

Abstract
Genetic kidney disease comprises a diverse group of disorders. These can roughly be divided in the phenotype groups congenital anomalies of the kidney and urinary tract, ciliopathies, glomerulopathies, stone disorders, tubulointerstitial kidney disease, and tubulopathies. Many etiologies can lead to chronic kidney disease that can progress to end‐stage kidney disease. Despite each individual disease being rare, together these genetic disorders account for a large proportion of kidney disease cases. With the introduction of massively parallel sequencing, genetic testing has become more accessible, but a comprehensive analysis of the diagnostic yield is lacking. This review gives an overview of the diagnostic yield of genetic testing across and within the full range of kidney disease phenotypes through a systematic literature search that resulted in 115 included articles. Patient, test, and cohort characteristics that can influence the diagnostic yield are highlighted. Detection of copy number variations and their contribution to the diagnostic yield is described for all phenotype groups. Also, the impact of a genetic diagnosis for a patient and family members, which can be diagnostic, therapeutic, and prognostic, is shown through the included articles. This review will allow clinicians to estimate an a priori probability of finding a genetic cause for the kidney disease in their patients.
Keywords: CKD, CNV, diagnostic yield, genetic testing, MPS, nephrogenetics, review
1. INTRODUCTION
Genetic kidney diseases form a heterogeneous group of disorders, which, although individually mostly rare, together are frequent and important to establish (Groopman et al., 2019). Identifying a genetic cause in kidney disease patients is essential for patients and their family members. For the patient, a genetic diagnosis can lead to knowledge about etiology, personalized treatment and the possibility to be counseled about prognosis and family planning options. Family members can be counseled about presymptomatic testing and screening options. A genetic diagnosis can also be important for living related kidney donation. Different categories of genetic kidney disease can be distinguished (Hildebrandt, 2010). An important clinical presentation is chronic kidney disease (CKD) that can progress to end‐stage kidney disease (ESKD), which has a high disease burden (Coresh, 2017). Many etiologies can lead to this final common endpoint (Groopman et al., 2019). Overall, in genetic kidney disease, pathogenic variants in multiple genes can cause a single phenotype (genetic heterogeneity) and pathogenic variants in a specific gene can have multiple clinical presentations (pleiotropy) (Stokman et al., 2016).
The introduction of massively parallel sequencing (MPS), previously referred to as next‐generation sequencing, has made testing for monogenic kidney diseases (MKD) more accessible and therefore diagnosing MKD easier. This has led to many new insights, but a comprehensive analysis of the diagnostic yield across and within phenotypes is lacking. Estimating the likely diagnostic yield of genetic testing, in a specific patient, is essential to determine the chance of diagnosing monogenic disease when performing genetic testing. This estimated yield, together with factors like cost, availability, but also specific needs of the patient and/or parents with regard to for instance family planning or living related kidney donation, informs shared decision‐making with regard to genetic testing. Multiple studies report varying diagnostic yields of genetic testing for potentially genetic kidney disease phenotypes. The varying reported diagnostic yields might be a consequence of differences in (a) patient characteristics, (b) genetic test characteristics, and/or (c) cohort characteristics. Patient factors that can influence diagnostic yield are a positive family history, extrarenal features, syndromal presentation, and severe disease, including early onset of disease and the presence of ESKD (Knoers et al., 2022). Test characteristics that can influence the reported yield are the number of analyzed genes (extending to all genes [exome] or genome wide as maximum), whether copy number‐variations (CNVs) are assessed, whether additional tests are performed for difficult genomic regions (e.g., MUC1 and exon 1 of PKD1), but also whether trio‐analyses (child and both parents tested) and segregation are performed for additional variant classification. Also, the specifics of the cohort can be of importance. In a clinical cohort (of consecutive cases), the phenotypes are often less homogeneous, and the population might be more representative for patients that are seen in one's own clinic. In a research cohort varying inclusion and exclusion criteria are applied, patients are often selected cases, most suspect for a genetic diagnosis, from many centers and results of diagnostic yield are not always generalizable to a diagnostic setting. Another relevant variable might be cohort size.
In this review, we aim to determine the diagnostic yield of genetic tests within and across the full range of potentially genetic kidney disease phenotypes. Patient, test, and cohort characteristics influencing the various yields are highlighted. We provide a detailed and nuanced overview that can help weighing the meaning of the different publications, with potential useful information for clinical practice. We also highlight the clinical impact of genetic diagnoses based on the gathered studies and we show how genetic testing approaches have evolved over the years.
2. METHODS
2.1. Study selection
A systematic literature search was performed to answer the review question: “What is the diagnostic yield of genetic testing in any kind of kidney disease and what patient, cohort, and test characteristics impact the diagnostic yield?” This review was not registered in a review database. The PubMed database was searched on April 2, 2021 using the terms displayed in Table 1. Two reviewers independently selected articles based on predetermined inclusion criteria using Rayyan (Ouzzani, Hammady, Fedorowicz, & Elmagarmid, 2016). Disagreements were resolved by consensus or with the help of a third reviewer.
TABLE 1.
The sets of terms used to conduct the PubMed search | Sets (1) and (2) were combined into one search using Boolean operator “AND,” the same was done for Sets (1) and (3)
| Set | Terms in title/abstract |
|---|---|
| (1) | Kidney OR Renal OR Alport OR ADPKD OR ARPKD OR CAKUT OR ciliopathy OR nephronophthisis OR nephrol* OR nephrog* OR nephrogenetic OR glomerul* OR urol* OR urinary tract OR tubulopath* OR nephrotic |
| (2) | Diagnostic yield OR yield OR genetic test OR WES OR WGS OR whole exome sequencing OR whole‐exome sequencing OR whole exome OR whole genome OR whole‐exome OR whole‐genome OR gene panel OR gene panels OR multigene panel OR multigene panels OR whole genome sequencing OR MPS OR massive parallel sequencing OR massively parallel sequencing OR NGS OR next generation sequencing OR next‐generation sequencing OR exomic sequencing OR genomic sequencing OR targeted gene sequenc* OR targeted sequenc* OR targeted panel* |
| (3) | CNV OR copy number variant OR copy number variation OR copy number variance OR SNP array OR SNP‐array OR array‐CGH OR array CGH |
All these inclusion criteria were mandatory: (a) original article published in last 10 years, (b) human participants, (c) diagnostic yield of genetic testing reported or deducible, (d) patients had some sort of kidney disease, and (e) cohort of at least 30 unrelated patients. Articles focusing on renal cancer were excluded. Additional articles were identified and included by snowballing.
2.2. Data extraction
Data extraction was performed using predefined data fields. Studies were grouped in the following kidney disease phenotype groups: autosomal dominant tubulointerstitial kidney disease (ADTKD), congenital anomalies of the kidney and urinary tract (CAKUT), ciliopathies (divided in autosomal dominant polycystic kidney disease [ADPKD] and other/mixed ciliopathies), glomerulopathies (nephrotic syndrome [NS] and other/mixed), nephrolithiasis/urolithiasis, tubulopathies, ESKD, and mixed kidney disease phenotypes. For all studies, details were extracted on patient characteristics, cohort characteristics and on the genetic test that was performed. Details of the cohort that were extracted were the number of patients included, phenotype, type of cohort (clinical cohort or research cohort). A cohort was considered a clinical cohort if the cohort was derived from a clearly defined clinical setting (e.g., all consecutive patients that received a kidney transplant) and/or reporting on genetic testing results from a diagnostic setting (e.g., all patients referred for gene panel testing). Patients' characteristics that were extracted included: the percentage of familial cases, percentage of consanguinity, percentage of cases presenting with extrarenal features, age of disease onset and percentage of people that had ESKD. For the genetic test, extracted details included: number of genes screened and if single nucleotide variant (SNV) and/or CNV analysis was performed. CNV analysis was considered to be performed, also if only one gene was assessed. CNVs were defined here as deletions or duplications that are too large to be picked up by traditional sequencing. Type of genetic test (e.g., MPS‐based multigene panel, single nucleotide polymorphism [SNP] array, whole exome sequencing [WES], whole genome sequencing [WGS], etc.), including type of CNV analysis were extracted. The diagnostic yield was either adopted from the paper or calculated from the data. Likely pathogenic variants and pathogenic variants were included in diagnostic yield, unless details on variant classification were not specified by the authors. In that case, the yield reported by the authors was extracted. If authors used an aberrant term to describe variants (i.e., other term than [likely] pathogenic, e.g., “probably disease causing” or “potentially pathogenic”) this term and the associated yield was extracted. Criteria used to assess variants in each article were also noted. Candidate genes were not included in the diagnostic yield, but highlighted under “noteworthy.” The diagnostic yield was calculated and noted per phenotype in cohorts with mixed phenotypes. When details were available with regard to the cohort from which the tested population was derived, a diagnostic yield extrapolated to the larger cohort was also calculated (e.g., when a specific subgroup of transplant patients was genetically tested, the diagnostic yield was extrapolated to the entire cohort of transplanted patients). If reported, percentage of cases with a variant of unknown significance was extracted. We also calculated the yield based solely on CNVs and determined what percentage of the reported diagnostic yield can be explained by CNVs. When the study reported a patient characteristic that positively impacted the diagnostic yield, this was noted. Any additional relevant details were also extracted. Finally, we established the number of genes that was responsible for the top 50% of the diagnostic yield. The genes responsible were noted, unless genes were responsible for only one positive case and/or multiple genes made up for the final percentages.
2.3. Data visualization
Data were summarized by combining the cohort, patient, and test characteristics and results on diagnostic yield per phenotype group in a summary table. More in detail overviews of all studies were summarized in separate tables per disease group. We first looked at patient characteristics positively impacting diagnostic yield within a single study and described this in the summary table. Next, we assessed the impact of patient, cohort, and test characteristics between the different studies. To visualize the influence of these characteristics scatter plots, box plots, and pie charts were made using SPSS (version 26, IBM, New York, NY) to look at the relation between two quantitative variables within a single study, across all studies. To avoid overinterpretation of the data derived from the highly variable studies, we decided to only portray visual representation of the data and to not perform statistical tests.
3. RESULTS
The PubMed search yielded 5,361 papers. After screening the title, abstract and full‐text, 98 papers remained, that met the inclusion criteria. Seventeen articles were additionally identified and included through snowballing.
Table 2 gives an overview of reported yields per disease group. Per disease group, 4 to 25 articles were included with varying patient, test, and cohort characteristics per study. The diagnostic yields differed widely within each disease group and between the different disease groups (Table 2; Figure 1a). We find the lowest diagnostic yield in the CAKUT disease group and the highest diagnostic yield in the ciliopathy group, and within this group an even higher yield for the ADPKD group. However, in the CAKUT disease group, we did find the highest diagnostic yield based solely on CNVs (Figure 1b). When we look at the yield extrapolated to the larger cohort, we often find a lower yield than initially reported, as we would expect (Table 2). These values represent a minimum diagnostic yield for the originating population.
TABLE. 2.
