

SUMMARY
Persistent neutrophil-dominated lung inflammation contributes to lung damage in cystic fibrosis (CF). However, the mechanisms that drive persistent lung neutrophilia and tissue deterioration in CF are not well characterized. Starting from the observation that, in patients with CF, c-c motif chemokine receptor 2 (CCR2)+ monocytes/macrophages are abundant in the lungs, we investigate the interplay between monocytes/macrophages and neutrophils in perpetuating lung tissue damage in CF. Here we show that CCR2+ monocytes in murine CF lungs drive pathogenic transforming growth factor β (TGF-β) signaling and sustain a pro-inflammatory environment by facilitating neutrophil recruitment. Targeting CCR2 to lower the numbers of monocytes in CF lungs ameliorates neutrophil inflammation and pathogenic TGF-β signaling and prevents lung tissue damage. This study identifies CCR2+ monocytes as a neglected contributor to the pathogenesis of CF lung disease and as a therapeutic target for patients with CF, for whom lung hyperinflammation and tissue damage remain an issue despite recent advances in CF transmembrane conductance regulator (CFTR)-specific therapeutic agents.
Graphical Abstract
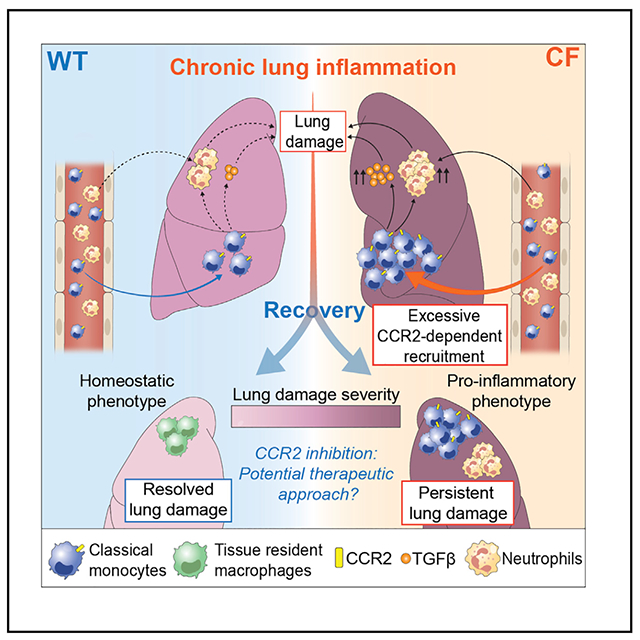
In brief
Lung damage starts early in life in people with cystic fibrosis (CF) and continuously progresses, ultimately leading to respiratory failure. Öz et al. show that recruited classical monocytes are the key drivers of signaling pathways that drive lung damage in CF and are thus a potential therapeutic target.
INTRODUCTION
Lung remodeling in people with cystic fibrosis (CF) starts early in life and progresses throughout their lifespan.1,2 It starts in the mucus-obstructed small airways, which become fibrotic over time, and proceeds toward alveoli, further limiting gas exchange.3–5 Lung remodeling in CF is caused by hyperinflammation, triggered by mucus airway obstruction, bacterial infections (especially Pseudomonas aeruginosa [PA]), and overwhelming and persistent neutrophil-dominated lung inflammation.6–8 This inflammatory microenvironment causes lung injury,9 which progressively disrupts homeostatic responses and tissue repair mechanisms, leading to irreversible lung damage and functional decline, ultimately requiring lung transplantation.6,10–12 CF transmembrane conductance regulator (CFTR) modulator therapies13,14 have improved the course of CF lung disease and the life expectancy of affected individuals. Nevertheless, long-term studies of patients suggest that these therapies are not sufficient to control lung hyperinflammation over time, which threatens maintenance of lung tissue integrity and functionality.15,16 Therefore, additional therapeutic targets are required to decrease neutrophilic inflammation and preserve lung function in the aging CF population.
Macrophages, key mediators of the immune response and tissue homeostasis17,18, contribute to CF lung hyperinflammation19,20 and impaired host response against major CF-associated pathogens.21–25 Increased numbers of monocytes/macrophages derived from the circulation are observed in CF airways, whereas tissue-resident alveolar macrophages (trAMs) are diminished.26,27 This is in line with the vast literature demonstrating that, with lung damage or infection, classic Ly6ChiCCR2+ monocytes (cMons) are recruited from the circulation in a C-C motif chemokine receptor 2 (CCR2) dependent manner in response to chemokines such as C-C motif chemokine ligand 2 (CCL2; MCP-1) and CCL7 (MCP-3)28,29 and, over time, replace alveolar macrophages and interstitial macrophages (IMs), defined as monocyte-derived macrophages.
Recruitment of cMons is a physiological response to perturbation of lung tissue homeostasis. However, uncontrolled regulation of cMon mobilization is detrimental to the host, leading to pathogenic responses that contribute to many lung diseases.30–32 In patients with CF, the pathogenic contribution of cMons to CF lung disease remains to be elucidated. The available studies on the topic are limited to characterization of immune cells in the CF airways and do not provide any insights regarding monocytes/macrophages that reside in the lung interstitium, which very likely also contribute to disease progression.33 Monocyte and monocyte-derived AM populations are key players in transforming growth factor β (TGF-β)-driven tissue fibrosis.34–36 Elevated TGF-β levels in the lungs of patients with CF are an indicator of lung disease progression4,37–40 and may exacerbate the disease.41–43
In this study, we investigate the interplay between cMons and neutrophils in perpetuating irreversible lung tissue scarring in a TGF-β-dependent manner in CF. We start from the observation that there are high numbers of CCR2+ monocytes in submucosal areas of lung explants from patients with CF. Next we demonstrate, in a CF mouse model of chronic inflammation, that these high numbers of CCR2+ monocyte-derived cells contribute to disease progression by driving pathological lung neutrophilia and TGF-β-dependent irreversible lung remodeling. We present foundational work showing that targeting the recruitment of CCR2+ monocytes can be of therapeutic value to prevent irreversible lung damage in patients with CF.
RESULTS
CCR2+ monocytes and monocyte-derived macrophages are abundant in CF lungs
We examined lung explants from patients with advanced CF disease who underwent lung transplantation for the presence of CCR2+ monocytes. In comparison with healthy donors, abundant CCR2+ cells were present in areas of remodeling (especially around bronchioles) in explanted lung tissues from CF patients (Figures 1A and S1A). The majority of the CCR2+ cells also expressed the monocyte/macrophage marker CD14 in CF lungs (Figure S1B). CCR2+ cells were also found in airway mucus (Figure 1A). A previous single-cell RNA sequencing (scRNA-seq) study from sputa of patients with CF and healthy donors (control [CTRL]) shows that AMs are very rare in CF lungs but monocytes are abundant (Figures 1B–1D, S1C, and S1D).26 Through analysis of these scRNA-seq datasets, we show that 4.3% of the monocytes from CF samples retained CCR2 expression (Figure 1E) and were scarce in healthy donors (CTRL) (Figures 1B–1E). The scRNA-seq data confirmed that CCR2 is expressed in lung monocytes and macrophages, with few exceptions among alveolar macrophages (AMs), neutrophils (polymorphonuclear [PMN]), and B cells (Figures 1B and 1D). These data suggest that CF airways are populated predominately by macrophages of monocytic origin, which are also abundant in the submucosal area of the lungs.
Figure 1. CCR2+ monocytes and monocyte-derived macrophages are abundant in CF lungs.
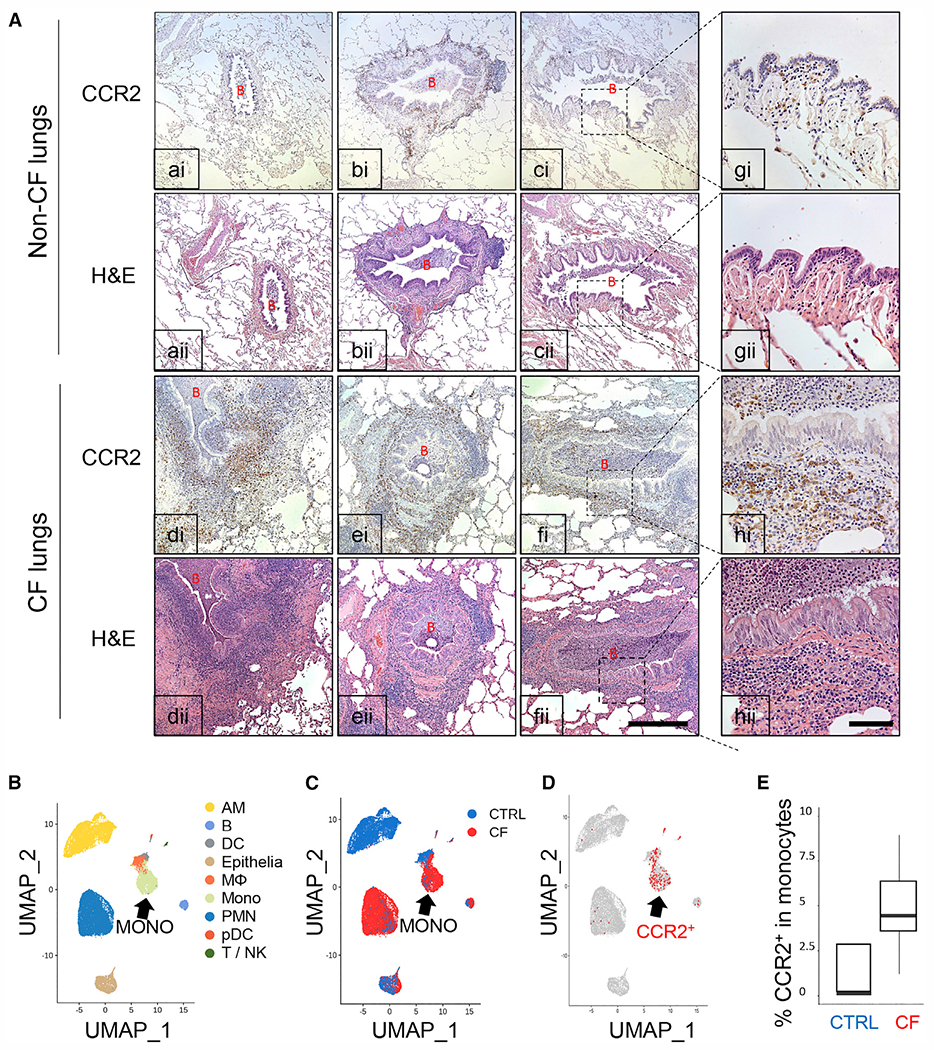
(A) Representative CCR2 immunohistochemistry and H&E staining in human lung tissue from healthy donors (non-CF, a–c) and patients with CF (CF, d–f) (n = 3). Greater magnification of c and f is shown in g and h, respectively. B, bronchiole. Magnification for a–f, 100×; magnification for g and h, 400×. Scale bars: 500 μm (a–f) and 100 μm (g and h).
(B-D) scRNA-seq data from cells isolated from induced sputum of healthy individuals (CTRL) versus patients with CF.
(B) Uniform manifold approximation and projection (UMAP) visualization of 20,095 sputum cells from nine patients with CF and five control (CTRL) subjects. Each dot represents a single cell, and cells are labeled by (B) cell type (AM, alveolar macrophage; B, B cell; DC, dendritic cell; Epithelia, epithelial cell; M, macrophage; Mono, monocyte; PMN, polymorphonuclear neutrophil granulocyte; pDC, plasmacytoid DC; T/NK, T and NK cells), (C) disease state (CTRL versus CF cells), and
(D) expression of at least one CCR2 mRNA molecule (red). Marker gene expression to identify cell populations is shown in Figure S1C.
(E) Boxplot of the percentage of CCR2-expressing cells in the monocyte population (CTRL versus CF).
Elevated numbers of classical monocytes and monocyte-derived macrophages promote and perpetuate neutrophilic pulmonary inflammation in CF
To identify aberrations of lung immune cell population dynamics in CF lungs in response to chronic inflammation, we used a CF mouse model of chronic pulmonary hyperinflammation caused by repeated exposure to PA lipololysaccharides (LPS), which recapitulates the irreversible lung remodeling and functional decline44 observed in patients with CF. To assess the role of recruited Ly6ChighCCR2pos cMons and monocyte-derived macrophages, we crossed CF knockout mice (B6.129P2-Cftrtm1Unc/J)45 with CCR2 knockout mice (CCR2−/−)46 (hereafter called double knockout [dKO]) and used CCR2−/− mice as additional controls (CTRLs). We confirmed the lack of CCR2 expression in CCR2−/− and dKO blood Ly6C+ cMons by flow cytometry (Figure S2A).
We used a flow cytometry panel47 to identify lung immune cell populations at steady state (T0), after 5 weeks of chronic nebulization with PA LPS (3 times a week for 5 weeks, chronic endpoint [T1]), and after recovery (6 weeks after the last LPS dose, recovery endpoint [T2]) (Figures 2A and 2B; see Figures S3A–S3C for a detailed description of the gating strategy and different composition of immune populations at T0, T1, and T2). At baseline (T0), CF mice did not show any statistically significant difference in lung immune cell numbers and composition compared with wild-type (WT) mice. As expected, untreated dKO and CCR2−/− mice had fewer cMons compared with WT and CF mice. Chronic exposure to LPS (T1) increased all assessed immune cell populations in WT and CF mice. However, compared with WT mice, CF mice had significantly more cMons and immune cells of monocytic origin, such as interstitial macrophages (IMs), monocyte-derived AMs (moAMs), and dendritic cells (DCs), but not trAMs, Ly6C− monocytes, or lymphocytes (Figures 2B and S2B). Importantly, CF mice had more neutrophilic lung inflammation compared with the WT, as we have reported previously.44 We identified that, in addition to SiglecF,47 moAMs can be clearly distinguished from trAMs by the pro-inflammatory macrophage marker CD38 (Figures S3A and S3B).48 CD38+SiglecFdim moAMs were absent in untreated lungs but became abundant (60.5% ± 10.7% of total AMs in lung tissues) at T1 and were not clearly distinguishable at T2 (Figure S3A). Bronchoalveolar lavage fluid (BALF) analysis confirmed that the CD38+SiglecFdim cell population resides in the alveolar space, excluding potential contamination from interstitial monocyte/macrophage populations (Figure S3B).
Figure 2. Elevated numbers of cMons and monocyte-derived macrophages promote and perpetuate neutrophilic pulmonary inflammation in CF.
