

Abstract
Tissue repair after myocardial infarction entails a vigorous angiogenic response involving as yet incompletely defined immune cell-endothelial cell interactions. We identify the monocyte and macrophage-derived cytokine meteorin-like (METRNL) as a driver of postinfarction angiogenesis and high-affinity ligand for the stem cell factor receptor KIT receptor tyrosine kinase (KIT). METRNL’s angiogenic effects on cultured human endothelial cells were mediated through KIT and its downstream signaling pathways. In a mouse model of myocardial infarction, METRNL promoted infarct repair by selectively expanding the KIT-expressing endothelial cell population in the infarct border zone. Metrnl-deficient mice failed to mount this KIT-dependent angiogenic response and developed severe postinfarction heart failure. Our data establish METRNL as a KIT receptor ligand in the context of ischemic tissue injury.
One-Sentence Summary:
Meteorin-like functions as a high-affinity ligand for the stem cell factor receptor KIT in endothelial cells.
Acute myocardial infarction (MI) is a common cardiac emergency and leading cause of heart failure (1). In acute MI, thrombotic occlusion of a coronary artery evokes ischemic cell death in the nonperfused myocardium. The adult mammalian heart has limited capacity for regeneration after ischemic injury and therefore heals by scar formation (2, 3).
Tissue repair after MI involves a vigorous angiogenic response that commences in the infarct border zone and extends into the necrotic infarct core. Neovessel formation after MI mitigates scarring and worsening of heart function and may represent a therapeutic target (4). Monocytes (Mo) and macrophages (Mph) accumulating in the infarct region drive postinfarction angiogenesis (5) by secreting proteins imparting signals to nearby endothelial cells (ECs) expressing their cognate receptors (6, 7). The full complexity of this intercellular crosstalk shaping angiogenesis and functional recovery after MI remains incompletely understood (8, 9).
We conducted a bioinformatic secretome analysis in a mouse model of acute MI to discover previously uncharacterized myeloid cell-derived growth factors driving infarct repair (fig. S1, A and B). We thus identified the 30 kDa protein meteorin-like (METRNL) as being strongly expressed by myeloid cells in the infarct region of the left ventricle (table S1). METRNL is known to be secreted by MPh during inflammation (10–12) and to promote metabolic adaptation and tissue protection under stressful conditions (12–14). The METRNL receptor is unknown.
METRNL was weakly expressed in the heart under sham-operated baseline conditions but strongly induced after MI (fig. S2A). METRNL was also abundantly expressed in myocardial tissue specimens from patients with acute MI (fig. S2B). As shown by confocal microscopy, METRNL-expressing cells co-expressed the myeloid cell marker CD11b and were often located in the vicinity of ECs in the infarct border zone (Fig. 1A). Quantitative reverse transcription polymerase chain reaction analysis identified Mo and Mph as the main Metrnl mRNA-expressing cell types in the infarct region, bone marrow, spleen, and peripheral blood (fig. S3A). Metrnl was broadly expressed in Mo and Mph clusters defined by single-cell RNA sequencing in the infarcted heart (fig. S3B). Delineating Mo and Mph subsets based on chemokine (C-C motif) receptor 2 (CCR2) and chemokine (C-X3-C motif) receptor 1 (CX3CR1) expression (15), Metrnl was more strongly expressed in CCR2high Mo and CCR2high CX3CR1high/low Mph than in CCR2low CX3CR1high Mph (fig. S3C).
Fig. 1. Myeloid cell-derived METRNL promotes angiogenesis after myocardial infarction.
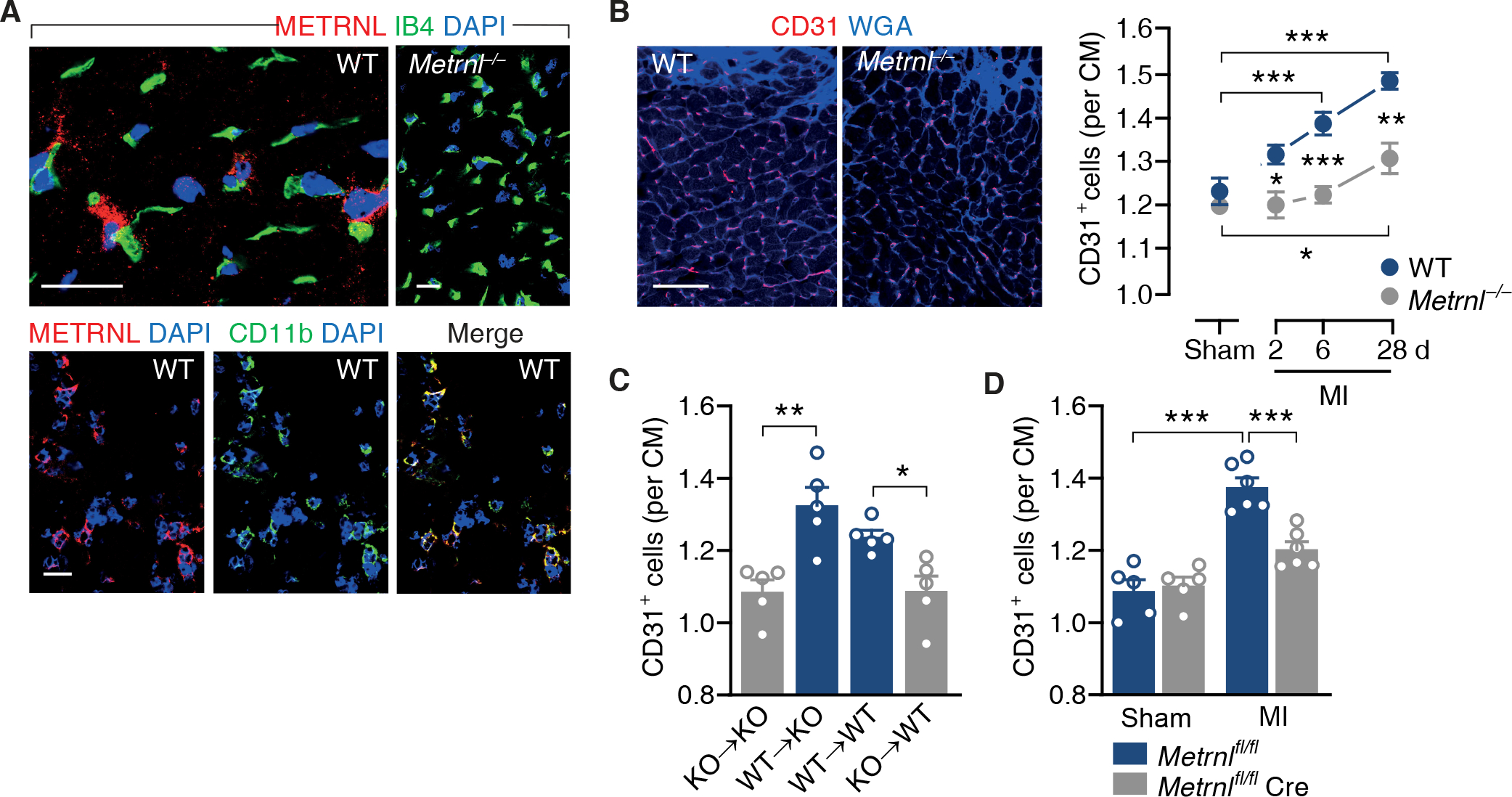
