

Summary
Clear cell renal cell carcinomas (ccRCCs) represent ~75% of RCC cases and account for most RCC-associated deaths. Inter- and intra-tumoral heterogeneity (ITH) results in varying prognosis and treatment outcomes. To obtain the most comprehensive profile of ccRCC, we perform integrative histopathologic, proteogenomic, and metabolomic analyses on 305 ccRCC tumor segments and 166 paired adjacent normal tissues from 213 cases. Combining histologic and molecular profiles reveals ITH in 90% of ccRCCs, with 50% demonstrating immune signature heterogeneity. High tumor grade, along with BAP1 mutation, genome instability, increased hypermethylation, and a specific protein glycosylation signature define a high-risk disease subset, where UCHL1 expression displays prognostic value. Single nuclei RNA-seq of the adverse sarcomatoid and rhabdoid phenotypes uncover gene signatures and potential insights into tumor evolution. In vitro cell line studies confirm the potential of inhibiting identified phosphoproteome targets. This study molecularly stratifies aggressive histopathologic subtypes that may inform more effective treatment strategies.
Keywords: clear cell renal cell carcinoma (ccRCC), proteomics, proteogenomics, glycoproteomics, phosphoproteomics, tumor heterogeneity, histology, methylation, metabolome, single nuclei RNA-seq, CPTAC, UCHL1
Graphical Abstract
eTOC Blurb
Li et al. integrate histopathologic, proteogenomic, and metabolomic data from 305 tumor segments and reveal intratumoral heterogeneity in at least 90% of clear cell renal cell carcinomas, signatures for sarcomatoid and rhabdoid features, and prognostic value of UCHL1. This study molecularly stratifies aggressive histopathologic subtypes to inform effective treatment strategies.
Introduction
Renal cell carcinoma (RCC) is among the ten most diagnosed cancers worldwide and comprises a wide array of histologic and genetic subtypes.1,2 and clear cell RCC (ccRCC) accounts for the majority (75%) of renal cancer-associated deaths.1 While treatment for localized ccRCC is surgical resection or ablation, therapeutic choices for advanced disease are limited due to chemotherapy resistance and lead to an emphasis on research-driven targeted therapies.3–7 Therapeutic combinations of tyrosine kinase inhibitors (TKIs), such as axitinib, cabozantinib, and lenvatinib, and immune checkpoint inhibitors (IMIs), such as pembrolizumab, nivolumab, and ipilimumab have proved their utility.7–9 However, these treatments have a variable impact on tumor inhibition and individual patient survival, meaning newer options are necessary to improve patient outcomes.
ccRCC is associated with a deregulation of hypoxia-inducible factor (HIF1) signaling, alteration of chromatin-modifying enzymes, metabolic reprogramming, and a distinct tumor immune microenvironment.10 Pseudohypoxic activation of HIF signaling is caused by the nearly ubiquitous bi-allelic loss of the VHL gene.11–13 Chromosome 3p loss also affects some or all three chromatin remodeling genes encoded within the same region, PBRM1, SETD2, and BAP1, the deficiency of which has been associated with disease progression and more aggressive phenotypes.14–17 The Cancer Genome Atlas (TCGA) ccRCC genomic analyses highlights tumor-specific shifts in multiple metabolic pathways associated with patient outcome, confirms the previous observation of high levels of intratumoral immune infiltration, and demonstrates significant heterogeneity across ccRCC patients.14,17–20
Significant intratumoral heterogeneity (ITH) encountered in ccRCC, results in confinement of several driver events to subclonal tumor populations.21–23 This observation suggests that multiple tumor subclones may reach metastatic potential and could independently influence response to therapies. Single-cell transcriptomic analyses now provide higher resolution insights into the tumor microenvironment (TME), cell of origin, and ITH within ccRCC and their relevance to therapeutic response,24,25 but the full extent of these factors remains unknown. Progress on these fronts will likely significantly impact treatment outcomes.
The initial Clinical Proteomic Tumor Analysis Consortium (CPTAC) investigation of ccRCC provides a landmark integrated proteogenomic characterization of 103 tumors that highlighted a variety of early chromosomal translocation alterations leading to chr3p loss, identifies tumor-specific proteomic and phosphoproteomic alterations that are independent of mRNA expression, and define specific immune-based subtypes based upon a combination of mRNA, proteome, and phosphoproteome markers.13 The current study expands CPTAC ccRCC cohort to 305 tumor segments from 213 cases. In addition, we evaluate ITH in multiple tumor areas from 40 cases and provide single-cell RNA expression analysis of 12 tumor segments from 4 cases. 5 RCC-derived cell line models were used to investigate the clinical translational relevance of kinase targets from initial observations. To investigate metabolic aberrations associated with patient outcomes in ccRCC, we first analyze metabolome profiles of 50 tumors and 7 normal adjacent tissues (NATs) and subsequently examine a validation set of 56 tumors and 15 NATs. Extensive tumor histopathologic reviews following the latest methodological recommendations for evaluating heterogeneity in ccRCCs were performed.26 Proteomic analyses were also expanded to include both mass spectrometry-based Data Independent Acquisition (DIA) methodology from global proteomics to phosphoproteomics and glycoproteomics.
These analyses collectively stress the importance of genetic instability and increased hypermethylation as markers of poor patient outcome, the relevance of proteomic changes, including phospho- and glycoproteins as specific indicators of differential ccRCC biology and patient outcome, and highlight the ITH of ccRCC, particularly concerning the immune landscape within a tumor. These discoveries should facilitate clinical translation, including elucidation of prospective patient outcome-dependent signatures and therapeutic targets to aid in personalized treatment of ccRCC patients.
Results
Overview of study design, cohort, and data types
CPTAC previously characterized 103 treatment-naïve ccRCC cases using Tandem Mass Tagging (TMT)-based global proteomics and phosphoproteomics platforms.13 This study increased the cohort size to 213 cases, with 40 cases being selected for multiple-segment profiling of an additional 92 segments to evaluate tumor evolution and ITH. The dataset contained 305 tumor samples, 165 paired NATs, and 213 blood normal samples among 16 different data types from the initial (INI), expanded (EXP), and ITH cohorts (Figure 1A; Table S1). Samples were genomically and epigenetically characterized as before,13 while DIA-based proteomic analysis was used to profile all samples for the global proteome and the newly added 110 cases for phosphoproteome and glycoproteome (Figure 1A). Integrating metabolomics of 106 selected cases, and single-nuclei RNA-seq (snRNA-seq) of 15 tumor specimens from 7 cases with the other multi-omics data helped investigate both tumor-intrinsic cell populations and TME (Figure 1A). In parallel, a comprehensive histopathologic evaluation was performed based on 21 parameters (STAR Methods) to define low- and high-grade features, spatial architecture, and TME. Molecular profiles and histopathologic annotations were integrated to characterize distinct histological features, understand molecular mechanisms that drive ccRCC, and provide a reference for selecting effective therapy (Table S1).
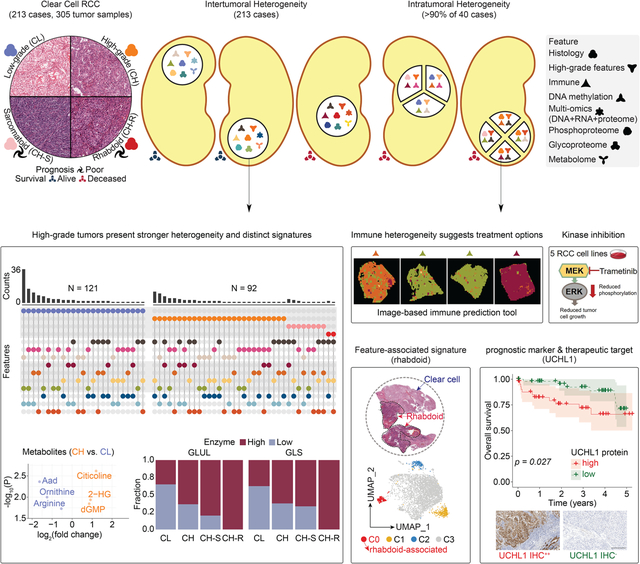
Figure 1. Molecular underpinnings of ccRCC histopathologic heterogeneity.
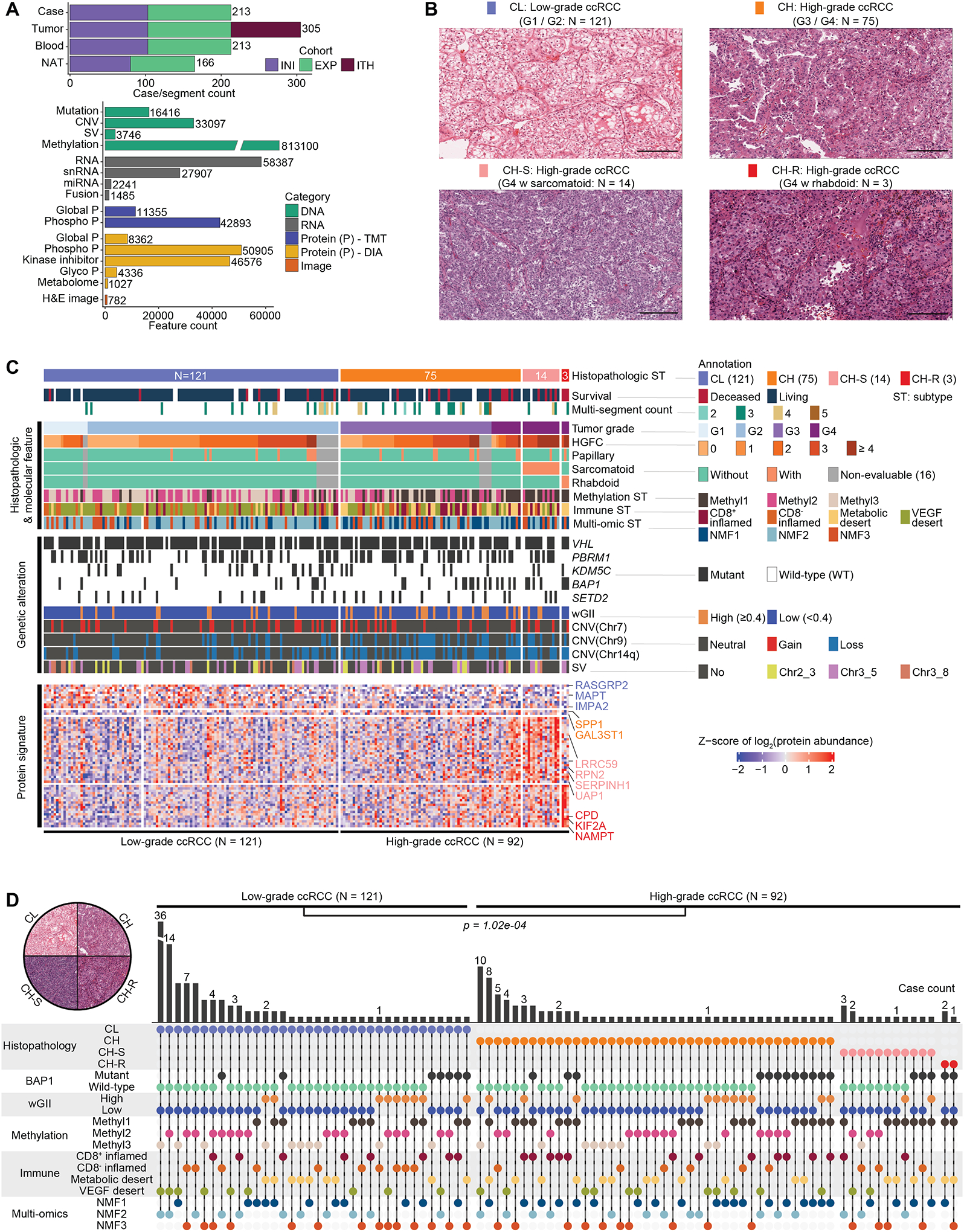
A) Sample cohorts and data type overview. Top: Numbers of cases, tumors, matched NATs, and peripheral blood samples profiled, and their cohort-wise distribution, namely INI- initial, EXP-expanded, and ITH- intratumoral heterogeneity sample cohorts. Bottom: Feature in genomics, proteomic, metabolomic, kinase inhibitor, and image data types.
B) Distribution of ccRCC cohort. Representative H&E based on nuclear grade and cytological features. Low-grade ccRCC (CL), High-grade ccRCC (CH) CH with sarcomatoid (CH-S), and CH with rhabdoid (CH-R). Scale bar = 200 microns.
C) Proteogenomic features associated with histopathologic subtypes. Key histological features, clinical parameters, genetic aberrations, and proteomic signatures are presented sequentially.
D) The distribution of histopathologic, BAP1 mutation, wGII, methylation, immune, and multi-omic subtypes among the 213 cases. Fisher’s exact test p = 1.02e-04; Pearson’s Chi-squared test p = 1.26e-04.
Molecular underpinnings of ccRCC histopathologic heterogeneity
ccRCC tissues display extensive histopathologic heterogeneity within the tumor epithelia manifesting as differences in nuclear/nucleolar features that form the basis of clinical Fuhrman grading.27,28 Heterogeneity also exists in tumor architecture, cytology, and changes in microenvironment.26 High-grade tumors are associated with higher post-surgical disease recurrence risk and may benefit from increased surveillance. Differences in cytological patterns have also recently been linked to aggressive disease.26,29 To identify underlying molecular changes associated with histopathologic heterogeneity in the first approach based on tumor grade and the presence of sarcomatoid or rhabdoid features (GSR), we classified the 213 ccRCC cases into four histopathologic subtypes and performed integrative multi-omic analysis (Figure 1B; Table S1). Low-grade ccRCC (CL) tumors (G1 / G2: N = 121) and high-grade ccRCC (CH) tumors (G3 / G4: N = 92) are fairly represented in the cohort. Among the CH group, 14 exhibited sarcomatoid features (CH-S), and 3 showed rhabdoid features (CH-R) that were linked to the distinct morphological pattern as shown in H&E (Figure 1B; Table S1). Overall, CH-R and CH-S were associated with worse prognosis compared to CL (Figures S1A–B). Differential expression (DE) analysis identified tumor markers associated with these four major histopathologic subtypes. Notably, LRRC59 and SERPINH1 were highly expressed in CH-S. KIF2A has been reported to be aberrantly expressed and correlated with patient survival,30 with significantly increased expression in CH-R (Figure 1C; Table S1). Methylation subtype Methyl1 was significantly enriched in high-grade tumors while VEGF immune desert was associated with CL (p = 2.15e-10; 1.69e-10) (Figure 1C). Although limited in number, all 3 CH-R tumors demonstrated BAP1 mutations and chr14 loss in addition to VHL mutation. Interestingly, high-grade tumors had significant enrichment of high weighted Genome Instability Index (wGII) scores (> 0.4, p = 0.0023), and enrichment for loss of chr9, and 14q (respective p = 0.00058; 0.0019; 6.36e-7) (Figure 1C). Following the initial clonal chr3p loss and acquisition of 3p driver gene mutations, a subset of ccRCC undergoes whole-genome duplication (WGD), resulting in tetraploidy. Following WGD, a significant subset of these tumors acquires several additional copy number changes (gains/losses) at an increased rate, resulting in genomic instability (GI). Distinguishing patient subsets with high GI may have clinical and therapeutic implications. GI we quantified here by wGII score correlated (r = 0.54) with ploidy in the wGII high group (Figure S1C).
In our second approach, we conducted a systematic histopathologic review of 197 tumors (with available H&E slides) for 21 morphological parameters (STAR Methods). To identify molecular changes associated with histopathologic heterogeneity, 7 high-grade morphologic features including eosinophilic/granular change, thick trabeculae, alveolar, solid, papillary/pseudopapillary patterns, and rhabdoid or sarcomatoid cytology (Figures 1B, S1D–E), were systematically assessed and quantified as High-Grade Feature Count (HGFC) per tumor. High-grade features contributing to histologic heterogeneity within tumors were specifically enriched among CH-S and CH-R tumors shown in the histopathologic annotation block of the heatmap (Figure 1C), with potential clinical implications.26,29
In addition to identifying sarcomatoid and rhabdoid feature-associated events, we ascertained the differentially expressed proteins (DEPs) of other high-grade features by comparing corresponding tumors to controls (tumors without any of the above-mentioned 7 high-grade features) (Figure S1F; Table S1). Papillary/pseudo-papillary feature noted in 10.2% of the tumors was associated with upregulation of HIGD1A and ROMO1 (Figures S1E–F). Some markers were not specific to a certain high-grade feature but generally overlapped with the high-grade-tumor DEPs (G3/4 tumors vs. G1/2 tumors). The top altered proteins included SQSTM1, GAL3ST1, and PLOD2 (Figure S1F). Protein abundances for LRRC59, RPN2, and SERPINH1 were converted into an integrative signature score that could serve as a potential prognostic indicator (Figures 1C, S1G). The group with a high signature score carries a statistically significant higher hazard ratio of 4.1 with a p-value of 0.049 adjusting by age, sarcomatoid feature status, tumor stage, and immune subtype in the Cox proportional hazards (Cox) models. Considering the status of all 7 high-grade features, an HGFC was determined (range from 0 to 7) for each tumor and evaluated for its prognostic value. Among the 197 tumors with evaluable H&E images and annotations, 68 (34.5%) presented an HGFC ≥3 that was associated with a worse prognosis (p = 0.003) (Figure S1H). By adjusting for other covariates (histopathologic subtype, age, sex, BAP1 mutation) in the Cox model, the hazard ratio of this group was 3.7 (p = 0.039) compared to (< 3) HGFC group (Figure S1H).
We evaluated associations between the omic layers and each of the seven high-grade histopathologic features mentioned above to identify the top three most informative omics layers that may be useful to describe the different phenotypes (Table S1). For example, methylation subtype, immune subtype, and BAP1 mutation showed strong associations with the sarcomatoid phenotype (Table S1). Tumors presented distinct features compared to NATs, as revealed by immune cell-type deconvolution analysis (Figures S1I–J). Abundances of macrophages and CD8+ T cells were significantly higher in tumors, while CD4+ T cells were enriched in NATs, a consistent feature across the ccRCC cohorts (Figure S1J). Among the 305 ccRCC specimens (Figure S1K), we detected four distinct immune subtypes (CD8+ inflamed with high immune infiltration; CD8− inflamed with high fibroblast; metabolic desert with high epithelial; and VEGF desert with high endothelial signature), which were largely consistent with the four previously reported immune subtypes.13 Tumors in the CD8+ inflamed group may be more likely to respond to immunotherapy than immune-desert tumors (Figure S1K; Table S1). Among the 19 patients who received adjuvant postoperative immunotherapy, four were classified as CD8+ inflamed subtype. We will follow up with their therapeutic and survival status for further investigation of this hypothesis. This immune subtyping approach provided an additional resolution to immune-inflamed and immune-desert tumors,31 identifying two distinct immune-desert subtypes, and shared some similarities with the unsupervised transcriptomic subtypes previously reported.32 By integrating CNV, gene expression, and global protein abundance in non-negative matrix factorization (NMF), we identified three major multi-omic subtypes, NMF1, NMF2, and NMF3 associated with metabolic desert, VEGF desert, and CD8− inflamed tumors, respectively (Figure S1L). These correlated with other molecular and clinical features such as wGII high and high-grade tumors that were enriched in NMF1 (Table S1). Moreover, a cluster membership score was calculated for each sample that defined the “cluster core”, a set of samples most representative of a given cluster (STAR Methods). Among the core samples in the three subtypes, overall survival differed significantly (p = 0.038) as NMF1 was associated with a worse prognosis, and compared with NMF3, carried a higher hazard ratio of 9.98 (p = 0.059) adjusting by age, sex, and tumor grade in the Cox model. In our comprehensive exploration of phenotype-genotype association, we integrated details of histopathologic heterogeneity in multi-omic analysis. Using this approach, we identified clinical and molecular features associated with high-risk disease, including Fuhrman grade, HGFC, genome instability (underexplored in the current literature), and proteomic markers. We further characterized UCHL1 protein expression as a prognostic biomarker associated with poor survival, BAP1 mutation, high wGII, and specific DNA methylation subtype. Detailed characterization of UCHL1 is presented in the DNA methylation section below. In summary, the results revealed a higher level of intertumoral heterogeneity in high-grade tumors compared with low-grade tumors (p = 1.02e-04) (Figure 1D; Table S1).
ccRCC proteogenomic and TME ITH characterization by multi-segment integrative analysis
To study the association between histopathologic features and molecular profiles for a deeper understanding of ccRCC ITH we generated multi-omic proteogenomic data on 132 tumor segments from 40 patients, and performed integrative analysis (Figure 2A; Table S2). Following the pathology review schema described in the previous section, we determined GSR and HGFC parameters for each segment from the corresponding H&E (N = 101). Briefly, each segment was scored against pre-decided 4 low and 7 high-grade features, including identifying areas of transition between phenotypes, broad features relatively prevalent in a subset of tumors (e.g., hyalinization and multi-nodularity), and some unique features in selected cases (Figure 2A; Table S2). In parallel, our ITH workflow generated bulk proteogenomic data that captured genomic and expression heterogeneities and snRNA-seq for select samples to characterize ITH at single-cell resolution (Figure 2A).
Figure 2. ccRCC proteogenomic and TME ITH characterization by multi-segment integrative analysis.
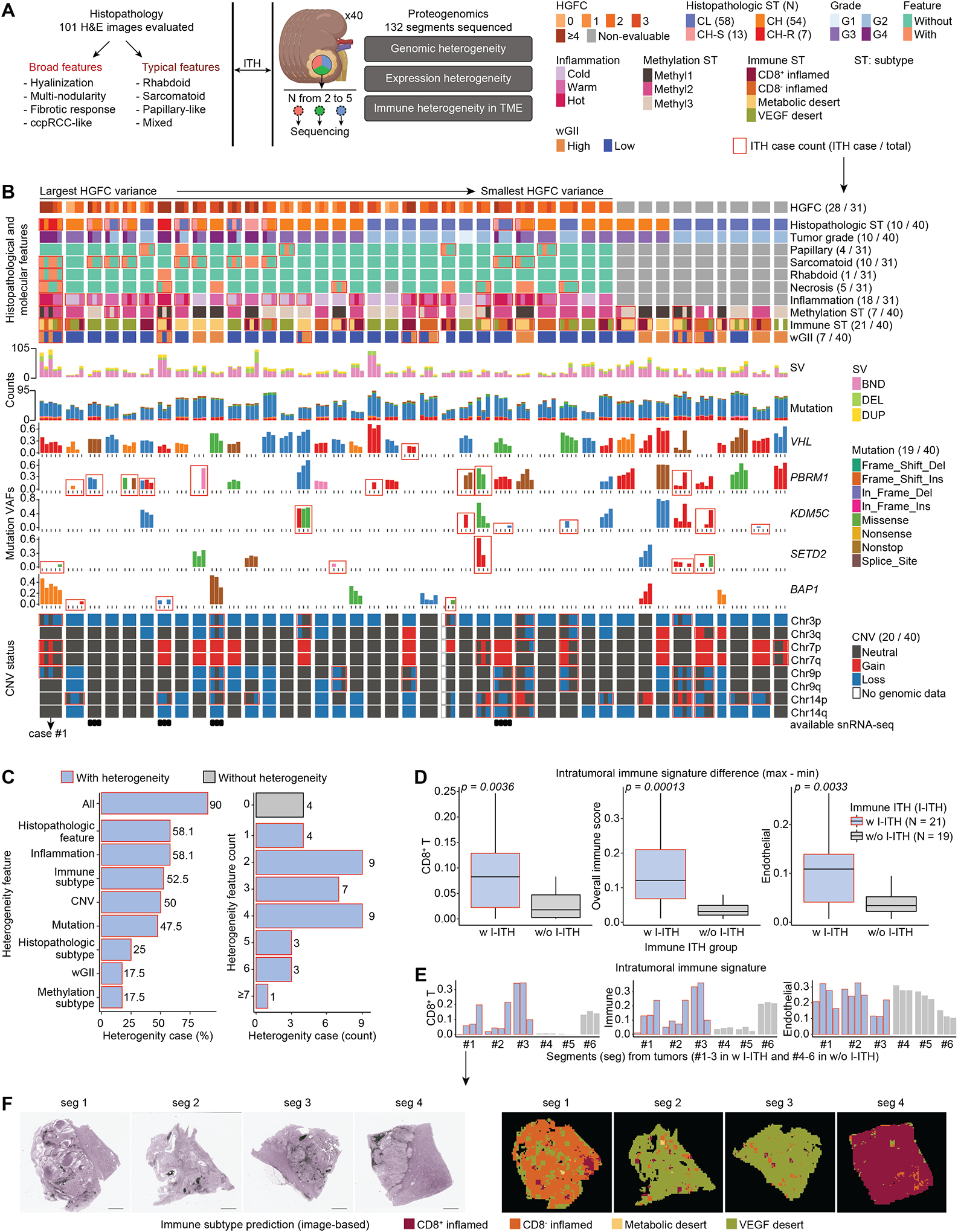
A) ITH cohort workflow showing multi-segment multi-omic molecular profiling strategy alongside extensive histopathologic assessment to enable integrative exploration of ITH.
B) Proteogenomic aberration and histological features landscape of ITH cohort samples. Multi-panel heatmap details the molecular and histological information of the 132 tumor segments from 40 patients.
C) Frequency of heterogeneity features and count in the ITH cohort.
D) Distributions of xCell CD8+ T signature, overall immune signature, and endothelial signature between the groups with (w I-ITH) and without immune heterogeneity (w/o I-ITH). Wilcoxon signed-rank test p is calculated. Boxes represent the interquartile range (IQR, e.g., median indicated by solid line in box, 0.25 and 0.75 quantiles) and whiskers represent the largest and smallest values within 1.5 × IQR range.
E) Comparison between 6 representative cases from w-ITH and w/o-ITH groups.
F) Panoptes-based multi-resolution neural network models were trained to predict immune subtypes (right) based on H&E (left).33 Scale bar = 3mm.
