

Abstract
With the advent of X-ray Free Electron Lasers (XFELs), new, high-throughput serial crystallography techniques for macromolecular structure determination have emerged. Serial femtosecond crystallography (SFX) and related methods provide possibilities beyond canonical, single-crystal rotation crystallography by mitigating radiation damage and allowing time-resolved studies with unprecedented temporal resolution. This primer aims to assist structural biology groups with little or no experience in serial crystallography planning and carrying out a successful SFX experiment. It discusses the background of serial crystallography and its possibilities. Microcrystal growth and characterization methods are discussed, alongside techniques for sample delivery and data processing. Moreover, it gives practical tips for preparing an experiment, what to consider and do during a beamtime and how to conduct the final data analysis. Finally, the Primer looks at various applications of SFX, including structure determination of membrane proteins, investigation of radiation damage-prone systems and time-resolved studies.
TOC blurb
Serial femtosecond crystallography (SFX) is a high-throughput, serial crystallography technique for studying macromolecular crystals at an X-ray free electron laser. This Primer gives an overview of microcrystallization methods, sample delivery and data analysis for applications including membrane proteins, radiation damage-prone systems and time-resolved experiments.
Introduction
Crystallography is a highly successful method to obtain atomic-level structural information about molecules. Unlike other structure determination methods, it has no restrictions on molecular size, but general limitations include the necessity to grow suitable well-ordered crystals and the sample-changing effects of X-rays used to collect the diffraction data. Radiation damage1, particularly in macromolecular crystals, was recognized early on as compromising structural studies. The impact of radiation on, for example, the resolution was mitigated by acquiring partial data sets from several crystals2 and then merging them into a complete data set. Cryocooling crystals during data collection can significantly slow radiation damage, enabling the acquisition of more data from each crystal and using smaller crystals. Consequently, data collection from a single crystal kept at 100 K became the norm3. However, despite many advantages, the problem of radiation damage remains. As a result, it is challenging to derive mechanistic insight from the structures due to photoreduction of redox-sensitive cofactors, for example, many metals or cofactors with large conjugated systems. The use of very small crystals, such as frequently observed for membrane protein crystals grown in lipidic cubic phase (LCP)4, is also limited. Furthermore, cryogenic temperatures can change the distribution of conformational substates5,6, which are mechanistically important, and preclude most time-resolved experiments. Obtaining high resolution, radiation-damage-free structures of molecules at ambient temperature, including those of short-lived reaction intermediates, is now possible using X-ray free-electron lasers (XFELs)7, TABLE 1. REF8 provides a detailed comparison of synchrotron sources and XFELs.
Table1.
Overview of XFEL facilities and instruments for SFX.
| Facility beamline | Beam parameters Photon energy, repetition rate, pulse length, photons per pulse, focus | Detector pixels, pixel size, frame rate, distance | Sample delivery options | Sample environ ment | Pump laser pulse duration, wavelength, timing | Remarks | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| keV | Hz | fs | ph/pulse | μm | 106 | μm2 | Hz | mm | fs | nm | fs | ||||
| LCLS, Menlo Park, USA, 2009 | |||||||||||||||
| MFX | 5 – 24 | 120 | 30 – 100 or <10 |
1·1012 or 1·1011 |
3×3 (2×2) | ePix10K-2.1M | SF-ROX with sample change robot MESH, GDVN, HVE, DoD, fixed targets |
90–278 K Ambient pressure Helium available, humidity controlled |
50 – 150 < 8ns |
410 – 2400 210 – 2200 |
δt0<200 spec. encod. | Protein Crystal Screening (PCS) beam time available Data analysis pipeline / support Screening beam time available at SSRL synchrotron with matching sample delivery options. Beamline website |
|||
| 2.1 | 100 × 100 | 120 | 50 – 1500 | ||||||||||||
| Rayonix 340-XFEL | |||||||||||||||
| 59 | 44 × 44 177 × 177 |
2.5 30 |
70 – 1100 | ||||||||||||
| CXI | 6 – 10.5 or 10.5 – 25 |
120 | 30 – 100 or <10 |
1·1012 or 1·1011 |
0.1 or 1 | Jungfrau | MESH, GDVN, HVE, fixed targets | Vacuum (10−5 Torr) Ambient pressure |
50 – 150 ns |
410 – 2400 200 – 266 |
δt0<200 | PCS available in parasitic mode Data analysis pipeline and support Beamline website |
|||
| 4 | 75 × 75 | 120 | 70 – 580 | ||||||||||||
| SACLA, Sayo, Hyogo, Japan, 2011 | |||||||||||||||
| BL3 | 4–20 | 30 (60) | <10 | 1011 | >1 | MPCCD | SF-ROX, fixed targets, GDVN, HVE226 and DoD84 (in DAPHNIS chamber) | 100K - RT Helium at ambient pressure |
30 | 200 – 2700 | δt0 <7 | Feasibility study beam time available. Data analysis pipeline and support Beamline details |
|||
| 4 | 50 × 50 | 60 | 50 – 150 | ||||||||||||
| BL2 | 4–15 | 30 (60) | <10 | 1011 | >1 | MPCCD | SF-ROX, GDVN, HVE226 and DoD (in DAPHNIS chamber) | 100K - RT, Helium at ambient pressure |
5 ns | 210 - 2600 | Feasibility study beam time available. Data analysis pipeline and support Beamline details |
||||
| 4 | 50 × 50 | 60 | 50 – 150 | ||||||||||||
| SwissFEL, Villigen, Switzerland, 2019 | |||||||||||||||
| Alvra | 2 −12.4 | 100 | - | 4·1011 | 1.5 | Jungfrau | HVE, GDVN (user supplied) | RT Helium at 5·10−4 - 800 mbar |
50 | 240 – 2500 | - | Data analysis pipeline and support Beamline website |
|||
| 4 or 16 | 75 × 75 | 100 | 100 | ||||||||||||
| EuXFEL, Schenefeld (near Hamburg), Germany, 2017 | |||||||||||||||
| SPB/SFX | 6–15 | 1.1 MHz or 4.5 MHz |
~25 | 1.5·1012 | 3 or <0.4 |
AGIPD | GDVN, aerosol injection HVE, fixted target at lower repetition rate (10 Hz) |
Vacuum, 1·10−6 mbar typical, 1.10−4 mbar maximum Ambient pressure at low rep rate (second interaction region) |
4.5 MHZ | Protein screening beamtime available Data analysis pipeline and support: EXtra-xwiz automated pipeline in beta-testing. Extra-xwiz documentation Up-to-date parameters |
|||||
| 15, 50 or 300 | 370 – 420 | - | |||||||||||||
| 1 | 200 × 200 | 4.5 MHz | 129–329 | 1.1 MHz | |||||||||||
| <100 | 420 – 2600 | - | |||||||||||||
| 4.5 MHz | |||||||||||||||
| 0.85 or 400 ps | 1030 | - | |||||||||||||
| Up to 20 Hz | |||||||||||||||
| 3–7 ns | 210 – 2400 | - | |||||||||||||
| PAL-XFEL, Pohang, Korea, 2017 | |||||||||||||||
| NCISFX | 2.2 – 15 | 60 | 25 | 3·1010 (monochrom.) or 1·1012 (pink) |
5×5 or 2×2 |
Rayonix MX225-HS | GDVN and HVE33,227, fixed targets | RT He at ambient pressure |
40 7 ns (15Hz) |
800/400/266 1064/532/355/266 |
δt0 < 20 | Protein screening beamtime available Beamline website |
|||
| 33 | 156×156 234×234 |
30 60 |
100 ~ 1000 | ||||||||||||
| Jungfrau | |||||||||||||||
| 4 | 75 × 75 | 60 | 100 ~ 1000 | - | |||||||||||
XFEL pulses typically have a duration in the femtosecond to tens of femtoseconds range, yet each pulse delivers as many photons as a synchrotron beam does per second8. Due to the XFEL’s high peak brilliance, data can be collected from small and/or weakly diffracting samples. In addition, the short XFEL pulse duration allows outrunning radiation damage effects. This is because X-ray scattering, which causes the desired diffraction signal, is almost instantaneous, occurring on attosecond (as) timescales, and only those changes that take place during the femtosecond pulse are observed in the collected data. The changes include those caused by photoionization (~10–100 as); the emission of Auger electrons (fs range); and the subsequent ionization cascades that result in random9,10 and correlated10–12 nuclear motions. Ultimately, the highly ionized sample explodes. Thus, for short pulse lengths, the diffraction pattern is generated before significant damage — reflected in changes in electron density or spectroscopic features — can accumulate and sample is destroyed. This forms the basis of the diffraction-before-destruction principle for data collection13. Therefore, XFEL-based crystallography requires a new crystal, or a fresh part thereof, for each exposure. Importantly, on the timescale of an XFEL pulse, the crystal cannot be rotated as it would be for synchrotron data collection, FIG. 1, and the resulting single-exposure snapshot corresponds to a thin slice through reciprocal space, called a still image. This approach to data collection is referred to as nanocrystallography14 or serial femtosecond crystallography (SFX)10,15 and involves use of specialized software to merge partial intensities from thousands of diffraction patterns from crystals intersecting the XFEL beam at random orientations, FIG. 1. SFX requires very high throughput serial data collection. For efficient data collection, the sample should be replenished with a rate commensurate with the repetition rate of the XFEL pulses or – if slower — the detector frame rate. Different methods to deliver microcrystals into the XFEL beam, with varying complexity, sample quantities and applicability, have been established over the years16,17.
Fig. 1 |. Differences in experimental crystallography setups.
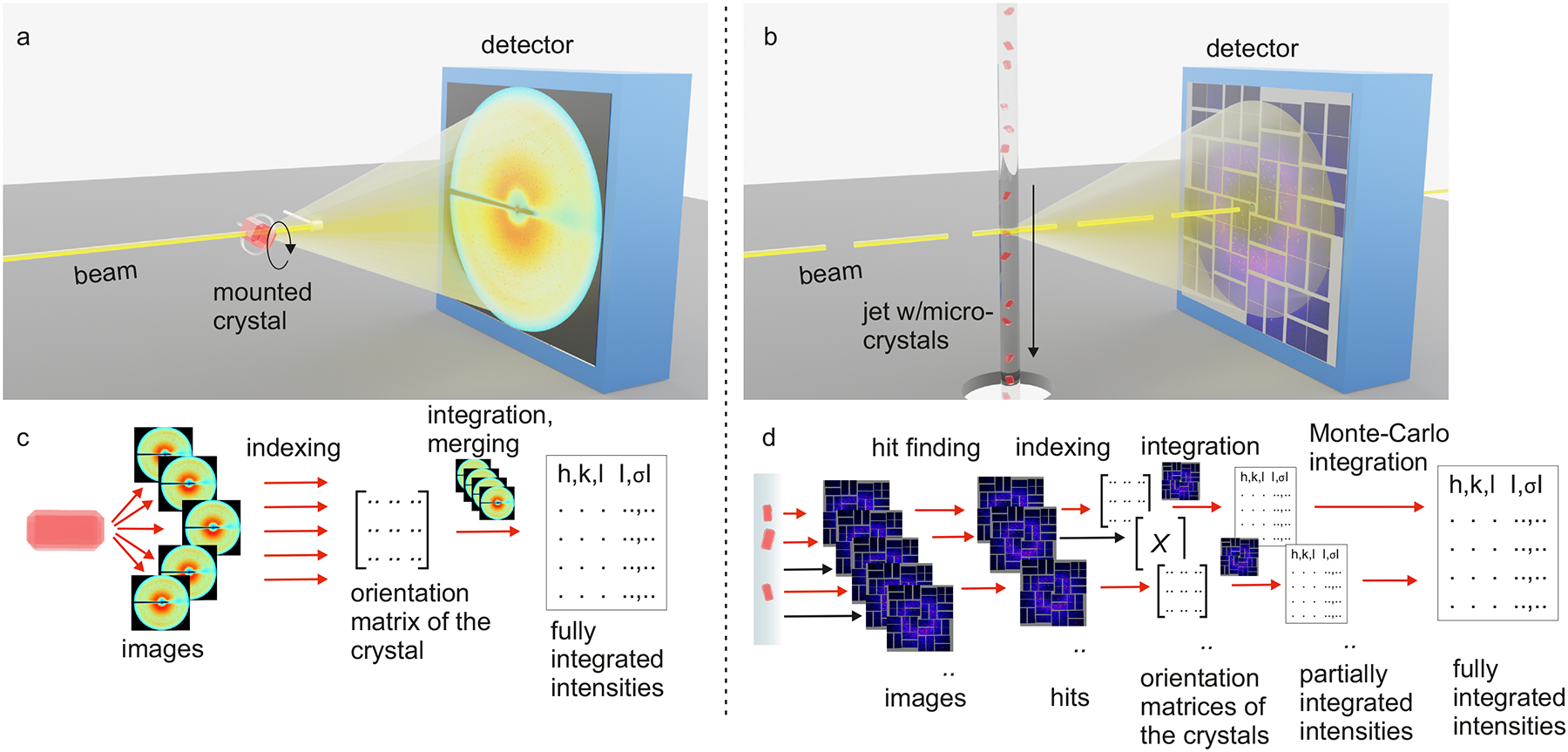
a, c | conventional rotation crystallography. b, d | SFX with sample delivery by high viscosity extrusion. In conventional MX, a single crystal (red) is mounted in a loop, kept at 100 K using a cryogenic nitrogen stream and rotated during sequential exposures, panel a. Consecutively acquired diffraction patterns are indexed, giving an orientation matrix of the crystal in the laboratory coordinate system, which is used to integrate the reflection intensities, panel c. In SFX, many microcrystals are sequentially delivered to the pulsed X-ray beam in random orientations, and a detector image is acquired for each XFEL pulse, panel b. A Bragg diffraction pattern containing partial intensities will only be produced when a crystal is present in the interaction region at the same time as an X-ray pulse arrives, panel d. Images containing a diffraction pattern are selected in a process called “hit finding”. The hit rate is the ratio of pattern-containing frames to the total number of collected frames. Hits are individually indexed, which is not always successful. The ratio of indexed patterns to the total number of hits is the indexing rate, panel d. Individual indexed diffraction patterns are integrated, and the resulting intensities are merged by Monte-Carlo integration.
Well-ordered crystals of appropriate dimensions are at the heart of any crystallographic study, and their diffraction quality and size often limit a project’s success. It is generally desirable to grow large single crystals rather than small ones. However, large crystal growth is challenging and forms a bottleneck in traditional macromolecular crystallography (MX). As SFX typically uses microcrystals, it can circumvent this bottleneck, but different challenges arise. It is a big step between observing microcrystals in 100 nanoliter crystallization drops in 96-well plates and producing large amounts of homogeneously sized microcrystals in tubes, syringes or plates. This requires a large quantity of protein and a trial and error approach, with less automation and experience than is available for growing large single-crystals. As a result, each project requires an assessment of whether SFX at an XFEL or MX at a synchrotron beamline is best suited to the investigation, BOX 1. Additional points to consider include whether a microfocus beamline is needed. For challenging projects, MX allows screening of hundreds of large crystals almost automatically to identify well-ordered regions suited to data collection, of which typically only a few are needed. By contrast, many thousands of well-ordered microcrystals are needed for SFX. However, despite the efforts involved, SFX is the method of choice for analyzing very small crystals, radiation damage-sensitive samples and performing time-resolved experiments that probe early reaction time points in general, and light-induced reactions in the single-photon regime specifically.
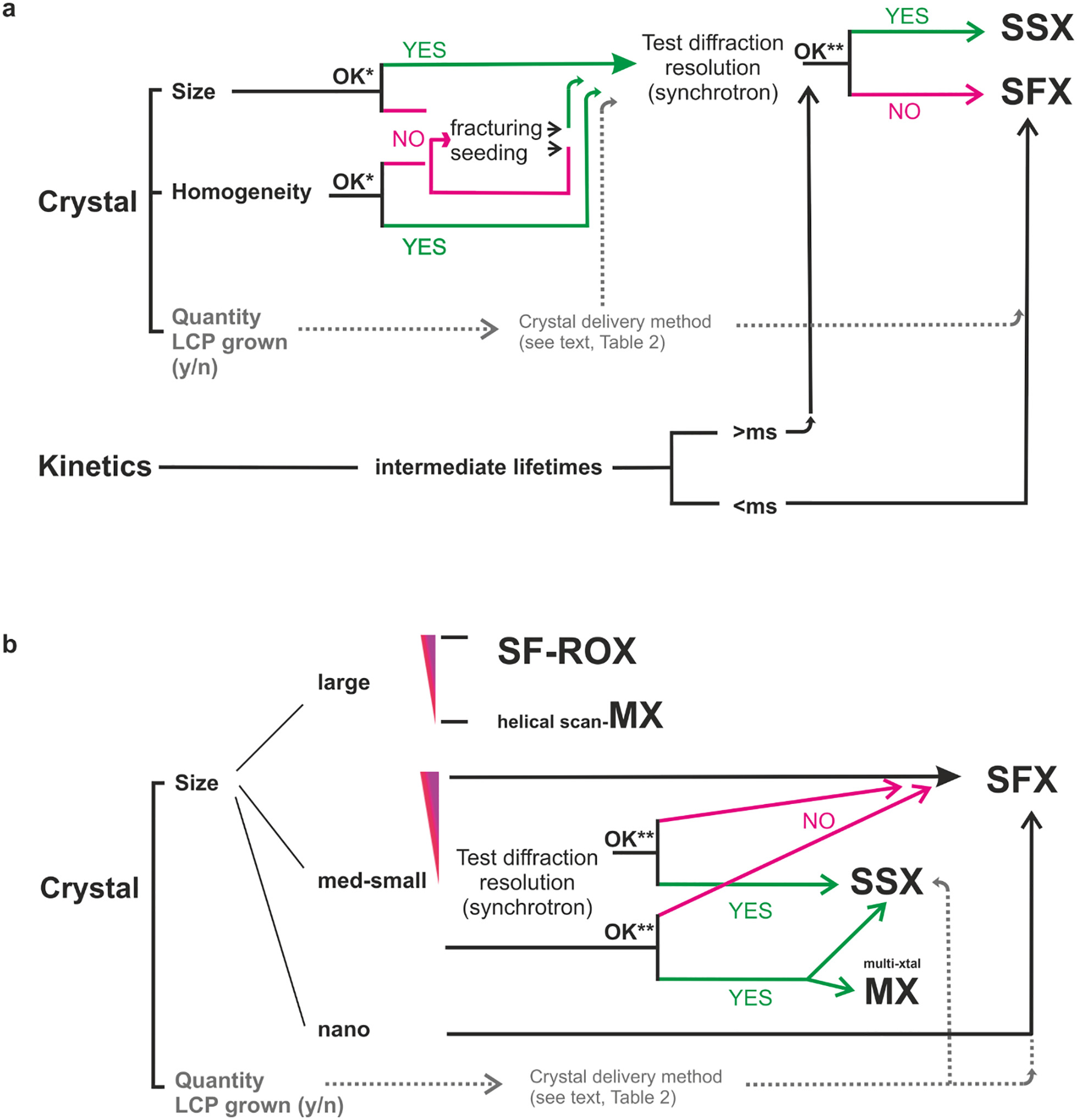
Box 1. Data collection flowcharts.
