

Abstract
RNA molecules adopt three-dimensional structures that are critical to their function and of interest in drug discovery. Few RNA structures are known, however, and predicting them computationally has proven challenging. We introduce a machine learning approach that enables identification of accurate structural models without assumptions about their defining characteristics, despite being trained with only 18 known RNA structures. The resulting scoring function, the Atomic Rotationally Equivariant Scorer (ARES), substantially outperforms previous methods and consistently produces the best results in community-wide blind RNA structure prediction challenges. By learning effectively even from a small amount of data, our approach overcomes a major limitation of standard deep neural networks. Because it uses only atomic coordinates as inputs and incorporates no RNA-specific information, this approach is applicable to diverse problems in structural biology, chemistry, materials science, and beyond.
RNA molecules, like proteins, fold into well-defined three-dimensional (3D) structures to perform a wide range of cellular functions, such as catalyzing reactions, regulating gene expression, modulating innate immunity, and sensing small molecules (1). Knowledge of these structures is extremely important for understanding the mechanisms of RNA function, designing synthetic RNAs, and discovering RNA-targeted drugs (2, 3). Our knowledge of RNA structure lags far behind that of protein structure: The fraction of the human genome transcribed to RNA is ~30 times as large as that coding for proteins (4), but the number of available RNA structures is <1% of that for proteins (5). Computational prediction of RNA 3D structure is thus of substantial interest (6).
Despite decades of intense effort, predicting the 3D structure of RNAs remains a grand challenge, having proven more difficult than prediction of protein structure. For proteins, state-of-the-art prediction methods leverage sequences or structures of related proteins (7–9). Such methods succeed much less frequently for RNA, both because template structures of closely related RNAs are available far less often and because sequence coevolution information provides less information about tertiary contacts in RNAs (10). Moreover, designing a scoring function that reliably distinguishes accurate structural models of RNA from less accurate ones has proven difficult, because the characteristics of energetically favorable RNA structures are not sufficiently well understood.
This problem raises the question of whether an algorithm could learn from known RNA structures to assess the accuracy of structural models of unrelated RNAs. Such a machine learning task poses two major challenges: (i) avoiding assumptions about which structural characteristics might distinguish accurate models from less accurate ones, and (ii) learning from the limited number of RNA structures that have been determined experimentally. Deep learning methods that do not require predefined features have led to notable recent advances in many fields, but their success has largely been restricted to domains where data are plentiful (11).
