

SUMMARY
Translating human genetic findings (genome-wide association studies [GWAS]) to pathobiology and therapeutic discovery remains a major challenge for Alzheimer’s disease (AD). We present a network topology-based deep learning framework to identify disease-associated genes (NETTAG). We leverage non-coding GWAS loci effects on quantitative trait loci, enhancers and CpG islands, promoter regions, open chromatin, and promoter flanking regions under the protein-protein interactome. Via NETTAG, we identified 156 AD-risk genes enriched in druggable targets. Combining network-based prediction and retrospective case-control observations with 10 million individuals, we identified that usage of four drugs (ibuprofen, gemfibrozil, cholecalciferol, and ceftriaxone) is associated with reduced likelihood of AD incidence. Gemfibrozil (an approved lipid regulator) is significantly associated with 43% reduced risk of AD compared with simvastatin using an active-comparator design (95% confidence interval 0.51–0.63, p < 0.0001). In summary, NETTAG offers a deep learning methodology that utilizes GWAS and multi-genomic findings to identify pathobiology and drug repurposing in AD.
Graphical abstract
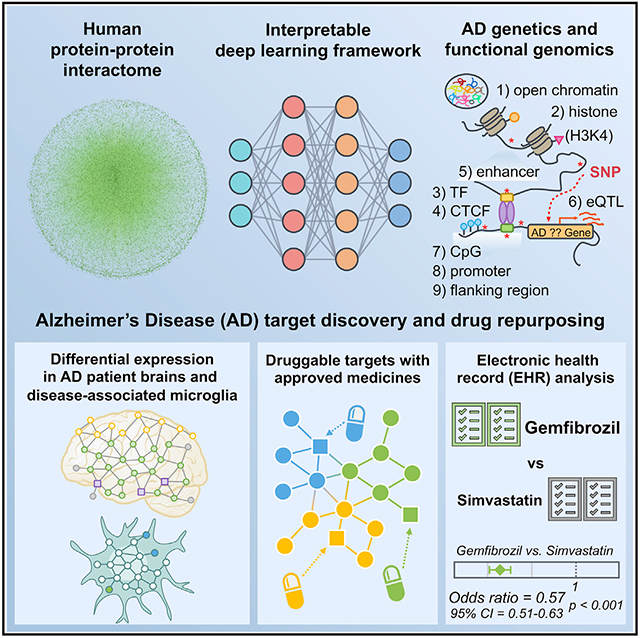
In brief
Xu et al. develop an interpretable deep learning framework (termed NETTAG) to identify Alzheimer’s disease (AD) risk genes and drug targets through leveraging GWAS, multimodal genomics, and human protein-protein interactome network data. Combining human interactome network-based prediction and EHR-based patient data validation identifies gemfibrozil’s effect on reduced incidence of AD.
INTRODUCTION
Alzheimer’s disease (AD), first described in 1907 by Alois Alzheimer, is the most common type of dementia with gradual cognitive decline and memory loss.1 AD and AD-related dementias (AD/ADRD) are a major global health challenge, and the number of affected individuals is expected to double by 2050,2,3 affecting more than 150 million people worldwide.2,4 The prevalence of AD in the United States is expected to double by 2050,5,6 while the attrition rate for AD clinical trials (2002–2012) is estimated at 99%.7 High-throughput DNA/RNA sequencing technologies have rapidly led to a robust body of genetic and genomic data in multiple national AD genome projects, including the Alzheimer’s Disease Sequencing Project (ADSP)8 and the Alzheimer’s Disease Neuroimaging Initiative (ADNI).9 Genome-wide association studies (GWAS) have also identified ~100 AD susceptibility loci.10-14 Despite this progress in understanding AD genetic risk, the complex, polygenic, and pleiotropic genetic architecture has precluded the development of therapeutics. This is partly because the growing mass of genetic datasets has not been effectively explored for AD drug discovery and development.
Recent advances in genetics and systems biology have shown that AD is governed by network-associated molecular determinants (termed disease modules) of common endotypes or endophenotypes.15,16 Approaching AD with a simplistic single-target approach has been demonstrated as effective for developing symptomatic therapies and for monoclonal antibody development, and specifically modulating genetic risk genes may lead to disease-modifying treatments in AD.16 However, more effective means of advancing therapies for disease modification are urgently needed.15 A recent study showed that selecting genetically supported targets can double the success rate in clinical development.17 However, existing data, including genomics, transcriptomics, proteomics, and interactomics (protein-protein interactions), has not yet been fully utilized and integrated for targeted therapeutic development for AD.18-20
Understanding AD from the point of view of human interactome perturbations is the essence of network medicine.15,16 The central hypothesis of AD network medicine is that cellular networks disturbed by genetic variants gradually rewire throughout disease pathogenesis and progression.15,16 Systematic characterization and identification of underlying pathobiology can provide a foundation for identifying disease-modifying targets for AD. Integration of the genome, transcriptome, proteome, and human interactome is essential for such identification. Several network-based analytic techniques have recently been developed to address the myriad different types of inputs of omics layers. Weighted gene co-expression network analysis (WGCNA)21 and multiscale embedded gene co-expression network analysis (MEGENA)22 are two commonly used network-based methods that are based on brain gene/protein expression profiles of AD patients. Random walk with restart (RWR)23 has also been used to predict AD-associated genes, based on input data from literature-reported genes. However, in contrast to NETTAG, these existing network-based approaches do not leverage the integration of multi-omics profiles, such as genetics, functional genomics, transcriptomics, and proteomics, for risk gene prediction and drug target identification.
This study introduces a network topology-based deep learning framework to identify disease-associated genes (NETTAG) and drug targets from genetic and multi-omics discoveries for AD. The fundamental premise of NETTAG is that disease risk genes (1) exhibit distinct functional characteristics compared with non-risk genes and therefore can be distinguished by their aggregated genomic features, (2) converge to a limited number of pathobiological pathways captured by the human protein-protein interactome, and (3) include multiple AD pathobiological modulators and potential therapeutic targets. In addition to pinpointing well-known AD-risk genes, (e.g., APOE, BIN1, and PICALM), NETTAG identified potential AD-associated genes, such as MEF2D and CPLX2. Importantly, we demonstrated the putative risk genes identified by NETTAG offer actionable drug targets for drug repurposing, and we further validated drug findings in a large real-world patient database.
RESULTS
An interpretable deep learning framework
Here, we present NETTAG, a network-based deep learning framework to identify likely risk genes from GWAS and brain-specific multi-omics findings in AD. NETTAG integrates multi-omics data and the human protein-protein interaction (PPI) network to infer likely risk genes and potential drug targets impacted by GWAS loci. The fundamental premise of NETTAG is that disease risk genes exhibit distinct functional characteristics compared with non-risk genes and that they can be distinguished by their aggregated genomic features under the human protein interactome. Specifically, we assembled non-coding GWAS loci effects on expression quantitative trait loci (eQTLs), histone QTLs, transcription factor binding QTLs, enhancers and CpG islands, promoter regions, open chromatin, and promoter flanking regions from GTEx,24,25 NIH RoadMap,26 Ensembl Regulatory Build,27 SNPnexus,28 and ENCODE.29 The procedure for NETTAG (Figure 1) is divided into five components. First, we utilized a deep learning model to cluster PPIs into multiple functional network modules by capturing the topological structures within the human protein-protein interactome (STAR Methods). We then characterized each functional network module by linking its nodes (genes) with protein annotation from the Gene Ontology (GO) knowledgebase.30 Second, we quantified a node’s (gene’s) score by integrating its functional similarity with each gene identified with multiple brain-specific gene regulatory evidences via influencing GWAS loci. Third, we prioritized likely risk genes in AD by their aggregated gene regulatory features. Fourth, we prioritized repurposable drugs for potential treatment of AD by evaluating network proximities between NETTAG-inferred AD-risk genes (alzRGs) and known drug targets under the human protein-protein interactome network model. Fifth, we identified supportive information for drug-AD outcomes using a large-scale patient longitudinal, patient electronic health record (EHR) database (Figure 1).
Figure 1. A diagram illustrating NETTAG.

We first applied a deep learning model to capture the topological structure of the PPIs and divided it into multiple subnetwork modules (STAR Methods). Then we discovered that the divided subnetwork module could approximate protein functions annotated by the Gene Ontology (GO) knowledge portal (STAR Methods). Next, we predicted likely AD-risk genes (alzRGs), which are functionally similar to genes that have been identified by different gene regulatory elements, i.e., CpG island, CCCTC-binding factor (CTCF), enhancer, expression quantitative trait loci (eQTL), histone, open chromatin, promoter, promoter flanking region, and transcriptional factor(TF). Finally, we prioritize repurposed drugs (e.g., gemfibrozil) for potential AD treatment and identified supportive information with the large-scale patient longitudinal database (STAR Methods).
A gene regulatory landscape of GWAS loci in AD
After mapping AD loci (p < 1.0 × 10−5) from multiple brain-specific gene regulatory elements (STAR Methods), we pinpointed 23 genes with CpG islands (e.g., APOE, PVR, and STK11), 19 genes with CTCF binding sites (e.g., BIN1, JPH1, and SYK), 13 genes with enhancers (e.g., BIN1, FARP1, and MARK4), 21 genes with eQTL (e.g.,CD2AP, IL6, and PVR), 169 genes with histone modifications including H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me2, H3K4me3, H3K9ac, and H4K20me1 (such as APOE, CKAP5, DST, and NECTIN2), 48 genes with open chromatin (e.g., BIN1, CLU, and INPP5D), 23 genes with promoters (e.g., APOE, IL6, and STK11), 59 genes with promoter flanking regions (e.g., BIN1, CLU, and MARK4), and 20 genes (e.g., BCAM, CLU, and VSNL1) with transcriptional factor binding sites, respectively (Figure 2A and Table S1). As shown in Figure 2A, 69 genes have AD loci with multiple gene regulatory evidence, such as apolipoprotein E (APOE), BIN1, CLU, IL6, and PTK2B (Table S1). Specifically, APOE loci have regulatory influence with CpG islands (rs429358 and rs7412), histones (rs405509 and rs769449), promoters (rs769449), and promoter flanking regions (rs75627662) (Figure 2A and S1A). Bridging integrator 1 (BIN1), another risk factor of late-onset Alzheimer’s disease (LOAD),31,32 is associated with multiple regulatory elements, including CTCF binding sites (rs12989701), enhancers (rs10207628), histones (rs6431219, rs10194375, rs72838215, rs10207628), open chromatin (rs6733839), and promoter flanking regions (rs10194375). Phosphatidylinositol binding clathrin assembly protein (PICALM) loci in AD display regulatory activity involving histones (rs867611, rs527162, rs639012, and rs17817600), promoters (rs867611), and promoter flanking regions (rs10792832). PICALM regulates AD pathology with Aβ generation and disordered lipid metabolism.33 Inositol polyphosphate-5-phosphatase D (INPP5D), a LOAD-risk gene,14 is associated with open chromatin regulation (rs10933431). Furthermore, a mouse model (5xFAD) study found that expression of Inpp5d was elevated in microglia as the disease progressed.34 We also found that spleen-associated tyrosine kinase (SYK) (rs1172922) is linked with a CTCF binding site. Activation of SYK boosts inflammation35 and modulates both Aβ- and tau-induced pathologies.36 Together, these multi-omics analyses highlight the crucial roles of gene regulation involving various AD GWAS loci. We therefore developed NETTAG to infer gene regulatory variants and putative risk genes in AD using network-based multi-omics evidence aggregation analyses.
Figure 2. Gene regulatory landscape of AD GWAS loci.

(A) Overview of AD GWAS loci across different chromosomes after considering nine gene regulatory elements: GpG island, CCCTC-binding factor (CTCF), enhancer, expression quantitative trait loci (eQTL), histone, open chromatin, promoter, promoter flanking region, and transcriptional factor (Table S1).
(B) Proteins’ cluster numbers are positively correlated with their gene ontology (GO) annotation. We divide proteins into 10groups according to their GO terms. For example, G1 group include the proteins that have at least one, but less than ten GO annotation (Table S2). Error bars denote 1,000 randomly replicated experiments.
(C) Receiver operating characteristic (ROC) analyses of NETTAG based on four collected AD-association gene sets, i.e., AlzGene, DistiLD, DISEASES (knowledge), and TIGA (STAR Methods; Table S2).
NETTAG-based prediction of likely risk genes in AD
Through NETTAG, we first clustered PPIs into functional subnetwork modules using a topology-based self-supervised learning framework. We found that these subnetwork modules could reflect biological relationships (Figure 2B). Specifically, proteins with more GO terms (not used in training) tend to have more network clusters in the human PPI interactome (Figure 2B and Table S2). We found that proteins in the same subnetwork module tend to have more shared GO annotation (Wilcoxon signed rank test, p < 2.2 × 10−16, STAR Methods), indicating that network-based fingerprints of the PPI module overlay among genes and can characterize functional modularities and similarities.37 We inferred likely risk genes by integrating PPI-derived network modules and multimodal analyses of nine types of gene regulatory elements implicated by AD GWAS loci. Taking CpG islands as an example, the predicted score for one particular gene regarding CpG islands could estimate its functional overlap (Spearman correlation, r = 0.44, p = 1.86 × 10−24, Figure S1B and STAR Methods) with all 23 AD CpG island-linked genes (Table S1). Finally, we inferred likely risk genes by integrating (summing) all nine types of gene regulatory elements (STAR Methods). The area under the curve (AUC) values for receiver operating characteristic curve (ROC) using reported AD-associated genes collected from AlzGene,38 DistiLD,39 TIGA,40 and DISEASES41 (Figures 2C and S2A) are 0.81, 0.80, 0.72, and 0.78 respectively, suggesting reasonable accuracy of NETTAG. We further compared NETTAG with three classical approaches: RWR,23 spectral clustering,42 and k-means clustering.43 We found that NETTAG predicted more genes non-overlapped with the mapped input genes associated with different regulatory elements (80%) compared with RWR (less than 50%, Figure S3A). Furthermore, genes predicted by NETTAG were more significantly enriched in AD-relevant functional pathways compared with those identified by RWR (Figure S3B). In addition, we found that the overlapping clustering method implemented in NETTAG outperformed traditional spectral clustering and k-means clustering (Figures S3C and S3D).