Structures of reaction intermediates are typically determined using time-resolved experiments (a). This places stringent constraints on crystal sizes, including the size distribution to provide well-defined reaction initiation (indicated by OK*). The actual range of appropriate crystal sizes depends on the properties of the crystals, for example, protein concentration, and the triggering approach, that is the extinction coefficient for photoexcitation and diffusion time for chemical mixing. Depending on the lifetime of the intermediate, time-resolved data can be collected at XFELs or synchrotron sources using either monochromatic (SSX) or polychromatic (Laue) diffraction. The resolution needs to be high enough (OK**) to resolve the structural changes occurring during the reaction. Appropriate exposure conditions — flux, exposure time — need to be chosen. (b) Protein crystals differ in their susceptibility to radiation damage. Systems can either be inherently sensitive to radiation or have small crystal size, meaning that the dose of radiation required for the collection of a full data set cannot be distributed over as many molecules as in a large crystal. In such cases, standard MX data collection might not be viable. In the figure, the red triangle indicates inherent radiation sensitivity, with the wider side indicating higher sensitivity. Radiation damage can be minimized by serial data collection (SFX, SSX), or adjusted MX data collection strategies, for example helical scan or combining partial data sets from multiple crystals. Samples with very high radiation sensitivity — for example, photosystem II which has a large metal cluster — require XFELs for data collection. For other, less radiation sensitive systems, diffraction data may be acquired by distributing the dose over several to many crystals using synchrotron radiation122,222,223. Given the relative ease and high automation of MX, it is advisable to perform extensive crystal diffraction screening at a synchrotron before committing to SFX219. The resolution should be high enough (OK**) to resolve important structural features. Appropriate exposure conditions — flux, exposure time — need to be chosen. The size of the crystals determines the data collection approach. For SF-ROX, the crystals are often >100 μm132,136. Medium-to-small crystals are typically 5–50 μm. Nanocrystals are crystals that have at least one dimension <1μm112,151,152. The quantity of crystalline material available and its crystallization conditions — for example, crystallization in LCP — strongly influence the choice of sample delivery methods into the X-ray beam (dotted gray lines). Since sample delivery is an integral part of SFX or SSX data collection, it should be considered early in experiment planning. This applies to both static and time-resolved experiments.
SFX is maturing as a technique, but the approach is still far from routine and lacks the stability, predictability and automation available for MX at synchrotron beamlines. SFX experiments take significantly longer, typically require several people, and data collection and evaluation requires expertise beyond standard MX. It is thus advisable that small research groups inexperienced with very high throughput serial crystallography collaborate closely with facility staff or experienced groups. This primer is aimed at such less experienced groups, focusing on what needs to be considered when applying for XFEL beamtime, how to plan and perform an SFX experiment, discusses potential advantages, and provides an overview of common pitfalls and misconceptions. Specific examples include time-resolved experiments and radiation damage-prone systems, such as metalloproteins and small membrane protein crystals. The primer will provide neither step-by-step descriptions of large volume microcrystallization, sample delivery approaches or data processing, nor detailed structural or mechanistic assessments of previous investigations. Instead, the relevant literature will be cited.
High throughput serial data collection is an intrinsic feature of crystallography at XFELs which necessitated the development of novel approaches in microcrystallization, sample delivery, data collection and evaluation. Subsequently, these serial data collection techniques were ported to synchrotrons as serial synchrotron crystallography (SSX)18–21, BOX 1, supplementary information, and to electron microscopes22, resulting in conceptually similar data collection approaches, for which this Primer is also helpful.
Experimentation
Experimental strategies
Successful crystal structure determination depends on the diffraction properties of the crystal(s), that is the sample quality. This also applies to SFX. When planning an XFEL experiment, initial questions relate to how much and what kind of crystalline sample is needed, and what crystal delivery method to use. Both depend on the amount of protein available, its general crystallization properties and the experiment type, such as collection of a static data set from crystals that are too radiation sensitive for synchrotron data collection or a series of data sets in a time-resolved experiment. Static data sets require significantly less material than multi-data point time-resolved experiments; they also pose no restrictions on crystal size or distribution, as long as the crystals provide sufficient diffraction signal. The collection of static data sets enables greater flexibility, including synchrotron-like data collection schemes using numerous very large, often cryocooled, crystals — serial femtosecond rotation crystallography (SF-ROX)23,24, akin to helical scans — alongside typical SFX approaches using microcrystals. The least technical and often most efficient sample presentation mode is a fixed target approach, with the most straightforward being a sheet-on-sheet (SOS) chip25. Although non-patterned chips such as the SOS chip are less efficient in terms of sample usage than patterned chips26–31, they have the significant advantage of being universally applicable. They can handle all crystalline sample types, irrespective of crystal size, distributions and medium viscosity, in addition to being readily available, TABLE 2. Other sample delivery approaches, such as low32 and high33 viscosity jets16,17,34 or droplet delivery35,36 can also be used for static data collection but have more boundary conditions related to crystal size, homogeneity and amount of material needed, TABLE 2.
Table 2:
Comparison of the most frequently used sample delivery approaches
| Delivery method | Crystal size | Mother liquor restrictions | Background scattering | Triggering compatibility | Technical Complexity | Efficient use of | Sample need for testing/static data set | Comments and Caveats | |
|---|---|---|---|---|---|---|---|---|---|
| sample | beamtime | ||||||||
| Fixed targets/chips | |||||||||
| SF-ROX (Goniometer mounted single crystals) | Several 100 μm | None. Typically cryocooled |
low | Only electric field jumps | Low | High (but ~ 50 μm translations between exposures) | medium due to frequent crystal changes | Few crystals/50–100 crystals | SF-ROX, similar to helical scans at synchrotron sources |
| Fixed targets (Non-patterned chip) | Any size | None, LCP possible | Low for thin sample thickness and thin films | Pump probe (light scattering may be an issue) Slow chemical mixing in humidity-controlled environment |
low | Lower than patterned | high | 5 μl conc. cystal suspension/1–2 chips | Loading must be fast or in humid atmosphere to prevent sample dehydration which can result in nonisomorphism or other changes in diffraction properties. Samples and foils must be thin (few μm) for low background. Evaporation through thin foils or XFEL generated shot holes may be an issue, in particular when using vacuum chambers. The latter as well as diffusion of X-ray induced radicals affects spacing between exposures. Spacing between adjacent exposures in a row can be shorter than between rows. Frequent chip changes required (typically 10–15 min/chip) |
| Fixed targets (Patterned chip) | Should fit hole size | Not too viscous | Low for thin sample thickness and thin films | Pump probe (light scattering may be an issue in transparent chips); Chemical mixing in humidity-controlled environment long time delays possible | low | High | high | 5–50 μl/1–2 chips | Loading and foil considerations as above. Moreover, blotting of mother liquor via perforated foils may result in crystal dehydration. The well depth determines the sample thickness (liquid film) in bottomless chips. |
| Droplet | |||||||||
| DoD DoT (tape) |
Big enough for good signal, 5–200 μm, but smaller than ID of capillary | Not too viscous (<40% PEG5000 or 35% glycerol) | High (due to droplet size and when hitting tape | Pump probe, chemical mixing 0.1 s - ~12s time delays | high | High | high | 100 μl, ideally 30 % crystals (v/v) / 200 μl | ID of capillary sets upper limit on crystal size (Standard is 200 μm ID (which works fine for up to 80 μm longest crystal dimension) |
| Jets | |||||||||
| GDVN | <20 μm | Not too viscous | Very low | Pump probe (fs-few μs) Chemical mixing, long time delays challenging |
high | Very low | Very high, including MHz | 300 μl, ideally 10–20 % crystals (v/v)/1–2 ml | Clogging of nozzles -> filtration of samples, prior and during injection, larger ID capillaries can help (50–100 μm) Settling of crystals -> anti-settling devices Fast video analysis to ensure jetting (and not spraying) Do not collect in the breakup region of the jet -> low, unreliable hit rate or very close to nozzle Test injection before beam time Compatible with MHz data collection |
| High viscosity extruders | Big enough to yield good signal but smaller than ID of the nozzle | None for grease-like matrix (but incompatible with some membrane protein crystals, causes dehydration in some crystals), LCP, cellulose, agarose, … limits on salt concentration, pH (not highly acidic) | high | Pump probe | Medium | High | high | 5 μl/50 μl | ID of nozzle (50–100 μm) sets upper limit on crystal size Clogging Uneven flow rates, complicating in particular time-resolved experiments The flow can be disrupted by unattenuated XFEL beam Efficient sample use for XFELs with pulse repetition rate of 120 Hz and lower, as well as synchrotrons |
Time-resolved experiments rely on fast, efficient, uniform and synchronous reaction initiation in a sufficiently large number of molecules in the crystal. This restricts the crystal size. For photoexcitation, the crystal thickness should not exceed the 1/e penetration depth of the excitation light at the pump wavelength. For chemical mixing, the diffusion time into the crystal must be significantly shorter than the lifetime of the first intermediate. Consequently, photoexcitation in the UV-Vis to near IR region and chemical mixing on the millisecond timescale require microcrystals with a narrow size distribution of ~5 μm. Triggering the reaction also places boundary conditions on the sample delivery approach, TABLE 2.
The sample quantity needed depends on how efficiently the microcrystalline sample is used for XFEL data collection, TABLE 2, and the required accuracy of the integrated diffraction intensities. To detect small intensity differences between Friedel mates (anomalous signal) — for example, for de novo phasing — or between entire data sets — for example, between subsequent reaction time-points — requires significantly more indexed diffraction patterns than are needed for static structure determination by molecular replacement methods37.
XFEL beam time is scarce and samples are often expensive. As a result, it is important to use both efficiently. When applying for XFEL beamtime, it is advisable to match the repetition rates of the chosen sample delivery approach the considered XFEL, or detector frame rate if slower, TABLES 1, 2. Since it is difficult to predict how well microcrystals will diffract in an XFEL beam, applying for Protein Crystal Screening (PCS) beamtime is highly recommended. PCS is typically one shift and enables testing of diffraction properties as a function of crystallization conditions or crystal size and establishing sample delivery approaches. This provides feedback on the XFEL experiment requirements and the likelihood of success, allowing informed decisions to be made about whether to undertake an experiment or not, and how to prepare for it.
Sample preparation and delivery
Crystallization
SFX is sometimes referred to as nanocrystallography38,39 or serial femtosecond nanocrystallography40. This is because the first seminal SFX experiment performed in 2009 used nanocrystals of photosystem I, showing that they give useful diffraction14. The experimental setup limited the resolution of the data to 8.7 Å. Subsequent measurements used 5×5×15 μm3 crystals that diffracted to 2.9 Å41, which, while impressive, is still lower than the resolution obtained at a synchrotron using large cryocooled crystals (2.5 Å)42. General observations made on both real and model systems suggest that small crystals are not necessarily better than large ones and that there appears to be a minimum, system-specific crystal size needed to yield sufficient signal for high-resolution diffraction. For example, 4–5 μm photosystem II crystals diffract to 4–5 Å43 and 20–50 μm crystals to ~2 Å44. Indeed, most SFX experiments to date have used microcrystals of 5–20 μm. Crystals of this size look crystalline — they show facets — when viewed under a high magnification light microscope. It is thus a misconception, borne out of the nanocrystallography notion, to aim for precipitate-like looking material when growing tiny crystals for SFX.
SFX experiments aimed at determining structures of short-lived intermediates or of radiation damage free molecules typically start from known crystallization conditions for macroscopic crystals. The task is then to grow appropriate crystals in the required amount for XFEL data collection. This section focuses on microcrystallization, distinguishing between crystals that grow in a liquid solution phase and those that grow in LCP.
Growing microcrystals in liquid phase
Crystals for applications with a low to medium sample requirement can be grown by increasing the drop volume and adjusting conditions slightly in, for example, Linbro or Cryschem plates, in hanging or sitting drop setups, which are then combined for data acquisition45,46. This approach is not feasible for time-resolved experiments using liquid jets that require large sample quantities, TABLE 2. Instead, batch crystallization approaches are used. The setup volumes range from 100s of microliters in Eppendorf tubes to 10s of milliliters in Falcon tubes47,48, corresponding to protein quantities up to the gram scale49,50. It is important to note that batch crystallization takes a different path through the phase diagram than vapour diffusion approaches. In principle, the latter can take any path, whereas batch conditions need to match the nucleation zone closely51. Therefore, to increase crystallization efficiency, microseeding is frequently used48,52–54. Seeding can also be used to control crystal size via the number of seeds added. Alternatively, crystal growth can be quenched by exchanging the protein-containing crystallization solution with a solution containing a higher concentration of precipitant55.
The LCP method
LCP is a lipid mesophase that spontaneously forms when a particular host lipid, such as monoolein, and an aqueous buffer are mixed at a certain ratio56. Lipids assemble in a single, continuous lipid bilayer convoluted in 3D space, dividing it into two non-intersecting networks of aqueous channels. Integral membrane proteins can be reconstituted in the lipid bilayer of LCP, which stabilizes them and allows their long-range diffusion. At the same time, the two non-intersecting water channels accommodate soluble protein parts and enable the diffusion of ligands, cofactors, and precipitants. Crystals grown in LCP have type I packing — stacks of 2D crystals — with contacts forming between hydrophilic and hydrophobic protein parts, making them, in general, better ordered. As a result, they typically diffract to a higher resolution than their type II counterparts grown in detergent solutions, in which only hydrophilic parts interact. LCP is therefore ideally suited as a crystal growth matrix for membrane proteins with relatively small soluble domains, such as G protein-coupled receptors (GPCRs) and many types of ion channels, transporters, and enzymes.
Crystallization in LCP has been miniaturized and automated, allowing the screening of thousands of precipitant conditions using only a few microliters of purified protein sample57. Initial crystal hits in LCP are typically observed as a large number of very small crystals because of a higher nucleation rate due to 2D confinement and a slower protein diffusion rate inside the lipid bilayer. Nucleation and crystal growth are competitive processes, and their mutual relationship can be modulated by choosing optimal precipitant conditions.
Traditional data collection at synchrotron beamlines requires relatively large (>20 μm) crystals. Optimizing for crystal size, however, can increase mosaicity and introduce growth defects, reducing their diffraction power, and for systems challenging to crystallize, obtaining large crystals is often impossible. However, SFX enables data collection from smaller (<10 μm) crystals, providing an alternative objective for crystal optimization, focusing on high density rather than large size. There is a natural synergy between LCP-grown crystals, which tend to be small, and SFX data collection. This has led to notable advances in membrane protein structure determination, particularly GPCRs.
To sufficiently sample the crystallization space and increase throughput, LCP crystallization experiments are often conducted in a 96-well, nanoliter-volume format using robotic liquid handling and imaging systems58. Glass sandwich plates prevent sample dehydration and provide excellent optical properties, enabling in situ crystal detection and growth monitoring by cross-polarization or UV fluorescence. If sufficiently large crystals can be grown that are suitable for synchrotron data collection, they can be harvested from these plates using MiTeGen dual thickness micromounts. Alternatively, if large crystals are not attainable, crystallization conditions can be optimized to produce a large number of uniformly sized microcrystals for SFX, for which the initial nanoliter volume setup needs to be scaled up ~1,000-fold. Different scale-up methods exist, each requiring careful optimization separate from the initial crystallization screening since crystallization depends on the geometry of the setup, in particular, the volume and shape of the LCP bolus. For GPCRs and other membrane proteins, the most successful method has been to use gas-tight syringes for large volume crystallization59. This approach mimics the geometry of an LCP bolus in a glass sandwich plate but at a much higher volume, and allows an easy separation of precipitant solution from the LCP and for sample consolidation. Additional rounds of optimization are often required to achieve the desired crystal density and uniformity.
Alternative scale-up methods of LCP crystallization for serial crystallography have been developed. In particular, batch crystallization in Eppendorf tubes using a needle-cleaning wire60 was successfully used to crystallize microbial rhodopsins for time-resolved SFX studies. This method is convenient for preparing large volumes of samples. However, it requires precise knowledge of the crystallization conditions, as it is not well-suited for optimization because of the large volumes used and the difficulty of observing small, colourless crystals. This approach was further modified to use 9-well glass plates61 with better optical properties for crystal observation and compatibility with condition optimization. This setup, however, can lead to losses during sample harvesting, consolidation, and removal of the precipitant solution.
