

Abstract
Introduction
Saturated fatty acids (FAs) are the main component of high‐fat diets (HFDs), and high consumption has been associated with the development of insulin resistance, endoplasmic reticulum stress and mitochondrial dysfunction in neuronal cells. In particular, the reduction in neuronal insulin signaling seems to underlie the development of cognitive impairments and has been considered a risk factor for Alzheimer's disease (AD).
Methods
This review summarized and critically analyzed the research that has impacted the field of saturated FA metabolism in neurons.
Results
We reviewed the mechanisms for free FA transport from the systemic circulation to the brain and how they impact neuronal metabolism. Finally, we focused on the molecular and the physiopathological consequences of brain exposure to the most abundant FA in the HFD, palmitic acid (PA).
Conclusion
Understanding the mechanisms that lead to metabolic alterations in neurons induced by saturated FAs could help to develop several strategies for the prevention and treatment of cognitive impairment associated with insulin resistance, metabolic syndrome, or type II diabetes.
Keywords: energy metabolism, insulin resistance, mitochondrial dysfunction, neurodegeneration, palmitic acid, saturated fatty acids
Saturated fatty acids induce insulin resistance in neurons. Neuronal metabolism of palmitic acid alters mitochondrial function.
1. INTRODUCTION
Obesity is a growing health problem that has reached critical levels in recent decades. Epidemiologic studies from 2016 indicate that 13% of adults and 18% of children and adolescents under 19 years are obese worldwide, 1 and it is estimated that obesity can affect 1.12 billion persons by 2030. 2 This condition is primarily caused by nutritional imbalance associated with the high consumption of diets rich in fat and sugars and a sedentary lifestyle. Several studies have shown that HFD intake leads to elevated levels of saturated FAs in plasma and contributes to various health problems that reduce life expectancy and quality due to the development of chronic diseases such as insulin resistance, type II diabetes, metabolic syndrome and cardiovascular disease. 3 , 4 , 5 , 6 , 7 , 8 The HFD is characterized by a high content of saturated FA from animal fat, including myristate (C14:0), palmitate (C16:0), stearate (C18:0) and laurate (C12:0). 9 , 10 , 11 Currently, it is recognized that high intake of saturated fat causes metabolic alterations not only in the peripheral organs, but also in the central nervous system (CNS). Although the mechanisms and signalling pathways affected in the brain by exposure to different concentrations of saturated FAs are not completely known, there is evidence of changes in energy metabolism, 12 , 13 reduced insulin sensitivity, 14 , 15 , 16 increased ceramide production, 17 , 18 neuroinflammation 19 and reduced neuronal viability. 20 , 21 , 22 , 23 Therefore, the chronic consumption of a HFD is considered a significant risk factor for cognitive decline, pathological brain aging and even AD. 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31
Due to the complex nature of FA effects on the brain, in this review, we describe the current knowledge of the mechanisms involved in their transport from the circulation to the brain as well as the main metabolic routes activated in neurons, including evidence for neuronal β‐oxidation. Finally, we highlight the major mechanisms by which exposure to high levels of saturated FA can lead to neuronal dysfunction.
2. FATTY ACID UPTAKE IN THE BRAIN
Fatty acids are made up of a hydrocarbon chain with a terminal carboxyl group. They are classified as saturated when composed of single aliphatic chain bonds, monounsaturated with one double bond, and polyunsaturated with two or more double bonds. According to the chain length, FAs are divided into short‐chain FAs (2–4 carbon atoms), medium‐chain FAs (6–12 carbon atoms), and long‐chain FAs (14–18 carbon atoms). 32 , 33 , 34 The physiological role of FAs depends on their length: short‐chain FAs are immediately available as an energy source, medium‐chain FAs can act as growth factors, and long‐chain FAs can act as structural components of cellular membranes and energy stores. The high levels of circulating long‐chain FA have been largely associated with atherogenic and thrombogenic diseases. 35 , 36 , 37 In circulation, saturated FA concentrations are significantly increased in diabetic patients (350 μmol/L) compared with normal subjects (230 μmol/L). 38 Additionally, PA is also increased in the cerebrospinal fluid in obese patients with poor cognitive performance. 39
FAs travel in circulation mostly bound to albumin (95%), esterified into lipoproteins, and as unbound FAs (a very small proportion). 40 Several studies have shown that FAs are able to cross the blood–brain barrier (BBB) and enter the brain to be taken up by endothelial cells, glial cells and neurons. 41 Some experiments have shown that radio‐labelled FAs injected into the carotid artery of rats can be traced to neuronal cells, 42 supporting the notion that plasma levels of FAs impact the type and concentration of lipid contents in the brain. How FAs cross the BBB and, in general, how they traverse the plasma membranes remains an open question that has generated two lines of evidence: one is that FAs can cross by simple diffusion, and the other proposes that they require a transport system mediated by specific proteins. It is recognized that both mechanisms can operate, but the protein‐dependent mechanism seems to be the prominent one. 43 A number of FA transporters have been identified, including the FA transporter protein, which is composed of six isoforms with high homology between species (FATPs 1–6), 44 , 45 , 46 and the FA translocase CD36, which has a high affinity for long‐chain FA transport and is also a receptor for lipoproteins. 47 , 48 The main proteins of the FATP family are members 1–4, which are located at the membranes of endothelial cells at the BBB and in grey matter. The transport through these proteins is dependent on chain length and the presence of double bonds. 46 , 47 , 49 Once in the brain, short‐ and medium‐chain FAs enter the cell by a flip‐flop mechanism, while long‐chain FAs need FATPs to enter in a nonionized form. Similar to the BBB, neuronal FA uptake is specific and chain length‐dependent transport. Once FAs are transported to the cells, they bind to membrane or cytoplasmic FA binding proteins (FABPs) that help to organize them into specific intracellular domains for further utilization in different metabolic routes. 50 For example, cytosolic fatty acid‐binding protein 3 (FABP3) is involved in arachidonic acid neuronal uptake but not in PA transport. 51 , 52 , 53 CD36, now designated scavenger receptor B2, 54 , 55 has been found in endothelial, glial, and neuronal cells not only at the cell membrane, but also in intracellular compartments. It is thought that CD36 participates in FA dissociation from albumin, and it has been placed in the context of various mechanisms related to physiological and pathological lipid metabolism. In hypothalamic neurons, CD36 acts as part of a lipid‐sensing mechanism for the control of food intake 56 and participates in the neurovascular dysfunction associated with AD. 57
3. ARE NEURONS ABLE TO METABOLIZE FATTY ACIDS TO PRODUCE ENERGY?
It is known that 20% of the total energy requirement in the adult brain is obtained from the oxidation of FA. 58 It was also long believed that astrocytes are the only brain cells able to metabolize FA and produce 14CO2 as an indicator of the β‐oxidation process. 59 , 60 , 61 , 62 , 63 , 64 However, it was reported that isolated neuronal mitochondria can oxidize palmitoyl carnitine in the presence of metabolic substrates. 65 Neurons possess the necessary machinery for fatty acid β‐oxidation, and they also express long‐chain fatty acid acyl‐CoA synthetase, carnitine palmitoyltransferase Ia and c (CPT1a and CPT1c), and mitochondrial uncoupling protein 2 (UCP2). In fact, the CPT1c form is highly expressed in neurons. 66 , 67 Although the function of CPT1c is not yet clear, metabolomic analysis has revealed that it could play an alternative role in neuronal oxidative metabolism. 68 , 69 , 70 , 71 , 72 Interestingly, some studies have shown that neuronal deficiency of this enzyme is associated with neurodegenerative diseases. 68 , 69 , 70 , 71 , 72 Currently, it is proposed that neurons present low levels of β‐oxidation due to the limited activity of their mitochondrial enzymes. Comparing neurons with other cells of the periphery, the subsequent β‐oxidation enzymes have low activity. For example, the 3‐ketoacyl‐coenzyme A thiolase has only 0.7% activity, acyl‐CoA dehydrogenase 50%, and enoyl‐CoA‐dehydrogenase 19%. At the same time, the activity of the mitochondrial respiratory chain in neurons is lower than that in other brain cells, such as astrocytes. For this reason, ATP production from FAs is scarce in neurons, and when FAs must be oxidized in the mitochondria by a metabolic situation, this metabolic route produces high ROS levels, making neurons susceptible to oxidative damage. 73 , 74 , 75 However, in some specific neurons, such as photoreceptors in the retina, FA oxidation seems to be the major source of energy to satisfy high metabolic demands. 76 It was also reported in cultured rat cortical neurons that the inhibition of fatty acid synthase (FAS) leads to a decrease in the levels of ATP and activation of AMP‐dependent kinase (AMPK) as well as CPT1; such effects result in increased fatty acid oxidation to restore ATP levels sufficient to sustain neuronal activity and survival. 77 FAS catalyses the condensation of acetyl‐CoA and malonyl‐CoA to generate long‐chain fatty acids and is highly expressed in neurons in different brain regions, including hypothalamic neurons that regulate feeding behaviour and systemic glucose levels. 78 In a recent transcriptomic study, it was demonstrated that neurons can respond to high but not toxic concentrations of PA, increasing the expression of several genes involved in lipid and energy metabolism. 79 Together, these data indicate that astrocytes are not only brain cells able to metabolize saturated FAs through β‐oxidation but also neurons can metabolize them under certain conditions.
