

Abstract
The advent of SARS-CoV-2, the causative agent of COVID-19, and its worldwide impact on global health, have provided the impetus for the development of effective countermeasures that can be deployed against the virus, including vaccines, monoclonal antibodies, and direct-acting antivirals (DAAs). Despite these efforts, the current paucity of DAAs has created an urgent need for the creation of an enhanced and diversified portfolio of broadly acting agents with different mechanisms of action that can effectively abrogate viral infection. SARS-CoV-2 3C-like protease (3CLpro), an enzyme essential for viral replication, is a validated target for the discovery of SARS-CoV-2 therapeutics. In this report, we describe the structure-guided utilization of the cyclopropane moiety in the design of highly potent inhibitors of SARS-CoV-2 3CLpro, SARS-CoV-1 3CLpro, and MERS-CoV 3CLpro. High-resolution cocrystal structures were used to identify the structural determinants associated with the binding of the inhibitors to the active site of the enzyme and unravel the mechanism of action. Aldehydes 5c and 11c inhibited SARS-CoV-2 replication with EC50 values of 12 and 11 nM, respectively. Furthermore, the corresponding aldehyde bisulfite adducts 5d and 11d were equipotent with EC50 values of 13 and 12 nM, respectively. The safety index (SI) values for compounds 5c/11c and 5d/11d ranged between 7692 and 9090. Importantly, aldehydes 5c/11c and bisulfite adducts 5d/11d potently inhibited MERS-CoV 3CLpro with IC50 values of 80 and 120 nM, and 70 and 70 nM, respectively. Likewise, compounds 5c/11c and 5d/11d inhibited SARS-CoV-1 with IC50 values of 960 and 350 nM and 790 and 240 nM, respectively. Taken together, these studies suggest that the inhibitors described herein have low cytotoxicity and high potency and are promising candidates for further development as broad-spectrum direct-acting antivirals against highly pathogenic coronaviruses.
Keywords: SARS-CoV-2, SARS-CoV-1, MERS-CoV, coronavirus, 3-chymotrypsin-like protease (3CLpro), broad-spectrum inhibitors, cyclopropane-derived inhibitors
The morbidity and mortality associated with the COVID-19 pandemic caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) are continuing to have a major impact on public health and the global economy.1−3 The timely discovery and introduction into the clinic of effective vaccines and monoclonal antibodies have ameliorated the impact and threat of the disease;4,5 however, there is a pressing need for the development of effective and complementary countermeasures, including an expanded and diversified portfolio of direct-acting antivirals (DAAs)6−11 that can be rapidly deployed against SARS-CoV-2 and variants, as well as emerging and re-emerging coronaviruses.3,12
In addition to host targets,9,13−15 the SARS-CoV-2 life cycle offers a wealth of choke points, including virus entry and virus replication, that can serve as a fruitful avenue of investigation for the discovery of DAA therapeutics. SARS-CoV-2 is a β-coronavirus with a large genome (∼30,000 nucleotides) that encodes two polyproteins (designated pp1a and pp1ab) which are processed by two viral-encoded cysteine proteases, 3CL protease (3CLpro), also called the Main protease (Mpro), and the papain-like protease (PLpro) to generate replicase complex nonstructural proteins, including the RNA-dependent RNA polymerase (RdRp), which are essential for viral replication.16 Importantly, the 3CL proteases of coronaviruses are highly conserved and, when coupled with their unique primary substrate specificity for a P1 Gln residue17−19 and the lack of a homologous human enzyme with the same substrate specificity, offer distinct advantages in terms of identifying inhibitors with broad-spectrum activity, enhanced selectivity, and low toxicity. Furthermore, the availability of multiple high-resolution cocrystal structures has greatly accelerated discovery efforts targeting SARS-CoV-2 3CLpro, and numerous covalent and noncovalent peptidyl or non-peptidyl inhibitors of the enzyme have been reported.20−34 Notably, the FDA approval of Paxlovid for clinical use, a combination therapy composed of Nirmatrelvir (a 3CLpro inhibitor) and Ritonavir (a Cyp3A4 inhibitor used to suppress the metabolism of Nirmatrelvir), provides a strong measure of confidence regarding the clinical efficacy of 3CLpro inhibitors.35,36 In continuing our foray in this area,37−44 we describe herein the structure-guided design and evaluation of inhibitors of SARS-CoV-2 3CLpro that incorporate in their structure a cyclopropyl moiety as a design element45 well-suited for optimizing potency by exploiting favorable hydrophobic and H-bonding interactions with the S4 subsite of the enzyme. Furthermore, it was anticipated that the high conservation of the active site topography across coronaviral proteases would make possible the design of pan-coronavirus DAAs.
Results and Discussion
Inhibitor Design
The design of the inhibitors (Figure 1, general structure (I)) entailed appending a Leu-Gln surrogate recognition component corresponding to the known P2-P1 substrate preference of the protease to a cyclopropyl moiety intended to access new chemical space encompassing the hydrophobic S4 subsite of the enzyme (the S3 subsite is solvent exposed). Earlier studies by our group37−39,44 revealed that binding interactions with the S4 subsite and vicinity increase potency significantly, yielding compounds with demonstrated efficacy in mouse models of MERS-CoV and SARS-CoV-2 infection.37,40 Thus, it was envisaged that potency would be enhanced by (a) attaching appropriate elements to the cyclopropyl moiety that extend toward the S4 pocket and are capable of engaging in H-bonding and hydrophobic interactions and (b) by assuming an entropically more favorable conformation via the rigorous control of stereochemistry afforded by the cyclopropane fragment. Importantly, the cyclopropane derivatives are synthetically tractable and amenable to multiparameter optimization. The design of the inhibitors was greatly facilitated by the availability of high-resolution cocrystal structures.
Figure 1.

General structure of inhibitor (I).
Chemistry
Inhibitors 1c/d–17c/d were synthesized using cyclopropyl alcohol inputs 1–17 (Figure S1). The latter were prepared using well-precedented methodologies46,47 (Scheme S1). Then, an appropriate cyclopropyl alcohol was activated with disuccinimidyl carbonate,48 followed by coupling with peptidyl amino alcohol, and the product was subsequently oxidized with Dess–Martin periodinane reagent. The generated aldehydes were readily transformed to the corresponding bisulfite adducts, as described previously (Scheme S2).49
Biochemical Studies
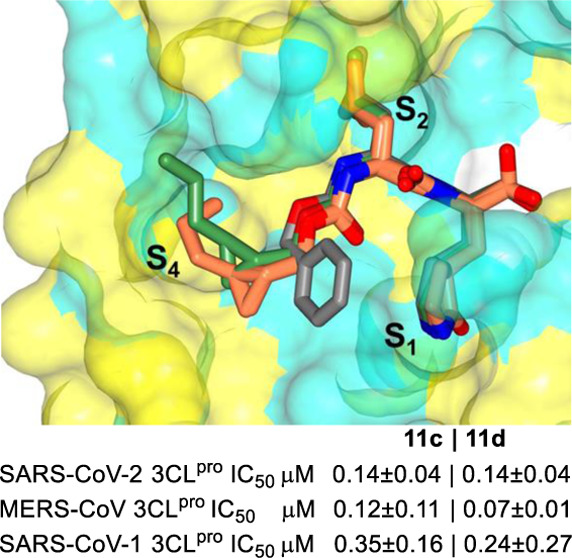
The inhibitory activity of compounds 1–17c/d toward SARS-CoV-2 3CLpro in biochemical and cell-based assays,37,38,44 as well as the cytotoxicity of the compounds, were determined, and the results are listed in Table 1. For comparative purposes, the interaction of a select number of compounds with MERS-CoV-2 3CLpro and SARS-CoV-1 3CLpro was also investigated (Table 1). Selected compounds were tested in a cell-based assay against SARS-CoV-2, as described in the Experimental Section. The IC50 values, EC50 values for a select number of inhibitors, and the CC50 values in 293T cells40 are summarized in Table 1, and they are the average of at least three determinations. All tested compounds were active against all 3CL proteases, and the IC50 values ranged between 0.14–0.46 μM, 0.24–2.56 μM, and 0.05–0.41 μM for SARS-CoV-2, SARS-CoV-1, and MERS-CoV 3CL proteases, respectively. Fourteen compounds were selected for EC50 determination against SARS-CoV-2. These were found to be potent inhibitors with EC50 values in the range 0.011–0.25 μM. The CC50 values of these compounds were >50 μM with safety indices >250. Representative dose-dependent inhibition curves with inhibitors 11c and 11d in enzyme and cell-based assays are shown in Figure 2.
Table 1. IC50, EC50, and CC50 Values of Compounds 1c/d–17c/d against SARS-CoV-2, MERS-CoV 3CL, and SARS-CoV-1 3CL Proteasesa.