Summary of included studies. Patient, cohort, and test characteristics are summarized per phenotype category. When data were missing for one of these characteristics, this was not included in the table. When >50% of studies had a missing value, this was reported in the table, next to a summary of the values that were present. Diagnostic yield is presented in tested cohort and extrapolated to larger (clinical) cohort; in this N/A means not applicable because already entire clinical cohort is reported. Also, the percentage of diagnostic yield that is explained by CNVs is summarized. References are listed in Supplementary Table 3.
| Phenotype | Number of studies* | Number of population (range) | Pediatric/adult (time of study) | Family history (% range) | Consang. (% range) | Extrarenal features (% range) | Mean/median age of onset | Adult‐onset (% range) | ESKD (% range) | Number of studies per variant type | Number of genes analyzed | Diagnostic yield (% range) | Extrapolated larger cohort (% range) | Percentage explained by CNV (% range) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADKTD | 4 | 56–585 | Adult or NR (in 2/4) | 54.4–72.4% | NR | NR | 45 y, rest NR | NR | 42.9–67.4% | 2 ‐ SNVs 2 ‐ both | 1–5 | 31.0–62.5% | 0.9–30.3%, NR in 1 | 0–1.3% |
| Positively impacting diagnostic yield within one study: Family history; ESKD | ||||||||||||||
| CAKUT | 25 | 30–650 | Mostly fetuses | 0.6–50% | 0–100%, NR in 18/25 | 0–100% | N/A | 0% | 15.6–42.6, NR in 21/25 | 7 ‐ SNVs 11 ‐ CNVs 7 ‐ both | 2–404 or genome/exome wide | 1.3–72.2% | 2.2–27.3%, N/A in 6/12; NR in 3/12 | 0–100%, 100% in 11/25 |
| Positively impacting diagnostic yield within one study: Family history; consanguinity; extrarenal features; CKD5; syndromic and specific phenotypes: MCDK, ciliopathies, multiple urinary tract infections, bilateral anomalies | ||||||||||||||
| Ciliopathies | ||||||||||||||
| ADPKD | 13 | 36–220 | Both | 0–88.4% | NR | 32.6–80.5%, NR in 9/13 | 25–56.7 y | 0–100%, NR in 9/13 | 18.9–57.5% | 3 ‐ SNVs 10 ‐ both | 2–69 or exome wide | 47.9–93.1% | 65.3–70.6%, N/A in 2/6; NR in 1/6 | 1.6–7.1% |
| Positively impacting diagnostic yield within one study: ESKD; disease severity; polycystic liver disease; typical ADPKD | ||||||||||||||
| Other/mixed | 15 | 12–384 | Both | 6.5–77.5% | 0–86.4% | 30–100% | Perinatal ‐ 36 y | 0% in 7/15; NR in 8/15 | 13.6–67.4% | 6 ‐ SNVs 9 ‐ both | 1–4,813 or exome wide | 18.5–97.2% | 22.4–66.7%, NR in 1/2 | 3.0–100% |
| Positively impacting diagnostic yield within one study: Family history; consanguinity; age at diagnosis; phenotype: ARPKD, MKS | ||||||||||||||
| Glomerulopathies | ||||||||||||||
| Nephrotic syndrome | 15 | 36–1,783 | Both, most pediatric | 0–31.0% | 0–75.5% | 0–32.4% | 3–30.3 y | 0–100%, often 0% (in 8/15) | 0–49.6% | 12 ‐ SNVs 3 ‐ both | 3–446 | 11.1–51.0% | 2.0–16.7%, N/A in 1/5; NR in 2/5 | 0–2.7%, NP in 11/15 |
| Positively impacting diagnostic yield within one study: Family history; consanguinity; age of onset; ESKD; lack of response to immunosuppressives; lower posttransplant recurrence | ||||||||||||||
| Other/ mixed | 11 | 40–441 | Both, most adult | 22.3–80.2%, NR in 7/11 | 0–15.5% | 27.8–46.5%, NR in 9/11 | 11–80.2 y | 0–86.4%, NR in 6/11 | 23.8–49.0%, NR in 8/11 | 12 ‐ SNVs 5 ‐ both | 1–109 | 14.1–90.0% | N/A in 3/5; NR in 2/5 | 2.7–21.8%, NP in 5/11 |
| Positively impacting diagnostic yield within one study: Family history; consanguinity; age of onset; number of AS criteria; phenotype: C3G, SRNS | ||||||||||||||
| Nephrolithiasis/urolithiasis | 4 | 48–235 | Both | 47.6–58.3% | 1.9–53.3% | NR | 2–8 y | 0% or NR (in 2/4) | NR | 4 ‐ SNVs | 30–117 | 7.2–37.5% | 6.6–19.1%, N/A in 1/3 | NP |
| Positively impacting diagnostic yield within one study: Family history; consanguinity; age of onset | ||||||||||||||
| Tubulopathies | 5 | 70–1,033 | Both | NR | 4.5%, rest NR | 77.5%, rest NR | 2.2–47.1 y | 0–100% | NR | 1 ‐ SNVs 4 ‐ both | 1–168 | 26.0–71.9% | N/A in 2/2 | 0–63.5% |
| Positively impacting diagnostic yield within one study: Extrarenal features; age of onset; absence of hypomagnesemia | ||||||||||||||
| ESKD | 5 | 50–5,606 | Mostly adult | 22.1%, rest NR (4/5) | 8.7%, rest NR (4/5) | 52.9% rest NR (4/5) | 23 y, rest NR | 30–43.4 y ESKD onset | 0–100% | 100% | 1 ‐ SNVs 4 ‐ both | 20–600 | 0.5–50.9% | 12.5–24.6% | 0–100% |
| Positively impacting diagnostic yield within one study: Family history; consanguinity; extrarenal features; age of onset ESKD | ||||||||||||||
| Mixed kidney disease phenotypes | 18 | 62–3,315 | Both | 9.8–92.0%, NR in 6/18 | 0.3–5.7%, NR in 14/18 | 0–35.9%, NR in 12/18 | 5–42 y, NR in 15/18 | 0–91.6%, NR in 10/18 | 6.3–69.3%, NR in 11/18 | 7 ‐ SNVs 10 ‐ both 1 ‐ NR | 1–4,000, some exome wide | 4.0–72.8% | 6.4–48.8%, N/A in 6/14 | 2.1–20.0% |
| Positively impacting diagnostic yield within one study: Family history; extrarenal features; age of onset; phenotypes: Congenital/cystic disease, unknown origin, ADTKD, TIKD, cystinosis, hematuria, glomerular disease | ||||||||||||||
Abbreviations: aCGH, array comparative genomic hybridization; ADPKD, autosomal dominant polycystic kidney disease; ADTKD, autosomal dominant tubulointerstitial kidney disease; ARPKD, autosomal recessive polycystic kidney disease; AS, Alport syndrome; C3GN, C3 glomerulopathy; CAKUT, congenital anomalies of kidney and urinary tract; CKD, chronic kidney disease; CNV, copy number variations; consang., consanguinity; ESKD, end‐stage kidney disease; MCDK, multicystic dysplastic kidney; MKS, Meckel syndrome; n, number; N/A, not applicable; NP, not performed; NR, not reported; SNV, single nucleotide variant; SRNS, steroid‐resistant nephrotic syndrome; TIKD, tubulointerstitial kidney disease.
References studies: ADTKD: [Ayasreh et al., 2018; Gast et al., 2018; Bleyer et al., 2020; Olinger et al., 2020]; CAKUT: [Sanna‐Cherchi et al., 2012; Hwang et al., 2014; Kohl et al., 2014; Caruana et al., 2015; Nicolaou et al., 2016; Xi et al., 2016; Faure et al., 2016; Fu et al., 2016; Vivante et al., 2017; Bekheirnia et al., 2017; Heidet et al., 2017; Lei et al., 2017; Rasmussen et al., 2018; Unzaki et al., 2018; Boissel et al., 2018; Van Der Ven et al., 2018; Li et al., 2019; Verbitsky et al., 2019; Lin et al., 2019; Cai et al., 2020a; Ahn et al., 2020; Lei et al., 2020; Zhou et al., 2020; Cai et al., 2020b; Zhou et al., 2021]; ciliopathies ADPKD: [Rossetti et al., 2012; Hwang et al., 2016; Jin et al., 2016; Kinoshita et al., 2016; Xu et al., 2018; Fujimaru et al., 2018; Zhang et al., 2019; Mochizuki et al., 2019; Mantovani et al., 2020; Schönauer et al., 2020; Mallawaarachchi et al., 2021; Nielsen et al., 2021; Durkie et al., 2021]; ciliopathies other/mixed: [Bachmann‐Gagescu et al., 2015; Knopp et al., 2015; Braun et al., 2016; Schueler et al., 2016; Al‐Hamed et al., 2016; Lindstrand et al., 2016; Vilboux et al., 2017; Stokman et al., 2018; Szabó et al., 2018; Al Alawi et al., 2019; Liang et al., 2020; Obeidova et al., 2020; Yue et al., 2020; Al Alawi et al., 2020; Benson et al., 2021]; glomerulopathies nephrotic syndrome: [McCarthy et al., 2013; Al‐Hamed et al., 2013; Kari et al., 2013; Giglio et al., 2015; Trautmann et al., 2015; Sadowski et al., 2015; Bierzynska et al., 2017; Wang et al., 2017a, 2017b; Warejko et al., 2018; Tan et al., 2018; Bezdíčka et al., 2018; Gribouval et al., 2018; Landini et al., 2020; Nagano et al., 2020]; glomerulopathies other/mixed: [Fallerini et al., 2014; Morinière et al., 2014; Nabais Sá et al., 2015a, 2015b; Gast et al., 2016; Bu et al., 2016; Sen et al., 2017; Yao et al., 2019; Schapiro et al., 2019; Yamamura et al., 2019; Ozdemir et al., 2020]; nephrolithiasis/urolithiasis: [Daga et al., 2018; Amar et al., 2019; Ziyadov et al., 2021; Zhao et al., 2021]; tubulopathies: [Palazzo et al., 2017; Ashton et al., 2018; Adalat et al., 2019; Hureaux et al., 2019; Mori et al., 2021]; ESKD: [Snoek et al., 2018; Mann et al., 2019; Ottlewski et al., 2019; Schrezenmeier et al., 2021; Snoek et al., 2022]; mixed kidney disease phenotypes: [Alkanderi et al., 2017; Mallett et al., 2017; Lata et al., 2018; Bullich et al., 2018; Groopman et al., 2019; Connaughton et al., 2019; Rao et al., 2019; Thomas et al., 2020; Benson et al., 2020; Riedhammer et al., 2020; Murray et al., 2020; Jayasinghe et al., 2021; Mansilla et al., 2021; Domingo‐Gallego et al., 2022; Oh et al., 2021; Tanudisastro et al., 2021; Amlie‐Wolf et al., 2021; Vaisitti et al., 2021].
FIGURE 1.
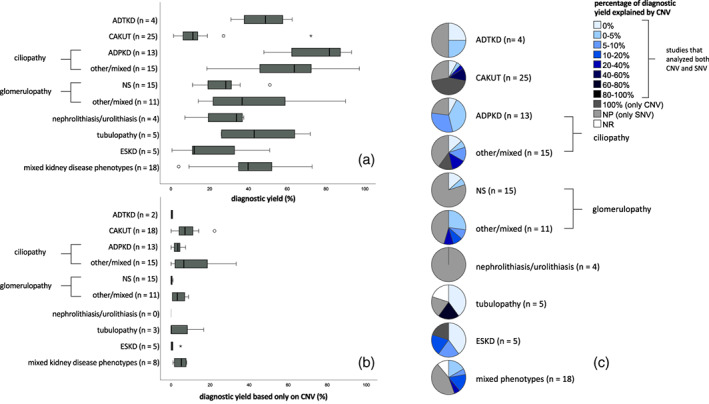
Diagnostic yield across disease categories. (a) Diagnostic yield per disease category. Subdivision of the specific phenotypes within the “mixed kidney disease phenotypes” group is not added up to the corresponding separate disease groups since patient/test/cohort characteristics are not available per phenotype within the studies in this group. Between brackets the number of included studies. (b) Diagnostic yield based only on copy number variation (CNV) detection. Between brackets the number of studies that performed CNV analysis. (c) Percentage of overall diagnostic yield (a) explained by CNVs (b). Between brackets the total number of studies. This is a ratio between subpanels (b) and (a), showing how much CNV testing contributed to the diagnostic yield per study.
In Supplementary Table 1, the disease groups are presented in more detail. Patient, cohort, and test characteristics, including noteworthy details, are described per individual study. When available diagnostic yield per phenotype (especially in Supplementary Table 1h covering mixed kidney disease phenotypes) and diagnostic yield extrapolated to a larger originating population are included.
Five studies reported on an ESKD population that was waitlisted for transplant surgery or had received a kidney transplant (Supplementary Table 1g). The diagnostic yield in these studies ranged from 0.5% (only full gene deletions in 20 genes assessed) to 50.9% in the genetically tested patients at time of study. When extrapolating this to the individual extrapolated cohorts of kidney transplant (waitlisted) patients the yield ranged from 12.5 to 24.6%.
Ten studies from varying disease groups reported on kidney disease of unknown origin (patients in whom the clinical phenotype does not point toward a specific diagnosis). The phenotype groups comprise ADTKD (1), ESKD (3), and mixed kidney disease phenotypes (6). The diagnostic yield in a population of patients with familial nephropathy of unknown cause, investigated for ADTKD, was 29.5%. In studies investigating ESKD of unsolved etiology, the diagnostic yield ranged from 11.6 to 44.4%. In the six studies with mixed kidney disease phenotypes the yield in patients with nephropathy of unknown origin ranged from 17.1 to 56.3% across five studies and one study reported a yield of 0% (only five patients had nephropathy of unknown origin in a cohort of 204 patients). Table 3 gives an overview of the clinical impact of genetic diagnoses highlighted by the different studies, divided over diagnostic impact, therapeutic impact, and prognostic impact.