(A) Confocal immunofluorescence microscopy images taken from the infarct border zone 3 days after myocardial infarction (MI) in wild-type (WT) or M etrnl−/− mice. Sections were stained with DAPI, fluorescein-labeled isolectin B4 (IB4), and antibodies against METRNL and CD11b. 92 ± 5% of the METRNL-expressing cells co-expressed CD11b (data from 4 mice). Scale bars, 25 μm. (B) WT and Metrnl−/− mice underwent sham or MI surgery and were followed for 6 (sham) or up to 28 days (MI). Fluorescent images depict CD31+ endothelial cells in the infarct border zone on day 28. Extracellular matrix and cardiac myocyte (CM) borders are highlighted by WGA staining. Scale bar, 50 μm. Summary data are from 5 mice per group. One-way ANOVA with Dunnett test (MI vs. same genotype sham), independent-samples t test (WT vs. Metrnl−/−). (C) Bone marrow cells from Metrnl−/− (knockout, KO) or WT mice were transplanted into (→) lethally irradiated KO or WT recipients. MI was induced after bone marrow reconstitution and CD31+ capillary density in the infarct border zone was determined on day 28. Independent-samples t test. D) CD31+ capillary density in the infarct border zone in Metrnlfl/fl mice and Metrnlfl/fl LysMCre/+ (Metrnlfl/fl Cre) mice on day 28. Two-way ANOVA with Tukey test. (B to D) Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
We subjected Metrnl-deficient (Metrnl−/−) mice to acute MI to explore METRNL’s function during infarct repair. Metrnl−/− mice breed normally and display no apparent phenotype (11, 12). Indeed, Metrnl−/− mice had a normal heart size and preserved left ventricular (LV) function under sham-operated baseline conditions (table S2). After MI, however, Metrnl−/− mice developed larger infarct scars than their wild-type littermates (fig. S4A), with more pronounced LV dilatation and contractile dysfunction, as demonstrated by microcatheterization (fig. S4B and table S2) and high-resolution echocardiography (fig. S4, C and D). This deleterious response was not related to differences in infarct size or cardiomyocyte apoptosis in the infarct border zone 24 hours after injury (fig. S4, E and F) or altered inflammatory cell accumulation in the infarct region (fig. S4G). Yet, new capillary formation in the infarct border zone was impaired in Metrnl−/− mice (Fig. 1B and fig. S4H). Myocardial capillary density under baseline conditions (Fig. 1B) or remote from the infarct region (fig. S4I) was not reduced in Metrnl−/− mice, indicating that Metrnl deletion specifically impaired angiogenesis after ischemic injury.
Transplanting Metrnl−/− mice with wild-type bone marrow cells rescued the angiogenic defect (Fig. 1C) and ameliorated LV scarring and remodeling after MI (fig. S5, A to C). Conversely, transplanting wild-type mice with Metrnl−/− bone marrow cells or deleting Metrnl selectively in myeloid cells by crossing Metrnlfl/fl mice with LysMCre/+ mice (fig. S6, A and B) impaired postinfarction angiogenesis (Fig. 1, C and D) and worsened LV scarring and remodeling (fig. S5, A to C, and fig. S6, C to E). Together, these findings show that myeloid cell-derived METRNL promotes angiogenesis, tissue repair, and functional adaptation after MI.
METRNL dose-dependently stimulated human coronary artery EC proliferation and migration after scratch injury (fig. S7, A and B), indicating that the protein directly acts on ECs. The sigmoidal dose-response relationships and METRNL’s low half-maximal effective concentration (EC50 below 1 ng/mL) suggested that METRNL signals through a high-affinity receptor. Using chemical cross-linking mass spectrometry, we identified KIT receptor tyrosine kinase (KIT) as a METRNL cell surface receptor candidate in ECs (fig. S8, A and B). KIT is the receptor for stem cell factor (SCF, also known as KIT ligand). KIT is expressed by various cell types, including hematopoietic stem and progenitor cells and germ cells. SCF signaling via KIT plays multifaceted roles during development and is important for normal hematopoiesis and fertility (16). The majority of KIT-expressing cells in the adult mouse myocardium are ECs that retain their endothelial identity after MI (17–20).
