

Abstract
Decarboxylative protonation is a general deletion tactic to replace polar carboxylic acid groups with hydrogen or its isotope. Current methods rely on the pre-activation of acids, non-sustainable hydrogen sources, and/or expensive/highly oxidizing photocatalysts, presenting challenges to their wide adoption. Here we show that a cooperative iron/thiol catalyst system can readily achieve this transformation, hydrodecarboxylating a wide range of activated and unactivated carboxylic acids and overcoming scope limitations in previous direct methods. The reaction is readily scaled in batch configuration and can be directly performed in deuterated solvent to afford high yields of d-incorporated products with excellent isotope incorporation efficiency; characteristics not attainable in previous photocatalyzed approaches. Preliminary mechanistic studies indicate a radical mechanism and kinetic results of unactivated acids (KIE = 1) are consistent with a light-limited reaction.
Keywords: Decarboxylation, Earth-Abundant Metal, Radicals, Synthetic Methods, Hydrogen Atom Transfer
Graphical Abstract
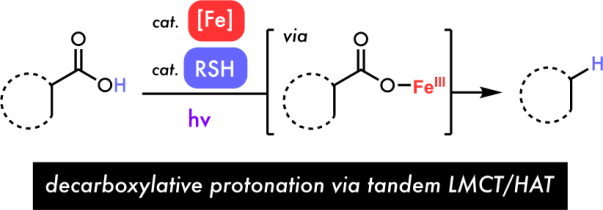
Combining simple iron and thiol catalysts allows carboxylic acids to be directly converted to C–H bonds under visible light irradiation. The cocatalytic system exhibits a wide scope, is scalable, and preliminary mechanistic studies suggest it functions via a tandem ligand-to-metal charge transfer (LMCT)/Hydrogen Atom Transfer (HAT) mechanism.
Introduction
Decarboxylative functionalization is an attractive class of chemical transformations for many reasons, including that carboxylic acids are abundant, stable and non-toxic feedstock building blocks and the reaction releases non-flammable and easy-to-remove CO2 as its byproduct.1 Within this class, decarboxylative protonation is the simplest, installing a hydrogen upon CO2 extrusion, making it an ideal model to study the decarboxylation step and its intersection with other catalytic cycles, providing fundamental insights useful for designing other valuable decarboxylative coupling reactions such as arylation and Heck-type olefination.1 From a synthetic perspective, decarboxylative protonation allows carboxylic acids to be the traceless directing groups and is often applied at the late stage of natural product syntheses.2 The replacement of hydrogen source by deuterium further serves as a chemoselective approach to synthesize isotopically labeled compounds valuable in materials and medicinal chemistry.3 Such utilities have inspired several decarboxylative protonation strategies, starting with Kochi’s seminal work in 1970 using silver and persulfate to oxidatively activate C(sp3)–COOH bonds.4 While able to decarboxylate simple acids, harsh conditions led to the formation of undesired products with poor selectivity in this approach. Similarly, Greaney and co-workers developed a radical decarboxylative protonation of more challenging aromatic acids using silver/persulfate system.5 Complementary to the radical process, a series of organometallic approaches have also been disclosed to decarboxylate C(sp2)–COOH bonds using copper6, silver6a,6d,7, palladium8 or gold9 followed by the hydrogen exchange of aryl–metal complexes to form the reduced product. Despite continuous progress in the catalytic thermo-decarboxylation of aromatic acids, these protocols required high temperature and possessed limited scope. More recent efforts by Yoshimi and Glorius successfully realized the light-mediated direct and/or two-step strategies to decarboxylate aromatic acids, allowing the reactions to proceed under milder conditions.10
While direct protodecarboxylation of C(sp2)–COOH bonds has been well-known, one-step removal of aliphatic acids remains challenging. Barton’s pioneering works demonstrated a general carboxylic acid deletion tactic involving two elementary steps: (1) converting carboxylic acid into the corresponding thiohydroxamate ester and (2) radical decarboxylation via photo- or thermo-cleavage followed by a hydrogen atom abstraction.11 Later, a series of advances have been made by different research groups towards milder and greener reaction conditions in a radical or organometallic fashion.12 However, most of these methods still required the pre-activation of aliphatic acids as redox-active esters or carboxylates, superstoichiometric amounts of hydrogen atom donors such as dithiothreitol (DTT), Hantzsch ester or phenylsilane, and/or precious metals (Figure 1a). Significant strides have been made toward direct decarboxylative protonation to avoid the need for pre-activation; however, the high potential of carboxylic acids remains a significant obstacle, requiring strong oxidants, harsh conditions, or stoichiometric photosensitizers to activate this otherwise stable functional group.12i,12j In 2015, Nicewicz and co-workers developed an elegant direct photocatalytic decarboxylative protonation leveraging the high reduction potential (E1/2 > 2 vs SCE) of an acridinium photoredox catalyst in its excited state (Figure 1b).13a Despite the ability to drive radical decarboxylation, the extremely oxidizing nature of its excited state leads to non-selective oxidation in the presence of other oxidatively-labile functional groups, including electron-rich arenes14, olefins15 and non-electron-deficient amines16, limiting the scope of this method to simple and/or highly activated carboxylic acids13. Additionally, this photocatalytic method suffers from low reaction efficiency likely due to the short excited state lifetime of these photocatalysts, resulting in extended reaction time, the need for multiple LED lamps to have sufficient photon flux, and poor scalability. Taking this together, a general and direct decarboxylative protonation tolerant of oxidatively-labile functionalities remains elusive.