Via NETTAG (Figure 1), we identified 156 likely alzRGs, such as APOE, APP, BIN1, FYN, and STK11. Products (proteins) of 139 alzRGs out of 156 formed the largest connected component within 294 PPIs (Figure 3A and Table S3) in the human protein-protein interactome. Via gene and functional enrichment analyses, we found that the NETTAG-predicted alzRGs are significantly enriched by gene regulatory elements (Figures 3B and S4A), compared with the same number of randomly selected genes with a similar degree distribution in the human interactome network. We assembled AD-associated genes from the GWAS Catalog,44 UK Biobank GWAS45 and DisGeNET with published experimental evidence from animal models and human studies.46 We found that alzRGs were significantly enriched in all three AD-associated gene sets: GWAS Catalog (adjusted p value [q] = 2.25 × 10−7), UK Biobank GWAS (q = 8.59 × 10−3), DisGeNET (q = 1.19 × 10−8, Fisher’s exact test, Table S3). Pathway enrichment analyses47 showed that alzRGs are significantly enriched in multiple immune pathways (Table S3 and Figure S4B), including B cell (q = 5.32 × 10−4), T cell receptor (q = 1.17 × 10−2), and cytokine signaling pathways (IL-2: q = 6.99 × 10−3, IL-7: q = 1.43 × 10−2, IL-18: q = 1.65 × 10−2). In summary, NETTAG achieved a high accuracy (AUC = 0.81) in predicting likely AD-risk genes with diverse functional pathways, including key immune pathways. We next performed multi-omics validation for NETTAG-predicted alzRGs, including single-cell/nuclei transcriptomics in disease-associated microglia (DAM) and astrocytes (DAA) isolated from transgenic mouse and human brains with well-established AD neuropathology.
Figure 3. Observations of 156 prioritized AD-risk genes (alzRGs) by NETTAG.

(A) Network-based visualization of 156 predicted alzRGs. 139 alzRGs are non-isolated and form a subnetwork with 294 protein-protein interactions (PPIs). Prioritized alzRGs are colored with various evidence. Green genes are the ones identified by GWAS Catalog but with no gene regulatory element evidence. Blue genes are the ones identified by GWAS Catalog and simultaneously with single or multiple gene regulatory element evidence. Yellow genes are the predicted genes with other types of evidence, e.g., multi-omics or literature evidence. Gray genes are the rest of predicted alzRGs (Table S3).
(B) Cumulative distributions of predicted scores with alzRGs and the same amount of random non-alzRGs with similar degree distribution (the human protein-protein interactome network) for expression quantitative trait loci (eQTLs), histones, and promoters, respectively.
NETTAG-predicted genes are differentially expressed in AD brains
We found 95 alzRGs (p = 2.67 × 10−7, Fisher’s exact test) that are differentially expressed regarding at least one type of transcriptomic study in AD brains. Specifically, 29 (p = 0.0185), 67 (p = 2.36 × 10−3), and 39 (p = 2.96 × 10−7) alzRGs are differentially expressed genes (DEGs) according to microarray (human AD patients and controls), bulk RNA sequencing (human AD patients and controls), and single-cell/nucleus RNA sequencing (AD transgenic mouse model and human postmortem brain samples) analyses, respectively (STAR Methods). Nine genes (ACTL6B, ATP2B1, EPB41L3, ABCA1, CPLX2, P2RX7, PDE1A, SLC38A2, and VSNL1) are DEGs based on all three types of differential expression evidence (Figure S5A). Additionally, the purinergic receptor P2X7 (P2RX7) inhibitor was found to reduce tau accumulation in P301Stau transgenic mice,48 and P2X7R antagonists were supported as potential therapeutic AD options.49 Also, visinin-like 1 (VSNL1) is co-expressed with multiple genes involved in molecular mechanisms of AD, including amyloid beta precursor protein (APP).50 Moreover, nineteen genes (ABCA1, APOE, BCL3, BIN1, CKAP5, CLU, FARP1, HSPG2, MADD, MAPK7, MARK4, NCS1, PICALM, PTK2B, SPRED2, TGFB2, TOMM40, TOP1, and VSNL1) have been identified by gene regulatory elements and AD GWAS studies44 (Figure S5B). More explicitly, microtubule affinity regulating kinase 4 (MARK4) is the protein linked with most gene regulatory elements, including CpG island (rs28469095), CTCF (rs12463049), enhancer (rs536518226), eQTL (rs8100183), histone (rs9653111 and rs10421247), open chromatin (rs138137383), promoter flanking region (rs10421247 and rs138137383), and TF-binding site (rs12463049) (Table S1 and Figure S5B). Furthermore, MARK4 has been suggested as a potential target for AD via binding with acetylcholinesterase (AChE) inhibitors, such as donepezil (an AChE inhibitor used for patients with AD).51 In addition to APOE, BIN1, and PICALM, CLU loci are linked with four types of regulatory elements, including histones (rs9331896 and rs1532278), open chromatin (rs2279590), promoter flanking regions (rs9331888), and TF-binding sites (rs1532278) (Table S1 and Figure S5B). Clusterin (CLU) is another risk gene for LOAD52 by multiple pathologies, including neuroinflammation, Aβ accumulation, and lipid metabolism.53
Among the 67 alzRGs that are differentially expressed based on bulk RNA-seq studies, 42 alzRGs form a subnetwork within the human interactome network (Figure 4A). Within the 23 top differentially expressed alzRGs such as HOMER2, ABCA1, and HSPG2 (∣log2FC∣ > 1, q < 0.05, Table S4), homer scaffold protein 2 (HOMER2) (downregulated log2FC = −1.65, q = 2.39 × 10−3, Table S4) was found to inhibit APP production and secretion of Aβ peptide together with HOMER3.54 ATP binding cassette sub-family A member 1 (ABCA1) was among the most upregulated genes according to bulk RNA-seq studies (log2FC = 1.21, q = 1.37 × 10−3, Table S4), and its mutation has been associated with an elevated risk of AD.55 Heparan sulfate proteoglycan 2 (HSPG2) was another highly upregulated gene according to bulk RNA-seq studies (Table S4). Correlation studies suggested that people with an APOEϵ4 allele have increased risk of AD if also carrying an HSPG2 A allele.56
Figure 4. Transcriptomics-based observation of 156 prioritized AD-risk genes (alzRGs) by NETTAG.

(A) Visualization of 67 predicted alzRGs that are also DEGs according to human bulk RNA-seq studies with both late-stage AD (LAD) and control donors (STAR Methods).
(B) Violin plots show alzRGs are more likely differentially expressed in disease-associated microglia (DAM) according to mouse single-nucleus (GEO: GSE140511; three mice for each group; 5xFAD, wild-type, Trem2 knockout 5xFAD, and Trem2 knockout wild type) RNA-seq datasets (unpaired t test, t test statistic = 34.65, p = 2.59 × 10−206, numbers of replicates = 1,000).
(C) Violin plot shows alzRGs are more likely differentially expressed in disease-associated astrocyte (DAA) according to human prefrontal cortex single-nucleus (GEO: GSE157827; nine normal control and 12 AD human postmortem brain samples) RNA-seq dataset (unpaired t test, t test statistic = 52.01, p = 0.00, numbers of replicates = 1,000).
(D) Violin plot shows alzRGs are more likely differentially expressed in DAA according to human entorhinal cortex (EC) single-nucleus (GEO: GSE138852; six control and six AD human postmortem brain samples) RNA-seq dataset (unpaired t test, t test statistic = 15.18, p = 2.33 × 10−49, numbers of replicates = 1,000).
(E) Visualization of 32 predicted alzRGs that are also differentially expressed genes (DEGs) according to single-cell/nucleus RNA-seq studies collected from both mouse models and human postmortem brain tissues in two disease-associated immune subtypes, i.e., DAM and DAA (STAR Methods).
We collected and analyzed six sets of proteomics data from transgenic mouse AD models (STAR Methods). Among the 156 predicted AD-associated genes, protein products of 29 genes (e.g., HSPA5, ENO1, FERMT2, and VAV1) are differentially expressed (p = 3.98 × 10−7, Fisher’s exact test, Table S4). Heat shock protein family A (Hsp70) member 5 (HSPA5) is important in tau phosphorylation and has been proposed as a potential target for AD treatment.57 Another study showed that oxidative inactivation of enolase 1 (ENO1) can significantly accelerate AD progression from mild cognitive impairment (MCI).58 Additionally, FERM domain containing kindlin 2 (FERMT2) was identified as an AD-risk gene by GWAS studies.14 It is observed that FERMT2 can modulate APP metabolism and Aβ formation, therefore linking its mechanism with AD pathology.59 Another mouse model study found that targeting vav guanine nucleotide exchange factor 1 (VAV1) could rescue neuronal death by inhibiting JNK signaling pathway.60
alzRGs are differentially expressed in AD-associated microglia and astrocytes
Neuroinflammation plays a crucial role in pathogenesis and progression of AD.61 We found that predicted alzRGs are significantly enriched by multiple immune pathways, including B cell receptor, T cell receptor, and cytokine (IL-2, IL-7, and IL18) signaling pathways (Table S3 and Figure S4B). We then investigated how neuroinflammatory pathways were impacted by predicted alzRGs using DAM and disease-associated astrocytes (DAA) as two examples. We found that 14 alzRGs were differentially expressed (∣log2FC∣ > 0.25, q < 0.05) in DAM from a 5xFAD mouse-model-derived single-cell RNA-seq dataset (one-sided t test: statistic = 33.85, p = 9.76 × 10−183, Figure S5C) and a 5xFAD mouse-model-derived single-nucleus RNA-seq dataset (one-sided t test: statistic = 34.65, p = 2.59 × 10−206, Figure 4B). For DAA, 25 alzRGs are differentially expressed (∣log2FC∣ > 0.25, q < 0.05) across three single-nucleus RNA-seq datasets from human postmortem brains with several brain regions, including prefrontal cortex (p < 1.0 × 10−3, Figure 4C), entorhinal cortex (p = 2.33 × 10−49, Figure 4D), and super frontal gyrus (p = 8.30 × 10−27) (Figure S5D, Table S4, and STAR Methods). Among 39 differentially expressed alzRGs in DAM or DAA, 28 alzRG-coding proteins form a subnetwork within the human interactome network (Figure 4E). Activating transcriptional factor 3 (ATF3) overexpressed in DAM (one top DEGs log2FC = 0.62, q = 9.73 × 10−16 with 5xFAD mouse models, Table S4), was observed to have elevated expression in another mouse model study.62 An elevated expression level of ATF3 is positively correlated with Aβ accumulation.62 Microtubule-associated protein RP/EB family member 2 (MAPRE2) overexpressed in DAA (one top DEG with log2FC = 0.51, q = 7.03 × 10−20in human postmortem brain tissues, Table S4) was identified as an AD-associated gene based on GWAS from the ADNI cohort.63 In summary, NETTAG-predicted alzRGs are differentially expressed in AD-associated microglia and astrocytes.
NETTAG-based discovery of potential risk genes in AD
We further identified potential risk genes in AD through combining multiple types of evidence: (1) top predicted genes by NETTAG (156 alzRGs), (2) NETTAG-predicted genes that are supported with at least three types of multi-omics evidence (Table S4), and (3) NETTAG-predicted genes that have not previously been identified by the GWAS Catalog.44 In total, we have identified 25 potential AD-risk genes, e.g., CPLX2, FYN, MAPKAPK2, MEF2D, KLF4, P2RX7, BACE1, and HK2. (Figure 5 and Table S4). Myocyte enhancer factor 2D (MEF2D), an alzRG with currently non-existing AD-associated DNA regulatory or GWAS evidence, has the highest NETTAG-predicted score. MEF2D is differently expressed in both mouse single-nucleus RNA-seq and human microarray data analyses (Figures 5 and S5E and Table S4). A previous study showed that protocatechuic acid rescued a cell model from okadaic acid-induced cytotoxicity (tau hyperphosphorylation) by modulating the Akt/GSK-3β/MEF2D pathway, and it also exhibited neuroprotective effects for AD.64 Complexin 2 (CPLX2) is the second top predicted alzRG with currently non-existing AD-associated DNA regulatory or GWAS evidence. CPLX2 was identified as AD associated with five types of multi-omics evidence and was differentially expressed according to both transcriptome (DAA) and proteome studies (Figures 5 and S5F and Table S4). Experimental data with hippocampus from 3x-Tg AD mice showed abnormally lower proteomic levels of CPLX2,65 which is consistent with the observed downregulation in DAA. Another independent study based on the memory and aging project (MAP) cohort found that lower levels of CPLX2 were significantly associated with cognitive decline.66 Together, these potential risk genes by NETTAG identify disease-associated genes and potential drug targets in AD.