Proteomic impact of ccRCC somatic aberrations ITH is underexplored. Upon sorting the cases by variances of HGFC (Figure 2B) we observed heterogeneities at various levels (features enclosed with red rectangles) (Figure 2B; Table S2). ITH at histopathologic and genomic levels was more prevalent in a subset of cases (Figures 2B–C). Among the five segments profiled from case#1, two lacked sarcomatoid or rhabdoid features, placing them into a different histopathologic subtype, and while SETD2 mutation was found in only one segment VHL and BAP1 mutations were common to all. Furthermore, 2 segments from case#1 showed additional distinct patterns such as high wGII, Methyl1, metabolic desert, high structural variation (SV) counts, copy number variation (CNV) gain and loss in chr7, and chr9p (Figure 2B; Table S2). Overall, heterogeneity in at least 1 of the 8 features was noted in 90% (36/40) of the cases and more than half showed immune or histologic feature heterogeneity (Figure 2C). Among ccRCC driver genes, VHL mutations were largely clonal, while PBRM1 contained frequent subclonal events (Figure S2A). The fractions of segment-specific, shared-subclonal, and shared-clonal events varied across tumors or segments of a given tumor (Figure S2B). Additionally, CNV heterogeneities (Figure S2C) will contribute to significant variation in the proteo-transcriptomic expression milieu in the tumor epithelia as evidenced in Figures S2D–E.
Using data-driven approaches and histopathologic review, we classified the immune heterogeneity level at segment level for a given case (Figure 2B). By comparing signature distributions (e.g., CD8+ T, endothelial cell, overall immune score) between groups with (w I-ITH) and without (w/o I-ITH) intratumoral immune heterogeneity, the signature difference tended to be higher in the w I-ITH group (p < 0.05) (Figure 2D), and 6 representative tumors (3 in w I-ITH and 3 in w/o I-ITH) were presented in Figure 2E. Overall, the w I-ITH group showed a high level of immune ITH. Heterogeneity in immune presentation could affect immunotherapy response and ultimately treatment failure or inappropriate therapy choices. Panoptes-based multi-resolution neural network models trained to predict immune subtypes based on H&E33 were also provided with transcriptomic immune subtyping data. They showed high consistency of immune subtype prediction (Figure 2F; Table S2). Tiles with similar histopathologic features related to immune subtypes clustered together (Figure S2F) and we also confirmed the consistency between the histopathologic review and data-driven delineation of the immune signature (Figure S2G). Heterogeneities in wGII status or mutations in ccRCC driver genes were associated with worse prognosis with hazard ratios of 16.03 (p = 0.003) and 8.09 (p = 0.012) (Figure S2H), respectively, after adjusting by age, sex, and tumor grade in the Cox model. Our ITH analysis showed that regional histologic and proteogenomic variations including somatic driver clonality and CNVs within a patient’s tumor are common in ccRCC and might play a significant role in shaping the observed regional TME heterogeneity. We next explored ITH and how some histologic features such as CH-S and CH-R associated with aggressive diseases can be characterized by snRNA-seq.
Single-cell analysis identified ITH, sarcomatoid and rhabdoid expression signatures
We studied transcriptomic ITH by snRNA-seq in 12 tumor segments from 4 cases selected based on certain features, including rhabdoid, sarcomatoid, multi-nodularity, and hyalinization (Figures 3A–B; Table S3). Among the 104,654 nuclei sequenced, 62% formed a main tumor cluster that contained case-specific sub-clusters, expressed tumor-intrinsic markers associated with certain features and the corresponding enriched pathways at the case level (Table S3) and 38% represented TME including, T, NK, B, macrophages, fibroblasts, and endothelial cells that formed cell-type-specific clusters (Figures 3A). Collectively, these data captured the cellular ITH in both tumor and TME compartments (Figures 3A–C, S3A–B). Cell-type fractions reflected molecular and pathologic annotations, for instance, case C3N-00149 containing fibrotic features was distinct from the others and concordantly showed higher abundance of fibroblasts (Figures 3A–B, S3A). Comparing the four segments from C3N-00148, revealed extensive ITH in TME compartment as CD8+ T cells were significantly enriched in segment 4 (seg 4), the only region classified as CD8+ inflamed with a higher immune infiltration post tumor content adjustment (Figures 3B–C; Table S3). −305.7207 pt. 392.3809 pt
Figure 3. Single-nuclei RNA-seq atlas identifies distinct intra-tumor epithelial populations.
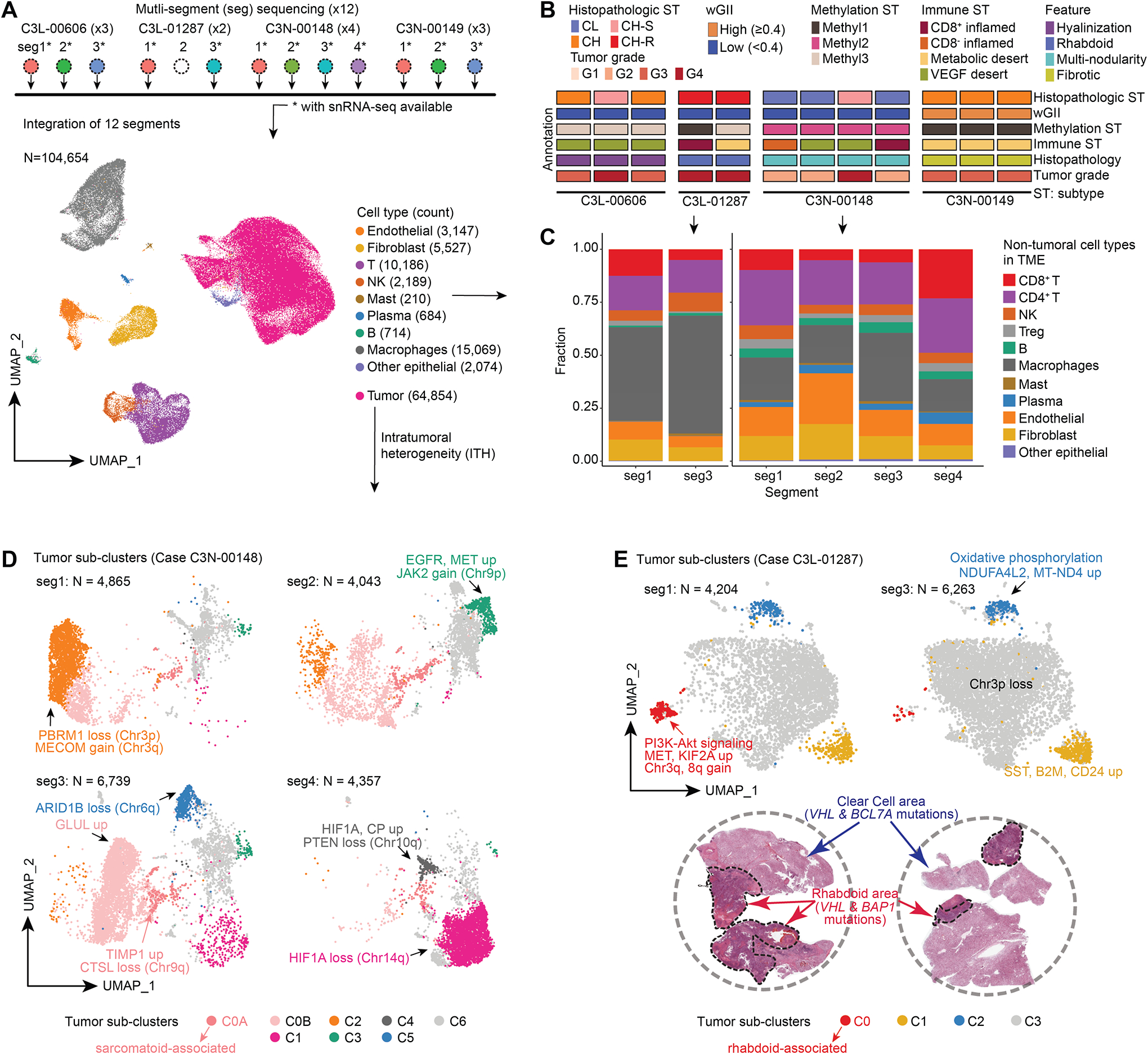
A) snRNA-seq analysis workflow schematic (Top). snRNA-seq cell atlas generated from 12 segments obtained from 4 cases (Bottom). The UMAP displays 26 cell clusters that were subsequently annotated as 10 different cell types.
B) Schematic tracks present the heterogeneity observed in the 12 segments at the histological and molecular characterizations.
C) Frequency and composition of non-tumoral cell types found in the TME in C3L-01287 and C3N-00148 with immune ITH.
D) UMAP shows the tumor sub-clusters and the corresponding ITH found in 4 segments obtained from C3N-00148 with critical molecular feature annotations indicated.
E) UMAP representation of tumor subclusters ITH in the 2 segments of C3L-01287 (Top). H&E reveals the presence of classic clear cell and mutually exclusive rhabdoid regions in this tumor (bottom). WES using dissected regions showed VHL mutation in clear cell region, and VHL and BAP1 mutations in rhabdoid region.
Sarcomatoid differentiation, a poor-prognosis feature, was variably distributed across segments in C3N-00148. Trajectory analysis (Figure S3C) of C3N-00148 revealed enrichment differences of segments in distinct branches and predicted a later evolution of tumor subpopulations in seg3 labeled as C0 with high expression of GLUL as a high-grade-tumor DEP (Figure S1F), chr9q loss (associated with sarcomatoid changes in RCC),34 and enriched Hippo signaling pathway corresponding to the trajectory branches (Figures 3D, S3D–E). Furthermore, we captured two subpopulations (e.g., C0A, C0B) in C0 as C0A, showed unique expression signatures (Figures 3D, Table S3). In agreement with the histopathologic review, the sarcomatoid and fibroblastic proliferations were mainly observed in seg3 (25–30%), while the others had little or focal fibroblastic proliferation mainly in high-grade areas (< 10%) (Figures S3F).
Rhabdoid phenotype, another poor prognosis ccRCC histology noted in C3N-01287 was juxtaposed next to clear cell area, and snRNA-seq captured both tumor features as distinct cell clusters (Figure 3E). We further annotated these tumor subclusters, with inferred CNV from snRNA-seq and WES-based CNV obtained from microdissected rhabdoid and clear cell regions (Figures 3E, S3G–H). Rhabdoid cell cluster (C0)/region contained BAP1 mutation, chr3q and 8q copy gains, and enrichment of PI3K-AKT and Rho GTPase signaling. Clear cell cluster/region contained BCL7A mutation and chr2 and 5 gains, while VHL mutation was common to both regions (Figures 3E, S3G–H).
We used such representative genomic alterations and marker expressions to render additional evidence for our feature-associated subcluster annotation. C0A in C3N-00148 showed significantly higher expressions in TIMP1, C1R, and TGFBI (Figures 4A; Table S3), and interestingly TIMPs overexpression in sarcomatoid RCCs has been reported.35 To further strengthen and validate our observations we sequenced two additional sarcomatoid cases for snRNA-seq integration (Figure 4B). ClusterC0A which highly expressed TIMP1, C1R, and TGFBI both at integration and case levels was found in all sarcomatoid cases (Figure 4C; Table S3). We validated TGFBI expression in two representative cases by immunohistochemistry (IHC) where we saw signal (high and diffuse staining) only in the sarcomatoid areas and not in conventional nested clear cell regions (Figure 4D). As for the subcluster with rhabdoid features in C3N-01287, it presented higher expression profiles of KIF2A, NAMPT, and GALNT2 (Figure 4E; Table S3) and was confirmed by another independent case with rhabdoid features (Figures 4F–G; Table S3). We independently validated KIF2A expression by IHC and saw strong positive staining intensity specifically in the rhabdoid area while the nested clear cell region was negative (Figure 4H). These markers also showed consistent patterns in bulk RNA expression and global protein abundance, such as high KIF2A in rhabdoid cases compared with control low-grade tumors (Figure S3I; Table S3).
Figure 4. snRNA-seq atlas further refines sarcomatoid and rhabdoid histology-associated gene expression signatures.
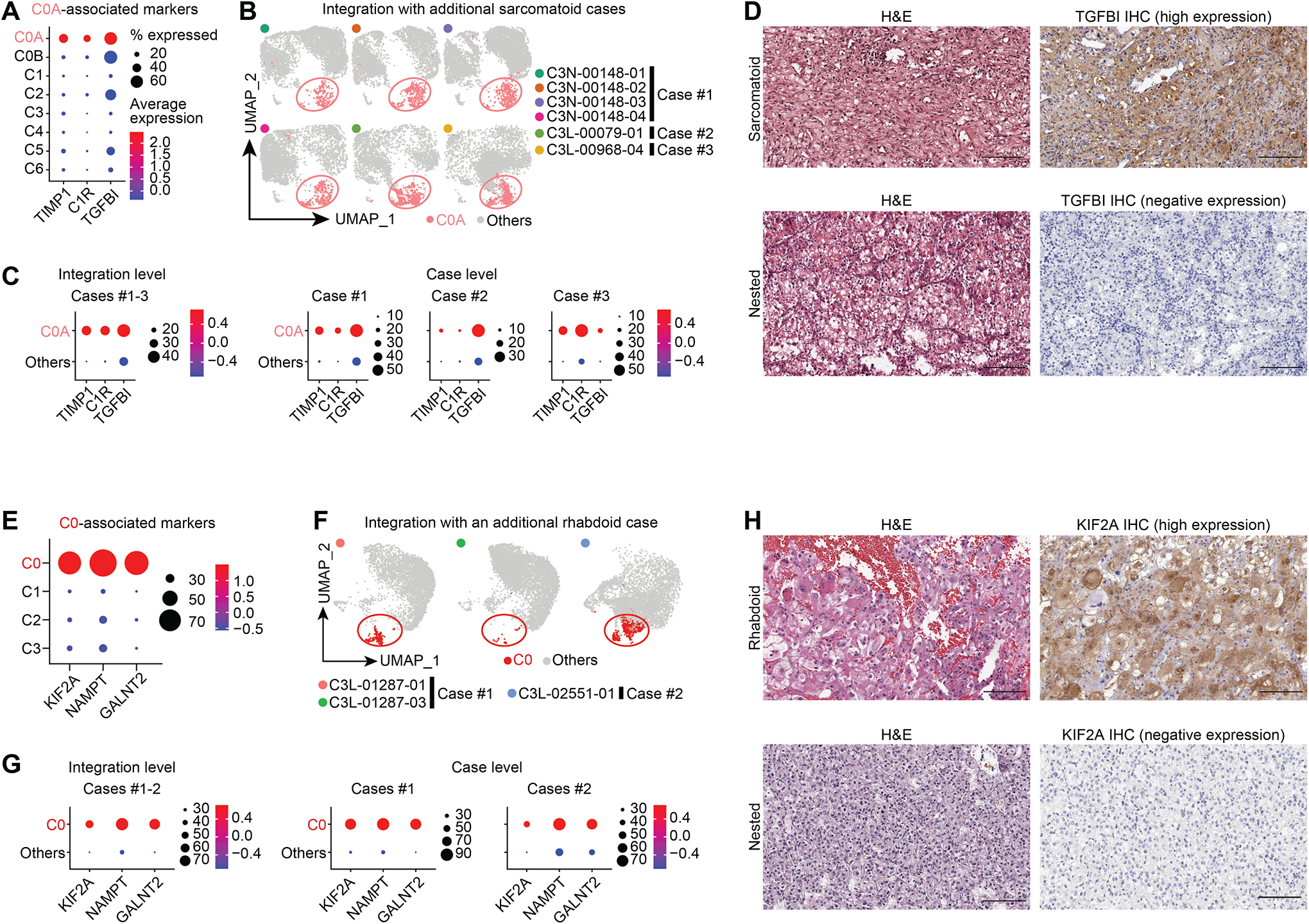
A) Expression profiles of the top markers associated with the C0A tumor cluster in C3N-00148, where bubble diameter represents fraction of cells with expression in a given cluster. Color blue to red- expression down to up.
B) UMAPs show the integration of three sarcomatoid cases.
C) Bubble plots show expression of top markers associated with C0A tumor cluster in snRNA-seq at integration and individual levels.
D) Corresponding high and low expression of TGFBI protein in two representative cases with strong staining positivity noted in sarcomatoid area and its absence in nested clear cell area. Scale bar = 200 microns.
E) Expression profiles of the top markers associated with C0.
F) UMAPs show the integration two rhabdoid cases.
G) Expression profiles of the top markers associated with C0 tumor cluster in snRNA-seq at integration and individual levels.
H) Corresponding high and low expression of KIF2A IHC with strong staining intensity noted in the rhabdoid area with no staining in the nested clear cell area. Scale bar = 200 microns.
DNA hypermethylated Methyl1 subtype is associated with BAP1 mutations and various other features linked to poor survival
Dysregulation of the epigenetic DNA methylation marks is considered an early event in carcinogenesis.36–39 Previous pan-RCC genomic studies have noted a strong association between increased DNA methylation and worse prognosis in ccRCC.17 Identification of specific prognostic markers to distinguish this patient subset remains an unmet clinical need and can now be explored with our extended cohort. Among the 8,000 most variable CpG sites (probes) that distinguished tumors from NATs, we identified the signature probes and related genes associated with histopathologic subtypes (Table S4; STAR Methods). For instance, we noticed 7 probes in the RNF39 CpG island were hypermethylated in CH-S (FDR < 0.05 & beta value difference > 0.1 & in CpG) as a part of an altered methylation profile (Table S4). Three methylation subtypes (Methyl1–3) were detected in both CPTAC ccRCC and TCGA KIRC cohorts by applying consensus clustering on the 8000 probes (Figures 5A; Table S4). Methyl1 was significantly associated with samples containing higher tumor grades, higher stemness score, and worse prognosis, as well as metabolic desert followed by CD8+ inflamed, and molecular features such as high ploidy, high wGII, losses of chr9,14q, and BAP1 mutations (Figures 5A–B, S4A–B). Panoptes-based models were trained to predict methylation subtypes from H&E (Figure S4C). The best-performing model achieved a macro-averaged multi-class per-slide area under the receiver-operating characteristic (ROC) curve of 0.836 (95% CI: 0.830–0.841) on the test set (Table S4). We further connected the prediction with histopathologic annotations. For example, in C3N-00148, classified into Methyl2, we observed heterogeneous features where the immuneinfiltrate, fibroblastic-rich area was called Methyl3, while the majority of conventional ccRCC area with marked trabecular change was labeled Methyl2 (Figure S4C).
Figure 5. DNA hypermethylated Methyl1 subtype is associated with BAP1 mutations and various other features linked to poor survival.
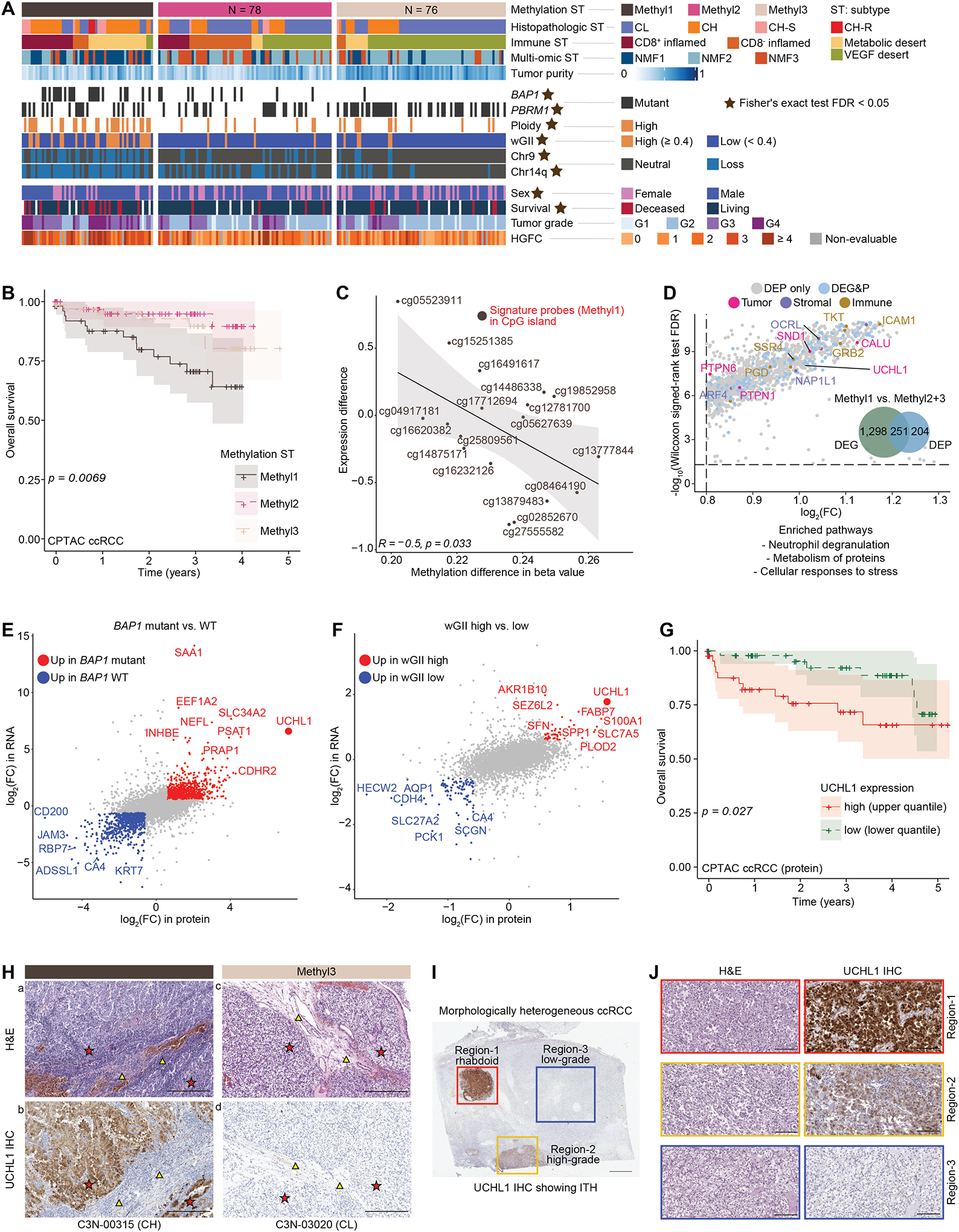
A) Patient classification by three DNA methylation subtypes (heatmap). Annotation tracks below indicate molecular, genetic, and clinical/histological categories. Star sign indicates the statistically significant association (Fisher’s exact test FDR < 0.05).
B) Kaplan Meier plot indicates the association between overall survival and the three methylation subtypes in CPTAC ccRCC cohort. Log-rank test p is 0.0069.
C) Correlation between methylation difference (beta value) and RNA expression difference of the top Methyl1 signature probes (genes).
D) DEPs associated with Methyl1 (vs. Methyl2 + Methyl3) and enriched pathways based on DEPs upregulated in Methyl1.
E) DEGs and DEPs associated with BAP1 mutation status. UCHL1 was amongst the markers up in BAP1 mutants.
F) DEGs and DEPs associated with wGII category. UCHL1 was amongst the markers up in high wGII high group.
G) Kaplan Meier plot indicates the association between overall survival and UCHL1 protein abundance in CPTAC ccRCC cohort. Log-rank test p is 0.027.
H) Matched topographical comparison (asterisks) of uniform high expression of UCHL1 in a Methyl1 ccRCC (a, b), and absence of UCHL1 expression in a Methyl3 ccRCC (c, d). TME (triangles) in corresponding H&E and UCHL1 staining to match the topography. Scale bar = 300 microns.
I) Characterization of UCHL1 in a morphologically heterogeneous ccRCC. Scale bar = 3mm.
J) Regions 1 and 2 correspond to rhabdoid (red panel) and high-grade (yellow panel) nodules demonstrating strong and moderate UCHL1 expression, respectively. Region 3, the low-garde area (blue panel) shows absence of UCHL1. Scale bar = 200 microns.
As Methyl1 was significantly associated with worse disease prognosis (Figure 5B), we captured the differentially methylated (DM) probes in both CPTAC ccRCC and TCGA KIRC cohorts14,17 and prioritized them as signature probes if (1) common DM probes were significant in both cohorts; (2) beta value differences were > 0.2; (3) probes were located in CpG island followed by shelf and shore regions; (4) corresponding genes identified as tumor-intrinsic were more highly expressed in tumor/epithelial cells than in immune or stromal cells. In total, we found 235 common significant DM probes corresponding to 198 genes showing an overall negative correlation (R = −0.5, p = 0.033) with cognate gene expressions (Figure 5C; Table S4). The top Methyl1 signature probes, located in CpG islands, include cg04917181 (TSPYL5), cg05523911 (TCHH), cg14875171 (NRXN1), cg16232126 (SLC5A7), and cg25809561 (MYO1D) (Figure 5C). To learn the characteristics of each methylation subtype, we conducted the DE analysis on both RNA level and protein abundance. Methyl1 showed significant upregulation of 251 candidates as both DEGs and DEPs, including UCHL1. While 204 markers significantly up only as DEPs contributed to pathways including cellular responses to stress (Figure 5D; Table S4). Methyl3, being enriched with VEGF desert, PBRM1 somatic mutations, and high tumor purity, carried 60 markers as both DEGs and DEPs and 116 additional DEPs contributing to the pathways including glycolysis/gluconeogenesis (Table S4).
