

SUMMARY
Due to the current climate change, many studies have described main drivers in abiotic stress. Recent findings suggest that alternative splicing (AS) has a critical role in controlling plant responses to high temperature. AS is a mechanism that allows organisms to create an assortment of RNA transcripts and proteins using a single gene. However, the most important roles of AS in stress could not be rigorously addressed because research has been focused on model species, covering only a narrow phylogenetic and lifecycle spectrum. Thus, AS degree of diversification among more dissimilar taxa in heat response is still largely unknown. To fill this gap, the present study employs a systems biology approach to examine how the AS landscape responds to and ‘remembers’ heat stress in conifers, a group which has received little attention even though their position can solve key evolutionary questions. Contrary to angiosperms, we found that potential intron retention may not be the most prevalent type of AS. Furthermore, our integrative analysis with metabolome and proteome data places splicing as the main source of variation during the response. Finally, we evaluated possible acquired long‐term splicing memory in a diverse subset of events, and although this mechanism seems to be conserved in seed plants, AS dynamics are divergent. These discoveries reveal the particular way of remembering past temperature changes in long‐lived plants and open the door to include species with unique features to determine the extent of conservation in gene expression regulation.
Keywords: acclimation, heat stress, integrative approach, long‐term memory, Pinus, splicing
Significance Statement
Splicing memory seems to be an evolutionarily conserved mechanism in distant seed plants, and long‐lived conifers appear to have developed a particular way of remembering high temperature changes over a longer period of time.
INTRODUCTION
Due to the high sensitivity of plants to environmental fluctuations together with the current climate change, many studies have described main drivers implied in abiotic stress adaptation and acclimation. Recent findings suggest that temperature‐responsive alternative splicing (AS) has a critical role in controlling plant responses to high temperature at the molecular level (Capovilla et al., 2018). AS is a mechanism that allows organisms to create an assortment of RNA transcripts and proteins by using information from a single gene and functions in a wide range of physiological processes, such as growth and development (Huang et al., 2020; Smith et al., 2018). During the last decade, analysis of high‐throughput RNA sequencing (RNA‐seq) data facilitated the characterization of processing patterns even in non‐sequenced species. Many of these works have contributed to general insights into the role of RNA biology in stress, and intron retention (IR) was found to be the most prevalent AS type in plants (Laloum et al., 2018). Although IR has no functional impact, mainly because introns have a high frequency of premature termination codons (PTCs), which disrupt protein translation through non‐sense‐mediated mRNA decay (NMD), several systems have been proposed linking this AS type with the acquisition and maintenance of stress tolerance like thermomemory, buffers to reduce the metabolic cost of translation, NMD resistance by post‐transcriptional splicing of introns, and exitron‐driven diversification of protein substrate specificity and enhanced regulatory capacity (Chaudhary et al., 2019; Jia et al., 2020; Ling et al., 2018). These systems have an incredible potential to improve plant resource management and breeding programs. However, splicing interplay with other regulatory layers and its degree of diversification among taxa in a process as complex as heat response is still largely unknown.
Evolution and acclimation are processes requiring coordinated changes in many traits according to environmental selective pressures. Considerable adaptation and acclimation may be mediated by modifications in AS, although the patterns of gene expression and splicing variation in response to abiotic stress are not conserved across species or even between genotypes within the same species (Hanemian et al., 2020; Meng et al., 2021; Smith et al., 2018). These reports highlight the importance of understanding the degree of splicing divergence among more dissimilar groups. Currently seed plants are represented by three lineages: one corresponding to the species‐rich angiosperms and two corresponding to gymnosperms, among which conifers are the most widely distributed group (One Thousand Plant Transcriptomes Initiative, 2019; Wan et al., 2018). Conifers have received little attention even though their phylogenetic position can solve several key evolutionary and environmental acclimation questions. Pinus, the largest genus of conifers and arguably the most important genus of trees in the world, provides an ideal example to explore splicing differentiation due to its long evolutionary history and potentially unique genomic features (De La Torre et al., 2014; De La Torre et al., 2020; Jin et al., 2021).
Some of the most important previously suggested general insights could not be rigorously addressed because research has mostly been focused on the study of model species. These organisms have been crucial for in‐depth study of cellular and molecular machinery but these findings might be far from being generalizable from an ecological and evolutionary point of view due to the narrow phylogenetic and lifecycle spectrum covered by model species (Valledor et al., 2018). In contrast to this narrow but deep knowledge bottleneck, a broad but shallow approach could be adopted using orphan species to not only directly ask big questions but also give simple large‐scale answers to derive ground truths that we would never have contemplated asking based on our understanding of the molecular biology of well‐known organisms (Kliebenstein, 2019). In the present study, considering this knowledge trade‐off, we comprehensively characterized the high temperature response in the conifer Pinus radiata and described long‐term splicing memory and wide stress‐responsive AS patterns in gymnosperms. Furthermore, the integration of splicing data with other layers revealed important sources of additional regulatory variation between distinct response stages.
RESULTS
Heat‐responsive differential splicing and/or differential expression characterization
Splicing and expression changes induced by high temperature were analyzed from previously generated RNA‐seq data of P. radiata needles subjected to 40°C stress pulses (Figure 1; Figure S2; see Experimental Procedures). Briefly, heat stress was applied for three consecutive days during the central hours of the day, employing a temperature ramp from 15°C to 40°C over 5 h, holding the high temperature for 6 h, and then decreasing the temperature back to night values. De novo calling of splicing events was performed using the KisSplice pipeline (Sacomoto et al., 2012), enabling the description of the splicing landscape without using a reference genome, which is a major bottleneck in the study of non‐model organisms like P. radiata. For each AS event (represented by a pair of isoforms), differential splicing (DS) was first analyzed using the KissDE algorithm. We found that out of the 13 980 isoforms, 5105 were differentially spliced (FDR‐adjusted P < 0.05 in at least one comparison; Figure 2a). Secondly, differential expression (DE) was analyzed for each isoform using the DESeq2 algorithm (Love et al., 2014). We detected 1568 DE isoforms (FDR‐adjusted P < 0.05 in at least one comparison), of which 944 were also DS (regulated by both transcription and splicing; double differential [DD]) and 624 were not DS (only regulated by transcription). Consequently, 5729 isoforms exhibited significantly altered splicing or expression patterns (Figure 2a).
Figure 1.
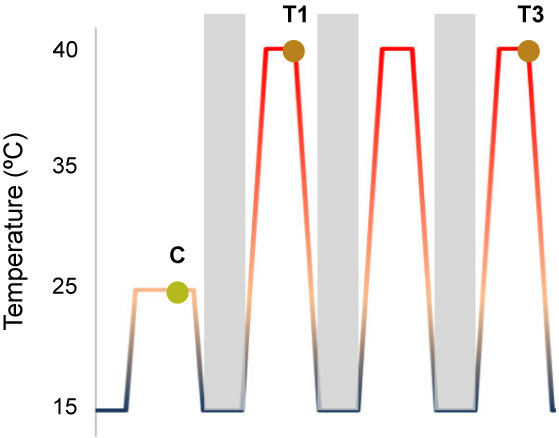
RNA‐seq experimental design. Temperature ranks from 15°C to 25°C in control conditions and to 40°C in stress conditions. Plants were stressed and sampled on day 1 (T1) and day 3 (T3) to characterize short and medium responses. Then, RNA extracted from treated plants was sequenced in order to perform splicing analysis. C, Control; T1 and T3, plant samples after 1 or 3 days of stress.
Figure 2.
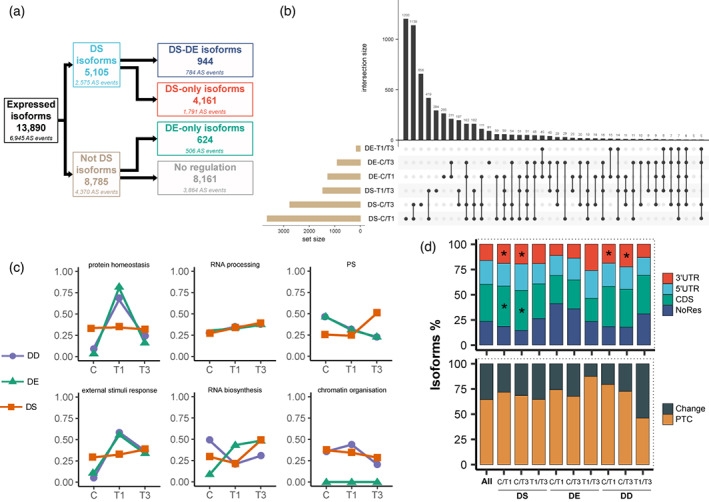
Summary of alternative splicing events and differential expression analysis of the P. radiata response to high temperature. (a) Flow chart showing the distribution of the 5105 DS and 1568 DE isoforms detected. DS, differential splicing; DE, differential expression; DD, double differential. (b) Matrix layout for all intersections of DS and DE comparisons, sorted by decreasing number of isoforms. Dark circles in the matrix indicate sets that are part of the intersection. C/T1, C versus T1 comparison; C/T3, C versus T3 comparison; T1/T3, T1 versus T3 comparison. (c) Differential expression trends as determined using Mercator4 categorization terms. The numbers indicate scaled expression according to the Mercator4 functional bin. (d) The distribution of distinct types of AS events by differential level and comparison. From bottom to top, 100% refers to all CDS AS events and all AS events. Change, make protein sequence changes; NoRes, neither full models nor full transcript sequences were predicted/mapped so events could not be classified. Significant enrichments are marked with ‘*’. The sampling times correspond to the 40°C assay shown in Figure 1.
