly advance their utility. Constructing MOCs with more than one ligand framework offers the potential to precision engineer both the shape and functionality of the cavity environment. Previous approaches to the assembly of heteroleptic metal‐organic architectures rely on integrative self‐assembly of multiple ligands, either resulting from geometric complementarity, or coordination sphere engineering strategies. Whilst these have proven effective to a certain extent, there are inherent limitations to these approaches.
In this work, strategies have been outlined towards the development of pseudo‐heteroleptic MOCs—cages that, although they are assembled from single ligands, possess cavity spaces described by multiple ligand environments. These rely on covalent tethering of ligand fragments to constrain their relative orientations. By controlling the geometry of the tethering—either bent or linearly fused—MOCs have been realised that incorporate two inequivalent, unsymmetrical ligand scaffolds in a defined manner, generating cavities of C 2 and C s symmetry, respectively—a feat that has so far not been achieved via integrative self‐assembly of ligand mixtures.
The delicate balance of entropic and enthalpic factors can be easily disturbed to shift thermodynamic minima away from desired products. Removing the reliance on integrative self‐sorting greatly simplifies the self‐assembly process, whilst enhancing the structural and functional complexity of the cavity space. As such, pseudo‐heteroleptic design strategies will allow fine‐tuning of the cavity shape and functionality of discrete, porous MOCs, under facile conditions, to an extent not previously possible.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Acknowledgements
Dr. Andrew J. P. White (Imperial College London) is thanked for collection and analysis of SCXRD data. The Imperial College Research Fellowship programme is acknowledged for funding. Professor Matthew J. Fuchter is thanked for discussions and access to resources.
J. E. M. Lewis, Angew. Chem. Int. Ed. 2022, 61, e202212392; Angew. Chem. 2022, 134, e202212392.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Fujita M., Chem. Soc. Rev. 1998, 27, 417–425; [Google Scholar]
- 1b. Holliday B. J., Mirkin C. A., Angew. Chem. Int. Ed. 2001, 40, 2022–2043; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2076–2097; [Google Scholar]
- 1c. Cook T. R., Stang P. J., Chem. Rev. 2015, 115, 7001–7045. [DOI] [PubMed] [Google Scholar]
- 2. Forgan R. S., Lloyd G. O., Reactivity in confined spaces, Royal Society of Chemistry, London, 2021. [Google Scholar]
- 3.
- 3a. Smulders M. M. J., Riddell I. A., Browne C., Nitschke J. R., Chem. Soc. Rev. 2013, 42, 1728–1754; [DOI] [PubMed] [Google Scholar]
- 3b. Harris K., Fujita D., Fujita M., Chem. Commun. 2013, 49, 6703–6712; [DOI] [PubMed] [Google Scholar]
- 3c. Debata N. B., Tripathy D., Sahoo H. S., Coord. Chem. Rev. 2019, 387, 273–298; [Google Scholar]
- 3d. Martín Díaz A. E., Lewis J. E. M., Front. Chem. 2021, 9, 706462; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Tateishi T., Yoshimura M., Tokuda S., Matsuda F., Fujita D., Furukawa S., Coord. Chem. Rev. 2022, 467, 214612; [Google Scholar]
