

Abstract
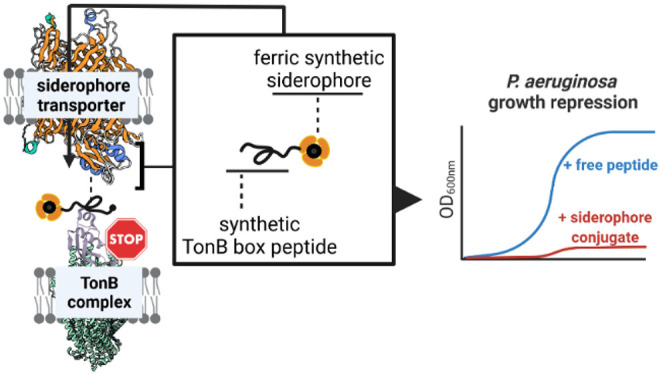
Rising infection rates with multidrug-resistant pathogens calls for antibiotics with novel modes of action. Herein, we identify the inner membrane protein TonB, a motor of active uptake in Gram-negative bacteria, as a novel target in antimicrobial therapy. The interaction of the TonB box of outer membrane transporters with TonB is crucial for the internalization of essential metabolites. We designed TonB box peptides and coupled them with synthetic siderophores in order to facilitate their uptake into bacteria in up to 32 synthetic steps. Three conjugates repressed the growth of Pseudomonas aeruginosa cells unable to produce their own siderophores, with minimal inhibitory concentrations between 0.1 and 0.5 μM. The transporters mediating uptake of these compounds were identified as PfeA and PirA. The study illustrates a variant of cellular suicide where a transporter imports its own inhibitor and demonstrates that artificial siderophores can import cargo with molecular weights up to 4 kDa.
Introduction
Due to antibiotic overuse and a shallow antibiotic pipeline, nosocomial infections with multidrug-resistant (MDR) Gram-negative bacteria, e.g., Pseudomonas aeruginosa or Escherichia coli, have become increasingly difficult to treat.1−3 Moreover, newly approved antimicrobials are largely derivatives of existing classes, while novel modes of action are scarce.4,5 “Critical” pathogens on the WHO’s priority list are all Gram-negative bacteria, mainly because they effectively prevent the accumulation of antibiotics using their double-layered cell membranes, which operate as tight biological barriers.6 The “Trojan horse” strategy outsmarts this hurdle by hijacking prokaryotic nutrient transport systems to increase the penetration of a variety of payloads (e.g., dyes, radioactive labels, and antibiotics) to image and treat infections.7,8 A key nutrient of prokaryotes is iron, which fulfills numerous enzymatic and metabolic functions, enabling bacterial growth and pathogenicity. Bacteria evade iron limitation in the host organism through the import of heme and ferric iron. In the latter case, this implies the synthesis and secretion of small organic iron chelators, so-called siderophores (Greek for “iron carriers”).9 After iron sequestration from host proteins, the ferric siderophore complexes are recaptured via specific outer-membrane transporters called TonB-dependent transporters (TBDTs, Figure 1).10 Ferric siderophores constitute cargo for TBDTs, but other nutrients such as heme, carbohydrates, nickel complexes, and vitamin B12 are also transported by TBDTs.11 Interestingly, TBDTs have also been parasitized by bacteriophages and colicins to board the bacterial cell.12 These transporters are unique to prokaryotes and present an unparalleled gateway to shuttle antibiotics inside bacterial pathogens.13 Natural iron-containing growth factors (sideramines) and their antibiotic iron-containing counterparts, the sideromycins, were first described by Prelog, Zähner, and coauthors in 1960, with many subsequent studies.14,15 Braun conducted research on the TonB-dependent internalization and intracellular activation of, e.g., the natural sideromycin albomycin and the TonB-dependent transport of colicins and microcins in E. coli and Salmonella.16−19 Later, the Miller group expanded on this work and prepared many synthetic covalent or cleavable conjugates linked to a variety of antibiotic payloads (e.g., β-lactams, daptomycin, or ciprofloxacin).20−24 The clinical applicability of this concept was demonstrated by the recently approved siderophore–cephalosporin antibiotic cefiderocol (Fetroja).25 The ability to transport even large cargo into bacteria is best illustrated by microcin MccE492, a natural product that consists of an 84 amino acid peptide chain attached via a sugar linkage to a triscatecholate chelator originating from enterobactin.26 MccE492 exerts its antimicrobial effect at the cytoplasmic membrane after import. The application of natural siderophores as molecular targeting entities is in part hampered by their challenging syntheses, their chemical lability, and in some cases their limited bacterial spectrum.27−30 Fortunately, much like piracy, prokaryotes seize so-called “xenosiderophores” (siderophores produced by other organisms or even synthetic siderophore mimetics) to satisfy their iron demands.31 Along those lines, we established enterobactin analogues based on synthetic DOTAM and MECAM scaffolds as robust, readily accessible, and variable vectors for bacterial imaging and antibacterial therapy in Gram-negative and Gram-positive bacteria.32−34
Figure 1.

Iron delivery by enterobactin (ENT) and envisioned suicide TonB siderophore strategy. (Left) Iron chelators such as heme, enterobactin (ENT), and pyoverdine (PYO) are recognized by their corresponding TonB-dependent transporters (TBDTs) HasR, PfeA, and FpvA in P. aeruginosa. In the case of ferri-enterobactin, after internalization, the esterase PfeE hydrolyzes the siderophore to release the iron. A fraction of ferri-enterobactin interacts with the two-component system PfeS/PfeR to regulate the transcription of pfeA and pfeE. (Right) Schematic depiction of the competitive inhibition of the TonB–TonB box interaction by peptide siderophore conjugates. Synthetic siderophores DOTAM and MECAM are coupled to TonB box peptides originating from the FpvA, PfeA, or HasR transporter amino acid sequence. The bacteria-specific vectors are transported into the periplasm and interfere with TonB function, thereby inhibiting the uptake of additional iron or heme and consequently repressing bacterial growth.
TBDTs are composed of a β-barrel of 22 β-strands inserted into the outer membrane, a plug domain that closes the channel formed by the barrel, and the N-terminal TonB box.10,35,36 The incoming ferric siderophore from the extracellular space binds to a specific site on the plug domain, promoting the protein–protein interaction (PPI) between the TBDT’s TonB box sequence protruding into the periplasm and the periplasmic domain of the TonB protein, which is anchored in the inner membrane.37 Located at the N-terminus of the TBDTs, the TonB box is a semiconserved stretch of five to seven amino acids that serves as a signature sequence for this transporter family. The TonB protein is in complex with two other proteins ExbB and ExbD in the inner membrane (stoichiometry of 1:5:2 for TonB/ExbB/ExbD) and forms a molecular motor that uses the proton gradient of the inner membrane to convey energy to TBDTs, allowing the active transport of nutrients into the periplasmic space.38 Similar to a lock-and-key principle, the PPI between TonB and the TonB box of the TBDT promotes a conformational change in the transporter and permits the internalization of an iron–siderophore complex into the periplasm.37P. aeruginosa has three genes in its genome that encode for TonB proteins (TonB1, TonB2, and TonB3), with only TonB1 interacting with the TBDTs involved in iron or heme acquisition.39 Moreover, the number of TonB proteins is very limited in relation to the different TBDTs in the outer membrane, implying the strong competition of TBDTs for TonB binding.40 In previous studies, bacterial virulence, TonB-mediated colicin killing, and φ80 phage infection could be reduced significantly by treating E. coli with a species-specific small TonB box consensus peptide (ETVIV), which was small enough to be internalized by polypeptide transporters.41 However, this effect was only observed for high concentrations (>100 μM). The overexpression of the periplasmic domain of TonB also inhibited the import of iron by the TBDTs in E. coli.42 Similar effects were observed in P. aeruginosa, with the overexpression of the N-terminal domain of the ferri-pyoverdine TBDT affecting the import of iron by this siderophore.43 Knowing that iron is a key nutrient for bacterial growth, all these data point out that the protein interaction between the periplasmic domain of TonB and the TonB boxes could be a promising target for new antibiotics.
In this study, we aimed to explore the disruption of the TBDT–TonB interaction as a novel principle in antimicrobial therapy, with a proof-of-concept in the challenging pathogen P. aeruginosa. A pharmacological approach was pursued by conjugating TonB box peptides of varying lengths to siderophore mimics to enable accumulation at their target site inside the bacterial cell. Similar to the natural microcins, an essential cellular machinery was preyed to import its own destroyer (Figure 1B).
Results and Discussion
Design and Synthesis of TonB Box Peptide DOTAM and MECAM Siderophore Conjugates
Based on previous work showing that an alteration in the vital interaction between the TonB protein and the TonB box of TBDTs induces the inhibition of bacterial growth,41,43,42 we designed synthetic TonB box peptides to compete for the protein–protein interaction with the TBDTs FpvA, HasR, and PfeA (Supplementary Figure S1). FpvA is the outer-membrane transporter of pyoverdine, the major siderophore produced by P. aeruginosa. HasR recognizes heme an important iron source during infection, and PfeA recognizes catechol-type siderophores such as DOTAM and MECAM used for antibiotic vectorization. The conservation among the curated sequences of FpvA (FPVA_PSEAE), PfeA (PFEA_PSEAE), and HasR (Q9HYJ7_PSEAE) retrieved from the UniProt database was low (score ≤0.3, black frame, Supplementary Figure S2).44 Consequently we opted for the separate syntheses of the three corresponding peptides. Each peptide was afforded in a long and a short form with either from four to eight (long) or one framing amino acid (short) around the TonB box (Figure 3). The peptides were too large for passive permeation through porins or the lipid bilayer and thus required the conjugation to siderophore mimics as molecular “Trojan horses” to allow their penetration into the bacterial periplasm. Siderophores were attached to the N- or C-terminus of the peptide by covalent or cleavable linkers. The artificial siderophores based on DOTAM (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic amide) or MECAM (1,3,5-N,N′,N″-tris(2,3-dihydroxybenzoyl)-triaminomethylbenzene) scaffolds were equipped with an alkyne handle for copper-catalyzed click chemistry according to established procedures (Figure 3 and Schemes S1–S3).33,34
Figure 3.
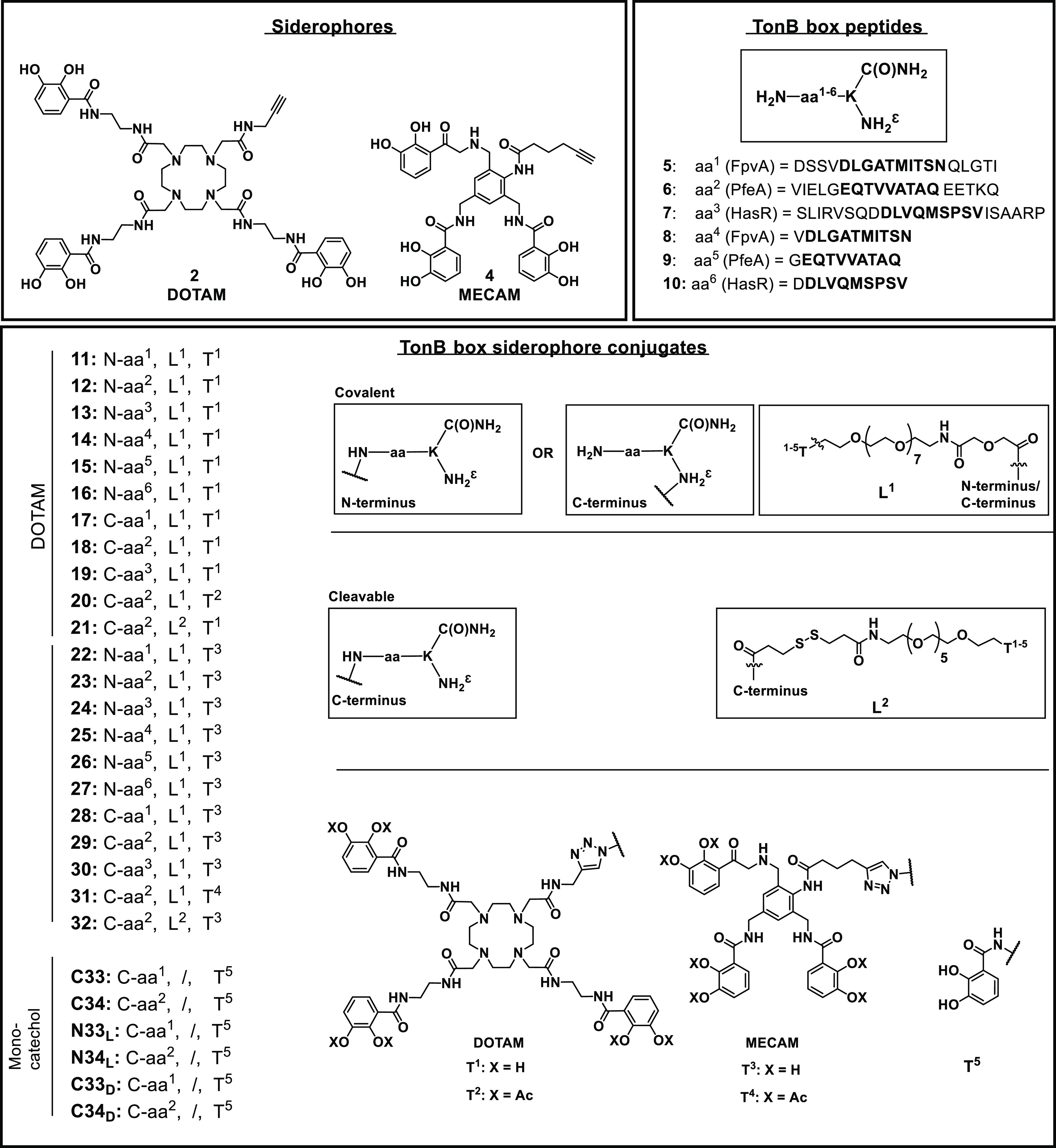
Structures of the DOTAM and MECAM siderophores 2 and 4, TonB box peptides 5–10, and siderophore-peptide conjugates 11–34. The conjugates consist of a siderophore unit (Tx) that is linked via a covalent or a cleavable disulfide unit (Lx) to the N-terminus or the C-terminus of the peptide (aax). Bold amino acids in 5–10 indicate the amino acids composing the TonB box, and framing amino acids are depicted roman.
Figure 2.
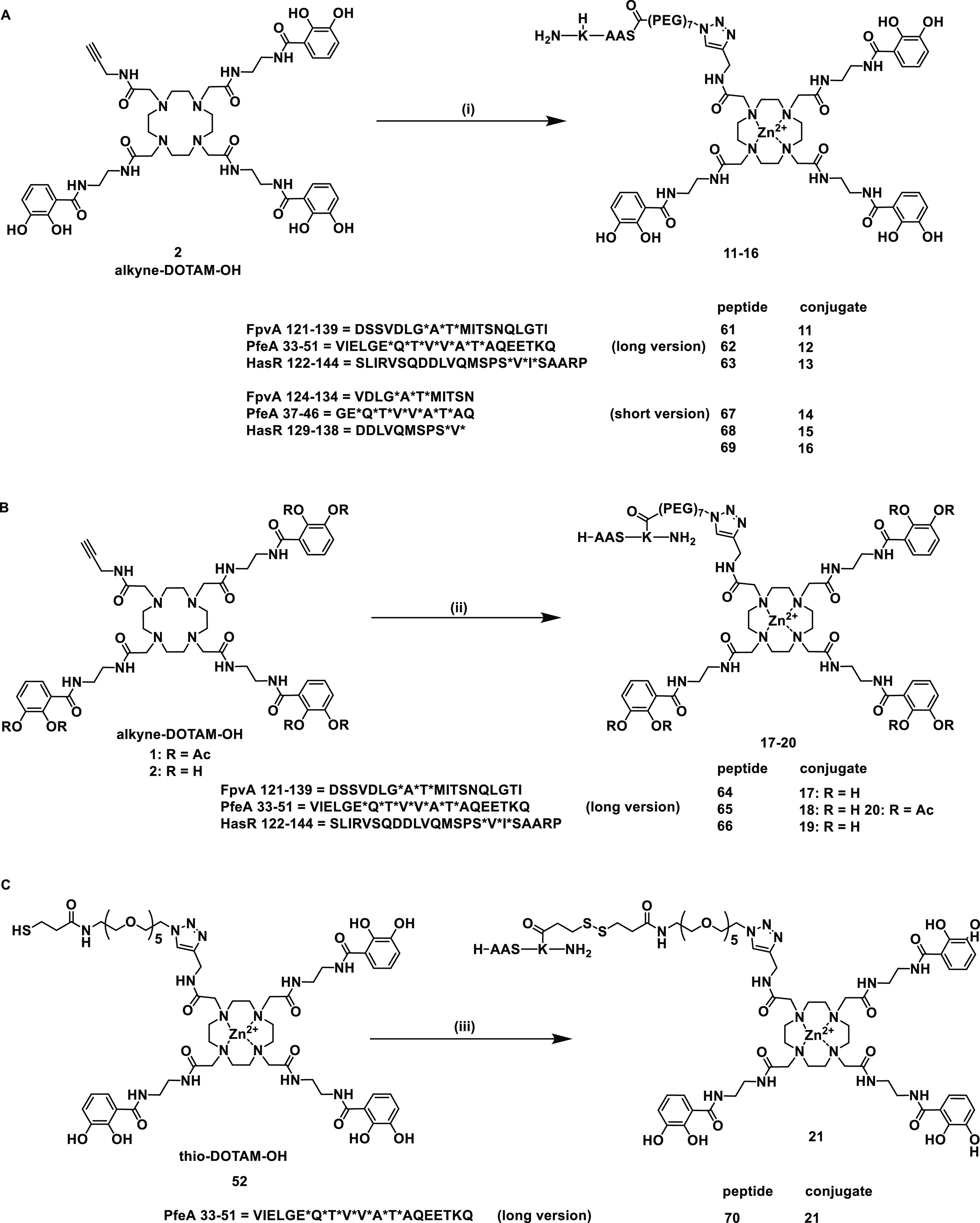
Synthesis of the siderophore TonB box peptide conjugates. (A) Conditions for the synthesis of long and short N-terminal peptide DOTAM conjugates are as follows: (i) Zn(OAc)2, DMSO/H2O, 23 °C, 5 min, 61/62/63/67/68/69 in DMSO, 5 min, 23 °C, CuSO4, sodium ascorbate, THPTA, PBS pH 7.4, 23 °C, 3 h, 81–99%. (B) Conditions for the synthesis of long C-terminal peptide DOTAM conjugates are as follows: (ii) Zn(OAc)2, DMSO/H2O, 23 °C, 5 min, 64/65/66 in DMSO, 5 min, 23 °C, CuSO4, sodium ascorbate, THPTA, PBS pH 7.4, 23 °C, 3 h, 79–99%. (C) Conditions for the synthesis of long disulfide C-terminal peptide DOTAM conjugates are as follows: (iii) 70, HEPES buffer pH 7.4, DMF/DMSO, 23 °C, 72 h, 76%. An asterisk (*) indicates complicated coupling reactions.
Starting from cyclen 43, DOTAM siderophores 1 and 2 were prepared over four (38%) and five (30%) steps for the longest linear sequence, respectively (Schemes S1 and S2). MECAM siderophores 3 and 4 were obtained from trisbromo-methylbenzene 47 over five (37%) and six steps (34%) in the longest linear sequence, respectively (Scheme S3). The peptides were synthesized on the solid phase using Fmoc chemistry. More side product formation was observed for the HasR and FpvA peptides than for the PfeA peptides. This was attributed to the aspartic acid (D) and methionine (M) aa’s in their sequences, which are known to display succinamide formation45 or oxidation to the sulfoxide under the reaction conditions.46 In addition, the unconjugated control peptides 5–10 were a also fforded (Schemes S5–S7 and Table S1). For the synthesis of covalent conjugates, a N3-PEG7-CO2H linker was attached at the C- or N-terminus of the peptide to permit the conjugation to 1–4 by copper-catalyzed azide alkyne addition (CuAAC). The azide PEG linker at the C-terminus was introduced using an orthogonal Dde projecting group (2-(1-aminoethylidene)-5,5-dimethylcyclohexane-1,3-dione) at the side-chain amino function of the terminal lysine. Following Fmoc-SPPS, the Dde group was cleaved selectively by the addition of hydrazine in THF/MeOH.47 The free amino group was modified in an on-resin HATU coupling with the CO2H-PEG7-N3 linker, followed by resin cleavage, global deprotection, and HPLC purification to yield the pure C-terminally modified azidopeptides (Schemes S6b and S7b). This yielded the DOTAM conjugates 11–21 and the MECAM conjugates 22–30 (Figure 3 and Scheme S8–S10). In the case of 20 and 31, the catechols were masked as acetylated prodrugs in order to avoid in vivo deactivation of the iron chelating units by catechol-O-methyltransferases. A combination of Dde protecting group chemistry and pyridyl disulfide chemistry was applied for the synthesis of the cleavable conjugates 21 and 32 that allowed the intracellular reductive release of the peptide, thereby reducing the steric hindrance of the siderophore (Scheme S10 and 11). In particular, the peptide’s Dde group was cleaved on the resin, and the free amino group was modified with 3-(pyridin-2-yldisulfanyl)propanoic acid (SPDP acid, Scheme S8a). Following global deprotection and resin cleavage, the pyridyl disulfide peptide 70 was obtained. Disulfide exchange with thio DOTAM 52 and thio MECAM 53 under basic reaction conditions yielded the disulfide conjugates 21 and 32, respectively. The monocatechol-modified peptides were constructed to reduce the overall molecular weight and still retain a TBDT-based internalization through a single chelator unit (Scheme S11). In total, we obtained six monocatechol peptides with a catechol unit C- or N-terminally attached to the respective FpvA or PfeA peptides composed of l- or d-amino acids, yielding C33, C34, N33L, N34L, N33D, and N34D.
Taken together, 28 covalent or cleavable siderophore conjugates with molecular weights of 2–4 kDa could be constructed in up to 32 synthetic steps. All conjugates were characterized by 1H NMR and high-resolution mass spectrometry, and exemplary molecules were also characterized by 13C NMR, HSQC-NMR, and tandem mass spectrometry (see the Supporting Information).
Peptide Siderophore Conjugates Show Antimicrobial Effects in a P. aeruginosa Strain Unable to Produce Its Own Siderophores
The siderophores 2 and 4, the peptides 5–10, and the siderophore conjugates 11–34 were evaluated for their antimicrobial activity on the P. aeruginosa wild-type (PAO1) and a siderophore-deficient P. aeruginosa mutant strain (ΔpvdFΔpchA). The ΔpvdFΔpchA mutant cannot synthesize its endogenous siderophores pyoverdine (PYO) and pyochelin (PCH) to access iron but probably uses low-affinity iron uptake pathways such as iron assimilation via citrate or an iron reduction process with the uptake of Fe2+ by the FeoAB system. During an infection, bacteria face iron-starvation as the host restricts the iron access of the pathogens by nutritional immunity.48 In order to test close to the in vivo infection conditions, all assays were conducted in an iron-depleted CAA medium (20 nM iron).49,50 Along those lines, differential proteomics has shown that iron uptake pathways is solely expressed in P. aeruginosa cells under iron-restricted conditions, which involve our compounds using analogues of siderophores as vectors have the highest chance to be active on the pathogen only in iron-restricted growth conditions.51 Consequently, the compounds were first evaluated for the lowest compound concentration preventing visible bacterial growth (minimal inhibitory concentration, MIC) with bacteria grown in iron-restricted conditions, and only the compounds with a growth inhibition activity were also tested in a CAA growth medium supplemented with iron.
In iron-restricted conditions, MICs were determined after 24 and 48 h of culture, as bacterial growth in iron-restricted conditions is slower than that in an iron-rich medium. While the positive control gentamicin exhibited an activity in the expected range, the free peptides 5–10 did not induce any growth inhibition (24 and 48 h). Additionally, the free DOTAM and MECAM siderophores 2 and 4, all MECAM conjugates, and the monocatechols C33, C34, N33L, N34L, N33D, and N34D remained inactive (Table 1). In contrast, several DOTAM conjugates inhibited bacterial growth. In particular, the long N-terminally linked conjugates 11 (FpvA), 12 (PfeA), and 13 (HasR) showed MICs of 0.5, 0.5, and 4 μM, respectively, at 24 h. The most potent growth inhibition (MIC = 0.1 μM) was observed for 17, which differs from 11 by its C-terminal (rather than N-terminal) linkage of the long FpvA-derived peptide. A moderate MIC of 8 μM was obtained for 21, which carries a cleavable linker connected to a long PfeA-derived peptide. At 48 h, growth was recovered for conjugates 14, 17, and 21 but not for 11 and 12. The peptide payloads potentially exhibit antimicrobial activity due an interaction with TonB, but over time either an adaptation of the bacteria or a degradation of the peptide payload by proteases takes place.52 An attempt to increase the antimicrobial efficacy through a combination treatment of N- or C-terminal peptide siderophore conjugates did not result in a potency boost (data not shown). Since our test is conducted under highly iron restricted conditions (20 nM iron in the CAA medium), most of the conjugates will stay in their apo form. Consequently the concentration of active ferric conjugates able to stop bacterial growth is much lower than suggested by the MIC value, which reflects the sum of metal-bound and unbound siderophores. Thus, the potency of the conjugates is rather underestimated.
Table 1. MIC Values (μM) in P. aeruginosa ΔpvdFΔpchA Strain for Siderophores 2 and 4, Peptides 5–10, and Peptide–Siderophore Conjugates 11–34a.