Similar to other cells, neurons have another option to metabolize FAs. While short‐, medium‐ and long‐chain FAs are β‐oxidized in mitochondria, very‐long‐chain FAs (26:0) are metabolized in peroxisomes. 80 The transport of FAs toward peroxisomes is mediated by the ABC transporter family through cycles of ATP binding and hydrolysis. 81 There is also another mechanism of carnitine‐dependent transport of FAs in peroxisomes with less participation. 82 FA oxidation in peroxisomes produces acetyl‐CoA or propionyl‐CoA, which are further metabolized in the mitochondria. Peroxisome‐mediated β‐oxidation has been proposed to be part of a homeostatic mechanism for hyperactive neurons to produce enough ATP to sustain neuronal functions. In summary, there is growing evidence that neurons can metabolize FAs by mitochondria or peroxisomes, depending on the chain length and energy requirements. In addition, if neurons increase their energy demands, neuron‐astrocyte metabolic coupling results in the best option to maintain energy homeostasis. Interestingly, neuron‐astrocyte coupling‐dependent FA detoxification has been also reported to prevent injury in the CNS. In cultured hippocampal neurons during excitotoxic stimulation with N‐methyl‐D‐aspartate (NMDA), saturated FAs are oxidized, and the resulting damaging oxidized FAs are released from neurons and taken up by astrocytes through a carrier protein. Then, these FAs are stored in lipid droplets and consumed for energy supply to neurons. 83 However, the neuronal energy imbalance is sometimes not resolved by the astrocyte metabolic support, as has been recently demonstrated after CNS damage, wherein reactive astrocytes can release toxic FAs that may contribute to neuronal damage. 84
4. ALTERATIONS IN NEURONAL METABOLISM ASSOCIATED WITH INTAKE OF SATURATED FAT
There is a strong correlation between chronic intake of HFD and the development of neuroinflammation and brain insulin resistance. The last effect was reported to be dependent on the increased phosphorylation of residue S307 of insulin receptor substrate‐2 in the hypothalamus 85 and a reduction in the activation of residue Y608 of insulin receptor substrate‐1 in hippocampal neurons. 25 According to positron emission tomography measurements using [11C]‐palmitate and [18F]‐fluoro‐6‐thia‐heptadecanoic acid in obese patients with metabolic syndrome, increased uptake and accumulation of FAs in different brain regions was found in comparison with healthy subjects. 86 The brain accumulation of FAs can be explained by the obesity‐induced increase in the transporter FTP1, as was found in the prefrontal cortex in rats fed a HFD. 87 Experiments in rats have shown that the increased levels of FAs in the brain result in metabolic changes consisting of lower brain glucose uptake and glucose transporters and alterations in glycolytic and acetate metabolism and central insulin resistance. 88 , 89 Insulin resistance could interfere with brain glucose utilization, resulting in a compensatory increase in saturated FA uptake and oxidation. The described metabolic abnormalities impact neuronal morphology and physiology, manifesting as decreased long‐term potentiation and reduced markers of synaptic plasticity. 25 , 89 To explain some of the brain alterations produced by the high intake of saturated fat, several mechanisms have been analysed and proposed as potential drivers of brain pathology. Among them, mitochondrial dysfunction, neuroinflammation and oxidative damage are prominent. 90 In fact, HFD intake directly enhances ROS generation 91 accompanied with elevated expression of NADPH oxidase enzyme. 92 It was reported that consumption of a HFD reduces the levels of the mitochondrial fusion protein mitofusin 2 (MFN2) in hypothalamic neurons, resulting in loss of mitochondrial‐ER contacts and leading to ER stress and the development of leptin resistance. 93 Mitochondrial‐ER contacts regulate mitochondrial shape and motility; thus, the loss of these contacts produce mitochondrial dysfunction and alters energy metabolism and the cellular redox state, inducing autophagy and inflammasome signalling. 94 , 95 These effects were corroborated in C57BL/6 mice fed a HFD, which presented a decrease in MFN2 expression in the arcuate nucleus. 96 In addition, the mitochondrial dysfunction caused by the content of saturated FAs in the HFD also reduced the mitochondrial‐dependent Ca2+ uptake capacity that was accompanied by a decrease in hypothalamic neuronal excitability and consequent impaired function of energy control in the hypothalamus during obesity. 97 Moreover, HFD alters the hypothalamus‐dependent regulation of body weight, changing the brain expression of diverse neuromodulators, such as neuropeptide Y (NPY), orexins and proopiomelanocortin (POMC). 98 , 99
5. PALMITIC ACID: THE MAIN SATURATED FATTY ACID UNDERLYING THE ADVERSE EFFECTS OF HFD IN NEURONS
PA is a 16‐carbon long‐chain saturated FA that is the most abundant saturated FA in the human body (65% of saturated FAs) and represents the main component of HFD. PA can be provided by the diet from vegetables (10%–40%) and animal fat (20–30%) 100 or synthesized from amino acids, carbohydrates and other fatty acids in peripheral cells. 101 , 102 , 103 An increase in circulating levels of PA has been considered the responsible factor for the development of several conditions, such as type II diabetes, cardiovascular diseases, pro‐metastatic activity and cognitive decline. 38 , 102 , 104 , 105 , 106 In the brain, PA follows different metabolic routes, some of which could be associated with neuronal dysfunction. Among these, ceramide synthesis has been proven to have a causal role in insulin resistance and neuroinflammation.
5.1. Ceramide synthesis and neurotoxicity
Ceramides are signalling molecules involved in neuronal development, neuronal death and cellular senescence. De novo ceramide synthesis is controlled by the availability of palmitoyl‐CoA, which activates the rate‐limiting enzyme serine‐palmitoyl transferase. 107 Depending on chain length, different species of ceramides are produced and serve different roles in cellular homeostasis, with ceramide 16 (C16) being the most involved in apoptosis. 108 Exposure to a high concentration of PA induced the intracellular accumulation of C16 accompanied by proinflammatory cytokine production in cultured neurons. 18 Similarly, PA activates the enzyme serine‐palmitoyl transferase, which is involved in the accumulation of ceramides in astrocytes, enhancing the release of cytokines and activating a signalling cascade in neurons that upregulates the pro‐amyloid enzyme BACE‐1. 109 In peripheral cells, C24 and C16 also participate in the development of insulin resistance through PP2A‐dependent dephosphorylation and inactivation of Akt. 110 Thus, the production of ceramides can be a critical mechanism linking the development of neuroinflammation and insulin resistance under high PA concentrations in the CNS.
5.2. Neuronal insulin resistance
The role of insulin in neuronal function as a metabolic, growth, synaptic and survival modulator has been extensively validated. 111 , 112 , 113 Although neuronal glucose uptake is not dependent on insulin, the insulin/PI3K/Akt pathway regulates the expression of GLUT3 transporter and the glycolytic enzyme phosphofructokinase‐1 in neurons. 114 After high neuronal firing rates, the activation of insulin signalling induces GLUT4 translocation to the membrane to increase glucose transport in hippocampal neurons. Thus, alterations in the insulin/PI3K/Akt pathway can also be associated with dysregulation of the energy balance and glucose homeostasis. 115 Similarly, as it occurs in the peripheral organs, PA also contributes to the development of insulin resistance in neurons. 13 , 16 , 116 , 117 , 118 , 119 As previously stated, there is evidence that neurons can metabolize saturated FAs to produce energy, although it is still unknown under which conditions they can be used for this purpose. Recently, it has been reported for both, cultured rat cortical neurons and differentiated human neuroblastoma cells that there is a significant reduction in NAD+ contents and an increase in ATP levels after exposure to high but not toxic doses of PA, suggesting that neurons can utilize PA as an energy substrate when exposed to high concentrations of saturated FAs. 13 , 16 , 120 The reduction in NAD+ was correlated with the blunted effects of PA on insulin‐induced metabolic activation as well as with the inhibition of the insulin/PI3K/Akt pathway. The participation of ROS production in this effect was demonstrated by the inhibition of mitochondrial ROS with the mitochondria‐targeted antioxidant mitoTEMPO, which prevented PA‐dependent insulin resistance. 16 PA‐dependent insulin resistance was also associated with the translocation of protein kinase C‐θ (PKC‐θ) toward the cell membrane, which leads to the phosphorylation of the insulin receptor in inhibitory residues in hypothalamic neurons. 14 Another mechanism involved in PA‐induced insulin resistance in neurons is the activation of MyD88, an essential signalling adaptor for most toll‐like receptors (TLRs) and members of the interleukin‐1 (IL‐1) receptor family. The activation of MyD88 by PA leads to a TLR‐4‐dependent inflammatory response that induces not only insulin resistance but also leptin resistance, which exacerbates the energy imbalance in neurons. 121 , 122 , 123 The PA‐induced loss of insulin sensitivity seems to also be associated with PA‐dependent ATP production that mediates the opening of voltage‐dependent Ca2+ channels. The resulting increase in intraneuronal Ca2+ concentrations activates Ca2+‐dependent cPKC, which phosphorylates Akt in inhibitory residues, leading to its inactivation. 13 The loss of insulin sensitivity induced by PA may explain the strong association between the intake of saturated fat and the increased risk of neuronal dysfunction (Figure 1).
