

SUMMARY
Obesity-induced chronic liver inflammation is a hallmark of nonalcoholic steatohepatitis (NASH)—an aggressive form of nonalcoholic fatty liver disease. However, it remains unclear how such low grade, yet persistent inflammation is sustained in the liver. Here we show that macrophage phagocytic receptor TREM2, induced by hepatocyte-derived sphingosine-1-phosphate, was required for efferocytosis of lipid-laden apoptotic hepatocytes and thereby maintained liver immune homeostasis. However, prolonged hypernutrition led to the production of proinflammatory cytokines TNF and IL-1β in the liver to induce TREM2 shedding through ADAM17-dependent proteolytic cleavage. Loss of TREM2 resulted in aberrant accumulation of dying hepatocytes, thereby further augmenting proinflammatory cytokine production. This ultimately precipitated a vicious cycle that licensed chronic inflammation to drive simple steatosis transition to NASH. Therefore, impaired macrophage efferocytosis is a previously unrecognized key pathogenic event that enables chronic liver inflammation in obesity. Blocking TREM2 cleavage to restore efferocytosis may represent an effective strategy to treat NASH.
eTOC/In Brief
Obesity-induced chronic liver inflammation drives nonalcoholic steatohepatitis (NASH) development, however it remains unclear how such low grade inflammation is sustained in the liver. Wang et al. now show that prolonged hypernutrition dismantles TREM2-dependent macrophage efferocytosis of lipid-laden apoptotic hepatocytes, resulting in their aberrant accumulation to precipitate chronic liver inflammation and NASH progression.
Graphical Abstract
INTRODUCTION
Nonalcoholic steatohepatitis (NASH) is an aggressive form of nonalcoholic fatty liver disease (NAFLD)—a spectrum of liver disorders originating from benign fatty liver (simple steatosis) that can further progress into NASH, cirrhosis, and even hepatocellular carcinoma (HCC)1–5. Chronic liver inflammation plays an essential role in promoting simple steatosis transition into NASH and beyond1,4. Through low-grade yet persistent production of proinflammatory cytokines, including tumor necrosis factor (TNF), interleukin-6 (IL-6) and IL-1β6,7, chronic liver inflammation can perpetuate hepatic wound healing processes, resulting in fibrosis that compromises immunosurveillance in the liver, thereby causing NASH progression and ultimately hepatic failure6–11. However, the molecular mechanism underlying obesity-induced chronic liver inflammation has remained unclear, making it a big hurdle for developing anti-NASH therapies.
Recent single-cell transcriptomic studies in both humans and mice have revealed the emergence of a liver macrophage population characterized by expression of triggering receptor expressed in myeloid cells 2 (TREM2), which is tightly associated with the pathogenic progression of many liver disorders, including NASH12–15, liver cirrhosis16, and HCC17. TREM2 is a single-pass transmembrane receptor of the immunoglobulin superfamily that was initially identified in monocyte-derived dendritic cells and macrophages18. TREM2 interacts with a wide array of ligands19, and recruits adaptor proteins DNAX activation protein 12 (DAP12) or DAP10 to activate spleen tyrosine kinase (Syk) and phosphatidylinositol 3-kinase (PI3K)20–22, thereby directing a signaling network responsible for sensing and restricting tissue damage23. Moreover, TREM2 shedding is a well-documented physiological event that occurs through αsecretases disintegrin and metalloproteinase domain-containing protein 17 (ADAM17) and ADAM10 mediated cleavage at the stalk region, resulting in production and release of soluble TREM2 (sTREM2). The truncated C-terminal fragment of TREM2 is subject to further cleavage by γ-secretase, leading to its dissociation from DAP12 and subsequent signaling blockage24–26. Although recent studies attempted to assess the role of TREM2 in hepatic disorders, the precise function of TREM2 and how it is regulated during NASH progression remains to be defined27–29.
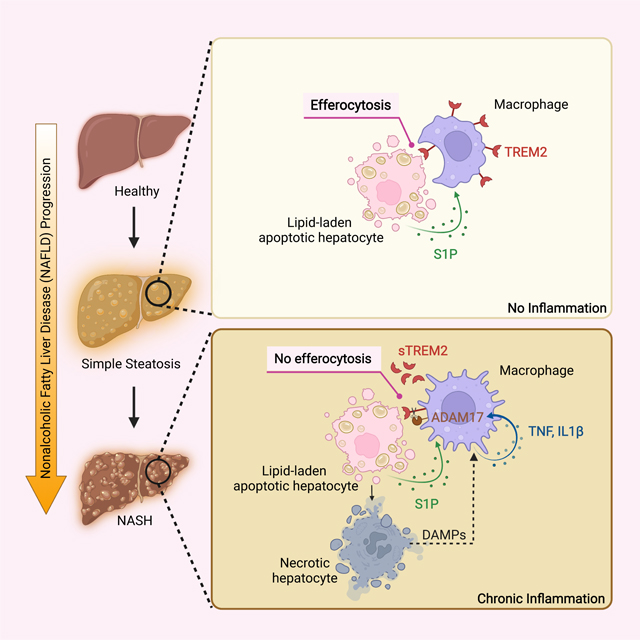
In this study, we found that TREM2 was a gatekeeper that maintained liver immune homeostasis to prevent NASH development. Upregulated by sphigosine-1-phosphate (S1P), TREM2 promoted macrophage-dependent clearance (efferocytosis) of lipid-laden apoptotic hepatocytes, thereby preserving immune silence in simple steatosis. However, prolonged hypernutrition led to the production of TNF and IL-1β in the liver which in turn induced TREM2 shedding through ADAM17-mediated proteolytic cleavage. Loss of TREM2 resulted in the aberrant accumulation of dying hepatocytes, and thereby further augmented proinflammatory cytokine production. This ultimately created a feedforward loop that licensed chronic liver inflammation to drive NASH progression. In summary, our study demonstrates that impaired macrophage efferocytosis is a previously unrecognized key pathogenic event that enables chronic liver inflammation and therefore sheds light on anti-NASH therapy development.
RESULTS
Genetic ablation of Trem2 in macrophages exacerbates NASH pathology
To investigate the role of macrophage TREM2 in NASH, we crossed Trem2F/F with Lyz2-Cre mice30 and generated myeloid cell-specific Trem2 deficient mice (referred as Trem2ΔMye). Since Lyz2-Cre is expressed in both macrophages and neutrophils, we assessed the efficacy of Trem2 deletion in both cell types. Relative to bone marrow derived macrophages (BMDMs) from Trem2F/F mice, Trem2ΔMye BMDMs had minimal TREM2 (Figure S1A). In contrast, TREM2 was not detected in bone marrow neutrophils (Figure S1B)—an observation consistent with an earlier report showing that Trem2 expression is restricted to macrophages within the myeloid cell compartment31. Additionally, deletion of Trem2 in myeloid cells did not affect the baseline systemic nor liver metabolism as shown by comparable body and liver weights, serum alanine aminotransferase (ALT), and aspartate aminotransferase (AST), and serum triglyceride (TG) when mice were fed with a normal chow diet (ND) (Figure S1C).
To recapitulate human NASH pathogenesis, we fed C57BL/6 mice with a western diet (WD) containing high-fat and high-cholesterol and supplemented with high fructose in drinking water32. After 8 weeks of WD feeding, mice displayed increased body weight, liver weight, ALT, AST, serum TG, and liver TG (Figure S1D). Lipid droplets started to accumulate in the hepatocytes of these mice, but no signs of NASH-specific hallmarks, such as liver inflammation and fibrosis, were detected (Figure S1E and S1F; Table S1). In contrast, after 24 weeks of WD feeding, mice began to exhibit key features of metabolic syndrome and NASH, such as weight gain, insulin resistance, elevated ALT and AST, pronounced hepatocellular ballooning, hepatic steatosis, inflammation and progressive fibrosis32,33 (Figure S1D, S1E, and S1F; Table S1). Based on these results, we conclude that WD feeding of C57BL/6 mice for 8 and 24 weeks represents simple steatosis and NASH, respectively.
Next, we assessed the role of macrophage TREM2 in NASH pathogenesis by subjecting Trem2F/F and Trem2ΔMye mice to WD feeding. First, Trem2F/F and Trem2ΔMye mice had similar food intake and exhibited comparable body weight gain throughout 24 weeks of WD feeding (Figure S1G). Consistently, when simple steatosis develops after 8 weeks of WD feeding, Trem2F/F and Trem2ΔMye mice had similar body weights, liver weights, serum TG, total cholesterol, serum NEFA, serum glucose, and serum insulin (Figure S1H). Also, hepatic steatosis (measured by Oil Red O and liver TG) and expression of lipid metabolism related genes, were comparable between Trem2F/F and Trem2ΔMye mice (Figure S1I). However, Trem2ΔMye mice had elevated ALT and AST, enhanced proinflammatory cytokine production, and exacerbated liver fibrosis measured by Sirius red staining, collagen gene expression, liver hydroxyproline amounts, and α-SMA positive areas (Figure 1A and 1B). Of note, Trem2ΔMye mice had a reduction of TREM2 expression in total liver extracts (Figure 1B), suggesting that Trem2 was predominantly expressed in macrophages (Figure S1A and S1B). To further confirm the inhibitory role of TREM2 in NASH progression, we extended WD feeding of Trem2F/F and Trem2ΔMye mice to 24 weeks at which point wild-type (WT) animals with a C57BL/6 genetic background had developed classical hallmarks of NASH (Figure S1D, S1E, and S1F; Table S1). As expected, relative to Trem2F/F mice, Trem2ΔMye mice had exacerbated NASH pathology, as evidenced by enhanced liver inflammation, fibrosis and elevated liver injury (Figure 1C and 1D), with no differences in systemic and hepatic lipid metabolism (Figure S1J, S1K and S1L). Notably, 1 out of 12 Trem2ΔMye mice even developed hepatic tumors (Figure S1M), which was not seen in any of the Trem2F/F mice after 24 weeks of WD feeding. Together, these results indicate that macrophage TREM2 restricts NASH pathogenic progression.
Figure 1. Genetic ablation of Trem2 in macrophages exacerbates NASH pathology.

(A and C) Representative H&E, Sirius Red/Fast Green, and α-SMA staining of liver sections from Trem2F/F and Trem2ΔMye mice fed with western diet (WD) for 8 (A) and 24 (C) weeks. Sirius Red/Fast Green staining was detected under polarized light. WD_8w, n = 8 mice per group. WD_24w Trem2F/F, n = 12 mice; WD_24w Trem2ΔMye, n = 14 mice.
(B and D) Serum ALT, AST, hepatic hydroxyproline, and TNF amounts in liver tissue from mice as in (A and C) were measured. Relative mRNA expression of fibrosis-related genes, inflammatory genes, and Trem2 in liver tissue were analyzed by RT-qPCR.
Scale bar, 100 μm. Data are shown as mean ± s.e.m.. *p<0.05; **p<0.01; ***p<0.001. See also Figure S1 and Table S1.
Hepatocyte-derived S1P upregulates TREM2 in infiltrated liver macrophages
Recent studies showed that TREM2 expression in liver macrophages is elevated in patients with NASH, cirrhosis, and HCC12,16,17, which is reproduced in relevant mouse models13,27,34. However, the molecular mechanism governing TREM2 upregulation remains unclear. To bridge this knowledge gap, we treated AML12 cells and primary hepatocytes with palmitic acid (PA)—the dominant form of saturated free fatty acid found in hypernutritional diets—to mimic lipid-induced hepatic stress and injury35,36. As shown in Figure S2A, treatment of hepatocytes with 800 μM PA, a concentration within the pathophysiological range37, led to early apoptosis within 24 hrs. Importantly, co-culturing of these PA-treated apoptotic AML12 cells or primary murine hepatocytes with BMDMs in a transwell system resulted in TREM2 upregulation in macrophages (Figure 2A and 2B), suggesting that certain soluble factor(s) released from apoptotic hepatocytes is responsible for upregulating TREM2 in macrophages. Consistently, BMDMs directly stimulated with the conditioned medium from PA-treated apoptotic AML12 cells (ACM) also upregulated TREM2 in a time-dependent manner (Figure 2C). In contrast, conditioned medium from either viable (LCM) or necrotic (NCM) hepatocytes, achieved by varying the doses of PA (Figure S2A), failed to do so (Figure 2D), suggesting that a soluble factor(s) capable of upregulating TREM2 in macrophages is specifically released from apoptotic, but not live nor necrotic, hepatocytes. Of note, we did not detect TREM2 upregulation in Kupffer cells upon coculturing with PA-treated apoptotic hepatocytes or direct ACM stimulation (Figure S2B). Lastly, neither PA nor cholesterol alone had any stimulatory effect on Trem2 expression in BMDMs (Figure S2C).
Figure 2. Obesity upregulates TREM2 in infiltrated liver macrophages via hepatocyte derived S1P.

(A and B) RT-qPCR analysis of Trem2 mRNA and immunoblot analysis of TREM2 in BMDMs after coculturing with palmitic acid (PA)-treated AML12 cells (A) or murine primary hepatocytes (B) in a transwell system.
(C) RT-qPCR analysis of Trem2 mRNA and immunoblot analysis of TREM2 in BMDMs after stimulation with cell culture medium collected from 800 μM PA-treated AML12 cells.
(D) RT-qPCR analysis of Trem2 mRNA and immunoblot analysis of TREM2 in BMDMs after stimulation with fresh culture medium (Ctrl) or culture medium collected from AML12 cells treated with PA of various concentrations for 24 hours. LCM, live cell culture medium; ACM, apoptotic cell culture medium; NCM, necrotic cell culture medium.
(E) RT-qPCR analysis of Trem2 mRNA and immunoblot analysis of TREM2 in BMDMs treated with S1P for 24 hours.
(F) RT-qPCR analysis of Trem2 mRNA and immunoblot analysis of TREM2 in BMDMs treated with S1P.
(G and H) S1P concentrations were quantified by ELISA in cell culture media collected from PA-treated AML12 cells (G) or liver tissue from C57BL/6 mice (H).
(I and J) RT-qPCR analysis of Trem2 mRNA, immunoblot analysis of TREM2, and flow cytometry analysis of cell surface TREM2 in BMDMs stimulated with S1P. BMDMs were either pretreated with VPC (VPC23019, an S1PR1/3 inhibitor), JTE (JTE-013, an S1PR2 inhibitor) (I), or transduced with lentiviral constructs expressing two different shRNAs against S1pr1 (J).
(K-M) Immunoblot analysis of TREM2 in BMDMs stimulated with cell culture medium of AML12 cells. BMDMs were either pretreated with VPC (K) or transduced with lentiviral constructs expressing shS1pr1 (L). LCM and NCM were collected from AML12 cells transduced with lentiviral constructs expressing indicated sgRNAs (M).
(N) RT-qPCR analysis of Trem2 mRNA and immunoblot analysis of TREM2 in liver tissue from C57BL/6 mice that were treated with VPC or PBS. n=6 mice per group.
Data are shown as mean ± s.e.m.. *p<0.05; **p<0.01; ***p<0.001; NS, not significant. All in vitro experiments were repeated independently at least three times. See also Figure S2.
Prompt clearance of the dying cells is key to maintaining tissue homeostasis38,39. This process starts with the release of ‘find-me’ signals from apoptotic cells that attract phagocytes, a process mediated by binding of ‘find-me’ signals to their receptors on the phagocyte surface38–40. In addition to chemotaxis, ‘find-me’ signals also prepare phagocytes for engulfment through upregulating cell surface phagocytic receptors41–43. To date, at least four classical dying cell released ‘find-me’ signals have been identified, including lysophosphatidylcholine (LPC)44,45, sphingosine-1-phosphate (S1P)46,47, CX3C motif chemokine ligand 1 (CX3CL1)45, and nucleotides (ATP and UTP)48. To test whether any of these ‘find-me’ signals induces Trem2 expression in macrophages, we treated BMDMs with individual ‘find-me’ signals and found that S1P, but not others, increased Trem2 mRNA and protein in a dose- and time-dependent manner (Figure 2E, 2F, and S2D). Moreover, RNA-seq analysis confirmed upregulation of Trem2 mRNA by S1P and further revealed that several of the previously identified cellular markers enriched in Trem2 expressing macrophages (including Cd36, Cd9, and Gpnmb) were also slightly upregulated upon S1P stimulation (Figure S2E). However, S1P did not upregulate TREM2 in Kupffer cells isolated from healthy mouse livers (Figure S2F). Notably, although minimally detected in LCM and NCM, the concentration of S1P increased to ~500 nM in ACM (Figure 2G). Consistent with these in vitro results, S1P amounts in liver tissue were much higher in simple steatosis and NASH, relative to healthy animals (Figure 2H), which positively correlated with the upregulated Trem2 mRNA during NAFLD progression (Figure 3A).