We designed a neural network, the Atomic Rotationally Equivariant Scorer (ARES), to address these challenges (Fig. 1). Given a structural model, specified by the 3D coordinates and chemical element type of each atom, ARES predicts the model’s root mean square deviation (RMSD) from the unknown true structure. ARES is a deep neural network: It consists of many processing layers, with each layer’s outputs serving as the next layer’s inputs (11). This network has a distinctive architecture that enables it to learn directly from 3D structures and to learn effectively given a very small amount of experimental data.
Fig. 1. The ARES network.
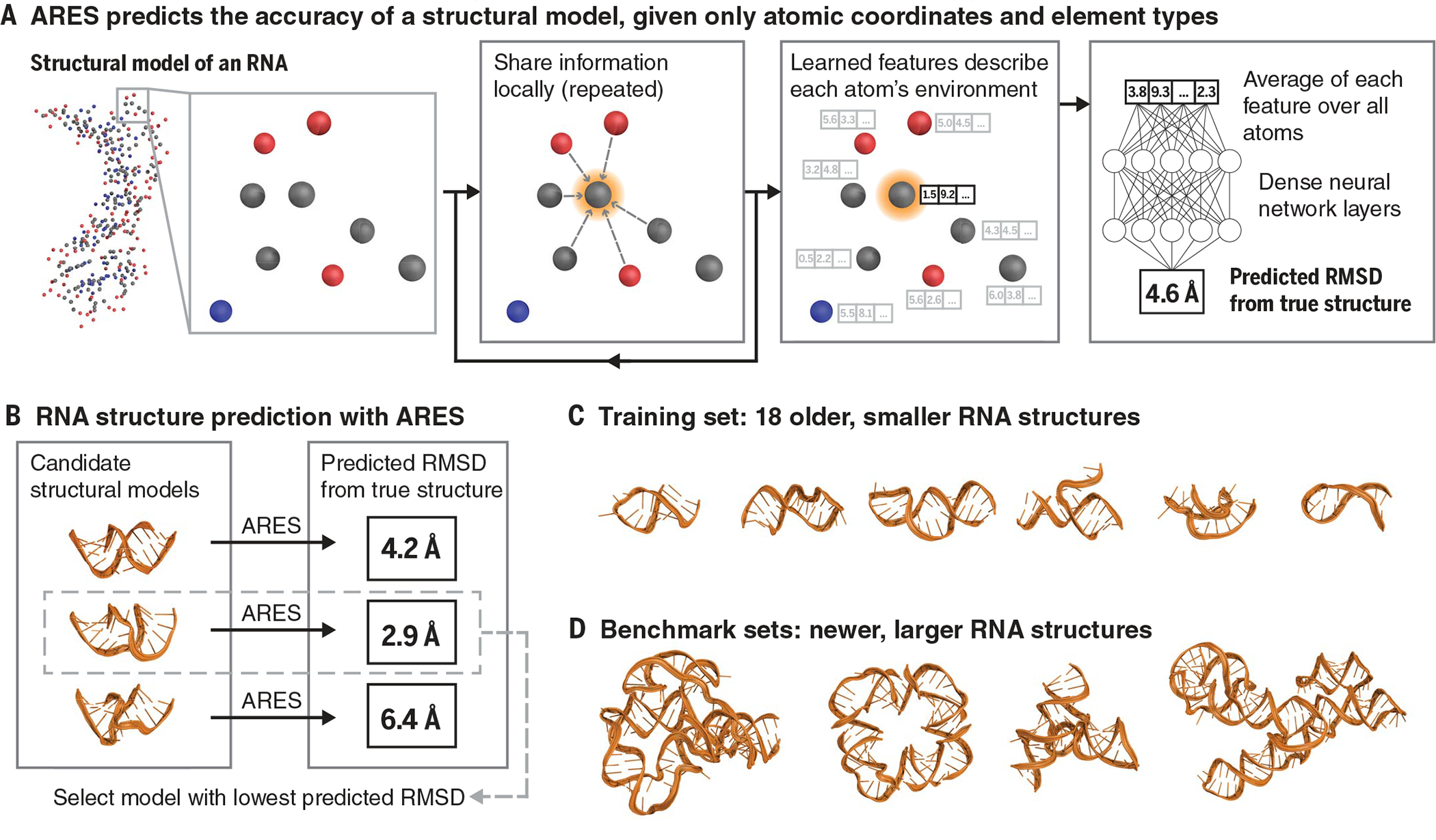
(A) ARES takes as input a structural model, specified by each atom’s element type and 3D coordinates. Atom features are repeatedly updated based on the features of nearby atoms. This process results in a set of features encoding each atom’s environment. Each feature is then averaged across all atoms, and the resulting averages are fed into additional neural network layers, which output the predicted RMSD of the structural model from the true structure of the RNA molecule. Figure S1 illustrates the ARES architecture in more detail. (B) To perform structure prediction, we use ARES to score candidate structural models (e.g., those generated by the FARFAR2 sampling software), selecting the models that ARES predicts to be most accurate (i.e., lowest RMSD). (C) ARES is trained using 18 RNA structures solved before 2007. (D) We benchmark ARES using more recently solved RNA structures, most of which are much larger than any of those used for training. Representative examples of structures used for training and benchmarking are shown in this figure, with the remainder in fig. S2.
ARES does not incorporate any assumptions about which features of a structural model are relevant to assessing its accuracy. For example, ARES has no preconceived notion of double helices, base pairs, nucleotides, or hydrogen bonds. The approach behind ARES is not at all specific to RNA and is thus applicable to any type of molecular system.
The initial layers of the ARES network are designed to recognize structural motifs, the identities of which are learned during the training process rather than specified in advance. Each of these layers computes several features for each atom based on the geometric arrangement of surrounding atoms and the features computed by the previous layer. The first layer’s only inputs are the 3D coordinates and chemical element type of each atom.
The architecture of these initial network layers recognizes that instances of a given structural motif are typically oriented and positioned differently from one another and that coarser-scale motifs (e.g., helices) often comprise particular arrangements of finer-scale motifs (e.g., base pairs). Each layer is rotationally and translationally equivariant—that is, rotation or translation of its input leads to a corresponding transformation of its output (12). This property captures the invariance of physics to rotation or translation of the frame of reference but ensures that orientation and position of an identified motif are passed on to the network’s next layer, which can use this information to recognize coarser-scale motifs. The design of these layers builds on recently developed machine learning techniques that capture rotational as well as translational symmetries (13–15)—particularly tensor field networks (16) and the PAUL method (17).