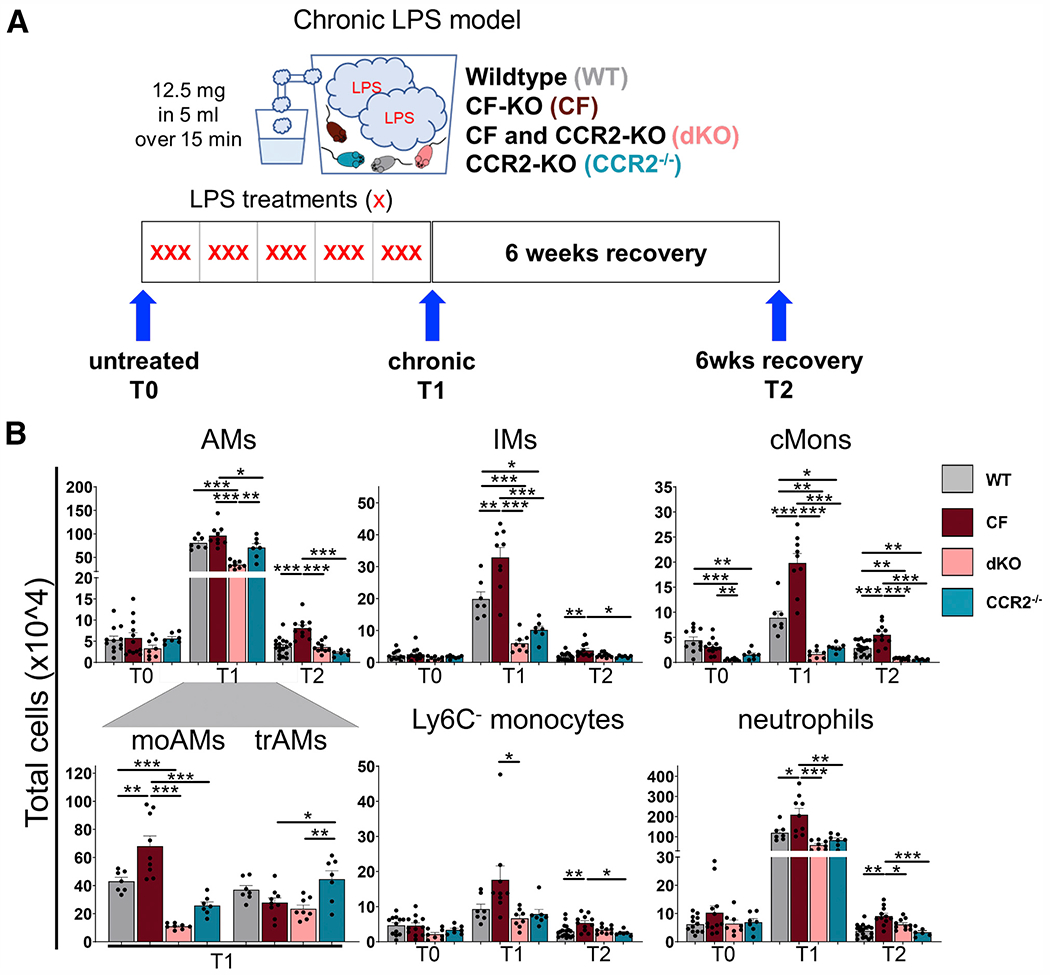
(A) Chronic LPS model. WT, CF, dKO, and CCR2−/− mice were nebulized with LPS from PA 3 times per week over 5 weeks (15 doses total) and sacrificed 24 h after the last nebulization (chronic endpoint [T1]) or left to recover for 6 weeks (recovery endpoint [T2]). At each time point, BALF and lung tissue were collected for flow cytometry, histology, RNA, and protein analysis. T0 indicates untreated mice.
(B) Total cell numbers of total AMs, moAMs, trAMs, IMs, cMons, Ly6C− Monos, and neutrophils were quantified by flow cytometry as percentage of viable cells multiplied by the total cell count in the inferior lung lobe. The gating strategy is described in Figure S3.
Data are generated from three independent experiments with 2–6 mice per genotype and time point. Each biological replicate is represented by a dot. Bars are depicted as means ± SEM, and p values were calculated using one-way ANOVA and Tukey’s test for multiple comparisons between the genotypes for each time point separately (*p < 0.05, **p < 0.01, ***p < 0.001). See also Figures S2 and S3.
In comparison with CF mice, dKO mice (and CCR2−/− CTRLs) had decreased cMons and monocyte-derived macrophage populations in lungs at T1, whereas trAM numbers were not altered (Figure 2B). dKO and CCR2−/− mice also had lower numbers of LPS-induced IMs, indicating that IMs originate at least partially from recruited monocytes during chronic LPS treatment.33,49 Recruitment of neutrophils and T cells was not affected in CCR2−/− mice because their counts were in line with WT mice at T1. We clearly observed that CCR2 deficiency in CF mice (dKO) lowered the exuberant neutrophil and T cell numbers observed in CF mice to levels comparable with WT mice (Figures 2B and S2B).
After six weeks of recovery from LPS exposure (T2), the number of immune cells in WT mice decreased to baseline. The two AM populations (CD38+ SiglecFdim; CD38dim SiglecF+) cannot be clearly distinguished at T2. However, at T2, the AM population has a shift toward lowered SiglecF expression compared with T0 AMs (Figures S3A–S3C). In contrast to WT mice, CF mice had significantly increased numbers of lung immune cells, including cMons, monocyte-derived cells, and neutrophils at T2 compared with T0 (Figures 2B and S2B), consistent with impaired resolution of the inflammatory response.44 At T2, CF lungs had more T and B cells than WT lungs (Figure S2B), suggesting a dysregulated adaptive immune response. This may be reflective of induced bronchially associated lymphoid tissues, which is observed in patients with CF50 and recapitulated in CF mice chronically challenged with LPS.44 Compared with CF mice, dKO mice had reduced numbers of cMons, AMs, and neutrophils at T2. AM and neutrophil numbers in dKO mice did not differ from WT or CCR2−/− CTRL groups at T2. No differences in the number of lung lymphocytes were observed between CF and dKO mice at T2 (Figures 2B and S2B).
Complete blood count (CBC) analysis showed that CF mice, compared with WT, dKO, and CCR2−/− mice, have significantly more monocytes and lymphocytes in the circulation at T0. The numbers of neutrophils were increased in CF compared with dKO mice at T1 and with CCR2−/− mice at T0. dKO mice also had increased numbers of lymphocytes compared with CCR2−/− mice at T2 (Figure S2C). Contrary to our expectation, we did not observe a significant reduction of monocytes in CCR2−/− and dKO mice by CBC analysis. However, consistent with previous studies,51 flow cytometry analysis of blood showed a reduction of cMons (Ly6C+CD11b+Ly6G−) in CCR2−/− and dKO mice compared with WT and CF mice at all assessed time points (Figure S2D).
In summary, these data demonstrate that CCR2+ monocyte recruitment and monocyte-derived macrophages are increased in CF lungs in response to chronic LPS and that the number of immune cells remains elevated after discontinuing LPS treatment. These data suggest that CCR2+ monocytes and monocyte-derived macrophages promote and prolong lung neutrophilia in CF lungs in response to chronic inflammatory triggers.
Enhanced CCR2-mediated monocyte recruitment drives pathogenic tissue remodeling in CF lungs
With the central hypothesis that increased numbers of CCR2+-derived monocytes/macrophages contribute to pathogenic lung tissue remodeling, we investigated the histology of WT, CF, dKO, and CCR2−/− mice to assess the degree of lung damage (Figure S4A; STAR Methods). All genotypes experience a pattern of alveolar and interstitial inflammation that peaks with chronic LPS treatment and wanes to varying degrees upon recovery (Figure 3). CF mice, compared with WT mice, had distal airspace enlargement, which developed in the absence of external inflammatory triggers at T0, as reported previously,52 but the difference did not reach statistical significance (Figures 3Aa–3Ad, 3Ba–3Bd, and 3C). Consistent with our previous findings,44 LPS-treated CF mice suffered from additional profound alveolar septal disruption accompanied by expanded and distorted alveolar architecture (enlargement and destruction of the air space) compared with the WT (Figures 3Ae–3Ah, 3Be–3Bh, and 3C). At T2, WT mice showed complete repair with reestablishment of normal physiological lung architecture. In contrast, CF lungs showed persistent severe lung damage with enlarged alveolar spaces and distorted alveolar structure (Figures 3Ai–3Al, 3Bi–3Bl, and 3C). This suggests that CF lungs have an impaired capability to repair the lungs, which is associated with a decline in lung function.44 We also observed more prominent collagen deposition (Masson’s trichrome staining) and airway wall thickening (α-smooth muscle actin staining) in CF versus WT lungs (Figures S5 and S6). CCR2−/− mice were protected from lung tissue damage in response to chronic LPS with minimal disruption of alveolar integrity at T1. Lung integrity was retained after recovery (Figures 3Ah, 3Al, 3Bh, 3Bl, 3C, S5, and S6). Lung remodeling in dKO mice was less severe compared with CF mice at T2 and more closely resembled WT mice. At T1, the alveolar structural damage in dKO mice compared with CF mice was also reduced but did not reach statistical significance (Figures 3A–3C, S5, and S6). These data show that decreasing CCR2+ cMon recruitment and reestablishing balanced immune cell populations in CF lungs can ameliorate the progression of lung remodeling driven by pro-inflammatory triggers such as bacterial colonization in CF.
Figure 3. Enhanced CCR2-mediated monocyte recruitment drives pathogenic tissue remodeling in CF lungs.
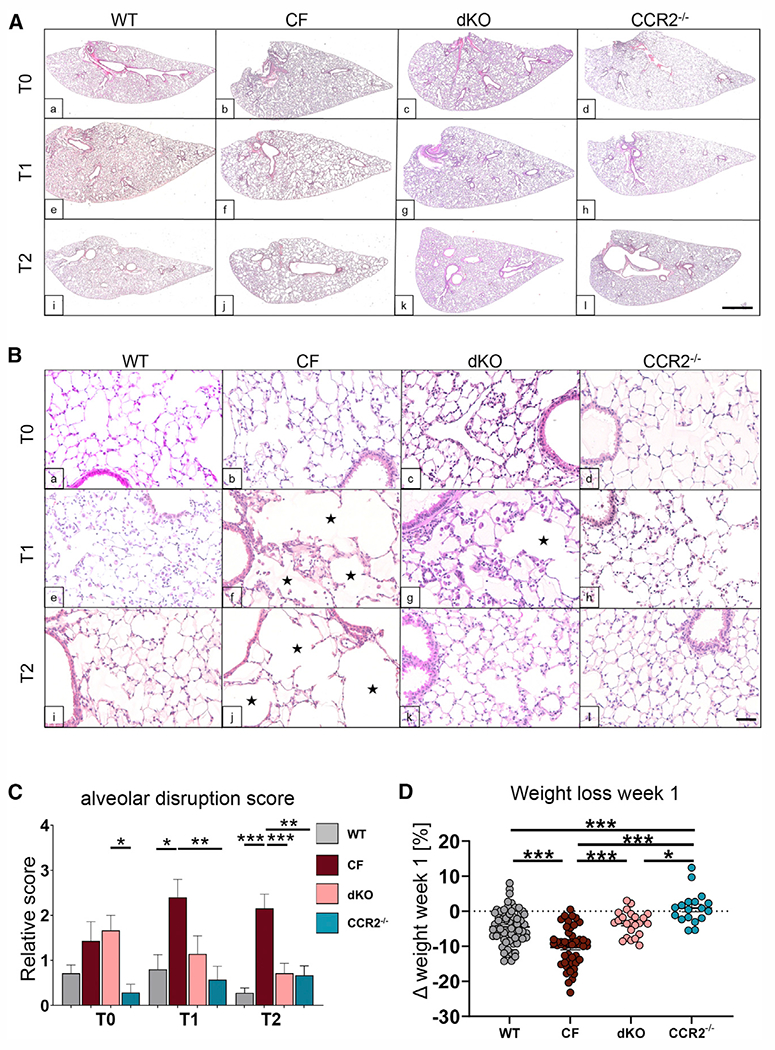
(A and B) Representative subgross H&E images of lung from WT, CF, dKO, and CCR2−/− mice at T0, T1, and T2. Black stars indicate enlarged alveolar spaces. Scale bars: 500 μm (A) and 50 μm (B).
(C) Relative alveolar disruption score from semiquantification analysis of lung histology. Bars are depicted as means ± SEM for 4 or more mice per genotype and time point.
(D) Relative weight loss of mice after week 1 of LPS administration. Each dot represents one biological replicate.
r multiple comparisons between the genotypes for each time point separately (*p < 0.05, **p < 0.01, ***p < 0.001). See also Figures S4, S5, and S6.
Weight loss is associated with significant mortality in patients with CF.53 This is recapitulated in CF animal models, including the mouse.54,55 CF mice had substantial weight loss compared with WT mice during the first week of chronic inflammation (on average −11% in CF versus −5% in WT mice of the initial weight at T0). CCR2−/− mice did not lose weight and, notably, dKO mice were protected from severe weight loss (−5% from the initial weight) (Figure 3D). After the first week of LPS challenges, all genotypes started gaining weight (Figure S4B), suggesting that immune tolerance toward LPS was acquired independent of the genotype. We show that exuberant cMon recruitment contributes to severe weight loss in CF mice in response to LPS.
CCR2-derived lung monocytes/macrophages drive pathogenic TGF-β signaling in CF lungs
Chronic LPS treatment induced TGF-β activation in lung tissue (Figure 4A). In line with severe lung tissue remodeling, at T1, the active TGF-β levels were higher in the BALF of CF than WT mice. At T2, total and active TGF-β levels remained elevated in CF airways (Figure 4A). Tgfβ expression was also increased in the lung lysates of CF mice (Figure 4B) compared with WT mice. Consistent with impaired cMon recruitment, reduced monocyte-derived macrophage populations, and lack of lung remodeling, CCR2−/− mice had significantly lower TGF-β levels in BALF and Tgfβ expression in the lungs compared with WT and CF mice (Figures 4A and 4B). Active TGF-β levels in BALF and Tgfβ expression in the lungs of dKO mice were similar to WT mice and significantly lower than in CF mice at T1 and T2 (Figures 4A and 4B).
Figure 4. CCR2-derived lung monocytes/macrophages drive pathogenic TGF-β signaling in CF lungs.
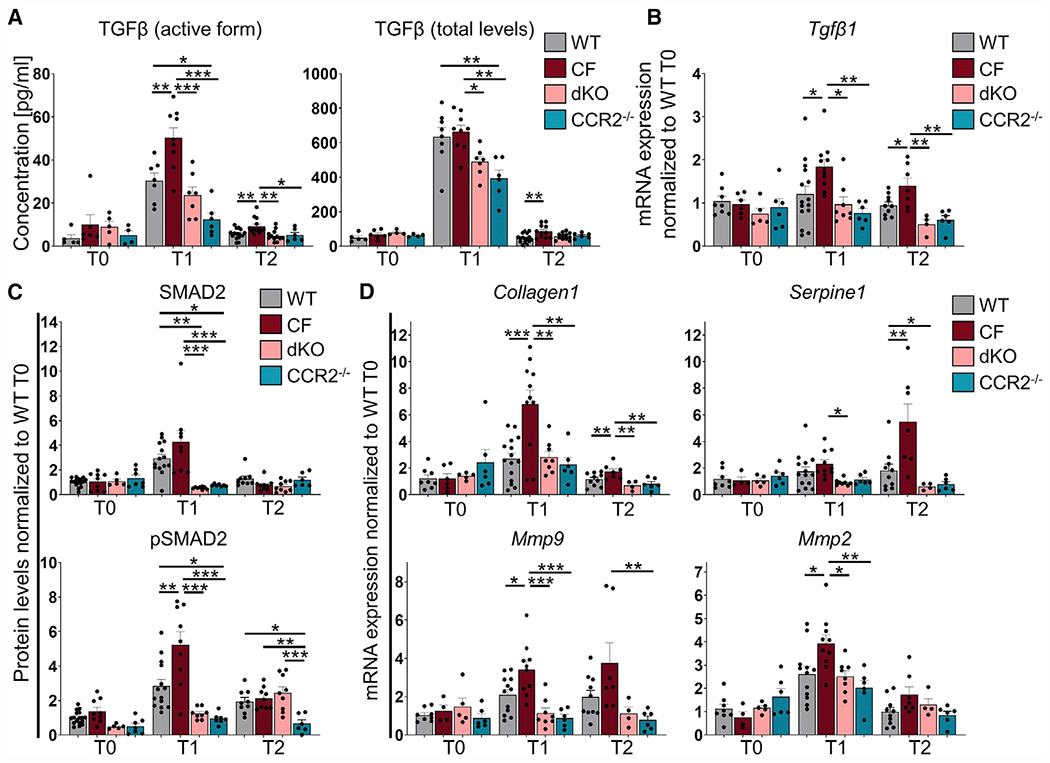
(A) Quantification of active (left) and total (right) TGF-β protein in BALF by ELISA.