Figure 3. Uncoupled regulation of Trem2 mRNA and its protein during NASH development.

(A and B) RT-qPCR analysis of Trem2 mRNA (A) and immunoblot analysis of TREM2 (B) in liver tissue from C57BL/6 mice. ND, n = 8 mice; WD_8w, n = 8 mice; WD_24w, n= 7 mice.
(C) Representative immunofluorescent staining of F4/80 (Red) and TREM2 (Green) in liver sections. The percentage of double positive cells (F4/80+TREM2+) in total number of macrophages (F4/80+) was quantified.
(D and E) RT-qPCR analysis of Trem2 mRNA (D) and immunoblot analysis of TREM2 (E) in primary liver macrophages isolated from C57BL/6 mice. ND, n = 3 mice; WD_8w, n = 10 mice; WD_24w, n = 6 mice.
(F) Flow cytometry analysis of CLEC4F and TIM-4 in F4/80+ liver macrophages isolated from C57BL/6 mice using anti-CD11b magnetic beads. Cell surface TREM2 in TIM-4−CLEC4F− recruited macrophages and TIM-4+CLEC4F+ resident macrophages was further quantified by MFI.
(G) Database (GSE89632, n = 63 and GSE130970, n = 78) analysis of TREM2 mRNA in two independent clinical cohorts of individuals with healthy, simple steatosis (SS), and NASH livers.
(H) Representative immunofluorescent staining of CD68 and TREM2 in human liver sections.
(I and J) immunoblot analysis of TREM2 (I) and RT-qPCR analysis of Trem2 mRNA (J) in BMDMs treated with TNF or IL-1β for 24 hours, or with 1 ng/ml of TNF or IL-1β for different time durations (hours).
(K) Flow cytometry analysis of cell surface TREM2 in BMDMs treated with 1 ng/ml TNF or IL-1β for 24 hours.
Scale bar, 100 μm. Data are shown as mean ± s.e.m.. *p<0.05; ***p<0.001. All in vitro experiments were repeated independently at least three times. See also Figure S3 and Table S2.
S1P is a lipid secondary messenger that signals through S1P receptors, S1PR1–549, albeit expression of these receptors appears to vary among different animal species, strains, and macrophage subtypes50. Consistent with a previous study51, we found that S1PR1 and S1PR2 were predominantly expressed in primary macrophages from C57BL/6 mice (Figure S2G). To determine whether S1PR1 or S1PR2 (or both) is responsible for S1P-induced TREM2 upregulation in macrophages, we treated BMDMs with S1PR1 and S1PR2 inhibitors prior to S1P stimulation. Pretreatment with VPC23019 (an S1PR1 and S1PR3 specific antagonist), but not JTE-013 (an S1PR2 specific antagonist), dampened S1P-induced TREM2 upregulation (Figure 2I and S2H). Similar results were obtained using shRNAs to silence S1pr1 and S1pr2 in BMDMs (Figure 2J and S2I). Furthermore, an S1PR1 selective agonist CYM-544252 was also capable of inducing TREM2 in BMDMs (Figure S2J). Next, we tested if S1P plays a dominant role in mediating ACM-induced TREM2 upregulation in macrophages. Indeed, ACM failed to upregulate TREM2 in the presence of an S1PR1-specific antagonist or shRNA (Figure 2K and 2L), but not S1PR2-specific ones (Figure S2K and S2L), suggesting that the S1P-S1PR1 signaling axis was responsible for TREM2 upregulation. Lastly, since S1P is generated from sphingosine by sphingosine kinases SphK146 and SphK247, and the expression of both kinases in hepatocytes can be induced by PA treatment (Figure S2M), we further tested whether deletion of SphK1 and SphK2 would affect the release of S1P when these cells undergo apoptosis. As expected, stable deletion of SphK1 and SphK2 in apoptotic AML12 cells completely abrogated S1P production (Figure S2N), resulting in the failure of TREM2 upregulation in BMDMs (Figure 2M). In line with these in vitro findings, weekly injection of an S1PR1 and S1PR3 antagonist (VPC23019) in WT mice dampened WD-induced TREM2 upregulation, demonstrating that S1P-S1PR1 signaling axis is responsible for TREM2 upregulation in liver macrophages in vivo (Figure 2N). Taken together, these results suggest that in response to dietary hypernutrition, lipid-laden apoptotic hepatocytes release S1P that signals through S1PR1 on liver macrophages to upregulate TREM2.
TREM2 protein, but not its mRNA, sharply declines in NASH-associated macrophages
Although both publicly available scRNA-seq results and our own findings obtained from the WD-induced NASH model support the notion that Trem2 mRNA in liver macrophages is continuously elevated in simple steatosis and NASH12 (Figure 3A), TREM2 protein has never been carefully examined. Unexpectedly, we found that although elevated in simple steatosis (WD_8w), TREM2 protein was drastically declined in NASH (WD_24w) (Figure 3B). Consistently, immunofluorescent staining of TREM2 and F4/80 confirmed that TREM2 was highly expressed in macrophages from simple steatosis livers, but barely detected in NASH livers (Figure 3C). Moreover, we confirmed that the uncoupled regulation of Trem2 mRNA and its protein was also evident in freshly isolated primary liver macrophages (Figure 3D, 3E, S3A and S3B). Furthermore, flow cytometry analysis suggested that Kupffer cells (F4/80+TIM4+CLEC4F+) minimally expressed TREM2 during the entire pathogenic progression of NASH (Figure 3F) and most of TREM2+ liver macrophages found in simple steatosis (WD_8w) were CX3CR1high (Figure S3C)—a well-defined marker of infiltrated monocyte-derived macrophages53,54. These results, consistent with previous studies13,15,55, suggest that dietary obesity-induced TREM2 upregulation in liver is largely restricted to the infiltrated monocyte-derived macrophage population.
To rule out TREM2 protein decline may tie specifically to the WD-induced NASH model, we analyzed Trem2 mRNA and its protein amounts in another well-established transgenic NASH model that is achieved by high-fat diet (HFD) feeding of MUP-uPA mice36. Consistent with the WD model, TREM2 protein was drastically decreased in NASH (HFD MUP-uPA) relative to simple steatosis livers (HFD WT), despite a continuous elevation of its mRNA throughout the course of NASH pathogenesis (Figure S3D). Lastly, we assessed whether liver macrophages in NASH patients exhibited a similar uncoupled regulation of TREM2 mRNA and its protein. Consistent with previous findings12, analysis of the transcriptome profile of two independent cohorts of NAFLD patients with full histological spectrum of NAFLD (GSE89632 and GSE130970)56,57 indicated that TREM2 mRNA were elevated in livers from simple steatosis and NASH patients relative to healthy donors (Figure 3G). Furthermore, we found that human TREM2 was largely expressed in liver macrophages, as evidenced by the overlay between CD68 (a human macrophage marker) and TREM2 on cryosections of human liver steatosis biopsies (Figure 3H; Table S2). In sharp contrast, TREM2 positive staining was barely detected in livers from NASH patients (Figure 3H; Table S2), indicating that TREM2 protein also sharply declines in human NASH-associated liver macrophages. The specificity of the antibodies against mouse and human TREM2 used for immunofluorescent staining was tested and confirmed in BMDMs from Trem2ΔMye mice and human THP-1 cells in which TREM2 was deleted using the CRISPR/Cas9 method (Figure S3E).
TNF and IL-1β reduce TREM2 protein in NASH-associated macrophages
A key hallmark that distinguishes NASH from simple steatosis is the onset of chronic liver inflammation, characterized by persistent yet low-grade production of proinflammatory cytokines, such as TNF, IL-6, and IL-1β10,36,58,59. We therefore sought to determine whether any of these proinflammatory cytokines is involved in promoting TREM2 protein decline. To this end, we treated WT BMDMs with TNF, IL-6, and IL1-β individually and found that TNF and IL-1β, but not IL-6, were able to decrease TREM2 protein in a dose-dependent manner (Figure 3I and S3F) while having no effects on its mRNA (Figure 3J). Consistently, flow cytometry analysis confirmed that surface TREM2 in BMDMs was reduced when stimulated with either TNF or IL-1β (Figure 3K). Moreover, TNF and IL-1β were still capable of reducing TREM2 protein abundance even after S1P pretreatment that upregulates Trem2 expression (Figure S3G, S3H, and S3I). Together, these results suggest that the presence of TNF and IL-1β leads to TREM2 protein decline in NASH-associated macrophages.
TNF and IL-1β induce TREM2 proteolytic cleavage by activating ADAM17
Next, we sought to delineate the molecular mechanism responsible for TREM2 protein decline in NASH-associated macrophages. Previous studies indicate that the full-length TREM2 protein is subject to proteolytic cleavage by ADAM10 and ADAM17, leading to the production of soluble TREM2 (sTREM2) and a C-terminal fragment (CTF) prone to subsequent proteolysis by γ secretase24,26,60. Therefore, we first quantified sTREM2 amounts in the circulation of mice with simple steatosis and NASH. We found that sTREM2 was increased in the sera of NASH mice (WD_24w) relative to those from simple steatosis (WD_8w) or healthy mice (ND) (Figure 4A), suggesting that TREM2 cleavage was specifically enhanced in NASH. Consistent with the WD-induced NASH model, circulating sTREM2 was also increased when NASH developed in the HFD fed MUP-uPA mice (Figure S4A), ruling out the possibility that sTREM2 elevation in the circulation ties to a specific mouse model of NASH. In further support of our findings, sTREM2 elevation in NASH was also reported in two recent studies that were published while our paper was in revision29,61.
Figure 4. TNF and IL-1β induce TREM2 proteolytic cleavage by activating ADAM17.

(A) ELISA analysis of sTREM2 in mouse sera of C57BL/6 mice. ND, n = 8 mice; WD_8w, n = 8 mice; or WD_24w, n = 7 mice.
(B) ELISA analysis of sTREM2 in cell culture medium from BMDMs treated with indicated stimuli for 24 hours. S1P (500 nM), TAPI (ADAM17 inhibitor, 10 μM) TNF or IL-1β (1 ng/ml). **p < 0.01 vs. Ctrl group; ##p < 0.01 vs. TNF treated group; &&p < 0.01 vs. IL-1β treated group.
(C and D) RT-qPCR analysis of Adam17 mRNA and immunoblot analysis of ADAM17 in BMDMs stimulated with TNF (C) or IL-1β (D).
(E) Relative ADAM17 activity was quantified in BMDMs after 24 hours of TNF (1 ng/ml) or IL-1β (1 ng/ml) stimulation.
(F) Immunoblot analysis of TREM2 in BMDMs treated with 10 μM TAPI (an ADAM17 inhibitor) or 20 μM GI (an ADAM10 inhibitor) in combination with 1 ng/ml of TNF or IL-1β for 24 hours.
(G) Flow cytometry analysis of cell surface TREM2 in BMDMs pretreated with 10 μM TAPI followed by stimulation with 1 ng/ml of TNF or IL-1β for 24 hours. MFI was used for flow cytometry analysis.
(H) Immunoblot analysis of ADAM10 and ADAM17 in primary liver macrophages isolated from C57BL/6 mice.
(I) ELISA analysis of sTREM2 in human plasma from individuals with healthy, steatosis, and NASH livers (n = 20 subjects per group).
(J) Relative mRNA expression of ADAM17, IL-1B, and TNF in human liver tissue in a clinical cohort (GSE130970, n = 78). Healthy, n = 6; simple steatosis (SS), n = 14; NASH, n = 58.
(K and L) Association of TREM2 expression in human liver tissue with their pathohistological annotations, SPHK1 and SPHK2 expression, Sphingosine biosynthesis, and S1P receptor activity in a clinical cohort of NAFLD patients (GSE130970, n = 78).
Data are shown as mean ± s.e.m.. *p<0.05; **p<0.01; ***p<0.001; NS, not significant. All in vitro experiments were repeated independently at least three times. See also Figure S4 and Table S3.
Based on the above results, we hypothesized that TNF and IL-1β may reduce TREM2 protein by inducing its proteolytic cleavage. Indeed, both TNF and IL-1β promoted TREM2 shedding in BMDMs, as shown by drastically elevated sTREM2 amounts in the culture medium after cytokine stimulation (Figure 4B). Furthermore, we observed that TREM2 CTF started to accumulate in the presence of an γ secretase inhibitor (DAPT), and the accumulation was further enhanced upon TNF or IL-1β stimulation (Figure S4B), confirming that TNF and IL-1β induce TREM2 cleavage, leading to the production of sTREM2 and TREM2 CTF. When assessing whether ADAM10 or ADAM17, or both, is involved in full length TREM2 cleavage, we found that TNF and IL-1β increased the expression of ADAM17 (Figure 4C and 4D), but not ADAM10 (Figure S4C and S4D). More importantly, the enzymatic activity of ADAM17, but not ADAM10, was elevated upon TNF or IL-1β treatment (Figure 4E and S4E). Consistently, pretreatment of the ADAM17 specific inhibitor (TAPI), but not the ADAM10 inhibitor (GI), greatly attenuated TNF and IL-1β-induced decline of TREM2 full-length protein (Figure 4F). In support of this finding, TAPI treatment almost completely abolished TNF and IL-1β-induced sTREM2 production (Figure 4B). Similar results were seen when analyzing the abundance of cell surface TREM2 by flow cytometry (Figure 4G). We noticed that TAPI treatment alone was able to increase cell surface TREM2 abundance, indicating that TREM2 may constantly undergo ADAM17-mediated proteolytic cleavage in BMDMs (Figure 4F and 4G). In further support of our results obtained using pharmacological approaches, lentivirus-mediated shRNA silencing of ADAM17 (Figure S4F, S4G, and S4H), but not ADAM10 (S4I), prevented TNF and IL-1β-induced TREM2 cleavage and sTREM2 production. Lastly, we found that ADAM17 protein was increased in freshly isolated NASH-associated macrophages (WD_24w) compared to liver macrophages from healthy (ND) or simple steatosis (WD_8w) livers (Figure 4H).
To validate our mice findings in humans, we first examined sTREM2 concentration in the circulation of healthy donors and NAFLD patients. Consistent with our mouse results, sTREM2 was also specifically elevated in sera of NASH patients compared to those from simple steatosis patients or healthy individuals (Figure 4I; Table S3). Moreover, sTREM2 elevation in the circulation correlated with an enhanced liver tissue expression of ADAM17 in NASH patients (Figure 4J, GSE130970). Next, we analyzed the association between hepatic TREM2 gene expression and S1P signaling pathway activity using the above-mentioned patient cohort56. We observed a statistically significant positive correlation between TREM2 and SPHK1 gene expression during NAFLD progression (Figure 4K and 4L). Consistently, GSEA pathway enrichment analysis also confirmed a strong positive correlation between TREM2 gene expression and S1P biosynthesis pathway as well as its receptor activity (Figure 4K and 4L). Moreover, a positive correlation between TNF and IL-1 signaling pathway activities and ADAM17 expression was also observed (Figure S4J). Taken together, these human results, which are consistent with our mouse data, suggest that proinflammatory cytokines, namely TNF and IL-1β, promote TREM2 proteolytic cleavage by activating ADAM17 in NASH patients, and that sTREM2 may represent a NASH biomarker.