Whereas the initial layers of ARES gather information locally, the remaining layers aggregate information across all atoms. This combination allows ARES to predict a global property (in this case, the accuracy of the structural model) while capturing local structural motifs and interatomic interactions in detail.
To train ARES, we used 18 RNA molecules for which experimentally determined structures were published between 1994 and 2006 (18). We generated 1000 structural models of each RNA with the Rosetta FARFAR2 sampling method (19), without making any use of the known structure. We then optimized the parameters of the ARES neural network such that its output matches as closely as possible the RMSD of each model from the corresponding structure.
To assess ARES’s ability to identify accurate structural models of previously unseen RNAs, we used a benchmark consisting of all RNAs that were included in the RNA-Puzzles structure prediction challenge and for which experimentally determined structures were published between 2010 and 2017 (20). For each of these RNAs, we generated at least 1500 structural models using FARFAR2 (12). To ensure that some models were near native (i.e., within a 2-Å RMSD of the experimentally determined native structure), we included energetic restraints to the native structure’s coordinates when generating 1% of the models for each RNA (12). We used the trained ARES network to produce a score for each model (i.e., the predicted RMSD of each model from the native structure). We also scored each model using three state-of-the-art scoring functions: the most recent (2020) version of Rosetta (19), Ribonucleic Acids Statistical Potential (RASP) (21), and 3dRNAscore (22).
ARES substantially outperforms the other three scoring functions on this first benchmark (Fig. 2, A to C, and figs. S3 and S4). The single best-scoring structural model is near native (<2 Å RMSD) for 62% of the benchmark RNAs when using ARES, compared with 43, 33, and 5% for Rosetta, RASP, and 3dRNAscore, respectively. The 10 best-scoring models include at least one near-native model for 81% of the benchmark RNAs when using ARES, compared with 48, 48, and 33% for Rosetta, RASP, and 3dRNAscore, respectively. Each of the best-scoring near-native models was generated with energetic restraints to the native structure.
Fig. 2. ARES substantially outperforms previous scoring functions at identifying accurate structural models.
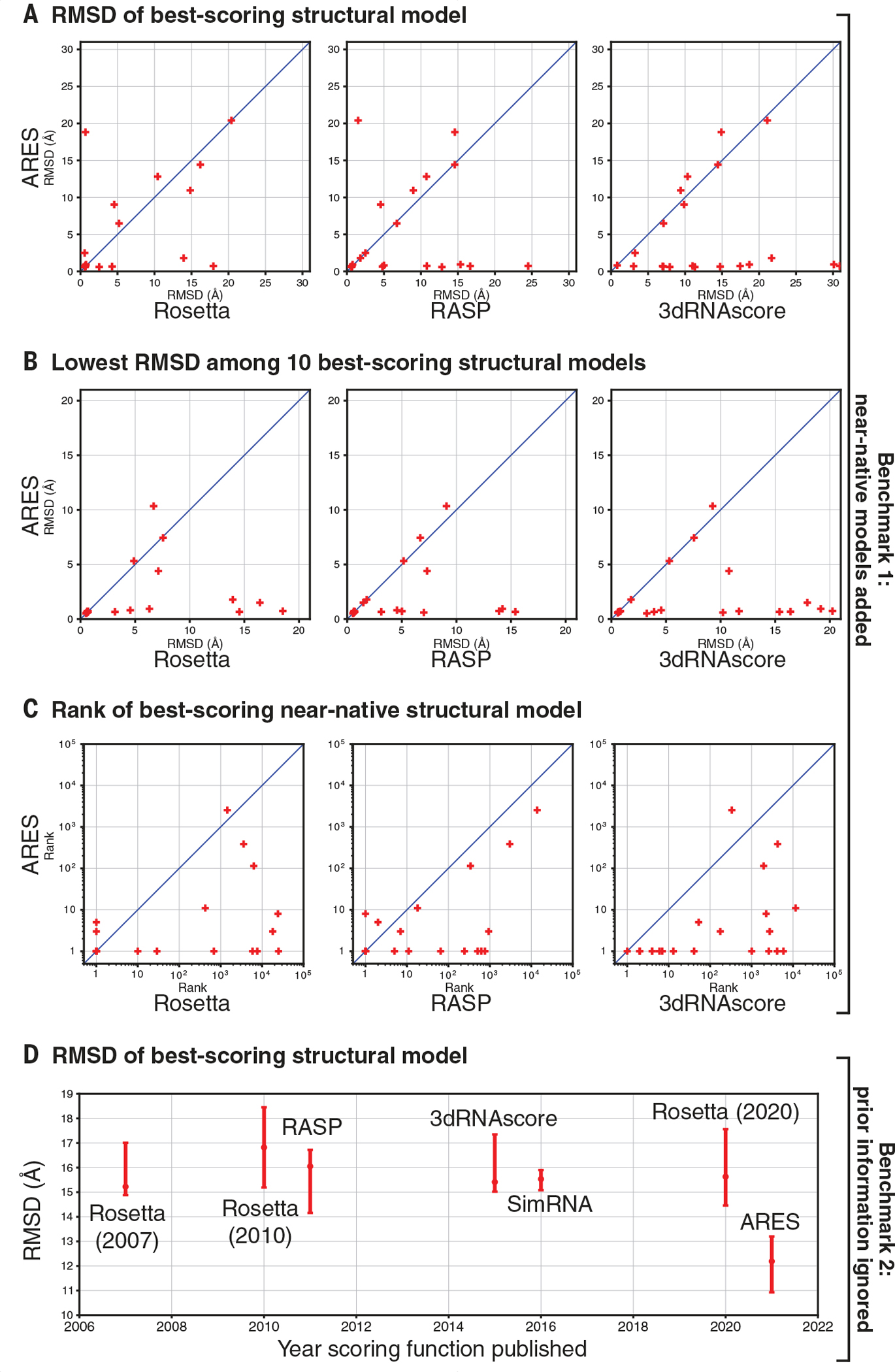
(A) Given a large set of candidate structural models for each RNA in benchmark 1—which includes some models restrained to be close to the experimentally determined (native) structure—we rank the models using ARES and three leading scoring functions. The model scored as best by ARES is usually more accurate (as assessed by RMSD from the native structure) than the model scored as best by the other scoring functions. Each cross corresponds to one RNA. “Rosetta” indicates the most recent (2020) version of the Rosetta scoring function. (B) When using ARES, the 10 best-scoring structural models for each RNA in benchmark 1 include an accurate model more frequently than when using the other