MIC values were determined after 24 and 48 h of growth and are the mean of three independent experiments. (l) = long peptide, 20–24 aa; (s) = short peptide, 11–12 aa; C/N = C- or N-terminal; and DS = disulfide. Generally, peptides are composed of l-amino acids; XD indicates a d-amino acid peptide, and XL indicates l-amino acids for discriminatory purposes. Values are given in μM, n = 3. A conversion of the MIC values to μg/mL can be found in Table S9.
All compounds were inactive in the wild-type P. aeruginosa strain PAO1 grown in iron-restricted conditions. This is consistent with previous studies of competition for iron between the natural siderophore PYO produced by P. aeruginosa and DOTAM, MECAM, enterobactin (siderophore produced by E. coli), ferrichrome (siderophore produced by fungi), and PCH (second siderophore produced by P. aeruginosa), showing that MECAM and DOTAM are less efficient in scavenging iron from PYO than enterobactin and even ferrichrome but more efficient than PCH.53,31 We cannot exclude that the high steric demand of the peptide payload might even reduce more the chelator’s affinity for iron, thereby reducing uptake and antimicrobial activity on a wild-type P. aeruginosa strain. Therefore, the ability of the conjugates to sequester ferric iron was tested with chrome azurol S (CAS), whose color shifts from blue (iron bound state) to bright red upon iron decomplexation. A color change was not observed for the free peptides, while the siderophores 2 and 4 showed a clear shift in the range from 300 to 800 nm, in line with previous reports (Figure S3 and Table S7).32,33 Notably, all siderophore–peptide conjugates retained their ability to complex iron. From the experiments, it becomes apparent that iron binding is possible, but the conjugates’ affinity is probably not sufficient to compete with the wild-type siderophore PYO for ferric iron and to confer growth inhibition on the PAO1 strain.
The DOTAM siderophore 2; the long peptide DOTAM conjugates 11, 12, and 13; the short peptide DOTAM conjugates 14, 15, 16, and 17; and the cleavable DOTAM conjugate 21 were also subjected to a MIC assay in a CAA medium supplemented with ferric iron (100 μM). As expected, all tested compounds lost their antimicrobial effect in the presence of excess ferric iron. In the presence of iron, the TBDTs involved in iron acquisition are not expressed and consequently the conjugates are no longer internalized.31
Peptide–Siderophore Conjugates Enter the Bacterial Periplasm of a P. aeruginosa Strain Unable to Produce Its Own Siderophores
In order to demonstrate the ability of our compounds to enter P. aeruginosa cells, we exploited the property of ferri-siderophores to interact with the two component system PfeS/PfeR and induce pfeA transcription. Upon binding of periplasmic ferri-siderophore to the PfeS sensor at the inner membrane, the transcriptional regulator PfeR is released, upregulating the transcription of the pfeA gene.54,55 Induction of pfeA transcription in the presence of the conjugates indicates the penetration of the compounds, since an interaction with PfeS can occur only in the periplasm. We have recently shown that unconjugated MECAM induced the transcription and expression of pfeA by interacting with PfeS.53 This induction was accompanied by a transcription repression of the genes involved in the acquisition of iron by the siderophore pyochelin (PCH).53
The PYO- and PCH-deficient ΔpvdFΔpchA strain was grown over 8 h in an iron-restricted CAA medium in the absence or presence of the peptides, free DOTAM and MECAM cores, and a selection of conjugates (11, 12, 13, 17, 22, and 28, each at 10 μM). The expression of the genes encoding pfeA (ferri-ENT TBDT),56pirA (ferri-ENT and ferri-catecholamines TBDT),57,58fpvA (ferri-PYO TBDT),35 and fptA (ferri-PCH TBDT)59−61 was analyzed by differential quantitative real-time PCR (qRT-PCR) (Figure 4). As expected, the cores 2 and 4 showed a one- to six(log2)fold induction of pfeA transcription in the PAO1 wild-type and the ΔpvdFΔpchA mutant.53 The free peptides 5–10 had no effect. For all tested MECAM and DOTAM conjugates, an induction of pfeA transcription was observed. This verifies the translocation of the conjugates into the periplasm and their interaction with the sensor PfeS at the inner membrane. The MECAM conjugates induced the transcription of pfeA with the same efficiency as the MECAM vector 4 alone. The DOTAM conjugates displayed an even stronger pfeA transcription induction than the free DOTAM vector 2. None of the vectors or conjugates had a significant effect on pirA transcription. The transcription of pirA, such as that of pfeA, is regulated by a two-component system, namely PirS/PirR. Apparently the MECAM and DOTAM conjugates were able to bind to PfeS and induce the release of PfeR but not to PirS because of different binding specificities of these two sensors.
Figure 4.

Modulation of TBDT gene expression by conjugates. P. aeruginosa PAO1 and ΔpvdFΔpchA cells were grown for 8 h in the presence or absence of test compounds (10 μM). The transcription of pfeA, pirA, fpvA, and fptA was followed by qRT-PCR.49pfeA encodes the ferri-ENT TBDT,62,56pirA encodes the ferri-ENT and ferri-catecholamines TBDT,57,58fpvA encodes the ferri-PYO TBDT,35 and fptA encodes the ferri-PCH TBDT.59,61
All compounds, vectors, and conjugates also induced a repression of fptA transcription. Interestingly, the highest-fold changes concerning the induction of pfeA and the repression of fptA transcription were seen for the inactive MECAM conjugates, while the DOTAM conjugates generally had lower effects. This is probably due to a higher iron affinity of the MECAM compounds compared to the DOTAM compounds. This adjustment of pfeA and fptA transcription indicates that the ΔpvdFΔpchA strain adapted its TBDT expression for optimal iron acquisition via the conjugates in its surroundings. We also checked the ability of 11 to modulate the transcription of pfeA, pirA, fpvA, and fptA in the PAO1 wild-type strain. However, their transcription was unaffected, possibly due to the greater ability of PYO and PCH to access iron than the larger 11. In summary, both MECAM and DOTAM conjugates were able to cross the outer membrane of P. aeruginosa unable to produce its own siderophores because they induced a phenotypic adaptation of the bacteria (↑ pfeA transcription, ↓ fptA transcription). Consequently, we conclude that the antibiotic activity of the DOTAM conjugates was not due to iron sequestration by the compounds outside bacterial cells but was instead caused by internalized peptides and probably an interaction of the peptides with TonB. Altogether, these data indicate that the self-internalizing suicide “Trojan horse” mechanism is operational with the vectors DOTAM in a P. aeruginosa strain unable to produce its own siderophores PYO and PCH.
The Transporters PfeA and PirA Mediate the Entry of Conjugates in P. aeruginosa
Next, we aimed to elucidate the TBDT(s) involved in conjugate uptake. For this purpose, the growth of the P. aeruginosa ΔpvdFΔpchA strain was compared to strains that carried additional TBDT deletions of (i) pfeA, (ii) pirA, or (iii) pfeA and pirA. For compounds that do not exert antimicrobial activities, a growth reduction after TBDT knockout proves that the missing siderophore transporter is crucial for the internalization of ferric chelates and thus permits bacterial growth.63,53 The free peptides 5–7 exerted no effect (Figure 5). The growth of DOTAM-based conjugates was hardly impaired by the lack of PfeA, but a strong growth inhibition was observed for all mutants lacking PirA, indicating that all these compounds enter the cells through this TBDT. For MECAM-based conjugates, the single deletion of PirA had no significant effect, whereas a lack of PfeA delayed growth slightly, in particular for the free MECAM 4 and the conjugate 29. Interestingly, the dual knockout of pfeA and pirA genes led to a complete growth inhibition for all MECAM compounds, indicating that these compounds entered P. aeruginosa cells by both PfeA and PirA. This result also indicates that one transporter was able to rescue when the other transporter was absent or not functional. The data are in agreement with our recent finding that the MECAM core and a MECAM–ampicillin conjugate are transported into P. aeruginosa cells via PfeA and PirA, whereas DOTAM is solely internalized via PirA.53 This demonstrates that PfeA and PirA have different binding specificities. The data on the induction of transcription of TBDT genes (Figure 4) indicate that the specificity of the sensors PfeS and PirA is different from that of their associated transporters PfeA and PirA. PirS, unlike PirA, does not seem to recognize DOTAM conjugates. Conversely, PfeS has a larger binding specificity than its associated transporter PfeA, since this internal membrane sensor recognizes both the MECAM and DOTAM conjugates.
Figure 5.

Growth kinetics of P. aeruginosa pfeA and pirA mutants in the presence of the conjugates. The PYO- and PCH-deficient strain of P. aeruginosa (ΔpvdFΔpchA) and its corresponding pfeA and pirA deletion mutants were used. Strains were grown in a CAA medium in the absence or presence of 10 μM peptides 5–7, DOTAM 2, MECAM 4, or the conjugates 11–13, 17–19, 22–24, and 28–30. Growth was followed by monitoring the optical density (OD) at 600 nm. Errors bars were calculated from three independent biological replicates.
We conclude that the transport mechanisms of the artificial siderophores were not altered by the conjugation of large peptidic cargo in the kilodalton range and that the MECAM conjugates enter bacteria by either PfeA or PirA and the DOTAM conjugates by PirA.
Conclusion
In this study, we propose the disturbance of the interaction between TBDTs involved in the uptake of nutrients across the outer membrane of Gram-negative bacteria and the TonB protein as a novel strategy to inhibit bacterial growth. A systematic attachment of siderophores to TonB box polypeptides from three TBDTs, which were coupled via cleavable or covalent linkers at the N- or C-terminus of the peptide to various of the targeting vectors (DOTAM and MECAM), yielded a diverse compound collection of two monocatechol peptides and 24 full siderophore conjugates. With molecular weights of up to 4 kDa, these are, to the best of our knowledge, among the largest synthetic siderophore conjugates. Their iron complexation capabilities were demonstrated by the CAS test. Growth recovery assays and the ability to induce the transcription of pfeA via the two-component PfeS/PfeR system proved that all compounds were able to enter the periplasm of P. aeruginosa unable to produce its on siderophores PYO and PCH. All MECAM conjugates were internalized by PfeA and PirA TBDTs, and DOTAM conjugates were internalized by PirA. A notable growth delay or inhibition, attributed to ferric siderophore internalization, was observed solely for DOTAM conjugates. The five conjugates 11, 12, 13, 17, and 21 displayed antimicrobial activity in siderophore-deficient P. aeruginosa strains, and the most potent analogs 11, 12, and 17 reached MICs of 0.5, 0.5, and 0.1 μM, respectively. Based on the above data, we derive the following preliminary structure–activity relationships (SAR) (Supplementary Figure S4). Because all active compounds were based on the triscatecholate DOTAM vector, it was obviously better suited than MECAM. Conjugates with FpvA-originating peptides were slightly more active compared to the equivalent congeners carrying sequences from PfeA and HasR. The active congeners had a N-terminal linkage rather than a C-terminal disulfide linker, and the longer peptides were superior to shorter ones.
Together, these results demonstrate the capability of MECAM and DOTAM to transport large cargo in the kilodalton range into bacteria. While first evidence in this direction has been obtained for the prototype natural product microcin MccE492, the data suggest that artificial siderophores, coupled to synthetic linkers and peptides, can also be employed. We realized the first siderophore “Trojan horse” antibiotics that target and disrupt a protein–protein interaction in the bacterial periplasm. Instead of satiating the pathogen’s appetite for iron, the TonB box peptide payload competes with the TBDTs for TonB and thereby prevents the internalization of further ferric chelates. Thus, the study illustrates a variant of cellular suicide where a transporter imports its own inhibitor.
Perturbing the TonB-TBDT interaction is a novel principle for interfering with the pathogen’s iron homeostasis and growth, yielding a decreased metabolism and fitness, and the conjugates described herein support the validity of the target. At this stage, structural information on the TonB box–TonB interaction at a molecular level would be beneficial to improve the understanding of why only a subset of the conjugates were active. From here, it appears attractive to initiate a search for less complex small molecules that interfere with the PPI in future studies.
Experimental Section
Chemical Synthesis
Chemicals and reagents were purchased from commercial vendors (TCI, Carl Roth, Baker, and Sigma-Aldrich), if not stated otherwise, and employed without further purification in the below synthetic procedures. For synthesis, solvents with purity grade 99.5% (extra dry, absolute, AcroSeal, ACROS Organics) were used. Work-up procedures and purifications solvent were either HPLC or p.a. grade. Glassware was oven-dried prior to synthesis. Reaction progress was controlled by thin layer chromatography (TLC) or liquid chromatography-coupled mass spectrometry (LCMS). Detailed information regarding the HPLC, NMR, and MS setups and equipment can be found in “General chemical information” in the Supporting Information. A detailed general procedure for the preparation and purification of the unmodified peptides 5–10 can be found in “General Procedure for Peptide Synthesis” and “General Procedure for Global Deprotection and Resin Cleavage” below. Since all unmodified peptides displayed similar retentions on the HPLC column, the same gradient was employed for all purifications. A detailed general procedure for the preparation and purification of the N- or C-terminally modified peptides can be found in “General Procedure for the N- or C-Terminal Modification, Global Deprotection, and Resin Cleavage” below. Since all modified peptides displayed similar retentions on the HPLC column, the same gradient was employed for all purifications. All compounds had purities ≥95% as determined by high-performance liquid chromatography (UV detection), 1H- and 13C NMR, and MS/MS analysis.
Siderophore Synthesis
tert-Butyl (2-(2-Bromoacetamido)ethyl)carbamate (36)64,34
tert-Butyl N-(2-aminoethyl)carbamate 35 (6.0 g, 37.4 mmol., 5.9 mL, 1.0 equiv) was dissolved in DCM (150 mL), and to the solution was added K2CO3 (20.7 g, 149.8 mmol, 4.0 equiv) dissolved in Milli-Q H2O (100 mL). The two-phase solution was cooled to 0 °C, and 2-bromoacetyl bromide (5.2 mL, 59.9 mmol, 1.6 equiv) in DCM (150 mL) was added to the mixture dropwise over 10 min at 0 °C under vigorous stirring. The reaction mixture stirred at 0 °C for 30 min, then was allowed to warm to room temperature and stirred for 90 min at 23 °C. The phases were separated, and the aqueous phase was extracted with DCM (2 × 100 mL). The combined organic phases were washed with 1 M HCl (2 × 100 mL), sat. NaHCO3 (2 × 100 mL), and brine (2 × 100 mL) and dried over Na2SO4. The solvent was removed by rotary evaporation to yield product 36 as a white solid (10.0 g, 35.6 mmol, 95%).
1H NMR (500 MHz, CDCl3): δ [ppm] = 7.14 (bs, 1H), 4.97 (bs, 1H), 3.84 (s, 2H), 3.37 (q, J = 5.4 Hz, 2H), 3.29 (q, J = 4.7 Hz, 2H), 1.43 (s, 9H).
13C NMR (126 MHz, CDCl3) δ [ppm] = 166.5, 156.9, 80.1, 41.6, 40.0, 29.0, 28.5.
HRMS (ESI) calculated for C9H18BrN2O3+ ([M + H]+): m/z = 281.0495, experimental 281.0493, δ [ppm] = 0.2. Calculated for C9H17BrN2NaO3+ ([M + Na]+): m/z = 303.0315, experimental 303.0313, δ [ppm] = 0.2.
2-Bromo-N-(prop-2-yn-1-yl)acetamide (38)64
Prop-2-yn-1-amine 37 (5.8 mL, 90.8 mmol, 1.0 equiv) was dissolved in DCM (150 mL), and to the solution was added K2CO3 (50.2 g, 363.1 mmol, 4.0 equiv) dissolved in Milli-Q H2O (100 mL). The two-phase solution was cooled to 0 °C, and 2-bromoacetyl bromide (12.7 mL, 145.2 mmol, 1.6 equiv) in DCM (150 mL) was added dropwise to the mixture over 10 min at 0 °C under vigorous stirring. The reaction mixture was stirred at 0 °C for 30 min. After this time, the mixture was allowed to warm to room temperature and stirred for another 90 min at 23 °C. The phases were separated, and the aqueous phase was extracted with DCM (2 × 100 mL). The combined organic extracts were washed with 1 M HCl (2 × 100 mL), sat. NaHCO3 (2 × 100 mL), and brine (2 × 100 mL) and dried over Na2SO4. The solvent was removed by rotary evaporation to yield product 38 as a brown solid (11.5 g, 65.1 mmol, 72%).
1H NMR (500 MHz, CDCl3): δ [ppm] = 6.87 (bs, 1H), 4.07 (dd, J = 5.4 Hz, 2.6 Hz, 2H), 3.87 (s, 2H), 2.27 (t, J = 2.6 Hz, 1H).
13C NMR (126 MHz, CDCl3) δ [ppm] = 165.5, 78.7, 72.2, 30.0, 28.7.
HRMS (ESI) calculated for C5H7BrNO+ ([M + H]+): m/z = 175.9706, experimental 175.9705, δ [ppm] = 0.1.
2,3-Diacetoxybenzoic acid (40)64
2,3-Dihydroxybenzoic acid 39 (25.0 g, 162.2 mmol, 1.0 equiv) was suspended in THF (200 mL), and to the solution were sequentially added Ac2O (46.0 mL, 486.6 mmol, 3.0 equiv), TEA (90.0 mL, 648.8 mmol, 4.0 equiv), and DMAP (1.98 g, 16.2 mmol, 0.1 equiv). The precipitate dissolved after a few minutes of stirring at 23 °C, then the reaction mixture was heated to 60 °C and stirred for 20 h at that temperature. The next morning, as much of the solvent as possible was removed by rotary evaporation (clear beige solution), and the residue was dissolved in DCM (500 mL). The organic phase was washed with 1 M HCl (4 × 100 mL) and brine (1 × 100 mL) and then dried over Na2SO4. The solvent was removed in vacuo to yield product 40 as a crude beige solid (37.0 g, 155.3 mmol, 96%).
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 13.23 (bs, 1H), 7.81 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 7.51 (dd, J = 8.0 Hz, 1.7 Hz, 1H), 7.41 (t, J = 8.0 Hz, 1H), 2.30 (s, 3H), 2.26 (s, 3H).
13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.3, 168.1, 165.1, 143.2, 142.1, 128.5, 127.7, 126.3, 125.8, 20.4, 20.3.
HRMS (ESI) calculated for C11H11O6+ ([M + H]+): m/z = 239.0550, experimental 239.0550, δ [ppm] = 0.0. Calculated for C11H14NO6+ ([M + NH4]+): m/z = 256.0816, experimental 256.0816, δ [ppm] = 0.0. Calculated for C11H10NaO6+ ([M + Na]+): m/z = 261.0370, experimental 261.0370, δ [ppm] = 0.0.
N-(17-Azido-3,6,9,12,15-pentaoxaheptadecyl)-3-(tritylthio)propanamide (42)33,65
3-Tritylsulfanylpropanoic acid (300.0 mg, 0.861 mmol, 1.0 equiv) was dissolved in anhydrous DCM (25 mL) and DMF (5 mL). Then to the solution was added HATU (818.4 mg, 2.152 mmol, 2.5 equiv) dissolved in anhydrous DMF (10 mL). The solution was cooled to 0 °C, and 17-azido-3,6,9,12,15-pentaoxaheptadecan-1-amine 41 (263.8 μL, 0.947 mmol, 1.1 equiv) in dry DMF (10 mL) was added to the mixture at 0 °C. Dry TEA (477.4 μL, 3.444 mmol, 4.0 equiv) was added dropwise to the mixture over 10 min at 0 °C. The reaction mixture continued to stir at 0 °C for 30 min, then allowed to warm to room temperature and continued to stir for an additional 20 h at 23 °C. After the completion of the reaction, controlled by LCMS, the solution was concentrated by rotary evaporation. The residue was taken up in EtOAc (50 mL); washed with 1 M HCl (2 × 25 mL), sat. NaHCO3 (2 × 25 mL), and brine (2 × 25 mL); and dried over Na2SO4. The solvent was removed by rotary evaporation, and the residue was dissolved in ACN/Milli-Q H2O/MeOH and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 65–95% ACN/Milli-Q H2O, 0.1% HCOOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield product 42 as a white solid (418.0 mg, 0.656 mmol, 76%).
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 7.88 (t, J = 5.6 Hz, 1H), 7.34–7.31 (m, 12H), 7.24 (dd, J = 8.8 Hz, 4.5 Hz, 3H), 3.59 (t, J = 4.8 Hz, 2H), 3.56–3.48 (m, 12H), 3.40–3.35 (m, 4H), 3.18 (q, J = 5.6 Hz, 2H), 2.26 (t, J = 7.3 Hz, 2H), 2.17 (t, J = 7.3 Hz, 2H).
13C NMR (126 MHz, DMSO-d6) δ [ppm] = 170.1, 144.5, 129.1, 128.0, 126.7, 69.9, 69.8, 69.7, 69.7, 69.6, 69.3, 69.1, 66.0, 50.0, 38.6, 33.9, 27.5.
HRMS (ESI) calculated for C34H45N4O6S+ ([M + H]+): m/z = 637.3054, experimental 637.3054, δ [ppm] = 0.0. Calculated for C34H48N5O6S+ ([M + NH4]+): m/z = 654.3320, experimental 654.3319, δ [ppm] = 0.1. Calculated for C34H44N4NaO6S+ ([M + Na]+): m/z = 659.2874, experimental 659.2873, δ [ppm] = 0.1.
Di-tert-butyl(((2,2′-(4-(2-((2-((tert-butoxycarbonyl)amino)ethyl)amino)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,7-diyl)bis(acetyl))bis(azanediyl))bis(ethane-2,1-diyl))dicarbamate (44)64,34
1,4,7,10-Tetraazacyclododecane 43 (186.0 mg, 1.080 mmol, 1.0 equiv) and NaOAc (354.3 mg, 4.319 mmol, 4.0 equiv) were suspended in ACN (75 mL), and the solution was stirred at 23 °C for 5 min. 36 (1.00 g, 3.563 mmol, 3.3 equiv) was added dropwise to the solution with a syringe pump (6 mL/h, 24 mL, 4 h total) under vigorous stirring at 23 °C over 4 h. The syringe pump was removed ,and the reaction continued to stir at 23 °C for 16 h. The reaction progress was monitored by LCMS. After the completion of the reaction, the suspension was filtered, and the solvent was removed by rotary evaporation and in vacuo to yield product 44 as a colorless oil in a quantitative yield without the need for further purification.
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 8.75 (s, 1H), 8.21–7.94 (m, 2H), 6.80 (s, 3H), 3.34–2.52 (m, 34H), 2.07 (s, 1H), 1.37 (s, 27H).
13C NMR (126 MHz, DMSO-d6): δ [ppm] = 170.5, 166.1, 155.7, 155.6, 77.7, 57.8, 55.6, 54.2, 50.8, 49.2, 45.0, 38.8, 38.7, 29.5, 28.2.
HRMS (ESI) calculated for C35H69N10O9+ ([M + H]+): m/z = 773.5244, experimental 773.5249, δ [ppm] = 0.5. Calculated for C35H68N10NaO9+ ([M + Na]+): m/z = 795.5063, experimental 795.5066, δ [ppm] = 0.3.
Di-tert-butyl(((2,2′-(4-(2-((2-((tert-butoxycarbonyl)amino)ethyl)amino)-2-oxoethyl)-10-(2-oxo-2-(prop-2-yn-1-ylamino)ethyl)-1,4,7,10-tetraazacyclododecane-1,7-diyl)bis(acetyl))bis(azanediyl))bis(ethane-2,1-diyl))dicarbamate (45)64,34
44 (834.6 mg 1.080 mmol, 1.0 equiv) and K2CO3 (597.0 mg, 4.319 mmol, 4.0 equiv) were suspended in ACN (75 mL) ,and the suspension was stirred at 23 °C for 5 min. 20 (342.1 mg, 1.943 mmol, 1.8 equiv) was dissolved in ACN (25 mL) and added dropwise over 10 min to the suspension. The reaction mixture was stirred vigorously at 23 °C for 20 h, and the reaction progress was monitored by LCMS. The reaction mixture was filtered after completeness, and the solvent was removed by rotary evaporation and in vacuo to yield product 45 as a light brown solid in a quantitative yield without the need for further purification.
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 8.72 (s, 3H), 8.34–7.87 (m, 3H), 6.81 (s, 1H), 4.04 (s, 1H), 3.91–2.80 (m, 38H), 1.38 (s, 27H).
13C NMR (126 MHz, DMSO-d6): δ [ppm] = 166.0, 165.8, 155.7, 80.4, 77.7, 73.4, 42.6, 42.3, 29.0, 28.5, 28.2.
HRMS (ESI) calculated for C40H74N11O10+ ([M + H]+): m/z = 868.5615, experimental 868.5612, δ [ppm] = 0.3. Calculated for C40H75N11O102+ ([M + 2H]2+): m/z = 434.7844, experimental 434.7845, δ [ppm] = 0.1.
2,2′,2″-(10-(2-Oxo-2-(prop-2-yn-1-ylamino)ethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)tris(N-(2-aminoethyl)acetamide) (46)64,34
45 (937.2 mg, 1.080 mmol, 1.0 equiv) was dissolved in DCM (75 mL) and cooled to 0 °C. TFA (25 mL) was added to the soltuion slowly to yield a 25% TFA solution in DCM, which changed the color of the solution to a neon yellow. The reaction mixture was stirred at 23 °C for 4 h, and the reaction progress was monitored by LCMS. After the completion of the reaction, the solvent was removed by rotary evaporation and in vacuo to yield product 46 (500.0 mg, 0.881 mmol, 82% crude) as a transparent yellow to brown oil. The residue was used crude directly in the next step without further purification.