In order to assess the specificity of heat‐induced changes, we next explored all differences by comparing which isoforms were DE and DS. Around 22% (1139) and 13% (211) of DS and DE isoforms, respectively, were common to both stress treatment durations (i.e., the initial response [day 1, T1] and acclimation [day 3, T3]). Major changes in AS and isoform‐level expression occurred during the initial shock and throughout the heat period (Figure 2b). These dynamics reflected crucial aspects of the stress response such as high temperature perception, initial response (T1), and acclimation (T3). It is important to note that isoforms, depending on their regulation level (DE, DS, or DD), presented diverse patterns in functional categories previously described as important in the stress response (Figure 2c) (Escandón et al., 2017; Lamelas et al., 2022). Thus, this functional discordance indicates that the role of isoforms and the variation produced by splicing may change according to their regulation or behavior.
AS can change transcript structures in different ways and have multiple molecular consequences (Laloum et al., 2018). Thus, to verify that the splicing variation effect was a driver of regulation and behavior, we investigated transcript models using CodAn (Nachtigall et al., 2020). This analysis was restricted to full models to ensure only one possible reading frame. Of all isoforms, 36.5% (5067) AS events occurred in coding sequences (CDS), among which 65% (3297) were predicted to introduce PTCs, probably leading to NMD, while the remaining 35% (1817) could result in protein sequence changes. Alternatively, 39.8% (5530) appeared in untranslated regions (UTRs), which might produce translation efficiency alterations, and 23.4% (3246) could not be classified because neither full models nor full transcript sequences were predicted or mapped (Figure 2d). Furthermore, changes in this proportion were tested among regulation levels and non‐supervised behavior clusters using hypergeometric tests (P < 0.05). Comparison of control versus stress DS and DD isoforms showed a significant increase in 3′UTR AS events, showing DS isoforms enrichment in CDS AS events as well (Figure 2d). At the behavior level, two clear non‐mutually exclusive trends were found in k‐means clusters: (i) 3′UTR AS events are enriched in clusters where isoforms presented lower expression in T3 than C (2, 4, 5, 8, 11, and 12) and (ii) 5′UTR AS events are enriched in clusters in which isoforms had higher expression in T3 than T1 (1, 3, 4, 6, 9, and 15) (Figures S3b and S4a). In most cases, CDS AS event enrichment was due to an increase in the proportion of PTCs (Figure 2d; Figure S4b; Table S4). Hence, we propose that isoform behavior and regulation are substantially influenced by the variation produced by splicing.
Intron morphology comparative analysis provides insight into intron retention splicing
Previous studies of stress‐induced AS in seed plants are consistent with a higher IR (Laloum et al., 2018). Consequently, potential introns were sought in the variable sequences introduced by AS. As the lack of reference genome forced us to employ a mapping‐free approach, variable sequences longer than 50 nucleotides (nt) and flanked by the conserved di‐nt GT‐AG were considered as potential introns. In contrast to aforementioned studies, we observed that only approximately 8% of AS events could be IR.
We next tested whether this low potential IR percentage could be an artifact of our analysis (biases in flanking di‐nts and presence of minor U12 introns which are surrounded by non‐canonical sites) by comparing intron morphologies between model species (mainly angiosperms) and gymnosperms. Pinus taeda was selected as a proxy for P. radiata since it is the closest species with largest scaffolds and better high‐quality gene models. In both U2‐ and U12‐type introns, the most predominant flanking sites were those used in our analysis (Figure 3; interactive version can be accessed at https://rocesv.github.io/introncompara/IntronCompara_finalfig2.html). Interestingly and in agreement with other studies (Wan et al., 2018), we noticed an increase in intron length effect size between angiosperms and gymnosperms. This trend remained constant even in Gnetum montanum despite its small introns/genome. Unlike other model species, our findings suggest a low IR percentage in P. radiata and places intron size as a possible key feature to understand splicing divergence between both groups of seed plants (Figure S5).
Figure 3.
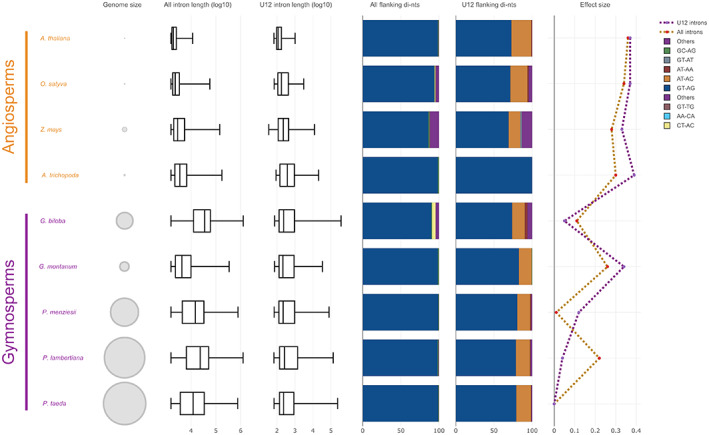
Comparative analysis of intron morphologies. Nine representative seed plants species were included: A. thaliana, Arabidopsis thaliana; O. sativa, Oryza sativa; Z. mays, Zea mays; A. trichopoda, Amborella trichopoda; G. biloba, Ginkgo biloba; G. montanum, Gnetum montanum; P. menziesii, Pseudotsuga menziesii; P. lambertiana, Pinus lambertiana; P. taeda, Pinus taeda. For all isoforms of each species we computed (from left to right): genome size (bp, scaled to the P. taeda genome), the all‐intron length distribution, the U12 intron length distribution, the all‐intron flanking di‐nt proportions, the U12 intron flanking di‐nt proportions, and the all‐intron/U12 intron length effect size, using P. taeda as reference. Different colors in flanking di‐nt proportion bar plots indicate distinct types of di‐nts. Gold and purple in effect size line plots indicate all introns and U12 introns, respectively.
Biological functions regulated at the splicing and transcription levels
To investigate general heat‐induced dynamics, we executed a heatmap/hierarchical clustering of total isoform expression levels according to Mercator4 functional annotation (Figure 4a). Four main expression profiles were revealed: (i) isoforms whose expression decreased during heat stress reflected homeostasis loss of control related to cell wall, chromatin, and cell cycle organization, solute transport, nutrient uptake, amino acid metabolism, and vesicle trafficking; (ii) isoforms whose expression increased during the treatments were associated with polyamine metabolism and external stimuli responses that illustrated heat perception; (iii) isoforms whose expression peaked at T1 displayed stress damage mostly linked to protein homeostasis; (iv) isoforms that presented the highest expression at T3 were connected to the DNA damage response, photosynthesis, secondary metabolism, phytohormone action, and RNA processing/biosynthesis functions and showed response or acclimation.
Figure 4.
Global dynamics and DD modulation functional description. (a) Heatmap/hierarchical clustering using Mercator4 categorization terms. The numbers indicate scaled expression, by rows, according to the Mercator4 functional bin. Relevant terms are highlighted. (b) Volcano analysis of DD event isoforms. Not significant, FDR‐adjusted P > 0.05 and absolute log2(fold change) < 1.8; Significant, FDR‐adjusted P < 0.05 and absolute log2(fold change) < 1.8; Fold Change, FDR‐adjusted P > 0.05 and absolute log2(fold change) > 1.8; Significant & Fold Change, FDR‐adjusted P < 0.05 and absolute log2(fold change) > 1.8. (c) Gene set enrichment analysis of DD event isoforms using Mercator4 functional categories. Blue and gold indicate significantly and non‐significantly enriched terms, respectively. The sampling times correspond to the 40°C assay shown in Figure 1.
Further, to identify genes displaying large changes and discover biological functions regulated by splicing and transcription, volcano analysis and Mercator4 annotation enrichment were performed on isoforms of DD events, respectively (Figure 4b,c; Figure S6a,b). In line with the previously described modulation, we observed enrichments in RNA processing/phytohormone action and protein homeostasis at T3 and T1, respectively. According to the first isoform expression profile we also detected significative alterations in homeostasis‐like categories for both stress versus control comparisons (Figure 4c). Stress‐specific comparison (T3 versus T1) exhibited enrichments in secondary metabolism, phytohormone action, and RNA biosynthesis congruent with the acclimation/response profile (Figure S6b). These insights were supported by some of the most relevant isoforms for each comparison, such as protein homeostasis HEAT SHOCK PROTEIN 21, CHLOROPLASTIC (HSP21) required for chloroplast development under heat stress, probably maintaining plastid‐encoded RNA polymerase‐dependent transcription; phytohormone action ABCISIC STRESS‐RIPENING PROTEIN 5 (ASR5) involved in the common cross heat‐drought responses playing a positive role by regulating abcisic acid signaling, stomatal closure and possibly preventing stress‐related proteins from inactivation; cell wall organisation GLYCINE‐RICH CELL WALL PROTEIN (EMB31) and secondary metabolism FLAVONOID 3’,5’‐HYDROXYLASE (CYP75A2) which is part of anthocyanins biosynthesis that are known to be associated to stress damage photoprotection as antioxidants (Figure 4b; Figure S6a) (Bateman et al., 2021).