TABLE 3.
Impact of genetic diagnosis highlighted by reviewed articles
| Diagnostic impact | Total cohort size | Phenotype | Correction of clinical diagnosis | Diagnosis established in patients with unknown origin of disease | Obviating need for diagnostic renal biopsy |
|---|---|---|---|---|---|
| Groopman et al. (2019) | n = 3,315 | CKD | 11% | 17% | |
| Connaughton et al. (2019) | n = 114 | CKD | 22% of families | 47% | |
| Mansilla et al. (2021) | n = 127 | Various phenotypes | 10% | ||
| Domingo‐Gallego et al. (2021) | n = 460 | CKD <30 y | 7% | ||
| Al Alawi et al. (2019) | n = 53 | Ciliopathy | 6% | ||
| Obeidova et al. (2020) | n = 31 | Ciliopathy | 16% | ||
| Snoek et al. (2022) | n = 110 | Transplant cohort |
6% of kidney transplant cohort 11% cases with a genetic diagnosis |
25% | |
| Jayasinghe et al. (2021) | n = 204 | Renal genetics clinic | 39% | 13% | |
| Gast et al. (2018) | n = 113 | CKD3‐5/ADTKD | 29.5% of familial nephropathy patients | ||
| Rao et al. (2019) | n = 1,001 | Suspected MKD | 26% | ||
| Lata et al. (2018) | n = 92 | CKD | 56% | ||
| Vaisitti et al. (2021) | n = 138 | Suspected MKD | 53% | ||
| Schrezenmeier et al. (2021) | n = 126 | KTx waitlisted | 12% of undetermined ESKD <40 patients | ||
| Mann et al. (2019) | n = 104 | KTx <25 | 44% |
| Therapeutic impact | Total cohort size | Phenotype | Inform therapy | Referral for extrarenal features |
|---|---|---|---|---|
| Groopman et al. (2019) | n = 3,315 | CKD | 50% | 53% |
| Jayasinghe et al. (2021) | n = 204 | Suspected MKD | 59% including obviating the need for diagnostic renal biopsy in 13%, changing surveillance in 44%, and changing the treatment plan in 20%. | |
| Daga et al. (2018) | n = 51 | Nephrolithiasis | 60% may have specific implications for stone management/prevention | |
| Murray et al. (2020) | n = 47 | Undergone renal biopsy | 26% treatment would have been altered based on established diagnosis | |
| Giglio et al. (2015) | n = 69 | Children with SRNS | 0% with vs. 58% without alterations responded to immunosuppressive drugs | |
| Wang et al. (2017) | n = 60 | Children with SRNS | 56% with vs. 44% without alterations did not respond to immunosuppressive therapy | |
| Kari et al. (2013) | n = 44 | Children with SRNS | 0% with identified genetic cause responded immunosuppressive drugs |
| Prognostic impact | Total cohort size | Phenotype | Posttransplant disease recurrence in patients with genetic diagnosis vs. patient without genetic diagnosis | ||
|---|---|---|---|---|---|
| Trautmann et al. (2015) | n = 1,174 | SRNS | 4.5 vs. 28.5% | ||
| Bierzynska et al. (2017) | n = 187 | SRNS | 0 vs. 52% | ||
| Yao et al. (2019) | n = 179 | FSGS | 0 vs. 13% | ||
| Gast et al. (2016) | n = 76 | FSGS/SRNS | 0 vs. 17% | ||
| Landini et al. (2020) | n = 111 | SRNS | 0 vs. 43% | ||
Abbreviations: CKD, chronic kidney disease; ESKD, end‐stage kidney disease; FSGS, focal segmental glomerulosclerosis; KTx, kidney transplantation; MKD, monogenic kidney disease; n, number; SRNS, steroid‐resistant nephrotic syndrome.
3.1. Characteristics influencing diagnostic yield
3.1.1. Patient characteristics
Looking at patient characteristics influencing diagnostic yield within a certain study we find that multiple studies reported a positive impact on the diagnostic yield based on positive family history (n = 18 studies), consanguinity (n = 12), extrarenal features (n = 16), early onset of disease (n = 14), and ESKD (n = 8) as described in Table 2 and Supplementary Table 1. Some additional features are mentioned as well, such as specific phenotypes, lower posttransplant recurrence and lack of response to immunosuppressives for the steroid resistant NS (SRNS) phenotype (Table 2).
When we assess these same variables (family history, extrarenal features, early onset of disease, and ESKD) between studies, we clearly see that in cohorts with a high percentage of familial cases and in cohorts with a high percentage of extrarenal cases, the diagnostic yield is higher (Figure 2a,b). However, we do not see this same clear pattern in cohorts with a high percentage of consanguinity or high percentage of ESKD (Figure 2c,d). When we look at cohorts with a high percentage of adult‐onset cases, we find both low and high percentages of diagnostic yield (Figure 2e). In the cohorts that have no (0%) adult‐onset cases we see the same, with a large proportion of the low yield studies being explained by the CAKUT group. The importance of CNV analysis is highlighted in Figure 2f where a high diagnostic yield is noticed in childhood onset disease as compared to adult‐onset disease.
FIGURE 2.
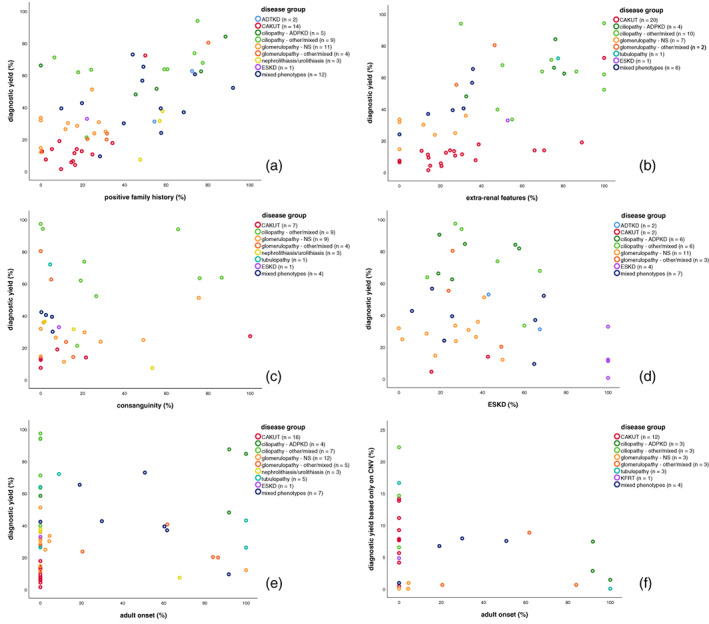
Scatterplots showing relationship between diagnostic yield and patient characteristics. Legend describes number of studies for which data on this specific patient characteristic was available. Colors indicate disease group from which study was derived. Each dot represents for one study what the percentage of a specific feature was in that cohort and what diagnostic yield was obtained from that same study. (a) Percentage of cases with a positive family history in relation to diagnostic yield. (b) Percentage of cases with extrarenal features in relation to diagnostic yield. (c) Percentage of cases from a consanguineous family in relation to diagnostic yield. (d) Percentage of cases with end‐stage kidney disease (ESKD) in relation to diagnostic yield. (e) Percentage of cases with adult onset of disease in relation to diagnostic yield. (f) Percentage of cases with adult onset of disease in relation to diagnostic yield only based on copy number variations (CNVs).
3.1.2. Cohort characteristics
In Figure 3a, we show the number of tested patients in a cohort in relation to the diagnostic yield. We find that the diagnostic yield decreases in larger cohorts. When we compare the boxplots representing the diagnostic yield in clinical cohorts versus research cohorts no clear difference seems to be present (Figure 3b). We used the diagnostic yield in the tested clinical cohort and not the extrapolated cohort, since data were not always available for the extrapolated cohort.
FIGURE 3.
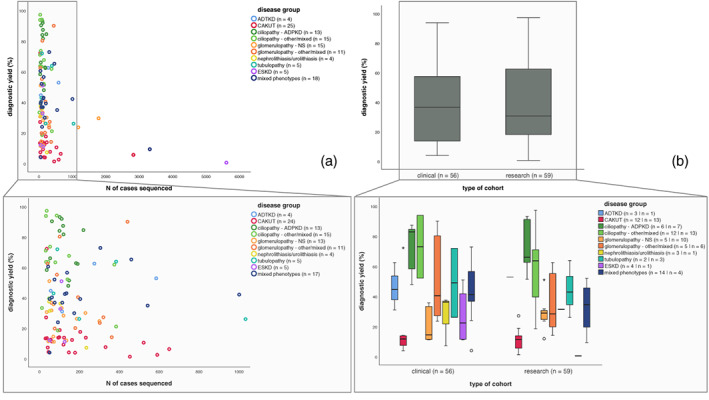
Scatterplots and box plots showing relation between diagnostic yield and cohort characteristics. (a) Scatterplots showing relationship between diagnostic yield and number of cases sequenced within a specific study. Legend describes number of studies for which data on this cohort characteristic was available. Colors indicate disease group from which studies were derived. Each dot represents for one study what the number of sequenced cases was in that cohort and what diagnostic yield was obtained from that same study. (b) Boxplots representing the diagnostic yield in clinical cohorts versus research cohorts. Lower half shows clinical versus research cohorts across the different disease groups. Legend describes number of studies for which data on this cohort characteristic was available.
3.1.3. Test characteristics
Figure 4 represents the test characteristics between the studies. It appears that the higher the number of analyzed genes the lower the diagnostic yield (Figure 4a). When relating sequencing approaches to diagnostic yield, we found that single gene testing had a lower diagnostic yield than when several genes (2–10) were tested, and we found the same with small gene panels (<100) versus large gene panels (Supplementary Figure 1). However, the categories WES/WGS and SNP‐array had a lower yield than the previously mentioned categories. 47/115 studies assessed SNVs, 12/115 assessed only CNVs and 55/115 assessed both (Figure 4b). As shown in Figure 4c, we found that the diagnostic yield was highest in cohorts where both SNVs and CNVs were assessed, and lowest in cohorts that only assessed CNVs. We saw that over the years there has been a shift from focusing either on SNVs or CNVs to including both in genetic testing (Supplementary Figure 2). There has also been a shift from enrichment‐based panels to an exome‐based approach. Noticeably in CNV testing, there is a shift toward the use of MPS‐based CNV‐calling.
FIGURE 4.
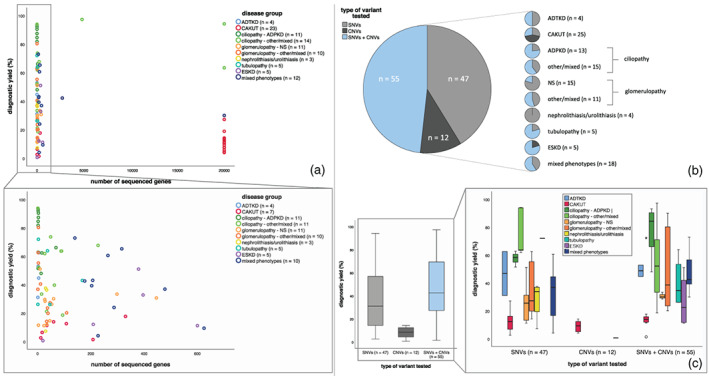
Scatterplots and box plots showing relation between diagnostic yield and test characteristics. (a) Scatterplots showing relationship between diagnostic yield and number of genes sequenced within a specific study. Legend describes number of studies for which data on this specific test characteristic was available. Not all studies were included since in some studies the number of genes sequenced differed within the study. Colors indicate disease group from which study was derived. Each dot represents for one study what the number of sequenced cases was in that cohort and what diagnostic yield was obtained from that same study. (b) Pie charts visualizing number of studies that performed either single nucleotide variant (SNV) or copy number variation (CNV) testing or both. (c) Boxplots representing the diagnostic yield in relation to type of variants that were tested. In the right panel, this is split in the different disease groups.