UCHL1, in addition to being enriched in Methyl1, was significantly associated with BAP1 mutants and the wGII-high category based on RNA expression, protein abundance, quantified UCHL1 immunohistochemistry (IHC) score, and IHC staining (Figures 5E–F, S4D; Table S4). UCHL1, a deubiquitinase, could serve as a prognostic marker of ccRCC whose high expression is associated with worse prognosis in both the CPTAC ccRCC and TCGA KIRC cohorts (Figures 5G, S4E). We evaluated 32 representative cases by a panel of IHC markers (UCHL1, BAP1, and CA9) to validate UCHL1 associations with BAP1 mutated and Methyl1 subgroups. IHC-based UCHL1 proteome abundance assessment showed a high correlation between quantified UCHL1 protein abundance and UCHL1 IHC score, where BAP1 mutants frequently displayed higher levels of UCHL1 (Figures S4F–G). BAP1 IHC is currently used in the clinic as a diagnostic marker to evaluate BAP1 protein loss. In this context, all 14 BAP1 deleterious mutants we tested showed loss of BAP1 staining, and 12 of these cases were positive for UCHL1. Among the 7 BAP1 missense mutants we examined, only 3 were negative for BAP1, and 1 positive for UCHL1. Contrariwise among 4 BAP1 missense mutants with BAP1 positivity, only 1 was UCHL1 positive (Figure S4D). ccRCC clinical marker CA9 was positive in all the ccRCC cases evaluated (Figure S4G). When these data were analyzed for methylation subtypes, 68.7% (11/16) of Methyl1 showed UCHL1 positivity and was significantly different from the Methyl3 group (Table S4). In addition, UCHL1 staining of a matched RCC primary (renal mass) and metastatic RCC (ovarian tubular mass) tumor from a patient with pathogenic germline BAP1 mutation40 also showed strong UCHL1 positivity (Figure S4H). Thus, UCHL1 positivity appears to be associated with BAP1 mutation, wGII high, worse survival, and Methyl1 (Figures 5E–H, S4D–H) in a collective manner, making it an important candidate prognostic marker that warrants additional validation in independent cohorts. Examination of an independent RCC primary tumor patient cohort (n = 16) who subsequently developed metastatic RCC, indicated that 68% (11/16 cases) of the primary tumors showed strong UCHL1 positivity. This was a dramatic increase compared to the 10–15% cases with UCHL1 expression noted in unenriched RCC primary tumor cohorts (CPTAC and TCGA). We also observed several different histopathologic features in these tumors (Figure S4I). Finally, we characterized UCHL1 staining topographically in one of the 16 independent clinically aggressive cases which showed morphological heterogeneity (Figure 5I), where the rhabdoid nodule and high-grade tumor showed strong and moderate UCHL1 staining, respectively, while the staining was negative in the low-grade clear cell area (Figure 5J). Hence, we were able to demonstrate alignment of UCHL1 expression with ITH. Panoptes-based models were trained to classify BAP1 mutated and WT samples. Overall, the cluster of BAP1 mutated tiles showed higher-grade aggressive-looking phenotypes, while the BAP1 WT tiles contained predominantly low-grade tumor components, such as acinar and tubular with areas of hemorrhage and hemosiderin-laden macrophages and hyalinization (Table S4). Encouraged by the availability of UCHL1 small molecule inhibitor (CAS 668467–91-2, also known as LDN-57444) and preliminary studies on its targetability from triple negative breast and neuroendocrine lung cancer models,41 we performed cell viability assays in RCC cell line models. Renal cancer cell lines Caki-1 and 786-O showed dose-dependent inhibition of cell viability with CAS 668467–91-2, while the normal kidney HK-2 cell line was resistant to the treatment (Figure S4J; STAR Methods). CAS 668467–91-2 treatment in 786-O renal cancer cells resulted in altered morphology being elongated and stressed (Figure S4K). Western blot analysis in 786-O cells demonstrated that UCHL1 inhibition suppressed activation of the Akt signaling pathway in a dose-dependent manner (Figure S4L).
Key phosphorylation signaling pathways and kinase-substrate interactions in ccRCC
To identify key phosphorylation signaling pathways in ccRCC, we investigated altered phosphosignaling networks based on the association of kinase-substrate (K-S) pairs. Our phosphoproteomic datasets contained 110 EXP cohort cases by DIA-based and 103 INI cohort cases by TMT-based methods13 (Figures 6A, S5A; Table S5). Approximately 80% of the K-S pairs with the highest phospho-substrate abundance (tumors vs NATs) identified, including signaling networks involving EGFR, MEK, ERK, and WEE1, were from both DIA or TMT-based analysis thereby providing good cross-verification. Furthermore, phosphorylations of PRKCZ and PARD3 were positively associated, and both proteins are involved in the Rap1 signaling pathway. Likewise, an association between phosphorylations of RPS6KA3 and RPS6 proteins was intriguing as they belong to the mTOR signaling pathway.
Figure 6. Identification of key phospho signaling pathways, kinase-substrate (K-S) interactions in ccRCC tissues, and integration of ex-vivo kinase drug inhibition data from RCC cell lines.

A) Top 50 signaling pathways of K-S pairs with the highest phospho-substrate abundance (tumors vs. NATs). The labeled cancer drugs are not exhaustive and they are either under investigation or FDA-approved.
B) Phosphoproteomic subtypes (P1–4) are overlaid with 11 variables from molecular, genetic, and histologic features shown as annotation tracks immediately below.
C) Pathways and kinase activities are inferred from phosphoproteomic data for each phospho subtype. NES: normalized enrichment score from PTM-SEA.
D) Schematic representation summarizing the kinase inhibition experiment conducted in five RCC cell lines targeting the kinases identified in our initial cohort. The phosphorylation level changes in the downstream substrates of targeted kinases indicate the response to the corresponding treatments relative to the control. Each cell line with one control and treated with five different kinase inhibitors.
To examine ccRCC inter-tumor phosphoproteomic heterogeneity, we used phosphorylation events with coefficient of variation (CV) in > 25% quartile from 110 tumors to construct unbiased phosphoproteomic clustering whereby four major ccRCC phosphoproteomic subtypes emerged (P1 to P4) (Figures 6B, S5B–C; Table S5). Among these subtypes, tumors in P1 had higher grades and stages and were enriched in BAP1 mutation, Methyl1, CD8+ inflamed, and metabolic desert. Groups P2 and P3 had lower grade tumors, with a higher percentage classified as Methyl2 and VEGF desert, respectively, while P4 showed a more mixed profile. PTM-SEA42 analysis of the tumor phosphoproteomics based on the changes in phosphosite abundance revealed distinct signatures for the phospho subtypes (Figure 6C; Table S5). MAPK14 and MAPKAPK2 were significantly enriched in P1. MAPK14 and downstream pathways are activated in response to various stresses and inflammation; moreover, activation of MAPKAPK2 by MAPK14 associates with biological processes, such as apoptosis and cell cycle; MAPK14 and MAPKAPK2 potentially play a role in cancer cell survival.43,44 P2 tumors showed phosphosite-driven activation of the leptin pathway, and leptin is associated with ccRCC progression and poor clinical outcome.45 P3 subtype was associated with the EGFR pathway and other kinases such as ROCK1, MAPK3 (VEGF/angiogenesis signaling), and MAPK9, GSK3B (focal adhesion). ROCK could be a potential target for P3 tumors since P3 was enriched with VEGF-desert samples, and ROCK inhibitors can reduce VEGF-induced angiogenesis.46,47 Both P1 and P4 showed enrichment in the TIE2 pathway, whose activity is associated with the activation of MAPK14, ERK1/2, and PI3K/AKT pathways.48,49
Our previous work13 used case-matched ccRCC tumors and NATs to examine the differentially-expressed K-S pairs. We found elevated levels in the majority of ccRCC tumors for K-S pairs, including cell cycle regulator WEE1 and ERK signaling. Similar results were found in the EXP cohort. The current study investigated the functional impacts of select kinases using inhibitors focusing on a panel of six K-S pairs prioritized previously (Figure 6D; Table S5). We characterized the phosphoproteome of 5 RCC cell lines treated with inhibitors targeting MAPK, EGFR, mTOR signaling, and WEE1 via DIA-MS. Variations in the inhibitory effects among the cell lines were observed based on the phosphorylation level of the downstream targeted substrates. Among the five drugs, inhibitors of WEE1 (AZD-1775), dual mTOR complex (TAK-228), and MEK (Trametinib) showed better responses. AZD-1775 reduced CDK1 phosphorylation levels in all five cell lines, with the highest reduction observed in CAKI-2 relative to the others. TAK-228 reduced phosphorylation of the mTOR complex component, AKT1S1, and its downstream phospho-substrate target, EIF4EBP1, while the MEK inhibitor reduced phosphorylation of both MAPK1 and MAPK3. In contrast, Everolimus (mTORC1 inhibitor) and gefitinib (EGFR inhibitor) showed minimal impact on their signaling-related phosphorylation events.
In order to identify BAP1 mutation-related events we examined phospho-substrates levels in BAP1 mutant 769-P versus the remaining cell lines and compared our observations with results from BAP1 mutant versus wildtype tumors in the EXP cohort (Figure S5D; Table S5). The identified events were related to biological functions, such as cell cycle (e.g., ANKRD17, SMC4) and DNA-binding (e.g., KLF3), that showed distinct expression profiles in the clinical cohort and drug-treated cell lines (Figure S5D). Intriguingly, ANKRD17 is a known interactor of BAP1 protein.50 ROC analysis of ANKRD17-S2400, KLF3-S92, and MAP1B-S1785 demonstrated the ability to distinguish BAP1 mutation and wild-type, with the area under the curve (AUC) of 0.80, 0.81, and 0.77, respectively. AUC was further improved to 0.87 when combining the three phospho-substrates (Figure S5E). The phosphoproteomic analysis identified multiple signaling pathways activated in tumors and revealed four major phosphoproteomic groups in ccRCC linked to unique K-S pairs. A subsequent kinase inhibition study and ROC analysis suggested additional targets, especially targets involving MAPK signaling. Among the drugs tested, MEK inhibitor showed superior performance in reducing phosphorylation of downstream phospho-substrates and inducing death at a low IC50. Taken together, the current results indicated a possibility of expanding treatment options beyond the current FDA-approved therapies targeting VEGF and mTOR.51
Alteration of protein glycosylation specific to ccRCC and high-grade ccRCC
Aberrant glycosylation of cell surface proteins observed in cancers can affect various biological functions, e.g., cell signaling.52,53 Glycoproteomic DE analysis of ccRCC tumors versus NATs identified 51 upregulated and 131 downregulated intact glycopeptides (IGPs) (Figure 7A; Table S6). Among them, four IGPs from four glycoproteins (FN1, FBLN5, BGN, and TNC) demonstrated an ability to differentiate tumor and non-tumor tissues with the AUCs ranging from 0.75 to 0.86 (Figure 7B). AUC increased to 0.89 when combining the four glycopeptides into a panel. Additionally, ECM-receptor interaction, focal adhesion, and PI3K-Akt signaling pathways were enriched from the glycoproteins of positively-regulated IGPs. On the other hand, renin-angiotensin system, glycosaminoglycan degradation, and lysosome pathways were enriched from negatively-regulated IGPs (Figure S6A).
Figure 7. Alteration of protein glycosylation specific to ccRCC and high-grade ccRCC.
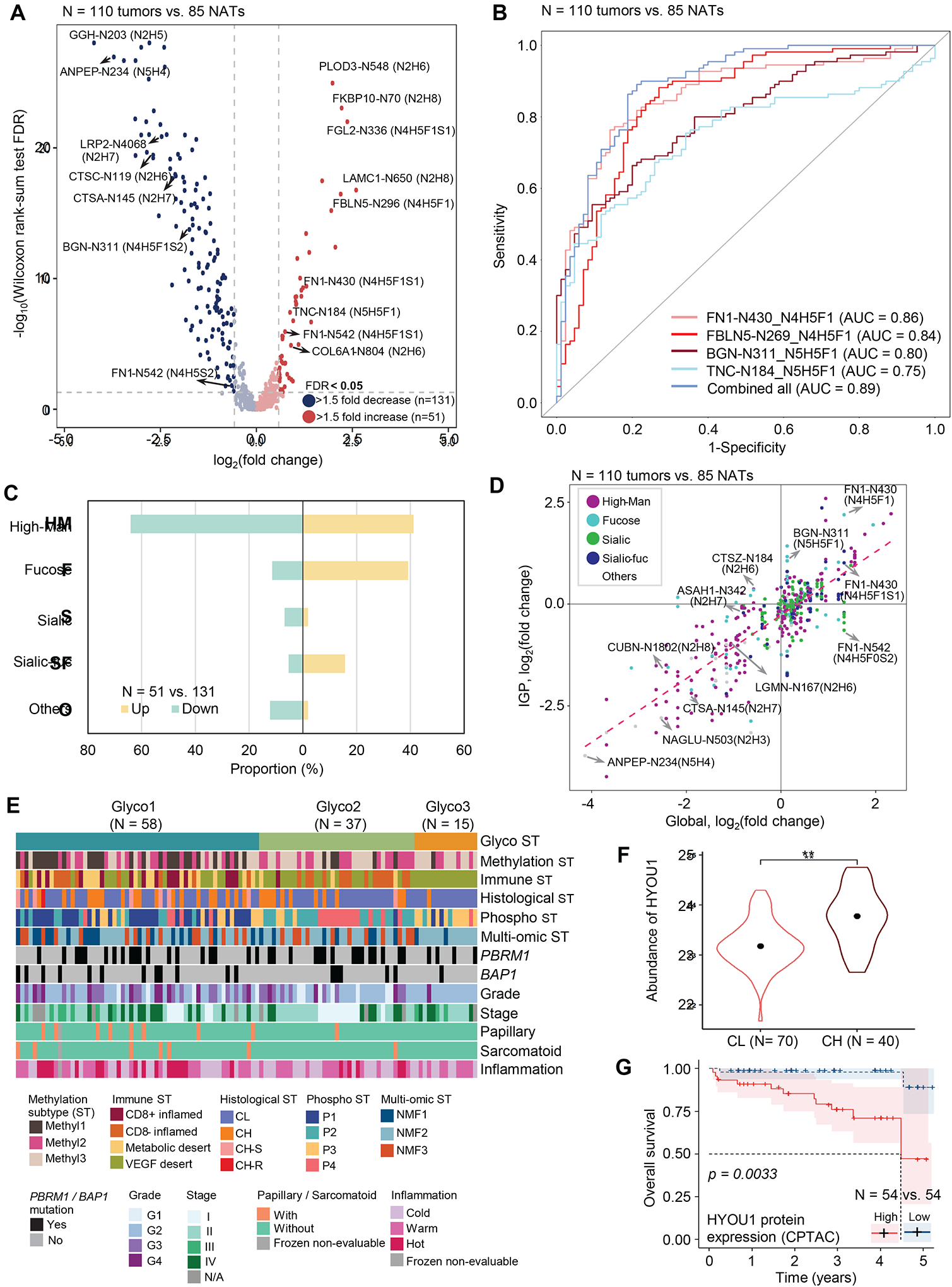
A) Volcano plot shows Intact glycopeptides (IGPs) differentially expressed between tumors and NATs.
B) Performance of glyco-signatures individually and as a multi-signature panel for differentiating tumor and non-tumor tissues.
C) Glycan type distribution of differentially expressed IGPs (tumors vs. NATs).
D) Glycosylation changes (y-axis) compared to global protein expression (x-axis) changes in tumors vs NATs.
E) Glycoproteomic subtypes (Glyco1–3) are overlaid with 12 variables represented by individual tracks immediately below.
F) Violin plot shows HYOU1 protein abundance between CL and CH tumors. ** indicates that Wilcoxon rank-sum test FDR < 0.01. Dots in the violin plots correspond to median abundance. The violin plot outlines demonstrate the kernel probability.
G) Kaplan Meier plot compares HYOU1 protein expression between High (upper quartile) and Low (lower quartile) groups in the CPTAC cohort. Log-rank test p is 0.0033.
According to the monosaccharide composition of the identified glycopeptides, five glycan types were investigated: glycans containing only oligomannose (High-Man), sialic acid containing glycans (Sialic), glycans containing sialic acid and fucose (Sialic-fuc), fucosylated glycans only (Fucose), and other glycans (Others). Fucose or Sialic-fuc glycans were enriched for the upregulated glycopeptides, whereas most of the downregulated glycopeptides were High-Man, Sialic, or other glycans (Figure 7C; Table S6). The glycopeptide abundance was regulated in both protein level and glycosylation by different glycans (Figure 7D). The alteration of IGPs was positively correlated to the cognate global protein expression. However, heterogeneities were noted in IGP abundances from the same proteins due to different glycan types.
Glycans that modify glycoproteins are regulated by glycan biosynthesis enzymes. We found upregulation of glycosylation enzymes, including MAN1C1, MGAT1, and ST6GAL1, in tumors relative to NATs at protein level (Figure S6B; Table S6). MAN1C1 and MGAT1 regulate the synthesis of complex glycans, while ST6GAL1 is responsible for transferring sialic acid from CMP-sialic acid to galactose-containing acceptor substrates. The altered glycosylation enzymes could be used as potential therapeutic targets, which would require further investigation.
While exploring intertumoral heterogeneity in glycoproteomics data, we observed three major ccRCC glycoproteomic subtypes (Glyco1–3, Figure 7E) with three intact glycopeptide clusters (IPC 1–3, Figure S6C). Among the three glycoproteomic subtypes, tumors in Glyco1 were associated with higher grade, BAP1 mutation, Methyl1 subtype, CD8+ inflamed, and IPC 1 compared to the other glyco subtypes (Figures 7E, S6D). The significantly upregulated IGPs in Glyco1 were mostly occupied by High-Man and Fucose type glycans (Figure S6E), and there were glycopeptides from glycoproteins (e.g., HYOU1) that influence metastasis of various cancers.54 Comparison between CL and CH tumors suggested that HYOU1 elevation in the latter hence could serve as a prognostic marker with an AUC of 0.76 (Figures 7F, S6F; Table S6). Immunohistochemistry evaluation of HYOU1 protein expression showed higher HYOU1 expression in CH tumors where the strongest signal came from immune cells (Figure S6G). Moreover, we examined the association between HYOU1 expression and survival using CPTAC ccRCC and TCGA KIRC cohorts. HYOU1 abundance could serve as a potential prognostic indicator only at the protein level in the CPTAC cohort but not at the RNA level in both cohorts (Figures 7G, S6H–I). HYOU1 protein abundance also showed a significant association with high-grade (G3/G4) tumors (p = 1.81e-7, Figure S6J). Furthermore, Glyco2 had an association mainly with IPC 2 (Figure S6D). The significantly upregulated IGPs in Glyco2 were occupied by sialylated glycans (Figure S6E). Since Glyco2 and 3 were dominated by low-grade and immune-desert tumors, targeted therapy against sialylated glycans could be a potential alternative approach for Glyco2 and 3 subtypes.
Metabolic signatures of high-grade ccRCC and low-grade ccRCC
Reprogrammed tumor metabolism is a hallmark of cancers, manifested through alterations in metabolite abundances and composition, and kidney cancer is strongly associated with metabolomic alterations.18,55 Herein, we quantified 183 metabolites across various metabolic pathways with high confidence from 50 ccRCCs and 7 NATs (STAR Methods; Table S7). PCA analysis found definitive separation between tumors and NATs, and distribution among the 50 tumors by histopathologic subtypes (Figure 8A; Table S7). We detected 55 metabolites with significantly higher tumor-specific abundance (FC > 2 and FDR < 0.05) that contributed to arginine biosynthesis, alanine, aspartate/glutamate metabolism, pyrimidine metabolism, and purine metabolism, while 35 were reduced in tumors compared to NATs (Figure S7A).
Figure 8. Dysregulated metabolism in high-grade and low-grade ccRCC.
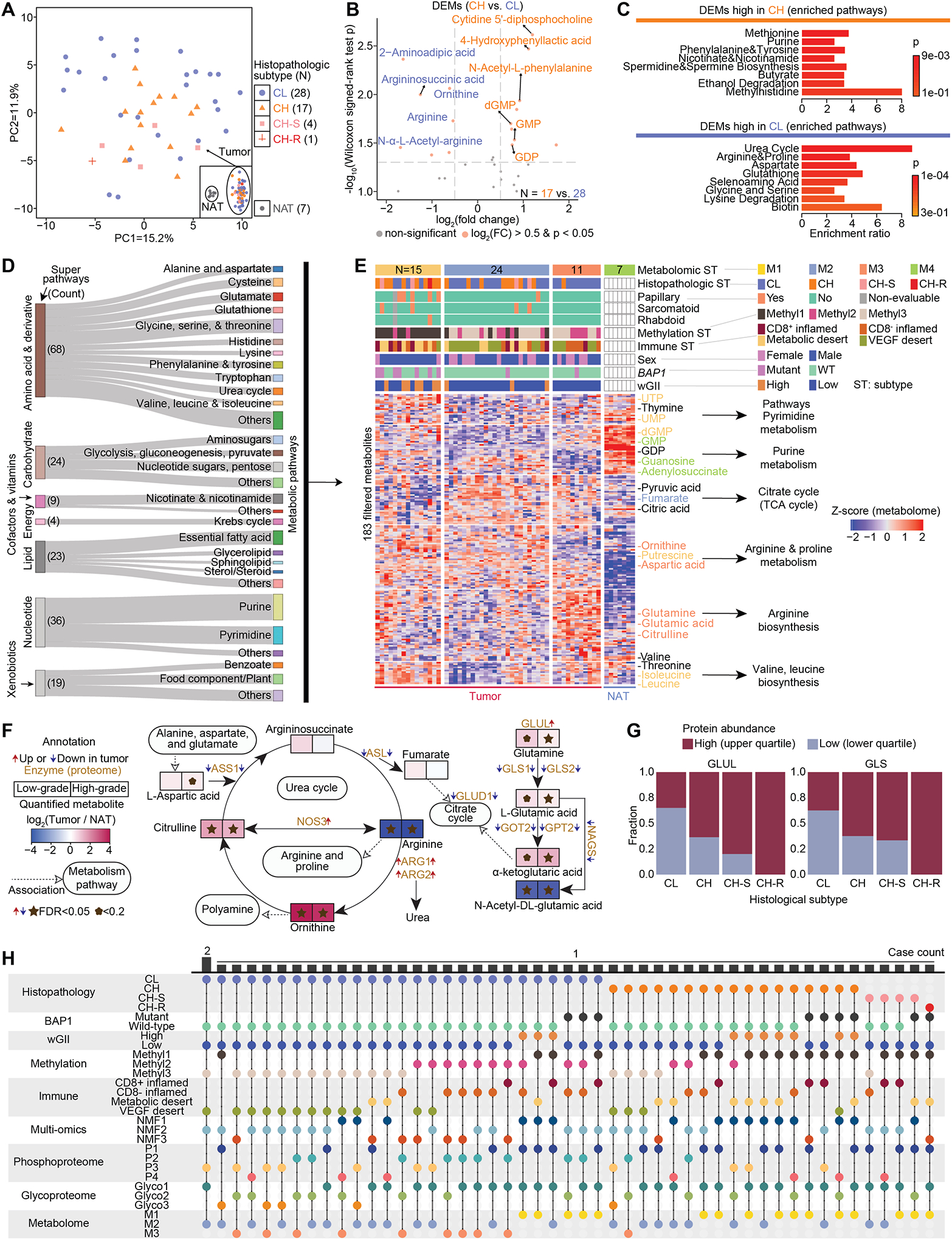
A) PCA plot shows the distribution of 50 tumors by metabolome characterization. The separation with 7 NATs is shown in the inset box.
B) DEMs between high-grade tumors and low-grade tumors. The significantly upregulated DEMs are in orange if their Wilcoxon signed-rank test p < 0.05 and absolute fold change > 2.
C) Enriched metabolic pathways corresponding to CH and CL, respectively. Hypergeometric test p is calculated.
D) Sankey diagram visualizes the distribution of metabolic pathways and super pathways for the 183 metabolites used for metabolomic subtyping.
E) Heatmap shows the four metabolomic subtypes that were identified among the 50 tumors and 7 NATs. The signature metabolites were annotated next to the heatmap and colored by the metabolomic subtype if they are significantly higher in one subtype.
F) Network plot of Arginine biosynthesis, Urea cycle, and Citrate cycle demonstrates the connection of metabolites and enzymes, and the expression fold change of metabolites and direction of enzymes in tumors compared with NATs. Wilcoxon signed-rank test FDR is calculated.
G) Among the 213 cases, the fractions of high expression of GLUL and GLS are higher in high-grade tumors compared with those of low-grade tumors as indicated by these stacked bar plots.
H) The distribution of 50 tumors with multi-level profiling of histological, BAP1 mutation, wGII, methylation, immune, multi-omic, phospho, glyco, and metabolome subtypes.
Further, CH and CL tumors differed dramatically in their metabolic profiles (Figures 8A–C; Table S7). Arginine, used in protein biosynthesis,56 was decreased in tumors compared with NATs, but significantly higher in CL compared to CH tumors (Figure 8B). The top 10 enriched pathways displayed distinct patterns between CH and CL tumors (Figure 8C). We used CL as a control group to capture the metabolic signature associated with CH-S (N = 4 with the sarcomatoid feature) (Figures S7B–C; Table S7). Differentially expressed metabolites (DEMs) such as GMP, N-acetyl-L-phenylalanine, and dGMP are high in CH, whereas inosine, and hypoxanthine were elevated in CH-S tumors (Figure S7B). To identify distinct high- and low-grade subsets associated with molecular and histological features, we identified four well-defined metabolomic subtypes (Figures 8D–E). M4 represented the 7 NATs, while M1 was significantly enriched with high-grade histopathologic subtypes, Methyl1, BAP1 mutants, wGII-high status, a mostly mutual exclusivity from the VEGF desert, and female patients (Figures 8E, S7D). The three metabolomic subtypes related to tumors with similar features were confirmed using the validation set (Table S7) and investigated DEMs among the three metabolomic subtypes. As Methyl1 was significantly enriched in M1, we found a considerable overlap of M1-associated and Methy1-associated metabolites such as 4-Hydroxyphenyllactic acid (Figure S7E; Table S7).
Combining metabolomic and proteogenomics analyses, we correlated expression of metabolites with their enzymes, and associated the metabolomics with pathways, molecular and histopathologic features, and clinical information. Dramatic changes in arginine and proline metabolism were observed, including arginine biosynthesis and urea cycle for both metabolites and related enzymes (Figure 8F). Glutamine, α-ketoglutaric acid, ornithine, and citrulline were significantly high in tumors, while L-glutamic acid, and argininosuccinic acid were higher in NATs (Figure 8F). Correspondingly, homologous trends between metabolites and their enzymes were seen, such as elevated GLUL and reduced ASS1 in tumors (Figures 8F, S7F). This correlated with previous studies showing that argininosuccinate synthase 1 (ASS1), was strongly repressed in ccRCCs compared with non-tumorous kidney tissues, and re-expression of ASS1 in ccRCC xenograft models reduced tumor growth.57–59
Higher fractions of GLUL-high and GLS-high samples were present in the higher-grade tumors (Figure 8G). Inhibition of glutaminolysis is a potential cancer therapy and GLUL could be a therapeutic target.60 By performing inhibitor treatment (L-Methionine sulfoximine) and a cell viability assay on GLUL, skrc42.EV responded to the anti-GLUL treatment, while the normal kidney HK-2 cell line was not sensitive to the treatment (Figure S7G). Moreover, tumors among the 3 metabolomic subtypes (M1–3) displayed a strong heterogeneity. Argininosuccinic acid and Fumarate were significantly elevated in M2, whereas Citrulline and Glutamine were high in M3 (Figure S7F), suggesting that any response to directed therapies, such as glutaminolysis inhibition may be dependent upon metabolomic subtype (Figure S7F). MYC-driven accumulation of 2-hydroxyglutarate (2-HG) has been associated with breast cancer prognosis,61 and 2-HG and MYC expression were significantly increased in Methyl1, with a worse prognosis (Figure S7H). Based on the comprehensive characterization depicting all available omics layers, we found that 48 of 50 tumors presented unique characterization profiles, demonstrating the strong intertumoral heterogeneity in ccRCC (Figure 8H).
