

Abstract
Polygenic risk scores (PRS) estimate an individual’s genetic likelihood of complex traits and diseases by aggregating information across multiple genetic variants identified from genome-wide association studies. PRS can predict a broad spectrum of diseases and have therefore been widely used in research settings. Some work has investigated their potential applications as biomarkers in preventative medicine, but significant work is still needed to definitively establish and communicate absolute risk to patients for genetic and modifiable risk factors across demographic groups. However, the biggest limitation of PRS currently is that they show poor generalizability across diverse ancestries and cohorts. Major efforts are underway through methodological development and data generation initiatives to improve their generalizability. This review aims to comprehensively discuss current progress on the development of PRS, the factors that affect their generalizability, and promising areas for improving their accuracy, portability, and implementation.
Keywords: polygenic risk scores (PRS), genetic risk prediction, diverse ancestry populations, PRS generalizability, clinical translation of PRS
GENOME-WIDE ASSOCIATION STUDIES AND GENETIC PREDICTION OF COMPLEX TRAITS
Genome-wide association studies (GWAS) of complex traits have grown explosively over the last decade (1, 2). In GWAS, researchers typically test millions of associations between the genetic variants included in the study [usually single-nucleotide polymorphisms (SNPs)] and the phenotype of interest, using a multiple testing significance threshold of p < 5 × 10−8 genome-wide. GWAS have been enormously helpful in two areas of biomedical research: providing unbiased insights into the molecular etiology of diseases and comorbidities and predicting genetic risk of diseases to further enable investigations into epidemiology and intervention strategies in preventative medicine.
To assess an individual’s genetic predisposition to a common disease, researchers use polygenic risk scores (PRS) created from GWAS and individual genotype data in an independent target cohort. In their simplest form, PRS are individual-level scores that aggregate the number of risk alleles across the genome weighted by their effect sizes. The theoretical underpinnings of this model have roots in concepts of complex trait genetics and genetic prediction that date back over a century (3). Many first applications of this model emerged in agriculture, particularly with estimated breeding values (BVs) in livestock genetics (4–6).Similar to challenges with transferring predicted BVs across purebred lines in animal models (7), such as the observed decrease in accuracy of estimated BVs in more genetically distant breeds, there are challenges with the transferability and thus translation of PRS developed across diverse human populations. We focus on generalizability of PRS in this review.
Factors That Influence Heritability in the Context of Polygenic Risk Scores
The goal of most prediction models in biomedical research is to predict whether a person will develop a disease or the age of onset in individuals who do not yet have the disease. The prediction accuracy of a model with genetic predictors, such as PRS, is bounded by the heritability of the phenotype. This limit theoretically refers to broad-sense heritability: the proportion of a trait’s variance attributable to all genetic variants (8). In practicality, however, it is almost impossible to estimate the broad-sense heritability of a phenotype because, by definition, it considers the effects of all genetic variants and interactions among them. In contrast, narrow-sense heritability, defined as the proportion of a trait’s phenotypic variance explained by the additive genetic variation, can be estimated in twin- and family-based studies (9). The majority of current PRS models are based solely on genotyped or high-quality imputed variants. Therefore, the upper limit of PRS is determined by the proportion of a trait’s variance captured by the additive effects of these SNPs, also known as SNP-based heritability , and tends to be a lower bound for narrow-sense heritability (4, 10). The expected performance of PRS as measured by R2 can be shown as
| 1. |
where is the proportion of phenotypic variance explained by genotyped and imputed SNPs, M is the effective number of genetic markers (e.g., independent SNPs), and N is the sample size (4, 10). It follows that as N goes to infinity, M/N approaches 0, and R2 approaches . Thus, can be used to guide how much predictive power to expect from PRS based on typical GWAS. Commonly used heritability estimation methods include linkage disequilibrium score regression (LDSC), which uses GWAS summary statistics (11), and genomic relatedness matrix restricted maximum likelihood (GREML), which uses individual-level genotype data (12).
While heritability estimates provide a helpful guide, it is important to note that they are not absolute bounds, as they are not fixed properties. Rather, they are specific to the context and population in which they are measured. Estimates may vary depending on differences in environmental exposures and genetic ancestries (8, 13, 14). Even within a population, they may change over time. Characteristics like age, sex, and socioeconomic status have been shown to influence heritability estimates for a range of phenotypes in the UK Biobank (14). Differences in heritability may to some extent contribute to disparities in PRS accuracy across populations, although sample size differences currently play a much larger role (15, 16). Further investigations into the phenotypes for which heritability estimates are particularly variable across populations will help guide expectations for PRS transferability when sample sizes are more comparable across populations.
Partitioning Heritability into Functional Categories for Enrichment Analysis
The advent of GWAS has also accelerated large-scale efforts to define corresponding functions across the genome. Some common examples of functional annotations include contributions to protein structure and function, potential gene regulatory roles, and sensitivity to evolutionary changes (17). These functional annotations are particularly useful in differentiating SNPs that potentially have larger effects, may be causal (i.e., mutating the genetic variant directly alters the trait), and may explain a larger portion of heritability than other SNPs. Altogether, they can help increase the accuracy of SNP heritability estimates (17–19). Several methods have been developed to partition SNP heritability by these annotations, such as stratified LDSC (S-LDSC) (20) and GREML-based methods (12, 19, 21). These in turn have been leveraged to improve PRS accuracy and transferability.
POLYGENIC RISK SCORE CONSTRUCTION METHODS
Given the rapid expansion of available GWAS summary statistics [e.g., see the Polygenic Score Catalog (22)], there has been a recent flurry of new PRS construction methods that improve upon methods originally applied in animal breeding to increase accuracy, computational efficiency, and generalizability (23). Each method has advantages and disadvantages with varying accuracies and computational burdens across different traits and cohorts. The main differences between PRS methods are in their assumptions about which variants are included in the predictor and what effect sizes or weights correspond to them. PRS methods that use individual-level data, such as LASSO (least absolute shrinkage and selection operator) and BLUP (best linear unbiased prediction), can predict the genetic component of multiple complex phenotypes with high accuracy (24, 25). However, access to individual-level genotype data is still currently limited because of logistical, data security, and ethical considerations. Furthermore, it is computationally challenging to implement those methods on current biobank-scale data. We therefore focus primarily on methods that only require GWAS summary statistics and a reference panel of linkage disequilibrium (LD) information in this review, although approaches have also been developed that combine both inputs when individual-level data are partially available (26). The implicit assumption for such methods is that the reference sample should be from the same population in which GWAS is performed, thus allowing unbiased estimation of LD from the reference panel. Discrepancies in LD structure between the GWAS summary statistics and reference panel are likely to reduce prediction accuracy. Additionally, the reference panel sample size balances computational burden and LD estimation accuracy.
Summary statistics–based PRS methods can be further categorized by variant selection strategy, i.e., SNP preselection methods or genome-wide methods. A widely used preselection method is pruning and thresholding (P+T), which usually applies multiple p-value thresholds together with a fixed LD r2 threshold to remove highly correlated SNPs. The LD window size is typically chosen arbitrarily, and SNPs are pruned through a process called LD clumping (23). P+T is then optimized by choosing the p-value threshold that produces the highest prediction accuracy in a validation or tuning cohort with both genotype and phenotype information available. P+T assumes that the selected SNPs are nearly independent from each other and thus can be fit additively. Extended models have been developed that correct winner’s curse effects or incorporate functional annotations (27, 28). More sophisticated genome-wide methods can model all markers simultaneously by rescaling or shrinking estimated effect sizes. One major advantage of such methods is that they account for LD between SNPs using a reference panel in a principled manner, and thus genome-wide SNPs can be fit simultaneously with a reduced risk of overfitting. Some examples include LDpred (29), SBLUP (30), lassosum (31), SBayesR (32), PRS-CS (33), and LDpred2 (34) (Table 1).
Table 1.
Overview of existing PRS methods
| Typea | Method (Ref.) | Description | Tuning parametersb | Extensions |
|---|---|---|---|---|
| Single trait, single ancestry | P+T (23) | Selects independent trait-associated SNPs within a specified LD window | Usually just p-value threshold; additional LD window and LD r2 tuning has the potential to improve accuracy | 2D PRS (27) (integrate P+T and functional annotations); doubly weighted GRS (28) (correct for winner’s curse); SCT (155) (stack multiple PRS built from P+T with varying parameters using penalized regression) |
| LDpred (29) | Uses a Bayesian multiple regression framework; LDpred-inf assumes an infinitesimal model | Proportion of SNPs with nonzero effects and LD radius for grid model | LDPred2 (34) (faster and more robust, automated model without tuning parameters implemented); LDpred-funct (49) (leverages functional annotations) | |
| SBLUP (30) | Assumes an infinitesimal model, approximates BLUP effects | NA | wMT-SBLUP (39) | |
| Lassosum (31) | Uses a penalized regression framework with a LASSO-type penalty | Penalty parameter and shrinkage parameter for the LD correlation matrix; pseudo-validation applicable | NA | |
| SBayesR (32) | Uses a Bayesian multiple regression framework; an approximation of BayesR | NA | SBayesS, SBayesRS (156) | |
| PRS-CS (33) | Uses a Bayesian multiple regression framework with continuous mixture shrinkage priors | Proportion of SNPs with nonzero effects for grid model | PRS-CSx (46) | |
| NPS (157) | Uses a partitioning-based nonparametric shrinkage framework | NA | NA | |
| DBSLMM (158) | Assumes all SNPs have nonzero effects, with some having larger effects; an approximation of BSLMM | NA | NA | |
| SDPR(159) | Uses a Bayesian nonparametric model through Dirichlet process regression | NA | NA | |
| Meta-PRS (26) | Uses a linear combination of one PRS derived from individual-level data using BOLT-LMM and another derived from GWAS summary statistics using LDpred/P+T | Weight for each PRS and LDpred/P+T-related hyperparameters | NA | |
| Single trait and single ancestry, with functional annotations | AnnoPred (50) | Leverages genomic and epigenomic functional annotations based on a Bayesian framework; AnnoPred-inf assumes infinitesimal models | Proportion of SNPs with nonzero effects | PleioPred (41) |
| JAMpred (160) | Uses a two-step Bayesian variable selection framework | Sparsity parameter reflecting the proportion of SNPs with nonzero effects | NA | |
| IMPACT (52) | Uses regulation annotations to prioritize nearly independent variants selected by P+T in Europeans and generalized in East Asians | Same as P+T; the proportion of SNPs explaining the closest 50% SNP-based heritability | NA | |
| Multitrait | wMT-SBLUP (39) | Combines genetically correlated traits in a weighted index; an approximation of MT-BLUP | NA | NA |
| MTAG (40) | Meta-analyzes genetically correlated traits accounting for sample overlap; usually the outputs are further used for other PRS construction methods | Dependent on the downstream PRS construction methods | NA | |
| CTPR(161) | Uses a cross-trait penalized regression framework with the LASSO and minimax concave penalty | Penalty parameters | NA | |
| PANPRS (162) | Uses a penalized regression framework integrating pleiotropy and functional annotations | Penalty and sparsity parameters | NA | |
| PDR(163) | Identifies shared pleiotropic components underlying genetically correlated traits to estimate posterior mean effect sizes | Dependent on the downstream PRS construction methods | NA | |
| Multitrait with functional annotations | PleioPred (41) | Leverages pleiotropy and functional annotations based on a Bayesian framework; PleioPred-inf/PleioPred-anno-inf assume infinitesimal models | Covariance within the overlapping individuals; not required for noninfinitesimal models | NA |
| Multiancestry | XP-BLUP (88) | Uses large-scale trans-ancestry auxiliary GWAS (usually European GWAS) to select trait-associated SNPs as a variance component and evaluates ancestry-specific effect sizes using linear mixed models | Same as P+T | NA |
| MultiPRS (43) | Uses a weighted combination of PRS trained from different populations | Weight for each PRS | NA | |
| XPASS (164) | Leverages trans-ancestry genetic correlation; XPASS+incorporates population-specific effects | Same as P+T for XPASS+ | NA | |
| PRS-CSx (46) | Jointly models multiple GWAS from diverse ancestries using a Bayesian framework assuming continuous effect size shrinkage | Proportion of SNPs with nonzero effects for grid model; weight for each PRS | NA | |
| shaPRS (165) | Utilizes shared genetic effects across ancestries using a modified meta-analysis from two GWAS (one is from target ancestries); also applies to two genetically correlated traits in the same ancestry | Dependent on the downstream PRS construction methods | NA | |
| Multiancestry with functional annotations/fine-mapping | Polypred, Polypred+ (44) | Uses a linear combination of predictors from functionally informed fine-mapping and BOLT-LMM/SBayesR/PRS-CS in large-scale European GWAS; Polypred+additionally incorporates predictors from large-scale data in target ancestry if available | Weight for each PRS | NA |
The listed PRS methods are categorized as single- or multiancestry and single- or multitrait, with some incorporating additional information such as functional annotations and fine-mapping (a detailed example is shown in Figure 1b).