TREM2 is required for macrophage-dependent efferocytosis of lipid-laden apoptotic hepatocytes
To understand how macrophage TREM2 prevents NASH development, we focused on pathways involved in clearance of dying cells (also known as “efferocytosis”) because analysis of gene sets from the KEGG enrichment database62 revealed that TREM2-dependent genes are highly involved in phagosome and lysosome functions (Figure 5A and 5B). Additionally, KEGG enrichment analysis also indicated that ACM treatment resulted in upregulated phagosome and lysosome-related genes in macrophages (Figure S5A). Consistently, Ingenuity Pathways Analysis (IPA)63 predicted a strong enhancement in biological processes related to phagocytosis upon ACM treatment of macrophages (z-score>1.5, P<0.01, Figure S5B). In line with these results, analysis of freshly isolated liver macrophages from WD-fed Trem2F/F and Trem2ΔMye mice revealed that TREM2 deficiency resulted in changes in the expression of genes involved in phagocytosis and phagosome functions (Figure 5C, 5D, and S5C). These results together prompted us to evaluate dying hepatocyte turnover during NASH progression. Although barely detected in healthy or simple steatosis livers, apoptotic hepatocytes started to accumulate in NASH livers (WD_24 weeks) (Figure S5D). This observation is in full agreement with earlier reports conducted in NASH patients64,65 and an independent murine NASH model36. To further validate our results, we measured the amounts of active caspase-3 (αCasp3) in livers from healthy (ND), simple steatosis (WD_8w), and NASH (WD_24w) mice and observed a sharp elevation of αCasp3 in NASH livers (Figure S5E). Based on these data, we speculated that TREM2 may promote phagocytosis of apoptotic hepatocytes to restrict their aberrant accumulation in the liver. Indeed, 8 weeks of WD feeding led to the detection of TUNEL positive cells in Trem2ΔMye livers, but not in their WT counterparts (Figure 5E), and we further confirmed that these TUNEL positive cells were hepatocytes (Figure S5F). Consistently, αCasp3 was also increased in livers from Trem2ΔMye mice after 8 weeks of WD feeding (Figure 5F). In further support, flow cytometry analysis of BMDMs or freshly isolated liver macrophages from WT obese mice (WD_8w) suggested that only TREM2high macrophages were able to engulf pHrodo-Red labeled apoptotic hepatocytes, whereas TREM2low cells or liver macrophages freshly isolated from Trem2ΔMye obese mice were incapable of doing so (Figure 5G). Lastly, relative to simple steatosis (WD_8w), although the full length TREM2 sharply declined in NASH (WD_24w), its amount in NASH was still higher compared to healthy livers (Figure 3B and 3E). This may explain why we could still detect an enhanced accumulation of TUNEL positive cells (Figure S5G) and exacerbated NASH pathology in the livers of Trem2ΔMye mice after 24 weeks of WD feeding (Figure 1C and 1D).
Figure 5. TREM2 is essential for macrophage efferocytosis of lipid-laden apoptotic hepatocytes.

(A) KEGG pathway enrichment analysis of differentially expressed genes in Trem2−/− (n = 3) and WT (n = 4) BMDMs.
(B) Heatmap of phagocytosis related genes (GO:0006909) that were downregulated in Trem2−/− (n = 3) BMDMs compared to WT (n = 4) BMDMs.
(C) IPA of phagocytosis related functions in liver macrophages isolated from WD fed Trem2F/F and Trem2ΔMye mice. n = 4 mice per group.
(D) GSEA reveals phagosome related genes are enriched in primary liver macrophages isolated from Trem2F/F mice compared to Trem2ΔMye mice after WD feeding. n = 4 mice per group.
(E) Representative TUNEL staining of liver sections from Trem2F/F and Trem2ΔMye mice fed with WD for 8 weeks (n = 8 mice per group).
(F) Immunoblot analysis of aCasp3 (cleaved caspase-3), Casp3 (caspase-3), and GAPDH in liver tissue from Trem2F/F and Trem2ΔMye mice fed with WD for 8 weeks (n = 3 mice per group).
(G) BMDMs (left panel) or primary liver macrophages (right panel) from Trem2F/F and Trem2ΔMye mice were cocultured for 4 hours with AML12 cells that were labeled with pHrodo™ Red and treated with PA to induce apoptosis. Flow cytometry analysis of TREM2 and pHrodo™ Red was then performed for assessing the efficacy of macrophage efferocytosis of apoptotic hepatocytes.
(H) PA-treated apoptotic AML12 cells were cocultured with Trem2F/F and Trem2ΔMye BMDMs for 2 hours. n = 6 per group.
(I) PA-treated apoptotic AML12 cells were cocultured with WT BMDMs that were pretreated with either TREM2 neutralizing antibody (Anti-TREM2, 200 ng/ml) or an isotype control antibody (IgG2B). n = 6 per group.
(J) PA-treated primary hepatocytes were cocultured with primary liver macrophages from C57BL/6 mice. n = 6 per group.
(K) WT BMDMs were cocultured with AML12 cells that were treated with PA to induce apoptosis followed by incubation with recombinant sTREM2 (200 ng/ml). n = 6 per group.
Scale bar, 100μm. Efferocytosis was quantified as the percentage of BMDMs engulfing apoptotic AML12 cells. Data are shown as mean ± s.e.m.. **p<0.01; ***p<0.001. All in vitro experiments were repeated independently at least three times. See also Figure S5, Video S1, and Video S2.
To recapitulate these in vivo and ex vivo findings in tissue culture dishes, we treated AML12 cells (labeled in red) with 800 μM PA for 24 hours to induce apoptosis. Then these cells were cocultured with BMDMs (labeled in green) isolated from Trem2F/F or Trem2ΔMye mice. Although green fluorescence labeled Trem2F/F BMDMs were able to engulf apoptotic hepatocytes within 4 hours, Trem2ΔMye BMDMs were largely defective at engulfing these dying cells (Figure 5H; Video S1 and S2). Similar results were seen when pretreating BMDMs with a TREM2 inhibitory antibody (Figure 5I) or repeating the experiment using human hepatocyte (THLE-3) and macrophage (THP-1) cell lines (Figure S5H and S5I). In line with these results, coculture of apoptotic primary hepatocytes with freshly isolated CD11b+ liver macrophages from healthy (ND), simple steatosis (WD_8w) and NASH (WD_24w) livers showed that macrophages from NASH livers were defective at clearing apoptotic hepatocytes when compared with cells from simple steatosis livers (Figure 5J). We also found that S1P promoted the engulfment of apoptotic hepatocytes by macrophages, which is consistent with its recently reported role in microglia in the context of ischemic brain injury66, whereas TNF and IL-1β compromised such engulfment, even in the presence of S1P (Figure S5J). Additionally, S1PR1 inhibitor (VPC) and ADAM17 inhibitor (TAPI) were able to block S1P- or TNF and IL-1β-induced effects on phagocytosis respectively (Figure S5J), consistent with their respective roles in regulating TREM2 abundance shown above. Taken together, these results suggest that TREM2 is required for macrophage efferocytosis of lipid-laden apoptotic hepatocytes. Lastly, pretreatment of sTREM2 inhibited the engulfment of apoptotic hepatocytes by macrophages from both mice and humans (Figure 5K and S5K), indicating that sTREM2 may act as a decoy receptor to competitively inhibit the function of cell surface full-length TREM2.
Loss of full-length, but not soluble, TREM2 is responsible for the exacerbated NASH pathology in Trem2ΔMye mice
We have demonstrated that Trem2ΔMye mice exhibited greatly exacerbated NASH pathology upon WD feeding, suggesting an inhibitory role of TREM2 in NASH development (Figure 1 and S1). However, it should be noted that genetic deletion of Trem2 also eliminates sTREM2 that is generated upon full-length TREM2 cleavage by ADAM17 (Figure 4 and S4). It is therefore possible that the absence of sTREM2, rather than full-length TREM2, may contribute to the exacerbated NASH pathology in WD-fed Trem2ΔMye mice, especially given that sTREM2 is biologically active in various circumstances67,68. To test this possibility, we conducted a proof-of-concept “rescue” experiment by putting back sTREM2 into TREM2-deficient mice and their WT control littermates and determined if sTREM2 administration would influence NASH development in these mice. Weekly injection (i.v.) of sTREM2 (1 μg/mouse) led to an average serum concentration of ~ 100 ng/ml during the treatment course (Figure S6A). However, sTREM2 administration failed to rescue NASH development in Trem2ΔMye mice (Figure 6A, 6B, and 6C). Of note, although a comparable NASH-associated pathology was seen between PBS and sTREM2 treated WD-fed Trem2ΔMye mice, sTREM2 administration resulted in NASH development in Trem2F/F mice after 8 weeks of WD feeding, as shown by increased serum ALT and AST, enhanced proinflammatory cytokine production and exacerbated hepatic fibrosis (Figure 6A, 6B, and 6C), with no changes in body and liver weights and hepatic steatosis (Figure 6D). Consistently, sTREM2 administration promoted the accumulation of TUNEL-positive apoptotic hepatocytes in Trem2F/F, but not Trem2ΔMye, mice (Figure 6E). Lastly, we repeated the same sTREM2 rescue experiment using TREM2 germline KO (Trem2−/−) mice and obtained similar results as seen in Trem2ΔMye mice (Figure S6B, S6C, S6D, and S6E). In summary, these results together with the inhibitory effect of sTREM2 on efferocytosis (Figure 5K and S5K) suggest that the absence of full-length TREM2, but not sTREM2, is responsible for the exacerbated NASH pathology in Trem2ΔMye mice, and that sTREM2 promotes NASH progression likely through competitively inhibiting full-length TREM2 mediated engulfment of lipid-laden apoptotic hepatocytes.
Figure 6. Absence of sTREM2 did not contribute to the exacerbated NASH pathology in WD-fed Trem2ΔMye mice.

Trem2F/F and Trem2ΔMye mice were fed with WD for 8 weeks and treated with PBS or sTREM2 (weekly i.v. injection, 1 μg/mouse). n = 8 mice per group.
(A) Representative histological results of liver sections stained with H&E, Sirius Red/Fast Green, and α-SMA. Sirius Red/Fast Green staining was detected under polarized light.
(B) Serum ALT, AST, and hepatic hydroxyproline amounts were measured.
(C) Relative mRNA expression of fibrosis-related genes, inflammatory genes, and Trem2 was measured by RT-qPCR in liver tissue.
(D) Liver weight, body weight, serum TG, and liver TG were analyzed.
(E) Apoptotic cells in liver tissue were stained with TUNEL and quantified.
Scale bar, 100 μm. Data are shown as mean ± s.e.m.. *p<0.05; **p<0.01; ***p<0.001; NS, not significant. See also Figure S6.
TREM2 is a gatekeeper that maintains liver immune homeostasis to restrict NASH development
Given that TREM2 is required for macrophage efferocytosis of lipid-laden apoptotic hepatocytes (Figure 5) and that Trem2ΔMye, but not Trem2F/F, mice exhibited classical NASH hallmarks, including chronic hepatic inflammation and fibrosis, after 8 weeks WD feeding (Figure 1 and S1), we hypothesized that TREM2 may function as a gatekeeper that maintains immune homeostasis in simple steatosis to restrict NASH development. Consistent with earlier studies36,69, 18 or 24 weeks HFD feeding only led to simple steatosis development in WT animals (Figure 7A, 7B, S7A, and S7B). However, Trem2ΔMye mice developed NASH pathology as shown by the enhanced ALT and AST, upregulated proinflammatory cytokine production, and hepatic fibrosis 18 weeks post HFD feeding (Figure 7A and 7B). Such NASH phenotype was further exacerbated 24 weeks after HFD feeding (Figure S7A and S7B), despite no differences in systemic and hepatic lipid metabolism (Figure 7C and S7C). Moreover, accumulation of apoptotic hepatocytes was evident in Trem2ΔMye, but absent in Trem2F/F mice after 18 or 24 weeks HFD feeding (Figure 7D and S7D). Furthermore, RNA-seq analysis of liver macrophages freshly isolated from ND and HFD fed WT mice revealed that phagocyte-related functions were upregulated in response to HFD feeding (Figure 7E). Consistently, Trem2 was among the most upregulated genes associated with phagocytosis-related functions (GO:0006909) after HFD feeding (Figure 7F). Together, these results establish TREM2 as a gatekeeper that restricts aberrant accumulation of lipid-laden apoptotic hepatocytes to maintain liver immune homeostasis, thereby preventing simple steatosis transition to NASH.
Figure 7. HFD feeding is sufficient to induce NASH development in Trem2ΔMye mice.

(A) Representative H&E, Sirius Red/Fast Green, and α-SMA staining of liver sections from Trem2F/F and Trem2ΔMye mice fed with HFD for 18 weeks, n = 6 mice per group. Sirius Red/Fast Green staining was detected under polarized light.
(B) Serum ALT, AST, hepatic hydroxyproline, and TNF amounts in liver tissue from mice as in (A) were measured. Relative mRNA expression of fibrosis-related genes, inflammatory genes, and Trem2 was analyzed by RT-qPCR using liver tissue, n = 6 mice per group.
(C) Representative Oil Red O staining of liver sections from mice as in (A). Scale bar, 100 μm. Body weight, liver weight, serum TG, liver TG, and total cholesterol from these mice were also analyzed. Relative mRNA expression of lipid-associated genes in liver tissue was measured by RT-qPCR. n = 6 mice per group.
(D) Apoptotic cells in liver tissue from mice as in (A) were stained with TUNEL and quantified. n = 6 mice per group.
(E) IPA on phagocytosis related functions in primary liver macrophages isolated from WT C57BL/6 mice that were fed with HFD (n=3) or ND (n=3) for 18 weeks.
(F) Heatmap of upregulated phagocytosis related genes (GO:0006909) in primary liver macrophages isolated from 18 weeks HFD (n=3) compared to ND (n=3) fed WT mice.
Scale bar, 100 μm. Data are shown as mean ± s.e.m.. *p<0.05; **p<0.01; ***p<0.001; NS, not significant. See also Figure S7.
DISCUSSION
Previous studies have shown that dietary obesity can induce Kupffer cell depletion followed by replenishment of infiltrated monocyte-derived macrophages with a hematopoietic origin13–15. Although barely expressed in Kupffer cells, we found that dietary obesity upregulates TREM2 in recruited liver macrophages. Importantly, we further identified S1P, released from lipid-injured apoptotic hepatocytes, as the key molecule responsible for inducing Trem2 expression in liver macrophages via S1PR1. In line with this finding, S1P was detected in extracellular vesicles released from PA-treated hepatocytes, which induces macrophage chemotaxis, thereby replenishing liver macrophage reservoir after obesity-induced Kupffer cell depletion70. In addition to the chemotaxis function, our study illustrated a role of S1P, which is to upregulate Trem2 expression in recruited liver macrophages. TREM2 upregulation ensures optimal efficiency of removing lipid-laden apoptotic hepatocytes by macrophages—an essential event restricting chronic liver inflammation. TREM2 upregulation is also evident in DEN-, acetaminophen- and CCl4 -treated mice27,34 and various liver diseases unrelated to lipid overload, such as virus-induced hepatitis71, cirrhosis16, and HCC17. Moreover, as a “find-me” signal released from apoptotic cells, S1P is elevated in distinct liver injury models and patients72–75. Based on these observations, we reason that the S1P-S1PR1 signaling axis likely represents a common pathway to cope with various types of liver injuries independent of disease etiology.
In support of the anti-NASH role of macrophage TREM2 revealed in our study, recent work, published while our paper was in revision, has also demonstrated a similar protective effect of TREM2 using a different NASH model induced by the methionine- and choline- deficient (MCD) diet29. Our work is also in line with earlier reports in which Trem2 germline deleted mice, relative to their WT littermates, exhibited much more severe liver pathology when subjected to acute liver injury inducing agents27,28,34,55. Moreover, earlier studies showed that Trem2−/− mice had exacerbated systemic metabolic alteration76,77, although it remains controversial as to whether TREM2 exerts such beneficial effects by functioning in the hematopoietic cells78 and whether gut microbiota determine such effects79.