Figure 1.

Current state of decarboxylative protonation. (a) decarboxylative protonation of activated redox ester. (b) direct decarboxylative protonation using acridinium photoredox catalyst. (c) this work: iron catalyzed chemoselective decarboxylative protonation.
Building on pioneering reports from Kochi17, Yoon18, Ritter19, Rovis20, and others21 recently developed methods taking advantage of the bond homolysis process using salts of earth abundant elements such as copper and iron. Such photo-induced Ligand-to-Metal-Charge-Transfer (LMCT) homolysis has provided a new opportunity to circumvent the non-selective and low-efficiency issues encountered in the outer-spheric oxidation of Fukuzumi-type photoredox catalysts (Figure 1c). While iron has been shown to catalyze decarboxylation reactions,22 many of these are thermally driven, require activated carboxylic acids, and a strong stoichiometric oxidant to proceed, with redox-neutral LMCT approaches relatively unexplored. We recently found iron catalysts to be compatible with thiol in our cooperative radical hydrogenation of olefins23, inspiring us to propose a new generation decarboxylative protonation via a photo-induced LMCT mechanism (Figure 2). First, the irradiation of an Fe(III) carboxylate from I leads to a photoexcited state that can homolyze to release carboxyl radical II and generate an Fe(II) species. Intermediate II can then rapidly decarboxylate to generate carbon-centered radical III which can be reduced by HAT from the thiol cocatalyst (step B) to form the desired product IV. Finally, the resultant thiyl, Fe(II) species, and another molecule of acid substrate I can combine to close their respective cycles and regenerate the Fe(III) carboxylate and thiol (step A). Importantly, we hypothesized that this inner-sphere LMCT process could significantly expand the scope of decarboxylative protonation reactions by avoiding unselective outer-sphere electron transfer from electron rich functional groups, allowing for incompatible substrates from the acridinium method to be engaged. Further, covalent pre-association of substrate with the iron photocatalyst should obviate the excited state lifetime limitations seen in outer-sphere approaches that depend on bimolecular reaction with an excited molecule, potentially allowing for this reaction to be scaled more easily.
Figure 2.

Proposed mechanism for the iron/thiol cocatalyzed decarboxylative protonation
Results and Discussion
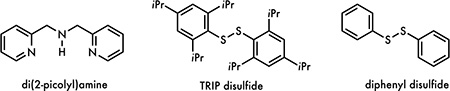
For our initial test, we chose 4-oxo-4-phenylbutanoic acid (1) as our substrate of study (Table 1). Gratifyingly, the reaction successfully generated 95% of propiophenone (2) using 2.5 mol% Fe(NO3)3·9H2O, 2.5 mol% di(2-picolyl)amine, 10 mol% Na2CO3 and 5 mol% TRIP disulfide in DCE/H2O under 390 nm LED irradiation (entry 1). We subsequently examined the reaction with lower Fe(NO3)3·9H2O/di(2-picolyl)amine loadings, furnishing 88% and 59% of 2 when 1 mol% and 0.5 mol% of Fe(NO3)3·9H2O/di(2-picolyl)amine were applied, respectively (entry 2 and 3). Control experiments showed both Fe(NO3)3·9H2O and TRIP disulfide to be essential, with only trace amount of 2 obtained in their absence (entry 4 and 5). The yield dropped to 25% when diphenyl disulfide was used as the HAT catalyst, potentially due to the in-situ coordination of iron with less hindered thiol (entry 6). It is worth noting that the reaction only generated 38% of 2 without the use of Na2CO3, suggesting its crucial role on the reaction efficiency (entry 7). Replacing 390 nm light source with 427 nm light led to 53% of the product formation (entry 8). No decarboxylation product was obtained when the reaction was conducted in the dark under the otherwise identical conditions (entry 9). Notably, DCE itself produced 77% of 2 with the formation of trace 1-phenylprop-2-en-1-one, indicating superior reactivity of the biphasic DCE/H2O solvent system (entry 10). Finally, we investigated the feasibility of ligand-free conditions and found the reaction smoothly produced 72% of 2 in the absence of di(2-picolyl)amine (entry 11). The yield further improved to 81% when 15 mol% of TRIP disulfide was used, making this ligand-free reaction a simple, economical and efficient photocatalytic decarboxylation approach (entry 12; see SI).