Pre-beam time crystal characterization
Microcrystallization often starts from similar crystallization conditions as macroscopic crystal growth. This suggests that the crystalline properties are retained, namely the space group and unit cell parameters. However, this assumption is frequently incorrect and must be tested45. Although microcrystals are often too small to collect a synchrotron data set, the diffraction limit can be determined, enabling a comparison of different crystal batches. Additionally, it is often possible to deduce unit cell constants. This allows identifying and discarding crystal forms that have unfavorable unit cell combinations — for example, where the unit cell lengths are very close to each other or are multiples of each other or of diagonals — which would impact unique indexing. Similarly, any changes in crystal environment and their effects on diffraction resolution or other crystal parameters should be tested at a synchrotron beamline prior to XFEL data collection. Relevant parameters include pH for embedding in viscous media or for reaction initiation; hydration upon embedding; or precipitant, such as malonate and ammonium sulphate exchange. The aim is to provide a fully tested microcrystalline sample for XFEL data collection and online analysis. The only property that cannot be tested before beamtime is the diffraction resolution on exposure to an intense fs XFEL pulse.
Sample delivery into the XFEL beam
XFELs are pulsed X-ray sources with repetition rates from tens of hertz to several Megahertz. For efficient beam time and sample usage, the rates of sample replenishment and XFEL pulses (the latter are typically set to the frame rate of the available detector) should roughly match, TABLE 1. There are different methods to deliver microcrystals into the XFEL beam, FIG. 2. They can be divided into jet, droplet-based methods, and fixed targets, FIG. 3. The approaches differ in the amount of material needed, limitations on crystal size and uniformity, scattering background, technical complexity and suitability for different applications or XFEL sources, TABLE 2. The data quality depends on recording diffraction intensities with a high signal-to-noise ratio. As a result, any background signal from the sample delivery medium should be reduced as much as possible62,63 by matching the crystal size with the beam size and surrounding medium thickness.
Fig. 2 |. SFX sample delivery methods.
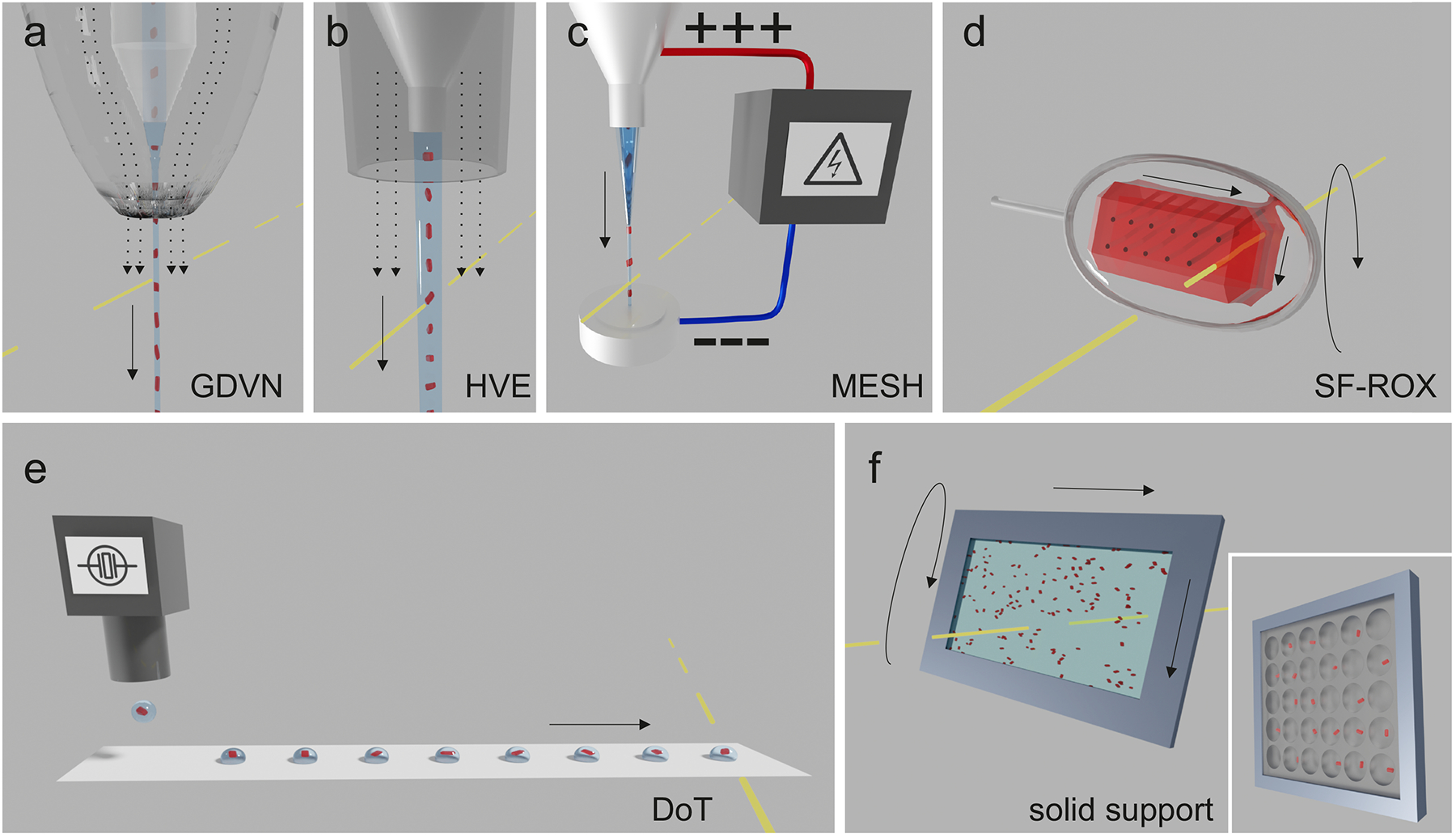
a | Gas dynamic virtual nozzle (GDVN)64. A suspension of crystal-containing liquid is pumped through an inner capillary, surrounded by an outer capillary through which a gas flows (dashed arrows). At the end of the GDVN, the outer capillary constricts to narrow the gas stream, accelerating both gas- and liquid. The liquid stream is focused into a narrow jet that breaks up into droplets. b | High viscosity extrusion (HVE) injector. Crystals are dispersed in a highly viscous medium and slowly extruded into a stream. A gas sheath prevents the stream from curling back onto itself. c | Microfluidic Electrokinetic Sample Holder (MESH)224,225. A high voltage (several kV) stretches a thin, slow-flowing stream of crystal-containing liquid between two electrodes. d | Serial Femtosecond Rotation crystallography (SF-ROX). A goniometer-mounted large crystal is translated and rotated between XFEL exposures. e | Drop-on-demand (DoD)83. Droplets of a crystal suspension are generated with, for example, a piezoelectric device. The droplet can be synchronized with and intersected by the XFEL pulse in free fall83, immersed in an oil stream85, or as shown in panel d, be deposited onto a tape and moved through the XFEL beam, Drop-on-Tape35 (DoT). f | Solid support methods. Crystals are deposited onto an X-ray transparent substrate, often referred to as a fixed target or chip, and scanned through the beam. Both unpatterned chips, on which crystals will assume random positions, and patterned chips (inset), which have wells for crystal location, are used.
Fig. 3 |. Distribution of sample delivery techniques used for published SFX experiments resulting in PDB codes.
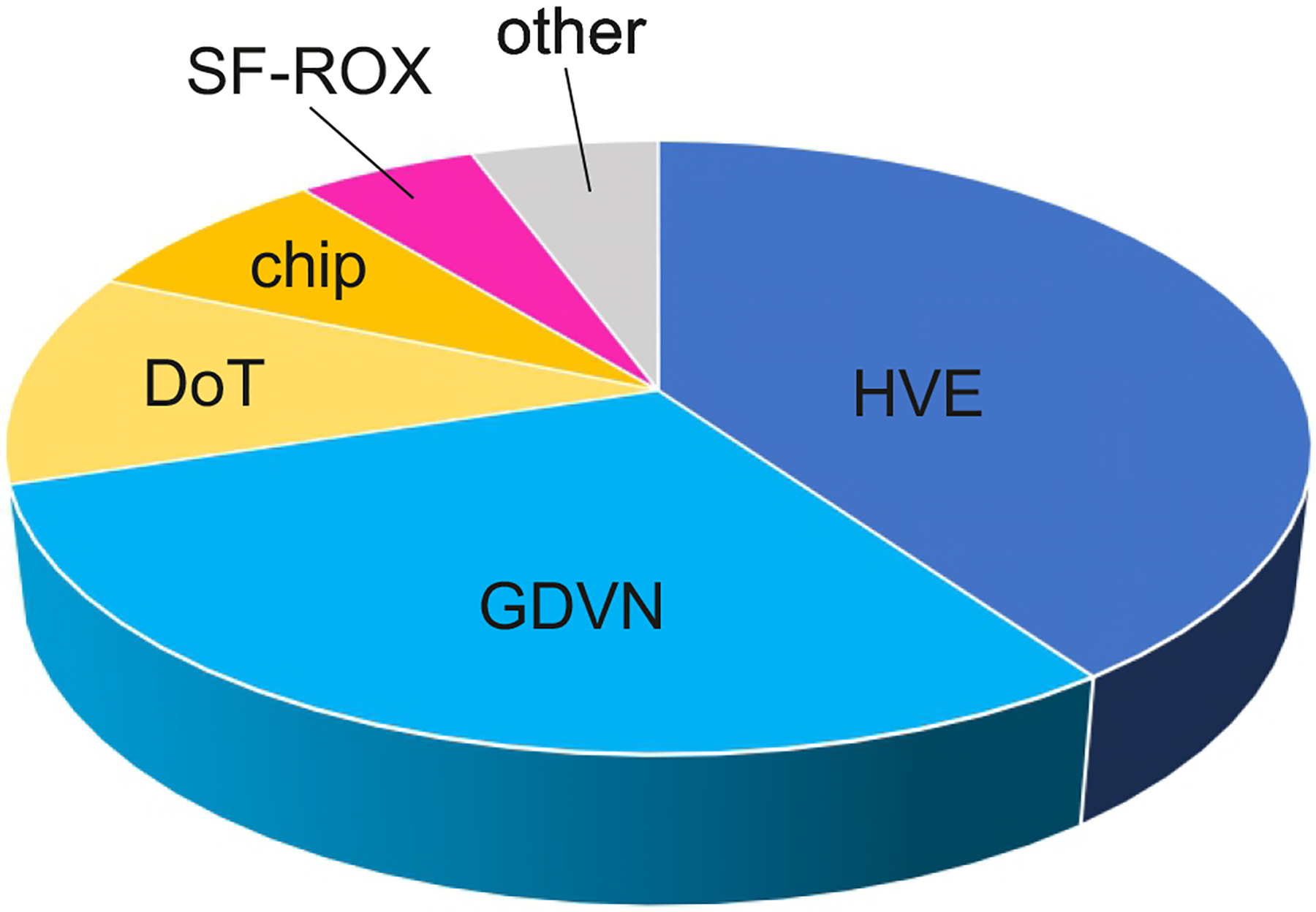
Data up to December 2021. Of a total of 417, 71% used jets — 41% high viscosity extrusion (HVE), 30% gas dynamic virtual nozzles (GDVN) — 10% used drop-on-tape (DoT), 7% employed fixed targets or chips, 5% SF-ROX136 and 5% MESH224,225. Jet-type techniques are shown in shades of blue. Other includes MESH (~ 5%), free droplets and segmented flow. Conceptually, SF-ROX and MESH can be considered to belong to fixed target and injection sample delivery, respectively. However, both approaches have distinct and unique features: SF-ROX136 uses very large single crystals and MESH uses high electric fields.
Gas dynamic virtual nozzles (GDVNs)
Gas dynamic virtual nozzles (GDVNs)64 are jet-based delivery methods. A suspension of crystals is pumped through a capillary at high flow rates (generally 30–60 μl/min). A helium gas sheath focuses the resulting stream to produce a thin (typically 3–6 μm diameter) fast-flowing jet32,64, well suited for tiny crystals, FIG. 2a. Gas-focusing is used to reduce scattering background by affording very thin jets without clogging narrow capillaries by flowing crystals. Jet imaging before and during XFEL beam time is required to establish jetting instead of spraying conditions. Moreover, it allows optimization of the length of the free-standing jet by adjusting the sample composition and flow conditions to avoid shooting into the Rayleigh breakup region downstream of the actual jet. In this region the continuous jet falls apart into droplets, FIG. 2a, resulting in reduced hit rates. It is also best to avoid shooting the jet close to the nozzle exit because this facilitates debris deposition from X-ray-induced jet explosion, promoting clogging and increased background scatter. Jet explosion results in transient formation of a gap in the jet65. Eventually, the gap will close due to material flowing from upstream. This, however, takes time and may limit the maximal XFEL repetition rate. Jet healing is fast enough to enable MHz data collection, but only for high jet speeds66–68, typically 80–100 m/s68,69. Such fast jets are very thin and often difficult to visualize. Jet speed can be determined using stroboscopic illumination65,70 and fast cameras, setups that were first developed to image jet explosions65. So far, GDVNs are the only approach shown to support SFX sample replenishment for full MHz data collection66–68. Disadvantages include high sample consumption — up to many tens of milligrams of crystallized protein to collect a data set — because most of the flowing material is not probed at low repetition rate XFELs and the EuXFEL71. The latter delivers its photons with a maximal 4.15 MHz intra-burst repetition rate, but in pulse trains of 10 Hz frequency, separated by 99.4 ms idle time. A variation of GDVNs is to use double-flow focusing nozzles where an inner crystal-containing liquid jet72 is focused by a coaxial faster outer liquid jet (typically ethanol) that is itself focused by gas as in the traditional GDVN72. This setup reduces flow rates of the inner jet, lowering sample consumption, while also reducing background scattering, and increasing jet stability, particularly for solutions containing high salt concentrations. To produce a stable GDVN jet, the mother liquor should have relatively low viscosity, and crystal thicknesses should not exceed the jet diameter significantly. Since large crystals, aggregates, or clusters of microcrystals can clog the lines, prefiltering of the sample and the use of inline filters, such as stainless steel with 20 μm or 40 μm pore size, is strongly recommended. It is critical to determine a suitable crystal concentration to ensure efficient injection. The concentration should be high enough to produce a good hit rate but low enough to provide a stable jet without clogging, and wiggling that leads to poor jet-XFEL-intersection. This concentration is highly sample-dependent and needs to be determined experimentally. Frequently, crystal concentration is reported as the number of crystals per ml, as determined by a Neubauer cell counting chamber. An alternative, faster way to determine the concentration is to use the percentage (v/v) of settled crystalline material73. A good starting concentration for injecting crystals < 5 μm is 15 % (v/v) gravity-settled material. Microcrystal settling also occurs during XFEL data collection, depleting the crystal concentration in the injected suspension over time, resulting in a steadily decreasing crystal hit rate. This can be prevented by using an anti-settling device74. Moreover, for most samples, regular rinsing of the lines and injector — for example washing for one minute per every 10 minutes of injection — with water can alleviate clogging problems. Instead of using gas, liquid jets can be focused by electric fields, as in MESH (microfluidic electrokinetic sample holder) injectors, FIG. 2c.
High viscosity extrusion
High viscosity extrusion (HVE) is a jet-based approach that relies on the extrusion of a highly viscous delivery matrix in which crystals are either grown or incorporated through a narrow capillary20,33 (FIG. 2b). A helium or nitrogen gas sheath is used in HVE to stabilize the jet rather than focus it. Because the flow rates are very low — tens of nanoliters to several microliters per minute — the sample is used efficiently at low pulse repetition rate XFELs and synchrotrons. However, the stream diameter is large, often greater than the capillary’s 50–100 μm inner diameter, resulting in a relatively high background. Using capillary diameters below 50 μm is not practical because of the extremely high pressure required for extrusion and greatly increased capillary clogging rates. HVE injection is the method of choice to deliver LCP grown crystals into the beam since they are intrinsically embedded in a highly viscous matrix. However, solution-grown crystals may also be embedded20,75,76 in viscous material. A number of viscous matrices have been described62,63,77,78, all of which have limitations in terms of tolerated pH values and salt concentrations. The exceptions are grease40,75 and similar materials, such as vaseline20, which are largely insensitive to buffer composition, but display a relatively strong X-ray background, scatter pump laser light significantly79, may cause crystal dehydration80 and are generally unsuited for grease-or-oil-sensitive protein crystals77. Regardless of the matrix, constant jet speed is critical for well-defined X-ray data collection conditions. This is particularly important when analyzing radiation-sensitive samples or performing time-resolved experiments. A lower jet speed may result in undesired multiple exposures of radiation-sensitive crystals, or, in time-resolved light-triggered experiments, doubly photoexcited crystals. Increased jet speed may transport crystals featuring long-lived intermediates out of the X-ray focus before the next XFEL pulse arrives16. The speed of viscous jets often fluctuates strongly. While it is relatively straightforward to omit jet curling and stop-and-go periods from data acquisition or discard the respective data afterward, it is more difficult to detect changes in jet speed. Therefore, in particular for time-resolved experiments16, jet speed needs to be measured simultaneously with data acquisition, as calculations based on jet diameter and flow rates are not sufficiently accurate. To derive the jet speed, moving features in the crystal carrying jet can be tracked over time. Using this approach, data can be excluded where the variation in jet speed is too high for reliable pump excitation conditions81. At XFELs this method can be facilitated by making use of X-ray pulse induced changes in the consistency of the viscous matrix in a localized region around the impact site, appearing as a dark stripe on the jet. Consecutive pulses each leave a stripe, resulting in dark ladder steps, with a step size dependent on jet speed and X-ray repetition rate82. The distance between stipes can be determined from images of the jet, providing together with the pulse repetition rate information about jet speed.