(B and D) The mRNA expression levels of Tgfβ (B) and TGF-β signaling target genes (D) in lung tissues of WT, CF, dKO, and CCR2−/− mice at T0, T1, and T2. The average expression from two technical replicates was normalized to 18S and WT at T0.
(C) Densitometric analysis of Western blot for SMAD2 and pSMAD2 from lung tissues of WT, CF, dKO, and CCR2−/− mice at T0, T1, and T2. The expression was normalized to VINCULIN and the WT at T0.
Data are generated from three independent experiments with 2–4 mice per genotype and time point. Each biological replicate is represented by a dot. Bars are depicted as means ± SEM, and p values were calculated using one-way ANOVA and Tukey’s test for multiple comparisons between the genotypes for each time point separately (*p < 0.05, **p < 0.01, ***p < 0.001). See also Figure S7.
We next assessed whether increased active TGF-β levels in CF lungs correlated with altered TGF-β signaling. At baseline (T0), no differences in TGF-β expression and signaling were observed among experimental groups (Figure 4). At T1, the lung tissues of CF (versus WT) mice had more phosphorylated SMAD2 (pSMAD2), a downstream mediator of the canonical TGF-β signaling pathway (Figure 4C), recapitulating late-stage CF lung disease.4 At T2, pSMAD2 levels were close to baseline, and no differences were observed between WT, CF, and dKO mice, whereas pSMAD2 levels were lower in CCR2−/− lungs (Figure 4B). Activation of non-canonical TGF-β signaling via ERK1/2 was similar between WT and CF lungs at T1 (Figure S7A). In line with our previous findings,55 we observed alteration of the miR-199a-5p/caveolin 1 axis in CF lungs, which is associated with increased SMAD2 signaling in idiopathic pulmonary fibrosis (Figure S7B).56 At T1, the lung tissue of CF (versus WT) mice had greater mRNA expression of SMAD2 target genes such as Collagen 1 (Col1) and matrix metallopeptidase (Mmp) 9 but not Mmp2 and Serpine1 (Figure 4D). Serpine1 was specifically upregulated in CF lungs at recovery (T2). High expression of Serpine1 is strongly associated with pro-fibrotic tissue remodeling57 and, thus, may contribute to the lack of resolution from damage in CF lungs.
Consistent with low active TGF-β levels and moderate lung remodeling, lung tissues from dKO mice (and CCR2−/− CTRLs) displayed reduced TGF-β expression, activation, and signaling (SMAD2 levels) at T1 compared with CF (Figure 4C). The levels of Col1, Mmp9, Mmp2, and Serpine1 were significantly lower in dKO than in CF lungs and at levels similar to WT lungs at T1 (Figure 4D). At T2, the lungs of dKO mice, compared with CF mice, maintained the reduced expression of Col1 and Serpine1 (Figure 4D).
To test whether the lack of CCR2 in immune cells (and not epithelial/parenchymal cells) protects lung tissue from recruitment of monocytes and monocyte-derived macrophages and the subsequent activated TGF-β levels in response to chronic LPS, we created WT and CCR2−/− chimeric mice by bone marrow transplantation. Total-body-irradiated WT or CCR2−/− mice were transplanted with donor WT or CCR2−/− bone marrow (BM) (WTBM → WT; WTBM → CCR2−/−; CCR2−/−BM→ WT; CCR2−/−BM → CCR2−/−) (Figure S8A). Engraftment, which exceeded 95%, was representatively measured by flow cytometry in blood and lung tissues of simultaneously assessed CD45.1BM → CD45.2 CTRL mice (Figure S8B). After chronic LPS, lungs of WT mice receiving CCR2−/−BM (CCR2−/−BM→ WT) had significantly less lung monocyte/macrophage populations (which were all of BM origin in this model) than mice receiving WT BM (WTBM→WT and WTBM→CCR2−/−) and a trend toward reduced neutrophils (Figure S8C). The weight loss 1 week after the start of chronic LPS challenge was comparable among BM chimera mice (Figure S8D). CCR2−/−BM→WT chimera had lower active TGF-β levels in BALF compared with WTBM→WT (p = 0.046) and a trend toward lower levels compared with WTBM→CCR2−/− (p = 0.089) and were not different from CCR2−/−BM→CCR2−/− mice (Figure S8E).
These data support the contribution of CCR2+ cMons and cMon-derived macrophage populations to pathogenic TGF-β levels and SMAD2-dependent signaling in CF lungs exposed to chronic inflammatory triggers.
cMons in CF perpetuate immune activation and neutrophil recruitment
To investigate whether lung monocyte and macrophage populations in CF lungs were also altered in function, we sorted cMons, IMs, moAMs, and trAMs from WT and CF mice at T1 and T2 via fluorescence-activated cell sorting (FACS) (Figure S9A) and analyzed their gene expression profile by RNA sequencing (RNA-seq). We kept the same gating strategy that was established at T0 and T1 and sorted moAMs as a SiglecFdim cell population at T2 (Figure S9A). Quantification of gene levels (17,025 genes) identified population marker genes, specifically upregulated in individual monocytes and macrophage lung populations, independent of the different genotypes and time points (Figure S10A). We confirmed the gene expression of marker genes used to distinguish and sort the different lung monocyte/macrophage populations (e.g., IMs had higher major histocompatibility complex [MHC] class II expression compared with cMons, and moAMs had lower SiglecF expression but higher CD38 expression compared with trAMs) (Figure S9B), confirming the robustness of our approach. Consistent with other published data,34 cMons had the highest expression of receptors such as Ccr2, Cd14, Csf1r, and Infgr1. Compared with cMons, IMs expressed higher levels of genes related to adhesion and cross-talk with the lung extracellular matrix (ECM) (integrins and Mmps; e.g., Adam19, Mmp9, and Mmp13), ECM remodeling genes (e.g., Col1a1, Eln, Fgf1), MHC class II genes (e.g., H2 -Ab1), and anti-inflammatory markers (e.g., Arg1 and Chil1). MoAMs and trAMs had similar expression profiles compared with the other populations (Figures 5A, S9B, and S10A). However, compared with moAMs, trAMs expressed high levels of Tgfb2 and its receptor (Tgfbr2), which promotes AM development and homeostasis,58 and genes involved in cell division regulators (e.g., Chek2), which is consistent with their self-renewal capability (Figure S10A).59
Figure 5. cMons in CF perpetuate immune activation and neutrophil recruitment.
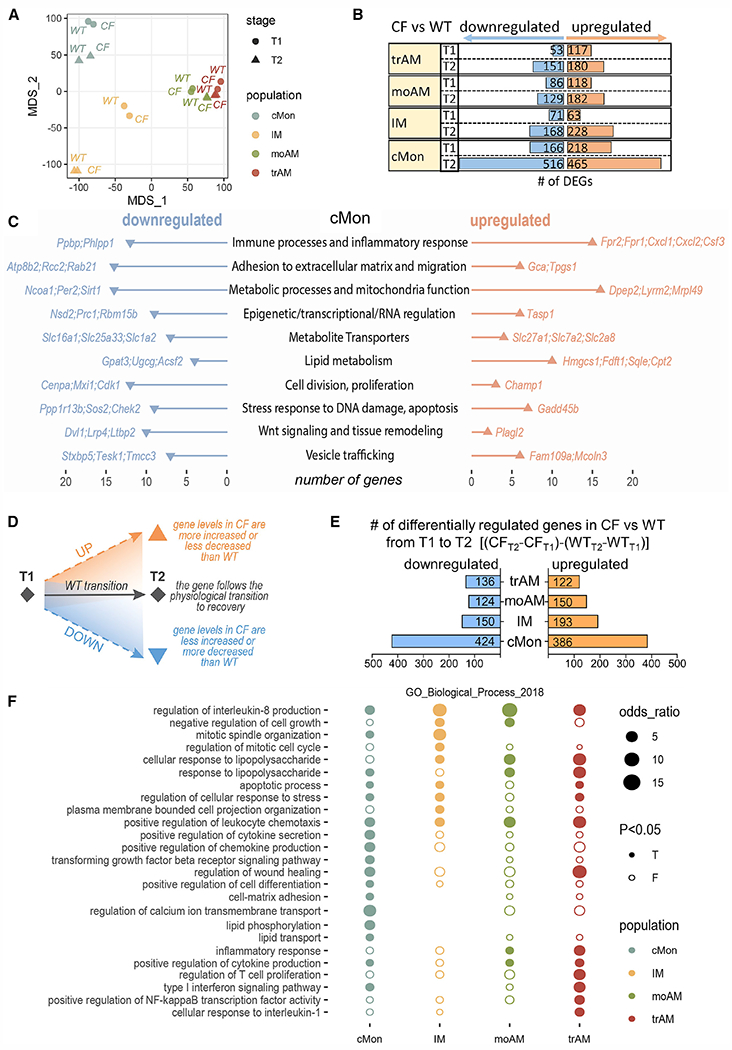
(A) MDS plot based on RNA expression profiles of cMons, IMs, moAMs, and trAMs at T1 and T2.
(B) The number of differentially expressed genes (DEGs) when comparing CF with the WT in each cell population at T1 or T2.
(C) Pathway analysis of genes differentially expressed in CF versus WT cMons at T2. The plot shows the number of up- or downregulated genes associated with each pathway. Two samples per genotype and time point were used for analysis.
(D) Schematic of the differential analysis depicted in (E).
(E) Numbers of differentially up- or downregulated genes between T1 and T2 in each population.
(F) Gene Ontology (GO) biological process enrichment analysis of differentially regulated genes in CF between T1 and T2 compared with the WT for each population (cMons, IMs, moAMs, and trAMs). Node size: odds ratio associated with enrichment; p value for Fisher’s exact test.
Data were generated from 2 samples per genotype and time point. To ensure sufficient cell counts at T2, lungs from 2 WT or 2 CF mice were pooled for one sample. See also Figures S9, S10, and S11.
Multidimensional scaling (MDS) analysis visualized the similarity between each condition, defined by cell population, genotype, and stage, based on the levels of all expressed genes (Figure 5A). Of the four monocyte/macrophage populations, cMons and IMs form distinct clusters, whereas trAMs and moAMs have similar expression profiles. Especially at T2, moAMs and trAMs have high similarity, in line with the more uniform AM population observed by flow cytometry analysis at recovery (Figures S3A, S3B, and S9A). The expression profile of IMs and cMons changed the most from T1 to T2 (Figure 5A), suggesting that these populations are significantly altered during recovery from chronic LPS. In contrast, the expression profiles of moAMs and trAMs did not change as much between stages (Figure 5A). The greatest difference between WT and CF populations was observed in cMons at T2. This population also had the most differentially expressed genes (DEGs) (Figure 5B; Data S1A and S1B). Of these, CF cMons (relative to the WT) had increased expression of genes driving immune cell activation and chemotaxis (summarized in Figure 5C; Data S1C), such as receptors involved in monocyte response to bacterial formylated peptides (e.g., Fpr1 and Fpr2) and production of chemokines/cytokines that stimulate chemotaxis, proliferation, and differentiation of neutrophils (e.g., Cxcl1, Cxcl2, and Csf3). CF cMons also showed profound changes in genes involved in metabolic regulation compared with WT CTRLs and increased expression of genes involved in fatty acid oxidation and cholesterol synthesis (e.g., Hmgcs1, Sqle, Fdft1, and Cpt2), metabolic pathways that are altered in patients with CF.60,61 CF cMons at T2 also had altered expression of genes involved in adhesion to the ECM (e.g., Gca and Tpgs1), cell division, and Wnt signaling, suggesting that cMons may have dysregulated differentiation capabilities. In line with previous55 and current work (Figure S7), we observed that, upon inflammatory stimulation, CF macrophages and lung tissues fail to downregulate miR-199a-5p expression, which targets many genes with reduced expression in CF cMons, such as Sirt162 and Wnt signaling-related genes.63 qRT-PCR on sorted cMons at T2 validated the RNA-seq results for key genes involved in neutrophil activity (e.g., Fpr1, Fpr2, Cxcl1, Cxcl2, and Csf3) as well as in metabolic responses (e.g., Sqle and Sirt1) (Figure S10B). Among these, Cxcl2 was the highest mediator expressed in CF cMons, as assessed by RNA-seq (Data S1A). We confirmed increased CXCL2 levels in BALF of CF mice at T2 compared with WT, dKO, and CCR2−/− mice. Importantly, CXCL2 concentrations in BALF of dKO mice were comparable with WT and CCR2−/− mice (Figure S10C). This could account for the lowered neutrophil numbers seen in dKO at T2 compared with CF mice (Figure 2B). At T2, CF lung submucosae were abundantly populated by CCR2+CXCL2+ cells (Figure S10D), which were rarely seen in WT mice. Although all monocyte/macrophage populations expressed TGF-β (counts per million [CPM] > 200 in all populations), the expression levels were similar among genotypes. This suggests that greater levels of active TGF-β in CF lungs (Figure 4A) are caused by an excessive number of TGF-β-producing cells and/or enhanced TGF-β activation. In summary, these data show that the expression profile of CF cMons at T2 diverges the most from WT CTRLs. cMons, even without an active inflammatory trigger, retains a pro-inflammatory phenotype with increased expression of genes that can propagate recruitment of neutrophils.
During recovery, pulmonary CF monocyte/macrophage populations feature a skewed pro-inflammatory gene signature
To identify genes that are dynamically dysregulated in CF during recovery from chronic exposure to LPS, we compared the changes in gene expression in CF macrophages from T1 to T2 to the “reference” changes in WT macrophages (STAR Methods). We thus identified genes with an altered recovery process with respect to the WT, being upregulated or downregulated from T1 to T2 (Figure 5D). Of the four assessed populations, the cMon population had the most differentially regulated genes, with 424 genes having lowered expression and 386 genes having elevated expression compared with the WT. This was followed by IMs (150 lower and 193 elevated expressed genes), moAMs (124 lower and 150 elevated expressed genes), and trAMs (136 lower and 122 elevated expressed genes) (Figure 5E). Gene Ontology analysis was performed to identify pathways enriched in individual cell populations. All four CF monocyte/macrophage lung populations retained a differential expression profile (compared with the WT) related to the response to LPS exposure. In line with the previous analysis (Figure 5C), during recovery from inflammation, CF (versus WT) cMons have an increased migratory signature as well as expression profile suggesting active proliferation64 and failure to acquire a tissue macrophage phenotype. These alterations can account for the increase in numbers of cMons in CF lungs (Figures 2B and 5). They retained elevated expression of chemokines (including Cxcl1 and Cxcl2) and dysregulated expression of genes related to the TGF-β signaling pathway and ECM adhesion (Figures 5F and S11A). BioPlanet pathway analysis (Figure S11D; Data S1D) identified differentially regulated ECM-receptor interaction and integrin signaling pathways, which have been reported to be altered in CF monocytes.65 Consistent with higher levels of miR-199a-5p and increased TGF-β levels at T2 (Figure S6), CF cMons have reduced Akt1 and Cav1 expression.55,56 CF cMons also retain high expression of Gdf15 , which has been associated with lung tissue fibrosis66 and tissue tolerance to infections.67 Calcium transport and lipid transport pathways were also affected in CF cMons during recovery (Figures 5F and S11A; Data S1E–S1F).