Relative to primary liver macrophages from mice with simple steatosis, we demonstrated that NASH-associated macrophages are defective at engulfing lipid-laden apoptotic hepatocytes. Consistent with this finding, several animal and human studies utilizing superparamagnetic iron oxide imaging also suggest that phagocytic activity of macrophages is impaired in NASH80,81. To further define the molecular mechanism underlying this efferocytosis defect in NASH-associated macrophages, we revealed that TNF and IL-1β, two key proinflammatory cytokines driving NASH progression7,36,82, can upregulate and activate ADAM17 to induce proteolytic cleavage of TREM2 in liver macrophages. This ultimately results in the failure of macrophage-dependent removal of lipid-laden apoptotic hepatocytes, in which TREM2 plays a dominant role. Although TNF and IL-1β are essential proinflammatory cytokines that promote NASH development in both humans and mice10,36,58,59, it remains unclear how these cytokines are initially produced in response to dietary obesity. Previous studies suggest that WD can induce intestinal dysbiosis in mice, resulting in the translocation of gut microbial products (e.g. LPS) to the liver through portal circulation, which might contribute to the initial TNF and IL-1β production83,84. However, whether the alteration of gut permeability is a cause or a consequence of NAFLD remains unclear85. Alternatively, WD contains high fructose and high cholesterol, which can lead to chronic ER and mitochondrial stress in the liver that might gradually augment the rate of hepatocyte apoptosis, especially at the late stage of simple steatosis86–88. The increased rate of hepatocyte apoptosis may eventually outcompete phagocyte efferocytosis efficacy, resulting in the accumulation of dying hepatocytes that in turn release DAMPs, such as HMGB1, oxidized mtDNA, and phospholipids89,90, to induce TNF and IL-1β production from liver macrophages. However, whether different liver macrophage subpopulations may have different capacities in producing these cytokines remains an open question. Nonetheless, once produced in response to dietary obesity, TNF and IL-1β trigger ADAM17-dependent proteolytic cleavage of TREM2 to disarm macrophage efferocytosis of lipid-laden apoptotic hepatocytes, thereby setting the stage for chronic liver inflammation that ultimately drives NASH progression.
Lastly, we revealed that TREM2 is a master regulator in macrophages by directing the removal of lipid-laden apoptotic hepatocytes both in vitro and in vivo. However, the underlying mechanism remains to be further investigated. Among the phagocytosis-related genes that are upregulated in liver macrophages upon HFD feeding, some of them, namely Anxa1, Cd36, Ccr2 and Itgam, are regulated in a TREM2-dependent manner. Consistent with the protective role of TREM2 in NASH, Annexin A1 (encoded by Anxa1)—an important factor involved in phagocytosis of particulate substances, pathogens, and dying host cells91,92—was shown to be specifically expressed in liver macrophages and exerted a protective effect by preventing MCD diet induced NASH development in mice93. Moreover, in addition to its well-established role in fatty acid uptake by hepatocytes94,95, CD36—a scavenging receptor involved in efferocytosis of apoptotic cells—may also contribute to TREM2-dependent clearance of apoptotic hepatocytes, similar to what has been suggested during the process of Aβ phagocytosis by microglia96. In contrast, TREM2-dependent upregulation of Ccr2 and Itgam expression may instead increase myeloid cell migration and their hepatic infiltration in steatotic livers15,97, thereby setting the stage for macrophage efferocytosis of lipid-laden apoptotic hepatocytes. Given that TREM2 is capable of binding to phosphatidylserine and phosphatidylethanolamine, the classic tags for apoptotic cells98, it is also possible that TREM2 itself may serve as a scavenger receptor to directly mediate the engulfment of lipid-laden apoptotic hepatocytes.
In summary, our study reveals a protective role of TREM2 and its unexpected dynamic regulation during NASH development. Although initially upregulated by S1P in simple steatosis, prolonged hypernutrition leads to TREM2 shedding via TNF and IL-1β induced, ADAM17 mediated proteolytic cleavage. More importantly, we further demonstrated that impaired TREM2-dependent efferocytosis is a previously unrecognized key pathogenic event that licenses chronic liver inflammation to ultimately drive simple steatosis transition to NASH. Therefore, we propose that therapeutic strategies capable of preventing TREM2 cleavage to restore efferocytosis should be developed to treat NASH.
Limitation of the Study
Our study illuminates an essential role of macrophage TREM2 in preventing NASH development and further reveals an uncoupled regulation of TREM2 mRNA and protein abundance during NAFLD progression. However, it would be important to spatiotemporally define the heterogeneity and distinct functions of different subgroups of liver macrophages during NAFLD progression and identify the TREM2+ liver macrophage subpopulation(s) responsible for clearance of lipid-laden apoptotic hepatocytes. Additionally, it should be noted that CD11b+ magnetic beads were used in our study for liver macrophage isolation. This method may preferentially enrich monocyte-derived liver macrophages over resident Kupffer cells, so it remains to be determined if TREM2low Kupffer cells may also contribute to efferocytosis of dying hepatocytes in vivo.
STAR* METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and fulfilled by the lead contact, Zhenyu Zhong (zhenyu.zhong@utsouthwestern.edu).
Materials Availability
All reagents generated in this study are available upon request, which should be directed to Zhenyu Zhong (zhenyu.zhong@utsouthwestern.edu).
Data Availability
Data generated in this study are available in the Gene Expression Omnibus (GEO) database under the accession numbers GSE193577, GSE197695 and GSE198595.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients
In the study cohort for TREM2 expression analysis by immunofluorescent assay, 26 healthy controls (20 from NJDH, 5 from MSSM, 1 from UTSW), 28 steatosis (20 from NJDH, 6 from MSSM, 2 from UTSW) and 26 steatohepatitis (20 from NJDH, 5 from MSSM, 1 from UTSW) patient samples were collected during surgical operation between 2015 and 2021. The histological grading and staging of NAFLD and fibrosis were determined according to the published criteria99 and all specimens were examined by experienced hepatological pathologists blinded to the clinical and biochemical data of the patients. The detailed information of the patients involved is listed in Table S2 and S3. NJDH, Nanjing Drum Tower Hospital, Nanjing, China; MSSM, Mount Sinai Hospital, New York, NY, USA; UTSW, University of Texas Southwestern Medical Center, Dallas, TX, USA.
In vivo animal studies
Wild type (WT), Lyz2-Cre, Trem2F/F and Trem2−/− mice (all on the C57BL/6J background) were purchased from Jackson laboratory (Bar Harbor). Trem2ΔMye mice were generated by crossing Trem2F/F with Lyz2-Cre mice. MUP-uPA mice were from Dr. Michael Karin. All mice were bred and maintained under specific pathogen free conditions in the animal facility at the University of Texas Southwestern Medical Center (UTSW). Only littermate mice were used throughout the study, and they were kept in the same cage (except for the food intake measurement described below) throughout the study to reduce confounding impacts of differences in gut microbiota. The High Fat Diet (HFD, 60% kcals from fat) soft pellets were purchased from Bio-Serv (S3282). Western Diet (WD) that contains high-fat, high-sucrose, and high-cholesterol (21.1% fat, 41% sucrose, and 1.25% cholesterol by weight, Teklad diets, TD. 120528) supplemented with a high sugar solution (23.1g/L d-fructose and 18.9 g/L d-glucose) provided in the form of drinking water was used to feed mice of various genotypes as described in the study. Mice were fed and weighed every week on the same day. To calculate food intake, mice with the same genotype were maintained in the same cage and the total food consumed each week was recorded. The food intake per mouse was used for the analysis. All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at UTSW. 6~8 weeks old male mice were used for all studies.
sTREM2 in vivo study
Trem2+/− mice (on the C57BL/6 background) were bred to each other to generate Trem2+/+ and Trem2−/− mice, respectively. 8-week-old Trem2+/+ male mice were used for sTREM2 i.v. injection. During the initial experiment, Trem2+/+ and Trem2−/− mice were treated weekly with sTREM2 (R&D, i.v. injection, 1 μg/mouse in 100μl PBS) starting at 8 weeks (referred as week 0) after birth, then sacrificed 1, 3, and 7 weeks respectively post injection. Mouse sera was collected for sTREM2 detection by ELISA. These results showed that weekly i.v. injection of sTREM2 at 1 μg/mouse can maintain a serum sTREM2 concentration higher than 100 ng/ml after 3 weeks of treatment. In the subsequent experiments, 8-week-old Trem2+/+, Trem2−/−, Trem2F/F and Trem2ΔMye mice were fed with normal diet (ND) or WD and were treated with sTREM2 (i.v. injection, 1 μg/mouse in 100 μl PBS) or PBS as a control. All mice were sacrificed 8 weeks after sTREM2 treatment for the collection of sera and liver tissues for further pathological analysis.
S1PR1 inhibitor (VPC) in vivo study
8-week-old ND or WD fed C57BL/6 mice were treated with VPC (Cayman Chemical, i.p. injection, 0.5mg/kg in 100 μl PBS) or PBS (control). All mice were sacrificed 8 weeks after VPC treatment for the collection of sera and liver tissue for further pathological analysis.
Cells
Mouse normal liver hepatocyte line, AML12, and human cell lines, THLE-3 and THP-1, were purchased from ATCC. AML12 cells were cultured in DMEM-F12 medium (VWR) supplemented with 10% FBS (Corning), 1% ITS (Sigma) and 40 ng/ml dexamethasone (Sigma). THLE3 cells were cultured in BEGM medium (Lonza) supplemented with 5 ng/mL EGF (Corning), 70 ng/mL Phosphoethanolamine (Sigma) and 10% FBS. THP-1 cells were cultured in RPMI1640 (Gibco) medium supplemented with 10% FBS, 0.1% β-mercaptoethanol (Invitrogen) and 1% penicillin and streptomycin (Gemini). All cells were maintained in 5% CO2, 37°C atmosphere. All cells used for experiments were within 10 passages from thawing, and they were mycoplasma-free based on routine examination using a mycoplasma detection kit (Lonza).
METHOD DETAILS
Liver macrophage and hepatocyte isolation
Primary liver macrophages and hepatocytes were isolated from male mice by perfusing the liver with collagenase as previously described100,101. In brief, mice were anesthetized with ketamine/xylazine, and the perfusion was performed via portal vein. HBSS solution (without Ca2+/Mg2+) was used to clear the blood in the liver. Then, HBSS solution (with Ca2+/Mg2+) supplemented with Collagenase (Collagenase D for macrophage isolation, Collagenase D and Collagenase P for hepatocyte isolation) was used. After perfusion, liver was collected and digested in HBSS solution (with Ca2+/Mg2+) supplemented with pronase and DNase I for 1 hour (macrophage isolation) or 15 minutes (hepatocyte isolation). The digested liver was filtered through 70 μm cell strainer (BD). For cell culture purpose, the cells obtained from anti-CD11b microbeads isolation were placed in 6-well plates or 12-well glass bottom plates in RPMI1640 medium supplemented with 10% FBS, after 1-hour culture, non-adherent cells were discarded. Hepatocytes were collected through centrifugation at 500 rpm for 1 min and resuspended in Waymouth’s MB medium supplemented with 10% FBS. Cells were then seeded onto 6-well plates (105 cells/well) that were precoated with Rat tail collagen type I. Nycodenz solution was used for the density gradient centrifugation to get the mononuclear cells, and anti-CD11b microbeads were used for the isolation of macrophages.
Neutrophil isolation
6- to 8-week-old C57BL/6 mice were used for the collection of bone marrow cells. A neutrophil isolation kit (Miltenyi Biotec) was used as previously described102. In brief, freshly isolated bone marrow cells were resuspended in buffer solution containing phosphate-buffered saline (PBS), pH7.2, 0.5% bovine serum albumin (BSA), and 2 mM EDTA, which were then incubated with Neutrophil Biotin-Antibody Cocktail for 10 mins at 4°C. After 3 times of washing, these antibody-labeled cells were incubated with anti-biotin microbeads for 15 mins at 4°C. Magnetic separation with columns was performed to collect neutrophils per manufacturer’s instructions.
Serum alanine transaminase (ALT) and aspartate aminotransferase (AST) and Glucose analysis
Serum ALT and AST measurements were performed using Infinity ALT, AST (Thermo Fisher Scientific) and a VALIDATE calibration verification kit (Maine Standards Company LLC) as previously described100. In brief, 200 μl ALT/AST substrate was added to 96-well plates and preheated to 37°C. Serum samples were diluted by 5 to 200 folds, and a volume of 20 μl of each diluted sample was added into the substrate, after which the reaction was immediately measured for absorbance at 340 nm using a Cytation 5 Cell Imaging Multimode Reader (BioTek). Serum glucose measurements were performed by UT Southwestern Metabolic Phenotyping Core using VITROS® 350 (Fisher).
Serum triglyceride (TG) and liver TG analysis
Serum triglyceride (TG) was measured using Serum Triglyceride Determination Kit (Sigma) as previously described103. In brief, 0.8 ml of the Free Glycerol Reagent was added into each cuvette; then 10 μl of water, Glycerol Standard, and samples were added into cuvettes labeled with Blank, Standard, and Sample, respectively. The initial absorbances (IA) of Blank, Standard, and Sample at 540 nm versus water as the reference were measured after 5 minutes incubation at 37°C. Then, 0.2 ml of the reconstituted Triglyceride Reagent was added to each cuvette, mixed, and incubated at 37°C for additional 5 minutes. The final absorbances (FA) of Blank, Standard, and Sample at 540 nm versus water as the reference were then measured and recorded for calculating concentrations of glycerol, true triglycerides, and total triglycerides in the samples. Liver TG was measured using Triglyceride Assay Kit (Abcam) as instructed. Briefly, 100mg fresh liver tissue was lysed with NP40, and 50 μl of tissue lysate was used for the colorimetric analysis and measured for absorbance at 570 nm using a Cytation 5 Cell Imaging Multimode Reader (BioTek).
Serum cholesterol analysis and Non-esterified Free Fatty Acids (NEFA) Assay
Serum cholesterol was measured using Cholesterol Quantitation Kit (Sigma) per manufacturer’s instructions. In brief, serum samples were diluted 1: 20 to 1:100 with Cholesterol Assay Buffer, and a final volume of 50 μl was used for the assay. The reaction mixture was prepared per manufacturer’s instructions. A volume of 50 μl of the appropriate reaction mix was added into each well, mixed well and incubated for 60 minutes at 37°C in the dark. Finally, the absorbance at 570 nm (A570) was measured using a Cytation 5 Cell Imaging Multimode Reader (BioTek). Serum NEFA was measured using a Non-esterified Free Fatty Acids (NEFA) Colorimetric Assay Kit (Elabscience, Wuhan, China). In brief, 10 μl of serum was used for the colorimetric analysis and measured for absorbance at 546 nm using a Cytation 5 Cell Imaging Multimode Reader (BioTek).
Hydroxyproline Assay
Fresh liver tissue was collected and washed with PBS, 10 mg of which was used for the detection of hydroxyproline using Hydroxyproline Assay Kit (Sigma) per manufacturer’s instructions. In brief, for the preparation of samples, 10 mg liver tissue was homogenized in 100 μl of water followed by transfer to a pressure-tight polypropylene vial with a PTFE-lined cap. Then, 100 μl of concentrated hydrochloric acid (HCl, 12 M) was added and samples were hydrolyzed at 120°C for 3 hours. After mixture and centrifugation at 10,000 g for 3 minutes, 10~50 μl of supernatants were transferred to a 96-well plate, which was placed in a 60°C oven to dry. To measure hydoxyproline, 100 μl of the Chloramine T/Oxidation Buffer Mixture was added to each well followed by incubation at room temperature for 5 minutes. Then, 100 μl of the Diluted DMAB Reagent was added, and further incubated for 90 minutes at 60°C. The absorbance at 560 nm (A560) was measured using a Cytation 5 Cell Imaging Multimode Reader (BioTek).