For methods requiring additional validation/tuning cohorts, the corresponding tuning parameters are also briefly described.
Abbreviations: BayesR, Bayesian multiple regression model; BLUP, best linear unbiased prediction; CTPR, cross-trait penalized regression; DBSLMM, deterministic Bayesian sparse linear mixed model; GRS, genetic risk score; GWAS, genome-wide association study; IMPACT, inference and modeling of phenotype-related active transcription; LASSO, least absolute shrinkage and selection operator; LD, linkage disequilibrium; MTAG, multitrait analysis of GWAS; NA, not any; P+T, pruning and thresholding; PANPRS, Pleiotropy and ANnotation information into PRS; PRS, polygenic risk scores; PRS-CSx, PRS continuous shrinkage extension; SBayesR, summary statistics Bayesian multiple regression model; SBayesRS, SBayesS extension following the multicomponent mixture model of SBayesR; SBayesS, summary data-based BayesS; SBLUP, summary statistics–based BLUP; SCT, stacked clumping and thresholding; SDPR, summary data–based Dirichlet process regression model; SNP, single-nucleotide polymorphism; wMT, weighted multitrait; XP-BLUP, cross-population BLUP; XPASS, cross-population analysis with summary statistics.
PRS methods typically make different assumptions about the prior distribution of SNP effect sizes, that is, the proportion of causal SNPs across the genome (ρ) and their effect sizes. For example, LDpred uses a Bayesian framework to infer the posterior mean SNP effects by assuming a point-normal mixture distribution. One key parameter that needs to be optimized is ρ. When this parameter is set to 1 (i.e., all SNPs are causal), the method assumes an infinitesimal genetic architecture; this is the same assumption made in SBLUP. Data-driven methods such as SBayesR, LDpred2-auto, and PRS-CS-auto can estimate such parameters without post hoc tuning, which reduces computational burden. Comprehensive comparisons of prediction performance using these methods have been reported in different traits, and a standardized benchmarking framework called GenoPred has been developed to enable fair comparisons across methods (35–37). A recent comprehensive review connects most PRS methods through a multiple linear regression framework and thus compares their advantages and shortcomings from a statistical perspective (38). The optimal prediction method depends heavily on the trait-specific genetic architecture, and thus Bayesian or nonparametric methods that can adapt to different genetic architectures are expected to perform more robustly across phenotypes. However, some of these methods are also computationally burdensome. There are ongoing efforts in this active research area to develop methods that improve both prediction accuracy and computational efficiency in current biobank-scale datasets.
Increasing Polygenic Risk Score Accuracy and Generalizability Through Multitrait, Multiancestry, and Functional Annotation Extensions
There are several potential approaches for extending single-trait PRS methods to improve accuracy and transferability (Figure 1). For example, multitrait methods leverage abundant genetic correlations (rg) among complex traits by aggregating GWAS information across related traits (39–41). Previous studies have reported extensive genetic correlations among related traits, such as between schizophrenia and bipolar disorder [rg = 0.79, standard error (SE) = 0.04] and between type 2 diabetes and body mass index (BMI) (rg = 0.36, SE = 0.04) (11). By modeling the genetic correlations between related traits, multitrait PRS methods such as wMT-SBLUP (42) can estimate more accurate SNP effect sizes because of their shared genetic basis. Some methods, such as the multitrait analysis of GWAS (MTAG) method, boost power by modeling genetic correlation and GWAS summary statistics from related traits to produce trait-specific GWAS effect size estimates that can then be used as input to PRS methods (40). These approaches typically significantly increase prediction accuracy, especially for underpowered GWAS due to limited sample sizes or heritability; however, they inherently trade off interpretability of the estimates by combining multiple correlated traits for a single PRS construction.
Figure 1.
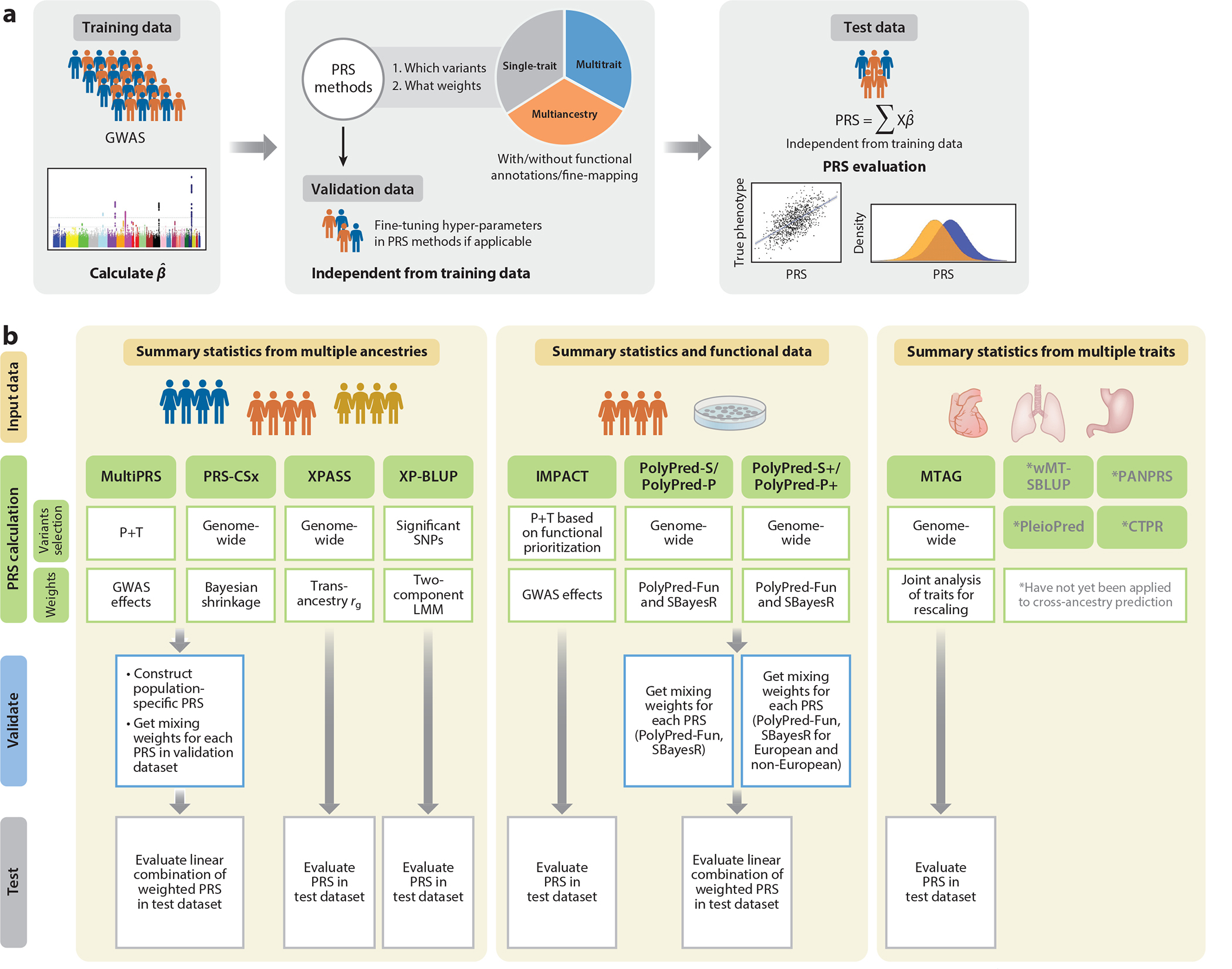
(a) PRS analysis steps. First, we obtain the estimated effect sizes of genetic markers from the training data. Second, we use different PRS construction methods to rescale or reshrink the estimated effect sizes. We optimize the hyperparameters of those methods requiring fine-tuning in the validation/tuning cohort. Finally, we construct the PRS and then validate their performance in the independent test data. (b) Extensions of PRS methods based on GWAS summary statistics that incorporate multitrait, multiancestry, and functional annotation data. Abbreviations: BLUP, best linear unbiased prediction; CTPR, cross-trait penalized regression; GWAS, genome-wide association study; IMPACT, inference and modeling of phenotype-related active transcription; LMM, linear mixed model; MTAG, multitrait analysis of GWAS; P+T, pruning and thresholding; PANPRS, Pleiotropy and ANnotation information into PRS; PRS, polygenic risk scores; PRS-CSx, PRS continuous shrinkage extension; SBayesR, summary statistics Bayesian multiple regression model; SBLUP, summary statistics–based BLUP; SNP, single-nucleotide polymorphism; wMT, weighted multitrait; XP-BLUP, cross-population BLUP; XPASS, cross-population analysis with summary statistics.
In addition to multitrait approaches, PRS approaches that incorporate information from ancestrally diverse populations improve prediction performance especially in underrepresented non-European populations by leveraging well-powered GWAS from European populations (43–46) (Table 1), typically with little if any decrease in accuracy for majority populations. Multiancestry PRS methods typically assume that genetic architecture is largely shared across populations. Indeed, an analysis of 31 complex traits identified high cross-population genetic correlations of 0.85 (SE = 0.01) on average between East Asians and Europeans (47), replicating earlier findings (48). Furthermore, multiancestry PRS methods such as PRS-CSx (46), which linearly combines PRS computed from GWAS of multiple ancestries, enable more accurate PRS construction by sharing information across multiple ancestry populations and leveraging differences in allele frequencies and LD. While methodological challenges remain stemming from cross-ancestry differences in biology (e.g., heterogeneous effect sizes), the environment [e.g., gene–environment (GxE) interaction effects], and technology (e.g., different phenotyping, genotyping, and imputation strategies), multiancestry PRS methods are promising approaches for improving PRS accuracy and transferability across populations, especially until we reach well-powered and comparable sample sizes of GWAS in underrepresented populations.
Additional extensions of PRS methods have been developed that incorporate functional annotations to improve the accuracy of PRS, such as LDpred-funct (49) and AnnoPred (50). LDpred-funct leverages trait-specific functional priors using a baseline-LD model (49, 51). AnnoPred estimates per-SNP heritability using S-LDSC to more heavily weight SNPs with greater potential functionality in PRS (50). These two methods have performed comparably in analyses despite differences in their inclusion of imputed variants and in how they model polygenicity. Relatedly, IMPACT is a resource of regulatory annotations from epigenetic and transcription factor binding datasets across a wide range of cell types that has been used in PRS to select SNPs by prioritizing functional variants with more ancestrally portable genetic effects in GWAS data (52). Other methods that attempt to identify and prioritize causal variants in PRS through (functionally informed) fine-mapping, such as PolyPred, more accurately assign causal effect sizes that are more transferable via their shared causal mechanisms of biology; these have also been shown to outperform standard PRS approaches (44).
Each category of PRS construction method discussed here—those that incorporate multitrait, multiancestry, or functional annotations—has separately been shown to improve prediction. However, most approaches do not link multiple extensions, such as using multitrait and multiancestry data together. Hypothetically, one approach would be to apply a method like MTAG for multiple traits within several populations, then use a multiancestry method such as PRS-CSx to combine results across populations. A limitation of this multimethod approach, however, is that MTAG requires all GWAS to have high statistical power, which is unlikely to be available for all populations and phenotypes. Approaches are therefore still clearly needed to model and include multiple data modalities to improve PRS portability.
Pleiotropy
The extensive genetic correlation discovered among traits is in part due to pleiotropy, a phenomenon where genetic variants affect multiple traits (53, 54). A systematic analysis of 558 publicly available well-powered GWAS demonstrated that 90% of trait-associated loci have pleiotropic effects (54). As previously discussed, we can leverage such pleiotropic effects to improve prediction accuracy, particularly in clinical research. For example, type 2 diabetes subtypes can be inferred by modeling polygenic risk from waist-hip ratio, BMI, lipids, and other traits (55). Theoretically, the proportion of variance explained R2 for the target trait yi by the PRS of its correlated trait can be written as
| 2. |
This equation can be approximated as
| 3. |
using approximations and where and are respectively SNP-based heritability for traits i and j, rg is their genetic correlation, and Nj and Mj are respectively numbers of samples and independent variants for PRS estimation in trait j (56). When Nj goes to infinity, R2 approaches . As SNP-based heritability is the theoretical upper bound of R2 for the PRS of any target disease, this equation provides an analogous upper bound of PRS derived from correlated traits, which is proportional to the squared genetic correlation of the target and correlated trait (Figure 2a).
Figure 2.