Coimmunoprecipitation experiments in HEK-293 cells expressing METRNL and KIT confirmed the physical interaction between the two proteins (fig. S9, A and B). After incubating human umbilical vein EC lysates containing endogenous KIT (21) with METRNL or SCF, we could copurify both ligands by KIT immunoprecipitation (fig. S9C). To assess whether METRNL binds to KIT’s extracellular domain, we prepared conditioned supernatants from HEK-293 cells expressing METRNL, SCF, vascular endothelial growth factor A (VEGFA), and/or a secreted KIT-extracellular domain-Fc fragment fusion protein (KIT-ECD-Fc). Pulling-down KIT-ECD-Fc from the supernatants, we copurified METRNL and SCF, but not VEGFA (Fig. 2A). Microscale thermophoresis showed that METRNL and SCF bind to KIT’s extracellular domain with similarly high affinity (Fig. 2B and fig. S9D).
Fig. 2. METRNL binds to the extracellular domain of KIT.
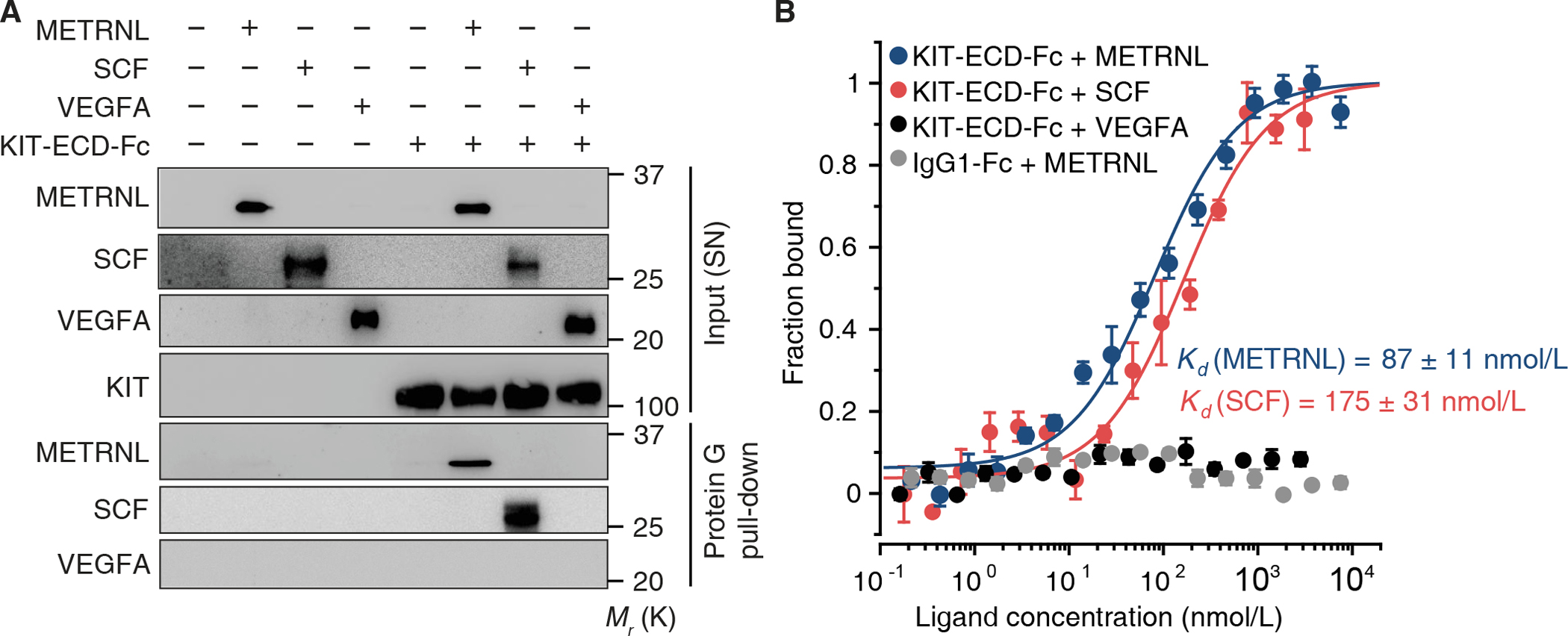
(A) Conditioned supernatants (SN) were prepared from HEK-293 cells transfected with expression plasmids encoding human KIT-extracellular domain-Fc fragment fusion protein (KIT-ECD-Fc), METRNL, stem cell factor (SCF), and/or vascular endothelial growth factor A (VEGFA). Pull-down of KIT-ECD-Fc using protein G-coated beads copurified METRNL and SCF. Exemplary results from 3 experiments. (B) Protein-protein interaction analysis by microscale thermophoresis. Binding curves illustrate the interaction of human METRNL and SCF with human KIT-ECD-Fc. Human VEGFA did not interact with KIT-ECD-Fc, nor did METRNL with an IgG1-Fc control protein. Data are means ± SEM from 3–4 technical replicates; 2 additional experiments yielded similar results.
We next explored whether METRNL mediates its angiogenic effects via KIT. In line with previous studies linking endothelial KIT activation to angiogenesis (21, 22), SCF stimulated human coronary artery EC proliferation and migration after scratch injury (fig. S7, C and D) as we had observed with METRNL (fig. S7, A and B). Small interfering RNA-mediated downregulation of KIT curtailed METRNL and SCF’s angiogenic effects but did not affect EC responses to VEGFA (fig. S7, E and F).
SCF stimulation induces KIT homodimerization and transphosphorylation of tyrosine residues (Y568/Y570) in the juxtamembrane region of the receptor (23), thus creating a docking site for SRC family kinases (24). Treating human coronary artery ECs with METRNL or SCF induced KIT (Y568/Y570) phosphorylation with similar kinetics (fig. S10A). Adding recombinant KIT-ECD-Fc to the culture medium prevented METRNL and SCF from inducing KIT (Y568/Y570) phosphorylation (Fig. 3A), consistent with the idea that transmembrane KIT and KIT-ECD-Fc compete for METRNL and SCF binding. Indeed, recombinant KIT-ECD-Fc dose-dependently inhibited the angiogenic effects of METRNL and SCF, but not those of VEGFA (Fig. 3B).