Drop-on-demand and drop-on-tape
An alternative to continuous jets is to use droplets for crystal delivery (FIG. 2f). The crystals can be resuspended, for example, from Eppendorf tubes or crystallization plates; droplets are generated by piezo drivers or acoustic ejection systems83,84. Droplet ejection has been synchronized with 120 Hz XFEL pulses, referred to as drop-on-demand (DoD), and shot in free fall83, reducing sample consumption significantly compared to GDVNs. Alternatively, the droplets can be deposited on a mylar tape, an approach referred to as drop-on-tape (DoT) delivery35. Typical droplet volumes are 4 nl, resulting in a drop on the tape with 250 μm diameter. The tape is translated in a chamber with a defined atmosphere that can be adjusted to the requirements of the experiment — for example, high or low oxygen concentration — presenting the crystals containing drops to the X-ray beam. Compared to jets and chips, the DoD and DoT delivery approaches are used often for relatively large crystals (>20 μm) which is also advantageous in view of the high background scattering from the drop. However, smaller drop sizes and crystals <20 μm are possible for DoD. The approach also allows to deliver bigger crystals, up to ~100 μm, that cannot easily be run in a jet. Drops-on-demand can also be generated by dispensing aqueous droplets into an oil phase using microfluidics36,85.
Fixed targets
In addition to free-standing jets and droplets, fixed targets can be used for microcrystal delivery into the XFEL beam (FIG. 2e). Such devices include various forms of chips. These approaches enable precise temporal and spatial presentation of crystals to the X-ray beam, in contrast to the stochastic delivery in jets or droplets. Patterned chips containing indentations or wells for trapping crystals have been made of silicon26,29,86, silicon nitride87 or polymers25,31. Chips are typically blotted after loading the crystal suspension to reduce their background, resulting in a thin liquid film around the crystals. Chip loading and blotting should be done in a humid environment to prevent dehydration. In most cases, chips are sealed on both sides by X-ray transparent membranes, for example, mylar, to prevent crystal dehydration during data collection, resulting in non-isomorphism88. High precision fast motors have been used to raster the chips through the X-ray beam at first-generation XFELs. Data collection in a helium atmosphere, rather than a vacuum, is highly advisable to reduce the setup complexity and slow desiccation around holes burned by the XFEL beam through the films used to seal off the chip25.
Results
Diffraction data analysis
Serial- versus rotation data processing
The analysis of XFEL diffraction data is essentially the same as the analysis of synchrotron data: diffraction spots are indexed and integrated, and the resulting structure factor amplitudes are used to calculate electron density maps. However, there are several important differences. First, there are fundamental complications caused by the fact that XFEL pulses are so short that the crystal cannot be rotated during the pulse, meaning only still images can be collected. Second, the as-yet experimental nature of XFEL-based crystallography often causes practical complications that need to be addressed.
We will first address the fundamental challenges in data processing caused by the short duration of XFEL pulses. Diffraction processes can be conveniently modelled using an Ewald construction as shown in FIG. 4a. During conventional rotation crystallography, as practiced at synchrotrons and home sources (FIG. 1a, 4a), the crystal is rotated during every exposure. Eventually, this allows all the reciprocal lattice points to pass through the Ewald sphere in their entirety, enabling the integration of full diffraction intensities (FIG. 4b). However, when femtosecond pulses are used, the crystals are essentially stationary during exposure, so only a very small slice of reciprocal space is sampled (FIG. 1, FIG. 4c,d). To obtain full intensities, a Monte-Carlo integration scheme is used, in which large numbers of partial intensities are averaged. In this way, all of reciprocal space is eventually sampled89. However, the crystals will not all be of the same size, affecting the intensities collected from them. Moreover, because most XFEL experiments use radiation generated by self-amplified spontaneous emission7, the spectrum and intensity of the incoming XFEL pulses will vary from shot to shot, also affecting the diffracted intensities. As the Monte-Carlo scheme also averages out all these sources of variation, typically, data from tens of thousands of diffraction patterns needs to be combined.
Fig. 4 |. Integration of intensities in rotation- and serial crystallography.
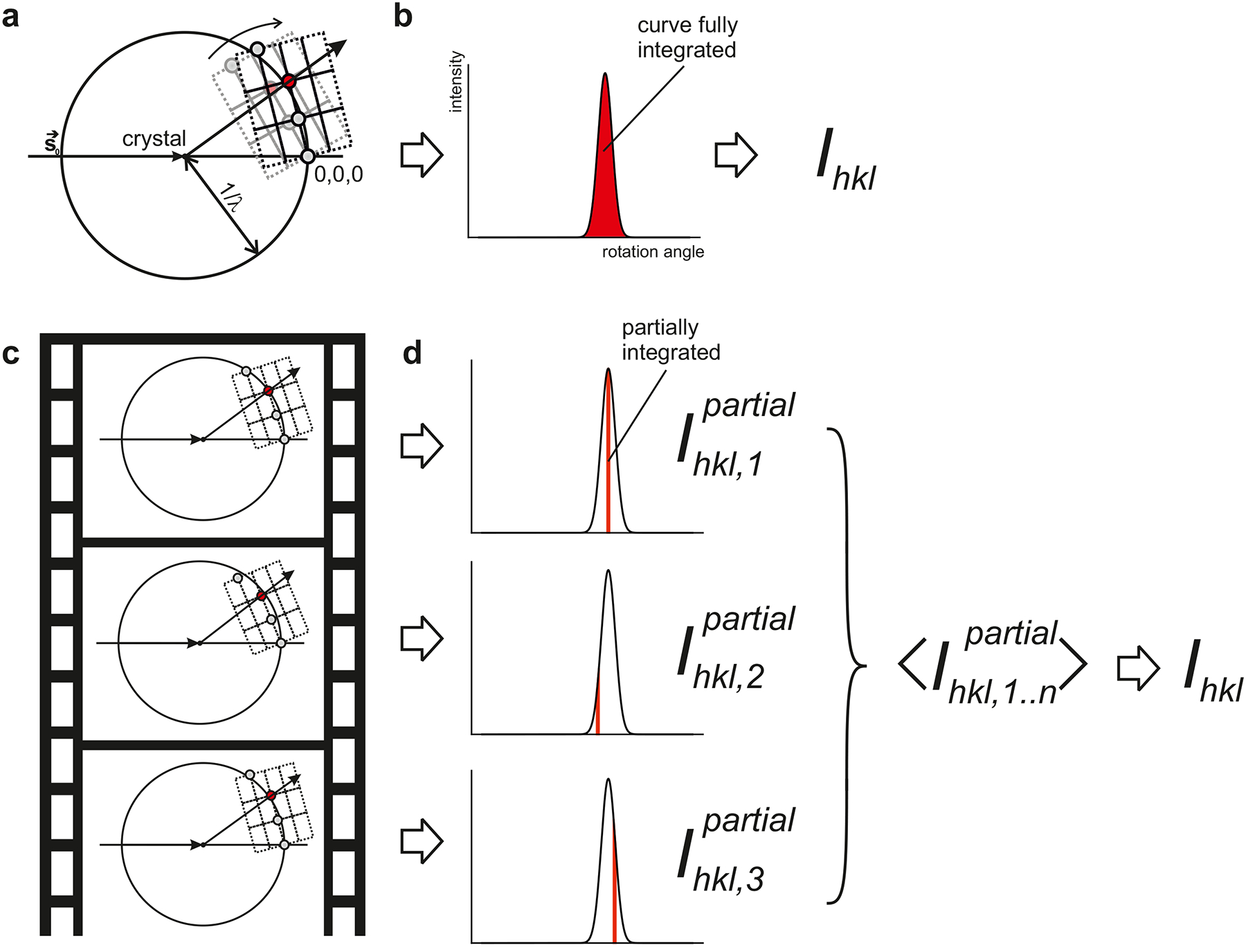
a | The Ewald construction can be used to visualize diffraction geometries. The incoming radiation hits the crystal located at the center of the Ewald sphere, which has radius 1/λ. The reciprocal lattice is drawn, with the origin at the position where the direct beam intersects the Ewald sphere behind the crystal. In conventional rotation crystallography, the crystal is rotated, and the reciprocal lattice rotates (curved arrow), causing reciprocal lattice points (spheres) to move through the Ewald sphere, rotating into and out of reflection condition. b | The observed intensity of diffraction from a certain reciprocal lattice point (such as the red sphere in panel a.) increases, then decreases with rotation angle. Integrating this rocking curve yields the intensity Ihkl. c | In SFX there is no a priori control of the crystal’s orientation, and intersection of the red reciprocal lattice point with the Ewald sphere is a stochastic process. d | The crystals are effectively motionless during the extremely brief exposure. As a result, the observed diffraction corresponds to a thin slice of the rocking curve of each reflection. Partial intensities from many exposures need to be averaged to give Ihkl for each reflection.
Collection of a diffraction pattern is not, however, sufficient to obtain partial intensities from it. First, the diffraction pattern needs to be indexed, that is the orientation of the crystal and its unit cell parameters must be determined to assign the correct Miller indices to the diffraction peaks. In conventional rotation crystallography, a range of diffraction patterns is available for each crystal, and each one usually contains many diffraction spots. Additionally, the rotational relationship between the patterns is known exactly. Modern software makes indexing a trivial exercise in most instances. In SFX, however, this is not the case. First, each diffraction pattern stems from an individual crystal, with no orientation relationship between the various crystals. Second, there may be relatively few diffraction spots to determine crystal orientation because of the narrow sampling of reciprocal space caused by the lack of crystal rotation. As a result, many diffraction patterns may be unindexable, increasing the number of crystals that need to be injected to obtain sufficient indexed diffraction patterns for Monte Carlo convergence. Thus, in the end, a Monte-Carlo-integrated data set will typically have a very high multiplicity, the average number of observations of a unique reflection I(hkl), which is usually in the hundreds. Importantly, this multiplicity should not be confused with the term redundancy, which is reported in rotation crystallography. Redundancy counts the average number of fully integrated observations of a unique reflection I(hkl), which all come from the same or several crystals and are measured with practically the same incident intensity. Thus, redundancy is therefore typically much lower. Redundancy and multiplicity can be used interchangeably in both SFX and rotation crystallography, but it is not useful to compare the two numbers between methods.
At present, SFX data processing is mainly performed with one of three software packages: CrystFEL90, cctbx.xfel91,92, and nXDS93. A detailed step-by-step guide for processing serial data using CrystFEL was recently published94, and the use of the cctbx.xfel GUI was described in REF95. The packages now offer algorithms beyond simple Monte-Carlo averaging, such as post-refinement96–98 and partiality analysis97,99, which can greatly reduce the number of images required for a data set of a certain quality.
An additional complication of processing single-exposure serial diffraction data arises when the symmetry of the crystal lattice exceeds that of the space group. There are two or more equally valid ways to define the unit cell in such cases. For instance, for a crystal with P6x space group symmetry, there are two ways to index the crystal, which differ only in the choice of axis defined as a and b. This is called an indexing ambiguity14. When data from two or more such crystals are to be combined in a meaningful way, they must all be indexed using the same choice of indexing. In conventional crystallography, this is easily done by comparing fully integrated intensities. By comparison, in SFX, fully integrated intensities are not available and in such a case, indiscriminate merging has the same result as (pseudo)merohedral twinning: the averaging of reflection intensities related by the excess symmetry of the lattice. Indeed, the standard tests for twinning will detect this issue, and in early SFX studies14, data affected by such an indexing ambiguity were treated as twinned. However, it was later shown that correlations between diffraction patterns could be used to assign the correct indexing choice to a set of indexed diffraction images93,100, which helped resolve this issue.
In a similar way as in conventional crystallography, data quality in SFX can be assessed in terms of precision, specifically by looking at the spread of individual observations of symmetry-related reflections. To this end, White90 defined a new R-factor, Rsplit, which is related to the conventional Rpim101 and behaves similarly102. Moreover, the signal-to-noise ratio as a function of resolution can be inspected as can CC1/2103 to obtain a measure of the resolution of the data. However, as has been pointed out previously, these simple measures of data quality are not necessarily good indicators of the success of an experiment; for one thing, a three-dimensional representation of data quality metrics in reciprocal space would be far more useful than any single measure describing the quality of all data104. The ultimate quality measure, however, is, of course, the quality of the (unbiased) electron density maps. Ideally, when studying structural changes, difference maps should show these as density features that are clearly above the local noise levels37,105, and in general, omit maps should always show clear density for side chains whose conformation is being studied. In time resolved studies, structural changes should develop smoothly over time when time delays are closely spaced. Therefore, it is worth considering to not only collect data for time delays where peak occupancies of certain intermediates are expected, but also one or more intermediate time points. In addition, one could collect time points close by other time points as a control on reproducibility.
Pre-beam time preparation and online feedback
Alongside the challenges inherent to the method of SFX, there are practical issues that affect data analysis. These stem from the still largely experimental nature of XFEL facilities. In contrast to the situation at dedicated MX beamlines at synchrotrons, many different types of experiments are performed at XFEL instruments, meaning setups are often changed between experiments. As a result, important parameters, such as the sample-to-detector distance and the direct beam position may not be known to the accuracy a crystallographer is used to at a synchrotron. This is even true for the geometry of the detector itself, that is the exact position of each pixel on the detector. Some detectors used at XFELs consist of panels that can be moved relative to each other, changing the shape of the detector. Since indexing relies entirely on the accurate determination of diffraction angles, the experimental geometry, such as detector distance, direct beam position, and detector geometry, must be determined as accurately as possible before meaningful data can be obtained, and this usually requires action on the part of the user. In fact, one can observe a strong dependence of the success rate of indexing on the quality of the detector geometry description, and one approach to quickly refine the detector distance is by optimizing this rate.
Given all the points described above, it is clear that the team members assigned to data analysis during an SFX experiment need to prepare carefully. Prior to the beam time, they need to obtain as much information as possible on the crystal system that will be used. Important parameters and properties include expected crystal symmetry; cell dimensions, and their potential to cause an indexing ambiguity; required resolution for an experiment to be successful; and necessary data quality. Moreover, the analysis team should contact the facility early on to obtain access to the on-site computing systems, ensure that all required software is installed, check that sufficient computing resources are set aside for online analysis and perhaps even test the software with trial data if available. Unless the goal is de novo structure determination, phasing models for the various crystal systems likely to be encountered are best prepared beforehand, too.
At the start of the beam time, or shortly before, a reliable detector geometry should be obtained from the beamline scientist, including a reasonable estimate of the detector distance. If this is not available, or if there is any reason to suspect that the geometry has changed since the previous experiment, the detector geometry must be checked and if required, corrected. A good way to confirm (and if necessary improve) the description of the experimental geometry is to collect data from a reference sample, such as lysozyme. The reference sample should be available in large quantities, inject relatively easily and diffract to high resolution. The relative orientation and position of detector panels can be checked using a large number of diffraction patterns that are summed to obtain a virtual powder pattern. This should show perfectly circular diffraction rings centered on the assumed direct beam position. Any discrepancies in the panel or direct beam position description can then be manually corrected. Next, one should attempt to index the reference sample data, while monitoring the cell parameters obtained. Histograms of the cell parameters derived from a large number of crystals should be inspected to ensure they match the expected values. The detector distance assumed during indexing may be corrected by optimizing the indexing rate and/or the shape of the peaks in the unit cell histograms53. When an initial, reasonable geometrical description of the experiment is obtained, software is available to further refine the detector geometry47,106. When this is completed, processing of the experimental data can begin.
During the actual experiment, data must be continuously analyzed to provide feedback to the experimental team. Different online data analysis packages exist107–110 (see also TABLE 1 for data analysis support) that allow to monitor crystal hit rate and resolution in relation to the goals of the experiment; whether a reaction has successfully been initiated; and if sufficiently many indexed patterns have been accumulated for a particular data set. This online data analysis involves not only the indexing and integration of data and the monitoring of quality indicators such as Rsplit, but we highly recommend that it extends to map calculation and interpretation. Thus, the data analysis team must be constantly available and work in close concert with the experimental team. During long beam times, shift work may be required to ensure constant high-quality feedback on data quality, which in turn requires excellent communication between shifts.
An important question to the analysis team is how many indexed images are needed for a data set. This seemingly simply question can be difficult to answer since it depends on the signal-to-noise ratio that is required for the particular experiment being performed. This ratio increases with the square root of the number of images used in Monte-Carlo integration. Simple molecular replacement phasing typically does not require a very high signal-to-noise ratio and may be possible with only a few thousand images or fewer111. De novo phase determination, however, requires accurate measurements of small differences between intensities of just a few percent or less of the total intensity. This requires a very high signal-to-noise ratio, and thus tens of thousands of images may be required for a serial crystallography phasing experiment102,112. The same is true when structural changes in response to an external trigger are to be observed as in a time-resolved study. Such experiments also depend on being able to accurately measure small intensity differences, the magnitudes of which scale with the size of the structural change, and depend on the fraction of molecules that were successfully triggered. A small change in the position of a few light atoms will only affect the diffracted intensities very slightly, particularly if that structural change has only occurred in a small fraction of molecules. To measure such differences requires a high signal-to-noise ratio and thus many thousands of images47,81,113–115. Importantly, this applies to both the data collected before and after the trigger – both data sets will require large numbers of images in such a case.