Table 1:
Development of iron-catalyzed decarboxylative protonation[a]

| |||||
|---|---|---|---|---|---|
| entry | Fe (a mol%) | ligand (b mol%) | disulfide (c mol%) | Na2CO3 (d mol%) | yield of 2 (%) |
| 1 | 2.5 | 2.5 | 5 | 10 | 95 |
| 2 | 1 | 1 | 5 | 10 | 88 |
| 3 | 0.5 | 0.5 | 5 | 10 | 59 |
| 4 | - | 2.5 | 5 | 10 | 4 |
| 5 | 2.5 | 2.5 | - | 10 | 6 |
| 6[b] | 2.5 | 2.5 | 5 | 10 | 25 |
| 7 | 2.5 | 2.5 | 5 | - | 38 |
| 8[c] | 2.5 | 2.5 | 5 | 10 | 53 |
| 9[d] | 2.5 | 2.5 | 5 | 10 | N.R. |
| 10[e] | 2.5 | 2.5 | 5 | 10 | 77 |
| 11 | 2.5 | - | 5 | 10 | 72 |
| 12 | 2.5 | - | 15 | 10 | 81 |
All reactions were conducted on a 0.4 mmol scale and the yields were determined by 1H NMR spectroscopy using trimethoxybenzene as an internal standard. TRIP disulfide = Bis(2,4,6-triisopropylphenyl) disulfide.
Diphenyl disulfide was used instead of TRIP disulfide.
427 nm LED light was used instead of 390 nm light.
No light; N.R. = no reaction.
DCE (0.1 M) was used as the solvent; Trace amount of α,β-unsaturated ketone was observed.
With the optimized reaction conditions in hand, we turned to examine substrates that were incompatible in previous direct decarboxylative protonations (Figure 3 and 4). Nicewicz and co-workers have demonstrated the wide applications of acridinium photoredox catalysts in cation-radical accelerated-nucleophilic aromatic substitutions (CRA-SNAr) via single electron oxidation of electron-rich arenes.14 Similar aromatic radical cation intermediates were also used to form high energy alkoxy radical species integrating proton-coupled electron transfer (PCET) for C–C bond cleavage.24 While powerful for these alternative reactions, such competing outer-spheric electron transfer processes render the generation of the key carboxylic radical needed for decarboxylation inefficient in the presence of electron-rich arenes (Figure 3).13 To our delight, the reactions of our iron-catalyzed decarboxylative protonation were found to proceed smoothly to afford product 3, 4, 7 and 8 in good to excellent yields despite bearing various electron-rich aromatics. In addition, the reaction similarly decarboxylated electron neutral and poor arene substrates to produce 5 and 6, showing an unmatched tolerance of our method toward a wide range of aromatic molecules.
Figure 3.

Decarboxylative protonation of arenes with different electronic properties: acridinium photoredox shows low reactivity in the presence of electron-rich arenes whereas iron-catalyzed decarboxylative protonation has broad arene scope.
Figure 4.

Decarboxylation in the presence of electron-rich olefins: acridinium photoredox selectively oxidizes alkene over carboxylic acid whereas iron catalysis enables decarboxylation in the presence of electron-rich olefins.
Furthermore, acridinium photoredox catalysts have been used to generate another important class of cation-radical intermediates from olefins for radical transformations including inter- and intra-molecular hydroesterifications (Figure 4).15 Under the acridinium-catalyzed decarboxylative protonation conditions, the presence of electron-rich olefins was found to generate high yields of hydroesterification products while suppressing the desired decarboxylation process. Since the same reaction system was used for both pathways, a direct strategy to decarboxylate these molecules remains elusive. To see if our iron-cocatalytic system could fill up this gap, we tested γ,δ-unsaturated carboxylic acids and successfully generated good yields of decarboxylation products 9a, 10a and 11a with only a minor amount of hydroesterification product (9b, 10b and 11b) formation, potentially due to competitive cyclization of the carboxylic radical onto the styrene to form a stabilized benzylic radical as opposed to direct styrene oxidation. The same protocol was also found to be feasible for intermolecular decarboxylative crossing coupling with styrene, a reaction that would otherwise give hydroesterification product using acridinium photo-catalysts (see SI). Together, these results show our method to be capable of chemo-selective decarboxylative protonation in the presence of previous incompatible functional groups, considerably expanding the scope of this chemistry to more general and synthetically-important applications.