Figure 5. Multi-omics observations of 156 prioritized AD-risk genes (alzRGs) by NETTAG.

Summary of multi-omics validations for all 156 predicted alzRGs (Table S3 and S4). The genes are sorted in predicted score decreasing order (clockwise direction). We have collected seven types of evidence, including drug target, differentially expressed genes (DEG) by microarray studies, DEG by bulk RNA-seq studies, DEG in disease-associated microglia (DAM), DEG in disease-associated astrocyte (DAA), DEG by proteome studies, and literature evidence. There are 126 predicted alzRGs that could be proved as associated with AD with at least one type of evidence.
Discovery of repurposable drugs via targeting NETTAG-predicted genes
Among the 156 predicted alzRGs, 38 proteins (gene products of alzRGs) have been identified as known drug targets with FDA-approved or clinically investigational medicines (p = 8.78 × 10−4, Fisher’s exact test, Table S4 and Figure 5). In total, nine targets (e.g., BACE1, CDK5, FYN, GSK3B, MARK4, MKL1, and PTK2B; Table S4) have been widely investigated as therapeutic approaches for treating AD. FYN proto-oncogene, Src family tyrosine kinase (FYN), which contributes to Aβ production and tau phosphorylation,67 has been suggested as one potential target for AD. Glycogen synthase kinase 3 beta (GSK3B) has been found to have a role in hyperphosphorylation of tau and Aβ production.68 A previous study showed that thiadiazolidinone (a GSK3B inhibitor) decreased tau phosphorylation and improved neuronal survival.68 Beta-secretase 1 (BACE1), a β-secretase enzyme involved in Aβ peptide generation, has also been demonstrated to be a promising target in AD.69 We next turned to identify repurposable drugs by specifically targeting protein products of alzRGs (Figure 3A).
Using network proximity approaches70 to evaluate the closest distance between disease module and a drug’s targets within the human protein-protein interactome network (STAR Methods), we computationally identified 118 candidate drugs using Z scores (Z) < −2 and false discovery rate (q) < 0.05 from the 2,938 US FDA-approved or clinically investigational drugs (Table S5 and STAR Methods). As shown in Figure 6A, we grouped these top predicted 118 candidate drugs into 14 pharmacological categories based on the first level of the Anatomical Therapeutic Chemical (ATC) code. Gemfibrozil, a lipid regulator used to treat hyperlipidemias, is one of these top predictions (Table S5). Gemfibrozil reduced the amyloid plaque burden in a mouse model of AD.71 Mechanistically, gemfibrozil’s targets (e.g., LPL, PPARA, and SLCO1B1) have physical interactions with proteins encoded by several predicted alzRGs (e.g., APOE, APP, HSPG2, and TOMM40) (Figure 6B). Cholecalciferol (vitamin D3) is another top predicted candidate (Table S5). Multiple cohort studies found that vitamin D deficiency was associated with elevated risk of AD.72,73 The drug target network analysis showed that cholecalciferol’s targets (e.g., CDC25A, VDR, and GLRA1) have physical interactions with proteins encoded by several predicted alzRGs (e.g., GSK3B and MAPKAPK2) (Figure 6C). Choline, a nutrient found in many vitamins, is our fifth ranked predicted drug (Table S5). Experiments with APP/PS1 mouse models showed that dietary choline reduced Aβ production and improved spatial memory by suppressing overactivation of DAM.74 While choline does not directly target any alzRGs, its targets interact with multiple protein products of predicted alzRGs, including BIN1, CDK5, and FYN (Figure S6A). Ibudilast, an anti-inflammatory drug used to attenuate multiple sclerosis, is another top predicted drug (Table S5). Ibudilast inhibited pro-inflammatory cytokine production and blocked neuroinflammation to prevent Aβ-induced cognitive impairment.75 Mechanistically, ibudilast’s targets (e.g., PDE3A, PDE4B, and PDE4D) have physical interactions with proteins encoded by several predicted alzRGs (e.g., BIN1, FYN, and GSK3B) (Figure S6B). Ceftriaxone (Table S5), an antibiotic, was found to be neuroprotective by inhibiting amyloid deposition and neuroinflammation in an AD mouse model.76 Ceftriaxone’s targets interact with multiple protein products of predicted alzRGs, such as CSNK2A1 and TOP1 (Figure S6C). Ibuprofen, one commonly used non-steroidal anti-inflammatory drug (Table S5), was capable of suppressing interleukin 1 beta (IL1B) protein level and amyloid deposition in an AD transgenic mouse model.77 Ibuprofen’s targets interact with multiple protein products of predicted alzRGs, such as MARK4 and FYN (Figure S6D). In summary, risk genes identified by NETTAG offer potential drug targets for AD therapeutic discovery, including drug repurposing (such as gemfibrozil and cholecalciferol). We next turned to test AD drug outcomes from large-scale patient EHR data using well-established pharmacoepidemiologic approaches.70,78
Figure 6. Network-based discovery of repurposable drug candidates for AD.

Drug-AD associations were evaluated by the network proximity between predicted alzRGs and drug-target networks.
(A) 118 prioritized drugs for AD treatment (Table S5). Drugs are grouped by fourteen different classes (e.g., immunological, respiratory, neurological, cardiovascular, and cancer) defined by the first level of the Anatomical Therapeutic Chemical (ATC) codes.
(B) Proposed mechanism of actions (MOAs) for gemfibrozil by drug-target network analysis.
(C) Proposed MOAs for cholecalciferol by drug-target network analysis.
Validating possible causal AD associations of candidate drugs in patient data
We used subject matter expertise based on a combination of prespecified factors to select candidate drugs: (1) strength of network proximity measures via quantifying relationships between drug targets and proteins encoded by NETTAG-predicted alzRGs; (2) novelty of the predicted associations through exclusion of drugs currently being tested in AD clinical trials; (3) availability of sufficient patient data for meaningful evaluation (exclusion of infrequently used medications); (4) published positive in vitro or in vivo experimental data in AD models; and (5) ideal brain penetration (STAR Methods). Applying these criteria resulted in identifying four top candidate drugs, including gemfibrozil, cholecalciferol, ceftriaxone, and ibuprofen (Table S5), for patient data analysis. We identified a total of 3,180,111 valid subject records from the Northwestern Medicine Enterprise Data Warehouse (NMEDW) database between 2011 and 2021 after screening 10 million patient records. We then adjusted various confounding factors (including age, race, sex, ethnicity, and disease comorbidities) based on previous studies.70,78 Overall, we found that use of four drugs was significantly associated with reduced incidence of AD compared with propensity score (PS)-matched non-drug exposure (STAR Methods, Table S6A): (1) ibuprofen vs. PS-matched non-ibuprofen users (odds ratio [OR] = 0.80, 95% confidence interval [CI] 0.74–0.87, p < 0.001); (2) gemfibrozil vs. matched non-gemfibrozil users (OR = 0.76, 95% CI 0.70–0.84, p < 0.001); (3) cholecalciferol vs. matched non-cholecalciferol users (OR = 0.87, 95% CI 0.82–0.91, p < 0.001); and (4) ceftriaxone vs. matched non-ceftriaxone users (OR = 0.86, 95% CI 0.78–0.95, p = 0.003).
We next performed additional active-comparator analyses based on an active-comparator design approach allowing comparison of the agent of interest to an agent used for the same medical indication but not identified as a candidate by NETTAG-predicted alzRGs (STAR Methods): (1) 712,103 patients in the ibuprofen cohort vs. 474,110 patients in an aspirin cohort; (2) 72,691 patients in the gemfibrozil cohort vs. 119,949 patients in a simvastatin cohort; (3) 447,846 patients in the cholecalciferol cohort vs. 235,993 patients in an ergocalciferol cohort; and (4) 91,192 patients in the ceftriaxone cohort vs. 248,724 patients in a ciprofloxacin cohort. Table S6A summarizes clinical characteristics of the patients across each drug cohort. More details for each drug cohort design are provided in Table S6D. Among four active-comparator design analyses, (1) gemfibrozil use was significantly associated with a reduced risk of AD compared with simvastatin (an approved anti-lipid medicine under the phase II AD trials [ClinicalTrials.gov identifier: NCT00053599 and NCT00939822]) users (OR = 0.57, 95% CI 0.51–0.63, p< 0.001); (2) ibuprofen usage was significantly associated with a reduced risk of AD compared with aspirin (a drug failed in a large phase II AD trial79) users (OR = 0.78, 95% CI 0.66–0.91, p = 0.002); (3) cholecalciferol was slightly associated with a reduced risk of AD compared with ergocalciferol users (OR = 0.85, 95% CI 0.73–0.99, p = 0.039); and (4) ceftriaxone use was not associated with a reduced risk of AD compared with ciprofloxacin (OR = 1.08, 95% CI 0.94–1.25, p = 0.263).
We further performed sex- and race-specific patient subgroup analyses (Figure 7), and we found that ibuprofen usage was significantly associated with a reduced incidence of AD compared with aspirin users in only male individuals; and ibuprofen usage was slightly associated with a reduced incidence of AD compared with PS-matched non-ibuprofen users in White Americans (OR = 0.82, 95% CI 0.68–0.97, p = 0.023) but not for Black Americans (OR = 0.90, 95% CI 0.56–1.42, p = 0.639). Gemfibrozil usage was significantly associated with a reduced risk of AD compared with simvastatin users and PS-matched non-gemfibrozil users, across all four sex- and race-specific subgroups (OR ranging from 0.49 to 0.58, p ≤ 0.002 for simvastatin users and OR ranging from 0.71 to 0.82, p ≤ 0.04 for PS-matched non-gemfibrozil users). Cholecalciferol usage was associated with reduced AD incidence compared with ergocalciferol users for females (OR = 0.79, 95% CI 0.65–0.94, p = 0.01) and White Americans (OR = 0.80, 95% CI 0.67–0.95, p = 0.01) but not for males (OR = 1.07, 95% CI 0.82–1.39, p = 0.636) and Black Americans (OR = 0.75, 95% CI 0.49–1.16, p = 0.196). Ceftriaxone use was not associated with reduced AD incidence compared with ciprofloxacin users across all four PS-matching subgroups.
Figure 7. Longitudinal patient data observations reveal that usage of four candidate drugs is associated with reduced incidence of Alzheimer’s disease (AD).

Odds ratios (ORs) and 95% confidence intervals (CIs) for eight drug cohort comparator studies are presented. Within each study, besides the overall group analysis (all), we also conducted four additional subgroup analyses by considering sex (female and male) and race (Black and White). Propensity score (PS)-stratified Cox proportional hazards models were used for statistical inference (Fisher’s exact test) of the ORs. For overall group analysis, the patient numbers in each group are ibuprofen (n = 712,103) vs. non-ibuprofen (n = 2,468,008); ibuprofen (n = 712,103) vs. aspirin (n = 474,110); gemfibrozil (n = 72,691) vs. non-gemfibrozil (n = 3,107,420); gemfibrozil (n = 72,691) vs. simvastatin (n = 119,949); cholecalciferol (n = 447,846) vs. non-cholecalciferol (n = 2,732,265); cholecalciferol (n = 447,846) vs. ergocalciferol (n = 235,993); ceftriaxone (n = 91,192) vs. non-ceftriaxone (n = 3,088,919); ceftriaxone (n = 91,192) vs. ciprofloxacin (n = 248,724); Table S6D summarizes more detailed patient counts and event counts in each drug cohort design. Table S6A summarizes detailed clinical characteristics of patients used for each subgroup comparison.
In summary, among the four NETTAG-predicted drugs, these comprehensive pharmacoepidemiologic observations revealed that gemfibrozil was a strong candidate drug for potential prevention and treatment of AD. The association between gemfibrozil use and decreased incidence of AD will require a randomized controlled trial with diverse population to establish the causality.
DISCUSSION
In this study, we presented the development of a deep learning framework known as NETTAG that integrates multi-omics data to infer putative risk genes in AD. To avoid “black-box” deep learning models, we utilized the human protein-protein interactome network to make NETTAG more transparent and interpretable in inferring AD-risk genes from GWAS and multi-omics findings. Specifically, there are 16,720 proteins in the human protein-protein interactome network according to the GO knowledgebase30 (Table S2). We found that 97% (16,214) of annotated proteins in the human interactome have multiple GO terms, ranging from two to around 200 (Table S2 and Figure S7A), and our NETTAG-predicted subnetwork modules were highly correlated with GO-annotated protein function (Figure 2B). We performed additional spearman (r) correlation analyses to evaluate the relationship between NETTAG-predicted scores (cumulative overlays of divided subnetwork modules) and cumulative overlays of protein functions by considering each gene regulatory element and found that deep-learning-predicted scores showed significant correlations across all gene regulatory elements (Figure S1B). For example, CpG islands (r = 0.44, p= 1.86 × 10−24), enhancer (r = 0.34, p = 9.07 × 10−13), histones (r= 0.50, p = 6.49 × 10−37), and TF-binding sites (r = 0.36, p = 3.50 × 10−17) show strong correlations, whereas promoter flanking regions (r = 0.26, p = 2.60 × 10−12), open chromatin (r = 0.22, p = 2.91 × 10−10), promoters (r = 0.20, p = 2.42 × 10−15), CTCFs (r = 0.09, p = 1.45 × 10−2), and eQTLs (r = 0.14, p = 8.62 × 10−10) show weak to moderate correlations. In addition, we scored genes according to their clustering similarities with genes associated with each regulatory element as shown in heatmaps (Figure S8). We found that top genes ranked by NETTAG can be attributed to their protein functional overlap with the input proteins associated with each regulatory element. Consistent with correlation analyses (Figure S1), CpG islands, enhancers, histone QTLs, and TF-binding sites play more important gene regulatory roles of GWAS loci in AD compared with promoter flanking regions, open chromatin, promoters, CTCF binding, and eQTLs. We also found that the AUC values for NETTAG-predicted gene scores, generated by integrating a total of nine genome regulatory elements, are 23% (AlzGene), 23% (DistiLD), 18% (DISEASES-knowledge), and 17% (TIGA) higher than the average AUC value when considering each single gene regulatory element alone (Figures S2B-S2F). Taken together, NETTAG offers an interpretable deep learning framework to identify AD-risk genes from GWAS loci by leveraging multi-omics findings.