Although various terms converged between general dynamics and coupled splicing/transcription modulation, other enriched categories largely differed. Thus, while general RNA processing isoform expression changes were not highlighted at T1 (Figure 4a), at the DD level we saw a significative enrichment in this term, exemplified by MULTIPLE ORGANELLAR RNA EDITING FACTOR 3, MITOCHONDRIAL (MORF3), which coordinates mitochondrial expression via epi‐ and post‐transcriptional regulation (Figure 4b,c). General photosynthesis‐associated expression increased at T3 (Figure 4a), exemplified by INNER MEMBRANE PROTEIN ALBINO 3, CHLOROPLASTIC (ALB3) in stress versus control volcano analysis (Figure 4b). ALB3 plays a key role in the insertion of some light‐harvesting chlorophyll‐binding proteins into the thylakoid membrane and, therefore, recovering thylakoid integrity associated with potential stress acclimation (Bateman et al., 2021). Notwithstanding the above, DD functional analysis revealed higher expression of isoforms related to this category in control plants (Figure 4c). Similarly, solute transport also appeared as inconsistent between general dynamics and DD regulation in stress‐specific comparisons (Figure S6a,b). The emergence of several isoforms such as PROBABLE AQUAPORIN PIP1‐4 (PIP1.4) and REF/SRPP‐LIKE PROTEIN (At3G05500) pointed towards water deprivation during the heat assay, possibly connecting high temperature to drought stress signaling (Figure 4b; Figure S6a). Collectively, these results reveal a wide range of stress‐responsive functional categories and elucidate the most important pathways involved in the heat response because of global and DD modulation discrepancies.
Relative contribution of multiple regulatory layers to the heat stress response
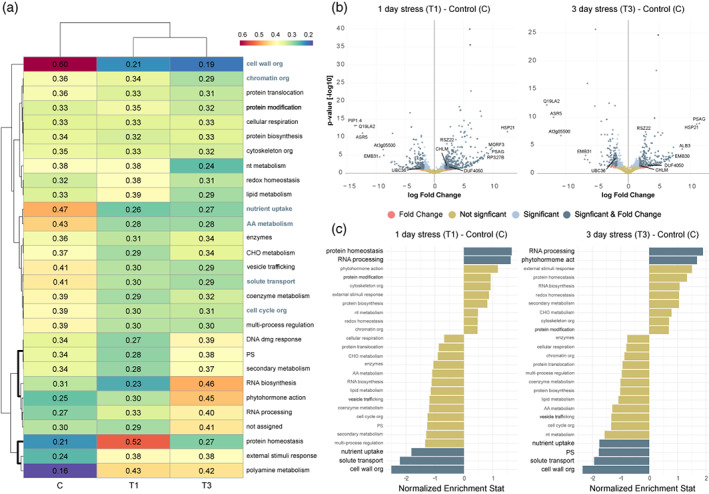
Heat is a critical and extensively studied type of abiotic stress. A growing body of literature reports the use of single omics analyses, yet the molecular determinants underlying high temperature responses are still not well understood for a lack of a holistic point of view. To fill this gap, we adopted a systems biology approach and expanded our splicing analysis with metabolome and proteome data previously generated by our group (Figure 5) (Escandón et al., 2017). To further research the relationships between all three regulatory layers during the heat response, we next applied multiomics factor analysis (MOFA2) (Argelaguet et al., 2018), a statistical framework for unsupervised identification of principal sources of variation from multiple data types. We sought to identify distinct response stages based on one or more underlying molecular levels. By querying the latent factor scores and loadings calculated by MOFA2, the variation explained by each latent factor could be annotated and deciphered as reported below.
Figure 5.
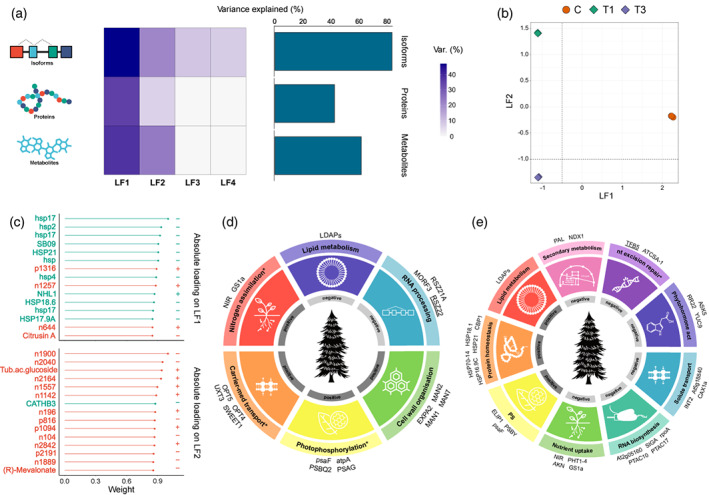
Relative contributions of distinct regulatory layers to the definition of the heat response. (a) Percentage of variance explained (R 2) by each MOFA2 factor (columns) across regulatory layers (rows) (left panel). Total contributions of each layer are summarized in the bar plot (right panel). (b) Scatter plot of latent factor 1 (x‐axis) and latent factor 2 (y‐axis) illustrating the variation described. Samples are colored according to treatment (C, T1, T3). (c) Lollipop plots showing top loading latent factor 1 (top) and latent factor 2 (bottom) features in descending order. Red and green indicate metabolome and proteome features, respectively. The sampling times correspond to the 40°C assay shown in Figure 1. (d,e) Enriched Mercator4 functional categories and top weight isoforms associated with latent factor 1 (e) and latent factor 2 (d). Functional categories marked with ‘*’ were renamed to subterms as follows because all top weight isoforms displayed belong to the same Mercator4 functional subterm: PS > photophosphorylation, solute transport > carrier‐mediated transport, nutrient uptake > nitrogen assimilation, DNA damage response > nucleotide excision repair. Positive, functional terms linked to samples with scores of >0; Negative, functional terms linked to samples with scores of <0.
MOFA inferred four latent factors (LF1, LF2, LF3, and LF4) with common and unique contributions at each molecular level (Figure 5a). Overall, isoforms represented the majority of total variance (R 2 = 84%), followed by metabolites (R 2 = 62%) and proteins (R 2 = 43%). We further inspected the top two factors (LF1 and LF2) sorted by variance explained, which defined control versus stress and stress intensity differences, respectively (Figure 5b).
We performed in‐depth characterization of LF1, which allowed to distinguish between control and stress conditions and explained 47% of isoform, 35% of protein, and 38% of metabolite variance. Despite the greater splicing variance explained, top scoring features associated with LF1 were predominantly proteins (Figure 5c). To investigate if these LF1 top scoring features were enriched for functional terms, we performed Mercator4 enrichment analysis for isoform and protein layers using as background all input features (FDR‐adjusted P < 0.1). We tested for both positive and negative enrichments, which means categories linked to samples with positive or negative factor values (Figure 5d; Figure S7a). Interestingly, we observed not only some common terms with global dynamics and DD regulation (RNA processing, solute transport, nutrient uptake, photosynthesis, and cell wall organization), but also new ones only revealed by the integration such as lipid metabolism at the isoform level (Figure 5d) and protein translocation at the protein level (Figure S7a).
On the other hand, LF2 enabled discrimination of all treatments and explained 21% of isoform, 7% of protein, and 23% of metabolite variance. In this case, top scoring features were principally metabolites, which is consistent with the variation explained (Figure 5c). At the isoform level, the functional profile displayed was a mix between heat response (lipid metabolism and protein homeostasis) and acclimation (phytohormone action, solute transport, nutrient uptake, RNA biosynthesis, DNA damage response, and lipid metabolism) terms. Additionally, we detected an enrichment in secondary metabolism functions which confirmed the prior role of metabolism layer in this latent factor (Figure 5e). This is for instance illustrated by the relevance of splicing in PHENYLALANINE AMMONIA‐LYASE (PAL), which catalyzes the production of an important variety of well‐known stress‐responsive secondary metabolites like the aforementioned flavonoids and anthocyanins (Junli et al., 2010). Proteins related to the heat response and acclimation were enriched in enzyme/protein translocation and lipid metabolism, respectively (Figure S7a). Intriguingly, despite both enrichments of lipid metabolism, at the isoform level it was related with heat response (positive), while at the protein level it was linked to acclimation (negative).
It is worth mentioning that despite the abovementioned top absolute loading features, all regulatory layers had strong biomarkers for each biologically relevant latent factor, as shown in Figure S7(b). Our findings put splicing as a crucial regulatory layer. These analyses underline the need for an integrative approach since the heat response more generally depends on the shared contributions of multiple molecular levels, with the relative contribution of each level possibly depending on the response stage. To examine the common belief that AS contributes to proteome diversity, we analyzed differential proteins and isoforms by volcano analysis (Figure S6c). Only stress versus control comparisons showed medium, strong, or very strong isoform–protein relations. These few direct or indirect isoform–protein relations could not be associated with variation produced by splicing and no relation was found between different proteins translated from the same splicing event isoforms. Protein projection over isoforms confirmed these results because no overlap was seen between both molecular levels (Figure S6d).