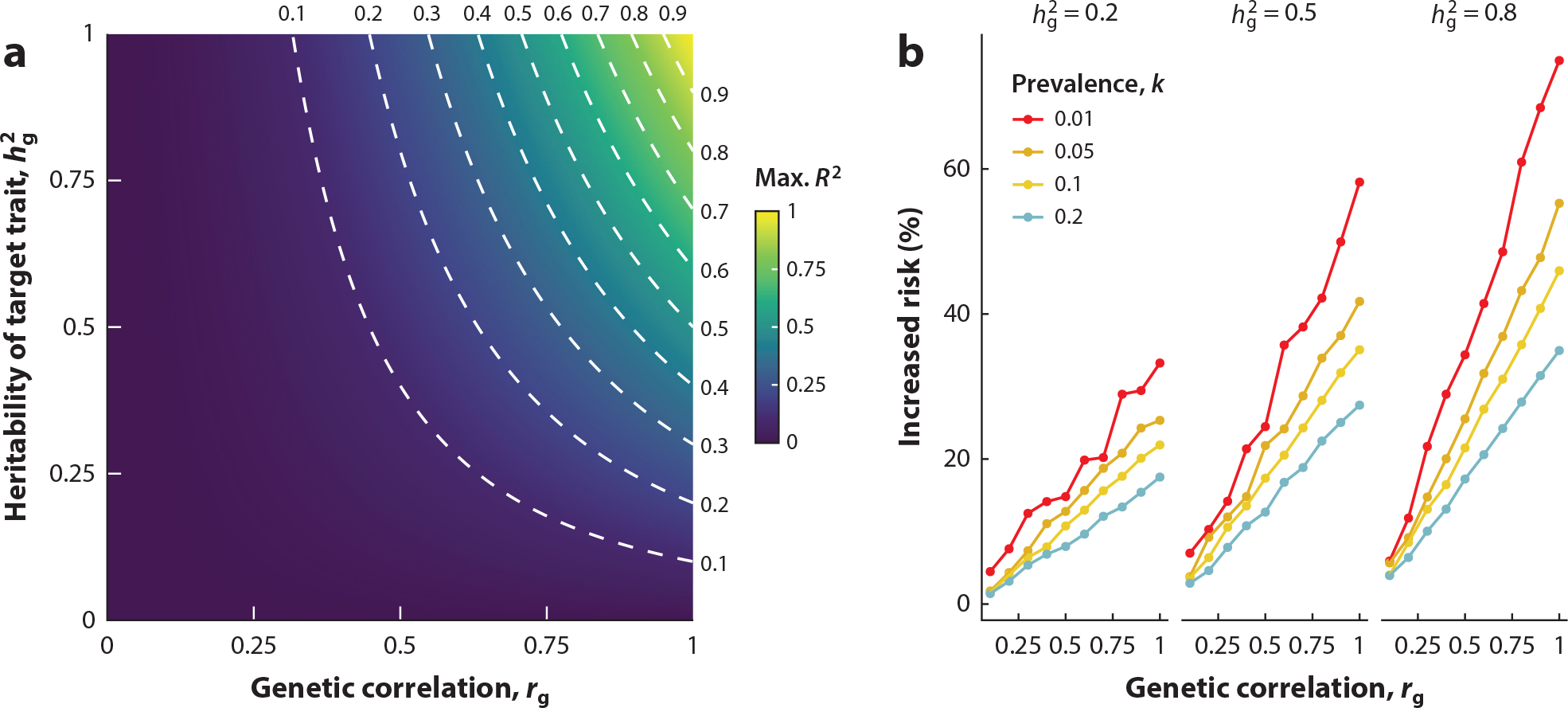
(a) Theoretical upper bound of the proportion of variance explained, R2, based on Equation 3. For a given target trait (with heritability ) and its correlated trait (with genetic correlation to the target trait), the plot shows distributions of theoretical maximum R2. Dotted lines represent contours of maximum R2 = 0.1,. . ., 0.9. (b) The relative risk increases for correlated traits by selecting embryos based on higher polygenic scores of educational attainment. With different levels of heritability and prevalence of correlated traits, the plot demonstrates how the relative risk increase changes with regard to genetic correlation with the target trait (educational attainment). We set the number of embryos for selection to be 10; broad-sense and SNP-based heritabilities of educational attainment to be 0.4 and 0.1, respectively; and standard deviation of educational attainment to be 3.2. To generate this figure, we modified the simulation framework developed by Turley et al. (57).
While potentially useful for increasing the accuracy of PRS for certain traits, leveraging the pervasive pleiotropy for prediction also raises serious concerns about unintended consequences of PRS in many settings, such as embryo selection. For example, a previous study reported that embryo selection based on higher polygenic scores for educational attainment would increase the risk of bipolar disorder by 16% from an absolute risk of 1% to 1.16% (57). The magnitude of this unintended consequence depends on many parameters, including heritability, genetic correlation, PRS predictive performance, the number of embryos for selection, and the prevalence of the traits in the population (Figure 2b). Other work has demonstrated how these parameters affect the expected risk reduction for the target disease and risk increase for correlated diseases under different simulation settings (58). With a typical number of embryos (n = 5) and prevalence (k = 1%), the relative risk increase ranges from ~6% for weakly correlated diseases (rg = −0.1) to ~22% for strongly correlated diseases (rg = −0.3). These observations indicate that careful consideration is required to prevent unintended and potentially harmful consequences of selecting embryos for pleiotropic traits.
EVALUATING POLYGENIC RISK SCORE ACCURACY
There are several metrics for evaluating PRS accuracy, each measuring various aspects of model performance. Typically, the performance of PRS is evaluated in an independent target dataset. Using linear regression for quantitative traits or logistic regression for binary traits, we test the relationship between the genetically predicted and measured phenotype. For PRS methods that require tuning of hyperparameters, an additional validation/tuning dataset may be required to avoid overfitting. In these cases, both target datasets (validation and testing cohorts) require individual-level genotype and phenotype data. Previous work has shown that pseudo-validation can be an alternative strategy to determine optimal hyperparameters when the validation phenotypes are not available (31).
While the squared correlation (R2) between true phenotype and PRS is an intuitive evaluation of phenotypic variance explained for quantitative traits, incremental or partial R2 is most often used to quantify the specific contribution of PRS after accounting for appropriate covariates in the regression. For binary traits, pseudo-R2 metrics serve as conceptual proxies, of which Nagelkerke’s R2 is one of the most widely used statistics. To improve interpretability and comparability across cohorts, however, pseudo-R2 metrics on the liability scale adjust the metric by case–control ratios so that it is comparable to trait heritability. This conversion typically requires disease population prevalence (59), which may require careful consideration if prevalences vary across populations. To assess the discriminative power of the model to correctly predict individuals with and without a disease, researchers most commonly use the metric known as area under the ROC (receiver operating characteristic) curve (AUC) (sometimes referred to as Concordance statistics, C-index, or C-statistics for survival models). AUC values of 0.5 and 1 indicate no and perfect discriminatory ability between cases and controls, respectively. PRS can be modeled both with and without other risk factors to understand the performance of specific risk factors, as well as the overall combined model. Additional evaluations based on the PRS distribution between cases and controls are often required in order to assess the clinical utility of predictive models. As a continuous score, PRS enable the use of any threshold to stratify individuals at different levels of risk. Recent studies have measured odds ratios (ORs) by comparing top ranked PRS values relative to other PRS strata (e.g., middle stratum or the remaining strata combined), but such comparisons can be misleading. Within such comparisons, ORs between extreme PRS strata (usually top versus bottom, e.g., 10%, 5%, or even 1%) are often inflated relative to OR estimates comparing groups with and groups without a disease (60). With information such as AUC and prevalence of binary traits, the relative risks in a reference group can be transformed to absolute scales in the general population using online tools (61) and R packages (62). Conversion from relative to absolute risk scales (e.g., lifetime remaining risk or five-year risk) aids interpretability, is necessary for clinical decision-making, and leaves less room for overly optimistic interpretations of model utility (63). Relatively few absolute risk models are used in clinical medicine, and an open challenge arises when PRS can add significantly to a disease area for which no absolute risk models exist beyond simple risk modifiers such as age and sex.
The exciting potential applications of PRS in the clinic should be accompanied by careful considerations of best practices for PRS construction and evaluation. PRS models for individual-level risk prediction are currently somewhat unstable; for example, when PRS are developed for the same trait and ancestry with different discovery GWAS, typically only small to modest proportions of individuals in the upper tails of the PRS distribution in the same target cohort overlap (64). This highlights the uncertainty of PRS estimates for individual-level risk stratification (65). Moreover, PRS performance is related to trait-specific genetic architecture. Therefore, systematically exploring the absolute risk of an individual developing a particular disease is necessary, particularly for the translation of PRS. Additionally, recent PRS evaluations have been highly inconsistent between studies, making it difficult to compare utility across studies. There are ongoing efforts to improve the reproducibility of PRS studies and benchmarking against other PRS, such as PRS repositories to encourage data-sharing and transparency (66). These efforts propose guidelines and protocols for performing PRS analyses and improving reporting standards (60, 63, 67). However, guidance on multiancestry PRS construction and best practices in all these efforts are lacking. Specifically, while multiancestry GWAS are currently critical for overcoming vast Eurocentric biases, they raise further challenges in PRS construction and evaluation practices. Furthermore, as PRS methods improve and GWAS attain larger sample sizes, corresponding effect sizes used as weights in PRS methods will change over time. Therefore, more sophisticated methods are needed to keep PRS updated or ensure that PRS are stable over time, with equitable benefits in different contexts.
POLYGENIC RISK SCORE TRANSFERABILITY ACROSS ANCESTRIES
Given vast Eurocentric biases in genetic studies, PRS have wide-ranging accuracies across populations. Specifically, PRS constructed from current Eurocentric GWAS are most accurate for European ancestry populations (68). PRS are typically constructed using common variants [with a minor allele frequency (MAF) of at least 1%]; these common variants typically arose long ago in human history and are thus expected to be shared across ancestries. While some degree of transferability of PRS across ancestries might be expected, such accuracy can be greatly attenuated, especially in more genetically divergent populations relative to the discovery population (16, 69). For example, predictions of PRS in African ancestry populations are only ~20–40% as accurate as in European populations when using European-based GWAS (70, 71) (Figure 3). This limited portability of PRS across ancestries is relatively consistent regardless of the PRS methods used (72, 73). Instead, GWAS discovery cohort composition has the largest impact on prediction accuracy. Many additional factors can contribute to such limited PRS transferability, such as (a) between-ancestry MAF and LD differences, (b) fine-scale population structures, (c) portability differences of indirect and direct effects, and (d) differences of cohort characteristics in the discovery and target populations. This has motivated data generation and methods development for improving PRS transferability across ancestrally diverse populations.
Figure 3.