The differential expression profile in CF (versus WT) IMs during recovery included genes controlling biological pathways associated with plasma membrane and cell projection organization, suggesting that IMs may have altered plasma membrane organization and cross-talk with the ECM and other lung cells. Genes associated with mitosis, microtubule organization, and the cell cycle were highly altered in CF IMs (Figure 5F). Thus, IM proliferation in CF may contribute to the greater IM numbers observed at T2 in CF lungs (Figure 2B). CF IMs retained higher expression of IL-10 and altered expression of genes regulating MHC class II antigen presentation (Figures 5F, S11B, and S11D; Data S1D–S1F), which may affect lymphocyte activation, polarization, and immunomodulatory functions in CF lungs.68
The differentially regulated pathways of CF (versus WT) moAMs during recovery from chronic exposure to LPS overlapped with trAMs, except for regulation of genes involved in wound healing, cell growth, and cellular response to interleukin-1 signaling (Figure 5F; Data S1E-S1F). Similar to cMons and IMs, and in contrast to their homeostatic anti-inflammatory phenotype,69,70 CF trAMs were pro-inflammatory, with increased expression of genes controlling inflammatory pathways (e.g., Nucleotide-binding, oligomerization domain (NOD) and interleukin-1 signaling, tumor necrosis factor alpha [TNF-α], and nuclear factor κB [NF-κB) (Figures 5F, S11C, and S11D; Data S1D–S1F). The expression of distinct type I interferon signaling genes, such as Oas2, Mx2, and Irf1, remains elevated in CF trAMs, which may increase susceptibility to bacterial lung infection71,72 and airway remodeling73 in CF lungs. Irf1 overexpression has been associated with dysfunctional efferocytosis and phagocytosis in AMs from a mouse model of muco-obstructive lung disease.74 Moreover, CF trAMs retained high expression of the interferon-inducible cytokine Cxcl10 (IP-10), which drives activation and recruitment of monocytes, T cells, eosinophils, and natural killer (NK) cells. We have shown previously that CXCL10 is elevated in the BALF of CF mice chronically exposed to LPS and remains elevated during recovery.44 Notably, CXCL10 is a biomarker for acute pulmonary exacerbations in patients with CF.75 CF trAMs had downregulated expression of genes for co-stimulatory molecules (Cd80 and Cd6) involved in activation and proliferation of T cells and elevated expression of Cd40, which is required for AM activation in response to interferon γ (IFNγ). This suggests that CF trAMs have altered cross-talk with adaptive immune cells. We also identified a strong signature in the enrichment of genes regulating TGF-β signaling and interaction with the ECM (Figures 5F and S11; Data S1E–S1F). Consistent with observations in CF lung tissue (Figure 4D), Serpine1 expression was elevated from T1 to T2 in all monocyte/macrophage populations compared with the WT (Figures 5F and S11A–S11C).
These data suggest that, after LPS withdrawal, CF lung monocytes/macrophages preserved an immune response signature to LPS that perpetuated immune cells chemotaxis, altered interaction with the ECM, and promoted tissue remodeling in CF lungs.
Cell-autonomous CFTR deficiency in CCR2-expressing cells drives excess accumulation of lung monocytes/macrophages and neutrophils in response to chronic LPS
To test whether the CF lung environment or cell-autonomous CFTR dysfunction in CCR2-expressing cells (e.g., cMons) is responsible for the enhanced recruitment of cMons during chronic pulmonary inflammation, we generated Ccr2cre/+Cftrfl/fl mice to knock out Cftr in CCR2-expressing cells76,77 and exposed these mice to 5 weeks of LPS (Figure 6A). We used the Cftrfl/fl CTRL as well as the Ccr2cre/+Cftrfl/+ line to exclude off-target effects of Cre. A few differences (e.g., blood neutrophil numbers at T0 and trAM and B cell numbers and total TGF-β levels at T1) were observed between the two CTRL lines. Ccr2cre/+Cftrfl/+ mice were used for comparison with the Ccr2cre/+Cftrfl/fl experimental group. At baseline, Ccr2cre/+Cftrfl/fl mice had increased lung CD8 T and B lymphocyte numbers compared with CTRL mice. In response to chronic LPS, in Ccr2cre/+Cftrfl/fl mice, all myeloid cells (except for Ly6C− Monos) as well as CD4 T cells were increased in number compared with Ccr2cre/+Cftrfl/+ mice, whereas CD8 T and B cells were not altered (Figure 6B). Active and total TGF-β levels, weight loss, as well as CBCs were not affected by the conditional CFTR deficiency in CCR2-expressing cells (Figures 6C–6E). These data indicate that the excessive recruitment of lung monocytes/macrophages and neutrophils in response to chronic LPS is driven by the intrinsic lack of CFTR function in CCR2+ cells. However, the elevated TGF-β activation and excessive weight loss during the first weeks of LPS exposure also required the concomitant absence of CFTR in the lung tissue.
Figure 6. CFTR deficiency in CCR2-expressing cells drives excessive accumulation of lung immune cells in response to chronic LPS.
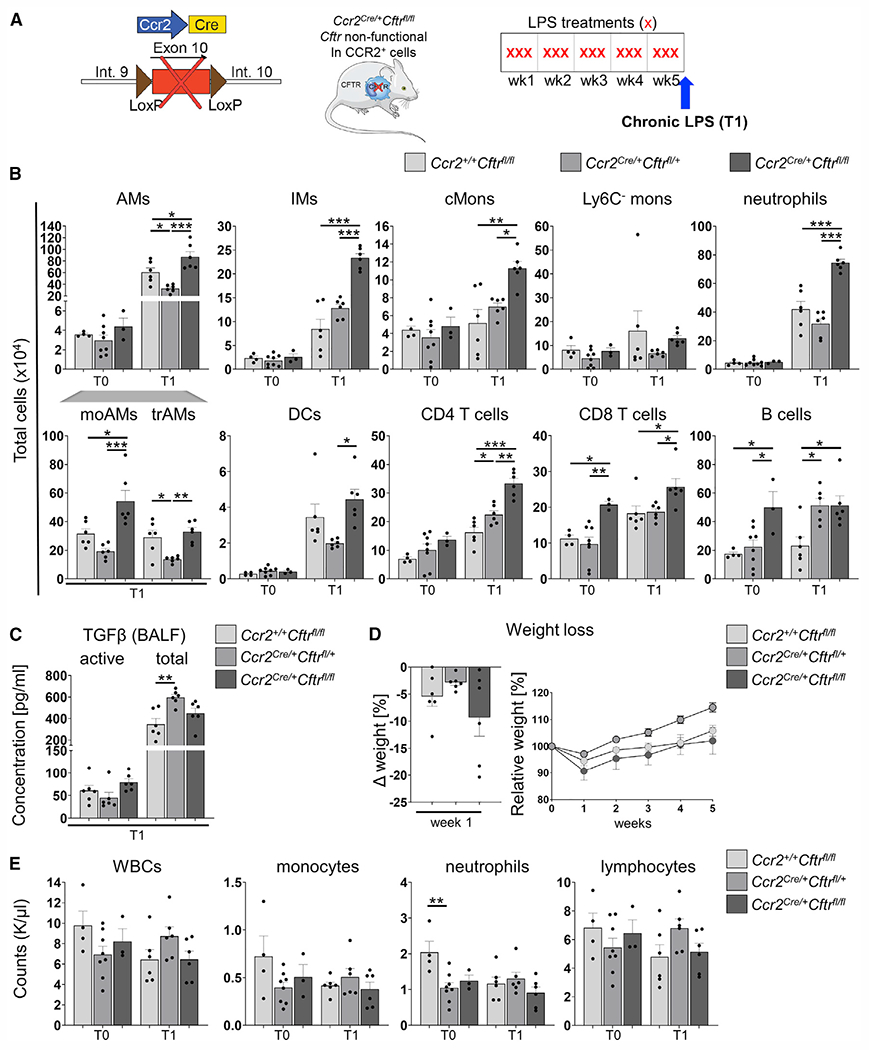
(A) Schematic of the chronic LPS model and Ccr2cre/+Cftrfl/fl mice.
(B) Total cell numbers of AMs, moAMs, trAMs, IMs, cMons, Ly6C− Monos, neutrophils, DCs, CD4 T cells, CD8 T cells, and B cells in lung tissue homogenates were quantified by flow cytometry as percentage of viable cells multiplied by the total cell count in the inferior lung lobe. The gating strategy is described in Figure S3.
(C) Quantification of active and total TGF-β levels in BALF at T1 by ELISA.
(D) Weight loss during 5 weeks of chronic LPS (right) and after the first week of LPS (left).
(E) CBCs at T0 and T1.
Data are generated from two independent experiments with 2–3 mice per genotype and time point. Each biological replicate is represented by a dot. Bars are depicted as means ± SEM, and p values were calculated using one-way ANOVA and Tukey’s test for multiple comparisons between the genotypes for each time point separately (*p < 0.05, **p < 0.01, ***p < 0.001).
Pharmacological targeting of CCR2 mitigates cMon recruitment and normalizes TGF-β levels in CF lungs
Last, we investigated whether pharmacologically targeting CCR2 to decrease cMon recruitment in a CF background can prevent excessive TGF-β signaling and may thus be a target to preserve lung function in patients with CF. CF mice were treated with a small-molecule CCR2 inhibitor (RS 102895) at 10 mg/kg/day in drinking water (as in Teng et al.78), starting 3 days before the first LPS treatment and maintained throughout the LPS challenge (Figure 7A). Inhibition of CCR2 in CF mice lowered the numbers of lung AMs (moAMs and trAMs), IMs, cMons, neutrophils, DCs, and CD4 T cells (but not Ly6C− monocytes, CD8 T cells, or B cells) to levels comparable with WT mice after chronic LPS administration (Figure 7B). CBCs were not altered by the CCR2 inhibitor treatment (Figure S9A). Pharmacological inhibition of CCR2 in CF mice also lowered the concentration of active TGF-β in BALF (Figure 7C) and expression of Tgfβ in lung tissue lysates of CF mice (Figure 7E) to levels comparable with WT mice. Although differences in pSMAD2 levels between inhibitor-treated CF mice and CF CTRLs did not reach statistical significance (p = 0.08), downstream expression of target genes (Col1 and Mmp9) was significantly lower (Figures 7D and 7E). Unlike in dKO mice, pharmacological inhibition of CCR2 in CF mice did not reduce weight loss severity during the first week of LPS exposure (Figure S9B). These data indicate that pharmacological inhibition of CCR2 is sufficient to control exuberant recruitment of cMons, neutrophils, and elevated pathological TGF-β signaling in CF lungs.
Figure 7. Pharmacological targeting of CCR2 mitigates cMon recruitment and normalizes TGF-β levels in CF lungs.
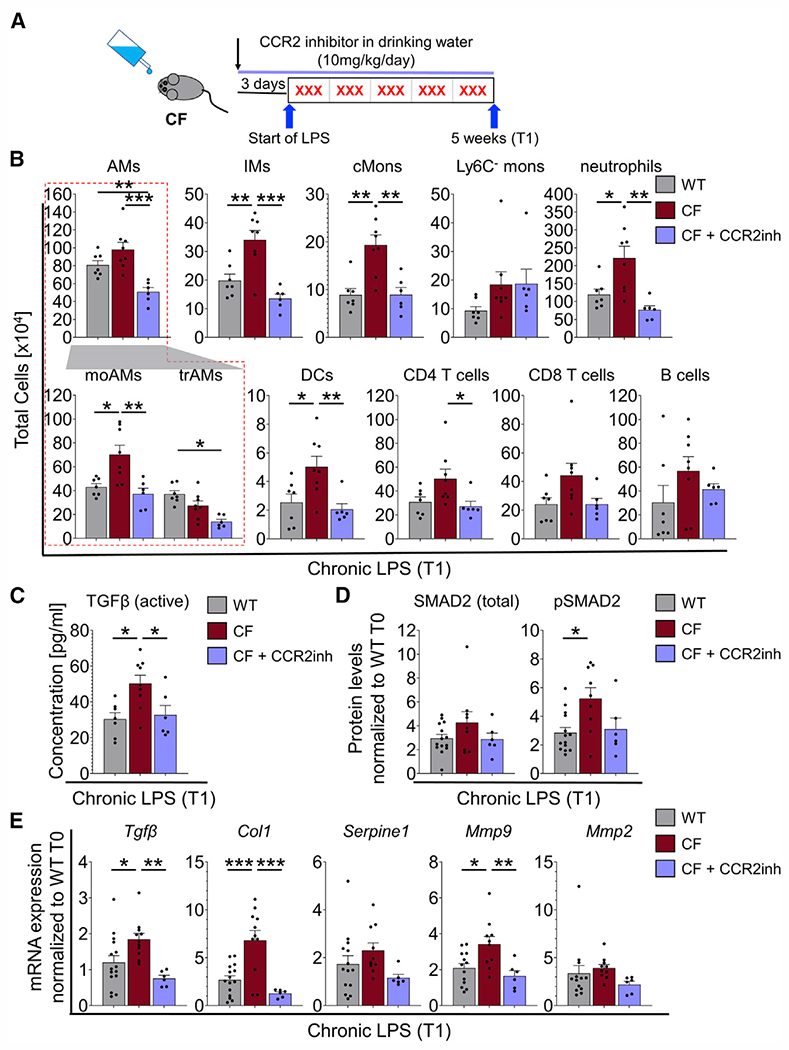
(A) Chronic infection model. CF mice were treated with CCR2 inhibitor (CCR2inh) starting 3 days before the first nebulization and throughout the experiment.
(B) Quantification of flow cytometry analysis of lung immune cells (see Figures 1B and 1C) of CF + CCR2inh-treated mice compared with WT and CF mice at T1.
(C) Active levels of TGF-β at T1.
(D) Densitometric analysis of western blot bands from lung tissues of WT, CF, and CF + CCR2inh mice at T1. The expression levels were normalized to VINCULIN and the WT at T0.
(E) mRNA expression levels in lung tissues of WT, CF, and CF + CCR2inh mice at T1. The expression was normalized to 18S and the WT at T0.
Each biological replicate per genotype and experimental condition is represented by a dot. Bars are depicted as means ± SEM, and p values were calculated using one-way ANOVA and Tukey’s test for multiple comparisons between the genotypes for each time point separately (*p < 0.05, **p < 0.01, ***p < 0.001). See also Figure S12.
DISCUSSION
Fibrotic scarring and emphysematous changes in the aging CF population3–5 are driven by tissue hyperinflammation, which may not be well controlled by CFTR modulator therapies.15,16 We observed a high abundance of CCR2+ cMons in parenchymal and submucosal areas on human lungs with late-stage CF, which are detected in much lower numbers in healthy donor lungs (Figure 1A). This neglected immune population in CF lung disease might play a key role in the progression of lung tissue remodeling in these patients.34–36 cMons in the CF airway submucosa may fuel pathogenic activity in the repair processes of basal cells in response to injury,79 contributing to TGF-β-driven basal cell hyperplasia.80 A small percentage of CCR2+ monocytes are also observed in CF sputum (Figures 1B–1D). The low number of CCR2+ cells in this compartment compared with the lung interstitium might be attributed to the fact that, when recruited into the airways, monocytes downregulate CCR2 expression and acquire a tissue-resident expression signature.81 Thus, the presence of CCR2+ monocytes in CF secretions suggests that the recruited monocytes fail to efficiently acquire a trAM phenotype in the CF airway environment. Alternatively, the increased numbers may be due to arrival of actively recruited cMons that have not yet downregulated CCR2. Consistent with this observation, the CCR2 ligand CCL2 is increased in BALF, sputum, and blood samples of patients with CF82,83 and in the lungs of CF mice exposed to LPS.19 Functional alterations of CCR2+ cMons might be already present in the circulation of patients with CF. Ex vivo studies of monocytes and monocyte-derived macrophages of patients with CF suggest that these cells are hyperinflammatory, have pathological activation of the NLRP3 inflammasome,84,85 and respond to CFTR modulators.85–88 In a previous study, we showed that priming blood monocytes to express the anti-inflammatory/antioxidant mediator heme oxygenase-1 ameliorates pathological lung inflammation in CF.89 Additional investigations are needed to understand the specific contribution of circulating monocytes to lung disease in patients with CF in response to CFTR modulator or immunomodulatory therapies.