To evaluate effects of modifications implemented in NETTAG (STAR Methods), we compared NETTAG with three additional experiments: (1) without any modifications, (2) subsampled topological similar subgraphs (Modification 2 in STAR Methods), and (3) coupling second-order term (Modification 1 in STAR Methods). Each group contained 10 random experiments with fixed seeds. We evaluated the performance of four experiments using the GWAS Catalog for validation, and we found superior efficacy of NETTAG with either modification, compared with the other groups (Figure S7C). In detail, we compared NETTAG with existing network-based approaches, including RWR, spectral clustering, and k-means (STAR Methods). We found that the percentage of predicted genes (not overlapped with input genes) by NETTAG was much higher compared with RWR (Figure S3A and Table S3). In addition, clustering approaches implemented in NETTAG outperformed classical spectral and k-means clustering methods (Figures S3C and S3D) as well. This indicates that overlapping clustering-based approaches could better coincide with the multiple biologic functions of any given protein.
We found that NETTAG-predicted drugs could guarantee consistency with forecasts from other methodologies. For example, pioglitazone was proposed as one potential AD treatment from another artificial intelligence-based approach.80 Sildenafil was suggested as promising AD treatment based on Aβ-tau synergistic endophenotype study.78 Our approach also stably highlights both drugs as top-ranked AD candidate options (Table S5). Combining network-based prediction and large-scale patient data observations, we identified four candidate drugs for treating AD: gemfibrozil, cholecalciferol, ceftriaxone, and ibuprofen. The beneficial effect of ceftriaxone with AD was demonstrated with both Aβ (APP1)81 and tau (3xTg-AD)82 mouse models as well. In both APP181 and 3xTg-AD mouse models,82 ceftriaxone restored glutamate-transporter-1 (GLT-1) expression levels and ameliorated cognitive decline. Ibuprofen’s treatment effects with AD were consistent with a mouse model study that ibuprofen could inhibit neuroinflammation and amyloid deposition.77
Gemfibrozil is a lipid regulator used to treat hyperlipidemia.71 Recent mouse (5xFAD) model studies have shown that oral gemfibrozil treatment reduces amyloid plaque pathology.71 The same team found that gemfibrozil reduced neuroinflammation by inhibiting microgliosis and astrogliosis.71 We selected gemfibrozil to test its possible association with AD using a large-scale patient longitudinal database (STAR Methods). With PS-matching, we found that gemfibrozil was significantly associated with a decreased risk of AD in the general population group after adjusting various cofounding factors, including age, gender, race, ethnicity, MCI, hypertension (HT), type-2 diabetes (T2D), and coronary artery disease (CAD) diagnoses, based on our sizable efforts (Figure 7). Compared with non-gemfibrozil users, gemfibrozil was significantly associated with reduced AD across all four subgroup (i.e., sex and race) analyses as well (Figure 7). The active-comparator design analyses showed that gemfibrozil was associated with greater risk reduction for AD compared with simvastatin in both general and all subgroup studies (Figure 7). Simvastatin is an HMG-CoA reductase inhibitor used for cardiovascular-related disease treatment, which is under several phase II AD trials (ClinicalTrials.gov identifier: NCT01439555 and NCT00486044). Altogether, gemfibrozil offers a promising candidate drug to be tested in future randomized controlled trial in AD patients with diverse populations to establish the causality.
In summary, we established a deep learning framework (NETTAG) that incorporates multi-genomic information along with human protein interactome to infer AD-associated genes. We showed that predicted genes are enriched with drug targets, differently expressed in DAM and astrocytes, and most importantly significantly associated with AD. We demonstrated that the predicted genes offer potential targets for drug repurposing, and we validated one NETTAG-predicted drug, gemfibrozil, in reducing risk of AD using large-scale, longitudinal patient data. We believe that the NETTAG presented here, if broadly applied, could significantly catalyze innovation in drug discovery for AD and other neurodegenerative diseases.
Limitations of the study
We acknowledge several potential limitations in this study. For example, incompleteness of the human protein-protein interactome and GWAS loci by limited population size may influence the model performance. More brain-specific functional genomics data should be integrated in the future studies,83 and the latest GWAS data (Bellenguez et al., 2022)10 should be integrated in the future. While considering all protein pairs sharing at least one common GO annotation, we found that shortest path-based distances among those protein pairs are 2 (53.97%), 3 (39.05%), and 4 (5.05%) in decreasing order (Figure S7B). Therefore, in NETTAG, we have two hidden layers to indirectly aggregate features from second-order neighbors. We postulate that reformulating the model to sample second- and third-order neighbors directly may improve the model performance further.
The EHR-based observational studies may have limitations. First, since true drug administration data were not available, for validation of the NMEDW data, we considered the patients that had been prescribed the drugs. Although patients are likely to adhere to their prescriptions, this is not always the case. Second, pharmacoepidemiologic studies could be biased due to unavailable confounding factors. Although we adjusted for age, sex, and disease comorbidities, including CAD, HT, T2D, and MCI, other factors not included in the database may also be associated with risk of AD, such as education level, socioeconomic status, and genotyping information (such as APOE).84-87 Finally, EHR-based observational studies cannot build causal relationships between drug use and beneficial clinical response of AD.78,88 Our results therefore warrant rigorous clinical trial testing of the treatment efficacy in patients with AD, inclusive of both sexes and controlled by placebo.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and software should be directed to and will be fulfilled by the lead contact, Feixiong Cheng (chengf@ccf.org).
Materials availability
This study did not generate new materials.
Data and code availability
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
The Python implementation of NETTAG is publicly available at https://github.com/ChengF-Lab/NETTAG and archived on Zenodo (https://doi.org/10.5281/zenodo.7159120). The Github repository contains the regulatory element data and NETTAG default parameters used in the manuscript.
Any additional information required for this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| GWAS Catalog | Buniello et al.44 | https://www.ebi.ac.uk/gwas/ |
| SNPnexus | Oscanoa et al.28 | https://www.snp-nexus.org/v4/ |
| GTEx | Lonsdale et al.24; GTEx Consortium25 | https://gtexportal.org/home/datasets |
| Ensembl Regulatory Build | Zerbino et al.27 | https://grch37.ensembl.org/index.html |
| ENCODE | Kundaje et al.29 | https://www.encodeproject.org/ |
| NIAGADS | NA | https://www.niagads.org |
| The Alzheimer’s disease Knowledge portal | NA | https://adknowledgeportal.synapse.org/ |
| Human protein-protein interactome | Zhou et al.89 | https://alzgps.lerner.ccf.org |
| Gene Ontology | Mi et al.30 | http://geneontology.org |
| AlzGene | Bertram et al.38 | http://www.alzgene.org |
| DistiLD | Palleja et al.39 | http://distild.jensenlab.org |
| TIGA | Yang et al.40 | https://unmtid-shinyapps.net/shiny/tiga/ |
| DISEASES | Pletscher-Frankild et al.41 | https://diseases.jensenlab.org/Search |
| Human microarray: 31 late-stage AD and 32 controls | Miller et al.90; Zhou et al.89 | GEO: GSE29378; https://alzgps.lerner.ccf.org |
| Human microarray: 42 late-stage AD and 173 controls |
Berchtold et al.91; Zhou et al.89 | GEO: GSE48350; https://alzgps.lerner.ccf.org |
| Human microarray: 328 late-stage AD and 214 controls |
Wang et al.92; Zhou et al.89 | GEO: GSE84422; https://alzgps.lerner.ccf.org |
| Human bulk-RNA seq: 4 late-stage AD and 4 controls |
Zhou et al.89; Magistri et al.93 | https://alzgps.lerner.ccf.org |
| Human bulk-RNA seq: 6 late-stage AD and 6 controls |
Zhou et al.89; Annese et al.94 | https://alzgps.lerner.ccf.org |
| Human bulk-RNA seq: 20 late-stage AD and 10 controls |
Zhou et al.89; van Rooij, et al.95 | https://alzgps.lerner.ccf.org |
| Mouse single-cell RNA seq: 6-month-old 16 5xFAD and16 WT mouses | Keren-Shaul et al.96 | GEO: GSE98696; http://taca.lerner.ccf.org/ |
| Mouse single-nucleus RNA seq: 7-month-old 3 TREM2-KO WT, 3 TREM2-KO 5xFAD, 3 5xFAD, and 3 WT mouses |
Zhou et al.97 | GEO: GSE140511; http://taca.lerner.ccf.org/ |
| Human single-nucleus RNA seq: 3 Braak stage 0, 4 Braak stage and 3 Braak stage brain samples | Leng et al.98 | GEO: GSE147528; http://taca.lerner.ccf.org/ |
| Human single-nucleus RNA seq: 6 AD and 6 controls | Grubman et al.99 | GEO: GSE138852; http://taca.lerner.ccf.org/ |
| Human single-nucleus RNA seq: 12 AD and 9 controls |
Lau et al.100 | GEO: GSE157827; http://taca.lerner.ccf.org/ |
| Mouse proteomic data (7- and 10-month-old ADLP mouse models [JNPL3 mouse model cross with 5xFAD mouse model]) | Zhou et al.89; Kim et al.101 | https://alzgps.lerner.ccf.org |
| Mouse proteomic data (7- and 10-month-old 5xFAD mouse model) | Zhou et al.89; Kim et al.101 | https://alzgps.lerner.ccf.org |
| Mouse proteomic data (12-month-old 5xFAD mouse model) |
Zhou et al.89; Savas et al.102 | https://alzgps.lerner.ccf.org |
| Mouse proteomic data (12-month-old hAPP mouse model) | Zhou et al.89; Savas et al.102 | https://alzgps.lerner.ccf.org |
| Software and algorithms | ||
| NETTAG | This paper | https://github.com/ChengF-Lab/NETTAG |
| diffusr R package | Valdeolivas et al.23 | https://github.com/dirmeier/diffusr |
| Seurat R package | Butler et al.103 | https://github.com/satijalab/seurat |
| MAST R package | Finak et al.104 | https://github.com/RGLab/MAST |
| scikit-learn Python package: spectral clustering, k-means, SVD | Pedregosa et al.105 | https://scikit-learn.org/stable/index.html |
| Enrichment analysis | Chen et al.106; Kuleshov et al.107 | https://maayanlab.doud/Enrichr/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient electronic health records (EHR) database
The de-identified patient EHR database is from Northwestern Medicine Enterprise Data Warehouse (NMEDW) which covers clinical and medical data from approximately 10 million individuals. The more detailed information is in the method details - pharmacoepidemiologic methods section.
METHOD DETAILS
Construction of multi-genomic features
In this study, we collected 1,047,489 SNPs across multiple genetic traits from GWAS catalog,44 such as Alzheimer’s disease, cerebral amyloid deposition measurement. Next, we performed web server SNPnexus28 to annotate all SNPs in human genome (GRCh38) and collected the regulatory elements information from five databases, including CpG Islands,28 Ensembl Regulatory Build,27 ENCODE,29 the Genotype-Tissue Expression (GTEx) portal24,25 and Roadmap.26 Finally, nine regulatory elements (histone, open chromatin, CpG Island, TF, CTCF, eQTL, enhancer, promoter and promoter flanking region) were used as features to evaluate AD disease-associated genes. To be more specific, step 1: for SNPs with respect to each regulatory elements, e.g., CpG island, we merge them with SNPs curated by GWAS Catalog44 with AD as the mapped traits. Step 2: For each AD related SNPs, the corresponding genes were identified by the “MAPPED GENE(S)” column as provided by GWAS Catalog.44 For SNPs with no mapped genes, if there were any reported genes (REPORTED GENE(S) column in GWAS Catalog44) associated with this SNP, we then map the SNP to its reported genes. For ENCODE29 and Ensembl Regulatory Build27 databases, we only consider epigenomes from brain and neuron tissues and normal karyotype. For eQTLs from GTEx,24,25 we only consider those from human brain tissues. When mapping eQTLs with GWAS Catalog, we only counted significant eSNP (q < 0.05, LD r2 < 0.1) associated eGenes as input features for downstream analysis.24,25 To enhance the statistical analysis from eQTL feature, all eGenes were counted as eQTL features when an eSNP associated with multiple genes. However, we only counted one most significant eQTL feature when an eGene associated with multiple SNPs. The specific epigenomes with brain and neurons for each database are presented in Table S1, separately. The final mapped genes for each regulatory element are provided in Table S1.