3.1.4. CNV analysis
CNVs were most often assessed in the disease groups CAKUT, ciliopathy, tubulopathies, ESKD, and mixed kidney disease phenotypes. In nephrolithiasis/urolithiasis, CNVs were never assessed. The diagnostic yield based solely on CNV testing was highest in CAKUT patients, followed by the disease group with mixed phenotypes and ciliopathies. Contribution of CNVs to the diagnostic yield was also highest in the CAKUT and ciliopathy group. When studies assessed both CNVs and SNVs, we found the highest percentage of CNVs in CAKUT and tubulopathies (Figure 1c). HNF1B deletions causing Gitelman(−like) tubulopathy explained the high percentage of CNVs in the latter one. Various CNV testing approaches were used, with the highest yield obtained by single gene testing and array‐based CNV testing (Supplementary Figure 3).
3.2. Core genes
In all the included studies, a limited number of genes were responsible for a large percentage of explained cases as shown in Figure 5. We found that a maximum of 10 genes was responsible for at least 50% of solved cases with often only one to four genes responsible. In phenotype groups ADTKD and APDKD, only one gene (UMOD or MUC1 and PKD1, respectively) was responsible for 50% of reported yield in a single study. But also in the phenotype groups with diverse kidney disease phenotypes, we found that variants in a maximum of 10 genes attributed to 50% of the diagnostic yield. The genes that are responsible for the top yield of ~50% are displayed in panel (b). Details per study can be found in Supplementary Table 3.
FIGURE 5.
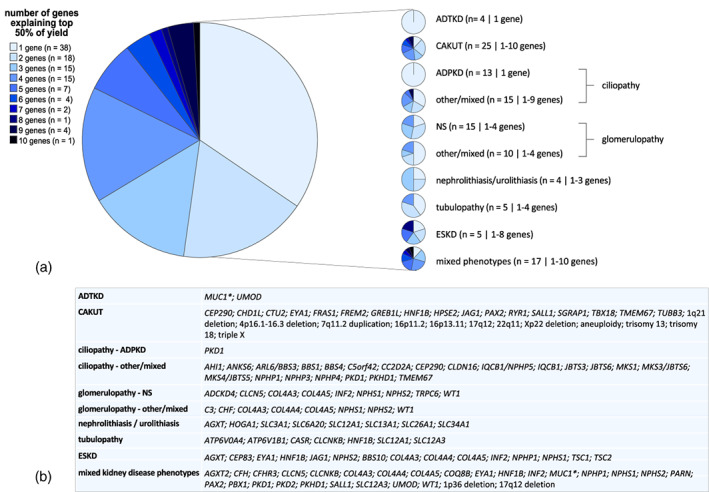
Top 50% of diagnostic yield explained by limited number of genes and/or copy number variations (CNVs). (a) Pie charts visualizing number of genes responsible for top 50% of yield. The legend on the left describes for each category in the pie chart, the number of studies identified with this number of genes. In between brackets on the right are the number of studies that reported on top 50% causal genes and separated by “|” the number of genes responsible for the top 50% for that specific phenotype. (b) The genes that made up this ~50% in each study per phenotype group are displayed here, unless genes were responsible for only one positive case and/or multiple genes made up for the final percentages. * Variants in the large variable number tandem repeats region of MUC1 are usually missed by massively parallel sequencing.
4. DISCUSSION
Determining true diagnostic yield from the papers studied is difficult because of the variability in patient, cohort, and test characteristics. The diagnostic yield for the different phenotype groups should therefore be thought of in terms of ranges, and recommendations should not be based on single studies. Given the enormous variation in key‐parameters, we wanted to avoid overinterpretation and therefore did not perform statistical analyses. However, the comprehensive overview presented, will help weigh the possible relevant factors with potential useful information for clinical practice. This overview allows clinicians to judge which studies are most relevant for their specific patients/patient groups and estimate an a priori probability of finding a genetic cause for their patients.
Not surprisingly, we found that diagnostic yield was higher based on expected patient characteristics (e.g., family history, consanguinity, extrarenal features, and young age of onset) within studies. When we assessed these same characteristics plotted against the diagnostic yield between the different studies (instead of within one study), this pattern was not seen for consanguinity and young age of onset. This might be explained by a combination of other characteristics having a larger impact on the diagnostic yield. Also, within the adult‐onset group there is a large variation in disease severity (e.g., ESKD at age 20 has a higher diagnostic yield than ESKD at age 70) and the likelihood of finding a monogenic cause (i.e., in a typical ADPKD cohort a high diagnostic yield is expected (Figure 2e)). We did find an indication that young age of onset is related to a higher diagnostic yield based only on CNVs (Figure 2f). This is likely explained by genome‐wide CNV analysis being performed more often in this group (Supplementary Table 3).
This review includes an overview of CNV yield per phenotype. CNVs that are not picked up by regular sequencing can be assessed using a separate test (e.g., SNP‐array) or by a CNV calling tool based on sequencing data (Knoers et al., 2022). The different types of tests that were used in the included studies varied greatly, as did the number of genes that were analyzed (i.e., covering one gene, the requested multigene panel or exome/genome wide and the impact this has on the diagnostic yield [Supplementary Table 3, Supplementary Figure 3]). We found the highest contribution of CNVs to diagnostic yield in CAKUT, ciliopathies, and tubulopathies. However, across all phenotype groups where CNV testing was performed, CNVs did contribute to the diagnostic yield and CNV analysis should be considered when genetic testing is performed. The high contribution of CNVs to diagnostic yield in CAKUT patients confirms previous reports (Knoers et al., 2022). CNVs were extensively investigated in CAKUT patients as SNV yield is relatively low in this group. It is yet to be established for some other phenotypes whether the diagnostic yield based on CNVs is underestimated hitherto as we found that in many phenotype groups CNVs were not investigated (Figure 1c).
Surprisingly, a higher number of genes tested did not always correlate with higher yield. In theory, this would always be the case in comparable cohorts. However, the cohorts we studied vary distinctly. On the one hand we describe cohorts with a highly likely monogenic cause (such as ADPKD suspected patients) requiring only a small number of tested genes to result in a high diagnostic yield. On the other hand, we find a lower diagnostic yield and an increase in number of tested genes in less highly suspected cohorts. Cohort size did not appear to correlate with number of tested genes (data not shown). Another explanation for this finding is an increase in number of tested genes in patients where testing of common known disease genes did not result in a genetic diagnosis. Since it is possible that a proportion of these unsolved cases have either a genetic diagnosis in a not yet discovered gene, or a non‐monogenic cause explaining their disease, a lower yield in this group can be hypothesized. Also, in some cohorts patients with known mutations were excluded, but the total number of these patients with a mutation were not reported (Bekheirnia et al., 2017; Braun et al., 2016; Faure et al., 2016; Heidet et al., 2017; Kohl et al., 2014; Schueler et al., 2016; Vivante et al., 2017; Ziyadov et al., 2021). Finally, in some studies, genetic testing in specific known genes was not performed (e.g., known CAKUT genes (Caruana et al., 2015; Sanna‐Cherchi et al., 2012)). For translation of the reported diagnostic yield to clinical practice, it is important to know these details. It was beyond the scope of this review to analyze whether for all cohorts the panel composition included all known causal genes, including appropriate phenocopy genes, for each phenotype at the time that that specific study was performed.
We found that the diagnostic yield decreased in larger cohorts. This might be explained by smaller cohorts being more clearly defined and therefore having a higher suspicion of a monogenic cause. In addition to selection bias within a study, publication bias might also explain this finding. The larger cohorts probably give a more reliable estimation of the true diagnostic yield in a relatively unselected kidney disease population. We would like to point out that a published cohort with a high diagnostic yield is not necessarily the “better” cohort to use for clinical decision‐making. When applying tightly restricted criteria genetic diagnoses are likely missed. When interpreting published cohorts for clinical practice, one should also take into account that there are many local differences in clinical phenotyping and diagnosing, especially when seen form an international perspective. For example, a patient with unknown CKD in one center might differ distinctly from a patient with unknown CKD in another center.
We were curious to find out if we would find a higher yield in research cohorts or diagnostic cohorts, although the distinction between these two was not always clearly described. One could assume that selection bias is higher in research cohorts resulting therefore in a higher yield. However, research cohorts are often gathered over the course of multiple years and phenotyping is not always on point. When interpreting publications on research cohorts, one should take into account that access to clinical information and variant interpretation, including opportunities for variant segregation, might differ from a clinical diagnostic setting and are likely less individualized. Clinical cohorts might be more reliable for obtaining a diagnostic yield that is generalizable to a diagnostic setting. However, clinicians might also miss cases that should be offered genetic testing. Furthermore, information on the originating population from which a cohort was derived from can be lacking in clinical cohorts, especially when the clinical cohort describes the diagnostic yield in a genetic center. We found that the diagnostic yield between the two types of cohorts did not differ distinctly. For studies from either cohort type, it remains unknown whether there is an overrepresentation or an underrepresentation of genetic cases. Performing a study that actually tests all kidney disease patients in a clinical setting would answer this question.
To derive a minimum diagnostic yield for a specific diagnostic setting, we extrapolated the diagnostic yield to the larger cohort from which the tested (clinical) cohort was derived from. An interesting example of this is presented by the studies of Snoek et al. and Schrezenmeier et al., that both report a diagnostic yield of 21% in a kidney transplant (waitlisted) cohort (Schrezenmeier et al., 2021; Snoek et al., 2022).
5. IMPACT OF GENETIC DIAGNOSIS
A genetic diagnosis can have a diagnostic, prognostic and treatment impact. The studies we selected for the review highlight this (Table 3). Multiple studies reported on the molecular genetic diagnosis resulting in correction of the clinical diagnosis. While percentages vary between these different studies, all highlight the potential importance of establishing the correct diagnosis through genetic testing. Therapeutic impact varies from referral and evaluation for previously unrecognized extrarenal features to changing treatment plan. A clear example of therapeutic consequences of genetic testing is SRNS; most genetic forms of SRNS do not respond to immunosuppressive drugs and can therefore be spared the potential toxicity of these ineffective medications. A clear example of prognostic impact is an extremely low disease recurrence in many genetic kidney diseases following kidney transplantation as opposed to kidney diseases with a nongenetic cause.
Importantly, identifying a genetic cause can be crucial for the patient and/or parents of a patient with regard to genetic counseling; it informs recurrence risks, and can support patients and parents' decision‐making regarding reproductive options such as prenatal and pre‐implantation genetic diagnosis. In addition, family members can be counseled about disease risk, presymptomatic testing and screening options for secondary signs in first degree family members whilst not performing genetic testing. A genetic diagnosis can also be of importance for living kidney donation by family members. What impact a genetic diagnosis has, including therapeutic impact, will depend on the phenotype, but also on the individual circumstances of a patient and family, and the local/regional availability of treatment and family planning options.
6. CONSIDERATIONS FOR GENETIC TESTING
It is important to note that the type of test that is chosen for genetic testing in a patient can have a big impact on the chance of finding a genetic cause. The technological advancements in genetic testing approaches have made (MPS‐based) CNV testing and exome‐based sequencing possible and the advantages are being recognized over the years (Supplementary Figure 2). Gene panel composition, number of tested genes and CNV analysis, can influence the likelihood of finding a genetic cause. In addition, in ADTKD suspected cases, it is important to consider MUC1 testing or additional PKD1 testing in ADPKD suspected cases after MPS‐based multigene panel or exome testing. In only 6 studies (including 3/4 ADTKD cohorts) additional testing was performed to detect a cytosine insertion in the variable number tandem repeats region of MUC1 that is usually missed by MPS (Supplementary Table 3) (Kirby et al., 2013). Nineteen studies reported additional tests to reach sufficient coverage of all PKD1 exons, which is challenging because of the existence of six pseudogenes (PKD1P1‐6) with 97.7% sequence identity. Fifteen of these studies focused on ciliopathy phenotypes and four included mixed kidney disease phenotypes. One study reported exclusion of ADPKD patients because PKD1 is not well‐captured by WES (Lata et al., 2018). Clinicians that request genetic testing need to be aware of what disease‐causing variants in what genes can be detected by what genetic test and when additional genetic tests should be requested (Knoers et al., 2022; Köttgen et al., 2022).