Alkyne-DOTAM-OAc (1)64,34
40 (740.1 mg, 3.107 mmol, 3.6 equiv) was dissolved under an argon atmosphere in dry DCM (8 mL) and dry DMF (2 mL), and the solution was cooled to 0 °C. Oxalyl chloride (666.3 μL, 7.768 mmol, 9.0 equiv) was added dropwise to the solution over the septum at 0 °C, and the reaction mixture was stirred for 10 more minutes after the addition was finished. The reaction mixture was allowed to warm to room temperature and stirred at 23 °C for an additional 3 h, and the reaction progress was checked via TLC. For this, an aliquot of the reaction was quenched with MeOH to form the corresponding methyl ester. The differential running behavior on TLC indicated the formation of the desired acid chloride compared to the starting material at the bottom of the TLC plate. After completion, the solvent was removed by rotary evaporation, the purity was checked via NMR, and the residue was dried overnight in vacuo. The next morning, 46 (490.0 mg, 0.863 mmol, 1.0 equiv) was dissolved in Milli-Q H2O (50 mL), and the pH was carefully adjusted with a NaHCO3 solution (0.5 M) to ca. 8.5. The reaction mixture was cooled to 0 °C. The formed acid chloride 40 was dissolved in 1,4-dioxane (50 mL) and added dropwise to the reaction mixture at 0 °C for 60 min. The pH was monitored during the addition and adjusted with a NaHCO3 solution (0.5 M) to 8.5 during the addition and more to pH 7.0 toward the end of the addition. After the addition was complete, the reaction progress was monitored by LCMS, while the reaction mixture was allowed to equilibrate to room temperature and stirred at 23 °C for an additional 4 h. After completion, the suspension was extracted with DCM (4 × 100 mL or until the organic phase became colorless), and the combined organic phases were washed with sat. NaHCO3 (2 × 100 mL) and brine (2 × 100 mL) and dried over Na2SO4. The solvent was removed by rotary evaporation. The residue was dissolved in ACN/Milli-Q H2O with 1% AcOH to delay further deacetylation and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 30–60% ACN/Milli-Q H2O, 1% AcOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield compound 1 as a white solid (485.0 mg, 0.395 mmol, 46%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 8.50 (bs, 1H), 8.39 (bs, 3H), 8.22–8.10 (m, 3H), 7.46 (dd, J = 7.4 Hz, 1.8 Hz, 3H), 7.39–7.32 (m, 6H), 3.90–3.89 (m, 2H), 3.45–3.34 (m, 12H), 3.28–2.94 (m, 24H), 2.52 (t, J = 1.9 Hz, 1H), 2.28 (s, 9H), 2.22 (s, 9H).
13C NMR (176 MHz, DMSO-d6): δ [ppm] = 170.6, 170.0, 169.4, 168.5, 149.8, 146.3, 118.7, 117.7, 117.2, 115.0, 81.0, 72.9, 62.2, 57.7, 54.9, 38.9, 38.0, 27.9, 20.4.
HRMS (ESI) calculated for C58H74N11O19+ ([M + H]+): m/z = 1228.5157, experimental 1228.5160, δ [ppm] = 0.3. Calculated for C58H75N11O192+ ([M + 2H]2+): m/z = 614.7614, experimental 614.7615, δ [ppm] = 0.1.
Alkyne-DOTAM-OH (2)64,34
1 (485.0 mg, 0.395 mmol, 1.0 equiv) was dissolved in MeOH (1.6 mL), and the solution was cooled to 0 °C. To the solution was added DIPEA (0.4 mL). The reaction mixture was allowed to warm to room temperature and continued to stir at 23 °C for 3 h. The reaction progress was controlled by LCMS. After the completion of the reaction, the solvent was removed by rotary evaporation, and the residue was taken up in ACN/Milli-Q and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 5–35% ACN/Milli-Q H2O, 0.1% HCOOH). The product0containing fractions were identified by LCMS and lyophilized to dryness to yield product 2 as a white solid (310.0 mg, 0.318 mmol, 80%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 8.92 (s, 3H), 8.50 (s, 1H), 8.31–8.05 (m, 3H), 7.23 (dd, J = 8.0 Hz, 1.2 Hz, 3H), 6.89 (ddd, J = 7.7 Hz, 3.5 Hz, 1.3 Hz, 3H), 6.66–6.62 (m, 3H), 3.90 (d, J = 1.9 Hz, 2H), 3.37 (s, 6H), 3.24–3.05 (m, 12H), 3.04 (t, J = 2.5 Hz, 1H), 3.04–2.50 (m, 18H).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 170.3, 169.9, 164.5, 164.4, 150.0, 150.0, 146.4, 118.6, 117.6, 117.6, 117.3, 115.0, 81.1, 72.9, 57.3, 57.1, 52.5, 38.9, 38.0, 27.9.
HRMS (ESI) calculated for C46H62N11O13+ ([M + H]+): m/z = 976.4523, experimental 976.4525, δ [ppm] = 0.2. Calculated for C46H61N11NaO13+ ([M + Na]+): m/z = 998.4343, experimental 998.4347, δ [ppm] = 0.4.
1,3,5-Tris(bromomethyl)-2-nitrobenzene (48)66
HNO3 (50%, 18 mL) and H2SO4 (95%, 18 mL) were mixed, and the solution was cooled to 0 °C. 1,3,5-Tris(bromomethyl)benzene 47(3.0 g, 8.406 mmol, 1.0 equiv) was added in small portions to the mixture at 0 °C over 10 min. The reaction mixture was stirred vigorously at 0 °C for 6 h, then subsequently allowed to warm to room temperature and stirred at 23 °C for additional 42 h. The reaction progress was controlled by TLC. The reaction was quenched with Milli-Q H2O (100 mL) after completeness, and the reaction mixture was extracted with EtOAc (3 × 150 mL). The combined organic phases were washed with sat. NaHCO3 (3 × 100 mL) and brine (3 × 100 mL) and dried over Na2SO4. The solvent was removed by rotary evaporation in vacuo to yield product 48 as a light yellow solid (3.3 g, 8.211 mmol, 98%).
TLC: Rf (47/PE/EtOAc = 10:1) = 0.90, Rf (48/PE/EtOAc = 10:1) = 0.72.
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 7.81 (s, 2H), 4.75 (s, 2H), 4.70 (s, 4H).
13C NMR (126 MHz, DMSO-d6) δ [ppm] = 148.4, 142.3, 132.8, 131.4, 31.5, 27.5.
(2-Nitrobenzene-1,3,5-triyl)trimethanamine (49)66
48 (3.3 g, 8.211 mmol, 1.0 equiv) was dissolved in THF (30 mL) and EtOH (30 mL), then NH4OH (30%, 30 mL) was added dropwise to the solution with a syringe pump (30 mL/h, 30 mL, 1 h total) under vigorous stirring at 23 °C for 1 h. The reaction mixture continued to stir at 23 °C for 20 h, and the reaction progress was controlled by LCMS. After the completion of the reaction, the solvent was removed by rotary evaporation and in vacuo to yield product 49 as a light brown solid in a quantitative yield without the need for further purification.
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 7.99 (s, 2H), 4.15 (s, 2H), 4.14 (s, 4H).
13C NMR (126 MHz, DMSO-d6): δ [ppm] = 148.7, 138.1, 132.1, 128.4, 41.6, 38.8.
HRMS (ESI) calculated for C9H15N4O2+ ([M + H]+): m/z = 211,1190, experimental 211.1190, δ [ppm] = 0.0.
Nitro-MECAM-OAc (50)66
40 (7.0 g, 29.6 mmol, 3.6 equiv) was dissolved under an argon atmosphere in anhydrous DCM (40 mL) and DMF (10 mL), then the mixture was cooled to 0 °C. Oxalyl chloride (6.3 mL, 73.903 mmol, 9.0 equiv) was added to the mixture at 0 °C, and the reaction mixture continued to stir at 0° for 10 min after the addition was finished. The reaction was equilibrated to 23 °C and stirred at that temperature for 3 h. The reaction progress was checked via TLC. For this, an aliquot of the reaction was quenched with MeOH to form the corresponding methyl ester. The differential running behavior on TLC indicated the formation of the desired acid chloride compared to the starting material at the bottom of the TLC plate. After completion, the solvent was removed by rotary evaporation, the purity was checked via NMR, and the residue was dried overnight in vacuo.
The next morning, 49 (1.7 g, 8.211 mmol, 1.0 equiv) was dissolved in Milli-Q H2O (200 mL), the pH was adjusted with a NaHCO3 solution (0.5 M) to 8.5, and the reaction mixture was cooled to 0 °C. The vacuum-dried acid chloride was dissolved in anhydrous 1,4-dioxane (100 mL) and added dropwise to the reaction mixture at 0 °C for 60 min. The pH was controlled during the addition and adjusted with a NaHCO3 solution (0.5 M) to 8.5 during the addition and more to pH 7.0 toward the end of the addition. After the addition was completed, the reaction was equilibrated to 23 °C and continued to stir for an additional 4 h. The reaction progress was controlled by LCMS. After completion, the suspension was extracted with EtOAc (4 × 200 mL or until the organic phase became colorless), and the combined organic extracts were washed with sat. NaHCO3 (2 × 200 mL) and brine (2 × 200 mL) and dried over Na2SO4. After the addition of 1% AcOH, the solvent was removed by rotary evaporation and purified by RP-Flash (EcoFlex, C18 particle size: 50 μm spherical, 20 g, 220 nm, collect all, 30–80% ACN/Milli-Q H2O, 1% AcOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield product 50 as a light brown solid (4.8 g, 5.57 mmol, 68%).
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 9.07 (t, J = 6.0 Hz, 1H), 9.02 (t, J = 5.8 Hz, 2H), 7.48 (dd, J = 7.7 Hz, 1.6 Hz, 2H), 7.46 (dd, J = 7.7 Hz, 1.5 Hz, 1H), 7.42 (s, 2H), 7.39 (dd, J = 8.1 Hz, 1.6 Hz, 2H), 7.37 (dd, J = 8.1 Hz, 1.6 Hz, 1H), 7.33 (t, J = 7.9 Hz, 2H), 7.25 (t, J = 7.9 Hz, 1H), 4.45 (d, J = 5.7 Hz, 2H), 4.42 (d, J = 5.7 Hz, 4H), 2.29 (s, 6H), 2.29 (s, 3H), 2.20 (s, 6H), 2.18 (s, 3H).
13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.3, 167.9, 167.9, 164.9, 164.8, 147.0, 142.9, 142.7, 140.2, 131.5, 130.3, 130.1, 126.3, 126.2, 126.1, 126.1, 125.8, 125.7, 42.0, 38.8, 20.4, 20.2.
HRMS (ESI) calculated for C42H39N4O17+ ([M + H]+): m/z = 871.2305, experimental 871.2304, δ [ppm] = 0.1. Calculated for C42H42N5O17+ ([M + NH4]+): m/z = 888.2570, experimental 888.2569, δ [ppm] = 0.1. Calculated for C42H38N4NaO17+ ([M + Na]+): m/z = 893.2124, experimental 893.2125, δ [ppm] = 0.1.
NH2-MECAM-OAc (51)66
50 (3.0 g, 3.445 mmol, 1.0 equiv) was dissolved under an argon atmosphere in anhydrous THF (12 mL), anhydrous EtOH (9.6 mL), and AcOH (2.4 mL), then Zn dust (3.4 g, 51.7 mmol, 15.0 equiv) was added to the mixture at 0 °C. The reaction mixture was stirred at 0 °C for 10 min, then allowed to warm to room temperature and continued to stir at 23 °C for 45 min. The reaction progress was controlled by LCMS. After completion, the reaction mixture was filtered over a pad of Celite, which was washed with EtOAc until the organic phase become colorless. The combined organic phases were washed with sat. NaHCO3 (2 × 150 mL) and brine (2 × 150 mL) and dried over Na2SO4. The solvent was removed by rotary evaporation and in vacuo to yield product 51 as a light brown solid in a quantitative yield without the need for further purification.
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 8.84 (t, J = 6.1 Hz, 2H), 8.75 (t, J = 6.0 Hz, 1H), 7.48 (dd, J = 7.5 Hz, 1.8 Hz, 2H), 7.45 (dd, J = 7.6 Hz, 1.7 Hz, 1H), 7.37 (dd, J = 8.1 Hz, 1.9 Hz, 2H), 7.35 (dd, J = 8.2 Hz, 1.8 Hz, 1H), 7.33 (t, J = 7.8 Hz, 2H), 7.30 (t, J = 7.8 Hz, 1H), 7.01 (s, 2H), 5.12 (s, 2H), 4.28 (d, J = 6.0 Hz, 4H), 4.25 (d, J = 5.9 Hz, 2H), 2.27 (s, 9H), 2.16 (s, 3H), 2.14 (s, 6H).
13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.3, 168.3, 167.8, 167.8, 164.8, 164.3, 142.8, 142.6, 140.1, 140.0, 131.0, 130.8, 127.8, 126.3, 126.1, 126.1, 126.0, 125.9, 125.5, 125.3, 122.0, 42.1, 30.4, 20.8, 20.3, 20.2, 20.1.
HRMS (ESI) calculated for C42H41N4O15+ ([M + H]+): m/z = 841.2563, experimental 841.2562, δ [ppm] = 0.1. Calculated for C42H40N4NaO15+ ([M + Na]+): m/z = 863.2382, experimental 863.2381, δ [ppm] = 0.1.
Alkyne-MECAM-OAc (3)66
Hex-5-ynoic acid (81.7 μL, 0.758 mmol, 2.2 equiv) was dissolved under an argon atmosphere in dry THF (3.6 mL) and cooled to 0 °C. NMM (229.8 μL, 2.067 mmol, 6.0 equiv) and iso-butyl chloroformate (89.6 μL, 0.689 mmol, 2.0 equiv) were added dropwise to the solution at 0 °C, which immediately resulted in the formation of a white precipitate. The reaction mixture was stirred 10 more minutes at 0 °C, then it was allowed to warm to room temperature and stirred at 23 °C for an additional 90 min. 51 (289.7 mg, 0.345 mmol, 1.0 equiv) was dissolved in dry THF (6 mL), then added dropwise at 0 °C to the reaction mixture, which continued to stir for 10 min at 0 °C. The reaction was equilibrated to 23 °C and stirred for 5 h at that temperature, while the reaction progress was controlled by LCMS. After completion or when overreaction was increasingly observed by LCMS, the suspension was quenched by the addition of H2O (5 mL) and sat. NaHCO3 (5 mL). The phases were separated, and the aqueous phase was extracted with EtOAc (3 × 25 mL). The combined organic extracts were washed with 0.1 M HCl (2 × 50 mL), sat. NaHCO3 (2 × 50 mL), and brine (2 × 50 mL) and dried over Na2SO4. The solvent was removed by rotary evaporation. The residue was taken up in ACN/Milli-Q H2O with 1% AcOH to delay further deacetylation and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 30–70% ACN/Milli-Q H2O, 1% AcOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield product 3 as a white solid (180.0 mg, 0.193 mmol, 56%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.54 (s, 1H), 8.94 (t, J = 6.1 Hz, 1H), 8.77 (t, J = 6.0 Hz, 2H), 7.52 (dd, J = 7.7 Hz, 1.6 Hz, 2H), 7.44 (dd, J = 7.7 Hz, 1.6 Hz, 1H), 7.38 (dd, J = 8.1 Hz, 1.6 Hz, 2H), 7.34 (dd, J = 8.1 Hz, 1.6 Hz, 1H), 7.31 (t, J = 7.8 Hz, 2H), 7.23 (t, J = 7.9 Hz, 1H), 7.20 (s, 2H), 4.38 (d, J = 5.9 Hz, 2H), 4.33 (bs, 4H), 2.81 (t, J = 2.6 Hz, 1H), 2.52–2.51 (m, 2H), 2.28 (s, 6H), 2.28 (s, 3H), 2.25 (td, J = 7.1 Hz, 2.7 Hz, 2H), 2.17 (s, 6H), 2.16 (s, 3H), 1.80 (p, J = 7.2 Hz, 2H).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.0, 171.2, 168.3, 167.9, 167.9, 164.7, 164.6, 142.9, 140.2, 137.6, 135.9, 131.8, 130.7, 130.6, 126.2, 126.1, 126.1, 126.0, 125.6, 125.4, 124.6, 84.0, 83.8, 71.7, 71.6, 48.6, 42.2, 34.2, 32.4, 24.1, 23.5, 20.4, 20.2, 17.5, 17.1.
HRMS (ESI) calculated for C48H47N4O16+ ([M + H]+): m/z = 935.2982, experimental 935.2979, δ [ppm] = 0.3. Calculated for C48H46N4NaO16+ ([M + Na]+): m/z = 957.2801, experimental 957.2797, δ [ppm] = 0.4. Calculated for C48H48N4O162+ ([M + 2H]2+): m/z = 468.1527, experimental 468.1527, δ [ppm] = 0.0. Calculated for C48H46N4Na2O162+ ([M + Na2]2+): m/z = 490.1347, experimental 490.1345, δ [ppm] = 0.2.
Alkyne-MECAM-OH (4)66
3 (500.0 mg, 0.535 mmol, 1.0 equiv) was dissolved in MeOH (4.8 mL), and the solution was cooled to 0 °C. To the solution was added DIPEA (1.2 mL), then the reaction was allowed to warm to room temperature and stirred at 23 °C for 3 h. The reaction progress was controlled by LCMS. After completion, the solvent was removed by rotary evaporation, and the residue was taken up in ACN/Milli-Q and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 25–65% ACN/Milli-Q H2O, 0.1% HCOOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield product 4 as a white solid (330.0 mg, 0.483 mmol, 90%).
1H NMR (500 MHz, DMSO-d6): δ [ppm] = 12.54 (s, 3H), 9.58 (s, 1H), 9.33 (t, J = 6.2 Hz, 1H), 9.16 (t, J = 6.1 Hz, 2H), 9.14 (bs, 3H), 7.29 (dd, J = 8.2 Hz, 1.3 Hz, 2H), 7.21 (s, 2H), 7.19 (dd, J = 8,2 Hz, 1.4 Hz, 1H), 6.92 (dd, J = 7.8 Hz, 1.2 Hz, 2H), 6.89 (dd, J = 7.8 Hz, 1.2 Hz, 1H), 6.67 (t, J = 7.9 Hz, 2H), 6.61 (t, J = 7.9 Hz, 1H), 4.46 (d, J = 6.1 Hz, 2H), 4.41 (d, J = 6.0 Hz, 4H), 2.81 (t, J = 2.6 Hz, 1H), 2.54–2.50 (m, 2H), 2.25 (td, J = 7.1 Hz, 2.6 Hz, 2H), 1.79 (p, J = 7.2 Hz, 2H).
13C NMR (126 MHz, DMSO-d6) δ [ppm] = 171.2, 169.8, 169.8, 149.7, 149.5, 146.2, 146.2, 137.5, 135.9, 132.1, 124.8, 118.9, 118.9, 118.1, 118.0, 117.3, 117.1, 115.1, 114.9, 84.0, 71.7, 42.2, 39.0, 34.1, 24.2, 17.5.
HRMS (ESI) calculated for C36H35N4O10+ ([M + H]+): m/z = 683.2348, experimental 683.2346, δ [ppm] = 0.2. Calculated for C36H34N4NaO10+ ([M + Na]+): m/z = 705.2167, experimental 705.2164, δ [ppm] = 0.3.
TrtS-PEG7-(Zn2+)-DOTAM-OH (52a)64
2 (92.0 mg, 0.094 mmol, 1.5 equiv) was dissolved under an argon atmosphere in DMSO (2 mL). To the solution was added zinc acetate (41.4 mg, 0.188 mmol, 3.0 equiv) dissolved in Milli-Q H2O (2 mL), and the solution was stirred at 23 °C for 5 min. 26 (40.0 mg, 0.063 mmol, 1.0 equiv) dissolved in DMSO (2 mL) was added to the solution. CuSO4 (5.0 mg, 0.031 mmol, 0.5 equiv in 0.5 mL PBS) and sodium ascorbate (12.4 mg, 0.063 mmol, 1.0 equiv in 1 mL PBS) was added to the CuSO4 solution, whereupon a white precipitate formed immediately. THPTA (6.8 mg, 0.016 mmol, 0.25 equiv in 0.5 mL PBS) was added to the suspension. The suspension was added to the reaction mixture under an argon atmosphere. The reaction mixture was stirred at 23 °C for 1 h, and the reaction progress was controlled by LCMS. After completion, the solution was filtered over cotton wool and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 5–35% ACN/Milli-Q H2O, 0.1% HCOOH). The product containing fractions were identified by LCMS and lyophilized to dryness to yield product 52a as a white solid (80.0 mg, 0.042 mmol, 76%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 10.32–9.39 (m, 3H), 9.04 (bs, 3H), 8.49 (bs, 1H), 8.21 (bs, 1H), 7.97–7.82 (m, 3H), 7.35–7.33 (m, 1H), 7.33–7.29 (m, 12H), 7.25–7.24 (m, 3H), 7.23–7.21 (m, 3H), 6.84–6.70 (m, 3H), 6.54–6.32 (m, 3H), 4.43 (t, J = 5.0 Hz, 2H), 4.38 (s, 2H), 3.75 (t, J = 4.8 Hz, 2H), 3.47–3.45 (m, 16H), 3.36 (t, J = 5.9 Hz, 2H), 3.31–3.24 (m, 12H), 3.14 (q, J = 5.7 Hz, 2H), 2.99–2.52 (m, 24H), 2.23 (t, J = 7.4 Hz, 2H), 2.15 (t, J = 7.4 Hz, 2H).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 172.3, 171.5, 170.1, 169.7, 169.6, 165.0, 147.5, 147.2, 144.5, 143.6, 129.1, 128.0, 126.7, 123.7, 118.0, 117.9, 116.6, 115.7, 115.4, 115.3, 114.4, 69.7, 69.7, 69.6, 69.5, 69.0, 68.7, 66.0, 55.6, 50.5, 49.4, 40.4, 40.0, 38.6, 38.0, 37.6, 34.6, 33.9, 27.5.
HRMS (ESI) calculated for (C80H107N15O19SZn2+)2+ ([M + 2H]2+): m/z = 838.8429, experimental 838.8427, δ [ppm] = 0.2. Calculated for (C80H106N15NaO19SZn2+)2+ ([M + H+Na]2+): m/z = 849.8339, experimental 849.8337, δ [ppm] = 0.2.
HS-PEG7-(Zn2+)-DOTAM-OH (52)
52a (15.0 mg, 8.938 μmol, 1.0 equiv) was dissolved in dry DCM (1.5 mL), then TFA (1.5 mL) and TIPS (75 μL) were added to the solution (immediate shift from slightly yellow to neon yellow/orange was observed) and the reaction mixture was stirred at 23 °C for 2 h. The reaction progress was controlled by LCMS. After completion, the solvent was removed by rotary evaporation. The residue was washed with ice-cold diethyl ether (2 × 50 mL), centrifuged (4500 rpm, 15 min, −20 °C), and decanted after each washing step. The residue was dried in vacuo to yield product 52 as a light brown solid (12.0 mg, 8.357 mmol, 94%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.45 (bs, 3H), 9.17 (t, J = 5.2 Hz, 1H), 8.91 (t, J = 5.3 Hz, 1H), 8.87 (t, J = 5.4 Hz, 2H), 8.80 (t, J = 5.4 Hz, 3H), 7.98–7.95 (m, 1H), 7.23 (dd, J = 8.2 Hz, 1.1 Hz, 3H), 7.12–6.99 (m, 1H), 6.91 (ddd, J = 7.8 Hz, 4.4 Hz, 1.3 Hz, 3H), 6.67 (ddd, J = 7.9 Hz, 4.3 Hz, 2.1 Hz, 3H), 4.45 (t, J = 5.2 Hz, 2H), 4.42 (s, 2H), 3.77 (t, J = 5.3 Hz, 2H), 3.48–3.46 (m, 16H), 3.45–3.40 (m, 12H), 3.38 (t, J = 6.9 Hz, 2H), 3.34 (t, J = 6.7 Hz, 2H), 3.31–2.78 (m, 24H), 2.73–2.72 (m, 2H), 2.63 (q, J = 7.0 Hz, 2H), 2.37 (t, J = 7.0 Hz, 1H).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 171.4, 171.3, 170.7, 170.6, 170.4, 170.2, 170.0, 149.5, 149.4, 149.4, 146.2, 143.3, 136.1, 135.8, 134.6, 130.0, 129.4, 129.1, 128.4, 126.3, 126.2, 123.7, 118.9, 118.3, 118.0, 117.4, 117.4, 116.6, 115.2, 115.0, 113.3, 69.8, 69.7, 69.6, 69.5, 69.1, 69.1, 69.1, 68.7, 65.0, 56.2, 55.9, 55.8, 55.7, 54.9, 50.9, 50.8, 50.4, 49.5, 40.4, 40.0, 39.1, 39.0, 38.6, 38.6, 38.6, 38.2, 38.2, 36.0, 35.8, 35.3, 35.2, 34.9, 34.7, 34.6, 34.0, 33.9, 30.8, 30.7, 26.6, 25.7, 23.3, 20.0, 19.4, 19.3, 19.0, 17.9, 17.3, 15.2, 12.7, 12.1, 9.7.