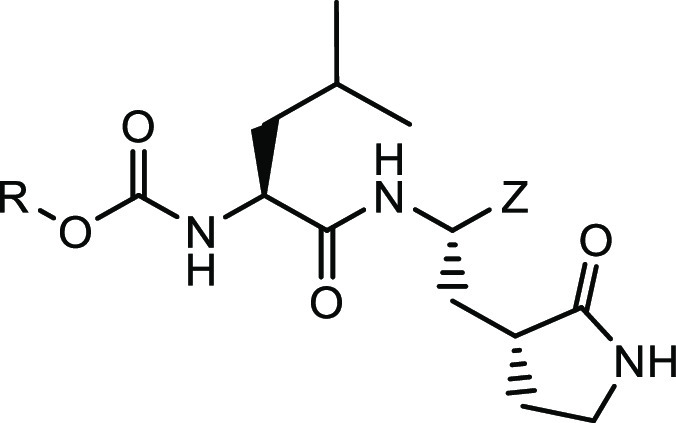
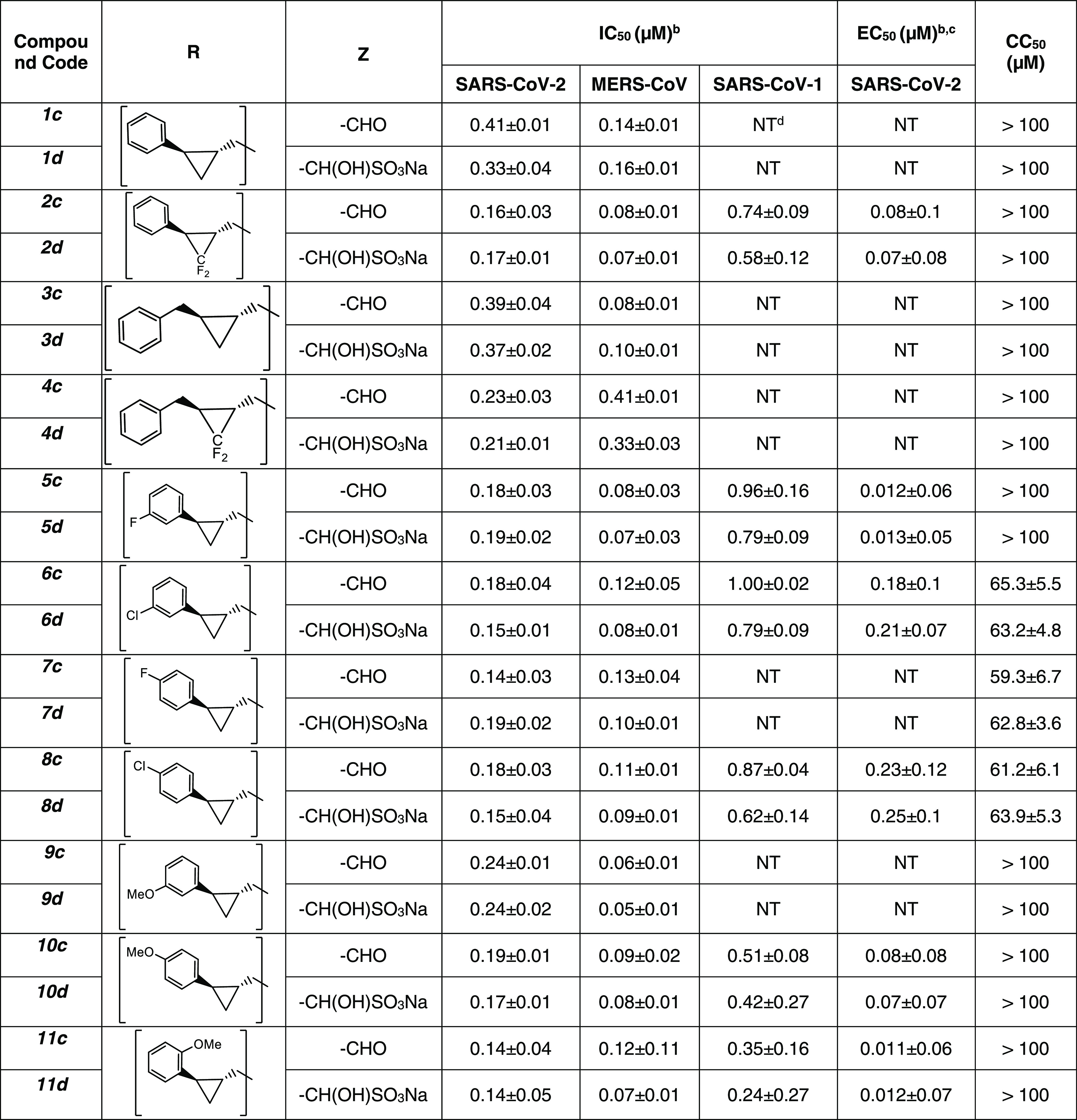
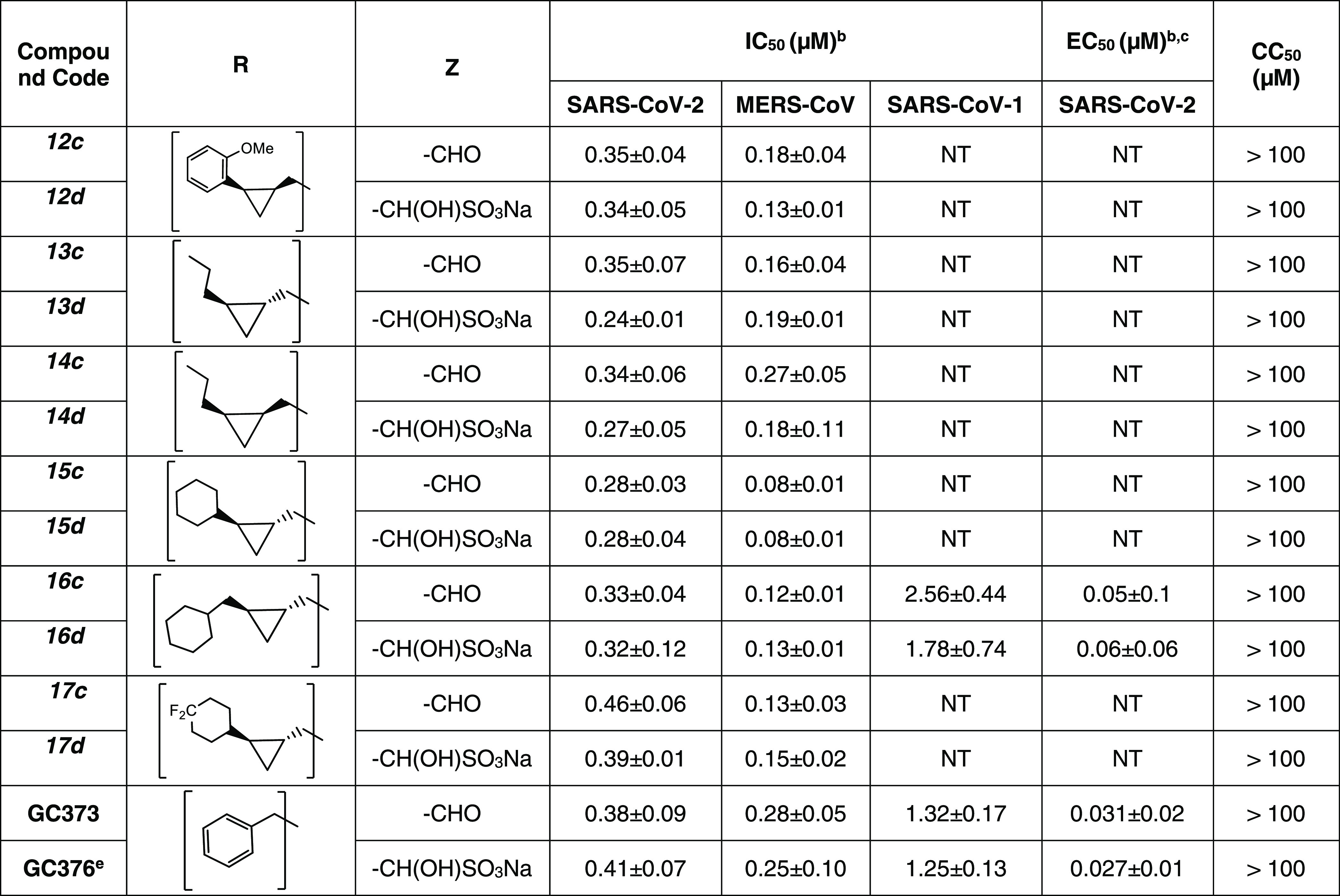
Figure 2.

Inhibition plots of compounds 11c and 11d in enzyme assay (panel A) and cell-based SARS-CoV-2 replicon assay (panel B).
X-ray Crystallography Studies
Determination of high-resolution cocrystal structures provided valuable insights into the structural determinants associated with the binding of the inhibitors to the active site of SARS-CoV-2 3CLpro and MERS-CoV 3CLpro. For some of the structures described below, the electron density was consistent with both the R and S enantiomers at the stereocenter formed by covalent attachment of the Sγ atom of Cys 145 or Cys 148 in SARS-CoV-2 3CLpro and MERS-CoV 3CLpro, respectively. Therefore, the alternate conformations for these cases were modeled with each enantiomer with 0.5 occupancy. Notably, these inhibitors are based on the GC376 scaffold and display a similar binding mode in the S1 and S2 subsites. However, GC376 adopts a “hairpin” conformation, which prevents the phenyl ring from interacting with residues in the hydrophobic ridge of the S4 subsite (vide infra). As such, the structures of inhibitors bound to SARS-CoV-2 3CLpro described here dramatically differ from GC376 in their conformations in the S4 subsite, which was the focus of this series of compounds.