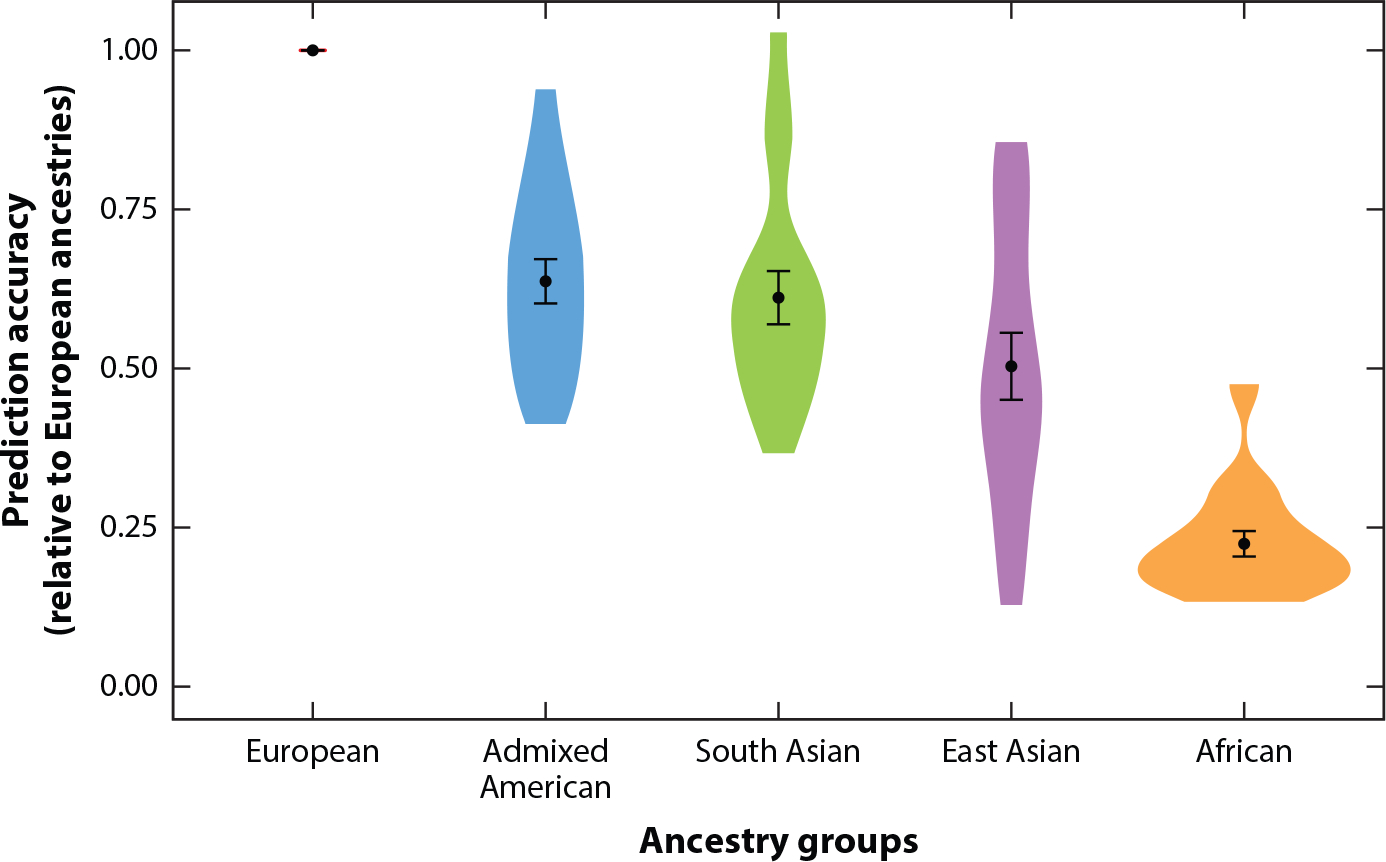
Prediction accuracy of polygenic risk scores relative to European ancestry individuals across 17 quantitative traits and 5 continental ancestry groups in the UK Biobank. Violin plots show the distributions of relative prediction accuracies, points show mean values, and error bars show standard error of the mean. Figure adapted from Reference 15.
Minor Allele Frequency and Linkage Disequilibrium Differences Across Ancestries
GWAS have the highest statistical power to identify common genome-wide significant variants, resulting in higher frequency variants in the discovery population. Therefore, variants that are rare or less common in Eurocentric GWAS can be more easily identified in other non-European ancestry populations when they are at intermediate frequencies. Relatedly, LD is dependent on the variant’s MAF (74). The LD statistics, r or r2, quantify the taggability of genotyped or imputed SNPs and vary markedly among ancestries with different demographic histories. Genetic variants with intermediate MAF (i.e., ~0.3) are likely to produce higher LD correlations, and thus have the highest power to be detected in the discovery population (75). LD differences in turn impact effect size estimation, which is proportional to the LD r between tag SNPs and causal variants. Accounting for MAF and LD differences across ancestries can largely explain the limited portability of PRS generated with European-based GWAS under the simplest assumption that causal variants underlying the trait are shared (72). Therefore, PRS transferability is expected to improve when modeling between-ancestry LD and MAF differences. For example, the highest genetic differentiation among continental ancestries is between African and out-of-Africa populations due to the out-of-Africa migration; consequently, extensive computer simulations have shown that using African ancestry cohorts as discovery GWAS will generate more generalizable associations, with more similar allele frequencies across continental populations from less genetic drift (76). Furthermore, the resolution of fine-mapping studies greatly benefits from multiancestry cohorts due to between-ancestry LD differences, especially when including African populations (77). This also has the potential to improve PRS transferability, as recent studies suggest that common causal variants tend to be shared across ancestries (45, 78, 79).
Fine-Scale Population Structure
The limits of PRS transferability have been less well studied within finer-scale population structures, such as among subpopulations of the same or similar ancestry groups. Geographic distributions or novel dimensionality-reduction methods can uncover discrete clusters of individuals within the same ancestry, such as in Finland (80), Japan (81) and the United Kingdom (73). Inconsistencies between PRS and observed phenotype differences across continental ancestry groups have also been identified in relatively homogeneous populations. These findings can pose problems in the interpretation and clinical translation of PRS. For example, previously identified polygenic adaptation signals were shown to be confounded by cryptic population structures in the UK Biobank (82, 83). Although cryptic population structures could potentially increase the prediction accuracy of PRS, they may simultaneously limit generalizability and interpretability (Figure 1). Therefore, understanding the full extent and consequences of uncorrected population structure (Figure 4) will be critical for improving PRS transferability.
Figure 4.
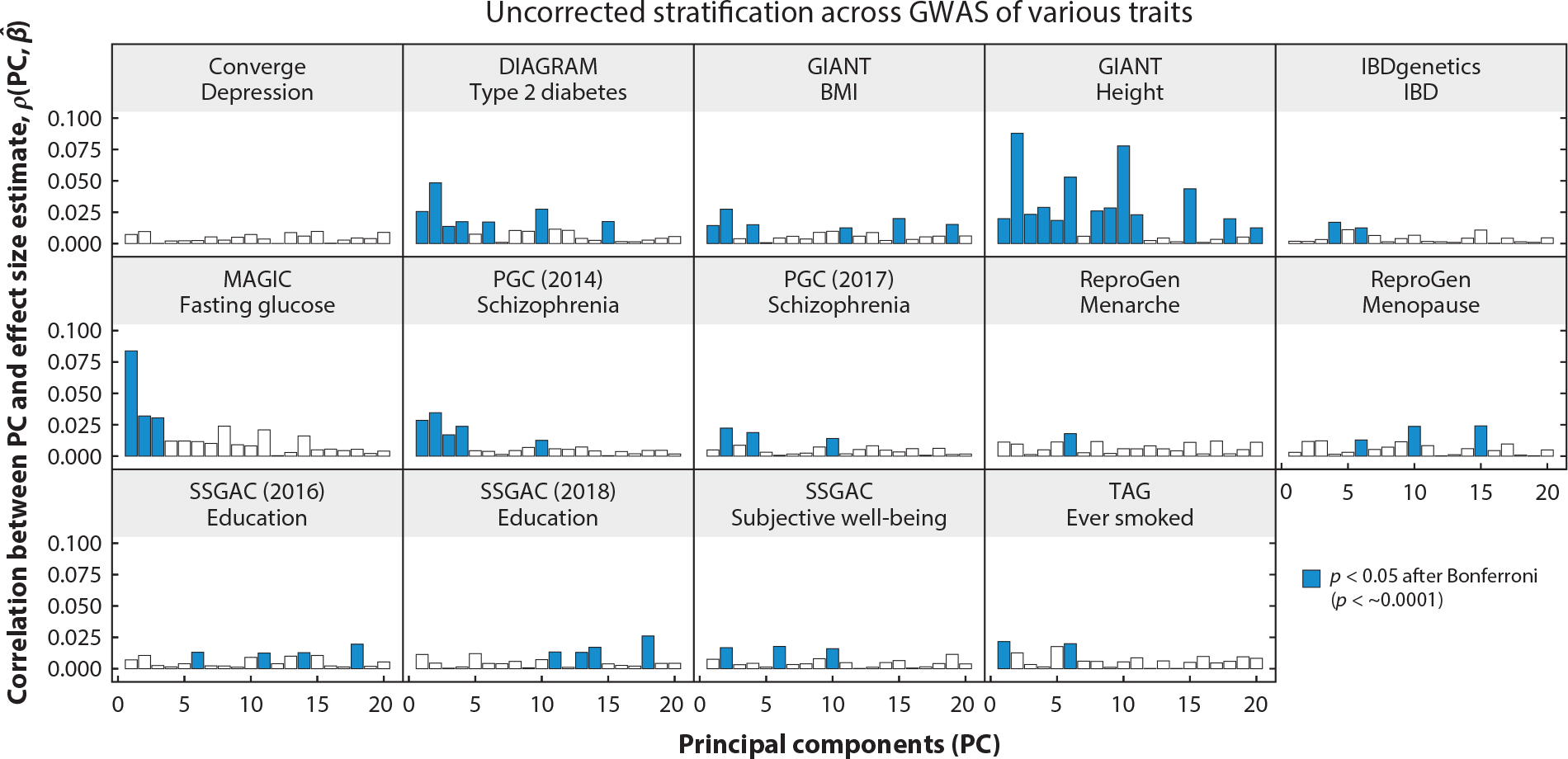
Uncorrected population stratification in GWAS is pervasive. Across several studies, there are significant correlations between PCs in the 1000 Genomes Project and effect size estimates from GWAS for a range of phenotypes. Abbreviations: BMI, body mass index; DIAGRAM, Diabetes Genetics Replication and Meta-analysis; GIANT, Genetic Investigation of Anthropometric Traits; GWAS, genome-wide association studies; IBD, inflammatory bowel disease; MAGIC, Meta-Analysis of Glucose and Insulin-related traits Consortium; PC, principal component; PGC, Psychiatric Genomics Consortium; ReproGen, Reproductive Genetics; SSGAC, Social Science Genetic Association Consortium; TAG, Tobacco and Genetics.
Admixed Populations
PRS transferability issues are especially evident in recently admixed populations, for which two or more ancestral components (typically originating from different continents) are present in each genome. Recently admixed populations are also largely underrepresented. However, these populations provide unique opportunities to explore approaches for improving PRS transferability. First, they provide some (albeit imperfect) level of control for environmental differences across groups given the disparate continental ancestral components within their genomes. Second, studies have shown that the prediction performance of PRS from European-derived GWAS decays with increasing admixture proportions from underrepresented ancestries, especially African ancestries (84, 85). Therefore, local ancestry-specific effect size estimates can boost GWAS power (86) and have the potential to improve prediction performance in admixed populations, particularly for traits with relatively sparse genetic architectures (84, 87). Some methods that model local ancestry have also been developed but not yet widely applied (88, 89). Linearly combining PRS from large-scale European ancestry GWAS and smaller underrepresented non-European target ancestries has improved prediction in mixed ancestry populations (43, 84). A schizophrenia study has shown that more diverse GWAS discovery cohorts can also improve prediction performance in recently admixed populations (90). These results highlight the utility of admixed populations for better understanding how PRS transferability could be improved with a larger effective sample size, a larger fraction of participants included with diverse ancestries, and more contributing genetically distant source populations.
Family History and Direct Versus Indirect Genetic Effects on Phenotypes
Differences in the contribution of direct versus indirect genetic effects may also influence effect size differences across populations and thus limit PRS transferability. Direct and indirect effects both refer to causal genetic variants. However, direct effects are the effects of inherited genetic variation on the phenotype of the individual that carries that variant. In contrast, indirect genetic effects denote effects of a relative’s genotype on the phenotype of an individual through a shared environment (91, 92). Examples of indirect genetic effects include variants that affect parental or sibling behaviors (93). GWAS typically only include unrelated samples without family data, and therefore capture the combination of direct and indirect effects (93). This maximizes predictive power for PRS, so in many cases effect size estimates from standard GWAS without family data is preferred and sufficient for PRS construction (93). Within-family genetic association studies (for example, within-sibship GWAS) can be used to obtain more precise estimates of direct genetic effects. Studies that have compared within- and between-family PRS prediction have indeed found that for a number of traits, standard GWAS have greater prediction accuracy than sib-GWAS, indicating that some degree of genotype-environment correlation typically impacts PRS accuracy (94–96). However, as noted by these studies, a consequence of this increased predictive power may be decreased portability, even within the same ancestry. Therefore, decomposing direct from indirect effects may guide more generalizable prediction models, particularly for phenotypes in which indirect effects may have outsized contributions, such as behavioral traits. This will require datasets with genotyped siblings and/or parents at a much larger scale than currently available to reach sufficient statistical power to parse direct versus indirect effects. Polygenic transmission disequilibrium tests (pTDT) in family data can also illuminate how common and rare genetic risk factors contribute to liability (97). In the absence of large-scale genetic studies with pedigree data, family history information alone can boost PRS accuracy (98). This builds on prior work showing that PRS informs risk somewhat independently of self-reported family history (99).
Collider Bias, Gene–Environment Interaction Effects, and Nongenetic Factors
Interpretation of PRS generalizability requires consideration of the GWAS cohort study design. Nearly all GWAS are subject to some degree of ascertainment bias. For example, the volunteer-based ascertainment of the UK Biobank study means that participants tend to be healthier, wealthier, and higher educated than average (100), whereas hospital-ascertained cases may be sicker on average. The latter may also introduce collider bias, in which even if two variables were initially unrelated, they become correlated through a downstream effect of the two variables. In the hospital ascertainment example, two unrelated diseases that cause hospitalization may become correlated in a dataset due to the study design. This may induce phenotypic correlation and some degree of GWAS effect size correlation between the two unrelated phenotypes.