HRMS (ESI) calculated for (C61H92N15O19SZn2+)+ ([M + H]+): m/z = 1434.5690, experimental 1434.5695, δ [ppm] = 0.5. Calculated for (C61H91N15NaO19SZn2+)+ ([M + Na]+): m/z = 1456.5509, experimental 1456.5504, δ [ppm] = 0.5. Calculated for (C61H93N15O19SZn2+)2+ ([M + 2H]2+): m/z = 717.78812, experimental 717.7880, δ [ppm] = 0.2. Calculated for (C61H92N15NaO19SZn2+)2+ ([M + H+Na]2+): m/z = 728.7791, experimental 728.7793, δ [ppm] = 0.2. Calculated for (C61H94N15O19SZn2+)3+ ([M + 3H]3+): m/z = 478.8612, experimental 478.8612, δ [ppm] = 0.0.
TrtS-PEG7-MECAM-OH (53a)66
4 (80.4 mg, 0.118 mmol, 1.5 equiv) was dissolved under an argon atmosphere in DMSO (2 mL) and Milli-Q H2O (2 mL). 26 (50.0 mg, 0.079 mmol, 1.0 equiv) dissolved in DMSO (2 mL) was added to the solution. CuSO4 (5.0 mg, 0.031 mmol, 0.5 equiv in 0.5 mL PBS) and sodium ascorbate (12.4 mg, 0.063 mmol, 1.0 equiv in 1 mL PBS) were combined, whereupon a white solid precipitated immediately. THPTA (6.8 mg, 0.016 mmol, 0.25 equiv in 0.5 mL PBS) was added to the suspension. The suspension was then added under an argon atmosphere to the reaction mixture. The reaction mixture was stirred at 23 °C for 1 h, and the reaction progress was controlled by LCMS. After completion, the solution was filtered over cotton wool and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 50–90% ACN/Milli-Q H2O, 0.1% HCOOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield product 53a as a white solid (85.0 mg, 0.064 mmol, 82%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.57 (s, 1H), 12.52 (s, 2H), 9.57 (s, 1H), 9.33 (t, J = 5.9 Hz, 1H), 9.19 (s, 1H), 9.16 (t, J = 5.9 Hz, 2H), 9.12 (s, 1H), 7.87 (t, J = 5.6 Hz, 1H), 7.83 (s, 1H), 7.36–7.27 (m, 15H), 7.25 (t, J = 1.4 Hz, 1H), 7.24–7.23 (m, 1H), 7.23 (t, J = 1.6 Hz, 1H), 7.22–7.21 (m, 2H), 7.21 (dd, J = 8.3 Hz, 1.2 Hz 1H) 6.92 (d, J = 7.8 Hz, 2H), 6.89 (d, J = 7.8 Hz, 1H), 6.67 (t, J = 7.9 Hz, 2H), 6.61 (t, J = 8.0 Hz, 1H), 4.46–4.45 (m, 4H), 4.42 (d, J = 5.7 Hz, 2H), 3.78 (t, J = 5.2 Hz, 2H), 3.50–3.49 (m, 2H), 3.46–3.45 (m, 16H), 3.36 (t, J = 5.6 Hz, 2H), 3.15 (q, J = 5.6 Hz, 2H), 2.70 (t, J = 7.3 Hz, 2H), 2.47 (t, J = 7.3 Hz, 2H), 2.24 (t, J = 7.4 Hz, 2H), 2.16 (t, J = 7.4 Hz, 2H), 1.95 (p, J = 7.3 Hz, 2H).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 171.6, 170.2, 169.8, 169.8, 149.7, 149.6, 146.2, 146.2, 144.5, 137.5, 136.0, 132.8, 132.2, 129.1, 128.1, 126.8, 124.9, 122.3, 118.9, 118.1, 118.0, 117.4, 117.2, 115.1, 115.0, 69.8, 69.7, 69.7, 69.6, 69.1, 68.8, 66.0, 49.3, 42.2, 39.1, 38.6, 34.9, 33.9, 27.5, 25.3, 24.8.
HRMS (ESI) calculated for C70H79N8O16S+ ([M + H]+): m/z = 1319.5329, experimental 1319.5332, δ [ppm] = 0.3. Calculated for C70H78N8NaO16S+ ([M + Na]+): m/z = 1341.5149, experimental 1341.5149, δ [ppm] = 0.0.
HS-PEG7-MECAM-OH (53)
53a (25.0 mg, 18.9 μmol, 1.0 equiv) was dissolved in dry DCM (1.5 mL), and to the solution were added TFA (1.5 mL) and TIPS (75 μL) were added, where an immediate shift from slight yellow to neon yellow/orange was observed. The reaction was stirred at 23 °C for 2 h. After complete conversion, the solvent was removed by rotary evaporation. The residue was washed with ice-cold diethyl ether (2 × 50 mL), centrifuged (4500 rpm, 15 min, −20 °C), and dried in vacuo to yield product 53 as a light yellow solid (18.3 mg, 17.0 mmol, 90%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.52 (s, 2H), 9.56 (s, 1H), 9.32 (t, J = 6.1 Hz, 1H), 9.16 (t, J = 6.0 Hz, 2H), 7.94 (t, J = 5.4 Hz, 1H), 7.84 (s, 1H), 7.29 (dd, J = 8.2 Hz, 1.1 Hz, 2H), 7.21 (s, 2H), 7.20 (dd, J = 8.3 Hz, 1.2 Hz, 1H), 6.92 (dd, J = 7.8 Hz, 1.3 Hz, 2H), 6.89 (dd, J = 7.8 Hz, 1.3 Hz, 1H), 6.67 (t, J = 7.9 Hz, 2H), 6.61 (t, J = 7.9 Hz, 1H), 4.51–4.44 (m, 4H), 4.40 (d, J = 5.8 Hz, 2H), 3.79 (t, J = 5.4 Hz, 2H), 3.53–3.49 (m, 2H), 3.48–3.44 (m, 16H), 3.39 (t, J = 5.9 Hz, 2H), 3.20 (q, J = 5.8 Hz, 2H), 2.70 (t, J = 7.6 Hz, 2H), 2.63 (dd, J = 14.9 Hz, 7.0 Hz, 2H), 2.47 (t, J = 7.5 Hz, 2H), 2.37 (t, J = 7.0 Hz, 2H), 2.23 (t, J = 8.0 Hz, 1H), 1.94 (p, J = 7.6 Hz, 2H).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 171.6, 170.4, 169.8, 169.8, 149.7, 149.6, 146.3, 146.2, 146.2, 137.5, 136.0, 132.2, 124.9, 122.4, 118.9, 118.9, 118.1, 118.0, 117.4, 117.2, 115.1, 115.0, 69.8, 69.8, 69.8, 69.7, 69.6, 69.1, 68.8, 49.3, 42.2, 39.2, 39.1, 38.6, 34.9, 25.3, 24.8, 20.0, 17.4.
HRMS (ESI) calculated for C51H65N8O16S+ ([M + H]+): m/z = 1077.4234, experimental 1077.4234, δ [ppm] = 0.0. Calculated for C51H64N8NaO16S+ ([M + Na]+): m/z = 1099.4053, experimental 1099.4055, δ [ppm] = 0.2. Calculated for C51H66N8O16S2+ ([M + 2H]2+): m/z = 539.2153, experimental 539.2154, δ [ppm] = 0.1. Calculated for C51H65N8NaO16S2+ ([M + H+Na]2+): m/z = 550.2063, experimental 550.2065, δ [ppm] = 0.2. Calculated for C51H67N8O16S+ ([M + 3H]+): m/z = 359.8126, experimental 359.8127, δ [ppm] = 0.1.
General Procedure for Peptide Synthesis
Solid-phase peptide synthesis (SPPS) was performed in a 50.0 μmol scale in 5 mL tubes with on an automated Syro Multiple Peptide Synthesizer (MultiSynTech, Witten, Germany) on Rapp TentaGel S RAM resin 54 (Rapp Polymere, Tübingen, Germany). Couplings were performed with HCTU (206.9 mg, 500.0 μmol, 10.0 equiv in 1.0 mL DMF)/DIPEA (176.0 μL, 1.035 mmol, 20.7 equiv). The C-terminal amino acid for all peptides was either Fmoc-Lys(Dde)-OH or Fmoc-Lys(Boc)–0OH and was coupled manually to the resin, then manually deprotected with first 40% piperidine in DMF (1.2 mL/tube, 2 min) and second 20% piperidine in DMF (1.2 mL/tube, 8 min). The remaining SPPS was performed automated on the previously mentioned peptide synthesizer. For the automated procedure, amino acids were dissolved fresh with the help of ultrasound sonication in NMP (500.0 μmol, 10.0 equiv, 0.5 M). HCTU (25.1 g, 60.0 mmol, 1200.0 equiv) was dissolved in DMF (120.0 mL), DIPEA (25.0 mL, 147.0 mmol, 2940.0 equiv) was bottled, piperidine (80.0 mL, 808.0 mmol, 16160.0 equiv) was prepared in DMF (120 mL), and acetic anhydride (5 mL, 52.9 mmol, 1058.0 equiv) and DMF (5000 mL) were bottled separately and were provided to the peptide synthesizer. Complicated couplings, indicated by an asterisk in the amino acid sequence above (*), were performed through a double coupling followed by capping with acetic anhydride (75.0 μL, 793.4 μmol, 15.9 equiv)/DIPEA (15.0 μL, 88.2 μmol, 1.8 equiv) in DMF (1410 μL) of potentially unreacted free α-amino groups. Side-chain protection groups of the amino acids were as follows: Arg, Pmc; Asn, Cys, Gln, and His, Trt; Asp, Glu, Ser, Thr, and Tyr, tBu; and Lys and Trp, Boc. For the peptides with C-terminal PEG derivatization, amino acids with a Boc-protecting group at the α-amino function were incorporated at the N-terminus to allow for simultaneous resin cleavage and the deprotection of side chains and the N-terminalinus.
General Procedure for Global Deprotection and Resin Cleavage
For C-terminal peptides, a Dde group was present at the ε-amino function at the C-terminal lysine, which was removed on the resin (50.0 μmol, 1.0 equiv) by treatment with hydrazine (8.0 equiv) in THF (1.0 M, 0.4 mL) further diluted in DMF (1.6 mL) at 23 °C for 3 h. The crude peptides were simultaneously cleaved from the resin and deprotected globally by a 3 h treatment with 2.0 mL of a deprotection solution (1.9 mL TFA, 60 μL tri-iso-propylsilane, 40 μL of Milli-Q H2O) at 23 °C. The cleaved and deprotected peptides were precipitated with ice-cold tert-butylmethyl ether, centrifuged (4500 rpm, 30 min, −20 °C) and the supernatant was discarded. The remaining pellets were taken up in ACN/Milli-Q H2O containing 0.1% TFA and were purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 5–50% ACN/Milli-Q H2O, 0.1% TFA). The product containing fractions were identified by LCMS and lyophilized to dryness to yield the peptides as white solids.
FpvA 121–139 Peptide (5)
5 (16.7 mg, 8.145 μmol, 16%) was prepared following the general procedures for peptide synthesis and global deprotection and resin cleavage.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.64 (s), 8.62 (d, J = 7.9 Hz), 8.29 (d, J = 7.4 Hz), 8.20–8.10 (m), 8.07 (t, J = 5.7 Hz), 8.05 (t, J = 5.7 Hz), 7.97 (t, J = 7.6 Hz), 7.93 (d, J = 7.7 Hz), 7.90–7.85 (m), 7.84 (d, J = 8.4 Hz), 7.80 (d, J = 7.8 Hz), 7.76 (d, J = 8.2 Hz), 7.73 (d, J = 8.6 Hz), 7.67 (d, J = 8.2 Hz), 7.63 (t, J = 6.8 Hz), 7.45 (s), 7.22 (s), 7.19 (s), 7.03 (s), 6.98 (s), 6.80 (s), 5.23–4.74 (m), 4.66–4.64 (m), 4.48–4.09 (m), 4.07–4.01 (m), 4.00–3.94 (m), 3.81 (d, J = 5.8 Hz), 3.79 (d, J = 5.6 Hz), 3.76–3.51 (m), 2.87 (d, J = 3.9 Hz), 2.84 (d, J = 3.8 Hz), 2.78–2.70 (m), 2.68 (d, J = 8.8 Hz), 2.65 (d, J = 8.8 Hz), 2.60–2.52 (m), 2.49–2.35 (m), 2.19–2.02 (m), 2.00–1.82 (m), 1.81–1.69 (m), 1.68–1.57 (m), 1.55–1.39 (m), 1.36–1.22 (m), 1.21 (d, J = 7.0 Hz), 1.15–0.99 (m), 0.90–0.76 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.2, 173.5, 172.6, 172.5, 172.3, 172.0, 171.9, 171.4, 171.3, 171.2, 171.2, 171.1, 170.9, 170.8, 170.6, 170.1, 170.0, 169.9, 169.9, 169.8, 169.7, 169.1, 168.4, 168.1, 69.9, 66.8, 66.7, 66.5, 61.8, 61.7, 61.4, 58.3, 58.0, 57.7, 57.4, 57.4, 57.1, 55.4, 55.2, 55.1, 52.8, 52.2, 51.8, 51.5, 51.5, 50.0, 49.5, 49.1, 48.2, 42.2, 42.0, 40.7, 40.5, 40.0, 38.8, 36.7, 36.4, 36.3, 35.8, 32.3, 31.5, 31.2, 30.8, 29.3, 27.6, 27.2, 26.6, 24.3, 24.2, 24.0, 23.1, 23.1, 22.3, 21.5, 21.5, 19.7, 19.5, 19.4, 19.1, 18.2, 17.7, 15.4, 15.4, 14.7, 11.2, 11.0.
HRMS (ESI) calculated for C85H150N24O32S2+ ([M + 2H]2+): m/z = 1025.5279, experimental 1025.5278, δ [ppm] = 0.1. Calculated for C85H149N24NaO32S2+ ([M + H+Na]2+): m/z = 1036.5189, experimental 1036.5188, δ [ppm] = 0.1. Calculated for C85H151N24O32S+ ([M + 3H]3+): m/z = 684.0210, experimental 684.0210, δ [ppm] = 0.0.
PfeA 33–51 Peptide (6)
6 (29.3 mg, 13.315 μmol, 27%) was prepared following the general procedures for peptide synthesis and global deprotection and resin cleavage.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.23 (s), 8.36 (d, J = 8.5 Hz), 8.23–8.14 (m), 8.08–8.01 (m), 8.00 (d, J = 7.3 Hz), 7.98–7.90 (m), 7.88 (d, J = 8.1 Hz), 7.80 (d, J = 7.9 Hz), 7.76 (d, J = 7.7 Hz), 7.73 (d, J = 7.8 Hz), 7.71–7.61 (m), 7.29 (d, J = 7.2 Hz), 7.26 (s), 7.05 (s), 6.79 (s), 6.77 (s), 5.10–4.80 (m), 4.38–4.09 (m), 4.07 (t, J = 7.6 Hz), 4.04–3.94 (m), 3.81 (d, J = 5.9 Hz), 3.78 (d, J = 5.7 Hz), 3.69–3.65 (m), 3.63 (d, J = 5.5 Hz), 2.79–2.71 (m), 2.34–2.06 (m), 2.05–1.83 (m), 1.82–1.40 (m), 1.39–1.24 (m), 1.22 (t, J = 7.2 Hz), 1.15–1.06 (m), 1.05–0.97 (m), 0.93–0.77 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.1, 174.1, 174.0, 173.9, 173.9, 173.5, 172.6, 172.3, 171.7, 171.5, 171.4, 171.1, 171.1, 171.1, 170.8, 170.7, 170.5, 170.0, 169.9, 168.7, 167.8, 66.6, 66.5, 58.3, 58.3, 58.0 57.7, 57.1, 57.0, 52.7, 52.4, 52.2, 52.1, 51.8, 51.7, 51.0, 48.6, 48.4, 41.9, 41.0, 40.0, 38.7, 36.6, 31.5, 31.4, 31.4, 31.0, 30.6, 30.2, 30.1, 30.1, 30.1, 30.0, 27.7, 27.6, 27.6, 27.5, 27.2, 27.1, 26,7, 26.7, 24.4, 24.1, 23.1, 22.2, 22.2, 21.5, 19.6, 19.5, 19.4, 19.2, 18.3, 18.3, 18.1, 17.9, 17.9, 17.6, 15.1, 11.0.
HRMS (ESI) calculated for C94H164N26O34+ ([M + 2H]2+): m/z = 1100.5941, experimental 1100.5940, δ [ppm] = 0.1.
HasR 122–144 Peptide (7)
7 (15.6 mg, 6.007 μmol, 12%) was prepared following the general procedures for peptide synthesis and global deprotection and resin cleavage.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.36 (s), 8.52 (d, J = 8.1 Hz), 8.17 (d, J = 7.4 Hz), 8.13 (d, J = 7.8 Hz), 8.11–8–08 (m), 8.06 (d, J = 7.3 Hz), 8.03–7.85 (m), 7.81 (d, J = 8.0 Hz), 7.77 (d, J = 8.2 Hz), 7.73–7.43 (m), 7.25 (s), 7.21–7.17 (m), 7.01 (s), 6.81 (s), 6.76 (s), 5.48 (bs), 5.15–4.77 (m), 4.58–4.01 (m), 3.88–3.83 (m), 3.74 (d, J = 4.2 Hz), 3.72 (d, J = 4.1 Hz), 3.70–3.49 (m), 3.15–3.04 (m), 2.80–2.73 (m), 2.72–2.65 (m), 2.53–2.34 (m), 2.19–2.02 (m), 1.99–1.02 (m), 0.93–0.68 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.1, 173.9, 173.9, 173.6, 172.1, 171.9, 171.9, 171.8, 171.8, 171.4, 171.1, 171.0, 171.0, 170.9, 170.8, 169.9, 169.8, 169.2, 166.6, 156.7, 61.6, 61.4, 61.4, 60.6, 59.7, 59.6, 57.8, 57.6, 57.0, 55.4, 55.0, 54.2, 53.3, 52.2, 51.6, 51.3, 50.0, 49.8, 49.7, 48.3, 48.1, 47.1, 46.9, 40.6, 40.5, 40.4, 40.0, 39.9, 39.8, 39.6, 39.5, 39.4, 39.3, 39.2, 38.8, 36.7, 36.2, 36.0, 32.2, 31.5, 31.1, 30.5, 30.4, 29.4, 29.2, 29.1, 28.2, 27.7, 27.2, 27.0, 26.6, 24.5, 24.3, 24.0, 23.2, 23.2, 23.1, 22.5, 22.3, 21.6, 21.5, 19.2, 19.2, 18.2, 18.2, 17.9, 17.8, 17.8, 15.3, 14.7, 11.0.
HRMS (ESI) calculated for C111H198N34O35S + ([M + 4H]+): m/z = 649.8614, experimental 649.8607, δ [ppm] = 0.7.
FpvA 124–134 Peptide (8)
8 (20.3 mg, 16.260 μmol, 33%) was prepared following the general procedures for peptide synthesis and global deprotection and resin cleavage.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.56 (s), 8.69 (d, J = 7.3 Hz), 8.24 (d, J = 7.7 Hz), 8.20 (d, J = 7.5 Hz), 8.14–8.07 (m), 8.06 (t, J = 5.9 Hz), 7.99 (d, J = 8.5 Hz), 7.92–7.85 (m), 7.82–7.77 (m), 7.72 (s), 7.44 (s), 7.26 (s), 7.23 (s), 7.20–7.16 (m), 7.11 (s), 7.08 (s), 6.98 (s), 4.97 (s), 4.67–4.61 (m), 4.52–4.47 (m), 4.44–4.36 (m), 4.34–4.32 (m), 4.29 (dd, J = 8.3 Hz, 3.8 Hz), 4.26–4.21 (m), 4.18 (dd, J = 8.3 Hz, 4.3 Hz), 4.10–4.04 (m), 4.03–3.95 (m), 3.72 (d, J = 5.8 Hz), 3.69 (d, J = 5.7 Hz), 3.67 (d, J = 5.9 Hz), 3.65–3.62 (m), 3.59–3.42 (m), 2.80–2.71 (m), 2.60–2.51 (m), 2.48–2.35 (m), 2.11–2.02 (m), 1.95–1.87 (m), 1.81–1.69 (m), 1.66–1.58 (m), 1.53–1.38 (m), 1.35–1.23 (m), 1.21 (d, J = 7.0 Hz), 1.12–1.06 (m), 1.02 (dd, J = 11.1 Hz, 6.3 Hz), 0.91 (dd, J = 10.2 Hz, 6.9 Hz), 0.86 (d, J = 6.6 Hz), 0.83–0.77 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 173.6, 172.4, 172.3, 171.8, 171.7, 171.1, 170.9, 170.7, 170.3, 170.2, 169.8, 169.8, 168.3, 167.7, 66.6, 66.5, 61.8, 58.3, 57.7, 57.2, 57.1, 54.8, 52.3, 51.8, 51.4, 50.1, 49.6, 48.1, 41.9, 40.5, 40.0, 38.8, 36.6, 36.2, 35.9, 32.2, 30.7, 29.9, 29.3, 26.5, 24.3, 24.0, 23.2, 22.2, 21.3, 19.7, 19.3, 18.3, 18.1, 17.5, 15.4, 14.6, 10.9.
HRMS (ESI) calculated for C52H94N15O18S+ ([M + H]+): m/z = 1248.6617, experimental 1248.6610, δ [ppm] = 0.7. Calculated for C52H94N15NaO18S2+ ([M + H+Na]2+): m/z = 635.8254, experimental 635.8249, δ [ppm] = 0.5. Calculated for C52H93N15Na2O18S2+ ([M + 2Na]2+): m/z = 646.8164, experimental 646.8158, δ [ppm] = 0.6. Calculated for C52H96N15O18S3+ ([M + 3H]3+): m/z = 416.8921, experimental 416.8918, δ [ppm] = 0.3.
PfeA 37–46 Peptide (9)
9 (31.6 mg, 25.957 μmol, 52%) was prepared following the general procedures for peptide synthesis and global deprotection and resin cleavage.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.14 (s), 8.54 (d, J = 7.9 Hz), 8.31 (d, J = 7.5 Hz), 8.06 (d, J = 6.8 Hz), 8.05–7.97 (m), 7.94–7.88 (m), 7.82–7.68 (m), 7.63 (d, J = 8.6 Hz), 7.29 (s), 7.25 (s), 7.05 (s), 6.78 (d, J = 5.4 Hz), 4.93 (s), 4.45–4.38 (m), 4.37–4.31 (m), 4.28–4.23 (m), 4.21–4.07 (m), 4.02–3.97 (m), 3.60–3.56 (m), 2.88 (s), 2.80–2.70 (m), 2.28–2.19 (m), 2.18–2.06 (m), 1.99–1.84 (m), 1.78–1.64 (m), 1.54–1.48 (m), 1.33–1.24 (m), 1.21 (dd, J = 11.4 Hz, 7.1 Hz), 1.02 (dd, J = 10.1 Hz, 6.4 Hz), 0.90–0.75 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 173.9, 173.9, 173.4, 172.5, 172.4, 171.3, 171.0, 171.0, 170.7, 170.6, 169.9, 169.7, 165.9, 162.4, 66.5, 66.5, 58.1, 58.1, 57.8, 57.6, 52.6, 52.3, 52.1, 51.8, 48.4, 48.3, 40.2, 40.0, 38.8, 31.6, 31.4, 31.4, 30.7, 30.3, 30.0, 27.9, 27.7, 27.6, 26.7, 22.2, 19.6, 19.5, 19.2, 18.2, 18.0, 18.0, 18.0.
HRMS (ESI) calculated for C47H84N15O17+ ([M + H]+): m/z = 1130.6164, experimental 1130.6154, δ [ppm] = 1.0. Calculated for C47H83N15NaO17+ ([M + Na]+): m/z = 1152.5984., experimental 1152.5980, δ [ppm] = 0.4. Calculated for C47H85N15O172+ ([M + 2H]2+): m/z = 565.8119, experimental 565.8118, δ [ppm] = 0.1. Calculated for C47H86N15O173+ ([M + 3H]3+): m/z = 377.5437, experimental 377.5436, δ [ppm] = 0.1.
HasR 129–138 Peptide (10)
10 (21.7 mg, 17.825 μmol, 36%) was prepared following the general procedures for peptide synthesis and global deprotection and resin cleavage.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.67 (s), 8.77 (d, J = 7.7 Hz), 8.20 (s), 8.10 (d, J = 6.8 Hz), 8.05 (t, J = 8.5 Hz), 8.00–7.95 (m), 7.84 (d, J = 8.1 Hz), 7.75 (s), 7.68 (d, J = 8.5 Hz), 7.63 (d, J = 8.1 Hz), 7.26 (s), 7.19 (s), 7.15 (s), 7.12 (s), 7.03 (s), 6.77 (s), 4.61 (td, J = 8.2 Hz, 4.7 Hz), 4.56 (q, J = 6.8 Hz), 4.44–4.33 (m), 4.30–4.26 (m), 4.23–4.19 (m), 4.16–4.03 (m), 3.97–3.34 (m), 3.15–2.99 (m), 2.84 (dd, J = 17.8 Hz, 3.3 Hz), 2.78–2.73 (m), 2.70 (dd, J = 16.9 Hz, 4.5 Hz), 2.66 (dd, J = 17.9 Hz, 9.0 Hz), 2.56–2.35 (m), 2.12–2.01 (m), 1.98–1.80 (m), 1.78–1.40 (m), 1.35–1.22 (m), 1.10 (s), 0.87 (dd, J = 6.5 Hz, 5.4 Hz), 0.84–0.78 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 173.9, 173.5, 171.8, 171.8, 171.4, 171.1, 171.0, 170.8, 170.6, 170.4, 170.0, 169.3, 169.1, 167.9, 61.5, 61.4, 59.6, 58.1, 57.5, 55.3, 53.3, 52.3, 52.2, 51.6, 51.4, 49.8, 48.8, 47.1, 43.7, 40.4, 40.0, 38.7, 36.0, 35.6, 32.2, 31.5, 31.1, 30.6, 30.2, 29.5, 29.1, 27.8, 26.6, 24.4, 24.2, 23.1, 22.4, 21.6, 19.2, 19.2, 18.2, 17.8.
