

Keywords: arthritis, chondroitin sulfate, Duchene muscular dystrophy
Abstract
Chondroitin sulfate (CS) is a ubiquitous glycosaminoglycan covalently attached to the core proteins of cell surface, extracellular, and intracellular proteoglycans. The multistep and highly regulated biosynthesis of chondroitin sulfate and its degradation products give rise to a diverse species of molecules with functional regulatory properties in biological systems. This review will elucidate and expand on the most recent advances in understanding the role of chondroitin sulfate and its associate proteoglycans, in arthritis and Duchenne muscular dystrophy (DMD), two different and discrete pathologies. Highlighting not only the biodiverse nature of this family of molecules but also the utilization of CS proteoglycans, CS, and its catabolic fragments as biomarkers and potential therapeutic targets for disease pathologies.
INTRODUCTION
Chondroitin sulfate (CS) is a ubiquitous glycosaminoglycan (GAG), that is covalently attached to the protein core of proteoglycans (PGs). The diverse forms and functions of CS and its associated PGs (CSPGs) in many tissue types and disease pathologies, have been well documented, most notable in cartilage and in arthritis (1–3). The role of CS and its counter ions with their water-imbibing properties within articular cartilage provides this tissue with self-lubricative and the ability to withstand compressive stress is well documented, CS also has a diverse range of functional properties in other tissues (4, 5). Early studies pioneered this narrative, using a range of CS-specific antibodies to distinct structural features encoded within CS chains (6, 7) further layering a level of complexity in a tissue- and/or spatio-temporal manner in tissue morphogenesis. Recent collaborative efforts to collate and genotype tissues from RNA-Seq data have provided valuable collated resources to overview tissue-specific CSPGs (Fig. 1A) and CS/dermatan sulfate (DS) expression (Fig. 1B) in tissues, as well as CSPG core proteins (Fig. 2A) and CS/DS biosynthetic enzymes (Fig. 2B) from single cell transcriptomics, demonstrating the scope and breadth of CS heterogeneity and its cell instructive roles contributing to the complexity of organismal biology. This review describes the role of CS in arthritis and Duchenne muscular dystrophy (DMD). Although both are established diseases, the role of CS is well-known and associated with cartilage and arthritis, whereas the association between CS/CSPGs and DMD is less known. In this review, we detail new insights into CS and its catabolic fragments in arthritis and review the role of CS and associated CSPGs as pathological biomarkers and potential therapeutic targets in DMD. Highlighting the diverse roles this family of molecules has on different and discrete pathologies.
Figure 1.
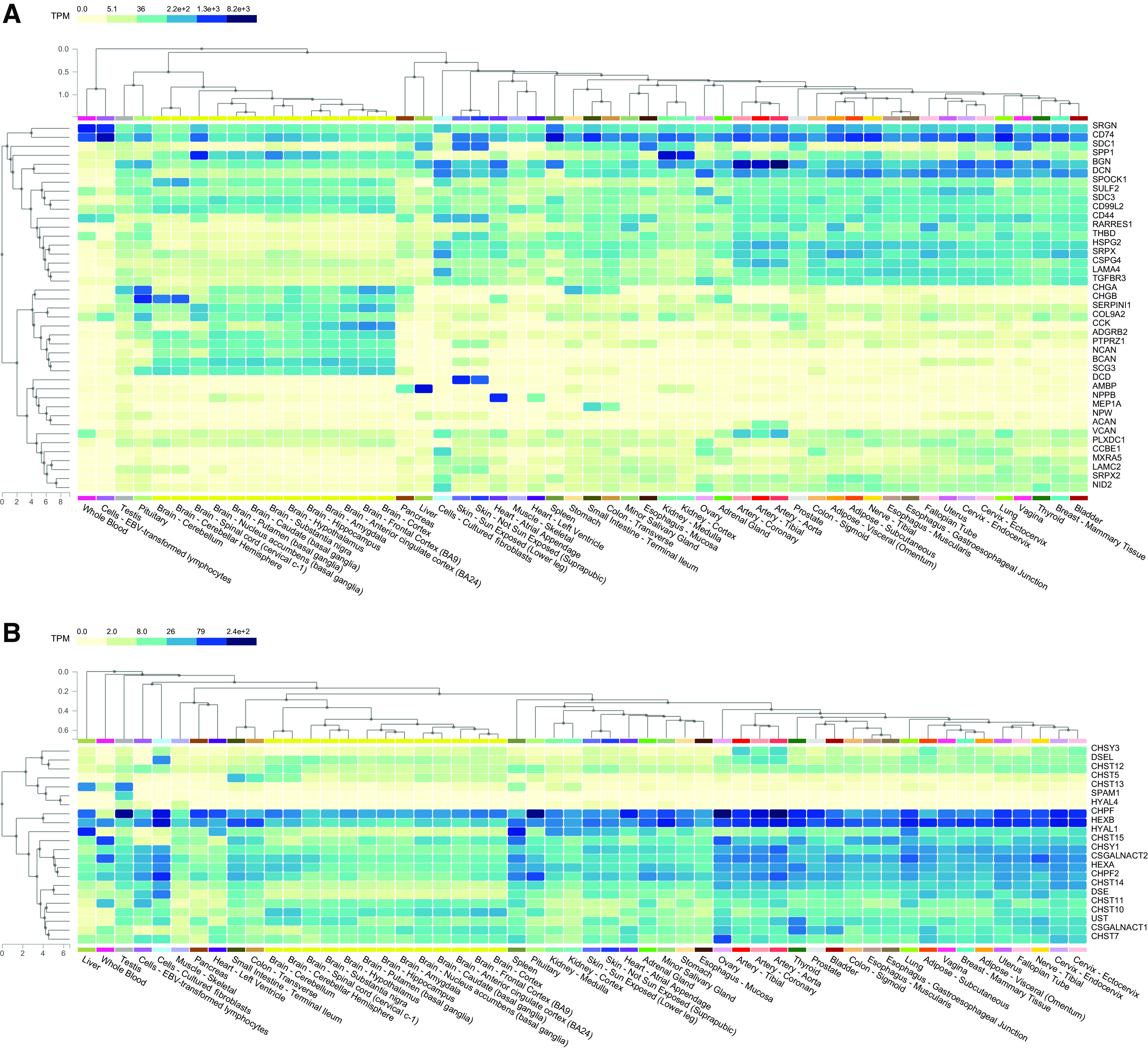
Tissue expression heat map of CSPG core proteins (A) and CS/DS biosynthetic enzymes (B). Under the CSPG superfamily umbrella, a number of subfamilies cluster particularly cell surface CSPGs, small leucine-rich PGs, and large extracellular CSPGs found in the extracellular matrix. Data publicly available from the Genotype-Tissue Expression (GTEx) project (V8) (https://www.gtexportal.org). CS/DS, chondroitin sulfate/dermatan sulfate; CSPG, chondroitin sulfate proteoglycan.
Figure 2.