The structures of MERS-CoV 3CLpro with 13c and 14d yielded well-defined electron density overall (Figure 3A–D), and the inhibitors form the typical array of hydrogen bond interactions with the protein, including Gln 167, Glu 169, Gln 192 His 41, His 166 Glu 169, and Phe 143 and the NH of the δ-lactam ring of the inhibitor. The n-propyl groups attached to the cyclopropyl rings are positioned in a similar manner within the S4 subsite, although the terminal carbon atoms are oriented in opposite directions, which is likely due to the differences in chirality at the cyclopropyl ring (Figure 3E,F).
Figure 3.

Binding mode of inhibitors 13c (A, C) and 14d (B, D) in complex with MERS-CoV 3CLpro. Fo-Fc omit map (green mesh) contoured at 3σ (A, B). Hydrogen bond interactions (dashed lines) (C, D). Surface representation showing the orientation of cyclopropyl group of 13c (E) and 14d (F) in the MERS S4 subsite. Neighboring residues are colored yellow (nonpolar), cyan (polar), and white (weakly polar).
The binding modes of inhibitors within the SARS-CoV-2 3CLpro active site containing an aromatic substituent connected to the cyclopropyl group were compared and included inhibitors 1c, 5c, 6c, and 10d (Figure 4). The typical hydrogen bonds were observed for these compounds, although the interaction with Gln 189 is either absent or weaker (one contact) in the case of 1c and 6c, respectively (Figure 4E–H). This is likely due to the orientation of the aryl rings in 1c and 6c, which are positioned over the hydrophobic ridge near the S4 subsite, whereas 5c and 10d are directed toward the S4 subsite pocket (Figure 5). Notably, 5c forms a polar contact between the fluorine atom and the backbone carbonyl of Gln 189. However, the aryl ring of 6c with the larger chlorine atom is rotated approximately 180° relative to 5c, which results in a small conformational change (∼2 Å) in the nearby loop spanning Gln 189 to Gly 195 (Figure 6).
Figure 4.

Binding modes of inhibitors containing an aromatic substituent to the SARS-CoV-2 3CLpro active site. 1c (A, E), 5c (B, F), 6c (C, G), and 10d (D, H). The Fo-Fc omit maps (green mesh, A–D) are contoured at 3σ, and hydrogen bond interactions (E–H) are drawn as dashed lines.
Figure 5.

Orientation of the inhibitors containing an aromatic substituent to SARS-CoV-2 3CLpro highlighting the S4 subsite: 1c (A), 5c (B), 6c (C), and 10d (D). Surface representation showing the orientation of the inhibitors near the S4 subsite of SARS-CoV-2 3CLpro with neighboring residues colored yellow (nonpolar), cyan (polar), and white (weakly polar).
Figure 6.

Superposition of the SARS-CoV-2 3CLpro inhibitor bound structures containing m-fluoro and m-chloro aryl rings. Differences in the orientation of 5c (cyan, fluoro) and 6c (coral, chloro) (A). Alternate view of panel A showing the conformational change in the SARS-CoV-2 3CLpro loop spanning Gln 189-Gly 195 associated with 5c (magenta) and 6c (light green) (B).
The binding mode of inhibitor 13d, which contains a bisulfite warhead but forms an identical covalent complex with 13c, has an n-propyl group connected to the cyclopropane ring. The binding mode of 13d to SARS-CoV-2 3CLpro positions the n-propyl group into the pocket of the S4 subsite and contains a variety of hydrogen bond interactions, as depicted in Figure 7A–C. The orientation of 13d is very similar to that observed as 13c complexed with MERS-CoV 3CLpro (Figure 7D).
Figure 7.

Binding mode of inhibitor 13d to the SARS-CoV-2 3CLpro active site. Fo-Fc omit maps (green mesh) contoured at 3σ (A), hydrogen bond interactions (dashed lines) (B), surface representation showing the orientation of the inhibitors near the S4 subsite with neighboring residues colored yellow (nonpolar), cyan (polar), and white (weakly polar) (C), and superposition of 13c bound to MERS-CoV 3CLpro (gold) and 13d bound to SARS-CoV-2 3CLpro (gray) (D).
While inhibitors 1c, 5c, 6c, 10d, and 13d adopt similar binding modes overall to SARS-CoV-2 3CLpro, distinct differences are observed in the S4 subsite. Notably, inhibitors 1c and 6c are directly away from the S4 pocket, and the aryl rings are positioned over the hydrophobic ridge, which results in a conformational change in the loop spanning Gln 189-Gly 195 (Figure 8A). Conversely, the aryl rings of inhibitors 5c and 10d and the propyl group of 13d are positioned within the S4 pocket. This results in conformations spanning approximately 6.8 Å within the planes and 4.6 Å perpendicular to the planes of the aryl rings (Figure 8B,C).
Figure 8.
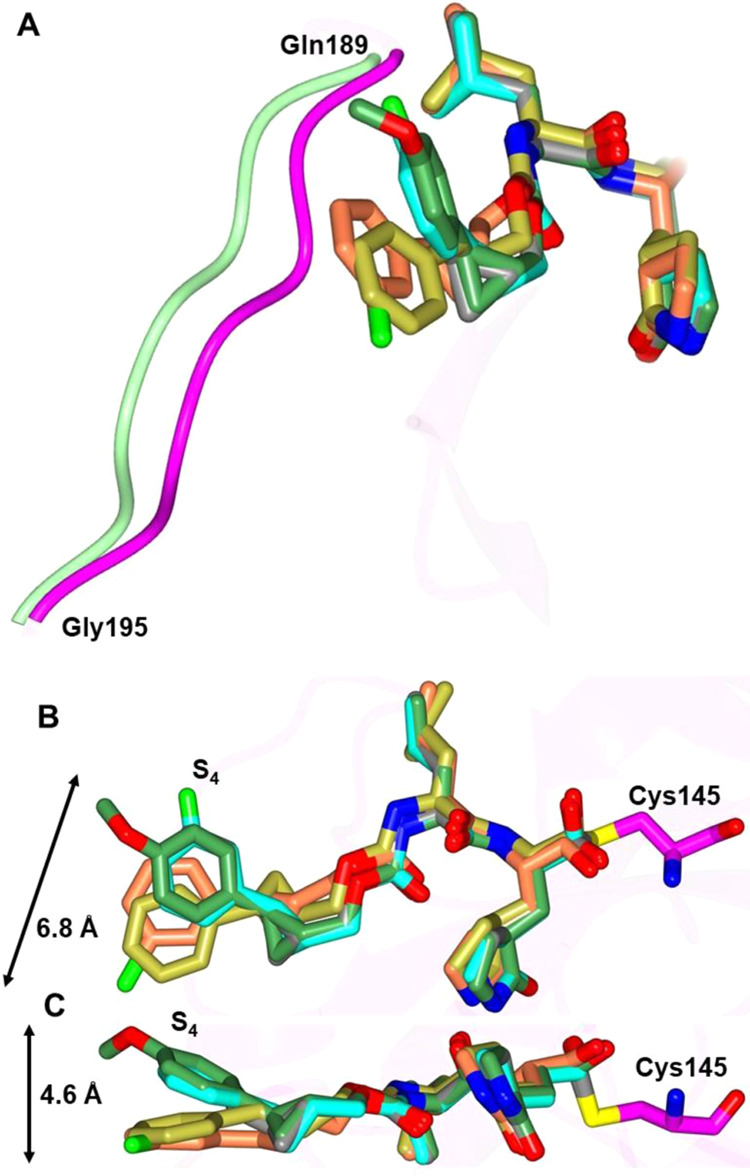
Superposition of the SARS-CoV-2 3CLpro inhibitor bound structures. Superposition of 1c (gold), 5c (cyan), 6c (coral), 10d (green), and 13d (gray) showing the conformational differences in the loop spanning Gln 189-Gly 195 (A). Inhibitors 1c and 6c move the loop (green) away from the active site relative to 5c, 10d, and 13d (magenta). Two views showing the superposition of 1c (gold), 5c (cyan), 6c (coral), 10d (green), and 13d (gray) bound to SARS-CoV-2 3CLpro highlighting the conformational differences in S4 subsite (B, C). The range of motion for the inhibitors is noted by the arrows.
The structures of inhibitors 15d, 16d, and 17d that contain cyclohexyl substituents connected to the cyclopropyl group were also obtained with MERS-CoV 3CLpro and are provided in Figure 9. The compounds adopt nearly identical binding modes in which the cyclohexyl groups are positioned deep within the S4 subsite, as shown in Figure 10. The main difference is observed for compound 16d, which has an extra methylene linker between the cyclopropyl and cyclohexyl rings. For this compound, the cyclohexyl ring is rotated approximately 90o relative to 15d and 17d.
Figure 9.

Structures of MERS-CoV 3CLpro in complex with cyclohexyl-containing compounds. Panels (A–C) are Fo-Fc polder omit maps (green mesh) contoured at 3σ, and hydrogen bond interactions are shown in panels D–F. The compounds are as follows: 15d (A, D), 16d (B, E), and 17d (C, F).