FIGURE 1.
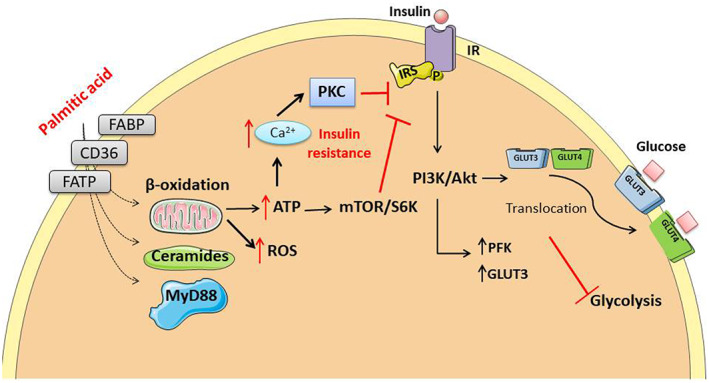
Palmitic acid causes insulin resistance by several mechanisms that include ROS and ceramide production as well as activation of MyD88 through PKCθ translocation to the cell membrane, which leads to internalization of the insulin receptor. Insulin resistance contributes to the impairment of glycolysis by decreasing the synthesis and activity of phospho‐fructose‐kinase and GLUT3 and impairing the translocation to the membrane of the glucose transporter, GLUT4.
5.3. Impairment of mitochondrial function in neurons
In addition to the above effects, PA exerts damaging consequences through enhances mitochondrial oxidative stress. PA treatment increased the production of superoxide dismutase after 8 h of exposure and decreased the production of ATP at 24 h, suggesting mitochondrial damage that correlates with the development of insulin resistance in neuroblastoma Neuro‐2a cells. 15 A similar effect was observed in differentiated human neuroblastoma cells, in which treatment with PA for 1 h generated sustained ROS production that inhibited insulin signalling and insulin‐dependent mitochondrial activation. 16 On the contrary, the exposure of hypothalamic cells to PA decreased the levels of the mitochondrial protein MFN2, ER stress and insulin resistance. 96 Furthermore, in dorsal root ganglion (DRG) neuronal cultures exposed to PA for 24 h, a marked reduction in mitochondrial membrane potential and changes in ATP production were observed and accompanied by altered mitochondrial morphology and impaired mitochondrial trafficking. 124 , 125 , 126 Overall, PA exposure affects neuronal metabolism, impairs mitochondrial function by loss of membrane potential and induces changes in mitochondrial morphology that result in inhibition of mitochondrial dynamics (Figure 2).
FIGURE 2.
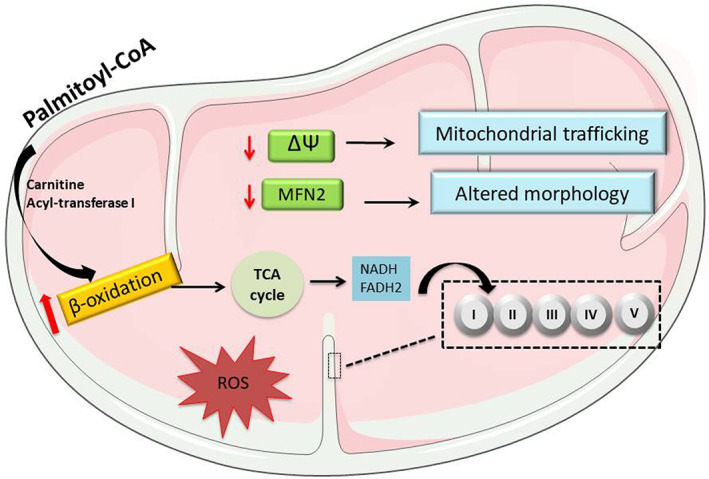
The possible route for β‐oxidation of palmitic acid in neurons is associated with the reduction of the mitochondrial fusion protein MFN2, decreased membrane potential, and ATP production as well as an increment in ROS levels. These conditions lead to decreased mitochondrial dynamics and bioenergetic capacity.
How might lipid‐overloaded mitochondria induce mitochondrial stress and insulin resistance? Although this question has not been fully resolved, it has been proposed that chronic elevation of fatty acids leads to persistent pressure on the electron transport chain, resulting in disruption of redox balance and ROS‐signalling. 127 Since most studies have been conducted in peripheral cells, further research is necessary to understand the connection between mitochondrial overload, redox imbalance and insulin resistance in neurons chronically exposed to saturated fatty acids.
5.4. Endoplasmic reticulum stress
It has been demonstrated that HFDs trigger excessive endoplasmic reticulum (ER) stress and exert opposite influences on the expression of plasticity‐related proteins such as BDNF, synaptophysin and NMDA receptors in the rat prefrontal cortex. 87 It is possible that many deleterious effects of HFD are exerted through its PA content, in view of the fact that in vivo and in vitro models have demonstrated that PA elicits the unfolded protein response and ER stress, is involved in the downregulation of leptin and insulin‐like growth factor receptor 1 expression in neurons 128 and activates autophagy and apoptotic pathways. Accordingly, in cultured hypothalamic neurons, lipotoxicity induced by high doses of PA (0.7–1 mM) was accompanied by the activation of the ER stress pathway leading to the phosphorylation of the initiator of translation, eIF2α, and activation of the cleaved enzyme caspase‐3, an apoptotic effector molecule. 114 Interestingly, some of the toxic effects of PA are prevented by the coadministration of the monounsaturated FA oleate. 15 The protective role of oleate against PA‐induced apoptosis is probably mediated by conducting PA to incorporate into triglycerides and in this way to form a storage pool of lipid droplets, as shown in CHO cells and embryonic fibroblasts. 129
6. ROLE OF GPR40 SIGNALLING ACTIVATION BY FATTY ACIDS
In addition to the consequences of the metabolic oxidation of PA on neurons, other effects can also be carried out through the activation of free fatty acid metabotropic receptors (FFARs). FFARs are G‐protein coupled receptors (GPCRs) located in the cell membrane that are mainly involved in metabolic regulation. FFARs are a family of four members: FFAR2 (also called GPR43) and FFAR3 (GPR41) are activated by short‐chain FFAs, whereas FFAR1 (GPR40) and FFAR4 (GPR120) are activated by medium‐ and long‐chain FFAs. 130 , 131 , 132 , 133 , 134 Interestingly, GPR40 is highly expressed in neurons and is activated by docosahexaenoic acid (DHA) and selective agonists, but few studies have shown that PA activates these receptors in neurons. 13 , 135 , 136
Activation of GPR40 regulates insulin secretion in pancreatic β‐cells, but its role in the CNS is not yet clear. 137 , 138 , 139 , 140 , 141 It has been found that GPR40 activation by DHA protects against the adverse effects of neuroinflammation and insulin resistance in the brain. 142 Additionally, signalling through GPR40 was found to be decreased in mice fed a HFD that developed cognitive deficits, but when GPR40 was activated by DHA or by its synthetic agonist, GW9508, improvements in cognitive functions resulted. 143 Conversely, in hypothalamic neurons, the activation of GPR40 by PA contributes to the development of insulin resistance. 136 In one study, in a human neuroblastoma model (SK‐N‐MC), it was found that PA‐mediated GPR40 signalling increased the expression of amyloid precursor protein (APP) and the catalytic enzyme BACE1, producing Aβ peptide through mTOR/p70S6K1‐mediated HIF‐1α expression and NF‐κB activation. 144
In general, polyunsaturated FAs prevent and/or reverse the metabolic alterations induced by saturated FAs and manifest beneficial effects in neurodegenerative diseases. 145 , 146 , 147 , 148 Unlike PA, DHA is considered a positive modulator of the insulin pathway and promotes neuronal protection and survival. 149 Due to its widely recognized influence on neuronal protection, DHA presents a compelling opportunity for the use of this nutritional therapy to counter the deleterious effects of saturated fat (Figure 3).
FIGURE 3.