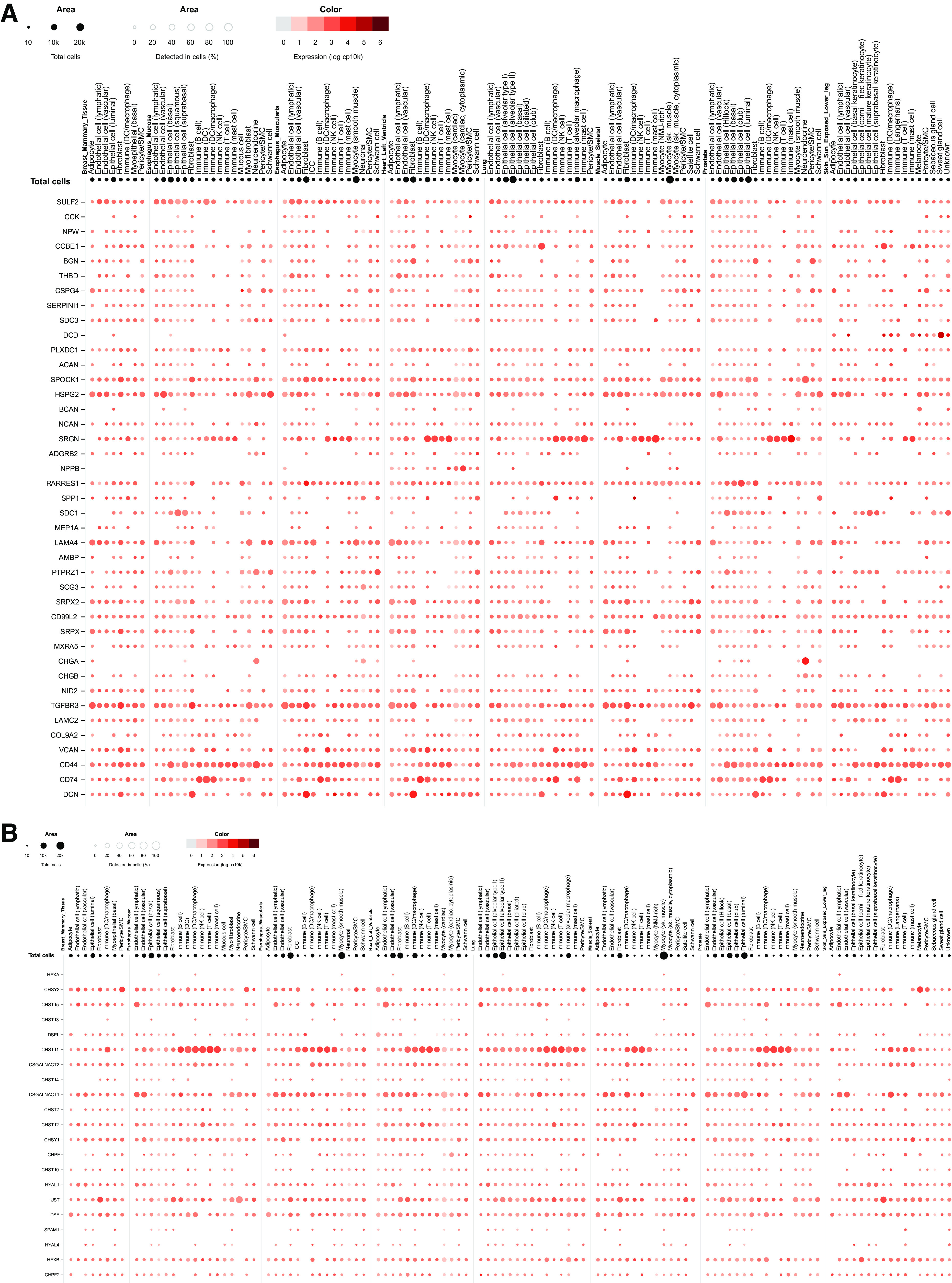
Expression of CSPG core proteins (A) and CS/DS biosynthetic enzymes detected in single cell analysis using RNA-Seq (B). Data publicly available from the Genotype-Tissue Expression (GTEx) project (V8) (https://www.gtexportal.org). CS/DS, chondroitin sulfate/dermatan sulfate; CSPG, chondroitin sulfate proteoglycan.
BIOSYNTHESIS AND CATABOLISM OF CHONDROITIN SULFATE
The biosynthesis of CS is a multistep process that produces a long polysaccharide chain covalently attached to the protein core of a CSPG (8). Similar to other members of the GAG family, heparan sulfate/heparin, keratan sulfate, CS, and its DS form are always produced as part of a CSPG. Biosynthesis of CS (Fig. 3A) (9) begins with the formation of the linkage tetrasaccharide; xylose-galactose-galactose-glucuronic acid (Xyl-Gal-Gal-GlcA) initiated through the coupling of xylose to a serine moiety along the protein core, catalyzed by xylosyltransferases (XylT-1 and XylT-2). Galactosyltransferases (GalT) I and II catalyze the addition of the two galactose residues, followed by the addition GlcA via glucuronic acid transferase I (GlcAT-I), completing the tetrasaccharide linkage sequence (10). The formation of the linkage tetrasaccharide sequence is the same for both the biosynthesis of CS/DS and HS/heparin, and it is after the formation of this sequence that the biosynthesis pathways of the two GAG families diverge. Following the linkage tetrasaccharide, addition of the first N-acetylgalactosamine (GalNAc) residue via GalNAc transferase I (GalNAcT-I) initiates the synthesis of the chondroitin backbone, followed by alternative addition of GlcA and GalNAc, which forms the repeating chondroitin disaccharide backbone, added via GlcA transferase II (GlcAT-II) and GalNAc transferase II (GalNAcT-II), respectively (10–12). Various types of CS disaccharide structures are formed during the polymerization of the chondroitin chain. Some GlcA residues can be epimerized to iduronic acid (IdoA) by two GlcA C-5 epimerases, (dermatan sulfate epimerase (DSE)1, DSE2/DSEL), forming the DS disaccharide unit. Furthermore, hydroxyl groups on GalNAc or GlcA/IdoA can be modified by several different sulfotransferases resulting in different CS and DS disaccharides isomers, with common CS structures shown in Fig. 3B, and all possible sulfation modifications for both CS and DS in Fig. 3C (13).
Figure 3.

Biosynthesis and disaccharide structures of CS/DS. The biosynthesis pathway of CS/DS (A) is a multistep highly regulated process, resulting in an extensive range of disaccharide structures [the more common disaccharide CS structures (B) and all possible sulfate modifications (C)]. CS, chondroitin sulfate; CS/DS, CS/dermatan sulfate.
Given the complexity and inherent heterogeneity of CS, there is a multitude of degradation enzymes that work together to catabolize CS chains, in addition to enzymes that also cleave and breakdown the protein core of the associated CSPGs. Degradation of CS predominantly occurs intracellularly within the lysosomes (8, 14), where the initial step involves the CS chain being fragmented via endo-type hydrolases. Following this step, the fragmented CS is broken down further from the nonreducing end by a series of glycosidases and sulfatases cleaving the CS oligosaccharides into disaccharide units, and removing sulfate moieties, respectively, depending on the glycosidic linkage and the location of sulfate. Although the degradation of CS is a highly ordered process, the specific enzymes associated with CS catabolism are still relatively unexplored. The family of hyaluronidases (HYALs), named due to their ability to degrade hyaluronan, comprises six members; HYAL1, HYAL2, HYAL3, HYAL4, SPAM1 (PH-20), and HYALP1 (15), though a number of them also recognize CS as a substrate (16). Human HYAL1 and SPAM1 catabolize CS and hyaluronan to a similar extent (17), whereas CS is the predominant substrate for HYAL4 (16), also referred to as CS hydrolase (CHSE). Furthermore, while HYAL1, SPAM1, and HYAL4 are capable of CS catabolism, the CS substrate they prefer differs from HYAL1 and SPAM1 degrading chondroitin 4-O-sulfate (C4S, CS-A) (17), and HYAL4/CHSE chondroitin 2-O-, 6-O-disulfate (C2,6S, CS-D) and to a lesser extent chondroitin 6-O-sulfate (C6S, CS-C) (16) (Fig. 4). Although HYAL4 expression was originally shown to be limited to placenta, testis, and skeletal muscle (16, 18, 19), a recent study from Yamada and Mizumoto (20) has demonstrated ubiquitous expression in various mouse organs, in addition to, demonstrating overexpression of HYAL4 resulted in a decrease in the total amount of CS produced by CHO cells. Further suggesting the involvement of HYAL4 in cellular catabolism of CS.
Figure 4.