Figure 10.

Superposition of 15d (pink), 16d (gray), and 17d (coral) in complex with MERS-CoV 3CLpro. Surface representations with neighboring residues are colored yellow (nonpolar), cyan (polar), and white (weakly polar) (A). Zoomed-in view showing the orientation of the cyclohexyl rings in the S4 subsite (B).
Similarly, the structures of SARS-CoV-2 3CLpro with 15d and 17d adopt very similar orientations with the active site (Figure 11A–D). The main difference is the presence of a water-mediated contact between a fluorine atom in 17d and the backbone N-atom of Ala 191. In addition, the superposition of 15d and 17d revealed nearly identical orientations of the inhibitors (Figure 11E) in which the cyclohexyl rings are positioned near Thr 190/Ala 191 in the S4 subsite.
Figure 11.

Structures of SARS-CoV-2 3CLpro in complex with cyclohexyl-containing compounds 15d and 17d. Panels (A, B) are Fo-Fc polder omit maps (green mesh) contoured at 3σ, and hydrogen bond interactions are shown in panels (C, D). The compounds are as follows15d (A, C) and 17d (B, D). Superposition of 15d (gray) and 17d (coral) showing the position of the cyclohexyl rings in the S4 subsite. Surface representation with residues colored yellow (nonpolar), cyan (polar), and white (weakly polar) (E).
In summary, these structures demonstrate that subtle changes to the substituents that interact with the S4 subsite can dramatically affect the binding affinity to SARS-CoV-2 3CLpro and MERS-CoV 3CLpro and serve as a guide for subsequent inhibitor design. Notably, all of these structures contain groups in the P4 position that interact with the S4 region of the proteases, which is markedly different from GC376, which served as the inhibitor template (Figure 12). It is clearly evidenced from Figure 12 that the phenyl ring in GC376 cannot participate in any binding interaction with the S4 pocket. Importantly, the results are congruent with recent crystallographic studies highlighting the plasticity in substrate binding orientation and adaptability between the S3 and S4 subsites of SARS-CoV-2 3CLpro and the significant contribution of the P4 group to binding affinity.50
Figure 12.
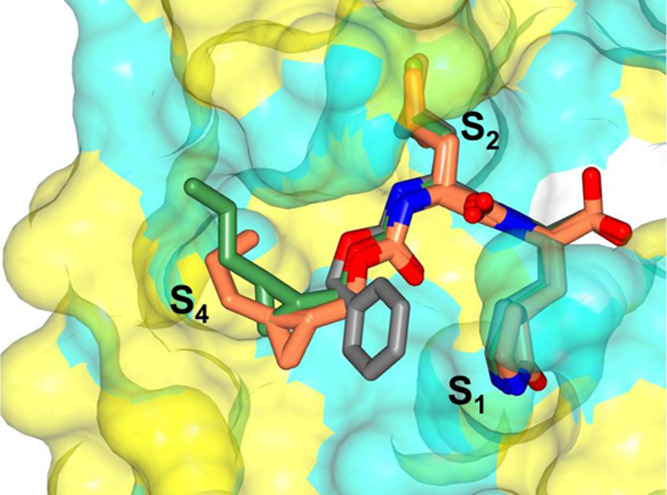
Superposition of GC376 (gray) bound to SARS-CoV-2 3CLpro (PDB 7K0G) with compound 13c bound to MERS 3CLpro (coral) and compound 13d bound to SARS-CoV-2 3CLpro (green). Surface representation of SARS-CoV-2 3CLpro with neighboring residues are colored yellow (nonpolar), cyan (polar), and white (weakly polar).
Structure–Activity Relationships
In the present study, the manifold features and advantages inherent in the cyclopropyl fragment,45 including enhancement in potency by a conformationally constrained and entropically more favorable binding to the active site of the target protease, were exploited in tandem with X-ray crystallography in the design of inhibitors of SARS-CoV-2 3CLpro capable of accessing new chemical space in the active site of the viral protease. The results shown in Table 1 indicate that all of the cyclopropane-derived inhibitors display high potency against SARS-CoV-2, SARS-CoV-1, and MERS-CoV 3CL proteases (for example, compounds 2c/d, 5c/d, 10c/d, 11c/d) in biochemical and cellular assays. The results suggest that these compounds behave as broad-spectrum coronavirus inhibitors. Replacement of the methylene group in the cyclopropane ring with a gem-difluoro group increased potency (1c/d vs 2c/d). Among the halogen-substituted compounds, potency was invariant to the nature and position of halogen substitution in the phenyl ring (compounds 5c/d through 8c/d); however, these compounds were ∼2-fold more potent than the corresponding unsubstituted compound (1c/d). Furthermore, the isomeric methoxy-substituted phenyl compounds (9c/d, 10c/d, and 11c/d) were fairly effective against both SARS-CoV-2 and MERS-CoV 3CL proteases in biochemical assays, as well as in cell-based assays in the case of SARS-CoV-2 with a safety index (SI), defined as CC50/EC50, of 1250. The transo-OCH3-substituted compound was moderately more potent than the corresponding cis isomer (11c/d vs 12c/d). No dramatic variations in potency were observed when the phenyl ring was replaced with an alkyl (13c/d, 14c/d), cyclohexyl (15c/d), cyclohexyl methyl (16c/d), or 4,4-dlifuorocyclohexyl (17c/d) group; however, potency remained consistently high with all compounds (submicromolar range). Collectively, the results indicate that the incorporation of a judiciously decorated cyclopropyl design element into the inhibitors enhances potency. The most potent compounds in this series, 5d and 11d, were found to be ∼2.3-fold more potent than GC376 in the cell-based assay using SARS-CoV-2 replicon system and ∼3.5-fold more potent against MERS-CoV 3CLpro in the FRET enzyme assay. Overall, the potency of the inhibitors also compares favorably with many aldehyde SARS-CoV-2 3CLpro inhibitors reported in the literature (Figure 13).
Figure 13.

IC50 values of representative aldehyde inhibitors against SARS-CoV-2 3CLpro and cathepsin L.
Notably, and in accord with the results of previous studies,37−39,43,44,51 the aldehyde bisulfite adducts are equipotent to the precursor aldehydes. Importantly, in previous studies, we described a general approach toward the optimization of the PK characteristics of aldehyde bisulfite adducts of peptidyl and non-peptidyl transition state inhibitors via the synthesis of the ester or carbamate prodrugs of aldehyde bisulfite adducts (Figure 14).52 The rate of ester cleavage is dependent on the alkyl chain length (R), and in blood plasma, the derivatized bisulfite adducts yield the aldehydes via a chemical and/or enzymatic process. Consequently, the therapeutic potential of the cyclopropane-derived series of inhibitors can be further augmented by exploiting this approach to optimize pharmacokinetics.
Figure 14.
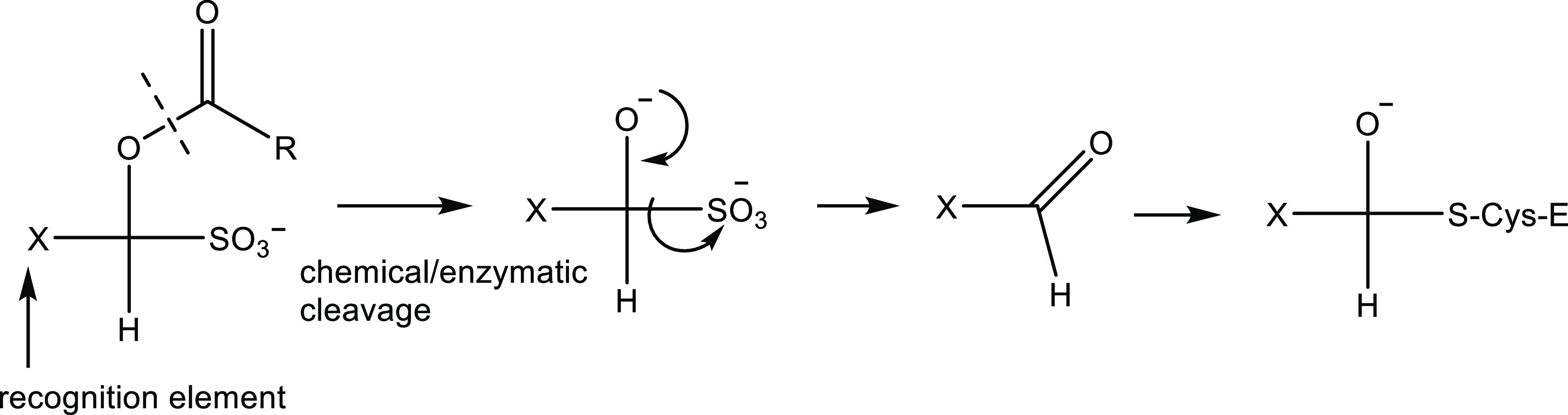
Postulated mechanism of action of prodrugs.