Fig. 3. METRNL and SCF signaling through KIT in endothelial cells.
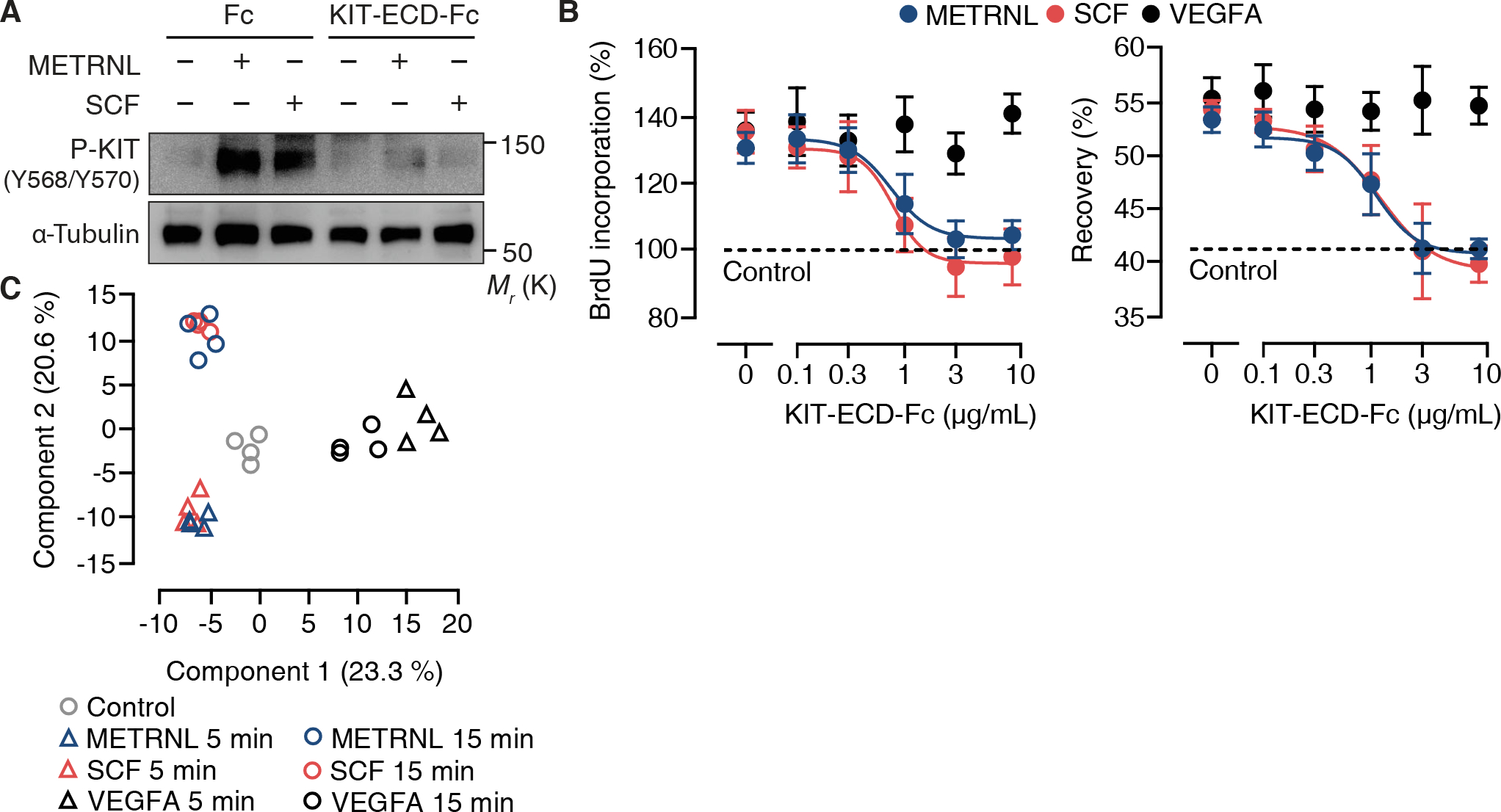
(A) Expression of P-KIT (Y568/Y570) and α-tubulin in human coronary artery endothelial cells (HCAECs) stimulated for 5 min with METRNL (10 ng/mL) or stem cell factor (SCF, 10 ng/mL) in the absence or presence of recombinant Fc fragment (10 μg/mL) or KIT-extracellular domain-Fc fragment fusion protein (KIT-ECD-Fc, 10 μg/mL). Exemplary results from 3 experiments. (B) BrdU incorporation and recovery after scratch injury of HCAECs stimulated with METRNL, SCF, or vascular endothelial growth factor (VEGFA, 25 ng/mL) in the absence or presence of KIT-ECD-Fc. Data are means ± SEM from 4 experiments. (C) Principal component analysis of phosphoproteome data sets from HCAECs stimulated with METRNL, SCF, or VEGFA (4 technical replicates per condition).
Downstream of KIT, both METRNL and SCF induced SRC (Y416) and AKT1 (S473) phosphorylation, again following comparable signaling kinetic trajectories (fig. S10, B and C). To gain a broader perspective, we applied high-resolution mass spectrometry to examine protein phosphorylation dynamics in human coronary artery ECs stimulated with METRNL, SCF, or VEGFA. In a principal component analysis based on 4,750 phosphosites distributed among 1,891 different proteins, METRNL and SCF’s phosphoproteome signatures clustered together and were segregated from VEGFA’s signatures in principal component space (Fig. 3C). Unsupervised hierarchical clustering and plotting the data in a correlation matrix confirmed that the phosphoproteomic responses to METRNL and SCF were closely related and distinct from the response to VEGFA (fig. S11, A and B). METRNL and SCF’s angiogenic effects are thus mediated through KIT and shared downstream signaling pathways.