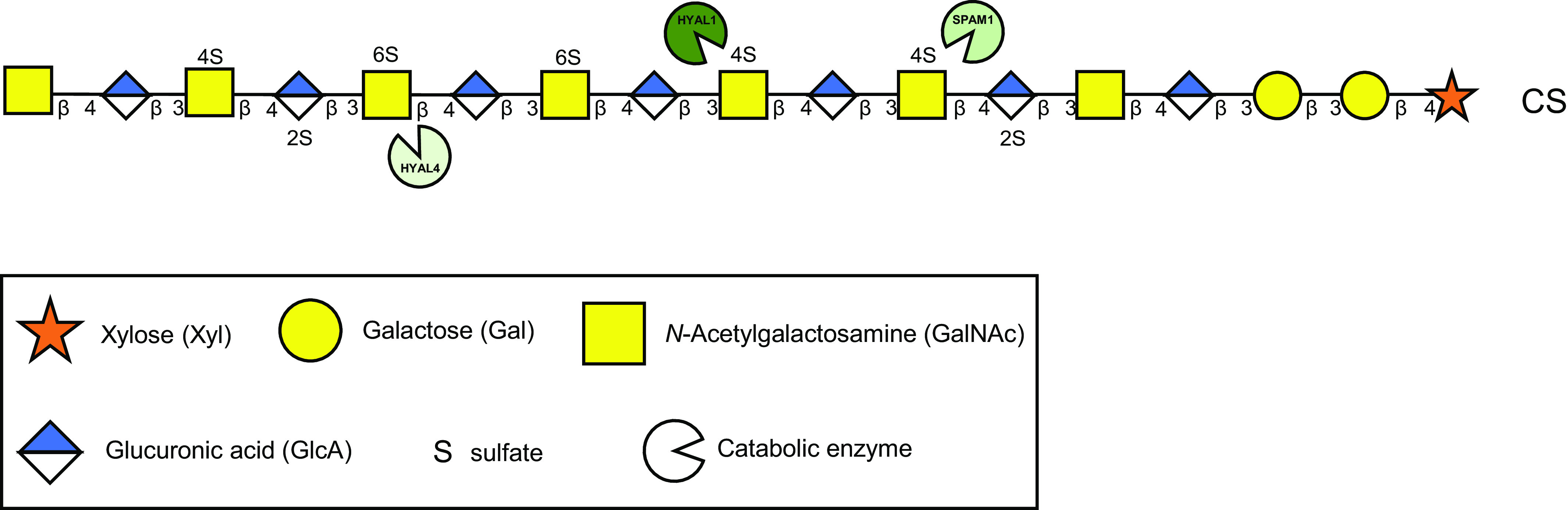
Endoglycosidase catabolism of CS chains. Several endoglycosidase degradation enzymes can cleave CS chains. Each enzyme has preferred CS substrates with HYAL1 and SPAM1 typically cleaving C4S (CS-A) units, HYAL4/CHSE cleaving at C2,6S (CS-D) units, and less avidly C6S (CS-C). No specific endoglycosidase for DS has been discovered to date. CS, chondroitin sulfate; DS, dermatan sulfate.
ARTHRITIS
Arthritis is a condition that impacts articular cartilage and diminishes the weight-bearing properties of the joint causing impaired articulation, joint swelling, and pain. There are two main types of arthritis, osteoarthritis (OA) and rheumatoid arthritis (RA), and while both have divergent causes, they are both characterized by gradual cartilage breakdown and loss of articular cartilage particularly in weight-bearing regions of the joint, eventually leading to erosion of cartilage down to the underlying bone (eburnation) (21). Arthritis is now recognized as a global disease of the joint and while changes in the articular cartilage have been focused on in the past it is now recognized that changes in the synovium, subchondral bone, ligaments, menisci, and infra-patellar fat pad all contribute to degenerative pathology in the joint and diminished joint function (22, 23).
The organization of the extracellular matrices of hyaline, fibro, and elastic cartilages differ providing them with the ability to resist tensional and compressive stresses. Hyaline cartilage is found in weight-bearing joints, including the hip, knee, and shoulder. With aging, arthritis commonly affects these joints resulting in degenerative changes compromising joint function. The extracellular matrix of hyaline cartilage is composed primarily of a network of type II fibrillar collagen that constrains aggrecan-hyaluronan aggregates that hydrate and provide weight-bearing properties to cartilage. Along with type II collagen and aggrecan, hyaline cartilage ECM components include decorin, biglycan, fibronectin, and multiple types of collagens (I, IV, V, VI, IX, and XI) (24). The predominant PG in cartilage is the multimodular CSPG, aggrecan. Aggrecan’s 220–250 kDa core protein contains three globular domains, the N-terminal G1 domain (37 kDa) interacts with link protein and hyaluronan. The remainder of the core protein contains a keratan sulfate-rich region containing ∼20 keratan sulfate chains of 5–15 kDa in size and a more extensive C1 and C2 region extensively substituted with ∼100 CS chains each ∼20 kDa in size. The C-terminus of the aggrecan core protein contains a G3 globular domain (36 kDa). (25). Within cartilage, aggrecan forms massive space-filling link protein stabilized macro-aggregate networks that provide dynamic hydration, viscoelastic properties, and the ability of cartilage to resist compressive forces. Aggrecan is also attached to collagen VI networks, through the G3 domain. Hyaluronan-aggrecan-collagen networks have mechanotransductive properties sending instructive cues to chondrocytes to maintain a homeostatic balance in cartilaginous tissues, which maintains their optimal functional properties (26).
A characteristic of an OA joint is the catabolic process that degrades the hyaluronan backbone that stabilizes the hyaluronan/aggrecan complex, as well as the core protein of aggrecan. A topic that has been well reported and reviewed within the literature (2). Although aggrecan is the predominant cartilage PG, a number of small leucine-rich PGs (SLRPs) including decorin, biglycan, lumican, and keratocan may also play an important role in OA (27). Breakdown products of these cartilage ECM SLRPs (28) were found to be elevated in patients with OA, indicating their potential for use as OA biomarkers. Studies specifically on the meniscus have shown how inflammatory cytokines TNFα and IL-1 drive this degenerative process and the zonal differential responsiveness of meniscal cell populations and how they generate catabolic enzymes that degrade ECM components in other tissues in the joint complex (23). This emphasizes the interdependence of the constituent joint tissues in a global multitissue structure (22).
The presence of CS degradation products, as well as CS biosynthesis or degradation enzymes, have a potential role as a biomarker for OA progression (29). An early study evaluating CS in OA found both a decrease in CS concentration as well as decreased CS chain length in patients (30). Similar results demonstrating a decrease in CS chain length as well as amount of CS in OA legions were also shown by Ishimaru et al. (31). Through probing for the presence of enzymes FAM20B (Family with sequence similarity 20, member B), and β-1,3-galactosyltransferase 6 (B3GALT6), that are involved in the biosynthesis of CS (32, 33), a study by Lei et al. (34) demonstrated decreased expression of these enzymes in tissue sections (human articular cartilage) with OA, where expression varied between the superficial, middle, and deep cartilage zones. In addition, CS encoding glycosyltransferases (chondroitin polymerizing factor, CS N-acetylgalactosaminyltransferase 1, and 2), and chondroitin sulfate synthase 3 (CSS3) were shown to be reduced in OA cartilage samples (31), and enzymes involved with CS sulfation by chondroitin 6-O-sulfotransferase 1, chondroitin 4-O-sulfotransferase 2, N-acetylgalactosamine-4-sulfate 6-O-sulfotransferase, and uronyl 2-O-sulfotransferase, which are encoded by carbohydrate sulfotransferase-3 (CHST3), CHST12, CHST15, and UST, respectively, were significantly reduced in patients with OA, with differing levels observed within the superficial, middle, and deep cartilage zones (35). Decreases in CHST2, CHST3, CHST7, CHST15, N-acetylgalactosamine-6-sulfatase, 3′-phosphoadenosine 5′-phosphosulfate synthases 1 and 2, CHST8, CHST12, CHST13, CHST14, and arylsulfatase B/N-acetylgalactosamine-6-sulfatase were also demonstrated in OA cartilage samples (36). Although changes in levels of CS biosynthetic enzymes may indicate a role in OA pathogenesis, the structure of CS, particularly the biochemical composition is also a critical element to take into consideration. Early studies evaluated the structure of CS in synovial fluid of patients with various forms of arthritis (37). Treatment of samples with the bacteria-derived lyase, chondroitinase ABC, followed by high-performance liquid chromatography analysis to determine the CS disaccharide content demonstrated that the structure of CS between different arthritis types varied, particularly the ratio of C4S to C6S, with this ratio being the highest in traumatic arthritis followed by OA, and lastly RA. Similar findings were presented in an ovine osteoarthritic model (38), where it was demonstrated that with increasing OA score (as an indication of disease progression), the level of C4S, C6S, and C6S/C4S in synovial fluid increased. Ishimaru et al. (31) demonstrated that decreased amount of nonsulfated chondroitin (C0S) and C4S in OA cartilage specimens, and the amount of C6S and the ratio of C6S/C4S did not significantly change. Furthermore, Lin et al. (36) evaluated specimens collected from the knees of 60 patients with end-stage OA and CS compositional analysis of the samples showed that in addition to there being a decrease in the total CS of the OA cartilage samples, the amount of C4S decreased. More recent studies have shown that fragments of CS, in an in vitro model, are capable of upregulating catabolic phenotypes of chondrocytes, through the upregulation of Mmp13, Adamts5, as well as oxidative associated genes (Fth1, Hmox1, and Txn) (39).
Even though the composition of CS in OA has been explored, a significant gap remains in our understanding of the presence and mechanistic role(s) of CS-degrading enzymes. Due to its well-known role in hyaluronan degradation, understanding the role of HYAL1 in disease pathologies has focused primarily on hyaluronan rather than CS. The Hyal1 knockout mouse has been reported to exhibit an osteoarthritic phenotype (40), however, characterization of CS structure in the absence of Hyal1 was not evaluated. Interestingly, the CS terminal structure that remains following the cleavage of CS by either HYAL1 and HYAL4 has been demonstrated to be detected using a suite of CS antibodies (41), and it was proposed that these terminal CS structures that are also detected in OA (42) may be due to the presence of HYAL1 or HYAL4 activities.