Meta‐network analysis reveals isoform co‐function modules
In view of the importance of splicing, we focused on DD isoform interactions and performed regulatory network analysis to identify co‐function modules and their master regulators. We employed a ‘wisdom of crowds’ approach using the Seidr toolkit (Schiffthaler et al., 2021) to avoid method bias towards certain types of biological interactions (Marbach et al., 2012). The resulting weighted undirected meta‐network was mainly constituted by five highly connected modules (Figure 6a). An interactive version of the meta‐network can be accessed at https://rocesv.github.io/network/index.html.
Figure 6.
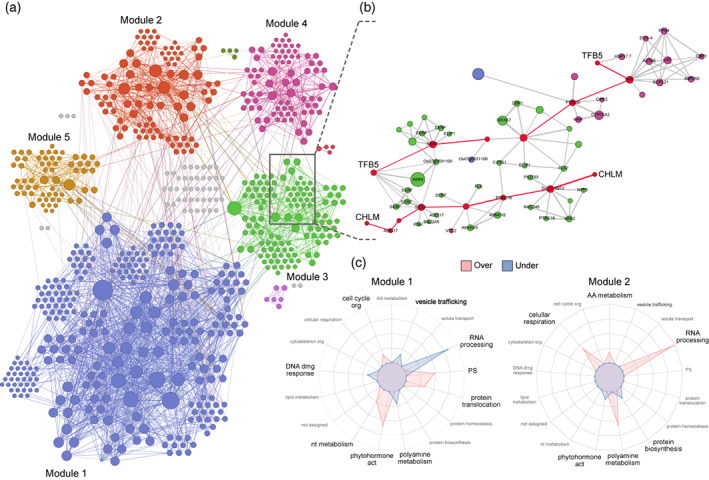
Meta‐network regulatory analysis. (a) Meta‐network constituted by five highly connected modules (different colors). Nodes represent isoforms and node size reflects Katz centrality. Network edges indicate biological interactions. (b) Local interaction network connecting both isoforms from two AS events. Nodes and edges highlighted in red illustrate the shortest path between intra‐event isoforms. (c) Radar plots showing topology enrichment (red) and reduction (blue) of Mercator4 functional terms for the two biggest modules of the meta‐network. Peak size = −log10(adjusted P).
Module nodes were ranked by Katz centrality to search the isoforms with the strongest regulatory effects. Interestingly, we found well‐known regulators such as SPLICING FACTOR U2AF SMALL SUBUNIT B (U2AF35B) (module 2) (Laloum et al., 2018); isoforms earlier proposed in volcano and integrative analysis (Figures 4b and 5d,e) like ALB3 (module 1), METALLOTHIONEIN‐LIKE PROTEIN (EMB30) (modules 1 and 5), SERINE/ARGININE‐RICH SPLICING FACTOR RSZ22 (RSZ22) (module 2), CYP75A2 (module 4), and ASR5 (module 3); and new ones, for instance 50S RIBOSOMAL PROTEIN 5, CHLOROPLASTIC (PSRP5) (module 1). In agreement with previous highlighted isoforms, PSRP5 plays a critical role in the translation of chloroplast‐encoded genes and it is specially related to thylakoid membrane proteins (Bateman et al., 2021). Our analysis also enabled the inspection of the connections between the same splicing event isoforms (intra‐event), for example, a local interaction network of GENERAL TRANSCRIPTION AND DNA REPAIR FACTOR IIH SUBUNIT TFB5 (TFB5) and MAGNESIUM PROPORPHYRIN IX METHYLTRANSFERASE, CHLOROPLASTIC (CHLM) (Figure 6b). The functions of both these isoforms and those involved in connecting them pointed to a nucleus–chloroplast coordination via regulation of transcription and the response to heat by detoxification of reactive species (Bateman et al., 2021).
Topology enrichments of the two biggest modules (Figure 6c) revealed that both modules manifested different or even opposite functional profiles, especially when looking at key response and acclimation terms like RNA processing, polyamine metabolism, and phytohormone action. These enrichments improve our understanding of the possible categories involved in the rewiring of inter−/intra‐event interactions.
Stress‐responsive AS events participate in the establishment of long‐term thermomemory
Finally, in order to validate identified stress‐responsive AS patterns and explore the role of splicing in long‐term memory, two assays were performed and several AS events were checked by reverse transcriptase‐PCR (RT‐PCR) (Figures 1 and 8a; Figure S2). Eight events were selected based on the following criteria: differential trends, integrative analysis relevance, meta‐network importance, and event length of at least 80 nt. As expected, five examined events, including RSZ22, GLYCINE‐RICH RNA‐BINDING PROTEIN RZ1A (RZ1A), UNCHARACTERIZED PROTEIN DUF4050 (DUF4050), HISTONE DEACETYLASE HDT2 (HDT2), and UBIQUITIN‐CONJUGATING ENZYME E2 36 (UBC36) (Figure 7), showed consistent splicing patterns with their profiles revealed by 40°C RNA‐seq data, which therefore corroborated the accuracy of our bioinformatics workflow. Curiously, two events, TFB5 and CHLM, presented the exact opposite trend. Lastly, although ESCRT‐RELATED PROTEIN CHMP1B (CHMP1B) had a clear heat‐specific pattern, it was only slightly congruent with the stress‐induced long isoform dominance (Figure 7).
Figure 8.
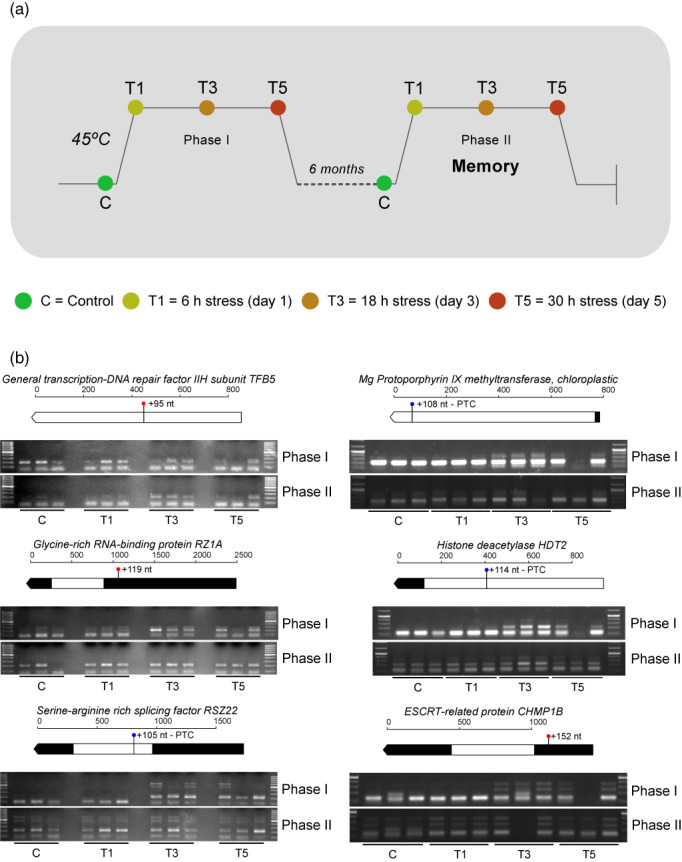
Memory experimental design and validation of splicing memory by RT‐PCR. (a) A further characterization of validated AS events was performed in a 45°C assay in order to check AS pattern coherence at a more lethal temperature, test the potential implication of AS in stress memory, and identify high temperature‐specific expression. The 45°C assay was divided in two phases. In Phase I, plants were stressed and sampled on day 1 (T1), day 3( T3), and day 5 (T5) to characterize long responses. At 6 months after the end of Phase I, the plants were subjected to another round of treatment (Phase II) to evaluate the potential acquisition of long‐term splicing memory. (b) Each AS event is represented by a block constituted by two panels. Top panel: transcript model of the small isoform displaying event length, PTC introduction, and where the variation is produced. Bottom panel: RT‐PCR AS patterns in the sampling times corresponding to the 45°C memory assay. The primers used allow the amplification of both splice variants. C, Control; T1, T3, and T5, stressed plant samples after 1, 3, or 5 days of stress.
Figure 7.
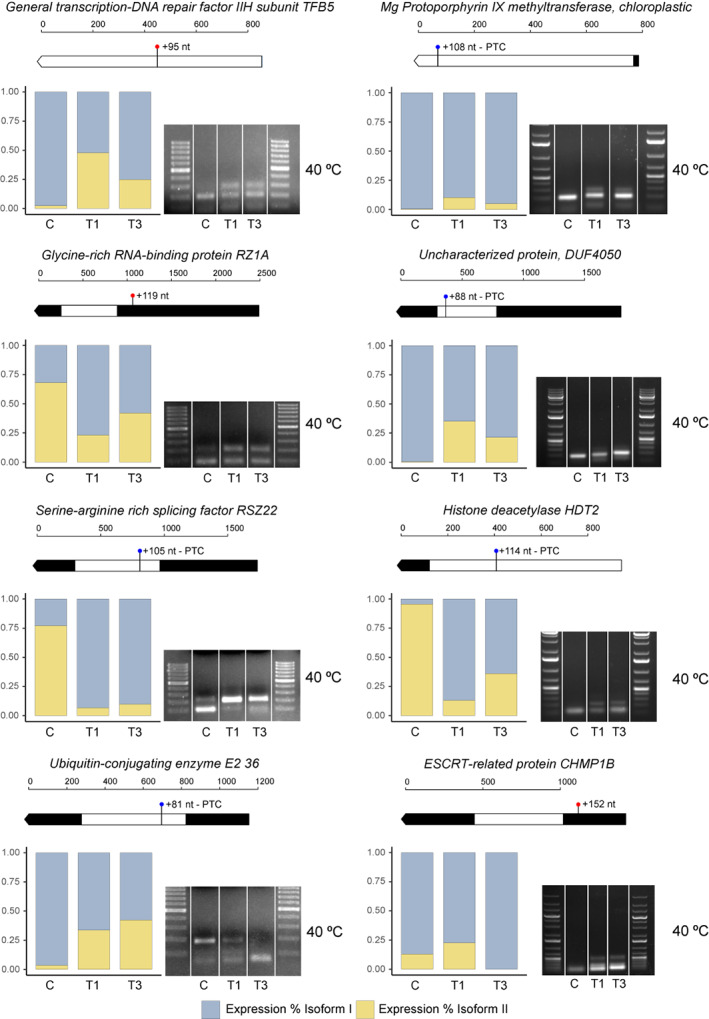
Experimental validation of heat‐induced AS events by RT‐PCR. Each AS event is represented by a block constituted by two panels. Top panel: transcript models of the small isoform displaying event length, PTC introduction, and where the variation is produced. Bottom left and bottom right panels: bar plots showing relative expression of isoform 1 (blue) and isoform 2 (yellow) of AS events revealed by RNA‐seq and RT‐PCR AS patterns at the sampling times corresponding to the 40°C assay shown in Figure 1. The primers used allow the amplification of both splice variants.