Liver histology
Fresh liver tissue was collected and fixed with formalin and then embedded with paraffin or OCT compound. The paraffin sections were stained with hematoxylin and eosin (H&E) for assessment of liver histology, and Sirius Red (Sigma, 365548)/Fast Green (Sigma, F258) for assessment of fibrosis. The frozen sections were used for Oil-Red O staining. The NAFLD activity and fibrosis stage was evaluated and scored by experienced pathologists based on the NASH CRN scoring system. The histological scoring was performed in a blind manner, with no sample identification information provided to the pathologists. The images with H&E and Oil-Red O staining were processed using a Cytation 5 Cell Imaging Multimode Reader (BioTek). The images with Sirius Red/Fast Green staining were detected using a Zeiss Axioscope (Zeiss, CA, USA) under polarized light.
Immunohistochemistry (IHC), immunofluorescence (IF), TUNEL and BODIPY staining
For IHC staining, formalin-fixed, paraffin-embedded liver sections were deparaffinized, hydrated and subjected to antigen retrieval, then incubated with primary antibodies against α-smooth muscle actin (α-SMA) overnight at 4°C. On the second day, after washing 3 times with 0.1% PBST, the slides were then incubated with HRP-labeled secondary antibodies for 1 hour at room temperature. A DAB kit (BD Bioscience) was used for the colorimetric reaction. For IF staining, formalin-fixed, OCT-embedded frozen sections were incubated with primary anti-mouse MPO, anti-mouse F4/80, anti-mouse TREM2, anti-mouse CX3CR1, anti-human TREM2 or anti-human CD68 antibodies overnight at 4°C. After washing 3 times with 0.1% PBST, the sections were then incubated with fluorescence-labeled second antibody for 1 hour at room temperature in the dark and DAPI was used for nuclear staining. For TUNEL staining, formalin-fixed, OCT-embedded frozen sections were incubated with TUNEL reagents at 37oC for 1 hour in the dark and DAPI was used for nuclear staining. For BODIPY staining, cells pretreated with 800 μM palmitic acid for 24 hours, washed with PBS, fixed in 4% paraformaldehyde for 15 minutes, and then stained with BODIPY™ 493/503 (Thermo Fisher Scientific, D3922), according to instructions, and Hoechst was used for nuclear staining. The images were recorded and processed using a Cytation 5 Cell Imaging Multimode Reader (BioTek). For LysoTracker staining, cells were washed with PBS, fixed in 4% paraformaldehyde for 15 minutes, and then stained with LysoTracker (Thermo fisher) according to instruction and Hoechst was used for nuclear staining. The images were taken using a Zeiss LSM880 microscope (Zeiss, CA, USA).
Immunoblot assay
Liver tissue and cells were lysed in RIPA buffer (R&D) supplemented by a protease inhibitor (Thermo Scientific) and a phosphatase inhibitor cocktail (Thermo Scientific). Protein concentrations were quantified using BCA Protein Assay Kit (Pierce). Equal amounts of protein were separated by SDS–PAGE and transferred onto nitrocellulose membranes. After blocking with 5% milk for 1 hour at room temperature, the membranes were incubated with primary antibodies (anti-mouse TREM2, anti-mouse GAPDH, anti-mouse Capase-3, anti-mouse Cleavage Capase-3, anti-mouse ADAM10, anti-mouse ADAM17, anti-mouse S1PR1, anti-mouse S1PR2, anti-mouse SPHK1, anti-mouse SPHK2, anti-mouse Akt, anti-mouse Phospho-Akt (Ser473), anti-human TREM2 and anti-human GAPDH), followed by incubation with the appropriate HRP-conjugated secondary antibodies and developed with ECL. Images were recorded using a ChemiDoc™ Touch Imaging System Surpasses (Bio-Rad).
Generation of bone marrow derived macrophages (BMDMs) and treatments
The femurs and tibias from 6-to 8-week-old male C57BL/6 mice were used for the collection of bone marrow cells. In brief, bone marrow cells were flushed out with sterile DMEM (Gibco), followed by removing the red blood cells using the Red Blood Cell Lysis Buffer (Thomas Scientific). Cells were then incubated for 7 days in the presence of 20% (v/v) L929 conditional medium supplemented with 10% FBS (Gibco), 100 mg/mL streptomycin, and 100 U/mL penicillin (Gibco) as previously described102. The medium was replaced on the 4th day and the non-adherent cells were discarded. After 7 days, BMDMs were collected and seeded in 6/12/24/48-well plates respectively overnight in DMEM without FBS. These cells were then treated with different reagents as detailed in each experiment.
Generation of apoptotic cell culture medium (ACM) and necrotic cell culture medium (NCM)
Primary hepatocytes or AML12 cells were treated with 800 or 2000 μM palmitic acid (PA) for 24 hours. Flow cytometry analysis was performed on these cells after PA treatment using an Apoptosis Detection Kit (BD Bioscience). Based on Annexin V and PI staining results, 800 and 2000 μM concentrations were determined to be capable of inducing apoptosis and necrosis of these hepatocytes, respectively. Cells were then placed in fresh FBS-free medium for 24 hours to collect apoptotic cell culture medium (ACM) or necrotic cell culture medium (NCM) which was filtered through 0.45 μm filters before proceeding with next steps as described in each experiment.
Transwell assay
In the transwell system (Thomas Scientific), differentiated BMDMs (2 × 106 cells/well) were seeded at the bottom of the chamber, and hepatocytes (1 × 106 cells/well) were placed at the upper layer of the chamber and treated with 800 μM PA to induce apoptosis. After 24-hour treatment, BMDMs were collected for analysis.
Phagocytosis assay
Apoptotic primary murine hepatocytes, AML12 cells (mouse hepatocyte cell line), and THLE-3 cells (human hepatocyte cell line) were generated by treatment of 800 μM PA for 24 hours. CellTracker-Red labeled apoptotic cells (105 cells/well) were incubated with CellTracker-Green labeled murine BMDMs or THP-1 cells (2 × 105 cells/well) in a 24-well glass bottomed plate (Greiner Bio-One). For experiments involving macrophage pretreatment, THP-1 cells and BMDMs were handled as follows: THP-1 cells were pretreated with 100 ng/ml PMA for 24 hours to induce their differentiation into macrophages. BMDMs were pretreated with S1P, TNF or IL-1β for 24 hours before coculturing with apoptotic hepatocytes. BMDMs or THP-1 cells were pretreated with TREM2 blocking antibody for 2 hours before coculturing with apoptotic hepatocytes.
Moreover, for sTREM2 in vitro experiment, 2 × 105 cells/well of BMDMs or THP-1 cells were plated in 24-well plates. Apoptotic AML12 cells or THLE-3 cells were pretreated with 200 ng/ml sTREM2 before coculturing with BMDMs for phagocytosis assay. In the coculture system containing both macrophages and apoptotic hepatocytes, sTREM2 was also added to make a final concentration of 200 ng/ml. Videos were recorded in a cell incubation system and images were taken after 2 hours incubation by a Cytation 5 Cell Imaging Multimode Reader (BioTek).
Lentivirus packaging and infection
293T cells were used for lentivirus packaging and production. The recombinant lentiviruses were collected and filtered with 0.45 μm filters. Cell lines (THP-1 cells, AML12 cells) were seeded in 6-well plates to reach 30–50% confluency for infection. For lentivirus infection, 200 μl virus-containing medium was added to each well in the presence of 6–8 μg/ml polybrene (Millipore). 12~24 hours post infection; the culture medium was replaced with fresh medium. After 36 hours of incubation, 2 μg/ml puromycin was added to selectively enrich shRNA/sgRNA-positive cells. For the infection of BMDMs, freshly isolated mouse bone-marrow cells were seeded in 6-well plates (1~2 × 105 cells/well) on day 0, followed by lentiviral infection on day 4. Infected BMDMs were then collected at day 8~9 for further experiments. Detailed information of the lentiviral plasmids and sequences of shRNAs and sgRNAs used in this study were as described above.
ADAM10 and ADAM17 activity assay
The ADAM10 and ADAM17 activities in BMDMs were assessed using an ADAM10 Activity Assay Kit and a TACE (α - Secretase) Activity Assay Kit, respectively. In brief, BMDMs were seeded in 6-well plates that were pretreated with/without TAPI 0 ADAM-17 (TACE) inhibitor (R&D) or GI 254023X Selective ADAM10 metalloprotease inhibitor (R&D) for 2 hours, followed by IL-1β or TNF treatment for additional 24 hours. Cells were then collected, and total lysates were prepared per the manufacturer’s instructions. A volume of 50 μl lysate was added into each well, followed by addition of 50 μl TACE or ADAM10 substrate. The reaction mixture was then incubated for 30~60 min in the dark, followed by addition of 50 μl of stop solution to each well. Fluorescence intensity at Ex/Em=490 nm/520 nm was then measured. All analyses were performed using a Cytation 5 Cell Imaging Multimode Reader (BioTek).
Enzyme-linked immunosorbent assay (ELISA)
The amounts of S1P in cell culture medium, serum, or tissue (fresh liver tissues were lysed with RIPA buffer) were quantified by an S1P ELISA kit (Cloud-Clone). Sandwich ELISAs were used to determine the amounts of TNF in fresh liver tissue and sTREM2 in cell culture medium. In brief, flat-bottomed 96-well plates were coated with a TREM2 antibody (R&D, 1:1000), or a TNF (eBioscience™, 2 μg/ml) capture antibody in coating buffer (0.05 M Carbonate buffer, pH 9.6) overnight at 4°C. The plates were then blocked in 5% milk (dissolved in 0.05% Tween 20 in PBS) for 4 hours at room temperature, followed by 3 washes with 0.05% PBS-T solution and incubated with samples diluted in assay buffer (0.05% PBST) overnight at 4°C. After washes with 0.05% PBST, human or mouse TREM2 biotinylated antibody (R&D Systems, 1:3000), or mouse TNF biotinylated antibody (2 μg/ml) were added and incubated for 2 hours at room temperature. After washing, Streptavidin Poly-HRP40 Conjugate (Fitzgerald, 65R-S104PHRP, 1:3000) was added and incubated for 1 hour at room temperature in the dark. After five additional washing steps, the plates were developed by adding the TMB substrate (Sigma-Aldrich, T5569) and the absorbance was read at 620 nm using a BioTek Cytation 5 Cell Imaging Multimode Reader (BioTek). Serum insulin concentrations were measured using Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem) as instructed. In brief, 5 μl of sample was used for the colorimetric analysis and measured for absorbance at 450 nm using a Cytation 5 Cell Imaging Multimode Reader (BioTek).
Quantitate Real-time PCR
Total RNA from cells or fresh liver tissue was extracted using TRIzol reagent and a RNeasy Mini Kit (QIAGEN) per the manufacturers’ instructions. RNA templates were treated with DNase (QIAGEN) to avoid genomic DNA contamination. RNA concentration was measured using NanoDrop™ One/OneC Microvolume UV-Vis Spectrophotometer (ThermoFisher). cDNAs were synthesized using the SuperScript First-Strand Synthesis System (Bio-Rad). Transcript amounts were measured by real-time PCR in SYBR Green reactions (Bio-Rad) using the CFX Real-Time PCR Detection Systems (Bio-Rad). Gene expression was calculated relative to GAPDH. The sequences of primer pairs used in the study were listed above.
RNA-Sequencing
BMDM cells after treatments or macrophages isolated from mouse livers were resuspended in TRIzol reagent (Life Technologies) and then total RNA was extracted and treated with DNase I (Qiagen, Dusseldorf). Library preparation, quality control and sequencing were performed by Novogene (CA, USA). RNA sequencing was performed via Illumina platform (Illumina, CA), based on the mechanism of SBS (sequencing by synthesis). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment and Ingenuity Pathways Analysis (IPA) analysis (Qiagen) were performed to assess the gene expression profile in different groups. Raw data were deposited in the Gene Expression Omnibus (GEO) database under the accession numbers GSE193577, GSE197695 and GSE198595.
Flow cytometry
Cultured cells or isolated liver macrophages were collected and washed in the buffer containing PBS and 2% BSA. Cells were stained with specific antibodies for 30 min at 4°C in the dark, washed three times with cold PBS, and analyzed by flow cytometry using a FACS LSR II flow cytometer (BD Biosciences). The fluorochromes and antibodies are listed above, and data were analyzed using FlowJo software (TreeStar).
Data analysis from Gene Expression Omnibus (GEO) database
Transcriptome profiles of 78 NAFLD-affected liver tissues and their histological annotations were obtained from NCBI Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo, accession number GSE130970)56. Genes of sphingosine biosynthesis (GOBP_SPHINGOSINE_BIOSYNTHETIC_PROCESS) and S1P receptor activity (GOMF_SPHINGOSINE_1_PHOSPHATE_RECEPTOR_ACTIVITY) as well as transcriptional targets of TNFα pathway (HALLMARK_TNFA_SIGNALING_VIA_NFKB) were obtained from Molecular Signature Database (MSigDB) (www.broadinstitute.org/msigdb)104,105. Modulation of the gene sets in each individual sample was determined by the previously described algorithm106,107. Transcriptome profiles of 63 NAFLD-affected liver tissues (24 from healthy controls, 20 with steatosis and 19 with steatohepatitis) from NCBI Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo, accession number GSE89632) were used for the analysis of TREM2 mRNA expression.
Statistical analysis
No statistical methods were applied to predetermine sample size. The experiments were not randomized except for the in vivo studies in which the age-matched mice were randomly allocated to different experimental groups based on their genotypes. Investigators were not blinded to allocation during experiments and outcome assessment except for microscopic analysis of H&E, IHC, IF and other staining results. Data are presented as the mean ± SD or ± SEM as indicated in the figure legends. Differences in means were analyzed by Student’s t-test and one-way ANOVA. Differences with p values < 0.05 were considered significant (*: p < 0.05, **: p < 0.01, ***: p < 0.001).
Data and code availability
All data reported in this paper will be shared by the lead contact upon reasonable request. This paper does not report any original code.
Supplementary Material
Video S1. Efferocytosis of lipid-laden apoptotic hepatocytes (green) by Trem2F/F BMDMs (red), Related to Figure 5 Apoptotic AML12 cells (mouse hepatocyte cell line) were generated by treatment of 800 μM PA for 24 hours. CellTracker-Green labeled apoptotic cells (105 cells/well) were incubated with CellTracker-Red labeled Trem2F/F BMDMs (2 × 105 cells/well) in a 24-well glass bottomed plate (Greiner Bio-One). Videos were recorded in a cell incubation system and images were taken every 15 min during incubation by a Cytation 5 Cell Imaging Multimode Reader (BioTek).
Video S2. Efferocytosis of lipid-laden apoptotic hepatocytes (green) by Trem2ΔMye BMDMs (red), Related to Figure 5 Apoptotic AML12 cells were generated as in Video S1. CellTracker-Green labeled apoptotic cells (105 cells/well) were incubated with CellTracker-Red labeled Trem2 ΔMye BMDMs (2 × 105 cells/well) in a 24-well glass bottomed plate (Greiner Bio-One). Videos were recorded as described in Video S1.