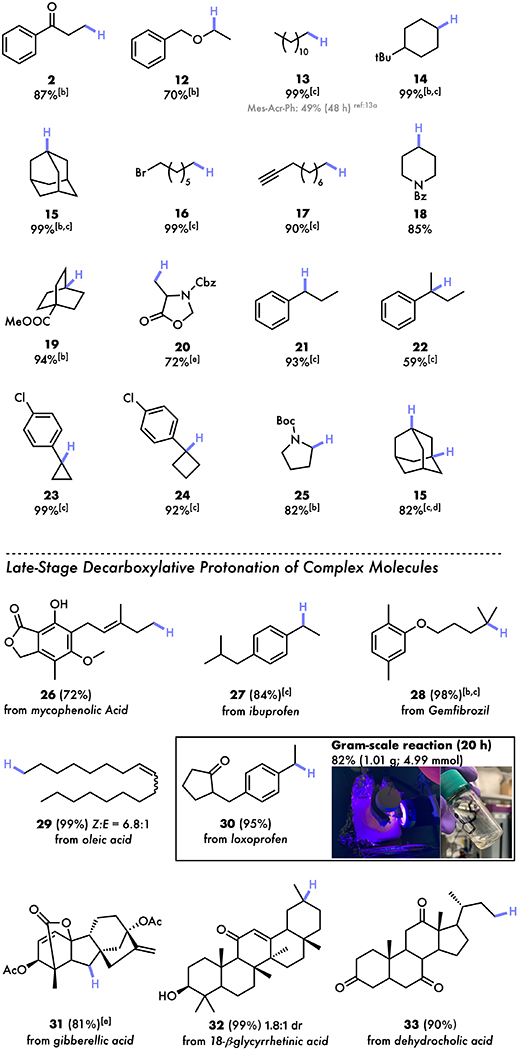
With exceptional chemo-selectivity demonstrated, we sought to assess the scope of our reaction (Table 2). First, we tested the substrates bearing ketone and ether functional groups, successfully obtaining 2 and 12 in 87 and 70% yields, respectively. We next investigated previously-challenging aliphatic acids and found the reactions efficiently formed 99% of primary, secondary and tertiary alkane products 13 – 15, presenting a potential approach to green fuel synthesis.25 The high compatibility with reducible functional groups including halide and alkyne (16 and 17) further highlighted the tolerance of our cooperative approach. Substrates with amide, ester or N-carbobenzyloxy functionalities also gave the corresponding products 18, 19 and 20 in 72 – 94% yields. The decarboxylation of more reactive benzylic substrates and an amino acid derivative similarly furnished 21 – 25 in good to high yields without the formation of undesired homocoupling products observed in the Lee’s decarboxylation method using a strong oxidant.12i Remarkably, a dicarboxylic acid substrate can be doubly-decarboxylated using these conditions, providing 82% of 15, albeit with the use of more solvent and 30% TRIP disulfide to address its reduced solubility.
Table 2:
Scope of Iron-catalyzed decarboxylative protonation[a]
|
All reactions were conducted on a 0.4 mmol scale.
2.5 mol% Fe(NO3)3·9H2O and 2.5 mol% di(2-picolyl)amine were used.
The yields were determined by 1H NMR spectroscopy using trimethoxybenzene as an internal standard due to the volatile properties or poor traceability on thin layer chromatography.
Reaction concentration = 0.05 M; 30 mol% TRIP disulfide were used.
15 mol% TRIP disulfide were used.
Encouraged by the success of small molecules, we then examined the viability of complex nature products and drugs. For mycophenolic acid, possessing alkene and alcohol functional groups, we obtained decarboxylation product 26 in a 72% yield. In analogy to the reaction of 21, the benzylic acid in ibuprofen decarboxylated to furnish 84% of 27. Gemfibrozil also transformed into the corresponding tertiary C–H product 28 in an outstanding 98% yield. The reaction of oleic acid bearing an internal Z-olefin provided 99% of 29 with the formation of minor E-isomer in 6.8:1 ratio. Loxoprofen was subjected to our conditions on a gram scale using the typical round bottom flask setup with one Kessil LED lamp, generating 82% of 30 overnight without special modifications. Notably, this result suggested that our method could address the light-dependent issues encountered in the previous photocatalytic decarboxylative protonation, where the reactions had drastically decreased initial rate without using multiple LED lamps on even moderate scales.13 Toward testing the more complex and steric demanding polycyclic structures, the reaction of a gibberellic acid derivative generated 81% of 31. 18-β-glycyrrhetinic acid and dehydrocholic acid similarly produced 32 and 33 in 99 and 90% yields. Furthermore, jasmonic acid decarboxylated smoothly to afford 90% of 34 with the formation of minor isomer. The reaction of indomethacin showed excellent tolerance of the oxidatively-labile indole moiety, providing 96% of 35. Bezofibrate and zaltoprofen possessing thioether and amide functional groups also exhibited good reactivities to form 36 and 37 in 98 and 81% yields. Interestingly, chlorambucil readily decarboxylated to 38 in a 79% yield despite having a N,N-dialkylaniline moiety that would otherwise be susceptible to highly-oxidizing acridinium and iridium photoredox catalysts16, showing iron catalyst to be selective for decarboxylation. Finally, the reaction of dichlofenac with a N,N-diaryl amine group was conducted to obtain 82% of 39.
Emboldened by the wide substrate tolerance of our reaction, we turned to explore the plausible reaction mechanism (Figure 5). First, the standard reactions performed in DCE/D2O led to high (95 and 98%) deuterium incorporations of 24´ and 2´, suggesting excess water to be the main hydrogen source in this reaction, likely via proton exchange with the thiol (5a). Lack of significant kinetic isotope effect (KIE ~ 1) of 4-oxo-4-phenylbutanoic acid obtained through separate reactions using H2O and D2O is consistent with the reaction being light-limited for aliphatic acids and is consistent with zero order behavior in all reaction components and a rate acceleration using multiple LED lamps (see SI).26 Interestingly, decarboxylative protonation of benzylic acid exhibits a primary isotopic effect (KIE = 3), suggesting the HAT from the thiol to be rate-determining in this case, presumably due to formation of a more stable benzylic carbon-radical intermediate. These observations are also notable as this one-step protocol has simplified the state-of-art decarboxylative deuteration, where the pre-salification with stoichiometric CsOH was essential to reach good d-incorporations and yields.12f The inhibition of 2 formation upon treatment with 50 mol% TEMPO suggests involvement of radical intermediates in our reaction (Figure 5b) as does a radical clock experiment which successfully furnished 72% of radical-induced ring-opening product 40 as an isomeric mixture with only trace amount of 41 generated (Figure 5c).