Many aspects of the biology and pathogenesis of SARS-CoV-2 are currently poorly defined. SARS-CoV-2 entry into host cells is multifactorial and involves the interplay of multiple pathways and mediators.53 The latter includes the host proteases transmembrane serine protease 2 (TSPRSS2), furin, and cathepsins B and L.54−57 Inhibitors bearing an aldehyde or masked aldehyde warhead are known to also inhibit cathepsin L,15,58−61 a cysteine protease present at elevated levels in SARS-CoV-2 infection.55,61 Consequently, the inhibitory activity of a select number of inhibitors was evaluated against cathepsin L. A select number of compounds (2c/d, 5c/d, and 11c/d) were found to inhibit cathepsin L, and the IC50 values ranged between 0.01 and 0.05 μM, comparable to MDL28170 (IC50 0.01 μM/Figure 13). The entry inhibition assays with pseudotyped virus expressing SARS-CoV-2 S protein in 293T cells expressing ACE2 alone or ACE2 plus TMPRSS262 were performed with the selected inhibitors and MDL28170. The selected compounds inhibited SARS-CoV-2 S-mediated entry with EC50 values in the range 0.06–0.1 μM in ACE2 expressing cells. The EC50 of MDL28170 in ACE2-expressing cells was 0.03 μM. However, neither the selected compounds nor MDL28170 inhibited SARS-CoV-2 S-mediated entry in cells expressing both ACE2 and TMPRSS2 up to 10 μM. These results are consistent with reports that SARS-CoV-2 internalizes cells through the outer membrane when TMPRSS2 is present.63 Because most susceptible cells in the respiratory system express both ACE2 and TMPRSS2, our findings indicate that cathepsin L inhibitors may not display significant antiviral effects in the major target tissues in humans.
Conclusions
SARS-CoV-2 3CLpro plays a critical role in viral replication and, as such, is an attractive druggable target that can be used in the discovery of direct-acting antivirals. A series of dipeptidyl inhibitors of SARS-CoV-2 3CLpro incorporating in their structure a variously embellished cyclopropane ring were synthesized and evaluated in biochemical and cell-based assays. The generated compounds were found to be highly potent toward SARS-CoV-2 3CLpro, SARS-CoV-1, and MERS-CoV 3CLpro, and were devoid of cytotoxicity. A noteworthy feature of the inhibitors is the broad spectrum of activity displayed by these compounds. Evaluation of a select number of compounds in mouse models of SARS-CoV-2 and MERS-CoV infections is envisaged, and the results will be reported in due course.
Experimental Section
Synthesis of Cyclopropane-Based Inhibitors
Inhibitors 1c/d through 17c/d were prepared and fully characterized, as described in detail in the Supporting Information.
Cloning and Expression of 3CL Proteases
The codon-optimized cDNA of full length of 3CLpro of SARS-CoV-2 (GenBank number MN908947.3) fused with sequences encoding 6 histidine at the N-terminal was synthesized by Integrated DNA (Coralville, IA). The synthesized gene was subcloned into the pET-28a(+) vector. The expression and purification of SARS-CoV-2 3CLpro were conducted following a standard procedure described previously.38 The expression and purification of the 3CL protease of MERS-CoV and SARS-CoV-1 were performed by standard methods described previously by our lab.37
Biochemical FRET Assays
Briefly, a stock solution of an inhibitor was prepared in DMSO and diluted in an assay buffer composed of 20 mM HEPES buffer, pH 8, containing NaCl (200 mM), EDTA (0.4 mM), glycerol (60%), and 6 mM dithiothreitol (DTT). The SARS-CoV-2 3CLpro (or MERS-CoV 3CLpro or SARS-CoV-1 3CLpro) was mixed with serial dilutions of inhibitors 1–17c/d or with DMSO in 25 μL of assay buffer and incubated at 37 °C for 1 h, followed by the addition of 25 μL of assay buffer containing substrate (FAM-SAVLQ/SG-QXL520, AnaSpec, Fremont, CA). The substrate was derived from the cleavage sites on the viral polyproteins of SARS-CoV (or MERS-CoV). Fluorescence readings were obtained using an excitation wavelength of 480 nm and an emission wavelength of 520 nm on a fluorescence microplate reader (FLx800; Biotec, Winooski, VT) 1 h following the addition of the substrate. Relative fluorescence units (RFU) were determined by subtracting background values (substrate-containing well without protease) from the raw fluorescence values, as described previously.37 Selected compounds, including 2c/d, 5c/d, and 11c/d, were also tested against cathepsin L using the cathepsin L inhibitor kit from Abscam (Waltham, MA) per the manufacturer’s procedure. A known cathepsin L aldehyde inhibitor, MDL28170 (Sigma-Aldrich, St. Louis, MO), was included as a positive control. The dose-dependent FRET inhibition curves were fitted with a variable slope using GraphPad Prism software (GraphPad, La Jolla, CA) to determine the IC50 values of the compounds.
Cell-Based Inhibition Assays
To assess the antiviral effects of selected compounds (dissolved in DMSO) in cell culture, the SARS-CoV-2 replicon system with pSMART-T7-scv2-replicon (pSMART BAC V2.0 Vector Containing the SARS-CoV-2, Wuhan-Hu-1 Noninfectious Replicon) was used.64 The clone was obtained from BEI Resources, and experiments were performed in a BSL-2 setting. The synthetic SARS-CoV-2 replicon RNA was prepared from the pSMART-T7-scv2-replicon as described,64 and the Neon Electroporation system (Thermo Fisher, Chicago, IL) was used for the RNA electroporation to 293T cells. After the electroporation, cells were incubated with DMSO (0.1%) or each compound at 2, 0.5, 0.1, and 0.02 μM for 30 h, and luciferase activities were measured for antiviral effects. The entry inhibition assay with pseudotyped virus expressing SARS-CoV-2 S protein in 293T cells expressing ACE2 alone or ACE2 plus TMPRSS2 was performed with the selected inhibitors 2c/d, 5c/d, 11c/d, and MDL28170 using a recently-established assay in our lab.62 The dose-dependent inhibition curve for each compound was prepared, and the 50% effective concentration (EC50) values were determined by GraphPad Prism software using a variable slope (GraphPad, La Jolla, CA).
Measurement of In Vitro Cytotoxicity (Nonspecific Cytotoxic Effects)
Confluent cells grown in 96-well plates were incubated with various concentrations (1 to 100 μM) of each compound for 72 h. Cell cytotoxicity was measured in 293T cells by a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI), and the CC50 values were calculated using a variable slope by GraphPad Prism software. The in vitro Safety Index was calculated by dividing the CC50 by the EC50.
Crystallization and Data Collection
Purified MERS-CoV 3CLpro and SARS-CoV-2 3CLpro in 100 mM NaCl, 20 mM Tris pH 8.0 were concentrated to 10 mg/mL (0.3 mM) for crystallization screening. Stock solutions of the inhibitors were prepared in DMSO at 100 mM, and the complexes with the 3CL proteases were prepared by adding 2 mM of each compound and incubating the complexes on ice for 1 h. All crystallization experiments were set up using an NT8 drop-setting robot (Formulatrix, Inc.) and UVXPO MRC (Molecular Dimensions) sitting drop vapor diffusion plates at 18 °C. Briefly, 100 nL of protein and 100 nL of crystallization solution were dispensed and equilibrated against 50 μL of the latter. Crystals for the 3CLpro:inhibitor complexes were obtained in 1–2 days from the following conditions. SARS-CoV-2 3CLpro: 1c Proplex HT screen (Molecular Dimensions) condition E2 (8% (w/v) PEG 8000, 100 mM sodium citrate pH 5.0), PACT screen (Molecular Dimensions) 5c condition C9 (20% (w/v) PEG 6000, 100 mM Hepes pH 7.0, 200 mM lithium chloride), 6c condition D6 (25% (w/v) PEG 1500, 100 MMT pH 9.0), 10d condition G4 (20% (w/v) PEG 3350, 100 Bis-Tris propane pH 7.5, 200 mM potassium thiocyanate), and 13d Index HT screen (Hampton Research) condition D10 (20% (w/v) PEG 5000 MME, 100 Bis-Tris pH 6.5). 15d and 17d PACT screen (Molecular Dimensions) condition B6 (25% (w/v) PEG 1500, 100 MIB pH 9.0). MERS-CoV 3CLpro: 13c Index screen (Hampton Research) condition F5 (17% (w/v) PEG 10000, 100 mM bis-Tris pH 5.5, 100 mM ammonium acetate), 14d Proplex HT screen (Molecular Dimensions) condition G3 (25% (w/v) PEG 3350, 100 mM Tris pH 8.5, 200 mM Lithium Sulfate). 15d and 17d Index HT screen (Hampton Research) condition F6 (25% (w/v) PEG 3350, 100 mM Bis-Tris pH 5.5, 200 mM Ammonium Sulfate), and 16d Index HT screen (Hampton Research) condition H4 (20% (w/v) PEG 3350, 200 ammonium citrate tribasic pH 7.0). Samples were transferred to a fresh drop composed of 80% crystallization solution and 20% (v/v) PEG 200 and stored in liquid nitrogen. X-ray diffraction data were collected at the Advanced Photon Source beamline 17-ID (IMCA-CAT) and the National Synchrotron Light Source II (NSLS-II) NYX beamline 19-ID.