PRS models, in their most common and basic form, do not consider effects of nongenetic, environmental variables on phenotypes. This may hinder PRS transferability in contexts where GxE interactions exist. GxE interaction effects can be defined as phenomena whereby the effect of a genotype on a phenotype depends on the environment (101, 102). Consequently, the effect of the variant on the phenotype can differ in magnitude across populations depending on the environment, so PRS will not necessarily transfer consistently (93). There are few reproducible examples of GxE interactions. One example is the attenuation of obesity risk from FTO variants as a function of multiple lifestyle factors, including physical activity and alcohol consumption (103, 104). Insufficient statistical power, multiple testing burden, and lack of reproducible environmental measures are major barriers to pinpointing GxE interactions (93). However, with the advent of PRS, studies have been able to move beyond candidate GxE interactions to genome-wide studies. This approach has been particularly prevalent in the neuropsychiatric field, where several investigations have been conducted into the effects of interactions between PRS and various relevant exposures on depression, psychosis, and neuroticism (105–107). Additionally, studies have investigated the effects of interactions among PRS for education on obesity (108, 109). The consistency of these effects and other interactions in non-European ancestry populations remains an open question. It is also unclear how these interactions may attenuate the power and generalizability of PRS in different populations.
Studying GxE interactions across populations is a considerable challenge that will require rigorous evaluations of the contribution of nongenetic factors to disease risk in different populations. The immense scale of possible environmental exposures and the difficulties in systematically and reproducibly collecting and defining these exposures have so far impeded investigations into the effects of nongenetic factors in aggregate. Some have considered coarse environmental risk scores in the context of multipollutants (110, 111). More recently, studies have more broadly evaluated polyexposome scores alongside PRS from insurance billing and zip code data (112); such analyses can be challenging to interpret, as environmental effects that are causal rather than a consequence of disease cannot be determined without further longitudinal measures. With the establishment of the UK Biobank, some progress has been made on quantifying the effects of a wide range of modifiable environmental factors on disease risk alongside PRS (113). However, we need more investment in the systematic collection of environmental variables, as well as novel analytic approaches, to fully elucidate environmental contributions to phenotypes.
POTENTIAL TRANSLATIONAL USES
Utility of Polygenic Risk Scores in Population Risk Stratification and Screening, Not Diagnostic Tests
A growing number of studies have identified significant associations between PRS and disease status, highlighting interest in their potential for clinical translation. While PRS hold clear promise in research settings and are increasingly studied in preventative medicine contexts, their clinical utility is neither definitive nor clear (114, 115). Currently, PRS enthusiasts, skeptics, and researchers along this spectrum disagree on the strength of evidence needed for risk stratification in clinical settings. Varying opinions notwithstanding, establishing clinical value requires an evidence base akin to existing biomarkers already used in preventative medicine—i.e., showing that incorporating PRS into current clinical models significantly improves patient outcomes, and in which specific contexts and areas of medicine (116, 117). Testing these models also requires shifting from a relative risk distribution to an absolute disease risk estimate (116, 118).
While not unique to PRS, an especially pernicious issue when evaluating their accuracy is context dependence. Interpreting the predictive value of PRS for individuals with ancestries from multiple disparate origins is particularly challenging with current scales of data and methods. Clinical models for risk factors in other areas of medicine over- and underestimate risk for certain populations as well. For example, including pooled cohort equations used in atherosclerotic cardiovascular diseases (CVD) may systematically underestimate risk in minorities or overestimate risk in patients with higher socioeconomic status (119). However, ancestral study composition has a far more direct impact on PRS prediction accuracy (15) than it does on the validity of biomarkers used in most areas of medicine.
A potential benefit of PRS beyond other biomarkers is their informativeness relatively early in life, before other biomarkers typically show increased risk (120, 121). This complicates clinical trial designs, which tend to be relatively short, however, because disease diagnoses are rarer early in life and, thus, more time is required to observe the potential benefits of early interventions. Recent studies show that PRS can be used as biomarkers before lab tests are elevated in presymptomatic patients, and higher PRS may be associated with an earlier age of onset for complex diseases (122, 123). Conversely, one benefit of the utility of PRS early in life is their potential to prevent overscreening (124, 125).
The most promising areas of medicine where PRS may be beneficial soonest are for diseases that have actionable interventions, are significantly heritable, have large-scale GWAS available, and improve current clinical models or facilitate the development of clinical models where they are lacking. Some examples include breast and prostate cancer (121, 124, 126, 127), type 1 and 2 diabetes (128, 129), and CVD (116, 120, 130, 131). As PRS offer probabilistic insights into disease risk and trait likelihood, they are not diagnostic tools. Therefore, communicating risk for complex traits distilled by PRS requires particular care to avoid perceptions of genetic determinism (57, 132). PRS for schizophrenia and psychosis have higher predictive power than most other common diseases but have shown limited prognostic value compared to features captured in a structured clinical interview (133) and unclear actionability. Potential applications for PRS beyond assessing disease risk include predicting response to treatment, such as for antipsychotic medications (134). In contrast to these promising areas of medicine, PRS have been considered for screening outside typical preventative medicine contexts, such as embryo selection offered by direct-to-consumer companies. This use of PRS is unregulated, remains ethically problematic and scientifically dubious, and is overshadowed by other considerations with in vitro fertilization (57, 135).
Use Cases of Polygenic Risk Scores Alongside Clinical Risk Factors and Demographic Information
In risk stratification models, PRS often perform poorly alone but comparatively well in combination with existing risk factors (114, 120). Their clinical utility should therefore be evaluated in concert with other disease-specific risk factors (114, 120). Perhaps the most obvious near-term use case for clinical implementation of PRS is in breast cancer; PRS have outperformed current existing clinical risk models for breast cancer in European ancestry populations for several years and are becoming increasingly precise and nuanced (121, 124, 126, 127). One clinical model used for breast cancer risk analysis, the Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm (BOADICEA), provides a flexible framework that can include rare truncating variants in BRCA1, BRCA2, PALB2, CHEK2, and ATM; PRS; family history; and mammographic density (136, 137). The effects of various risk factors are typically combined multiplicatively (126, 137). Incorporating genetic risk factors that are enriched in disease subtypes, for example, in estrogen receptor (ER)-positive versus ER-negative breast cancer, may be possible in further model extensions, informing prevention programs based on risk-reducing medication (136). While most PRS inform relative risk, a critical step in clinical utility adopted by BOADICEA is transforming to absolute risks and providing clear clinical thresholds for screening and prevention. Figure 5 combines information from previous proposals for implementation (136–138). In practice, however, guidelines for screening and management vary enormously across a wide range of professional organizations (e.g., American Cancer Society and US Preventive Services Task Force). Even when recommendations are clearer, for example, regarding the interpretation of pathogenic genetic variants from the American College of Medical Genetics and Genomics (139), health risk is communicated to patients who make the ultimate decisions about screening or prevention.
Figure 5.
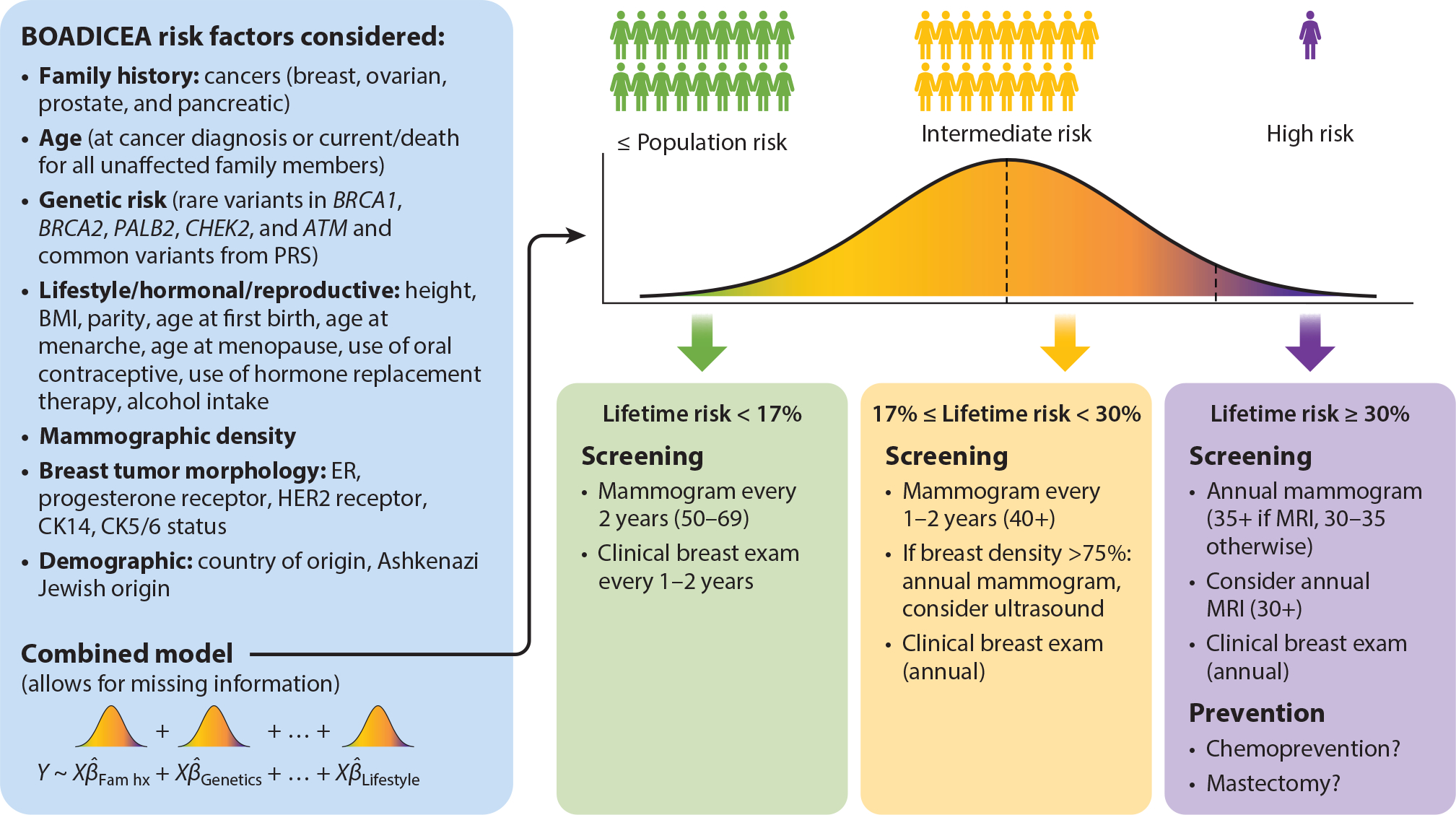
A clinical translated model for breast and ovarian cancer risk that incorporates PRS alongside other clinical information. While BOADICEA includes sex information, this was simplified in the diagram given the higher prevalence among women. The model and screening strategies summarized in this figure have been described more fully previously (136, 138, 145). Abbreviations: BMI, body mass index; BOADICEA, Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm; ER, estrogen receptor; Fam hx, family history; MRI, magnetic resonance imaging; PRS, polygenic risk scores.
Another clinical area where PRS show promise is the prevention of CVD. PRS in European ancestry populations aggregate effects similar in magnitude to monogenic risk variants (131). They also predict risk more accurately than other individual risk factors routinely used in clinical models including smoking, diabetes, family history of heart disease, BMI, hypertension, and high cholesterol; intuitively, the best performing model combines PRS with these conventional risk factors (120) (Figure 6). Evidence of PRS utility from retrospective studies of CVD in the United States and United Kingdom has so far been absent or modest (140, 141). However, a major challenge with these early interpretations and PRS evaluation is their utility earlier in life, requiring studies that are longer term than those used for typical risk factors. Prospective studies offer more real-world insights into the question of clinical utility, and an observational follow-up study in Finland has shown early promise, along with a clinical trial in the United States in which PRS motivated positive changes in health behavior (142, 143). Relatedly, the contributions of monogenic and polygenic risk factors to QT interval duration have also been jointly investigated in the UK Biobank and Trans-Omics for Precision Medicine (TOPMed) Consortium; in both studies, monogenic variants and PRS contribute to risk of long QT syndrome, but most patients do not have elevated risk from either risk factor (144). Implementing PRS more generally will likely require multiple large-scale follow-up studies, as notable differences abound in social cohesion and healthcare systems across countries participating in existing studies. Cost also needs to be evaluated to determine the economic burden for implementing such a system versus the amount recouped through early diagnosis or disease prevention (127).
Figure 6.