HRMS (ESI) calculated for C51H89N14O18S+ ([M + H]+): m/z = 1217.6195, experimental 1217.6194, δ [ppm] = 0.1. Calculated for C51H88N14NaO18S+ ([M + Na]+): m/z = 1239.6014, experimental 1239.6006, δ [ppm] = 0.8. Calculated for C51H90N14O18S2+ ([M + 2H]2+): m/z = 609.3134, experimental 609.3136, δ [ppm] = 0.2. Calculated for C51H89N14NaO18S2+ ([M + H+Na]2+): m/z = 620.3043, experimental 620.3047, δ [ppm] = 0.4. Calculated for C51H88N14Na2O18S2+ ([M + 2Na]2+): m/z = 631.2953, experimental 631.2949, δ [ppm] = 0.4.
General Procedure for N- or C-Terminal Modification, Global Deprotection, and Resin Cleavage
N-Terminal coupling with N3-(PEG)7-COOH (83.2 mg, 150 μmol, 3.0 equiv) was performed after manual deprotection of the N-terminal Fmoc group and cleavage of the peptide chain (50.0 μmol, 1.0 equiv) on the resin with HOBt (30.6 mg, 200.0 μmol, 4.0 equiv), HATU (85.6 mg, 225.0 μmol, 4.5 equiv), and NMM (0.2 mL) activation in threefold excess at 23 °C for 21 h in DMF (1.8 mL). The Dde group, if present, was removed on the resin by a treatment with hydrazine (8.0 equiv) in THF (1.0 M) (0.4 mL) in DMF (1.6 mL) at 23 °C for 3 h.
C-terminal coupling with N3-(PEG)7-COOH (83.2 mg, 150 μmol, 3.0 equiv), PDTP (32.3 mg, 150 μmol, 3.0 equiv), or 40 (35.7 mg, 150 μmol, 3.0 equiv) was performed after the Dde deprotection procedure described in “General Procedure for Global Deprotection and Resin Cleavage” after the same procedure as stated above for the N-terminal modification. For C-terminal coupling with 40, the reagents were added dropwise at 0 °C. After the coupling with 40, deacetylation was performed with DIPEA (0.4 mL) in MeOH (1.6 mL) at 0 °C for 3 h. The crude peptides were cleaved from the resin and globally deprotected as described in “General Procedure for Global Deprotection and Resin Cleavage”. After precipitation with ice-cold tert-butylmethyl ether and centrifugation (4500 rpm, 30 min, −20 °C), the resulting crude peptides were taken up in ACN/Milli-Q H2O containing 0.1% TFA and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 5–50% ACN/Milli-Q H2O, 0.1% TFA). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield the PEG-modified peptides as white solids.
Long N-Terminal Azido FpvA Peptide (61)
61 (4.8 mg, 5.721 μmol, 11%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the FpvA 121–139 C-term Dde peptide or FpvA 121–139 C-terminal Boc peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.33 (s), 8.28 (d, J = 8.1 Hz), 8.23 (s), 8.16 (d, J = 7.6 Hz), 8.11–8.01 (m), 7.99 (t, J = 5.6 Hz), 7.96 (d, J = 7.3 Hz), 7.93 (d, J = 7.4 Hz), 7.88 (d, J = 8.0 Hz), 7.87–7.82 (m), 7.80 (d, J = 7.4 Hz), 7.76 (d, J = 8.0 Hz), 7.70 (d, J = 8.2 Hz), 7.67 (d, J = 7.9 Hz), 7.60 (s), 7.44 (s), 7.22 (s), 7.19 (s), 7.04 (s), 7.02 (s), 6.99 (s), 6.80 (s), 5.12–4.80 (m), 4.68 (dd, J = 13.5 Hz, 7.7 Hz), 4.57–4.50 (m), 4.45–3.91 (m), 3.83–3.77 (m), 3.75–3.47 (m), 3.44 (t, J = 6.0 Hz), 3.40–3.14 (m), 3.09 (dd, J = 14.4 Hz, 7.2 Hz), 2.78–2.52 (m), 2.47–2.36 (m), 2.27 (s), 2.15–2.02 (m), 2.00–1.19 (m), 1.17 (t, J = 7.3 Hz), 1.15–1.05 (m), 1.02 (dd, J = 9.5 Hz, 5.9 Hz), 0.94 (s), 0.86 (dd, J = 11.9 Hz, 6.5 Hz), 0.84–0.75 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.0, 173.4, 173.3, 172.8, 172.4, 172.4, 172.2, 171.9, 171.9, 171.8, 171.3, 171.1, 171.0, 170.8, 170.8, 170.7, 170.5, 170.4, 170.0, 169.9, 169.8, 169.0, 168.8, 168.3, 70.3, 70.1, 69.8, 69.8, 69.7, 69.6, 69.3, 68.9, 66.7, 66.7, 66.6, 66.5, 61.7, 61.4, 58.2, 57.9, 57.6, 57.4, 57.2, 57.2, 57.0, 55.0, 55.0, 54.9, 52.7, 52.1, 51.7, 51.4, 51.4, 51.3, 50.0, 49.9, 49.4, 49.0, 48.6, 48.1, 45.8, 42.6, 42.2, 41.9, 40.6, 40.4, 40.2, 38.7, 38.1, 36.6, 36.4, 36.2, 32.2, 31.5, 31.2, 30.6, 29.7, 29.3, 27.9, 27.5, 27.1, 26.5, 24.2, 24.1, 23.9, 23.1, 23.0, 22.2, 21.5, 19.6, 19.5, 19.3, 19.1, 19.0, 18.2, 17.7, 17.3, 15.4, 15.3, 14.6, 11.2, 11.2, 10.9, 8.7.
HRMS (ESI) calculated for C107H190N28O43S2+ ([M + 2H]2+): m/z = 1293.6626, experimental 1293.6622, δ [ppm] = 0.4. Calculated for C107H191N28O43S2+ ([M + 3H]3+): m/z = 862.7775, experimental 862.7777, δ [ppm] = 0.2.
Long N-Terminal Azido PfeA Peptide (62)
62 (29.1 mg, 10.632 μmol, 21%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the PfeA 33–51 C-terminal Dde peptide or PfeA 33–51 C-terminal Boc peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.08 (s, 1H), 8.20–8.15 (m), 8.08–8.02 (m), 8.02–7.98 (m), 7.97 (d, J = 7.2 Hz), 7.95–7.85 (m), 7.79 (d, J = 8.0 Hz), 7.76 (d, J = 7.7 Hz), 7.73 (d, J = 7.9 Hz), 7.69 (bs), 7.65 (d, J = 8.3 Hz), 7.29 (s), 7.27 (s), 7.24 (s), 7.05 (s), 6.79 (s), 6.77 (s), 4.99–4.86 (m), 4.39–4.07 (m), 4.05–3.92 (m), 3.79 (dd, J = 16.5 Hz, 5.5 Hz), 3.67–3.51 (m), 3.50 (s), 3.43 (t, J = 6.0 Hz), 3.40–3.38 (m), 3.26 (q, J = 5.9 Hz), 3.19–3.15 (m), 2.76 (t, J = 7.3 Hz), 2.34–2.05 (m), 2.02–1.83 (m), 1.82–1.25 (m), 1.23–1.19 (m), 1.04 (dd, J = 11.7 Hz, 6.3 Hz), 1.01 (d, J = 6.3 Hz), 0.86 (d, J = 6.6 Hz), 0.85–0.77 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.1, 174.1, 174.0, 174.0, 173.9, 173.9, 173.4, 172.7, 172.3, 171.7, 171.5, 171.5, 171.4, 171.1, 171.1, 170.8, 170.7, 170.6, 170.0, 170.0, 168.9, 168.8, 168.7, 70.3, 70.2, 69.8, 69.8, 69.7, 69.7, 69.6, 69.3, 68.9, 66.5, 66.4, 58.3, 58.3, 58.0, 57.8, 57.2, 56.8, 52.7, 52.7, 52.6, 52.4, 52.2, 52.1, 51.7, 51.7, 51.0, 50.0, 48.6, 48.4, 41.9, 41.0, 40.0, 38.7, 38.1, 38.0, 36.4, 31.5, 31.5, 31.4, 31.3, 31.0, 30.6, 30.5, 30.2, 30.1, 30.0, 29.0, 27.7, 27.6, 27.4, 27.2, 27.2, 27.0, 26.7, 26.6, 24.4, 24.0, 23.1, 22.4, 22.2, 21.5, 19.6, 19.5, 19.4, 19.2, 19.2, 18.3, 18.2, 18.1, 17.9, 17.9, 15.2, 14.0, 11.0.
HRMS (ESI) calculated for C116H204N30O452+ ([M + 2H]2+): m/z = 1368.7292, experimental 1368.7312, δ [ppm] = 1.0. Calculated for C116H205N30O453+ ([M + 3H]3+): m/z = 912.8220, experimental 912.8232 δ [ppm] = 1.2.
Long N-Terminal Azido HasR Peptide (63)
63 (16.0 mg, 5.106 μmol, 10%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the HasR 122–144 C-terminal Dde peptide or HasR 122–144 C-term Boc peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.36 (s), 8.52 (d, J = 8.1 Hz), 8.48 (d, J = 8.3 Hz), 8.40–7.27 (m), 7.25 (s), 7.21 (s), 7.19 (s), 7.12 (s), 7.05 (s), 7.00 (s), 6.81 (s), 6.79 (s), 6.76 (s), 5.21–4.06 (m), 4.00 (s), 3.98 (s), 3.74 (d, J = 4.2 Hz), 3.72 (d, J = 4.3 Hz), 3.66–3.24 (m), 3.17 (s), 3.15–3.00 (m), 2.96 (s), 2.89–2.51 (m), 2.49–2.33 (m), 2.27 (s), 2.16–2.02 (m), 1.99–1.14 (m), 1.12 (s), 1.10–1.01 (m), 0.94 (s), 0.90–0.75 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.0, 173.9, 173.6, 172.8, 172.0, 171.7, 171.7, 171.5, 171.4, 171.4, 171.2, 171.0, 171.0, 170.8, 170.8, 170.7, 170.6, 170.3, 170.2, 170.2, 170.1, 170.0, 169.9, 169.8, 169.7, 169.7, 169.5, 169.4, 169.2, 169.1, 168.9, 166.6, 166.1, 157.5, 156.7, 73.6, 73.0, 72.9, 72.8, 70.3, 70.2, 69.8, 69.8, 69.7, 69.6, 69.3, 68.9, 61.6, 61.6, 61.5, 61.4, 60.6, 60.5, 59.6, 59.6, 59.5, 57.6, 57.3, 57.3, 56.9, 56.8, 56.7, 55.3, 54.9, 54.5, 54.2, 53.3, 52.7, 52.5, 52.3, 52.2, 52.1, 52.0, 51.6, 51.3, 51.2, 51.1, 50.0, 50.0, 49.9, 49.6, 49.5, 48.2, 48.1, 48.1, 47.0, 46.9, 46.8, 42.6, 42.5, 41.2, 40.6, 40.6, 40.5, 40.5, 40.4, 38.7, 38.2, 36.7, 36.6, 36.4, 36.3, 36.3, 36.0, 35.8, 32.2, 31.5, 31.3, 31.3, 31.1, 30.9, 30.8, 30.5, 30.4, 30.4, 29.8, 29.4, 29.2, 29.1, 28.3, 28.3, 28.2, 28.1, 27.9, 27.7, 27.1, 27.0, 26.6, 26.5, 25.0, 24.5, 24.3, 24.2, 24.1, 24.1, 24.1, 24.0, 24.0, 23.9, 23.2, 23.1, 22.6, 22.5, 22.5, 22.3, 22.2, 22.1, 21.8, 21.6, 21.5, 21.5, 19.2, 19.1, 19.0, 18.2, 18.0, 17.9, 17.8, 17.8, 17.6, 17.4, 15.3, 15.2, 14.6, 12.3, 10.9.
HRMS (ESI) calculated for C133H237N38O46S3+ ([M + 3H]3+): m/z = 1044.90261, experimental 1044.9013, δ [ppm] = 1.25. Calculated for C133H238N38O46S4+ ([M + 4H]4+): m/z = 783.92878, experimental 783.9286 δ [ppm] = 0.23.
Long C-Terminal Azido FpvA Peptide (64)
64 (15.7 mg, 6.069 μmol, 12%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the FpvA 121–139 N-terminal Boc C-terminal Dde peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.64 (s), 8.62 (d, J = 7.7 Hz), 8.36 (d, J = 7.5 Hz), 8.29 (d, J = 7.4 Hz), 8.19–8.10 (m), 8.09–8.02 (m), 7.98–7.94 (m), 7.92 (d, J = 7.4 Hz), 7.90–7.83 (m), 7.81–7.78 (m), 7.76 (d, J = 8.1 Hz), 7.74 (d, J = 8.6 Hz), 7.71–7.64 (m), 7.62 (s), 7.44 (s), 7.25–7.15 (m), 7.05–6.94 (m), 6.82–6.75 (m), 5.17–4.80 (m), 4.59–4.09 (m), 4.06–3.89 (m), 3.83–3.48 (m), 3.44 (t, J = 5.9 Hz), 3.42–3.38 (m), 3.28 (q, J = 6.0 Hz), 3.17 (s), 3.08 (dd, J = 13.3 Hz, 6.8 Hz), 2.88–2.82 (m), 2.77–2.63 (m), 2.60–2.53 (m), 2.50–2.35 (m), 2.27 (s, 1H), 2.14–2.03 (m), 2.00–1.82 (m), 1.81–1.17 (m), 1.16–0.98 (m), 0.94 (s), 0.90–0.76 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.0, 174.0, 173.5, 173.4, 173.4, 172.8, 172.4, 172.4, 172.4, 172.2, 171.8, 171.8, 171.3, 171.2, 171.1, 171.0, 171.0, 170.8, 170.7, 170.6, 170.6, 170.4, 170.0, 170.0, 169.9, 169.8, 169.8, 169.7, 169.5, 169.0, 168.9, 168.7, 168.4, 168.3, 167.8, 73.2, 72.9, 70.3, 70.2, 69.8, 69.8, 69.7, 69.6, 69.3, 68.9, 66.7, 66.7, 66.6, 66.5, 61.8, 61.7, 61.7, 61.4, 58.2, 57.8, 57.6, 57.2, 57.2, 57.0, 55.1, 55.0, 55.0, 52.7, 52.4, 52.3, 52.1, 52.1, 51.7, 51.3, 51.3, 50.0, 49.9, 49.4, 48.9, 48.8, 48.6, 48.1, 42.6, 42.5, 42.2, 42.1, 41.9, 40.7, 40.5, 40.4, 40.1, 40.0, 38.7, 38.1, 38.0, 36.7, 36.5, 36.5, 36.4, 36.3, 35.7, 35.5, 32.3, 31.5, 31.2, 30.8, 29.7, 29.3, 28.9, 28.1, 27.9, 27.6, 27.5, 27.1, 26.5, 24.3, 24.2, 24.1, 24.0, 23.9, 23.1, 23.0, 22.8, 22.7, 22.2, 21.5, 21.5, 19.6, 19.5, 19.3, 19.3, 19.1, 18.2, 17.6, 17.3, 15.4, 15.3, 14.6, 11.2, 11.2, 10.9.
HRMS (ESI) calculated for C107H190N28O43S2+ ([M + 2H]2+): m/z = 1293.6626, experimental 1293.6626, δ [ppm] = 0.0. Calculated for C107H189N28NaO43S2+ ([M + H+Na]2+): m/z = 1304.6535, experimental 1304.6535, δ [ppm] = 0.0. Calculated for C107H191N28O43S3+ ([M + 3H]3+): m/z = 862.7775, experimental 862.7777, δ [ppm] = 0.2. Calculated for C107H192N28O43S4+ ([M + 4H]4+): m/z = 647.3349, experimental 647.3351, δ [ppm] = 0.2.
Long C-Terminal Azido PfeA Peptide (65)
65 (31.3 mg, 11.436 μmol, 23%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the PfeA 33–51 N-terminal Boc C-terminal Dde peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.08 (s), 8.36 (d, J = 8.5 Hz), 8.21 (d, J = 8.0 Hz), 8.19–8.15 (m), 8.07–8.01 (m), 8.00 (d, J = 7.3 Hz), 7.96–7.86 (m), 7.78 (d, J = 7.7 Hz), 7.76 (d, J = 7.8 Hz), 7.73 (d, J = 7.9 Hz), 7.66–7.62 (m), 7.32 (s), 7.27 (s), 7.23 (s), 7.01 (s), 6.81–6.74 (m), 4.92 (bs), 4.89 (bs), 4.40–4.08 (m), 4.03–3.90 (m), 3.81 (d, J = 5.8 Hz), 3.79 (d, J = 5.7 Hz), 3.70–3.65 (m), 3.63 (d, J = 5.4 Hz), 3.62–3.58 (m), 3.57–3.48 (m), 3.44 (t, J = 6.0 Hz), 3.39–3.38 (m), 3.28 (q, J = 5.9 Hz), 3.17 (s), 3.09 (dd, J = 13.8 Hz, 7.1 Hz), 2.76 (dd, J = 12.7 Hz, 6.6 Hz), 2.33–1.83 (m), 1.80–1.24 (m), 1.21 (t, J = 6.2 Hz), 1.11–0.99 (m), 0.90 (t, J = 7.6 Hz), 0.86 (d, J = 6.6 Hz), 0.85–0.79 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.1, 174.0, 174.0, 173.9, 173.8, 173.5, 172.5, 172.4, 172.3, 171.5, 171.4, 171.3, 171.2, 171.0, 171.0, 171.0, 170.7, 170.6, 170.5, 169.8, 169.8, 169.8, 168.7, 168.7, 168.5, 167.6, 70.2, 70.2, 69.8, 69.8, 69.7, 69.6, 69.3, 68.9, 66.6, 66.5, 58.1, 58.1, 58.1, 57.8, 57.6, 57.1, 56.9, 52.5, 52.5, 52.4, 52.3, 52.0, 51.9, 51.7, 51.6, 51.0, 50.0, 48.6, 48.4, 48.3, 41.8, 41.0, 40.0, 38.8, 38.1, 38.1, 36.6, 31.7, 31.5, 31.5, 31.4, 31.1, 30.6, 30.3, 30.2, 30.1, 30.0, 30.0, 30.0, 29.0, 27.8, 27.7, 27.6, 27.5, 27.3, 27.2, 27.2, 26.6, 24.4, 24.0, 23.1, 22.7, 22.1, 21.5, 19.5, 19.4, 19.2, 18.3, 18.2, 18.0, 18.0, 17.9, 17.6, 15.1, 11.0.
HRMS (ESI) calculated for C116H204N30O452+ ([M + 2H]2+): m/z = 1368.7293, experimental 1368.7299, δ [ppm] = 0.6. Calculated for C116H205N30O453+ ([M + 3H]3+): m/z = 912.8220, experimental 912.8232, δ [ppm] = 1.2.
Long C-Terminal Azido HasR Peptide (66)
66 (18.6 mg, 5.936 μmol, 12%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the HasR 122–144 N-terminal Boc C-terminal Dde peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.33 (s), 8.53 (d, J = 8.1 Hz), 8.48 (d, J = 8.2 Hz), 8.38–7.45 (m), 7.40–7.17 (m), 7.03–6.95 (m), 6.83–6.72 (m), 5.48 (t, J = 4.7 Hz), 5.40 (s), 5.10–4.89 (m), 4.79 (d, J = 7.7 Hz), 4.58–4.05 (m), 3.95–3.83 (m), 3.76–3.20 (m), 3.17 (s), 3.15–2.51 (m), 2.47–2.33 (m), 2.27 (s), 2.20–1.01 (m), 0.94 (s), 0.91–0.74 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.0, 173.8, 173.6, 173.6, 172.8, 172.0, 171.7, 171.7, 171.7, 171.4, 171.4, 171.2, 171.0, 171.0, 171.0, 170.8, 170.8, 170.8, 170.8, 170.7, 170.6, 170.2, 170.2, 170.0, 170.0, 169.8, 169.8, 169.7, 169.7, 169.2, 169.1, 168.7, 168.5, 166.6, 166.1, 156.7, 73.6, 73.0, 72.8, 70.3, 70.2, 69.8, 69.8, 69.7, 69.6, 69.3, 68.9, 61.6, 61.6, 61.5, 61.4, 60.6, 60.5, 59.6, 59.6, 59.5, 59.5, 59.5, 57.7, 57.6, 57.2, 57.2, 56.9, 56.8, 56.7, 55.3, 55.3, 54.9, 54.9, 54.2, 53.3, 52.7, 52.5, 52.4, 52.3, 52.2, 52.2, 52.0, 51.6, 51.2, 51.2, 51.1, 50.0, 49.9, 49.6, 49.5, 48.1, 48.1, 47.0, 46.9, 46.8, 42.6, 41.2, 40.6, 40.6, 40.5, 40.5, 40.4, 40.0, 38.7, 38.2, 38.1, 36.7, 36.3, 36.0, 35.8, 32.2, 31.5, 31.3, 31.3, 31.1, 30.9, 30.9, 30.5, 30.4, 29.8, 29.4, 29.2, 29.1, 29.1, 29.0, 29.0, 29.0, 28.7, 28.3, 28.3, 28.2, 28.1, 27.9, 27.7, 27.7, 27.2, 27.1, 27.0, 26.6, 25.0, 24.5, 24.3, 24.2, 24.1, 24.1, 24.0, 23.1, 22.8, 22.6, 22.5, 22.3, 22.1, 21.7, 21.6, 21.5, 21.5, 19.2, 19.1, 18.2, 18.1, 18.0, 17.9, 17.8, 17.8, 17.8, 17.4, 15.3, 15.3, 14.6, 14.0, 10.9.
HRMS (ESI) calculated for C133H237N38O46S3+ ([M + 3H]3+): m/z = 1044.9026, experimental 1044.9043, δ [ppm] = 1.7. Calculated for C133H238N38O46S4+ ([M + 4H]4+): m/z = 783.9288, experimental 783.9303, δ [ppm] = 1.5. Calculated for C133H239N38O46S5+ ([M + 5H]5+): m/z = 627.3445, experimental 627.3454, δ [ppm] = 0.9.
Short N-Terminal Azido FpvA Peptide (67)
67 (21.2 mg, 11.876 μmol, 24%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the FpvA 124–134 C-terminal Boc peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.39 (s), 8.40 (d, J = 7.5 Hz), 8.18 (d, J = 7.5 Hz), 8.08 (t, J = 5.8 Hz), 8.05 (t, J = 5.8 Hz), 7.98 (d, J = 8.5 Hz), 7.92 (d, J = 7.6 Hz), 7.89–7.82 (m), 7.82 (d, J = 7.5 Hz), 7.80 (dd, J = 8.0 Hz, 3.9 Hz), 7.67 (s), 7.44 (s), 7.24 (s), 7.08 (s), 6.99 (s), 5.11 (t, J = 5.5 Hz), 4.98 (d, J = 4.8 Hz), 4.87 (d, J = 5.1 Hz), 4.57 (dd, J = 13.8 Hz, 7.7 Hz), 4.50 (dd, J = 13.9 Hz, 6.6 Hz), 4.44–4.36 (m), 4.33 (dd, J = 13.1 Hz, 5.7 Hz), 4.30 (dd, J = 8.3 Hz, 3.9 Hz), 4.26–4.16 (m), 4.10–4.05 (m), 4.04–3.96 (m), 3.94 (s), 3.72–3.62 (m), 3.61–3.59 (m), 3.57–3.46 (m), 3.43 (t, J = 6.0 Hz), 3.40–3.38 (m), 3.26 (q, J = 6.0 Hz), 2.79–2.70 (m), 2.58 (dd, J = 15.7 Hz, 6.6 Hz), 2.54–2.51 (m), 2.49–2.35 (m), 1.97 (q, J = 6.7 Hz), 1.95–1.88 (m), 1.82–1.70 (m), 1.64–1.56 (m), 1.54–1.38 (m), 1.36–1.23 (m), 1.21 (d, J = 7.0 Hz), 1.16–1.05 (m), 1.02 (dd, J = 10.6 Hz, 6.3 Hz), 0.95–0.77 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 173.6, 172.4, 172.2, 171.9, 171.8, 171.1, 170.9, 170.7, 170.6, 170.5, 170.1, 169.8, 169.8, 168.9, 168.3, 70.4, 70.2, 69.8, 69.8, 69.7, 69.6, 69.3, 68.9, 66.5, 66.5, 61.8, 58.2, 57.7, 57.1, 57.0, 54.8, 52.3, 51.8, 51.4, 50.0, 50.0, 49.4, 48.1, 41.9, 40.6, 40.0, 38.7, 38.1, 36.6, 36.2, 35.7, 32.2, 30.8, 30.7, 29.3, 27.2, 26.5, 24.3, 24.0, 23.1, 22.2, 21.4, 19.6, 19.3, 19.1, 18.2, 18.0, 15.3, 14.6, 10.9.