Importantly, as explained above, these numbers vary with application and even within an experiment. In a time-resolved experiment, for instance, it may be that for early time points with small structural changes more images are required than for later time points, when changes have grown in magnitude and so has the signal. The fact that it is difficult to predict how many images are required (see Fig. 3 in REF37) is another important reason for ongoing rolling data analysis during SFX data acquisition.
Offline analysis
The first step in offline analysis after the beam time is to ensure all hits are extracted from the available data, and then to maximize the number of indexed crystal lattices, which often involves final optimization of the detector geometry and detector distance. The latter may need to be optimized for individual stretches of data collection, for example when a sample injector is exchanged, the new injector is likely to be in a slightly different position than the previous one. After final integration, structure determination and interpretation follow. Many of the challenges involved were discussed recently37, particularly regarding the complications associated with determining structures with less than 100% occupancies, as frequently observed in time-resolved studies. Dealing with low occupancy states is never straightforward, even if the underlying diffraction intensities are highly accurately determined which is rarely the case for SFX. Moreover, deriving models of protein-ligand structures is inherently problematic, as the resolution is often not sufficient for unambiguous interpretation of the chemistry of the species and its stereochemical configuration116–118. So far, there is no consensus in the field on best practices for the whole process of data analysis in such cases37, starting from the integration and scaling of the diffraction intensities all the way to the calculation of extrapolated structure factors116,119, in which the fraction of the low-occupancy excited state is extrapolated to 100%. Thus, it seems prudent to apply a range of approaches in data analysis and interpretation (see REF37 for a discussion of the various approaches), checking for consistency and quantifying - or at least estimating - uncertainties. In particular, resampling techniques such as bootstrapping may be used to estimate error bars on refinement results37. Moreover, where enzymatic reaction mechanisms are concerned, collaborating with a quantum chemist experienced in the study of the system being investigated, or similar systems, is invaluable in guiding the interpretation of the experimental data, and helps avoid situations when chemically impossible structural models are built and published.
Applications
Ionization is an inherent by-product of any analysis using X-rays, but causes radiation damage. The initial ionization causes electronic rearrangements that can ultimately result in conformational rearrangements. This can affect the diffraction quality — global damage — and can cause localized structural changes — local damage — which is often detrimental to meaningful mechanistic interpretation. In crystallography, radiation damage is typically mitigated by using large cryocooled crystals for data acquisition, as the number of photons a crystal will diffract before degrading is proportional to its volume1. However, it is often not possible to grow large well-ordered crystals that diffract to high resolution. Moreover, even in cases where high quality large crystals can be grown, applications exist that require use of small crystals, for example, in time-resolved crystallography. Additionally, many metalloproteins are highly radiation-sensitive and are photo-reduced within the first few X-ray exposures120,121. SFX at XFELs offers a solution to these issues, enabling the acquisition of high-quality diffraction data that is essentially damage-free. This is not because of a lack of absorbed dose122,123, but rather the result of typically very short XFEL pulse lengths during which the extent of nuclear motion is limited. During the past decade, structural biology at XFELs has moved from method development and showcasing examples14,15,102 to providing novel scientific insight into challenging questions, such as the mechanism of water oxidation in photosystem II124.
Radiation-damage prone systems
Metalloproteins
Metalloproteins are prone to radiation damage. The presence of heavy atom(s) results in higher X-ray absorption — the interaction cross-section is roughly proportional to Z2.7, where Z is the atomic number — and thus a higher dose. The photoelectrons and subsequently liberated electrons ionize the sample, and the radical chemistry ensuing during conventional crystallographic data acquisition results in reduction of catalytic centers in redox enzymes. Consequently, the coordination geometries of metal centers, flavins and other cofactors with large conjugated systems in crystal structures determined using synchrotron radiation are often considered questionable, especially if they differ from spectroscopic insight. This concerns in particular the catalytically-important oxy, peroxo and oxo ferryl complexes of heme proteins, as evidenced by the X-ray-driven catalytic reduction of a bound dioxygen species in cytochrome P450125 and horseradish peroxidase126. Thus, an increasing number of structural investigations of metalloenzymes use SFX in the quest for a molecular understanding of the enzymatic activation of oxygen127, peroxide122,128,129, nitric oxide130,131 and nitrite132,133. Other investigations focus on, for example, understanding the metal-catalyzed oxidation of water by photosystem II43,44,134–142 and of methane by methane monooxygenase46. Oxidation states, particularly of metalloproteins, can change during purification, crystallization or crystal handling. Since knowledge of the oxidation state is crucial for a mechanistic interpretation of the structural data, it is worthwhile to combine SFX data acquisition with X-ray emission spectroscopy134,143,144.
Many heme enzymes use ferryl heme intermediates during catalysis. In these intermediates, the heme iron is highly oxidized, with an oxidation state that is either one equivalent (FeIV, Compound II) or two equivalents (formally FeV, Compound I) above the resting ferric oxidation state (FeIII). The second oxidizing equivalent in Compound I resides on the porphyrin ring or a protein radical. The nature of the ferryl heme — FeIV=O versus FeIV-OH — is important for the catalytic outcome, and in many cases, highly debated. Most studies have been performed on peroxidases since their ferryl intermediates are significantly more stable than the corresponding species in other enzymes, such as P450s. SFX data of different peroxidases were used to determine structures of the ferric state — dye decolorising peroxidase122 — of Compound I — cytochrome c peroxidase128, dye decolorising peroxidase122 — and of Compound II — cytochrome c peroxidase, soybean ascorbate peroxidase129. The structures highlight the importance of specific hydrogen bonding in the active site for the generation and conformation of ferryl intermediates and indicate fine-tuned functional differences related to substrate specificity129.
Cytochrome c oxidases (CcO) are integral membrane complexes in mitochondria and many bacteria that catalyze the reduction of molecular oxygen to water and use the released energy to translocate protons across the membrane. In the oxidized state, the oxygen reduction site of CcO consists of a heme a3 iron (Fea3), copper (CuB) and a putative peroxide ligand. Structures from synchrotron data could not identify the active site ligand: peroxide or hydroxide. Three SFX studies addressed this question by analyzing the oxidized form of the enzyme. Bovine CcO was investigated by either SF-ROX using large crystals kept at cryogenic temperature23 or microcrystals in an ambient temperature liquid jet127, whereas the microbial form (ba3 type) of CcO was studied using microcrystals injected at room temperature in an LCP stream145. Interestingly, an elongated electron density close to the heme was observed in bovine CcO and was modelled by a peroxide23,127 in line with resonance Raman studies146. In contrast, for the microbial form, the corresponding electron density was round and modelled by a water or hydride. It is unclear whether these differences are related to structural differences around the proton loading site145.
In-vivo grown crystals
Protein crystals can grow spontaneously inside living cells, in cellulo147,148. Depending on the size of the surrounding cellular structure, the crystals are often too small for synchrotron structure determination — exceptions include the relatively large crystals formed by proteins overexpressed in insect cells that often exceed cellular dimensions18,149 — and are thus perfect candidates for structure determination using SFX. Examples include naturally occurring proteinaceous112,150,151 and viral152 insecticides, alongside proteins heterologously overexpressed in insect153–155 or mammalian cells156. Conveniently, it is not necessary to purify these crystals. In cellulo diffraction is possible150,155 and can even be advantageous for preserving crystal quality149 and retaining natural ligands154. In addition, structures derived from in vivo grown crystals may contain posttranslational modifications, such as glycosylation153, that are often removed to improve homogeneity when the protein is crystallized in vitro.
Membrane proteins
Structure determination of membrane proteins — in particular, metalloproteins involved in energy metabolism, reaction centers157, photosystems41,43,44,134–142,158, cytochrome c oxidases23,127,145, microbial rhodopsins60,81,114,115,159,160, G protein-coupled receptors (GPCRs), and other membrane proteins161,162 — has greatly benefitted from the development of SFX techniques82. GPCRs constitute a large superfamily of heptahelical cell-surface transmembrane receptors with over 800 members in the human proteome. These receptors translate extracellular signals into intracellular responses by coupling to transducers such as the eponymous G proteins. GPCRs have been traditionally considered difficult to crystallize because of low expression yields, dynamic nature and instability outside their native membrane environment. A number of technical advances in the early 2000s addressed these issues and enabled GPCR crystallization163. The advances include engineering of recombinantly expressed GPCRs by truncating and/or substituting flexible segments by stabilizing fusion partner domains. Additional approaches include locking receptors in specific conformational states with point mutations; application of binding partners such as antibodies, nanobodies or high affinity ligands; and growing crystals in the lipidic environment of LCP. Crystallization in LCP enables better packing and crystal quality for membrane proteins but often suffers from small crystal size. The advent of SFX alleviated the bottleneck of optimizing crystal size for data acquisition using synchrotron sources: the high peak brilliance of XFELs allows collection of diffraction data from smaller, potentially better ordered crystals kept at room-temperature. For GPCRs, this possibility was realized in 2013 when the structure of the serotonin receptor 5HT-2B164 was solved by SFX. Microcrystals were delivered into the XFEL beam using a high viscosity injector33, which has become the standard for delivery of membrane protein crystals grown in LCP because of its synergy with the gel-like nature of LCP matrix and the minimization of sample waste given the low extrusion speed. Since then, the SFX of GPCRs has rapidly outgrown its proof-of-concept phase. The structure of a cancer target — the human smoothened receptor bound to the teratogen cyclopamine — was solved at an XFEL source33, whereas at a synchrotron, larger crystals displayed high mosaicity and poor diffraction, rendering structure determination unsuccessful. Similarly, SFX substantially improved the resolution of the delta opioid receptor bound to a peptide ligand, from 3.3 Å using cryocooled crystals at a synchrotron source to 2.7 Å165. The resolution increase was important since it enabled unambiguous modelling of the flexible peptide ligand that offers efficient pain relief without associated tolerance and dependancy. Shortly after, the structure of the angiotensin II receptor type 1 (AT1R)166 and the structure of the visual rhodopsin bound to arrestin167 represented the first time SFX enabled solving structures of an unknown GPCR and a unknown complex of a GPCR with its signaling partner. That high quality SFX data can be obtained from GPCR microcrystals was illustrated by native sulphur phasing of the adenosine A2A receptor168. Since then, SFX has enabled the determination of several additional novel GPCR structures, including the angiotensin II type 2 receptor (AT2R)169, which is implicated in neuropathic pain relief and tissue protection, revealing the molecular foundation for its unusual activation mechanism. Several structures of lipid receptors solved by SFX, such as the prostaglandin EP3 receptor receptor170, the prostaglandin D2 receptor (CRTH2)171, and cysteinyl-leukotriene receptor type 1 (CysLT1)172 provided insights in receptor activation by signaling lipid molecules. More recently, SFX structures of melatonin receptors MT1173 and MT2174, revealed an unexpected ligand entry path from within the lipid bilayer that explains their selectivity towards their cognate ligands. All of these investigations benefitted from powerful XFEL sources, as crystals of sufficient size or quality could not be obtained at the time for high-resolution data collection. We note that individual GPCRs and their ligands differ widely in their ability to form sufficiently large and well-diffracting crystals, making it necessary to use the appropriate structure determination technique for a given target-ligand combination and for the primary objective of the structural study. We cannot exclude that in principle, and with continued optimization, crystals suitable for synchrotron data collection could be attainable for some of these systems in the future. However, given the high cost of GPCR expression and the value of their structures for the community, SFX offers the ability to bypass months or even years of crystal optimization.
GPCRs are among the most important drug targets owing to their exposed location on the cell surface and involvement in (patho)physiological processes. The value of GPCR structures for structure based drug discovery critically depends on the ability of researchers to quickly and efficiently obtain structures of receptors bound to multiple ligands and to rationalize structure-activity relationship (SAR) data or test binding hypotheses. Alongside this, most GPCR crystals are not amenable to traditional ligand soaking experiments because they typically cannot be crystallized in their apo form. To this end, a method that enables the determination of multiple co-crystal structures from the same receptor preparation was developed175. The approach employs a transient ligand that can be exchanged during crystallization and takes advantage of SFX data collection from small crystals.
Time-resolved studies
Time-resolved studies aim to characterize transient states to elucidate reaction mechanisms. For crystallographic characterization, the reaction must occur in the crystalline state. Additionally, the intermediates need to accumulate to high enough occupancy, structural changes must be sufficiently large for detection, and feasible within the constraints of the crystal lattice, BOX 2. If the latter is an issue, particularly for intermediates occurring late along the reaction coordinate, alternative approaches can be considered, such as time-resolved solution scattering (SAXS/WAXS)176–178 or time-resolved cryo-EM179,180.
Box 2. Checklist for planning SFX experiments, including time-resolved studies.
Experiment cost – benefit analysis:
Consider the scientific impact or technical advances of the proposed research in view of the resources and efforts involved in SFX data collection.
Use of SFX data collection:
Decide whether SFX is needed or if another method — MX or SSX — can be used instead. This depends on the sample to be studied, the required spatial resolution and in case of time-resolved experiments, the timescale and structural changes expected.
If SFX is the best approach, choose an appropriate facility and instrument for data collection.
Sample properties and availability:
Sample-related considerations include the material availability; stability; aggregation; crystallization conditions, as this affects the choice of sample delivery; and crystal properties, including space group, unit cell, diffraction resolution, and physical characteristics.
Sample delivery method:
Choice of method is dependent on the available crystal quantity, crystal size and size homogeneity, crystals growth medium, and scattering strength.
For time-resolved experiments the choice of delivery method is additionally influenced by the triggering method and time delay.
Available options include jets (GVDN, HVE, MESH); drop-on-demand or drop-on-tape methods; and chips or fixed targets.
Risk assessment:
The risk assessment should evaluate what assumptions are involved in the experiment, what can go wrong, how to respond, and steps that can be taken to rescue the beamtime if the planned experiment cannot be performed (Plan B).
Additional points to consider for time-resolved experiments:
The reaction scheme, including the kinetic rates and thus time scales and — in case of mixing experiments — affinities.
The observables to assess the expected signal. This includes the magnitude of the anticipated conformational changes and the expected occupancies taking into account the yield of reaction initiation, forward and backward reactions.
The reaction initiation. This includes the method —photoexcitation, chemical mixing, and jumps in pH, temperature, pressure, or electric field — the time scale, which needs to be faster than the subsequent process investigated, the yield, and the permissible crystal size.
It needs to be established that the reaction proceeds in crystallo and the kinetic rates need to be determined. If the latter is not possible, appropriate data collection schemes must be devised, such as using logarithmically spaced time-delays.
Precise reaction initiation is key to a successful time-resolved experiment181,182. Three goals must be met. First, only the intended reaction should be initiated, which is often an issue when using high-intensity lasers for photoexcitation79,183. Second, the desired intermediate should be observed. This is related to the first point, but can also become an issue when the in crystallo reaction kinetics differ from those in solution or if reaction initiation is too slow, for example, in mixing experiments, resulting in an undefined combination of reaction intermediates. The third goal is to obtain the highest possible yield. Addressing these points requires the use of very small crystals. The reason is that the dense molecular packing in crystals entails high protein concentration (typically 5–30 mM) complicating reaction initiation by light due to very high optical density and by chemical mixing because of the length of diffusion pathways to the active sites, respectively. The crystal size d determines whether a significant fraction can be photoexcited by a single photon (d ≤1/e light penetration depth) instead of by multiple photons (d >1/e)79. In chemical mixing experiments, d sets the minimal time for diffusion of ligand molecules to binding sites184, often limiting the effective time resolution. The need for small crystals — typically around 5 μm — in time-resolved crystallography and the unique ability of XFELs to use such crystals make them an excellent experimental match181,182.
For reaction initiation, a suitable reaction trigger needs to be identified. The most commonly used approaches are light (photoexcitation) and chemical mixing (diffusion), for which it is important to establish the reaction kinetics in the crystalline state, ideally by spectroscopic methods. Often the kinetics are slow compared to in-solution conditions due to crystalline confinement and/or the chemical composition of the crystallization solution. Changes can extend to the ultrafast time scale185. Knowing the in crystallo kinetics enables determination of the correct time delay after reaction initiation, that is when to collect data for a given reaction intermediate. In addition, it is highly desirable to establish online monitoring to determine what SFX is probing. A good example is the simultaneous SFX data collection and monitoring of metal oxidation states by X-ray emission spectroscopy134,143,144.
Examples using photoexcitation
The fs pulse length afforded by XFELs has extended the time-resolution of photocrystallography by at least three orders of magnitude from ~100 ps limited by the electron bunch length at synchrotron sources. As a result, femtochemistry and femtobiology studies are now accessible with X-ray analysis. In particular, XFELs enable probing of system dynamics47,81,177 because the initial synchronization by an fs pump laser pulse is preserved on the ultrafast timescale. For longer time delays (> a few ps), the statistically distributed onset of thermally activated processes results in exponential-like kinetics.
Time-resolved optical pump X-ray probe SFX complements spectroscopic data by providing structural information to understand the biologically relevant reaction, initiated by absorption of a single photon. In spectroscopy, this implies performing the experiment in the linear photoexcitation regime, established by performing a power titration where the magnitude of the photoproduct signal is plotted as a function of laser energy. Deviation from a straight line through the origin indicates too much energy was absorbed by the system, populating non-productive pathways, possibly resulting in artefacts that may be mistaken for physiological structural changes. In contrast to spectroscopy, there is currently a debate about appropriate photoexcitation in the structural biology community. Published studies used extremely high photoexcitation power densities79,182,183, which are particularly problematic when using fs laser pulses. The problem originates from the desire to maximize the magnitude of the light state signal, initially determined by calculating Fobs(light)-Fobs(dark) difference electron density maps. However, the light intensity inside the crystals decreases according to the Lambert-Beer law, meaning fewer molecules on the far side the crystal are exposed to a photon. This can apparently be overcome by increasing the pump laser energy, however, this results in molecules on the near side of the crystal absorbing more than one photon. Crystal thickness should, therefore, not exceed the pump laser penetration depth. A power titration should be performed, taking spectroscopic information as a starting point. The time delay should be chosen such that a relatively stable intermediate (photoproduct) has formed. Fast online SFX data analysis during beamtime is necessary to calculate Fobs(light)-Fobs(dark) difference electron density maps. Difference peaks should be integrated and plotted against laser energy. Visual examination of electron densities is not helpful160 in establishing a functionally relevant excitation regime. The difference signal is likely to be smaller when using the correct regime compared to using laser energies that are too high. This means, a lot of data needs to be collected to achieve a good signal-to-noise ratio37. So far, all pump-probe studies used very high laser energies182, outside the linear range used by spectroscopy183.