Sometimes clinicians who do not yet have expertise in (nephro)genetics seem to assume that “performing WES” will cover any disease‐causing variant. One aspect of this false assumption is CNV detection. While WES‐based CNV analysis is upcoming, this is not yet always included. It is therefore good to consider whether for a specific patient additional CNV testing is needed. In the studies included in this review, many different tools were used, with differences in yield per tool being reported (Moreno‐Cabrera et al., 2020; Yao et al., 2017). Some studies covered CNVs in all genes, while others focused only on genes within a gene panel (Supplementary Figure 3). Also, separate tests (e.g., MLPA covering one gene or genome‐wide SNP‐array) were performed for CNV detection. Other aspects of this false assumption are variations in gene coverage, difficult to sequence regions (e.g., repeat regions, pseudogenes), and noncoding variants.
In the past, gene panels were always enrichment‐based, meaning that only the set of genes which were selected prior sequencing would be sequenced and analyzed. Today, diagnostic labs are often using exome‐based gene panels (Supplementary Figure 2b). With this approach, the complete exome is sequenced, but only the genes of interested from a specific gene panel are analyzed. An advantage of WES and usage of exome‐based gene panels is the efficient method in which data is derived including the possibility to analyze additional genes without having to resequence the patient's DNA (Knoers et al., 2022). WES also makes it possible to reanalyze or identify phenocopies. The studies included in this review highlight this. Warejko et al. identified phenocopies in 4% of patients in a SRNS cohort (Warejko et al., 2018). Also in the NS cohort of Landini et al., reverse phenotyping of patients let to the diagnosis of phenocopies in 28% of cases (Landini et al., 2020). Szabó et al. found phenocopies in 22% of patients with ARPKD (Szabó et al., 2018). In a cohort with various phenotypes, Riedhammer et al. discovered that 19% of diagnosed cases was a phenocopy (Riedhammer et al., 2020). Phenocopies and local differences in clinical phenotyping are arguments for a broader gene panel composition. Broad genetic testing also has significant challenges, including a higher chance of incidental findings, and the difficulties in interpretation of variants of unknown significance (VUS) (Bertier, Hétu, & Joly, 2016). This should be taken into account when choosing an initial smaller gene panel or a broad multigene panel or exome wide analysis. Also, counselors should be comfortable with, and skilled in counseling these findings and when to refer to a clinical geneticist. Availability of different genetic tests, genetic care and agreement on what test can be requested by nongeneticists will differ per country.
In all the phenotypes described in this review, only a limited number of genes are responsible for the top 50% of established diagnoses, highlighting the relevance of core genes for phenotypes (Martin et al., 2019). Even though only a limited number of genes are responsible for the top 50% of genetic diagnoses, the study of Groopman et al. found that 39/66 detected monogenic disorders were detected in only a single patient (Groopman et al., 2019). In this same study, four genes were responsible for 54% of confirmed diagnoses. Rao et al. reported that 15 genes accounted for 61% of genetic diagnosis, but in total, 106 distinct monogenic disorders were detected in a cohort of 1,001 pediatric patients with clinical suspicion of genetic kidney disease (Rao et al., 2019). Therefore, we recommend considering analysis of a complete set of known kidney disease genes after a first negative focused exome‐based panel result, also in light of the possibility of phenocopies. We recommend not starting with this large set of genes to avoid unnecessary variants of unknown significance and incidental findings. This recommendation is again dependent on availability and costs of reanalysis per country. While this review focuses primarily on diagnostic yield across and within kidney phenotype groups, additional genetic testing considerations for kidney disease patients can be found in recently published recommendations (Knoers et al., 2022; Köttgen et al., 2022).
7. STRENGTHS AND LIMITATIONS
Our systematic approach for selection and data‐extraction resulted in the most extensive overview of diagnostic yield in nephrogenetics yet. Given the challenges in comparing the different studies, we only summarized and visualized the data but did not perform a statistical meta‐analysis. One important variable was variant classification that was not identical in all studies. In 59/115 articles only the American College of Medical Genetics and Genomics (ACMG) criteria were used and in 4/115 ACMG criteria were used together with other filtering steps (Supplementary Table 3). Some studies that used other variant classifications than the ACMG criteria did use the same variant descriptions (i.e., pathogenic and likely pathogenic variants) as shown in the pivot table in Supplementary Table 3. We did find that most recently published papers used ACMG criteria (Supplementary Figure 2d). Benson et al. highlight the impact that different classification criteria can have on the reported yield by reporting a diagnostic yield of 68% using ACMG criteria and a yield of 81% using Mayo clinic pathogenicity guidelines. Furthermore, it was not always reported whether both likely pathogenic and pathogenic variants were included. In some cases, VUS were included in the reported diagnostic yield and it was not possible to subtract these, which might give an unjustly high yield. With WES‐based panels and ACMG criteria being used more often, comparing future studies might be easier. We chose to include studies published in the last 10 years, but even with this limitation, we expect that the diagnostic yield was likely higher in the more recent articles because of an increase in number of known kidney disease genes and novel techniques. Although our data do not clearly support this notion (data not shown), this is likely explained by other factors masking this effect. Future reporting on diagnostic yield would also benefit from reporting of the population of which study population was derived from. This was not always reported in the included articles in this review, rendering interpretation of the minimum diagnostic yield in these (clinical) cohorts challenging.
8. WHAT IS NEXT?
Although this review gives an extensive overview of reported diagnostic yield, there are still gaps in knowledge regarding the true prevalence of hereditary kidney disease in the general CKD population. To get an estimate of this, a study would need to be set‐up in which all kidney disease patients seen in different clinical settings would receive genetic testing. Since this might not be feasible, we would like this to be at least a call for action to report in detail on the population reported in diagnostic yield studies, to report on the patient, cohort, and test characteristics mentioned in this review and to use standard variant classification protocols. Also, opportunities for segregation of variants (of unknown significance) and CNV analysis should not be missed. Another opportunity for diagnosing additional patients is WGS, which makes it possible to sequence the entire genome. For ultimately determining the true diagnostic yield there is still a long road ahead since there are likely still many coding and noncoding genetic causes involved in kidney disease that need to be discovered.
9. CONCLUSION
This review gives an overview of the diagnostic yield of genetic testing across and within kidney disease phenotypes. The most important findings and key take home messages are summarized in Figure 6. We confirm that patient characteristics (e.g., family history, consanguinity, extrarenal features, and young age of onset) can positively impact the diagnostic yield. Furthermore, we emphasize the impact of the specific genetic test requested, including its ability to reveal CNVs. We also show the importance of considering the kind of cohort in which a study was performed, for interpreting the reported yield. We show that a genetic diagnosis can have a diagnostic, therapeutic and prognostic impact. Considering reclassifications based on genetic findings and the possibility to obviate the need for a diagnostic renal biopsy in many cases, a genetics first approach can be considered in clinical practice for establishing the patient's diagnosis. The number of genes to examine, whether and how to perform CNV analysis and, in ADTKD/ADPKD additional tests to cover for MUC1 and PKD1 need to be weighed when requesting a genetic test. Of course, it is important to note that patient and family specific situations can also influence the decision to do a genetic test, and also what genetic test is chosen. In addition, the availability of genetic testing in different countries can have in impact on accessibility of genetic testing. This review gives clinicians guidance on estimating an a priori probability of finding a genetic cause for the kidney disease in their patients.
FIGURE 6.
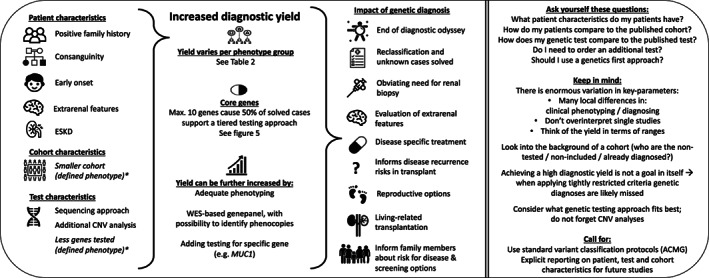
Summary of key findings from literature review (n = 115 articles). Left side of figure summarizes the characteristics influencing diagnostic yield and the impact of a genetic diagnosis. The right side of this figure summarizes the additional key take home messages. *Estimated by authors of this review that this is due to tightly restricted phenotype criteria.
AUTHOR CONTRIBUTIONS
Rozemarijn Snoek, Laura R. Claus, and Albertien M. van Eerde set up the design for the review. Nine V. A. M. Knoers provided structural feedback on the study design and progress. Rozemarijn Snoek extracted data from a subset of articles to define the variables of interest. Laura R. Claus extracted, evaluated, and analyzed the data and drafted the paper. Albertien M. van Eerde and Nine V. A. M. Knoers critically assessed the paper. All authors approved the final version of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
SUPPLEMENTARY TABLE 1 Summary of individual studies
SUPPLEMENTARY TABLE 2 Abbreviations
SUPPLEMENTARY TABLE 3 Additional data from individual studies
SUPPLEMENTARY FIGURE 1 Summary of sequencing approach. A. Boxplots show diagnostic yield in relation to sequencing approaches. B. Pie charts show whether the approaches in A were based on enrichment or exome‐based approach.
SUPPLEMENTARY FIGURE 2 Pie charts show shift in genetic testing approaches over the years. A. Type of variants tested: combination of CNVs + SNVs in more recent years. B. Shift of enrichment‐based panels to exome‐based approach over the years. C. CNV testing approach is shifting toward MPS‐based CNV calling (blue). D. Moving toward standard variants classification protocol using ACMG criteria.
SUPPLEMENTARY FIGURE 3 CNV testing approach and impact on diagnostic yield. A. Diagnostic yield based only on CNV detection per CNV testing approach. Between brackets the number of studies that used a particular approach. B. Percentage of overall diagnostic yield explained by the CNVs. This is an extended version of Figure 1c. On top: between brackets the total number of studies per disease group and on the right: between brackets the number of studies that used a particular approach. The table on the bottom shows how this is divided over the different phenotypes.
ACKNOWLEDGMENTS
The authors acknowledge and thank Rieko Haring and Richard van Kemenade who ran the literature database searches and screened articles for eligibility as part of their bachelor thesis. This work was supported by the Dutch Kidney Foundation (18OKG19 to A. M. v. E.). The authors of this publication are members of the European Reference Network for Rare Kidney Diseases (ERKNet).
