

Abstract
Introduction:
Gestational diabetes (GDM) is traditionally thought to emerge from placental endocrine dysregulations, but recent evidence suggests that fetal sex can also impact GDM development. Understanding the molecular mechanisms through which sex modulates placenta physiology can help identify novel molecular targets for future clinical care. Thus, we investigated the nutrient-sensing O-GlcNAc pathway as a potential mediator of sex-specific placenta dysfunction in GDM.
Methods:
Expression levels of O-GlcNAc enzymes were measured in male and female (n=9+/gender) human placentas based on the maternal diagnosis of GDM. We then simulated the observed differences in both BeWo cells and human syncytiotrophoblasts primary cells (SCT) from male and female origins (n=6/gender). RNA sequencing and targeted qPCR were performed to characterize the subsequent changes in the placenta transcriptome related to gestational diabetes.
Results:
O-GlcNAc transferase (OGT) expression was significantly reduced only in male placenta collected from mothers with GDM compared to healthy controls. Similar downregulation of OGT in trophoblast-like BeWo male cells demonstrated significant gene expression deregulations that overlapped with known GDM-related genes. Notably, placental growth hormone (GH) production was significantly elevated, while compensatory factors against GH-related insulin resistance were diminished. Inflammatory and immunologic factors with toxic effects on pancreatic β cell mass were also increased, altogether leaning toward a decompensatory diabetic profile. Similar changes in hormone expression were confirmed in male human primary SCTs transfected with siOGT. However, down-regulating OGT in female primary SCTs did not impact hormone production.
Conclusion:
Our study demonstrated the significant deregulation of placental OGT levels in mothers with GDM carrying a male fetus. When simulated in vitro, such deregulation impacted hormonal production in BeWo trophoblast cells and primary SCTs purified from male placentas. Interestingly, female placentas were only modestly impacted by OGT downregulation, suggesting that the sex-specific presentation observed in gestational diabetes could be related to O-GlcNAc-mediated regulation of placental hormone production.
Keywords: O-GlcNAcylation, Placenta, RNA-sequencing, gestational diabetes, syncytiotrophoblast
Graphical Abstract
INTRODUCTION
The placenta is a fetal secretory organ responsible for producing hormones, cytokines, and regulatory proteins and adapting maternal metabolism for pregnancy. Aberrant placenta function leads to various endocrine diseases, the most common being gestational diabetes (GDM) estimated to affect one in ten pregnancies globally[1]. The diagnosis of GDM poses immediate risks to the pregnancy, including a high likelihood of preeclampsia (preE), stillbirth, and preterm delivery. Furthermore, GDM carries long-term health risks for the mother and offspring [1–4]. Up to 70% of women with a history of GDM will develop type 2 diabetes (T2DM) within 10 years following pregnancy [5], while the offspring is at increased risk for fetal adiposity, obesity, and early-onset T2DM as young adults [6–10].
Recent clinical evidence suggests that fetal sex is an additional risk factor for GDM. Indeed, women carrying a male fetus have up to a 39% increased odds risk of developing GDM [11]. This sexual dimorphic risk for GDM provides a unique comparative framework to study inherent metabolic drivers for glucose and insulin deregulation and, by extension, identify targets for metabolic therapy.
We believe that females could be protected from adverse metabolic outcomes from diseases like GDM due to sexual dimorphism in the regulation of key placental signaling. In this study, we focused on the O-GlcNAc pathway as a nutrient-sensing X-linked mediator of cell signaling. O-GlcNAcylation is a highly conserved modification consisting of post-translational addition of a single residual of N-acetylglucosamine onto serine or threonine moieties of intracellular proteins (Figure 1A) [12]. Because this modification is highly dependent on glucose level, the O-GlcNAc pathway is considered a key nutrient sensor, capable of regulating a wide range of physiologic processes in response to nutritional environment [13–15].
Figure 1: GDM correlates with downregulation of O-GlcNAc enzymes in male placentas. O-GlcNAcylation of proteins.
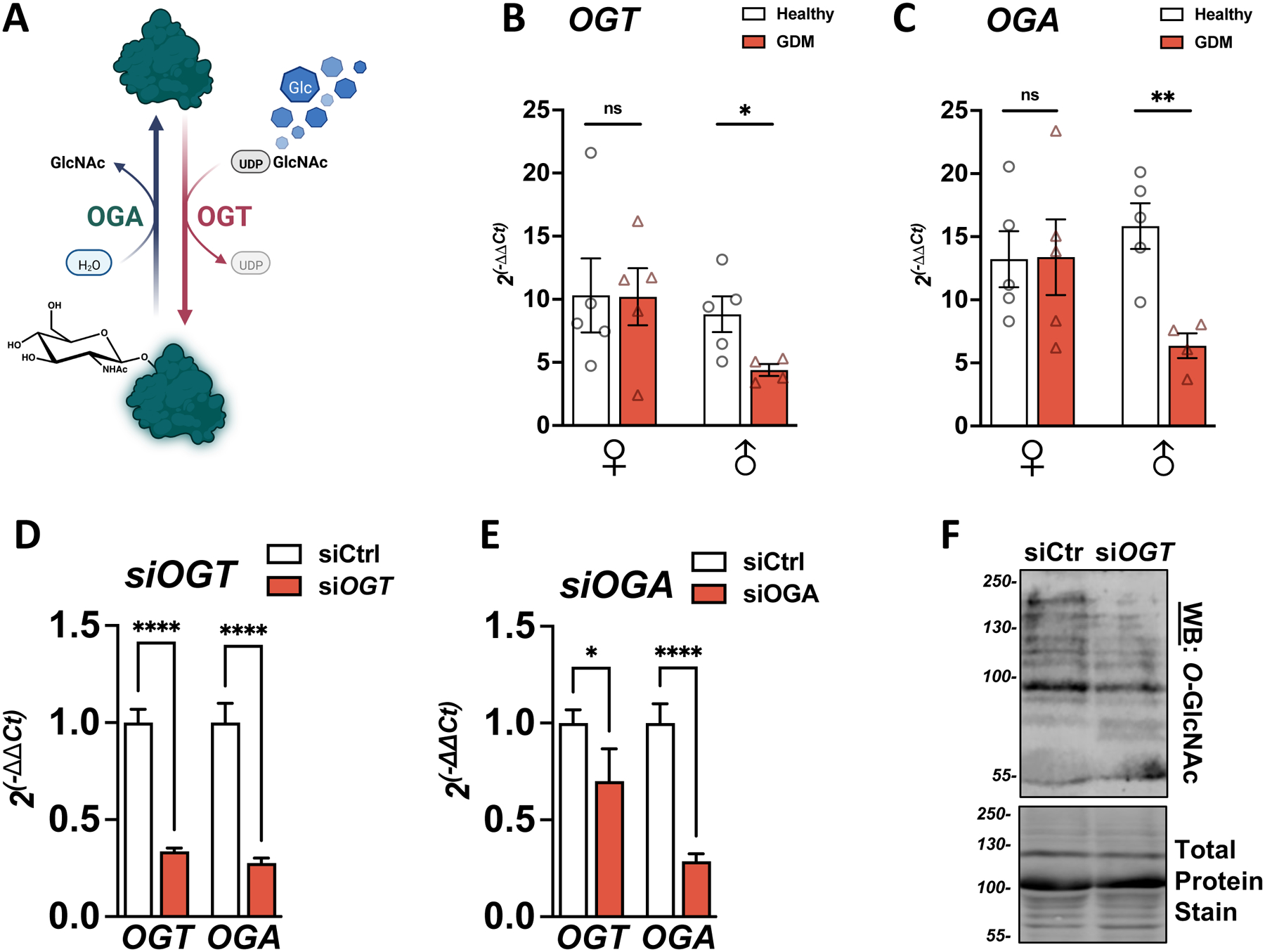
(A) 2–3% of glucose (Glc) entering cells is shuttled through the Hexosamine Biosynthesis Pathway (HBP) to produce UDP-GlcNAc, the nucleotide donor for O-GlcNAcylation. O-GlcNAc transferase (OGT) adds the GlcNAc residue to serine or threonine (S/T) residues. O-GlcNAcase (OGA) removes the sugar residue. (B/C) Expressions of OGT (B) or OGA (C) were measured in healthy or Gestational diabetes (GDM) placentas by qPCR and reported on ACTIN (n=4–5/group). (D/E) OGT and OGA expression were measured by qPCR in BeWo cells transfected with siRNA against OGT (D) or OGA (E) for 48h. Relative expression was calculated using the −2ΔΔCt method (n=3/group). Significance was calculated by student t-test; ns ≥ 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. (F) O-GlcNAcylation levels in BeWo cells transfected with siRNA against OGT for 48h. Total protein stain was used as the loading control.
O-GlcNAcylation has been associated with X-linked diseases [16–20] due to the localization of the main enzyme O-GlcNAc transferase (OGT) on the X-chromosome (Xq13.1) [21,22]. In pregnant murine models, Ogt has been found to escape X-chromosome inactivation, potentially resulting in differential protein dosages [23–26] and increased plasticity against environmental stress factors[27]. Furthermore, animal phenotypes resulting from O-GlcNAc modulation, often vary based on sex [27–34], emphasizing the link between sexual dimorphism and O-GlcNAcylation.
Herein, we investigated the role of OGT in the development of GDM. We first demonstrated a decrease in OGT expression and the complementary enzyme O-GlcNAcase (OGA) (Figure 1A) specific to human male placenta samples from GDM patients. We next modeled the decreased OGT expression through transfection of siOGT in trophoblast-like BeWo cells and performed a comprehensive transcriptomic profile using RNA sequencing. We identified significant changes in the production of placenta hormones and inflammatory markers implicated in insulin dysregulation of GDM. Using primary syncytiotrophoblast (SCT) cultures from human placentas, we confirmed similar hormonal changes following downregulation of OGT in male cells but noted the limited impact on female SCT cultures. Altogether, this study demonstrates a role for OGT and O-GlcNAcylation in placenta endocrine deregulation that has sex dimorphic clinical implications.
MATERIALS AND METHODS
Frozen human placenta acquirement
Placenta samples were obtained from the Medical College of Wisconsin Maternal Research and Placenta and Cord Blood Bank (MCW MRPCB) under IRB approval. Written informed consent was received before participation. Gestational diabetes (n = 5 males, 5 females) was diagnosed by the 100g oral glucose tolerance test following the Carpenter-Coustan criteria or if the 1-hour 50g glucose challenge test is > 200mg/dl. Placental samples were selected for this study on the following criteria (1) GDM diagnosis (2) use of insulin or oral hypoglycemic medication during pregnancy (3) no maternal history of hypertension (4) single gestation with known fetal sex without congenital anomalies. Placenta samples used as the control group (n= 4 males, 5 females) were comprised of women without GDM but with the same exclusion criteria applied.
BeWo Cell culture
Bewo cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L (Standard, Std), 10% (v/v) fetal bovine serum (FBS), and 1% penicillin/streptomycin at 37°C in a 5% (v/v) CO2-enriched humidified atmosphere.