Discussion
Clear cell RCC is a complex disease defined by a histopathologic feature that has demonstrated ever-increasing levels of variability and heterogeneity through the increased use of diverse analytic methodologies. Building on the foundation of the initial CPTAC study,13 this study expanded both the patient cohort and analyses to include an enhanced histopathologic review, proteogenomic and single-nuclei analysis of multi-sampled tumors, in vitro analysis of therapeutically targeted tumor-specific phosphoproteomic events, tumor metabolomics, and the first analysis of tumor-specific glycoprotein signatures.
The multi-omic nature of this study highlighted the intertumoral heterogeneity present in ccRCC. Despite the heterogeneity, we demonstrated enrichment of specific proteogenomic features within the high-grade tumors, including distinct phospho- and glycoprotein signatures, and confirmed several features, such as BAP1 mutation, tumor hypermethylation, and sarcomatoid features, previously associated with poor patient outcomes.14,15,17,62 An increased understanding of accurate and easily evaluable biomarkers for these specific proteogenomic features will be fundamental in producing effective and adaptable combination therapies. This study highlights UCHL1 expression, measurable by IHC, as one such potential biomarker for high-grade tumors with BAP1 mutation, genome instability, or increased tumor hypermethylation that could influence the clinical and therapeutic management of these patients.
Our comprehensive histopathologic review of the ccRCCs, based upon recent advances in the morphological evaluation of ccRCC,26 immediately highlighted the intratumoral heterogeneity present in this cohort and demonstrated the correlation of either sarcomatoid or rhabdoid features with significantly poorer patient outcomes. Protein markers were identified for each histopathologic feature, as well as for high-grade tumors in general, including SQSTM1, GPNMB, and GAL3ST1 which have previous associations with RCC.14,63–66 Notably, GPNMB is a cell surface protein for which an antibody-drug conjugate-based targeted agent, glembatumumab vedotin, is in clinical trials for advanced melanoma and breast cancer.67,68
Proteogenomic analysis of a subset of 40 multi-sampled cases expanded the expected subclonal nature of specific somatic mutation and copy number events to include ITH of genome stability and DNA methylation, both shown to correlate with patient outcomes.21–23 At least one feature demonstrated heterogeneity in 90% of these tumors. The ITH observed in immune subtyping is particularly important, as ccRCC is considered an immunotherapy-sensitive tumor with increasing use of checkpoint inhibitors as first-line therapy for metastatic ccRCC and the degree of immune ITH could influence response.69 Furthermore, single-cell-based analysis within this study confirmed the differences in tumor, stromal, and immune components between segments and highlighted the ability to isolate specific subgroups of histopathologically distinct cells within a segment, such as those with rhabdoid or sarcomatoid features.70 Consequently, multi-segment profiling and single-cell analysis could better capture the full picture of the TME immune component to predict immunotherapy response to support precision oncology.
While refinement of combination therapies and the development of a specific HIF2 inhibitor (Belzutifan/MK-6482) represent great advances in the treatment of ccRCC, therapeutic options with biological targets are necessary to further improve patient outcomes.7–9,71–73 This study highlighted several potential avenues for targeted therapy. The increased tumor-specific hypermethylation is potentially therapeutically susceptible to demethylating agents such as decitabine (5-Aza-2’-deoxycytidine) or guadecitabine (SGI-110), and therapeutically induced demethylation has been proposed to potentially improve response to common immune-based advanced ccRCC therapies.74 Tumor-specific phosphorylation of the MAPK pathway was targeted in several ccRCC cell line models with MEK inhibitors that induced both effective on-target dephosphorylation and significant antiproliferative effects. MEK inhibitors have been previously considered for treating ccRCC, and this study strengthens support for their potential application.75–77 The increased expression of the glycoprotein HYOU1 in high-grade tumors ccRCCs and its correlation with poor survival makes it a potential therapeutic target and biomarker, and these correlations have been seen in other cancer types.54 Notably, this observation was not detectable at the RNA expression level and highlights the power of multi-omic protein analysis to identify potential IHC-based biomarkers. Altered tumor-specific expressions of glycoproteins have been associated with invasiveness and metastatic potential in various cancers and have been considered therapeutic targets.78–81 Finally, metabolome analysis confirmed the increased abundance of glutamine and metabolites in the urea cycle in higher-grade ccRCC and both of these observations are currently being considered therapeutic targets.57,60,82
In summary, this study enhances our understanding of this complex and heterogeneous disease by utilizing a multitude of analytical approaches. Observations of histologic heterogeneity correlate closely with underlying molecular heterogeneity, but molecular heterogeneity also extends beyond that observed at the visualized tissue level. This study provides a wealth of data that will serve as an invaluable resource for further study and delineates proteogenomic features that can drive the translation of therapeutic research with the aim of improving the outcomes of ccRCC patients.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Li Ding (lding@wustl.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Clinical data and proteomic data (raw MS files and processed data files) reported in this paper can be accessed via the Proteomic Data Commons (PDC) at: https://pdc.cancer.gov/ (Project: CPTAC3 Discovery and Confirmatory, Disease Type: Clear Cell Renal Cell Carcinoma). Genomic, transcriptomic, and snRNA-seq data files can be accessed via Genomic Data Commons (GDC) at: https://portal.gdc.cancer.gov/projects/CPTAC-3 (Project: CPTAC-3, Primary Site: Kidney). Processed data used in this publication can also be found in the PDC, the Python package and LinkedOmics.108 Pathology and radiology images can be accessed via Imaging Data Commons (IDC) at https://portal.imaging.datacommons.cancer.gov/explore/filters/?collection_id=cptac_ccrcc (Collection: CPTAC-CCRCC), and The Cancer Imaging Archive at https://doi.org/10.7937/K9/TCIA.2018.OBLAMN27 (Collection: CPTAC-CCRCC).109 In addition, other data including TCGA KIRC83 at https://portal.gdc.cancer.gov/ (Project: TCGA-KIRC), OmniPath84 at https://omnipathdb.org/#faq, NetworKIN85 at https://networkin.info/, DEPOD86 at http://www.depod.bioss.uni-freiburg.de/, and SIGNOR87 at https://signor.uniroma2.it/.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
A total of 213 participants, with an age range of 30–90, were included in this study. This cohort contained males (n = 149) and females (n = 64) and reflects the gender distribution of clear cell renal cell carcinoma (ccRCC).83 Only histopathologically defined adult ccRCC tumors were only included in the analysis. Institutional review boards at each Tissue Source Site (TSS) reviewed protocols and consent documentation, in adherence to Clinical Proteomic Tumor Analysis Consortium (CPTAC) guidelines.
Clinical Data Annotation
Clinical data were obtained from TSS and aggregated by the Biospecimen Core Resource (BCR, Van Andel Research Institute (Grand Rapids, MI)). Data forms were stored as Microsoft Excel files (.xls). Clinical data can be accessed and downloaded from the CPTAC Data Portal. Patients with any prior history of other malignancies within twelve months or any systemic treatment (chemotherapy, radiotherapy, of immune-related therapy) were excluded from this study. Demographics, histopathologic information, and treatment details were collected and summarized in Table S1. The characteristics of the CPTAC ccRCC cohort reflect the general incidence of ccRCC.83
Cell Lines
The ccRCC cell line Caki-1 and a control cell line HK-2 were maintained in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) culture medium (Gibco - 11320033) supplemented with 10% FBS (Sigma, F-9665) and 1% Pen Strep (Gibco, 10,000 U/mL - 15140122). 786-O cells were maintained in Gibco RPMI-1640 supplemented with 10% FBS. In addition, 769-P, A-498, and Caki-2 were used for in vitro experiments assessing the impact of select kinase inhibition.
METHOD DETAILS
Sample Processing
The CPTAC Biospecimen Core Resource (BCR) at the Pathology and Biorepository Core of the Van Andel Research Institute in Grand Rapids, Michigan manufactured and distributed biospecimen kits to the Tissue Source Sites (TSS) located in the US, Europe, and Asia. Each kit contains a set of pre-manufactured labels for unique tracking of every specimen respective to TSS location, disease, and sample type, used to track the specimens through the BCR to the CPTAC proteomic and genomic characterization centers.
Tissue specimens averaging 200 mg were snap-frozen by the TSS within a 30 min cold ischemic time (CIT) (CIT average = 13 min) and an adjacent segment was formalin-fixed paraffin-embedded (FFPE) and H&E stained by the TSS for quality assessment to meet the CPTAC ccRCC requirements. Routinely, several tissue segments for each case were collected. Tissues were flash-frozen in liquid nitrogen (LN2) and then transferred to a liquid nitrogen freezer for storage until approval for shipment to the BCR.
Specimens were shipped using a cryoport that maintained an average temperature of under −140°C to the BCR with a time and temperature tracker to monitor the shipment. Receipt of specimens at the BCR included a physical inspection and review of the time and temperature tracker data for specimen integrity, followed by barcode entry into a biospecimen tracking database. Specimens were again placed in LN2 storage until further processing. Acceptable ccRCC tumor tissue segments were determined by TSS pathologists based on the percent viable tumor nuclei (> 60%), total cellularity (> 50%), and necrosis (< 50%). Segments received at the BCR were verified by BCR and Leidos Biomedical Research (LBR) pathologists and the percent of the total area of tumor in the segment was also documented. Additionally, disease-specific working group pathology experts reviewed the morphology to clarify or standardize specific disease classifications and correlation to the proteomic and genomic data.
Specimens selected for the discovery set were determined on the maximal percent in the pathology criteria and best weight. Specimens were pulled from the biorepository using an LN2 cryocart to maintain specimen integrity and then cryopulverized. The cryopulverized specimen was divided into aliquots for DNA (30 mg) and RNA (30 mg) isolation and proteomics (50 mg) for molecular characterization. Nucleic acids were isolated and stored at −80°C until further processing and distribution; cryopulverized protein material was returned to the LN2 freezer until distribution. Shipment of the cryopulverized segments used cryoports for distribution to the proteomic characterization centers and shipment of the nucleic acids used dry ice shippers for distribution to the genomic characterization centers; a shipment manifest accompanied all distributions for the receipt and integrity inspection of the specimens at the destination.
Comprehensive Schematic Histopathology Evaluation
A comprehensive evaluation of the hematoxylin and eosin (H&E) stained histopathologic samples was undertaken with a focus on the tumor epithelial component and the surrounding tumor microenvironmental alterations including the immune cell characterization. The overall grading of the tumor samples was based on the findings noted from the previous histopathologic patient reports which were re-confirmed. Broadly the epithelial cell assessment was done under three categories of recognition of nodular areas/distinct sudden transitional areas and sub-dividing the morphologic patterns and cytology under low-grade and high-grade parameters. Every histopathologic tissue section was annotated for recognized low-grade features (nested, tubular/acinar, microcystic, and bleeding follicles) and high-grade features (eosinophilic/granular, thick trabecular, alveolar, solid, papillary/pseudo-papillary, sarcomatoid, and rhabdoid).26,29 Apart from the detailed spatial architecture and cytological assessment, tumor microenvironment evaluation was also performed detailing immune characterization (semi-quantitative scoring, type of infiltration-intra-tumoral, intratumoral septal, peri-tumoral and stromal, and immune subpopulation types) and the presence or absence of necrosis. In addition, specialized histopathologic annotations such as the presence of hyalinization, fibrotic response, extensive multi-nodularity, or histopathologic resemblance to other renal cell carcinoma subtypes were also noted. Thus, in each tumor sample instead of focusing on the higher grade or aggressive spatial topography, the whole tissue area was evaluated against the entire spectrum of morphological parameters as described above. These findings were recorded and tabulated. A semi-quantitative score for each tumor was rendered based on the presence (scored as 1) or absence (scored as 0) of the individual histologic parameters. This way a detailed assessment of histologic tumor heterogeneity was assessed (Table S1).
Sample Processing for Genomic DNA and Total RNA Extraction
Our study sampled a single site of the primary tumor from surgical resections, due to the internal requirement to process a minimum of 125 mg of tumor issue and 50 mg of adjacent normal tissue. DNA and RNA were extracted from tumor and blood normal specimens in a co-isolation protocol using Qiagen’s QIAsymphony DNA Mini Kit and QIAsymphony RNA Kit. Genomic DNA was also isolated from peripheral blood (3–5 mL) to serve as matched normal reference material. The Qubit™ dsDNA BR Assay Kit was used with the Qubit® 2.0 Fluorometer to determine the concentration of dsDNA in an aqueous solution. Any sample that passed quality control and produced enough DNA yield to go through various genomic assays was sent for genomic characterization. RNA quality was quantified using both the NanoDrop 8000 and quality assessed using Agilent Bioanalyzer. A sample that passed RNA quality control and had a minimum RIN (RNA integrity number) score of 7 was subjected to RNA sequencing. Identity match for germline, normal adjacent tissue, and tumor tissue was assayed at the BCR using the Illumina Infinium QC array. This beadchip contains 15,949 markers designed to prioritize sample tracking, quality control, and stratification.
Whole Exome Sequencing
Library Construction
Library construction was performed as described in,110 with the following modifications: initial genomic DNA input into shearing was reduced from 3 μg to 20–250 ng in 50 μL of solution. For adapter ligation, Illumina paired-end adapters were replaced with palindromic forked adapters, purchased from Integrated DNA Technologies, with unique dual-indexed molecular barcode sequences to facilitate downstream pooling. Kapa HyperPrep reagents in 96-reaction kit format were used for end repair/A-tailing, adapter ligation, and library enrichment PCR. In addition, during the post-enrichment SPRI cleanup, elution volume was reduced to 30 μL to maximize library concentration, and a vortexing step was added to maximize the amount of template eluted.
In-solution Hybrid Selection
After library construction, libraries were pooled into groups of up to 96 samples. Hybridization and capture were performed using the relevant components of Illumina’s Nextera Exome Kit and following the manufacturer’s suggested protocol, with the following exceptions. First, all libraries within a library construction plate were pooled prior to hybridization. Second, the Midi plate from Illumina’s Nextera Exome Kit was replaced with a skirted PCR plate to facilitate automation. All hybridization and capture steps were automated on the Agilent Bravo liquid handling system.
Preparation of Libraries for Cluster Amplification and Sequencing
After post-capture enrichment, library pools were quantified using qPCR (automated assay on the Agilent Bravo) using a kit purchased from KAPA Biosystems with probes specific to the ends of the adapters. Based on qPCR quantification, libraries were normalized to 2 nM.
Cluster Amplification and Sequencing
Cluster amplification of DNA libraries was performed according to the manufacturer’s protocol (Illumina) using exclusion amplification chemistry and flowcells. Flowcells were sequenced utilizing sequencing-by-synthesis chemistry. The flow cells were then analyzed using RTA v.2.7.3 or later. Each pool of whole-exome libraries was sequenced on paired 76 cycle runs with two 8 cycle index reads across the number of lanes needed to meet coverage for all libraries in the pool. Pooled libraries were run on HiSeq 4000 paired-end runs to achieve a minimum of 150x on target coverage per each sample library. The raw Illumina sequence data were demultiplexed and converted to fastq files; adapter and low-quality sequences were trimmed. The raw reads were mapped to the hg38 human reference genome and the validated BAMs were used for downstream analysis and variant calling.
PCR-free Whole Genome Sequencing
Preparation of Libraries for Cluster Amplification and Sequencing
An aliquot of genomic DNA (350 ng in 50 μL) was used as the input into DNA fragmentation (aka shearing). Shearing was performed acoustically using a Covaris focused-ultrasonicator, targeting 385bp fragments. Following fragmentation, additional size selection was performed using a SPRI cleanup. Library preparation was performed using a commercially available kit provided by KAPA Biosystems (KAPA Hyper Prep without amplification module) and with palindromic forked adapters with unique 8-base index sequences embedded within the adapter (purchased from IDT). Following sample preparation, libraries were quantified using quantitative PCR (kit purchased from KAPA Biosystems), with probes specific to the ends of the adapters. This assay was automated using Agilent’s Bravo liquid handling platform. Based on qPCR quantification, libraries were normalized to 1.7 nM and pooled into 24-plexes.
Cluster Amplification and Sequencing (HiSeq X)
Sample pools were combined with HiSeq X Cluster Amp Reagents EPX1, EPX2, and EPX3 into single wells on a strip tube using the Hamilton Starlet Liquid Handling system. Cluster amplification of the templates was performed according to the manufacturer’s protocol (Illumina) with the Illumina cBot. Flow cells were sequenced to a minimum of 15x on HiSeq X utilizing sequencing-by-synthesis kits to produce 151bp paired-end reads. Output from Illumina software was processed by the Picard data processing pipeline to yield BAMs containing demultiplexed, aggregated, and aligned reads. All sample information tracking was performed by automated LIMS messaging.
Illumina Infinium MethylationEPIC Beadchip Array
The MethylationEPIC array uses an 8-sample version of the Illumina Beadchip capturing > 850,000 DNA methylation sites per sample. 250 ng of DNA was used for the bisulfite conversation using Infinium MethylationEPIC BeadChip Kit. The EPIC array includes sample plating, bisulfite conversion, and methylation array processing. After scanning, the data was processed through an automated genotype calling pipeline. Data generated consisted of raw idats and a sample sheet.
RNA Sequencing
Quality Assurance and Quality Control of RNA Analytes
All RNA analytes were assayed for RNA integrity, concentration, and fragment size. Samples for total RNA-seq were quantified on a TapeStation system (Agilent, Inc. Santa Clara, CA). Samples with RINs > 8.0 were considered high quality.
Total RNA-seq Library Construction
Total RNA-seq library construction was performed from the RNA samples using the TruSeq Stranded RNA Sample Preparation Kit and bar-coded with individual tags following the manufacturer’s instructions (Illumina, Inc. San Diego, CA). Libraries were prepared on an Agilent Bravo Automated Liquid Handling System. Quality control was performed at every step and the libraries were quantified using the TapeStation system.
Total RNA Sequencing
Indexed libraries were prepared and run on HiSeq 4000 paired-end 75 base pairs to generate a minimum of 120 million reads per sample library with a target of greater than 90% mapped reads. Typically, these were pools of four samples. The raw Illumina sequence data were demultiplexed and converted to FASTQ files, and adapter and low-quality sequences were quantified. Samples were then assessed for quality by mapping reads to the hg38 human genome reference, estimating the total number of reads that mapped, amount of RNA mapping to coding regions, amount of rRNA in sample, number of genes expressed, and relative expression of housekeeping genes. Samples passing this QA/QC were then clustered with other expression data from similar and distinct tumor types to confirm expected expression patterns. Atypical samples were then SNP typed from the RNA data to confirm the source analyte. FASTQ files of all reads were then uploaded to the GDC repository.
miRNA-seq Library Construction
miRNA-seq library construction was performed from the RNA samples using the NEXTflex Small RNA-Seq Kit (v3, PerkinElmer, Waltham, MA) and bar-coded with individual tags following the manufacturer’s instructions. Libraries were prepared on Sciclone Liquid Handling Workstation Quality control was performed at every step, and the libraries were quantified using a TapeStation system and an Agilent Bioanalyzer using the Small RNA analysis kit. Pooled libraries were then size selected according to NEXTflex Kit specifications using a Pippin Prep system (Sage Science, Beverly, MA).
miRNA Sequencing
Indexed libraries were loaded on the Hiseq 4000 to generate a minimum of 10 million reads per library with a minimum of 90% reads mapped. The raw Illumina sequence data were demultiplexed and converted to FASTQ files for downstream analysis. Resultant data were analyzed using a variant of the small RNA quantification pipeline developed for TCGA.111 Samples were assessed for the number of miRNAs called, species diversity, and total abundance. Samples passing quality control were uploaded to the GDC repository.
Single-nuclei RNA Library Preparation and Sequencing
About 20–30 mg of cryopulverized powder from ccRCC specimens was resuspended in Lysis buffer (10 mM Tris-HCl (pH 7.4); 10 mM NaCl; 3 mM MgCl2; and 0.1% NP-40). This suspension was pipetted gently 6–8 times, incubated on ice for 30 seconds, and pipetted again 4–6 times. The lysate containing free nuclei was filtered through a 40 μm cell strainer. We washed the filter with 1 mL Wash and Resuspension buffer (1X PBS + 2% BSA + 0.2 U/μL RNase inhibitor) and combined the flow through with the original filtrate. After 6-minute centrifugation at 500 × g and 4°C, the nuclei pellet was resuspended in 500 μL of Wash and Resuspension buffer. After staining by DRAQ5, the nuclei were further purified by Fluorescence-Activated Cell Sorting (FACS). FACS-purified nuclei were centrifuged again and resuspended in a small volume (about 30 μL). After counting and microscopic inspection of nuclei quality, the nuclei preparation was diluted to about 1,000 nuclei/μL. About 20,000 nuclei were used for single-nuclei RNA sequencing (snRNA-seq) by the 10X Chromium platform. We loaded the single nuclei onto a Chromium Chip B Single Cell Kit, 48 rxns (10x Genomics, PN-1000073), and processed them through the Chromium Controller to generate GEMs (Gel Beads in Emulsion). We then prepared the sequencing libraries with the Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3, 16 rxns (10x Genomics, PN-1000075) following the manufacturer’s protocol. Sequencing was performed on an Illumina NovaSeq 6000 S4 flow cell. The libraries were pooled and sequenced using the XP workflow according to the manufacturer’s protocol with a 28×8×98bp sequencing recipe. The resulting sequencing files were available as FASTQs per sample after demultiplexing.
MS Sample Processing and Data Collection
Sample Processing for Protein Extraction and Tryptic Digestion
All samples for the current study were prospectively collected as described above and processed for mass spectrometric (MS) analysis at the PCC. Tissue lysis and downstream sample preparation for global proteomic and phosphoproteomic analysis were carried out as previously described.13 Approximately 25–120 mg of each cryopulverized renal tumor tissues or NATs were homogenized separately in an appropriate volume of lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris, pH 8.0, 1 mM EDTA, 2 g/mL aprotinin, 10 g/mL leupeptin, 1 mM PMSF, 10 mM NaF, Phosphatase Inhibitor Cocktail 2 and Phosphatase Inhibitor Cocktail 3 [1:100 dilution], and 20 mM PUGNAc) by repeated vortexing. Lysates were clarified by centrifugation at 20,000 × g for 10 min at 4°C, and protein concentrations were determined by BCA assay (Pierce). Lysates were diluted to a final concentration of 8 mg/ml with lysis buffer, and 800 g of protein was reduced with 5 mM dithiothreitol (DTT) for 1 h at 37°C and subsequently alkylated with 10 mM iodoacetamide for 45 min at RT (room temperature) in the dark. Samples were diluted 1:3 with 50 mM Tris-HCl (pH 8.0) and subjected to proteolytic digestion with LysC (Wako Chemicals) at 1 mAU:50 g enzyme-to-substrate ratio for 2h at RT, followed by the addition of sequencing-grade modified trypsin (Promega) at a 1:50 enzyme-to-substrate ratio and overnight incubation at RT. The digested samples were then acidified with 50% trifluoroacetic acid (TFA, Sigma) to a pH value of approximately 2.0. Tryptic peptides were desalted on reversed-phase C18 SPE columns (Waters), followed by aliquoting 20 g of digested peptides for global proteomic analysis, dried in a Speed-Vac, and resuspended in 3% ACN/0.1% formic acid prior to ESI-LC-MS/MS analysis. The remaining sample was dried down in a Speed-Vac and utilized for phosphopeptide and intact glycopeptide enrichment.
Enrichment of Phosphopeptides by Fe-IMAC.
A 450 g aliquot of digested peptide material was subjected to phosphopeptide enrichment using immobilized metal affinity chromatography (IMAC) as previously described.112 In brief, Ni-NTA agarose beads were used to prepare Fe3+-NTA agarose beads, and 450 g of peptides were reconstituted in 80% ACN/0.1% trifluoroacetic acid and incubated with 10 L of the Fe3+-IMAC beads for 30 min. Samples were then centrifuged, and the supernatant containing unbound peptides was removed. The beads were washed twice and then transferred onto equilibrated C-18 Stage Tips with 80% ACN/0.1% trifluoroacetic acid. Tips were rinsed twice with 1% formic acid and eluted from the Fe3+-IMAC beads onto the C-18 Stage Tips with 70 L of 500 mM dibasic potassium phosphate, pH 7.0 a total of three times. C-18 Stage Tips were then washed twice with 1% formic acid, followed by elution of the phosphopeptides from the C-18 Stage Tips with 50% ACN/0.1% formic acid twice. Samples were dried down and resuspended in 3% ACN/0.1% formic acid prior to ESI-LC-MS/MS analysis.
Enrichment of Intact Glycopeptides by MAX Columns from Tryptic Peptides
The glycopeptides were enriched from 350 μg C18 cleaned up tryptic peptides using 30 mg MAX columns (Waters). 350 μg tryptic peptides were first dried down in SpeedVac and reconstituted in 50% ACN/0.1% TFA, then constituted to 95% ACN/1% TFA. MAX columns were sequentially conditioned with 1ml 100% ACN 3 times, then 1 ml 100 mM triethylammonium acetate buffer 3 times and 1 ml 95% ACN/1% TFA 3 times. Tryptic peptides were conditioned to bind onto the MAX columns 2 times and then washed with 1 ml 95% ACN/1% TFA 3 times. Non-intact glycopeptides were eluted/washed off the MAX columns, while intact glycopeptides were bound onto the MAX column during the process. Intact glycopeptides were then eluted using 50% ACN/0.1% TFA, dried down, and reconstituted in 3% ACN/ 0.1% FA prior to ESI-LC-MS/MS analysis.