Claus, L. R. , Snoek, R. , Knoers, N. V. A. M. , & van Eerde, A. M. (2022). Review of genetic testing in kidney disease patients: Diagnostic yield of single nucleotide variants and copy number variations evaluated across and within kidney phenotype groups. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 190C:358–376. 10.1002/ajmg.c.31995
Funding information Nierstichting, Grant/Award Number: 18OKG19
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Adalat, S. , Hayes, W. N. , Bryant, W. A. , Booth, J. , Woolf, A. S. , Kleta, R. , … Bockenhauer, D. (2019). HNF1B mutations are associated with a Gitelman‐like tubulopathy that develops during childhood. Kidney International Reports, 4, 1304–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, Y. H. , Lee, C. , Kim, N. K. D. , Park, E. , Kang, H. G. , Ha, I. S. , … Cheong, H. I. (2020). Targeted exome sequencing provided comprehensive genetic diagnosis of congenital anomalies of the kidney and urinary tract. Journal of Clinical Medicine, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Alawi, I. , Al Salmi, I. , Al Rahbi, F. , Al Riyami, M. , Al Kalbani, N. , Al Ghaithi, B. , … Sayer, J. A. (2019). Molecular genetic diagnosis of Omani patients with inherited cystic kidney disease. Kidney International Reports, 4, 1751–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Alawi, I. , Molinari, E. , Al Salmi, I. , Al Rahbi, F. , Al Mawali, A. , & Sayer, J. A. (2020). Clinical and genetic characteristics of autosomal recessive polycystic kidney disease in Oman. BMC Nephrology, 21, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Hamed, M. H. , Al‐Sabban, E. , Al‐Mojalli, H. , Al‐Harbi, N. , Faqeih, E. , Al Shaya, H. , … Meyer, B. F. (2013). A molecular genetic analysis of childhood nephrotic syndrome in a cohort of Saudi Arabian families. Journal of Human Genetics, 58, 480–489. [DOI] [PubMed] [Google Scholar]
- Al‐Hamed, M. H. , Kurdi, W. , Alsahan, N. , Alabdullah, Z. , Abudraz, R. , Tulbah, M. , … Albaqumi, M. (2016). Genetic spectrum of Saudi Arabian patients with antenatal cystic kidney disease and ciliopathy phenotypes using a targeted renal gene panel. Journal of Medical Genetics, 53, 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkanderi, S. , Yates, L. M. , Johnson, S. A. , & Sayer, J. A. (2017). Lessons learned from a multidisciplinary renal genetics clinic. QJM, 110, 453–457. [DOI] [PubMed] [Google Scholar]
- Amar, A. , Majmundar, A. J. , Ullah, I. , Afzal, A. , Braun, D. A. , Shril, S. , … Hildebrandt, F. (2019). Gene panel sequencing identifies a likely monogenic cause in 7% of 235 Pakistani families with nephrolithiasis. Human Genetics, 138, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlie‐Wolf, L. , Baker, L. , Hiddemen, O. , Thomas, M. , Burke, C. , Gluck, C. , … Gripp, K. W. (2021). Novel genetic testing model: A collaboration between genetic counselors and nephrology. American Journal of Medical Genetics A, 185, 1142–1150. [DOI] [PubMed] [Google Scholar]
- Ashton, E. J. , Legrand, A. , Benoit, V. , Roncelin, I. , Venisse, A. , Zennaro, M. C. , … Bockenhauer, D. (2018). Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies. Kidney International, 93, 961–967. [DOI] [PubMed] [Google Scholar]
- Ayasreh, N. , Bullich, G. , Miquel, R. , Furlano, M. , Ruiz, P. , Lorente, L. , … Torra, R. (2018). Autosomal dominant tubulointerstitial kidney disease: Clinical presentation of patients with ADTKD‐UMOD and ADTKD‐MUC1. American Journal of Kidney Diseases, 72, 411–418. [DOI] [PubMed] [Google Scholar]
- Bachmann‐Gagescu, R. , Dempsey, J. C. , Phelps, I. G. , O'Roak, B. J. , Knutzen, D. M. , Rue, T. C. , … Doherty, D. (2015). Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. Journal of Medical Genetics, 52, 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekheirnia, M. R. , Bekheirnia, N. , Bainbridge, M. N. , Gu, S. , Akdemir, Z. H. C. , Gambin, T. , … Lamb, D. J. (2017). Whole‐exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genetics in Medicine, 19, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, K. A. , Murray, S. L. , Doyle, R. , Doyle, B. , Dorman, A. M. , Sadlier, D. , … Conlon, P. J. (2020). Diagnostic utility of genetic testing in patients undergoing renal biopsy. Cold Spring Harbor Molecular Case Studies, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, K. A. , Murray, S. L. , Senum, S. R. , Elhassan, E. , Conlon, E. T. , Kennedy, C. , … Conlon, P. (2021). The genetic landscape of polycystic kidney disease in Ireland. European Journal of Human Genetics, 29, 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertier, G. , Hétu, M. , & Joly, Y. (2016). Unsolved challenges of clinical whole‐exome sequencing: A systematic literature review of end‐users' views Donna Dickenson, Sandra Soo‐Jin Lee, and Michael Morrison. BMC Medical Genomics, 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezdíčka, M. , Štolbová, Š. , Seeman, T. , Cinek, O. , Malina, M. , Šimánková, N. , … Zieg, J. (2018). Genetic diagnosis of steroid‐resistant nephrotic syndrome in a longitudinal collection of Czech and Slovak patients: A high proportion of causative variants in NUP93. Pediatric Nephrology, 33, 1347–1363. [DOI] [PubMed] [Google Scholar]
- Bierzynska, A. , McCarthy, H. J. , Soderquest, K. , Sen, E. S. , Colby, E. , Ding, W. Y. , … Saleem, M. A. (2017). Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney International, 91, 937–947. [DOI] [PubMed] [Google Scholar]
- Bleyer, A. J. , Kidd, K. , Robins, V. , Martin, L. , Taylor, A. , Santi, A. , … Kmoch, S. (2020). Outcomes of patient self‐referral for the diagnosis of several rare inherited kidney diseases. Genetics in Medicine, 22, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissel, S. , Fallet‐Bianco, C. , Chitayat, D. , Kremer, V. , Nassif, C. , Rypens, F. , … Michaud, J. L. (2018). Genomic study of severe fetal anomalies and discovery of GREB1L mutations in renal agenesis. Genetics in Medicine, 20, 745–753. [DOI] [PubMed] [Google Scholar]
- Braun, D. A. , Schueler, M. , Halbritter, J. , Gee, H. Y. , Porath, J. D. , Lawson, J. A. , … Hildebrandt, F. (2016). Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood‐onset increased renal echogenicity. Kidney International, 89, 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu, F. , Borsa, N. G. , Jones, M. B. , Takanami, E. , Nishimura, C. , Hauer, J. J. , … Smith, R. J. H. (2016). High‐throughput genetic testing for thrombotic microangiopathies and C3 glomerulopathies. Journal of the American Society of Nephrology, 27, 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullich, G. , Domingo‐Gallego, A. , Vargas, I. , Ruiz, P. , Lorente‐Grandoso, L. , Furlano, M. , … Ars, E. (2018). A kidney‐disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney International, 94, 363–371. [DOI] [PubMed] [Google Scholar]
- Cai, M. , Lin, N. , Su, L. , Wu, X. , Xie, X. , Li, Y. , … Xu, L. (2020a). Detection of copy number disorders associated with congenital anomalies of the kidney and urinary tract in fetuses via single nucleotide polymorphism arrays. Journal of Clinical Laboratory Analysis, 34, e23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, M. , Lin, N. , Su, L. , Wu, X. , Xie, X. , Li, Y. , … Xu, L. (2020b). Copy number variations associated with fetal congenital kidney malformations. Molecular Cytogenetics, 13, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruana, G. , Wong, M. N. , Walker, A. , Heloury, Y. , Webb, N. , Johnstone, L. , … Bertram, J. F. (2015). Copy‐number variation associated with congenital anomalies of the kidney and urinary tract. Pediatric Nephrology, 30, 487–495. [DOI] [PubMed] [Google Scholar]
- Connaughton, D. M. , Kennedy, C. , Shril, S. , Mann, N. , Murray, S. L. , Williams, P. A. , … Hildebrandt, F. (2019). Monogenic causes of chronic kidney disease in adults. Kidney International, 95, 914–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coresh, J. (2017). Update on the burden of CKD. Journal of the American Society of Nephrology, 28, 1020–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daga, A. , Majmundar, A. J. , Braun, D. A. , Gee, H. Y. , Lawson, J. A. , Shril, S. , … Hildebrandt, F. (2018). Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney International, 93, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo‐Gallego, A. , Pybus, M. , Bullich, G. , Furlano, M. , Ejarque‐Vila, L. , Lorente‐Grandoso, L. , … Ars, E. (2022). Clinical utility of genetic testing in early‐onset kidney disease: Seven genes are the main players. Nephrology Dialysis Transplantation, 37, 687–696. [DOI] [PubMed] [Google Scholar]
- Durkie, M. , Chong, J. , Valluru, M. K. , Harris, P. C. , & Ong, A. C. M. (2021). Biallelic inheritance of hypomorphic PKD1 variants is highly prevalent in very early onset polycystic kidney disease. Genetics in Medicine, 23, 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallerini, C. , Dosa, L. , Tita, R. , Del Prete, D. , Feriozzi, S. , Gai, G. , … Ariani, F. (2014). Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clinical Genetics, 86, 252–257. [DOI] [PubMed] [Google Scholar]
- Faure, A. , Bouty, A. , Caruana, G. , Williams, L. , Burgess, T. , Wong, M. N. , … Heloury, Y. (2016). DNA copy number variants: A potentially useful predictor of early onset renal failure in boys with posterior urethral valves. Journal of Pediatric Urology, 12, 227.e1–227.e7. [DOI] [PubMed] [Google Scholar]
- Fu, F. , Chen, F. , Li, R. , Zhang, Y. , Pan, M. , Li, D. , & Liao, C. (2016). Prenatal diagnosis of fetal multicystic dysplastic kidney via high‐resolution whole‐genome array. Nephrology Dialysis Transplantation, 31, 1693–1698. [DOI] [PubMed] [Google Scholar]
- Fujimaru, T. , Mori, T. , Sekine, A. , Mandai, S. , Chiga, M. , Kikuchi, H. , … Sohara, E. (2018). Kidney enlargement and multiple liver cyst formation implicate mutations in PKD1/2 in adult sporadic polycystic kidney disease. Clinical Genetics, 94, 125–131. [DOI] [PubMed] [Google Scholar]
- Gast, C. , Marinaki, A. , Arenas‐Hernandez, M. , Campbell, S. , Seaby, E. G. , Pengelly, R. J. , … Venkat‐Raman, G. (2018). Autosomal dominant tubulointerstitial kidney disease‐UMOD is the most frequent non polycystic genetic kidney disease. BMC Nephrology, 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gast, C. , Pengelly, R. J. , Lyon, M. , Bunyan, D. J. , Seaby, E. G. , Graham, N. , … Ennis, S. (2016). Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrology Dialysis Transplantation, 31, 961–970. [DOI] [PubMed] [Google Scholar]
- Giglio, S. , Provenzano, A. , Mazzinghi, B. , Becherucci, F. , Giunti, L. , Sansavini, G. , … Romagnani, P. (2015). Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. Journal of the American Society of Nephrology, 26, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribouval, O. , Boyer, O. , Hummel, A. , Dantal, J. , Martinez, F. , Sberro‐Soussan, R. , … Servais, A. (2018). Identification of genetic causes for sporadic steroid‐resistant nephrotic syndrome in adults. Kidney International, 94, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Groopman, E. E. , Marasa, M. , Cameron‐Christie, S. , Petrovski, S. , Aggarwal, V. S. , Milo‐Rasouly, H. , … Gharavi, A. G. (2019). Diagnostic utility of exome sequencing for kidney disease. The New England Journal of Medicine, 380, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidet, L. , Morinière, V. , Henry, C. , De Tomasi, L. , Reilly, M. L. , Humbert, C. , … Jeanpierre, C. (2017). Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. Journal of the American Society of Nephrology, 28, 2901–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt, F. (2010). Genetic kidney diseases. Lancet, 375, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hureaux, M. , Ashton, E. , Dahan, K. , Houillier, P. , Blanchard, A. , Cormier, C. , … Vargas‐Poussou, R. (2019). High‐throughput sequencing contributes to the diagnosis of tubulopathies and familial hypercalcemia hypocalciuria in adults. Kidney International, 96, 1408–1416. [DOI] [PubMed] [Google Scholar]