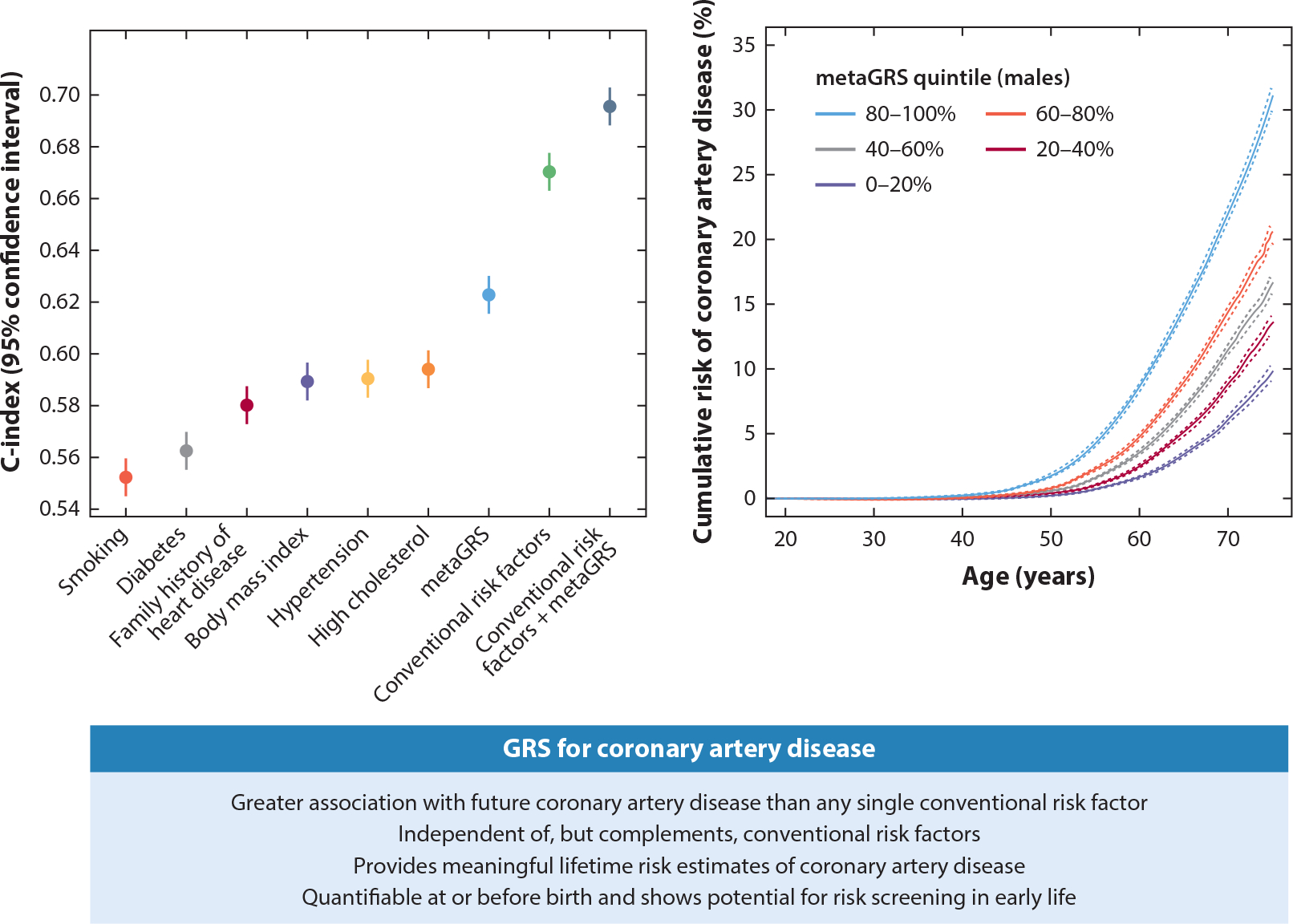
Current clinical risk factors for cardiovascular disease alongside PRS. Figure adapted from Reference 120 (CC BY 4.0). Abbreviations: GRS, genetic risk score; PRS, polygenic risk scores.
Current Deployment Examples
Clinical translation of PRS has already begun, with initial efforts in a limited number of health systems, although studies to evaluate efficacy and considerations are still needed. A prominent area where PRS are being integrated and tested is in breast and ovarian cancer, for example, with the BOADICEA approach. This model is implemented as a web interface in the CanRisk Tool (145), which is already publicly available to medical practitioners and researchers for breast cancer screening and risk stratification research. PRS are also being deployed and evaluated in the context of CVD risk screening. For example, a report recently described a framework for how policymakers and healthcare systems could incorporate PRS for CVD into NHS (National Health Service) health checks in the United Kingdom (146). Similarly, the Electronic Medical Records and Genomics (eMERGE) Network has also evaluated PRS for CVD risk across some ancestries and healthcare settings, with consistent findings to those described previously (130).
There are also already several early examples of PRS available directly to consumers, through their healthcare providers via for-profit and nonprofit companies, and in research settings. For example, consumers who already have their own genotyping data can upload their genetic data to Impute.me (147) to calculate their PRS and their contextualized meaning with respect to a broader population. 23andMe already provides PRS for type 2 diabetes (148). Additionally, Myriad Genetics markets the Myriad RiskScore, which combines an 86-SNP-based PRS for breast cancer with a clinical risk score (149). Recently, Ambry Genetics discontinued AmbryScore, a similar product, in part because of limited data across ancestry groups (117). Genomics PLC has developed PRS as well as predictive models for CVD and other disease areas that integrate PRS (150, 151).
Communicating risk remains a major challenge for integrated polygenic and traditional risk models across all disease areas. Current evidence on the effect of communicating genetic risk alongside lifestyle factors in, for example, CVD is mixed, with caveats regarding who communicates the information and how (i.e., a trusted doctor versus a web-based form) (142, 152).
FUTURE DIRECTIONS
As PRS have become increasingly powerful with the exponential growth in GWAS, attention has shifted toward new research directions and clinical translation. Far beyond their original human applications in biology, more recent proposals include applications in social sciences such as personalized education. Some researchers and groups have called for a society-wide conversation on acceptable uses of PRS accompanied by potential regulation and oversight particularly with more controversial uses such as embryo selection (57, 153). Simultaneously, enthusiasm has ramped up for implementation in some areas of healthcare as PRS have become increasingly available and predictive.
A major ethical and scientific concern with all of these use cases of PRS currently is that they have uneven accuracies across populations due to Eurocentric genetic study biases (15). Therefore, major efforts are underway to increase the generalizability of PRS across diverse cohorts, ancestries, and populations. In addition to rapidly expanding the diversity of GWAS data, several promising areas of exploration are underway for improving the accuracy of PRS across all populations. These include extending PRS methods to integrate multiple ancestries, traits, and functional annotations.
The lack of PRS generalizability across ancestries is easily measurable and has been well documented. While genetic studies are especially prone to issues of stratification, other epidemiological risk factors are likely to suffer from the same study biases in medicine more broadly but may be less reproducibly measured and therefore less obvious. Already, some evidence of racial biases has been identified in algorithms that have been designed to coordinate care among patients (154). Therefore, the lack of PRS generalizability should be a warning that many other algorithms used to assess risk and care for patients are also likely to suffer from biases that could exacerbate health disparities but are fixable.
ACKNOWLEDGMENTS
We thank Anna C.F. Lewis and Robert Maier for helpful discussions. A.R.M. and Y.W. are in part supported by funding from the NIH/National Institute of Mental Health (K99/R00MH117229). Additional support for this work to A.R.M., Y.W., and B.M.N. also comes from the European Union’s Horizon 2020 research and innovation program under grant agreement 101016775 (INTERVENE Consortium).
Footnotes
DISCLOSURE STATEMENT
A.R.M. has consulted for Illumina and received speaker fees from Genentech, Pfizer, and Illumina. B.M.N. is a member of the Deep Genomics Scientific Advisory Board. He also serves as a consultant for the Camp4 Therapeutics Corporation, Takeda Pharmaceutical, and Biogen. The other authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Uffelmann E, Huang QQ, Munung NS, de Vries J, Okada Y, et al. 2021. Genome-wide association studies. Nat. Rev. Methods Primers 1:59 [Google Scholar]
- 2.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, et al. 2017. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101(1):5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher RA. 1919. The correlation between relatives on the supposition of Mendelian inheritance. Trans. R. Soc. Edinb. 52(2):399–433 [Google Scholar]
- 4.Wray NR, Kemper KE, Hayes BJ, Goddard ME, Visscher PM. 2019. Complex trait prediction from genome data: contrasting EBV in livestock to PRS in humans: genomic prediction. Genetics 211(4):1131–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meuwissen TH, Hayes BJ, Goddard ME. 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lande R, Thompson R. 1990. Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 124(3):743–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wientjes YCJ, Bijma P, Calus MPL. 2020. Optimizing genomic reference populations to improve crossbred performance. Genet. Sel. Evol. 52:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visscher PM, Hill WG, Wray NR. 2008. Heritability in the genomics era—concepts and misconceptions. Nat. Rev. Genet. 9(4):255–66 [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Zeng J, Goddard ME, Wray NR, Visscher PM. 2017. Concepts, estimation and interpretation of SNP-based heritability. Nat. Genet. 49(9):1304–10 [DOI] [PubMed] [Google Scholar]
- 10.Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. 2013. Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 14(7):507–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, et al. 2015. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47(11):1236–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, et al. 2010. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42(7):565–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tropf FC, Lee SH, Verweij RM, Stulp G, van der Most PJ, et al. 2017. Hidden heritability due to heterogeneity across seven populations. Nat. Hum. Behav. 1(10):757–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge T, Chen C-Y, Neale BM, Sabuncu MR, Smoller JW. 2017. Phenome-wide heritability analysis of the UK Biobank. PLOS Genet. 13(4):e1006711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. 2019. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51(4):584–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, et al. 2017. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 100(4):635–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu H, Zhou X. 2020. Statistical methods for SNP heritability estimation and partition: a review. Comput. Struct. Biotechnol. J. 18:1557–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kichaev G, Yang W-Y, Lindstrom S, Hormozdiari F, Eskin E, et al. 2014. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLOS Genet. 10(10):e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gusev A, Lee SH, Trynka G, Finucane H, Vilhjálmsson BJ, et al. 2014. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 95(5):535–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, et al. 2015. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47(11):1228–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. 2012. Estimation of pleiotropy between complex diseases using SNP-derived genomic relationships and restricted maximum likelihood. Bioinformatics 28(19):2540–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambert SA, Gil L, Jupp S, Ritchie SC, Xu Y, et al. 2021. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 53(4):420–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Int. Schizophr. Consort. 2009. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460(7256):748–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kooperberg C, LeBlanc M, Obenchain V. 2010. Risk prediction using genome-wide association studies. Genet. Epidemiol. 34(7):643–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Speed D, Balding DJ. 2014. MultiBLUP: improved SNP-based prediction for complex traits. Genome Res. 24(9):1550–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Albiñana C, Grove J, McGrath JJ, Agerbo E, Wray NR, et al. 2021. Leveraging both individual-level genetic data and GWAS summary statistics increases polygenic prediction. Am. J. Hum. Genet. 108(6):1001–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi J, Park J-H, Duan J, Berndt ST, Moy W, et al. 2016. Winner’s curse correction and variable thresholding improve performance of polygenic risk modeling based on genome-wide association study summary-level data. PLOS Genet. 12(12):e1006493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Läll K, Mägi R, Morris A, Metspalu A, Fischer K. 2017. Personalized risk prediction for type 2 diabetes: the potential of genetic risk scores. Genet. Med. 19(3):322–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, et al. 2015. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97(4):576–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson MR, Kleinman A, Graff M, Vinkhuyzen AAE, Couper D, et al. 2017. Genetic evidence of assortative mating in humans. Nat. Hum. Behav. 1:16 [Google Scholar]
- 31.Mak TSH, Porsch RM, Choi SW, Zhou X, Sham PC. 2017. Polygenic scores via penalized regression on summary statistics. Genet. Epidemiol. 41(6):469–80 [DOI] [PubMed] [Google Scholar]
- 32.Lloyd-Jones LR, Zeng J, Sidorenko J, Yengo L, Moser G, et al. 2019. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat. Commun. 10:5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge T, Chen C-Y, Ni Y, Feng Y-CA, Smoller JW. 2019. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10:1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Privé F, Arbel J, Vilhjálmsson BJ. 2020. LDpred2: better, faster, stronger. Bioinformatics 36(22–23):5424–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ni G, Zeng J, Revez JA, Wang Y, Zheng Z, et al. 2021. A comparison of ten polygenic score methods for psychiatric disorders applied across multiple cohorts. Biol. Psychiatry 90(9):611–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pain O, Glanville KP, Hagenaars SP, Selzam S, Fürtjes AE, et al. 2021. Evaluation of polygenic prediction methodology within a reference-standardized framework. PLOS Genet. 17(5):e1009021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulm S, Marderstein A, Mezey J, Elemento O. 2021. A systematic framework for assessing the clinical impact of polygenic risk scores. medRxiv 10.1101/2020.04.06.20055574. 10.1101/2020.04.06.20055574 [DOI] [Google Scholar]