Although the evaluation of CS biosynthesis enzymes has demonstrated differences between OA pathologies and normal cartilage tissue, specific CS structures and fragments of CS, and their role in both mechanistic events in OA and their potential as biomarkers remains relatively unexplored, demonstrating a significant gap in our understanding of this disease and highlights the potential role of CS structures as diagnostic markers.
DUCHENNE MUSCULAR DYSTROPHY
Duchenne muscular dystrophy (DMD) is part of a group of genetic conditions characterized by progressive muscle weakness and wasting (atrophy). DMD is the most common type of muscular dystrophy, severely affecting males, in rare cases females too, due to loss-of-function mutations in the X-linked dystrophin (DMD) gene (43). DMD encodes a muscle cell membrane-stabilizing protein that connects actin filaments to the sarcolemma and maintains muscle strength, primarily in skeletal and cardiac muscles (44). However, in patients with DMD, the absence of functional dystrophin protein diminishes muscle durability, such that as muscles contract and relax with use, the fibers damage over time.
Under normal circumstances, muscles are regenerated from satellite cells, a mitotically quiescent population that resides beneath the basal lamina, next to the plasma membrane of muscle fibers in the satellite cell niche. When muscle tissue is injured, the satellite cells activate and begin to proliferate, forming myoblasts. These myoblasts differentiate into myocytes that then fuse with each other to produce new myofibers or fuse with existing myofibers to repair the tissue. A minor proportion of satellite cells self-renew and return to a quiescent state, awaiting the next injury stimuli. However, in DMD, the muscle regeneration capacity of satellite (muscle stem) cells is diminished, and continuous tissue damage is thought to pressurize satellite cells to generate new muscle, which leads to decreased cell populations, progressive weakness, and chronic degeneration (45). Optimal tissue regeneration is also directed by cross talk with the muscle extracellular matrix. In DMD, persistent inflammation results in the accumulation of fibrotic extracellular matrix in muscle tissue around skeletal muscle fibers and interstitial space, particularly of CSPGs such as versican, biglycan, and decorin (46). Fibrosis causes dysfunction of the skeletal muscle and contributes to the lethal phenotype of DMD.
Of the 41 members in the CSPG family, 22 CSPGs have been detected in healthy skeletal muscle by RNA-Seq (Table 1) using 17 CS/DS-associated enzyme genes to build and modify the CS/DS chains. However, analysis of RNA-Seq data from DMD muscle revealed significant upregulation of seven CSPGs and three CS/DS biosynthetic enzymes, priming changes in the CS/DS chain structures expressed: namely, CHPF that polymerizes the backbone of the chain, one of the specific 4-O-sulfotransferases, CHST13, and dermatan sulfate epimerase-2/dermatan sulfate epimerase-like, one of two epimerases that switches GlcA residues to IdoA, creating DS. A separate study also noted a significant increase in another C4ST enzyme, CHST11, and a decrease in HYAL4 mRNA expression (47), suggesting altered CS/DS degradation and CSPG turnover may also affect DMD pathology. Compositional analysis of CS disaccharides also supported changes in CS/DS chain biochemical structure, with a shift toward more sulfated CS chains in DMD skeletal muscle accompanied by a general increase in CS/DS content in the tissue.
Table 1.
CSPG and CS/DS associated enzyme RNA-Seq transcripts detected in healthy skeletal muscle (with a medium TPM above 1) and in DMD muscle [upregulated 5-fold, italic; 1.5-fold, bold; downregulated, underlined; *data from (47)]
| Healthy Skeletal Muscle Genes |
DMD Muscle Genes |
||
|---|---|---|---|
| CSPGs | CS/DS Enzymes | CSPGs | CS/DS Enzymes |
| CD99L2 | CSGALNACT1 | CD74 | CHPF |
| CD74 | CSGALNACT2 | LAMA4 | DES2/DSEL |
| DCN | CHSY1 | VCAN | CHST13 |
| BGN | CHPF | CD44 | CHST11* |
| SRGN | CHPF2 | BGN | HYAL4/CHSE* |
| CSPG4 | CHST7 | SRPX | |
| HSPG2 | CHST10 | SPP1 | |
| SULF2 | CHST12 | SULF2 | |
| CD44 | CHST13 | ||
| TGFBR3 | CHST14 | ||
| THBD | CHST15 | ||
| SPOCK1 | UST | ||
| SRPX | DSE | ||
| SRPX2 | HYAL1 | ||
| LAMA4 | HYAL4/CHSE | ||
| SDC3 | HEXA | ||
| PLXDC1 | HEXB | ||
| LAMC2 | |||
| NID2 | |||
| RARRES1 | |||
| VCAN | |||
Healthy skeletal muscle data were obtained from Genotype-Tissue Expression (GTEx) project (V8). DMD skeletal muscle data obtained from previous studies (47–49). CS/DS, chondroitin sulfate/dermatan sulfate; CSPGs, chondroitin sulfate proteoglycans; DMD, Duchenne muscular dystrophy; TPM, transcripts per million.
Several skeletal muscle CSPGs (Fig. 1, Table 1) have been studied, especially the extracellular matrix CSPGs biglycan, decorin, and versican. Versican plays multiple roles in skeletal muscle including satellite cell proliferation (50), myogenesis, and myofiber formation, as well as in the process of inflammation (51, 52). In healthy skeletal muscle, versican expression is relatively low and expressed mainly by fibroblasts, dendritic cells, macrophages, and mast cells (Fig. 2A). Accumulation of V0/V1 versican forms is associated with DMD pathology and decreased physical activity of skeletal muscle. Both the V0 and V1 forms of versican display multiple CS chains, which participate in ligand interactions [e.g., transforming growth factor-β (TGF-β)] that modulate cell signaling and cell behavior. Moreover, dystrophic muscle has upregulated TGF-β that promotes fibrosis and further versican synthesis (53), augmenting versican accumulation. Using the mdx DMD mouse model, genetic reduction of versican showed improved physical activity and attenuated dystrophic pathology in the diaphragm muscle, demonstrating that the accumulation of versican contributes to the pathophysiological state of DMD muscles (54). Decorin and biglycan are members of the SLRP family. Decorin is predominantly synthesized by skeletal muscle fibroblasts, although all skeletal muscle cell types can express it (Fig. 2). Interestingly, expression of decorin varies in DMD muscle (55, 56) although multiple positive roles for decorin in muscle are known: 1) decorin interacts with collagen fibers, regulates collagen fibrillogenesis, and reduces collagen I expression; 2) decorin interacts with myostatin that negatively regulates myogenesis; and 3) decorin binds TGF-β and inhibits its activity, protecting myoctyes from differentiating into fibrotic cells. (57–60). Together, these positive activities potentiate the use of decorin as a therapeutic agent for DMD, primarily to combat TGF-β-induced fibrosis. The other SLRP, biglycan, interacts with dystroglycans and contributes to synapse stability. Although biglycan is increased in DMD muscle in relation to increased fibrosis (55, 56), it does not always carry CS chains, with the non-GAG form displaying differential behavior to the CSPG form. Non-GAG biglycan has pivotal roles in the recruitment of utrophin, a paralog of dystrophin, and NOS signaling at the muscle cell membrane, which improves the health and function of muscles (61). Hence, non-GAG biglycan is being explored as a potential therapeutic for DMD (62).
Notably, CD74, which performs important functions in injury, inflammation, and repair (63), had significantly increased expression in DMD muscle (Table 1). CD74 is known in other contexts for its regulation of cell survival (64, 65), but has not been closely studied in DMD. Interestingly, the upregulation of CD44 (Table 1) may be entangled with CD74 as CD44 is required to transduce CD74/MIF-induced protection from apoptosis via intracellular ERK signaling (66). CD44 also regulates myoblast migration and differentiation (67). However, CD44-activated T cells (both CD4+ and CD8+) have been demonstrated to play detrimental roles in DMD muscle of mdx mice, contributing largely to muscle pathology (48, 68, 69), indicating that CD44 functions could contribute to both positive and negative aspects of DMD.