Building the human protein-protein interactome
To build the comprehensive human interactome from the most contemporary data available, we assembled commonly used PPI databases with experimental evidence and in-house systematic human PPIs: (i) binary PPIs assessed by high-throughput yeast-two-hybrid (Y2H) experiments18; (ii) kinase-substrate interactions via literature-derived low-throughput and high-throughput experiments from Human Protein Resource Database (HPRD),108 DbPTM 3.0109, Phospho.ELM,110 KinomeNetworkX,111 PhosphoNetworks,112 and PhosphositePlus113; (iii) binary PPIs from 3D protein structures from Instruct114; (iv) signaling networks by literature-derived low-throughput experiments from the SignaLink2.0115; (v) protein complex data (~56,000 candidate interactions) identified by a robust affinity purification-mass spectrometry collected from BioPlexV2.0116; and (vi) literature-curated PPIs identified by affinity purification followed by mass spectrometry from HPRD,117 PINA,118 MINT,119 InnateDB,120 IntAct,121 and BioGRID.122 In total, 351,444 PPIs connecting 17,706 distinctive proteins are now freely available at https://alzgps.lerner.ccf.org. In this study, we consider only the largest connected components of this dataset, which includes 17,456 proteins and 336,549 PPIs.
Description of NETTAG
NETTAG involves 3 steps. Step 1: we build up a graph neural network (GNN) model to capture PPI’s topology structure and establish the appropriate overlapping clustering. The GNN model is motivated by NOCD-G123 which is one GNN-based overlapping community detection framework. And in NETTAG, we made 2 modifications with respect to NOCD-G.123
Modification 1: The model architecture used in NETTAG is defined below (Equation 1)
| (Equation 1) |
Here: , , is the elementwise square of and the sub/super-script l is the layer index. A is the adjacency matrix of the PPI, D is the corresponding diagonal degree matrix. X in generate denotes the node feature matrix, and here we set X = A as implemented by NOCD-G.123 In classical graph convolution network (GCN) models,124 nodes with various degrees share the same weight matrix, the normalized matrix could alleviate this. In NETTAG, coupling into the model is to enhance this, i.e., to strengthen different weight matrices for nodes with high and low degrees. Finally, the dimension of the final output layer interprets the clustering number.
The output matrix F is then feed into Bernoulli-Poisson model125 (Equation 2.1) to learn PPI’s topology (Equation 2.2). The output matrix F has N (number of total nodes in PPI) rows and C (clustering numbers) columns. Locally minimal communities126 estimate the lower bound of the clustering number and we tested different clustering number with increment 100. The final clustering number was determined as the one with lowest akaike information criterion (AIC) value (Equation 2.3, Figure S7F). Each specific row (vector) denotes the node’s weights for being assigned to each cluster. Therefore, we can interpret the loss as follows: if two nodes have multiple commonly shared clusters ( is large), then there should exist an edge connecting each other ( is close to 1) and vice versa.
| (2.1) |
| (2.2) |
| (2.3) |
Modification 2: The PPI network is a highly sparse network which implies number of connected edges is far less than that of non-connected edges (For our PPI, the ratio between number of connected edges over number of non-connected edges = 1/451). To address this imbalanced training problem, the authors in123 first uniformly subsampled certain amounts of connected edges and then subsampled the equal amounts of non-connected edges for training. Instead of uniformly sampling edges directly, we first group nodes according to their degrees into multiple bins. And in each training iteration, we first uniformly subsample same amounts of nodes from each bin, then we extract an adjacency matrix Asub which compromising only selected nodes. In this way, we can keep the topology similarities among sampled subgraphs from different iterations. Next, we use the connected and non-connected edges in Asub to compute the train loss, and the rest connected and non-connected edges in A Asub for test loss evaluation. With this graph subsampling scheme, we find that we are capable to maintain similar connected and non-connected training edge percentages (Figure S7G).
After learning the clustering affinity matrix F, we used the threshold defined in127 as the cutoff for F to determine the node (gene) clustering membership (Equation 3.1).
| (Equation 3.1) |
The rationality of the threshold can be explained from the Bernoulli-Poisson model (Equation 2.1). In detail, we have:
| (Equation 3.2) |
This calculation indicates that a given node could be designated as belonging to a given cluster because it was connected with one or more nodes in the same cluster, rather than being randomly selected alone.
Step 2 of NETTAG: After clustering the entire PPI into overlapping communities, we next scored each node with respect to each gene regulatory element. The detailed steps are described below: Step 2.1. Compute node (gene, e.g., ‘APP’) score regarding one particular regulatory element , here Ci denotes the cluster assignments of node i, denotes the cluster assignments of gene g identified with this regulatory element. The node score equals the sum of clustering overlaps with all genes identified regarding this regulatory element. ∣feat∣ denotes the numbers of genes associated with this regulatory element.
Step 2.2 Construct background distribution to evaluate the significance for the score computed in Step 2.1. For node (gene, e.g., ‘APP’), we run 1000 random experiment, and in each random experiment, we do two things as listed below:
Select ∣feat∣ of genes randomly from GenePPI {feat∪i}
(Equation 4)
Compute the Z score for as , with and , and the corresponding p value () with respect to a standard normal distribution. The final node (gene, e.g., ‘APP’) score regarding the current regulatory element after considering statistical significance is:
| (Equation 5) |
Here, it means we consider the gene score to be meaningful only if its clustering overlap with input genes is statistically significant.
Step 2.3. integrating multiple regulatory elements
The final node (gene, e.g., ‘APP’) score after integrating all regulatory elements is:
| (Equation 6) |
Step 3of NETTAG: After inferring the nodes’ score, we extracted the disease module by considering only genes inferred as significantly associated with AD. We collected all positive gene scores {Si > 0}, and computed the mean μ, standard deviation σ and the Z score. We considered genes with p value ≤0.01 as the predicted AD associated genes and mapped those genes to the background PPI network to generate disease module.
Other existing network-based approaches for risk genes prediction
Random walk with restart (RWR)
We used R package ‘diffusr’ to implement risk gene prediction based on RWR.23 We used genes associated with each regulatory element as the starting distribution for RWR, and gene scores regarding each specific regulatory element equaled to their stationary distribution. The integrated gene scores, same as NETTAG, equaled to summation after considering scores with all gene regulatory elements. We prioritized genes predicted by RWR with the same Z score cutoff as with NETTAG. We conducted disease enrichment analyses using GWAS Catalog44 and DisGeNET46 (Enrichment analysis).
Spectral clustering.
We used the function ‘SpectralClustering’ from Python package ‘scikit-learn’ to implement risk gene prediction based on spectral clustering.42 We used the adjacency matrix of our PPI as the precomputed affinity matrix, and we set the clustering number to be 1200 because this was the same as we set with NETTAG. The subsequent steps of gene scoring were the same as NETTAG (STAR Methods - Step 2 of NETTAG).
K-means.
We used function ‘KMeans’ from Python package ‘scikit-learn’ to implement risk gene prediction based on k-means.43 Considering the curse of dimensionality, we performed singular value decomposition (SVD)128 with function ‘TruncatedSVD’ from Python package ‘scikit-learn’ to the adjacency matrix of our PPI first. The selection of desired dimensionality (parameter ‘n_components’) was described in Table S3. The reduced version of adjacency matrix was then used as the input for k-means clustering. We set the clustering number to be 1200 because this was the same as we set with NETTAG. The subsequent steps of gene scoring were the same as NETTAG (STAR Methods - Step 2 of NETTAG).
Network proximity for drug prediction
We assembled drugs from the DrugBank database relating 2,938 FDA-approved drugs or clinically investigated molecules.129 To predict drugs using the extracted disease module from NETTAG, we adopted the closest-based network proximity measure70 shown below.
| (Equation 7) |
where d(x,y) is the shortest path length between protein x and y from protein sets X and Y, respectively. In our work, X denotes the disease module from NETTAG, and Y denotes the drug targets (protein/gene set) for each drug. To evaluate whether such proximity was significant, the computed network proximity is transferred into Z score form as shown below:
| (Equation 8) |
Here, μd and σd are the mean and standard deviations of permutation tests with 1,000 random experiments. In each random experiment, two random subnetworks Xr and Yr are constructed with the same numbers of nodes and degree distribution as the given 2 subnetworks X and Y, separately, in the PPI network. This reduces any literature bias associated with well-studied proteins.
Pharmacoepidemiologic methods
Dataset source
The data for validation is from Northwestern Medicine Enterprise Data Warehouse (NMEDW),130 which contains medical and clinical data from approximately 10 million patients across 11 hospitals in Illinois.131 We extracted the data from NMEDW between 2011 and 2021 for analysis.
Study design
We evaluated the effect of four target drugs on AD, i.e., Ibuprofen, gemfibrozil, cholecalciferol and ceftriaxone. For each drug, to minimize confounding factors, we conducted two types of drug cohort-based observational studies: (1) users versus non-users of a certain drug, denoted as drug vs. non-drug, such as Ibuprofen vs. non-Ibuprofen; (2) active-comparator design132 that constituted users of a drug versus users of a comparator drug with similar U.S. FDA-approved indications. The target drugs and comparator drugs we used are listed in Table S6B. The primary outcome variable was incidence of AD defined by the International Classification of Diseases (ICD-9/ICD-10) codes (Table S6C).
For each comparison, we executed 3 steps to generate the final cohort, i.e., data extraction, filtering and matching. For drug vs. non-drug comparisons, in the data extraction step, we extracted all the patients who had a drug order record from NMEDW for the target drug. The earliest drug order date was recorded as the index date based on the new-user design.132 All other patients in the NMEDW database were extracted from the non-drug group. In the filtering step, we excluded patients who had been diagnosed with AD before the index date for the target drug group. Patients in both groups whose age was missing were also excluded. Finally, in the matching step, we performed a propensity score matching algorithm133 to mitigate potential confounding variables, including age, gender, race, and ethnicity, as well as diagnosis of mild cognitive impartment (MCI), hypertension (HT), type 2 diabetes (T2D), and coronary artery disease (CAD). All diagnoses were defined by ICD9/10 codes, which are listed in Table S6C. In propensity score matching,134-136 the propensity score was estimated by fitting a logit model in which the outcome variable was a binary variable indicating whether the target drug was used, and the predictors were the potential confounding variables of age, gender, race, ethnicity, and diagnosis of MCI, HT, T2D, and CAD. By propensity score matching, each patient in the target drug group was paired with a patient from the non-drug group with a similar propensity score.
For drug vs. comparator drug comparisons, in the data extraction step we extracted all patients who had been prescribed the corresponding drug. According to the new-user design (Ho et al., 2011,2007; Stuart, 2010), we indicated the earliest drug order date as the index date for both the target group and the comparator. In the filtering step, besides excluding patients diagnosed with AD before the index date and patients with missing age, we also excluded patients who had been prescribed both the target drug and the comparator drug. Finally, in the matching step, we applied a propensity score matching algorithm with confounding variables, including age, gender, race, ethnicity, and diagnosis of MCI, HT, T2D, and CAD. For comparisons in which the target drug group had more patients than the corresponding comparator group, we matched each patient in the comparator group to a patient with similar confounding factors from the target group. In our dataset, for the comparison between ibuprofen and its comparator drug aspirin, we identified more younger patients and fewer older patients, causing failure of the propensity score matching with age. Thus, we excluded young patients with age<65 in the filtering step.
Collections of known AD-associated genes
We collected AD-associated genes from four databases, including AlzGene, DistiLD, TIGA and DISEASES. AlzGene38 collected AD-associated genes via genetic association studies. Thirty-two genes supported by genetic evidence are collected from AlzGene. DistiLD made the existing GWAS studies easier for accessing disease-associated SNPs and genes.39 We collected 19 genes (p < 5.0 × 10−8) with AD GWAS from DistiLD. TIGA40 was a web application that helps drug discovery scientists to prioritize targets by leveraging gene-trait association with multiple studies and evidence. Fifty-one genes with scores greater than 80 defined by TIGA specifically for AD (EFO_0000249) were collected. DISEASES41 was a text-mining founded database that collected disease-gene associations with manual curations, cancer mutation data and existing databases. We picked up twenty-seven genes associated with AD (DOID:10,652) which were curated from multiple sources, including MedlinePlus, AmyCo, UniProtKB-KW. The complete lists of genes we used for ROC analyses are presented in Table S2.
Gene Ontology
All proteins’ gene ontology annotation (human, gaf-version 2.1) are extracted from The Gene Ontology (GO) Knowledgebase.30 For our PPI with 17,706 proteins, there are 16,736 proteins with total 268,241 GO annotation.
QUANTIFICATION AND STATISTICAL ANALYSIS
Alignments between clustered subnetwork modules and protein functions
With respect to each clustered subnetwork modules, we considered all possible protein-protein pairs by counting the protein-protein pairs that shared at least one common GO annotation, and the protein-protein pairs that shared no common GO annotation. We then conducted a paired statistical test with null hypothesis of proteins in the same clustered subnetwork modules having no protein functional similarities and the alternative hypothesis that the proteins in the same clustered subnetwork module possessed common protein functions. We used Wilcoxon signed rank test with R (4.2.0). For each cluster and each possible protein pair we checked whether they shared common GO annotation. Therefore, for each cluster, we could have 2 values: a) numbers of protein pairs that share at least one common GO annotation, and b) numbers of protein pairs that share no common GO annotation. Two groups of data (Table S2) were generated after considering all clusters, and we performed the paired Wilcoxon signed rank test.