scoring functions. (C) For each RNA in benchmark 1, we determine the rank of the best-scoring near-native structural model—that is, how far down the ranked list we need to go to include one near-native structural model (RMSD < 2 Å). This rank is usually lower (better) for ARES than for the other scoring functions. Across the RNAs, the mean rank of the best-scoring near-native model is 3.6 for ARES, compared with 73.0, 26.4, and 127.7 for Rosetta, RASP, and 3dRNAscore, respectively (geometric means). (D) For each of the 16 RNAs in benchmark 2—for which all structural models were generated without using any template structures or other experimental data that could provide information on local tertiary structure—we determine the RMSD of the model scored as best by each of seven scoring functions. For each scoring function, we plot the median across RNAs, with a 95% confidence interval determined by bootstrapping (12). ARES significantly outperforms each of the other scoring functions [P values 0.001 to 0.016 (12)]. Of the other scoring functions, none significantly outperforms any other [P values 0.24 to 0.66].
Current methods for sampling candidate structural models often fail to generate near-native models in a reasonable amount of computation time. We therefore compiled a second benchmark that includes no near-native models. When predicting RNA structure, experts can often find some known structures that can be used as local templates, or other published experimental data that provide information on local tertiary structure—but we ignored all such prior information when generating candidate models, to simulate a difficult modeling scenario. We selected 16 structurally diverse RNAs, all substantially different from any of those used to train ARES or those in our previous benchmark, and each including one or more of the following hallmarks of structural complexity: ligand binding sites, multiway junctions, and tertiary contacts. We scored all models using ARES as well as six other scoring functions that have been used widely over the past 14 years.
On this second benchmark, ARES again outperforms all the other scoring functions (Fig. 2D and fig. S5). The median RMSD across RNAs of the best-scoring structural model is significantly lower for ARES than for any other scoring function. The same is true when considering the most accurate of the 10 best-scoring structural models for each RNA.
Next, we turned to blind prediction of 3D RNA structure, participating in four rounds of RNA-Puzzles, a long-running community-wide challenge in which newly determined experimental structures are held in confidence until all participants have submitted their structural predictions (20). For each RNA molecule, we generated candidate structural models using the FARFAR2 sampling protocol and then selected among these models using the ARES scoring function. We describe the exact inputs to FARFAR2 in table S1. No template information was available for one of these structure prediction challenges, an intricate adenoviral RNA. Limited template information was available for the other three, which are distinct T-box–tRNA complexes (fig. S6 and table S1).