Recent research in Arabidopsis linked splicing memory to the ability of plants to survive subsequent and otherwise lethal heat stress (Ling et al., 2018). To further explore the potential acquisition of long‐term stress memory, we reviewed these AS patterns in 45°C heat‐primed and non‐primed plants (Phase II and Phase I; Figure 8a; Figure S2). Of eight candidate events (Figure 7), the existence of acquired memory could only be checked in seven as UBC36 was not expressed at 45°C. It is important to note that the interpretation of T1 differs considerably according to stress intensity. In the RNA‐seq‐based assay at 40°C we could already observe a heat response at T1; however, in the memory assay at 45°C this sampling time reflected essentially damage. The response in the memory assay started at Phase I T3. Five AS events, comprising CHLM, RZ1A, RSZ22, HDT2, and CHMP1B, displayed clear stress splicing memory due to the appearance of response‐specific isoforms (Phase I T3) in Phase II control (Figure 8b). The disappearance of the large TFB5 isoform at Phase II control seems to indicate thermomemory. However, the expression of the two isoforms at both T3 phases made this trend too complex to be able to undoubtedly confirm splicing memory. Detection of more than two isoforms reflected the effect of our stringent k‐mer (CHMP1B) and other 45°C‐exclusive AS events (RSZ22). These findings provided evidence of stable acquired splicing memory in heat‐stressed P. radiata seedlings, this mechanism being a general feature of heat stress responses that could be particularly relevant in long‐lifecycle plants.
DISCUSSION
RNA splicing is an essential process for plant development and intersects with many hallmarks of environmental responses. The link between AS and abiotic stress is well stablished (Laloum et al., 2018), but we only recently started to understand the molecular networks that drive RNA processing alterations in the acquisition and maintenance of heat stress tolerance, highlighting sophisticated mechanisms like splicing memory, protein‐to‐RNA trans‐regulation, and NMD avoidance (Jia et al., 2020; Ling et al., 2018).
A major focus of this work was the analysis of splicing and expression differences between plants subjected to a moderate high temperature stress treatment and control plants. We show that more than 4000 isoforms exhibiting a significant change in AS are not differentially expressed during heat stress. Moreover, functional categories covered by DS, DE, and DD isoforms are not interlinked (Figure 2). Our results together with other works underline the importance of wide AS reprogramming as an additional layer of regulation for stress responses (Calixto et al., 2018). In line with earlier research (Escandón et al., 2017), short‐term stressed plants (T1) present the most relevant changes and display a functional profile defined by loss of control and damage categories like protein homeostasis (Figures 2, 4, and 5), which is mainly involved in protein protection against heat‐induced denaturation and aggregation through HSPs (Jacob et al., 2017). Interestingly, stress‐specific comparison shows T3 expression recovery of photosynthesis‐ and water‐related isoforms and enrichments in functions associated with control, such as solute transport (Figure S6), thus suggesting some degree of acclimation in mid‐term stressed plants (T3). Nevertheless, these changes are not enough to detect a control‐like recovery since homeostasis functions are still diminished in comparison with non‐stressed plants (Figures 4c and 5e). Collectively, these observations establish T3 as an essential point required to reveal key players and pathways in both the initial response and acclimation such as potential AS feedback loops through RNA processing of several trans‐acting RNA‐binding proteins, for instance, RSZ22 and RZ1A, which have previously been described to play roles in stress tolerance and may be connected with splicing specificity (Laloum et al., 2018; Reddy et al., 2013). By the same token, coordinated control of RNA biosynthesis (Figures 5e and 6b; Figure S6b) between nucleus and chloroplast is also emphasized including probably retrograde signaling acting as mediator of nuclear photosynthesis‐related gene expression and straightforward regulation of nuclear transcription through control of RNA polymerase II dynamics and bivalent epigenetic signals (both active and repressive), respectively, represented by CHLM, TFB5, and HDT2‐UBC36 events (Bateman et al., 2021).
An important goal of stress systems biology is to understand how different regulatory layers are integrated to contribute to shared or unique phases of the response. We observed that the underlying variation between control and stress involves coordinated programs at all molecular levels but it is mainly linked to splicing, while top loadings are principally proteomic features (Figure 5a–c). This result could reflect greater participation of splicing and proteins in early stages of the response probably due to their implication in perception and initial signaling (Lin et al., 2020). On the other hand, metabolite variation tends to discriminate between all response stages. We propose that this metabolome phase‐specific contribution might be justified by distinct timescales for turnover between all molecular levels (Shamir et al., 2016). Metabolites have the highest turnover rate; hence they are more dynamic and accurately reflect the current state of the plant. Despite greater splicing variance explained, we suggest that the recurrent presence of proteome and metabolome data in top loading features (Figure 5a,c) could be a consequence of two main reasons: (i) proteins and metabolites are directly functional components, and therefore they are subjected to more layers of down‐/upstream regulation that shift their abundances towards qualitative patterns and optimize energy investment in expensive processes such as metabolism or translation; (ii) splicing, because of its co‐transcriptional nature, is one of the closest regulatory levels to the genome, lacking direct functional effects, and its activity is not as controlled as the other molecular levels. Thus, splicing trends not only reveal effects and consequences, but also part of the artifacts and stochastic reprogramming that occur during the stress response (Chaudhary et al., 2019). Assuming trypsin digestion limitations, we detected little evidence of splicing contributions to the proteome and poor coordination at the functional, abundance, and variation effect levels (Figure 2d; Figures S6c,d and S7a). These analyses may constitute further evidence against AS as a key mechanism to expand proteome complexity. More generally, our findings suggest that response stages are correlated to contributions of molecular levels and place splicing as the main driver of heat stress variation.
The evolutionary history of gymnosperms involves a series of functional and morphological traits accompanied by well‐defined genomic features (De La Torre et al., 2020) such as rarity of whole‐genome duplications, slow mutation rates, high repeat content, big genomes, and long introns (Figure 3). Although these organisms are a clear hotspot of plant molecular diversity, they remain underrepresented and unexplored in gene regulation research as a result of their long lifecycle. Contrary to angiosperms, we found that IR may not be the most prevalent type of AS in conifers. Taking all the above into account, together with the limited capacity of KisSplice to assemble long introns, which requires their full coverage by reads to be correctly assembled (Ashraf et al., 2020), and earlier described accelerated RNA Polymerase II kinetics and nuclear DNA hypomethylation during high temperature treatment (Jonkers & Lis, 2015; Lamelas et al., 2020), we propose that simultaneous availability of weak and strong splice sites due to loss of transcription control (Luco et al., 2011) could result in favored retention of introns flanked by weak splice sites. However, short introns are significantly more retained because their flanking splice sites are recognized as a unit (Monteuuis et al., 2019). We hypothesize that the low potential IR exhibited could be caused by the fact that long introns in gymnosperms might hinder intron recognition as single unit or could even prompt the appearance of strong cryptic splice sites, thus probably forcing the retention of fragmented introns instead of complete sequences (Figure S5). Considering the small body of evidence of splicing in gymnosperms, IR is reported as dominant in Gnetum but as a non‐majority processing type in Ginkgo biloba (Deng et al., 2019; Sun et al., 2020). Although we cannot directly extrapolate these reports because they are not stress studies, these findings may fit with our hypothesis since the case of Gnetum could be justified due to its particularly small introns/genome compared to the rest of the group (De La Torre et al., 2020; Wan et al., 2018). Further full‐length analyses become a priority to validate this fragmented IR hypothesis. In summary, this insight pinpoints the importance of understanding the influence of clade‐specific genomic features if we aim to generalize our knowledge about molecular mechanisms in model plants.