Primary human syncytiotrophoblast model from third-trimester placentas
Patients delivering at a tertiary birth center were consented to placenta donation if they met the criteria of having a singleton pregnancy unaffected by fetal anomalies, hypertensive orders of pregnancy or diabetes of any type. Approximately 100 grams of full-thickness placenta sections were excised 2–3cm from the periphery of the placenta at random. The placenta sample was washed with phosphate-buffered saline (PBS) followed by trimming of the chorionic and basal plates as previously shown [35]. Placenta villi were diced into small pieces (1–3mm). Three digestion steps were performed in 50mL conical tubes at 37°C in Hanks balanced salt solution (HBSS) containing 0.25% trypsin (Gibco) and DNase I (Sigma-Aldrich) for 30 min each step. Digestion was stopped using 10% fetal bovine serum (FBS) (PAA Laboratories), and cells from each digestion were pooled and purified using a Percoll gradient [10–70% (vol/vol)]. Villous cytotrophoblasts were collected between 35 and 50% of Percoll layers and contaminating red blood cells were removed by incubating cells with erythrocyte lysis buffer (Thermo Fisher Scientific) for 10 min at room temperature. Purification yield was confirmed by flow cytometry using a cytokeratin-7 antibody (Thermo Fisher Scientific). Cells were then washed with PBS and cultured in media of 50% DMEM (Sigma) and Ham’s F12 (Gibco) supplemented with 10% (v/v) FBS, and 1% penicillin/streptomycin and maintained at 37°C in a 5% (v/v) CO2-enriched humidified atmosphere. Media was changed daily. After 48 hours (h) from plating, syncytialization was observed, and any planned experimental treatment was performed at this time.
To confirm the purification of trophoblasts, cells were fixed in 4% paraformaldehyde solution for 30 minutes at room temperature, then washed 3 times with PBS and incubated with primary mouse antibodies to cytokeratin-7 (1:100; Thermo Fisher Scientific) for 1 hour at room temperature. The monolayer was washed with PBS and incubated with fluorescent secondary antibodies (anti-mouse IgG; Sigma-Aldrich) for 1 hour in the dark. The nuclei were co-stained with DAPI and analyzed by confocal microscopy. Specific primers for syncytiotrophoblast-specific genes were used to confirm a proper transcriptional profile.
OGT small interfering RNA (siRNA)
MISSION siRNA targeting Human OGT were ordered at Sigma. Silencer™ Negative Control No. 1 siRNA (Thermo Fisher Scientific) was used as a control. Cells were transfected with Lipofectamine RNAiMax (Thermo Fisher Scientific) according to the manufacturer protocol.
RNA extraction
mRNA was isolated with the PureLink RNA Mini Kit (Thermo Fisher Scientific) following the manufacturer’s instruction. RNA integrity was verified by visual inspection of ribosomal RNA on agarose gels. RNA concentrations were measured with the LVis microplate on a FLUOstar Omega plate reader (BMG Labtech).
cDNA preparation and qPCR
cDNA was then synthesized with SuperScript™ IV VILO™ Master Mix with ezDNase (Thermo Fisher Scientific) according to the manufacturer’s instructions. qPCR reaction was then performed using PowerSYBR qPCR Master Mix (Thermo Fisher Scientific). Specific primers for each reaction are as follows: ACTIN-F: GATTCCTATGTGGGCGACGA; ACTIN-R: AGGTCTCAAACATGATCTGGGT; OGT-F: GGTGACTATGCCAGGAGAGACTCTTGC; OGT-R: CGAACTTTCTTCAGGTATTCTAGATC; OGA-F: CGGTGTGGTGGAAGGATTTTA; OGA-R: GTTGCTCAGCTTCCTCCACTG; HCG-F: GCTACTGCCCCACCATGACC; HCG-R: ATGGACTCGAAGCGCACATC; GH2-F: AAACGCTGATGTGGAGGCTG; GH2-R: GCCCGTAGTTCTTGAGCAGT; LEP-F: CTGTGCGGATTCTTGTGGCT; LEP-R: GAGGAGACTGACTGCGTGTGT; PL-F: TGACACCTACCAGGAGTTTGAAG; PL-R: GGGGTCACAGGATGCTACTC. qPCR was run on a QuantStudio 3 instrument (Applied Biosystems) with the recommended settings. The data were collected and processed on DataConnect (Thermo Fisher Scientific), and 2−DDCq were calculated and plotted using Prism 9 (GraphPad Software).
Protein lysis, SDS-PAGE, and Western Blotting
Samples were lysed in RIPA lysis buffer [10mM Tris-HCl, 150mM NaCl, 1% Triton X-100 (v/v), 0.5% NaDOC (w/v), 0.1% SDS (w/v), and protease inhibitors; pH7.5], vortexed and centrifuged at 18,000 g for 10 minutes at 4C. Sample lysates were resolved on 8% tris-glycine and transferred onto nitrocellulose. Membranes were then washed with ultra-purified water and labeled with No-Stain Protein Labeling Reagent (Thermo Fisher Scientific) according to kit instructions. Next, the membranes were blocked for 45 minutes with 5% (w/v) non-fat milk in Tris-buffered saline-Tween 20 buffer (TBS-T). Primary antibodies were added to the blocking solution, and the blots were incubated overnight at 4C with gentle agitation. Following primary incubation, blots were washed three times with 10mL of TBS-T for 10 minutes and incubated with anti-mouse and anti-rabbit fluorescent-conjugated secondary antibodies in a 1:20,000 dilution for 1 hour at room temperature. Three additional TBS-T washes with 10mL in 10 minutes were performed, and the blot signal was captured using Odyssey Fc (LI-COR).
Antibodies
Antibodies for western blotting are used as follow: anti-O-GlcNAc (#ab2739, Abcam): 1:1000; Anti-actin (#A2066, Sigma Aldrich), 1:1000.
RNA sequencing
Total RNA samples were quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA), and RNA integrity was checked using Agilent TapeStation 4200 (Agilent Technologies, Palo Alto, CA, USA). RNA sequencing libraries were prepared using the NEBNext Ultra RNA Library Prep Kit for Illumina, following the manufacturer’s instructions (NEB, Ipswich, MA, USA). Briefly, mRNAs were first enriched with Oligo(dT) beads. Enriched mRNAs were fragmented for 15 minutes at 94 °C. First-strand and second strand cDNAs were subsequently synthesized. cDNA fragments were end-repaired and adenylated at 3’ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by limited-cycle PCR. The sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA) and quantified by using Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA). Samples were processed on an Illumina HiSeq platform by GENEWIZ (Azenta Life Science).
Sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the Homo sapiens GRCh38 reference genome available on ENSEMBL using the STAR aligner v.2.5.2b. The STAR aligner is a splice aligner that detects splice junctions and incorporates them to help align the entire read sequences. BAM files were generated as a result of this step. Statistics of mapping the reads to the reference genome can be found in Table S1. Unique gene hit counts were calculated using featureCounts from the Subread package v.1.5.2. Only unique reads that fell within exon regions were counted. After extraction of gene hit counts, the gene hit counts table was used for downstream differential expression analysis. Using DESeq2, a comparison of gene expression between the various groups was performed. The Wald test was used to generate p-values and log2 fold changes. Genes with an adjusted p-value < 0.05 were called differentially expressed genes for each comparison.
Gene Ontology (GO) enrichment analysis
BinGO app within Cytoscape was used to perform GO enrichment analysis. The goa_human GO list was used to cluster the genes based on their biological processes and determine their statistical significance.
Statistics
Statistical analyses were performed on Prism 9 (Graphpad) and R version 4.1.0 (R Core Team). Error bars represented mean ± standard error of the mean (SEM). For a clinical sample, Mann–Whitney U test was performed to measure significance (unpaired, non-parametric) [36], and Pearson R was computed for correlation analysis. For RNA sequencing, the Log 2 fold change (Log2FoldChange) was calculated as follows: Log2(Group 2 mean normalized counts/Group 1 mean normalized counts). The Wald test was used to generate p-values, and the Benjamin-Hochberg correction was used to obtain adjusted p-values [37]. For all others, a classic student t-test (unpaired, parametric) was performed. Statistical significance was always represented as follows: ns ≥ 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Data availability
RNA-sequencing raw (*R1.fastq and *R2.fastq) and processed (*.bam and counts.txt) files were deposited in Gene Expression Omnibus (GEO) repository under GSE200983.
RESULTS
OGT is selectively downregulated in male fetal placenta from GDM mothers
To examine the pathological relevance of O-GlcNAcylation in GDM, OGT and OGA expression were measured by qPCR in human placentas grouped by fetal sex and maternal diagnosis of GDM (n=4+/group) (Table 1). OGT and OGA were significantly downregulated in male GDM placenta compared to controls (OGT: p=0.03, OGA p=0.004, Figure 1 B/C). In contrast, this difference was not observed in the female group.
Table 1:
Demographics and clinical characteristics of healthy and GDM placentas used.
| Group | Race | GCT | Medication | |||
|---|---|---|---|---|---|---|
| MALE | ||||||
| Health | ||||||
| #1 | 19 | Black | 26.4 | 89 | - | - |
| #2 | 25 | Black | 33.2 | 90 | - | - |
| #3 | 23 | Black | 26.9 | 130 | - | - |
| #4 | 28 | White | 28.3 | 97 | - | - |
| #5 | 25 | White | 29.5 | 91 | - | - |
| Average | 24 | 28.86 | 99.4 | |||
| GDM | ||||||
| #6 | 32 | White | 32.6 | 153 | 27 | NPH |
| #7 | 30 | White | 38.1 | 246 | 24 | Detemir |
| #8 | 31 | Asian | 25.7 | 211 | 25 | NPH |
| #9 | 40 | Black | 28.4 | 234 | 25 | NPH |
| Average | 33.25 | 31.2 | 211 | 25.25 | ||
| Male Healthy vs. GDM | p=0.0159(*) | p=0.7302(ns) | p= 0.0159(*) | |||
| FEMALE | ||||||
| Healthy | ||||||
| #10 | 23 | Black | 24.9 | 46 | - | - |
| #11 | 25 | Black | 20.5 | 116 | - | - |
| #12 | 27 | Black | 28.8 | 83 | - | - |
| #13 | 29 | White | 24.1 | 94 | - | - |
| #14 | 29 | White | 26.9 | 114 | - | - |
| Average | 26.6 | 25.04 | 90.6 | |||
| GDM | ||||||
| #15 | 30 | White | 30.3 | 189 | 26 | NPH |
| #16 | 39 | White | 33.8 | 173 | 15 | NPH |
| #17 | 34 | White | 29.1 | 187 | 29 | Metformin |
| #18 | 35 | Asian | 25.4 | 190 | 26 | NPH |
| #19 | 38 | Black | 45.3 | 170 | 27 | Glargine |
| Average | 34.5 | 29.65 | 184.75 | 24 | ||
| Female Healthy vs. GDM | 0.0079(**) | p=0.0317(*) | p=0.0079(**) | |||
GCT = 1-hour glucose challenge test; GA = gestational age; NPH: neutral protamine Hagedorn Insulin. Significance was calculated between healthy and GDM values with a Mann-Whitney test.
There were significant differences in maternal age and body mass index (BMI) between GDM and control subgroups within each sex (Table 1). However, OGT and OGA levels are poorly correlated with these maternal characteristics (Figure S1), suggesting that the significant decrease in OGT in the male group was related to GDM pathology.
To determine the physiologic impact of the observed downregulation O-GlcNAc enzymes in GDM, we used trophoblast-like BeWo cells to model downstream transcriptional changes. BeWo cells are male choriocarcinoma trophoblast cells commonly used in the literature as surrogates for placenta syncytiotrophoblasts (SCT) due to similar secretory profiles [38]. We separately downregulated OGT and OGA expression with small interfering RNA (siOGT or siOGA) (Figure 1 D/E) and found that OGT downregulation led to a more substantial decrease in OGA levels, closer to the profile observed in GDM male placentas. This crosstalk between OGT and OGA expression levels correlated with previous observations in the literature and is explained by the regulation of OGA intron retention by OGT [39]. Additionally, siOGT led to a strong reduction in the total O-GlcNAcylation level observed by Western Blot (Figure 1 F). We subsequently focused on the siOGT model as it closely simulated our findings from the GDM placenta samples.