We used several mouse lines and a model of chronic lung inflammation44 that recapitulates elevated TGF-β levels and signaling and enlargement and distortion of the air sacs observed in patients with CF5,38,40 to investigate the role of CCR2+ monocytes in CF lung remodeling. We modeled a persistent hyperinflammatory lung environment with repeated exposure to LPS, each one inducing a lung inflammatory flare, which can be seen like exacerbations in patients with CF. Our data demonstrate that cMons and monocyte-derived lung macrophages contribute to excessive and prolonged recruitment of neutrophils, pathogenic TGF-β signaling, and tissue remodeling in CF lungs. Because CF mice do not develop CF-like airway mucus obstruction or suffer from spontaneous bacterial infections, our data also show that chronic inflammation is sufficient to drive severe and irreversible lung structural changes in CF. We found that blocking recruitment of cMons is systemically beneficial in CF mice because it prevented excessive weight loss (a strong indicator of mortality in patients with CF53) because of LPS exposure.
RNA-seq analysis shows that CF (versus WT) lung monocytes/macrophages preserve altered expression profiles after six weeks of recovery from chronic LPS exposure, suggesting failure to adapt in the CF lung environment, leading to a more active immune response signature. Because of the lengthy recovery period, this altered immune response signature might be driven by aberrant epigenetic memory formation after chronic inflammation.90 The most prominent differentially regulated pathways that persist in CF cMons after 6 weeks of recovery from chronic LPS were related to increased production of leukocyte chemoattractants. Our data suggest that prolonged production of CXCL2 by CF cMons may play a central role in driving prolonged recruitment of neutrophils in CF (Figures 5C, S10C–S10D, and S11A). In a model of cardiac transplantation-mediated ischemia reperfusion injury, tissue-resident CCR2+ macrophages were found to be the major producers of CXCL2 and CXCL5, which promoted extravasation of neutrophils into hearts by guiding neutrophil adhesion and crawling on the vasculature.91 Thus, CXCL2 potentially mediates upregulation of adhesion molecules in the pulmonary vascular endothelium to drive lung neutrophilia.
Our study, using a small molecule CCR2 inhibitor, suggests that targeting recruitment of cMons by blocking CCR2 is sufficient to reduce pathological neutrophil inflammation and active TGF-β levels in CF lungs (Figure 7). However, in contrast to the genetic model (dKO mice), we did not observe reduced weight loss in response to LPS. This may be due to short-term use of the drug (started 3 days before LPS), which may be too short to modulate the systemic pathology in CF mice.
We establish the CCR2+ cMon population as a potential therapeutic target in CF. There is a strong clinical interest in developing selective CCR2, CCR2/CCL2, and CCR2/CCR5 antagonists to treat inflammatory diseases, atherosclerosis, tissue fibrosis, cancer, and, more recently, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection.92 Several small molecules have entered phase I/II clinical trials (ClinicalTrials.gov: NCT00689273, NCT02388971, NCT02996110, NCT04123379, NCT03767582, and NCT03496662). We propose that CCR2 inhibitors combined with CFTR modulators may ameliorate long-term lung remodeling caused by chronic inflammation. We suggest targeting CCR2 instead of its ligand CCL2 to avoid chemokine redundancy because CCR2 is also activated by CCL7, CCL8, and CCL13.29
In conclusion, we have shown that a chronic lung inflammatory environment causes a pathogenic CCR2-dependent monocytic response that drives exuberant neutrophilia, pathological TGF-β signaling, and non-resolving tissue remodeling in CF lungs. Impairing recruitment of cMons to CF lungs by targeting CCR2 prevents lung tissue damage and, thus, may be a potential therapeutic target to preserve lung function in patients with CF (Figure S13).
Limitations of the study
Future studies should assess the safety and efficacy of CCR2 inhibitors during lung infection. CCR2+ monocytes contribute to innate host defense against many pathogens. However, the outcomes can be positive or negative depending on infection type and bacterial load.93–95 To the best of our knowledge, the susceptibility of CCR2−/− mice to common CF pathogens such as PA has not yet been reported and is an ongoing investigation in our group. We anticipate that, as with drugs hampering neutrophil migration in CF,96 modulating monocyte recruitment may require fine calibration to prevent lung tissue damage without impairing the host response against bacteria. In mouse models (i.e., dKO and CF mice treated with a CCR2 inhibitor), we observed fewer neutrophils recruited to the lungs in response to LPS compared with CF mice, which, however, were similar to numbers observed in WT lungs (Figures 2 and 7). Our study identified cMons as a major dysregulated cell population in CF lungs. However, more investigations are needed to assess how they contribute to lung pathogenesis over time in patients with CF. This is a general limitation in this field because submucosal immune cell populations are difficult to investigate in the context of the human disease because available specimens from individuals with CF are sputum/BALF samples or explanted lung tissues from late-stage CF lung disease.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Emanuela M Bruscia (emanuela.bruscia@yale.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
ScRNAseq and bulk RNAseq have been deposited on GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti mouse CD45 BUV395 clone 30-F11 | BD | Cat# 565967; RRID: AB_2651134 |
| Rat anti mouse CD38 FITC clone 90 | Invitrogen | Cat# 11-0381-82; RRID: AB_465024 |
| Rat anti mouse CCR2 PE clone SA3203G11 | BioLegend | Cat# 150610; RRID: AB_2616982 |
| Rat anti mouse CD11b PE-Cy7 clone M1/70 | Invitrogen | Cat# 25-0112-82; RRID: AB_469588 |
| Rat anti mouse CD64 APC clone X54-5/7.1 | BioLegend | Cat#139306; RRID: AB_11219391 |
| Rat anti mouse CD45 eFluor450 clone M1/69 | ThermoFisher Scientific | Cat# 48-0242-82; RRID: AB_1311169 |
| Rat anti mouse Ly6C clone AL-21 | BD | Cat# 563011; RRID: AB_2737949 |
| Rat anti mouse CD11c BV711 clone HL3 | BD | Cat# 563048; RRID: AB_2734778 |
| Rat anti mouse Ly6G AF700 clone 1A8 | BD | Cat# 551459; RRID: AB_394206 |
| Rat anti mouse Siglec-F PerCP-Cy5.5 clone E50-2440 | BD | Cat# 565526; RRID: AB_2739281 |
| Rat anti mouse I-A/I-E (MHC-II) APC/Fire 750 clone M5/114.15.2 | BioLegend | Cat# 107652; RRID: AB_2616729 |
| Rat anti mouse CD4 PerCP-Cy5.5 clone RM4-5 | BD | Cat# 561115; RRID: AB_393977 |
| Rat anti mouse CD8a FITC clone 53–6.7 | BioLegend | Cat# 100706; RRID: AB_312745 |
| Rat anti mouse CD45R/B220 PE-Cy7 clone RA3-6B2 | BD | Cat# 552772; RRID: AB_394458 |
| mouse anti-alpha smooth muscle actin clone 1A4 | Abcam | Cat# ab7817; RRID: AB_2865422 |
| rabbit mAB anti-SMAD2 clone D43B4 | Cell Signaling | Cat# 5339 |
| rabbit mAB anti-phospho-SMAD2 (Ser465/467) clone 138D4 | Cell Signaling | Cat# 3107 |
| rabbit anti-VINCULIN | Cell Signaling | Cat# 4650 |
| rabbit anti-p44/42 MAPK (Erk1/2) clone 137F5 | Cell Signaling | Cat# 4695 |
| rabbit anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) clone D13.14.4E | Cell Signaling | Cat# 4370 |
| rabbit mAB anti-CXCL2 clone 16H3L10 | invitrogen | Cat# 701126 |
| mouse anti-CCR2 clone 3G7 | Novus Biologicals | Cat# NBP2-35334 |
| mouse anti-CCR2 clone 7A7 | abcam | Cat# ab176390 |
| FcBlock | BD | Cat# 553141; RRID: AB_394656 |
| goat anti-rabbit IgG-HRP | Santa Cruz | Cat# sc-2004 |
| mouse anti-alpha smooth muscle Actin (aSMA) clone 1A4 | Abcam | Cat# ab7817 |
| rabbit Cy5 Tyramide | Akoya | Cat# SAT705A001 EA |
| Biotin Tyramide | Akoya | Cat# NEL705A001KT |
| rabbit envision secondary antibody | Agilent/Dako | Cat# K4001-8 |
| mouse envision secondary antibody | Agilent/Dako | Cat# K4005 |
| Alexa 750-Streptavidin | Thermofisher/Life Technologies | Cat# S21384 |
| Chemicals, peptides, and recombinant proteins | ||
| cOmplete Mini EDTA-free protease inhibitor cocktail | Roche | Cat# 11836170001 |
| PBS | Gibco | Cat# 10010023 |
| UltraPure Low Melting Point agarose | Invitrogen | Cat# 16520–050 |
| Lipopolysaccharides from Pseudomonas aeruginosa | Millipore Sigma | Cat# L9143 |
| CCR2 inhibitor RS 102895 | Cayman Chemical | Cat# 1173022-16-6 |
| Cell Lysis Buffer | Cell Signaling | Cat# 9803 |
| PhosSTOP | Roche | Cat# 4906837001 |
| 4–15% Mini PROTEAN TGX Gels | Bio-Rad | Cat# 4561086 |
| ECL Plus Western blotting system | GE Healthcare | Cat# RPN2132 |
| Superscript II Reverse Transcriptase | ThermoFisher | Cat# 18064–022 |
| RBC lysis buffer | eBioscience | Cat# 00-4300-54 |
| Critical commercial assays | ||
| active TGF-beta ELISA kit | BioLegend | Cat# 437707 |
| total TGF-beta ELISA kit | BioLegend | Cat# 436707 |
| V-PLEX Proinflammatory Panel 1 (mouse) | Mesoscale Discovery | Cat# K15048G-1 |
| V-PLEX Cytokine Panel 1 (mouse) | Mesoscale Discovery | K15245D-1 |
| Lung dissociation kit (mouse) | Mitenyi Biotec | Cat# 130-095-927 |
| miRNEasy Micro kit | Qiagen | Cat# 217084 |
| Agilent RNA6000 Pico Kit | Agilent | Cat# G2938-90046 |
| SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing | Takara Bio | Cat# 634890 |
| Nextera XT DNA Sample Preparation kit | Illumina | Cat# FC-131-1096 |
| miScript II RT kit | Qiagen | Cat# 218161 |
| Mouse CXCL2/MIP-2 DuoSet ELISA | R&D systems | Cat# DY452-05 |
| Deposited data | ||
| scRNAseq data | Schupp et al.26 | GEO: GSE145360 |
| Bulk RNAseq data | GEO | GEO: GSE217102 |
| Experimental models: Organisms/strains | ||
| Cftr knockout mice B6.129P2-KOCftrtm1UNC | Jackson Laboratory | Stock No. 002196 |
| CCR2 knockout mice B6.129S4-Ccr2tm1Ifc/J | Jackson Laboratory | Stock No. 004999 |
| Ccr2cre mice | Dr. Tobias Hohl; Heung and Hohl77 | N/A |
| cftrflox/flox mice | CF animal core facility at Case Western Reserve University | N/A |
| CD45.1 mice B6.SJL-Ptprca Pepcb/BoyJ | Jackson Laboratory | Stock No. 002014 |
| Software and algorithms | ||
| FACSDiva | BD | N/A |
| FlowJo | BD | N/A |
| Prism | GraphPad | N/A |
| enrichR package | https://maayanlab.cloud/Enrichr | N/A |
| edgeR package | https://bioconductor.org | N/A |
| Other | ||
| Nebulizer Pulmo-Aide Compressor | Natallergy | Cat# DRV5650D |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study design
The objective of this study was to clarify the underlying cellular mechanisms of inflammation-driven lung tissue remodeling in CF by analyzing mouse models. Lung remodeling was achieved by repeated administration of LPS. Sample size was determined by previous experience and preliminary data. The numbers of animals or samples and repeated experiments are indicated in the figure legends. All assessed data points are shown depicted unless indicated otherwise.
Mouse models
CFTR−/− (B6.129P2-KOCftrtm1UNC; CF)and CCR2−/− (B6.129S4-Ccr2tm1Ifc/J; CCR2−/−) mice were purchased from the Jackson Laboratory. Drinking water for CF mice was supplemented with Colyte (Braintree Labs), to prevent gastrointestinal obstruction at weaning. Non-CF littermate controls were maintained on identical dietary conditions to the CF mice to eliminate nutritional confounding. CF and CCR2−/− mice were crossed to generate CFTR−/−CCR2−/− double knockout mice (dKO). Ccr2cre mice were generated and provided by Dr. Tobias Hohl77 and were crossed with Cftrflox10/flox10 (Cftrfl/fl) mice (provided by Case Western Reserve University76) to generate CCR2-specific CFTR knockouts (Ccr2cre/+Cftrfl/fl) and heterozygous control mice (Ccr2cre/+Cftrfl/+). All mouse colonies were bred in the Yale University Animal Facility with food and water ad libitum. To control for microbiome effects, experimental groups were co-housed. All procedures were performed in compliance with relevant laws and institutional guidelines and were approved by the Yale University Institutional Animal Care and Use Committee. All mice were aged 8–12 weeks at the start of experimentation and both female and male mice were used equally in the study.
METHOD DETAILS
ScRNAseq
Spontaneously expectorated sputum from nine patients with CF and induced sputum from five healthy control subjects was collected and processed into a single-cell suspension as previously described.26 Patient demographic data is available as Table S1. Single cells were barcoded using the 10x Chromium Single Cell platform and cDNA libraries were prepared according to the manufacturer’s protocol (Single Cell 3′ Reagent Kits v3, 10x Genomics, USA). cDNA was analyzed on an Agilent Bioanalyzer High Sensitivity DNA chip. cDNA libraries were analyzed on a HiSeq 4000 Illumina platform aiming for 150 million reads per library. Full de-identified sequencing data for all subjects is available in the gene expression omnibus (GEO) under accession number GEO: GSE145360. Detailed description of data processing and computational analyses can be found in the original manuscript.26 Cell type annotation was adopted from the original publication.26 15 principal components were used to generate a revised 2-dimensional Uniform Manifold Approximation and Projection (UMAP) for visualization (with 50 nearest neighbors).
Chronic LPS model
The chronic LPS model was adapted from our previous work.44 Briefly, mice were exposed to PA-LPS (L9143 Millipore Sigma) 3 times a week for 5 weeks (Every Monday, Wednesday, and Friday) using a nebulizer (Pulmo-Aide Compressor; Natallergy, Duluth, GA). 8–12-week-old female and male mice were equally used in experiments. Mice were nebulized with 12.5 mg of LPS in 5 mL PBS for 15 min, three times per week for five weeks. Mice were sacrificed 24 h after the last nebulization (T1) or after 6 weeks of recovery (T2) (Figure 1A). At each time point, Blood, BALF and lung tissues were collected. The CCR2 inhibitor RS 102895 (Cayman chemical) was dissolved in DMSO at 25 mg/mL and added to the drinking water of animals to a final concentration of 10 mg/kg/day/mouse (Figure 7A). Drinking behavior was monitored daily by weighing of the water bottles and drug concentration was adjusted accordingly.