Structure Solution and Refinement
Intensities were integrated using XDS65,66 via autoPROC67 and the Laue class analysis, and data scaling was performed with AIMLESS.68 Structure solution was conducted by molecular replacement with Phaser69 using a previously determined inhibitor bound structures of MERS-CoV 3CLpro (5WKK) and SARS-CoV-2 3CLpro (PDB 6XMK) as the search models. Structure refinement and manual model building were conducted with Phenix70 and Coot,71 respectively. Disordered side chains were truncated to the point for which electron density could be observed. Structure validation was conducted with MolProbity,72 and figures were prepared using the CCP4MG package.73 Crystallographic data are provided in Tables S1 and S2.74−78
Acknowledgments
This research was supported in part by grants from the National Institutes of Health (NIH) (Grants R01 AI130092 and AI161085 to K.O.C.). Use of the University of Kansas Protein Structure Laboratory was supported by a grant from the National Institute of General Medical Sciences (P30GM110761) of the NIH. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract No. DE-AC02-06CH11357. This research used the NYX beamline 19-ID of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The NYX detector instrumentation was supported by Grant S10OD030394 through the Office of the Director of the National Institutes of Health. The qNMR analysis was performed at the University of Kansas Nuclear Magnetic Resonance Laboratory, and support for the NMR instrumentation was provided by NIH Shared Instrumentation Grant # S10RR024664 and NSF Major Research Instrumentation Award # 1625923. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010). The following reagent was obtained through BEI Resources, NIAID, NIH: pSMART BAC V2.0 Vector Containing the SARS-Related Coronavirus 2, Wuhan-Hu-1 Noninfectious Replicon, NR-54972.
Glossary
Abbreviations
- CC50
50% cytotoxic concentration in cell-based assays
- CDI
carbonyl diimidazole
- CPE
cytopathic effects
- DAAs
direct-acting antivirals
- DMSO
dimethyl sulfoxide
- DMP
Dess–Martin periodinane
- DSC
N,N′-disuccinimidyl carbonate
- DTT
dithiothreitol
- EC50
50% effective concentration in cell culture
- GESAMT
general efficient structural alignment of macromolecular targets
- IC50
50% inhibitory concentration in the enzyme assay
- MME
monomethyl ether
- MNV
murine norovirus
- MOI
multiplicity of infection
- ORF
open reading frame
- PK
pharmacokinetics
- RMSD
root-mean-square deviation
- TCID50
50% tissue culture infectious dose
- TEA
triethyl amine
- XDS
X-ray detector software
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00206.
Synthesis and characterization of inhibitors 1–17b/c and crystallographic data for MERS 3CLpro and SARS-CoV-2 3CLpro inhibitor complexes (PDF)
Accession Codes
Coordinates and structure factors for complexes with the following inhibitors were deposited to the Worldwide Protein Databank (wwPDB) with the accession codes: MERS-CoV 3CLpro complexes 13c (7TQ7), 14d (7TQ8), 15d (8CZT), 16d (8DGY), and 17d (8CZV). SARS-CoV-2 3CLpro complexes 1c (7TQ2), 5c (7TQ3), 6c (7TQ4), 10d (7TQ5), 13d (7TQ6), 15d (8CZW), and 17d (8CZX).
The authors declare no competing financial interest.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
Supplementary Material
References
- accessed Dec 7
- Wu F.; Zhao S.; Yu B.; Chen Y.-M.; Wang W.; Song Z.-G.; Hu Y.; Tao Z.-W.; Tian J.-H.; Pei Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanet A.; Autran B.; Lina B.; Kieny M. P.; Karim S. S. A.; Sridhar D. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021, 397, 952–954. 10.1016/S0140-6736(21)00370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Koganti R.; Lambe U. P.; Yadavalli T.; Nandi S. S.; Shukla D. J. Vaccines and therapies in development for SARS-CoV-2 infections. J. Clin. Med. 2020, 9, 1885 10.3390/jcm9061885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Li W.; Drabek D.; Okba N. M. A.; van Haperen R.; Osterhaus A. D. M. E.; van Kuppeveld F. J. M.; Haagmans B. L.; Grosveld F.; Bosch B-J. A human monoclonal antibody blocking SARS-CoV-2. Nat. Commun. 2020, 11, 2251 10.1038/s41467-020-16256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Mishevich J. L.; Mesecar A. D.; Mitsuya H. Recent drug development and medicinal approaches for the treatment of SARS-CoV-2 infection and COVID-19. ChemMedChem 2022, 17, e202200440 10.1002/cmdc.202200440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monica G. L.; Bono A.; Lauria A.; Martorana A. Targeting SARS-CoV-2 main protease for treatment of COVID-19: Covalent inhibitors structure-activity relationship insights and evolution perspectives. J. Med. Chem. 2022, 65, 12500–12534. 10.1021/acs.jmedchem.2c01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson C. B.; Golden J. E.; Meibohm B. Time to “Mind the Gap” in novel small molecule drug discovery for direct-acting antivirals for SARS-CoV-2. Curr. Opin. Virol. 2021, 50, 1–7. 10.1016/j.coviro.2021.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil C.; Ginex T.; Maestro I.; Nozal V.; Barrado-Gil L.; Cuesta-Geijo M. A.; Urquiza J.; Ramírez D.; Alonso C.; Campillo N. E.; Martinez A. COVID-19: Drug targets and potential treatments. J. Med. Chem. 2020, 63, 12359–12386. 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Tiwari S.; Deb M. K.; Marty J. L. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): A global pandemic and treatments strategies. Int. J. Antimicrob. Agents 2020, 56, 106054 10.1016/j.ijantimicag.2020.106054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannalire R.; Cerchia C.; Beccari A. R.; Di Leva F. S.; Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: State of the art and future opportunities. J. Med. Chem. 2022, 65, 2716–2746. 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim S. S. A.; de Oliveira T. New SARS-CoV-2 variants – clinical, public health, and vaccine implications. N. Engl. J. Med. 2021, 384, 1866–1868. 10.1056/NEJMc2100362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y-W.; Chao T-L.; Li C-L.; Chiu M-F.; Kao H-C.; Sheng-Han W.; Yu-Hao P.; Chih-Hui L.; Ya-Min T.; Wen-Hau L.; Mi-Hua T.; Tung-Ching H.; Ping-Yi W.; Li-Ting J.; Pei-Jer C.; Sui-Yuan C.; Shiou-Hwei Y. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. 2020, 33, 108254 10.1016/j.celrep.2020.108254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T.; Laufer S. Kinases as potential therapeutic targets for anti-coronaviral therapy. J. Med. Chem. 2022, 65, 955–982. 10.1021/acs.jmedchem.1c00335. [DOI] [PubMed] [Google Scholar]
- Sacco M. D.; Ma C.; Lagarias P.; Gao A.; Townsend J. A.; Meng X.; Dube P.; Zhang X.; Hu Y.; Kitamura N.; Hurst B.; Tarbet B.; Marty M. T.; Kolocouris A.; Xiang Y.; Chen Y.; Wang J. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020, 6, eabe0751 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman S.; Masters P. S.. Coronaviridae: The Viruses and Their Replication. In Fields Virology: Emerging Viruses, 7th ed.; Howley P. M.; Knipe D. M.; Whelan S., Eds.; Wolters Kluwer: Wichita, 2020; Vol. 1, pp 410–448. [Google Scholar]
- Berger A. Reprint of “On the Size of the Active Site in Proteases. I. Papain”. Biochem. Biophys. Res. Com. 2012, 425, 497–502. 10.1016/j.bbrc.2012.08.015. [DOI] [PubMed] [Google Scholar]; where the residues on the N-terminus side of the peptide bond that is cleaved are designated as P1-Pn and those on the C-terminus side are designated P1-Pn’. The corresponding active site subsites are designated S1-Sn and S1-Sn’.; S1 is the primary substrate specificity subsite and P1-P1′ is the scissile bond
- Rut W.; Groborz K.; Zhang L.; Sun X.; Zmudzinski M.; Hilgenfeld R.; Drag M.. Substrate specificity profiling of SARS-CoV-2 Mpro protease provides basis for anti-COVID-19 drug design bioRxiv preprint, 2020.