ESI-LC-MS/MS for Global Proteome, Phosphoproteome, and Glycoproteome Using DIA-MS Analysis
Individual global proteome and phosphoproteome samples were analyzed using the same instrumentation and methodology; albeit with varied gradient settings. Individual glycoproteomic samples were analyzed using the same MS instrument and gradient settings as phosphoproteome, except the MS settings, which used the methodology as previously described.113 Unlabeled, digested peptide material from individual tissue samples (ccRCC and NAT) was spiked with index Retention Time (iRT) peptides (Biognosys) and subjected to data-independent acquisition (DIA) analysis. Peptides (~0.8 g; ~1 ug for glycopeptides) were separated on an Easy nLC 1200 UHPLC system (Thermo Scientific) on an in-house packed 20 cm × 75 m diameter C18 column (1.9 m Reprosil-Pur C18-AQ beads (Dr. Maisch GmbH); Picofrit 10 m opening (New Objective). The column was heated to 50°C using a column heater (Phoenix-ST). The flow rate was 0.200 μl/min with 0.1% formic acid and 3% acetonitrile in water (A) and 0.1% formic acid, 90% acetonitrile (B). For global proteomic characterization of ccRCC tumors and NATs, the peptides were separated using the following LC gradient: 0–3 min (2% B, isocratic), 3–103 min (7%-20% B, linear), 103–121 min (20–30% B, linear), 121–125 min (30–60% B, linear), 125–126 min (60–90% B, linear), 126–130 min (90% B, isocratic), 130–131 min (90–50% B, linear), 131–140 min (50% B, isocratic). For global proteomic characterization of samples annotated as intra-tumor heterogeneity segments, phosphoproteomic, and glycoproteomic characterization, the peptides were separated using the following LC gradient: 0–3 min (2% B, isocratic), 3–93 min (7%-25% B, linear), 93–121 min (25–30% B, linear), 121–125 min (30–60% B, linear), 125–126 min (60–90% B, linear), 126–130 min (90% B, isocratic), 130–131 min (90–50% B, linear), 131–140 min (50% B, isocratic). Samples were analyzed using the Thermo Fusion Lumos mass spectrometer (Thermo Scientific). For global and phosphoproteome, the DIA segment consisted of one MS1 scan (350–1650 m/z range, 120K resolution) followed by 30 MS2 scans (variable m/z range, 30K resolution) as described previously.114 Additional parameters were as follows: MS1: RF Lens – 30%, AGC Target 4.0e5, Max IT – 50 ms, charge state include - 2–6; MS2: isolation width (m/z) – 0.7, AGC Target – 3.0e6, Max IT – 120 ms. For glycoproteome, the DIA segment consisted of one MS1 scan (450–1650 m/z range, 120K resolution) followed by 50 MS2 scans (variable m/z range within 120–2000 m/z, 15K resolution) as described previously.113 Additional parameters were as follows: MS1: RF Lens – 30%, AGC Target 3.0e6, Max IT – 60 ms, charge state include - 2–6; MS2: AGC Target – 5.3e5, Max IT – 44 ms.
Kinase Inhibition Assessment in Renal Cell Carcinoma Cell Models
For in vitro experiments assessing the impact of select kinase inhibition on renal cancer cell models (786-O, 769-P, A-498, Caki-1, and Caki-2), the kinase inhibitors, Adaversotib, Everolimus, Sapanisterib, Gefitinib, and Trametinib were dissolved in dimethyl sulfoxide (DMSO) and subjected to sonication in a water bath at room temperature. Following an assessment of individual cell line growth rates to enable calculation of half maximal inhibitory concentration (IC50), cells were seeded in triplicate at concentrations of either 1,000cells/well (786-O, 769-P, A-498, Caki-2) or 10,000 cells/well (Caki-1). Post-24 hour seeding, cells were subjected to kinase inhibitors at final concentrations of 1 nm, 10 nM, 50 nM, 100 nM, 500 nM, 1 mM, 10 mM, with non-treated cells and DMSO treated cells included as controls. Cell growth was measured on day 1 (kinase inhibitor treatment), day 4, and day 6 using the colorimetric CellTiter 96® Aqueous One Cell Proliferation Solution Assay (MTS) following the manufacturer’s instructions. IC50 for each cell line in response to single kinase inhibitor exposure was determined by plotting inhibitor concentration against percent activity relative to DMSO-treated controls and calculating the x-intercept of the linear logarithmic trend line. For phosphoproteomic characterization of renal cell models treated with individual kinase inhibitors, six treatment conditions were devised – control, Adaversotib treatment, Everolimus treatment, Sapanisterib treatment, Gefitinib treatment, and Trametinib treatment - and cells were seeded at ~5E6 cells/15 cm plate and allowed to reach ~80% confluency. 30 minutes prior to kinase inhibitor treatment, fresh media was exchanged. Cells were then treated with kinase inhibitors at calculated IC50 values for 1.5 hours. Media was removed and cells were three times with a volume of ice-cold PBS. Cells were scraped using 1.5 mL of ice-cold PBS, transferred to Eppendorf tubes, and spun at 3,000 × g for 5 minutes at 4°C. A volume of lysis buffer (8 M urea, 75 mM NaCl, 50 mM Tris, pH 8.0, 1 mM EDTA, 2 g/mL aprotinin, 10 g/mL leupeptin, 1 mM PMSF, 10 mM NaF, Phosphatase Inhibitor Cocktail 2 and Phosphatase Inhibitor Cocktail 3 [1:100 dilution], and 20 mM PUGNAc) was added and cells lysed. Subsequent sample preparation and ESI-LC-MS/MS analysis for global proteomic and phosphoproteomic characterization for DIA analysis were performed as described for tissue samples.
Spectral Library Generation for DIA-MS Analysis of Intact Glycopeptides
For spectral library generation, an aliquot (5 g) of unlabeled glycopeptides from individual tissue samples (ccRCC and NAT) was pooled and subjected to bRPLC as previously described.13 In brief, the desalted, pooled sample was reconstituted in 900 L of 20 mM ammonium formate (pH 10) and 2% acetonitrile (ACN) and loaded onto a 4.6 mm × 250 mm RP Zorbax 300 A Extend-C18 column with 3.5 m size beads (Agilent). Peptides were separated at a flow-rate of 0.2 mL/min using an Agilent 1200 Series HPLC instrument via bHPLC with Solvent A (2% ACN, 5 mM ammonium formate, pH 10) and a non-linear gradient of Solvent B (90% ACN, 5 mM ammonium formate, pH 10) as follows: 0% Solvent B (7 min), 0% to 16% Solvent B (6 min), 16% to 40% Solvent B (60 min), 40% to 44% Solvent B (4 min), 44% to 60% Solvent B (5 min), then holding at 60% Solvent B for 14 min, 60% to 98% Solvent B (14 min). Collected fractions were concatenated into 12 fractions previously described115 and dried down in a Speed-Vac. For glycoproteomic characterization, a 5% aliquot each of the 12 fractions was resuspended in 3% ACN, 0.1% formic acid, and was spiked with index Retention Time (iRT) peptides (Biognosys) prior to ESI-LC-MS/MS analysis. Data acquisition using the same instrumentation for DIA-based analyses was employed using the same corresponding LC gradient, with the following Thermo Fusion Lumos mass spectrometer (Thermo Scientific) parameters: MS1: resolution – 60K, mass range – 350 to 2000 m/z, RF Lens – 30%, AGC Target 4.0e5, Max IT – 50 ms, charge state include - 2–6, dynamic exclusion – 45 s, top 20 ions selected for MS2; MS2: resolution – 15K, high-energy collision dissociation activation energy (HCD) – 34, isolation width (m/z) – 0.7, AGC Target – 2.0e5, Max IT – 105 ms.
Metabolome Analysis of Tissue Samples
To extract metabolites, a solution consisting of 80% (vol/vol) mass spectrometry-grade methanol and 20% (vol/vol) mass spectrometry-grade water were used to extract the metabolites from the tissue samples as described previously.116–118 The metabolite samples then underwent speed vacuum processing to evaporate the methanol and lyophilization to remove the water. The dried metabolites were re-suspended in a solution consisting of 50% (vol/vol) acetonitrile and 50% (vol/vol) mass spectrometry-grade water before data acquisition. Data acquisition was performed using a Vanquish ultra-performance liquid chromatography (UPLC) system and a Thermo Scientific Q Exactive Plus Orbitrap Mass Spectrometer.
The samples were kept at 4° C inside the Vanquish UPLC auto-sampler. The injection volume for each sample was 2 uL. A Discovery® HSF5 reverse phase HPLC column (Sigma) kept at 35° C with a guard column was used for reverse-phase chromatography. The mobile aqueous phase was mass spectrometry-grade water containing 0.1% formic acid, while the mobile organic phase was acetonitrile containing 0.1% formic acid. Mass calibration was performed prior to data acquisition to ensure the sensitivity and accuracy of the system. The total run time for each sample was 15 minutes, for which 11 minutes was used for data acquisition. Full MS data were acquired to quantify the metabolites while Full MS/ddMS2 data were also acquired to identify the metabolites based on fragmentation matching.
Immunohistochemistry Analysis
Immunohistochemistry (IHC) was performed on 4-micron formalin-fixed, paraffin-embedded (FFPE) tissue sections. The antibodies characterized include CA9 (Carbonic anhydrase IX) rabbit polyclonal primary antibody (Cat No. NB100–417, Novus Biologicals, Centennial, CO), BAP1 (BRCA1 associated protein 1) mouse monoclonal primary antibody (Cat No. sc-28382, Santa Cruz Biotechnology, Dallas, TX), UCHL1 (Ubiquitin C-terminal hydrolase 1) rabbit polyclonal primary antibody (Cat No. HPA005993, Sigma-Aldrich (Atlas), St. Louis, Mo), HYOU1 (Hypoxia up-regulated 1) rabbit polyclonal primary antibody (Cat No. HPA049296, Atlas Antibodies, Bromma, Sweden), IFI30 (Interferon gamma-inducible protein 30) rabbit polyclonal primary antibody (Cat No. HPA026650, Atlas Antibodies, Bromma, Sweden), CTSA (Cathepsin A) rabbit polyclonal primary antibody (HPA031068, Atlas Antibodies, Bromma, Sweden), GAL3ST1 (Galactose-3-O-sulfotransferase 1) rabbit polyclonal primary antibody (Cat No. HPA001220, Atlas Antibodies, Bromma, Sweden), KIF2A (Kinesin heavy chain member 2A) rabbit polyclonal primary antibody (Cat No. HPA004716, Atlas Antibodies, Bromma, Sweden), PLXDC2 (Plexin domain containing 2) rabbit polyclonal primary antibody (Cat No. HPA017268, Atlas Antibodies, Bromma, Sweden) and TGFBI (Transforming growth factor beta induced) rabbit polyclonal primary antibody (Cat No. HPA008612, Atlas Antibodies, Bromma, Sweden). IHC was carried out on the Benchmark XT automated slide staining system using the UltraView Universal DAB detection kit for CA9 and UCHL1 and OptiView DAB detection kit for BAP1 (Cat No. 760–500 and 760–700 respectively, Roche-Ventana Medical Systems, Oro Valley, AZ). IHC for HYOU1, IFI30, CTSA, GAL3ST1, KIF2A, PLXDC2 and TGFBI was performed using an automated platform Dako Autostainer Link 48 and EnVision FLEX visualizing kit (cat. no. K800221–2; Dako, Agilent Technologies Inc., Carpinteria, CA). Appropriate known positive and negative control tissue were run in each assay batch.
A semi-quantitative product score was determined for BAP1 and UCHL1 where the presence and intensity of BAP1 nuclear and UCHL1 cytoplasmic/membranous staining were scored by the study pathologists. This product score represents the percentage of positive neoplastic cells and the staining intensity (none, 0; weak, 1; moderate, 2; strong, 3) which were recorded for each tumor as described previously.119
In Vitro Cell Line Drug Treatment and Growth Inhibition Assessment
The ccRCC cell line Caki-1 and a control cell line HK-2 were maintained in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) culture medium (Gibco - 11320033) supplemented with 10% FBS (Sigma, F-9665) and 1% Pen Strep (Gibco, 10,000 U/mL - 15140122). All cell lines were seeded at 50,000 cells/well in duplicates in 24-well plates at day 0 and were treated with either UCHL1 inhibitor (CAS 668467–91-2 - Calbiochem, Sigma Aldrich - 662086–10MG) or GLUL inhibitor (L-Methionine sulfoximine, Sigma Aldrich -M5379–500MG) upon reaching 50–60% confluency at day 3 in culture. For both UCH-L1inhibitor or GLUL inhibitor treatment, the working concentrations were used at 1μM, 5μM, and 25μM. Treatment was maintained in culture for a total of 7 days and growth inhibition assessment was performed using AlamarBlue™ Cell Viability Reagent (Invitrogen - DAL1025) at a ratio of 1:10 for 4 hours according to the manufacturer’s protocol. Plots and IC50 concentrations were produced in Prism GraphPad (version 9.2.0) by plotting the percent growth inhibition on the y-axis and the Log(concentration) on the x-axis. The corresponding IC50 was extracted from the nonlinear regression curve fitting analysis using Prism GraphPad. Cells treated with only a growth medium without any drugs were used as negative controls.
786-O cells were maintained in Gibco RPMI-1640 supplemented with 10% FBS. CAS 668467–91-2 (UCHL-1 inhibitor) was purchased from Sigma-Aldrich (L4170) and its impact on cell viability was evaluated. Briefly, 2000 cells were seeded on white flat bottom 96 well plates and were treated with increasing concentrations of the inhibitor for a week. CellTiter-Glo Luminescent Cell Viability Assay (Promega) was used to assess cell viability and IC-50 was calculated using a graph pad. Impact of UCHL-1 inhibitor on cell morphology was evaluated using IncuCyte ZOOM assay. For western blot analysis, cell lysates were harvested from control and UCHL-1 treated 786-O cells. Following protein quantification, lysates were resolved in NuPAGE Bis-Tris Protein Gel (ThermoFisher Scientific), transferred on to nitrocellulose membranes, blocked with 5% milk, and incubated overnight with UCHL-1 antibody (HPA005993, Sigma-Aldrich). Following day, membranes were washed with TBST buffer, incubated with HRP-conjugated secondary antibodies, washed, and imaged using Odyssey Fc imager (LiCOR Biosciences).
QUANTIFICATION AND STATISTICAL ANALYSIS
Genomic Data Analysis
Harmonized Genome Alignment
WGS, WES, RNA-Seq sequence data were harmonized by NCI Genomic Data Commons (GDC) https://gdc.cancer.gov/about-data/gdc-data-harmonization, which included alignment to GDC’s hg38 human reference genome (GRCh38.d1.vd1) and additional quality checks. All the downstream genomic processing was based on the GDC-aligned BAMs to ensure reproducibility.
Somatic Mutation Calling
Somatic mutations were called by the Somaticwrapper pipeline v1.6 (https://github.com/ding-lab/somaticwrapper), which includes four different callers, i.e., Strelka v.2,102 MUTECT v1.7,97 VarScan v.2.3.8,104 and Pindel v.0.2.598 from WES. We kept the exonic SNVs called by any two callers among MUTECT v1.7, VarScan v.2.3.8, and Strelka v.2 and indels called by any two callers among VarScan v.2.3.8, Strelka v.2, and Pindel v.0.2.5. For the merged SNVs and indels, we applied a 14X and 8X coverage cutoff for tumor and normal, separately. We also filtered SNVs and indels by a minimal variant allele frequency (VAF) of 0.05 in tumors and a maximal VAF of 0.02 in normal samples. We filtered any SNV, which was within 10bp of an indel found in the same tumor sample. Finally, we rescued the rare mutations with VAF of [0.015, 0.05) in ccRCC driver genes based on the gene consensus list.120
DNP Calling
In step 12 of Somaticwrapper pipeline v1.6 (https://github.com/ding-lab/somaticwrapper), it combined adjacent SNVs into DNP by using COCOON (https://github.com/ding-lab/COCOONS): As input, COCOON takes a MAF file from standard variant calling pipeline. First, it extracts variants within a 2bp window as DNP candidate sets. Next, suppose the corresponding BAM files used for variant calling are available. In that case, it extracts the reads (denoted as n_t) spanning all candidate DNP locations in each variant set, and then counts the number of reads with all the co-occurring variants (denoted as n_c) to calculate the co-occurrence rate (r_c=n_c/n_t); If r_c ≥ 0.8, the nearby SNVs will be combined into DNP and it also updates annotation for the DNPs from the same codon based on the transcript and coordinates information in the MAF file.
Mutational Signature Analysis
Non-negative matrix factorization algorithm (NMF) was used in deciphering mutation signatures in cancer somatic mutations stratified by 96 base substitutions in tri-nucleotide sequence contexts. To obtain a reliable signature profile, we used the Somaticwrapper pipeline to call mutations from WES data. SignatureAnalyzer exploited the Bayesian variant of the NMF algorithm and enabled an inference for the optimal number of signatures from the data itself at a balance between the data fidelity (likelihood) and the model complexity (regularization).91 As decomposed into signatures, signatures are compared against known signatures derived from COSMIC,121 and cosine similarity is calculated to identify the best match (parameters: --cosmic cosmic3_exome -objective Poisson -n 200).
Germline Variant Calling
Germline variant calling was performed using the GermlineWrapper v1.1 pipeline, which implements multiple tools for the detection of germline INDELs and SNVs. Germline SNVs were identified using VarScan v2.3.8 (with parameters:-min-var-freq 0.10, -p-value 0.10, -min-coverage 3, -strand-filter 1) operating on a mpileup stream produced by samtools v1.2 (with parameters:-q 1 -Q 13) and GATK v4.0.0.0122 using its haplotype caller in single-sample mode with duplicate and unmapped reads removed and retaining calls with a minimum quality threshold of 10. All resulting variants were limited to the coding region of the full-length transcripts obtained from Ensembl release 95 plus additional two base pairs flanking each exon to cover splice donor/acceptor sites. We required variants to have allelic depth ≥ 5 reads for the alternative allele in both tumor and normal samples. We used bam-readcount v0.8 for reference and alternative alleles quantification (with parameters: -q 10 -b 15) in both normal and tumor samples. Additionally, we filtered all variants with ≥ 0.05% frequency in gnomAD v2.1123 and The 1000 Genomes Project.124 To predict the pathogenicity of germline variants, we annotate each variant with Variant Effect Predictor (VEP) and process them using the CharGer pipeline with the parameters from a previous pan-cancer TCGA study.89,125 Briefly, the CharGer pipeline considers pathogenic peptide changes from ClinVar, hotspot variants, minor allele frequency from ExAC, and several in silico analyses (such as Sift and PolyPhen). Each predicted pathogenic variant was then manually reviewed.
Copy Number Variant Calling
Copy-number analysis was performed jointly leveraging both whole-genome sequencing (WGS) and whole-exome sequencing data of the tumor and germline DNA. To perform the analysis, we used CNVEX (https://github.com/mctp/cnvex), a comprehensive copy number analysis tool that has been used previously in our ccRCC studies.13,24 CNVEX uses whole-genome aligned reads to estimate coverage within fixed genomic intervals and whole-exome variant calls to compute B-allele frequencies (BAFs) at variant positions (called by Sentieon DNAscope algorithm). Coverages were computed in 10kb bins, and the resulting log coverage ratios between tumor and normal samples were adjusted for GC bias using weighted LOESS smoothing across mappable and non-blacklisted genomic intervals within the GC range 0.3–0.7, with a span of 0.5 (the target and configuration files are provided with CNVEX). The adjusted log coverage ratios (LR) and BAFs were jointly segmented by a custom algorithm based on Circular Binary Segmentation (CBS). Alternative probabilistic algorithms were implemented in CNVEX, including algorithms based on recursive binary segmentation (RBS), as implemented in the R-package jointseg.126 For the CBS-based algorithm, first LR and mirrored BAF were independently segmented using CBS(parameters alpha = 0.01, trim = 0.025) and all candidate breakpoints were collected. The resulting segmentation track was iteratively “pruned” by merging segments that had similar LR and BAFs, short lengths, were rich in blacklisted regions, and had a high coverage variation in coverage among whole cohort germline samples. For the RBS- and DP-based algorithms, joint-break-points were “pruned” using a statistical model selection method (https://hal.inria.fr/inria-00071847). For the final set of CNV segments, we chose the CBS-based results as they did not require specifying a prior number of expected segments (K) per chromosome arm, were robust to unequal variances between the LR and BAF tracks, and provided empirically the best fit to the underlying data. The resulting segmented copy-number profiles were then subject to the joint inference of tumor purity and ploidy and absolute copy number state, implemented in CNVEX, which is most similar to the mathematical formalism of ABSOLUTE127 and PureCN (http://bioconductor.org/packages/PureCN/). Briefly, the algorithm inputs the observed log-ratios (of 10kb bins) and BAFs of individual SNPs. LRs and BAFs are assigned to their joint segments and their likelihood is determined given a particular purity, ploidy, absolute segment copy number, and the number of minor alleles. To identify candidate combinations with a high likelihood, we followed a multi-step optimization procedure that includes grid-search (across purity-ploidy combinations), greedy optimization of absolute copy numbers, and maximum-likelihood inferences of minor allele counts. Following optimization, CNVEX ranks candidate solutions. Because the copy-number inference problem can have multiple equally likely solutions, further biological insights are necessary to choose the most parsimonious result. The solutions have been reviewed by independent analysts following a set of guidelines. Solutions implying whole-genome duplication must be supported by at least one large segment that cannot be explained by a low-ploidy solution, inferred purity must be consistent with the variant-allele-frequencies of somatic mutations, and large homozygous segments are not allowed.
In parallel, we used BIC-seq2,128 a read-depth-based CNV calling algorithm to detect somatic copy number variation (CNVs) from the WGS data of tumors. Briefly, BIC-seq2 divides genomic regions into disjoint bins and counts uniquely aligned reads in each bin. Then, it combines neighboring bins into genomic segments with similar copy numbers iteratively based on Bayesian Information Criteria (BIC), a statistical criterion measuring both the fitness and complexity of a statistical model. We used paired-sample CNV calling that takes a pair of samples as input and detects genomic regions with different copy numbers between the two samples. We used a bin size of ~100 bp and a lambda of 3 (a smoothing parameter for CNV segmentation). We recommend calling segments as copy gain or loss when their log2 copy ratios were larger than 0.2 or smaller than −0.2, respectively (according to the BIC-seq publication).
Structural Variant Calling
Structural variants (SVs) were called by Manta v1.6.096 from WGS tumor and normal paired BAMs. We ran Manta on canonical chromosomes with the default record- and sample-level filters., retaining variants where sample site depth is less than 3x the median chromosome depth near one or both variant breakends, the somatic score is greater than 30, and for small variants (< 1000 bases) in the normal sample, the fraction of reads with MAPQ0 around either breakend does not exceed 0.4. It is optimized for the analysis of somatic variation in tumor/normal sample pairs. The paired and split-read evidence were combined during the SV discovery and scoring to improve accuracy. We prioritized the variants by the number of spanning read pairs that strongly (Q30) support the variants (> 5 as the high confidence level). Lastly, we manually reviewed all the SV calls in the genes of interest.
Instability (wGII Calculation)
To estimate the chromosomal instability, we used a modified version of the Genome Instability Index (GII).129 We calculated GII scores for each sample as the portion of the autosome that has an absolute copy-number unequal to the weighted median absolute copy-number across the autosomal chromosomes. To account for the variation in chromosome size and avoid the overrepresentation of larger chromosomes in the CIN estimation, we used a modified version of GII called weighted Genome Instability Index (wGII).130 To generate wGII, we first calculated the GII for each autosomal chromosome, then took the mean of all the GII scores for all 22 chromosomes.
DNA Methylation Microarray Processing
Raw methylation idat files were downloaded from CPTAC DCC and GDC. Beta values of CpG loci were reported after functional normalization, quality check, common SNP filtering, and probe annotation using Li Ding Lab’s methylation pipeline v1.1 https://github.com/ding-lab/cptac_methylation.
RNA Quantification and Analysis
RNA Quantification
We obtained the gene-level read count, Fragments Per Kilobase of transcript per Million mapped reads (FPKM), and FPKM Upper Quartile (FPKM-UQ) values by following the GDC’s RNA-Seq pipeline (Expression mRNA Pipeline) https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/, except running the quantification tools in the stranded mode. We used HTSeq v0.11.294 to calculate the gene-level stranded read count (parameters: -r pos -f bam -a 10 -s reverse -t exon -i gene_id -m intersection-nonempty --nonunique=none) using GENCODE v22 (Ensembl v79) annotation downloaded from GDC (gencode.gene.info.v22.tsv). The read count was then converted to FPKM and FPKM-UQ using the same formula described in GDC’s Expression mRNA Pipeline documentation.
miRNA Quantification
miRNA-Seq FASTQ files were downloaded from GDC. We reported the mature miRNA and precursor miRNA expression in TPM (Transcripts Per Million) after adapter trimming, quality check, alignment, annotation, reads counting using Li Ding Lab’s miRNA pipeline https://github.com/ding-lab/CPTAC_miRNA. The mature miRNA expression was calculated irrespective of its gene of origin by summing the expression from its precursor miRNAs.
RNA Fusion Detection
We used three callers, STAR-Fusion v1.5.0,101 INTEGRATE v0.2.6,95 and EricScript v0.5.5,93 to call consensus fusion/chimeric events in our samples. Calls by each tool using tumor and normal RNA-Seq data were then merged into a single file and extensive filtering was done. As STAR-Fusion has higher sensitivity, calls made by this tool with higher supporting evidence (defined by fusion fragments per million total reads, or FFPM > 0.1) were required, or a given fusion must be reported by at least 2 callers. We then removed fusions present in our panel of blacklisted or normal fusions, which included uncharacterized genes, immunoglobulin genes, mitochondrial genes, and others, as well as fusions from the same gene or paralog genes and fusions reported in TCGA normal samples,131 GTEx tissues (reported in STAR-Fusion output), and non-cancer cell studies.132 Finally, we removed normal fusions from the tumor fusions to curate the final set.
snRNA-seq Quantification and Analysis
snRNA-seq Data Preprocessing
For each sample, we obtained the unfiltered feature-barcode matrix per sample by passing the demultiplexed FASTQs to Cell Ranger v3.1.0 ‘count’ command using default parameters, and a customized pre-mRNA GRCh38 genome reference was built to capture both exonic and intronic reads. The customized genome reference modified the transcript annotation from the 10x Genomics pre-built human genome reference 3.0.0 (GRCh38 and Ensembl 93).
Seurat v3.1.2133,134 was used for all subsequent analyses. We constructed a Seurat object using the unfiltered feature-barcode matrix for each sample. A series of quality filters were applied to the data to remove those cell barcodes which fell into any one of these categories recommended by Seurat: too few total transcript counts (< 300); possible debris with too few genes expressed (< 200) and too few UMIs (< 1,000); possibly more than one cell with too many genes expressed (> 10,000) and too many UMIs (> 10,000); possible dead cell or a sign of cellular stress and apoptosis with a too high proportion of mitochondrial gene expression over the total transcript counts (> 10%).