Light-induced functional changes were followed using pump-probe SFX in carboxy-myoglobin47 and carboxy-cytochrome c oxidase186 by photodissociating the heme-iron CO bond, in photosystem II137–142, in P450nor upon release of NO from caged NO131,187, in a photosynthetic reaction center157 and fatty acid photodecarboxylase188. Chromophore isomerization was investigated in photoactive yellow protein50,113, fluorescent protein48,189, bacterial phytochromes190,191 and rhodopsins. Light-triggered ion translocation was investigated in bacteriorhodopsin, a proton pump60,81,114, chloride ion–pumping rhodopsin115,160, sodium-pumping rhodopsin KR2159 and channelrhodopsin2192.
Light can be used not only to start reactions in inherently light-sensitive systems, but also to trigger the activation of caged compounds. Caged compounds are molecules whose biochemical functionality is blocked by a photolabile protecting group, allowing a rapid, localized increase in the concentration of a certain species upon exposure to light. Examples of caged molecules include substrates, cofactors, allosteric ligands and amino acids in proteins193. The approach has been used mainly in cellular applications but also in crystallography131,194–196. A number of cage groups exist. When selecting a cage group, important variables to consider include the release rate, which needs to be faster than the reaction being studied, for example, the commonly used nitrobenzyl derivatives are relatively slow (ms range) due to reactions downstream of photochemistry; excitation wavelength; extinction coefficient; quantum yield; solubility, which needs to be high due to the high protein concentration in crystals; and availability. The reactivity of the caged compound and the extent of release in the dark need to be examined experimentally to ensure it does not react with the system.
Examples using chemical mixing
Most biochemical reactions involve structural changes of a macromolecule induced by binding and/or chemical transformation of a small molecule ligand. Such reactions can be initiated by rapid concentration changes of ligands or other mechanistically relevant molecules. Rapid concentration changes can be achieved by chemical mixing, the mass transfer between one or more liquid streams or phases. Microfluidic devices197,198 can be used for chemical mixing, for example, by coaxial flow focusing197, to rapidly combine protein crystals and ligands. Alternatively, crystal containing nanolitre drops, either confined in a chip199 or deposited on a tape via acoustic droplet ejection35, can be injected with picolitre droplets of highly concentrated ligand solutions199,200. The best-reported time resolution using a microfluidic mixer201 is 5 ms202, whereas for DoT35,144 and DoD200 it is in the 100s of milliseconds range due to the large droplet size, ~ 100 μm radius for a spherical drop. Reaction times can be adjusted by using microfluidic devices with appropriate geometries, reaction channel lengths, changing the speed of the tape drive or changing the time between triggering and probing when using fixed targets. For the latter two approaches, dehydration may be an issue for longer reaction delays.
Chemical mixing may be considered the most generic triggering method. While this is correct in principle, the approach is complicated by the crystal lattice in crystallographic studies. Diffusion is restricted to solvent channels and is often slowed with a time resolution limited by the crystal size. Additionally, binding sites may be shielded and structural changes can be restrained by neighboring molecules or crystal contacts. These issues are reflected in the relatively few time-resolved mix-and-inject serial crystallography (MISC) studies that have been performed so far. Examples include ligand binding to a riboswitch with a 10 s delay to observe an intermediate203; binding and reaction of ceftriaxone with beta lactamase using two differently behaving crystal forms204; and the observation of a thioimidate covalent intermediate in isocyanide hydratase205. Oxygen complexes of ribonucleotide reductase35 and isopenicillin N synthase144 were obtained by diffusion of oxygen gas into crystals contained in droplets on tape, or by mixing oxygen-saturated and crystal-containing fluids, respectively (8 s delay), in case of bovine cytochrome c oxidase127. Spectroscopy was used to confirm intermediate formation which is important in view of the unexpectedly long reaction times required.
Reproducibility and data deposition
Establishing standards for structure validation and requiring the deposition of the diffraction data on which a publication is based are major achievements of the macromolecular crystallography community. As XFEL-based structural biology is maturing, it is time not only to extend these invaluable practices to this field, but where necessary to develop methods of validation and standards for deposition specific to SFX-derived or other serial data37. To validate the geometry of structures determined from serial crystallographic data, existing software tools can be used, and as outlined above, the data quality metrics used for serial data are largely the same as those for conventional crystallographic data, with few exceptions37. In the classical data collection statistics table, Rsplit replaces Rmerge, and the number of successfully indexed lattices should be reported alongside the other standard indicators of data quality, such as I/σ(I) and CC1/2. However, the ultimate quality measure is, of course, the quality of the unbiased electron density maps. The derived intensities should therefore be deposited together with the structure coordinates as usual, as this allows anyone to reproduce electron density maps to see if they support the paper’s conclusions. For a static structure determination of, for instance, a GPCR or a radiation-prone metalloenzyme, deposition in the Protein Data Bank (PDB) is sufficient. However, serial data collection protocols such as SFX are often used for nonstandard experiments such as time-resolved studies that make use of exotic or experimental methods of data analysis. The results of some of these methods, such as the use of extrapolated structure factors81, can be reproduced using deposited Monte-Carlo intensities, but the resampling methods described above for the estimation of coordinate errors require unmerged intensities from individual images. Other analyses can only be reproduced using raw diffraction image data. It is desirable to have databases where raw image data can be deposited for such cases. The databases CXIDB206 or IRRMC207,208, for instance, can be used to deposit raw images. Additionally, any custom software used to arrive at the reported results should be made available via repositories such as Github. Groups performing serial data collection experiments that depend on complicated analysis techniques are encouraged to deposit as much data, intermediate results and software as possible. We therefore argue for a dedicated repository for the deposition of all types of data, code, metadata and results from such non-standard serial crystallography experiments, ideally at one of the established sites (see above).
Apart from the crystallographic details of an experiment, adequate reporting is required for many other, equally crucial aspects of the experiment. Past experience shows that, in particular, the excitation parameters used in a pump-probe experiment are often poorly reported, leading to debates about illumination efficiency and photodamage, and the often very high intensities used in such experiments have led to their biological relevance being questioned79. Recently, a minimal set of excitation parameters that should be reported was proposed37. This set includes, but is not limited to, excitation laser parameters such as focus size and the definition thereof wavelength, pulse energy, pulse duration but also parameters such as sample extinction coefficient, thickness, number of chromophores per asymmetric unit, and others.
Limitations and optimisations
For all the exciting, high-impact science that has been enabled by high-throughput serial crystallography methods in the last dozen years and more, it is important to remain mindful of the limitations of these techniques. First and foremost, the success of any diffraction experiment depends on the quality of the crystals used. Even the most intense X-rays cannot produce high-resolution diffraction with poorly ordered crystals. Therefore, it is essential to perform as much characterization as possible before applying for beamtime. For microcrystals, this often includes characterization by powder diffraction. However, since screening can rarely give accurate information on how a particular batch of crystals will behave at an XFEL, it is advisable not to combine multiple batches of crystals. Instead, they should be tested separately, and afterward batches of similar properties and quality can be combined. Most facilities offer crystal screening opportunities to test samples.
Obtaining beamtime is another major constraint to prospective XFEL experiments. With the opening of new facilities and the upgrade of older ones to higher repetition rates, beamtime is becoming more available209, although access still requires a high-quality proposal. Considerations for an SFX experiment listed in BOX 2 are a good starting point, while BOX 1 may aid decision-making relating to experimental design. A successful XFEL beamtime proposal should outline what will be studied, why it is scientifically or technically interesting, and, most importantly, should state convincingly why the proposed experiments require an XFEL. SFX is an exciting new tool for structural biology, but it is not the only recent development. Where structure determination is concerned, cryo-EM may be a more suitable technique, particularly for poorly expressing proteins or those hard to crystallize210. A recent example is the structure determination of a GPCR-arrestin complex initially achieved by SFX167. Several years later, cryo-EM became the preferred technique for structure determination of GPCRs in complex with their signaling partners, including arrestins211. Furthermore, structure prediction has improved greatly, with AlphaFold2212 and similar AI-based approaches predicting reliable structures that can be used to design for example mutations to test interaction interfaces between different proteins using functional assays, or for phasing by molecular replacement (MR). Such predictions may be able to replace material-hungry de novo phasing approaches.
Once it is established that XFEL experiments are required, a beamtime proposal should demonstrate that suitable crystals are available. It should also make realistic projections of the amount of beam time needed. This depends on many factors, such as the crystal delivery method, the data quality required, the symmetry of the crystal lattice and the expected hit- and indexing rates (FIG. 1). Time estimations need to include overhead time for loading injectors or fixed-target chips, mounting them, as well as for washing lines, etc. The amount of extra time needed due to samples clogging injectors depends on the nature of the sample and the experience of the people in charge of injection. Another important consideration is the availability of suitable lasers for triggering time-resolved conformational changes. Obtaining this information and deciding the best experimental strategy is a good reason to involve the XFEL instrument scientists early on and to apply for crystal screening beamtime, from which lessons can be learned ahead of preparing a full proposal. Offline testing of injection conditions is also beneficial to avoid loss of sample and beam-time.
Outlook
SFX remains a relatively new and complex technique. So far, most experiments have been conducted by a small number of groups who have become experts in the various SFX-specific aspects of the experiment, such as microcrystallization, sample delivery and data analysis. This situation is highly unsatisfactory, and in order to expand the use of SFX within the structural biology community some hurdles need to be removed. The largest difficulty for new groups are arguably sample delivery and serial data analysis. The most pragmatic way to extend the use of SFX would be to transfer sample delivery expertise from user groups to facility staff and to put them in charge and to automate data processing as much as possible. In our opinion, making these two changes is the only viable way for SFX to become generally available. Having injection performed by highly experienced beamline scientists avoids the current situation, where each new user group has to learn for themselves how to deliver samples to the XFEL beam, wasting beam time. This would also enable mail-in crystallography, a process routinely practiced at synchrotrons. It could also help make XFEL-based crystallography more accessible, in particular for investigations aiming to collect a static, damage-free data set. Standardization and automation have facilitated synchrotron-based crystallography and will likely do the same for XFEL-based experiments. Standardized setups will reduce the time needed to exchange samples and injectors. Automatic data processing pipelines that provide the user with ready-for-use intensity data will help users with limited or no experience in running SFX-specific software on high-performance computing clusters. Steps in this direction have been taken at most XFEL facilities, but data storage and high performance computing power is often insufficient (TABLE 1). Detectors have advanced greatly over the last decade213. However, there is room for further improvements, particularly in increasing the dynamic range, for example by making gain switching more reliable at higher frame rates. The latter is particularly relevant for the MHz repetition rate XFELs.
A joint refinement scheme for time-resolved crystallography data series would be an interesting new development. When a reaction is initiated, concentrations of intermediates and products rise and fall smoothly. There is rarely a time when a single species is present, even if 100% of the molecules were successfully triggered. A time-resolved crystallography experiment always yields a series of snapshots of mixed states. A refinement scheme in which all data are explained simultaneously in terms of mixtures of these states could be highly beneficial to interpret such data. In spectroscopy, global fitting of a data series is routinely performed, but a similar scheme has yet to be developed for macromolecular crystallography. Developing such schemes for time-resolved crystallography will be much more difficult than for spectroscopy. This is because there is no one-to-one correspondence between macromolecular species and features in a diffraction pattern. Structural changes affect an entire diffraction pattern in crystallography, whereas a single species may often be exclusively correlated with a single spectral feature in spectroscopy.
Serial crystallography has been ported to synchrotron beamlines, and several serial synchrotron or serial millisecond experiments have been reported at 3rd generation sources19–21,214–219. Given the current and planned upgrades of most major synchrotron sources to diffraction-limited storage rings220 with highly increased brilliance, it is conceivable that serial synchrotron crystallography (SSX) will become much more popular than it currently is (see supplementary information). However, the behavior of crystals in the extremely high brilliance beams of new, diffraction-limited storage rings remains unknown. In particular, it needs to be determined to what extent the diffraction data are affected by radiation damage, which is also relevant for time-resolved experiments and their need for small crystals. Best practices for data collection need to be established, for example, whether to use monochromatic or pink beam. Similar issues as for XFELs, concerning automation and standardization of sample shipping, handling and data analysis, will need to be solved. In any event, the future is bright.
Supplementary Material
Acknowledgements
We thank the Heidelberg FEL group and the instrument scientists at the various XFEL facilities for numerous stimulating discussions over the years. This work was funded by the National Institutes of Health grant R35 GM127086 and the Max Planck Society.
Glossary
- Drop-on-demand (DoD)
In Drop-on-Demand sample injection, a piezoelectric device generates small droplets of a crystal suspension synchronously with the arrival of the X-ray pulses
- Drop-on-tape (DoT)
In the Drop-on-Tape method, the droplets are deposited on a tape that moves the droplets through the beam as on a conveyor belt
- Free Electron Laser
X-ray Free-Electron Lasers or XFELs are radiation sources based on a linear electron accelerator and a very long undulator, a periodic array of magnets. Electrons travel through the undulator at relativistic speed and experience an alternating magnetic field, causing them to emit X-ray radiation. This emitted radiation acts back on the electrons, causing some to accelerate and others to decelerate, grouping them into microbunches spaced at the wavelength of the radiation. Because of this, the electrons emit in phase, resulting in extremely intense pulses of coherent radiation. This process is known as self-amplified spontaneous emission (SASE)
- Repetition rate
XFELs produce radiation in pulses. The number of pulses per second is called the repetition rate, which varies from 10–120 Hz for early XFELs to several MHz for XFELs using superconducting accelerators
- Photoionization
Photoionization is the near-instantaneous emission of an electron, typically from an inner shell, caused by the absorption of a photon
- Auger electrons
Inner-shell vacancies in atoms, caused by, for example, photoionization, may be filled by an electron from a higher shell falling into the vacancy. For low Z-atoms, the energy released by this is transferred to another electron, which is then emitted as an Auger electron
- Hit rate
In SFX, crystal delivery is typically not synchronized with the X-ray pulses and therefore, not every exposure results in a diffraction pattern being generated. The fraction of images containing a diffraction pattern is the hit rate
- Indexing rate
Typically, in SFX not every collected diffraction pattern can be indexed, often for lacking sufficient suitable diffraction spots. The indexing rate is the fraction of indexable diffraction patterns of all hits
- Injector
A device used to introduce crystals into the X-ray beam in a thin liquid jet, gas-dynamic virtual nozzle injector, or viscous stream, high-viscosity extrusion injector, for serial data collection
- Pump-probe
A method for collecting time-resolved SFX data, in which a laser flash pump is used to trigger a reaction and then its structure is probed after a specified time delay by the XFEL beam. The wavelength range of the pump spans the X-ray region to THz
- Fixed targets
A crystal delivery method, in which crystals are grown inside of a chip or deposited on a solid support, typically also a chip. The solid support is rastered in the X-ray beam using fast X-Y stages
- Gas dynamic virtual nozzles (GDVN)
A gas dynamic virtual nozzle or GDVN is a device for jet-type crystal delivery that consists of an inner nozzle, through which a crystal suspension is pumped, surrounded by an outer nozzle through which a sheath gas is delivered. A constriction at the end of the outer nozzle causes the gas stream to narrow and accelerate. This virtual nozzle focuses the liquid stream into a narrow jet
- High viscosity extrusion (HVE) injector
HVE injector is a special device for the delivery of crystals grown inside viscous media such as lipidic cubic phase or embedded into a viscous matrix. The HVE injector consists of a reservoir filled with viscous matrix containing crystals, a hydraulic or mechanical plunger and a narrow capillary through which the viscous medium is streamed. The stream is supported by a sheath gas, typically nitrogen or helium, to keep it straight and avoid curling back and sticking to the nozzle
- Extrapolated structure factors
Triggering a reaction inside a crystal often results in activated state occupancies below 100%, meaning that activation is not achieved in all unit cells, complicating map interpretation. Extrapolated structure factor amplitudes can be estimated corresponding to 100% occupancy using a linear extrapolation method
- Rsplit
The precision of serial data can be assessed using a special R-factor, Rsplit, calculated by randomly splitting the available images into two halves (A and B), integrating both to prepare two half data sets and calculating the R-factor between them, corrected for the reduction in multiplicity
- CC1/2
A measure of the precision of X-ray crystallography data that can be applied to serial crystallography. To calculate it for serial data, the available images are divided randomly into two subsets. Each subset is subjected to Monte-Carlo integration to obtain two sets of integrated intensities. The Pearson correlation between the two is then calculated
- Friedel mates
Pairs of reflections F(h,k,l) and F(-h,-k,-l) that are not related by space group point symmetry have the same magnitude unless anomalous signal is present. Highly precise quantification of intensity differences facilitates certain techniques for experimental phasing
- Lipidic cubic phase
A gel-like membrane-mimetic mesophase suitable for stabilization and crystallization of membrane proteins in lipidic environment
- Fo-Fc maps
Electron density difference map that highlights areas where the atomic model disagrees with the experimental diffraction data, be it by absent, superfluous, or incorrect model features
- Omit maps
An electron density map calculated from observed intensities and phases from a model that excludes certain atoms, or groups of atoms, with the objective of removing the contribution of these atoms to minimize model phase bias
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s415XX-XXX-XXXX-X
Useful links
Alvra at SwissFEL: https://www.psi.ch/en/swissfel/alvra
BL2 and BL3 at SACLA: http://xfel.riken.jp/eng/users/bml04-11.html
CXI at LCLS: https://lcls.slac.stanford.edu/instruments/cxi/specifications
CXIDB: https://cxidb.org/
MFX at LCLS: https://lcls.slac.stanford.edu/instruments/mfx/specifications
NCI SFX at PAL-XFEL: https://pal.postech.ac.kr/paleng/Menu.pal?method=menuView&pageMode=paleng&top=7&sub=2&sub2=2&sub3=4
IRRMC: https://proteindiffraction.org/
SPB/SFX at EuXFEL: https://www.xfel.eu/facility/instruments/spb_sfx/index_eng.html
References
- 1.Holton JM A beginner’s guide to radiation damage. J. Synchrotron Radiat 16, 133–142 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kendrew JC et al. Structure of myoglobin: A three-dimensional Fourier synthesis at 2 A. resolution. Nature 185, 422–7 (1960). [DOI] [PubMed] [Google Scholar]
- 3.Garman EF & Schneider TR Macromolecular cryocrystallography. J. Appl. Crystallogr 30, 211–237 (1997). [Google Scholar]
- 4.Liu W, Wacker D, Wang C, Abola E & Cherezov V Femtosecond crystallography of membrane proteins in the lipidic cubic phase. Philos. Trans. R. Soc. Lond. B Biol. Sci 369, 20130314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Austin RH, Beeson KW, Eisenstein L, Frauenfelder H & Gunsalus IC Dynamics of ligand binding to myoglobin. Biochemistry 14, 5355–73 (1975). [DOI] [PubMed] [Google Scholar]
- 6.Fraser JS et al. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci U S A 108, 16247–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang N, Deng H, Liu B, Wang D & Zhao Z Features and futures of X-ray free-electron lasers. Innovation (N Y) 2, 100097 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwu Y & Margaritondo G Synchrotron radiation and X-ray free-electron lasers (X-FELs) explained to all users, active and potential. J. Synchrotron Radiat 28, 1014–1029 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barty A et al. Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nat. Photonics 6, 35–40 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lomb L et al. Radiation damage in protein serial femtosecond crystallography using an x-ray free-electron laser. Phys. Rev. B 84, 214111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nass K et al. Indications of radiation damage in ferredoxin microcrystals using high-intensity X-FEL beams. J. Synchrotron Radiat 22, 225–38 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Nass K et al. Structural dynamics in proteins induced by and probed with X-ray free-electron laser pulses. Nat. Commun 11, 1814 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman HN, Caleman C & Timneanu N Diffraction before destruction. Philos. Trans. Royal Soc. B 369, 20130313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chapman HN et al. Femtosecond X-ray protein nanocrystallography. Nature 470, 73–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the first serial femtosecond crystallography experiment, which was then called “nanocrystallography”.