- 3f. McConnell A. J., Chem. Soc. Rev. 2022, 51, 2957–2971. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Maurizot V., Yoshizawa M., Kawano M., Fujita M., Dalton Trans. 2006, 2750–2756; [DOI] [PubMed] [Google Scholar]
- 4b. McTernan C. T., Davies J. A., Nitschke J. R., Chem. Rev. 2022, 122, 10393–10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Samanta S. K., Isaacs L., Coord. Chem. Rev. 2020, 410, 213181. For selected examples, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Therrien B., Süss-Fink G., Govindaswamy P., Renfrew A. K., Dyson P. J., Angew. Chem. Int. Ed. 2008, 47, 3773–3776; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3833–3836; [Google Scholar]
- 5c. Lewis J. E. M., Gavey E. L., Cameron S. A., Crowley J. D., Chem. Sci. 2012, 3, 778–784; [Google Scholar]
- 5d. Schmitt F., Freudenreich J., Barry N. P. E., Juillerat-Jeanneret L., Süss-Fink G., Therrien B., J. Am. Chem. Soc. 2012, 134, 754–757; [DOI] [PubMed] [Google Scholar]
- 5e. Zheng Y.-R., Suntharalingam K., Johnstone T. C., Lippard S. J., Chem. Sci. 2015, 6, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Percástegui E. G., Chem. Commun. 2022, 58, 5055–5071. For selected examples, see: [DOI] [PubMed] [Google Scholar]
- 6b. Riddell I. A., Smulders M. M. J., Clegg J. K., Nitschke J. R., Chem. Commun. 2011, 47, 457–459; [DOI] [PubMed] [Google Scholar]
- 6c. Preston D., White K. F., Lewis J. E. M., Vasdev R. A. S., Abrahams B. F., Crowley J. D., Chem. Eur. J. 2017, 23, 10559–10567; [DOI] [PubMed] [Google Scholar]
- 6d. Wright J. S., Metherell A. J., Cullen W. M., Piper J. R., Dawson R., Ward M. D., Chem. Commun. 2017, 53, 4398–4401; [DOI] [PubMed] [Google Scholar]
- 6e. Zhang D., Ronson T. K., Lavendomme R., Nitschke J. R., J. Am. Chem. Soc. 2019, 141, 18949–18953; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6f. de Jesús Valencia-Loza S., López-Olvera A., Martínez-Ahumada E., Martínez-Otero D., Ibarra I. A., Jancik V., Percástegui E. G., ACS Appl. Mater. Interfaces 2021, 13, 18658–18665. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Custelcean R., Chem. Soc. Rev. 2014, 43, 1813–1824. For selected examples, see: [DOI] [PubMed] [Google Scholar]
- 7b. Zhang D., Ronson T. K., Mosquera J., Martinez A., Nitschke J. R., Angew. Chem. Int. Ed. 2018, 57, 3717–3721; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3779–3783; [Google Scholar]
- 7c. Plajer A. J., Percástegui E. G., Santella M., Rizzuto F. J., Gan Q., Laursen B. W., Nitschke R J. R., Angew. Chem. Int. Ed. 2019, 58, 4200–4204; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4244–4248; [Google Scholar]
- 7d. Andrews R., Begum S., Clemett C. J., Faulkner R. A., Ginger M. L., Harmer J., Molinari M., Parkes G. M. B., Qureshi Z. M. H., Rice C. R., Ward M. D., Williams H. M., Wilson P. B., Angew. Chem. Int. Ed. 2020, 59, 20480–20484; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 20660–20664; [Google Scholar]
- 7e. Lee H., Kim D., Oh H., Jung O.-S., Chem. Commun. 2020, 56, 2841–2844; [DOI] [PubMed] [Google Scholar]
- 7f. Preston D., Patil K. M., O'Neil A. T., Vasdev R. A. S., Kitchen J. A., Kruger P. E., Inorg. Chem. Front. 2020, 7, 2990–3001. [Google Scholar]
- 8.