The next question to solve is: Is AS a key factor in the maintenance of heat stress memory in P. radiata? The maintenance of acquired thermotolerance is crucial for successful tolerance to subsequent exposure to otherwise lethal temperatures (Ling et al., 2018). We validated the acquisition of heat stress‐linked splicing memory in a diverse subset of genes associated to aforementioned pathways and others previously described (Lamelas et al., 2020), such as protein degradation illustrated by the CHMP1B event, which is particularly required for autophagic degradation of plastid proteins to the vacuole, again highlighting the importance of chloroplast proteome turnover during acclimation as one of the main targets of heat stress damage (Spitzer et al., 2015). The present report solves the initial question and address new ones such as: 1) Heat stress‐induced splicing memory seems to be conserved between both groups of seed plants, therefore, underlining the importance of this mechanism to provide plants a versatile way to cope with changing environmental conditions. 2) Moreover, all splicing events validated are intron complete retention‐independent. Despite this conservation, these results suggest that the processes that drive splicing memory are substantially different between angiosperms and gymnosperms, probably because of distinct IR relevance due to genome architecture divergence (Figures 3 and 8b). 3) Finally, this work provides further evidence for both long‐term splicing memory and its presence in long‐lifecycle species. The extension of memory maintenance from 1 month, which was the period originally described in Arabidopsis thaliana (Ling et al., 2018), to 6 months has an even greater impact for the development of tolerant plants in order to ensure food security and forest resources in the current context of climate change. Given the realization that chromatin structure can affect AS and the memory detected in HDT2, it is attractive to speculate about coupled epigenetic processes and splicing memory as a system to maintain acclimation‐associated AS patterns over a longer period of time, which could be potentially implicated in transgenerational tolerance (Luco et al., 2011). So far, all validated events that impact CDS introduce PTCs, but they could be equally responsible for thermotolerance through earlier proposed models like buffering against the stress‐responsive transcriptome to reduce the metabolic cost of translating all AS transcripts and targeted decay stability that may underpin memory and resetting (Chaudhary et al., 2019; Crisp et al., 2016). The concept of environmental acclimation by transcript dysfunction and splicing memory has great potential to uncover the molecular diversity underlying stress responses, which will deepen our understanding of how the phenotypic plasticity of plants has fascinatingly evolved. Our study reveals how long‐living conifers have developed a low IR relevance way of remembering past temperature changes over a longer period of time and opens the door to include plant species with unique features, like gymnosperms, to determine the extent of conservation in gene expression regulation.
EXPERIMENTAL PROCEDURES
In silico analysis of AS events raw data
In this work we studied the response of P. radiata to heat (40°C) employing transcriptomics, proteomics, and metabolomics datasets previously generated by our group (Escandón et al., 2017; Escandón et al., 2022; see Data Availability Statement).
RNA‐seq data pre‐processing and AS event identification
An overview of the bioinformatic workflow used in this work is shown in Figure S1. Data pre‐processing was performed following the recommendations of Sacomoto et al. (2012). De novo AS events were identified using KisSplice (v.2.4.0‐p1) with stringent parameters ‘‐C 0.05’ and testing different k‐mer values between 51 and 61 nucleotides; we selected 51 nucleotides as this represented a good specificity–sensitivity balance.
Differential splicing and differential expression analysis
The KisSplice raw counts values were imported into R (v.3.6.0) using the Bioconductor (v.3.9) kissDE package (v.1.4.0). For visualization and quality assessment, raw counts were normalized using a variance‐stabilizing transformation as implemented in DESeq2 (v.1.24.0) (Love et al., 2014). The biological importance of the data was evaluated by principal component analysis and other visualizations using the pRocessomics R package (github.com/Valledor/pRocessomics), developed in our lab, and custom R scripts.
Statistical analysis of event‐level DS and isoform‐level DE were carried out in R using the Bioconductor KissDE and DESeq2 packages, respectively. For both differential analyses, all possible two‐by‐two comparisons were performed (T1 versus C, T3 versus C, and T3 versus T1), allowing to reveal the contributions of (i) stress damage and (ii) acclimation/response. A threshold of FDR‐adjusted P ≤ 0.05 was used to assess significance. Only isoforms showing log2(fold change) > 1.8 were considered as biologically relevant for volcano plots.
Annotation and functional enrichment analysis
The whole annotation pipeline is shown in Figure S3(a). KisSplice isoform sequences were mapped to Trinity, Transabyss, and Consensus assemblies (see Data Availability Statement) using bowtie2 (v.2.4.2) (Langmead & Salzberg, 2012) with the following parameters: ‘‐‐end‐to‐end ‐N 1 ‐t ‐‐no‐unal ‐p 6’. Mapped transcript sequences were analyzed with InterPro (v.5.45–80.0) (Jones et al., 2014) and dammit (v.1.2.0) using all default databases, Uniref90, and a custom database containing all gymnosperm data from the 1KP transcriptomes project (One Thousand Plant Transcriptomes Initiative, 2019).
Gene Ontology (GO) (Carbon et al., 2021) annotations were obtained using the RSQLite package (v.2.2.0) with custom R scripts (see Data Availability Statement). GO tree maps were generated using REVIGO (Supek et al., 2011) changing the background database to ‘Arabidopsis thaliana’. Mapped transcript sequences were also annotated using Mercator4 (Schwacke et al., 2019) in order to obtain functional bin information. Gene set enrichment analysis was performed using the Bioconductor fgsea package (v.1.10.0). Not identified/annotated isoforms were completed with results of other assemblies (complementary assembler) and the information of the other isoform from the same AS event (complementary splicing).
Transcript model prediction, splicing variation effect and protein–isoform analysis
Identification of transcript CDS was performed using CodAn (v.1.0.0) (Nachtigall et al., 2020) with the PLANTS_full model and protein sequences of the 40°C assay proteome and transcriptome (Escandón et al., 2017) as blast database. Splicing variation effects were determined using custom R scripts with the following program: (i) bowtie2 coordinates and CodAn gtf transcript models were used to know if the variation was produced in CDS or UTRs, and (ii) CDS‐affecting isoforms were examined to detect if the variation led to PTCs or CDS changes.
Statistical analysis of protein‐level differential abundance was carried out in R using the Bioconductor limma (v.3.40.0) (Ritchie et al., 2015) package. All possible two‐by‐two comparisons were performed (T1 versus C; T3 versus C, and T3 versus T1) and a threshold of FDR‐adjusted P ≤ 0.05 was used to assess significance. Isoforms corresponding to proteins were identified using BLASTp with an E‐value cutoff of 1e−5 and an 80% identity threshold. Protein–isoform combinations showing log2(fold change) values of more than 0.5, 1, and 1.8 were considered as weak, strong, and very strong associations, respectively. To reveal isoform–protein relationships, UMAP training and projection were performed with isoform/protein data using the uwot (v.0.1.8) R package.
Multiomics factor analysis
Inference of shared sources of variation from combined data types was performed in R (v.4.0.3) using the Bioconductor (v.3.12) MOFA2 (v.1.0.1) package. To prepare for model training, three sets of 40°C heat stress assay regulatory layers were used: isoforms, proteins, and metabolites (Escandón et al., 2017). One replicate for each treatment was imputed for isoform data using the pRocessomics R package. Training of the model was carried out using the following options: maxiter = 60 000, convergence_mode = ‘slow’. Each biologically relevant latent factor underwent overrepresentation analysis to determine the magnitude to which factors represented different Mercator4 functional categories.
Meta‐network analysis
To avoid bias towards specific interaction patterns, a meta‐network was created using the Seidr network toolkit (v.0.14.2) (Schiffthaler et al., 2021). Double differential VST‐normalized isoform expression data were used as input for network construction, keeping the same number of samples as in MOFA. Inference methods applied were ARACNE, CLR, Elastic Net and SVM ensemble, Partial Correlation, NARROMI, GENIE3, PLSNET, TIGRESS, TOM similarity, and Spearman correlation. The outputs were aggregated into a meta‐network using the irp method and the resulting network was filtered executing network backboning with ‐F 1. Infomap (v.1.3.0) (Rosvall & Bergstrom, 2008) was used to detect highly connected modules. The Neat (v.1.1.3) R package was used to discover network modules enriched for Mercator4 functional categories (alpha = 0.05).
Intron comparisons
All genome data sources are shown in Table S1. Among all sequenced conifer genomes, P. taeda was selected as the most appropriate reference species for genomic comparisons with A. thaliana, Zea mays, Oryza sativa, Amborella trichopoda, Ginkgo biloba, G. montanum, Pseudotsuga menziesii, and Pinus lambertiana. IntronIC (v.1.1.2) (Moyer et al., 2020) was applied to classify U12/U2 introns and obtain information with respect to intron length and terminal dinucleotides taking into account all isoforms. Boxplot construction and effect size calculation were performed according to Wan et al. (2018).
Experimental validation of AS events
Plant material and growth conditions
Plant material was generated for (i) validating RNA‐seq‐based results in a 40°C assay and (ii) further characterizing our validated AS events in a 45°C assay in order to check AS pattern coherence at a more lethal temperature, test the potential implication of AS in stress memory, and elucidate high temperature‐specific expression (Figures 1 and 8a; Figure S2).
One‐year‐old P. radiata seedlings (height 33 ± 4 cm) were kept in 1‐L pots under a photoperiod of 16 h (400 μmol m−2 sec−1) at 25°C and 50% relative humidity (RH) and 8 h at 15°C and 60% RH during the night in a climate chamber under controlled conditions (Fitoclima 1200, Aralab, Albarraque, Portugal) where stress assays were also conducted. Prior to assays, plants were acclimatized for a >1‐month period inside the controlled chamber, watered at field capacity every day, and fertilized weekly with nutritive solution (N:P:K 5:8:10).