Differential expression analyses of brain transcriptomic data
Transcriptome analyses were performed based on microarray, bulk RNA-seq, and single-cell/nucleus (sc/sn) RNA-seq datasets. We utilized three sets of human brain microarray transcriptome data collected from late-stage AD and control donors. The original data are available from Gene Expression Omnibus database,137 i.e., (GEO:GSE29378 with 31 late-stage AD and 32 controls),90 (GEO:GSE48350 with 42 late-stage AD and 173 controls),91 and (GEO:GSE84422 with 328 late-stage AD and 214 controls),92 and the results are available from our recently developed AD knowledgebase AlzGPS.89 We also included human brain bulk RNA-seq transcriptome data collected from the hippocampus of late-stage AD and control donors with three studies, including 4 late-stage AD versus 4 controls,93 6 late-stage AD versus 6 controls,94 and 20 late-stage AD versus 10 controls.95 The complete sc/sn RNA-seq datasets used for differentially expressed genes (DEGs) analyses in this study are available from the Gene Expression Omnibus database137 under accession numbers: GSE98969,96 GSE140511,97 GSE147528,138 GSE138 8 5299 and GSE157827.100 The DEGs between DAM and non-DAM were based on mouse scRNA-seq datasets GEO:GSE98969 and mouse snRNA-seq dataset GEO:GSE140511. The DEGs between DAA and non-DAA were based on the other three human snRNA-seq datasets GEO:GSE147528, GEO:GSE138852 and GEO:GSE157827. We used DEG analytical pipeline in our previous study88 for datasets GEO:GSE98969, GEO:GSE140511, GEO:GSE147528 and GEO:GSE138852. For the additionally added dataset GEO:GSE157827 which contained 21 prefrontal cortex tissue postmortem samples with 12 diagnosed as AD and 9 controls, we performed bioinformatics analyses according to the parameters used in the original manuscript100 with Seurat (4.0.6)103. Nuclei with ≤200 genes and ≥20,000 unique molecular identifiers and ≥20% mitochondrial genes were removed. Then, the raw count was log-normalized and the top 1000 most variable genes were detected with the function FindVariableFeatures with selection.method = ‘Vst’. Next, all 21 samples were integrated by functions FindIntegrationAnchors and InegrateData with parameter dims = 1:20. Principal component analysis (PCA) was performed with parameter npcs = 50, and clustering was performed with the first 20 pcs and resolution 1. After identifying astrocytes with marker genes (ADGRV1, GPC5 and RYR3) provided by the original manuscript,100 we continued with the subcluster analysis. We ran the subcluster with the first 20 pcs and resolution 0.3. Next, we identified disease associated astrocytes (DAA) and homeostasis astrocytes by established marker genes.138 DEGs were calculated between DAA and HAA with MAST R package.104 For all DEGs generated from sc/sn RNA-seq datasets, we applied uniform criterion with q < 0.05 and ∣log2FC∣ ≥ 0.25. We have also included the complete DEG results based on microarray, bulk RNA-seq, sc/sn-RNA-seq aforementioned in Table S4.
Differential expression analyses from brain proteomic data
Differential expression analyses of brain proteomic data were conducted based on six mouse model datasets: (1) 7-month old ADLP mouse models (JNPL3 mouse model cross with 5xFAD mouse model),101 (2) 10-month old ADLP mouse models (JNPL3 mouse model cross with 5xFAD mouse model),101 (3) 7-month old 5xFAD mouse models,101 (4) 10-month old 5xFAD mouse models,101 (5) 12-month old 5xFAD mouse models,102 and (6) 12 month old hAPP mouse models.102 We obtained the differentially expressed proteins (DEPs) for each mouse brain proteomic dataset from the supplemental tables of the original manuscripts for the first four datasets101 with p value < 0.05 regarding the Student’s t-test, and last two datasets102 with FDR <0.05. All lists of DEPs across different mouse brain datasets are available from AlzGPS.89 The complete lists of DEPs are provided in Table S4.
Enrichment analysis
All pathway and disease enrichment analyses were conducted using WikiPathways,139 GWAS Catalog 2019,44 UK Biobank GWAS v145 and DisGeNET46 from Enrichr,106,107 respectively. The combined score defined in Enrichr106 equaled the product of log of p value from the Fisher’s test and Z score which characterized the departure from the expected rank. The GWAS Catalog44 contributes as a freely and easily assessable database of SNP-trait associations which were identified by literatures. The DisGeNET46 platform integrates information about disease-associated genes. Specifically, it assembles disease-associated genes from several sources: expert curated repositories, animal models, and the scientific literatures. Our prediction was enriched with 43 AD-associated genes from DisGeNET (Alzheimer’s disease [C0002395]).
Supplementary Material
Highlights.
NETTAG identifies Alzheimer’s disease (AD) risk genes from multi-omics and network data
NETTAG-predicted risk genes are differentially expressed in AD brains and microglia
NETTAG-predicted AD-risk genes are enriched in known druggable targets
Large-scale patient health record data support gemfibrozil use for reduced AD risk
ACKNOWLEDGMENTS
We thank members from The National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site and The Alzheimer’s disease Knowledge portal to provide technical support for multi-omics data access and analysis.
This work was primarily supported by the National Institute on Aging (NIA) under Award Number U01AG073323, R01AG066707 and R01AG076448 to F.C. This work was supported in part by NIA grants 3R01AG066707-01S1, 3R01AG066707-02S1, and R56AG074001 to F.C. This work was supported in part by the Translational Therapeutics Core of the Cleveland Alzheimer’s Disease Research Center (NIH/NIA: P30AG072959) to F.C., A.A.P., J.B.L., and J.C. This work was supported in part by the Brockman Foundation, Project 19PABH134580006-AHA/Allen Initiative in Brain Health and Cognitive Impairment, the Elizabeth Ring Mather & William Gwinn Mather Fund, S. Livingston Samuel Mather Trust, and the Louis Stokes VA Medical Center resources and facilities to A.A.P. This work was supported in part by Keep Memory Alive (KMA), National Institute of General Medical Sciences (NIGMS) grant P20GM109025, National Institute of Neurological Disorders and Stroke (NINDS) grant U01NS093334, NIA grant R01AG053798, and R35AG071476, and the Alzheimer’s Disease Drug Discovery Foundation (ADDF) to J.C.
Footnotes
DECLARATION OF INTERESTS
J.C. has provided consultation to AB Science, Acadia, Alkahest, AlphaCognition, ALZPathFinder, Annovis, AriBio, Artery, Avanir, Biogen, Biosplice, Cassava, Cerevel, Clinilabs, Cortexyme, Diadem, EIP Pharma, Eisai, GatehouseBio, GemVax, Genentech, Green Valley, Grifols, Janssen, Karuna, Lexeo, Lilly, Lundbeck, LSP, Merck, NervGen, Novo Nordisk, Oligomerix, Ono, Otsuka, PharmacotrophiX, PRODEO, Prothena, ReMYND, Renew, Resverlogix, Roche, Signant Health, Suven, Unlearn AI, Vaxxinity, VigilNeuro pharmaceutical, assessment, and investment companies. J.B.L. has received consulting fees from consulting fees from Vaxxinity, grant support from GE Healthcare and serves on a Data Safety Monitoring Board for Eisai.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111717.
REFERENCES
- 1.Zilka N, and Novak M (2006). The tangled story of Alois Alzheimer. Bratisl. Lek. Listy 107, 343–345. [PubMed] [Google Scholar]
- 2.GBD 2019 Dementia Forecasting Collaborators (2022). Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health 7, e105–e125. 10.1016/S2468-2667(21)00249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hebert LE, Beckett LA, Scherr PA, and Evans DA (2001). Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis. Assoc. Disord 15, 169–173. 10.1097/00002093-200110000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Alzheimer’s Association (2016). 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 12, 459–509. 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Kodamullil AT, Zekri F, Sood M, Hengerer B, Canard L, McHale D, and Hofmann-Apitius M (2017). Trial watch: tracing investment in drug development for Alzheimer disease. Nat. Rev. Drug Discov 16, 819. 10.1038/nrd.2017.169. [DOI] [PubMed] [Google Scholar]
- 6.Be Open about Drug Failures to Speed up Research. https://www.nature.com/articles/d41586-018-07352-7. [DOI] [PubMed]
- 7.Cummings JL, Morstorf T, and Zhong K (2014). Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimer’s Res. Ther 6, 37. 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beecham GW, Bis JC, Martin ER, Choi S-H, DeStefano AL, van Duijn CM, Fornage M, Gabriel SB, Koboldt DC, Larson DE, et al. (2017). The Alzheimer’s disease sequencing project: study design and sample selection. Neurol. Genet 3, e194. 10.1212/NXG.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR, Jagust WJ, Shaw LM, Toga AW, et al. (2010). Alzheimer’s disease neuroimaging initiative (ADNI). Neurology 74, 201–209. 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, et al. (2022). New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet 54, 412–436. 10.1038/s41588-022-01024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuyvers E, and Sleegers K (2016). Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. Lancet Neurol. 15, 857–868. 10.1016/S1474-4422(16)00127-7. [DOI] [PubMed] [Google Scholar]
- 12.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet 51, 404–413. 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung YJ, Kim YH, Bhalla M, Lee SB, and Seo J (2018). Genomics: new light on Alzheimer’s disease research. Int. J. Mol. Sci 19, 3771. 10.3390/ijms19123771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. (2013). Meta-analysis of 74, 046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458. 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang J, Pieper AA, Nussinov R, Lee G, Bekris L, Leverenz JB, Cummings J, and Cheng F (2020). Harnessing endophenotypes and network medicine for Alzheimer’s drug repurposing. Med. Res. Rev 40, 2386–2426. 10.1002/med.21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swarup V, Hinz FI, Rexach JE, Noguchi KI, Toyoshiba H, Oda A, Hirai K, Sarkar A, Seyfried NT, Cheng C, et al. (2019). Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat. Med 25, 152–164. 10.1038/s41591-018-0223-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, Floratos A, Sham PC, Li MJ, Wang J, et al. (2015). The support of human genetic evidence for approved drug indications. Nat. Genet 47, 856–860. 10.1038/ng.3314. [DOI] [PubMed] [Google Scholar]
- 18.Luck K, Kim D-K, Lambourne L, Spirohn K, Begg BE, Bian W, Brignall R, Cafarelli T, Campos-Laborie FJ, Charloteaux B, et al. (2020). A reference map of the human binary protein interactome. Nature 580, 402–408. 10.1038/s41586-020-2188-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng F, Lu W, Liu C, Fang J, Hou Y, Handy DE, Wang R, Zhao Y, Yang Y, Huang J, et al. (2019). A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun 10, 3476. 10.1038/s41467-019-10744-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng F, Zhao J, Wang Y, Lu W, Liu Z, Zhou Y, Martin WR, Wang R, Huang J, Hao T, et al. (2021). Comprehensive characterization of protein–protein interactions perturbed by disease mutations. Nat. Genet 53, 342–353. 10.1038/s41588-020-00774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol 4, Article17, Article17. 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 22.Song W-M, and Zhang B (2015). Multiscale embedded gene Co-expression network analysis. PLoS Comput. Biol 11, e1004574. 10.1371/journal.pcbi.1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valdeolivas A, Tichit L, Navarro C, Perrin S, Odelin G, Levy N, Cau P, Remy E, and Baudot A (2019). Random walk with restart on multiplex and heterogeneous biological networks. Bioinformatics 35, 497–505. 10.1093/bioinformatics/bty637. [DOI] [PubMed] [Google Scholar]
- 24.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, et al. (2013). The genotype-tissue expression (GTEx) project. Nat. Genet 45, 580–585. 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium GTEx (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. (2010). The NIH Roadmap epigenomics mapping consortium. Nat. Biotechnol 28, 1045–1048. 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerbino DR, Wilder SP, Johnson N, Juettemann T, and Flicek PR (2015). The Ensembl regulatory build. Genome Biol. 16, 56. 10.1186/s13059-015-0621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oscanoa J, Sivapalan L, Gadaleta E, Dayem Ullah AZ, Lemoine NR, and Chelala C (2020). SNPnexus: a web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 48, W185–W192. 10.1093/nar/gkaa420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. ; The ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franzmeier N, Rubinski A, Neitzel J, and Ewers M; Alzheimer’s Disease Neuroimaging Initiative ADNI (2019). The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat. Commun 10, 1766. 10.1038/s41467-019-09564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan M-S, Yu J-T, and Tan L (2013). Bridging integrator 1 (BIN1): form, function, and Alzheimer’s disease. Trends Mol. Med 19, 594–603. 10.1016/j.molmed.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 33.Xu W, Tan L, and Yu J-T (2015). The role of PICALM in Alzheimer’s disease. Mol. Neurobiol 52, 399–413. 10.1007/s12035-014-8878-3. [DOI] [PubMed] [Google Scholar]