In every case, this procedure yielded the most accurate model submitted by any participant, as measured by both RMSD and deformation index (Fig. 3, fig. S8, and table S2). For each RNA, competing submissions were produced by at least nine other methods, including methods that used the same sets of candidate FARFAR2 structural models but selected among them using the judgment of human experts in the Das lab or the Rosetta (2020) scoring function. We also found that the ARES scoring function outperforms a variety of other scoring functions applied to the same sets of candidate models, including a recent machine learning approach based on a convolutional neural network (23) (table S3).
Fig. 3. ARES achieves state-of-the-art results in blind RNA structure prediction.
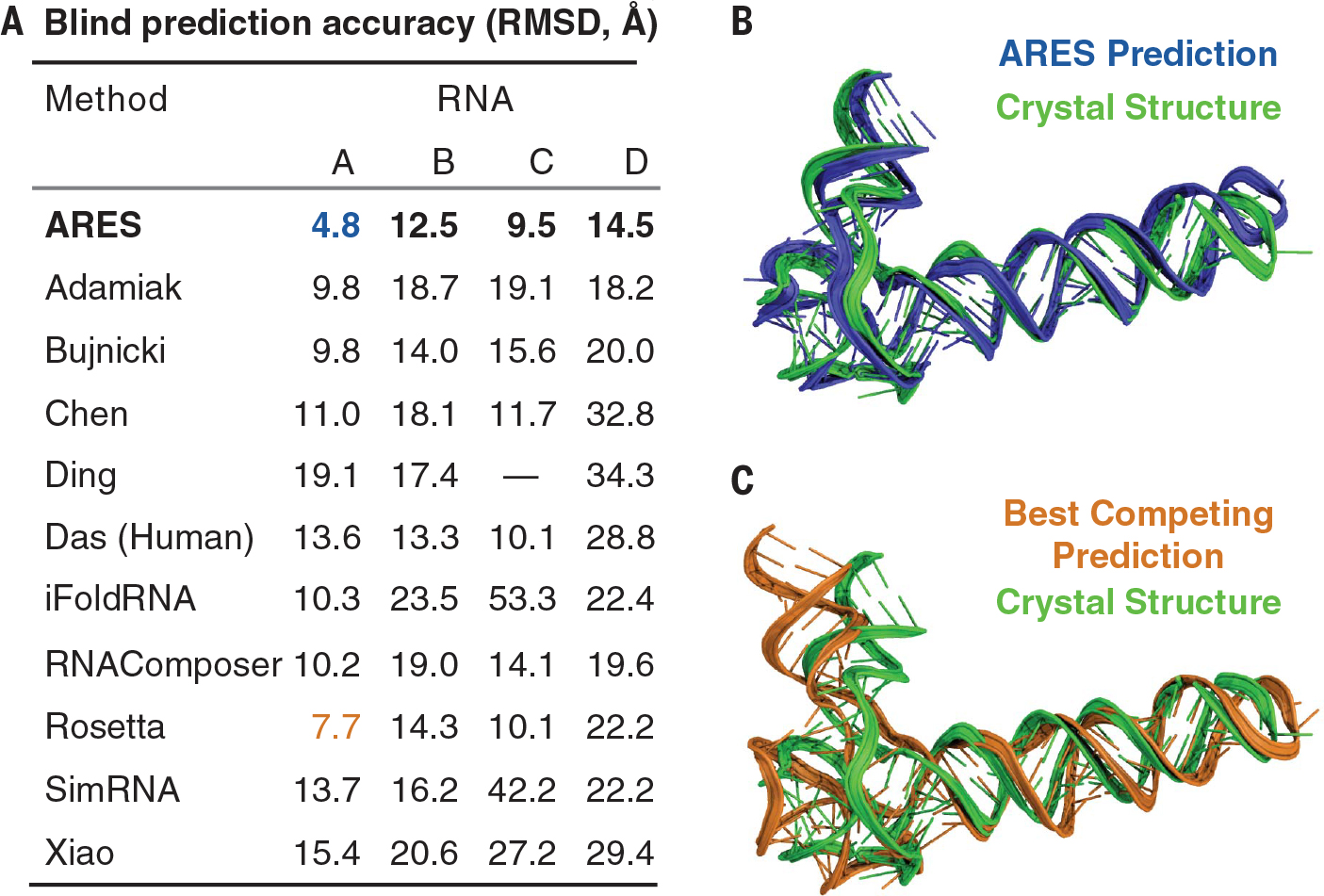
(A) We submitted structural models, which ARES selected from sets of candidates generated by FARFAR2, to four recent rounds of the RNA-Puzzles blind structure prediction challenge: RNA A (the adenovirus VA-I RNA), RNA B (the Geobacillus kaustophilus T-box discriminator–tRNAGly), RNA C (the Bacillus subtilis T-box–tRNAGly), and RNA D (the Nocardia farcinic T-box–tRNAIle), whose structures are now in the Protein Data Bank with IDs 6OL3, 6PMO, 6POM, and 6UFM, respectively. For all four RNAs, ARES produced the most accurate structural model of any method. The dash indicates no submission. (B) For RNA A, the adenovirus VA-I RNA, ARES selected a structural model (blue) with a 4.8-Å RMSD to the experimentally determined structure (green), which was not available at prediction time. (C) For RNA A, the most accurate structural model produced by any another method (orange; produced by Rosetta) had an RMSD of 7.7 Å. ARES predicts the 3D geometry of the hinge motif at lower left much more accurately than Rosetta (fig. S7). Figure S8 illustrates results for the other RNAs.