- Hwang, D. Y. , Dworschak, G. C. , Kohl, S. , Saisawat, P. , Vivante, A. , Hilger, A. C. , … Hildebrandt, F. (2014). Mutations in 12 known dominant disease‐causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney International, 85, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, Y.‐H. , Conklin, J. , Chan, W. , Roslin, N. M. , Liu, J. , He, N. , … Pei, Y. (2016). Refining genotype‐phenotype correlation in autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology, 27, 1861–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe, K. , Stark, Z. , Kerr, P. G. , Gaff, C. , Martyn, M. , Whitlam, J. , … Quinlan, C. (2021). Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genetics in Medicine, 23, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, M. , Xie, Y. , Chen, Z. , Liao, Y. , Li, Z. , Hu, P. , … Chen, X. (2016). System analysis of gene mutations and clinical phenotype in Chinese patients with autosomal‐dominant polycystic kidney disease. Scientific Reports, 6, 35945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari, J. A. , El‐Desoky, S. M. , Gari, M. , Malik, K. , Vega‐Warner, V. , Lovric, S. , & Bockenhauer, D. (2013). Steroid‐resistant nephrotic syndrome: Impact of genetic testing. Annals of Saudi Medicine, 33, 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, M. , Higashihara, E. , Kawano, H. , Higashiyama, R. , Koga, D. , Fukui, T. , … Nutahara, K. (2016). Technical evaluation: Identification of pathogenic mutations in PKD1 and PKD2 in patients with autosomal dominant polycystic kidney disease by next‐generation sequencing and use of a comprehensive new classification system. PLoS One, 11, e0166288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby, A. , Gnirke, A. , Jaffe, D. B. , Barešová, V. , Pochet, N. , Blumenstiel, B. , … Daly, M. J. (2013). Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nature Genetics, 45, 299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoers, N. , Antignac, C. , Bergmann, C. , Dahan, K. , Giglio, S. , Heidet, L. , … for the ERA Working Group on Inherited Kidney Disorders (WGIKD) and the Molecular Diagnostics Taskforce of the European Rare Kidney Disease Reference Network (ERKNet) which is an official body of the ERA (European RA) . (2022). Genetic testing in the diagnosis of chronic kidney disease: Recommendations for clinical practice. Nephrology Dialysis Transplantation, 37, 239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopp, C. , Rudnik‐Schöneborn, S. , Eggermann, T. , Bergmann, C. , Begemann, M. , Schoner, K. , … Ortiz, B. N. (2015). Syndromic ciliopathies: From single gene to multi gene analysis by SNP arrays and next generation sequencing. Molecular and Cellular Probes, 29, 299–307. [DOI] [PubMed] [Google Scholar]
- Kohl, S. , Hwang, D.‐Y. , Dworschak, G. C. , Hilger, A. C. , Saisawat, P. , Vivante, A. , … Hildebrandt, F. (2014). Mild recessive mutations in six Fraser syndrome‐related genes cause isolated congenital anomalies of the kidney and urinary tract. Journal of the American Society of Nephrology, 25, 1917–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köttgen, A. , Cornec‐Le Gall, E. , Halbritter, J. , Kiryluk, K. , Mallett, A. J. , Parekh, R. S. , … Gharavi, A. G. (2022). Genetics in chronic kidney disease: Conclusions from a kidney disease: Improving global outcomes (KDIGO) controversies conference. Kidney International, 101, 1126–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landini, S. , Mazzinghi, B. , Becherucci, F. , Allinovi, M. , Provenzano, A. , Palazzo, V. , … Romagnani, P. (2020). Reverse phenotyping after whole‐exome sequencing in steroid‐resistant nephrotic syndrome. Clinical Journal of the American Society of Nephrology, 15, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata, S. , Marasa, M. , Li, Y. , Fasel, D. A. , Groopman, E. , Jobanputra, V. , … Gharavi, A. G. (2018). Whole‐exome sequencing in adults with chronic kidney disease: A pilot study. Annals of Internal Medicine, 168, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, T.‐Y. , Fu, F. , Li, R. , Wang, D. , Wang, R.‐Y. , Jing, X.‐Y. , … Liao, C. (2017). Whole‐exome sequencing for prenatal diagnosis of fetuses with congenital anomalies of the kidney and urinary tract. Nephrology Dialysis Transplantation, 32, 1665–1675. [DOI] [PubMed] [Google Scholar]
- Lei, T.‐Y. , Fu, F. , Li, R. , Yu, Q.‐X. , Du, K. , Zhang, W.‐W. , … Liao, C. (2020). Whole‐exome sequencing in the evaluation of fetal congenital anomalies of the kidney and urinary tract detected by ultrasonography. Prenatal Diagnosis. [DOI] [PubMed] [Google Scholar]
- Li, S. , Han, X. , Wang, Y. , Chen, S. , Niu, J. , Qian, Z. , … Xu, C. (2019). Chromosomal microarray analysis in fetuses with congenital anomalies of the kidney and urinary tract: A prospective cohort study and meta‐analysis. Prenatal Diagnosis, 39, 165–174. [DOI] [PubMed] [Google Scholar]
- Liang, N. , Jiang, X. , Zeng, L. , Li, Z. , Liang, D. , & Wu, L. (2020). 28 novel mutations identified from 33 Chinese patients with cilia‐related kidney disorders. Clinica Chimica Acta, 501, 207–215. [DOI] [PubMed] [Google Scholar]
- Lin, S. , Shi, S. , Huang, L. , Lei, T. , Cai, D. , Hu, W. , … Luo, Y. (2019). Is an analysis of copy number variants necessary for various types of kidney ultrasound anomalies in fetuses? Molecular Cytogenetics, 12, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrand, A. , Frangakis, S. , Carvalho, C. M. B. , Richardson, E. B. , McFadden, K. A. , Willer, J. R. , … Katsanis, N. (2016). Copy‐number variation contributes to the mutational load of Bardet‐Biedl syndrome. American Journal of Human Genetics, 99, 318–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallawaarachchi, A. C. , Lundie, B. , Hort, Y. , Schonrock, N. , Senum, S. R. , Gayevskiy, V. , … Furlong, T. J. (2021). Genomic diagnostics in polycystic kidney disease: An assessment of real‐world use of whole‐genome sequencing. European Journal of Human Genetics, 29, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallett, A. J. , McCarthy, H. J. , Ho, G. , Holman, K. , Farnsworth, E. , Patel, C. , … Alexander, S. I. (2017). Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney International, 92, 1493–1506. [DOI] [PubMed] [Google Scholar]
- Mann, N. , Braun, D. A. , Amann, K. , Tan, W. , Shril, S. , Connaughton, D. M. , … Hildebrandt, F. (2019). Whole‐exome sequencing enables a precision medicine approach for kidney transplant recipients. Journal of the American Society of Nephrology, 30, 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansilla, M. A. , Sompallae, R. R. , Nishimura, C. J. , Kwitek, A. E. , Kimble, M. J. , Freese, M. E. , … Thomas, C. P. (2021). Targeted broad‐based genetic testing by next‐generation sequencing informs diagnosis and facilitates management in patients with kidney diseases. Nephrology, Dialysis, Transplantation, 36, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani, V. , Bin, S. , Graziano, C. , Capelli, I. , Minardi, R. , Aiello, V. , … La Manna, G. (2020). Gene panel analysis in a large cohort of patients with autosomal dominant polycystic kidney disease allows the identification of 80 potentially causative novel variants and the characterization of a complex genetic architecture in a subset of families. Frontiers in Genetics, 11, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, A. R. , Williams, E. , Foulger, R. E. , Leigh, S. , Daugherty, L. C. , Niblock, O. , … McDonagh, E. M. (2019). PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nature Genetics, 51, 1560–1565. [DOI] [PubMed] [Google Scholar]
- McCarthy, H. J. , Bierzynska, A. , Wherlock, M. , Ognjanovic, M. , Kerecuk, L. , Hegde, S. , … Saleem, M. A. (2013). Simultaneous sequencing of 24 genes associated with steroid‐resistant nephrotic syndrome. Clinical Journal of the American Society of Nephrology, 8, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki, T. , Teraoka, A. , Akagawa, H. , Makabe, S. , Akihisa, T. , Sato, M. , … Nitta, K. (2019). Mutation analyses by next‐generation sequencing and multiplex ligation‐dependent probe amplification in Japanese autosomal dominant polycystic kidney disease patients. Clinical and Experimental Nephrology, 23, 1022–1030. [DOI] [PubMed] [Google Scholar]
- Moreno‐Cabrera, J. M. , del Valle, J. , Castellanos, E. , Feliubadaló, L. , Pineda, M. , Brunet, J. , … Gel, B. (2020). Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. European Journal of Human Genetics, 28, 1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori, T. , Chiga, M. , Fujimaru, T. , Kawamoto, R. , Mandai, S. , Nanamatsu, A. , … Uchida, S. (2021). Phenotypic differences of mutation‐negative cases in Gitelman syndrome clinically diagnosed in adulthood. Human Mutation, 42, 300–309. [DOI] [PubMed] [Google Scholar]
- Morinière, V. , Dahan, K. , Hilbert, P. , Lison, M. , Lebbah, S. , Topa, A. , … Heidet, L. (2014). Improving mutation screening in familial hematuric nephropathies through next generation sequencing. Journal of the American Society of Nephrology, 25, 2740–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, S. L. , Dorman, A. , Benson, K. A. , Connaughton, D. M. , Stapleton, C. P. , Fennelly, N. K. , … Conlon, P. J. (2020). Utility of genomic testing after renal biopsy. American Journal of Nephrology, 51, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabais Sá, M. J. , Sampaio, S. , Oliveira, A. , Alves, S. , Moura, C. P. , Silva, S. E. , … Oliveira, J. P. (2015a). Collagen type IV‐related nephropathies in Portugal: Pathogenic COL4A5 mutations and clinical characterization of 22 families. Clinical Genetics, 88, 462–467. [DOI] [PubMed] [Google Scholar]
- Nabais Sá, M. J. , Storey, H. , Flinter, F. , Nagel, M. , Sampaio, S. , Castro, R. , … Oliveira, J. P. (2015b). Collagen type IV‐related nephropathies in Portugal: Pathogenic COL4A3 and COL4A4 mutations and clinical characterization of 25 families. Clinical Genetics, 88, 456–461. [DOI] [PubMed] [Google Scholar]
- Nagano, C. , Yamamura, T. , Horinouchi, T. , Aoto, Y. , Ishiko, S. , Sakakibara, N. , … Nozu, K. (2020). Comprehensive genetic diagnosis of Japanese patients with severe proteinuria. Scientific Reports, 10, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou, N. , Pulit, S. L. , Nijman, I. J. , Monroe, G. R. , Feitz, W. F. J. , Schreuder, M. F. , … Renkema, K. Y. (2016). Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney International, 89, 476–486. [DOI] [PubMed] [Google Scholar]
- Nielsen, M. L. , Lildballe, D. L. , Rasmussen, M. , Bojesen, A. , Birn, H. , & Sunde, L. (2021). Clinical genetic diagnostics in Danish autosomal dominant polycystic kidney disease patients reveal possible founder variants. European Journal of Medical Genetics, 64, 104183. [DOI] [PubMed] [Google Scholar]
- Obeidova, L. , Seeman, T. , Fencl, F. , Blahova, K. , Hojny, J. , Elisakova, V. , … Stekrova, J. (2020). Results of targeted next‐generation sequencing in children with cystic kidney diseases often change the clinical diagnosis. PLoS One, 15, e0235071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, J. , Shin, J. I. , Lee, K. , Lee, C. , Ko, Y. , & Lee, J.‐S. (2021). Clinical application of a phenotype‐based NGS panel for differential diagnosis of inherited kidney disease and beyond. Clinical Genetics, 99, 236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olinger, E. , Hofmann, P. , Kidd, K. , Dufour, I. , Belge, H. , Schaeffer, C. , … Devuyst, O. (2020). Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1. Kidney International, 98, 717–731. [DOI] [PubMed] [Google Scholar]
- Ottlewski, I. , Münch, J. , Wagner, T. , Schönauer, R. , Bachmann, A. , Weimann, A. , … Halbritter, J. (2019). Value of renal gene panel diagnostics in adults waiting for kidney transplantation due to undetermined end‐stage renal disease. Kidney International, 96, 222–230. [DOI] [PubMed] [Google Scholar]
- Ouzzani, M. , Hammady, H. , Fedorowicz, Z. , & Elmagarmid, A. (2016). Rayyan—A web and mobile app for systematic reviews. Systematic Reviews, 5, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir, G. , Gulhan, B. , Atayar, E. , Saygılı, S. , Soylemezoglu, O. , Ozcakar, Z. B. , … Topaloglu, R. (2020). COL4A3 mutation is an independent risk factor for poor prognosis in children with Alport syndrome. Pediatric Nephrology, 35, 1941–1952. [DOI] [PubMed] [Google Scholar]
- Palazzo, V. , Provenzano, A. , Becherucci, F. , Sansavini, G. , Mazzinghi, B. , Orlandini, V. , … Giglio, S. (2017). The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney International, 91, 1243–1255. [DOI] [PubMed] [Google Scholar]
- Rao, J. , Liu, X. , Mao, J. , Tang, X. , Shen, Q. , Li, G. , … Xu, H. (2019). Genetic spectrum of renal disease for 1001 Chinese children based on a multicenter registration system. Clinical Genetics, 96, 402–410. [DOI] [PubMed] [Google Scholar]