HRMS (ESI) calculated for C74H135N19O29S2+ ([M + 2H]2+): m/z = 892.9691, experimental 892.9687, δ [ppm] = 0.4. Calculated for C74H134N19NaO29S2+ ([M + H + Na]2+): m/z = 903.9601, experimental 903.9599, δ [ppm] = 0.2. Calculated for C74H133N19Na2O29S2+ ([M + 2Na]2+): m/z = 914.9511, experimental 914.9512, δ [ppm] = 0.1. Calculated for C74H136N19O29S3+ ([M + 3H]3+): m/z = 595.6485, experimental 595.6484, δ [ppm] = 0.1.
Short N-Terminal Azido PfeA Peptide (68)
68 (27.8 mg, 16.678 μmol, 33%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the PfeA 37–46 C-terminal Boc peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.08 (s), 8.25 (t, J = 6.0 Hz), 8.15 (d, J = 7.6 Hz), 8.10 (d, J = 7.9 Hz), 8.05 (dd, J = 12.8 Hz, 6.6 Hz), 7.99 (d, J = 7.5 Hz), 7.92 (d, J = 7.0 Hz), 7.90 (d, J = 8.5 Hz), 7.87 (d, J = 8.3 Hz), 7.76 (d, J = 8.1 Hz), 7.73–7.68 (m), 7.62 (d, J = 8.6 Hz), 7.31–7.24 (m), 7.06 (s), 6.78 (s), 6.76 (s), 4.98 (d, J = 4.7 Hz), 4.89 (d, J = 4.7 Hz), 4.36 (p, J = 7.1 Hz), 4.33–4.23 (m), 4.19–4.08 (m), 4.03–3.94 (m), 3.80 (d, J = 6.1 Hz), 3.61–3.58 (m), 3.56–3.48 (m), 3.44 (t, J = 6.1 Hz), 3.40–3.38 (m), 3.28 (q, J = 6.0 Hz), 2.78–2.71 (m), 2.27–2.19 (m), 2.16–2.05 (m), 2.01–1.84 (m), 1.78–1.64 (m), 1.56–1.47 (m), 1.34–1.25 (m), 1.23 (d, J = 7.1 Hz), 1.21 (d, J = 7.0 Hz), 1.03 (d, J = 6.3 Hz), 1.01 (d, J = 6.4 Hz), 0.84 (d, J = 2.4 Hz), 0.83 (d, J = 2.4 Hz), 0.82 (d, J = 6.8 Hz).
HRMS (ESI) calculated for C59H125N19O282+ ([M + 2H]2+): m/z = 833.9465, experimental 833.9459, δ [ppm] = 0.6. Calculated for C59H124N19NaO282+ ([M + H + Na]2+): m/z = 844.9375, experimental 844.9372, δ [ppm] = 0.3. Calculated for C59H123N19Na2O282+ ([M + 2Na]2+): m/z = 855.9285, experimental 855.9285, δ [ppm] = 0.0. Calculated for C59H126N19O283+ ([M + 3H]3+): m/z = 556.3001, experimental 556.3002, δ [ppm] = 0.1.
Short N-Terminal Azido HasR Peptide (69)
69 (12.8 mg, 7.298 μmol, 15%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the HasR 129–138 C-terminal Boc peptide and N3-(PEG)7-COOH.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.33 (s), 8.28 (t, J = 8.0 Hz), 8.09 (d, J = 6.8 Hz), 8.04–7.98 (m), 7.96 (d, J = 8.0 Hz), 7.82 (d, J = 8.1 Hz), 7.73 (d, J = 8.0 Hz), 7.71–7.65 (m), 7.64 (d, J = 8.5 Hz), 7.59 (d, J = 8.2 Hz), 7.26 (s), 7.15 (s), 7.04 (s), 6.76 (s), 5.10–5.04 (m), 4.62 (td, J = 8.2 Hz, 5.0 Hz), 4.56 (q, J = 6.8 Hz), 4.52 (dd, J = 13.4 Hz, 7.6 Hz), 4.41–4.33 (m), 4.30–4.25 (m), 4.23–4.10 (m), 3.99–3.92 (m), 3.73–3.65 (m), 3.64–3.49 (m), 3.44 (t, J = 6.1 Hz), 3.40–3.38 (m), 3.27 (q, J = 6.1 Hz), 2.79–2.67 (m), 2.61–2.51 (m), 2.46–2.36 (m), 2.13–2.01 (m), 1.97–1.82 (m), 1.79–1.63 (m), 1.60–1.38 (m), 1.37–1.21 (m), 1.14–1.07 (m), 0.89–0.79 (m).
HRMS (ESI) calculated for C73H130N18O29S2+ ([M + 2H]2+): m/z = 877.4480, experimental 877.4486, δ [ppm] = 0.6. Calculated for C73H133N19O29S2+ ([M + H + NH4]2+): m/z = 885.9613, experimental 885.9613, δ [ppm] = 0.0. Calculated for C73H129N18NaO29S2+ ([M + H+Na]2+): m/z = 888.4390, experimental 888.4395, δ [ppm] = 0.5. Calculated for C73H128N18Na2O29S2+ ([M + 2Na]2+): m/z = 899.4300, experimental 899.4305, δ [ppm] = 0.5. Calculated for C73H131N18O29S3+ ([M + 3H]3+): m/z = 585.3011, experimental 585.3018, δ [ppm] = 0.7.
Long N-Terminal Pyridyl Disulfide-Modified PfeA Peptide (70)
70 (12.7 mg, 5.297 μmol, 11%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the PfeA 33–51 N-terminal Boc C-terminal Dde peptide and PDTP.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.10 (s), 8.47–8.43 (m), 8.37 (d, J = 8.4 Hz), 8.20 (d, J = 8.0 Hz), 8.18–8.14 (m), 8.09–8.02 (m), 7.99 (d, J = 7.3 Hz), 7.97–7.90 (m), 7.88 (d, J = 8.1 Hz), 7.82 (td, J = 8.0 Hz, 1.7 Hz), 7.79 (d, J = 7.7 Hz), 7.76–7.74 (m), 7.72 (d, J = 8.1 Hz), 7.69 (bs), 7.63 (d, J = 8.3 Hz), 7.31 (s), 7.28 (s), 7.25–7.23 (m), 7.01 (s), 6.78 (s), 6.77 (s), 4.93 (bs), 4.71–4.65 (m), 4.45 (d, J = 5.8 Hz), 4.42 (d, J = 5.8 Hz), 4.39–4.05 (m), 4.04–3.99 (m), 3.97 (dd, J = 10.9 Hz, 6.1 Hz), 3.80 (d, J = 5.8 Hz), 3.78 (d, J = 5.5 Hz), 3.71–3.66 (m), 3.64 (d, J = 5.4 Hz), 3.20–3.11 (m), 3.03–2.97 (m), 2.80–2.73 (m), 2.60–2.52 (m), 2.48 (s), 2.47 (s), 2.41–2.07 (m), 2.04–1.83 (m), 1.81–1.25 (m), 1.24–1.18 (m), 1.11–0.97 (m), 0.90 (t, J = 6.9 Hz), 0.86 (d, J = 6.6 Hz), 0.84–0.78 (m).
HRMS (ESI) calculated for C102H171N27O6S22+ ([M + 2H]2+): m/z = 1199.0930, experimental 1199.0925, δ [ppm] = 0.5. Calculated for C102H172N27O6S23+ ([M + 3H]3+): m/z = 799.7311, experimental 799.7309, δ [ppm] = 0.20. Calculated for C102H173N27O6S24+ ([M + 4H]4+): m/z = 600.0502 experimental 600.0500, δ [ppm] = 0.2. Calculated for C102H174N27O6S25+ ([M + 5H]5+): m/z = 480.2416, experimental 480.2415, δ [ppm] = 0.1.
Long C-Terminal Monocatechol FpvA (C33)
C33 (9.6 mg, 4.391 μmol, 9%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the FpvA 121–139 N-terminal Boc C-terminal Dde peptide and catechol 40.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.82 (s), 9.13 (s), 8.75 (t, J = 5.5 Hz), 8.64–8.55 (m), 8.37–8.31 (m), 8.28 (d, J = 7.4 Hz), 8.27–8.25 (m), 8.16 (d, J = 7.4 Hz), 8.14 (d, J = 7.6 Hz), 8.11–7.94 (m), 7.92 (d, J = 7.7 Hz), 7.90–7.76 (m), 7.72 (d, J = 8.5 Hz), 7.68 (d, J = 8.1 Hz), 7.45 (s), 7.25 (dd, J = 8.2 Hz, 1.4 Hz), 7.23 (s), 7.18 (s), 7.17 (s), 7.03–6.94 (m), 6.89 (dd, J = 7.8 Hz, 1.3 Hz), 6.79 (s), 6.66 (t, J = 7.9 Hz), 5.20–5.06 (m), 4.98 (s), 4.88 (s), 4.66–3.92 (m), 3.83–3.50 (m), 3.24 (dd, J = 12.9 Hz, 7.0 Hz), 2.97 (dd, J = 12.8 Hz, 7.0 Hz), 2.91–2.81 (m), 2.72 (dd, J = 16.7 Hz, 5.9 Hz), 2.69–2.62 (m), 2.61–2.55 (m), 2.54 (s), 2.48–2.34 (m), 2.18 (t, J = 7.3 Hz), 2.14–2.02 (m), 2.00 (s), 1.99–1.81 (m), 1.78 (s), 1.76–1.70 (m), 1.68–1.19 (m), 1.15–1.04 (m), 1.02–0.98 (m), 0.94–0.75 (m).
HRMS (ESI) calculated for C92H154N24O35S2+ ([M + 2H]2+): m/z = 1093.5359, experimental 1093.5365, δ [ppm] = 0.6. Calculated for C92H153N24NaO35S2+ ([M + H + Na]2+): m/z = 1104.5269, experimental 1104.5264, δ [ppm] = 0.5. Calculated for C92H155N24O35S3+ ([M + 3H]3+): m/z = 729.3597, experimental 729.3597, δ [ppm] = 0.0.
Long C-Terminal Monocatechol PfeA (C34)
C34 (17.4 mg, 7.447 μmol, 15%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the PfeA 33–51 N-terminal Boc C-terminal Dde peptide and catechol 40.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.80 (s), 12.09 (s), 9.12 (s), 8.76 (t, J = 5.6 Hz), 8.36 (d, J = 8.5 Hz), 8.21 (d, J = 8.0 Hz), 8.19–8.15 (m), 8.03 (dd, J = 14.7 Hz, 7.6 Hz), 8.00 (d, J = 7.3 Hz), 7.97–7.87 (m), 7.79 (d, J = 7.6 Hz), 7.72 (d, J = 8.0 Hz), 7.63 (d, J = 8.3 Hz), 7.33 (s), 7.29–7.21 (m), 7.02 (s), 6.90 (dd, J = 7.8 Hz, 1.4 Hz), 6.81–6.74 (m), 6.67 (t, J = 7.9 Hz), 4.93 (s), 4.89 (s), 4.40–4.08 (m), 4.03–3.94 (m), 3.80 (d, J = 5.9 Hz), 3.78 (d, J = 4.4 Hz), 3.69–3.65 (m), 3.63 (d, J = 5.5 Hz), 3.50 (s), 3.25 (dd, J = 13.0 Hz, 7.1 Hz), 2.76 (t, J = 7.3 Hz), 2.61–2.52 (m), 2.47 (s), 2.41–2.08 (m), 2.06–1.83 (m), 1.80–1.42 (m), 1.39–1.19 (m), 1.11–0.99 (m), 0.94 (s), 0.90 (d, J = 7.3 Hz), 0.89 (d, J = 7.1 Hz), 0.86 (d, J = 6.6 Hz), 0.85–0.78 (m).
HRMS (ESI) calculated for C101H168N26O372+ ([M + 2H]2+): m/z = 1168.6026, experimental 1168.6046, δ [ppm] = 2.0. Calculated for C101H167N26NaO372+ ([M + H + Na]2+): m/z = 1179.5936, experimental 1179.5953, δ [ppm] = 1.7. Calculated for C101H166N26Na2O372+ ([M + 2Na]2+): m/z = 1190.5846, experimental 1190.5853, δ [ppm] = 0.7. Calculated for C101H169N26O373+ ([M + 3H]3+): m/z = 779.4042, experimental 779.4054, δ [ppm] = 1.2. Calculated for C101H170N26O374+ ([M + 4H]4+): m/z = 584.8050, experimental 584.8062, δ [ppm] = 1.2.
Long N-Terminal Monocatechol FpvA with l-Amino Acids (N33L)
N33L (4.1 mg, 1.756 μmol, 1%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the FpvA 121–139 N-terminal Boc C-terminal Dde peptide and catechol 40.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.36 (s, 1H), 8.23 (m, 1H), 8.16 (d, J = 7.53 Hz, 1H), 8.05 (m, 2H), 7.96 (d, J = 7.34 Hz, 2H), 7.93 (d, J = 7.89 Hz, 1H), 7.87 (m, 6H), 7.81 (d, J = 7.89, 1H), 7.76 (d, J = 7.34 Hz, 1H), 7.72 (8.08 Hz, 1H), 7.68 (d, J = 7.89 Hz, 1H), 7.62 (m, 1H), 7.45 (s, 1H), 7.35 (d, J = 8.63 Hz, 1H), 7.22 (s, 1H), 7.19 (s, 1H), 7.04 (s, 1H), 6.99 (s, 1H), 6.93 (d, J = 7.89 Hz, 1H), 6.80 (s, 1H), 6.71 (t, J = 8.08 Hz, 1H)), 5.07 (t, J = 4.77 Hz, 1H), 4.97 (d, J = 4.77 Hz, 1H), 4.94 (d, J = 4.77 Hz, 1H), 4.86 (m, 1H), 4.54 (t, J = 6.98 Hz, 2H), 4.41 (m, 2H), 4.37 (m, 2H), 4.31 (m, 3H), 4.28 (m, 3H)), 4.23 (m, 2H), 4.19 (m, 4H), 4.15 (m, 4H), 4.03 (m, 1H), 3.97 (m, 2H), 3.81 (m, 1H), 3.74 (m, 1H), 3.71 (m, 1H), 3.69 (m, 1H), 3.63 (m, 5H), 3.56 (m, 4H), 3.51 (s, 8H), 3.30 (s, 5H), 2.74, (m, 4H), 2.58 (m, 2H), 2.46 (m, 2H), 2.40 (m, 4H), 2.09 (m, 4H), 1.96 (m, 2H), 1.90 (m, 2H), 1.84 (s, 1H), 1.76 (m, 4H), 1.63 (m, 4H), 1.50 (m, 4H), 1.46 (m, 4H), 1.29 (m 2H), 1.23 (s, 2H), 1.20 (d, J = 6.98 Hz, 4H), 1.02 (m, 6H), 0.88 (m, 4H), 0.86 (m, 4H), 0.82 (m, 6H), 0.80 (m, 4H).
HRMS (ESI) calculated for C92H154N24O35S2+ ([M + 2H]2+): m/z = 1093.5359, experimental 1093.5350, δ [ppm] = 0.9. Calculated for C92H153N24NaO35S2+ ([M + H + Na]2+): m/z = 1104.5269, experimental 1104.5252, δ [ppm] = 1.7. Calculated for C92H155N24O35S3+ ([M + 3H]3+): m/z = 729.3597, experimental 729.3593, δ [ppm] = 0.4.
Long N-Terminal Monocatechol PfeA with l-Amino Acids (N34L)
N34L (7.5 mg, 3.432 μmol, 7%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the PfeA 33–51 N-terminal Boc C-terminal Dde peptide and catechol 40.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.06 (m, 3H), 8.67 (d, J = 8.27 Hz, 1H), 8.17 (m, 2H), 8.06 (m, 5H), 7.93 (m, 8H), 7.76 (m, 4H), 7.65 (d, J = 8.27 Hz, 2H), 7.39 (d, J = 7.75 Hz, 1H), 7.29 (d, J = 12.66 Hz, 2H), 7.24 (s, 2H), 7.06 (s, 1H), 6.92 (d, J = 8.27 Hz, 1H), 6.79 (m, 3H), 6.70 (t, J = 7.88 Hz, 1H), 4.92 (m, 2H), 4.41 (m, 1H), 4.36 (t, J = 6.85 Hz, 1H), 4.29 (m, 4H), 4.25 (m, 3H), 4.21 (m, 4H), 4.15 (m, 4H), 4.09 (m, 1H), 4.02 (m, 1H), 3.98 (m, 2H), 3.78 (dd, J = 5.94, 17.18 Hz, 1H), 3.66 (dd, J = 4.52,), 3.51 (s, 2H), 2.76 (t, J = 7.10 Hz, 4H), 2.31 (m, 1H), 2.22 (m, 6H), 2.10 (m, 5H), 1.96 (m, 2H), 1.88 (m, 6H), 1.74 (m, 6H), 1.61 (m, 1H), 1.53 (m, 5H), 1.45 (m, 2H), 1.32 (m, 2H), 1.22 (m, 4H), 1.02 (m, 10H), 0.89 (m, 7H), 0.86 (m, 5H), 0.83 (m, 21H), 0.79 (m, 10H).
HRMS (ESI) calculated for C101H168N26O372+ ([M + 2H]2+): m/z = 1168.6026, experimental 1168.6030, δ [ppm] = 0.4. Calculated for C101H169N26O373+ ([M + 3H]3+): m/z = 779.4042, experimental 779.4047, δ [ppm] = 0.5.
Long N-Terminal Monocatechol FpvA with l-Amino Acids (N33D)
N33D (6 mg, 2.569 μmol, 4%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the FpvA 121–139 N-terminal Boc C-terminal Dde peptide and catechol 40.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 8.24 (m, 1H), 8.17 (d, J = 7.45 Hz, 1H), 8.05 (m, 2H), 7.96 (dd, J = 7.43, 24.55 Hz, 4H), 7.92 (m, 2H), 7.89 (m, 2H), 7.86 (m, 2H), 7.79–7.75 (m, 2H), 7.66 (d, J = 8.02 Hz, 1H), 7.60 (m, 2H), 7.45 (m, 1H), 7.22 (M, 1H), 7.19 (M, 1H), 7.04 (m, 1H), 6.99 (m, 1H), 6.92 (d, J = 7.40 Hz, 1), 6.80 (m, 1H), 6.52 (m, 2H), 5.75 (m, 1H), 5.07 (t, J = 2 Hz, 2H), 4.97 (d, J = 5.04 Hz, 1H), 4.94 (d, J = 5.06 Hz, 2H), 4.86 (m, 1H), 4.54 (m, 3H), 4.39 (m, 3H), 4.32 (m, 2H), 4.25 (m, 2H), 4.19 (m, 3H), 4.15 (m, 3H), 4.03 (m, 1H), 3.97 (m, 2H), 3.81 (m, 2H), 3.71 (m, 4H), 3.66 (m, 1H), 3.63 (m, 2H), 3.56 (m, 3H), 3.51 (s, 6H), 2.75 (m, 2H), 2.08 (m, 2H), 2.01 (m, 4H), 1.91 (m, 2H), 1.84 (s, 2H), 1.75 (m, 4H), 1.63 (m, 4H), 1.48 (m, 4H), 1.29 (m, 4H), 1.23 (s, 2H), 1.20 (d, J = 7.02 Hz, 4H), 1.09 (m, 4H), 1.02 (m, 8H), 0.88–0.87 (d, J = 6.18 Hz, 6H), 0.85 (d, J = 6.57 Hz, 6H), 0.81 (m, 26HZ).
HRMS (ESI) calculated for C92H154N24O35S2+ ([M + 2H]2+): m/z = 1093.5359, experimental 1093.5338, δ [ppm] = 2.2. Calculated for C92H155N24O35S3+ ([M + 3H]3+): m/z = 729.3597, experimental 729.3588, δ [ppm] = 0.9.
Long N-Terminal Monocatechol PfeA with l-Amino Acids (N34D)
N34L (1.5 mg, 0,6865 μmol, 0.5%) was prepared following the general procedures for peptide synthesis, N- or C-terminal modification, and global deprotection with the PfeA 33–51 N-terminal Boc C-terminal Dde peptide and catechol 40.
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 8.17 (m, 2H), 8.06 (m, 2H), 8.02 (m, 2H), 7.93 (m, 9H), 7.76 (m, 4H), 7.65 (m, 2H), 7.26 (m, 4H), 7.06 (s, 1H), 6.92 (d, J = 8.12 Hz, 1H), 6.79 (m, 4H), 6.70 (t, J = 7.31 Hz, 1H), 4.40 (m, 1H), 4.35 (m, 1H), 4.29 (m, 4H), 4.24 (m, 4H), 4.15 (m, 1H), 4.08 (m, 1H), 4.00 (m, 4H), 3.77 (m, 1H), 3.66 (m, 1H), 3.51 (s, 8H), 2.76 (t, J = 7.73 Hz, 5H), 2.23 (m, 6H), 2.11 (m, 6H), 1.98 (m, 3H), 1.89 (m, 3H), 1.73 (m, 10H), 1.61 (m, 2H), 1.52 (m, 6H), 1.44 (m, 2H), 1.23 (m, 14H), 1.13 (d, J = 6.84 Hz, 1H), 1.03 (m, 14H), 0.88 (m, 4H), 0.86 (m, 4H), 0.84 (m, 22H), 0.79 (m, 6H).
HRMS (ESI) calculated for C101H168N26O372+ ([M + 2H]2+): m/z = 1168.6026, experimental 1168.6029, δ [ppm] = 0.3. Calculated for C101H167N26NaO372+ ([M + H+Na]2+): m/z = 1179.5936, experimental 1179.5943, δ [ppm] = 1.0. Calculated for C101H169N26O373+ ([M + 3H]3+): m/z = 779.4042, experimental 779.4045, δ [ppm] = 0.3.
General Procedure for CuAAC with DOTAM 1 or 2
11–21 were prepared via an adaptation of an established procedure reported by Ferreira et al.641 or 2 (2.5 equiv) was dissolved under an argon atmosphere in DMSO (1 mL). To the solution was added zinc acetate (5.0 equiv) dissolved in Milli-Q H2O (1 mL), and the solution was stirred at 23 °C for 5 min. In the case where 2 was used, acetic acid (1%, 40 μL) was added to the reaction mixture to prevent deacetylation. PEG-modified peptide (61–69) (1.0 equiv) dissolved in DMSO (1 mL) was added to the solution. CuSO4 (0.5 equiv) dissolved in PBS (pH 7.4, 0.3 mL) and sodium ascorbate (1.0 equiv) dissolved in PBS (pH 7.4, 0.4 mL) were added to the CuSO4 solution, whereupon a white solid precipitated immediately. THPTA (0.25 equiv) dissolved in PBS (pH 7.4, 0.3 mL) was added to the suspension under an Ar atmosphere. The reaction was stirred at 23 °C for 1 h, and the reaction progress was controlled by LCMS. After the completion of the reaction, the solution was filtered over cotton wool and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 5–35% ACN/Milli-Q H2O, 0.1% HCOOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield product 11–21 as white solids.
FpvA 121–139 N-Terminal (PEG)7-Zn2+-DOTAM-OH (11)
11 (36.0 mg, 9.9 μmol, 91%) was prepared following the general procedure for CuAAC with DOTAM 2 (26.7 mg, 27.4 μmol, 2.5 equiv) and peptide 61 (30.0 mg, 11.0 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.36 (s), 9.02 (s), 8.62–8.57 (m), 8.55–8.51 (m), 8.44–8.39 (m), 8.37–8.29 (m), 8.26 (s), 8.22–8.10 (m), 8.09–7.75 (m), 7.72 (d, J = 7.8 Hz), 7.55 (d, J = 7.3 Hz), 7.51–7.44 (m), 7.28–7.20 (m), 7.18 (s), 7.02 (s), 6.98 (s), 6.87–6.81 (m), 6.80 (s), 6.56 (t, J = 7.6 Hz), 4.61 (q, J = 6.7 Hz), 4.56–4.47 (m), 4.44 (t, J = 5.1 Hz), 4.40 (s), 4.39–4.36 (m), 4.34–4.27 (m), 4.26–4.20 (m), 4.17–4.11 (m), 4.09–3.96 (m), 3.94 (d, J = 5.1 Hz), 3.92 (s), 3.85–3.58 (m), 3.55 (dd, J = 10.6 Hz, 5.6 Hz), 3.52–3.20 (m), 3.14 (q, J = 6.3 Hz), 3.02–2.85 (m), 2.74 (t, J = 7.4 Hz), 2.71–2.55 (m), 2.54 (s), 2.48–2.35 (m), 2.15–2.02 (m), 1.95–1.87 (m), 1.84–1.71 (m), 1.68–1.58 (m), 1.56–1.40 (m), 1.37 (s), 1.35–1.26 (m), 1.25 (d, J = 7.0 Hz), 1.15–0.98 (m), 0.87 (d, J = 6.6 Hz), 0.85–0.76 (m).
HRMS (ESI) calculated for (C153H252N39O56SZn2+)3+ ([M + 3H]3+): m/z = 1209.2352, experimental 1209.2347, δ [ppm] = 0.5. Calculated for (C153H253N39O56SZn2+)4+ ([M + 4H]4+): m/z = 907.1782, experimental 907.1777, δ [ppm] = 0.5. Calculated for (C153H254N39O56SZn2+)5+ ([M + 5H]5+): m/z = 725.9440, experimental 725.9441, δ [ppm] = 0.1.