During the development of new muscle, the expression and/or localization of several cell surface CSPGs change, including syndecan 1 and 3. These hybrid HS/CSPGs are both localized to myoblasts and muscle fibers in embryonic tissue (70). Postnatal tissue, however, no longer expresses syndecan 1, and expression of syndecan 3 is restricted to satellite cells (and maybe vascular cells) (70). Sdc3−/− satellite cells display impaired self-renewal abilities and quiescence maintenance, favoring myoblastic states instead (71). Interestingly, DMD muscle shows augmented expression of syndecan 3, suggesting that defective satellite cell dynamics may underpin progressive disease pathology (72). Other CSPGs, such as thrombomodulin, a part-time CSPG, CSPG4, and sulfatase-2 (SULF2), are specifically decreased during DMD satellite cell activation (72). Thrombomodulin is a cofactor for thrombin and reduces blood coagulation via activation of protein C in the anticoagulation pathway (73). Thrombomodulin also regulates Factor I C3b inactivation, but its role in muscle biology has not yet been elucidated. CSPG4 is normally expressed in the sarcolemma and neuromuscular junction, with expression declining gradually with age. However, CSPG4 was reported to be overexpressed in biopsies of patients with DMD, suggesting an important pathological role in DMD aside from satellite cells (74). CSPG4 expression marks fibrogenic/adipogenic differentiation induced by TGF-β and basic fibroblast growth factor in skeletal muscle, indicating that CSPG4-targeted therapeutics could prevent fibrotic tissue formation in DMD (75). SULF2 activity is known to enhance myoblast fusion via promotion of the HS/Wnt canonical signaling pathway (76, 77). Thus, decreased expression of active SULF2 may skew the balance of Wnt canonical/noncanonical signaling to favor myogenic differentiation over myoblast fusion, thereby contributing to the reduced regeneration capacity of satellite cells observed in DMD muscle. Recently, the CS chain on recombinant human SULF2 was reported to modulate enzyme activity, with removal of the CS chain increasing SULF2 desulfation of HS chains (78). Notably, mRNA expression of the CS degradation enzyme, HYAL4/CHSE, was significantly downregulated in human DMD biopsies (47), suggesting that a lack of CS degradation (i.e., removal of the SULF2 CS chain) may further compound the problem.
Alongside the recent addition of SULF2 to the CSPG family, several other novel CSPGs have also been identified including two basement membrane proteins, laminin subunit α 4 (lama4) and nidogen 2 (NID2) (79). Lama4-mediated interactions are important for extracellular matrix-muscle synergy. Lama4 is localized and upregulated in damaged fibers, contributing to fiber survival by modulating muscle integrity through integrin signaling (80). NID2, collagen IV and VI, laminin, collagen XVII, agrin, and perlecan form a stabilizing network in basement membranes (81). No biochemical information on the lama4 or NID2 CS chain types, length, and/or sulfation patterns have been gathered yet, although these data may provide further insight into the role of CSPGs in muscle preservation and highlight potential new mechanistic pathways for targeted treatment. Sushi repeat-containing protein X-linked (SRPX) is another relatively new member of the CSPG family. First described as a suppressor of v-Src transformation that inhibits its myogenic differentiation (82), SRPX localizes with the cytoskeleton and interacts with pelota mRNA surveillance and ribosome rescue factor, a protein that detects and degrades aberrant mRNAs (48). This suggests that DMD SRPX overexpression may be involved in muscle regeneration, although little else is known about SRPX biology in healthy or DMD skeletal muscle except that SRPX expression is upregulated (Table 1). Finally, osteopontin is predominantly known as a secreted phosphoprotein displaying pleiotropic functions with matrix, chemoattractant, and cytokine properties. However, in 2015, two CS chain assembly sites were identified in osteopontin (serines 234 and 308) (83). Phosphorylation of those serines has also been reported (84), suggesting that CS chains may not always form on those sites. Markedly, levels of osteopontin are upregulated in DMD muscle (Table 1) (48, 85). Studies in the mdx mouse model pinpointed osteopontin as a profibrotic cytokine, modulating the type of immune cells that infiltrated the dystrophic muscle. Both muscle and immune cells were significant sources of secreted phosphoprotein-1 (SPP1) expression, with a particular subset of T cells (Vβ8.1/8.2+) responsible for most expression (85). Ablation of SPP1 expression significantly improved fibrosis via infiltration of regulatory T cells rather than neutrophils and natural killer-like cells, and a reduction in TGF-β at later stages of the disease (85). In a follow-up study, Capote et al. (86) investigated the effects of SPP1 ablation on macrophages, revealing the removal of osteopontin. Together, these studies highlight osteopontin as another therapeutic target for patients with DMD, promising a reduction in inflammation and fibrosis that drive disease lethality and promotion of muscle longevity, strength, and improved repair. Whether a CSPG form of osteopontin drives DMD pathology remains to be verified, and what potential roles the CS chains might play can only be surmised.
There is no cure for DMD, and care consists of pharmacological treatments to improve short-term muscle strength (e.g., corticosteroids, β2 agonists), physical therapy to maintain muscle strength, flexibility and function, corrective surgery, reliance on a pacemaker to stabilize cardiac muscle, and eventually, respiration assistance when breathing muscles are affected. Methods for targeting CSPG accumulation in DMD have shown beneficial outcomes for disease progression. Prevention of CSPG accumulation by glucocorticoid treatment, which suppresses CSPG synthesis and matrix accumulation (54), slows disease progression (87), and is currently recommended as a standard of care. However, better (or complementary) treatments are still desired. Clearance of CSPG accumulation by intramuscular injection of bacterial CS-degradation enzyme (88) in the DMD mdx mouse model also improved skeletal muscle regeneration and pathology, suggesting administration of CS degradation as another therapeutic option for patients with DMD. Of the human enzymes that degrade CS, HYAL4/CHSE is the only CS-specific enzyme, which coincidently is also lacking expression in DMD muscle biopsies compared with healthy muscle tissue (47, 89). HYAL1, with dualistic CS and hyaluronan degradation activities, also contributes to degradation of CS in muscle. Notably, Hyal1 expression was coexpressed with Myog in mouse mdx muscle cells and knockdown of Hyal1 in differentiating C2C12 cells led to the accumulation of matrix CS, suggesting that HYAL1 activity may regulate myoblast fusion (88). Due to their discrete CS degradation abilities (Fig. 4), both recombinant human HYAL1 and HYAL4 enzymes may possess therapeutic potential to aid clearance and turnover of accumulated CS and maintain correct turnover of CS, ameliorating muscle regeneration for patients with DMD.
CONCLUSIONS
The heterogeneity of CS/DS structures along with its ubiquitous expression in tissues enables this glycan module to regulate and modulate a diverse range of disease pathologies. Although several CSPGs have established roles in disease pathology, many CSPGs accompanied by CS/DS oligosaccharide structure and their catabolic fragments have not been explored to date, demonstrating that substantial gaps remain in our knowledge. This aside, data gleaned from both arthritis and DMD studies thus far clearly demonstrate that CS serves both as a useful biomarker for pathology and as a therapeutic target. Connecting the CSPG(s) with detailed analysis of specific CS/DS chains, oligosaccharides, and their degradation process will further our understanding of underlying mechanisms that drive disease pathology and guide the development of a new generation of treatment for patients.
GRANTS
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund (https://commonfund.nih.gov/GTEx) of the Office of the Director of the National Institutes of Health, and by the National Cancer Institute (NCI), National Human Genome Research Institute (NHGRI), National Heart, Lung, and Blood Institute (NHLBI), National Institute on Drug Abuse (NIDA), NIMH, and the National Institute of Neurological Disorders and Stroke (NINDS).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.L.M.-H. and B.L.F. prepared figures; M.L.M.-H., J.M., and B.L.F. drafted manuscript; M.L.M.-H., J.M., and B.L.F. edited and revised manuscript; M.L.M.-H., J.M., and B.L.F. approved final version of manuscript.
ACKNOWLEDGMENTS
The data used for the analyses described in this review were obtained from GTex Multigene Query of RNA-Seq data in the GTEx Portal on 02/04/2022 and/or dbGaP Accession No. phs000424.vN.pN on 02/04/2022.
This article is part of the special collection “Deciphering the Role of Proteoglycans and Glycosaminoglycans in Health and Disease.” Liliana Schaefer, served as Guest Editor of this collection.