For the first experiment, P. radiata seedlings were subjected to 40°C heat stress as described by Escandón et al. (2017). Briefly, heat stress was applied during the central hours of the day, employing a temperature ramp from 15°C to 40°C over 5 h. The high temperature was held for 6 h and then the temperature was decreased again to night values. The stress was applied for three consecutive days, and plants were sampled at the end of the 40°C period on the first (T1) and the last day (T3) (Figure 1).
In the second experiment, P. radiata seedlings were subjected to 45°C heat stress as described by Lamelas et al. (2020). Briefly, to test long‐lasting memory acquisition related to an acclimation process, the assay was divided into two similar phases 6 months apart from each other. In each phase, treatment was applied during the central hours of the day, employing a temperature ramp from 15°C to 45°C over 5 h. The high temperature was held for 6 h, and then the temperature was decreased again to night values. This procedure was repeated for five consecutive days, and plants were sampled at the end of the 45°C period on day 1 (T1), day 3 (T3), and day 5 (T5). Thus, Phase I was established as the first 45°C treatment and Phase II as the second treatment after 6 months (Figure 8a).
Control samples (C) were collected on the first day of each experiment at the same time before starting heat exposure. Three biological replicates for each treatment were pools of three plants each. The individuals constituting each pool were kept during the experiments. All samples were immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction.
RT‐PCR analysis
RNA was extracted as described by Valledor et al. (2014) and then quantified in a Nabi UV/Vis Nano Spectrophotometer. RNA integrity was checked by agarose gel electrophoresis, and potential DNA contamination was checked by PCR employing a GLYCERALDEHYDE‐3‐212 PHOSPHATE DEHYDROGENASE (GADPH) primer pair (Table S2). cDNA was obtained from 500 ng of RNA using the RevertAid kit (ThermoFisher Scientific), where random hexamers were used as primers following the manufacturer's instructions. RT‐PCR was performed with DreamTaq DNA Polymerase under the following conditions: 95°C for 4 min, followed by 40 cycles of 95°C for 30 sec, 60°C or 61°C for 30 s, and 72°C for 1 min, a final extension step at 72°C for 10 min, and an 8°C hold until the product was used. Primers for each AS event (Table S2) were designed to amplify both two splice variants in a single reaction. Sequences of different splice variants were validated by Sanger sequencing (Stab Vida, Lda; Portugal) (Table S3).
AUTHOR CONTRIBUTIONS
MM and MJC conceived the study. MM, LL, and VR designed the research. LV and LL collected the data. MM supervised the study. VR performed computational analyses, analyzed/interpreted the data, and wrote the manuscript. VR and MC performed experiments. All authors revised, read, and approved the final manuscript.
CONFLICT OF INTEREST
The authors declare there is no conflict of interest.
Supporting information
Table S1. All genome data sources for the species used in intron comparative analysis.
Table S2. List of primers used for RT‐PCR.
Table S3. Sanger sequencing data validating RT‐PCR results.
Table S4. K‐means cluster data.
Table S5. Isoform sequences, expression, annotation, statistics, and potential event type data.
Table S6. Multiomics integration data.
Figure S1. Overview of the bioinformatics workflow.
Figure S2. General experimental design.
Figure S3. Annotation pipeline and non‐supervised behavior clusters.
Figure S4. Splicing variation effect proportions across non‐supervised behavior clusters.
Figure S5. Heat stress‐responsive intron retention hypothesis in gymnosperms.
Figure S6. Functional description of stress‐specific isoforms and isoform–protein relationships.
Figure S7. Functional characterization of proteins and all regulatory layers for each MOFA2 latent factor.
ACKNOWLEDGMENTS
Funding for this work was provided by the Spanish Ministry of Economy and Competitiveness (PID2019‐1071076B‐100). VR was supported by a fellowship from the Spanish Ministry of Science, Innovation and Universities (FPU18/02953). LL was supported by a fellowship from the Spanish Ministry of Science, Innovation and Universities (BES‐2017‐082092). MC was supported by the Severo Ochoa Predoctoral Program (BP19‐137). LV was supported by the Spanish Ministry of Economy and Competitiveness (RYC‐2015‐17871).
Contributor Information
María Jesús Cañal, Email: mjcanal@uniovi.es.
Mónica Meijón, Email: meijonmonica@uniovi.es.
DATA AVAILABILITY STATEMENT
All data, codes and gels/blots generated during the study are available in the GitHub repository (https://github.com/RocesV/AS_heat_Pra). The raw RNA‐seq data have been deposited in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA851373. The mass spectrometry proteomics data including RAW, msf, and pepXML files have been deposited in the ProteomeXchange Consortium database via the PRIDE (Perez‐Riverol et al., 2022) partner repository with the dataset identifier PXD032754. The metabolomics data have been deposited in the Metabolights repository (https://www.ebi.ac.uk/metabolights/) under dataset identifier MTBLS4567.
REFERENCES
- Argelaguet, R. , Velten, B. , Arnol, D. , Dietrich, S. , Zenz, T. , Marioni, J.C. et al. (2018) Multi‐omics factor analysis—a framework for unsupervised integration of multi‐omics data sets. Molecular Systems Biology, 14(6), e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf, U. , Benoit‐Pilven, C. , Navratil, V. , Ligneau, C. , Fournier, G. , Munier, S. et al. (2020) Influenza virus infection induces widespread alterations of host cell splicing. NAR Genomics and Bioinformatics, 2(4), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A. , Martin, M.J. , Orchard, S. , Magrane, M. , Agivetova, R. , Ahmad, S. et al. (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Research, 49(D1), D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calixto, C.P.G. , Guo, W. , James, A.B. , Tzioutziou, N.A. , Entizne, J.C. , Panter, P.E. et al. (2018) Rapid and dynamic alternative splicing impacts the arabidopsis cold response transcriptome[CC‐BY]. Plant Cell, 30(7), 1424–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capovilla, G. , Delhomme, N. , Collani, S. , Shutava, I. , Bezrukov, I. , Symeonidi, E. et al. (2018) PORCUPINE regulates development in response to temperature through alternative splicing. Nature Plants, 4(8), 534–539. [DOI] [PubMed] [Google Scholar]
- Carbon, S. , Douglass, E. , Good, B.M. , Unni, D.R. , Harris, N.L. , Mungall, C.J. et al. (2021) The gene ontology resource: enriching a GOld mine. Nucleic Acids Research, 49(D1), D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary, S. , Jabre, I. , Reddy, A.S.N. , Staiger, D. & Syed, N.H. (2019) Perspective on alternative splicing and proteome complexity in plants. Trends in Plant Science, 24(6), 496–506. [DOI] [PubMed] [Google Scholar]
- Crisp, P.A. , Ganguly, D. , Eichten, S.R. , Borevitz, J.O. & Pogson, B.J. (2016) Reconsidering plant memory: intersections between stress recovery, RNA turnover, and epigenetics. Science Advances, 2(2), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Torre, A.R. , Birol, I. , Bousquet, J. , Ingvarsson, P.K. , Jansson, S. , Jones, S.J.M. et al. (2014) Insights into conifer giga‐genomes. Plant Physiology, 166(4), 1724–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Torre, A.R. , Piot, A. , Liu, B. , Wilhite, B. , Weiss, M. & Porth, I. (2020) Functional and morphological evolution in gymnosperms: a portrait of implicated gene families. Evolutionary Applications, 13(1), 210–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, N. , Hou, C. , Ma, F. , Liu, C. & Tian, Y. (2019) Single‐molecule long‐read sequencing reveals the diversity of full‐length transcripts in leaves of gnetum (Gnetales). International Journal of Molecular Sciences, 20(24), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escandón, M. , Valledor, L. , Lamelas, L. , Manuel‐Alvarez, J. , Cañal, M.J. & Meijón, M. (2022) Multiomics analyses reveal the central role of nucleolus and nucleoid machinery during heat stress acclimation in Pinus radiata. bioRxiv: 2022.07.08.499117. 10.1101/2022.07.08.499117 [DOI] [PubMed] [Google Scholar]
- Escandón, M. , Valledor, L. , Pascual, J. , Pinto, G. , Cañal, M.J. & Meijón, M. (2017) System‐wide analysis of short‐term response to high temperature in Pinus radiata. Journal of Experimental Botany, 68(13), 3629–3641. [DOI] [PubMed] [Google Scholar]
- Hanemian, M. , Vasseur, F. , Marchadier, E. , Gy, I. , Violle, C. & Loudet, O. (2020) Natural variation at FLM splicing has pleiotropic effects modulating ecological strategies in Arabidopsis thaliana. Nature Communications, 11(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. , Lu, X. , Wu, H. , Xie, Y. , Peng, Q. , Gu, L. et al. (2020) Phytophthora effectors modulate genome‐wide alternative splicing of host mRNAs to reprogram plant immunity. Molecular Plant, 13(10), 1470–1484. [DOI] [PubMed] [Google Scholar]
- Jacob, P. , Hirt, H. & Bendahmane, A. (2017) The heat‐shock protein/chaperone network and multiple stress resistance. Plant Biotechnology Journal, 15(4), 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, J. , Long, Y. , Zhang, H. , Li, Z. , Liu, Z. , Zhao, Y. et al. (2020) Post‐transcriptional splicing of nascent RNA contributes to widespread intron retention in plants. Nature Plants, 6(7), 780–788. [DOI] [PubMed] [Google Scholar]