- 8a. Ueda Y., Ito H., Fujita D., Fujita M., J. Am. Chem. Soc. 2017, 139, 6090–6093; [DOI] [PubMed] [Google Scholar]
- 8b. Cullen W., Metherell A. J., Wragg A. B., Taylor C. G. P., Williams N. H., Ward M. D., J. Am. Chem. Soc. 2018, 140, 2821–2828; [DOI] [PubMed] [Google Scholar]
- 8c. Martí-Centelles V., Lawrence A. L., Lusby P. J., J. Am. Chem. Soc. 2018, 140, 2862–2868; [DOI] [PubMed] [Google Scholar]
- 8d. Ngai C., Sanchez-Marsetti C. M., Harman W. H., Hooley R. J., Angew. Chem. Int. Ed. 2020, 59, 23505–23509; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 23711–23715; [Google Scholar]
- 8e. Guo J., Fan Y.-Z., Lu Y.-L., Zheng S.-P., Su C.-Y., Angew. Chem. Int. Ed. 2020, 59, 8661–8669; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 8739–8747; [Google Scholar]
- 8f. Paul A., Shipman M. A., Onabule D. Y., Sproules S., Symes M. D., Chem. Sci. 2021, 12, 5082–5090; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8g. Bierschenk S. M., Pan J. Y., Settineri N. S., Warzok U., Bergman R. G., Raymond K. N., Toste F. D., J. Am. Chem. Soc. 2022, 144, 11425–11433. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Mal P., Breiner B., Rissanen K., Nitschke J. R., Science 2009, 324, 1697–1699; [DOI] [PubMed] [Google Scholar]
- 9b. Yamashina M., Sei Y., Akita M., Yoshizawa M., Nat. Commun. 2014, 5, 4662; [DOI] [PubMed] [Google Scholar]
- 9c. Hasegawa S., Meichsner S. L., Holstein J. J., Baksi A., Kasanmascheff M., Clever G. H., J. Am. Chem. Soc. 2021, 143, 9718–9723; [DOI] [PubMed] [Google Scholar]
- 9d. Sumida R., Tanaka Y., Niki K., Sei Y., Toyota S., Yoshizawa M., Chem. Sci. 2021, 12, 9946–9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Gemen J., Ahrens J., Shimon L. J. W., Klajn R., J. Am. Chem. Soc. 2020, 142, 17721–17729; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Tang X., Jiang H., Si Y., Rampal N., Gong W., Cheng C., Kang X., Fairen-Jimenez D., Cui Y., Liu Y., Chem 2021, 7, 2771–2786. [Google Scholar]
- 11. Pullen S., Tessarolo J., Clever G. H., Chem. Sci. 2021, 12, 7269–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Lewis J. E. M., Crowley J. D., ChemPlusChem 2020, 85, 815–827; [DOI] [PubMed] [Google Scholar]
- 12b. Tripathy D., Debata N. B., Naik K. C., Sahoo H. S., Coord. Chem. Rev. 2022, 456, 214396. [Google Scholar]
- 13.
- 13a. Bloch W. M., Clever G. H., Chem. Commun. 2017, 53, 8506–8516; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Pullen S., Clever G. H., Acc. Chem. Res. 2018, 51, 3052–3064; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Bardhan D., Chand D. K., Chem. Eur. J. 2019, 25, 12241–12269. [DOI] [PubMed] [Google Scholar]
- 14.For some recent examples of heteroleptic metal-organic cages, see:
- 14a. Sudan S., Li R.-J., Jansze S. M., Platzek A., Rudolf R., Clever G. H., Fadaei-Tirani F., Scopelliti R., Severin K., J. Am. Chem. Soc. 2021, 143, 1773–1778; [DOI] [PubMed] [Google Scholar]
- 14b. Li R.-J., Fadaei-Tirani F., Scopelliti R., Severin K., Chem. Eur. J. 2021, 27, 9439–9445; [DOI] [PubMed] [Google Scholar]
- 14c. Findlay J. A., Patil K. M., Gardiner M. G., MacDermott-Opeskin H. I., O'Mara M. L., Kruger P. E., Preston D., Chem. Asian J. 2022, 17, e202200093; [DOI] [PubMed] [Google Scholar]
- 14d. Li S.-C., Cai L.-X., Hong M., Chen Q., Sun Q.-F., Angew. Chem. Int. Ed. 2022, 61, e202204732; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202204732. [DOI] [PubMed] [Google Scholar]
- 15. Wu K., Tessarolo J., Baksi A., Clever G. H., Angew. Chem. Int. Ed. 2022, 61, e202205725; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202205725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. McMorran D. A., Steel P. J., Angew. Chem. Int. Ed. 1998, 37, 3295–3297; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 3495–3497; [Google Scholar]