Knocking down OGT leads to critical changes in the placental transcriptome
Using the cellular model described above, six RNA-Seq datasets were generated from two sample groups (siCtrl, siOGT) to conduct comparative pathway analysis that could elucidate the role of OGT downregulation in placental dysfunction leading to GDM. Each sample group was provided in biological replicates and obtained a total of 298,978,153 reads (44–55 million reads per sample) with ~92% were over 30 bases (Table S1). 68 genes (48 upregulated and 20 downregulated) had more than a 2-fold change with an adjusted p-value (AdjpValue) < 0.05 (Figure 2A, Table S2). An additional 4,383 genes were also significantly deregulated (AdjpValue <0.05) but with less than a 2-fold change (Table S2).
Figure 2: Modulating OGT in male placental cells leads to important changes in the transcriptome.
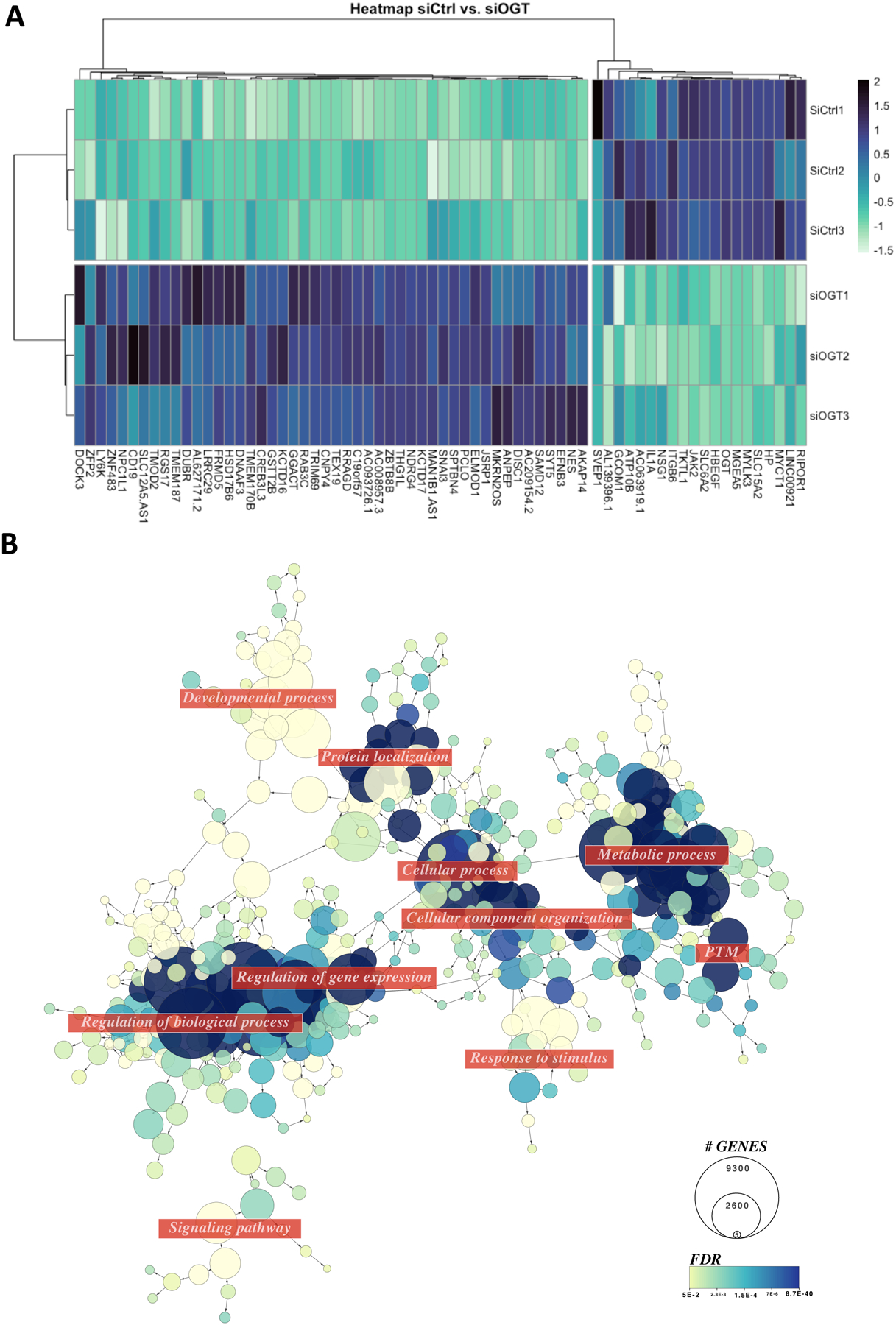
(A) Heatmap of the deregulated genes in siOGT vs. siCtrl Bewo cells. Genes shown have an adjPValue<0.05 and abs(Log2FC)>1 (n=3/group). (B) Network representation of enriched GO terms (FDR<0.05) using deregulated genes (AdjPValue<0.05) in siOGT vs. siCtrl Bewo Cells.
Gene Ontology (GO) analysis on the significatively deregulated genes demonstrated enrichment in cellular metabolic processing (GO:0044237, FDR=8.78E-40), protein post-translational modification (GO:0043687, FDR=2.12E-10), gene expression (GO:00010467, FDR=1.83E-14), and cellular process (GO:0050794, FDR=3.91E-11) (Figure 2B, Table S3). Organelle organization (GO:0006996, FDR=1.92E-22) was also enriched, driven by chromatin modification (GO:0016568, FDR= 1.46E-09). Macromolecule localization (GO:0033036, FDR=5.58E-13), including protein transport (GO:0015031, FDR=9.35E-11) and cellular localization (GO: 0051641, FDR= 3.27E-09) was associated with the significantly deregulated genes in the siOGT model. Overall, the analysis demonstrated the validity of our model as it highlighted the previously described roles of O-GlcNAcylation in regulating transcription [12].
Transcriptomic profile from siOGT BeWo model overlaps with gene expression changes observed in GDM placenta.
We further compared our RNA-sequencing data with previously published transcriptomes of human GDM placenta. Using the Alliance of Genome Resources database (https://www.alliancegenome.org) to query for the deregulated genes, we found that our model shared 32% of gene expression (10/ 31 genes) (Figure 3A) known to be associated with GDM. Interestingly, the siOGT model also overlapped in 21% of preE gene deregulations, highlighting potentially shared pathways between preE and GDM and the involvement of OGT in both pathologies for future consideration (Figure S2).
Figure 3: OGT siRNA in BeWo cells lead to transcriptional changes common with GDM placentas.
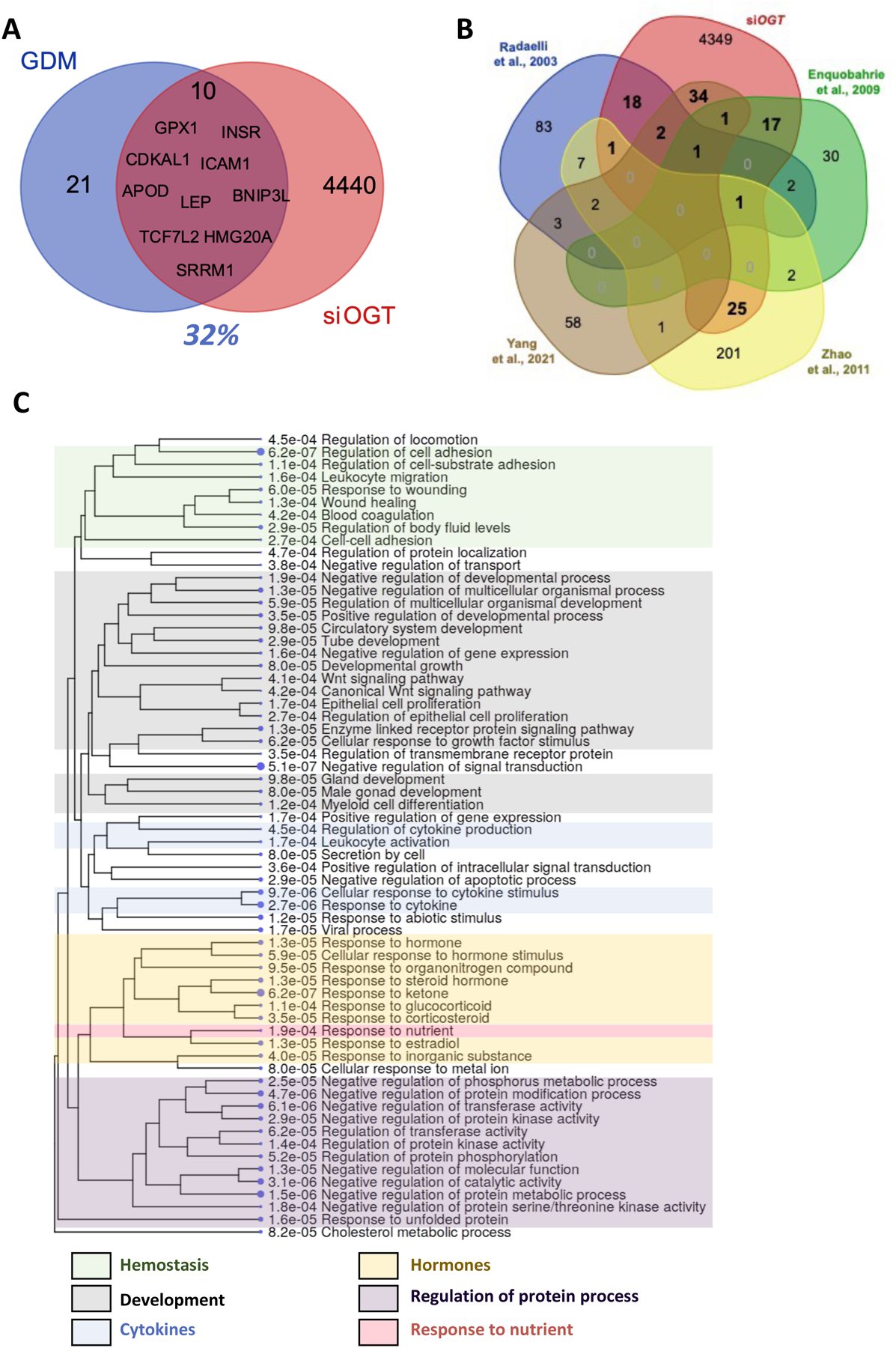
(A) GDM signature genes were found in the Alliance of Genome Resources under DOID: 11714. (B) Venn diagram representing commonly deregulated genes in placentas from reference GDM studies. (C) GO enrichment tree of the 100 deregulated genes found in our study and previously identified deregulated in GDM placentas.
The limitation to Alliance of Genome Resources database was the lack in reporting tissue-specificity for the GDM-related genes. Thus, we cross-referenced the siOGT deregulated genes with published studies specifically on GDM transcriptional changes in the human placenta [40–42] or purified trophoblasts [43]. While neither OGT nor OGA was significantly deregulated in any of the studies, none reported or stratified their analysis by fetal sex. Nevertheless, 100 shared deregulated genes (Figure 3B, Table S4) were commonly found between these studies and ours.
GO enrichment of the shared genes highlighted the following categories: hemostasis, cytokine signaling, peptide and steroid hormone signaling, developmental processes, and response to nutrients (Figure 3C). For each GO term, we mapped a complex web of transcriptional changes, integrating hormonal, immune, and inflammatory mechanisms all deregulated in siOGT BeWo cells (Figure S3).