Each sample was scaled and normalized using Seurat’s ‘SCTransform’ function to correct for batch effects (with parameters: vars.to.regress = c(“nCount_RNA”, “percent.mito”), variable.features.n = 3000). We then merged all samples and repeated the same scaling and normalization method. All cells in the merged Seurat object were then clustered and the top 30 PCA dimensions via Seurat’s ‘FindNeighbors’ and ‘FindClusters’ (with parameters: resolution = 0.5) functions. The resulting merged and normalized matrix was used for the subsequent analysis.
snRNA-seq Cell Type Annotation
Cell types were assigned to each cluster by manually reviewing the expression of marker genes.135,136 For instance, the marker genes used were AIF1, CD68, LST1, IFITM2 (Macrophages). CD8A, CD8B, CD3E, CD3D, PRF1, GZMA, GZMB, GZMK, GZMH, CD4, IL7R, LTB, LDHB, CD69, FAS, KLRG1, CD28, DPP4 (CD4/CD8 T-cells); CD19, CD79A, CD79B, MS4A1, SDC1, IGHG1, IGHG3, IGH4 (B-cells/Plasma); EMCN, FLT1, PECAM1, KDR, PLVAP, PLVAP, TEK, VWF, ACTA2, ANGPT2, COL1A1, COL3A1, COL5A1, COL12A1, EMILIN1, LUM (Stroma).
snRNA-seq Analysis
Differentially expressed genes within each cell type were identified by the FindMarkers function comparing cells belonging to one subtype (immune subtype or multi-omic subtype) to the rest. Wilcoxon statistical test was used. log2FC > 0.25 and FDR < 0.05 was used to filter DEGs.
Trajectory-based Analysis
We evaluated the relationships between the tumor subclusters observed across the different segments of each of the four cases by constructing their trajectories. Monocle-type analysis of ordering single cells in pseudotime placed the connections of multiple segments along the trajectory. We imported snRNA-seq into Monocle2.137 Parameters for the analysis were consistent with the tutorial (http://cole-trapnell-lab.github.io/monocle-release/docs/#constructing-single-cell-trajectories), except that (1) cell type is set as the variable for differential expression text and (2) to select genes used for ordering, we set 1e–10 as the q value cutoff. We used the function “plot_cell_trajectory” to visualize subcluster projection in the trajectory.
MS Data Interpretation
ccRCC Whole Proteome DIA Data (INI+EXP)
We used data independent acquisition (DIA) proteomics technology to perform protein quantification across the combined set of 487 samples: 199 samples from the confirmatory ccRCC cohort (acquired as part of this work), 94 ITH samples (acquired as part of this work), and 194 discovery ccRCC study samples.13 In addition, 16 DDA files were used as part of the spectral library building step. The DDA files were obtained from fractionated peptide samples (8 fractions from the pooled confirmatory ccRCC sample, and 8 fractions from the pooled discovery ccRCC sample).
Raw mass spectrometry files were converted into mzML file format. FragPipe computational platform (version 15) with MSFragger(version 3.2),138,139 Philosopher (version 3.4.13),140 and EasyPQP (version 0.1.9 doi: https://doi.org/10.1101/2021.03.08.434385) was used to build combined (DIA plus DDA) spectral libraries. DIA files were first processed using DIA-Umpire141 to extract the so-called pseudo-MS/MS spectra (3 mzML files for each input DIA file corresponding to MS/MS spectra assigned to precursors of different quality, indicated as Q1, Q2, and Q3 files). DDA mzML files and DIA-Umpire extracted DIA pseudo-MS/MS mzML files (using the highest quality, Q1, files only) were processed together through all subsequent stages of the spectral library building process. Peptide identification from MS/MS spectra was done using the MSFragger search engine against the CPTAC harmonized H. sapiens RefSeq protein sequence database13 (which included reversed protein sequences appended as decoys for subsequent false discovery rate, FDR, estimation). Both precursor and (initial) fragment mass tolerances were set to 20 ppm. Spectrum deisotoping,142 mass calibration, and parameter optimization139 were enabled. Enzyme specificity was set to ‘stricttrypsin’ (i.e. allowing cleavage before Proline). Up to two missed trypsin cleavages were allowed. Isotope error was set to 0/1/2. Peptide length was set from 7 to 50, and peptide mass was set from 500 to 5000 Da. Oxidation of methionine and acetylation of protein N-termini were set as variable modifications. Carbamidomethylation of Cysteine was set as a fixed modification. Maximum number of variable modifications per peptide was set to 3.
MSFragger search results (in pepXML format) were processed using the Philosopher toolkit.140 First, PeptideProphet143 (run with the high–mass accuracy binning and semi-parametric mixture modeling options) was run to compute the posterior probability of correct identification for each peptide to spectrum match (PSM). The resulting output files from PeptideProphet were processed together using ProteinProphet144 to perform protein inference (assemble peptides into proteins) and to create a combined file (protXML format) of high confidence proteins groups, encompassing both DDA and DIA-identified peptides. The minimum PeptideProphet probability for input to ProteinProphet was set to 0.9. The combined ProteinProphet file was further processed using Philosopher Filter command, which characterized each identified peptide as unique peptide to a particular protein (or protein group containing indistinguishable proteins) or assigned it as a razor peptide to a single protein (protein group) that had the most peptide evidence. Both unique and razor peptides were used for subsequent analysis. The data was filtered to 1% protein-level FDR using the picked FDR strategy.145 The peptide, PSM, and ion-level reports were then generated and filtered using the 2D FDR approach (i.e. 1% protein FDR plus 1% PSM/ion/peptide-level FDR for each corresponding PSM.tsv, ion.tsv, and peptide.tsv files).146
PSM.tsv files, filtered as described above, along with the spectral files (mzML files used as input to MSFragger) were used as input to EasyPQP for generation of the consensus spectrum library. As an additional filter in EasyPQP, only peptides contained in the Philosopher-generated peptide.tsv report file were used, ensuring that the resulting spectral library was filtered to global 1% FDR at both protein and peptide level. EasyPQP was run with the ‘RT selection option’ set to ‘Automatic selection of a run as reference run’. Thus, peptide retention times (RT) in each run were non-linearly aligned (using loess method) by EasyPQP to a reference run (which was one of the DIA runs in the dataset showing the best average correlation coefficient against all other runs in the experiment). Only y and b fragments ions were considered, and the fragment ion annotation tolerance was set to 15ppm. The final spectral library contained 178022 precursors representing 9245 proteins.
The spectral library described above was used for targeted extraction of precursor ion and protein intensities from the 487 DIA runs (samples) using DIA-NN (version 1.7.13)146 as previously described. Protein inference in DIA-NN was disabled to use peptide-protein grouping as provided by the spectral library. The MS1 and MS2 tolerances and the RT extraction window were automatically determined for each run by the algorithm. Quantification mode was set to “Robust LC (high precision)”. The output was filtered at experiment-specific precursor Q-value < 1%, global protein Q-value < 1%, and run-specific protein Q-value < 1%. Protein abundances were computed from the precursor ion intensities (summed to the unique gene symbol level) using the DIA-NN reimplementation of the MaxLFQ147 normalization method. The final table contained protein level quantification for 8363 genes.
ccRCC Phosphoproteomic Data (EXP)
Analysis of the phosphopeptide quantification data for the 199 samples from the confirmatory ccRCC cohort profiled using DIA was performed as described above, with the following changes. The spectral library was built from the 199 DIA runs supplemented with 9 DDA runs from the pooled fractionated phosphopeptide sample. All 3 sets of pseudo-MS/MS files extracted by DIA-Umpire for each run (i.e., Q1, Q2, and Q3 mzML files) were used. MSFragger search parameters included an additional variable modification - phosphorylation on STY. Isotope error was set to 0/1. After PeptideProphet and before ProteinProphet, PTMProphet148 was run to perform phosphosite localization, which was then propagated to the PSM.tsv reports by Philosopher. The resulting spectral library built with EasyPQP contained 7968 proteins and 121563 precursors (including non-phosphorylated proteins and peptides). When running DIA-NN, the PTM scoring option for phosphorylation was activated using the ‘--monitor-mod Unimod 21’ command. The precursor-level output table generated by DIA-NN was further processed using an R script available as part of the DIA-NN distribution to create a “sequence plus modification”-level report by summing precursor intensities based on the “Modified.Sequence” column. The data was filtered to global and run-specific precursor and protein Q-values < 0.01. MaxLFQ methods were used to roll-up and normalize precursor intensities to the “sequence plus modification” level. The resulting table was then additionally processed to remove non-phosphorylated peptides and to mark which sites were localized with confidence by PTMProphet (localization probability 0.75 or higher) at the spectral library building step. The final table contained quantitative information for 71913 phosphorylated peptide forms, representing 26998 peptides from 6467 proteins (6262 unique gene symbols).
Kinase Inhibitor Study, Proteomic and Phosphoproteomic Data
Analysis of the DIA data from the kinase inhibitor study (whole proteome and phosphopeptide-enriched data) was performed as described above. For each data type, the libraries were built from the corresponding 30 DIA runs (6 treatments × 5 cell lines) supplemented with 8 or 9 fractionated DDA files for whole proteome and phosphopeptide-enriched samples, respectively. The resulting whole proteome spectral library contained 8882 proteins and 173932 precursors; the phosphopeptide enriched sample library contained 7841 proteins and 101491 precursors (including non-phosphorylated proteins and peptides). After DIA-NN quantification, the final quantification table for the phosphopeptide-enriched dataset contained quantification information for 46577 phosphorylated peptides forms, representing 22161 peptide sequences from 5154 proteins. The whole proteome dataset table contained quantification for 7654 genes.
Quantification of Intact Glycopeptides
The DIA raw files of the intact glycopeptides were searched against the spectral library for the quantification of intact glycopeptides via Spectronaut (version 15.4, Biognosys). Mass tolerance of MS and MS/MS was set as dynamic with a correction factor of one. Source-specific iRT calibration was enabled with a local (non-linear) RT regression. All multi-channel interferences were excluded and the decoy method was set as “mutated”. The precursors were filtered by a Q value cutoff of 0.01 (which corresponds to an FDR of 1%). The quantity of a modified peptide was decided by summing the quantity of its precursors, whereas the quantity for a precursor was calculated by summing the area of its fragment ions at MS2 level. The reported quantification result was filtered as previously described113. In brief, the filtering criteria consisted of following: the FWHM of XIC of the fragment ions < 1 minute, the shape quality score for the XIC of the precursor transition groups > 0.6, S/N ratio of the fragment ions > 3, and cosine similarity between theoretical and measured isotopic patterns of precursors > 0.9. The missing values were imputed using DreamAI (https://github.com/WangLab-MSSM/DreamAI), which was the tool used in our previous study for the imputation of phosphoproteomic data. Only glycopeptides with a missing rate less than 50% across all samples were imputed.
Processing of Metabolomic Data
Acquired data were analyzed first using Thermo Scientific Compound Discoverer® software. The chromatographic peaks were integrated to obtain raw intensities of metabolites. Compounds with definite peaks and names in the software were selected. The data were then filtered based on the following criteria: m/z Cloud score greater than 60 (good fragmentation matching with compounds in the m/z Cloud database) or mass list match (mass lists include common pathways such as glycolysis, pentose phosphate pathway, hexosamine, and sialic acid pathway, purine and pyrimidine synthesis, and amino acid metabolism) and intensity > 10000. Thermo Scientific TraceFinder® software was then used to quantify compounds in common pathways not found using Compound Discoverer® where the retention time (RT) was determined using Freestyle® software based on mass accuracy and fragmentation match. The data from Thermo Scientific Compound Discoverer® and TraceFinder® software were combined to generate the final list of compounds.
Other Proteogenomic Analysis
Differential Abundance Analysis
Global proteomic data and gene expression were used to perform pairwise differential analysis between groups of samples. A Wilcoxon rank-sum test was performed to determine the differential abundance of proteins and gene expression. At least four samples in both groups were required to have non-missing values, and the p-value was adjusted using the Benjamini-Hochberg procedure, and features were considered significant with an adjusted p-value < 0.05. Proteomic features with at least a 2x fold increase in tumors were deemed to be tumor-associated markers. These markers were the DEGs/DEPs captured by our “level 1” DE analysis on the cohort level using the bulk proteogenomic data. To select the top feature-associated marker candidates, we performed DE analysis with utilizing the bulk proteogenomic data in the intratumoral heterogeneity (ITH) cohorts (e.g., given cases with multiple segments) on the case level as “level 2”; snRNA-seq on the segment level as “level 3”, specifically, among the tumor cell population; and last, snRNA-seq on the tumoral-cluster level as “level 4” with the resolution to identify specific tumor subpopulations.
Tumor Microenvironment Inference
The ESTIMATE scores reflecting the overall immune and stromal infiltration were calculated by the R package ESTIMATE113 using the normalized RNA expression data (FPKM-UQ).
Cell Type Enrichment Using Gene Expression
The abundance of each cell type was inferred by the xCell web tool,105 which performed the cell type enrichment analysis from gene expression data for 64 immune and stromal cell types (default xCell signature). xCell is a gene signatures-based method learned from thousands of pure cell types from various sources. We used the FPKM-UQ expression matrix as the input of xCell. xCell generated an immune score per sample that integrates the enrichment scores of B cells, CD4+ T-cells, CD8+ T-cells, DC, eosinophils, macrophages, monocytes, mast cells, neutrophils, and NK cells; a micro-environment score which was the sum of the immune score and stroma score. Besides, we applied CIBERSORTx106 to compute immune cell fractions from bulk gene expression data.
Immune Clustering Using Cell Type Enrichment Scores
Immune subtypes of each of the four cancer types were generated based on the consensus clustering90 of the cell type enrichment scores by xCell. Among the 64 cell types tested in xCell, we selected the cell types that were significant in at least 10% of the samples (xCell enrichment p < 0.05, which filtered out the cell types not typical in kidneys). We performed consensus immune clustering based on the z-score normalized xCell enrichment scores. The consensus clustering was determined by the R package ConsensusClusterPlus(parameters: reps = 2000, pItem = 0.9, pFeature = 0.9, clusterAlg = “kmdist”, distance = “spearman”).
Survival Analysis
The R package “survival” was used to perform survival analysis. The Kaplan-Meier curve of overall survival was used to compare the prognosis among subtypes (function survfit). Log-rank test (from the R package survminer) was used to test the differential survival outcomes between categorical variables. The standard multivariate Cox-proportional hazard modeling was applied to estimate the hazard ratio among subtypes (function coxph). Age, gender, histopathologic subtype, and BAP1 mutation status, as the covariates, were included in the model.
Panoptes-based multi-resolution Neural Network Imaging Models
The Panoptes-based multi-resolution neural network imaging models were trained with digitized H&E stained histopathologic slide images. Due to the size and the multi-resolution data structure of the whole slide images, they were cut into 299×299 pixel tiles at 10x, 5x, and 2.5x equivalent magnification of the scanned whole slide images. 10x, 5x, and 2.5x tiles covering the same regions were then grouped into tilesets and were treated as 1 sample following the Panoptes sample preparation protocol.33 The samples were split into training, validation, and testing set at 70:15:15 ratio at the per-patient level for BAP1 mutation prediction task, and per-slide level for immune and methylation subtype prediction tasks. The models were trained with a batch size of 24, the initial learning rate of 0.0001, the dropout rate of 0.5, and Adam optimizer with early stop criteria when the validation loss did not decrease for at least 10000 iterations and the state at which the lowest validation loss was achieved were recorded to be the final model for testing. 4 Panoptes architectures were trained simultaneously into models and the best performing models were selected based on various statistical metrics, particularly AUROC. The activations of the second-to-the-last layer of the test set were extracted and dimensionally reduced and plotted with tSNE for feature visualization. Example tiles were highlighted and sent to pathologists for a secondary review. Selected whole slide cases from the test set were fed into the trained model and per-tile level predictions were aggregated into heatmap layers to overlay onto the original slides for feature visualization and localization.
Ancestry Prediction Using SNPs from 1000 Genomes Project
We used a reference panel of genotypes and a clustering based on principal components to identify likely ancestry. We selected 107,765 coding SNPs with a minor allele frequency > 0.02 from the final phase release of The 1000 Genomes Project.149 From this set of loci, we measured the depth and allele counts of each sample in our cohort using bam-readcount v0.8.0. Genotypes were then called for each sample based on the following criteria: 0/0 if reference count ≥ 8 and alternate count < 4; 0/1 if reference count ≥ 4 and alternate count ≥ 4; 1/1 if reference count < 4 and alternate count ≥ 8; and ./. (missing) otherwise. After excluding markers with missingness > 5%, 70,968 markers were kept for analysis. We performed PCA on the 1000 Genomes samples to identify the top 20 principal components. We then projected our cohort onto the 20-dimensional space representing the 1000 Genomes data. We then trained a random forest classifier with the 1000 Genomes dataset using these 20 principal components. The 1000 Genomes dataset was split 80/20 for training and validation respectively. On the validation dataset, our classifier achieved 99.6% accuracy. We then used the fitted classifier to predict the likely ancestry of our cohort.
MSI Prediction
MSI scores were calculated by MSIsensor (https://github.com/ding-lab/msisensor) and interpreted as the percentage of microsatellite sites (with deep enough sequencing coverage) that have a lesion. Samples with an MSIscore > 3.5 are classified as “MSI-High” and the rest will be classified as “MSS.” An intermediate class with 1.0 <= score <= 3.5 can be defined as “MSI-Low.”
Unsupervised Multi-omic Clustering Using NMF
We used non-negative matrix factorization (NMF)-based multi-omic clustering using protein abundance, RNA transcript abundance, and log ratios of gene copy number variants (CNV).
Balancing Contribution of Data Types:
To mitigate the impact of a potential bias towards a particular data type in the multi-omic clustering (e.g. vastly different number of genomic and proteomic features), the following filtering approach was applied: Data matrices were concatenated and all rows containing missing values were removed. The resulting multi-omic data matrix was then standardized by z-scoring of the rows followed by z-scoring of columns. Principal component analysis (PCA) was applied to the resulting standardized multi-omic data matrix. The PCA-derived factors matrix was used to determine the number of principal components (PCs) cumulatively explaining 90% of the variance in the standardized multi-omic data matrix (PCs90). The PCA-derived loadings matrix was used to calculate the relative contribution of each feature to each PCs90, equivalent to the squared cosine described in (Abdi and Williams https://wires.onlinelibrary.wiley.com/doi/10.1002/wics.101), and the relative, cumulative contributions of each feature across all PCs90 was subsequently derived. The resulting vector of relative contributions of each feature (i.e. vector sums up to 1) was then used to balance the contribution of the different data types using the following procedure:
For each data type sum up the contributions of all features; this determines the overall contribution of each data type, which ideally should be equal across the data types within a given tolerance, i.e.: sumome ≈ 1/(No. data types)
Remove the feature with the lowest contribution that belongs to the data type with the largest overall contribution
Recalculate the overall contributions of each data type and repeat steps 1–2 until the deviation is within the specified tolerance (tol=0.01).
Non-negative Transformation:
The data matrix of z-scores was converted to a non-negative input matrix required by NMF as follows:
Create one data matrix with all negative numbers zeroed.
Create another data matrix with all positive numbers zeroed and the signs of all negative numbers removed.
Concatenate both matrices resulting in a data matrix twice as large as the original, but containing only positive values and zeros and hence appropriate for NMF.
Non-negative Matrix Factorization:
Given a factorization rank k, where k is the number of clusters, NMF decomposes a p × n data matrix V (p - number of features; n - number of samples) into two matrices W and H such that multiplication of W and H approximates V. Matrix H is a k × n matrix whose entries represent weights for each sample (1 to n) to contribute to each cluster (1 to k), whereas matrix W is a p × k matrix representing weights for each feature (1 to p) to contribute to each cluster (1 to k). Matrix H was used to assign samples to clusters by choosing the row (i.e. cluster) with the maximum score in each column of H.
Determination of Factorization Rank:
To determine the optimal factorization rank k (number of clusters) for the multi-omic data matrix, a range of clusters between k=2 and 8 was tested. For each value of the k matrix, V was subjected to NMF using 50 iterations with random initialization of W and H. To determine the optimal factorization rank two metrics for each value of k were calculated: 1) cophenetic correlation coefficient measuring how well the intrinsic structure of the data is recapitulated after clustering and 2) the dispersion coefficient of the consensus matrix as defined in150 measuring the reproducibility of the clustering across the 50 iterations. The optimal kopt is defined as kopt= max(dispK^(1-cophK) for cluster numbers between k=3 and 8.
Having determined the optimal factorization rank k, to achieve robust factorization of the multi-omic data matrix V, the NMF procedure described above was repeated using 500 iterations with random initializations of W and H. Due to the non-negative transformation applied to the z-scored data matrix as described above, matrix W of feature weights contained two separate weights for positive and negative z-scores of each feature, respectively. To revert the non-negative transformation and to derive a single signed weight for each feature, we first normalized each row in matrix W by dividing by the sum of feature weights in each row, aggregated both weights per feature and cluster by keeping the maximal normalized weight and multiplication with the sign of the z-score in the initial data matrix. Thus, the resulting transformed version of matrix Wsigned contained signed cluster weights for each feature in the input matrix.
Cluster Membership:
For each sample, a cluster membership score was calculated as the maximal fractional score of the corresponding column in matrix H. The score indicates how representative a sample is to each cluster and was used to define the “cluster core”, a set of samples most representative for a given cluster. Core samples were required to have a minimal membership score difference between all pairs of clusters to be greater than 1/k, where k is the total number of clusters.
The entire workflow described above has been implemented as a module for PANOPLY151 (https://github.com/broadinstitute/PANOPLY) which runs on Broad’s Cloud platform Terra (https://app.terra.bio/).
Unsupervised Clustering of DNA Methylation
Methylation subtypes were segregated based on the top 8,000 most variable probes using k-means consensus clustering as previously described.152 We first removed underperforming probes,153 and then the samples with more than 30% missing values. Remaining missing values were imputed using the mean of the corresponding probe value. We then performed clustering 1000 times using the ConsensusClusterPlus R package (parameters: maxK = 10 reps = 1000 pItem = 0.8 pFeature = 1 clusterAlg = “km” distance = “euclidean”). We choose k = 6 based on the delta area plot of consensus CDF.
Determination of Stemness Score
Stemness scores were calculated as previously described.154 Firstly, we used MoonlightR155 to query, download, and preprocess the pluripotent stem cell samples (ESC and iPSC) from the Progenitor Cell Biology Consortium (PCBC) dataset.156,157 Secondly, to calculate the stemness scores based on mRNA expression, we built a predictive model using one-class logistic regression (OCLR)158 on Progenitor Cell Biology Consortium (PCBC) dataset. For mRNA expression-based signatures, to ensure compatibility with our cohort, we first mapped the Ensembl IDs to Human Genome Organization (HUGO) gene names and dropped any genes that had no such mapping. The resulting training matrix contained 12,945 mRNA expression values measured across all available PCBC samples. To calculate the mRNA-based sternness index (mRNASi), we used FPKM-UQ mRNA expression values for all CPTAC ccRCC tumors. We used the TCGAanalyze_Stemness function from the R package TCGAbiolinks159 and following our previously described workflow,160 with “stemSig” argument set to PCBC stemSig.
Mutation Impact on RNA, Proteome, Phosphoproteome, and Metabolome
We aggregated a set of interacting proteins (e.g. kinase/phosphatase-substrate or complex partners) from OmniPath (downloaded on 2018–03-29),84 DEPOD (downloaded on 2018–03-29),161 CORUM (downloaded on 2018–06-29),162 Signor2 (downloaded on 2018–10-29),163 and Reactome (downloaded on 2018–11-01).164 We focused our analyses on ccRCC SMGs previously reported in the literature.120
For each interacting protein pair, we split samples with and without mutations in partner A and compare expression levels (RNA, protein, and phosphosites) both in cis (partner A) and in trans (partner B), calculating a median difference in expression and testing for significance with the Wilcoxon rank-sum test, with the Benjamini-Hochberg multiple test correction. For mutational impact analysis on metabolomes, all possible pairs between SMGs and metabolites were tested.
Kinase-substrate Pairs Regression Analysis
For each kinase-substrate protein pair supported by previous experimental evidence (OmniPath, NetworKIN, DEPOD, and SIGNOR), we tested the associations between all sufficiently detected phosphosites on the substrate and the kinase. For a kinase-substrate pair to be tested, we required both kinase protein/phosphoprotein expression and phosphosite phosphorylation to be observed in at least 20 samples in the respective datasets and the overlapped dataset. We then applied the linear regression model using lm function in R to test for the relation between kinase and substrate phosphosite. For the i-th trial for kinase phosphosite abundance in the cis associations, kinase phosphosite abundance Ai depends on kinase protein expression Si and error Ei,
For the i-th trial for kinase phosphosite abundance in the trans associations, substrate phosphosite abundance Ai depends on kinase phosphosite expression Ki substrate protein expression Si and error Ei,
where the regression slope M coefficients are determined by least-square calculation. Bs are y-axis intercepts. The resulting p-values were adjusted for multiple testing using the Benjamini-Hochberg procedure.
Phosphoproteomic and Glycoproteomic Subtyping
Phosphopeptides with CVs in the > 25% quartile were analyzed by CancerSubtypes165 for consensus clustering of tumor subtypes. The same procedure was carried out for glyco subtyping using intact glycopeptides as well. Specifically, 80% of the original sample pool was randomly subsampled without replacement and partitioned into four major clusters (phospho) and three major clusters (glyco) using hierarchical clustering, which was repeated 2000 times. The consensus-clustered samples were overlaid with other features (e.g., grade, stage) and other omics subtypes (e.g., methylation subtype, histopathologic subtype). Phosphopeptides and intact glycopeptides were grouped into four and three clusters using K-means clustering in ComplexHeatmap,166 respectively. The predictive models of phospho- and glyco-signatures were built using caret (https://doi.org/10.18637/jss.v028.i05) and ROC curves were generated using pROC.167 We performed KEGG pathway enrichment analysis via WebGestalt.168 We utilized PTM-SEA to find signatures (pathways and kinases) of the phospho subtypes. We first conducted the differential analysis between a phospho subtype to the remaining phospho subtypes (P1 vs Others, P2 vs Others etc.) as well as the pairwise comparison between phospho subtypes (P1 vs P2, P1 vs P3 etc.) on phosphosite level by calculating median log2 fold change and obtaining p-value from Wilcoxon rank-sum test. Next, we examined each differentially expressed phosphosites in one phospho subtype relative to the remaining phospho subtypes (p≤0.05 and fold change ≥1.5) to ensure that it was also differentially expressed (p≤0.05 and fold change ≥1.5) in the particular subtype from the pairwise comparison (at least compared to two out of three other phospho subtypes) in order to generate a list of phosphosites for PTM-SEA input. To obtain a single enrichment score from PTM-SEA and adequately account for variance in phosphosite abundance across subtypes, we utilized the differential analysis results from one subtype vs the remaining to calculate signed (according to the fold change between one subtype and the remaining), log-transformed p-value from Wilcoxon Rank Sum Test as input to PTM-SEA. Only pathways and kinases significantly enriched (FDR < 0.05) in at least one of the subtypes were plotted. The differential analysis between a glyco subtype to the remaining glyco subtypes was conducted by calculating median log2 fold change and using Wilcoxon rank-sum test (p-value was adjusted using Benjamini Hochberg method). The significance threshold was set as FDR < 0.05.