KEY RESOURCES TABLE
| REGENTS or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| V500 Rat Anti-Mouse CD45 Clone 30-F11 (RUO) | BD Biosciences |
561487 |
| CD11b Monoclonal Antibody (M1/70), eFIuor 660, | BD Biosciences |
50-0112-82 |
| MHC Class II (I-A/I-E) Monoclonal Antibody (M5/114.15.2), FITC, eBioscience™ | BD Biosciences |
11-5321-85 |
| Ly-6G Monoclonal Antibody (1A8-Ly6g), PerCP-eFluor 710, eBioscience™ | BD Biosciences |
46-9668-82 |
| F4/80 Monoclonal Antibody (BM8), PE, eBioscience™ | BD Biosciences |
12-4801-82 |
| Ly-6C Monoclonal Antibody (HK1.4), eFluor 450, eBioscience™ | BD Biosciences |
48-5932-82 |
| Fixable Viability Dye eFluor™ 780 | BD Biosciences |
65-0865-18 |
| CD16/CD32 Monoclonal Antibody (KT1632) | Invitrogen | MA5-18012 |
| CD11b MicroBeads, human and mouse | Miltenyi Biotec | 130-049-601 |
| TREM2 (E7P8J) Rabbit mAb (Carboxy-terminal Antigen, Mouse Specific) | Cell Signaling Technology | 76765S |
| Human/Mouse TREM2 APC-conjugated Antibody | R&D | FAB17291A |
| CD11b Monoclonal Antibody (M1/70), eFluor 450, eBioscience™ | BD Biosciences |
48-0112-82 |
| Anti-TREM2 antibody (ab95470) | Abcam | ab95470 |
| PE anti-mouse CX3CR1 Antibody | Biolegend | 149006 |
| ADAM17 Polyclonal Antibody | Invitrogen | PA5-27395 |
| Recombinant Anti-ADAM10 antibody [EPR5622] | Abcam | ab124695 |
| Mouse TREM2 Biotinylated Antibody | R&D | BAF1729 |
| TNF alpha Monoclonal Antibody (TN3-19.12), eBioscience™ | eBioscience | 14-7423-85 |
| TNF alpha Polyclonal Antibody, Biotin, eBioscience™ | eBioscience | 13-7341-85 |
| GAPDH (D16H11) XP® Rabbit mAb | CST | 5174S |
| F4/80 Monoclonal Antibody (BM8), eBioscience™ | eBioscience | 14-4801-82 |
| Anti-CD68 antibody [KP1] (ab955) | Abcam | ab955 |
| Anti-rabbit HRP for IHC | Thomas Scientific |
NC9027342 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | A21206 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A21207 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | A21208 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A21209 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | A-21203 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A-21202 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 | Thomas Scientific |
A32795 |
| Donkey anti-Sheep IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, HRP | Invitrogen | A16047 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Invitrogen | A-11004 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, DyLight 488 | Thomas Scientific |
SA5-10086 |
| Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 594 | Thermo Fisher | A32758 |
| Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 647 | Thermo Fisher | A32849 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 647 | Thermo Fisher | A48272 |
| Anti-TREM2 antibody [RM0139-5J46] (ab86491) | Abcam | ab86491 |
| Mouse TREM2 Antibody | R&D | AF1729 |
| FITC Annexin V Apoptosis Detection Kit with PI | BioLegend | 640914 |
| Human/Mouse TREM2 APC-conjugated Antibody | R&D | FAB17291A |
| Anti-rabbit IgG, HRP-linked Antibody | CST | 7074S |
| Human/Mouse TREM2 Antibody | R&D | MAB17291 |
| S1P1 Polyclonal Antibody (S1PR1/EDG1) | Invitrogen | PA1-1040 |
| S1PR2 Polyclonal Antibody | Invitrogen | PA5-23208 |
| F4/80 Monoclonal Antibody (SP115) (Rb antimouse) | Invitrogen | MA5-16363 |
| Rat IgG2B Isotype Control | Thomas Scientific |
MAB0061 |
| Cleaved Caspase-3 (Asp175) Antibody #9661 | CST | 9661 |
| Caspase-3 Antibody #9662 | CST | 9662 |
| Human/Mouse/Rat alpha -Smooth Muscle Actin Antibody | R&D | MAB1420 |
| SPHK1 Polyclonal Antibody | Invitrogen | PA5-87473 |
| SPHK2 Polyclonal Antibody | Invitrogen | 17096-1-AP |
| Mouse CLEC4F/CLECSF13 Antibody | R&D | AF2784 |
| BB700 Rat Anti-Mouse TIM-4 | BD Biosciences |
746108 |
| TREM2 (D8I4C) Rabbit mAb #91068 | CST | 91068 |
| Mouse IgG VisUCyte HRP Polymer Antibody | R&D | VC001-025 |
| Syk (D3Z1E) XP® Rabbit mAb #13198 | CST | 13198T |
| Phospho-Zap-70 (Tyr319)/Syk (Tyr352) (65E4) Rabbit mAb #2717 | CST | 2717 |
| a-Smooth Muscle Actin (D4K9N) XP® Rabbit mAb #19245 | CST | 19245S |
| Mouse IgM Isotype Control (F8) | Novus | NBP2-21945 |
| Purified Rat Anti-Mouse Ly-6G | VWR | 127602-BL |
| Anti-mouse F4/80 BV421 (clone BM8) | Biolegend | 123132 |
| CD45 MicroBeads, mouse | Miltenyi Biotec | 130-052-301 |
| Anti-mouse CD45 BB515 (clone 30-F11) | Thermo Fisher | 564590 |
| Anti-mouse/human CD11b PE (clone M1/70) | Biolegend | 101208 |
| Anti-mouse CD146 PE/Cy7 (clone ME-9F1) | Biolegend | 134714 |
| Anti-mouse Tim4 Alexa647 (clone RMT4-54) | Biolegend | 130008 |
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Fisher | L10119 |
| Human/Mouse Myeloperoxidase/MPO Antibody | R&D | AF3667 |
| Anti-rat IgG, HRP-linked Antibody | CST | 7077S |
| HRP-conjugated Anti-Goat IgG Secondary Antibody | R&D | HAF019 |
| Mouse Osteopontin/OPN Antibody | R&D | AF808 |
| Mouse Osteopontin/OPN PE-conjugated Antibody | R&D | IC808P |
| Recombinant Anti-HNF-4-alpha antibody [EPR16885-99] | Abcam | ab201460 |
| Phospho-Akt (Ser473) Antibody | CST | 9271 |
| Akt Antibody | CST | 9272 |
| PE-CF594 Rat Anti-Mouse Ly-6C | BD Biosciences |
BDB562728 |
| PerCP/Cyanine5.5 anti-mouse Ly-6C Antibody | Biolegend | 128012 |
| TIM4 Polyclonal Antibody | Bioss | BS-6197R |
| Human/Mouse CX3CR1 Antibody | R&D | AF5825 |
| Chemicals, peptides, and recombinant Proteins | ||
| Polybrene Infection | EMD Millipore | TR-1003-G |
| RBC Lysis Buffer | Thomas Scientific |
00-4300-54 |
| Pierce™ High Sensitivity Streptavidin-HRP | Thomas Scientific |
21130 |
| Recombinant Mouse IL-1 beta/IL-1F2 Protein | R&D | 401-ML-005 |
| 1-Step™ Ultra TMB-ELISA Substrate Solution | Thomas Scientific |
34029 |
| TNF Recombinant Mouse Protein | R&D | 410-MT-025/CF |
| DAPI (4‘,6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | D1306 |
| Trizol | Invitrogen | 15596018 |
| DNase I | BioRad | 7326828 |
| DMSO | Sigma | D2650-5X10ML |
| Cellsome - Phosphatidylserine (PS)- based cellsome made from DOPC: DOPS (70:30) molar ratio with 10mM lipid concentration and sized to 100nm in PBS | Encapsula | CPS-507 |
| Cellsome® made from Unsaturated Phosphatidylcholine (PC) Cellsome made from 100% 18:1 (Δ9-Cis) PC (DOPC) with 10 mM lipid concentration and sized to 100 nm in PBS | Encapsula | CUP-506 |
| IL6 Recombinant Mouse Protein | R&D | 406-ML-005/CF |
| Lipofectamine™ 3000 Transfection Reagent | Thomas Scientific |
L3000015 |
| Antibody diluent with background reducing components | Agilent | S302283 |
| Target Retrieval Solution | Agilent | S169984 |
| 2-Mercaptoethanol | Invitrogen | 21985023 |
| CellTracker™ CM-DiI Dye | Invitrogen | C7000 |
| Fixation Buffer | Biolegend | 420801 |
| BSA Fatty acid free | Sigma | A8806 |
| Collagenase D | Sigma | 11088866001 |
| ITS Liquid Media Supplement | Sigma | I3146 |
| Dexamethasone | Sigma | D4902 |
| Molecular Probes™ Hoechst 33342, Trihydrochloride, Trihydrate | Invitrogen | H1399 |
| Recombinant Mouse TREM2 His-tag Protein | R&D | 9228-T2-050 |
| Oil red O | Sigma | O0625-25G |
| Sirus Red F3BA 100mg (Direct Red 80) | Sigma | 365548-5G |
| Fast Green FCF | VWR | AAA16520-06 |
| Saturated Picric Acid Solution | VWR | 101410-760 |
| CellTracker™ Green CMFDA Dye | Invitrogen | C7025 |
| Sphingosine-1-Phosphate (d18:1) | Avanti Polar Lipids | 860492P-1mg |
| Sodium palmitate | Sigma | P9767-5G |
| TAPI 0 ADAM-17 (TACE) inhibitor | R&D | 5523/1 |
| GI 254023X Selective ADAM10 metalloprotease inhibitor | R&D | 3995/1 |
| VPC 23019 S1P1 and S1P3 antagonist | selleckchem | NC1529596 |
| VPC 23019 S1P1 and S1P3 antagonist | Cayman Chemical |
13240 |
| JTE 013 S1P2 receptor antagonist | selleckchem | NC1591300 |
| BD Collagen I Rat Tail Nat 100MG 354236 w/ Free Shipping | BD Bioscience | 354236 |
| Waymouth‘s Medium | Gibco | 11220035 |
| Pronase | Sigma | 10165921001 |
| DNase I | Sigma | 10104159001 |
| pHrodo™ Red Zymosan Bioparticles™ Conjugate for Phagocytosis | Invitrogen | NC0710404 |
| Recombinant Mousec/Fractalkine (Full Length), CF | R&D | 472-FF-025/CF |
| Rps19 (NM_023133) Mouse Recombinant Protein | Origene | TP519906 |
| Yars (NM_134151) Mouse Recombinant Protein | Origene | TP508462 |
| Flavopiridol (Alvocidib) | Selleck | S1230 |
| L-α-Lysophosphatidylcholine from bovine brain | Millipore | L1381 |
| Sucrose | Fisher | BP220212 |
| Activated charcoal | Sigma | 242276-250G |
| Concentrated (37%) Hydrochloric Acid | Sigma | 320331 |
| Glycerol Standard Solution | Sigma | G7793 |
| PF-543 (inhibitor of SphK1) | selleckchem | S7177 |
| Opaganib (ABC294640) (inhibitor of SphK2) | selleckchem | S7174 |
| Adenosine 5‘-triphosphate disodium salt 100 mM solution, ATP | Sigma | A6559-25UMO |
| Nycodenz AG | Accurate Chemical (NY) | 1002424 |
| Phorbol 12-myristate 13-acetate | Enzo | BML-PE160-0001 |
| Cholesterol-Water Soluble | Sigma | C4951 |
| PRT062607 (P505-15) HCl Syk inhibitor | Selleck | S8032 |
| EGF HUMAN RECOMBIANT | Corning | CB40052 |
| Recombinant Human TREM2 His-tag Protein, CF | R&D | 9256-T2-050 |
| Fibronectin Proteolytic Fragment from human plasma | Sigma | F0162 |
| Collagen I, bovine | Gibco™ | A1064401 |
| Bovine Serum Albumin | Sigma | A7030 |
| HANKS BSS W/OCA 1X | Corning | MT21022CV |
| HANKS BSS 1X W/O PR | Corning | MT21023CV |
| BEGM KIT TO MIX | Lonza Walkersville |
NC9202780 |
| pHrodo™ Red, succinimidyl ester (pHrodo™ Red, SE) | Invitrogen | P36600 |
| PHOSPHATASE INHIBITOR COCKTAIL | Thermo Scientific |
AAJ61022AA |
| PIERCE PROTEASE INH | Thermo Scientific |
PIA32953 |
| LysoTracker™ Deep Red | Invitrogen | L12492 |
| CYM-5442 | Sigma | C1997 |
| DAPT | Sigma | CYM-5442 |
| Critical commercial assays | ||
| ELISA Kit for Sphingosine-1-Phosphate (S1P) | cloud-clone | CEG031Ge |
| SensoLyte ® 520 TACE (a - Secretase) Activity Assay Kit *Fluorimetric* | AnaSpec | AS-72085 |
| Thermo Scientific™ ALT/GPT Reagent | Thermo Scientific |
TR71121 |
| Thermo Scientific™ AST/GOT Reagent | Thermo Scientific |
TR70121 |
| Validate GC3 Linearity Test Kit | Maine Standards |
1300sd |
| In Situ Cell Death Detection Kit, Fluorescein | Sigma | 11684795910 |
| Hydroxyproline Assay Kit | Sigma | MAK008-1KT |
| AlphaTRAK 2 Blood Glucose Monitoring System Kit | Sigma | S5134-6X100ML |
| Serum Triglyceride Determination Kit | Sigma | TR0100-1KT |
| DAB Substrate Kit | BD Bioscience | 550880 |
| Pierce™ BCA Protein Assay Kit | Fisher | PI23227 |
| SensoLyte ® 520 ADAM10 Activity Assay Kit *Fluorimetric* | AnaSpec | AS-72226 |
| Cholesterol Quantitation Kit | Sigma | MAK043-1KT |
| Neutrophil Isolation Kit, mouse | Miltenyi Biotec | 130-097-658 |
| BODIPY™ 493/503 | Thermo Scientific |
D3922 |
| Triglyceride Assay Kit - Quantification | Abcam | ab65336 |
| Non-esterified Free Fatty Acids (NEFA) Colorimetric Assay Kit | Elabscience | E-BC-K014 |
| Ultra Sensitive Mouse Insulin ELISA Kit | Crystal Chem | 501947920 |
| Diet | ||
| Western and Fast-Food diet | Teklad | TD.120528 |
| D-(−)-Fructose | Sigma-Aldrich | F0127 |
| Mouse diet high fat (60%) soft pellets | Bio-Serv | S3282 |
| D-(+)-Glucose | Sigma-Aldrich | G8270 |
| Sequencing data | ||
| RNA-Seq (BMDMs) | Gene expression omnibus (GEO) database | GSE193577 |
| RNA-Seq (Liver macrophages from HFD/ND mice) | Gene expression omnibus (GEO) database | GSE197695 |
| RNA-Seq (Liver macrophages from WD mice) | Gene expression omnibus (GEO) database | GSE198595 |
| Plasmids | ||
| pLKO.1-puro non-Mammalian shRNA Control Plasmid DNA | Sigma | SHC002 |
| psPAX2 | Addgene | 12260 |
| pMD2.G | Addgene | 12259 |
| lentiCRISPR v2 | Addgene | 52961 |
| Oligonucleotides | ||
| Hprt-F (5‘-3‘) TCAGTCAACGGGGGACATAAA Hprt-R (5‘-3‘) GGGGCTGTACTGCTTAACCAG | ||
| mSphk1-1F (5‘-3‘) AT GGAACCAGT AGAAT GCCCT | ||
| mSphk1-1R (5‘-3‘) TCCGTTCGGT GAGTAT CAGTTT A | ||
| mSphk2-F (5‘-3‘) CACGGCGAGTTT GGTT CCT A mSphk2-R (5‘-3‘) CTTCTGGCI I I GGGCGTAGT | ||
| musa-Acna2-F (5‘-3‘) GTCCCAGACATCAGGGAGTAA musa-Acna2-R (5‘-3‘) TCGGATACTTCAGCGTCAGGA | ||
| musAdam10-F (5‘-3‘) ATGGTGTTGCCGACAGTGTTA musAdam10-R (5‘-3‘) GTTTGGCACGCTGGTGTTTTT | ||
| musAdam17-F (5‘-3‘) GGAACACGTCGTGGGATAATG musAdam17-R (5‘-3‘) GGCAGACI I IGGATGCTTCTT | ||
| musApoe-F (5‘-3‘) CT GACAGGAT GCCT AGCCG musApoe-R (5‘-3‘) CGCAGGTAATCCCAGAAGC | ||
| musCol1a1-F (5‘-3‘) GCTCCTCTTAGGGGCCACT musCol1a1-R (5‘-3‘) CCACGT CT CACCATTGGGG | ||
| musIl-1b-F (5‘-3‘) G CAACT GTTCCT GAACT CAACT musIl-1b-R (5‘-3‘) ATCIIIIGGGGTCCGTCAACT | ||
| musIl-6-F (5‘-3‘) CCAAGAGGTGAGTGCTTCCC musIl-6-R (5‘-3‘) CT GTT GTT CAGACT CT CTCCCT | ||
| musLipa-F (5‘-3‘) TGTTCGTTTTCACCATTGGGA musLipa-R (5‘-3‘) CGCATGATTATCTCGGTCACA | ||
| musLpl-F (5‘-3‘) GGGAGTTTGGCTCCAGAGTTT musLpl-R (5‘-3‘) T GT GT CTT CAGGGGTCCTT AG | ||
| musPpara-F (5‘-3‘) AGAGCCCCATCTGTCCTCTC musPpara-R (5‘-3‘) ACTGGTAGTCTG CAAAACCAAA | ||
| musPparg-F (5‘-3‘) TCGCTGATGCACTGCCTATG musPparg-R (5‘-3‘) GAGAGGTCCACAGAGCTGATT | ||
| musS1pr1-F (5‘-3‘) AT GGT GTCCACTAGCATCCC musS1pr1-R (5‘-3‘) CGAT GTT CAACTT GCCTGT GT AG | ||
| musS1pr2-F (5‘-3‘) ATGGGCGGCTTATACTCAGAG musS1pr2-R (5‘-3‘) GCG CAGCACAAGAT GAT GAT | ||
| musS1pr3-F (5‘-3‘) ACTCTCCGGGAACATTACGAT musS1pr3-R (5‘-3‘) CAAG ACGAT GAAGCT ACAGGT G | ||
| musS1pr4-F (5‘-3‘) GTCAGGGACTCGT ACCTTCCA musS1pr4-R (5‘-3‘) GAT GCAGCCAT ACACACGG | ||
| musS1pr5-F (5‘-3‘) GCTTTGGTTTGCGCGTGAG musS1pr5-R (5‘-3‘) GGCGTCCTAAGCAGTTCCAG | ||
| musSrebf1-F (5‘-3‘) GCAGCCACCATCTAGCCTG musSrebf1-R (5‘-3‘) CAGCAGT GAGT CT GCCTT GAT | ||
| musTgfb1-F (5‘-3‘) CTCCCGTGGCTTCTAGTGC musTgfb1-R (5‘-3‘) GCCTTAG I I I GGACAGGATCTG | ||
| musTimp1-F (5‘-3‘) GCAACTCGGACCTGGTCATAA musTimp1-R (5‘-3‘) CGGCCCGT GAT GAGAAACT | ||
| musTnf-F (5‘-3‘) GACGTGGAACTGGCAGAAGAG musTnf-R (5‘-3‘) TTGGTGG I I I GTGAGTGTGAG | ||
| musTrem2-F (5‘-3‘) CT GGAACCGT CACCATCACT C musTrem2-R (5‘-3‘) CGAAACTCGATGACTCCTCGG | ||
| shAdam17-1 (mouse) (5‘-3‘) TGCTGTTGACAGTGAGCGCCAGAATG I I I GTGGCTACCAATAGTGAAGCCACAGATGTATT GGT AGCCACAAACATT CT GTT GCCT ACTGCCTCGGA | ||
| shAdam17-2 (mouse) (5‘-3‘) TGCTGTTGACAGTGAGCGCCAGAGCCGAGTTGACAGCAAATAGTGAAGCCACAGATGTATT TGCTGT CAACTCGGCT CT GATGCCT ACT GCCTCGGA | ||
| shAdam10-1 (mouse) (5‘-3‘) TGCT GTT GACAGT GAGCGACACAGT GTGCATT CAAGT CAAT AGT GAAGCCACAGAT GTATT GACTT GAAT GCACACT GTGCTGCCT ACT GCCTCGGA | ||
| shAdam10-2 (mouse) (5‘-3‘) | ||
| TGCTGTTGACAGTGAGCGCCAGCAGGTAGTTGAAATTGTATAGTGAAGCCACAGATGTATA CAA1 1 1 CAACT ACCT GCT GATGCCT ACT GCCTCGGA | ||