Figure 5.

(a) Isotope labeling of iron-catalyzed decarboxylative protonation provided high deuterium incorporation. (b) 50% TEMPO drastically hampered the formation of 2. (c) Radical clock experiment produced high yield of linear olefin as an isomeric mixture.
Conclusion
In conclusion, we have provided a chemoselective decarboxylative protonation using Earth-abundant iron photocatalysis. Our method features the synergistic combination of iron and hydrogen atom transfer catalysts, eliminating the use of expensive and complex photoredox catalysts, toxic hydrogen atom sources, high temperature, strong oxidants and the requirement to pre-activate acid substrates. Furthermore, this operationally simple protocol exhibits wide substrate tolerance including previously unattainable functionalities with high yields and great scalability using a simple batch setup. Critically, this LMCT approach allows for selective decarboxylation of substrates incompatible with previous photoredox methods, opening the door for design of new decarboxylative functionalization reactions. Mechanistic studies revealed our reaction to exhibit zero order behavior in unactivated acid substrate, iron and disulfide catalysts with a KIE = 1, suggesting the reaction to be photon flux limited. Additionally, the excellent d-incorporation can be achieved simply by swapping water for deuterium oxide without the requirement of pre-salification with strong base. Together, this reaction provided a powerful platform to decarboxylate complex chemical structures in a mild, efficient, economical, and catalytic manner and a clear demonstration of the versatility of iron-photocatalyzed cooperative catalyst systems.
Supplementary Material
Acknowledgements
We acknowledge financial support from CPRIT (RR190025) and NIH (R35GM142738). J.G.W. is a CPRIT Scholar in Cancer Research. Y.-C.L. is supported by a Government Scholarship to Study Abroad (GSSA) from the Ministry of Education of Taiwan. Dr. Yohannes H. Rezenom (TAMU/LBMS), Dr. Ian M Riddington (UT Austin Mass Spectrometry Facility), and Dr. Christopher L. Pennington (Rice University Mass Spectrometry Facility) are acknowledged for assistance with mass spectrometry analysis. Profs. John Swierk (Binghampton University) and Cortney Roberts (University of Minnesita, Twin Cities) are acknowledged for helpful discussions.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].For selected literature examples of decarboxylative functionalizations, see: Wei Y, Hu P, Zhang M, Su W, Chem. Rev. 2017, 117, 8864 – 8907. Schwarz J, König B, Green Chem. 2018, 20, 323 – 361. Chen H, Liu YA, Liao X, Synthesis 2021, 53, 1 – 29. Huang H, Jia K, Chen Y, ACS Catal. 2016, 6, 4983 – 4988. Jin Y, Fu H, Asian J Org. Chem. 2017, 6, 368 – 385. Rodríguez N, Goossen L, Chem. Soc. Rev. 2011, 40, 5030 – 5048. Varenikov A, Shapiro E, Gandelman M, Chem. Rev. 2021, 121, 412 – 484. Patra T, Maiti D, Chem. Eur. J. 2017, 23, 7382 – 7401.
- [2].For selected literature examples of carboxylic acid-directed transformations, see: Drapeau MP, Gooßen LJ, Chem. Eur. J. 2016, 22, 18654 – 18677.27730686 Das J, Mal DK, Maji S, Maiti D, ACS Catal. 2021, 11, 4205 – 4229. Engle KM, Mei T-S, Wasa M, Yu J-Q, Acc. Chem. Res. 2012, 45, 788 – 802. Chen Z, Wang B, Zhang J, Yu W, Liu Z, Zhang Y, Org. Chem. Front. 2015, 2, 1107 – 1295. Zhang F, Spring DR, Chem. Soc. Rev. 2014, 43, 6906 – 6919. Font M, Quibell JM, Perry GJP, Larrosa I, Chem. Commun. 2017, 53, 5584 – 5597. Sambiagio C, Schönbauer D, Blieck R, Dao-Huy T, Pototschnig G, Schaaf P, Wiesinger T, Zia MF, Wencel-Delord J, Besset T, Maes BUW, Schnürch M, Chem. Soc. Rev. 2018, 47, 6603 – 6743. For selected applications of decarboxylative protonation in total synthesis, see: Lathrop SP, Pompeo M, Chang W-TT, Movassaghi M, J. Am. Chem. Soc. 2016, 138, 7763 – 7769. Fang C, Shanahan CS, Paull DH, Martin SF, Angew. Chem. Int. Ed. 2012, 51, 10596 – 10599. Salahi F, Yao C, Norton JR, Snyder SA, Nat. Synthesis 2022, 1, 313 – 321.