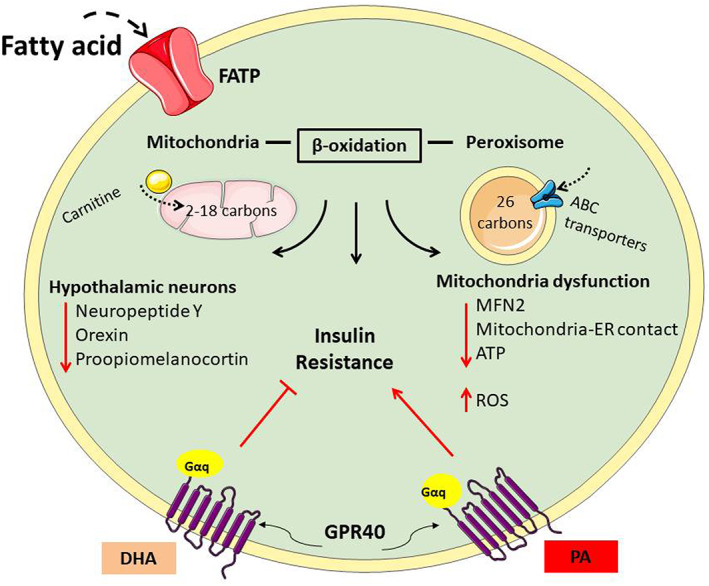
Fatty acids enter the brain and cells through FATPs. Within the cells, fatty acids (FAs) can be β‐oxidized by mitochondria (for FAs 2‐18 carbon atoms in length) or by peroxisomes (for 28‐carbon FAs). Palmitic acid metabolism can impact the hypothalamic energy sensing neurons, resulting in a loss of energy homeostasis. FAs can also activate GPR40 signalling associated with insulin resistance, but docosahexaenoic acid‐dependent activation of GPR40 improves insulin signalling.
7. ASSOCIATION BETWEEN FATTY ACID METABOLISM AND ALZHEIMER'S DISEASE
Comorbidities associated with a high intake of saturated fat, such as obesity and type II diabetes, are risk factors for the development of cognitive impairments and even AD. 150 , 151 , 152 , 153 , 154 , 155
In fact, new evidence supports the relationship between obesity and dementia from a review of 19 longitudinal studies including people aged 35–65 years. 156 AD is a neurodegenerative condition characterized by two hallmarks: the formation of amyloid plaques and the intraneuronal accumulation of neurofibrillary tangles by hyperphosphorylation of the cytoskeletal associated protein tau. 157 , 158 , 159 , 160 , 161 Amyloid‐β protein (Aβ) originates from APP by the sequential enzymatic actions of β‐secretase and γ‐secretase. 162 , 163 , 164 , 165 The presence of high levels of saturated FAs and cholesterol in membrane lipid rafts was observed to contribute to the amyloidogenic processing of APP in APP/PSEN1 transgenic mice 166 , 167 and in astrocytes. 168 Similarly, rats fed a HFD also showed increased APP and Aβ contents in the rat cerebral cortex, 169 increased APP and BACE‐1 expression in the mouse brain 144 and induced tau hyperphosphorylation in the rat hippocampus 25 through the formation of an enzymatic complex that activates the enzyme GSK3β. 170 Studies with 3xTg‐AD model have demonstrated that HFD treatment induces cognitive decline with or without increased Aβ levels. 171 , 172 In other AD model, the knock‐in mouse App NL−F/NL−F, long‐term HFD intake was associated with insulin resistance, poor cognitive performance, increased deposition of Aβ and the presence of neuroinflammation and oxidative stress markers. 173 Current research has shown an upregulation of BACE‐1 after PA exposure in cultured hippocampal neurons by a mechanism depending on the reduced activity of the deacetylase sirtuin 1 120 and through PA‐mediated transcriptional activation of BACE‐1 in neurons after exposure to astrocyte‐conditioned media. 109 Interestingly, a correlation between differential FA metabolism in specific brain areas and the development of some biochemical markers of AD has been shown. For example, cortical neurons exposed to conditioned medium obtained from PA‐treated cortical astrocytes expressed tau phosphorylation and BACE1, but not when neurons were exposed to cerebellar astrocyte media. 174
These data demonstrate the effects of brain exposure to high levels of saturated FAs and point to the connection between lipid dyshomeostasis and the risk for AD.
8. CONCLUSIONS
The consumption of saturated FAs is strongly associated with morphological and functional changes in neurons. Accumulating evidence describes consistent dysregulation of neuronal metabolism induced by PA that leads to insulin resistance, decreased glycolysis, altered mitochondrial function and ER stress. These effects seem to contribute to cognitive decline. Recent interesting evidence supports the notion that in certain conditions, neurons can metabolize saturated long‐chain FAs through a metabolic energy pathway that sustains part of their deleterious effect. Understanding the mechanisms that lead to metabolic alterations in neurons could help to develop several strategies for the prevention, early detection and treatment of cognitive impairment associated with high consumption of saturated FAs or associated with insulin resistance, metabolic syndrome or diabetes. Although a variety of potential mechanisms have been explored, the underlying molecular cascade responsible for dietary fat‐induced neuronal dysfunction and behavioural changes remain elusive, and more research is needed to understand the signalling pathways that are activated in a specific metabolic context. An interesting avenue of studies to clarify the mechanisms downstream of the activation of GPR40 in neurons by polyunsaturated and saturated fatty acids is now open, as well as interrogation of the conditions that determine the metabolic routes that FAs follow into the brain to induce or avoid lipotoxicity.
AUTHOR CONTRIBUTIONS
Karina Sánchez‐Alegría: Conceptualization (equal); data curation (equal); formal analysis (equal); funding acquisition (supporting); investigation (equal); methodology (equal); project administration (supporting); resources (supporting); software (equal); supervision (supporting); validation (supporting); visualization (supporting); writing – original draft (equal); writing – review and editing (supporting). Clorinda Arias: Conceptualization (equal); data curation (equal); formal analysis (equal); funding acquisition (lead); investigation (equal); methodology (equal); project administration (lead); resources (equal); software (equal); supervision (lead); validation (lead); visualization (lead); writing – original draft (equal); writing – review and editing (lead).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVAL
The authors declare that neither animals nor human subjects were involved in the creation of this manuscript.
ACKNOWLEDGEMENTS
This work was supported by Consejo Nacional de Ciencia y Tecnología, A1‐S‐9559.
Sánchez‐Alegría K, Arias C. Functional consequences of brain exposure to saturated fatty acids: From energy metabolism and insulin resistance to neuronal damage. Endocrinol Diab Metab. 2023;6:e386. doi: 10.1002/edm2.386
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analysed in this study.