- 16b. Steel P. J., McMorran D. A., Chem. Asian J. 2019, 14, 1098–1101. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Schmidt A., Casini A., Kühn F. E., Coord. Chem. Rev. 2014, 275, 19–36. For some recent examples of homoleptic Pd2L4 cages, see: [Google Scholar]
- 17b. Tsutsui T., Catti L., Yozac K., Yoshizawa M., Chem. Sci. 2020, 11, 8145–8150; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Birvé A. P., Patel H. D., Price J. R., Bloch W. M., Fallon T., Angew. Chem. Int. Ed. 2022, 61, e202115468; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202115468; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17d. Lee H., Tessarolo J., Langbehn D., Baksi A., Herges R., Clever G. H., J. Am. Chem. Soc. 2022, 144, 3099–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.R. A. S. Vasdev, D. Preston, C. A. Casey-Stevens, V. Martí-Centelles, P. J. Lusby, A. L. Garden, J. D. Crowley, Inorg. Chem. 2022, 10.1021/acs.inorgchem.2c00937. [DOI] [PubMed]
- 19. Samantray S., Krishnaswamy S., Chand D. K., Nat. Commun. 2020, 11, 880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Lewis J. E. M., Tarzia A., White A. J. P., Jelfs K. E., Chem. Sci. 2020, 11, 677–683; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Lewis J. E. M., Chem. Eur. J. 2021, 27, 4454–4460; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20c. Tarzia A., Lewis J. E. M., Jelfs K. E., Angew. Chem. Int. Ed. 2021, 60, 20879–20887; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 21047–21055. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Ogata D., Yuasa J., Angew. Chem. Int. Ed. 2019, 58, 18424–18428; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18595–18599; [Google Scholar]
- 21b. Mishra S. S., Kompella S. V. K., Krishnaswamy S., Balasubramanian S., Chand D. K., Inorg. Chem. 2020, 59, 12884–12894. [DOI] [PubMed] [Google Scholar]
- 22.Higher nuclearity PdII assemblies with unsymmetrical ditopic ligands have also been reported:
- 22a. Li R.-J., Marcus A., Fadaei-Tirani F., Severin K., Chem. Commun. 2021, 57, 10023–10026; [DOI] [PubMed] [Google Scholar]
- 22b. Mishra S. S., Chand D. K., Dalton Trans. 2022, 51, 11650–11657. [DOI] [PubMed] [Google Scholar]
- 23. Lisboa L. S., Findlay J. A., Wright L. J., Hartinger C. G., Crowley J. D., Angew. Chem. Int. Ed. 2020, 59, 11101–11107; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 11194–11200. [Google Scholar]
- 24.
- 24a. Bosch E., Barnes C. L., Inorg. Chem. 2001, 40, 3097–3100; [DOI] [PubMed] [Google Scholar]
- 24b. Hu Y.-Z., Chamchoumis C., Grebowicz J. S., Thummel R. P., Inorg. Chem. 2002, 41, 2296–2300. [DOI] [PubMed] [Google Scholar]
- 25. Fujita M., Tominaga M., Hori A., Therrien B., Acc. Chem. Res. 2005, 38, 369–378. [DOI] [PubMed] [Google Scholar]
- 26. Pereira F. A., Fallows T., Frank M., Chen A., Clever G. H., Z. Anorg. Allg. Chem. 2013, 639, 1598–1605. [Google Scholar]
- 27.
- 27a. Tashiro S., Tominaga M., Kusukawa T., Kawano M., Sakamoto S., Yamaguchi K., Fujita M., Angew. Chem. Int. Ed. 2003, 42, 3267–3270; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 3389–3392; [Google Scholar]
- 27b. Sun Q.-F., Murase T., Sato S., Fujita M., Angew. Chem. Int. Ed. 2011, 50, 10318–10321; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10502–10505; [Google Scholar]
- 27c. Bhat I. A., Samanta D., Mukherjee P. S., J. Am. Chem. Soc. 2015, 137, 9497–9502; [DOI] [PubMed] [Google Scholar]
- 27d. Escobar L., Escuedro-Adán E. C., Ballester P., Angew. Chem. Int. Ed. 2019, 58, 16105–16109; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16251–16255; [Google Scholar]
- 27e. Wu K., Zhang B., Drechsler C., Holstein J. J., Clever G. H., Angew. Chem. Int. Ed. 2021, 60, 6403–6407; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 6473–6478. [Google Scholar]
- 28.Deposition numbers 2202605 (C P4 ), 2202606 (C1 Xy ), and 2202607 (C P3Q ) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 29.