REFERENCES
- 1. Mort JS, Billington CJ. Articular cartilage and changes in arthritis: matrix degradation. Arthritis Res 3: 337–341, 2001. doi: 10.1186/ar325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roughley PJ, Mort JS. The role of aggrecan in normal and osteoarthritic cartilage. J Exp Orthop 1: 8, 2014. doi: 10.1186/s40634-014-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hayes A, Sugahara K, Farrugia B, Whitelock JM, Caterson B, Melrose J. Biodiversity of CS-proteoglycan sulphation motifs: chemical messenger recognition modules with roles in information transfer, control of cellular behaviour and tissue morphogenesis. Biochem J 475: 587–620, 2018. doi: 10.1042/BCJ20170820. [DOI] [PubMed] [Google Scholar]
- 4. Djerbal L, Lortat-Jacob H, Kwok JCF. Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconj J 34: 363–376, 2017. doi: 10.1007/s10719-017-9761-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hatano S, Watanabe H. Regulation of macrophage and dendritic cell function by chondroitin sulfate in innate to antigen-specific adaptive immunity. Front Immunol 11: 232, 2020. doi: 10.3389/fimmu.2020.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sorrell JM, Lintala AM, Mahmoodian F, Caterson B. Epitope-specific changes in chondroitin sulfate/dermatan sulfate proteoglycans as markers in the lymphopoietic and granulopoietic compartments of developing bursae of Fabricius. J Immunol 140: 4263–4270, 1988. [PubMed] [Google Scholar]
- 7. Sorrell JM, Mahmoodian F, Schafer IA, Davis B, Caterson B. Identification of monoclonal antibodies that recognize novel epitopes in native chondroitin/dermatan sulfate glycosaminoglycan chains: their use in mapping functionally distinct domains of human skin. J Histochem Cytochem 38: 393–402, 1990. doi: 10.1177/38.3.1689338. [DOI] [PubMed] [Google Scholar]
- 8. Prabhakar V, Sasisekharan R. The biosynthesis and catabolism of galactosaminoglycans. Adv Pharmacol 53: 69–115, 2006. doi: 10.1016/S1054-3589(05)53005-9. [DOI] [PubMed] [Google Scholar]
- 9. Mikami T, Kitagawa H. Biosynthesis and function of chondroitin sulfate. Biochim Biophys Acta 1830: 4719–4733, 2013. doi: 10.1016/j.bbagen.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 10. Sugahara K, Kitagawa H. Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr Opin Struct Biol 10: 518–527, 2000. doi: 10.1016/S0959-440X(00)00125-1. [DOI] [PubMed] [Google Scholar]
- 11. Uyama T, Kitagawa H, Sugahara K. Biosynthesis of glycosaminoglycans and proteoglycans. In: Comprehensive Glycoscience, edited by Kamerling H. Oxford: Elsevier, 2007, p. 79–104. [Google Scholar]
- 12. Silbert JE, Sugumaran G. Biosynthesis of chondroitin/dermatan sulfate. IUBMB Life 54: 177–186, 2002. doi: 10.1080/15216540214923. [DOI] [PubMed] [Google Scholar]
- 13. Hayes AJ, Melrose J. Neural tissue homeostasis and repair is regulated via CS and DS proteoglycan motifs. Front Cell Dev Biol 9: 696640, 2021. doi: 10.3389/fcell.2021.696640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamada S. Catabolism of chondroitin sulfate. Cell Mol Biol Lett 20: 196–212, 2015. doi: 10.1515/cmble-2015-0011. [DOI] [PubMed] [Google Scholar]
- 15. Stern R, Jedrzejas MJ. Hyaluronidases: their genomics, structures, and mechanisms of action. Chem Rev 106: 818–839, 2006. doi: 10.1021/cr050247k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaneiwa T, Mizumoto S, Sugahara K, Yamada S. Identification of human hyaluronidase-4 as a novel chondroitin sulfate hydrolase that preferentially cleaves the galactosaminidic linkage in the trisulfated tetrasaccharide sequence. Glycobiology 20: 300–309, 2010. doi: 10.1093/glycob/cwp174. [DOI] [PubMed] [Google Scholar]
- 17. Honda T, Kaneiwa T, Mizumoto S, Sugahara K, Yamada S. Hyaluronidases have strong hydrolytic activity toward chondroitin 4-sulfate comparable to that for hyaluronan. Biomolecules 2: 549–563, 2012. doi: 10.3390/biom2040549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaneiwa T, Miyazaki A, Kogawa R, Mizumoto S, Sugahara K, Yamada S. Identification of amino acid residues required for the substrate specificity of human and mouse chondroitin sulfate hydrolase (conventional hyaluronidase-4). J Biol Chem 287: 42119–42128, 2012. doi: 10.1074/jbc.M112.360693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Csóka AB, Scherer SW, Stern R. Expression analysis of six paralogous human hyaluronidase genes clustered on chromosomes 3p21 and 7q31. Genomics 60: 356–361, 1999. doi: 10.1006/geno.1999.5876. [DOI] [PubMed] [Google Scholar]
- 20. Yamada S, Mizumoto S. Characterization of hyaluronidase 4 involved in the catabolism of chondroitin sulfate. Molecules 27: 6103, 2022. doi: 10.3390/molecules27186103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pap T, Korb-Pap A. Cartilage damage in osteoarthritis and rheumatoid arthritis—two unequal siblings. Nat Rev Rheumatol 11: 606–615, 2015. doi: 10.1038/nrrheum.2015.95. [DOI] [PubMed] [Google Scholar]
- 22. Melrose J, Fuller ES, Little CB. The biology of meniscal pathology in osteoarthritis and its contribution to joint disease: beyond simple mechanics. Connect Tissue Res 58: 282–294, 2017. doi: 10.1080/03008207.2017.1284824. [DOI] [PubMed] [Google Scholar]
- 23. Fuller ES, Smith MM, Little CB, Melrose J. Zonal differences in meniscus matrix turnover and cytokine response. Osteoarthritis Cartilage 20: 49–59, 2012. doi: 10.1016/j.joca.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 24. Sophia Fox AJ, Bedi A, Rodeo SA. The basic science of articular cartilage: structure, composition, and function. Sports Health 1: 461–468, 2009. doi: 10.1177/1941738109350438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kiani C, Chen L, Wu YJ, Yee AJ, Yang BB. Structure and function of aggrecan. Cell Res 12: 19–32, 2002. doi: 10.1038/sj.cr.7290106. [DOI] [PubMed] [Google Scholar]
- 26. Aspberg A. The different roles of aggrecan interaction domains. J Histochem Cytochem 60: 987–996, 2012. doi: 10.1369/0022155412464376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ni GX, Li Z, Zhou YZ. The role of small leucine-rich proteoglycans in osteoarthritis pathogenesis. Osteoarthritis Cartilage 22: 896–903, 2014. doi: 10.1016/j.joca.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 28. Melrose J, Fuller ES, Roughley PJ, Smith MM, Kerr B, Hughes CE, Caterson B, Little CB. Fragmentation of decorin, biglycan, lumican and keratocan is elevated in degenerate human meniscus, knee and hip articular cartilages compared with age-matched macroscopically normal and control tissues. Arthritis Res Ther 10: R79, 2008. doi: 10.1186/ar2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lord MS, Farrugia BL, Rnjak-Kovacina J, Whitelock JM. Current serological possibilities for the diagnosis of arthritis with special focus on proteins and proteoglycans from the extracellular matrix. Expert Rev Mol Diagn 15: 77–95, 2015. doi: 10.1586/14737159.2015.979158. [DOI] [PubMed] [Google Scholar]
- 30. Bollet AJ, Nance JL. Biochemical findings in normal and osteoarthritic articular cartilage. II. Chondroitin sulfate concentration and chain length, water, and ash content. J Clin Invest 45: 1170–1177, 1966. doi: 10.1172/JCI105423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ishimaru D, Sugiura N, Akiyama H, Watanabe H, Matsumoto K. Alterations in the chondroitin sulfate chain in human osteoarthritic cartilage of the knee. Osteoarthritis Cartilage 22: 250–258, 2014. doi: 10.1016/j.joca.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 32. Koike T, Izumikawa T, Tamura J-I, Kitagawa H. FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem J 421: 157–162, 2009. doi: 10.1042/BJ20090474. [DOI] [PubMed] [Google Scholar]