REFERENCES
- 1. World Health Organization . World health statistics 2021: monitoring health for the SDGs, sustainable development goals; 2021. Accessed May 20, 2021. https://www.who.int/publications/i/item/9789240027053
- 2. Kelly T, Yang W, Chen C‐S, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes. 2008;32(9):1431‐1437. [DOI] [PubMed] [Google Scholar]
- 3. Blázquez E, Quijada CL. The effect of a high‐fat diet on glucose, insulin sensitivity and plasma insulin in rats. J Endocrinol. 1968;42(4):489‐494. [DOI] [PubMed] [Google Scholar]
- 4. Stern J, Johnson P, Batchelor B, Zucker L, Hirsch J. Pancreatic insulin release and peripheral tissue resistance in Zucker obese rats fed high‐ and low‐carbohydrate diets. Am J Physiol Content. 1975;228(2):543‐548. [DOI] [PubMed] [Google Scholar]
- 5. Lavau M, Fried SK, Susini C, Freychet P. Mechanism of insulin resistance in adipocytes of rats fed a high‐fat diet. J Lipid Res. 1979;20(1):8‐16. [PubMed] [Google Scholar]
- 6. Boden G. Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc Assoc Am Physicians. 1999;111(3):241‐248. [DOI] [PubMed] [Google Scholar]
- 7. Vessby B. Dietary fat, fatty acid composition in plasma and the metabolic syndrome. Curr Opin Lipidol. 2003;14(1):15‐19. [DOI] [PubMed] [Google Scholar]
- 8. Sheehy T, Sharma S. The nutrition transition in Barbados: trends in macronutrient supply from 1961 to 2003. Br J Nutr. 2010;104(8):1222‐1229. [DOI] [PubMed] [Google Scholar]
- 9. Wang L, Folsom AR, Zheng Z‐J, Pankow JS, Eckfeldt JH, ARIC Study Investigators . Plasma fatty acid composition and incidence of diabetes in middle‐aged adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Clin Nutr. 2003;78(1):91‐98. [DOI] [PubMed] [Google Scholar]
- 10. Austin GL, Ogden LG, Hill JO. Trends in carbohydrate, fat, and protein intakes and association with energy intake in normal‐weight, overweight, and obese individuals: 1971–2006. Am J Clin Nutr. 2011;93(4):836‐843. [DOI] [PubMed] [Google Scholar]
- 11. Chen B, Huang Y, Zheng D, Ni R, Bernards M. Dietary fatty acids alter lipid profiles and induce myocardial dysfunction without causing metabolic disorders in mice. Nutrients. 2018;10(1):106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takahashi S, Iizumi T, Mashima K, Abe T, Suzuki N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro. 2014;6(5):175909141455099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sánchez‐Alegría K, Bastián‐Eugenio C, Vaca L, Arias C. Palmitic acid induces insulin resistance by a mechanism associated with energy metabolism and calcium entry in neuronal cells. FASEB J. 2021;35(7):e21712. [DOI] [PubMed] [Google Scholar]
- 14. Benoit SC, Kemp CJ, Elias CF, et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC‐theta subcellular localization in rodents. J Clin Invest. 2009;119(9):2577‐2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kwon B, Lee H‐K, Querfurth HW. Oleate prevents palmitate‐induced mitochondrial dysfunction, insulin resistance and inflammatory signaling in neuronal cells. Biochim Biophys Acta. 2014;1843(7):1402‐1413. [DOI] [PubMed] [Google Scholar]
- 16. Calvo‐Ochoa E, Sánchez‐Alegría K, Gómez‐Inclán C, Ferrera P, Arias C. Palmitic acid stimulates energy metabolism and inhibits insulin/PI3K/AKT signaling in differentiated human neuroblastoma cells: the role of mTOR activation and mitochondrial ROS production. Neurochem Int. 2017;110:75‐83. [DOI] [PubMed] [Google Scholar]
- 17. Patil S, Melrose J, Chan C. Involvement of astroglial ceramide in palmitic acid‐induced Alzheimer‐like changes in primary neurons. Eur J Neurosci. 2007;26(8):2131‐2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sergi D, Morris AC, Kahn DE, et al. Palmitic acid triggers inflammatory responses in N42 cultured hypothalamic cells partially via ceramide synthesis but not via TLR4. Nutr Neurosci. 2020;23(4):321‐334. [DOI] [PubMed] [Google Scholar]
- 19. Amine H, Benomar Y, Taouis M. Palmitic acid promotes resistin‐induced insulin resistance and inflammation in SH‐SY5Y human neuroblastoma. Sci Rep. 2021;11(1):5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Almaguel F, Liu J, Pacheco F, Casiano C, De Leon M. Activation and reversal of lipotoxicity in PC12 and rat cortical cells following exposure to palmitic acid. J Neurosci Res. 2009;87(5):1207‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duffy CM, Nixon JP, Butterick TA. Orexin a attenuates palmitic acid‐induced hypothalamic cell death. Mol Cell Neurosci. 2016;75:93‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schommer J, Marwarha G, Nagamoto‐Combs K, Ghribi O. Palmitic acid‐enriched diet increases α‐synuclein and tyrosine hydroxylase expression levels in the mouse brain. Front Neurosci. 2018;12:552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beaulieu J, Costa G, Renaud J, et al. The neuroinflammatory and neurotoxic potential of palmitic acid is mitigated by oleic acid in microglial cells and microglial‐neuronal co‐cultures. Mol Neurobiol. 2021;58(6):3000‐3014. [DOI] [PubMed] [Google Scholar]
- 24. Grant WB. Dietary links to Alzheimer's disease: 1999 update. J Alzheimer's Dis. 1999;1(4–5):197‐201. [DOI] [PubMed] [Google Scholar]
- 25. Calvo‐Ochoa E, Hernández‐Ortega K, Ferrera P, Morimoto S, Arias C. Short‐term high‐fat‐and‐fructose feeding produces insulin signaling alterations accompanied by neurite and synaptic reduction and astroglial activation in the rat hippocampus. J Cereb Blood Flow Metab. 2014;34(6):1001‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arnold SE, Lucki I, Brookshire BR, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis. 2014;67:79‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Busquets O, Ettcheto M, Pallàs M, et al. Long‐term exposition to a high fat diet favors the appearance of β‐amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer's disease. Mech Ageing Dev. 2017;162:38‐45. [DOI] [PubMed] [Google Scholar]
- 28. Snowden SG, Ebshiana AA, Hye A, et al. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: a nontargeted metabolomic study. PLoS Med. 2017;14(3):e1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chudoba C, Wardelmann K, Kleinridders A. Molecular effects of dietary fatty acids on brain insulin action and mitochondrial function. Biol Chem. 2019;400(8):991‐1003. [DOI] [PubMed] [Google Scholar]
- 30. McLean FH, Campbell FM, Langston RF, et al. A high‐fat diet induces rapid changes in the mouse hypothalamic proteome. Nutr Metab. 2019;16(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakagawa K, Islam S, Ueda M, Nakagawa T. Endoplasmic reticulum stress contributes to the decline in doublecortin expression in the immature neurons of mice with long‐term obesity. Sci Rep. 2022;12(1):1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nelson D, Cox M. Lipid biosynthesis. In: Nelson D, Cox MM, eds. Principles of Biochemistry. 4th ed. W.F. Freeman and Company; 2005:787‐815. [Google Scholar]
- 33. Tvrzicka E, Kremmyda L‐S, Stankova B, Zak A. Fatty acids as biocompounds: their role in human metabolism, health and disease‐‐a review. Part 1: classification, dietary sources and biological functions. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2011;155(2):117‐130. [DOI] [PubMed] [Google Scholar]
- 34. Bond LM, Miyazaki M, O'Neill LM, Ding F, Ntambi JM. Chapter 6. Fatty acid desaturation and elongation in mammals. In: Biochemestry of Lipids, Lipoproteins and Membranes. 6th ed. Elsevier Science; 2016:185‐208. [Google Scholar]
- 35. Agostoni C, Bruzzese MG. Fatty acids: their biochemical and functional classification. Pediatr Med Chir. 1992;14(5):473‐479. [PubMed] [Google Scholar]
- 36. Salati LM, Goodridge AG. Fatty acid synthesis in eukaryotes. New Compr Biochem. 1996;31:101‐127. [Google Scholar]
- 37. Berg J, Tymoczko J, Stryer L. Fatty acid metabolism. In: Stryer L, ed. Biochemistry. 5th ed. W.H. Freeman and Company; 2002. [Google Scholar]
- 38. Clore JN, Allred J, White D, Li J, Stillman J. The role of plasma fatty acid composition in endogenous glucose production in patients with type 2 diabetes mellitus. Metabolism. 2002;51(11):1471‐1477. [DOI] [PubMed] [Google Scholar]
- 39. Melo H, Seixas da Silva G, Sant'Ana M, et al. Palmitate is increased in the cerebrospinal fluid of humans with obesity and induces memory impairment in mice via pro‐inflammatory TNF‐α. Cell Rep. 2020;30(7):2180‐2194.e8. [DOI] [PubMed] [Google Scholar]
- 40. Richieri G, Kleinfeld A. Unbound free fatty acid levels in human serum. J Lipid Res. 1995;36(2):229‐240. [PubMed] [Google Scholar]
- 41. Hofmann K, Rodriguez‐Rodriguez R, Gaebler A, Casals N, Scheller A, Kuerschner L. Astrocytes and oligodendrocytes in grey and white matter regions of the brain metabolize fatty acids. Sci Rep. 2017;7(1):10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rapoport S, Chang M, Spector A. Delivery and turnover of plasma‐derived essential PUFAs in mammalian brain. J Lipid Res. 2001;42(5):678‐685. [PubMed] [Google Scholar]