- Zhao Y.; Zhu Y.; Liu X.; Jin Z.; Duan Y.; Zhang Q.; Wu C.; Feng L.; Du X.; Zhao J.; Shao M.; Zhang B.; Yang X.; Wu L.; JI X.; Guddat L. W.; Yang K.; Rao Z.; Yang H. Structural basis for replicase polyprotein cleavage and substrate specificity of main protease from SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2117142119 10.1073/pnas.2117142119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agost-Beltrán L.; de la Hoz Rodriguez S.; Bou-Iserte L.; Rodriguez S.; Fernadez-de-la-Pradilla A.; Gonzalez F. V. Advances in the development of SARS-CoV-2 Mpro inhibitors. Molecules 2022, 27, 2523 10.3390/molecules27082523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K.; Wang R.; Chen J.; Tepe J. J.; Huang F.; Wei G-W. Perspectives on SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2021, 64, 16922–16955. 10.1021/acs.jmedchem.1c00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Hu L.; Huang X.; Wang C.; Zhang Z.; Wang Y.; Zhang D.; Ye W. Potential of coronavirus 3C-like protease inhibitors for the development of new anti-SARS-CoV-2 drugs: Insights from structures of protease and inhibitors. Int. J. Antimicrob. Agents 2020, 56, 106055 10.1016/j.ijantimicag.2020.106055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Dai W.; Jochmans D.; Xie H.; Yang H.; Li J.; Su H.; Chang D.; Wang J.; Peng J.; Zhu L.; Nian Y.; Hildenfeld R.; Jiang H.; Chen K.; Zhang L.; Xu Y.; Neyts J.; Liu H. Design, synthesis, and biological evaluation of peptidomimetic aldehydes as broad-spectrum inhibitors against enterovirus and SARS-CoV-2. J. Med. Chem. 2022, 65, 2794–2808. 10.1021/acs.jmedchem.0c02258. [DOI] [PubMed] [Google Scholar]
- Dai W.; Zhang B.; Jiang X.-M.; Su H.; Li J.; Zhao Y.; Xie X.; Jin Z.; Peng J.; Liu F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J.; Li; Zeng R.; Liu F-L.; Luo R-H.; Huang C.; Wang Y-F.; Zhang J.; Quan B.; Shen C.; Mao X.; Liu X.; Sun W.; Yang W.; Ni X.; Wang K.; Xu L.; Duan Z-L.; Zou Q-C.; Zhang H-L.; Qu W.; Peng Y-H.; Li M-H.; Yang R-C.; Liu X.; You J.; Zhou Y.; Yao R.; Li W-P.; Liu J-M.; Chen P.; Liu Y.; Lin G-F.; Yang X.; Zou J.; Li L.; Hu Y.; Lu G-W.; Li W-M.; Wei Y-Q.; Zheng Y-T.; Lei J.; Yang S. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B.; Belovodskiy A.; Hena M.; Kandadai A. S.; Joyce M. A.; Saffran H. A.; Shields J. A.; Khan M. B.; Arutyunova E.; Lu J.; Bajwa S. K.; Hockman D.; Fischer C.; Lamer T.; Vuong W.; van Belkum M. J.; Gu Z.; Lin F.; Du Y.; Xu J.; Rahim M.; Young H. S.; Vederas J. C.; Tyrrell L. D.; Lemieux J.; Nieman J. A. Peptidomimetic α-acyloxymethylketone warheads with six-membered lactam P1 glutamine mimic: SARS-CoV-2 3CL protease inhibition, coronavirus antiviral activity, and in vitro biological stability. J. Med. Chem. 2022, 65, 2905–2925. 10.1021/acs.jmedchem.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Raghavaiah J.; Shahabi D.; Yadav M.; Anson B. J.; Lendy E. K.; Hattori S. I.; Higashi-Kuwata N.; Mitsuya H.; Mesecar A. D. Indole chloropyridinyl ester-derived SARS-CoV-2 3CLpro inhibitors: enzyme inhibition, antiviral efficacy, structure-activity relationship and X-ray structural studies. J. Med. Chem. 2021, 64, 14702–14714. 10.1021/acs.jmedchem.1c01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno S.; Kobayashi K.; Senda M.; Funai Y.; Tamai I.; Schakel L.; Sakata K.; Pillaiyar T.; Taguchi A.; Taniguchi A.; Gutschow M.; Muller C. E.; Takeuchi K.; Hirohama M.; Kawaguchi A.; Kojima M.; Senda T.; Shirasaka Y.; Kamitani W.; Hayashi Y. 3CL protease inhibitors with an electrophilic acylketone moiety as anti-SARS-CoV-2 agents. J. Med. Chem. 2022, 65, 2926–2939. 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- Li L.; Chenna B. C.; Yang K. S.; Cole T. R.; Goodall Z. T.; Giardini M.; Moghadamchargari Z.; Hernandez E. A.; Gomez J.; Calvet C. M.; Bernatchez J. A.; Mellott D. M.; Zhu J.; Rademacher A.; Thomas D.; Blankenship L. R.; Drelich A.; Laganowsky A.; Tseng C-T. K.; Liu W. R.; Wand A. J.; Cruz-Reyes J.; Siquira-Neto J.; Meek T. D. Self-masked aldehyde inhibitors: a novel strategy for inhibiting cysteine proteases. J. Med. Chem. 2021, 64, 11267–11287. 10.1021/acs.jmedchem.1c00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S. H.; Goins C. M.; Arya T.; Shin W-J.; Maw J.; Hooper A.; Sonawane D. P.; Porter M. R.; Bannister B. E.; Crouch; Lindsey A. A.; Lakatos G.; Marttinez S. R.; Alvarado J.; Akers W. S.; Wang N. S.; Jung J. U.; Macdonald J. D.; Stauffer S. R. Structure-based optimization of ML300-derived, noncovalent inhibitors targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL protease (SARS-CoV-3CLpro). J. Med. Chem. 2022, 65, 2880–2904. 10.1021/acs.jmedchem.1c00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenbach J.; Lemke C.; Pillaiyar T.; Schakel L.; Al Hamwi G.; Diett M.; Gedschold R.; Geiger N.; Lopez V.; Mirza S.; Namasivayam V.; Schiedel A. C.; Sylvester K.; Thimm D.; Vielmuth C.; Vu L. P.; Zyulina M.; Bodem J.; Gutschow M.; Muller C. E. Targeting the main protease of SARS-CoV-2: from the establishment of a high throughput screening to the design of tailored inhibitors. Angew. Chem., Int. Ed. 2021, 60, 10423–10429. 10.1002/anie.202016961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura N.; Sacco M. D.; Ma C.; Hu Y.; Townsend J. A.; Meng X.; Zhang F.; Zhang X.; Ba M.; Szeto T.; Kukuljac A.; Marty M. T.; Schultz D.; Cherry S.; Xiang Y.; Chen Y.; Wang J. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2022, 65, 2848–2865. 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery L.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak R.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Boras B.; Jones R. M.; Anson B. J.; Arenson D.; Aschenbrenner L.; Bakowski M. A.; Beutler N.; Binder J.; Chen E.; Eng E.; Hammond H.; Hammond J.; Haupt R. E.; Hoffman R.; Kadar E. P.; Kania R.; Kimoto E.; Kirkpatrick M. G.; Lanyon L.; Lendy E. K.; Lillis J. R.; Logue J.; Luthra S. A.; Ma C.; Mason S. W.; McGrath M. E.; Noell S.; Obach R. S.; O’Brien M. N.; O’Connor R.; Ogilvie K.; Owen D.; Pettersson M.; Reese M. R.; Rogers T. F.; Rosales R.; Rossulek M. I.; Sathish J. G.; Shirai N.; Steppan C.; Ticehurst M.; Updyke L. W.; Weston S.; Zhu Y.; White K. M.; Garcia-Sastre A.; Wang J.; Chatterjee A. K.; Mesecar A. D.; Frieman M. B.; Anderson A. S.; Allerton C. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 6055 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathnayake A. D.; Zheng J.; Kim Y.; Perera K. D.; Mackin S.; Meyerholz D. K.; Kashipathy M. M.; Battaile K. P.; Lovell S.; Perlman S. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV–infected mice. Sci. Transl. Med. 2020, 12, eabc5332 10.1126/scitranslmed.abc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampalla C. S.; Kim Y.; Bickmeier N.; Rathnayake A. D.; Nguyen H. N.; Zheng J.; Kashipathy M. M.; Baird M. A.; Battaile K. P.; Lovell S.; Perlman S.; Chang K. O.; Groutas W. C. Structure-guided design of conformationally-constrained cyclohexane inhibitors of severe acute respiratory syndrome coronavirus-2 3CL protease. J. Med. Chem. 2021, 64, 10047–10058. 10.1021/acs.jmedchem.1c00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampalla C. S.; Rathnayake A. D.; Perera K. D.; Jesri A-R. M.; Nguyen H. N.; Miller M. J.; Thurman H. A.; Zheng J.; Kashipathy M. M.; Battaille K. P.; Lovell S.; Perlman S.; Kim Y.; Groutas W. C.; Chang K. O. Structure-guided design of potent inhibitors of SARS-CoV-2 3CL protease: structural, biochemical, and cell-based studies. J. Med. Chem. 2021, 64, 17846–17865. 10.1021/acs.jmedchem.1c01037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampalla C. S.; Zheng J.; Perera K. D.; Wong L-Y. R.; Meyerholz D. K.; Nguyen H. N.; Kashipathy M. M.; Battaile K. P.; Lovell S.; Kim Y.; Perlman S.; Groutas W. C.; Chang K-O. Post-infection treatment with a protease inhibitor increases survival of mice with a fatal SARS-CoV-2 infection. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2101555118 10.1073/pnas.2101555118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen N. C.; Kim Y.; Liu H.; Kankanamalage A. C. G.; Eckstrand C.; Groutas W. C.; Bannasch M.; Meadows J. M.; Chang K.-O. Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2018, 20, 378–392. 10.1177/1098612X17729626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Liu H.; Kankanamalage A. C. G.; Weerasekara S.; Hua D. H.; Groutas W. C.; Chang K.-O.; Pedersen N. C. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 2016, 12, e1005531 10.1371/journal.ppat.1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Lovell S.; Tiew K.-C.; Mandadapu S. R.; Alliston K. R.; Battaile K. P.; Groutas W. C.; Chang K.-O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampalla C. S.; Rathnayake A. D.; Kankanamalage A. C. G.; Kim Y.; Perera K. D.; Nguyen H. N.; Miller M. J.; Madden T. K.; Picard H. R.; Thurman H. A.; Kishipathy M. M.; Liu L.; Battaille K. P.; Lovell S.; Chang K. O.; Groutas W. C. Structure-guided design of potent spirocyclic inhibitors of severe acute respiratory syndrome coronavirus-2 3C-like protease. J. Med. Chem. 2022, 65, 7818–7832. 10.1021/acs.jmedchem.2c00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talele T. T. The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules. J. Med. Chem. 2016, 59, 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Z.; Benmohamed R.; Zhang W.; Kim J.; Edgerly C. K.; Zhu Y.; Morimoto R. I.; Ferrante R. J.; Kirsch D. R.; Silverman R. B. Chiral cyclohexane 1,3-diones as inhibitors of mutant SOD1-dependent protein aggregation for the treatment of ALS. ACS Med. Chem. Lett. 2012, 3, 584–587. 10.1021/ml3000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro K.; Morimoto Y.; Amii H. Sodium bromodifluoroacetate: a difluorocarbene source for the synthesis of gem-difluorocyclopropanes. Synthesis 2010, 12, 2080–2084. 10.1055/s-0029-1218754. [DOI] [Google Scholar]
- Ghosh A. K.; Duong T. T.; McKee S. P.; Thompson W. J. N, N′-Disuccinimidyl carbonate: A useful reagent for alkoxycarbonylation of amines. Tetrahedron Lett. 1992, 33, 2781–2784. 10.1016/S0040-4039(00)78856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjell D. P.; Slattery B. J.; Semo M. J. A novel, nonaqueous method for regeneration of aldehydes from bisulfite adducts. J. Org. Chem. 1999, 64, 5722–5724. 10.1021/jo990543v. [DOI] [PubMed] [Google Scholar]
- Lee J.; Kenward C.; Worrall L. J.; Vuckovic M.; Gentile F.; Ton A-T.; Ng M.; Cherkasov A.; Strynadka N. C. J.; Poetzel M. X-ray crystallographic characterization of the SARS-CoV-2 main protease polyprotein cleavage sites essential for viral processing and maturation. Nature Comm. 2022, 13, 5196 10.1038/s41467-022-32854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandadapu S. R.; Gunnam M. R.; Tiew K. C.; Uy R. A.; Prior A. M.; Alliston K. R.; Hua D. H.; Kim Y.; Chang K. O.; Groutas W. C. Inhibition of norovirus 3CL protease by bisulfite adducts of transition state inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 62–65. 10.1016/j.bmcl.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankanamalage A. C. G.; Kim Y.; Rathnayake A. D.; Alliston K. R.; Butler M. M.; Cardinale S. C.; Bowlin T. L.; Groutas W. C.; Chang K. O. Design, synthesis and evaluation of novel prodrugs of transition state inhibitors of norovirus 3CL protease. J. Med. Chem. 2017, 60, 6239–6248. 10.1021/acs.jmedchem.7b00497. [DOI] [PubMed] [Google Scholar]
- Shang J.; Wan Y.; Luo C.; Ye G.; Geng Q.; Auerbach A.; Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 11727–11734. 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestle D.; Heindl M. R.; Limburg H.; van Lam van T.; Pilgram O.; Moulton H.; Stein D. A.; Hardes K.; Eickmann M.; Dolnik O.; Rohde C.; Klenk H. D.; Garten W.; Steinmetzer T.; Bottcher-Friebertschauser E. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786 10.26508/lsa.202000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M-M.; Yang W-L.; Yang F-Y.; Zhang L.; Huang W-J.; Hou W.; Fan C-F.; Jin R-H.; Feng Y-M.; Wang Y-C.; Yang J-K. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduction Targeted Ther. 2021, 6, 134 10.1038/s41392-021-00558-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng B.; Abdullahi A.; Ferreira I. A. T. M.; Goonawardane N.; Saito A.; Kimura I.; Yamasoba D.; Gerber P. P.; Fatihi S.; Rathore S.; Zepeda S. K.; Papa G.; Kemp S. A.; Ikeda T.; Toyoda M.; Tan T. S.; Kuramochi J.; Mitsunaga S.; Ueno T.; Shirakawa K.; Takaori-Kondo A.; Brevini T.; Mallery D. L.; Charles O. J.; Bowen J. E.; Joshi A.; Walls A. C.; Jackson L.; Martin D.; Smith K. G. C.; Bradley J.; Briggs J. A. G.; Choi J.; Madissoon E.; Meyer K. B.; Micochova P.; Ceron-Gutierrez L.; Doffinger R.; Teichmann S. A.; Fisher A. J.; Pizzuto M. S.; de Marco A.; Corti D.; Hosmillo M.; Lee J. H.; James L. C.; Thukral L.; Veesler D.; Sigal A.; Sampaziotis F.; Goodfellow I. G.; Matheson N. J.; Sato K.; Gupta R. K.; et al. Altered TMPRSS2 usage of SARS-CoV-2 omicron impacts infectivity and furogenicity. Nature 2022, 603, 706–714. 10.1038/s41586-022-04474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G.; Gosalia D. N.; Rennekamp A. J.; Reeves J. D.; Diamond S. L.; Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 11876–11881. 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X. R.; Alugubelli Y. R.; Ma Y.; Vatansever E. C.; Scott D. A.; Qiao Y.; Yu G.; Xu S.; Liu W. R. MPI8 is potent against SARS-CoV-2 by inhibiting dually and selectively the SARS-CoV-2 main protease and the host cathepsin L. ChemMedChem 2022, 17, e2021100456 10.1002/cmdc.202100456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashhurst A. S.; Tang A. H.; Fajtova P.; Yoon M. C.; Aggarwal A.; Bedding M. J.; Stoye A.; Beretta L.; Pwee D.; Drelich A.; Skinner D.; Li L.; Meek T. D.; McKerrow J. H.; Hook V.; Tseng C-T.; Larance M.; Turville S.; Gerwick W. H.; O’Donoghue A. J.; Payne R. J. Potent anti-SARS-CoV-2 activity by the natural product gallinamide A and analogues via inhibition of cathepsin L. J. Med. Chem. 2022, 65, 2956–2970. 10.1021/acs.jmedchem.1c01494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Chen Y.; Lockbaum G. J.; Sen S.; Chaudhuri S.; Reyes A. C.; Lee J. M.; Kaur A. N.; Sultana N.; Cameron M. D.; Shaffer S. A.; Schiffer C. A.; Fitzgerald K. A.; Thomson P. R. Dual inhibitors of main protease (Mpro) and cathepsin L as potent antivirals against SARS-CoV-2. J. Am. Chem. Soc. 2022, 144, 21035–21045. 10.1021/jacs.2c04626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Ratia K. M.; Richner J. M.; Thatcher G. R. J.; Kadam R.; Smieszek S. P.; Przhycodzen B. P.; Koprivica V.; Birznieks G.; Polymeropoulos M. H.; Prabhakar B. S.. Dual inhibition of cathepsin L and and 3CL-pro by GC376 constrains SARS-CoV-2 infection including omicron variant bioRxiv Preprint, 2022.
- Kim Y.; Gaudreault N. N.; Meekins D. A.; Perera K. D.; Bold D.; Trujillo J. D.; Morozov I.; McDowell C. D.; Chang K-O.; Richt J. A. Effects of spike mutations in SARS-CoV-2 variants of concern on human or animal ACE2-mediated virus entry and neutralization. Microbiol. Spectrum 2022, 10, e0178921 10.1128/spectrum.01789-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J.; Uckeley Z. M.; Doldan P.; Stanifer M.; Boulant S.; Lozach P-Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821 10.15252/embj.2021107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.; Quan S.; Xu M.; Rodriguez S.; Goh S. L.; Wei J.; Fridman A.; Koeplinger K. A.; Carroll S. S.; Grobler J. A.; Espeseth A. S.; Olsen D. B.; Hazudas D. J.; Wang D. Generation of SARS-CoV-2 reporter replicon for high-throughput antiviral screening and testing. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2025866118 10.1073/pnas.2025866118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Automatic indexing of rotation diffraction patterns. J. Appl. Crystallogr. 1988, 21, 67–72. 10.1107/S0021889887009737. [DOI] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 293–302. 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 282–292. 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potterton L.; McNicholas S.; Krissinel E.; Gruber J.; Cowtan K.; Emsley P.; Murshudov G. N.; Cohen S.; Perrakis A.; Noble M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2288–2294. 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62, 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Diederichs K.; Karplus P. A. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct. Biol. 1997, 4, 269–275. 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- Weiss M. S. Global indicators of X-ray data quality. J. Appl. Crystallogr. 2001, 34, 130–135. 10.1107/S0021889800018227. [DOI] [Google Scholar]
- Karplus P. A.; Diederichs K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Biochemistry. Resolving some old problems in protein crystallography. Science 2012, 336, 986–987. 10.1126/science.1222162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.