- 33. Nakajima M, Mizumoto S, Miyake N, Kogawa R, Iida A, Ito H, Kitoh H, Hirayama A, Mitsubuchi H, Miyazaki O, Kosaki R, Horikawa R, Lai A, Mendoza-Londono R, Dupuis L, Chitayat D, Howard A, Leal Gabriela F, Cavalcanti D, Tsurusaki Y, Saitsu H, Watanabe S, Lausch E, Unger S, Bonafé L, Ohashi H, Superti-Furga A, Matsumoto N, Sugahara K, Nishimura G, Ikegawa S. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am J Hum Genet 92: 927–934, 2013. doi: 10.1016/j.ajhg.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lei J, Deng H, Ran Y, Lv Y, Amhare AF, Wang L, Guo X, Han J, Lammi MJ. Altered expression of aggrecan, FAM20B, B3GALT6, and EXTL2 in patients with osteoarthritis and Kashin-Beck disease. Cartilage 13: 818S–828S, 2021. doi: 10.1177/1947603520932199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han J, Li D, Qu C, Wang D, Wang L, Guo X, Lammi MJ. Altered expression of chondroitin sulfate structure modifying sulfotransferases in the articular cartilage from adult osteoarthritis and Kashin-Beck disease. Osteoarthritis Cartilage 25: 1372–1375, 2017. doi: 10.1016/j.joca.2017.02.803. [DOI] [PubMed] [Google Scholar]
- 36. Lin T-S, Hsieh C-H, Kuo C, Juang Y-P, Hsieh YSY, Chiang H, Hung S-C, Jiang C-C, Liang P-H. Sulfation pattern of chondroitin sulfate in human osteoarthritis cartilages reveals a lower level of chondroitin-4-sulfate. Carbohydr Polym 229: 115496, 2020. doi: 10.1016/j.carbpol.2019.115496. [DOI] [PubMed] [Google Scholar]
- 37. Shinmei M, Miyauchi S, Machida A, Miyazaki K. Quantitation of chondroitin 4—sulfate and chondroitin 6—sulfate in pathologic joint fluid. Arthritis Rheum 35: 1304–1308, 1992. doi: 10.1002/art.1780351110. [DOI] [PubMed] [Google Scholar]
- 38. Ishimaru JI, Ogi N, Mizuno S, Goss AN. Quantitation of chondroitin-sulfates, disaccharides and hyaluronan in normal, early and advanced osteoarthritic sheep temporomandibular joints. Osteoarthritis Cartilage 9: 365–370, 2001. doi: 10.1053/joca.2000.0397. [DOI] [PubMed] [Google Scholar]
- 39. Jung Y-K, Park H-R, Cho H-J, Jang J-A, Lee E-J, Han M-S, Kim G-W, Han S. Degrading products of chondroitin sulfate can induce hypertrophy-like changes and MMP-13/ADAMTS5 production in chondrocytes. Sci Rep 9: 15846, 2019. doi: 10.1038/s41598-019-52358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martin DC, Atmuri V, Hemming RJ, Farley J, Mort JS, Byers S, Hombach-Klonisch S, Csoka AB, Stern R, Triggs-Raine BL. A mouse model of human mucopolysaccharidosis IX exhibits osteoarthritis. Hum Mol Genet 17: 1904–1915, 2008. [Erratum in Hum Mol Genet 17: 2919, 2008]. doi: 10.1093/hmg/ddn088. [DOI] [PubMed] [Google Scholar]
- 41. Farrugia BL, Mizumoto S, Lord MS, O'Grady RL, Kuchel RP, Yamada S, Whitelock JM. Hyaluronidase-4 is produced by mast cells and can cleave serglycin chondroitin sulfate chains into lower molecular weight forms. J Biol Chem 294: 11458–11472, 2019. doi: 10.1074/jbc.RA119.008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Asari A, Akizaki S, Itoh T, Kominami E, Uchiyama Y. Human osteoarthritic cartilage exhibits the 2B6 epitope without pretreatment with chondroitinase ABC. Osteoarthritis Cartilage 4: 149–152, 1996. doi: 10.1016/S1063-4584(05)80324-3. [DOI] [PubMed] [Google Scholar]
- 43. Emery AEH. The muscular dystrophies. The Lancet 359: 687–695, 2002. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 44. Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the duchenne muscular dystrophy locus. Cell 51: 919–928, 1987. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 45. Dumont NA, Wang YX, von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, Rudnicki MA. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med 21: 1455–1463, 2015. doi: 10.1038/nm.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stupka N, Kintakas C, White JD, Fraser FW, Hanciu M, Aramaki-Hattori N, Martin S, Coles C, Collier F, Ward AC, Apte SS, McCulloch DR. Versican processing by a disintegrin-like and metalloproteinase domain with thrombospondin-1 repeats proteinases-5 and -15 facilitates myoblast fusion. J Biol Chem 288: 1907–1917, 2013. doi: 10.1074/jbc.M112.429647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Negroni E, Henault E, Chevalier F, Gilbert-Sirieix M, Van Kuppevelt TH, Papy-Garcia D, Uzan G, Albanese P. Glycosaminoglycan modifications in Duchenne muscular dystrophy: specific remodeling of chondroitin sulfate/dermatan sulfate. J Neuropathol Exp Neurol 73: 789–797, 2014. doi: 10.1097/NEN.0000000000000098. [DOI] [PubMed] [Google Scholar]
- 48. An HB, Zheng HC, Zhang L, Ma L, Liu ZY. Partial least squares based identification of Duchenne muscular dystrophy specific genes. J Zhejiang Univ Sci B 14: 973–982, 2013. doi: 10.1631/jzus.B1300060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ralston E, Gutierrez-Cruz G, Kenea A, Brooks S. Transcriptomic analysis of mdx mouse muscles reveals a signature of early human Duchenne muscular dystrophy. BioRxiv, 2021. doi: 10.1101/2021.07.16.452553. [DOI]
- 50. Velleman SG, Sporer KR, Ernst CW, Reed KM, Strasburg GM. Versican, matrix Gla protein, and death-associated protein expression affect muscle satellite cell proliferation and differentiation. Poult Sci 91: 1964–1973, 2012. doi: 10.3382/ps.2012-02147. [DOI] [PubMed] [Google Scholar]
- 51. Wight TN, Kang I, Evanko SP, Harten IA, Chang MY, Pearce OMT, Allen CE, Frevert CW. Versican-A critical extracellular matrix regulator of immunity and inflammation. Front Immunol 11: 512, 2020. doi: 10.3389/fimmu.2020.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hirose J, Kawashima H, Yoshie O, Tashiro K, Miyasaka M. Versican interacts with chemokines and modulates cellular responses. J Biol Chem 276: 5228–5234, 2001. doi: 10.1074/jbc.M007542200. [DOI] [PubMed] [Google Scholar]
- 53. Ceco E, McNally EM. Modifying muscular dystrophy through transforming growth factor-beta. FEBS J 280: 4198–4209, 2013. doi: 10.1111/febs.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McRae NL, Addinsall AB, Howlett KF, McNeill B, McCulloch DR, Stupka N. Genetic reduction of the extracellular matrix protein versican attenuates inflammatory cell infiltration and improves contractile function in dystrophic mdx diaphragm muscles. Sci Rep 10: 11080, 2020. doi: 10.1038/s41598-020-67464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fadic R, Mezzano V, Alvarez K, Cabrera D, Holmgren J, Brandan E. Increase in decorin and biglycan in duchenne muscular dystrophy: role of fibroblasts as cell source of these proteoglycans in the disease. J Cell Mol Med 10: 758–769, 2006. doi: 10.1111/j.1582-4934.2006.tb00435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zanotti S, Negri T, Cappelletti C, Bernasconi P, Canioni E, Di Blasi C, Pegoraro E, Angelini C, Ciscato P, Prelle A, Mantegazza R, Morandi L, Mora M. Decorin and biglycan expression is differentially altered in several muscular dystrophies. Brain 128: 2546–2555, 2005. doi: 10.1093/brain/awh635. [DOI] [PubMed] [Google Scholar]
- 57. Reed CC, Iozzo RV. The role of decorin in collagen fibrillogenesis and skin homeostasis. Glycoconj J 19: 249–255, 2002. doi: 10.1023/A:1025383913444. [DOI] [PubMed] [Google Scholar]
- 58. Geng J, Liu G, Peng F, Yang L, Cao J, Li Q, Chen F, Kong J, Pang R, Zhang C. Decorin promotes myogenic differentiation and mdx mice therapeutic effects after transplantation of rat adipose-derived stem cells. Cytotherapy 14: 877–886, 2012. doi: 10.3109/14653249.2012.688944. [DOI] [PubMed] [Google Scholar]
- 59. Li Y, Foster W, Deasy BM, Chan Y, Prisk V, Tang Y, Cummins J, Huard J. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am J Pathol 164: 1007–1019, 2004. doi: 10.1016/s0002-9440(10)63188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gosselin LE, Williams JE, Deering M, Brazeau D, Koury S, Martinez DA. Localization and early time course of TGF-beta 1 mRNA expression in dystrophic muscle. Muscle Nerve 30: 645–653, 2004. doi: 10.1002/mus.20150. [DOI] [PubMed] [Google Scholar]