- 15.Boutet S et al. High-Resolution Protein Structure Determination by Serial Femtosecond Crystallography. Science 337, 362–364 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration of high-resolution (1.9 Å) SFX using the model system lysozyme.
- 16.Grünbein ML & Kovacs GN Sample delivery for serial crystallography at free-electron lasers and synchrotrons. Acta Crystallogr. D 75, 178–191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martiel I, Muller-Werkmeister HM & Cohen AE Strategies for sample delivery for femtosecond crystallography. Acta Crystallogr. D Biol. Crystallogr 75, 160–177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gati C et al. Serial crystallography on in vivo grown microcrystals using synchrotron radiation. IUCrJ 1, 87–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nogly P et al. Lipidic cubic phase serial millisecond crystallography using synchrotron radiation. IUCrJ 2, 168–76 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Botha S et al. Room temperature serial crystallography at synchrotron X-ray sources. Acta Crystallogr. D 71, 387–397 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Diederichs K & Wang M Serial Synchrotron X-ray Crystallography (SSX). Methods and Protocols: Meth. Mol. Biol 1607, 239–272 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Bücker R et al. Serial protein crystallography in an electron microscope. Nat. Commun 11, 996 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirata K et al. Determination of damage-free crystal structure of an X-ray-sensitive protein using an XFEL. Nat. Methods 11, 734–6 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Cohen AE et al. Goniometer-based femtosecond crystallography with X-ray free electron lasers. Proc. Natl. Acad. Sci. U S A 111, 17122–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doak RB et al. Crystallography on a chip - without the chip: sheet-on-sheet sandwich. Acta Crystallogr. D 74, 1000–1007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roedig P et al. A micro-patterned silicon chip as sample holder for macromolecular crystallography experiments with minimal background scattering. Sci. Rep 5, 10451 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mueller C et al. Fixed target matrix for femtosecond time-resolved and in situ serial microcrystallography. Struct. Dyn 2, 054302 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baxter EL et al. High-density grids for efficient data collection from multiple crystals. Acta Crystallogr. D 72, 2–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oghbaey S et al. Fixed target combined with spectral mapping: approaching 100% hit rates for serial crystallography. Acta Crystallogr. D 72, 944–55 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorzny ML et al. Microfabricated silicon chip as lipid membrane sample holder for serial protein crystallography. Micro and Nano Engineering 3, 31–36 (2019). [Google Scholar]
- 31.Martiel I et al. Versatile microporous polymer-based supports for serial macromolecular crystallography. Acta Crystallogr. D 77, 1153–1167 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weierstall U, Spence JC & Doak RB Injector for scattering measurements on fully solvated biospecies. Rev.Sci.Instrum 83, 035108 (2012). [DOI] [PubMed] [Google Scholar]; Primary reference for the gas dynamic virtual nozzle (GDVN) sample injector.
- 33.Weierstall U et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun 5, 3309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Description of the high viscosity injector for LCP, grease and other viscous media.
- 34.Weierstall U Liquid sample delivery techniques for serial femtosecond crystallography. Phil.Trans.R.Soc.B 369, 20130337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuller FD et al. Drop-on-demand sample delivery for studying biocatalysts in action at X-ray free-electron lasers. Nat. Methods 14, 443–449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Echelmeier A et al. Segmented flow generator for serial crystallography at the European X-ray free electron laser. Nat. Commun 11, 4511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorel A, Schlichting I & Barends TRM Discerning best practices in XFEL-based biological crystallography - standards for nonstandard experiments. IUCrJ 8, 532–543 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fromme P & Spence JC Femtosecond nanocrystallography using X-ray lasers for membrane protein structure determination. Curr. Opin. Struct. Biol 21, 509–16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darmanin C et al. Protein crystal screening and characterization for serial femtosecond nanocrystallography. Sci. Rep 6, 25345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sugahara M et al. Viscosity-adjustable grease matrices for serial nanocrystallography. Sci. Rep 10, 1371 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gisriel C et al. Membrane protein megahertz crystallography at the European XFEL. Nat. Commun 10, 5021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jordan P et al. Three-dimensional structure of cyanobacterial photosystem I at 2.5 A resolution. Nature 411, 909–17 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Kupitz C et al. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature 513, 261–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young ID et al. Structure of photosystem II and substrate binding at room temperature. Nature 540, 453–457 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bublitz M et al. Structural studies of P-type ATPase-ligand complexes using an X-ray free-electron laser. IUCrJ 2, 409–420 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srinivas V et al. High-Resolution XFEL Structure of the Soluble Methane Monooxygenase Hydroxylase Complex with its Regulatory Component at Ambient Temperature in Two Oxidation States. J. Am. Chem. Soc 142, 14249–14266 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barends TR et al. Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation. Science 350, 445–50 (2015). [DOI] [PubMed] [Google Scholar]; First sub-ps time resolution SFX study, on carboxymyoglobin, showing both large-scale motions and coherent oscillations of side chains upon photolysis of the heme-CO bond. This work reached a time resolution orders of magnitude better than previously obtained at a synchrotron source.
- 48.Coquelle N et al. Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nat. Chem 10, 31–37 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Schlichting I Serial femtosecond crystallography: the first five years. IUCrJ 2, 246–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tenboer J et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 346, 1242–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; First pump-probe SFX study, on photoactive yellow protein, at nanosecond time resolution.
- 51.Beale JH et al. Successful sample preparation for serial crystallography experiments. J. Appl. Crystallogr 52, 1385–1396 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ibrahim M et al. Improvements in serial femtosecond crystallography of photosystem II by optimizing crystal uniformity using microseeding procedures. Struct. Dyn 2, 041705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nass K et al. Protein structure determination by single-wavelength anomalous diffraction phasing of X-ray free-electron laser data. IUCrJ 3, 180–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dods R et al. From Macrocrystals to Microcrystals: A Strategy for Membrane Protein Serial Crystallography. Structure 25, 1461–1468 e2 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Kupitz C et al. Microcrystallization techniques for serial femtosecond crystallography using photosystem II from Thermosynechococcus elongatus as a model system. Phil.Trans.R.Soc.B 369, 20130316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Landau EM & Rosenbusch JP Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. U S A 93, 14532–5 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cherezov V Lipidic cubic phase technologies for membrane protein structural studies. Curr. Opin. Struct. Biol 21, 559–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caffrey M & Cherezov V Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4, 706–31 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu W, Ishchenko A & Cherezov V Preparation of microcrystals in lipidic cubic phase for serial femtosecond crystallography. Nat. Protoc 9, 2123–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nango E et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 354, 1552–1557 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Andersson R et al. Well-based crystallization of lipidic cubic phase microcrystals for serial X-ray crystallography experiments. Acta Crystallogr. D 75, 937–946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kovacsova G et al. Viscous hydrophilic injection matrices for serial crystallography. IUCrJ 4, 400–410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sugahara M et al. Hydroxyethyl cellulose matrix applied to serial crystallography. Sci. Rep 7, 703 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DePonte DP et al. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J. Phys. D Appl. Phys 41, 195505 (2008). [Google Scholar]
- 65.Stan CA et al. Liquid explosions induced by X-ray laser pulses. Nat. Physics 12, 966–971 (2016). [Google Scholar]
- 66.Grünbein ML et al. Megahertz data collection from protein microcrystals at an X-ray free-electron laser. Nat. Commun 9, 3487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wiedorn MO et al. Megahertz serial crystallography. Nat. Commun 9, 4025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wiedorn MO et al. Rapid sample delivery for megahertz serial crystallography at X-ray FELs. IUCrJ 5, 574–584 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grünbein ML et al. Observation of shock-induced protein crystal damage during megahertz serial femtosecond crystallography. Phys. Rev. Res 3, 013046 (2021). [Google Scholar]
- 70.Grünbein ML, Shoeman RL & Doak RB Velocimetry of fast microscopic liquid jets by nanosecond dual-pulse laser illumination for megahertz X-ray free-electron lasers. Optics Express 26, 7190–7203 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Altarelli M The European X-ray Free-Electron Laser: toward an ultra-bright, high repetition-rate x-ray source. High Power Laser Science and Engineering 3(2015). [Google Scholar]
- 72.Oberthuer D et al. Double-flow focused liquid injector for efficient serial femtosecond crystallography. Sci Rep 7, 44628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schlichting I & Miao J Emerging opportunities in structural biology with X-ray free-electron lasers. Curr.Opin.Struct.Biol 22, 613–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lomb L et al. An Anti-Settling Sample Delivery Instrument for Serial Femtosecond Crystallography. J. Appl. Crystallogr 45, 674–678 (2012). [Google Scholar]
- 75.Sugahara M et al. Grease matrix as a versatile carrier of proteins for serial crystallography. Nat. Methods 12, 61–3 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Fromme R et al. Serial femtosecond crystallography of soluble proteins in lipidic cubic phase. IUCrJ 2, 545–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sugahara M et al. Oil-free hyaluronic acid matrix for serial femtosecond crystallography. Sci. Rep 6, 24484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Conrad CE et al. A novel inert crystal delivery medium for serial femtosecond crystallography. IUCrJ 2, 421–430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grünbein ML et al. Illumination guidelines for ultrafast pump-probe experiments by serial femtosecond crystallography. Nat. Methods 17, 681–684 (2020). [DOI] [PubMed] [Google Scholar]
- 80.Colletier JP et al. Serial Femtosecond Crystallography and Ultrafast Absorption Spectroscopy of the Photoswitchable Fluorescent Protein IrisFP. J. Phys. Chem. Lett 7, 882–7 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Nass Kovacs G et al. Three-dimensional view of ultrafast dynamics in photoexcited bacteriorhodopsin. Nat. Commun 10, 3177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stauch B & Cherezov V Serial Femtosecond Crystallography of G Protein-Coupled Receptors. Annu. Rev. Biophys 47, 377–397 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roessler CG et al. Acoustic Injectors for Drop-On-Demand Serial Femtosecond Crystallography. Structure 24, 631–640 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hutchison CDM et al. X-ray Free Electron Laser Determination of Crystal Structures of Dark and Light States of a Reversibly Photoswitching Fluorescent Protein at Room Temperature. Int J Mol Sci 18(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Echelmeier A et al. 3D printed droplet generation devices for serial femtosecond crystallography enabled by surface coating. J Appl Crystallogr 52, 997–1008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mehrabi P et al. The HARE chip for efficient time-resolved serial synchrotron crystallography. J. Synchrotron Radiat 27, 360–370 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hunter MS et al. Fixed-target protein serial microcrystallography with an x-ray free electron laser. Sci. Rep 4, 6026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stander N, Fromme P & Zatsepin N DatView: a graphical user interface for visualizing and querying large data sets in serial femtosecond crystallography. J Appl Crystallogr 52, 1440–1448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kirian RA et al. Structure-factor analysis of femtosecond microdiffraction patterns from protein nanocrystals. Acta Crystallogr. A 67, 131–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes how serial crystallography data can be integrated using Monte-Carlo methods.
- 90.White TA et al. CrystFEL: a software suite for snapshot serial crystallography. J. Appl. Crystallogr 45, 335–341 (2012). [Google Scholar]; Primary reference for the CrystFEL serial data processing package.
- 91.Sauter NK, Hattne J, Grosse-Kunstleve RW & Echols N New Python-based methods for data processing. Acta Crystallogr. D 69, 1274–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; Initial description of the cctbx.xfel package for serial data processing.
- 92.Hattne J et al. Accurate macromolecular structures using minimal measurements from X-ray free-electron lasers. Nat Methods 11, 545–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kabsch W Processing of X-ray snapshots from crystals in random orientations. Acta Crystallogr. D 70, 2204–2216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.White TA Processing serial crystallography data with CrystFEL: a step-by-step guide. Acta Crystallogr. D 75, 219–233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work is a beginner’s guide for serial data processing with the CrystFEL software suite.