- 29a. Bloch W. M., Abe Y., Holstein J. J., Wandtke C. M., Dittrich B., Clever G. H., J. Am. Chem. Soc. 2016, 138, 13750–13755; [DOI] [PubMed] [Google Scholar]
- 29b. Bloch W. M., Holstein J. J., Hiller W., Clever G. H., Angew. Chem. Int. Ed. 2017, 56, 8285–8289; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8399–8404; [Google Scholar]
- 29c. Preston D., Barnsley J. E., Gordon K. C., Crowley J. D., J. Am. Chem. Soc. 2016, 138, 10578–10585; [DOI] [PubMed] [Google Scholar]
- 29d. Zhu R., Bloch W. M., Holstein J. J., Mandal S., Schäfer L. V., Clever G. H., Chem. Eur. J. 2018, 24, 12976–12982; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29e. Chen B., Holstein J. J., Platzek A., Schneider L., Wu K., Clever G. H., Chem. Sci. 2022, 13, 1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schulte T. R., Holstein J. J., Krause L., Michel R., Stalke D., Sakuda E., Umakoshi K., Longhi G., Abbate S., Clever G. H., J. Am. Chem. Soc. 2017, 139, 6863–6866. [DOI] [PubMed] [Google Scholar]
- 31. Vasdev R. A. S., Preston D., Crowley J. D., Chem. Asian J. 2017, 12, 2513–2523. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Bandi S., Pal A. K., Hanan G. S., Chand D. K., Chem. Eur. J. 2014, 20, 13122–13126; [DOI] [PubMed] [Google Scholar]
- 32b. Johnstone M. D., Schwarze E. K., Clever G. H., Pfeffer F. M., Chem. Eur. J. 2015, 21, 3948–3955; [DOI] [PubMed] [Google Scholar]
- 32c. Preston D., Lewis J. E. M., Crowley J. D., J. Am. Chem. Soc. 2017, 139, 2379–2386; [DOI] [PubMed] [Google Scholar]
- 32d. Yazaki K., Akita M., Prusty S., Chand D. K., Kikuchi T., Sato H., Yoshizawa M., Nat. Commun. 2017, 8, 15914; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32e. Zhu R., Regeni I., Holstein J. J., Dittrich B., Simon M., Prévost S., Gradzielski M., Clever G. H., Angew. Chem. Int. Ed. 2018, 57, 13652–13656; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13840–13844. [Google Scholar]
- 33.
- 33a. Tominaga M., Suzuki K., Kawano M., Kusukawa T., Ozeki T., Sakamoto S., Yamaguchi K., Fujita M., Angew. Chem. Int. Ed. 2004, 43, 5621–5625; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 5739–5743; [Google Scholar]
- 33b. Sun Q.-F., Iwasa J., Ogawa D., Ishido Y., Sato S., Ozeki T., Sei Y., Yamaguchi K., Fujita M., Science 2010, 328, 1144–1147; [DOI] [PubMed] [Google Scholar]
- 33c. Fujita D., Ueda Y., Sato S., Mizuno N., Kumasaka T., Fujita M., Nature 2016, 540, 563–567. [DOI] [PubMed] [Google Scholar]
- 34.Crowley and co-workers have recently reported a heteronuclear, double-cavity [Pd2PtL4] cage using a sub-component self-assembly strategy. The unsymmetrical ligand is formed in situ using dynamic covalent chemistry, distinguishing it from the systems investigated in this work: Lisboa L. S., Preston D., McAdam C. J., Wright L. J., Hartinger C. G., Crowley J. D., Angew. Chem. Int. Ed. 2022, 61, e202201700; [Google Scholar]; Angew. Chem. 2022, 134, e202201700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.