Analysis of the trained ARES network indicates that it has spontaneously discovered certain fundamental characteristics of RNA structure. For example, ARES correctly predicts the optimal distance between the two strands in a double helix—i.e., the distance that allows for ideal base pairing (Fig. 4A). In addition, the high-level features ARES extracts from a set of RNA structures reflect the extent of hydrogen bonding and Watson-Crick base pairing in each structure (Fig. 4B), even though we never informed ARES that hydrogen bonding and base pairing are key drivers of RNA structure formation. We also observe that ARES is able to accurately identify complex tertiary structure elements, including ones that are not represented in the training dataset (figs. S7 and S9).
Fig. 4. ARES learns to identify key characteristics of RNA structure that are not specified in advance.
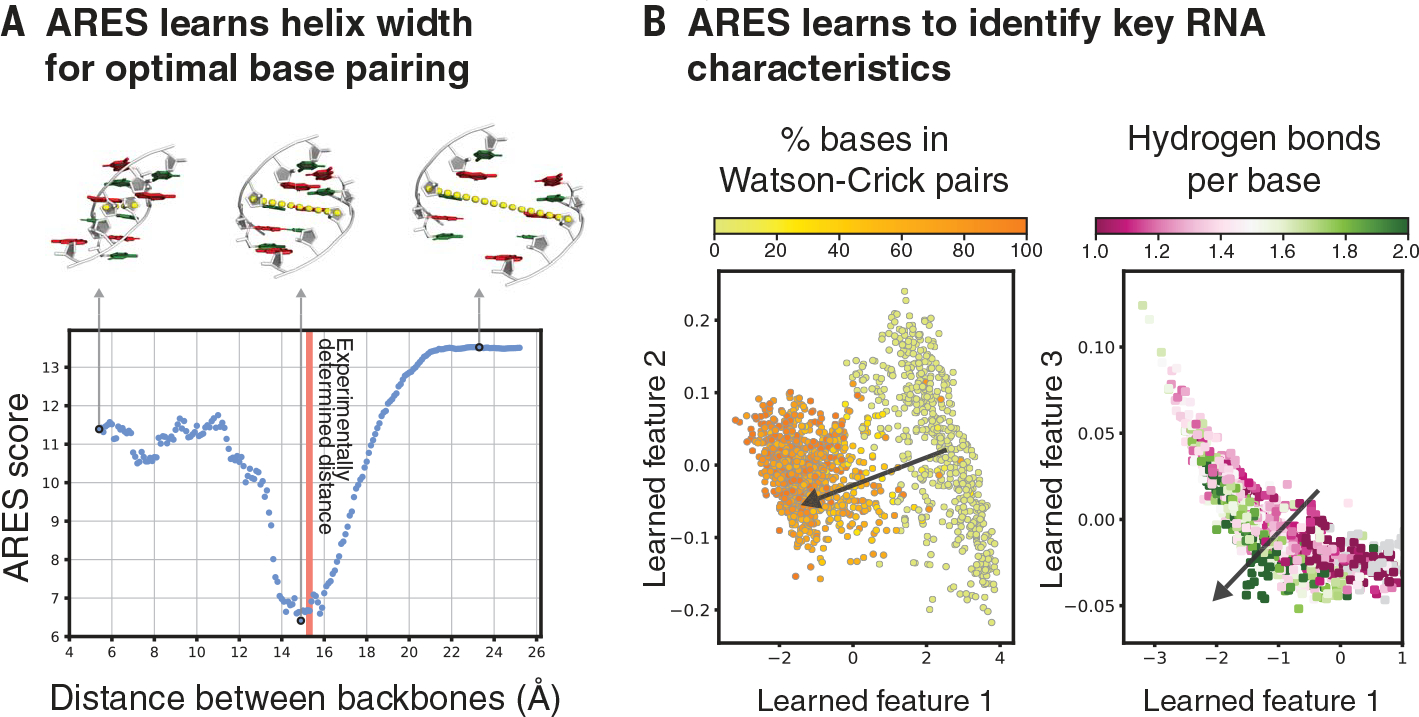
(A) As the distance between two complementary strands of an RNA double helix is varied, ARES assigns the best scores when the distance closely approximates the experimentally observed distance (red vertical line). The distance is measured between the C-4′ atoms of the central base pair (yellow dotted lines). (B) ARES’s learned features separate RNA structures according to the fraction of bases that form Watson-Crick pairs (left) and the average number of hydrogen bonds per base (right). The arrow in each plot indicates the direction of separation. Learned features 1, 2, and 3 are the first, second, and third principal components, respectively, of the activation values of the 256 nodes in ARES’s penultimate layer across 1576 RNA structures. Each dot corresponds to one of these structures (12).