- 95.Brewster AS, Young ID, Lyubimov A, Bhowmick A & Sauter NK Processing serial crystallographic data from XFELs or synchrotrons using the cctbx.xfel GUI Comp. Crystallogr. Newsletter 10, 22–39 (2019). [Google Scholar]
- 96.Sauter NK et al. Improved crystal orientation and physical properties from single-shot XFEL stills. Acta Crystallogr. D 70, 3299–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.White TA et al. Recent developments in CrystFEL. J. Appl. Crystallogr 49, 680–689 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.White TA Post-refinement method for snapshot serial crystallography. Philosophical Transactions of the Royal Society B-Biological Sciences 369, 20130330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ginn HM et al. A revised partiality model and post-refinement algorithm for X-ray free-electron laser data. Acta Crystallogr. D 71, 1400–1410 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brehm W & Diederichs K Breaking the indexing ambiguity in serial crystallography. Acta Crystallogr. D 70, 101–9 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Weiss MS Global indicators of X-ray data quality. J. Appl. Crystallogr 34, 130–135 (2001). [Google Scholar]
- 102.Barends TR et al. De novo protein crystal structure determination from X-ray free-electron laser data. Nature 505, 244–7 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Karplus PA & Diederichs K Assessing and maximizing data quality in macromolecular crystallography. Curr. Opin. Struct. Biol 34, 60–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rupp B Against Method: Table 1-Cui Bono? Structure 26, 919–923 (2018). [DOI] [PubMed] [Google Scholar]
- 105.Sauter NK et al. No observable conformational changes in PSII. Nature 533, E1–E2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yefanov O et al. Accurate determination of segmented X-ray detector geometry. Opt Express 23, 28459–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Foucar L CFEL–ASG Software Suite (CASS): usage for free-electron laser experiments with biological focus. J. Appl. Cryst 49, 1336–1346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Foucar L et al. CASS-CFEL-ASG software suite. Comp. Phys. Commun 183, 2207–2213 (2012). [Google Scholar]
- 109.Coquelle N et al. Raster-scanning serial protein crystallography using micro- and nano-focused synchrotron beams. Acta Cryst. D71, 1184–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mariani V et al. OnDA: online data analysis and feedback for serial X-ray imaging. J Appl Crystallogr 49, 1073–1080 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Uervirojnangkoorn M et al. Enabling X-ray free electron laser crystallography for challenging biological systems from a limited number of crystals. Elife 4(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Colletier JP et al. De novo phasing with X-ray laser reveals mosquito larvicide BinAB structure. Nature 539, 43–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pande K et al. Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein. Science 352, 725–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nogly P et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond x-ray laser. Science 361, eaat0094 (2018). [DOI] [PubMed] [Google Scholar]
- 115.Mous S et al. Dynamics and mechanism of a light-driven chloride pump. Science 375, 845–851 (2022). [DOI] [PubMed] [Google Scholar]
- 116.Genick UK et al. Structure of a protein photocycle intermediate by millisecond time-resolved crystallography. Science 275, 1471–5 (1997). [DOI] [PubMed] [Google Scholar]
- 117.Warren GL, Do TD, Kelley BP, Nicholls A & Warren SD Essential considerations for using protein-ligand structures in drug discovery. Drug Discov Today 17, 1270–81 (2012). [DOI] [PubMed] [Google Scholar]
- 118.Deller MC & Rupp B Models of protein-ligand crystal structures: trust, but verify. J Comput Aided Mol Des 29, 817–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Genick UK Structure-factor extrapolation using the scalar approximation: theory, applications and limitations. Acta Crystallogr. D 63, 1029–1041 (2007). [DOI] [PubMed] [Google Scholar]
- 120.Yano J et al. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. USA 102, 12047–12052 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beitlich T, Kuhnel K, Schulze-Briese C, Shoeman RL & Schlichting I Cryoradiolytic reduction of crystalline heme proteins: analysis by UV-Vis spectroscopy and X-ray crystallography. J. Synchrotron Radiat 14, 11–23 (2007). [DOI] [PubMed] [Google Scholar]
- 122.Lucic M et al. Serial Femtosecond Zero Dose Crystallography Captures a Water-Free Distal Heme Site in a Dye-Decolorising Peroxidase to Reveal a Catalytic Role for an Arginine in Fe(IV) =O Formation. Angew Chem Int Ed Engl 59, 21656–21662 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dickerson JL, McCubbin PTN & Garman EF RADDOSE-XFEL: femtosecond time-resolved dose estimates for macromolecular X-ray free-electron laser experiments. J. Appl. Crystallogr 53, 549–560 (2020). [Google Scholar]
- 124.Cox N, Pantazis DA & Lubitz W Current Understanding of the Mechanism of Water Oxidation in Photosystem II and Its Relation to XFEL Data. Annu Rev Biochem 89, 795–820 (2020). [DOI] [PubMed] [Google Scholar]
- 125.Schlichting I et al. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 287, 1615–1622 (2000). [DOI] [PubMed] [Google Scholar]
- 126.Berglund GI et al. The catalytic pathway of horseradish peroxidase at high resolution. Nature 417, 463–8 (2002). [DOI] [PubMed] [Google Scholar]
- 127.Ishigami I et al. Snapshot of an oxygen intermediate in the catalytic reaction of cytochrome c oxidase. Proc. Natl. Acad. Sci. U S A 116, 3572–3577 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chreifi G et al. Crystal structure of the pristine peroxidase ferryl center and its relevance to proton-coupled electron transfer. Proc. Natl. Acad. Sci. U S A 113, 1226–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kwon H et al. XFEL Crystal Structures of Peroxidase Compound II. Angew Chem Int Ed Engl 60, 14578–14585 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fukuda Y et al. Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography. Proc. Natl. Acad. Sci. U S A 113, 2928–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tosha T et al. Capturing an initial intermediate during the P450nor enzymatic reaction using time-resolved XFEL crystallography and caged-substrate. Nat. Commun 8, 1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Halsted TP et al. Catalytically important damage-free structures of a copper nitrite reductase obtained by femtosecond X-ray laser and room-temperature neutron crystallography. IUCrJ 6, 761–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rose SL et al. An unprecedented insight into the catalytic mechanism of copper nitrite reductase from atomic-resolution and damage-free structures. Sci. Adv 7(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kern J et al. Simultaneous Femtosecond X-ray Spectroscopy and Diffraction of Photosystem II at Room Temperature. Science 340, 491–495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; In this work, X-ray emission spectra are recorded concomitantly with SFX diffraction data collection, allowing on-line monitoring of the oxidation state of the metal ions of the oxygen-evolving complex of photosystem II, showing no X-ray photoreduction.
- 135.Kern J et al. Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nat. Commun 5, 4371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Suga M et al. Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X-ray pulses. Nature 517, 99–103 (2015). [DOI] [PubMed] [Google Scholar]; This work describes the high-resolution structure of photosystem II in the S1 state, determined by SF-ROX and therefore virtually free of radiation damage. The structure shows differences in the interatomic distances in the oxygen-evolving complex compared to structures determined using conventional, synchrotron-based crystallography.
- 137.Suga M et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 543, 131–135 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Kern J et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 563, 421–425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Suga M et al. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an x-ray free-electron laser. Science 366, 334–338 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Ibrahim M et al. Untangling the sequence of events during the S2 --> S3 transition in photosystem II and implications for the water oxidation mechanism. Proc. Natl. Acad. Sci. U S A 117, 12624–12635 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hussein R et al. Structural dynamics in the water and proton channels of photosystem II during the S2 to S3 transition. Nat. Commun 12, 6531 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li H et al. Capturing structural changes of the S1 to S2 transition of photosystem II using time-resolved serial femtosecond crystallography. IUCrJ 8, 431–443 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fransson T et al. X-ray Emission Spectroscopy as an in Situ Diagnostic Tool for X-ray Crystallography of Metalloproteins Using an X-ray Free-Electron Laser. Biochemistry 57, 4629–4637 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rabe P et al. X-ray free-electron laser studies reveal correlated motion during isopenicillin N synthase catalysis. Sci. Adv 7(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Andersson R et al. Serial femtosecond crystallography structure of cytochrome c oxidase at room temperature. Sci. Rep 7, 4518 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sakaguchi M, Shinzawa-Itoh K, Yoshikawa S & Ogura T A resonance Raman band assignable to the O-O stretching mode in the resting oxidized state of bovine heart cytochrome c oxidase. J. Bioenerg. Biomembr 42, 241–3 (2010). [DOI] [PubMed] [Google Scholar]
- 147.Doye JPK & Poon WCK Protein crystallization in vivo. Current Opinion in Colloid & Interface Science 11, 40–46 (2006). [Google Scholar]
- 148.Schonherr R, Rudolph JM & Redecke L Protein crystallization in living cells. Biol. Chem 399, 751–772 (2018). [DOI] [PubMed] [Google Scholar]
- 149.Schonherr R et al. Real-time investigation of dynamic protein crystallization in living cells. Struct. Dyn 2, 041712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sawaya MR et al. Protein crystal structure obtained at 2.9 A resolution from injecting bacterial cells into an X-ray free-electron laser beam. Proc. Natl. Acad. Sci. U S A 111, 12769–74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tetreau G et al. Serial femtosecond crystallography on in vivo-grown crystals drives elucidation of mosquitocidal Cyt1Aa bioactivation cascade. Nat. Commun 11, 1153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gati C et al. Atomic structure of granulin determined from native nanocrystalline granulovirus using an X-ray free-electron laser. Proc. Natl. Acad. Sci. U S A 114, 2247–2252 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Redecke L et al. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science 339, 227–230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nass K et al. In cellulo crystallization of Trypanosoma brucei IMP dehydrogenase enables the identification of genuine co-factors. Nat. Commun 11, 620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lahey-Rudolph JM et al. Fixed-target serial femtosecond crystallography using in cellulo grown microcrystals. IUCrJ 8, 665–677 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tsutsui H et al. A diffraction-quality protein crystal processed as an autophagic cargo. Mol. Cell 58, 186–93 (2015). [DOI] [PubMed] [Google Scholar]
- 157.Dods R et al. Ultrafast structural changes within a photosynthetic reaction centre. Nature 589, 310–314 (2021). [DOI] [PubMed] [Google Scholar]
- 158.Keable SM et al. Room temperature XFEL crystallography reveals asymmetry in the vicinity of the two phylloquinones in photosystem I. Sci Rep 11, 21787 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Skopintsev P et al. Femtosecond-to-millisecond structural changes in a light-driven sodium pump. Nature 583, 314–318 (2020). [DOI] [PubMed] [Google Scholar]
- 160.Yun JH et al. Early-stage dynamics of chloride ion-pumping rhodopsin revealed by a femtosecond X-ray laser. Proc. Natl. Acad. Sci. U S A 118(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Li D et al. Ternary structure reveals mechanism of a membrane diacylglycerol kinase. Nat Commun 6, 10140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thomaston JL et al. XFEL structures of the influenza M2 proton channel: Room temperature water networks and insights into proton conduction. Proc Natl Acad Sci U S A 114, 13357–13362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ghosh E, Kumari P, Jaiman D & Shukla AK Methodological advances: the unsung heroes of the GPCR structural revolution. Nat. Rev. Mol. Cell. Biol 16, 69–81 (2015). [DOI] [PubMed] [Google Scholar]
- 164.Liu W et al. Serial femtosecond crystallography of G protein-coupled receptors. Science 342, 1521–4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration of GPCR structure determination by SFX using microcrystals grown in LCP.
- 165.Fenalti G et al. Structural basis for bifunctional peptide recognition at human delta-opioid receptor. Nat. Struct. Mol. Biol 22, 265–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang H et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 161, 833–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kang Y et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Batyuk A et al. Native phasing of x-ray free-electron laser data for a G protein-coupled receptor. Sci. Adv 2, e1600292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang H et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature 544, 327–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Audet M et al. Crystal structure of misoprostol bound to the labor inducer prostaglandin E2 receptor. Nat. Chem. Biol 15, 11–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liu H et al. Molecular basis for lipid recognition by the prostaglandin D2 receptor CRTH2. Proc. Natl. Acad. Sci. U S A 118, e2102813118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Luginina A et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv 5, eaax2518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Stauch B et al. Structural basis of ligand recognition at the human MT1 melatonin receptor. Nature 569, 284–288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Johansson LC et al. XFEL structures of the human MT2 melatonin receptor reveal the basis of subtype selectivity. Nature 569, 289–292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ishchenko A et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ 6, 1106–1119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Arnlund D et al. Visualizing a protein quake with time-resolved X-ray scattering at a free-electron laser. Nat Methods 11, 923–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Levantino M et al. Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser. Nat. Commun 6, 6772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lee Y et al. Ultrafast coherent motion and helix rearrangement of homodimeric hemoglobin visualized with femtosecond X-ray solution scattering. Nat Commun 12, 3677 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dandey VP et al. Time-resolved cryo-EM using Spotiton. Nat Methods 17, 897–900 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Maeots ME et al. Modular microfluidics enables kinetic insight from time-resolved cryo-EM. Nat Commun 11, 3465 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Orville AM Recent results in time resolved serial femtosecond crystallography at XFELs. Curr. Opin. Struct. Biol 65, 193–208 (2020). [DOI] [PubMed] [Google Scholar]
- 182.Brändén G & Neutze R Advances and challenges in time-resolved macromolecular crystallography. Science 373, eaba0954 (2021). [DOI] [PubMed] [Google Scholar]
- 183.Miller RJD, Pare-Labrosse O, Sarracini A & Besaw JE Three-dimensional view of ultrafast dynamics in photoexcited bacteriorhodopsin in the multiphoton regime and biological relevance. Nat. Commun 11, 1240 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Schmidt M Mix and Inject: Reaction Initiation by Diffusion for Time-Resolved Macromolecular Crystallography. Adv. Cond. Matt. Phys (2013). [Google Scholar]
- 185.Konold PE et al. Confinement in crystal lattice alters entire photocycle pathway of the Photoactive Yellow Protein. Nat. Commun 11, 4248 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Shimada A et al. A nanosecond time-resolved XFEL analysis of structural changes associated with CO release from cytochrome c oxidase. Sci. Adv 3, e1603042 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nomura T et al. Short-lived intermediate in N2O generation by P450 NO reductase captured by time-resolved IR spectroscopy and XFEL crystallography. Proc. Natl. Acad. Sci. U S A 118, e2101481118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sorigue D et al. Mechanism and dynamics of fatty acid photodecarboxylase. Science 372, eabd5687 (2021). [DOI] [PubMed] [Google Scholar]
- 189.Woodhouse J et al. Photoswitching mechanism of a fluorescent protein revealed by time-resolved crystallography and transient absorption spectroscopy. Nat. Commun 11, 741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Claesson E et al. The primary structural photoresponse of phytochrome proteins captured by a femtosecond X-ray laser. eLife 9, e53514 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Carrillo M et al. High-resolution crystal structures of transient intermediates in the phytochrome photocycle. Structure 29, 743–754 e4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Oda K et al. Time-resolved serial femtosecond crystallography reveals early structural changes in channelrhodopsin. Elife 10, e62389 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Monteiro DCF, Amoah E, Rogers C & Pearson AR Using photocaging for fast time-resolved structural biology studies. Acta Crystallogr. D 77, 1218–1232 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Schlichting I et al. Time-resolved X-ray crystallographic study of the conformational change in Ha-Ras p21 protein on GTP hydrolysis. Nature 345, 309–315 (1990). [DOI] [PubMed] [Google Scholar]
- 195.Bolduc JM et al. Mutagenesis and Laue structures of enzyme intermediates: isocitrate dehydrogenase. Science 268, 1312–8 (1995). [DOI] [PubMed] [Google Scholar]
- 196.Mehrabi P et al. Time-resolved crystallography reveals allosteric communication aligned with molecular breathing. Science 365, 1167–1170 (2019). [DOI] [PubMed] [Google Scholar]
- 197.Calvey GD, Katz AM, Schaffer CB & Pollack L Mixing injector enables time-resolved crystallography with high hit rate at X-ray free electron lasers. Struct. Dyn 3, 054301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Monteiro DCF et al. 3D-MiXD: 3D-printed X-ray-compatible microfluidic devices for rapid, low-consumption serial synchrotron crystallography data collection in flow. IUCrJ 7, 207–219 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mehrabi P et al. Liquid application method for time-resolved analyses by serial synchrotron crystallography. Nat. Methods 16, 979–982 (2019). [DOI] [PubMed] [Google Scholar]
- 200.Butryn A et al. An on-demand, drop-on-drop method for studying enzyme catalysis by serial crystallography. Nat. Commun 12, 4461 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Calvey GD, Katz AM & Pollack L Microfluidic Mixing Injector Holder Enables Routine Structural Enzymology Measurements with Mix-and-Inject Serial Crystallography Using X-ray Free Electron Lasers. Anal. Chem 91, 7139–7144 (2019). [DOI] [PubMed] [Google Scholar]
- 202.Pandey S et al. Observation of substrate diffusion and ligand binding in enzyme crystals using high-repetition-rate mix-and-inject serial crystallography. IUCrJ 8, 878–895 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Stagno JR et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 541, 242–246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Olmos JL Jr. et al. Enzyme intermediates captured “on the fly” by mix-and-inject serial crystallography. BMC Biol 16, 59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dasgupta M et al. Mix-and-inject XFEL crystallography reveals gated conformational dynamics during enzyme catalysis. Proc. Natl. Acad. Sci. U S A 116, 25634–25640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Maia FR The Coherent X-ray Imaging Data Bank. Nat Methods 9, 854–5 (2012). [DOI] [PubMed] [Google Scholar]
- 207.Grabowski M et al. A public database of macromolecular diffraction experiments. Acta Crystallogr. D 72, 1181–1193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Grabowski M et al. The Integrated Resource for Reproducibility in Macromolecular Crystallography: Experiences of the first four years. Struct Dyn 6, 064301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Johansson LC, Stauch B, Ishchenko A & Cherezov V A Bright Future for Serial Femtosecond Crystallography with XFELs. Trends Biochem Sci 42, 749–762 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ishchenko A, Gati C & Cherezov V Structural biology of G protein-coupled receptors: new opportunities from XFELs and cryoEM. Curr. Opin. Struct. Biol 51, 44–52 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lee Y et al. Molecular basis of beta-arrestin coupling to formoterol-bound beta1-adrenoceptor. Nature 583, 862–866 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bergamaschi A, Mozzanica A & Schmitt B XFEL detectors. Nature Reviews Physics 2, 335–336 (2020). [Google Scholar]
- 214.Martin-Garcia JM et al. Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 4, 439–454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Weinert T et al. Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun 8, 542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Owen RL et al. Low-dose fixed-target serial synchrotron crystallography. Acta Crystallogr. D 73, 373–378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Weinert T et al. Proton uptake mechanism in bacteriorhodopsin captured by serial synchrotron crystallography. Science 365, 61–65 (2019). [DOI] [PubMed] [Google Scholar]
- 218.Martin-Garcia JM et al. High-viscosity injector-based pink-beam serial crystallography of microcrystals at a synchrotron radiation source. IUCrJ 6, 412–425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Shahsavar A et al. Structural insights into the inhibition of glycine reuptake. Nature 591, 677–+ (2021). [DOI] [PubMed] [Google Scholar]
- 220.Eriksson M, van der Veen JF & Quitmann C Diffraction-limited storage rings - a window to the science of tomorrow. J. Synchrotron Radiat 21, 837–842 (2014). [DOI] [PubMed] [Google Scholar]
- 221.Diederichs K & Karplus PA Better models by discarding data? Acta Crystallogr. D 69, 1215–1222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Pfanzagl V et al. X-ray-induced photoreduction of heme metal centers rapidly induces active-site perturbations in a protein-independent manner. J Biol Chem 295, 13488–13501 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ebrahim A et al. Dose-resolved serial synchrotron and XFEL structures of radiation-sensitive metalloproteins. IUCrJ 6, 543–551 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Sierra RG et al. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr. D 68, 1584–1587 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sierra RG et al. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat. Methods 13, 59–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Shimazu Y et al. High-viscosity sample-injection device for serial femtosecond crystallography at atmospheric pressure. J Appl Crystallogr 52, 1280–1288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Eom I et al. Recent Progress of the PAL-XFEL. Appl. Sci 12, 1010 (2022). [Google Scholar]
- 228.Schulz EC et al. The hit-and-return system enables efficient time-resolved serial synchrotron crystallography. Nat Methods 15, 901–904 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.