- 38.Ma Y, Zhou X. 2021. Genetic prediction of complex traits with polygenic scores: a statistical review. Trends Genet. 37(11):995–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maier RM, Zhu Z, Lee SH, Trzaskowski M, Ruderfer DM, et al. 2018. Improving genetic prediction by leveraging genetic correlations among human diseases and traits. Nat. Commun. 9:989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turley P, Walters RK, Maghzian O, Okbay A, Lee JJ, et al. 2018. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50(2):229–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Y, Lu Q, Liu W, Zhang Y, Li M, Zhao H. 2017. Joint modeling of genetically correlated diseases and functional annotations increases accuracy of polygenic risk prediction. PLOS Genet. 13(6):e1006836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maier RM, Zhu Z, Lee SH, Trzaskowski M, Ruderfer DM, et al. 2018. Improving genetic prediction by leveraging genetic correlations among human diseases and traits. Nat. Commun. 9:989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Márquez-Luna C, Loh P-R, South Asian Type 2 Diabetes (SAT2D) Consort., SIGMA Type 2 Diabetes Consort., Price AL. 2017. Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 41(8):811–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weissbrod O, Kanai M, Shi H, Gazal S, Peyrot W, et al. 2021. Leveraging fine-mapping and non-European training data to improve trans-ethnic polygenic risk scores. medRxiv 10.1101/2021.01.19.21249483. 10.1101/2021.01.19.21249483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Majara L, Kalungi A, Koen N, Zar H, Stein DJ, et al. 2021. Low generalizability of polygenic scores in African populations due to genetic and environmental diversity. bioRxiv 10.1101/2021.01.12.426453. 10.1101/2021.01.12.426453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruan Y, Lin Y-F, Feng Y-CA, Chen C-Y, Lam M, et al. 2021. Improving polygenic prediction in ancestrally diverse populations. medRxiv 10.1101/2020.12.27.20248738. 10.1101/2020.12.27.20248738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi H, Gazal S, Kanai M, Koch EM, Schoech AP, et al. 2021. Population-specific causal disease effect sizes in functionally important regions impacted by selection. Nat. Commun. 12:1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown BC, Asian Genetic Epidemiol. Netw. Type 2 Diabetes Consort., Ye CJ, Price AL, Zaitlen N. 2016. Transethnic genetic-correlation estimates from summary statistics. Am. J. Hum. Genet. 99(1):76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Márquez-Luna C, Gazal S, Loh P-R, Kim SS, Furlotte N, et al. 2021. Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets. Nat. Commun. 12:6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y, Lu Q, Powles R, Yao X, Yang C, et al. 2017. Leveraging functional annotations in genetic risk prediction for human complex diseases. PLOS Comput. Biol. 13(6):e1005589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gazal S, Finucane HK, Furlotte NA, Loh P-R, Palamara PF, et al. 2017. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49(10):1421–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amariuta T, Ishigaki K, Sugishita H, Ohta T, Koido M, et al. 2020. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat. Genet. 52(12):1346–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sivakumaran S, Agakov F, Theodoratou E, Prendergast JG, Zgaga L, et al. 2011. Abundant pleiotropy in human complex diseases and traits. Am. J. Hum. Genet. 89(5):607–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watanabe K, Stringer S, Frei O, Umićević Mirkov M, de Leeuw C, et al. 2019. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 51:1339–48 [DOI] [PubMed] [Google Scholar]
- 55.Udler MS, Kim J, von Grotthuss M, Bonàs-Guarch S, Cole JB, et al. 2018. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLOS Med. 15(9):e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daetwyler HD, Villanueva B, Woolliams JA. 2008. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLOS ONE 3(10):e3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turley P, Meyer MN, Wang N, Cesarini D, Hammonds E, et al. 2021. Problems with using polygenic scores to select embryos. N. Engl. J. Med. 385:78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lencz T, Backenroth D, Granot-Hershkovitz E, Green A, Gettler K, et al. 2021. Utility of polygenic embryo screening for disease depends on the selection strategy. eLife 10:e64716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee SH, Goddard ME, Wray NR, Visscher PM. 2012. A better coefficient of determination for genetic profile analysis. Genet. Epidemiol. 36(3):214–24 [DOI] [PubMed] [Google Scholar]
- 60.Choi SW, Mak TS-H, O’Reilly PF. 2020. Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc. 15(9):2759–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pain O, Gillett AC, Austin JC, Folkersen L, Lewis CM. 2022. A tool for translating polygenic scores onto the absolute scale using summary statistics. Eur. J. Hum. Genet. 10.1038/s41431-021-01028-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pal Choudhury P, Maas P, Wilcox A, Wheeler W, Brook M, et al. 2020. iCARE: an R package to build, validate and apply absolute risk models. PLOS ONE 15(2):e0228198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lewis ACF, Green RC, Vassy JL. 2021. Polygenic risk scores in the clinic: translating risk into action. Human Genet. Genom. Adv. 2(4):100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schultz LM, Merikangas AK, Ruparel K, Jacquemont S, Glahn DC, et al. 2022. Stability of polygenic scores across discovery genome-wide association studies. Hum. Genet. Genom. Adv. 3(2):100091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ding Y, Hou K, Burch KS, Lapinska S, Privé F, et al. 2021. Large uncertainty in individual PRS estimation impacts PRS-based risk stratification. Nat. Genet. 54:30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lambert SA, Gil L, Jupp S, Ritchie SC, Xu Y, et al. 2021. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 53(4):420–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wand H, Lambert SA, Tamburro C, Iacocca MA, O’Sullivan JW, et al. 2021. Improving reporting standards for polygenic scores in risk prediction studies. Nature 591(7849):211–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, et al. 2017. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 100(4):635–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scutari M, Mackay I, Balding D. 2016. Using genetic distance to infer the accuracy of genomic prediction. PLOS Genet. 12(9):e1006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. 2019. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 51(4):584–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Duncan L, Shen H, Gelaye B, Meijsen J, Ressler K, et al. 2019. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 10:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y, Guo J, Ni G, Yang J, Visscher PM, Yengo L. 2020. Theoretical and empirical quantification of the accuracy of polygenic scores in ancestry divergent populations. Nat. Commun. 11:3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Privé F, Aschard H, Carmi S, Folkersen L, Hoggart C, et al. 2022. Portability of 245 polygenic scores when derived from the UK Biobank and applied to 9 ancestry groups from the same cohort. Am. J. Hum. Genet. 109(1):12–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wray NR. 2005. Allele frequencies and the r2 measure of linkage disequilibrium: impact on design and interpretation of association studies. Twin Res. Hum. Genet. 8(2):87–94 [DOI] [PubMed] [Google Scholar]
- 75.VanLiere JM, Rosenberg NA. 2008. Mathematical properties of the r2 measure of linkage disequilibrium. Theor. Popul. Biol. 74(1):130–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim MS, Patel KP, Teng AK, Berens AJ, Lachance J. 2018. Genetic disease risks can be misestimated across global populations. Genome Biol. 19:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin AR, Teferra S, Möller M, Hoal EG, Daly MJ. 2018. The critical needs and challenges for genetic architecture studies in Africa. Curr. Opin. Genet. Dev. 53:113–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo J, Bakshi A, Wang Y, Jiang L, Yengo L, et al. 2021. Quantifying genetic heterogeneity between continental populations for human height and body mass index. Sci. Rep. 11:5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi H, Burch KS, Johnson R, Freund MK, Kichaev G, et al. 2020. Localizing components of shared transethnic genetic architecture of complex traits from GWAS summary data. Am. J. Hum. Genet. 106(6):805–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kerminen S, Martin AR, Koskela J, Ruotsalainen SE, Havulinna AS, et al. 2019. Geographic variation and bias in the polygenic scores of complex diseases and traits in Finland. Am. J. Hum. Genet. 104(6):1169–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sakaue S, Hirata J, Kanai M, Suzuki K, Akiyama M, et al. 2020. Dimensionality reduction reveals fine-scale structure in the Japanese population with consequences for polygenic risk prediction. Nat. Commun. 11:1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berg JJ, Harpak A, Sinnott-Armstrong N, Joergensen AM, Mostafavi H, et al. 2019. Reduced signal for polygenic adaptation of height in UK Biobank. eLife 8:e39725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sohail M, Maier RM, Ganna A, Bloemendal A, Martin AR, et al. 2019. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. eLife 8:e39702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cavazos TB, Witte JS. 2021. Inclusion of variants discovered from diverse populations improves polygenic risk score transferability. Hum. Genet. Genom. Adv. 2(1):100017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bitarello BD, Mathieson I. 2020. Polygenic scores for height in admixed populations. G3 10(11):4027–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Atkinson EG, Maihofer AX, Kanai M, Martin AR, Karczewski KJ, et al. 2021. Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat. Genet. 53(2):195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marnetto D, Pärna K, Läll K, Molinaro L, Montinaro F, et al. 2020. Ancestry deconvolution and partial polygenic score can improve susceptibility predictions in recently admixed individuals. Nat. Commun. 11:1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Coram MA, Fang H, Candille SI, Assimes TL, Tang H. 2017. Leveraging multi-ethnic evidence for risk assessment of quantitative traits in minority populations. Am. J. Hum. Genet. 101(2):218–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marnetto D, Pärna K, Läll K, Molinaro L, Montinaro F, et al. 2020. Ancestry deconvolution and partial polygenic score can improve susceptibility predictions in recently admixed individuals. Nat. Commun. 11:1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bigdeli TB, Genovese G, Georgakopoulos P, Meyers JL, Peterson RE, et al. 2019. Contributions of common genetic variants to risk of schizophrenia among individuals of African and Latino ancestry. Mol. Psychiatry 25:2455–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brandes N, Weissbrod O, Linial M. 2021. Open problems in human trait genetics. arXiv:2108.06684 [q-bio.PE] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Howe LJ, Nivard MG, Morris TT, Hansen AF. 2021. Within-sibship GWAS improve estimates of direct genetic effects. bioRxiv 10.1101/2021.03.05.433935. 10.1101/2021.03.05.433935 [DOI] [Google Scholar]
- 93.Young AI, Benonisdottir S, Przeworski M, Kong A. 2019. Deconstructing the sources of genotype-phenotype associations in humans. Science 365(6460):1396–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mostafavi H, Harpak A, Agarwal I, Conley D, Pritchard JK, Przeworski M. 2020. Variable prediction accuracy of polygenic scores within an ancestry group. eLife 9:e48376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kong A, Thorleifsson G, Frigge ML, Vilhjalmsson BJ, Young AI, et al. 2018. The nature of nurture: effects of parental genotypes. Science 359(6374):424–28 [DOI] [PubMed] [Google Scholar]
- 96.Selzam S, Ritchie SJ, Pingault J-B, Reynolds CA, O’Reilly PF, Plomin R. 2019. Comparing within- and between-family polygenic score prediction. Am. J. Hum. Genet. 105(2):351–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, et al. 2017. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 49(7):978–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hujoel MLA, Loh P-R, Neale BM, Price AL. 2021. Incorporating family history of disease improves polygenic risk scores in diverse populations. bioRxiv 10.1101/2021.04.15.439975. 10.1101/2021.04.15.439975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tada H, Melander O, Louie JZ, Catanese JJ, Rowland CM, et al. 2016. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur. Heart J. 37(6):561–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, et al. 2017. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am. J. Epidemiol. 186(9):1026–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Freeman GH. 1973. Statistical methods for the analysis of genotype-environment interactions. Heredity 31(3):339–54 [DOI] [PubMed] [Google Scholar]
- 102.Hunter DJ. 2005. Gene-environment interactions in human diseases. Nat. Rev. Genet. 6(4):287–98 [DOI] [PubMed] [Google Scholar]