- 61. Amenta AR, Yilmaz A, Bogdanovich S, McKechnie BA, Abedi M, Khurana TS, Fallon JR. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc Natl Acad Sci USA 108: 762–767, 2011. doi: 10.1073/pnas.1013067108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fallon JR, McNally EM. Non-glycanated biglycan and LTBP4: leveraging the extracellular matrix for duchenne muscular dystrophy therapeutics. Matrix Biol 68–69: 616–627, 2018. doi: 10.1016/j.matbio.2018.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Farr L, Ghosh S, Moonah S. Role of MIF cytokine/CD74 receptor pathway in protecting against injury and promoting repair. Front Immunol 11: 1273, 2020. doi: 10.3389/fimmu.2020.01273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Su H, Na N, Zhang X, Zhao Y. The biological function and significance of CD74 in immune diseases. Inflamm Res 66: 209–216, 2017. doi: 10.1007/s00011-016-0995-1. [DOI] [PubMed] [Google Scholar]
- 65. Gil-Yarom N, Radomir L, Sever L, Kramer MP, Lewinsky H, Bornstein C, Blecher-Gonen R, Barnett-Itzhaki Z, Mirkin V, Friedlander G, Shvidel L, Herishanu Y, Lolis EJ, Becker-Herman S, Amit I, Shachar I. CD74 is a novel transcription regulator. Proc Natl Acad Sci USA 114: 562–567, 2017. doi: 10.1073/pnas.1612195114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25: 595–606, 2006. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mylona E, Jones KA, Mills ST, Pavlath GK. CD44 regulates myoblast migration and differentiation. J Cell Physiol 209: 314–321, 2006. doi: 10.1002/jcp.20724. [DOI] [PubMed] [Google Scholar]
- 68. Tripodi L, Villa C, Molinaro D, Torrente Y, Farini A. The immune system in duchenne muscular dystrophy pathogenesis. Biomedicines 9: 1447, 2021. doi: 10.3390/biomedicines9101447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Spencer MJ, Montecino-Rodriguez E, Dorshkind K, Tidball JG. Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol 98: 235–243, 2001. doi: 10.1006/clim.2000.4966. [DOI] [PubMed] [Google Scholar]
- 70. Cornelison DDW, Filla MS, Stanley HM, Rapraeger AC, Olwin BB. Syndecan-3 and syndecan-4 specifically mark skeletal muscle satellite cells and are implicated in satellite cell maintenance and muscle regeneration. Dev Biol 239: 79–94, 2001. doi: 10.1006/dbio.2001.0416. [DOI] [PubMed] [Google Scholar]
- 71. Pisconti A, Cornelison DD, Olguín HC, Antwine TL, Olwin BB. Syndecan-3 and Notch cooperate in regulating adult myogenesis. J Cell Biol 190: 427–441, 2010. doi: 10.1083/jcb.201003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pisconti A, Bernet JD, Olwin BB. Syndecans in skeletal muscle development, regeneration and homeostasis. Muscles Ligaments Tendons J 2: 1–9, 2012. [PMC free article] [PubMed] [Google Scholar]
- 73. Esmon CT. The protein C pathway. Chest 124: 26S–32S, 2003. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 74. Petrini S, Tessa A, Carrozzo R, Verardo M, Pierini R, Rizza T, Bertini E. Human melanoma/NG2 chondroitin sulfate proteoglycan is expressed in the sarcolemma of postnatal human skeletal myofibers. Abnormal expression in merosin-negative and Duchenne muscular dystrophies. Mol Cell Neurosci 23: 219–231, 2003. doi: 10.1016/s1044-7431(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 75. Takeuchi S, Nakano S-I, Nakamura K, Ozoe A, Chien P, Yoshihara H, Hakuno F, Matsuwaki T, Saeki Y, Takahashi S-I, Yamanouchi K, Nishihara M. Roles of chondroitin sulfate proteoglycan 4 in fibrogenic/adipogenic differentiation in skeletal muscle tissues. Exp Cell Res 347: 367–377, 2016. doi: 10.1016/j.yexcr.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 76. Langsdorf A, Do AT, Kusche-Gullberg M, Emerson CP Jr, Ai X. Sulfs are regulators of growth factor signaling for satellite cell differentiation and muscle regeneration. Dev Biol 311: 464–477, 2007. doi: 10.1016/j.ydbio.2007.08.053. [DOI] [PubMed] [Google Scholar]
- 77. Tran TH, Shi X, Zaia J, Ai X. Heparan sulfate 6-O-endosulfatases (Sulfs) coordinate the Wnt signaling pathways to regulate myoblast fusion during skeletal muscle regeneration. J Biol Chem 287: 32651–32664, 2012. doi: 10.1074/jbc.M112.353243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. El Masri R, Seffouh A, Roelants C, Seffouh I, Gout E, Pérard J, Dalonneau F, Nishitsuji K, Noborn F, Nikpour M, Larson G, Crétinon Y, Friedel-Arboleas M, Uchimura K, Daniel R, Lortat-Jacob H, Filhol O, Vivès RR. Extracellular endosulfatase Sulf-2 harbors a chondroitin/dermatan sulfate chain that modulates its enzyme activity. Cell Rep 38: 110516, 2022. doi: 10.1016/j.celrep.2022.110516. [DOI] [PubMed] [Google Scholar]
- 79. Noborn F, Nikpour M, Persson A, Nilsson J, Larson G. Expanding the chondroitin sulfate glycoproteome - but how far? Front Cell Dev Biol 9: 695970, 2021. doi: 10.3389/fcell.2021.695970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sztal TE, Sonntag C, Hall TE, Currie PD. Epistatic dissection of laminin-receptor interactions in dystrophic zebrafish muscle. Hum Mol Genet 21: 4718–4731, 2012. doi: 10.1093/hmg/dds312. [DOI] [PubMed] [Google Scholar]
- 81. Zhou S, Chen S, Pei YA, Pei M. Nidogen: a matrix protein with potential roles in musculoskeletal tissue regeneration. Genes Dis 9: 598–609, 2022. doi: 10.1016/j.gendis.2021.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Falcone G, Ciuffini L, Gauzzi MC, Provenzano C, Strano S, Gallo R, Castellani L, Alemà S. v-Src inhibits myogenic differentiation by interfering with the regulatory network of muscle-specific transcriptional activators at multiple levels. Oncogene 22: 8302–8315, 2003. doi: 10.1038/sj.onc.1206915. [DOI] [PubMed] [Google Scholar]
- 83. Noborn F, Gomez Toledo A, Sihlbom C, Lengqvist J, Fries E, Kjellén L, Nilsson J, Larson G. Identification of chondroitin sulfate linkage region glycopeptides reveals prohormones as a novel class of proteoglycans. Mol Cell Proteomics 14: 41–49, 2015. doi: 10.1074/mcp.M114.043703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mateos B, Holzinger J, Conrad-Billroth C, Platzer G, Żerko S, Sealey-Cardona M, Anrather D, Koźmiński W, Konrat R. Hyperphosphorylation of human osteopontin and its impact on structural dynamics and molecular recognition. Biochemistry 60: 1347–1355, 2021. doi: 10.1021/acs.biochem.1c00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Vetrone SA, Montecino-Rodriguez E, Kudryashova E, Kramerova I, Hoffman EP, Liu SD, Miceli MC, Spencer MJ. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest 119: 1583–1594, 2009. doi: 10.1172/JCI37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Capote J, Kramerova I, Martinez L, Vetrone S, Barton ER, Sweeney HL, Miceli MC, Spencer MJ. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J Cell Biol 213: 275–288, 2016. doi: 10.1083/jcb.201510086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, Clemens PR, Hoffman EP, Cnaan A, Gordish-Dressman H; CINRG Investigators. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 391: 451–461, 2018. doi: 10.1016/S0140-6736(17)32160-8. [DOI] [PubMed] [Google Scholar]
- 88. Mikami T, Koyama S, Yabuta Y, Kitagawa H. Chondroitin sulfate is a crucial determinant for skeletal muscle development/regeneration and improvement of muscular dystrophies. J Biol Chem 287: 38531–38542, 2012. doi: 10.1074/jbc.M111.336925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Maciej-Hulme ML. New insights into human hyaluronidase 4/chondroitin sulphate hydrolase. Front Cell Dev Biol 9: 767924, 2021. doi: 10.3389/fcell.2021.767924. [DOI] [PMC free article] [PubMed] [Google Scholar]