- [3].For selected literature examples, see: Kopf S, Bourriquen F, Li W, Neumann H, Junge K, Beller M, Chem. Rev. 2022, 122, 6634 – 6718. Li N, Li Y, Wu X, Zhu C, Xie J, Chem. Soc. Rev. 2022, 51, 6291 – 6306. Prakash G, Paul N, Oliver GA, Werz DB, Maiti D, Chem. Soc. Rev. 2022, 51, 3123 – 3163.
- [4].(a) Anderson JM, Kochi JK, J. Am. Chem. Soc. 1970, 92, 1651 – 1659. [Google Scholar]; (b) Anderson JM, Kochi JK, J. Org. Chem. 1970, 35, 986 – 989. [Google Scholar]
- [5].Seo S, Taylor JB, Greaney MF, Chem. Commun. 2012, 48, 8270 – 8272. [DOI] [PubMed] [Google Scholar]
- [6].(a) Xue L, Su W, Lin Z, Dalton Trans. 2011, 40, 11926 – 11936. [DOI] [PubMed] [Google Scholar]; (b) Cahiez G, Moyeux A, Gager O, Poizat M, Adv. Synth. Catal. 2013, 355, 790 – 796. [Google Scholar]; (c) Gooßen LJ, Thiel WR, Rodríguez N, Linder C, Melzer B, Adv. Synth. Catal. 2007, 349, 2241 – 2246. [Google Scholar]; (d) Rudzki M, Alcalde-Aragonés A, Dzik WI, Rodríguez N, Gooßen LJ, Synthesis 2012, 44, 184 – 193. [Google Scholar]; (e) Zou Y, Huang Q, Huang T. -k., Ni Q. -c., Zhang E. -s., Xu T. -l., Yuan M, Li J, Org. Biomol. Chem. 2013, 11, 6967 – 6974. [DOI] [PubMed] [Google Scholar]; (f) Gooßen LJ, Rodríguez N, Linder C, Lange PP, Fromm A, ChemCatChem 2010, 2, 430 – 442. [Google Scholar]
- [7].(a) Gooßen LJ, Linder C, Rodríguez N, Lange PP, Fromm A, Chem. Commun. 2009, 7173 – 7175. [DOI] [PubMed] [Google Scholar]; (b) Grainger R, Nikmal A, Cornella J, Larrosa I, Org. Biomol. Chem. 2012, 10, 3172 – 3174. [DOI] [PubMed] [Google Scholar]; (c) Jafarpour F, Jalalimanesh N, Olia MBA, Kashani AO, Tetrahedron 2010, 66, 9508 – 9511. [Google Scholar]; (d) Toy XY, Roslan IIB, Chuah GK, Jaenicke S, Catal. Sci. Technol. 2014, 4, 516 – 523. [Google Scholar]; (e) Cisneros-Pérez PA, Martínez-Otero D, Cuevas-Yánez E, Uribe-Frontana BA, Synth. Commun. 2014, 44, 222 – 230. [Google Scholar]
- [8].(a) Dickstein JS, Mulrooney CA, O’Brien EM, Morgan BJ, Kozlowski MC, Org. Lett. 2007, 9, 2441 – 2444. [DOI] [PubMed] [Google Scholar]; (b) Xue L, Su W, Lin Z, Dalton Trans. 2010, 39, 9815 – 9822. [DOI] [PubMed] [Google Scholar]; (c) Al-Huniti MH, Perez MA, Garr MK, Croatt MP, Org. Lett. 2018, 20, 7375 – 7379. [DOI] [PubMed] [Google Scholar]
- [9].(a) Cornella J, Rosillo-Lopez M, Larrosa I, Adv. Synth. Catal. 2011, 353, 1359 – 1366. [Google Scholar]; (b) Dupuy S, Nolan SP, Chem. Eur. J. 2013, 19, 14034 – 14038. [DOI] [PubMed] [Google Scholar]; (c) Dupuy S, Crawford LE, Bühl M, Nolan SP, Chem. Eur. J. 2015, 21, 3399 – 3408. [DOI] [PubMed] [Google Scholar]
- [10].(a) Candish L, Freitag M, Gensch T, Glorius F, Chem. Sci. 2017, 8, 3618 – 3622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kubosaki S, Takeuchi H, Iwata Y, Tanaka Y, Osaka K, Yamawaki M, Morita T, Yoshimi Y, J. Org. Chem. 2020, 85, 5362 – 5369. [DOI] [PubMed] [Google Scholar]
- [11].(a) Barton DHR, Crich D, Motherwell WB, J. Chem. Soc., Chem. Commun. 1983, 939 – 941. [Google Scholar]; (b) Barton DHR, Crich D, Motherwell WB, Tetrahedron Lett. 1985, 41, 3901 – 3924. [Google Scholar]; (c) Barton DHR, Crich D, Motherwell WB, Tetrahedron, 1987, 43, 2733 – 2740. [Google Scholar]
- [12].(a) Ko EJ, Savage GP, Williams CM, Tsanaktsidis J, Org. Lett. 2011, 13, 1944 – 1947. [DOI] [PubMed] [Google Scholar]; (b) Qin T, Malins LR, Edwards JT, Merchant RR, Novak AJE, Zhong JZ, Mills RB, Yan M, Yuan C, Eastgate MD, Baran PS, Angew. Chem. Int. Ed. 2017, 56, 260 – 265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li Z, Wang K-F, Zhao X, Ti H, Liu X-G, Wang H, Nat. Commun. 