Blood, bronchoalveolar lavage fluid (BALF), and lung tissue collection
Mice were anesthetized and up to 500 μL of blood was recovered by retro-orbital puncture. Complete blood counts (CBCs) from 50 μL of blood were assessed by Hemavet 950 (Drew Scientific). 150 μL of blood was treated with 1 mL of RBC lysis buffer (eBioscience) for 45 min, then centrifuged at 2800RPM for 7 min, supernatants were discarded, and cells were resuspended in 1 mL RBC lysis buffer for 15 min, centrifuged at 2800 RPM for 7 min, supernatants were discarded and cleared cell suspension was collected in 400 μL of PBS and subsequently stained for flow cytometry. BALF was collected using standard methods with 2 mL BAL solution (PBS with 5mM EDTA and cOmplete Mini EDTA-free protease inhibitor cocktail, Roche; 1 tablet per 25 mL). BALF was first passed through 100-μm cell strainer and then centrifuged at 1200 rpm for 10 min. Supernatants were collected and stored in single-use aliquots at −80°C until analysis, while cell pellets were resuspended in PBS, counted, and used for flow cytometry for differential cell counting. Lung tissues: lungs were collected via a midline incision from sternum to diaphragm, and blood was removed from the pulmonary circulation by transcardial perfusion of PBS supplemented with heparin (1:1000). The right lobes were ligated, and the left lung lobes were inflated via intratracheal catheter with 0.5%UltraPure Low Melting Point agarose (Invitrogen) in PBS at constant pressure of 20 cmH2O, then harvested, fixed overnight in 10% neutral buffered formalin, and embedded in paraffin in longitudinal orientation. The right lobes were used as follows: superior lobes were collected in RNAlater and used for RNA; middle and postcaval lobes were snap-frozen in liquid nitrogen and used for protein isolation; inferior lobes were collected in PBS and used for flow cytometry. Paraffin-embedded tissues were stained with hematoxylin-eosin for morphological analysis or used for immunohistochemistry.
FACS and flow cytometry staining
Single cell suspensions were isolated from inferior lobes (or whole lungs for sorting) using C tubes, a lung dissociation kit (Miltenyi, CA) and a GentleMACS dissociator (Miltenyi, CA) following manufacturer’s instructions. Homogenized lungs were passed through a 100-μm cell strainer to obtain a single-cell suspension. Red blood cells were lysed using BD Pharm Lyse (BD Biosciences, CA). Cells were counted using a hemocytometer with trypan blue exclusion. 1 × 106 cells were used for FACS staining and up to 2 × 107 cells were used for sorting. Cells were stained with a fixable viability dye (Invitrogen, MA), incubated with FcBlock (BD Biosciences, CA), and then washed and stained with freshly prepared antibody cocktails for 30 min at 4°C following standard cell surface staining protocols. For cell sorting, cells were washed and sorted on a FACSAria II (BD Biosciences). For flow cytometry, cells were washed and fixed with BD Cytofix, (BD Biosciences, CA) and acquired on a BD LSR II using BD FACSDiva software. Data analyses were performed using FlowJo software (TreeStar, Ashland, OR). The percentage of each assessed population among singlet/live cells was multiplied by the total lung cell counts in the inferior lobes to obtain total live cell counts.
Flow cytometry gating strategy
The gating strategy is adjusted from Misharin et al. 201347 and is shown in Figure S3: Singlet events were gated followed by exclusion of dead cells stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (ThermoFisher Scientific). Small debris were excluded by exclusion of FSC-H SSC-H small events and from here CD45+ cells were gated. As a next step we gated alveolar macrophages (AMs) by first selecting for CD11c+ cells. From the CD11c+ fraction we distinguished dendritic cells (DCs) from AMs by CD64/CD24 expression levels with DCs being selected as CD24highCD64neg. Within the AM population, we were able to distinguish monocyte-derived AMs (moAMs) as SiglecFintermediateCD38high from tissue-resident AMs (trAMs) as SiglecFhighCD38intermediate at T1. At T0 and T2 moAMs are not clearly distinguishable from trAMs, although at T2 AMs skew toward lesser SiglecF expression compared to T0. This skewing toward SiglecFdim expression was used for sorting moAMs at T2 (as shown in Figures S9A, S3C). From the CD11c− fraction we gated the remaining myeloid cells by expression of CD11b. Granulocytes (gran) were then gated from the CD11b+ cells by high expression of CD24 and low expression of MHC-II. Granulocytes were then split into neutrophils as Ly6G+SiglecF− and eosinophils as Ly6G−SiglecF+. The non-granulocytic (not gran) fraction was then split into interstitial macrophages (IMs) as the MHC-II+ cells and monocytes as MHC-II− cells. Lastly, from the MHC-II− fraction we gated classical monocytes (cMons) as Ly6C+CD64+ cells and other, Ly6C− monocytes as Ly6C− cells. Lymphocytes were stained separately and identified as CD4+ CD4 T cells, CD8+ CD8 T cells and B220+ B cells among singlet viable CD45+ cells.
Circulating cMons were assessed as singlet viable CD45+CD11b+Ly6C+Ly6G− cells in blood (see Figure S2D).
Cellular markers used to identify lung monocyte/macrophage populations via flow cytometry/FACS were validated by RNAseq from FACS-sorted population (Figure S9B).
Bone marrow transplant
We used bone marrow transplant methods as previously described.19 Briefly, WT or CCR2−/− mice were total-body-irradiated using a cesium irradiator (1000 rads, split in two doses 12 h apart) and transplanted intravenously with donor WT or CCR2 BM (WTBM→WT; WTBM→CCR2−/−; CCR2−/−BM→WT; CCR2−/−BMCCR2−/−) 3 h after irradiation. Mice were administered Baytril for the first two weeks following irradiation for infection prevention and left to recover for additional 4 weeks before starting of LPS administrations. Percentage of donor vs host immune cells were representatively assessed by FACS in blood, BALF and lung tissues of CD45.1BM→CD45.2 mice.
RNA sequencing and data analysis
The RNA from FACS-sorted cMons, IMs, moAMs, and trAMs (Figure 1A) was extracted using miRNEasy Micro kit (Qiagen, MD). The RNA integrity (RIN) and quantity was determined on the Agilent 2100 Bioanalyzer (Agilent, CA) with Agilent RNA 6000 Pico Kit (Agilent, CA). Only samples with RIN ≥8.0 were used for library preparation. Reverse transcription and cDNA preamplification were performed using the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara Bio USA, MI), and sequencing libraries were prepared using Nextera XT DNA Sample Preparation kit (Illumina, CA, USA), according to manufacturers’ instructions. Libraries were pooled and sequenced on Illumina HiSeq4000 (Illumina, CA) using paired-end sequencing for 100 cycles. After quality control (FastQC), reads generated from each sample were aligned to the mouse genome (GRCm38.p5) with STAR (version 2.5.3a, -quantMode GeneCounts), using the Gencode M15 transcript annotation as transcriptome guide. Normalization with the TMM method and identification of differentially expressed genes were performed with the edgeR package in Bioconductor (https://bioconductor.org). Differentially expressed genes were identified with the glmQLFTest function using a double threshold on gene expression changes and associated statistical significance (absolute log2 fold change >0.75, p value < 0.05). To identify, within each cellular population, genes differentially regulated in CF between T1 and T2, the following contrast, based on 2 factors, was used: (CFT2 - WTT2) - (CFT1 - WTT1). Functional annotation and enrichment analysis were performed with the enrichR package (https://maayanlab.cloud/Enrichr). Seqdata was generated from 2 samples per genotype and time point. To ensure sufficient cell counts at T2, lungs from 2 WT or 2 CF mice were pooled for one sample. Sequencing data for all samples is available in the GEO under accession number GEO: GSE217102.
Immunohistochemistry
For human samples, paraffin slides of lung tissues from patients with late-stage CF (kindly provided by Dr. Randell at UNC; Tissue Procurement and Cell Culture Core) and healthy lung tissues (UNC and Yale Pathology Tissue Services) were stained with H&E, or for CCR2 (1:50, clone 7A7, ab176390, Abcam) by Yale Pathology Tissue Services following standard procedures. For murine samples left lobes were inflated, paraffin-embedded and sectioned. For alveolar damage score, the H&E-stained slides were examined blinded to the genotype and treatment. Qualitative histopathology was supplemented by a semiquantitative scoring system (Figure S2B) that assessed the degree of alveolar architectural disruption (Figure 2A). In addition, Masson’s trichrome staining was performed by Yale Pathology Tissue Services. αSMA staining was performed with an overnight antibody incubation (1:100, clone1A4, ab7817, Abcam) at 4°C. The images were taken using a Zeiss Axioskop light microscope and imaged using a Zeiss AxioCam MrC digital camera. Semi-quantitative scoring of alveolar architectural disruption was performed by a board-certified veterinary pathologist masked to the genotype and treatment status (see Figure S4 for scoring system).
Immunofluorescence
Human lung tissues were stained by the Yale Pathology Tissue Services for CCR2 (1:500, mouse monoclonal, clone 7A7, ab176390, Abcam) and CD14 (1:500, rabbit monoclonal, clone D7A2T, CST). Briefly, formalin-fixed, paraffin-embedded lung tissue sections were deparaffinized with xylene, rehydrated gradually with graded alcohol solutions, and then washed with deionized water. After antigen retrieval and blocking, sections were incubated overnight with primary antibodies, washed, and incubated with rabbit or mouse envision secondary antibody respectively. CD14 signal was boosted by incubation with envision secondary antibody (Agilent/Dako K4001-8) at RT for 1h, washed, incubated with Cy5 Tyramide 1:50 (Akoya, Cat#SAT705A001 EA) for 10 min. CCR2 signal was boosted by incubation with envision secondary antibody (Agilent/Dako K4005) for 1h, washed, incubated with Biotin Tyramide (Akoya, Cat#NEL705A001KT) for 10 min, washed, and incubated with Alexa 750-Streptavidin 1:100 (Thermofisher/Life Technologies S21384) for 1h. Slides were washed, incubated with DAPI 1:1000 for 5 min and mounted with Prolong Gold. Mouse lung sections were stained for CCR2 (1:100, mouse monoclonal, clone 3G7, Novus Biologicals) and CXCL2 (1:100, rabbit monoclonal, clone 16H3L1, Invitrogen) following antigen retrieval and blocking (as described above). Sections were washed and stained with a 1:500 dilution of fluorescent-labeled anti-mouse (Alexa 568) and anti-rabbit (Alexa 647) antibodies at room temperature for 2 h. Slides were washed, incubated with DAPI 1:100 for 5 min and mounted with Prolong Gold. Images were acquired with a Leica Thunder Imager 3D Microscope or with Leica TCS SP5 Spectral Confocal Microscope.
Quantitative RT-PCR analysis
Up to 20 mg of lung tissues were homogenized in trizol using a tissue homogenizer. Total RNA was isolated using miRNA Mini Kit (Qiagen), RNA concentration was measured by Nanodrop and 1 μg or RNA was reverse-transcribed using SuperScript™ II Reverse Transcriptase (Thermo Fisher) following the manufacturers’ instructions. Real-time PCR analysis was performed with a Bio-Rad iCycler using TaqMan technology with primers and probes purchased from Applied Biosystems (Life Technology). For microRNA studies miRNA primers were purchased from Qiagen and qPCR was performed using miScript SYBR green PCR kit (Qiagen) and RNA was reverse-transcribed using the miscript II RT Kit (Qiagen). Relative gene expression was normalized by 18S rRNA levels and copy number for miR199a-5p was normalized by RNU6B. Relative expression to control samples (WT T0) was calculated by ΔΔCt method.
Protein isolation and Western blot
Cold RIPA lysis buffer (Cell Signaling) containing 1mM phenylmethane sulfonyl fluoride (PMSF), protease and phosphatase inhibitor cocktails (Roche Diagnostics) were added to cells or lung tissue lysates. After 30 min of incubation in ice, the lysates were centrifuged at 12,000g for 15 min, and the supernatants recovered. An equal amount of protein was separated by electrophoresis on 4–15% Mini PROTEAN Gels (Bio-Rad Laboratories, CA), transferred to nitrocellulose membrane (Bio-Rad Laboratories, CA) and incubated with primary antibodies overnight at 4 °C. HRP conjugated to IgG secondary Abs (1:2000; Santa Cruz Biotechnology) and Amersham ECL Plus Western blotting system (GE Healthcare Bio-Sciences) were used for detection. The following primary antibodies were used: rabbit mAB anti-SMAD2 (1:1000, D43B4 Cell signaling) and rabbit mAB anti-pSMAD2 (1:1000, 138D4 Cell Signaling), rabbit anti-VINCULIN (1:500, #4650 Cell Signaling), rabbit anti-ERK (p44/42 MAP Kinase, 1:1000, #4695 Cell Signaling) rabbit anti-pERK (1:1000, #4370 Cell Signaling). The chemiluminescence imaging system ChemiDoc (Biorad) and the Image lab software (Biorad) were used for image acquisition and for signal quantification. Specific protein band intensities were normalized to VINCULIN and the fold increase to control samples (WT T0) was calculated.
ELISA
Active and total levels of TGFβ and CXCL2 in BALF supernatants were assessed by ELISA following manufacturer’s instruction (active free form: 437707 Biolegend; total levels: 436707 Biolegend; CXCL2: DY452-05 R&D Systems).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data presented as mean ± SEM and are plotted using GraphPad Prism software. Raw data are available in Data S2. The p values were calculated using one-way ANOVA and Tukey’s test for multiple comparisons or students’ T test. A p value <0.05 were considered significant: *p ≤ 0.05, **p < 0.01, and ***p < 0.001. Datasets used in Figures 2, 3 and 4 are from Experiments that were repeated at least three times with three or more samples per group each. Datasets used in Figure 6 are derived from one experiment with six samples per group, and in Figure seven from two experiments with 5 or more samples per group.
Supplementary Material
Highlights.
Chronic neutrophilic inflammation causes lung damage in patients with cystic fibrosis (CF)
CCR2+ monocytes (cMons) accumulate in inflamed human and murine CF lungs
cMons drive lung damage via neutrophil recruitment and TGF-β signaling in CF
Targeting cMon recruitment by CCR2 inhibition prevents lung damage in CF
ACKNOWLEDGMENTS
We thank Dr. Ravindra Gudneppanavar, Marco Arocha Lopez, Richard Nguyen, and Christina Barone for technical support; the Yale CF Research Center and the Yale Cooperative Center of Excellence in Hematology (NIH/NIDDK U54DK106857 to D.S.K.) for valuable support; the Yale FACS Facility and the Yale Pathology Core for outstanding service; Dr. Scott Randell at the UNC-Chapel Hill Tissue Procurement and Cell Culture Core (CFF-BOUCHE15R0 and NIH-DK065988) for provision of specimens; Dr. Tobias Hohl at Memorial Sloan Kettering Cancer Center for providing CCR2cre mice; the CF animal core facility at Case Western Reserve University for providing Cftrflox10/flox10 mice; and Dr. Dominik Hartl for reviewing and Grace Fox for copyediting the manuscript. This work was supported by the Cystic Fibrosis Foundation (OZ18F0 to H.H.Ö., BRUSCI20G0 to E.M.B., EGAN16G0 to M.E.E., and ESQUIB19H0 to S.S.E.). E.M.B. is supported by National Institutes of Health (NIH) grants R01HL157776 and R01AI153422. D.S.K. is partially supported by NIH/NIDDK U54DK106857. S.H. is partially supported by NIH/NIDDK R01DK124788. G.B. is supported by NIH/NIDDK Cooperative Centers of Excellence in Hematology (CCEH) U24DK126127.T.T. is supported by AIRC under MFAG 2020 (ID 24883 project).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111797.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
DATA AND MATERIALS AVAILABILITY
All data are available in the main text or the supplemental information.