Several deep learning techniques have recently been applied to problems in structural biology as well as quantum chemistry, leading to substantial advances in protein structure prediction and other areas (8, 24–30). ARES, however, tackles a particularly challenging geometric learning problem, in that it (i) learns entirely from atomic structure, using no other information (e.g., sequences of related RNAs or proteins), and (ii) makes no assumptions about what structural features might be important, taking inputs specified simply as atomic coordinates and without even being provided basic information such as the fact that RNAs comprise chains of nucleotides.
ARES’s performance is particularly notable given that all of the RNAs used for blind structure prediction (Fig. 3), and most of those used for systematic benchmarking (Fig. 2), are larger and more complex than those used to train ARES (Fig. 1). RNAs in the training set comprise 17 to 47 nucleotides (median: 26), whereas RNAs in the blind structure prediction challenges comprise 112 to 230 nucleotides (median: 152.5), and RNAs in the benchmark sets comprise 27 to 188 nucleotides (median: 75, with 31 of 37 RNAs comprising more nucleotides than any RNA in the training set).
A limitation of the current study is its reliance on a previously developed sampling method to generate candidate structural models. Future work might use ARES to guide sampling, so as to increase the accuracy of the best candidate models. ARES might be improved further by incorporating other types of experimental data, including low-resolution cryogenic electron microscopy and chemical mapping data (31).
ARES’s ability to outperform the previous state of the art despite using only a small number of structures for training suggests that similar neural networks could lead to substantial advances in other areas involving 3D molecular structure, where data are often limited and expensive to collect. In addition to structure prediction, examples might include molecular design (both for macromolecules such as proteins or nucleic acids and for small-molecule drugs), estimation of electromagnetic properties of nanoparticle semiconductors, and prediction of mechanical properties of alloys and other materials.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank R. Altman, R. Betz, T. Dao, H. Hanley, S. Hollingsworth, M. Jagota, B. Jing, Y. Laloudakis, N. Latorraca, J. Paggi, A. Powers, P. Suriana, and N. Thomas for discussions and advice.
Funding:
Funding was provided by National Science Foundation Graduate Research Fellowships (R.J.L.T. and R.R.); the U.S. Department of Energy, Office of Science Graduate Student Research program (R.J.L.T.); a Stanford Bio-X Bowes Fellowship (S.E.); the Army Research Office Multidisciplinary University Research Initiative program (R.D.); the U.S. Department of Energy, Office of Science, Scientific Discovery through Advanced Computing (SciDAC) program (R.O.D.); Intel (R.O.D.); a Stanford Bio-X seed grant (R.D. and R.O.D.); and National Institutes of Health grants R21CA219847 (R.D.) and R35GM122579 (R.D.).
Footnotes
Competing interests: Stanford University has filed a provisional patent application related to this work. R.J.L.T. is the founder of Atomic AI, an artificial intelligence–driven rational design company. R.D. has received honoraria for seminars at Ribometrix and Pfizer.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/373/6558/1047/suppl/DC1
MDAR Reproducibility Checklist
Data and materials availability:
Code for the ARES network and data analysis is available on Zenodo (32–34). All generated models, submitted blind predictions, and other ARES predictions that support the findings of this study are also available at the Stanford Digital Repository (35, 36). The trained ARES model is available at http://drorlab.stanford.edu/ares.html as a web server.