The setting in PTM-SEA is as follows.
sample.norm.type: rank
weight: 1
statistic: area.under.RES
output.score.type NES
nperm: 5000
min.overlap: 5
correl.type: rank
Metabolome Analysis
Metabolome data were used to perform pairwise differential analysis between groups of samples. A Wilcoxon rank-sum test was performed to determine the differential abundance of metabolites. At least four samples in both groups were required to have non-missing values and the p-value was adjusted using the Benjamini-Hochberg procedure. The metabolite annotations were based on HMDB (https://hmdb.ca/), MetaboAnalyst (https://www.metaboanalyst.ca/), and KEGG (https://www.genome.jp/kegg/).
Interactive Data Visualization and Exploration
We have developed a ProTrack web portal169 for interactive visualization and exploration of this data set. The ProTrack web app consists of two main views: a sample dashboard and an interactive heatmap. The sample dashboard visualizes the distribution of the cohorts along clinical, demographic, and molecular variables. The graphs can be reordered and hidden or shown according to user preference. The graphs can also be used to create custom cohorts, as users can filter samples into a custom cohort by toggling demographic features on and off. The filtered cohort can optionally be used to generate an interactive heatmap. On the heatmap view, users input a query list of genes of interest. A multi-omic heatmap is then generated for those genes, including protein, RNA, phosphoprotein, and glycoprotein data tracks when available. Additionally, using the interactive legend, users can add or remove top tracks to include immune subtype classification tracks, mutation information, chromosomal gains or losses, and clinical or demographic data such as BMI, hypertension, vital status. To facilitate the visualization of trends of interest, users can select any track and sort the entire heatmap along that axis. The underlying ordered data can then be downloaded as an Excel table and the heatmap can be exported as an image file. The ProTrack application is available at http://ccrcc-conf.cptac-data-view.org.
Supplementary Material
Table S1. CPTAC ccRCC cohort summary, ccRCC histopathological annotation, and feature-associated markers, related to Figure 1.
Table S2. CPTAC ccRCC ITH cohort summary, ccRCC ITH histopathological annotation, CNV, and imaging statistics, related to Figure 2.
Table S3. snRNA-seq meta table, cell type fraction, and sarcomatoid-associated and rhabdoid-associated markers, related to Figures 3–4.
Table S4. ccRCC methylation subtype, methylation signature probes, methylation DEG and DEP, and inhibition study of UCHL1, related to Figure 5.
Table S5. Kinase-substrate network, phosphoproteome subtype and associated signatures, and cell line results, related to Figure 6.
Table S6. Glycoproteome subtype and associated signatures, tumor and NAT comparison, and CH and CL comparison, related to Figure 7.
Table S7. Quantified metabolites, metabolome PCA, metabolome subtype and associated signatures, and metabolome feature validation, related to Figure 8.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti CA9 antibody | Novus Biologicals | Catalog: NB100-417, RRID: AB_10003398 |
| Rabbit polyclonal anti HYOU1 antibody | Atlas Antibodies | Catalog: HPA049296, RRID: AB_2680702 |
| Rabbit polyclonal anti GAL3ST1 antibody | Atlas Antibodies | Catalog: HPA001220, RRID: AB_1078933 |
| Rabbit polyclonal anti KIF2A antibody | Atlas Antibodies | Catalog: HPA004716, RRID: AB_1079211 |
| Rabbit polyclonal anti PLXDC2 antibody | Atlas Antibodies | Catalog: HPA017268, RRID: AB_10965928 |
| Rabbit polyclonal anti TGFBI antibody | Atlas Antibodies | Catalog: HPA008612, RRID: AB_1857970 |
| Rabbit polyclonal anti UCHL1 antibody | Sigma-Aldrich | Catalog: HPA005993, RRID: AB_1858560 |
| Mouse monoclonal anti BAP1 antibody | Santa Cruz Biotechnology | Catalog: sc-28382 |
| Biological samples | ||
| Primary tumor and normal adjacent tissue samples | This manuscript | See Table S1 |
| Critical commercial assays | ||
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold | Illumina | Catalog: RS-122-2301 |
| Infinium MethylationEPIC Kit | Illumina | Catalog: WG-317-1003 |
| Exome | Illumina | Catalog: 20020617 |
| KAPA Hyper Prep Kit, PCR-free | Roche | Catalog: 07962371001 |
| BCA Protein Assay Kit | ThermoFisher Scientific | Catalog: 23225 |
| RPMI Medium 1640 | ThermoFisher Scientific | Catalog:11875168 |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher Scientific | Catalog: 15140122 |
| Fetal bovine serum | ThermoFisher Scientific | Catalog: 16140071 |
| Adaversotib (AZD-1775) | MedChemExpress | Catalog: HY-10993 |
| Everolimus | Selleckchem | Catalog: S1120 |
| Sapanisterib (TAK-228) | Selleckchem | Catalog: S2811 |
| Gefitinib | Cell Signaling Technology | Catalog: 4765S |
| Tramenitib | Selleckchem | Catalog: S2673 |
| Dimethyl sulfoxide | ThermoFisher Scientific | Catalog: 85190 |
| CellTiter® 96 AQueuous One Solution Proliferation Assay (MTS) | ThermoFisher Scientific | Catalog: PR-G3580 |
| OptiView DAB detection kit | Roche-Ventana Medical Systems | Catalog: 760-700 |
| EnVision FLEX visualizing kit | Agilent Technologies Inc | Catalog: K800221-2 |
| AlamarBlue™ Cell Viability Reagent | Invitrogen | Catalog: DAL1025 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Catalog: G9241 |
| UltraView Universal DAB detection kit | Roche-Ventana Medical Systems | Catalog: 760-500 |
| Cell lines | ||
| Renal cancer cell line 769-P | ATCC | Catalog: CRL-1933 |
| Renal cancer cell line 786-O | ATCC | Catalog: CRL-1932 |
| Renal cancer cell line A498 | ATCC | Catalog: HTB-44 |
| Renal cancer cell line CAKI-1 | ATCC | Catalog: HTB-46 |
| Renal cancer cell line CAKI-2 | ATCC | Catalog: HTB-47 |
| Renal cell line HK-2 | ATCC | Catalog: CRL-2190 |
| Chemicals, peptides, and recombinant proteins | ||
| Calbiochem | Sigma-Aldrich | Catalog: 662086 |
| L-Methionine sulfoximine | Sigma-Aldrich | Catalog: M5379 |
| Phenylmethylsulfonyl fluoride | Sigma-Aldrich | Catalog:93482 |
| Sodium fluoride | Sigma | Catalog: S7920 |
| Phosphatase Inhibitor Cocktail 2 | Sigma | Catalog: P5726 |
| Phosphatase Inhibitor Cocktail 3 | Sigma | Catalog: P0044 |
| Urea | Sigma | Catalog: U0631 |
| Tris(hydroxymethyl)aminome thane | Invitrogen | Catalog: AM9855G |
| Ethylenediaminetetraacetic acid | Sigma | Catalog: E7889 |
| Sodium chloride | Santa Cruz Biotechnology | Catalog: sc-295833 |
| PUGNAc | Sigma | Catalog: A7229 |
| Dithiothretiol | ThermoFisher Scientific | Catalog: 20291 |
| Iodoacetamide | ThermoFisher Scientific | Catalog: A3221 |
| Sequencing grade modified trypsin | Promega | Catalog: V511X |
| Lysyl endopeptidase, aass spectrometry grade | Wako Chemicals | Catalog: 125-05061 |
| Formic acid | Fisher Chemical | Catalog: A117-50 |
| Reversed-phase C18 SepPak | Waters | Catalog: WAT054925 |
| 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid | Alfa Aesar | Catalog: J63218 |
| Ammonium formate | Sigma | Catalog: 70221 |
| Tandem mass tags - 10plex | ThermoFisher Scientific | Catalog: 90110 |
| Trifluoroacetic acid | Sigma | Catalog: 302031 |
| Ammonium Hydroxide solution | Sigma | Catalog: 338818 |
| Hydroxylamine solution | Aldrich | Catalog: 467804 |
| Ni-NTA agarose beads | Qiagen | Catalog: 30410 |
| Iron (III) chloride | Sigma | Catalog:451649 |
| Oasis MAX Cartridge | Waters | Catalog: 186000366 |
| Triethylammonium acetate buffer | Sigma | Catalog: 90358 |
| Data deposition | ||
| CPTAC ccRCC clinical data and proteomic data | This manuscript | https://pdc.cancer.gov/ |
| CPTAC ccRCC genomic, transcriptomic, and snRNA-seq data | This manuscript | https://portal.gdc.cancer.gov/projects/CPTAC-3 |
| CPTAC ccRCC pathology and radiology images | This manuscript |
https://portal.imaging.datacommons.cancer.gov/explore/filters/?collection_id=cptac_ccrcc
https://doi.org/10.7937/K9/TCIA.2018.OBLAMN27 |
| TCGA KIRC | Cancer Genome Atlas Research et al.83 | https://portal.gdc.cancer.gov/ |
| OmniPath | Turei et al.84 | https://omnipathdb.org/#faq |
| NetworKIN | Linding et al.85 | https://networkin.info/ |
| DEPOD | Damle et al.86 | http://www.depod.bioss.uni-freiburg.de/ |
| SIGNOR | Licata et al.87 | https://signor.uniroma2.it/ |
| Software and algorithms | ||
| BWA v0.7.17-r1188 | Li et al.88 | http://bio-bwa.sourceforge.net/ |
| CharGer v.0.5.4 | Scott et al.89 | https://github.com/ding-lab/CharGer |
| COCOON | Clark et al.13 | https://github.com/ding-lab/COCOONS |
| ConsensusClusterPlus v1.48.0 | Wilkerson et al.90 | https://bioconductor.org/packages/ConsensusClusterPlus/ |
| COSMIC Mutational Signatures v3 | Alexandrov et al.91 | https://cancer.sanger.ac.uk/cosmic/signatures/ |
| DEPO | Sun et al.92 | http://dinglab.wustl.edu/depo |
| EricScript v0.5.5 | Benelli et al.93 | https://sites.google.com/site/bioericscript/ |
| germlinewrapper v1.1 | Clark et al.13 | https://github.com/ding-lab/germlinewrapper |
| HTSeq v0.11.2 | Anders et al.94 | https://github.com/simon-anders/htseq |
| INTEGRATE v0.2.6 | Zhang et al.95 | https://sourceforge.net/projects/integrate-fusion/ |
| Manta v1.6.0 | Chen et al.96 | https://github.com/Illumina/manta |
| MuTect v1.1.7 | Cibulskis et al.97 | https://github.com/broadinstitute/mutect |
| Pindel v0.2.5 | Ye et al.98 | https://github.com/genome/pindel |
| Python v3.7 | Python Software Foundation | https://www.python.org/ |
| R v3.6 | R Development Core Team | https://www.R-project.org |
| R-rollup | Polpitiya et al.99 | https://omics.pnl.gov/software/danter |
| Samtools v1.2 | Li et al.100 | https://www.htslib.org/ |
| SignatureAnalyzer | Alexandrov et al.91 | https://github.com/broadinstitute/getzlab-SignatureAnalyzer |
| somaticwrapper v1.6 | Clark et al.13 | https://github.com/ding-lab/somaticwrapper |
| STAR-Fusion v1.5.0 | Haas et al.101 | https://github.com/STAR-Fusion/STAR-Fusion |
| Strelka v2.9.2 | Kim et al.102 | https://github.com/Illumina/strelka |
| UpSetR | Conway et al.103 | https://github.com/hms-dbmi/UpSetR/ |
| VarScan v2.3.8 | Koboldt et al.104 | https://dkoboldt.github.io/varscan/ |
| xCell v1.2 | Aran et al.105 | http://xcell.ucsf.edu/ |
| CIBERSORTx | Newman et al.106 | https://cibersortx.stanford.edu/ |
| MetaboAnalyst 5.0 | Pang et al.107 | https://www.metaboanalyst.ca/ |
Highlights.
Integrated multi-omics and histopathology reveal intratumoral heterogeneity in ccRCCs
Signatures of aggressive sarcomatoid and rhabdoid histology are uncovered by snRNA-seq
High-grade ccRCCs have specific glycoproteomic, metabolomic, and methylation signatures
UCHL1 correlates with methylation, genome instability, BAP1 mutation, and poor survival
Acknowledgments
The Clinical Proteomic Tumor Analysis Consortium (CPTAC) is supported by the National Cancer Institute of the National Institutes of Health under award numbers U24CA210955, U24CA210985, U24CA210986, U24CA210954, U24CA210967, U24CA210972, U24CA210979, U24CA210993, U01CA214114, U01CA214116, U01CA214125, and U24CA271114.
Clinical Proteomic Tumor Analysis Consortium
Eunkyung An, Andrzej Antczak, Meenakshi Anurag, Thomas Bauer, Jasmin Bavarva, Chet Birger, Michael J. Birrer, Melissa Borucki, Gabriel Bromiński, Shuang Cai, Anna Calinawan, Song Cao, Wagma Caravan, Steven A. Carr, Patricia Castro, Sandra Cerda, Daniel W. Chan, Feng Chen, Lijun Chen, Siqi Chen, Xi S. Chen, David Chesla, Arul M. Chinnaiyan, Hanbyul Cho, Kyung-Cho Cho, Seema Chugh, Marcin Cieslik, David J. Clark, Antonio Colaprico, Sandra Cottingham, Felipe da Veiga Leprevost, Aniket Dagar, Nataly Naser Al Deen, Saravana M. Dhanasekaran, Rajiv Dhir, Li Ding, Marcin J. Domagalski, Brian J. Druker, Elizabeth Duffy, Maureen Dyer, Jennifer Eschbacher, Mina Fam, David Fenyö, Brenda Fevrier-Sullivan, John Freymann, Alicia Francis, Jesse Francis, Stacey Gabriel, Gad Getz, Michael A. Gillette, Charles A. Goldthwaite, Jr., Anthony Green, Shenghao Guo, Jason Hafron, Ari Hakimi, Pushpa Hariharan, Sarah Haynes, David Heiman, Tara Hiltke, Barbara Hindenach, Katherine A. Hoadley, Jennifer Hon, Runyu Hong, Alex Hopkins, Noshad Hosseini,Galen Hostetter, James Hsieh, Yingwei Hu, Jasmine Huang, Michael M Ittmann, Scott D. Jewell, Xiaojun Jing, Corbin D. Jones, Karen A. Ketchum, Justin Kirby, Iga Kołodziejczak, Karsten Krug, Chandan Kumar-Sinha, Paweł Kurzawa, Alexander J. Lazar, Toan Le, Anne Le, Qing Kay Li, Yize Li, Ginny Xiaohe Li, Qin Li, Tung-Shing M. Lih, W. Marston Linehan, Wenke Liu, Tao Liu, Rita Jui-Hsien Lu, Jan Lubiński, Weiping Ma, D. R. Mani, Rahul Mannan, Sailaja Mareedu, Nicollette Maunganidze, Rohit Mehra, Mehdi Mesri, Rebecca Montgomery, Alexey I. Nesvizhskii, Chelsea J. Newton, Gilbert S. Omenn, Russell Pachynski, Oxana V. Paklina, Amanda G. Paulovich, Samuel H. Payne, Francesca Petralia, Olga Potapova, Barb Pruetz, Liqun Qi, Gabriela M. Quiroga-Garza, Melissa A. Reimers, Boris Reva, Shannon Richey, Christopher J. Ricketts, Ana I. Robles, Nancy Roche, Karin D. Rodland, Henry Rodriguez, Michael H. Roehrl, Daniel C. Rohrer, Shankha Satpathy, Eric E. Schadt, Michael Schnaubelt, Yvonne Shutack, Shilpi Singh, Michael Smith, Richard D. Smith, Cezary Szczylik, Darlene Tansil, Guo Ci Teo, Ratna R. Thangudu, Mathangi Thiagarajan, Matt Tobin, Shirley X. Tsang, Ki Sung Um, Pamela VanderKolk, Brian A. Van Tine, Negin Vatanian, Josh Vo, Pei Wang, Yuefan Wang, Joshua Wang, Michael C. Wendl, George D. Wilson, Maciej Wiznerowicz, Jason Wright, Yige Wu, Matthew A. Wyczalkowski, Yuanwei Xu, Birendra Kumar Yadav, Yi Hsiao, Kakhaber Zaalishvili, Bing Zhang, Cissy Zhang, Hui Zhang, Yuping Zhang, Xu Zhang, Zhen Zhang, Grace Zhao, Yanyan Zhao
Secondary authors
Alicia Francis, Amanda G. Paulovich, Andrzej Antczak, Anthony Green, Antonio Colaprico, Ari Hakimi, Barb Pruetz, Barbara Hindenach, Birendra Kumar Yadav, Boris Reva, Brenda Fevrier-Sullivan, Brian J. Druker, Cezary Szczylik, Charles A. Goldthwaite, Jr., Chet Birger, Corbin D. Jones, Daniel C. Rohrer, Darlene Tansil, David Chesla, David Heiman, Elizabeth Duffy, Eri E. Schadt, Francesca Petralia, Gabriel Bromiński, Gabriela M. Quiroga-Garza, George D. Wilson, Ginny Xiaohe Li, Grace Zhao, Yi Hsiao, James Hsieh, Jan Lubiński, Jasmin Bavarva, Jasmine Huang, Jason Hafron, Jennifer Eschbacher, Jennifer Hon, Jesse Francis, John Freymann, Josh Vo, Joshua Wang, Justin Kirby, Kakhaber Zaalishvili, Karen A. Ketchum, Katherine A. Hoadley, Ki Sung Um, Liqun Qi, Marcin J. Domagalski, Matt Tobin, Maureen Dyer, Meenakshi Anurag, Melissa Borucki, Michael A. Gillette, Michael J. Birrer, Michael M. Ittmann, Michael H. Roehrl, Michael Schnaubelt, Michael Smith, Mina Fam, Nancy Roche, Negin Vatanian, Nicollette Maunganidze, Olga Potapova, Oxana V. Paklina, Pamela VanderKolk, Patricia Castro, Paweł Kurzawa, Pushpa Hariharan, Qin Li, Qing Kay Li, Rajiv Dhir, Ratna R. Thangudu, Rebecca Montgomery, Richard D. Smith, Sailaja Mareedu, Samuel H. Payne, Sandra Cerda, Sandra Cottingham, Sarah Haynes, Shankha Satpathy, Shannon Richey, Shilpi Singh, Shirley X.Tsang, Shuang Cai, Song Cao, Stacey Gabriel, Steven A. Carr, Tao Liu, Thomas Bauer, Toan Le, Xi S. Chen, Xu Zhang, Yvonne Shutack, Zhen Zhang
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, and Ficarra V (2017). Renal cell carcinoma. Nat Rev Dis Primers 3, 17009. 10.1038/nrdp.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fuchs HE, and Jemal A (2021). Cancer Statistics, 2021. CA Cancer J Clin 71, 7–33. 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 3.Motzer RJ, Jonasch E, Boyle S, Carlo MI, Manley B, Agarwal N, Alva A, Beckermann K, Choueiri TK, Costello BA, et al. (2020). NCCN Guidelines Insights: Kidney Cancer, Version 1.2021. J Natl Compr Canc Netw 18, 1160–1170. 10.6004/jnccn.2020.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanco AI, Teh BS, and Amato RJ (2011). Role of radiation therapy in the management of renal cell cancer. Cancers (Basel) 3, 4010–4023. 10.3390/cancers3044010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diamond E, Molina AM, Carbonaro M, Akhtar NH, Giannakakou P, Tagawa ST, and Nanus DM (2015). Cytotoxic chemotherapy in the treatment of advanced renal cell carcinoma in the era of targeted therapy. Crit Rev Oncol Hematol 96, 518–526. 10.1016/j.critrevonc.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh JJ, Le VH, Oyama T, Ricketts CJ, Ho TH, and Cheng EH (2018). Chromosome 3p Loss-Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. J Clin Oncol, JCO2018792549. 10.1200/JCO.2018.79.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atkins MB, and Tannir NM (2018). Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat Rev 70, 127–137. 10.1016/j.ctrv.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Choueiri TK, and Motzer RJ (2017). Systemic Therapy for Metastatic Renal-Cell Carcinoma. N Engl J Med 376, 354–366. 10.1056/NEJMra1601333. [DOI] [PubMed] [Google Scholar]
- 9.Powles T, Plimack ER, Soulieres D, Waddell T, Stus V, Gafanov R, Nosov D, Pouliot F, Melichar B, Vynnychenko I, et al. (2020). Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol 21, 1563–1573. 10.1016/S1470-2045(20)30436-8. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez DJ, and Simon MC (2018). Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim Biophys Acta Rev Cancer 1870, 23–31. 10.1016/j.bbcan.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H, et al. (2013). Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 45, 860–867. 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, Martincorena I, Tarpey P, Angelopoulos N, Yates LR, et al. (2018). Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 173, 611–623 e617. 10.1016/j.cell.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih TM, Chang HY, et al. (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983 e931. 10.1016/j.cell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research, N., Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr., Laird PW, et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368, 2059–2074. 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, et al. (2013). Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res 19, 3259–3267. 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapur P, Pena-Llopis S, Christie A, Zhrebker L, Pavia-Jimenez A, Rathmell WK, Xie XJ, and Brugarolas J (2013). Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol 14, 159–167. 10.1016/S1470-2045(12)70584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, Bowlby R, Gibb EA, Akbani R, Beroukhim R, et al. (2018). The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep 23, 313–326 e315. 10.1016/j.celrep.2018.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W, et al. (2016). An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 29, 104–116. 10.1016/j.ccell.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senbabaoglu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, Miao D, Ostrovnaya I, Drill E, Luna A, et al. (2016). Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol 17, 231. 10.1186/s13059-016-1092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rooney MS, Shukla SA, Wu CJ, Getz G, and Hacohen N (2015). Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366, 883–892. 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. (2014). Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 46, 225–233. 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turajlic S, Xu H, Litchfield K, Rowan A, Horswell S, Chambers T, O’Brien T, Lopez JI, Watkins TBK, Nicol D, et al. (2018). Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 173, 595–610 e511. 10.1016/j.cell.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Narayanan SP, Mannan R, Raskind G, Wang X, Vats P, Su F, Hosseini N, Cao X, Kumar-Sinha C, et al. (2021). Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2103240118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu J, Chen Z, Bao L, Zhou L, Hou Y, Liu L, Xiong M, Zhang Y, Wang B, Tao Z, and Chen K (2020). Single-Cell Transcriptome Analysis Reveals Intratumoral Heterogeneity in ccRCC, which Results in Different Clinical Outcomes. Mol Ther 28, 1658–1672. 10.1016/j.ymthe.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai Q, Christie A, Rajaram S, Zhou Q, Araj E, Chintalapati S, Cadeddu J, Margulis V, Pedrosa I, Rakheja D, et al. (2020). Ontological analyses reveal clinically-significant clear cell renal cell carcinoma subtypes with convergent evolutionary trajectories into an aggressive type. EBioMedicine 51, 102526. 10.1016/j.ebiom.2019.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novara G, Martignoni G, Artibani W, and Ficarra V (2007). Grading systems in renal cell carcinoma. J Urol 177, 430–436. 10.1016/j.juro.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 28.Fuhrman SA, Lasky LC, and Limas C (1982). Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol 6, 655–663. 10.1097/00000478-198210000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Verine J, Colin D, Nheb M, Prapotnich D, Ploussard G, Cathelineau X, Desgrandchamps F, Mongiat-Artus P, and Feugeas JP (2018). Architectural Patterns are a Relevant Morphologic Grading System for Clear Cell Renal Cell Carcinoma Prognosis Assessment: Comparisons With WHO/ISUP Grade and Integrated Staging Systems. Am J Surg Pathol 42, 423–441. 10.1097/PAS.0000000000001025. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Shu K, Wang Z, and Ding D (2019). Prognostic significance of KIF2A and KIF20A expression in human cancer: A systematic review and meta-analysis. Medicine (Baltimore) 98, e18040. 10.1097/MD.0000000000018040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braun DA, Hou Y, Bakouny Z, Ficial M, Sant’ Angelo M, Forman J, Ross-Macdonald P, Berger AC, Jegede OA, Elagina L, et al. (2020). Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med 26, 909–918. 10.1038/s41591-020-0839-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motzer RJ, Banchereau R, Hamidi H, Powles T, McDermott D, Atkins MB, Escudier B, Liu LF, Leng N, Abbas AR, et al. (2020). Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell 38, 803–817 e804. 10.1016/j.ccell.2020.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong R, Liu W, DeLair D, Razavian N, and Fenyo D (2021). Predicting endometrial cancer subtypes and molecular features from histopathology images using multi-resolution deep learning models. Cell Rep Med 2, 100400. 10.1016/j.xcrm.2021.100400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito T, Pei J, Dulaimi E, Menges C, Abbosh PH, Smaldone MC, Chen DY, Greenberg RE, Kutikov A, Viterbo R, et al. (2016). Genomic Copy Number Alterations in Renal Cell Carcinoma with Sarcomatoid Features. J Urol 195, 852–858. 10.1016/j.juro.2015.10.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kallakury BV, Karikehalli S, Haholu A, Sheehan CE, Azumi N, and Ross JS (2001). Increased expression of matrix metalloproteinases 2 and 9 and tissue inhibitors of metalloproteinases 1 and 2 correlate with poor prognostic variables in renal cell carcinoma. Clin Cancer Res 7, 3113–3119. [PubMed] [Google Scholar]
- 36.Lasseigne BN, and Brooks JD (2018). The Role of DNA Methylation in Renal Cell Carcinoma. Mol Diagn Ther 22, 431–442. 10.1007/s40291-018-0337-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evelonn EA, Degerman S, Kohn L, Landfors M, Ljungberg B, and Roos G (2016). DNA methylation status defines clinicopathological parameters including survival for patients with clear cell renal cell carcinoma (ccRCC). Tumour Biol 37, 10219–10228. 10.1007/s13277-016-4893-5. [DOI] [PubMed] [Google Scholar]
- 38.Evelonn EA, Landfors M, Haider Z, Kohn L, Ljungberg B, Roos G, and Degerman S (2019). DNA methylation associates with survival in non-metastatic clear cell renal cell carcinoma. BMC Cancer 19, 65. 10.1186/s12885-019-5291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malouf GG, Su X, Zhang J, Creighton CJ, Ho TH, Lu Y, Raynal NJ, Karam JA, Tamboli P, Allanick F, et al. (2016). DNA Methylation Signature Reveals Cell Ontogeny of Renal Cell Carcinomas. Clin Cancer Res 22, 6236–6246. 10.1158/1078-0432.CCR-15-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell HN, Kumar-Sinha C, Mannan R, Zakalik D, Zhang Y, Mehra R, Jagtap D, Dhanasekaran SM, and Vaishampayan U (2022). Pathogenic ATM and BAP1 germline mutations in a case of early-onset, familial sarcomatoid renal cancer. Cold Spring Harb Mol Case Stud 8. 10.1101/mcs.a006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimada Y, Kudo Y, Maehara S, Matsubayashi J, Otaki Y, Kajiwara N, Ohira T, Minna JD, and Ikeda N (2020). Ubiquitin C-terminal hydrolase-L1 has prognostic relevance and is a therapeutic target for high-grade neuroendocrine lung cancers. Cancer Sci 111, 610–620. 10.1111/cas.14284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, Szucs M, Mundt F, Forestier D, Jane-Valbuena J, et al. (2019). A Curated Resource for Phosphosite-specific Signature Analysis. Mol Cell Proteomics 18, 576–593. 10.1074/mcp.TIR118.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koul HK, Pal M, and Koul S (2013). Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 4, 342–359. 10.1177/1947601913507951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez-Limon A, Joaquin M, Caballero M, Posas F, and de Nadal E (2020). The p38 Pathway: From Biology to Cancer Therapy. Int J Mol Sci 21. 10.3390/ijms21061913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan WL, Yeh YM, Liu TT, Lin WM, Yang TY, Lee CW, and Lin TC (2021). Leptin Is Associated with Poor Clinical Outcomes and Promotes Clear Cell Renal Cell Carcinoma Progression. Biomolecules 11. 10.3390/biom11030431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Wada Y, Katsura M, Tozawa H, Erwin N, Kapron CM, Bao G, and Liu J (2018). Rho-Associated Coiled-Coil Kinase (ROCK) in Molecular Regulation of Angiogenesis. Theranostics 8, 6053–6069. 10.7150/thno.30305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen W, Mao K, Liu Z, and Dinh-Xuan AT (2014). The role of the RhoA/Rho kinase pathway in angiogenesis and its potential value in prostate cancer (Review). Oncol Lett 8, 1907–1911. 10.3892/ol.2014.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makinde T, and Agrawal DK (2008). Intra and extravascular transmembrane signalling of angiopoietin-1-Tie2 receptor in health and disease. J Cell Mol Med 12, 810–828. 10.1111/j.1582-4934.2008.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim M, Allen B, Korhonen EA, Nitschke M, Yang HW, Baluk P, Saharinen P, Alitalo K, Daly C, Thurston G, and McDonald DM (2016). Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest 126, 3511–3525. 10.1172/JCI84871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baas R, J.v.d.W. F, Bleijerveld OB, van Attikum H, and Sixma TK (2021). Proteomic analysis identifies novel binding partners of BAP1. PLoS One 16, e0257688. 10.1371/journal.pone.0257688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khetani VV, Portal DE, Shah MR, Mayer T, and Singer EA (2020). Combination drug regimens for metastatic clear cell renal cell carcinoma. World J Clin Oncol 11, 541–562. 10.5306/wjco.v11.i8.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Y, Wang Y, Hoti N, Clark DJ, Chen SY, and Zhang H (2021). The next “sweet” spot for pancreatic ductal adenocarcinoma: Glycoprotein for early detection. Mass Spectrom Rev, e21748. 10.1002/mas.21748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meany DL, and Chan DW (2011). Aberrant glycosylation associated with enzymes as cancer biomarkers. Clin Proteomics 8, 7. 10.1186/1559-0275-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X, Zhang NX, Ye HY, Song PP, Chang W, Chen L, Wang Z, Zhang L, and Wang NN (2019). HYOU1 promotes cell growth and metastasis via activating PI3K/AKT signaling in epithelial ovarian cancer and predicts poor prognosis. Eur Rev Med Pharmacol Sci 23, 4126–4135. 10.26355/eurrev_201901_17914. [DOI] [PubMed] [Google Scholar]