- Jin, W. , Gernandt, D.S. , Wehenkel, C. , Xia, X. , Wei, X. & Wang, X.Q. (2021) Phylogenomic and ecological analyses reveal the spatiotemporal evolution of global pines. Proceedings of the National Academy of Sciences of the United States of America, 118(20), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H.Y. , Fraser, M. , Li, W. , McAnulla, C. et al. (2014) InterProScan 5: genome‐scale protein function classification. Bioinformatics, 30(9), 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers, I. & Lis, J.T. (2015) Getting up to speed with transcription elongation by RNA polymerase II. Nature Reviews Molecular Cell Biology, 16(3), 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junli, H. , Min, G. , Zhibing, L. , Baofang, F. , Kai, S. , Yan‐Hong, Z. et al. (2010) Functional analysis of the Arabidopsis PAL gene family in plant growth, development, and response to environmental stress. Plant Physiology, 153(4), 1526–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliebenstein, D.J. (2019) Questionomics: using big data to ask and answer big questions. Plant Cell, 31(7), 1404–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laloum, T. , Martín, G. & Duque, P. (2018) Alternative splicing control of abiotic stress responses. Trends in Plant Science, 23, 140–150. [DOI] [PubMed] [Google Scholar]
- Lamelas, L. , Valledor, L. , Escandón, M. , Pinto, G. , Cañal, M.J. & Meijón, M. (2020) Integrative analysis of the nuclear proteome in Pinus radiata reveals thermopriming coupled to epigenetic regulation. Journal of Experimental Botany, 71(6), 2040–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamelas, L. , Valledor, L. , López‐Hidalgo, C. , Cañal, M.J. & Meijón, M. (2022) Nucleus and chloroplast: a necessary understanding to overcome heat stress in Pinus radiata. Plant, Cell and Environment, 45(2), 446–458. [DOI] [PubMed] [Google Scholar]
- Langmead, B. & Salzberg, S.L. (2012) Fast gapped‐read alignment with bowtie 2. Nature Methods, 9(4), 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J. , Xu, Y. & Zhu, Z. (2020) Emerging plant Thermosensors: from RNA to protein. Trends in Plant Science, 25(12), 1187–1189. [DOI] [PubMed] [Google Scholar]
- Ling, Y. , Serrano, N. , Gao, G. , Atia, M. , Mokhtar, M. , Woo, Y.H. et al. (2018) Thermoprimming triggers splicing memory in Arabidopsis. Journal of Experimental Botany, 69(10), 2659–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. & Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15(12), 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco, R.F. , Allo, M. , Schor, I.E. , Kornblihtt, A.R. & Misteli, T. (2011) Epigenetics in alternative pre‐mRNA splicing. Cell, 144(1), 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach, D. , Costello, J.C. , Küffner, R. , Vega, N.M. , Prill, R.J. , Camacho, D.M. et al. (2012) Wisdom of crowds for robust gene network inference. Nature Methods, 9(8), 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, X. , Liang, Z. , Dai, X. , Zhang, Y. , Mahboub, S. , Ngu, D.W. et al. (2021) Predicting transcriptional responses to cold stress across plant species. Proceedings of the National Academy of Sciences of the United States of America, 118(10), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteuuis, G. , Wong, J.J.L. , Bailey, C.G. , Schmitz, U. & Rasko, J.E.J. (2019) The changing paradigm of intron retention: regulation, ramifications and recipes. Nucleic Acids Research, 47(22), 11497–11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer, D.C. , Larue, G.E. , Padgett, R.A. , Hershberger, C.E. & Roy, S.W. (2020) Comprehensive database and evolutionary dynamics of U12‐type introns. Nucleic Acids Research, 48(13), 7066–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachtigall, P.G. , Kashiwabara, A.Y. & Durham, A.M. (2020) CodAn : predictive models for precise identification of coding regions in eukaryotic transcripts. Briefings in Bioinformatics, 22(3), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- One Thousand Plant Transcriptomes Initiative . (2019) One thousand plant transcriptomes and the phylogenomics of green plants. Nature, 574, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol, Y. , Bai, J. , Bandla, C. , Hewapathirana, S. , García‐Seisdedos, D. , Kamatchinathan, S. et al. (2022) The PRIDE database resources in 2022: a hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Research, 50(D1), D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, A.S.N. , Marquez, Y. , Kalyna, M. & Barta, A. (2013) Complexity of the alternative splicing landscape in plants. The Plant Cell, 25(10), 3657–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, M.E. , Phipson, B. , Wu, D. , Hu, Y. , Law, C.W. , Shi, W. et al. (2015) Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Research, 43(7), e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosvall, M. & Bergstrom, C.T. (2008) Maps of random walks on complex networks reveal community structure. Proceedings of the National Academy of Sciences of the United States of America, 105(4), 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacomoto, G.A.T. , Kielbassa, J. , Chikhi, R. , Uricaru, R. , Antoniou, P. , Sagot, M.F. et al. (2012) KISSPLICE : de‐novo calling alternative splicing events from RNA‐seq data. BMC Bioinformatics, 13(Suppl 6), S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffthaler, B. , van Zalen, A. , Serrano, A. , Street, N. & Delhomme, N. (2021) Seidr:efficient calculation of robust ensemble gene networks. bioRxiv. 10.1101/250696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwacke, R. , Ponce‐soto, G.Y. , Krause, K. , Bolger, A.M. , Arsova, B. , Hallab, A. et al. (2019) MapMan4 : a refined protein classification and annotation framework applicable to multi‐omics data analysis. Molecular Plant, 12, 879–892. [DOI] [PubMed] [Google Scholar]
- Shamir, M. , Bar‐On, Y. , Phillips, R. & Milo, R. (2016) SnapShot: timescales in cell biology. Cell, 164(6), 1302–1302.e1. [DOI] [PubMed] [Google Scholar]
- Smith, C.C.R. , Tittes, S. , Mendieta, J.P. , Collier‐zans, E. , Rowe, H.C. , Rieseberg, L.H. et al. (2018) Genetics of alternative splicing evolution during sunflower domestication. Proceedings of the National Academy of Sciences of the United States of America, 115(26), 6768–6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer, C. , Li, F. , Buono, R. , Roschzttardtz, H. , Chung, T. , Zhang, M. et al. (2015) The endosomal protein CHARGED MULTIVESICULAR BODY PROTEIN1 regulates the autophagic turnover of plastids in Arabidopsis. Plant Cell, 27(2), 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, S. , Li, Y. , Chu, L. , Kuang, X. , Song, J. & Sun, C. (2020) Full‐length sequencing of ginkgo transcriptomes for an in‐depth understanding of flavonoid and terpenoid trilactone biosynthesis. Gene, 758, 144961. [DOI] [PubMed] [Google Scholar]
- Supek, F. , Bosnjak, M. , Skunca, N. & Smuc, T. (2011) REVIGO summarizes and visualizes Long lists of gene ontology terms. PLoS One, 6(7), e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valledor, L. , Carbó, M. , Lamelas, L. , Escandón, M. , Colina, F.J. , Cañal, M.J. et al. (2018) When the tree let us see the Forest: systems biology and natural variation studies in Forest species. In: Lüttge, U. , Cánovas, F.M. , Risueño, M.C. & Leuschner, C. (Eds.) Progress in botany, Vol. 81, Cham: Springer, pp. 353–375. [Google Scholar]
- Valledor, L. , Escandón, M. , Meijón, M. , Nukarinen, E. , Cañal, M.J. & Weckwerth, W. (2014) A universal protocol for the combined isolation of metabolites, DNA, long RNAs, small RNAs, and proteins from plants and microorganisms. Plant Journal, 79(1), 173–180. [DOI] [PubMed] [Google Scholar]
- Wan, T. , Liu, Z.M. , Li, L.F. , Leitch, A.R. , Leitch, I.L. , Lohaus, R. et al. (2018) A genome for gnetophytes and early evolution of seed plants. Nature Plants, 4, 82–89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. All genome data sources for the species used in intron comparative analysis.
Table S2. List of primers used for RT‐PCR.
Table S3. Sanger sequencing data validating RT‐PCR results.
Table S4. K‐means cluster data.
Table S5. Isoform sequences, expression, annotation, statistics, and potential event type data.
Table S6. Multiomics integration data.
Figure S1. Overview of the bioinformatics workflow.
Figure S2. General experimental design.
Figure S3. Annotation pipeline and non‐supervised behavior clusters.
Figure S4. Splicing variation effect proportions across non‐supervised behavior clusters.
Figure S5. Heat stress‐responsive intron retention hypothesis in gymnosperms.
Figure S6. Functional description of stress‐specific isoforms and isoform–protein relationships.
Figure S7. Functional characterization of proteins and all regulatory layers for each MOFA2 latent factor.
Data Availability Statement
All data, codes and gels/blots generated during the study are available in the GitHub repository (https://github.com/RocesV/AS_heat_Pra). The raw RNA‐seq data have been deposited in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA851373. The mass spectrometry proteomics data including RAW, msf, and pepXML files have been deposited in the ProteomeXchange Consortium database via the PRIDE (Perez‐Riverol et al., 2022) partner repository with the dataset identifier PXD032754. The metabolomics data have been deposited in the Metabolights repository (https://www.ebi.ac.uk/metabolights/) under dataset identifier MTBLS4567.