Knockdown of OGT in primary human syncytiotrophoblast culture reveals dimorphic expression of essential placental hormones based on fetal sex.
We specifically focused on hormonal expression as the placenta is a major endocrine organ responsible for secreting many hormones that drive metabolic adaptations in pregnancy. We found that the downregulation of OGT in the BeWo model led to the upregulation of human choriogonadotropin hormone (hCG), placental growth hormone (GH2) and an estrogen-conversion gene (HSD17B), all known to increase insulin resistance [44–51](Figure 4A, Figure S3F). Conversely, there was downregulation of Leptin (LEP) and the Leptin Receptor (LEPR), which have compensatory roles against insulin resistance [50,52].
Figure 4: Placental hormone expression is deregulated by siOGT, particularly in male cells.
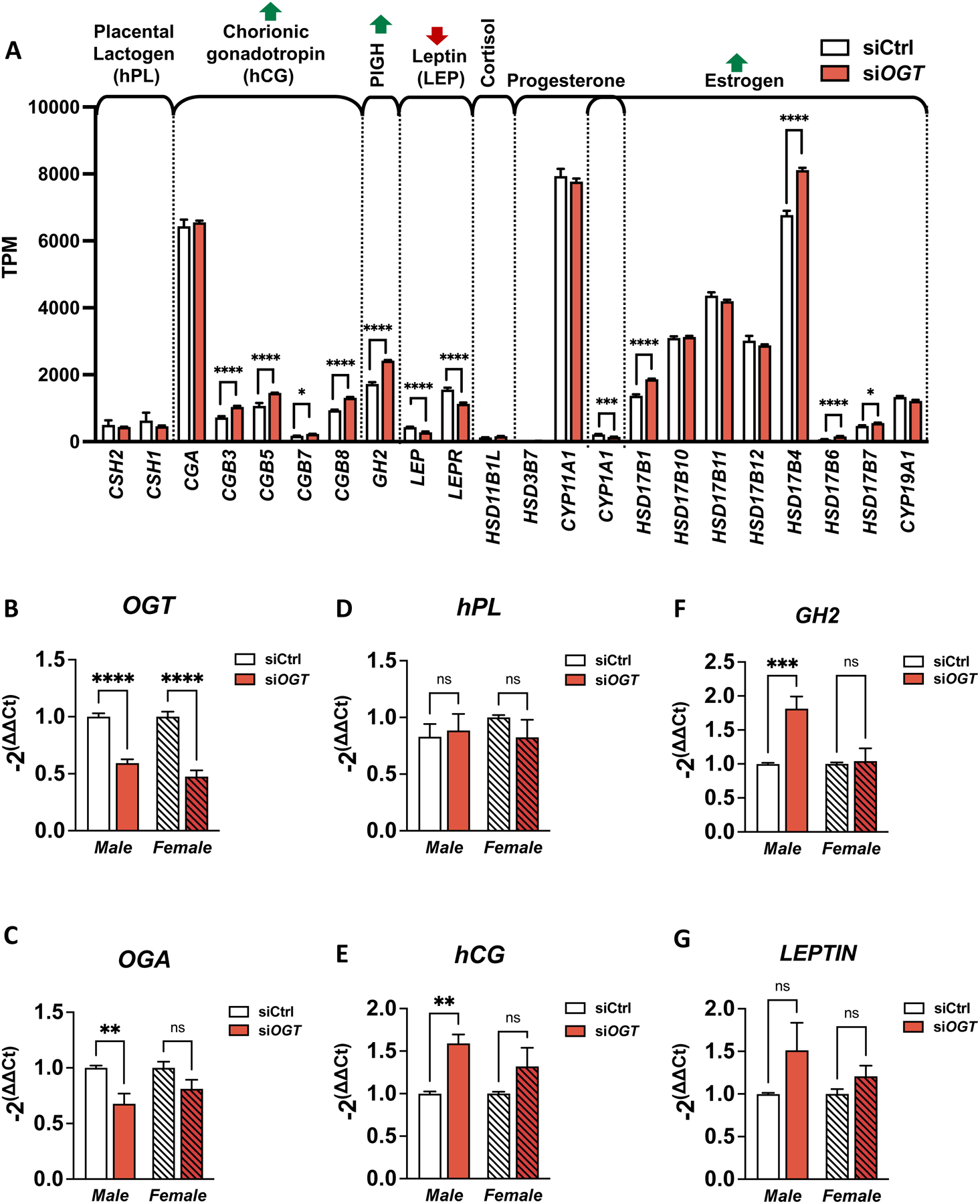
(A) Expression (TPM) of hormone-related genes from BeWo cells treated with siOGT measured by RNA-sequencing (n=3/group). (B-G) Transcript levels in primary syncytiotrophoblasts from male and female placentas. The following transcripts have been measured by qPCR and reported on ACTIN: (B) OGT, (C) OGA, (D) human placental lactogen (hPL), (E) human chorionic gonadotropin (hCG), (F) Placental Growth hormone (GH2) and (G) Leptin (n=3/group). Significance was calculated by student t-test; ns ≥ 0.05, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
However, major limitations to the BeWo cells are (1) they are only representative of male placental cells, and (2) they are trophoblast-like and thus may have inherent discrepancies with placental SCTs. Consequently, we aimed to confirm our findings in primary human male and female SCTs.
We purified cytotrophoblasts from human female and male placentas and allowed syncytialization within 48 hours of culture. Syncytiotrophoblast traits were confirmed by expression of CYP19A1, Galectins (LGALS), and Glial Cells Missing Transcription Factor 1 (GCM1) by qPCR and immunostaining of cytokeratin 7 (Figure S5). The cells were then transfected with siOGT or siCtrl, and knockdown efficiency was confirmed by qPCR (Figure 4B).
Gene expression in the siOGT-treated male SCT culture was similar to that in the BeWo model. In particular, GH2 and hCG followed the same significant upregulation (Figure 4E/F). No decrease in LEP was observed, likely due to the variability amongst the samples suggestive of additional confounding factors that influence basal Leptin levels.
Gene expression in the treated female SCT culture had notable differences in that all hormone levels (GH2, hCG, LEP) of interest were not impacted by downregulation of OGT (Figure 4E/F). Moreover, OGA expression in female cells did not trend down with siOGT, highlighting a possible sexual dimorphism in regulation of O-GlcNAc enzymes in primary SCTs (Figure 4C).
Transcriptomic changes in siOGT cells can be explained by common transcription factors.
To offer some mechanistic insight and potential clinical application as biomarkers, we further constructed a regulatory network from the deregulated genes in the siOGT BeWo model. We interrogated the TRRUST database with the list of significantly deregulated genes in siOGT (AdjpValue<0.05) associated with hormone-related GO terms (GO:0042445, GO:0005179, GO:0046879). Transcription factors such as SP1, STAT1, and NFKB1 were found to be responsible for a large portion of deregulated genes and were also themselves significantly deregulated in the siOGT BeWo model (Figure 5A/B). Other transcription factors like CEBPA/B, REST, CREB1, RELA, TFAP2A, and FOXO1 were responsible for regulating the transcription of many deregulated genes in siOGT but were themselves not significantly altered (Figure 5A/B). All but CEBPA were identified as O-GlcNAcylated (Figure 5A), while most were expressed in placentas (Figure 5C). Finally, NR5A1, JUN, and GATA4 were poorly represented in placentas despite their association with a small percentage of deregulated genes, suggesting that they are unlikely to be relevant to placental deregulation toward GDM (Figure 5).
Figure 5: Common transcription factors explained hormonal changes in expression levels after siOGT in BeWo cells.
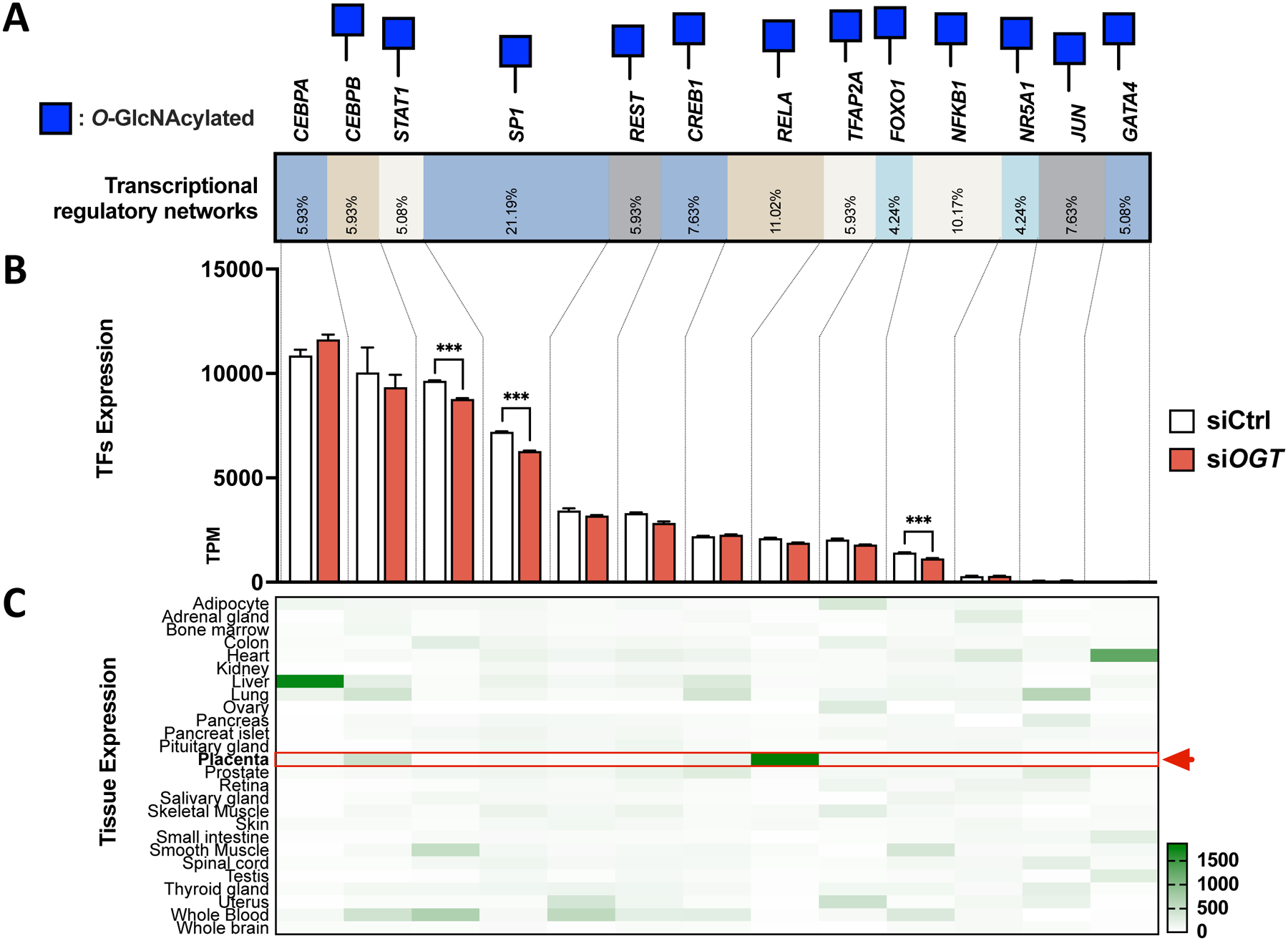
(A) TRRUST analysis highlighted the transcription factor responsible for regulating hormones-related genes deregulated in siOGT. Blue Square represents the O-GlcNAcylation status assessed with the O-GlcNAcylated database. (B) RNA-sequencing expression of transcription factors identified by the TRRUST database. TPMs are represented for biological replicate (n=3/group), and significance was analyzed by regular T-Test. *** p < 0.001. (C) Tissue expression for each transcription factor was assessed in BioGPS and normalized by transcription factors.