- 43. Schwenk RW, Holloway GP, Luiken JJFP, Bonen A, Glatz JFC. Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot Essent Fatty Acids. 2010;82(4–6):149‐154. [DOI] [PubMed] [Google Scholar]
- 44. Spector R. Fatty acid transport through the blood‐brain barrier. J Neurochem. 1988;50(2):639‐643. [DOI] [PubMed] [Google Scholar]
- 45. Edmond J. Essential polyunsaturated fatty acids and the barrier to the brain: the components of a model for transport. J Mol Neurosci. 2001;16(2–3):181‐194. [DOI] [PubMed] [Google Scholar]
- 46. Mitchell RW, On NH, Del Bigio MR, Miller DW, Hatch GM. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem. 2011;117(4):735‐746. [DOI] [PubMed] [Google Scholar]
- 47. Mitchell RW, Hatch GM. Fatty acid transport into the brain: of fatty acid fables and lipid tails. Prostaglandins Leukot Essent Fatty Acids. 2011;85(5):293‐302. [DOI] [PubMed] [Google Scholar]
- 48. Ioghen O, Chițoiu L, Gherghiceanu M, Ceafalan LC, Hinescu ME. CD36 – a novel molecular target in the neurovascular unit. Eur J Neurosci. 2021;53(8):2500‐2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ochiai Y, Uchida Y, Ohtsuki S, Tachikawa M, Aizawa S, Terasaki T. The blood‐brain barrier fatty acid transport protein 1 (FATP1/SLC27A1) supplies docosahexaenoic acid to the brain, and insulin facilitates transport. J Neurochem. 2017;141(3):400‐412. [DOI] [PubMed] [Google Scholar]
- 50. Jang S, Choi B, Lim C, Lee B, Cho KS. Roles of drosophila fatty acid‐binding protein in development and behavior. Biochem Biophys Res Commun. 2022;599:87‐92. [DOI] [PubMed] [Google Scholar]
- 51. Hamilton JA. Transport of fatty acids across membranes by the diffusion mechanism. Prostaglandins Leukot Essent Fatty Acids. 1999;60(5–6):291‐297. [DOI] [PubMed] [Google Scholar]
- 52. Murphy EJ, Owada Y, Kitanaka N, Kondo H, Glatz JFC. Brain arachidonic acid incorporation is decreased in heart fatty acid binding protein gene‐ablated mice. Biochemistry. 2005;44(16):6350‐6360. [DOI] [PubMed] [Google Scholar]
- 53. Kamp F, Hamilton JA. How fatty acids of different chain length enter and leave cells by free diffusion. Prostaglandins Leukot Essent Fatty Acids. 2006;75(3):149‐159. [DOI] [PubMed] [Google Scholar]
- 54. Prabhudas M, Bowdish D, Drickamer K, et al. Standardizing scavenger receptor nomenclature. J Immunol. 2014;192(5):1997‐2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Glatz J, Luiken J. Dynamic role of the transmembrane glycoprotein CD36 (SR‐B2) in cellular fatty acid uptake and utilization. J Lipid Res. 2018;59(7):1084‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Le Foll C. Hypothalamic fatty acids and ketone bodies sensing and role of FAT/CD36 in the regulation of food intake. Front Physiol. 2019;10:1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Park L, Wang G, Zhou P, et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid‐beta. Proc Natl Acad Sci USA. 2011;108(12):5063‐5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ebert D, Haller RG, Walton ME. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci. 2003;23(13):5928‐5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Robert J, Montaudon D, Hugues P. Incorporation and metabolism of exogenous fatty acids by cultured normal and tumoral glial cells. Biochim Biophys Acta. 1983;752(3):383‐395. [DOI] [PubMed] [Google Scholar]
- 60. Yim SH, Monsma S, Hertz L, Szuchet S. Lipid and glycolipid metabolism of cultured astrocytes: a time course study. J Neurosci Res. 1986;15(1):29‐37. [DOI] [PubMed] [Google Scholar]
- 61. Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res. 1987;18(4):551‐561. [DOI] [PubMed] [Google Scholar]
- 62. Auestad N, Korsak RA, Morrow JW, Edmond J. Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J Neurochem. 1991;56(4):1376‐1386. [DOI] [PubMed] [Google Scholar]
- 63. Murphy MG, Jollimore C, Crocker JFS, Her H. Beta‐oxidation of [1‐14C]palmitic acid by mouse astrocytes in primary culture: effects of agents implicated in the encephalopathy of Reye's syndrome. J Neurosci Res. 1992;33(3):445‐454. [DOI] [PubMed] [Google Scholar]
- 64. Amruthesh SC, Boerschel MF, McKinney JS, Willoughby KA, Ellis EF. Metabolism of arachidonic acid to epoxyeicosatrienoic acids. Hydroxyeicosatetraenoic acids, and prostaglandins in cultured rat hippocampal astrocytes. J Neurochem. 1993;61(1):150‐159. [DOI] [PubMed] [Google Scholar]
- 65. Panov A, Orynbayeva Z, Vavilin V, Lyakhovich V. Fatty acids in energy metabolism of the central nervous system. Biomed Res Int. 2014;2014:472459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nalecz K, Miecz D, Berezowski V, Cecchelli R. Carnitine: transport and physiological functions in the brain. Mol Aspects Med. 2004;25(5–6):551‐567. [DOI] [PubMed] [Google Scholar]
- 67. Sierra AY, Gratacós E, Carrasco P, et al. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J Biol Chem. 2008;283(11):6878‐6885. [DOI] [PubMed] [Google Scholar]
- 68. Januszewicz E, Bekisz M, Mozrzymas JW, Nałęcz KA. High affinity carnitine transporters from OCTN family in neural cells. Neurochem Res. 2010;35(5):743‐748. [DOI] [PubMed] [Google Scholar]
- 69. Carrasco P, Sahún I, McDonald J, et al. Ceramide levels regulated by carnitine palmitoyltransferase 1C control dendritic spine maturation and cognition. J Biol Chem. 2012;287(25):21224‐21232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee J, Wolfgang MJ. Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism. BMC Biochem. 2012;13(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tonazzi A, Mantovani C, Colella M, Terenghi G, Indiveri C. Localization of mitochondrial carnitine/acylcarnitine translocase in sensory neurons from rat dorsal root ganglia. Neurochem Res. 2013;38(12):2535‐2541. [DOI] [PubMed] [Google Scholar]
- 72. Virmani A, Pinto L, Bauermann O, et al. The carnitine palmitoyl transferase (CPT) system and possible relevance for neuropsychiatric and neurological conditions. Mol Neurobiol. 2015;52(2):826‐836. [DOI] [PubMed] [Google Scholar]
- 73. Yang SY, He XY, Schulz H. Fatty acid oxidation in rat brain is limited by the low activity of 3‐ketoacyl‐coenzyme a thiolase. J Biol Chem. 1987;262(27):13027‐13032. [PubMed] [Google Scholar]
- 74. Speijer D. Oxygen radicals shaping evolution: why fatty acid catabolism leads to peroxisomes while neurons do without it. Bioessays. 2011;33(2):88‐94. [DOI] [PubMed] [Google Scholar]
- 75. Schönfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab. 2013;33(10):1493‐1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fu Z, Kern T, Hellström A, Smith L. Fatty acid oxidation and photoreceptor metabolic needs. J Lipid Res. 2021;62:10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Landree LE, Hanlon AL, Strong DW, et al. C75, a fatty acid synthase inhibitor, modulates AMP‐activated protein kinase to Alter neuronal energy metabolism. J Biol Chem. 2004;279(5):3817‐3827. [DOI] [PubMed] [Google Scholar]
- 78. Moullé VS, Le Foll C, Philippe E, et al. Fatty acid transporter CD36 mediates hypothalamic effect of fatty acids on food intake in rats. PLoS One. 2013;8(9):e74021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Flores‐León M, Alcaraz N, Pérez‐Domínguez M, et al. Transcriptional profiles reveal deregulation of lipid metabolism and inflammatory pathways in neurons exposed to palmitic acid. Mol Neurobiol. 2021;58(9):4639‐4651. [DOI] [PubMed] [Google Scholar]
- 80. Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75(1):295‐332. [DOI] [PubMed] [Google Scholar]
- 81. Morita M, Imanaka T. Peroxisomal ABC transporters: structure, function and role in disease. Biochim Biophys Acta. 2012;1822(9):1387‐1396. [DOI] [PubMed] [Google Scholar]
- 82. Visser WF, van Roermund CWT, Ijlst L, Waterham HR, Wanders RJA. Metabolite transport across the peroxisomal membrane. Biochem J. 2007;401(2):365‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ioannou MS, Jackson J, Sheu S‐H, et al. Neuron‐astrocyte metabolic coupling protects against activity‐induced fatty acid toxicity. Cell. 2019;177(6):1522‐1535.e14. [DOI] [PubMed] [Google Scholar]
- 84. Guttenplan K, Weigel M, Prakash P, et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature. 2021;599(7883):102‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. De Souza CT, Araujo EP, Bordin S, et al. Consumption of a fat‐rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146(10):4192‐4199. [DOI] [PubMed] [Google Scholar]
- 86. Karmi A, Iozzo P, Viljanen A, et al. Increased brain fatty acid uptake in metabolic syndrome. Diabetes. 2010;59(9):2171‐2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li F, Liu B, Cai M, Li J, Lou S. Excessive endoplasmic reticulum stress and decreased neuroplasticity‐associated proteins in prefrontal cortex of obese rats and the regulatory effects of aerobic exercise. Brain Res Bull. 2018;140:52‐59. [DOI] [PubMed] [Google Scholar]