REFERENCES
- 1.Ramsey KA, Rosenow T, Turkovic L, Skoric B, Banton G, Adams AM, Simpson SJ, Murray C, Ranganathan SC, Stick SM, et al. (2016). Lung clearance index and structural lung disease on computed tomography in early cystic fibrosis. Am. J. Respir. Crit. Care Med 193, 60–67. 10.1164/rccm.201507-1409OC. [DOI] [PubMed] [Google Scholar]
- 2.Martínez TM, Llapur CJ, Williams TH, Coates C, Gunderman R, Cohen MD, Howenstine MS, Saba O, Coxson HO, and Tepper RS (2005). High-resolution computed tomography imaging of airway disease in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med 172, 1133–1138. 10.1164/rccm.200412-1665OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loeve M, van Hal PTW, Robinson P, de Jong PA, Lequin MH, Hop WC, Williams TJ, Nossent GD, and Tiddens HA (2009). The spectrum of structural abnormalities on CT scans from patients with CF with severe advanced lung disease. Thorax 64, 876–882. 10.1136/thx.2008.110908. [DOI] [PubMed] [Google Scholar]
- 4.Harris WT, Kelly DR, Zhou Y, Wang D, Macewen M, Macewen M, Hagood JS, Clancy JP, Ambalavanan N, and Sorscher EJ (2013). Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS One 8, e70196. 10.1371/journal.pone.0070196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mets OM, Roothaan SM, Bronsveld I, Luijk B, van de Graaf EA, Vink A, and de Jong PA (2015). Emphysema is common in lungs of cystic fibrosis lung transplantation patients: a histopathological and computed tomography study. PLoS One 10, e0128062. 10.1371/journal.pone.0128062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenow T, Mok LC, Turkovic L, Berry LJ, Sly PD, Ranganathan S, Tiddens HAWM, and Stick SM (2019). The cumulative effect of inflammation and infection on structural lung disease in early cystic fibrosis. Eur. Respir. J 54, 1801771. 10.1183/13993003.01771-2018. [DOI] [PubMed] [Google Scholar]
- 7.Rosen BH, Evans TIA, Moll SR, Gray JS, Liang B, Sun X, Zhang Y, Jensen-Cody CW, Swatek AM, Zhou W, et al. (2018). Infection is not required for mucoinflammatory lung disease in CFTR-knockout ferrets. Am. J. Respir. Crit. Care Med 197, 1308–1318. 10.1164/rccm.201708-1616OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruscia EM, and Bonfield TL (2016). Innate and adaptive immunity in cystic fibrosis. Clin. Chest Med 37, 17–29. 10.1016/j.ccm.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Genschmer KR, Russell DW, Lal C, Szul T, Bratcher PE, Noerager BD, Abdul Roda M, Xu X, Rezonzew G, Viera L, et al. (2019). Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell 176, 113–126.e15. 10.1016/j.cell.2018.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, and Riches DW (1995). Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med 151, 1075–1082. 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 11.Garratt LW, Sutanto EN, Ling KM, Looi K, Iosifidis T, Martinovich KM, Shaw NC, Kicic-Starcevich E, Knight DA, Ranganathan S, et al. (2015). Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur. Respir. J 46, 384–394. 10.1183/09031936.00212114. [DOI] [PubMed] [Google Scholar]
- 12.Margaroli C, Garratt LW, Horati H, Dittrich AS, Rosenow T, Montgomery ST, Frey DL, Brown MR, Schultz C, Guglani L, et al. (2019). Elastase exocytosis by airway neutrophils is associated with early lung damage in children with cystic fibrosis. Am. J. Respir. Crit. Care Med 199, 873–881. 10.1164/rccm.201803-0442OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F, et al. (2019). Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med 381, 1809–1819. 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shteinberg M, Haq IJ, Polineni D, and Davies JC (2021). Cystic fibrosis. Lancet 397, 2195–2211. 10.1016/S0140-6736(20)32542-3. [DOI] [PubMed] [Google Scholar]
- 15.Harris JK, Wagner BD, Zemanick ET, Robertson CE, Stevens MJ, Heltshe SL, Rowe SM, and Sagel SD (2020). Changes in airway microbiome and inflammation with ivacaftor treatment in patients with cystic fibrosis and the G551D mutation. Ann. Am. Thorac. Soc 17, 212–220. 10.1513/AnnalsATS.201907-493OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, Sagel SD, Khan U, Mayer-Hamblett N, Van Dalfsen JM, et al. (2014). Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med 190, 175–184. 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrne AJ, Mathie SA, Gregory LG, and Lloyd CM (2015). Pulmonary macrophages: key players in the innate defence of the airways. Thorax 70, 1189–1196. 10.1136/thoraxjnl-2015-207020. [DOI] [PubMed] [Google Scholar]
- 18.Okabe Y, and Medzhitov R (2016). Tissue biology perspective on macrophages. Nat. Immunol 17, 9–17. 10.1038/ni.3320. [DOI] [PubMed] [Google Scholar]
- 19.Bruscia EM, Zhang PX, Ferreira E, Caputo C, Emerson JW, Tuck D, Krause DS, and Egan ME (2009). Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− mice. Am. J. Respir. Cell Mol. Biol 40, 295–304. 10.1165/rcmb.2008-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonfield TL, Hodges CA, Cotton CU, and Drumm ML (2012). Absence of the cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection .J.Leukoc. Biol 92, 1111–1122. 10.1189/jlb.0412188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, et al. (2006). CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol 8, 933–944. 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- 22.Del Porto P, Cifani N, Guarnieri S, Di Domenico EG, Mariggiò MA, Spadaro F, Guglietta S, Anile M, Venuta F, Quattrucci S, and Ascenzioni F (2011). Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS One 6, e19970. 10.1371/journal.pone.0019970PONE-D-10-05437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DHA, Newland C, Rosales-Reyes R, Kopp B, McCoy K, et al. (2011). Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 7, 1359–1370. 10.4161/auto.7.11.17660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Assani K, Shrestha CL, Robledo-Avila F, Rajaram MV, Partida-Sanchez S, Schlesinger LS, and Kopp BT (2017). Human cystic fibrosis macrophages have defective calcium-dependent protein Kinase C activation of the NADPH oxidase, an effect augmented by burkholderia cenocepacia. J. Immunol 198, 1985–1994. 10.4049/jimmunol.1502609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riquelme SA, Hopkins BD, Wolfe AL, DiMango E, Kitur K, Parsons R, and Prince A (2017). Cystic fibrosis transmembrane conductance regulator attaches tumor suppressor PTEN to the membrane and promotes anti Pseudomonas aeruginosa immunity. Immunity 47, 1169–1181.e7. 10.1016/j.immuni.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schupp JC, Khanal S, Gomez JL, Sauler M, Adams TS, Chupp GL, Yan X, Poli S, Zhao Y, Montgomery RR, et al. (2020). Single cell transcriptional archetypes of airway inflammation in cystic fibrosis. Am. J. Respir. Crit. Care Med 202, 1419–1429. 10.1164/rccm.202004-09910C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Margaroli C, Horati H, Garratt LW, Giacalone VD, Schofield C, Dittrich AS, Rosenow T, Dobosh BS, Lim HS, Frey DL, et al. (2022). Macrophage PD-1 associates with neutrophilia and reduced bacterial killing in early cystic fibrosis airway disease. J. Cyst. Fibros 10.1016/j.jcf.2022.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Blériot C, Chakarov S, and Ginhoux F (2020). Determinants of resident tissue macrophage identity and function. Immunity 52, 957–970. 10.1016/j.immuni.2020.05.014. [DOI] [PubMed] [Google Scholar]
- 29.David BA, and Kubes P (2019). Exploring the complex role of chemokines and chemoattractants in vivo on leukocyte dynamics. Immunol. Rev 289, 9–30. 10.1111/imr.12757. [DOI] [PubMed] [Google Scholar]
- 30.Kaur M, Bell T, Salek-Ardakani S, and Hussell T (2015). Macrophage adaptation in airway inflammatory resolution. Eur. Respir. Rev 24, 510–515. 10.1183/16000617.0030-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morales-Nebreda L, Misharin AV, Perlman H, and Budinger GRS (2015). The heterogeneity of lung macrophages in the susceptibility to disease. Eur. Respir. Rev 24, 505–509. 10.1183/16000617.0031-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, Leach SM, Fingerlin TE, O’Connor BP, Reisz JA, et al. (2017). Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. Am. J. Respir. Cell Mol. Biol 57, 294–306. 10.1165/rcmb.2017-0061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, Zhang XM, Foo S, Nakamizo S, Duan K, et al. (2019). Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, eaau0964. 10.1126/science.aau0964. [DOI] [PubMed] [Google Scholar]
- 34.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, et al. (2017). Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med 214, 2387–2404. 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ, et al. (2011). Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med 184, 569–581. 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 36.Wynn TA, and Vannella KM (2016). Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462. 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, Zariwala M, Fargo D, Xu A, Dunn JM, et al. (2005). Genetic modifiers of lung disease in cystic fibrosis. N. Engl. J. Med 353, 1443–1453. 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 38.Harris WT, Muhlebach MS, Oster RA, Knowles MR, and Noah TL (2009). Transforming growth factor-beta(1) in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr. Pulmonol 44, 1057–1064. 10.1002/ppul.21079. [DOI] [PubMed] [Google Scholar]
- 39.Harris WT, Muhlebach MS, Oster RA, Knowles MR, Clancy JP, and Noah TL (2011). Plasma TGF-beta(1) in pediatric cystic fibrosis: potential biomarker of lung disease and response to therapy. Pediatr. Pulmonol 46, 688–695. 10.1002/ppul.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eickmeier O, Boom L.v.D., Schreiner F, Lentze MJ, NGampolo D, Schubert R, Zielen S, and Schmitt-Grohé S (2013). Transforming growth factor beta1 genotypes in relation to TGFbeta1, interleukin-8, and tumor necrosis factor alpha in induced sputum and blood in cystic fibrosis. Mediators Inflamm. 2013, 913135. 10.1155/2013/913135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicola T, Kabir FL, Coric T, Wall SB, Zhang W, James M, MacEwen M, Ren C, Halloran B, Ambalavanan N, and Harris WT (2019). CFTR dysfunction increases endoglin and TGF-beta signaling in airway epithelia. Physiol. Rep 7, e13977. 10.14814/phy2.13977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kramer EL, Madala SK, Hudock KM, Davidson C, and Clancy JP (2020). Subacute TGFbeta exposure drives airway hyperresponsiveness in cystic fibrosis mice through the PI3K pathway. Am. J. Respir. Cell Mol. Biol 62, 657–667. 10.1165/rcmb.2019-0158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kramer EL, and Clancy JP (2018). TGFbeta as a therapeutic target in cystic fibrosis. Expert Opin. Ther. Targets 22, 177–189. 10.1080/14728222.2018.1406922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bruscia EM, Zhang PX, Barone C, Scholte BJ, Homer R, Krause DS, and Egan ME (2016). Increased susceptibility of Cftr−/− mice to LPS-induced lung remodeling. Am. J. Physiol. Lung Cell Mol. Physiol 310, L711–L719. 10.1152/ajplung.00284.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, and Koller BH (1992). An animal model for cystic fibrosis made by gene targeting. Science 257, 1083–1088. [DOI] [PubMed] [Google Scholar]
- 46.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV Jr., Broxmeyer HE, and Charo IF (1997). Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest 100, 2552–2561. 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, and Perlman H (2013). Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol 49, 503–510. 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado J.d.D., Popovich PG, Partida-Sanchez S, and Guerau-de-Arellano M (2015). Novel markers to delineate murine M1 and M2 macrophages. PLoS One 10, e0145342. 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schyns J, Bai Q, Ruscitti C, Radermecker C, De Schepper S, Chakarov S, Farnir F, Pirottin D, Ginhoux F, Boeckxstaens G, et al. (2019). Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat. Commun 10, 3964. 10.1038/s41467-019-11843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frija-Masson J, Martin C, Regard L, Lothe MN, Touqui L, Durand A, Lucas B, Damotte D, Alifano M, Fajac I, and Burgel PR (2017). Bacteria-driven peribronchial lymphoid neogenesis in bronchiectasis and cystic fibrosis. Eur. Respir. J 49, 1601873. 10.1183/13993003.01873-2016. [DOI] [PubMed] [Google Scholar]
- 51.Serbina NV, and Pamer EG (2006). Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol 7, 311–317. 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 52.Darrah RJ, Mitchell AL, Campanaro CK, Barbato ES, Litman P, Sattar A, Hodges CA, Drumm ML, and Jacono FJ (2016). Early pulmonary disease manifestations in cystic fibrosis mice. J. Cyst. Fibros 15, 736–744. 10.1016/j.jcf.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma R, Florea VG, Bolger AP, Doehner W, Florea ND, Coats AJ, Hodson ME, Anker SD, and Henein MY (2001). Wasting as an independent predictor of mortality in patients with cystic fibrosis. Thorax 56, 746–750. 10.1136/thorax.56.10.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Heeckeren AM, Tscheikuna J, Walenga RW, Konstan MW, Davis PB, Erokwu B, Haxhiu MA, and Ferkol TW (2000). Effect of Pseudomonas infection on weight loss, lung mechanics, and cytokines in mice. Am. J. Respir. Crit. Care Med 161, 271–279. [DOI] [PubMed] [Google Scholar]
- 55.Zhang PX, Cheng J, Zou S, D’Souza AD, Koff JL, Lu J, Lee PJ, Krause DS, Egan ME, and Bruscia EM (2015). Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway ameliorates cystic fibrosis lung hyper-inflammation. Nat. Commun 6, 6221. 10.1038/ncomms7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lino Cardenas CL, Henaoui IS, Courcot E, Roderburg C, Cauffiez C, Aubert S, Copin MC, Wallaert B, Glowacki F, Dewaeles E, et al. (2013). miR-199a-5p Is upregulated during fibrogenic response to tissue injury and mediates TGFbeta-induced lung fibroblast activation by targeting caveolin-1. PLoS Genet. 9, e1003291. 10.1371/journal.pgen.1003291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghosh AK, and Vaughan DE (2012). PAI-1 in tissue fibrosis. J. Cell. Physiol 227, 493–507. 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu X, Buttgereit A, Lelios I, Utz SG, Cansever D, Becher B, and Greter M (2017). The cytokine TGF-beta promotes the development and homeostasis of alveolar macrophages. Immunity 47, 903–912.e4. 10.1016/j.immuni.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 59.Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, and Lambrecht BN (2013). Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med 210, 1977–1992. 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freedman SD, Blanco PG, Zaman MM, Shea JC, Ollero M, Hopper IK, Weed DA, Gelrud A, Regan MM, Laposata M, et al. (2004). Association of cystic fibrosis with abnormalities in fatty acid metabolism. N. Engl. J. Med 350, 560–569. 10.1056/NEJMoa021218. [DOI] [PubMed] [Google Scholar]