| shS1pr1-1 (mouse) (5‘-3’) TGCT GTT GACAGT GAGCGAT AGGT AGGTAGT AATT GT GAAT AGT GAAGCCACAGAT GT ATT CACAATT ACT ACCT ACCTACT GCCT ACTGCCTCGGA | ||
| shS1pr1-2 (mouse) (5‘-3’) TGCT GTT GACAGT GAGCGCCCGCAGCT CAGT CTCT GACT AT AGT GAAGCCACAGAT GTATA GTCAGAGACT GAGCTGCGGAT GCCT ACTGCCTCGGA | ||
| shS1pr2-1 (mouse) (5‘-3’) TGCT GTT GACAGT GAGCGCACGGTACAAACT GT A1 1 1 AT AT AGT GAAGCCACAGAT GT AT AT AAATACAG 1 1 1GTACCGTATGCCTACTGCCTCGGA | ||
| sgNT1-F (5‘-3’) CACCGCT GAAAAAGGAAGGAGTT GA sgNT1-R (5‘-3’) AAACT CAACTCCTTCCTTTTT CAGC | ||
| hTREM2 gRNA2 F (5‘-3’) CACCGGCCATCACAGACGATACCCT hTREM2_gRNA2_R (5‘-3’) AAACAGGGTATCGTCTGTGATGGCC | ||
| mSphk1 gRNA1 F (5‘-3’) CACCGCGGT GAT GGT CT GATGCAT G mSphk1_gRNA1_R (5‘-3’) AAACCATGCATCAGACCATCACCGC | ||
| mSphk2 gRNA1 F (5‘-3’) CACCGGCGCATTGCCCGATCCACAG mSphk2_gRNA1_R (5‘-3’) AAACCTGTGGATCGGGCAATGCGCC | ||
| Software and Algorithms | ||
| Prism version 8 | GraphPad | RRID:SCR_002798; https://www.graphpad.com/scientificsoftware/prism/ |
| Image Software | BioTek | BioTek Cytation 5 |
| FlowJo v10.4 or greater | Tree Star | RRID:SCR_008520; https://www.flowjo.com |
| Ingenuity Pathway Analysis | QIAGEN | RRID:SCR_008653; https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/content-exploration-and-databases/qiagen-ipa/ |
| R 3.6.1 | R Core Team (2020) | https://www.R-project.org/ |
| Image J | NIH | Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2018. |
| RGB Measure Plus ImageJ Plugin | University of Leiden | Dimiter Prodanov (D.Prodanov at lumc.nl) University of Leiden |
| UTSW BioHPC | UTSW | https://portal.biohpc.swmed.edu |
Highlights.
TREM2-dependent macrophage efferocytosis maintains immune homeostasis in fatty liver
Hepatocyte-derived S1P upregulates TREM2 in infiltrated liver macrophages
Persistent obesity causes TREM2 shedding via TNF and IL-1β induced proteolytic cleavage
Loss of TREM2 disables efferocytosis to license chronic liver inflammation and NASH
ACKNOWLEDGMEMTS
Y.X. is supported by CAGT fellowship from Center for Advanced Genomic Technologies at Duke University. S.L. is supported by a CPRIT Individual Scholar Research Award (RP200197) and an AASLD (AASLDF 50028) Pinnacle Research Award. Z.Z. is a CPRIT Scholar and supported by a CPRIT First-time Tenure-Track Recruitment Award (RR180014). Z.Z. is also supported by an AASLD Pinnacle Research Award and a UTSW Circle of Friends Award in Cancer Research. Y.H. is supported by grants from NIH (R01DK099558 and R01CA233794), European Commission (ERC-2014-AdG-671231 HEPCIR), and CPRIT (RR180016). This research was supported in part by the computational resources provided by the BioHPC supercomputing facility in the Lyda Hill Department of Bioinformatics at UTSW. We also thank Dr. David Farrar from the UTSW Flow Cytometry Core facility for technical assistance, and Drs. Lora Hooper and Hao Zhu for critical reading and feedbacks of the manuscript. Graphic abstract was made using BioRender.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Arrese M, Cabrera D, Kalergis AM, and Feldstein AE (2016). Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci 61, 1294–1303. 10.1007/s10620-016-4049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrne CD, and Targher G. (2015). NAFLD: a multisystem disease. J Hepatol 62, S47–64. 10.1016/j.jhep.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL, Neuschwander-Tetri BA, Rinella M, and Sanyal AJ (2018). Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24, 908–922. 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuster S, Cabrera D, Arrese M, and Feldstein AE (2018). Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 15, 349–364. 10.1038/s41575-018-0009-6. [DOI] [PubMed] [Google Scholar]
- 5.Arab JP., Arrese M., and Trauner M. (2018). Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu Rev Pathol 13, 321–350. 10.1146/annurev-pathol-020117-043617. [DOI] [PubMed] [Google Scholar]
- 6.Sun B, and Karin M. (2013). Inflammation and liver tumorigenesis. Front Med 7, 242–254. 10.1007/s11684-013-0256-4. [DOI] [PubMed] [Google Scholar]
- 7.Sun B, and Karin M. (2012). Obesity, inflammation, and liver cancer. J Hepatol 56, 704–713. 10.1016/j.jhep.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, and Karin M. (2019). Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab 29, 18–26. 10.1016/j.cmet.2018.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grivennikov SI, Greten FR, and Karin M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899. 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, and Karin M. (2010). Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208. 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, Vrbanac AF, Li W, Perkins A, Matsutani T, et al. (2017). Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345. 10.1038/nature24302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, Ji Y, Li C, Guo L, et al. (2019). Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol Cell 75, 644–660 e645. 10.1016/j.molcel.2019.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, Ouyang Z, Li RZ, Sun X, et al. (2020). Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 52, 1057–1074 e1057. 10.1016/j.immuni.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Remmerie A, Martens L, Thone T, Castoldi A, Seurinck R, Pavie B, Roels J, Vanneste B, De Prijck S, Vanhockerhout M, et al. (2020). Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 53, 641–657 e614. 10.1016/j.immuni.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, Liss KH, Klein S, Feldstein AE, Finck BN, et al. (2021). Dynamic Shifts in the Composition of Resident and Recruited Macrophages Influence Tissue Remodeling in NASH. Cell Rep 34, 108626. 10.1016/j.celrep.2020.108626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, et al. (2019). Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 10.1038/s41586-019-1631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D, et al. (2019). Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 179, 829–845 e820. 10.1016/j.cell.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Bouchon A, Dietrich J, and Colonna M. (2000). Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol 164, 4991–4995. 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- 19.Kober DL, and Brett TJ (2017). TREM2-Ligand Interactions in Health and Disease. J Mol Biol 429, 1607–1629. 10.1016/j.jmb.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamerman JA., Jarjoura JR., Humphrey MB., Nakamura MC., Seaman WE., and Lanier LL. (2006). Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 177, 2051–2055. 10.4049/jimmunol.177.4.2051. [DOI] [PubMed] [Google Scholar]
- 21.Paloneva J, Mandelin J, Kiialainen A, Bohling T, Prudlo J, Hakola P, Haltia M, Konttinen YT, and Peltonen L. (2003). DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med 198, 669–675. 10.1084/jem.20030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, and Humphrey MB (2010). TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal 3, ra38. 10.1126/scisignal.2000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deczkowska A, Weiner A, and Amit I. (2020). The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 181, 1207–1217. 10.1016/j.cell.2020.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, and Walter J. (2013). Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J Biol Chem 288, 33027–33036. 10.1074/jbc.M113.517540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feuerbach D, Schindler P, Barske C, Joller S, Beng-Louka E, Worringer KA, Kommineni S, Kaykas A, Ho DJ, Ye C, et al. (2017). ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci Lett 660, 109–114. 10.1016/j.neulet.2017.09.034. [DOI] [PubMed] [Google Scholar]
- 26.Schlepckow K, Kleinberger G, Fukumori A, Feederle R, Lichtenthaler SF, Steiner H, and Haass C. (2017). An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med 9, 1356–1365. 10.15252/emmm.201707672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esparza-Baquer A, Labiano I, Sharif O, Agirre-Lizaso A, Oakley F, Rodrigues PM, Zhuravleva E, O’Rourke CJ, Hijona E, Jimenez-Aguero R, et al. (2020). TREM-2 defends the liver against hepatocellular carcinoma through multifactorial protective mechanisms. Gut. 10.1136/gutjnl-2019-319227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou J, Zhang J, Cui P, Zhou Y, Liu C, Wu X, Ji Y, Wang S, Cheng B, Ye H, et al. (2021). TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J Clin Invest 131. 10.1172/JCI135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Milicevic N, Hoebinger C, Goderle L, Hladik A, Shaw LE, Horstmann H, et al. (2022). Soluble TREM2 levels reflect the recruitment and expansion of TREM2(+) macrophages that localize to fibrotic areas and limit NASH. J Hepatol. 10.1016/j.jhep.2022.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Forster I. (1999). Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8, 265–277. 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 31.Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, and Colonna M. (2006). Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 177, 3520–3524. 10.4049/jimmunol.177.6.3520. [DOI] [PubMed] [Google Scholar]
- 32.Tsuchida T., Lee YA., Fujiwara N., Ybanez M., Allen B., Martins S., Fiel MI., Goossens N., Chou HI., Hoshida Y., and Friedman SL. (2018). A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 69, 385–395. 10.1016/j.jhep.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, and Gores G. (2011). Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 301, G825–834. 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perugorria MJ, Esparza-Baquer A, Oakley F, Labiano I, Korosec A, Jais A, Mann J, Tiniakos D, Santos-Laso A, Arbelaiz A, et al. (2018). Non-parenchymal TREM-2 protects the liver from immune-mediated hepatocellular damage. Gut. 10.1136/gutjnl-2017-314107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, and Karin M. (2011). Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147, 173–184. 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, et al. (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343. 10.1016/j.ccr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song X, Huang Y, Neuhouser ML, Tinker LF, Vitolins MZ, Prentice RL, and Lampe JW (2017). Dietary long-chain fatty acids and carbohydrate biomarker evaluation in a controlled feeding study in participants from the Women’s Health Initiative cohort. Am J Clin Nutr 105, 1272–1282. 10.3945/ajcn.117.153072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morioka S, Maueroder C, and Ravichandran KS (2019). Living on the Edge: Efferocytosis at the Interface of Homeostasis and Pathology. Immunity 50, 1149–1162. 10.1016/j.immuni.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arandjelovic S, and Ravichandran KS (2015). Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16, 907–917. 10.1038/ni.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park SY, and Kim IS (2017). Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Exp Mol Med 49, e331. 10.1038/emm.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leonardi-Essmann F, Emig M, Kitamura Y, Spanagel R, and Gebicke-Haerter PJ (2005). Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. J Neuroimmunol 160, 92–101. 10.1016/j.jneuroim.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 42.Miksa M, Amin D, Wu R, Ravikumar TS, and Wang P. (2007). Fractalkine-induced MFG-E8 leads to enhanced apoptotic cell clearance by macrophages. Mol Med 13, 553–560. 10.2119/2007-00019.Miksa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo B, Gan W, Liu Z, Shen Z, Wang J, Shi R, Liu Y, Liu Y, Jiang M, Zhang Z, and Wu Y. (2016). Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 44, 287–302. 10.1016/j.immuni.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, Wesselborg S, and Lauber K. (2008). Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem 283, 5296–5305. 10.1074/jbc.M706586200. [DOI] [PubMed] [Google Scholar]
- 45.Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, et al. (2003). Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113, 717–730. 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 46.Gude DR., Alvarez SE., Paugh SW., Mitra P., Yu J., Griffiths R., Barbour SE., Milstien S., and Spiegel S. (2008). Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J 22, 2629–2638. 10.1096/fj.08107169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weigert A, Cremer S, Schmidt MV, von Knethen A, Angioni C, Geisslinger G, and Brune B. (2010). Cleavage of sphingosine kinase 2 by caspase-1 provokes its release from apoptotic cells. Blood 115, 3531–3540. 10.1182/blood-2009-10-243444. [DOI] [PubMed] [Google Scholar]
- 48.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, et al. (2009). Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286. 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, and Brown S. (2009). Sphingosine 1-phosphate receptor signaling. Annu Rev Biochem 78, 743–768. 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 50.Bryan AM, and Del Poeta M. (2018). Sphingosine-1-phosphate receptors and innate immunity. Cell Microbiol 20, e12836. 10.1111/cmi.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, and Hedrick CC (2008). Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res 102, 950–958. 10.1161/CIRCRESAHA.107.170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez-Cabrera PJ, Jo E, Sanna MG, Brown S, Leaf N, Marsolais D, Schaeffer MT, Chapman J, Cameron M, Guerrero M, et al. (2008). Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol Pharmacol 74, 1308–1318. 10.1124/mol.108.049783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krenkel O, and Tacke F. (2017). Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17, 306–321. 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- 54.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coelho I, Duarte N, Barros A, Macedo MP, and Penha-Goncalves C. (2020). Trem-2 Promotes Emergence of Restorative Macrophages and Endothelial Cells During Recovery From Hepatic Tissue Damage. Front Immunol 11, 616044. 10.3389/fimmu.2020.616044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoang SA, Oseini A, Feaver RE, Cole BK, Asgharpour A, Vincent R, Siddiqui M, Lawson MJ, Day NC, Taylor JM, et al. (2019). Gene Expression Predicts Histological Severity and Reveals Distinct Molecular Profiles of Nonalcoholic Fatty Liver Disease. Sci Rep 9, 12541. 10.1038/s41598-019-48746-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arendt BM, Comelli EM, Ma DW, Lou W, Teterina A, Kim T, Fung SK, Wong DK, McGilvray I, Fischer SE, and Allard JP (2015). Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 61, 1565–1578. 10.1002/hep.27695. [DOI] [PubMed] [Google Scholar]