- 88. Clegg DJ, Gotoh K, Kemp C, et al. Consumption of a high‐fat diet induces central insulin resistance independent of adiposity. Physiol Behav. 2011;103(1):10‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liu Z, Patil I, Sancheti H, Yin F, Cadenas E. Effects of lipoic acid on high‐fat diet‐induced alteration of synaptic plasticity and brain glucose metabolism: a PET/CT and 13C‐NMR study. Sci Rep. 2017;7(1):5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tracey TJ, Steyn FJ, Wolvetang EJ, Ngo ST. Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci. 2018;11:1‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Xu S, Nam SM, Kim J‐H, et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015;6(11):e1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang X, Dong F, Ren J, Driscoll MG, Culver B. High dietary fat induces NADPH oxidase‐associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol. 2005;191(2):318‐325. [DOI] [PubMed] [Google Scholar]
- 93. Schneeberger M, Dietrich MO, Sebastián D, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155(1):172‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843(10):2253‐2262. [DOI] [PubMed] [Google Scholar]
- 95. Carraro RS, Souza GF, Solon C, et al. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet‐induced obesity. Mol Cell Endocrinol. 2018;460:238‐245. [DOI] [PubMed] [Google Scholar]
- 96. Diaz B, Fuentes‐Mera L, Tovar A, et al. Saturated lipids decrease mitofusin 2 leading to endoplasmic reticulum stress activation and insulin resistance in hypothalamic cells. Brain Res. 2015;1627:80‐89. [DOI] [PubMed] [Google Scholar]
- 97. Paeger L, Pippow A, Hess S, et al. Energy imbalance alters Ca2+ handling and excitability of POMC neurons. Elife. 2017;6:e25641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dziedzic B, Szemraj J, Bartkowiak J, Walczewska A. Various dietary fats differentially change the gene expression of neuropeptides involved in body weight regulation in rats. J Neuroendocrinol. 2007;19(5):364‐373. [DOI] [PubMed] [Google Scholar]
- 99. Lemus MB, Bayliss JA, Lockie SH, et al. A stereological analysis of NPY, POMC, orexin, GFAP astrocyte, and Iba1 microglia cell number and volume in diet‐induced obese male mice. Endocrinology. 2015;156(5):1701‐1713. [DOI] [PubMed] [Google Scholar]
- 100. Murru E, Manca C, Carta G, Banni S. Impact of dietary palmitic acid on lipid metabolism. Front Nutr. 2022;9:861664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Carta G, Murru E, Lisai S, et al. Dietary triacylglycerols with palmitic acid in the sn‐2 position modulate levels of N‐acylethanolamides in rat tissues. Eckel J, ed. PLoS One. 2015;10(3):e0120424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mancini A, Imperlini E, Nigro E, et al. Biological and nutritional properties of palm oil and palmitic acid: effects on health. Molecules. 2015;20(9):17339‐17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Innis SM. Palmitic acid in early human development. Crit Rev Food Sci Nutr. 2016;56(12):1952‐1959. [DOI] [PubMed] [Google Scholar]
- 104. Trombetta A, Togliatto G, Rosso A, et al. Increase of palmitic acid concentration impairs endothelial progenitor cell and bone marrow–derived progenitor cell bioavailability: role of the STAT5/PPARγ transcriptional complex. Diabetes. 2013;62(4):1245‐1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Carta G, Murru E, Banni S, Manca C. Palmitic acid: physiological role, metabolism and nutritional implications. Front Physiol. 2017;8:902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pascual G, Domínguez D, Elosúa‐Bayes M, et al. Dietary palmitic acid promotes a prometastatic memory via Schwann cells. Nature. 2021;599(7885):485‐490. [DOI] [PubMed] [Google Scholar]
- 107. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9(2):139‐150. [DOI] [PubMed] [Google Scholar]
- 108. Czubowicz K, Strosznajder R. Ceramide in the molecular mechanisms of neuronal cell death. The role of sphingosine‐1‐phosphate. Mol Neurobiol. 2014;50(1):26‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu L, Martin R, Chan C. Palmitate‐activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol Aging. 2013;34(2):540‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ghosh N, Patel N, Jiang K, et al. Ceramide‐activated protein phosphatase involvement in insulin resistance via Akt, serine/arginine‐rich protein 40, and ribonucleic acid splicing in L6 skeletal muscle cells. Endocrinology. 2007;148(3):1359‐1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhao W, Chen H, Xu H, et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem. 1999;274(49):34893‐34902. [DOI] [PubMed] [Google Scholar]
- 112. Biessels GJ, Reagan LP. Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci. 2015;16(11):660‐671. [DOI] [PubMed] [Google Scholar]
- 113. Sánchez‐Alegría K, Flores‐León M, Avila‐Muñoz E, Rodríguez‐Corona N, Arias C. PI3K signaling in neurons: a central node for the control of multiple functions. Int J Mol Sci. 2018;19(12):3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Agostini M, Romeo F, Inoue S, et al. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016;23(9):1502‐1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pearson‐Leary J, Jahagirdar V, Sage J, McNay EC. Insulin modulates hippocampally‐mediated spatial working memory via glucose transporter‐4. Behav Brain Res. 2018;338:32‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mayer CM, Belsham DD. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: rescue of resistance and apoptosis through adenosine 5′ monophosphate‐activated protein kinase activation. Endocrinology. 2010;151(2):576‐585. [DOI] [PubMed] [Google Scholar]
- 117. Kim J, Park Y‐J, Jang Y, Kwon YH. AMPK activation inhibits apoptosis and tau hyperphosphorylation mediated by palmitate in SH‐SY5Y cells. Brain Res. 2011;1418:42‐51. [DOI] [PubMed] [Google Scholar]
- 118. Spinelli M, Fusco S, Mainardi M, et al. Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat Commun. 2017;8(1):2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Campana M, Bellini L, Rouch C, et al. Inhibition of central de novo ceramide synthesis restores insulin signaling in hypothalamus and enhances β‐cell function of obese Zucker rats. Mol Metab. 2018;8:23‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Flores‐León M, Pérez‐Domínguez M, González‐Barrios R, Arias C. Palmitic acid‐induced NAD+ depletion is associated with the reduced function of SIRT1 and increased expression of BACE1 in hippocampal neurons. Neurochem Res. 2019;44(7):1745‐1754. [DOI] [PubMed] [Google Scholar]
- 121. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88‐deficient mice to endotoxin. Immunity. 1999;11(1):115‐122. [DOI] [PubMed] [Google Scholar]
- 122. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. J Clin Invest. 2006;116(11):3015‐3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kleinridders A, Schenten D, Könner AC, et al. MyD88 signaling in the CNS is required for development of fatty acid‐induced leptin resistance and diet‐induced obesity. Cell Metab. 2009;10(4):249‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rumora AE, Lentz SI, Hinder LM, et al. Dyslipidemia impairs mitochondrial trafficking and function in sensory neurons. FASEB J. 2018;32(1):195‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Rumora AE, LoGrasso G, Haidar JA, Dolkowski JJ, Lentz SI, Feldman EL. Chain length of saturated fatty acids regulates mitochondrial trafficking and function in sensory neurons. J Lipid Res. 2019;60(1):58‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rumora AE, LoGrasso G, Hayes JM, et al. The divergent roles of dietary saturated and monounsaturated fatty acids on nerve function in murine models of obesity. J Neurosci. 2019;39(19):3770‐3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Muoio DM, Neufer PD. Lipid‐induced mitochondrial stress and insulin action in muscle. Cell Metab. 2012;15(5):595‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Marwarha G, Claycombe K, Schommer J, Collins D, Ghribi O. Palmitate‐induced endoplasmic reticulum stress and subsequent C/EBPα homologous protein activation attenuates leptin and insulin‐like growth factor 1 expression in the brain. Cell Signal. 2016;28(11):1789‐1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid‐induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100(6):3077‐3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Briscoe CP, Tadayyon M, Andrews JL, et al. The orphan G protein‐coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278(13):11303‐11311. [DOI] [PubMed] [Google Scholar]
- 131. Hirasawa A, Hara T, Katsuma S, Adachi T, Tsujimoto G. Free fatty acid receptors and drug discovery. Biol Pharm Bull. 2008;31(10):1847‐1851. [DOI] [PubMed] [Google Scholar]