- 103.Kilpeläinen TO, Qi L, Brage S, Sharp SJ, Sonestedt E, et al. 2011. Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLOS Med. 8(11):e1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Young AI, Wauthier F, Donnelly P. 2016. Multiple novel gene-by-environment interactions modify the effect of FTO variants on body mass index. Nat. Commun. 7:12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mullins N, Power RA, Fisher HL, Hanscombe KB, Euesden J, et al. 2016. Polygenic interactions with environmental adversity in the aetiology of major depressive disorder. Psychol. Med. 46(4):759–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mas S, Boloc D, Rodríguez N, Mezquida G, Amoretti S, et al. 2020. Examining gene-environment interactions using aggregate scores in a first-episode psychosis cohort. Schizophr. Bull. 46(4):1019–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Werme J, van der Sluis S, Posthuma D, de Leeuw CA. 2021. Genome-wide gene-environment interactions in neuroticism: an exploratory study across 25 environments. Transl. Psychiatry 11:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barcellos SH, Carvalho LS, Turley P. 2018. Education can reduce health differences related to genetic risk of obesity. PNAS 115(42):E9765–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tyrrell J, Wood AR, Ames RM, Yaghootkar H, Beaumont RN, et al. 2017. Gene-obesogenic environment interactions in the UK Biobank study. Int. J. Epidemiol. 46(2):559–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Park SK, Tao Y, Meeker JD, Harlow SD, Mukherjee B. 2014. Environmental risk score as a new tool to examine multi-pollutants in epidemiologic research: an example from the NHANES study using serum lipid levels. PLOS ONE 9(6):e98632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Park SK, Zhao Z, Mukherjee B. 2017. Construction of environmental risk score beyond standard linear models using machine learning methods: application to metal mixtures, oxidative stress and cardiovascular disease in NHANES. Environ. Health 16(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lakhani CM, Tierney BT, Manrai AK, Yang J, Visscher PM, Patel CJ. 2019. Repurposing large health insurance claims data to estimate genetic and environmental contributions in 560 phenotypes. Nat. Genet. 51(2):327–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.He Y, Lakhani CM, Rasooly D, Manrai AK, Tzoulaki I, Patel CJ. 2021. Comparisons of polyexposure, polygenic, and clinical risk scores in risk prediction of type 2 diabetes. Diabetes Care 44(4):935–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lewis CM, Vassos E. 2020. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Khan SS, Cooper R, Greenland P. 2020. Do polygenic risk scores improve patient selection for prevention of coronary artery disease? JAMA 323(7):614–15 [DOI] [PubMed] [Google Scholar]
- 116.Torkamani A, Wineinger NE, Topol EJ. 2018. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19(9):581–90 [DOI] [PubMed] [Google Scholar]
- 117.Shah PD. 2021. Polygenic risk scores for breast cancer—Can they deliver on the promise of precision medicine? JAMA Netw. Open 4(8):e2119333. [DOI] [PubMed] [Google Scholar]
- 118.Sugrue LP, Desikan RS. 2019. What are polygenic scores and why are they important? JAMA 321(18):1820–21 [DOI] [PubMed] [Google Scholar]
- 119.Lloyd-Jones DM, Braun LT, Ndumele CE, Smith SC, Sperling LS, et al. 2019. Use of risk assessment tools to guide decision-making in the primary prevention of atherosclerotic cardiovascular disease: a special report from the American Heart Association and American College of Cardiology. Circulation 139(25):e1162–77 [DOI] [PubMed] [Google Scholar]
- 120.Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, et al. 2018. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J. Am. Coll. Cardiol. 72(16):1883–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mars N, Widén E, Kerminen S, Meretoja T, Pirinen M, et al. 2020. The role of polygenic risk and susceptibility genes in breast cancer over the course of life. Nat. Commun. 11:6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dennis JK, Sealock JM, Straub P, Lee YH, Hucks D, et al. 2021. Clinical laboratory test-wide association scan of polygenic scores identifies biomarkers of complex disease. Genome Med. 13:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Feng Y-CA, Ge T, Cordioli M, Ganna A, Smoller JW, et al. 2020. Findings and insights from the genetic investigation of age of first reported occurrence for complex disorders in the UK Biobank and FinnGen. bioRxiv 10.1101/2020.11.20.20234302. 10.1101/2020.11.20.20234302 [DOI] [Google Scholar]
- 124.Gao C, Polley EC, Hart SN, Huang H, Hu C, et al. 2021. Risk of breast cancer among carriers of pathogenic variants in breast cancer predisposition genes varies by polygenic risk score. J. Clin. Oncol. 39(23):2564–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Esserman LJ, WISDOM Study Athena Investig. 2017. The WISDOM Study: breaking the deadlock in the breast cancer screening debate. npj Breast Cancer 3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mavaddat N, Michailidou K, Dennis J, Lush M, Fachal L, et al. 2019. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 104(1):21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maas P, Barrdahl M, Joshi AD, Auer PL, Gaudet MM, et al. 2016. Breast cancer risk from modifiable and nonmodifiable risk factors among white women in the United States. JAMA Oncol. 2(10):1295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Padilla-Martínez F, Collin F, Kwasniewski M, Kretowski A. 2020. Systematic review of polygenic risk scores for type 1 and type 2 diabetes. Int. J. Mol. Sci. 21(5):1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Udler MS, McCarthy MI, Florez JC, Mahajan A. 2019. Genetic risk scores for diabetes diagnosis and precision medicine. Endocr. Rev. 40(6):1500–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dikilitas O, Schaid DJ, Kosel ML, Carroll RJ, Chute CG, et al. 2020. Predictive utility of polygenic risk scores for coronary heart disease in three major racial and ethnic groups. Am. J. Hum. Genet. 106(5):707–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, et al. 2018. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50(9):1219–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lewis ACF, Green RC. 2021. Polygenic risk scores in the clinic: new perspectives needed on familiar ethical issues. Genome Med. 13:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Landi I, Kaji DA, Cotter L, Van Vleck T, Belbin G, et al. 2021. Prognostic value of polygenic risk scores for adults with psychosis. Nat. Med. 27:1576–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ruderfer DM, Charney AW, Readhead B, Kidd BA, Kähler AK, et al. 2016. Polygenic overlap between schizophrenia risk and antipsychotic response: a genomic medicine approach. Lancet Psychiatry 3(4):350–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Karavani E, Zuk O, Zeevi D, Barzilai N, Stefanis NC, et al. 2019. Screening human embryos for polygenic traits has limited utility. Cell 179(6):1424–35.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee A, Mavaddat N, Wilcox AN, Cunningham AP, Carver T, et al. 2019. BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 21(8):1708–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pashayan N, Antoniou AC, Ivanus U, Esserman LJ, Easton DF, et al. 2020. Personalized early detection and prevention of breast cancer: ENVISION consensus statement. Nat. Rev. Clin. Oncol. 17(11):687–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gagnon J, Lévesque E, Clin. Advis. Comm. Breast Cancer Screen. Prev., Borduas F, Chiquette J, et al. 2016. Recommendations on breast cancer screening and prevention in the context of implementing risk stratification: impending changes to current policies. Curr. Oncol. 23(6):e615–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Richards S, Aziz N, Bale S, Bick D, Das S, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17(5):405–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mosley JD, Gupta DK, Tan J, Yao J, Wells QS, et al. 2020. Predictive accuracy of a polygenic risk score compared with a clinical risk score for incident coronary heart disease. JAMA 323(7):627–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Elliott J, Bodinier B, Bond TA, Chadeau-Hyam M, Evangelou E, et al. 2020. Predictive accuracy of a polygenic risk score-enhanced prediction model versus a clinical risk score for coronary artery disease. JAMA 323(7):636–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Widén E, Junna N, Ruotsalainen S, Surakka I, Mars N, et al. 2020. Communicating polygenic and non-genetic risk for atherosclerotic cardiovascular disease—an observational follow-up study. medRxiv 10.1101/2020.09.18.20197137. 10.1101/2020.09.18.20197137 [DOI] [PubMed] [Google Scholar]
- 143.Kullo IJ, Jouni H, Austin EE, Brown S-A, Kruisselbrink TM, et al. 2016. Incorporating a genetic risk score into coronary heart disease risk estimates: effect on low-density lipoprotein cholesterol levels (the MI-GENES Clinical Trial). Circulation 133(12):1181–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nauffal V, Morrill VN, Jurgens SJ, Choi SH, Hall AW, et al. 2021. Monogenic and polygenic contributions to QTc prolongation in the population. medRxiv 10.1101/2021.06.18.21258578. 10.1101/2021.06.18.21258578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Carver T, Hartley S, Lee A, Cunningham AP. 2021. CanRisk Tool—a web interface for the prediction of breast and ovarian cancer risk and the likelihood of carrying genetic pathogenic variants. Cancer Epidemiol. 30(3):469–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brigden T, Sanderson S, Janus J, de Villiers CB, Moorthie S, et al. 2021. Implementing polygenic scores for cardiovascular disease into NHS health checks. Tech. Rep., phg Foundation, Cambridge, UK [Google Scholar]
- 147.Folkersen L, Pain O, Ingason A, Werge T, Lewis CM, Austin J. 2020. Impute.me: an open-source, nonprofit tool for using data from direct-to-consumer genetic testing to calculate and interpret polygenic risk scores. Front. Genet. 11:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Multhaup ML, Kita R, Krock B, Eriksson N, Fontanillas P, et al. 2019. The science behind 23andMe’s type 2 diabetes report. White Pap 23–19, 23andMe, Sunnyvale, CA [Google Scholar]
- 149.Hughes E, Tshiaba P, Wagner S, Judkins T, Rosenthal E, et al. 2021. Integrating clinical and polygenic factors to predict breast cancer risk in women undergoing genetic testing. JCO Precis. Oncol. 5:307–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Riveros-Mckay F, Weale ME, Moore R, Selzam S, Krapohl E, et al. 2021. Integrated polygenic tool substantially enhances coronary artery disease prediction. Circ. Genom. Precis. Med. 14(2):e003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Genom. plc. 2019. Polygenic risk scores. White Pap, Genomics plc, Oxford, UK. https://www.genomicsplc.com/wp-content/uploads/2020/11/Genomics-plc-PRS-details_White-Paper-April-2019.pdf [Google Scholar]
- 152.Silarova B, Sharp S, Usher-Smith JA, Lucas J, Payne RA, et al. 2019. Effect of communicating phenotypic and genetic risk of coronary heart disease alongside web-based lifestyle advice: the INFORM randomised controlled trial. Heart 105(13):982–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.ISPG. 2021. Advisory on the use of polygenic risk scores to screen embryos for adult mental health conditions. Statement, ISPG. https://ispg.net/ethics-statement/ [Google Scholar]
- 154.Obermeyer Z, Powers B, Vogeli C, Mullainathan S. 2019. Dissecting racial bias in an algorithm used to manage the health of populations. Science 366(6464):447–53 [DOI] [PubMed] [Google Scholar]
- 155.Privé F, Vilhjálmsson BJ, Aschard H, Blum MGB. 2019. Making the most of clumping and thresholding for polygenic scores. Am. J. Hum. Genet. 105(6):1213–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zeng J, Xue A, Jiang L, Lloyd-Jones LR, Wu Y, et al. 2021. Widespread signatures of natural selection across human complex traits and functional genomic categories. Nat. Commun. 12:1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chun S, Imakaev M, Hui D, Patsopoulos NA, Neale BM, et al. 2020. Non-parametric polygenic risk prediction via partitioned GWAS summary statistics. Am. J. Hum. Genet. 107(1):46–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang S, Zhou X. 2020. Accurate and scalable construction of polygenic scores in large biobank data sets. Am. J. Hum. Genet. 106(5):679–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhou G, Zhao H. 2021. A fast and robust Bayesian nonparametric method for prediction of complex traits using summary statistics. PLOS Genet. 17(7):e1009697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Newcombe PJ, Nelson CP, Samani NJ, Dudbridge F. 2019. A flexible and parallelizable approach togenome-wide polygenic risk scores. Genet. Epidemiol. 43(7):730–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chung W, Chen J, Turman C, Lindstrom S, Zhu Z, et al. 2019. Efficient cross-trait penalized regression increases prediction accuracy in large cohorts using secondary phenotypes. Nat. Commun. 10:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen T-H, Chatterjee N, Landi MT, Shi J. 2021. A penalized regression framework for building polygenic risk models based on summary statistics from genome-wide association studies and incorporating external information. J. Am. Stat. Assoc. 116(533):133–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ballard JL, O’Connor LJ. 2021. Shared components of heritability across genetically correlated traits. bioRxiv 10.1101/2021.11.25.470021. 10.1101/2021.11.25.470021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cai M, Xiao J, Zhang S, Wan X, Zhao H, et al. 2021. A unified framework for cross-population trait prediction by leveraging the genetic correlation of polygenic traits. Am. J. Hum. Genet. 108(4):632–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kelemen M, Vigorito E, Anderson CA, Wallace C. 2021. ShaPRS: leveraging shared genetic effects across traits or ancestries improves accuracy of polygenic scores. medRxiv 10.1101/2021.12.10.21267272 [DOI] [PMC free article] [PubMed] [Google Scholar]