DISCUSSION
Sex differences exist throughout life, particularly when gestational exposures can impact disease susceptibility [53]. Pregnancies with male and female fetuses have differing obstetric risk profiles, including miscarriage, preterm birth, and placenta insufficiency [54–56]. As the placenta mediates fetal growth and underlies many pregnancy complications, sex differences arising in utero can influence placenta development and function.
This study shows that X-linked genes can broadly impact placenta trophoblast functions to differentially activate hormonal, endocrine, and immunologic pathways diving toward insulin resistance and likely, maternal gestational diabetes. Our findings corroborate the clinical data supporting the risk of carrying a male fetus for gestational diabetes.
Our study focused on the X-linked gene OGT involved in the O-GlcNAcylation pathway, which can escape X-inactivation in the placenta and, consequently, results in higher levels of the enzyme in female versus male placentas [23–26]. In studies on murine prenatal stress, males’ placental OGT and O-GlcNAcylation levels were more impacted than females. With extra copies of OGT available, it was hypothesized that female offspring might have more flexibility to rapidly respond to maternal stress [27] or, in the case of gestational diabetes, prolonged exposure to hyperglycemic stress [57–60]. It also seems that murine O-GlcNAc-specific regulation of placental H3K27me3 establishes female resilience to prenatal stressors [61].
In this context, we hypothesized that male placental OGT and O-GlcNAcylation levels might be more affected by stressors leading to the induction of GDM. Using matched human placental tissue of male or female origins, we show that, as expected, OGT and OGA are downregulated in GDM placentas of male, but not female, origin. We are confident that this OGA decrease is driven by OGT-dependent intron retention of OGA as previously demonstrated in the literature [39]. Our novel observation of the sexual dimorphism in O-GlcNAc enzyme regulation further strengthens our hypothesis that O-GlcNAcylation plays a role in the sex-specific risk to GDM development.
Previous studies have investigated the function of O-GlcNAcylation in the placenta using deletion models of Ogt in trophoblast lineage in murine models. Consequences of this deletion included increased responsivity of the hypothalamic-pituitary-adrenal stress axis in male offspring and increased insulin resistance to high-fat-diet in females [34,62]. However, rodent models are imperfect simulations of human placental biology and pathophysiology due to the limited endocrine function inherent to mouse placenta. It secretes a significantly lower level of steroids, including a complete absence of GH2, while main hormone production remains in traditional endocrine organs, e.g., corpus luteum (progesterone) or the pituitary gland (GH) [63]. This lack of endocrine function in mice placentas accentuates the role of hormones in human pregnancy complications like GDM, a condition that mice never spontaneously develop [64]. However, mice supplemented with human placenta hormones develop similar insulin resistance as humans (GH2 [45,46], hPL [48,65], steroid hormones [66–71]). Thus, human-based models are needed to capture the placenta endocrine role in maternal disease accurately.
To this end, we not only showed that the human BeWo cell line could be an adequate surrogate to human SCT cells but also used it to study the downstream transcriptional impact of OGT downregulation as it would relate to GDM development. Using siOGT on BeWo cells to model the downregulation of OGT observed in human male GDM placenta, we found a 30% overlap in gene expression with previously published GDM-associated genes. Notable among the shared genes are the transcription factor 7-like 2(TCF7L2) and cdk5 regulatory associated protein 1-like 1 (CDKAL1), two of the most potent genes associated with T2DM, consistently replicated in multiple populations and implicated in impaired glucose-mediated insulin secretion [72,73].
Interestingly, cross-referencing the deregulated genes in our siOGT BeWo model with other transcriptomic profiling of GDM placentas [40–43] revealed altered placental hormone signaling. Indeed, while proper hormones secretion is necessary to reach a natural state of mild insulin resistance in pregnancy to meet fetal nutritional needs [44,74–87], dysregulation of hormonal secretion, particularly placental growth hormone [44–46], prolactin [48,65], estrogens and progesterone [47,88,89] and cytokines [40,90], are thought to influence the development of GDM. It is the placenta production of such hormones [44–48,65,67,79,87–89,91–94] unique to pregnancy, which has been suggested to contribute to the key clinical characteristic of transiency in GDM, vastly different from the slow progression in T2DM.
Following siOGT, we noted an increase in placental growth hormone, a potent insulin antagonist that stimulates maternal lipolysis and hepatic gluconeogenesis [95]. Its rise in the second half of gestation, corresponding to the emergence of insulin resistance during pregnancy, is thought to participate in GDM etiology. Physiologically, this rise in insulin resistance is countered by the upregulation of maternal insulin production driven by placenta-produced prolactin. The surge in hPL parallels an increase in beta-cell replication in pregnancy, which has been attributed to the role of hPL in prolonging beta-cell survival in isolated human and rodent islets [96,97]. However, siOGT cells did not compensate for the increased GH2 production with a change in hPL. Additionally, the significant decrease in Leptin and Leptin receptors in the siOGT BeWo model further contributed toward a decompensatory capacity to respond to GH2-driven insulin resistance. Leptin is consistently shown to be insulin-sensitizing [50,52], and its rise in pregnancy is driven by placental trophoblasts [98]. Thus, we demonstrated that downregulation of OGT may contribute to insulin resistance due to GH2 hyperproduction in placenta trophoblasts while limiting compensatory insulin sensitivity measures (hPL and leptin), altogether driving toward a hormonal diabetogenic profile.
Most of our findings were confirmed in primary human male SCTs, while female SCTs behaved significantly different, further demonstrating the downstream sex dimorphic expression when OGT was downregulated. We observed the increased expression of GH2 and hCG in male SCT after OGT downregulation, which was consistent with our siOGT BeWo model. However, expression levels of placental hormones were unchanged in human female SCT. This confirmed our suspicion that BeWo cells, being of male origin, might not be representative of the female placenta.
We also noticed significant upregulation of SCT markers following siOGT in male SCT cells not seen in female, suggesting that differences in SCT differentiation and by extension, morphology could contribute to the sexual dimorphism we observed. Interestingly, it was previously shown that O-GlcNAcylation stimulated trophoblast differentiation to invasive trophoblast [99] but comparison with our study is challenging. Indeed, this study was performed in mice, who have major differences in placental secretion, and conjointly in placental choriocarcinoma cells (BeWo) treated with OGA inhibitors, which present significant differences with our protocol.
Taken together, our data supports an evolutionary disadvantage in hormonal glucose homeostasis for the male through the O-GlcNAc pathway. In placentas affected by GDM, OGT/OGA expressions were differentially downregulated only in males. Even when we experimentally downregulate OGT expression in female SCT, placental hormonal expression remained unchanged, contrary to the deregulatory patterns in males.
In our constructed regulatory network of hormonal genes in trophoblast cells, we identified several critical O-GlcNAc-regulated transcriptional factors (TFs) for future focus. While some are widely studied and ubiquitous, like SP1[100], TFAP2A is an O-GlcNAcylated, highly placental-specific transcription factor responsible for 6% of hormonal gene regulations in our model [105,106]. Its role in regulating the expression of placental endocrine genes, including hCG [101], CYP11A1 [102], and HSD17s [103], is particularly relevant as they are altered by the downregulation of OGT. Interestingly, abnormal TFAP2A protein levels have been found in pathologic placentas, including preE, GDM, chronic hypertension, and fetal growth restriction [104]. Thus, the O-GlcNAc-dependent regulation of TFAP2A might lead to broad downstream placental hormonal changes characteristic of GDM.
We acknowledge the limitations of the current study, including the limited availability of matching samples for healthy and GDM placentas for each sex. However, the tight range in OGT/OGA expression, particularly in our population of interest (e.g. male GDM), lends confidence to the significance of our finding. We initially chose the BeWo cell line for ease of access to a large material quantity and increased statistical power from large experimental replicates. We suspected that using BeWo as an SCT surrogate could incorporate inherent differences between choriocarcinoma cells and natural SCT into our study findings. To overcome these limitations, we established proof-of-concept experiments on primary human male and female SCTs. Expectedly, results observed from the BeWo cells was only confirmed in human male SCT cultures. This shortcoming should serve as an important consideration for future studies based on cell lines, as results may not be generalizable not only due to in vitro limitations but also the sex origin of cell lines. Again, despite the limited sample size for the primary SCT cultures, we confirmed the differential impact of OGT downregulation based on sex. Thus, our hypothesis was validated in three different models and suggested that the O-GlcNAc pathway could be linked to GDM development through a sex-dimorphic regulation of placental hormones.
In summary, our study provides new insights into the sex dimorphic in O-GlcNAc-dependent signaling and how such differential responses can drive placental physiology toward pregnancy complications like GDM. Understanding the biological mechanisms underlying the sex differences in placenta pathophysiology becomes important as gestational diseases like preE and GDM—previously thought to be transient—have lasting effects on the health of the offspring and mother decades after the event. As medical practice evolves towards precision medicine, incorporating fetal sex and O-GlcNAcylation into clinical consideration can improve future diagnostic and targeted treatment for pregnancy complications.
Supplementary Material
Figure S1: OGT and OGA expression do not correlate with BMI in male and female placentas. Pearson correlations were computed between BMI(A/B) and Age(C/D) and OGT(A/C) or OGA (B/D) relative expression. ns p>0.05.
Figure S2: 28 genes deregulated in siOGT overlap with preeclampsia. Preeclampsia genes were found in the Alliance of Genome Resources under DOID: 10591.
Figure S3 Various pathways are deregulated in siOGT BeWo cells compared to Ctrl, including nutrient sensing and hormone signaling. Volcano plots for siCtrl vs. siOGT. Colored datapoints have an AdjpValue<0.05 (n=3/group). Genes are labeled when they are significant and found in related GO terms.
Figure S4: siOGT impacts hormonal levels in BeWo cells. GH, hCG, and LEP were measured by qPCR after 48h of siOGT or siCtrl. Relative expression was calculated using the −2ΔΔCt method (n=3/group). Significance was calculated by student t-test; ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Figure S5: Verification of OGT knockdown in primary male and female syncytiotrophoblast culture. (A-C) Expression CYP17A (A), LGALS (B), and GCM1 (C) by qPCR measured relative to ACTIN in male and female primary syncytiotrophoblast cultures (n=3/group). Relative expression was calculated using the −2ΔΔCt method. Significance was calculated by student t-test; ** p < 0.01, *** p < 0.001, **** p < 0.0001. (D) Immunostaining of primary syncytiotrophoblast culture with cytokeratin-7 (red) and DAPI (blue) at 48 hours-post plating demonstrating multinucleated cells.
Table S1: Quality control of RNA sequencing.
Table S2: Differentially expressed genes in siOGT vs. SiCtrl.
Table S3: GO enrichment analysis.
Table S4: Genes commonly deregulated with other GDM studies.
HIGHLIGHTS.
O-GlcNAc transferase (OGT) decreases in gestational diabetes (GDM) male placentas.
OGT downregulation in placenta cell models prompted transcriptional signatures of GDM.
Placental hormones involved in GDM development are deregulated following OGT knocked down.
O-GlcNAcylation links fetal sex to placental deregulation in GDM development.