We used single-cell RNA sequencing to identify Kit-expressing ECs in the adult mouse myocardium. Whereas few ECs, scattered across several EC clusters, expressed Kit under baseline conditions, the number of Kit-expressing ECs markedly increased after MI (Fig. 4A). Kit was the only gene that was differentially expressed (log2 ratio > 1, P < 0.05) in Kit-expressing vs. Kit-nonexpressing ECs under baseline conditions. After MI, however, genes related to cell proliferation and migration were highly enriched in Kit-expressing ECs, indicating that the cells had become activated (table S3).
Fig. 4. METRNL expands the KIT-expressing endothelial cell population after myocardial infarction.
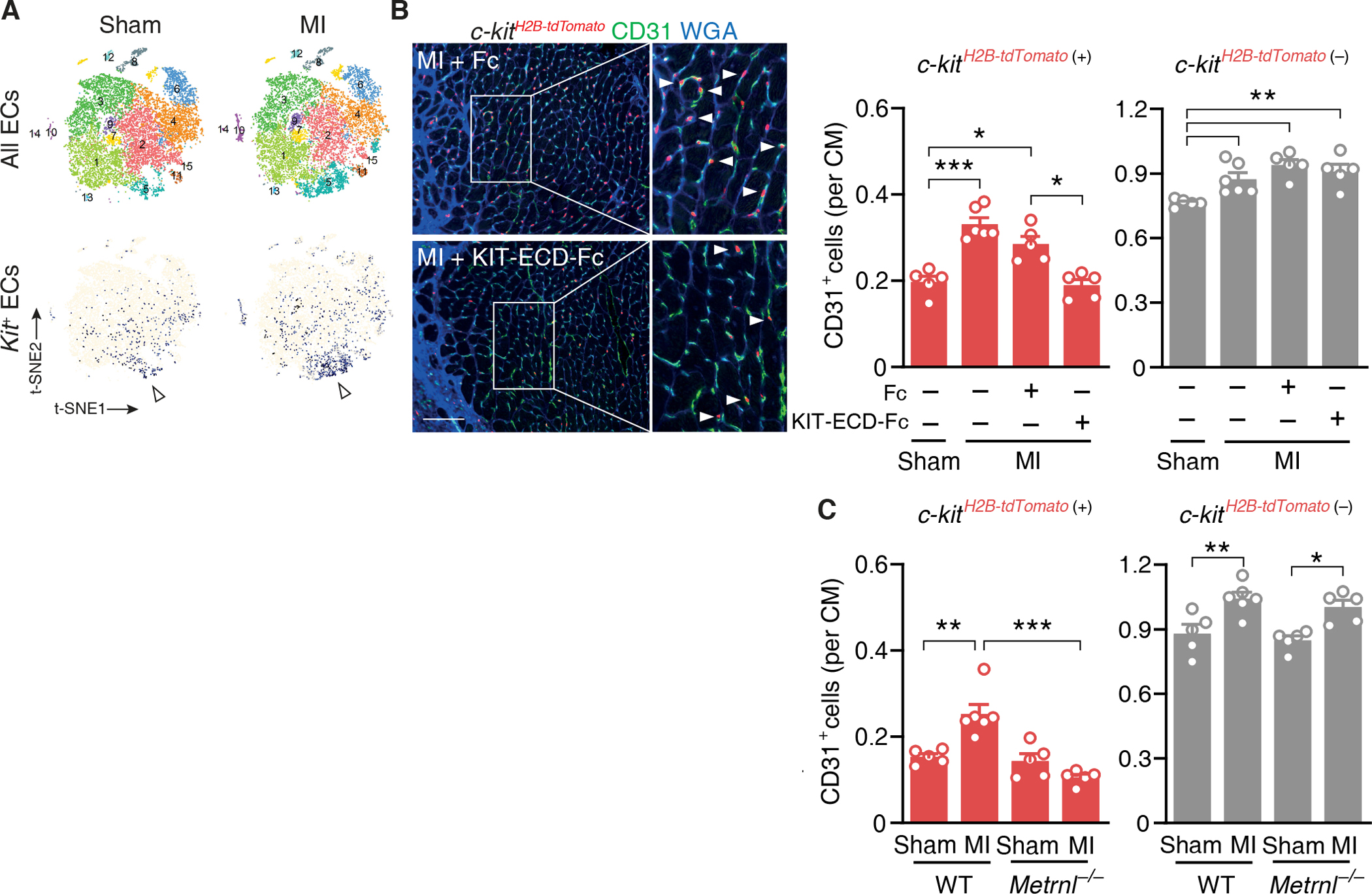
(A) t-SNE plots depicting all cardiac endothelial cell (EC) populations and Kit-expressing cardiac ECs (colored in blue) 3 days after sham or myocardial infarction (MI) surgery in wild-type mice. Cluster #5 comprised a high number of Kit-expressing ECs (arrowheads). (B) c-kitH2B-tdTomato/+ mice underwent sham or MI surgery and were treated with Fc fragment or KIT-extracellular domain-Fc fragment fusion protein (KIT-ECD-Fc, 100μg into the left ventricular cavity each). Fluorescent images and summary data depicting c-kitH2B-tdTomato/+-positive (arrow-heads) and c-kitH2B-tdTomato/+-negative CD31+ ECs in the infarct border zone on day 6. Extracellular matrix and cardimyocyte (CM) borders are highlighted by WGA staining. Scale bar, 100 μm. One-way ANOVA with Tukey test. (C) c-kitH2B-tdTomato/+-positive and c-kitH2B-tdTomato/+-negative CD31+ ECs in the infarct border zone in c-kitH2B-tdTomato/+ Metrnl+/+ (WT) and c-kitH2B-tdTomato/+ Metrnl−/− (Metrnl−/−) mice on day 6. (B and C) Summary data are means ± SEM. Two-way ANOVA with Tukey test. *P < 0.05, **P < 0.01, ***P < 0.001.