PfeA 33–51 N-Terminal (PEG)7-Zn2+-DOTAM-OH (12)
12 (34.0 mg, 9.0 μmol, 99%) was prepared following the general procedure for CuAAC with DOTAM 2 (22.3 mg, 22.8 μmol, 2.5 equiv) and peptide 62 (25.0 mg 9.1 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 8.31 (s), 8.24 (s), 8.21–8.13 (m), 8.11–7.75 (m), 7.59 (d, J = 8.0 Hz), 7.55 (s), 7.38 (s), 7.30 (s), 7.23 (d, J = 7.0 Hz), 7.20 (d, J = 7.3 Hz), 7.08 (s), 7.07 (s), 6.81–6.74 (m), 6.70 (s), 6.47 (t, J = 7.9 Hz), 6.42 (t, J = 7.4 Hz), 4.44 (t, J = 5.3 Hz), 4.38 (s), 4.35–4.24 (m), 4.23–3.92 (m), 3.89 (s), 3.84 (s), 3.76 (t, J = 5.2 Hz), 3.61–3.28 (m), 3.26 (q, J = 5.8 Hz), 3.00–2.80 (m), 2.73 (t, J = 7.5 Hz), 2.64–2.56 (m), 2.54 (s), 2.44 (s), 2.41–2.39 (m), 2.26–2.08 (m), 2.05–1.14 (m), 1.06 (dd, J = 12.6 Hz, 6.0 Hz), 1.02 (d, J = 6.1 Hz), 0.90–0.76 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.8, 174.8, 174.8, 174.3, 174.3, 173.9, 173.9, 173.9, 173.7, 173.7, 173.5, 172.7, 172.3, 172.0, 171.3, 171.2, 171.2, 170.9, 170.8, 170.7, 170.1, 169.9, 169.9, 168.9, 168.8, 168.7, 167.7, 164.0, 150.1, 150.0, 146.5, 146.5, 143.4, 123.7, 118.4, 118.4, 117.5, 117.5, 117.4, 117.4, 115.3, 115.3, 70.3, 70.2, 69.8, 69.7, 69.6, 69.5, 68.9, 68.7, 57.2, 56.9, 55.8, 55.7, 53.0, 52.8, 52.2, 49.4, 41.9, 40.4, 40.0, 38.1, 31.5, 30.6, 28.3, 27.4, 26.7, 26.6, 24.4, 24.1, 23.1, 22.2, 21.5, 19.6, 19.6, 19.5, 19.2, 19.2, 18.3, 18.2, 18.1, 17.9, 17.8, 15.2, 11.0.
HRMS (ESI) calculated for (C162H266N41O58Zn2+)3+ ([M + 3H]3+): m/z = 1259.2797, experimental 1259.2793, δ [ppm] = 0.4. Calculated for (C162H267N41O58Zn2+)4+ ([M + 4H]4+): m/z = 944.7116, experimental 944.7114, δ [ppm] = 0.2. Calculated for (C162H268N41O58Zn2+)5+ ([M + 5H]5+): m/z = 755.9707, experimental 755.9708, δ [ppm] = 0.1. Calculated for (C162H269N41O58Zn2+)6+ ([M + 6H]6+): m/z = 630.1435, experimental 630.1434, δ [ppm] = 0.1.
HasR 122–144 N-Terminal (PEG)7-Zn2+-DOTAM-OH (13)
13 (29.0 mg, 6.9 μmol, 87%) was prepared following the general procedure for CuAAC with DOTAM 2 (19.5 mg, 19.9 μmol, 2.5 equiv) and peptide 63 (25.0 mg, 8.0 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.27 (s), 9.05 (s), 8.63–8.36 (m), 8.29 (s), 8.24–7.47 (m), 7.40–7.18 (m), 7.05–6.95 (m), 6.90–6.83 (m), 6.76 (s), 6.72 (s), 6.66–6.55 (m), 6.40–6.31 (m), 5.07–4.76 (m), 4.55 (d, J = 6.3 Hz), 4.45 (d, J = 4.9 Hz), 4.42–4.09 (m), 4.04 (t, J = 7.3 Hz), 4.00 (s), 3.97 (s), 3.92 (d, J = 9.4 Hz), 3.80–3.54 (m), 3.52–3.40 (m), 3.39–3.30 (m), 3.27 (t, J = 5.9 Hz), 3.13 (q, J = 6.3 Hz), 3.11–3.02 (m), 2.99 (q, J = 6.1 Hz), 2.91–2.84 (m), 2.75 (bs), 2.67 (bs), 2.64 (s), 2.54 (s), 2.49–2.35 (m), 2.22–2.01 (m), 1.98–1.38 (m), 1.37 (s), 1.26–1.00 (m), 0.90–0.73 (m).
HRMS (ESI) calculated for (C179H298N49O59SZn2+)3+ ([M + 3H]3+): m/z = 1391.36031, experimental 1391.3564, δ [ppm] = 2.81. Calculated for (C179H299N49O59SZn2+)4+ ([M + 4H]4+): m/z = 1043.77205, experimental 1043.7701, δ [ppm] = 1.87. Calculated for (C179H300N49O59SZn2+)5+ ([M + 5H]5+): m/z = 835.21909, experimental 835.2184, δ [ppm] = 0.83. Calculated for (C179H301N49O59SZn2+)6+ ([M + 6H]6+): m/z = 696.18379, experimental 696.1829, δ [ppm] = 1.28.
FpvA 124–134 N-Terminal (PEG)7-Zn2+-DOTAM-OH (14)
14 (26.0 mg, 9.2 μmol, 91%) was prepared following the general procedure for CuAAC with DOTAM 2 (24.6 mg, 25.2 μmol, 2.5 equiv) and peptide 67 (18.0 mg, 10.1 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 8.36 (d, J = 8.5 Hz), 8.21 (d, J = 8.0 Hz), 8.17 (t, J = 5.9 Hz), 8.08–8.03 (m), 8.00 (d, J = 7.3 Hz), 7.97–7.91 (m), 7.88 (d, J = 8.3 Hz), 7.81–7.75 (m), 7.72 (d, J = 7.9 Hz), 7.70–7.60 (m), 7.31 (s), 7.27 (s), 7.24 (s), 7.20 (s), 7.13 (s), 7.06 (s), 7.04 (bs), 7.02 (s), 6.78 (bs), 6.77 (s), 6.43 (s), 6.38 (bs), 4.39–4.07 (m), 4.03–3.99 (m), 3.97 (dd, J = 10.8 Hz, 5.8 Hz), 3.80 (d, J = 5.9 Hz), 3.78 (d, J = 5.7 Hz), 3.68–3.62 (m), 3.10 (ddd, J = 8.6 Hz, 6.1 Hz, 4.6 Hz), 2.99 (dd, J = 14.5 Hz, 7.6 Hz), 2.82 (dd, J = 12.5 Hz, 5.1 Hz), 2.76 (dd, J = 13.0 Hz, 7.2 Hz), 2.58 (s), 2.57 (s), 2.33–1.83 (m), 1.80–1.24 (m), 1.21 (t, J = 6.5 Hz), 1.11–0.99 (m), 0.90 (t, J = 7.3 Hz), 0.86 (d, J = 6.6 Hz), 0.84–0.79 (m).
HRMS (ESI) calculated for (C120H196N30O42SZn2+)2+ ([M + 2H]2+): m/z = 1412.6557, experimental 1412.6552, δ [ppm] = 0.5. Calculated for (C120H197N30O42SZn2+)3+ ([M + 3H]3+): m/z = 942.1062, experimental 942.1065, δ [ppm] = 0.3. Calculated for (C120H198N30O42SZn2+)4+ ([M + 4H]4+): m/z = 706.8315, experimental 706.8318, δ [ppm] = 0.3. Calculated for (C120H199N30O42SZn2+)5+ ([M + 5H]5+): m/z = 565.6666, experimental 565.6665, δ [ppm] = 0.1.
PfeA 37–46 N-Terminal (PEG)7-Zn2+-DOTAM-OH (15)
15 (25.0 mg, 9.2 μmol, 96%) was prepared following the general procedure for CuAAC with DOTAM 2 (23.4 mg, 24.0 μmol, 2.5 equiv) and peptide 68 (16.0 mg, 9.6 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 10.03 (s), 9.86 (s), 9.29–8.78 (m), 8.47 (s), 8.36 (s), 8.30 (s), 8.19 (d, J = 5.4 Hz), 8.11 (d, J = 6.4 Hz), 8.04–7.99 (m), 7.96 (t, J = 5.3 Hz), 7.92 (s), 7.91–7.84 (m), 7.80 (d, J = 7.1 Hz), 7.77–7.64 (m), 7.35 (s), 7.27 (s), 7.26–7.14 (m), 7.10 (d, J = 7.7 Hz), 7.05 (s), 6.94–6.89 (m), 6.87 (d, J = 7.7 Hz), 6.83–6.69 (m), 6.66–6.59 (m), 6.51 (d, J = 6.5 Hz), 6.48–6.29 (m), 4.93–3.88 (m), 3.82–3.75 (m), 3.49–3.42 (m), 3.38–3.36 (m), 3.32–3.27 (m), 3.01–2.78 (m), 2.73 (t, J = 7.4 Hz), 2.63–2.51 (m), 2.19–2.08 (m), 2.03–1.85 (m), 1.80–1.71 (m), 1.70–1.63 (m), 1.57–1.46 (m), 1.35–1.25 (m), 1.23 (t, J = 7.7 Hz), 1.03 (dd, J = 8.9 Hz, 6.4 Hz), 0.87–0.78 (m).
HRMS (ESI) calculated for (C115H186N30O41Zn2+)2+ ([M + 2H]2+): m/z = 1353.6331, experimental 1353.6328, δ [ppm] = 0.3. Calculated for (C115H187N30O41Zn2+)3+ ([M + 3H]3+): m/z = 902.7578, experimental 902.7575, δ [ppm] = 0.3. Calculated for (C115H188N30O41Zn2+)4+ ([M + 4H]4+): m/z = 677.3202, experimental 677.3200, δ [ppm] = 0.2. Calculated for (C115H189N30O41Zn2+)5+ ([M + 5H]5+): m/z = 542.0576, experimental 542.0573, δ [ppm] = 0.3.
HasR 129–138 N-Terminal (PEG)7-Zn2+-DOTAM-OH (16)
16 (12.9 mg, 4.6 μmol, 81%) was prepared following the general procedure fur CuAAC with DOTAM 2 (13.9 mg, 14.3 μmol, 2.5 equiv) and peptide 69 (10.0 mg, 5.7 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.37 (s), 9.07 (s), 8.57–8.32 (m), 8.30 (d, J = 5.0 Hz), 8.20–8.12 (m), 8.04–7.90 (m), 7.82–7.65 (m), 7.56–7.46 (m), 7.40–7.15 (m), 7.07 (s), 7.02 (s), 6.88–6.62 (m), 6.56–6.29 (m), 5.22–5.00 (m), 4.62–3.57 (m), 3.49–3.48 (m), 3.17–2.77 (m), 2.73 (t, J = 6.6 Hz), 2.67–2.56 (m), 2.54 (s), 2.53–2.52 (m), 2.46–2.30 (m), 2.22–2.08 (m), 2.06–1.95 (m), 1.92–1.74 (m), 1.69–1.44 (m), 1.40–1.21 (m), 1.15–1.06 (m), 0.89–0.80 (m), 0.78 (d, J = 5.5 Hz).
HRMS (ESI) calculated for (C119H191N29O42SZn2+)2+ ([M + 2H]2+): m/z = 1397.1346, experimental 1397.1358, δ [ppm] = 0.8. Calculated for (C119H190N29NaO42SZn2+)2+ ([M + H+Na]2+): m/z = 1408.1256, experimental 1408.1230, δ [ppm] = 2.6. Calculated for (C119H189N29Na2O42SZn2+)2+ ([M + 2Na]2+): m/z = 1419.1165, experimental 1419.1149, δ [ppm] = 1.6. Calculated for (C119H192N29O42SZn2+)3+ ([M + 3H]3+): m/z = 931.7588, experimental 931.7571, δ [ppm] = 1.7.
FpvA 121–139 C-Terminal (PEG)7-Zn2+-DOTAM-OH (17)
17 (31.0 mg, 8.6 μmol, 94%) was prepared following the general procedure for CuAAC with DOTAM 2 (22.3 mg, 22.8 μmol, 2.5 equiv) and peptide 64 (25.0 mg, 9.1 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.42 (s), 9.09–8.80 (m), 8.60 (s), 8.52 (s), 8.42 (s), 8.38–8.27 (m), 8.22 (s), 8.20–8.14 (m), 8.10–7.78 (m), 7.70 (d, J = 7.7 Hz), 7.65 (d, J = 8.5 Hz), 7.60 (d, J = 6.4 Hz), 7.45 (s), 7.30–7.19 (m), 7.16 (s), 6.99 (s), 6.98 (s), 6.84 (d, J = 6.9 Hz), 6.78 (s), 6.69–6.50 (m), 4.52 (q, J = 6.9 Hz), 4.48 (q, J = 6.8 Hz), 4.44 (t, J = 5.1 Hz), 4.42–4.37 (m), 4.36–4.27 (m), 4.24 (q, J = 6.0 Hz), 4.16 (t, J = 7.5 Hz), 4.15–3.94 (m), 3.92 (s), 3.91 (s), 3.82–3.28 (m), 3.27 (q, J = 5.9 Hz), 3.26–3.09 (m), 3.07 (dd, J = 13.3 Hz, 6.9 Hz), 2.72–2.55 (m), 2.54 (s), 2.47–2.34 (m), 2.15–2.07 (m), 2.07 (s), 2.06–1.98 (m), 1.95–1.91 (m), 1.90 (s), 1.87–1.70 (m), 1.67–1.32 (m), 1.30–1.17 (m), 1.15–0.99 (m), 0.87 (d, J = 6.5 Hz), 0.85 (d, J = 6.7 Hz), 0.83–0.75 (m).
HRMS (ESI) calculated for (C153H252N39O56SZn2+)3+ ([M + 3H]3+): m/z = 1209.2352, experimental 1209.2338, δ [ppm] = 1.4. Calculated for (C153H253N39O56SZn2+)4+ ([M + 4H]4+): m/z = 907.1782, experimental 907.1775, δ [ppm] = 0.7. Calculated for (C153H254N39O56SZn2+)5+ ([M + 5H]5+): m/z = 725.9440, experimental 725.9435, δ [ppm] = 0.5.
PfeA 33–51 C-Terminal (PEG)7-Zn2+-DOTAM-OH (18)
18 (34.0 mg, 9.0 μmol, 99%) was prepared following the general procedure for CuAAC with DOTAM 2 (26.7 mg, 27.4 μmol, 2.5 equiv) and peptide 65 (25.0 mg, 9.1 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.37–8.73 (m), 8.30–8.16 (m), 8.08 (d, J = 7.7 Hz), 8.04–7.63 (m), 7.33 (s), 7.28 (d, J = 12.0 Hz), 7.24 (d, J = 9.0 Hz), 7.02 (s), 6.86 (d, J = 6.7 Hz), 6.76 (d, J = 8.9 Hz), 6.74 (s), 6.67–6.53 (m), 4.52–4.46 (m), 4.44 (t, J = 5.0 Hz), 4.40 (s), 4.35 (t, J = 6.8 Hz), 4.26 (dd, J = 13.5 Hz, 7.0 Hz), 4.24–3.96 (m), 3.92 (d, J = 8.1 Hz), 3.88–3.52 (m), 3.50–3.30 (m), 3.28 (q, J = 5.7 Hz), 3.08 (dd, J = 13.2 Hz, 6.8 Hz), 3.03–2.89 (m), 2.75 (t, J = 6.6 Hz), 2.72–2.58 (m), 2.54 (s), 2.45–2.39 (m), 2.31–2.08 (m), 2.03–1.94 (m), 1.90 (s), 1.84–1.18 (m), 1.12–0.98 (m), 0.88 (d, J = 6.9 Hz), 0.86 (d, J = 6.6 Hz), 0.85–0.77 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.9, 174.4, 173.9, 173.9, 173.7, 172.7, 172.4, 172.1, 171.9, 171.6, 171.3, 171.1, 171.0, 170.7, 170.5, 170.1, 170.0, 169.8, 169.7, 168.8, 168.5, 164.1, 150.7, 146.7, 130.4, 123.7, 117.7, 116.8, 115.4, 70.2, 69.8, 69.7, 69.6, 69.5, 68.9, 68.7, 66.5, 66.4, 66.3, 59.2, 58.3, 58.3, 58.2, 58.0, 56.8, 55.7, 55.7, 52.8, 52.7, 52.7, 52.5, 52.0, 51.2, 49.4, 48.6, 42.0, 40.8, 40.4, 40.0, 38.1, 38.1, 36.8, 31.5, 31.5, 31.4, 30.7, 30.7, 30.5, 30.4, 30.2, 29.0, 27.6, 27.5, 27.4, 27.3, 27.3, 27.3, 26.6, 26.6, 24.4, 24.1, 23.1, 22.8, 22.0, 21.5, 21.2, 19.7, 19.6, 19.5, 19.2, 18.9, 18.4, 18.2, 17.9, 17.7, 17.3, 15.2, 11.1.
HRMS (ESI) calculated for (C162H266N41O58Zn2+)3+ ([M + 3H]3+): m/z = 1259.2797, experimental 1259.2767, δ [ppm] = 3.0. Calculated for (C162H267N41O58Zn2+)4+ ([M + 4H]4+): m/z = 944.7116, experimental 944.7091, δ [ppm] = 1.5. Calculated for (C162H268N41O58Zn2+)5+ ([M + 5H]5+): m/z = 755.9707, experimental 755.9697, δ [ppm] = 1.0. Calculated for (C162H269N41O58Zn2+)6+ ([M + 6H]6+): m/z = 630.1435, experimental 630.1424, δ [ppm] = 1.1.
HasR 122–144 C-Termainl (PEG)7-Zn2+-DOTAM-OH (19)
19 (42.0 mg, 10.1 μmol, 79%) was prepared following the general procedure for CuAAC with DOTAM 2 (31.1 mg, 31.9 μmol, 2.5 equiv) and peptide 66 (40.0 mg, 12.8 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.53 (s), 9.45 (s), 8.91 (s), 8.53–8.39 (m), 8.34–8.24 (m), 8.22 (s), 8.05 (dd, J = 13.4 Hz, 5.9 Hz), 8.02–7.78 (m), 7.76–7.66 (m), 7.57 (d, J = 7.5 Hz), 7.40–7.28 (m), 7.25–7.17 (m), 6.98 (s), 6.82 (t, J = 6.6 Hz), 6.75 (s), 6.71 (s), 6.55–6.46 (m), 4.55 (dd, J = 12.6 Hz, 6.2 Hz), 4.48 (dd, J = 11.7 Hz, 5.1 Hz), 4.44 (t, J = 5.2 Hz), 4.42–4.12 (m), 4.06 (dd, J = 13.5 Hz, 8.4 Hz), 4.03 (t, J = 7.5 Hz), 3.93 (s), 3.92 (s), 3.80–3.43 (m), 3.28–2.84 (m), 2.71–2.58 (m), 2.54 (s), 2.48–2.32 (m), 2.19–2.01 (m), 1.98–1.35 (m), 1.31–1.00 (m), 0.87 (d, J = 6.7 Hz), 0.85–0.73 (m).
HRMS (ESI) calculated for (C179H299N49O59SZn2+)4+ ([M + 4H]4+): m/z = 1043.7721, experimental 1043.7742, δ [ppm] = 2.1. Calculated for (C179H300N49O59SZn2+)5+ ([M + 5H]5+): m/z = 835.2191, experimental 835.2201, δ [ppm] = 1.0. Calculated for (C179H301N49O59SZn2+)6+ ([M + 6H]6+): m/z = 696.1838, experimental 696.1848, δ [ppm] = 1.0.
PfeA 33–51 C-Terminal (PEG)7-Zn2+-DOTAM-OAc (20)
20 (18.0 mg, 4.5 μmol, 82%) was prepared following the general procedure for CuAAC with DOTAM 1 (16.8 mg, 13.7 μmol, 2.5 equiv) and peptide 65 (15.0 mg, 5.5 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 8.30 (s), 8.23–8.15 (m), 8.13–7.96 (m), 7.95–7.77 (m), 7.75–7.63 (m), 7.61–7.55 (m), 7.54–7.49 (m), 7.40–7.27 (m), 7.25 (s), 7.01 (s), 6.76 (s), 6.74 (s), 4.58–3.93 (m), 3.92 (d, J = 8.0 Hz), 3.82–3.67 (m), 3.53–3.43 (m), 3.27 (q, J = 6.1 Hz), 3.07 (dd, J = 13.3 Hz, 6.8 Hz), 2.98 (dd, J = 12.7 Hz, 6.6 Hz), 2.61–2.52 (m), 2.47–2.39 (m), 2.29–2.08 (m), 2.02–1.92 (m), 1.91 (s), 1.90–1.86 (m), 1.86 (s), 1.85–1.79 (m), 1.78 (s), 1.77–1.19 (m), 1.15 (s), 1.04 (dd, J = 11.0 Hz, 5.9 Hz), 1.02 (d, J = 6.2 Hz), 0.94 (s), 0.88 (d, J = 6.9 Hz), 0.86 (d, J = 6.6 Hz), 0.85–0.76 (m).
HRMS (ESI) calculated for (C174H278N41O64Zn2+)3+ ([M + 3H]3+): m/z = 1343.3008, experimental 1343.2967, δ [ppm] = 4.1. Calculated for (C174H279N41O64Zn2+)4+ ([M + 4H]4+): m/z = 1007.7274, experimental 1007.7240, δ [ppm] = 3.4. Calculated for (C174H280N41O64Zn2+)5+ ([M + 5H]5+): m/z = 806.3834, experimental 806.3806, δ [ppm] = 2.8. Calculated for (C174H281N41O64Zn2+)6+ ([M + 6H]6+): m/z = 672.1540, experimental 672.1522, δ [ppm] = 1.8. Calculated for (C174H282N41O64Zn2+)7+ ([M + 7H]7+): m/z = 576.2759, experimental 576.2750, δ [ppm] = 0.9.
PfeA 33–51 C-Terminal Disulfide-(PEG)5-Zn2+-DOTAM-OH (21)
21 was prepared via an adaptation of an established procedure reported by Song, Yang, Hall, Gurnani, and Perrier,67 which is a modified version of the procedures for disulfide bond formation by Bernatowicz, Matsueda, and Matsueda.68
52 (8.2 mg, 5.687 μmol, 1.0 equiv) was dissolved in DMF (1 mL) and HEPES buffer (1.0 M, 1 mL). 70 (15 mg, 6.256 μmol, 1.1 equiv) dissolved in DMF (1 mL), DMSO (1 mL), and HEPES buffer (1.0 M, 1 mL) was added to the solution. The reaction mixture was stirred at 23 °C for 72 h, and the reaction progress was controlled by LCMS. The reaction mixture was concentrated by rotary evaporation and filtered after completeness. The remains in the filter were washed with ACN (20 mL), and the solvent was removed by rotary evaporation. The residue was taken up, diluted with ACN/Milli-Q H2O, and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 5–45% ACN/Milli-Q H2O, 0.1% HCOOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield the product 21 as a white solid. Yield 16.0 mg (4.298 mmol, 76%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.60 (s), 8.47–8.36 (m), 8.29 (s), 8.27 (s), 8.23–8.00 (m), 7.97–7.54 (m), 7.42–7.12 (m), 7.02 (s), 6.84–6.79 (m), 6.77 (s), 6.73 (s), 6.54–6.42 (m), 4.50–3.95 (m), 3.80–3.70 (m), 3.48 (d, J = 3.8 Hz), 3.46–3.21 (m), 3.19 (q, J = 5.6 Hz), 3.06 (t, J = 7.1 Hz), 3.03–2.89 (m), 2.87 (t, J = 7.2 Hz), 2.79–2.70 (m), 2.66 (t, J = 7.4 Hz), 2.64–2.54 (m), 2.54 (s), 2.53–2.51 (m), 2.48–2.39 (m), 2.36–2.08 (m), 2.06–1.19 (m), 1.17 (d, J = 6.7 Hz), 1.14 (s), 1.09–0.98 (m), 0.88 (d, J = 6.9 Hz), 0.86 (d, J = 6.7 Hz), 0.85–0.77 (m).
HRMS (ESI) calculated for (C158H258N41O54S2Zn2+)3+ ([M + 3H]3+): m/z = 1240.5803, experimental 1240.5800, δ [ppm] = 0.3. Calculated for (C158H259N41O54S2Zn2+)4+ ([M + 4H]4+): m/z = 930.6870, experimental 930.6872, δ [ppm] = 0.2. Calculated for (C158H260N41O54S2Zn2+)5+ ([M + 5H]5+): m/z = 744.7511, experimental 744.7511, δ [ppm] = 0.0. Calculated for (C158H261N41O54S2Zn2+)6+ ([M + 6H]6+): m/z = 620.7938, experimental 620.7938, δ [ppm] = 0.0.
General Procedure for CuAAC with MECAM 3 or 4
Compounds 22–32 were prepared via an adaptation of an established procedure reported by Pinkert et al.333 or 4 (2.5 equiv) was dissolved under an argon atmosphere in DMSO (1 mL) and Milli-Q H2O (1 mL). In the case where 3 was employed as the starting material, acetic acid (1%, 40 μL) was added to the solution to prevent deacetylation. PEG-modified peptide (61-70) (1.0 equiv) dissolved in DMSO (1 mL) was added to the solution. CuSO4 (1.0 equiv) dissolved in PBS (0.3 mL) and sodium ascorbate (2.0 equiv) dissolved in PBS (0.4 mL) was added to the CuSO4 solution, whereupon a white solid precipitated immediately. THPTA (0.5 equiv) dissolved in PBS (0.3 mL) was added to the suspension. The suspension was added to the reaction under an argon atmosphere. The reaction mixture was stirred at 23 °C for 1 h, and the reaction progress was controlled by LCMS. After the completion of the reaction, the solution was filtered over a cotton wool and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 5–45% ACN/Milli-Q H2O, 0.1% HCOOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield products 22–32 as white solids.