ACKNOWLEDGMENTS
We thank John Corbett, PhD, Meredith Cruz, MD, and the other member of the OVS lab for their valuable feedback along this project. We also thank Jennifer McIntosh, DO and Mary Rau for their help with the MCW placental bank.
FUNDING:
Funding for this study has been provided by the Women’s Health Research Program at Medical College of Wisconsin, the National Institute of Child Health and Development (R01HD104808), and the Translational GlycOmics Program for Career Development in Glycoscience at Milwaukee, WI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST: The authors report no conflict of interest.
REFERENCES
- [1].Gunderson EP, Chiang V, Pletcher MJ, Jacobs DR, Quesenberry CP, Sidney S, Lewis CE, History of gestational diabetes mellitus and future risk of atherosclerosis in mid-life: the Coronary Artery Risk Development in Young Adults study, J. Am. Heart Assoc 3 (2014) e000490. 10.1161/JAHA.113.000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Xu Y, Shen S, Sun L, Yang H, Jin B, Cao X, Metabolic syndrome risk after gestational diabetes: a systematic review and meta-analysis, PloS One. 9 (2014) e87863. 10.1371/journal.pone.0087863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shah BR, Retnakaran R, Booth GL, Increased risk of cardiovascular disease in young women following gestational diabetes mellitus, Diabetes Care. 31 (2008) 1668–1669. 10.2337/dc08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bentley-Lewis R, Late cardiovascular consequences of gestational diabetes mellitus, Semin. Reprod. Med 27 (2009) 322–329. 10.1055/s-0029-1225260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Di Cianni G, Lacaria E, Lencioni C, Resi V, Preventing type 2 diabetes and cardiovascular disease in women with gestational diabetes - The evidence and potential strategies, Diabetes Res. Clin. Pract 145 (2018) 184–192. 10.1016/j.diabres.2018.04.021. [DOI] [PubMed] [Google Scholar]
- [6].Billionnet C, Mitanchez D, Weill A, Nizard J, Alla F, Hartemann A, Jacqueminet S, Gestational diabetes and adverse perinatal outcomes from 716,152 births in France in 2012, Diabetologia. 60 (2017) 636–644. 10.1007/s00125-017-4206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Burlina S, Dalfrà MG, Lapolla A, Short- and long-term consequences for offspring exposed to maternal diabetes: a review, J. Matern.-Fetal Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat. Obstet 32 (2019) 687–694. 10.1080/14767058.2017.1387893. [DOI] [PubMed] [Google Scholar]
- [8].Lowe WL, Scholtens DM, Lowe LP, Kuang A, Nodzenski M, Talbot O, Catalano PM, Linder B, Brickman WJ, Clayton P, Deerochanawong C, Hamilton J, Josefson JL, Lashley M, Lawrence JM, Lebenthal Y, Ma R, Maresh M, McCance D, Tam WH, Sacks DA, Dyer AR, Metzger BE, HAPO Follow-up Study Cooperative Research Group, Association of Gestational Diabetes With Maternal Disorders of Glucose Metabolism and Childhood Adiposity, JAMA. 320 (2018) 1005–1016. 10.1001/jama.2018.11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bianco ME, Josefson JL, Hyperglycemia During Pregnancy and Long-Term Offspring Outcomes, Curr. Diab. Rep 19 (2019) 143. 10.1007/s11892-019-1267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tam WH, Ma RCW, Ozaki R, Li AM, Chan MHM, Yuen LY, Lao TTH, Yang X, Ho CS, Tutino GE, Chan JCN, In Utero Exposure to Maternal Hyperglycemia Increases Childhood Cardiometabolic Risk in Offspring, Diabetes Care. 40 (2017) 679–686. 10.2337/dc16-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Retnakaran R, Kramer CK, Ye C, Kew S, Hanley AJ, Connelly PW, Sermer M, Zinman B, Fetal sex and maternal risk of gestational diabetes mellitus: the impact of having a boy, Diabetes Care. 38 (2015) 844–851. 10.2337/dc14-2551. [DOI] [PubMed] [Google Scholar]
- [12].Wulff-Fuentes E, Berendt RR, Massman L, Danner L, Malard F, Vora J, Kahsay R, Olivier-Van Stichelen S, The human O -GlcNAcome database and meta-analysis, Sci. Data 8 (2021) 25. 10.1038/s41597-021-00810-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Harwood KR, Hanover JA, Nutrient-driven O-GlcNAc cycling - think globally but act locally, J. Cell Sci 127 (2014) 1857–1867. 10.1242/jcs.113233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chang Y-H, Weng C-L, Lin K-I, O-GlcNAcylation and its role in the immune system, J. Biomed. Sci 27 (2020) 57. 10.1186/s12929-020-00648-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hart GW, Three Decades of Research on O-GlcNAcylation - A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism, Front. Endocrinol 5 (2014) 183. 10.3389/fendo.2014.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pravata VM, Muha V, Gundogdu M, Ferenbach AT, Kakade PS, Vandadi V, Wilmes AC, Borodkin VS, Joss S, Stavridis MP, van Aalten DMF, Catalytic deficiency of O-GlcNAc transferase leads to X-linked intellectual disability, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 14961–14970. 10.1073/pnas.1900065116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pravata VM, Gundogdu M, Bartual SG, Ferenbach AT, Stavridis M, Õunap K, Pajusalu S, Žordania R, Wojcik MH, van Aalten DMF, A missense mutation in the catalytic domain of O-GlcNAc transferase links perturbations in protein O-GlcNAcylation to X-linked intellectual disability, FEBS Lett. 594 (2020) 717–727. 10.1002/1873-3468.13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Selvan N, George S, Serajee FJ, Shaw M, Hobson L, Kalscheuer V, Prasad N, Levy SE, Taylor J, Aftimos S, Schwartz CE, Huq AM, Gecz J, Wells L, O-GlcNAc transferase missense mutations linked to X-linked intellectual disability deregulate genes involved in cell fate determination and signaling, J. Biol. Chem 293 (2018) 10810–10824. 10.1074/jbc.RA118.002583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vaidyanathan K, Niranjan T, Selvan N, Teo CF, May M, Patel S, Weatherly B, Skinner C, Opitz J, Carey J, Viskochil D, Gecz J, Shaw M, Peng Y, Alexov E, Wang T, Schwartz C, Wells L, Identification and characterization of a missense mutation in the O-linked β-N-acetylglucosamine (O-GlcNAc) transferase gene that segregates with X-linked intellectual disability, J. Biol. Chem 292 (2017) 8948–8963. 10.1074/jbc.M116.771030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Willems AP, Gundogdu M, Kempers MJE, Giltay JC, Pfundt R, Elferink M, Loza BF, Fuijkschot J, Ferenbach AT, van Gassen KLI, van Aalten DMF, Lefeber DJ, Mutations in N-acetylglucosamine (O-GlcNAc) transferase in patients with X-linked intellectual disability, J. Biol. Chem 292 (2017) 12621–12631. 10.1074/jbc.M117.790097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Olivier-Van Stichelen S, Hanover JA, X-inactivation normalizes O-GlcNAc transferase levels and generates an O-GlcNAc-depleted Barr body, Front. Genet 5 (2014) 256. 10.3389/fgene.2014.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Olivier-Van Stichelen S, Abramowitz LK, Hanover JA, X marks the spot: does it matter that O-GlcNAc transferase is an X-linked gene?, Biochem. Biophys. Res. Commun 453 (2014) 201–207. 10.1016/j.bbrc.2014.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dubois A, Deuve JL, Navarro P, Merzouk S, Pichard S, Commere P-H, Louise A, Arnaud D, Avner P, Morey C, Spontaneous reactivation of clusters of X-linked genes is associated with the plasticity of X-inactivation in mouse trophoblast stem cells, Stem Cells Dayt. Ohio 32 (2014) 377–390. 10.1002/stem.1557. [DOI] [PubMed] [Google Scholar]
- [24].Mugford JW, Starmer J, Williams RL Jr, Calabrese JM, Mieczkowski P, Yee D, Magnuson T, Evidence for Local Regulatory Control of Escape from Imprinted X Chromosome Inactivation, Genetics. (2014). 10.1534/genetics.114.162800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Calabrese JM, Sun W, Song L, Mugford JW, Williams L, Yee D, Starmer J, Mieczkowski P, Crawford GE, Magnuson T, Site-specific silencing of regulatory elements as a mechanism of X inactivation, Cell. 151 (2012) 951–963. 10.1016/j.cell.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Splinter E, de Wit E, Nora EP, Klous P, van de Werken HJG, Zhu Y, Kaaij LJT, van Ijcken W, Gribnau J, Heard E, de Laat W, The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA, Genes Dev. 25 (2011) 1371–1383. 10.1101/gad.633311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Howerton CL, Morgan CP, Fischer DB, Bale TL, O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 5169–5174. 10.1073/pnas.1300065110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu W, Wang H, Xue X, Xia J, Liu J, Qi Z, Ji L, OGT-related mitochondrial motility is associated with sex differences and exercise effects in depression induced by prenatal exposure to glucocorticoids, J. Affect. Disord 226 (2018) 203–215. 10.1016/j.jad.2017.09.053. [DOI] [PubMed] [Google Scholar]
- [29].Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, Marth JD, The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny, Proc. Natl. Acad. Sci. U. S. A 97 (2000) 5735–5739. 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mohan R, Jo S, Da Sol Chung E, Oribamise E, Lockridge A, Abrahante-Lloréns JE, Ruan H-B, Yang X-Y, Alejandro EU, Pancreatic β-Cell O-GlcNAc Transferase Overexpression Increases Susceptibility to Metabolic Stressors in Female Mice, Cells. 10 (2021) 2801. 10.3390/cells10102801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shi J-J, Liu H-F, Hu T, Gao X, Zhang Y-B, Li W-R, Wang Q, Zhang S-J, Tang D, Chen Y-B, Danggui-Shaoyao-San improves cognitive impairment through inhibiting O-GlcNAc-modification of estrogen α receptor in female db/db mice, J. Ethnopharmacol 281 (2021) 114562. 10.1016/j.jep.2021.114562. [DOI] [PubMed] [Google Scholar]
- [32].Pantaleon M, Steane SE, McMahon K, Cuffe JSM, Moritz KM, Placental O-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice, Sci. Rep 7 (2017) 2017. 10.1038/s41598-017-01666-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sinclair DAR, Syrzycka M, Macauley MS, Rastgardani T, Komljenovic I, Vocadlo DJ, Brock HW, Honda BM, Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc), Proc. Natl. Acad. Sci. U. S. A 106 (2009) 13427–13432. 10.1073/pnas.0904638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Moore M, Avula N, Jo S, Beetch M, Alejandro EU, Disruption of O-Linked N-Acetylglucosamine Signaling in Placenta Induces Insulin Sensitivity in Female Offspring, Int. J. Mol. Sci 22 (2021) 6918. 10.3390/ijms22136918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Simon B, Bucher M, Maloyan A, A Primary Human Trophoblast Model to Study the Effect of Inflammation Associated with Maternal Obesity on Regulation of Autophagy in the Placenta, J. Vis. Exp. JoVE (2017). 10.3791/56484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sundjaja JH, Shrestha R, Krishan K, McNemar And Mann-Whitney U Tests, in: StatPearls, StatPearls Publishing, Treasure Island (FL), 2022. http://www.ncbi.nlm.nih.gov/books/NBK560699/ (accessed May 11, 2022). [PubMed] [Google Scholar]