- Rasmussen, M. , Sunde, L. , Nielsen, M. L. , Ramsing, M. , Petersen, A. , Hjortshøj, T. D. , … Lildballe, D. L. (2018). Targeted gene sequencing and whole‐exome sequencing in autopsied fetuses with prenatally diagnosed kidney anomalies. Clinical Genetics, 93, 860–869. [DOI] [PubMed] [Google Scholar]
- Riedhammer, K. M. , Braunisch, M. C. , Günthner, R. , Wagner, M. , Hemmer, C. , Strom, T. M. , … Hoefele, J. (2020). Exome sequencing and identification of phenocopies in patients with clinically presumed hereditary nephropathies. American Journal of Kidney Diseases, 76, 460–470. [DOI] [PubMed] [Google Scholar]
- Rossetti, S. , Hopp, K. , Sikkink, R. A. , Sundsbak, J. L. , Lee, Y. K. , Kubly, V. , … Harris, P. C. (2012). Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. Journal of the American Society of Nephrology, 23, 915–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski, C. E. , Lovric, S. , Ashraf, S. , Pabst, W. L. , Gee, H. Y. , Kohl, S. , … Hildebrandt, F. (2015). A single‐gene cause in 29.5% of cases of steroid‐resistant nephrotic syndrome. Journal of the American Society of Nephrology, 26, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna‐Cherchi, S. , Kiryluk, K. , Burgess, K. E. , Bodria, M. , Sampson, M. G. , Hadley, D. , … Gharavi, A. G. (2012). Copy‐number disorders are a common cause of congenital kidney malformations. American Journal of Human Genetics, 91, 987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro, D. , Daga, A. , Lawson, J. A. , Majmundar, A. J. , Lovric, S. , Tan, W. , … Hildebrandt, F. (2019). Panel sequencing distinguishes monogenic forms of nephritis from nephrosis in children. Nephrology Dialysis Transplantation, 34, 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönauer, R. , Baatz, S. , Nemitz‐Kliemchen, M. , Frank, V. , Petzold, F. , Sewerin, S. , … Halbritter, J. (2020). Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genetics in Medicine, 22, 1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrezenmeier, E. , Kremerskothen, E. , Halleck, F. , Staeck, O. , Liefeldt, L. , Choi, M. , … Bergmann, C. (2021). The underestimated burden of monogenic kidney disease in adults waitlisted for kidney transplantation. Genetics in Medicine, 23, 2468. 10.1038/s41436-021-01127-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler, M. , Halbritter, J. , Phelps, I. G. , Braun, D. A. , Otto, E. A. , Porath, J. D. , … Hildebrandt, F. (2016). Large‐scale targeted sequencing comparison highlights extreme genetic heterogeneity in nephronophthisis‐related ciliopathies. Journal of Medical Genetics, 53, 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, E. S. , Dean, P. , Yarram‐Smith, L. , Bierzynska, A. , Woodward, G. , Buxton, C. , … Saleem, M. A. (2017). Clinical genetic testing using a custom‐designed steroid‐resistant nephrotic syndrome gene panel: Analysis and recommendations. Journal of Medical Genetics, 54, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoek, R. , van Jaarsveld, R. H. , Nguyen, T. Q. , Peters, E. D. J. , Elferink, M. G. , Ernst, R. F. , … van Eerde, A. M. (2022). Genetics‐first approach improves diagnostics of ESKD patients <50 years old. Nephrology Dialysis Transplantation, 37, 349–357. [DOI] [PubMed] [Google Scholar]
- Snoek, R. , Van Setten, J. , Keating, B. J. , Israni, A. K. , Jacobson, P. A. , Oetting, W. S. , … Van Eerde, A. M. (2018). NPHP1 (Nephrocystin‐1) gene deletions cause adult‐onset ESRD. Journal of the American Society of Nephrology, 29, 1772–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokman, M. F. , Renkema, K. Y. , Giles, R. H. , Schaefer, F. , Knoers, N. V. A. M. , & van Eerde, A. M. (2016). The expanding phenotypic spectra of kidney diseases: Insights from genetic studies. Nature Reviews. Nephrology, 12, 472–483. [DOI] [PubMed] [Google Scholar]
- Stokman, M. F. , van der Zwaag, B. , van de Kar, N. C. A. J. , van Haelst, M. M. , van Eerde, A. M. , van der Heijden, J. W. , … Lilien, M. R. (2018). Clinical and genetic analyses of a Dutch cohort of 40 patients with a nephronophthisis‐related ciliopathy. Pediatric Nephrology, 33, 1701–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó, T. , Orosz, P. , Balogh, E. , Jávorszky, E. , Máttyus, I. , Bereczki, C. , … Balogh, I. (2018). Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatric Nephrology, 33, 1713–1721. [DOI] [PubMed] [Google Scholar]
- Tan, W. , Lovric, S. , Ashraf, S. , Rao, J. , Schapiro, D. , Airik, M. , … Hildebrandt, F. (2018). Analysis of 24 genes reveals a monogenic cause in 11.1% of cases with steroid‐resistant nephrotic syndrome at a single center. Pediatric Nephrology, 33, 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanudisastro, H. A. , Holman, K. , Ho, G. , Farnsworth, E. , Fisk, K. , Gayagay, T. , … McCarthy, H. J. (2021). Australia and New Zealand renal gene panel testing in routine clinical practice of 542 families. npj Genomic Medicine, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, C. P. , Freese, M. E. , Ounda, A. , Jetton, J. G. , Holida, M. , Noureddine, L. , & Smith, R. J. (2020). Initial experience from a renal genetics clinic demonstrates a distinct role in patient management. Genetics in Medicine, 22, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann, A. , Bodria, M. , Ozaltin, F. , Gheisari, A. , Melk, A. , Azocar, M. , … Schaefer, F. (2015). Spectrum of steroid‐resistant and congenital nephrotic syndrome in children: The podoNet registry cohort. Clinical Journal of the American Society of Nephrology, 10, 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unzaki, A. , Morisada, N. , Nozu, K. , Ye, M. J. , Ito, S. , Matsunaga, T. , … Iijima, K. (2018). Clinically diverse phenotypes and genotypes of patients with branchio‐oto‐renal syndrome. Journal of Human Genetics, 63, 647–656. [DOI] [PubMed] [Google Scholar]
- Vaisitti, T. , Sorbini, M. , Callegari, M. , Kalantari, S. , Bracciamà, V. , Arruga, F. , … Deaglio, S. (2021). Clinical exome sequencing is a powerful tool in the diagnostic flow of monogenic kidney diseases: An Italian experience. Journal of Nephrology, 34, 1767–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Ven, A. T. , Connaughton, D. M. , Ityel, H. , Mann, N. , Nakayama, M. , Chen, J. , … Hildebrandt, F. (2018). Whole‐exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. Journal of the American Society of Nephrology, 29, 2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbitsky, M. , Westland, R. , Perez, A. , Kiryluk, K. , Liu, Q. , Krithivasan, P. , … Sanna‐Cherchi, S. (2019). Author Correction: The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nature Genetics, 51, 764. 10.1038/s41588-018-0281-y [DOI] [PubMed] [Google Scholar]
- Vilboux, T. , Doherty, D. A. , Glass, I. A. , Parisi, M. A. , Phelps, I. G. , Cullinane, A. R. , … Gunay‐Aygun, M. (2017). Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genetics in Medicine, 19, 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivante, A. , Hwang, D.‐Y. , Kohl, S. , Chen, J. , Shril, S. , Schulz, J. , … Hildebrandt, F. (2017). Exome sequencing discerns syndromes in patients from consanguineous families with congenital anomalies of the kidneys and urinary tract. Journal of the American Society of Nephrology, 28, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F. , Zhang, Y. , Mao, J. , Yu, Z. , Yi, Z. , Yu, L. , … Hildebrandt, F. (2017a). Spectrum of mutations in Chinese children with steroid‐resistant nephrotic syndrome. Pediatric Nephrology, 32, 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Dang, X. , He, Q. , Zhen, Y. , He, X. , Yi, Z. , & Zhu, K. (2017b). Mutation spectrum of genes associated with steroid‐resistant nephrotic syndrome in Chinese children. Gene, 625, 15–20. [DOI] [PubMed] [Google Scholar]
- Warejko, J. K. , Tan, W. , Daga, A. , Schapiro, D. , Lawson, J. A. , Shril, S. , … Hildebrandt, F. (2018). Whole exome sequencing of patients with steroid‐resistant nephrotic syndrome. Clinical Journal of the American Society of Nephrology, 13, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi, Q. , Zhu, X. , Wang, Y. , Ru, T. , Dai, C. , Wang, Z. , … Hu, Y. (2016). Copy number variations in multicystic dysplastic kidney: Update for prenatal diagnosis and genetic counseling. Prenatal Diagnosis., 36, 463–468. [DOI] [PubMed] [Google Scholar]
- Xu, D. , Ma, Y. , Gu, X. , Bian, R. , Lu, Y. , Xing, X. , & Mei, C. (2018). Novel mutations in the PKD1 and PKD2 genes of Chinese patients with autosomal dominant polycystic kidney disease. Kidney & Blood Pressure Research, 43, 297–309. [DOI] [PubMed] [Google Scholar]
- Yamamura, T. , Nozu, K. , Minamikawa, S. , Horinouchi, T. , Sakakibara, N. , Nagano, C. , … Iijima, K. (2019). Comparison between conventional and comprehensive sequencing approaches for genetic diagnosis of Alport syndrome. Molecular Genetics & Genomic Medicine, 7, e883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, R. , Zhang, C. , Yu, T. , Li, N. , Hu, X. , Wang, X. , … Shen, Y. (2017). Evaluation of three read‐depth based CNV detection tools using whole‐exome sequencing data. Molecular Cytogenetics, 10, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, T. , Udwan, K. , John, R. , Rana, A. , Haghighi, A. , Xu, L. , … Barua, M. (2019). Integration of genetic testing and pathology for the diagnosis of adults with FSGS. Clinical Journal of the American Society of Nephrology, 14, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, Z. , Lin, H. , Li, M. , Wang, H. , Liu, T. , Hu, M. , … Sun, L. (2020). Clinical and pathological features and varied mutational spectra of pathogenic genes in 55 Chinese patients with nephronophthisis. Clinica Chimica Acta, 506, 136–144. [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Liu, S. , Xia, X. , Cui, Y. , & Li, X. (2019). Identification of novel mutations and risk assessment of Han Chinese patients with autosomal dominant polycystic kidney disease. Nephrology, 24, 504–510. [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Fang, X. , Fan, Y. , Sun, Y. , He, L. , Xu, M. , … Geng, H. (2021). Integration of exome sequencing and metabolic evaluation for the diagnosis of children with urolithiasis. World Journal of Urology, 39, 2759–2765. [DOI] [PubMed] [Google Scholar]
- Zhou, X. , Wang, Y. , Shao, B. , Wang, C. , Hu, P. , Qiao, F. , & Xu, Z. (2020). Molecular diagnostic in fetuses with isolated congenital anomalies of the kidney and urinary tract by whole‐exome sequencing. Journal of Clinical Laboratory Analysis, 34, e23480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. Y. , Zheng, H. Y. , Han, L. , Wang, Y. , Zhang, L. , Shu, X. M. , … Ding, L. S. (2021). Copy number variations analysis identifies QPRT as a candidate gene associated with susceptibility for solitary functioning kidney. Frontiers in Genetics, 12, 575830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziyadov, E. , Bisgin, A. , Deger, M. , Akdogan, N. , Izol, V. , Aridogan, I. A. , & Satar, N. (2021). Determination of the etiology of pediatric urinary stone disease by multigene panel and metabolic screening evaluation. Journal of Pediatric Urology, 17, 476.e1–476.e7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY TABLE 1 Summary of individual studies
SUPPLEMENTARY TABLE 2 Abbreviations
SUPPLEMENTARY TABLE 3 Additional data from individual studies
SUPPLEMENTARY FIGURE 1 Summary of sequencing approach. A. Boxplots show diagnostic yield in relation to sequencing approaches. B. Pie charts show whether the approaches in A were based on enrichment or exome‐based approach.
SUPPLEMENTARY FIGURE 2 Pie charts show shift in genetic testing approaches over the years. A. Type of variants tested: combination of CNVs + SNVs in more recent years. B. Shift of enrichment‐based panels to exome‐based approach over the years. C. CNV testing approach is shifting toward MPS‐based CNV calling (blue). D. Moving toward standard variants classification protocol using ACMG criteria.
SUPPLEMENTARY FIGURE 3 CNV testing approach and impact on diagnostic yield. A. Diagnostic yield based only on CNV detection per CNV testing approach. Between brackets the number of studies that used a particular approach. B. Percentage of overall diagnostic yield explained by the CNVs. This is an extended version of Figure 1c. On top: between brackets the total number of studies per disease group and on the right: between brackets the number of studies that used a particular approach. The table on the bottom shows how this is divided over the different phenotypes.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.