- 34.Tsai AP, Lin PB-C, Dong C, Moutinho M, Casali BT, Liu Y, Lamb BT, Landreth GE, Oblak AL, and Nho K (2021). INPP5D expression is associated with risk for Alzheimer’s disease and induced by plaque-associated microglia. Neurobiol. Dis 153, 105303. 10.1016/j.nbd.2021.105303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh S, and Geahlen RL (2015). Stress granules modulate SYK to cause microglial cell dysfunction in Alzheimer’s disease. EBioMedicine 2, 1785–1798. 10.1016/j.ebiom.2015.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paris D, Ait-Ghezala G, Bachmeier C, Laco G, Beaulieu-Abdelahad D, Lin Y, Jin C, Crawford F, and Mullan M (2014). The spleen tyrosine kinase (syk) regulates alzheimer amyloid-β production and tau hyperphosphorylation. J. Biol. Chem 289, 33927–33944. 10.1074/jbc.M114.608091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, and Barabási AL (2015). Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 347, 1257601. 10.1126/science.1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertram L, McQueen MB, Mullin K, Blacker D, and Tanzi RE (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet 39, 17–23. 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 39.Pallejà A, Horn H, Eliasson S, and Jensen LJ (2012). DistiLD Database: diseases and traits in linkage disequilibrium blocks. Nucleic Acids Res. 40, D1036–D1040. 10.1093/nar/gkr899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang JJ, Grissa D, Lambert CG, Bologa CG, Mathias SL, Waller A, Wild DJ, Jensen LJ, and Oprea TI (2021). TIGA: target illumination GWAS analytics. Bioinformatics 37, 3865–3873. 10.1093/bioinformatics/btab427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pletscher-Frankild S, Pallejà A, Tsafou K, Binder JX, and Jensen LJ (2015). DISEASES: text mining and data integration of disease–gene associations. Methods 74, 83–89. 10.1016/j.ymeth.2014.11.020. [DOI] [PubMed] [Google Scholar]
- 42.Zare H, Shooshtari P, Gupta A, and Brinkman RR (2010). Data reduction for spectral clustering to analyze high throughput flow cytometry data. BMC Bioinformatics 11, 403. 10.1186/1471-2105-11-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lloyd SM Jr., and Johnson DG (1982). Least squares quantization in PCM. J. Natl. Med. Assoc 74, 129–141. 10.1109/TIT.1982.1056489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012. 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. (2015). UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779. 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F, and Furlong LI (2017). DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 45, D833–D839. 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slenter DN, Kutmon M, Hanspers K, Riutta A, Windsor J, Nunes N, Mélius J, Cirillo E, Coort SL, Digles D, et al. (2018). WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 46, D661–D667. 10.1093/nar/gkx1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruan Z, Delpech J-C, Venkatesan Kalavai S, Van Enoo AA, Hu J, Ikezu S, and Ikezu T (2020). P2RX7 inhibitor suppresses exosome secretion and disease phenotype in P301S tau transgenic mice. Mol. Neurodegener 15, 47. 10.1186/s13024-020-00396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Illes P, Rubini P, Huang L, and Tang Y (2019). The P2X7 receptor: a new therapeutic target in Alzheimer’sdisease. Expert Opin. Ther. Targets 23, 165–176. 10.1080/14728222.2019.1575811. [DOI] [PubMed] [Google Scholar]
- 50.Lin C-W, Chang L-C, Tseng GC, Kirkwood CM, Sibille EL, and Sweet RA (2015). VSNL1 Co-expression networks in aging include calcium signaling, synaptic plasticity, and Alzheimer’s disease pathways. Front. Psychiat 0. 6, 30. 10.3389/fpsyt.2015.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shamsi A, Anwar S, Mohammad T, Alajmi MF, Hussain A, Rehman MT, Hasan GM, Islam A, and Hassan MI (2020). MARK4 inhibited by AChE inhibitors, donepezil and rivastigmine tartrate: insights into Alzheimer’s disease therapy. Biomolecules 10, 789. 10.3390/biom10050789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet 49, 1373–1384. 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foster EM, Dangla-Valls A, Lovestone S, Ribe EM, and Buckley NJ (2019). Clusterin in Alzheimer’s disease: mechanisms, genetics, and lessons from other pathologies. Front. Neurosci 13, 164. 10.3389/fnins.2019.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parisiadou L, Bethani I, Michaki V, Krousti K, Rapti G, and Efthimiopoulos S (2008). Homer2 and Homer3 interact with amyloid precursor protein and inhibit Aβ production. Neurobiol. Dis 30, 353–364. 10.1016/j.nbd.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 55.Nordestgaard LT, Tybjarg-Hansen A, Nordestgaard BG, and Frikke-Schmidt R (2015). Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement. 11, 1430–1438. 10.1016/j.jalz.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 56.Iivonen S, Helisalmi S, Mannermaa A, Alafuzoff I, Lehtovirta M, Soininen H, and Hiltunen M (2003). Heparan sulfate proteoglycan 2 polymorphism in Alzheimer’s disease and correlation with neuropathology. Neurosci. Lett 352, 146–150. 10.1016/j.neulet.2003.08.041. [DOI] [PubMed] [Google Scholar]
- 57.Booth L, Roberts JL, and Dent P (2015). HSPA5/Dna K may Be a useful target for human disease therapies. DNA Cell Biol. 34, 153–158. 10.1089/dna.2015.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Butterfield DA, Poon HF, St. Clair D, Keller JN, Pierce WM, Klein JB, and Markesbery WR (2006). Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol. Dis 22, 223–232. 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 59.Chapuis J, Flaig A, Grenier-Boley B, Eysert F, Pottiez V, Deloison G, Vandeputte A, Ayral A-M, Mendes T, Desai S, et al. (2017). Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol. 133, 955–966. 10.1007/s00401-016-1652-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He B, Chen W, Zeng J, Tong W, and Zheng P (2020). MicroRNA-326 decreases tau phosphorylation and neuron apoptosis through inhibition of the JNK signaling pathway by targeting VAV1 in Alzheimer’s disease. J. Cell. Physiol 235, 480–493. 10.1002/jcp.28988. [DOI] [PubMed] [Google Scholar]
- 61.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet. Neurol 14, 388–405. 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu M, Allard JS, Zhang Y, Perez E, Spangler EL, Becker KG, and Rapp PR (2014). Age-related brain expression and regulation of the chemokine CCL4/MIP-1A in APP/PS1 double-transgenic mice. J. Neuropathol. Exp. Neurol 73, 362–374. 10.1097/NEN.0000000000000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim S, Nho K, Risacher SL, Shen L, Shaw LM, Trojanowski JQ, Weiner MW, and Saykin AJ (2015). P4-008: mapre2 as a novel Alzheimer’s disease target gene from gwas of CSF amyloid beta 1-42, tau and hyperphosphorylated tau in the ADNI cohort. Alzheimer’s. &. Dementia 11, P767–P768. 10.1016/j.jalz.2015.06.1712. [DOI] [Google Scholar]
- 64.Huang L, Zhong X, Qin S, and Deng M (2020). Protocatechuic acid attenuates β-secretase activity and okadaic acid-induced autophagy via the Akt/GSK-3β/MEF2D pathway in PC12 cells. Mol. Med. Rep 21, 1328–1335. 10.3892/mmr.2019.10905. [DOI] [PubMed] [Google Scholar]
- 65.Yu J, Luo X, Xu H, Ma Q, Yuan J, Li X, Chang RC-C, Qu Z, Huang X, Zhuang Z, et al. (2015). Identification of the key molecules involved in chronic copper exposure-aggravated memory impairment in transgenic mice of Alzheimer’s disease using proteomic analysis. J. Alzheimers Dis 44, 455–469. 10.3233/JAD-141776. [DOI] [PubMed] [Google Scholar]
- 66.Ramos-Miguel A, Sawada K, Jones AA, Thornton AE, Barr AM, Leurgans SE, Schneider JA, Bennett DA, and Honer WG (2017). Presynaptic proteins complexin-I and complexin-II differentially influence cognitive function in early and late stages of Alzheimer’s disease. Acta Neuropathol. 133, 395–407. 10.1007/s00401-016-1647-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang K, Belrose J, Trepanier CH, Lei G, Jackson MF, and Mac-Donald JF (2011). Fyn, a potential target for Alzheimer’s disease. J. Alzheimers Dis 27, 243–252. 10.3233/JAD-2011-110353. [DOI] [PubMed] [Google Scholar]
- 68.Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M, Avila J, Guardia-Laguarta C, et al. (2009). A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis 35, 359–367. 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 69.Vassar R (2014). BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res. Ther 6, 89. 10.1186/s13195-014-0089-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheng F, Kovács IA, and Barabási AL (2019). Network-based prediction of drug combinations. Nat. Commun 10, 1197. 10.1038/s41467-019-09186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chandra S, and Pahan K (2019). Gemfibrozil, a lipid-lowering drug, lowers amyloid plaque pathology and enhances memory in a mouse model of Alzheimer’s disease via peroxisome proliferator-activated receptor α. J. Alzheimers Dis. Rep 3, 149–168. 10.3233/ADR-190104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chai B, Gao F, Wu R, Dong T, Gu C, Lin Q, and Zhang Y (2019). Vitamin D deficiency as a risk factor for dementia and Alzheimer’s disease: an updated meta-analysis. BMC Neurol. 19, 284. 10.1186/s12883-019-1500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Littlejohns TJ, Henley WE, Lang IA, Annweiler C, Beauchet O, Chaves PHM, Fried L, Kestenbaum BR, Kuller LH, Langa KM, et al. (2014). Vitamin D and the risk of dementia and Alzheimer disease. Neurology 83, 920–928. 10.1212/WNL.0000000000000755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Velazquez R, Ferreira E, Knowles S, Fux C, Rodin A, Winslow W, and Oddo S (2019). Lifelong choline supplementation ameliorates Alzheimer’s disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell 18, e13037. 10.1111/acel.13037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang H, Mei Z.l., Zhong KL, Hu M, Long Y, Miao MX, Li N, Yan TH, and Hong H (2014). Pretreatment with antiasthmatic drug ibudilast ameliorates Aβ1–42-induced memory impairment and neurotoxicity in mice. Pharmacol. Biochem. Behav 124, 373–379. 10.1016/j.pbb.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 76.Tikhonova MA, Amstislavskaya TG, Ho Y-J, Akopyan AA, Tenditnik MV, Ovsyukova MV, Bashirzade AA, Dubrovina NI, and Aftanas LI (2021). Neuroprotective effects of ceftriaxone involve the reduction of Aβ burden and neuroinflammatory response in a mouse model of Alzheimer’s disease. Front. Neurosci 15, 736786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, and Cole GM (2000). Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci 20, 5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fang J, Zhang P, Zhou Y, Chiang C-W, Tan J, Hou Y, Stauffer S, Li L, Pieper AA, Cummings J, and Cheng F (2021). Endophenotypebased in silico network medicine discovery combined with insurance record data mining identifies sildenafil as a candidate drug for Alzheimer’s disease. Nat. Aging 1, 1175–1188. 10.1038/s43587-021-00138-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ryan J, Storey E, Murray AM, Woods RL, Wolfe R, Reid CM, Nelson MR, Chong TTJ, Williamson JD, Ward SA, et al. (2020). Randomized placebo-controlled trial of the effects of aspirin on dementia and cognitive decline. Neurology 95, e320–e331. 10.1212/WNL.0000000000009277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fang J, Zhang P, Wang Q, Chiang C-W, Zhou Y, Hou Y, Xu J, Chen R, Zhang B, Lewis SJ, et al. (2022). Artificial intelligence framework identifies candidate targets for drug repurposing in Alzheimer’s disease. Alzheimers Res. Ther 14, 7. 10.1186/s13195-021-00951-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hefendehl JK, LeDue J, Ko RWY, Mahler J, Murphy TH, and MacVicar BA (2016). Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Aβ plaques by iGluSnFR two-photon imaging. Nat. Commun 7, 13441. 10.1038/ncomms13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zumkehr J, Rodriguez-Ortiz CJ, Cheng D, Kieu Z, Wai T, Hawkins C, Kilian J, Lim SL, Medeiros R, and Kitazawa M (2015). Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer’s disease. Neurobiol. Aging 36, 2260–2271. 10.1016/j.neurobiolaging.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 83.Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. (2019). Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139. 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barnes DE, Zhou J, Walker RL, Larson EB, Lee SJ, Boscardin WJ, Marcum ZA, and Dublin S (2020). Development and validation of eRADAR: a tool using EHR data to detect unrecognized dementia. J. Am. Geriatr. Soc 68, 103–111. 10.1111/jgs.16182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maserejian N, Krzywy H, Eaton S, and Galvin JE (2021). Cognitive measures lacking in her prior to dementia or Alzheimer’s disease diagnosis. Alzheimers Dement. 17, 1231–1243. 10.1002/alz.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Russ TC, Stamatakis E, Hamer M, Starr JM, Kivimäki M, and Batty GD (2013). Socioeconomic status as a risk factor for dementia death: individual participant meta-analysis of 86 508 men and women from the UK. Br. J. Psychiatry 203, 10–17. 10.1192/bjp.bp.112.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tang AS, Oskotsky T, Havaldar S, Mantyh WG, Bicak M, Solsberg CW, Woldemariam S, Zeng B, Hu Z, Oskotsky B, et al. (2022). Deep phenotyping of Alzheimer’s disease leveraging electronic medical records identifies sex-specific clinical associations. Nat. Commun 13, 675. 10.1038/s41467-022-28273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu J, Zhang P, Huang Y, Zhou Y, Hou Y, Bekris LM, Lathia J, Chiang C-W, Li L, Pieper AA, et al. (2021). Multimodal single-cell/nucleus RNA sequencing data analysis uncovers molecular networks between disease-associated microglia and astrocytes with implications for drug repurposing in Alzheimer’s disease. Genome Res. 31, 1900–1912. 10.1101/gr.272484.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou Y, Fang J, Bekris LM, Kim YH, Pieper AA, Leverenz JB, Cummings J, and Cheng F (2021). AlzGPS: a genome-wide positioning systems platform to catalyze multi-omics for Alzheimer’s drug discovery. Alzheimers Res. Ther 13, 24. 10.1186/s13195-020-00760-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miller JA, Woltjer RL, Goodenbour JM, Horvath S, and Geschwind DH (2013). Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 5, 48. 10.1186/gm452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Berchtold NC, Coleman PD, Cribbs DH, Rogers J, Gillen DL, and Cotman CW (2013). Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol. Aging 34, 1653–1661. 10.1016/j.neurobiolaging.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ, De Luca GC, Crary JF, Casaccia P, Buxbaum JD, et al. (2016). Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 8, 104. 10.1186/s13073-016-0355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Magistri M, Velmeshev D, Makhmutova M, and Faghihi MA (2015). Transcriptomics profiling of Alzheimer’s disease reveal neurovascular defects, altered amyloid-β homeostasis, and deregulated expression of long noncoding RNAs. J. Alzheimers Dis 48, 647–665. 10.3233/JAD-150398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Annese A, Manzari C, Lionetti C, Picardi E, Horner DS, Chiara M, Caratozzolo MF, Tullo A, Fosso B, Pesole G, and D’Erchia AM (2018). Whole transcriptome profiling of Late-Onset Alzheimer’s Disease patients provides insights into the molecular changes involved in the disease. Sci. Rep 8, 4282. 10.1038/s41598-018-22701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Rooij JGJ, Meeter LHH, Melhem S, Nijholt DAT, Wong TH, Netherlands Brain Bank; Rozemuller A, Uitterlinden AG, van Meurs JG, and van Swieten JC (2019). Hippocampal transcriptome profiling combined with protein-protein interaction analysis elucidates Alzheimer’s disease pathways and genes. Neurobiol. Aging 74, 225–233. 10.1016/j.neurobiolaging.2018.10.023. [DOI] [PubMed] [Google Scholar]