- [37].Varet H, Brillet-Guéguen L, Coppée J-Y, Dillies M-A, SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data, PloS One. 11 (2016) e0157022. 10.1371/journal.pone.0157022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu J, Shao X, Qin W, Zhang Y, Dang F, Yang Q, Yu X, Li Y-X, Chen X, Wang C, Wang Y-L, Quantitative chemoproteomics reveals O-GlcNAcylation of cystathionine γ-lyase (CSE) represses trophoblast syncytialization, Cell Chem. Biol 28 (2021) 788–801.e5. 10.1016/j.chembiol.2021.01.024. [DOI] [PubMed] [Google Scholar]
- [39].Park S-K, Zhou X, Pendleton KE, Hunter OV, Kohler JJ, O’Donnell KA, Conrad NK, A Conserved Splicing Silencer Dynamically Regulates O-GlcNAc Transferase Intron Retention and O-GlcNAc Homeostasis, Cell Rep. 20 (2017) 1088–1099. 10.1016/j.celrep.2017.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Radaelli T, Varastehpour A, Catalano P, Hauguel-de Mouzon S, Gestational diabetes induces placental genes for chronic stress and inflammatory pathways, Diabetes. 52 (2003) 2951–2958. 10.2337/diabetes.52.12.2951. [DOI] [PubMed] [Google Scholar]
- [41].Zhao Y-H, Wang D-P, Zhang L-L, Zhang F, Wang D-M, Zhang W-Y, Genomic expression profiles of blood and placenta reveal significant immune-related pathways and categories in Chinese women with gestational diabetes mellitus, Diabet. Med 28 (2011) 237–246. 10.1111/j.1464-5491.2010.03140.x. [DOI] [PubMed] [Google Scholar]
- [42].Enquobahrie DA, Williams MA, Qiu C, Meller M, Sorensen TK, Global placental gene expression in gestational diabetes mellitus, Am. J. Obstet. Gynecol 200 (2009) 206.e1–13. 10.1016/j.ajog.2008.08.022. [DOI] [PubMed] [Google Scholar]
- [43].Yang Y, Guo F, Peng Y, Chen R, Zhou W, Wang H, OuYang J, Yu B, Xu Z, Transcriptomic Profiling of Human Placenta in Gestational Diabetes Mellitus at the Single-Cell Level, Front. Endocrinol 12 (2021) 474. 10.3389/fendo.2021.679582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Barbour LA, Shao J, Qiao L, Leitner W, Anderson M, Friedman JE, Draznin B, Human placental growth hormone increases expression of the p85 regulatory unit of phosphatidylinositol 3-kinase and triggers severe insulin resistance in skeletal muscle, Endocrinology. 145 (2004) 1144–1150. 10.1210/en.2003-1297. [DOI] [PubMed] [Google Scholar]
- [45].del Rincon J-P, Iida K, Gaylinn BD, McCurdy CE, Leitner JW, Barbour LA, Kopchick JJ, Friedman JE, Draznin B, Thorner MO, Growth hormone regulation of p85alpha expression and phosphoinositide 3-kinase activity in adipose tissue: mechanism for growth hormone-mediated insulin resistance, Diabetes. 56 (2007) 1638–1646. 10.2337/db06-0299. [DOI] [PubMed] [Google Scholar]
- [46].Dominici FP, Cifone D, Bartke A, Turyn D, Loss of sensitivity to insulin at early events of the insulin signaling pathway in the liver of growth hormone-transgenic mice, J. Endocrinol 161 (1999) 383–392. 10.1677/joe.0.1610383. [DOI] [PubMed] [Google Scholar]
- [47].Qi X, Gong B, Yu J, Shen L, Jin W, Wu Z, Wang J, Wang J, Li Z, Decreased cord blood estradiol levels in related to mothers with gestational diabetes, Medicine (Baltimore). 96 (2017) e6962. 10.1097/MD.0000000000006962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ryan EA, Enns L, Role of gestational hormones in the induction of insulin resistance, J. Clin. Endocrinol. Metab 67 (1988) 341–347. 10.1210/jcem-67-2-341. [DOI] [PubMed] [Google Scholar]
- [49].Liu Y, Guo F, Maraka S, Zhang Y, Zhang C, Korevaar TIM, Fan J, Associations between Human Chorionic Gonadotropin, Maternal Free Thyroxine, and Gestational Diabetes Mellitus, Thyroid. 31 (2021) 1282–1288. 10.1089/thy.2020.0920. [DOI] [PubMed] [Google Scholar]
- [50].Perry RJ, Zhang X-M, Zhang D, Kumashiro N, Camporez J-PG, Cline GW, Rothman DL, Shulman GI, Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis, Nat. Med 20 (2014) 759–763. 10.1038/nm.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yamashita H, Shao J, Ishizuka T, Klepcyk PJ, Muhlenkamp P, Qiao L, Hoggard N, Friedman JE, Leptin Administration Prevents Spontaneous Gestational Diabetes in Heterozygous Leprdb/+ Mice: Effects on Placental Leptin and Fetal Growth*, Endocrinology 142 (2001) 2888–2897. 10.1210/endo.142.7.8227. [DOI] [PubMed] [Google Scholar]
- [52].Guerre-Millo M, Extending the glucose/fatty acid cycle: a glucose/adipose tissue cycle, Biochem. Soc. Trans 31 (2003) 1161–1164. 10.1042/bst0311161. [DOI] [PubMed] [Google Scholar]
- [53].Barker DJ, The fetal and infant origins of adult disease, BMJ. 301 (1990) 1111. 10.1136/bmj.301.6761.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Di Renzo GC, Rosati A, Sarti RD, Cruciani L, Cutuli AM, Does fetal sex affect pregnancy outcome?, Gend. Med 4 (2007) 19–30. 10.1016/s1550-8579(07)80004-0. [DOI] [PubMed] [Google Scholar]
- [55].Vatten LJ, Skjaerven R, Offspring sex and pregnancy outcome by length of gestation, Early Hum. Dev 76 (2004) 47–54. 10.1016/j.earlhumdev.2003.10.006. [DOI] [PubMed] [Google Scholar]
- [56].Persson M, Fadl H, Perinatal outcome in relation to fetal sex in offspring to mothers with pre-gestational and gestational diabetes--a population-based study, Diabet. Med. J. Br. Diabet. Assoc 31 (2014) 1047–1054. 10.1111/dme.12479. [DOI] [PubMed] [Google Scholar]
- [57].OuYang H, Chen B, Abdulrahman A-M, Li L, Wu N, Associations between Gestational Diabetes and Anxiety or Depression: A Systematic Review, J. Diabetes Res 2021 (2021) 9959779. 10.1155/2021/9959779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Feng Y, Feng Q, Qu H, Song X, Hu J, Xu X, Zhang L, Yin S, Stress adaptation is associated with insulin resistance in women with gestational diabetes mellitus, Nutr. Diabetes 10 (2020) 1–4. 10.1038/s41387-020-0107-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li H, Shen L, Song L, Liu B, Zheng X, Xu S, Wang Y, Early age at menarche and gestational diabetes mellitus risk: Results from the Healthy Baby Cohort study, Diabetes Metab. 43 (2017) 248–252. 10.1016/j.diabet.2017.01.002. [DOI] [PubMed] [Google Scholar]
- [60].Zhang X, Zhao X, Huo L, Yuan N, Sun J, Du J, Nan M, Ji L, Risk prediction model of gestational diabetes mellitus based on nomogram in a Chinese population cohort study, Sci. Rep 10 (2020) 21223. 10.1038/s41598-020-78164-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nugent BM, O’Donnell CM, Epperson CN, Bale TL, Placental H3K27me3 establishes female resilience to prenatal insults, Nat. Commun 9 (2018) 2555. 10.1038/s41467-018-04992-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Howerton CL, Bale TL, Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 9639–9644. 10.1073/pnas.1401203111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Malassiné A, Frendo J-L, Evain-Brion D, A comparison of placental development and endocrine functions between the human and mouse model, Hum. Reprod. Update 9 (2003) 531–539. 10.1093/humupd/dmg043. [DOI] [PubMed] [Google Scholar]
- [64].Pasek RC, Gannon M, Advancements and challenges in generating accurate animal models of gestational diabetes mellitus, Am. J. Physiol. - Endocrinol. Metab 305 (2013) E1327–E1338. 10.1152/ajpendo.00425.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Beck P, Daughaday WH, Human Placental Lactogen: Studies of Its Acute Metabolic Effects and Disposition in Normal Man*, J. Clin. Invest 46 (1967) 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Leturque A, Burnol AF, Ferre P, Girard J, Pregnancy-induced insulin resistance in the rat: assessment by glucose clamp technique, Am. J. Physiol.-Endocrinol. Metab 246 (1984) E25–E31. 10.1152/ajpendo.1984.246.1.E25. [DOI] [PubMed] [Google Scholar]
- [67].Ryan EA, O’Sullivan MJ, Skyler JS, Insulin action during pregnancy. Studies with the euglycemic clamp technique, Diabetes. 34 (1985) 380–389. 10.2337/diab.34.4.380. [DOI] [PubMed] [Google Scholar]
- [68].Kim JK, Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo, Methods Mol. Biol. Clifton NJ 560 (2009) 221–238. 10.1007/978-1-59745-448-3_15. [DOI] [PubMed] [Google Scholar]
- [69].Ahmed-Sorour H, Bailey CJ, Role of ovarian hormones in the long-term control of glucose homeostasis. Interaction with insulin, glucagon and epinephrine, Horm. Res 13 (1980) 396–403. 10.1159/000179307. [DOI] [PubMed] [Google Scholar]
- [70].Takeda K, Toda K, Saibara T, Nakagawa M, Saika K, Onishi T, Sugiura T, Shizuta Y, Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency, J. Endocrinol 176 (2003) 237–246. 10.1677/joe.0.1760237. [DOI] [PubMed] [Google Scholar]
- [71].Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, Dahlman-Wright K, Nilsson S, Gustafsson J-A, Efendic S, Khan A, Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver, Diabetologia. 49 (2006) 588–597. 10.1007/s00125-005-0105-3. [DOI] [PubMed] [Google Scholar]
- [72].Scott LJ, Bonnycastle LL, Willer CJ, Sprau AG, Jackson AU, Narisu N, Duren WL, Chines PS, Stringham HM, Erdos MR, Valle TT, Tuomilehto J, Bergman RN, Mohlke KL, Collins FS, Boehnke M, Association of transcription factor 7-like 2 (TCF7L2) variants with type 2 diabetes in a Finnish sample, Diabetes. 55 (2006) 2649–2653. 10.2337/db06-0341. [DOI] [PubMed] [Google Scholar]
- [73].El-Lebedy D, Ashmawy I, Common variants in TCF7L2 and CDKAL1 genes and risk of type 2 diabetes mellitus in Egyptians, J. Genet. Eng. Biotechnol 14 (2016) 247–251. 10.1016/j.jgeb.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kirwan JP, Varastehpour A, Jing M, Presley L, Shao J, Friedman JE, Catalano PM, Reversal of insulin resistance postpartum is linked to enhanced skeletal muscle insulin signaling, J. Clin. Endocrinol. Metab 89 (2004) 4678–4684. 10.1210/jc.2004-0749. [DOI] [PubMed] [Google Scholar]
- [75].Bailey CJ, Ahmed-Sorour H, Role of ovarian hormones in the long-term control of glucose homeostasis, Diabetologia. 19 (1980) 475–481. 10.1007/BF00281829. [DOI] [PubMed] [Google Scholar]