- 55.Linehan WM, Schmidt LS, Crooks DR, Wei D, Srinivasan R, Lang M, and Ricketts CJ (2019). The Metabolic Basis of Kidney Cancer. Cancer Discov 9, 1006–1021. 10.1158/2159-8290.CD-18-1354. [DOI] [PubMed] [Google Scholar]
- 56.Wu G, and Morris SM Jr. (1998). Arginine metabolism: nitric oxide and beyond. Biochem J 336 (Pt 1), 1–17. 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ochocki JD, Khare S, Hess M, Ackerman D, Qiu B, Daisak JI, Worth AJ, Lin N, Lee P, Xie H, et al. (2018). Arginase 2 Suppresses Renal Carcinoma Progression via Biosynthetic Cofactor Pyridoxal Phosphate Depletion and Increased Polyamine Toxicity. Cell Metab 27, 1263–1280 e1266. 10.1016/j.cmet.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wettersten HI, Aboud OA, Lara PN Jr., and Weiss RH (2017). Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol 13, 410–419. 10.1038/nrneph.2017.59. [DOI] [PubMed] [Google Scholar]
- 59.Khare S, Kim LC, Lobel G, Doulias PT, Ischiropoulos H, Nissim I, Keith B, and Simon MC (2021). ASS1 and ASL suppress growth in clear cell renal cell carcinoma via altered nitrogen metabolism. Cancer Metab 9, 40. 10.1186/s40170-021-00271-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen YA, Chen CL, Huang YH, Evans EE, Cheng CC, Chuang YJ, Zhang C, and Le A (2021). Inhibition of glutaminolysis in combination with other therapies to improve cancer treatment. Curr Opin Chem Biol 62, 64–81. 10.1016/j.cbpa.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Terunuma A, Putluri N, Mishra P, Mathe EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, et al. (2014). MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest 124, 398–412. 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang B, Xia H, Xu C, Lu M, Zhang S, Wang G, and Ma L (2020). Impact of sarcomatoid differentiation and rhabdoid differentiation on prognosis for renal cell carcinoma with vena caval tumour thrombus treated surgically. BMC Urol 20, 14. 10.1186/s12894-020-0584-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, Cowley GS, Lizotte P, Liberzon E, Bair S, et al. (2013). SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell 24, 738–750. 10.1016/j.ccr.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Porubsky S, Nientiedt M, Kriegmair MC, Siemoneit JH, Sandhoff R, Jennemann R, Borgmann H, Gaiser T, Weis CA, Erben P, et al. (2021). The prognostic value of galactosylceramide-sulfotransferase (Gal3ST1) in human renal cell carcinoma. Sci Rep 11, 10926. 10.1038/s41598-021-90381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baba M, Furuya M, Motoshima T, Lang M, Funasaki S, Ma W, Sun HW, Hasumi H, Huang Y, Kato I, et al. (2019). TFE3 Xp11.2 Translocation Renal Cell Carcinoma Mouse Model Reveals Novel Therapeutic Targets and Identifies GPNMB as a Diagnostic Marker for Human Disease. Mol Cancer Res 17, 1613–1626. 10.1158/1541-7786.MCR-18-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhai JP, Liu ZH, Wang HD, Huang GL, and Man LB (2022). GPNMB overexpression is associated with extensive bone metastasis and poor prognosis in renal cell carcinoma. Oncol Lett 23, 36. 10.3892/ol.2021.13154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ott PA, Hamid O, Pavlick AC, Kluger H, Kim KB, Boasberg PD, Simantov R, Crowley E, Green JA, Hawthorne T, et al. (2014). Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patients with advanced melanoma. J Clin Oncol 32, 3659–3666. 10.1200/JCO.2013.54.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tray N, Adams S, and Esteva FJ (2018). Antibody-drug conjugates in triple negative breast cancer. Future Oncol 14, 2651–2661. 10.2217/fon-2018-0131. [DOI] [PubMed] [Google Scholar]
- 69.Koneru R, and Hotte SJ (2009). Role of cytokine therapy for renal cell carcinoma in the era of targeted agents. Curr Oncol 16 Suppl 1, S40–44. 10.3747/co.v16i0.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bakouny Z, Braun DA, Shukla SA, Pan W, Gao X, Hou Y, Flaifel A, Tang S, Bosma-Moody A, He MX, et al. (2021). Integrative molecular characterization of sarcomatoid and rhabdoid renal cell carcinoma. Nat Commun 12, 808. 10.1038/s41467-021-21068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, et al. (2016). Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539, 112–117. 10.1038/nature19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, et al. (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765. 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ, et al. (2021). Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med 385, 2036–2046. 10.1056/NEJMoa2103425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, Yang B, and Ding L (2020). Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B 10, 723–733. 10.1016/j.apsb.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Diaz-Montero CM, Mao FJ, Barnard J, Parker Y, Zamanian-Daryoush M, Pink JJ, Finke JH, Rini BI, and Lindner DJ (2016). MEK inhibition abrogates sunitinib resistance in a renal cell carcinoma patient-derived xenograft model. Br J Cancer 115, 920–928. 10.1038/bjc.2016.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chauhan A, Semwal DK, Mishra SP, Goyal S, Marathe R, and Semwal RB (2016). Combination of mTOR and MAPK Inhibitors-A Potential Way to Treat Renal Cell Carcinoma. Med Sci (Basel) 4. 10.3390/medsci4040016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Voss MH, Gordon MS, Mita M, Rini B, Makker V, Macarulla T, Smith DC, Cervantes A, Puzanov I, Pili R, et al. (2020). Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br J Cancer 123, 1590–1598. 10.1038/s41416-020-01041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hsieh CC, Shyr YM, Liao WY, Chen TH, Wang SE, Lu PC, Lin PY, Chen YB, Mao WY, Han HY, et al. (2017). Elevation of beta-galactoside alpha2,6-sialyltransferase 1 in a fructoseresponsive manner promotes pancreatic cancer metastasis. Oncotarget 8, 7691–7709. 10.18632/oncotarget.13845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suzuki O (2019). Glycosylation in lymphoma: Biology and glycotherapy. Pathol Int 69, 441–449. 10.1111/pin.12834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vajaria BN, Patel KR, Begum R, and Patel PS (2016). Sialylation: an Avenue to Target Cancer Cells. Pathol Oncol Res 22, 443–447. 10.1007/s12253-015-0033-6. [DOI] [PubMed] [Google Scholar]
- 81.Garnham R, Scott E, Livermore KE, and Munkley J (2019). ST6GAL1: A key player in cancer. Oncol Lett 18, 983–989. 10.3892/ol.2019.10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chakraborty S, Balan M, Sabarwal A, Choueiri TK, and Pal S (2021). Metabolic reprogramming in renal cancer: Events of a metabolic disease. Biochim Biophys Acta Rev Cancer 1876, 188559. 10.1016/j.bbcan.2021.188559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cancer Genome Atlas Research, N. (2013). Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49. 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Turei D, Korcsmaros T, and Saez-Rodriguez J (2016). OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat Methods 13, 966–967. 10.1038/nmeth.4077. [DOI] [PubMed] [Google Scholar]
- 85.Linding R, Jensen LJ, Pasculescu A, Olhovsky M, Colwill K, Bork P, Yaffe MB, and Pawson T (2008). NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Res 36, D695–699. 10.1093/nar/gkm902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Damle NP, and Kohn M (2019). The human DEPhOsphorylation Database DEPOD: 2019 update. Database (Oxford) 2019. 10.1093/database/baz133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Licata L, Lo Surdo P, Iannuccelli M, Palma A, Micarelli E, Perfetto L, Peluso D, Calderone A, Castagnoli L, and Cesareni G (2020). SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res 48, D504–D510. 10.1093/nar/gkz949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scott AD, Huang KL, Weerasinghe A, Mashl RJ, Gao Q, Martins Rodrigues F, Wyczalkowski MA, and Ding L (2019). CharGer: clinical Characterization of Germline variants. Bioinformatics 35, 865–867. 10.1093/bioinformatics/bty649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun SQ, Mashl RJ, Sengupta S, Scott AD, Wang W, Batra P, Wang LB, Wyczalkowski MA, and Ding L (2018). Database of evidence for precision oncology portal. Bioinformatics 34, 4315–4317. 10.1093/bioinformatics/bty531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Benelli M, Pescucci C, Marseglia G, Severgnini M, Torricelli F, and Magi A (2012). Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 28, 3232–3239. 10.1093/bioinformatics/bts617. [DOI] [PubMed] [Google Scholar]
- 94.Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang J, White NM, Schmidt HK, Fulton RS, Tomlinson C, Warren WC, Wilson RK, and Maher CA (2016). INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res 26, 108–118. 10.1101/gr.186114.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 97.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Polpitiya AD, Qian WJ, Jaitly N, Petyuk VA, Adkins JN, Camp DG 2nd, Anderson GA, and Smith RD (2008). DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24, 1556–1558. 10.1093/bioinformatics/btn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Haas BJ, Dobin A, Li B, Stransky N, Pochet N, and Regev A (2019). Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol 20, 213. 10.1186/s13059-019-1842-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, and Saunders CT (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594. 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 103.Conway JR, Lex A, and Gehlenborg N (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. 10.1093/bioinformatics/btx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576. 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18, 220. 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. (2019). Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 37, 773–782. 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pang Z, Chong J, Zhou G, de Lima Morais DA, Chang L, Barrette M, Gauthier C, Jacques PE, Li S, and Xia J (2021). MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res 49, W388–W396. 10.1093/nar/gkab382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vasaikar SV, Straub P, Wang J, and Zhang B (2018). LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res 46, D956–D963. 10.1093/nar/gkx1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Clark K, Vendt B, Smith K, Freymann J, Kirby J, Koppel P, Moore S, Phillips S, Maffitt D, Pringle M, et al. (2013). The Cancer Imaging Archive (TCIA): maintaining and operating a public information repository. J Digit Imaging 26, 1045–1057. 10.1007/s10278-013-9622-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fisher S, Barry A, Abreu J, Minie B, Nolan J, Delorey TM, Young G, Fennell TJ, Allen A, Ambrogio L, et al. (2011). A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol 12, R1. 10.1186/gb-2011-12-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chu A, Robertson G, Brooks D, Mungall AJ, Birol I, Coope R, Ma Y, Jones S, and Marra MA (2016). Large-scale profiling of microRNAs for The Cancer Genome Atlas. Nucleic Acids Res 44, e3. 10.1093/nar/gkv808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, and Carr SA (2013). Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods 10, 634–637. 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dong M, Lih TM, Ao M, Hu Y, Chen SY, Eguez RV, and Zhang H (2021). Data-Independent Acquisition-Based Mass Spectrometry (DIA-MS) for Quantitative Analysis of Intact N-Linked Glycopeptides. Anal Chem 93, 13774–13782. 10.1021/acs.analchem.1c01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cho KC, Clark DJ, Schnaubelt M, Teo GC, Leprevost FDV, Bocik W, Boja ES, Hiltke T, Nesvizhskii AI, and Zhang H (2020). Deep Proteomics Using Two Dimensional Data Independent Acquisition Mass Spectrometry. Anal Chem 92, 4217–4225. 10.1021/acs.analchem.9b04418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, Clauser KR, Clauss TR, Shah P, Gillette MA, et al. (2018). Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat Protoc 13, 1632–1661. 10.1038/s41596-018-0006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nguyen T, Kirsch BJ, Asaka R, Nabi K, Quinones A, Tan J, Antonio MJ, Camelo F, Li T, Nguyen S, et al. (2019). Uncovering the Role of N-Acetyl-Aspartyl-Glutamate as a Glutamate Reservoir in Cancer. Cell Rep 27, 491–501 e496. 10.1016/j.celrep.2019.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Elgogary A, Xu Q, Poore B, Alt J, Zimmermann SC, Zhao L, Fu J, Chen B, Xia S, Liu Y, et al. (2016). Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci U S A 113, E5328–5336. 10.1073/pnas.1611406113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Udupa S, Nguyen S, Hoang G, Nguyen T, Quinones A, Pham K, Asaka R, Nguyen K, Zhang C, Elgogary A, et al. (2019). Upregulation of the Glutaminase II Pathway Contributes to Glutamate Production upon Glutaminase 1 Inhibition in Pancreatic Cancer. Proteomics 19, e1800451. 10.1002/pmic.201800451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Skala SL, Wang X, Zhang Y, Mannan R, Wang L, Narayanan SP, Vats P, Su F, Chen J, Cao X, et al. (2020). Next-generation RNA Sequencing-based Biomarker Characterization of Chromophobe Renal Cell Carcinoma and Related Oncocytic Neoplasms. Eur Urol 78, 63–74. 10.1016/j.eururo.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385 e318. 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947. 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR (2015). A global reference for human genetic variation. Nature 526, 68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huang KL, Mashl RJ, Wu Y, Ritter DI, Wang J, Oh C, Paczkowska M, Reynolds S, Wyczalkowski MA, Oak N, et al. (2018). Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 173, 355–370 e314. 10.1016/j.cell.2018.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pierre-Jean M, Rigaill G, and Neuvial P (2015). Performance evaluation of DNA copy number segmentation methods. Brief Bioinform 16, 600–615. 10.1093/bib/bbu026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30, 413–421. 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xi R, Lee S, Xia Y, Kim TM, and Park PJ (2016). Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res 44, 6274–6286. 10.1093/nar/gkw491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chin SF, Teschendorff AE, Marioni JC, Wang Y, Barbosa-Morais NL, Thorne NP, Costa JL, Pinder SE, van de Wiel MA, Green AR, et al. (2007). High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol 8, R215. 10.1186/gb-2007-8-10-r215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. (2013). Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, Liao WW, Reynolds SM, Wyczalkowski MA, Yao L, et al. (2018). Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep 23, 227–238 e223. 10.1016/j.celrep.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Babiceanu M, Qin F, Xie Z, Jia Y, Lopez K, Janus N, Facemire L, Kumar S, Pang Y, Qi Y, et al. (2016). Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res 44, 2859–2872. 10.1093/nar/gkw032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hafemeister C, and Satija R (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296. 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Young MD, Mitchell TJ, Custers L, Margaritis T, Morales-Rodriguez F, Kwakwa K, Khabirova E, Kildisiute G, Oliver TRW, de Krijger RR, et al. (2021). Single cell derived mRNA signals across human kidney tumors. Nat Commun 12, 3896. 10.1038/s41467-021-23949-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Borcherding N, Vishwakarma A, Voigt AP, Bellizzi A, Kaplan J, Nepple K, Salem AK, Jenkins RW, Zakharia Y, and Zhang W (2021). Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun Biol 4, 122. 10.1038/s42003-020-01625-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982. 10.1038/nmeth.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, and Nesvizhskii AI (2017). MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods 14, 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yu F, Teo GC, Kong AT, Haynes SE, Avtonomov DM, Geiszler DJ, and Nesvizhskii AI (2020). Identification of modified peptides using localization-aware open search. Nat Commun 11, 4065. 10.1038/s41467-020-17921-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.da Veiga Leprevost F, Haynes SE, Avtonomov DM, Chang HY, Shanmugam AK, Mellacheruvu D, Kong AT, and Nesvizhskii AI (2020). Philosopher: a versatile toolkit for shotgun proteomics data analysis. Nat Methods 17, 869–870. 10.1038/s41592-020-0912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, and Nesvizhskii AI (2015). DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods 12, 258–264, 257 p following 264. 10.1038/nmeth.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Teo GC, Polasky DA, Yu F, and Nesvizhskii AI (2021). Fast Deisotoping Algorithm and Its Implementation in the MSFragger Search Engine. J Proteome Res 20, 498–505. 10.1021/acs.jproteome.0c00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74, 5383–5392. 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 144.Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75, 4646–4658. 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 145.Savitski MM, Wilhelm M, Hahne H, Kuster B, and Bantscheff M (2015). A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol Cell Proteomics 14, 2394–2404. 10.1074/mcp.M114.046995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Demichev V, Messner CB, Vernardis SI, Lilley KS, and Ralser M (2020). DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat Methods 17, 41–44. 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13, 2513–2526. 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shteynberg DD, Deutsch EW, Campbell DS, Hoopmann MR, Kusebauch U, Lee D, Mendoza L, Midha MK, Sun Z, Whetton AD, and Moritz RL (2019). PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J Proteome Res 18, 4262–4272. 10.1021/acs.jproteome.9b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, and McVean GA (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073. 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kim H, and Park H (2007). Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. Bioinformatics 23, 1495–1502. 10.1093/bioinformatics/btm134. [DOI] [PubMed] [Google Scholar]
- 151.Mani DR, Maynard M, Kothadia R, Krug K, Christianson KE, Heiman D, Clauser KR, Birger C, Getz G, and Carr SA (2021). PANOPLY: a cloud-based platform for automated and reproducible proteogenomic data analysis. Nat Methods 18, 580–582. 10.1038/s41592-021-01176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, et al. (2012). Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437. 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 153.Zhou W, Laird PW, and Shen H (2017). Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res 45, e22. 10.1093/nar/gkw967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, Kaminska B, Huelsken J, Omberg L, Gevaert O, et al. (2018). Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 173, 338–354 e315. 10.1016/j.cell.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Colaprico A, Olsen C, Bailey MH, Odom GJ, Terkelsen T, Silva TC, Olsen AV, Cantini L, Zinovyev A, Barillot E, et al. (2020). Interpreting pathways to discover cancer driver genes with Moonlight. Nat Commun 11, 69. 10.1038/s41467-019-13803-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Daily K, Ho Sui SJ, Schriml LM, Dexheimer PJ, Salomonis N, Schroll R, Bush S, Keddache M, Mayhew C, Lotia S, et al. (2017). Molecular, phenotypic, and sample-associated data to describe pluripotent stem cell lines and derivatives. Sci Data 4, 170030. 10.1038/sdata.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Salomonis N, Dexheimer PJ, Omberg L, Schroll R, Bush S, Huo J, Schriml L, Ho Sui S, Keddache M, Mayhew C, et al. (2016). Integrated Genomic Analysis of Diverse Induced Pluripotent Stem Cells from the Progenitor Cell Biology Consortium. Stem Cell Reports 7, 110–125. 10.1016/j.stemcr.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sokolov A, Paull EO, and Stuart JM (2016). One-Class Detection of Cell States in Tumor Subtypes. Pac Symp Biocomput 21, 405–416. [PMC free article] [PubMed] [Google Scholar]
- 159.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44, e71. 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Silva TC, Colaprico A, Olsen C, D’Angelo F, Bontempi G, Ceccarelli M, and Noushmehr H (2016). TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Res 5, 1542. 10.12688/f1000research.8923.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Duan G, Li X, and Kohn M (2015). The human DEPhOsphorylation database DEPOD: a 2015 update. Nucleic Acids Res 43, D531–535. 10.1093/nar/gku1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Mewes HW (2010). CORUM: the comprehensive resource of mammalian protein complexes--2009. Nucleic Acids Res 38, D497–501. 10.1093/nar/gkp914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Perfetto L, Briganti L, Calderone A, Cerquone Perpetuini A, Iannuccelli M, Langone F, Licata L, Marinkovic M, Mattioni A, Pavlidou T, et al. (2016). SIGNOR: a database of causal relationships between biological entities. Nucleic Acids Res 44, D548–554. 10.1093/nar/gkv1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, et al. (2020). The reactome pathway knowledgebase. Nucleic Acids Res 48, D498–D503. 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xu T, Le TD, Liu L, Su N, Wang R, Sun B, Colaprico A, Bontempi G, and Li J (2017). CancerSubtypes: an R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 33, 3131–3133. 10.1093/bioinformatics/btx378. [DOI] [PubMed] [Google Scholar]
- 166.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 167.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, and Muller M (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77. 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47, W199–W205. 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Calinawan AP, Song X, Ji J, Dhanasekaran SM, Petralia F, Wang P, and Reva B (2020). ProTrack: An Interactive Multi-Omics Data Browser for Proteogenomic Studies. Proteomics 20, e1900359. 10.1002/pmic.201900359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. CPTAC ccRCC cohort summary, ccRCC histopathological annotation, and feature-associated markers, related to Figure 1.
Table S2. CPTAC ccRCC ITH cohort summary, ccRCC ITH histopathological annotation, CNV, and imaging statistics, related to Figure 2.
Table S3. snRNA-seq meta table, cell type fraction, and sarcomatoid-associated and rhabdoid-associated markers, related to Figures 3–4.
Table S4. ccRCC methylation subtype, methylation signature probes, methylation DEG and DEP, and inhibition study of UCHL1, related to Figure 5.
Table S5. Kinase-substrate network, phosphoproteome subtype and associated signatures, and cell line results, related to Figure 6.
Table S6. Glycoproteome subtype and associated signatures, tumor and NAT comparison, and CH and CL comparison, related to Figure 7.
Table S7. Quantified metabolites, metabolome PCA, metabolome subtype and associated signatures, and metabolome feature validation, related to Figure 8.
Data Availability Statement
Clinical data and proteomic data (raw MS files and processed data files) reported in this paper can be accessed via the Proteomic Data Commons (PDC) at: https://pdc.cancer.gov/ (Project: CPTAC3 Discovery and Confirmatory, Disease Type: Clear Cell Renal Cell Carcinoma). Genomic, transcriptomic, and snRNA-seq data files can be accessed via Genomic Data Commons (GDC) at: https://portal.gdc.cancer.gov/projects/CPTAC-3 (Project: CPTAC-3, Primary Site: Kidney). Processed data used in this publication can also be found in the PDC, the Python package and LinkedOmics.108 Pathology and radiology images can be accessed via Imaging Data Commons (IDC) at https://portal.imaging.datacommons.cancer.gov/explore/filters/?collection_id=cptac_ccrcc (Collection: CPTAC-CCRCC), and The Cancer Imaging Archive at https://doi.org/10.7937/K9/TCIA.2018.OBLAMN27 (Collection: CPTAC-CCRCC).109 In addition, other data including TCGA KIRC83 at https://portal.gdc.cancer.gov/ (Project: TCGA-KIRC), OmniPath84 at https://omnipathdb.org/#faq, NetworKIN85 at https://networkin.info/, DEPOD86 at http://www.depod.bioss.uni-freiburg.de/, and SIGNOR87 at https://signor.uniroma2.it/.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.