To explore whether KIT expression determines EC susceptibility to METRNL and SCF stimulation, we isolated cardiac ECs from c-kitH2B-tdTomato/+ knock-in mice. The nuclear tdTomato signal in these mice faithfully recapitulates the expression pattern of endogenous KIT (17). Using click chemistry to fluorescently label EdU+ proliferating ECs, we found that METRNL and SCF stimulated c-kitH2B-tdTomato-positive but not c-kitH2B-tdTomato-negative EC proliferation (fig. S12). To survey KIT-expressing EC behavior in the infarct border zone, we subjected c-kitH2B-tdTomato/+ mice to acute MI. As shown by single-cell RNA sequencing, c-kitH2B-tdTomato-negative ECs outnumbered c-kitH2B-tdTomato-positive ECs under baseline conditions and after MI (Fig. 4B). Yet, expansion of both populations contributed about equally to the increase in capillary density in the infarct border zone (Fig. 4B). Treating c-kitH2B-tdTomato/+ mice with KIT-ECD-Fc (100 μg into the LV cavity immediately after MI) selectively prevented c-kitH2B-tdTomato-positive EC expansion in the infarct border zone (Fig. 4B) and worsened LV dysfunction after MI (table S4), indicating that growth factor(s) signaling via KIT stimulate KIT-expressing EC expansion and augment heart function after MI.
Unlike METRNL, SCF was expressed by ECs, fibroblasts, and cardiomyocytes in the infarct region (fig. S13A) and its expression did not increase after MI (fig. S13B). Further, we observed no upregulation of SCF in the infarct region of Metrnl−/− mice (fig. S13C). To explore therefore if METRNL drives KIT-expressing EC expansion after MI, we crossed the c-kitH2B-tdTomato allele into Metrnl−/− mice. Metrnl deletion did not impact c-kitH2B-tdTomato-positive or -negative EC densities in the myocardium under baseline conditions (Fig. 4C), indicating that METRNL is not necessary for establishing either cell population during development. After MI, however, c-kitH2B-tdTomato-positive EC numbers failed to increase in Metrnl−/− mice whereas c-kitH2B-tdTomato-negative EC expansion was not affected (Fig. 4C).
To explore METRNL’s therapeutic potential, we treated heart failure-prone FVB/N mice (25) with METRNL after MI (bolus injection into the LV cavity immediately after MI, followed by subcutaneous infusion) (fig. S14A). METRNL treatment enhanced infarct border zone capillarization (fig. S14B), limited scar formation (fig. S14C), and exerted sustained beneficial effects on LV function (table S5). In c-kitH2B-tdTomato/+ reporter mice, these therapeutic effects were associated with selective expansion of c-kitH2B-tdTomato-positive ECs in the infarct border zone (fig. S14D), showing that endogenous METRNL’s effects on angiogenesis and heart function after MI can be therapeutically enhanced. Infarct size and cardiomyocyte apoptosis in the infarct border zone 24 hours after injury (fig. S14, E and F) and cardiomyocyte cell cycle activity in the left ventricle during the first week after MI (fig. S14, G and H) were not affected by METRNL therapy. Of note, treating c-kitH2B-tdTomato/+ mice with SCF expanded the c-kitH2B-tdTomato-positive EC population in the infarct border zone and improved systolic function as did treating the mice with METRNL (fig. S14, I and J). Increasing the availability of either ligand therefore promotes similar beneficial effects after MI.
In summary, we define a METRNL-based crosstalk between myeloid cells and ECs promoting tissue repair after MI. We identify KIT as the cell surface receptor mediating METRNL’s angiogenic effects. While METRNL and SCF bind to KIT with high affinity, engendering concordant phosphoproteome signatures in cultured human ECs, both ligands exert non-redundant functions during development and after injury. In contrast to Scf-deficient mice, which die of severe anemia around birth (26), Metrnl-deficient mice develop normally with no overt hematological abnormalities (fig. S15 and table S6). After MI, myeloid cell-derived METRNL drives expansion of the KIT-expressing EC population in the infarct border zone. This KIT-dependent angiogenic response is severely blunted in Metrnl-deficient mice, indicating a limited role for SCF in this setting, in line with its persistently low cardiac expression after MI (fig. S13 and ref. 27). Myeloid cell production of METRNL may thus provide a mechanism for activating endothelial KIT in the context of tissue injury and inflammation. Further studies may reveal other cell types and pathological conditions in which METRNL-KIT signaling is important.
Supplementary Material
Acknowledgments:
We thank O. Dittrich-Breiholz for advice on the transcriptome and single-cell RNA sequencing analyses and M. Ballmaier for support with flow cytometry. We thank A. Kanwischer and E. Brinkmann for technical assistance.
Funding:
German Research Foundation grant WO 552/10–2 (KCW)
German Research Foundation grant KFO311 WO 552/11–2 (KCW)
German Research Foundation Excellence Cluster REBIRTH-2 grant EXC62 (AS, KCW)
Lower Saxony Ministry of Science and Culture grant Niedersächsisches Vorab VW-ZN3440 (AS, KCW)
China Scholarship Council grant 201606210134 (XW)
China Scholarship Council grant 202008080224 (LZ)
Footnotes
Data and materials availability:
c-kitH2B-tdTomato/+ mice were obtained from Icahn School of Medicine at Mount Sinai, New York City, under a materials transfer agreement. Microarray and single cell RNA sequencing raw data have been deposited in Gene Expression Omnibus (GEO; accession numbers, GSE198376 and GSE198401). All other data are available in the main text or the supplementary materials.