- 96.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A unique microglia type Associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 97.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, et al. (2020). Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med 26, 131–142. 10.1038/s41591-019-0695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, et al. (2021). Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci 24, 276–287. 10.1038/s41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci 22, 2087–2097. 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- 100.Lau S-F, Cao H, Fu AKY, and Ip NY (2020). Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 117, 25800–25809. 10.1073/pnas.2008762117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim DK, Park J, Han D, Yang J, Kim A, Woo J, Kim Y, and Mook-Jung I (2018). Molecular and functional signatures in a novel Alzheimer’s disease mouse model assessed by quantitative proteomics. Mol. Neurodegener 13, 2. 10.1186/s13024-017-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Savas JN, Wang Y-Z, DeNardo LA, Martinez-Bartolome S, McClatchy DB, Hark TJ, Shanks NF, Cozzolino KA, Lavallée-Adam M, Smukowski SN, et al. (2017). Amyloid accumulation drives proteome-wide alterations in mouse models of Alzheimer’s disease-like pathology. Cell Rep. 21, 2614–2627. 10.1016/j.celrep.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278. 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: machine learning in Python. J Mach Learn Res 12, 2825–2830. [Google Scholar]
- 106.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97, –W97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Peri S, Navarro JD, Kristiansen TZ, Amanchy R, Surendranath V, Muthusamy B, Gandhi TKB, Chandrika KN, Deshpande N, Suresh S, et al. (2004). Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 32, 497D–D501. 10.1093/nar/gkh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lu C-T, Huang K-Y, Su M-G, Lee T-Y, Bretaña NA, Chang W-C, Chen Y-J, Chen Y-J, and Huang H-D (2013). dbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 41, D295–D305. 10.1093/nar/gks1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, and Diella F (2011). Phospho.ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res. 39, D261–D267. 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cheng F, Jia P, Wang Q, and Zhao Z (2014). Quantitative network mapping of the human kinome interactome reveals new clues for rational kinase inhibitor discovery and individualized cancer therapy. Oncotarget 5, 3697–3710. 10.18632/oncotarget.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hu J, Rho H-S, Newman RH, Zhang J, Zhu H, and Qian J (2014). PhosphoNetworks: a database for human phosphorylation networks. Bioinformatics 30, 141–142. 10.1093/bioinformatics/btt627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520. 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Meyer MJ, Das J, Wang X, and Yu H (2013). INstruct: a database of high-quality 3D structurally resolved protein interactome networks. Bioinformatics 29, 1577–1579. 10.1093/bioinformatics/btt181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fazekas D, Koltai M, Türei D, Módos D, Pálfy M, Dúi Z, Zsákai L, Szalay-Bekő M, Lenti K, Farkas IJ, et al. (2013). SignaLink 2 – a signaling pathway resource with multi-layered regulatory networks. BMC Syst. Biol 7, 7. 10.1186/1752-0509-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440. 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goel R, Harsha HC, Pandey A, and Prasad TSK (2012). Human protein reference database and human proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst 8, 453–463. 10.1039/c1mb05340j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cowley MJ, Pinese M, Kassahn KS, Waddell N, Pearson JV, Grimmond SM, Biankin AV, Hautaniemi S, and Wu J (2012). PINA v2.0: mining interactome modules. Nucleic Acids Res. 40, D862–D865. 10.1093/nar/gkr967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, Galeota E, Sacco F, Palma A, Nardozza AP, Santonico E, et al. (2012). MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 40, D857–D861. 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock REW, Brinkman FSL, and Lynn DJ (2013). InnateDB: systems biology of innate immunity and beyond—recent updates and continuing curation. Nucleic Acids Res. 41, D1228–D1233. 10.1093/nar/gks1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, et al. (2014). The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 42, D358–D363. 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chatr-aryamontri A, Breitkreutz B-J, Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A, Kolas N, O’Donnell L, et al. (2015). The BioGRID interaction database: 2015 update. Nucleic Acids Res. 43, D470–D478. 10.1093/nar/gku1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shchur O, and Günnemann S (2019). Overlapping community detection with graph neural networks. Preprint at arXiv. 10.48550/arXiv.1909.12201. [DOI] [Google Scholar]
- 124.Kipf TN, and Welling M (2017). Semi-supervised classification with graph convolutional networks. Preprint at arXiv. 10.48550/arXiv.1609.02907. [DOI] [Google Scholar]
- 125.Yang J, and Leskovec J (2013). Overlapping community detection at scale: a nonnegative matrix factorization approach. In Proceedings of the sixth ACM international conference on Web search and data mining WSDM ’13 (New York: Association for Computing Machinery; ), pp. 587–596. https://dl.acm.org/doi/10.1145/2433396.2433471. [Google Scholar]
- 126.Gleich D, and Seshadhri C (2011). Neighborhoods are good communities. Preprint at arXiv. 10.48550/arXiv.1112.0031. [DOI] [Google Scholar]
- 127.Yang J, McAuley J, and Leskovec J (2013). Community detection in networks with node attributes. In 2013 IEEE 13th International Conference on Data Mining (IEEE), pp. 1151–1156. 10.1109/ICDM.2013.167. [DOI] [Google Scholar]
- 128.Halko N, Martinsson PG, and Tropp JA (2011). Finding structure with randomness: probabilistic algorithms for constructing approximate matrix decompositions. SIAM Rev. Soc. Ind. Appl. Math 53, 217–288. 10.1137/090771806. [DOI] [Google Scholar]
- 129.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082. 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Starren JB, Winter AQ, and Lloyd-Jones DM (2015). Enabling a learning health system through a unified Enterprise data Warehouse: the experience of the northwestern university clinical and translational sciences (NUCATS) Institute. Clin. Transl. Sci 8, 269–271. 10.1111/cts.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.About the Northwestern Medicine Health System. https://www.nm.org/about-us/northwestern-medicine-newsroom/media-relations/about-our-health-system.
- 132.Yoshida K, Solomon DH, and Kim SC (2015). Active-comparator design and new-user design in observational studies. Nat. Rev. Rheumatol 11, 437–441. 10.1038/nrrheum.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cheng F, Desai RJ, Handy DE, Wang R, Schneeweiss S, Barabási AL, and Loscalzo J (2018). Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun 9, 2691. 10.1038/s41467-018-05116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ho DE, Imai K, King G, and Stuart EA (2011). MatchIt: nonparametric preprocessing for parametric causal inference. J. Stat. Softw 42, 1–28. 10.18637/jss.v042.i08. [DOI] [Google Scholar]
- 135.Ho DE, Imai K, King G, and Stuart EA (2007). Matching as nonparametric preprocessing for reducing model dependence in parametric causal inference. Polit. Anal 15, 199–236. 10.1093/pan/mpl013. [DOI] [Google Scholar]
- 136.Stuart EA (2010). Matching methods for causal inference: a review and a look forward. Stat. Sci 25, 1–21. 10.1214/09-STS313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, et al. (2021). Molecular characterization of selectively vulnerable neurons in Alzheimer’s Disease (Neuroscience). Nat. Neurosci 24, 276–287. 10.1038/s41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, A Miller R, Digles D, Lopes EN, Ehrhart F, et al. (2021). WikiPathways: connecting communities. Nucleic Acids Res. 49, D613–D621. 10.1093/nar/gkaa1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
The Python implementation of NETTAG is publicly available at https://github.com/ChengF-Lab/NETTAG and archived on Zenodo (https://doi.org/10.5281/zenodo.7159120). The Github repository contains the regulatory element data and NETTAG default parameters used in the manuscript.
Any additional information required for this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| GWAS Catalog | Buniello et al.44 | https://www.ebi.ac.uk/gwas/ |
| SNPnexus | Oscanoa et al.28 | https://www.snp-nexus.org/v4/ |
| GTEx | Lonsdale et al.24; GTEx Consortium25 | https://gtexportal.org/home/datasets |
| Ensembl Regulatory Build | Zerbino et al.27 | https://grch37.ensembl.org/index.html |
| ENCODE | Kundaje et al.29 | https://www.encodeproject.org/ |
| NIAGADS | NA | https://www.niagads.org |
| The Alzheimer’s disease Knowledge portal | NA | https://adknowledgeportal.synapse.org/ |
| Human protein-protein interactome | Zhou et al.89 | https://alzgps.lerner.ccf.org |
| Gene Ontology | Mi et al.30 | http://geneontology.org |
| AlzGene | Bertram et al.38 | http://www.alzgene.org |
| DistiLD | Palleja et al.39 | http://distild.jensenlab.org |
| TIGA | Yang et al.40 | https://unmtid-shinyapps.net/shiny/tiga/ |
| DISEASES | Pletscher-Frankild et al.41 | https://diseases.jensenlab.org/Search |
| Human microarray: 31 late-stage AD and 32 controls | Miller et al.90; Zhou et al.89 | GEO: GSE29378; https://alzgps.lerner.ccf.org |
| Human microarray: 42 late-stage AD and 173 controls |
Berchtold et al.91; Zhou et al.89 | GEO: GSE48350; https://alzgps.lerner.ccf.org |
| Human microarray: 328 late-stage AD and 214 controls |
Wang et al.92; Zhou et al.89 | GEO: GSE84422; https://alzgps.lerner.ccf.org |
| Human bulk-RNA seq: 4 late-stage AD and 4 controls |
Zhou et al.89; Magistri et al.93 | https://alzgps.lerner.ccf.org |
| Human bulk-RNA seq: 6 late-stage AD and 6 controls |
Zhou et al.89; Annese et al.94 | https://alzgps.lerner.ccf.org |
| Human bulk-RNA seq: 20 late-stage AD and 10 controls |
Zhou et al.89; van Rooij, et al.95 | https://alzgps.lerner.ccf.org |
| Mouse single-cell RNA seq: 6-month-old 16 5xFAD and16 WT mouses | Keren-Shaul et al.96 | GEO: GSE98696; http://taca.lerner.ccf.org/ |
| Mouse single-nucleus RNA seq: 7-month-old 3 TREM2-KO WT, 3 TREM2-KO 5xFAD, 3 5xFAD, and 3 WT mouses |
Zhou et al.97 | GEO: GSE140511; http://taca.lerner.ccf.org/ |
| Human single-nucleus RNA seq: 3 Braak stage 0, 4 Braak stage and 3 Braak stage brain samples | Leng et al.98 | GEO: GSE147528; http://taca.lerner.ccf.org/ |
| Human single-nucleus RNA seq: 6 AD and 6 controls | Grubman et al.99 | GEO: GSE138852; http://taca.lerner.ccf.org/ |
| Human single-nucleus RNA seq: 12 AD and 9 controls |
Lau et al.100 | GEO: GSE157827; http://taca.lerner.ccf.org/ |
| Mouse proteomic data (7- and 10-month-old ADLP mouse models [JNPL3 mouse model cross with 5xFAD mouse model]) | Zhou et al.89; Kim et al.101 | https://alzgps.lerner.ccf.org |
| Mouse proteomic data (7- and 10-month-old 5xFAD mouse model) | Zhou et al.89; Kim et al.101 | https://alzgps.lerner.ccf.org |
| Mouse proteomic data (12-month-old 5xFAD mouse model) |
Zhou et al.89; Savas et al.102 | https://alzgps.lerner.ccf.org |
| Mouse proteomic data (12-month-old hAPP mouse model) | Zhou et al.89; Savas et al.102 | https://alzgps.lerner.ccf.org |
| Software and algorithms | ||
| NETTAG | This paper | https://github.com/ChengF-Lab/NETTAG |
| diffusr R package | Valdeolivas et al.23 | https://github.com/dirmeier/diffusr |
| Seurat R package | Butler et al.103 | https://github.com/satijalab/seurat |
| MAST R package | Finak et al.104 | https://github.com/RGLab/MAST |
| scikit-learn Python package: spectral clustering, k-means, SVD | Pedregosa et al.105 | https://scikit-learn.org/stable/index.html |
| Enrichment analysis | Chen et al.106; Kuleshov et al.107 | https://maayanlab.doud/Enrichr/ |