REFERENCES AND NOTES
- 1.Cech TR, Steitz JA, Cell 157, 77–94 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Warner KD, Hajdin CE, Weeks KM, Nat. Rev. Drug Discov. 17, 547–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Churkin A et al. , Brief. Bioinform. 19, 350–358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.ENCODE Project Consortium, Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman HM et al. , Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jain S, Richardson DC, Richardson JS, Methods Enzymol. 558, 181–212 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Marks DS et al. , PLOS ONE 6, e28766 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Senior AW et al. ,. Nature 577, 706–710 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Kamisetty H, Ovchinnikov S, Baker D, Proc. Natl. Acad. Sci. U.S.A. 110, 15674–15679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pucci F, Zerihun MB, Peter EK, Schug A, RNA 26, 794–802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LeCun Y, Bengio Y, Hinton G, Nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Materials and methods are available as supplementary materials.
- 13.Worrall DE, Garbin SJ, Turmukhambetov D, Brostow GJ, in Proceedings of IEEE Conference on Computer Vision and Pattern Recognition (IEEE, 2017), pp. 7168–7177. [Google Scholar]
- 14.Anderson B, Hy TS, Kondor R, in Advances in Neural Information Processing Systems 32 (NeurIPS; 2019), [Google Scholar]; Wallach H et al. , Eds. (Curran Associates, 2019), pp. 14537–14546. [Google Scholar]
- 15.Weiler M, Geiger M, Welling M, Boomsma W, Cohen TS, in Advances in Neural Information Processing Systems 31 (NeurIPS; 2018), [Google Scholar]; Bengio S et al. , Eds. (Curran Associates, 2018), pp. 10381–10392. [Google Scholar]
- 16.Thomas N et al. , arXiv:1802.08219 [cs.LG] (2018). [Google Scholar]
- 17.Eismann S et al. , Proteins 89, 493–501 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Das R, Baker D, Proc. Natl. Acad. Sci. U.S.A. 104, 14664–14669 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watkins AM, Rangan R, Das R, Structure 28, 963–976.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao Z et al. , RNA 26, 982–995 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capriotti E, Norambuena T, Marti-Renom MA, Melo F, Bioinformatics 27, 1086–1093 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Zhao Y, Zhu C, Xiao Y, Nucleic Acids Res. 43, e63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J et al. , PLOS Comput. Biol. 14, e1006514 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Behler J, Parrinello M, Phys. Rev. Lett. 98, 146401 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Proc. Natl. Acad. Sci. U.S.A. 116, 16856–16865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith JS, Isayev O, Roitberg AE, Chem. Sci. 8, 3192–3203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR, J. Chem. Inf. Model. 57, 942–957 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu K, Wang Z, Shi J, Li H, Zhang QC, in Proceedings of the AAAI Conference on Artificial Intelligence (AAAI Press, 2019), pp. 1230–1237. [Google Scholar]
- 29.Noé F, Olsson S, Köhler J, Wu H, Science 365, eaaw1147 (2019). [DOI] [PubMed] [Google Scholar]
- 30.AlQuraishi M, Cell Syst. 8, 292–301.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kutchko KM, Laederach A, WIREs RNA 8, e1374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Townshend RJL et al. , Training code for ARES neural network, Version 1.0, Zenodo (2021); 10.5281/zenodo.5088971. [DOI]
- 33.Townshend RJL et al. , ARES-specific adaptation of E3NN, Version 1.0, Zenodo (2021); 10.5281/zenodo.5090151. [DOI]
- 34.Townshend RJL et al. , Auxiliary code related to the publication “Geometric Deep Learning of RNA Structure,” Version 1.0, Zenodo (2021); 10.5281/zenodo.5090157. [DOI] [Google Scholar]
- 35.Townshend RJL et al. , Structural data used to train, test, and characterize a new geometric deep learning RNA scoring function, Stanford Digital Repository (2020); 10.25740/bn398fc4306. [DOI] [Google Scholar]
- 36.Watkins AM et al. , Structural data used to test a new geometric deep learning RNA scoring function emulating fully de novo modeling conditions, Stanford Digital Repository (2021); 10.25740/sq987cc0358. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code for the ARES network and data analysis is available on Zenodo (32–34). All generated models, submitted blind predictions, and other ARES predictions that support the findings of this study are also available at the Stanford Digital Repository (35, 36). The trained ARES model is available at http://drorlab.stanford.edu/ares.html as a web server.