- 58.Dixon LJ, Berk M, Thapaliya S, Papouchado BG, and Feldstein AE (2012). Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest 92, 713–723. 10.1038/labinvest.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wree A, McGeough MD, Pena CA, Schlattjan M, Li H, Inzaugarat ME, Messer K, Canbay A, Hoffman HM, and Feldstein AE (2014). NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J Mol Med (Berl) 92, 1069–1082. 10.1007/s00109-014-1170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thornton P, Sevalle J, Deery MJ, Fraser G, Zhou Y, Stahl S, Franssen EH, Dodd RB, Qamar S, Gomez Perez-Nievas B, et al. (2017). TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol Med 9, 1366–1378. 10.15252/emmm.201707673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Indira Chandran V., Wernberg CW., Lauridsen MM., Skytthe MK., Bendixen SM., Larsen FT., Hansen CD., Gronkjaer LL., Siersbaek MS., Caterino TD., et al. (2022). Circulating TREM2 as a noninvasive diagnostic biomarker for NASH in patients with elevated liver stiffness. Hepatology. 10.1002/hep.32620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanehisa M, and Goto S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kramer A, Green J, Pollard J Jr., and Tugendreich S. (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, and Gores GJ (2003). Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 125, 437–443. 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 65.Kahraman A, Schlattjan M, Kocabayoglu P, Yildiz-Meziletoglu S, Schlensak M, Fingas CD, Wedemeyer I, Marquitan G, Gieseler RK, Baba HA, et al. (2010). Major histocompatibility complex class I-related chains A and B (MIC A/B): a novel role in nonalcoholic steatohepatitis. Hepatology 51, 92–102. 10.1002/hep.23253. [DOI] [PubMed] [Google Scholar]
- 66.Xue T, Ji J, Sun Y, Huang X, Cai Z, Yang J, Guo W, Guo R, Cheng H, and Sun X. (2022). Sphingosine-1-phosphate, a novel TREM2 ligand, promotes microglial phagocytosis to protect against ischemic brain injury. Acta Pharm Sin B 12, 1885–1898. 10.1016/j.apsb.2021.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu K, Byers DE, Jin X, Agapov E, Alexander-Brett J, Patel AC, Cella M, Gilfilan S, Colonna M, Kober DL, et al. (2015). TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med 212, 681–697. 10.1084/jem.20141732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhong L, Xu Y, Zhuo R, Wang T, Wang K, Huang R, Wang D, Gao Y, Zhu Y, Sheng X, et al. (2019). Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat Commun 10, 1365. 10.1038/s41467-019-09118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asai A, Chou PM, Bu HF, Wang X, Rao MS, Jiang A, DiDonato CJ, and Tan XD (2014). Dissociation of hepatic insulin resistance from susceptibility of nonalcoholic fatty liver disease induced by a high-fat and high-carbohydrate diet in mice. Am J Physiol Gastrointest Liver Physiol 306, G496–504. 10.1152/ajpgi.00291.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liao CY, Song MJ, Gao Y, Mauer AS, Revzin A, and Malhi H. (2018). Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front Immunol 9, 2980. 10.3389/fimmu.2018.02980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kosack L, Gawish R, Lercher A, Vilagos B, Hladik A, Lakovits K, Bhattacharya A, Schliehe C, Mesteri I, Knapp S, and Bergthaler A. (2017). The lipid-sensor TREM2 aggravates disease in a model of LCMV-induced hepatitis. Sci Rep 7, 11289. 10.1038/s41598-017-10637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang R, Ding Q, Yaqoob U, de Assuncao TM, Verma VK, Hirsova P, Cao S, Mukhopadhyay D, Huebert RC, and Shah VH (2015). Exosome Adherence and Internalization by Hepatic Stellate Cells Triggers Sphingosine 1-Phosphate-dependent Migration. J Biol Chem 290, 30684–30696. 10.1074/jbc.M115.671735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sato M, Ikeda H, Uranbileg B, Kurano M, Saigusa D, Aoki J, Maki H, Kudo H, Hasegawa K, Kokudo N, and Yatomi Y. (2016). Sphingosine kinase-1, S1P transporter spinster homolog 2 and S1P2 mRNA expressions are increased in liver with advanced fibrosis in human. Sci Rep 6, 32119. 10.1038/srep32119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Geng T., Sutter A., Harland MD., Law BA., Ross JS., Lewin D., Palanisamy A., Russo SB., Chavin KD., and Cowart LA. (2015). SphK1 mediates hepatic inflammation in a mouse model of NASH induced by high saturated fat feeding and initiates proinflammatory signaling in hepatocytes. J Lipid Res 56, 2359–2371. 10.1194/jlr.M063511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miura K, Nagahashi M, Prasoon P, Hirose Y, Kobayashi T, Sakata J, Abe M, Sakimura K, Matsuda Y, Butash AL, et al. (2021). Dysregulation of sphingolipid metabolic enzymes leads to high levels of sphingosine-1-phosphate and ceramide in human hepatocellular carcinoma. Hepatol Res 51, 614–626. 10.1111/hepr.13625. [DOI] [PubMed] [Google Scholar]
- 76.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. (2019). Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698 e614. 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu C, Li P, Li H, Wang S, Ding L, Wang H, Ye H, Jin Y, Hou J, Fang X, and Shu Q. (2019). TREM2 regulates obesity-induced insulin resistance via adipose tissue remodeling in mice of high-fat feeding. J Transl Med 17, 300. 10.1186/s12967-019-2050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sharif O, Brunner JS, Korosec A, Martins R, Jais A, Snijder B, Vogel A, Caldera M, Hladik A, Lakovits K, et al. (2021). Beneficial Metabolic Effects of TREM2 in Obesity Are Uncoupled From Its Expression on Macrophages. Diabetes 70, 2042–2057. 10.2337/db20-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Winn NC, Wolf EM, Garcia JN, and Hasty AH (2022). Exon 2-mediated deletion of Trem2 does not worsen metabolic function in diet-induced obese mice. J Physiol. 10.1113/JP283684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tonan T, Fujimoto K, Qayyum A, Morita Y, Nakashima O, Ono N, Kawahara A, Kage M, Hayabuchi N, and Ueno T. (2012). CD14 expression and Kupffer cell dysfunction in non-alcoholic steatohepatitis: superparamagnetic iron oxide-magnetic resonance image and pathologic correlation. J Gastroenterol Hepatol 27, 789–796. 10.1111/j.1440-1746.2011.07057.x. [DOI] [PubMed] [Google Scholar]
- 81.Cheong H, Lee SS, Lee JS, Kim J, Kim SW, and Lee WJ (2015). Phagocytic function of Kupffer cells in mouse nonalcoholic fatty liver disease models: Evaluation with superparamagnetic iron oxide. J Magn Reson Imaging 41, 1218–1227. 10.1002/jmri.24674. [DOI] [PubMed] [Google Scholar]
- 82.Szabo G, and Petrasek J. (2015). Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol 12, 387–400. 10.1038/nrgastro.2015.94. [DOI] [PubMed] [Google Scholar]
- 83.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. (2012). Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185. 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Macpherson AJ, Heikenwalder M, and Ganal-Vonarburg SC (2016). The Liver at the Nexus of Host-Microbial Interactions. Cell Host Microbe 20, 561–571. 10.1016/j.chom.2016.10.016. [DOI] [PubMed] [Google Scholar]
- 85.Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, Motola DL, Luther S, Bohr S, Jeoung SW, et al. (2015). Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol Gastroenterol Hepatol 1, 222–232. 10.1016/j.jcmgh.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parlakgul G, Arruda AP, Pang S, Cagampan E, Min N, Guney E, Lee GY, Inouye K, Hess HF, Xu CS, and Hotamisligil GS (2022). Regulation of liver subcellular architecture controls metabolic homeostasis. Nature. 10.1038/s41586-022-04488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Choi Y, Abdelmegeed MA, and Song BJ (2017). Diet high in fructose promotes liver steatosis and hepatocyte apoptosis in C57BL/6J female mice: Role of disturbed lipid homeostasis and increased oxidative stress. Food Chem Toxicol 103, 111–121. 10.1016/j.fct.2017.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bellanti F., Villani R., Tamborra R., Blonda M., Iannelli G., di Bello G., Facciorusso A., Poli G., Iuliano L., Avolio C., et al. (2018). Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol 15, 86–96. 10.1016/j.redox.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gaskell H, Ge X, and Nieto N. (2018). High-Mobility Group Box-1 and Liver Disease. Hepatol Commun 2, 1005–1020. 10.1002/hep4.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang X, Wu X, Hu Q, Wu J, Wang G, Hong Z, Ren J, Lab for T, and Surgical I. (2019). Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci 236, 116464. 10.1016/j.lfs.2019.05.020. [DOI] [PubMed] [Google Scholar]
- 91.Yona S, Heinsbroek SE, Peiser L, Gordon S, Perretti M, and Flower RJ (2006). Impaired phagocytic mechanism in annexin 1 null macrophages. Br J Pharmacol 148, 469–477. 10.1038/sj.bjp.0706730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McArthur S, Cristante E, Paterno M, Christian H, Roncaroli F, Gillies GE, and Solito E. (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J Immunol 185, 6317–6328. 10.4049/jimmunol.1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Locatelli I, Sutti S, Jindal A, Vacchiano M, Bozzola C, Reutelingsperger C, Kusters D, Bena S, Parola M, Paternostro C, et al. (2014). Endogenous annexin A1 is a novel protective determinant in nonalcoholic steatohepatitis in mice. Hepatology 60, 531–544. 10.1002/hep.27141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miquilena-Colina ME, Lima-Cabello E, Sanchez-Campos S, Garcia-Mediavilla MV, Fernandez-Bermejo M, Lozano-Rodriguez T, Vargas-Castrillon J, Buque X, Ochoa B, Aspichueta P, et al. (2011). Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 60, 1394–1402. 10.1136/gut.2010.222844. [DOI] [PubMed] [Google Scholar]
- 95.Koonen DP, Jacobs RL, Febbraio M, Young ME, Soltys CL, Ong H, Vance DE, and Dyck JR (2007). Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 56, 2863–2871. 10.2337/db07-0907. [DOI] [PubMed] [Google Scholar]
- 96.Kim SM, Mun BR, Lee SJ, Joh Y, Lee HY, Ji KY, Choi HR, Lee EH, Kim EM, Jang JH, et al. (2017). TREM2 promotes Abeta phagocytosis by upregulating C/EBPalpha-dependent CD36 expression in microglia. Sci Rep 7, 11118. 10.1038/s41598-017-11634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Issekutz TB (1995). In vivo blood monocyte migration to acute inflammatory reactions, IL-1 alpha, TNF-alpha, IFN-gamma, and C5a utilizes LFA-1, Mac-1, and VLA-4. The relative importance of each integrin. J Immunol 154, 6533–6540. [PubMed] [Google Scholar]
- 98.Shirotani K, Hori Y, Yoshizaki R, Higuchi E, Colonna M, Saito T, Hashimoto S, Saito T, Saido TC, and Iwata N. (2019). Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Sci Rep 9, 7508. 10.1038/s41598-019-43535-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321. 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 100.Liang S, Ma HY, Zhong Z, Dhar D, Liu X, Xu J, Koyama Y, Nishio T, Karin D, Karin G, et al. (2019). NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 156, 1156–1172 e1156. 10.1053/j.gastro.2018.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, and Schwabe RF (2007). TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13, 1324–1332. 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 102.Zhong Z., Liang S., Sanchez-Lopez E., He F., Shalapour S., Lin XJ., Wong J., Ding S., Seki E., Schnabl B., et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen YH, Huang TY, Lin YT, Lin SY, Li WH, Hsiao HJ, Yan RL, Tang HW, Shen ZQ, Chen GC, et al. (2021). VPS34 K29/K48 branched ubiquitination governed by UBE3C and TRABID regulates autophagy, proteostasis and liver metabolism. Nat Commun 12, 1322. 10.1038/s41467-021-21715-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fujiwara N, Kobayashi M, Fobar AJ, Hoshida A, Marquez CA, Koneru B, Panda G, Taguri M, Qian T, Raman I, et al. (2021). A blood-based prognostic liver secretome signature and long-term hepatocellular carcinoma risk in advanced liver fibrosis. Med (N Y) 2, 836–850 e810. 10.1016/j.medj.2021.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nakagawa S, Wei L, Song WM, Higashi T, Ghoshal S, Kim RS, Bian CB, Yamada S, Sun X, Venkatesh A, et al. (2016). Molecular Liver Cancer Prevention in Cirrhosis by Organ Transcriptome Analysis and Lysophosphatidic Acid Pathway Inhibition. Cancer Cell 30, 879–890. 10.1016/j.ccell.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Efferocytosis of lipid-laden apoptotic hepatocytes (green) by Trem2F/F BMDMs (red), Related to Figure 5 Apoptotic AML12 cells (mouse hepatocyte cell line) were generated by treatment of 800 μM PA for 24 hours. CellTracker-Green labeled apoptotic cells (105 cells/well) were incubated with CellTracker-Red labeled Trem2F/F BMDMs (2 × 105 cells/well) in a 24-well glass bottomed plate (Greiner Bio-One). Videos were recorded in a cell incubation system and images were taken every 15 min during incubation by a Cytation 5 Cell Imaging Multimode Reader (BioTek).
Video S2. Efferocytosis of lipid-laden apoptotic hepatocytes (green) by Trem2ΔMye BMDMs (red), Related to Figure 5 Apoptotic AML12 cells were generated as in Video S1. CellTracker-Green labeled apoptotic cells (105 cells/well) were incubated with CellTracker-Red labeled Trem2 ΔMye BMDMs (2 × 105 cells/well) in a 24-well glass bottomed plate (Greiner Bio-One). Videos were recorded as described in Video S1.
Data Availability Statement
Data generated in this study are available in the Gene Expression Omnibus (GEO) database under the accession numbers GSE193577, GSE197695 and GSE198595.
All data reported in this paper will be shared by the lead contact upon reasonable request. This paper does not report any original code.