- 61.Fang D, West RH, Manson ME, Ruddy J, Jiang D, Previs SF, Sonawane ND, Burgess JD, and Kelley TJ (2010). Increased plasma membrane cholesterol in cystic fibrosis cells correlates with CFTR genotype and depends on de novo cholesterol synthesis. Respir. Res 11, 61. 10.1186/1465-9921-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, and Abdellatif M (2009). Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res 104, 879–886. 10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alexander MS, Kawahara G, Motohashi N, Casar JC, Eisenberg I, Myers JA, Gasperini MJ, Estrella EA, Kho AT, Mitsuhashi S, et al. (2013). MicroRNA-199a is induced in dystrophic muscle and affects WNT signaling, cell proliferation, and myogenic differentiation. Cell Death Differ. 20, 1194–1208. 10.1038/cdd.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pang J, Maienschein-Cline M, and Koh TJ (2021). Enhanced proliferation of Ly6C(+) monocytes/macrophages contributes to chronic inflammation in skin wounds of diabetic mice. J. Immunol 206, 621–630. 10.4049/jimmunol.2000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sorio C, Montresor A, Bolomini-Vittori M, Caldrer S, Rossi B, Dusi S, Angiari S, Johansson JE, Vezzalini M, Leal T, et al. (2016). Mutations of cystic fibrosis transmembrane conductance regulator gene cause a monocyte-selective adhesion deficiency. Am. J. Respir. Crit. Care Med 193, 1123–1133. 10.1164/rccm.201510-1922OC. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y, Jiang M, Nouraie M, Roth MG, Tabib T, Winters S, Chen X, Sembrat J, Chu Y, Cardenes N, et al. (2019). GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 317, L510–L521. 10.1152/ajplung.00062.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, Herzog EL, Kang I, Pisani MA, Yu S, et al. (2019). GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell 178, 1231–1244.e11. 10.1016/j.cell.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sabatel C, Radermecker C, Fievez L, Paulissen G, Chakarov S, Fernandes C, Olivier S, Toussaint M, Pirottin D, Xiao X, et al. (2017). Exposure to bacterial CpG DNA protects from airway allergic inflammation by expanding regulatory lung interstitial macrophages. Immunity 46, 457–473. 10.1016/j.immuni.2017.02.016. [DOI] [PubMed] [Google Scholar]
- 69.Hussell T, and Bell TJ (2014). Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol 14, 81–93. 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- 70.Joshi N, Walter JM, and Misharin AV (2018). Alveolar macrophages. Cell. Immunol 330, 86–90. 10.1016/j.cellimm.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 71.Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, Di YP, Clay ME, Enelow RI, Dubin PJ, and Alcorn JF (2015). Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am. J. Physiol. Lung Cell Mol. Physiol 309, L158–L167. 10.1152/ajplung.00338.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parker D, Planet PJ, Soong G, Narechania A, and Prince A (2014). Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLoS Pathog. 10, e1003951. 10.1371/journal.ppat.1003951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yun JH, Lee S, Srinivasa P, Morrow J, Chase R, Saferali A, Xu Z, Cho M, Castaldi P, and Hersh CP (2022). An interferon-inducible signature of airway disease from blood gene expression profiling. Eur. Respir. J 59, 2100569. 10.1183/13993003.00569-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hey J, Paulsen M, Toth R, Weichenhan D, Butz S, Schatterny J, Liebers R, Lutsik P, Plass C, and Mall MA (2021). Epigenetic reprogramming of airway macrophages promotes polarization and inflammation in muco-obstructive lung disease. Nat. Commun 12, 6520. 10.1038/s41467-021-26777-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Solomon GM, Frederick C, Zhang S, Gaggar A, Harris T, Woodworth BA, Steele C, and Rowe SM (2013). IP-10 is a potential biomarker of cystic fibrosis acute pulmonary exacerbations. PLoS One 8, e72398. 10.1371/journal.pone.0072398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hodges CA, Cotton CU, Palmert MR, and Drumm ML (2008). Generation of a conditional null allele for Cftr in mice. Genesis 46, 546–552. 10.1002/dvg.20433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heung LJ, and Hohl TM (2019). Inflammatory monocytes are detrimental to the host immune response during acute infection with Cryptococcus neoformans. PLoS Pathog. 15, e1007627. 10.1371/journal.ppat.1007627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Teng KY, Han J, Zhang X, Hsu SH, He S, Wani NA, Barajas JM, Snyder LA, Frankel WL, Caligiuri MA, et al. (2017). Blocking the CCL2-CCR2 Axis using CCL2-neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol. Cancer Ther 16, 312–322. 10.1158/1535-7163.MCT-16-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Engler AE, Ysasi AB, Pihl RMF, Villacorta-Martin C, Heston HM, Richardson HMK, Thapa BR, Moniz NR, Belkina AC, Mazzilli SA, and Rock JR (2020). Airway-associated macrophages in homeostasis and repair. Cell Rep. 33, 108553. 10.1016/j.celrep.2020.108553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rock JR, Randell SH, and Hogan BLM (2010). Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech 3, 545–556. 10.1242/dmm.006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, Hoffmann E, Beyaert R, Saeys Y, Lambrecht BN, and Guilliams M (2016). Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity 44, 755–768. 10.1016/j.immuni.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 82.Rao S, Wright AKA, Montiero W, Ziegler-Heitbrock L, and Grigg J (2009). Monocyte chemoattractant chemokines in cystic fibrosis. J. Cyst. Fibros 8, 97–103. 10.1016/j.jcf.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 83.Brennan S, Sly PD, Gangell CL, Sturges N, Winfield K, Wikstrom M, Gard S, Upham JW, and Arest CF (2009). Alveolar macrophages and CC chemokines are increased in children with cystic fibrosis. Eur. Respir. J 34, 655–661. 10.1183/09031936.00178508. [DOI] [PubMed] [Google Scholar]
- 84.Bruscia EM, Zhang PX, Satoh A, Caputo C, Medzhitov R, Shenoy A, Egan ME, and Krause DS (2011). Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J. Immunol 186, 6990–6998. 10.4049/jimmunol.1100396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gabillard-Lefort C, Casey M, Glasgow AMA, Boland F, Kerr O, Marron E, Lyons AM, Gunaratnam C, McElvaney NG, and Reeves EP (2022). Trikafta rescues CFTR and lowers monocyte P2X7R-induced inflammasome activation in cystic fibrosis. Am. J. Respir. Crit. Care Med 205, 783–794. 10.1164/rccm.202106-1426OC. [DOI] [PubMed] [Google Scholar]
- 86.Jarosz-Griffiths HH, Scambler T, Wong CH, Lara-Reyna S, Holbrook J, Martinon F, Savic S, Whitaker P, Etherington C, Spoletini G, et al. (2020). Different CFTR modulator combinations downregulate inflammation differently in cystic fibrosis. Elife 9, e54556. 10.7554/eLife.54556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bratcher PE, Rowe SM, Reeves G, Roberts T, Szul T, Harris WT, Tirouvanziam R, and Gaggar A (2016). Alterations in blood leukocytes of G551D-bearing cystic fibrosis patients undergoing treatment with ivacaftor. J. Cyst. Fibros 15, 67–73. 10.1016/j.jcf.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hisert KB, Schoenfelt KQ, Cooke G, Grogan B, Launspach JL, Gallagher CG, Donnelly SC, Welsh MJ, Singh PK, McKone EF, and Becker L (2016). Ivacaftor-induced proteomic changes suggest monocyte defects may contribute to the pathogenesis of cystic fibrosis. Am. J. Respir. Cell Mol. Biol 54, 594–597. 10.1165/rcmb.2015-0322LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Di Pietro C, Oz HH, Zhang PX, Cheng EC, Martis V, Bonfield TL, Kelley TJ, Jubin R, Abuchowski A, Krause DS, et al. (2022). Recruitment of monocytes primed to express heme oxygenase-1 ameliorates pathological lung inflammation in cystic fibrosis. Exp. Mol. Med 54, 639–652. 10.1038/s12276-022-00770-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, et al. (2020). Defining trained immunity and its role in health and disease. Nat. Rev. Immunol 20, 375–388. 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, Krupnick AS, Gelman AE, Lavine KJ, and Kreisel D (2016). Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1, 87315. 10.1172/jci.insight.87315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Merad M, and Martin JC (2020). Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol 20, 355–362. 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Espinosa V, Jhingran A, Dutta O, Kasahara S, Donnelly R, Du P, Rosenfeld J, Leiner I, Chen CC, Ron Y, et al. (2014). Inflammatory monocytes orchestrate innate antifungal immunity in the lung. PLoS Pathog. 10, e1003940. 10.1371/journal.ppat.1003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scott HM, and Flynn JL (2002). Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: influence of dose on disease progression. Infect. Immun 70, 5946–5954. 10.1128/iai.70.11.5946-5954.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gurczynski SJ, Nathani N, Warheit-Niemi HI, Hult EM, Podsiad A, Deng J, Zemans RL, Bhan U, and Moore BB (2019). CCR2 mediates increased susceptibility to post-H1N1 bacterial pneumonia by limiting dendritic cell induction of IL-17. Mucosal Immunol. 12, 518–530. 10.1038/s41385-018-0106-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Konstan MW, Döring G, Heltshe SL, Lands LC, Hilliard KA, Koker P, Bhattacharya S, Staab A, and Hamilton A; Investigators and Coordinators of BI Trial 54345 (2014). A randomized double blind, placebo controlled phase 2 trial of BIIL 284 BS (an LTB4 receptor antagonist) for the treatment of lung disease in children and adults with cystic fibrosis. J. Cyst. Fibros 13, 148–155. 10.1016/j.jcf.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ScRNAseq and bulk RNAseq have been deposited on GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti mouse CD45 BUV395 clone 30-F11 | BD | Cat# 565967; RRID: AB_2651134 |
| Rat anti mouse CD38 FITC clone 90 | Invitrogen | Cat# 11-0381-82; RRID: AB_465024 |
| Rat anti mouse CCR2 PE clone SA3203G11 | BioLegend | Cat# 150610; RRID: AB_2616982 |
| Rat anti mouse CD11b PE-Cy7 clone M1/70 | Invitrogen | Cat# 25-0112-82; RRID: AB_469588 |
| Rat anti mouse CD64 APC clone X54-5/7.1 | BioLegend | Cat#139306; RRID: AB_11219391 |
| Rat anti mouse CD45 eFluor450 clone M1/69 | ThermoFisher Scientific | Cat# 48-0242-82; RRID: AB_1311169 |
| Rat anti mouse Ly6C clone AL-21 | BD | Cat# 563011; RRID: AB_2737949 |
| Rat anti mouse CD11c BV711 clone HL3 | BD | Cat# 563048; RRID: AB_2734778 |
| Rat anti mouse Ly6G AF700 clone 1A8 | BD | Cat# 551459; RRID: AB_394206 |
| Rat anti mouse Siglec-F PerCP-Cy5.5 clone E50-2440 | BD | Cat# 565526; RRID: AB_2739281 |
| Rat anti mouse I-A/I-E (MHC-II) APC/Fire 750 clone M5/114.15.2 | BioLegend | Cat# 107652; RRID: AB_2616729 |
| Rat anti mouse CD4 PerCP-Cy5.5 clone RM4-5 | BD | Cat# 561115; RRID: AB_393977 |
| Rat anti mouse CD8a FITC clone 53–6.7 | BioLegend | Cat# 100706; RRID: AB_312745 |
| Rat anti mouse CD45R/B220 PE-Cy7 clone RA3-6B2 | BD | Cat# 552772; RRID: AB_394458 |
| mouse anti-alpha smooth muscle actin clone 1A4 | Abcam | Cat# ab7817; RRID: AB_2865422 |
| rabbit mAB anti-SMAD2 clone D43B4 | Cell Signaling | Cat# 5339 |
| rabbit mAB anti-phospho-SMAD2 (Ser465/467) clone 138D4 | Cell Signaling | Cat# 3107 |
| rabbit anti-VINCULIN | Cell Signaling | Cat# 4650 |
| rabbit anti-p44/42 MAPK (Erk1/2) clone 137F5 | Cell Signaling | Cat# 4695 |
| rabbit anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) clone D13.14.4E | Cell Signaling | Cat# 4370 |
| rabbit mAB anti-CXCL2 clone 16H3L10 | invitrogen | Cat# 701126 |
| mouse anti-CCR2 clone 3G7 | Novus Biologicals | Cat# NBP2-35334 |
| mouse anti-CCR2 clone 7A7 | abcam | Cat# ab176390 |
| FcBlock | BD | Cat# 553141; RRID: AB_394656 |
| goat anti-rabbit IgG-HRP | Santa Cruz | Cat# sc-2004 |
| mouse anti-alpha smooth muscle Actin (aSMA) clone 1A4 | Abcam | Cat# ab7817 |
| rabbit Cy5 Tyramide | Akoya | Cat# SAT705A001 EA |
| Biotin Tyramide | Akoya | Cat# NEL705A001KT |
| rabbit envision secondary antibody | Agilent/Dako | Cat# K4001-8 |
| mouse envision secondary antibody | Agilent/Dako | Cat# K4005 |
| Alexa 750-Streptavidin | Thermofisher/Life Technologies | Cat# S21384 |
| Chemicals, peptides, and recombinant proteins | ||
| cOmplete Mini EDTA-free protease inhibitor cocktail | Roche | Cat# 11836170001 |
| PBS | Gibco | Cat# 10010023 |
| UltraPure Low Melting Point agarose | Invitrogen | Cat# 16520–050 |
| Lipopolysaccharides from Pseudomonas aeruginosa | Millipore Sigma | Cat# L9143 |
| CCR2 inhibitor RS 102895 | Cayman Chemical | Cat# 1173022-16-6 |
| Cell Lysis Buffer | Cell Signaling | Cat# 9803 |
| PhosSTOP | Roche | Cat# 4906837001 |
| 4–15% Mini PROTEAN TGX Gels | Bio-Rad | Cat# 4561086 |
| ECL Plus Western blotting system | GE Healthcare | Cat# RPN2132 |
| Superscript II Reverse Transcriptase | ThermoFisher | Cat# 18064–022 |
| RBC lysis buffer | eBioscience | Cat# 00-4300-54 |
| Critical commercial assays | ||
| active TGF-beta ELISA kit | BioLegend | Cat# 437707 |
| total TGF-beta ELISA kit | BioLegend | Cat# 436707 |
| V-PLEX Proinflammatory Panel 1 (mouse) | Mesoscale Discovery | Cat# K15048G-1 |
| V-PLEX Cytokine Panel 1 (mouse) | Mesoscale Discovery | K15245D-1 |
| Lung dissociation kit (mouse) | Mitenyi Biotec | Cat# 130-095-927 |
| miRNEasy Micro kit | Qiagen | Cat# 217084 |
| Agilent RNA6000 Pico Kit | Agilent | Cat# G2938-90046 |
| SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing | Takara Bio | Cat# 634890 |
| Nextera XT DNA Sample Preparation kit | Illumina | Cat# FC-131-1096 |
| miScript II RT kit | Qiagen | Cat# 218161 |
| Mouse CXCL2/MIP-2 DuoSet ELISA | R&D systems | Cat# DY452-05 |
| Deposited data | ||
| scRNAseq data | Schupp et al.26 | GEO: GSE145360 |
| Bulk RNAseq data | GEO | GEO: GSE217102 |
| Experimental models: Organisms/strains | ||
| Cftr knockout mice B6.129P2-KOCftrtm1UNC | Jackson Laboratory | Stock No. 002196 |
| CCR2 knockout mice B6.129S4-Ccr2tm1Ifc/J | Jackson Laboratory | Stock No. 004999 |
| Ccr2cre mice | Dr. Tobias Hohl; Heung and Hohl77 | N/A |
| cftrflox/flox mice | CF animal core facility at Case Western Reserve University | N/A |
| CD45.1 mice B6.SJL-Ptprca Pepcb/BoyJ | Jackson Laboratory | Stock No. 002014 |
| Software and algorithms | ||
| FACSDiva | BD | N/A |
| FlowJo | BD | N/A |
| Prism | GraphPad | N/A |
| enrichR package | https://maayanlab.cloud/Enrichr | N/A |
| edgeR package | https://bioconductor.org | N/A |
| Other | ||
| Nebulizer Pulmo-Aide Compressor | Natallergy | Cat# DRV5650D |
All data are available in the main text or the supplemental information.