- [76].Costrini NV, Kalkhoff RK, Relative effects of pregnancy, estradiol, and progesterone on plasma insulin and pancreatic islet insulin secretion, J. Clin. Invest 50 (1971) 992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Brelje TC, Scharp DW, Lacy PE, Ogren L, Talamantes F, Robertson M, Friesen HG, Sorenson RL, Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy, Endocrinology. 132 (1993) 879–887. 10.1210/endo.132.2.8425500. [DOI] [PubMed] [Google Scholar]
- [78].Huang C, Snider F, Cross JC, Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy, Endocrinology. 150 (2009) 1618–1626. 10.1210/en.2008-1003. [DOI] [PubMed] [Google Scholar]
- [79].Fujinaka Y, Sipula D, Garcia-Ocaña A, Vasavada RC, Characterization of mice doubly transgenic for parathyroid hormone-related protein and murine placental lactogen: a novel role for placental lactogen in pancreatic beta-cell survival, Diabetes. 53 (2004) 3120–3130. 10.2337/diabetes.53.12.3120. [DOI] [PubMed] [Google Scholar]
- [80].N B, Jh N, The stimulatory effect of growth hormone, prolactin, and placental lactogen on beta-cell proliferation is not mediated by insulin-like growth factor-I., Endocrinology. 129 (1991) 883–888. 10.1210/endo-129-2-883. [DOI] [PubMed] [Google Scholar]
- [81].Nielsen JH, Effects of Growth Hormone, Prolactin, and Placental Lactogen on Insulin Content and Release, and Deoxyribonucleic Acid Synthesis in Cultured Pancreatic Islets, Endocrinology. 110 (1982) 600–606. 10.1210/endo-110-2-600. [DOI] [PubMed] [Google Scholar]
- [82].Brelje TC, Allaire P, Hegre O, Sorenson RL, Effect of prolactin versus growth hormone on islet function and the importance of using homologous mammosomatotropic hormones, Endocrinology. 125 (1989) 2392–2399. 10.1210/endo-125-5-2392. [DOI] [PubMed] [Google Scholar]
- [83].Weinhaus AJ, Stout LE, Sorenson RL, Glucokinase, hexokinase, glucose transporter 2, and glucose metabolism in islets during pregnancy and prolactin-treated islets in vitro: mechanisms for long term up-regulation of islets, Endocrinology. 137 (1996) 1640–1649. 10.1210/endo.137.5.8612496. [DOI] [PubMed] [Google Scholar]
- [84].Sorenson RL, Brelje TC, Adaptation of Islets of Langerhans to Pregnancy: β-Cell Growth, Enhanced Insulin Secretion and the Role of Lactogenic Hormones, Horm. Metab. Res 29 (1997) 301–307. 10.1055/s-2007-979040. [DOI] [PubMed] [Google Scholar]
- [85].Arumugam R, Fleenor D, Freemark M, Knockdown of prolactin receptors in a pancreatic beta cell line: effects on DNA synthesis, apoptosis, and gene expression, Endocrine. 46 (2014) 568–576. 10.1007/s12020-013-0073-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Dominici FP, Argentino DP, Muñoz MC, Miquet JG, Sotelo AI, Turyn D, Influence of the crosstalk between growth hormone and insulin signalling on the modulation of insulin sensitivity, Growth Horm. IGF Res. Off. J. Growth Horm. Res. Soc. Int. IGF Res. Soc 15 (2005) 324–336. 10.1016/j.ghir.2005.07.001. [DOI] [PubMed] [Google Scholar]
- [87].Napso T, Yong HEJ, Lopez-Tello J, Sferruzzi-Perri AN, The Role of Placental Hormones in Mediating Maternal Adaptations to Support Pregnancy and Lactation, Front. Physiol 9 (2018) 1091. 10.3389/fphys.2018.01091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Brănişteanu DD, Mathieu C, Progesterone in gestational diabetes mellitus: guilty or not guilty?, Trends Endocrinol. Metab. TEM 14 (2003) 54–56. 10.1016/s1043-2760(03)00003-1. [DOI] [PubMed] [Google Scholar]
- [89].Picard F, Wanatabe M, Schoonjans K, Lydon J, O’Malley BW, Auwerx J, Progesterone receptor knockout mice have an improved glucose homeostasis secondary to β-cell proliferation, Proc. Natl. Acad. Sci 99 (2002) 15644–15648. 10.1073/pnas.202612199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Li H-P, Chen X, Li M-Q, Gestational diabetes induces chronic hypoxia stress and excessive inflammatory response in murine placenta, Int. J. Clin. Exp. Pathol 6 (2013) 650–659. [PMC free article] [PubMed] [Google Scholar]
- [91].Rygaard K, Revol A, Esquivel-Escobedo D, Beck BL, Barrera-Saldaña HA, Absence of human placental lactogen and placental growth hormone (HGH-V) during pregnancy: PCR analysis of the deletion, Hum. Genet 102 (1998) 87–92. 10.1007/s004390050658. [DOI] [PubMed] [Google Scholar]
- [92].Le TN, Elsea SH, Romero R, Chaiworapongsa T, Francis GL, Prolactin receptor gene polymorphisms are associated with gestational diabetes, Genet. Test. Mol. Biomark 17 (2013) 567–571. 10.1089/gtmb.2013.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ekinci EI, Torkamani N, Ramchand SK, Churilov L, Sikaris KA, Lu ZX, Houlihan CA, Higher maternal serum prolactin levels are associated with reduced glucose tolerance during pregnancy, J. Diabetes Investig 8 (2017) 697–700. 10.1111/jdi.12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Buschur E, Stetson B, Barbour LA, Diabetes In Pregnancy, in: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP (Eds.), Endotext, MDText.com, Inc., South Dartmouth (MA), 2000. http://www.ncbi.nlm.nih.gov/books/NBK279010/ (accessed May 9, 2020). [Google Scholar]
- [95].Handwerger S, Freemark M, The roles of placental growth hormone and placental lactogen in the regulation of human fetal growth and development, J. Pediatr. Endocrinol. Metab. JPEM 13 (2000) 343–356. 10.1515/jpem.2000.13.4.343. [DOI] [PubMed] [Google Scholar]
- [96].Arumugam R, Horowitz E, Lu D, Collier JJ, Ronnebaum S, Fleenor D, Freemark M, The interplay of prolactin and the glucocorticoids in the regulation of beta-cell gene expression, fatty acid oxidation, and glucose-stimulated insulin secretion: implications for carbohydrate metabolism in pregnancy, Endocrinology. 149 (2008) 5401–5414. 10.1210/en.2008-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Fujinaka Y, Takane K, Yamashita H, Vasavada RC, Lactogens promote beta cell survival through JAK2/STAT5 activation and Bcl-XL upregulation, J. Biol. Chem 282 (2007) 30707–30717. 10.1074/jbc.M702607200. [DOI] [PubMed] [Google Scholar]
- [98].Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, Nishimura H, Yoshimasa Y, Tanaka I, Mori T, Nakao K, Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans, Nat. Med 3 (1997) 1029–1033. 10.1038/nm0997-1029. [DOI] [PubMed] [Google Scholar]
- [99].Ruane PT, Tan CMJ, Adlam DJ, Kimber SJ, Brison DR, Aplin JD, Westwood M, Protein O-GlcNAcylation Promotes Trophoblast Differentiation at Implantation, Cells. 9 (2020) 2246. 10.3390/cells9102246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Solomon SS, Majumdar G, Martinez-Hernandez A, Raghow R, A critical role of Sp1 transcription factor in regulating gene expression in response to insulin and other hormones, Life Sci. 83 (2008) 305–312. 10.1016/j.lfs.2008.06.024. [DOI] [PubMed] [Google Scholar]
- [101].LiCalsi C, Christophe S, Steger DJ, Buescher M, Fischer W, Mellon PL, AP-2 family members regulate basal and cAMP-induced expression of human chorionic gonadotropin, Nucleic Acids Res. 28 (2000) 1036–1043. 10.1093/nar/28.4.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Pena P, Reutens AT, Albanese C, D’Amico M, Watanabe G, Donner A, Shu IW, Williams T, Pestell RG, Activator protein-2 mediates transcriptional activation of the CYP11A1 gene by interaction with Sp1 rather than binding to DNA, Mol. Endocrinol. Baltim. Md 13 (1999) 1402–1416. 10.1210/mend.13.8.0335. [DOI] [PubMed] [Google Scholar]
- [103].Piao YS, Peltoketo H, Vihko P, Vihko R, The proximal promoter region of the gene encoding human 17beta-hydroxysteroid dehydrogenase type 1 contains GATA, AP-2, and Sp1 response elements: analysis of promoter function in choriocarcinoma cells, Endocrinology. 138 (1997) 3417–3425. 10.1210/endo.138.8.5329. [DOI] [PubMed] [Google Scholar]
- [104].Sheridan RM, Stanek J, Khoury J, Handwerger S, Abnormal expression of transcription factor AP-2α in pathologic placentas, Hum. Pathol 43 (2012) 1866–1874. 10.1016/j.humpath.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Xie X, Wu Q, Zhang K, Liu Y, Zhang N, Chen Q, Wang L, Li W, Zhang J, Liu Y, O-GlcNAc modification regulates MTA1 transcriptional activity during breast cancer cell genotoxic adaptation, Biochim. Biophys. Acta Gen. Subj 1865 (2021) 129930. 10.1016/j.bbagen.2021.129930. [DOI] [PubMed] [Google Scholar]
- [106].Wulff-Fuentes E, Berendt RR, Massman L, Danner L, Malard F, Vora J, Kahsay R, Olivier-Van Stichelen S, The human O-GlcNAcome database and meta-analysis, Sci. Data 8 (2021) 25. 10.1038/s41597-021-00810-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: OGT and OGA expression do not correlate with BMI in male and female placentas. Pearson correlations were computed between BMI(A/B) and Age(C/D) and OGT(A/C) or OGA (B/D) relative expression. ns p>0.05.
Figure S2: 28 genes deregulated in siOGT overlap with preeclampsia. Preeclampsia genes were found in the Alliance of Genome Resources under DOID: 10591.
Figure S3 Various pathways are deregulated in siOGT BeWo cells compared to Ctrl, including nutrient sensing and hormone signaling. Volcano plots for siCtrl vs. siOGT. Colored datapoints have an AdjpValue<0.05 (n=3/group). Genes are labeled when they are significant and found in related GO terms.
Figure S4: siOGT impacts hormonal levels in BeWo cells. GH, hCG, and LEP were measured by qPCR after 48h of siOGT or siCtrl. Relative expression was calculated using the −2ΔΔCt method (n=3/group). Significance was calculated by student t-test; ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Figure S5: Verification of OGT knockdown in primary male and female syncytiotrophoblast culture. (A-C) Expression CYP17A (A), LGALS (B), and GCM1 (C) by qPCR measured relative to ACTIN in male and female primary syncytiotrophoblast cultures (n=3/group). Relative expression was calculated using the −2ΔΔCt method. Significance was calculated by student t-test; ** p < 0.01, *** p < 0.001, **** p < 0.0001. (D) Immunostaining of primary syncytiotrophoblast culture with cytokeratin-7 (red) and DAPI (blue) at 48 hours-post plating demonstrating multinucleated cells.
Table S1: Quality control of RNA sequencing.
Table S2: Differentially expressed genes in siOGT vs. SiCtrl.
Table S3: GO enrichment analysis.
Table S4: Genes commonly deregulated with other GDM studies.
Data Availability Statement
RNA-sequencing raw (*R1.fastq and *R2.fastq) and processed (*.bam and counts.txt) files were deposited in Gene Expression Omnibus (GEO) repository under GSE200983.