- 132. Ichimura A, Hirasawa A, Hara T, Tsujimoto G. Free fatty acid receptors act as nutrient sensors to regulate energy homeostasis. Prostaglandins Other Lipid Mediat. 2009;89(3–4):82‐88. [DOI] [PubMed] [Google Scholar]
- 133. Hara T, Hirasawa A, Ichimura A, Kimura I, Tsujimoto G. Free fatty acid receptors FFAR1 and GPR120 as novel therapeutic targets for metabolic disorders. J Pharm Sci. 2011;100(9):3594‐3601. [DOI] [PubMed] [Google Scholar]
- 134. Hara T, Kimura I, Inoue D, Ichimura A, Hirasawa A. Free fatty acid receptors and their role in regulation of energy metabolism. Rev Physiol Biochem Pharmacol. 2013;164:77‐116. [DOI] [PubMed] [Google Scholar]
- 135. Zamarbide M, Etayo‐Labiano I, Ricobaraza A, et al. GPR40 activation leads to CREB and ERK phosphorylation in primary cultures of neurons from the mouse CNS and in human neuroblastoma cells. Hippocampus. 2014;24(7):733‐739. [DOI] [PubMed] [Google Scholar]
- 136. Hernández‐Cáceres MP, Toledo‐Valenzuela L, Díaz‐Castro F, et al. Palmitic acid reduces the autophagic flux and insulin sensitivity through the activation of the free fatty acid receptor 1 (FFAR1) in the hypothalamic neuronal cell line N43/5. Front Endocrinol. 2019;10:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Itoh Y, Kawamata Y, Harada M, et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature. 2003;422(6928):173‐176. [DOI] [PubMed] [Google Scholar]
- 138. Itoh Y, Hinuma S. GPR40, a free fatty acid receptor on pancreatic β cells, regulates insulin secretion. Hepatol Res. 2005;33(2):171‐173. [DOI] [PubMed] [Google Scholar]
- 139. Shapiro H, Shachar S, Sekler I, Hershfinkel M, Walker MD. Role of GPR40 in fatty acid action on the β cell line INS‐1 E. Biochem Biophys Res Commun. 2005;335(1):97‐104. [DOI] [PubMed] [Google Scholar]
- 140. Nolan CJ, Madiraju MSR, Delghingaro‐Augusto V, Peyot M‐L, Prentki M. Fatty acid signaling in the beta‐cell and insulin secretion. Diabetes. 2006;55(Suppl 2):S16‐S23. [DOI] [PubMed] [Google Scholar]
- 141. Schnell S, Schaefer M, Schöfl C. Free fatty acids increase cytosolic free calcium and stimulate insulin secretion from β‐cells through activation of GPR40. Mol Cell Endocrinol. 2007;263(1–2):173‐180. [DOI] [PubMed] [Google Scholar]
- 142. Sartorius T, Drescher A, Panse M, et al. Mice lacking free fatty acid receptor 1 (GPR40/FFAR1) are protected against conjugated linoleic acid‐induced fatty liver but develop inflammation and insulin resistance in the brain. Cell Physiol Biochem. 2015;35(6):2272‐2284. [DOI] [PubMed] [Google Scholar]
- 143. Sona C, Kumar A, Dogra S, Kumar BA, Umrao D, Yadav PN. Docosahexaenoic acid modulates brain‐derived neurotrophic factor via GPR40 in the brain and alleviates diabesity‐associated learning and memory deficits in mice. Neurobiol Dis. 2018;118:94‐107. [DOI] [PubMed] [Google Scholar]
- 144. Kim JY, Lee HJ, Lee S‐J, et al. Palmitic acid‐BSA enhances amyloid‐β production through GPR40‐mediated dual pathways in neuronal cells: involvement of the Akt/mTOR/HIF‐1α and Akt/NF‐κB pathways. Sci Rep. 2017;7(1):4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hainault I, Carlotti M, Hajduch E, Guichard C, Lavau M. Fish oil in a high lard diet prevents obesity, hyperlipemia, and adipocyte insulin resistance in rats. Ann N Y Acad Sci. 1993;683(1 Dietary Lipid):98‐101. [DOI] [PubMed] [Google Scholar]
- 146. Clarke SD. Polyunsaturated fatty acid regulation of gene transcription: a mechanism to improve energy balance and insulin resistance. Br J Nutr. 2000;83(Suppl 1):S59‐S66. [DOI] [PubMed] [Google Scholar]
- 147. Shamim A, Mahmood T, Ahsan F, Kumar A, Bagga P. Lipids: an insight into the neurodegenerative disorders. Clin Nutr Exp. 2018;20:1‐19. [Google Scholar]
- 148. Hachem M, Nacir H. Emerging role of phospholipids and lysophospholipids for improving brain docosahexaenoic acid as potential preventive and therapeutic strategies for neurological diseases. Int J Mol Sci. 2022;23(7):3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Akbar M, Calderon F, Wen Z, Kim H‐Y. Docosahexaenoic acid: a positive modulator of Akt signaling in neuronal survival. Proc Natl Acad Sci. 2005;102(31):10858‐10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Li L, Hölscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev. 2007;56(2):384‐402. [DOI] [PubMed] [Google Scholar]
- 151. Liu Y, Liu F, Grundke‐Iqbal I, Iqbal K, Gong C‐X. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol. 2011;225(1):54‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Talbot K, Wang H‐Y, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF‐1 resistance, IRS‐1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Barnard N, Bush A, Ceccarelli A, et al. Dietary and lifestyle guidelines for the prevention of Alzheimer's disease. Neurobiol Aging. 2014;35(Suppl 2):S74‐S78. [DOI] [PubMed] [Google Scholar]
- 154. Calvo‐Ochoa E, Arias C. Cellular and metabolic alterations in the hippocampus caused by insulin signalling dysfunction and its association with cognitive impairment during aging and Alzheimer's disease: studies in animal models. Diabetes Metab Res Rev. 2015;31(1):1‐13. [DOI] [PubMed] [Google Scholar]
- 155. Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet commission. Lancet. 2020;396(10248):413‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Albanese E, Launer LJ, Egger M, et al. Body mass index in midlife and dementia: systematic review and meta‐regression analysis of 589,649 men and women followed in longitudinal studies. Alzheimers Dement. 2017;8:165‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Davies P. The neurochemistry of Alzheimer's disease and senile dementia. Med Res Rev. 1983;3(3):221‐236. [DOI] [PubMed] [Google Scholar]
- 158. Gottfries CG. Alzheimer's disease and senile dementia: biochemical characteristics and aspects of treatment. Psychopharmacology. 1985;86(3):245‐252. [DOI] [PubMed] [Google Scholar]
- 159. Arai H. Biological markers for the clinical diagnosis of Alzheimer's disease. Tohoku J Exp Med. 1996;179(2):65‐79. [DOI] [PubMed] [Google Scholar]
- 160. Growdon JH. Biomarkers of Alzheimer disease. Arch Neurol. 1999;56(3):281. [DOI] [PubMed] [Google Scholar]
- 161. Neugroschl J, Davis KL. Biological markers in Alzheimer disease. Am J Geriatr Psychiatry. 2002;10(6):660‐677. [PubMed] [Google Scholar]
- 162. Haass C, Selkoe DJ. Cellular processing of beta‐amyloid precursor protein and the genesis of amyloid beta‐peptide. Cell. 1993;75(6):1039‐1042. [DOI] [PubMed] [Google Scholar]
- 163. Vassar R, Bennett BD, Babu‐Khan S, et al. Beta‐secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735‐741. [DOI] [PubMed] [Google Scholar]
- 164. De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113(Pt 11):1857‐1870. [DOI] [PubMed] [Google Scholar]
- 165. Takahashi RH, Nam EE, Edgar M, Gouras GK. Alzheimer beta‐amyloid peptides: normal and abnormal localization. Histol Histopathol. 2002;17(1):239‐246. [DOI] [PubMed] [Google Scholar]
- 166. Rushworth JV, Hooper NM. Lipid rafts: linking Alzheimer's amyloid‐β production, aggregation, and toxicity at neuronal membranes. Int J Alzheimers Dis. 2010;2011:603052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Fabelo N, Martín V, Marín R, et al. Evidence for premature lipid raft aging in APP/PS1 double‐transgenic mice, a model of familial Alzheimer disease. J Neuropathol Exp Neurol. 2012;71(10):868‐881. [DOI] [PubMed] [Google Scholar]
- 168. Avila‐Muñoz A, Arias C. Cholesterol‐induced astrocyte activation is associated with increased amyloid precursor protein expression and processing. Glia. 2015;63(11):2010‐2022. [DOI] [PubMed] [Google Scholar]
- 169. Mendoza‐Oliva A, Ferrera P, Fragoso‐Medina J, Arias C. Lovastatin differentially affects neuronal cholesterol and amyloid‐β production in vivo and in vitro. CNS Neurosci Ther. 2015;21(8):631‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Elahi M, Motoi Y, Shimonaka S, et al. High‐fat diet‐induced activation of SGK1 promotes Alzheimer's disease‐associated tau pathology. Hum Mol Genet. 2021;30(18):1693‐1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Vandal M, White PJ, Tremblay C, et al. Insulin reverses the high‐fat diet‐induced increase in brain Aβ and improves memory in an animal model of Alzheimer disease. Diabetes. 2014;63(12):4291‐4301. [DOI] [PubMed] [Google Scholar]
- 172. Sah SK, Lee C, Jang J‐H, Park GH. Effect of high‐fat diet on cognitive impairment in triple‐transgenic mice model of Alzheimer's disease. Biochem Biophys Res Commun. 2017;493(1):731‐736. [DOI] [PubMed] [Google Scholar]
- 173. Mazzei G, Ikegami R, Abolhassani N, et al. A high‐fat diet exacerbates the Alzheimer's disease pathology in the hippocampus of the AppNL‐F/NL‐F knock‐in mouse model. Aging Cell. 2021;20(8):e13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Patil S, Balu D, Melrose J, Chan C. Brain region‐specificity of palmitic acid‐induced abnormalities associated with Alzheimer's disease. BMC Res Notes. 2008;1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.