References and Notes
- 1.Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, Lewis TT, Liu J, Loop MS, Lutsey PL, Ma J, Mackey J, Martin SS, Matchar DB, Mussolino ME, Navaneethan SD, Perak AM, Roth GA, Samad Z, Satou GM, Schroeder EB, Shah SH, Shay CM, Stokes A, VanWagner LB, Wang NY, Tsao CW, on behalf of the American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee, Heart disease and stroke statistics–2021 update: A report from the American Heart Association. Circulation 143, e254–e743 (2021). doi: 10.1161/CIR.0000000000000950 [DOI] [PubMed] [Google Scholar]
- 2.Eschenhagen T, Bolli R, Braun T, Field LJ, Fleischmann BK, Frisén J, Giacca M, Hare JM, Houser S, Lee RT, Marbán E, Martin JF, Molkentin JD, Murry CE, Riley PR, Ruiz-Lozano P, Sadek HA, Sussman MA, Hill JA, Cardiomyocyte regeneration: A consensus statement. Circulation 136, 680–686 (2017). doi: 10.1161/CIRCULATIONAHA.117.029343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prabhu SD, Frangogiannis NG, The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ Res 119, 91–112 (2016). doi: 10.1161/CIRCRESAHA.116.303577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu X, Reboll MR, Korf-Klingebiel M, Wollert KC, Angiogenesis after acute myocardial infarction. Cardiovasc Res 117, 1257–1273 (2021). doi: 10.1093/cvr/cvaa287 [DOI] [PubMed] [Google Scholar]
- 5.Swirski FK, Nahrendorf M, Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat Rev Immunol 18, 733–744 (2018). doi: 10.1038/s41577-018-0065-8 [DOI] [PubMed] [Google Scholar]
- 6.Walter W, Alonso-Herranz L, Trappetti V, Crespo I, Ibberson M, Cedenilla M, Karaszewska A, Núñez V, Xenarios I, Arroyo AG, Sánchez-Cabo F, Ricote M, Deciphering the dynamic transcriptional and post-transcriptional networks of macrophages in the healthy heart and after myocardial injury. Cell Rep 23, 622–636 (2018). doi: 10.1016/j.celrep.2018.03.029 [DOI] [PubMed] [Google Scholar]
- 7.Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JWK, Nordon RE, Harvey RP, Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 8, e43882 (2019). doi: 10.7554/eLife.43882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korf-Klingebiel M, Reboll MR, Klede S, Brod T, Pich A, Polten F, Napp LC, Bauersachs J, Ganser A, Brinkmann E, Reimann I, Kempf T, Niessen HW, Mizrahi J, Schönfeld HJ, Iglesias A, Bobadilla M, Wang Y, Wollert KC, Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat Med 21, 140–149 (2015). doi: 10.1038/nm.3778 [DOI] [PubMed] [Google Scholar]
- 9.Reboll MR, Korf-Klingebiel M, Klede S, Polten F, Brinkmann E, Reimann I, Schönfeld HJ, Bobadilla M, Faix J, Kensah G, Gruh I, Klintschar M, Gaestel M, Niessen HW, Pich A, Bauersachs J, Gogos JA, Wang Y, Wollert KC, EMC10 (endoplasmic reticulum membrane protein complex subunit 10) is a bone marrow-derived angiogenic growth factor promoting tissue repair after myocardial infarction. Circulation 136, 1809–1823 (2017). doi: 10.1161/CIRCULATIONAHA.117.029980 [DOI] [PubMed] [Google Scholar]
- 10.Ushach I, Burkhardt AM, Martinez C, Hevezi PA, Gerber PA, Buhren BA, Schrumpf H, Valle-Rios R, Vazquez MI, Homey B, Zlotnik A, Meteorin-like is a cytokine associated with barrier tissues and alternatively activated macrophages. Clin Immunol 156, 119–127 (2015). doi: 10.1016/j.clim.2014.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ushach I, Arrevillaga-Boni G, Heller GN, Pone E, Hernandez-Ruiz M, Catalan-Dibene J, Hevezi P, Zlotnik A, Meteorin-like/meteorin-β is a novel immunoregulatory cytokine associated with inflammation. J Immunol 201, 3669–3676 (2018). doi: 10.4049/jimmunol.1800435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baht GS, Bareja A, Lee DE, Rao RR, Huang R, Huebner JL, Bartlett DB, Hart CR, Gibson JR, Lanza IR, Kraus VB, Gregory SG, Spiegelman BM, White JP, Meteorin-like facilitates skeletal muscle repair through a Stat3/IGF-1 mechanism. Nat Metab 2, 278–289 (2020). doi: 10.1038/s42255-020-0184-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar I, Jedrychowski MP, Ruas JL, Wrann CD, Lo JC, Camera DM, Lachey J, Gygi S, Seehra J, Hawley JA, Spiegelman BM, Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 157, 1279–1291 (2014). doi: 10.1016/j.cell.2014.03.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rupérez C, Ferrer-Curriu G, Cervera-Barea A, Florit L, Guitart-Mampel M, Garrabou G, Zamora M, Crispi F, Fernandez-Solà J, Lupón J, Bayes-Genis A, Villarroya F, Planavila A, Meteorin-like/Meteorin-β protects heart against cardiac dysfunction. J Exp Med 218, e20201206 (2021). doi: 10.1084/jem.20201206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D, Lavine KJ, Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res 124, 263–278 (2019). doi: 10.1161/CIRCRESAHA.118.314028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lennartsson J, Rönnstrand L, Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol Rev 92, 1619–1649 (2012). doi: 10.1152/physrev.00046.2011 [DOI] [PubMed] [Google Scholar]