2020, 11, 5036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen X, Luo X, Peng X, Guo J, Zai J, Wang P, Chem. Eur. J. 2020, 26, 3226 – 3230. [DOI] [PubMed] [Google Scholar]; (e) Patra T, Mukherjee S, Ma J, Strieth-Kalthoff F, Glorius F, Angew. Chem. Int. Ed. 2019, 58, 10514 – 10520. [DOI] [PubMed] [Google Scholar]; (f) Li N, Ning Y, Wu X, Xie J, Li W, Zhu C, Chem. Sci. 2021, 12, 5505 – 5510. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zheng C, Wang G-Z, Shang R, Adv. Synth. Catal. 2019, 361, 4500 – 4505. [Google Scholar]; (h) Beato E. d. P., Spinnato D, Zhou W, Melchiorre P, J. Am. Chem. Soc. 2021, 143, 12304 – 12314. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Mclean EB, Mooney DT, Burns DJ, Lee A-L, Org. Lett. 2022, 24, 686 – 691. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Yoshimi Y, Itou T, Hatanaka M, Chem. Commun. 2007, 5244 – 5246. [DOI] [PubMed] [Google Scholar]
- [13].(a) Griffin JD, Zeller MA, Nicewicz DA, J. Am. Chem. Soc. 2015, 137, 11340 – 11340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cassani C, Bergonzini G, Wallentin C-J, Org. Lett. 2014, 16, 4228 – 4231. [DOI] [PubMed] [Google Scholar]
- [14].(a) Chen W, Wang H, Tay NES, Pistritto VA, Li K-P, Zhang T, Wu Z, Nicewicz DA, Li Z, Nat. Chem. 2022, 14, 216 – 223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang L, White AR, Chen W, Wu Z, Nicewicz DA, Li Z, Org. Lett. 2020, 22, 7971 – 7975. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pistritto VA, Schutzbach-Horton ME, Nicewicz DA, J. Am. Chem. Soc. 2020, 142, 17187 – 17194. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tay NES, Chen W, Levens A, Pistritto VA, Huang Z, Wu Z, Li Z, Nicewicz DA, Nat. Catal. 2020, 3, 734 – 742. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Venditto NJ, Nicewicz DA, Org. Lett. 2020, 22, 4817 – 4822. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Holmberg-Douglas N, Onuska NPR, Nicewicz DA, Angew. Chem. Int. Ed. 2020, 59, 7425 – 7429. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Holmberg-Douglas N, Nicewicz DA, Org. Lett. 2019, 21, 7114 – 7118. [DOI] [PubMed] [Google Scholar]; (h) Chen W, Huang Z, Tay NES, Giglio B, Wang M, Wang H, Wu Z, Nicewicz DA, Li Z, Science, 2019, 364, 1179 – 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Cruz CL, Nicewicz DA, ACS Catal. 2019, 9, 3926 – 3935. [Google Scholar]; (j) Tay NES, Nicewicz DA, J. Am. Chem. Soc. 2017, 139, 16100 – 16104. [DOI] [PubMed] [Google Scholar]; (k) Margrey KA, Levens A, Nicewicz DA, Angew. Chem. Int. Ed. 2017, 56, 15644 – 15648. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Margrey KA, McManus JB, Bonazzi S, Zecri F, Nicewicz DA, J. Am. Chem. Soc. 2017, 139, 11288 – 11299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) McManus JB, Nicewicz DA, J. Am. Chem. Soc. 2017, 139, 2880 – 2883. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Romero NA, Margrey KA, Tay NE, Nicewicz DA, Science, 2015, 349, 1326 – 1330. [DOI] [PubMed] [Google Scholar]
- [15].(a) Hamilton DS, Nicewicz DA, J. Am. Chem. Soc. 2012, 134, 18577 – 18580. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Grandjean J-MM, Nicewicz DA, Angew. Chem. Int. Ed. 2013, 52, 3967 – 3971. [DOI] [PubMed] [Google Scholar]; (c) Wilger DJ, Gesmundo NJ, Nicewicz DA, Chem. Sci. 2013, 4, 3160 – 3165. [Google Scholar]; (d) Nguyen TM, Nicewicz DA, J. Am. Chem. Soc. 2013, 135, 9588 – 9591. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Perkowski AJ, Nicewicz DA, J. Am. Chem. Soc. 2013, 135, 10334 – 10337. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Nguyen TM, Manohar N, Nicewicz DA, Angew. Chem. Int. Ed. 2014, 53, 6198 – 6201. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wilger DJ, Grandjean J-MM, Lammert TR, Nicewicz DA, Nature Chem. 2014, 6, 720 – 726. [DOI] [PubMed] [Google Scholar]; (h) Zeller MA, Riener M, Nicewicz DA, Org. Lett. 2014, 16, 4810 – 4813. [DOI] [PubMed] [Google Scholar]; (i) Romero NA, Nicewicz DA, J. Am. Chem. Soc. 2014, 136, 17024 – 17035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Morse PD, Nicewicz DA, Chem. Sci. 2015, 6, 270 – 274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Gesmundo NJ, Grandjean J-MM, Nicewicz DA, Org. Lett. 2015, 17, 1316 – 1319. [DOI] [PubMed] [Google Scholar]; (l) Cavanaugh CL, Nicewicz DA, Org. Lett. 2015, 17, 6082 – 6085. [DOI] [PubMed] [Google Scholar]; (m) Wu F, Wang L, Chen J, Nicewicz DA, Huang Y, Angew. Chem. Int. Ed. 2018, 57, 2174 – 2178. [DOI] [PubMed] [Google Scholar]; (o) Wu F, Wang L, Ji Y, Zou G, Shen H, Nicewicz DA, Chen J, Huang Y, iScience 2020, 23, 101395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].(a) McManus JB, Onuska NPR, Jeffreys MS, Goodwin NC, Nicewicz DA, Org. Lett. 2020, 22, 679 – 683. [DOI] [PubMed] [Google Scholar]; (b) Nakajima K, Miyake Y, Nishibayashi Y, Acc. Chem. Res. 2016, 49, 1946 – 1956. [DOI] [PubMed] [Google Scholar]
- [17].(a) Kochi JK, J. Am. Chem. Soc. 1965, 87, 2500 – 2502. [Google Scholar]; (b) Bacha JD, Kochi JK, Tetrahedron 1968, 24, 2215 – 2226. [Google Scholar]
- [18].Li QY, Gockel SN, Lutovsky GA, DeGlopper KS, Baldwin NJ, Bundesmann MW, Tucker JW, Bagley SW, Yoon TP, Nat. Chem. 2022, 1 – 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].(a) Xu P, López-Rojas P, Ritter T, J. Am. Chem. Soc. 2021, 143, 5349 – 5354. [DOI] [PubMed] [Google Scholar]; (b) Su W, Xu P, Ritter T, Angew. Chem. Int. Ed. 2021, 60, 24012 – 24017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].(a) Kang YC, Treacy SM, Rovis T, ACS Catal. 2021, 11, 7442 – 7449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Treacy SM, Rovis T, J. Am. Chem. Soc. 2021, 143, 2729 – 2735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kang YC, Treacy SM, Rovis T, Synlett 2021, 32, 1767 – 1771. [Google Scholar]
- [21].Abderrazak Y, Bhattacharyya A, Reiser O, Angew. Chem. Int. Ed. 2021, 60, 21100 – 21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].For selected iron-catalyzed decarboxylation, see: Siegel B, Lanphear J, J. Am. Chem. Soc. 1979, 101, 2221 – 2222. Davis R, Schultz HP, J. Org. Chem. 1962, 27, 854 – 857. Komuro M, Nagatsu Y, Higuchi T, Hirobe M, Tetrahedron Lett. 1992, 33, 4949 – 4952. Bi H-P, Chen W-W, Liang Y-M, Li C-J, Org. Lett. 2009, 11, 3246 – 3249. Sandfort F, O’Neill MJ, Cornella J, Wimmer L, Baran PS, Angew. Chem. Int. Ed. 2017, 56, 3319 – 3323. Jiang Q, Jia J, Xu B, Zhao A, Guo C-C, J. Org. Chem. 2015, 80, 3586 – 3596. Exner B, Bayarmagnai B, Jia F, Goossen LJ, Chem. Eur. J. 2015, 21, 17220 – 17223. Quintard A, Rodriguez J, Chem. Eur. J. 2015, 21, 14717 – 14722. Tan HY, Xiang S, Leng WL, Liu X-W, RSC Adv. 2014, 4, 34816 – 34822. Li Z, Wang X, Xia S, Jin J, Org. Lett. 2019, 21, 4259 – 4265. Feng G, Wang X, Jin J, Eur. J. Org. Chem. 2019, 6728 – 6732. Gong P-X, Xu F, Cheng L, Gong X, Zhang J, Gu W-J, Han W, Chem. Commun. 2021, 57, 5905 – 5908.
- [23].(a) Kattamuri PV, West JG, J. Am. Chem. Soc. 2020, 142, 19316 – 19326. [DOI] [PubMed] [Google Scholar]; (b) Kattamuri PV, West JG, Synlett 2021, 32, 1179 – 1186. [Google Scholar]
- [24].(a) Huang L, Ji T, Zhu C, Yue H, Zhumabay N, Rueping M, Nat. Commun. 2022, 13, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X, Li Y, Wu X, ACS Catal. 2022, 12, 3710 – 3718. [Google Scholar]; (c) Wong TH-F, Ma D, Sanza RD, Melchiorre P, Org. Lett. 2022, 24, 1695 – 1699. [DOI] [PubMed] [Google Scholar]
- [25].Chen B-S, Zeng Y-Y, Liu L, Chen L, Duan P, Luque R, Ge R, Zhang W, Renew. Sust. Energ. Rev. 2022, 158, 112178. [Google Scholar]
- [26].Bloh JZ, Front. Chem. 2019, 7, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.