- 17.Sultana N, Zhang L, Yan J, Chen J, Cai W, Razzaque S, Jeong D, Sheng W, Bu L, Xu M, Huang GY, Hajjar RJ, Zhou B, Moon A, Cai CL, Resident c-kit+ cells in the heart are not cardiac stem cells. Nat Commun 6, 8701 (2015). doi: 10.1038/ncomms9701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SCJ, Middleton RC, Marbán E, Molkentin JD, c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509, 337–341 (2014). doi: 10.1038/nature13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He L, Li Y, Li Y, Pu W, Huang X, Tian X, Wang Y, Zhang H, Liu Q, Zhang L, Zhao H, Tang J, Ji H, Cai D, Han Z, Han Z, Nie Y, Hu S, Wang QD, Sun R, Fei J, Wang F, Chen T, Yan Y, Huang H, Pu WT, Zhou B, Enhancing the precision of genetic lineage tracing using dual recombinases. Nat Med 23, 1488–1498 (2017). doi: 10.1038/nm.4437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kretzschmar K, Post Y, Bannier-Hélaouët M, Mattiotti A, Drost J, Basak O, Li VSW, van den Born M, Gunst QD, Versteeg D, Kooijman L, van der Elst S, van Es JH, van Rooij E, van den Hoff MJB, Clevers H, Profiling proliferative cells and their progeny in damaged murine hearts. Proc Natl Acad Sci U S A 115, E12245–E12254 (2018). doi: 10.1073/pnas.1805829115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsui J, Wakabayashi T, Asada M, Yoshimatsu K, Okada M, Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J Biol Chem 279, 18600–18607 (2004). doi: 10.1074/jbc.M311643200 [DOI] [PubMed] [Google Scholar]
- 22.Kim KL, Seo S, Kim JT, Kim J, Kim W, Yeo Y, Sung JH, Park SG, Suh W, SCF (stem cell factor) and cKIT modulate pathological ocular neovascularization. Arterioscler Thromb Vasc Biol 39, 2120–2131 (2019). doi: 10.1161/ATVBAHA.119.313179 [DOI] [PubMed] [Google Scholar]
- 23.Mol CD, Lim KB, Sridhar V, Zou H, Chien EYT, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE, Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem 278, 31461–31464 (2003). doi: 10.1074/jbc.C300186200 [DOI] [PubMed] [Google Scholar]
- 24.Lennartsson J, Blume-Jensen P, Hermanson M, Pontén E, Carlberg M, Rönnstrand L, Phosphorylation of Shc by Src family kinases is necessary for stem cell factor receptor/c-kit mediated activation of the Ras/MAP kinase pathway and c-fos induction. Oncogene 18, 5546–5553 (1999). doi: 10.1038/sj.onc.1202929 [DOI] [PubMed] [Google Scholar]
- 25.van den Borne SWM, van de Schans VAM, Strzelecka AE, Vervoort-Peters HTM, Lijnen PM, Cleutjens JPM, Smits JFM, Daemen MJAP, Janssen BJA, Blankesteijn WM, Mouse strain determines the outcome of wound healing after myocardial infarction. Cardiovasc Res 84, 273–282 (2009). doi: 10.1093/cvr/cvp207 [DOI] [PubMed] [Google Scholar]
- 26.Ding L, Saunders TL, Enikolopov G, Morrison SJ, Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012). doi: 10.1038/nature10783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vandervelde S, van Luyn MJA, Rozenbaum MH, Petersen AH, Tio RA, Harmsen MC, Stem cell-related cardiac gene expression early after murine myocardial infarction. Cardiovasc Res 73, 783–793 (2007). doi: 10.1016/j.cardiores.2006.11.030 [DOI] [PubMed] [Google Scholar]
- 28.Antrobus R, Borner GH, Improved elution conditions for native co-immunoprecipitation. Plos One 6, e18218 (2011). doi: 10.1371/journal.pone.0018218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S, Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun 1, 100 (2010). doi: 10.1038/ncomms1093 [DOI] [PubMed] [Google Scholar]
- 30.Soonpaa MH, Koh GY, Klug MG, Field LJ, Formation of nascent intercalated disks between grafted fetal cardiomyocytes and host myocardium. Science 264, 98–101 (1994). doi: 10.1126/science.8140423 [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Dembowsky K, Chevalier E, Stüve P, Korf-Klingebiel M, Lochner M, Napp LC, Frank H, Brinkmann E, Kanwischer A, Bauersachs J, Gyöngyösi M, Sparwasser T, Wollert KC, C-X-C motif chemokine receptor 4 blockade promotes tissue repair after myocardial infarction by enhancing regulatory T cell mobilization and immune-regulatory function. Circulation 139, 1798–1812 (2019). doi: 10.1161/CIRCULATIONAHA.118.036053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ, The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204, 3037–3047 (2007). doi: 10.1084/jem.20070885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim YC, Luscinskas FW, Isolation and culture of murine heart and lung endothelial cells for in vitro model systems. Methods Mol Biol 341, 141–154 (2006). doi: 10.1385/1-59745-113-4:141 [DOI] [PubMed] [Google Scholar]
- 34.Junemann J, Birgin G, Erdmann J, Schröder A, Just I, Gerhard R, Pich A, Toxin A of the nosocomial pathogen Clostridium difficile induces primary effects in the proteome of HEp-2 cells. Proteomics Clin Appl 11, 1600031 (2017). doi: 10.1002/prca.201600031 [DOI] [PubMed] [Google Scholar]
- 35.Nel AJM, Garnett S, Blackburn JM, Soares NC, Comparative reevaluation of FASP and enhanced FASP methods by LC-MS/MS. J Proteome Res 14, 1637–1642 (2015). doi: 10.1021/pr501266c [DOI] [PubMed] [Google Scholar]
- 36.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J, The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740 (2016). doi: 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
c-kitH2B-tdTomato/+ mice were obtained from Icahn School of Medicine at Mount Sinai, New York City, under a materials transfer agreement. Microarray and single cell RNA sequencing raw data have been deposited in Gene Expression Omnibus (GEO; accession numbers, GSE198376 and GSE198401). All other data are available in the main text or the supplementary materials.