FpvA 121–139 N-Terminal (PEG)7-MECAM-OH (22)
22 (36.0 mg, 11.0 μmol, 95%) was prepared following the general procedure for CuAAC with MECAM 4 (19.8 mg, 29.0 μmol, 2.5 equiv) and peptide 61 (30.0 mg, 11.6 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.59 (s), 9.38 (t, J = 5.3 Hz), 9.23 (s), 8.49–8.12 (m), 8.08–7.99 (m), 7.96 (d, J = 6.4 Hz), 7.93 (d, J = 6.7 Hz), 7.88 (d, J = 6.0 Hz), 7.83 (s), 7.81 (dd, J = 14.8 Hz, 8.0 Hz), 7.72–7.50 (m), 7.46 (s), 7.28 (d, J = 7.9 Hz), 7.23 (s), 7.21 (s), 7.19 (dd, J = 8.2 Hz, 1.2 Hz), 7.03 (s), 6.99 (s), 6.91 (d, J = 7.7 Hz), 6.88 (d, J = 7.7 Hz), 6.82–6.74 (m), 6.65 (t, J = 7.9 Hz), 6.59 (t, J = 7.9 Hz), 5.08 (bs), 4.99 (bs), 4.63 (dd, J = 14.1 Hz, 6.7 Hz), 4.60–4.47 (m), 4.46 (t, J = 5.3 Hz), 4.43 (s), 4.40 (d, J = 5.8 Hz), 4.39–3.95 (m), 3.94 (d, J = 5.5 Hz), 3.92 (t, J = 4.9 Hz), 3.79 (t, J = 5.3 Hz), 3.77–3.53 (m), 3.52–3.41 (m), 3.28–3.23 (m), 2.75 (bs), 2.69 (t, J = 7.6 Hz), 2.67–2.51 (m), 2.47–2.34 (m), 2.27–2.22 (m), 2.15–2.02 (m), 1.99–1.87 (m), 1.84–1.70 (m), 1.68–1.58 (m), 1.56–1.39 (m), 1.37–1.26 (m), 1.23 (d, J = 6.8 Hz), 1.22–0.97 (m), 0.87 (d, J = 6.5 Hz), 0.85 (d, J = 6.4 Hz), 0.84–0.76 (m).
HRMS (ESI) calculated for C143H225N32O53S3+ ([M + 3H]3+): m/z = 1090.1866, experimental 1090.1872, δ [ppm] = 0.6. Calculated for C143H226N32O53S4+ ([M + 4H]4+): m/z = 817.8918, experimental 817.8917, δ [ppm] = 0.1.
PfeA 33–51 N-Terminal (PEG)7-MECAM-OH (23)
23 (36.2 mg, 10.6 μmol, 97%) was prepared following the general procedure for CuAAC with MECAM 4 (18.7 mg, 27.4 μmol, 2.5 equiv) and peptide 62 (30.0 mg, 11.0 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.62 (s), 9.42 (t, J = 5.3 Hz), 9.28 (s), 8.22 (s), 8.20–8.16 (m,), 8.10 (t, J = 6.2 Hz), 8.05 (t, J = 5.6 Hz), 8.04–7.94 (m), 7.92 (d, J = 7.0 Hz), 7.88 (d, J = 8.8 Hz), 7.86–7.80 (m), 7.75–7.67 (m), 7.41–7.33 (m), 7.31–7.24 (m), 7.21 (s), 7.20 (dd, J = 6.9 Hz, 1.2 Hz), 7.06 (s), 6.89 (dd, J = 7.7 Hz, 0.8 Hz), 6.87 (dd, J = 7.8 Hz, 1.0 Hz), 6.80–6.73 (m), 6.64 (t, J = 7.9 Hz), 6.58 (t, J = 7.9 Hz), 4.46 (t, J = 5.3 Hz), 4.43 (s), 4.41 (d, J = 5.7 Hz), 4.37–4.32 (m), 4.31–4.02 (m), 4.00 (d, J = 7.4 Hz), 3.99–3.98 (m), 3.95 (d, J = 3.9 Hz), 3.93 (s), 3.79 (t, J = 5.3 Hz), 3.76–3.58 (m), 3.52–3.44 (m), 3.43 (t, J = 6.0 Hz), 3.26 (q, J = 5.9 Hz), 2.74 (t, J = 6.9 Hz), 2.69 (t, J = 7.6 Hz), 2.64–2.58 (m), 2.54 (s), 2.48–2.43 (m), 2.41–2.33 (m), 2.28–2.10 (m), 2.08 (s), 2.03–1.35 (m), 1.33 (s), 1.30 (s), 1.28–1.13 (m), 1.09–0.99 (m), 0.89–0.68 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 173.9, 170.5, 169.7, 163.4, 153.4, 149.8, 149.8, 146.3, 140.7, 140.7, 135.9, 135.8, 133.5, 132.0, 126.9, 125.5, 125.5, 125.2, 124.8, 124.8, 124.6, 124.4, 124.3, 124.3, 123.3, 122.3, 119.0, 118.9, 118.7, 117.9, 117.9, 117.4, 115.2, 70.3, 69.8, 69.8, 69.6, 69.6, 68.9, 68.8, 66.4, 40.4, 40.0, 31.3, 31.2, 30.7, 30.4, 29.8, 29.0, 28.7, 26.5, 25.2, 22.1, 19.2, 14.0, 11.0.
HRMS (ESI) calculated for C152H239N34O553+ ([M + 3H]3+): m/z = 1140.2311, experimental 1140.2301, δ [ppm] = 1.0. Calculated for C152H240N34O554+ ([M + 4H]4+): m/z = 855.4252, experimental 855.4255, δ [ppm] = 0.3. Calculated for C152H241N34O555+ ([M + 5H]5+): m/z = 684.5416, experimental 684.5425, δ [ppm] = 0.9.
HasR 122–144 N-Terminal (PEG)7-MECAM-OH (24)
24 (21.0 mg, 5.5 μmol, 86%) was prepared following the general procedure for CuAAC with MECAM 4 (10.9 mg, 16.0 μmol, 2.5 equiv) and peptide 63 (20.0 mg, 6.4 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.68 (s), 9.55 (s), 9.44 (s), 8.50 (s), 8.31 (s), 8.27 (s), 8.16 (d, J = 6.5 Hz), 8.13–7.85 (m), 7.84 (s), 7.83–7.28 (m), 7.26 (dd, J = 8.1 Hz, 1.2 Hz), 7.22 (s), 7.19 (dd, J = 8.1 Hz, 1.4 Hz), 7.05–6.94 (m), 6.88 (dd, J = 7.4 Hz, 0.7 Hz), 6.85 (dd, J = 8.7 Hz, 1.0 Hz), 6.75 (s), 6.71 (s), 6.60 (t, J = 7.9 Hz), 6.56 (t, J = 7.9 Hz), 4.58–4.52 (m), 4.46 (t, J = 5.3 Hz), 4.44–4.02 (m), 4.00 (s), 3.97 (s), 3.96–3.90 (m), 3.79 (t, J = 5.3 Hz), 3.77–3.54 (m), 3.52–3.40 (m), 3.26 (q, J = 5.8 Hz), 3.17–2.95 (m), 2.78 (t, J = 2.7 Hz), 2.75 (s), 2.70 (t, J = 7.6 Hz), 2.68–2.59 (m), 2.54 (s), 2.47 (t, J = 7.5 Hz), 2.45–2.30 (m), 2.29 (t, J = 7.4 Hz), 2.27–2.20 (m), 2.18 (td, J = 7.1 Hz, 2.6 Hz), 2.16–2.07 (m), 2.06–2.01 (m), 2.00 (s), 1.99–1.93 (m), 1.90–1.25 (m), 1.24–1.17 (m), 1.14–1.00 (m), 0.86 (d, J = 6.5 Hz), 0.84–0.73 (m).
HRMS (ESI) calculated for C169H271N42O56S3+ ([M + 3H]3+): m/z = 1272.3118, experimental 1272.3114, δ [ppm] = 0.4. Calculated for C169H272N42O56S4+ ([M + 4H]4+): m/z = 954.4857, experimental 954.4850, δ [ppm] = 0.7. Calculated for C169H273N42O56S5+ ([M + 5H]5+): m/z = 763.7900, experimental 763.7890, δ [ppm] = 1.0. Calculated for C169H274N42O56S6+ ([M + 6H]6+): m/z = 636.6595, experimental 636.6587, δ [ppm] = 0.8.
FpvA 124–134 N-Terminal (PEG)7-MECAM-OH (25)
25 (22.0 mg, 8.9 μmol, 88%) was prepared following the general procedure for CuAAC with MECAM 4 (17.2 mg, 25.2 μmol, 2.5 equiv) and peptide 67 (18.0 mg, 10.1 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.62 (s), 9.43 (s), 9.29 (s), 8.37 (s), 8.33 (d, J = 7.5 Hz), 8.21 (s), 8.18–8.10 (m), 8.06 (t, J = 5.4 Hz), 7.85–7.78 (m), 7.83 (s), 7.73 (d, J = 7.9 Hz), 7.45 (s), 7.27 (dd, J = 8.2 Hz, 1.1 Hz), 7.22 (s), 7.21 (s), 7.19 (dd, J = 8.2 Hz, 1.2 Hz), 7.08 (s), 6.99 (s), 6.89 (dd, J = 7.8 Hz, 1.2 Hz), 6.86 (dd, J = 7.8 Hz, 1.3 Hz), 6.63 (t, J = 7.9 Hz), 6.58 (t, J = 7.9 Hz), 4.53 (dd, J = 14.2 Hz, 7.3 Hz), 4.49 (dd, J = 13.8 Hz, 6.7 Hz), 4.46 (t, J = 5.3 Hz), 4.43 (s), 4.41 (d, J = 5.8 Hz), 4.39–4.34 (m), 4.32–4.26 (m), 4.25–4.18 (m), 4.14 (dd, J = 15.1 Hz, 7.7 Hz), 4.12 (dd, J = 7.9 Hz, 4.2 Hz), 4.07 (td, J = 9.4 Hz, 4.4 Hz), 4.04–4.03 (m), 4.01 (d, J = 3.2 Hz), 4.00–3.98 (m), 3.96 (d, J = 6.8 Hz), 3.94 (s), 3.79 (t, J = 5.3 Hz), 3.75–3.69 (m), 3.67–3.61 (m), 3.56–3.44 (m), 3.43 (t, J = 6.1 Hz), 3.26 (q, J = 5.9 Hz), 2.75 (t, J = 7.1 Hz), 2.69 (t, J = 7.6 Hz), 2.65–2.51 (m), 2.48–2.35 (m), 2.01 (s), 1.99–1.88 (m), 1.85–1.70 (m), 1.65–1.58 (m), 1.56–1.39 (m), 1.36–1.28 (m), 1.26 (d, J = 7.0 Hz), 1.23–1.19 (m), 1.15–1.07 (m), 1.04 (d, J = 6.4 Hz), 1.02 (d, J = 6.3 Hz), 0.88 (d, J = 6.6 Hz), 0.87–0.77 (m).
HRMS (ESI) calculated for C110H170N23O39S3+ ([M + 3H]3+): m/z = 823.0577, experimental 823.0600, δ [ppm] = 2.3. Calculated for C110H171N23O39S4+ ([M + 4H]4+): m/z = 617.5451, experimental 617.5460, δ [ppm] = 0.9.
PfeA 37–46 N-Terminal (PEG)7-MECAM-OH (26)
26 (27.0 mg, 11.5 μmol, 96%) was prepared following the general procedure for CuAAC with MECAM 4 (20.5 mg, 30.0 μmol, 2.5 equiv) and peptide 68 (20.0 mg, 12.0 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.67 (s), 9.53 (s), 9.42 (s), 8.26 (t, J = 5.6 Hz), 8.17 (d, J = 7.3 Hz), 8.12–8.06 (m), 7.97 (d, J = 6.9 Hz), 7.93 (d, J = 6.4 Hz), 7.87 (d, J = 7.3 Hz), 7.84 (s), 7.74 (d, J = 7.7 Hz), 7.68 (d, J = 8.0 Hz), 7.30 (s), 7.27 (s), 7.26 (s), 7.25 (s), 7.22 (s), 7.19 (d, J = 8.0 Hz), 7.06 (s), 6.87 (d, J = 7.7 Hz), 6.85 (d, J = 7.8 Hz), 6.78 (s), 6.76 (s), 6.61 (t, J = 7.8 Hz), 6.56 (t, J = 7.9 Hz), 4.46 (t, J = 5.3 Hz), 4.43 (s), 4.41 (d, J = 5.7 Hz), 4.38–4.32 (m), 4.31–4.19 (m), 4.17–4.10 (m), 4.09 (t, J = 7.5 Hz), 4.04–3.99 (m), 3.98 (s), 3.96 (s), 3.79 (t, J = 5.5 Hz), 3.50–3.44 (m), 3.43 (t, J = 6.0 Hz), 3.27 (q, J = 5.9 Hz), 2.74 (t, J = 7.3 Hz), 2.70 (t, J = 7.6 Hz), 2.47 (t, J = 7.4 Hz), 2.23–2.07 (m), 2.02–1.85 (m), 1.79–1.63 (m), 1.57–1.46 (m), 1.35–1.25 (m), 1.23 (d, J = 7.2 Hz), 1.21 (d, J = 7.1 Hz), 1.03 (d, J = 6.3 Hz), 1.01 (d, J = 6.3 Hz), 0.83 (dd, J = 11.2 Hz, 6.5 Hz).
HRMS (ESI) calculated for C105H159N23O382+ ([M + 2H]2+): m/z = 1175.0603, experimental 1175.0617, δ [ppm] = 1.4. Calculated for C105H158N23NaO382+ ([M + H+Na]2+): m/z = 1186.0512, experimental 1186.0523, δ [ppm] = 1.1. Calculated for C105H157N23NaO382+ ([M + 2Na]2+): m/z = 1197.0422, experimental 1197.0430, δ [ppm] = 0.8. Calculated for C105H160N23O383+ ([M + 3H]3+): m/z = 783.7093, experimental 783.7104, δ [ppm] = 1.1.
HasR 129–138 N-Termianl (PEG)7-MECAM-OH (27)
27 (12.0 mg, 4.9 μmol, 86%) was prepared following the general procedure for CuAAC with MECAM 4 (9.7 mg, 14.3 μmol, 2.5 equiv) and peptide 69 (10.0 mg, 5.7 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.61 (s), 9.41 (t, J = 5.7 Hz), 9.26 (t, J = 7.0 Hz), 8.37 (s), 8.29 (s), 8.28 (s), 8.22 (bs), 8.09 (t, J = 5.2 Hz), 7.96 (d, J = 6.6 Hz), 7.83 (s), 7.76 (d, J = 7.7 Hz), 7.57 (d, J = 6.9 Hz), 7.41 (s), 7.38 (s), 7.27 (dd, J = 8.1 Hz, 1.0 Hz), 7.21 (s), 7.19 (dd, J = 8.2 Hz, 1.2 Hz), 7.10 (s), 7.04 (s), 6.90 (dd, J = 7.8 Hz, 0.9 Hz), 6.87 (dd, J = 7.8 Hz, 1.0 Hz), 6.76 (s), 6.73 (s), 6.64 (t, J = 7.9 Hz), 6.58 (t, J = 7.9 Hz), 4.78 (d, J = 8.3 Hz), 4.59–4.52 (m), 4.46 (t, J = 5.3 Hz), 4.43 (bs), 4.41 (d, J = 5.7 Hz), 4.38–4.30 (m), 4.25–4.18 (m), 4.17–4.10 (m), 4.09 (t, J = 7.2 Hz), 4.01 (t, J = 7.6 Hz), 3.99 (s), 3.97 (s), 3.95 (s), 3.93 (s), 3.79 (t, J = 5.3 Hz), 3.69 (t, J = 6.0 Hz), 3.66–3.55 (m), 3.54–3.44 (m), 3.43 (t, J = 6.1 Hz), 3.28–3.26 (m), 2.77–2.73 (m), 2.69 (t, J = 7.6 Hz), 2.63–2.52 (m), 2.46 (t, J = 7.5 Hz), 2.44–2.36 (m), 2.21–2.08 (m), 2.04 (dd, J = 13.7 Hz, 7.2 Hz), 2.01 (s), 2.00 (s), 1.94 (p, J = 7.6 Hz), 1.91–1.72 (m), 1.70–1.63 (m), 1.61–1.43 (m), 1.40–1.26 (m), 1.23 (s), 1.14 (s), 1.13 (d, J = 2.3 Hz), 1.12 (s), 1.11 (s), 1.10–1.06 (m), 0.90–0.82 (m), 0.80 (d, J = 6.4 Hz).
HRMS (ESI) calculated for C109H164N22O39S2+ ([M + 2H]2+): m/z = 1218.5618, experimental 1218.5614, δ [ppm] = 0.4. Calculated for C109H163N22NaO39S2+ ([M + H+Na]2+): m/z = 1229.5528, experimental 1229.5523, δ [ppm] = 0.5. Calculated for C109H165N22O39S3+ ([M + 3H]3+): m/z = 812.7103, experimental 812.7104, δ [ppm] = 0.1. Calculated for C109H166N22O39S4+ ([M + 4H]4+): m/z = 609.7845, experimental 609.7845, δ [ppm] = 0.0.
FpvA 121–139 C-Terminal (PEG)7-MECAM-OH (28)
28 (18.0 mg, 5.5 μmol, 95%) was prepared following the general procedure for CuAAC with MECAM 4 (9.9 mg, 14.5 μmol, 2.5 equiv) and peptide 64 (15.0 mg, 5.8 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 12.53 (bs), 9.55 (s), 9.33 (t, J = 6.0 Hz), 9.16 (t, J = 5.7 H), 8.57 (d, J = 5.7 Hz), 8.31 (d, J = 6.4 Hz), 8.16 (d, J = 7.5 Hz), 8.11 (t, J = 5.2 Hz), 8.07–8.00 (m), 7.95 (t, J = 8.9 Hz), 7.91 (d, J = 7.8 Hz), 7.89 (d, J = 8.0 Hz), 7.88–7.84 (m), 7.83 (s), 7.81–7.77 (m), 7.68 (d, J = 8.1 Hz), 7.61 (d, J = 8.0 Hz), 7.44 (s), 7.28 (dd, J = 8.2 Hz, 1.3 Hz), 7.21 (s), 7.20 (s), 7.19 (dd, J = 8.3 Hz, 1.4 Hz), 7.17 (s), 6.99 (s), 6.98 (s), 6.91 (dd, J = 7.8 Hz, 1.3 Hz), 6.88 (dd, J = 7.8 Hz, 1.4 Hz), 6.78 (s), 6.66 (t, J = 7.9 Hz), 6.60 (t, J = 7.9 Hz), 5.09 (s), 4.96 (s), 4.57–4.50 (m), 4.46 (t, J = 5.3 Hz), 4.44–4.38 (m), 4.36 (t, J = 7.0 Hz), 4.34–4.29 (m), 4.28–4.09 (m), 4.06–4.02 (m), 4.00–3.94 (m), 3.93 (s), 3.91 (s), 3.81 (d, J = 6.0 Hz), 3.79 (t, J = 5.3 Hz), 3.74–3.69 (m), 3.67 (d, J = 5.9 Hz), 3.65–3.59 (m), 3.56–3.53 (m), 3.51–3.43 (m), 3.27 (q, J = 6.0 Hz), 3.08 (dd, J = 13.3 Hz, 7.0 Hz), 2.71 (s), 2.69 (t, J = 7.6 Hz), 2.62–2.52 (m), 2.48–2.35 (m), 2.18 (t, J = 7.4 Hz), 2.13–2.07 (m), 2.06–2.01 (m), 2.01 (s), 2.00–1.87 (m), 1.81–1.70 (m), 1.67–1.57 (m), 1.55–1.35 (m), 1.32–1.18 (m), 1.15–1.05 (m), 1.02 (d, J = 6.4 Hz), 1.01 (dd, J = 6.3 Hz, 1.3 Hz), 0.87 (d, J = 6.6 Hz), 0.86 (d, J = 6.6 Hz), 0.84–0.75 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.1, 173.5, 172.5, 172.4, 172.4, 172.3, 171.9, 171.2, 171.0, 170.1, 170.0, 170.0, 169.9, 169.8, 169.7, 168.9, 168.8, 168.5, 168.4, 149.7, 149.6, 146.3, 146.2, 146.2, 137.5, 135.9, 132.2, 124.8, 122.3, 118.9, 118.8, 118.8, 118.0, 117.9, 117.3, 117.1, 115.1, 113.3, 70.3, 70.3, 70.2, 69.8, 69.7, 69.6, 69.6, 69.6, 68.9, 68.8, 57.8, 57.2, 55.0, 52.7, 52.4, 51.8, 51.5, 51.3, 49.2, 42.2, 42.1, 42.0, 40.5, 40.0, 39.0, 38.2, 38.0, 36.5, 36.3, 34.8, 32.2, 31.5, 29.3, 28.9, 27.6, 25.2, 24.8, 24.3, 24.2, 24.1, 24.0, 23.1, 23.0, 22.8, 21.5, 19.7, 19.3, 19.3, 19.1, 18.1, 17.8, 15.4, 15.3, 14.6, 11.3, 10.9.
HRMS (ESI) calculated for C143H225N32O53S3+ ([M + 3H]3+): m/z = 1090.1866, experimental 1090.1870, δ [ppm] = 0.4. Calculated for C143H226N32O53S4+ ([M + 4H]4+): m/z = 817.8918, experimental 817.8931, δ [ppm] = 1.4.
PfeA 33–51 C-Terminal (PEG)7-MECAM-OH (29)
29 (36.0 mg, 10.5 μmol, 96%) was prepared following the general procedure for CuAAC with MECAM 4 (18.7 mg, 27.4 μmol, 2.5 equiv) and peptide 65 (30.0 mg, 11.0 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.62 (s), 9.44 (t, J = 5.4 Hz), 9.31 (t, J = 5.7 Hz), 8.25 (d, J = 7.0 Hz), 8.23–8.15 (m), 8.12–8.09 (m), 8.07 (d, J = 6.6 Hz), 8.05 (dd, J = 12.4 Hz, 6.0 Hz), 7.99 (t, J = 5.0 Hz), 7.95 (d, J = 6.6 Hz), 7.92 (d, J = 8.0 Hz), 7.84 (bs), 7.84 (s), 7.78 (d, J = 7.4 Hz), 7.70 (d, J = 7.5 Hz), 7.31 (s), 7.29 (s), 7.27 (dd, J = 8.1 Hz, 1.4 Hz), 7.23 (bs), 7.21 (s), 7.19 (dd, J = 8.2 Hz, 1.4 Hz), 7.01 (s), 6.89 (dd, J = 7.8 Hz, 1.3 Hz), 6.86 (dd, J = 7.8 Hz, 1.4 Hz), 6.77 (d, J = 6.0 Hz), 6.75 (s), 6.63 (t, J = 7.9 Hz), 6.58 (t, J = 7.9 Hz), 4.46 (t, J = 5.4 Hz), 4.43 (bs), 4.41 (d, J = 5.8 Hz), 4.35 (p, J = 6.9 Hz), 4.31–4.22 (m), 4.19–3.97 (m), 3.93 (s), 3.91 (s), 3.79 (t, J = 5.4 Hz), 3.76 (d, J = 5.4 Hz), 3.74 (d, J = 5.0 Hz), 3.69 (d, J = 5.4 Hz), 3.67 (d, J = 5.3 Hz), 3.53–3.45 (m), 3.43 (t, J = 6.0 Hz), 3.28 (q, J = 6.0 Hz), 3.08 (dd, J = 13.3 Hz, 7.0 Hz), 2.75 (t, J = 6.8 Hz), 2.70 (t, J = 7.6 Hz), 2.46 (t, J = 7.5 Hz), 2.30–2.04 (m), 2.02–1.85 (m), 1.84–1.35 (m), 1.34–1.26 (m), 1.24 (d, J = 7.1 Hz), 1.23 (d, J = 6.9 Hz), 1.07–0.99 (m), 0.88 (d, J = 6.9 Hz), 0.86 (d, J = 6.6 Hz), 0.85–0.79 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.9, 174.9, 174.9, 174.5, 174.4, 173.9, 173.9, 173.9, 173.6, 172.8, 172.8, 172.8, 172.7, 172.3, 171.9, 171.8, 171.6, 171.6, 171.5, 171.2, 171.1, 171.0, 170.8, 170.7, 170.4, 170.2, 170.1, 169.6, 169.6, 168.8, 168.8, 168.5, 150.0, 150.0, 146.4, 146.4, 146.3, 137.5, 135.9, 132.2, 124.8, 122.3, 118.5, 117.6, 117.5, 117.3, 115.3, 70.2, 70.2, 69.8, 69.7, 69.6, 69.6, 68.9, 68.8, 66.5, 66.4, 66.3, 58.5, 56.7, 53.0, 52.9, 52.8, 52.7, 52.5, 52.0, 52.0, 51.1, 49.2, 48.8, 48.6, 42.1, 42.0, 40.8, 40.0, 39.0, 38.3, 38.1, 38.1, 36.8, 34.8, 31.6, 31.5, 31.5, 31.4, 31.3, 31.1, 30.6, 30.6, 30.5, 30.5, 30.5, 30.5, 30.2, 29.0, 27.6, 27.6, 27.6, 27.5, 27.4, 27.3, 27.2, 26.6, 25.2, 24.8, 24.4, 24.0, 23.1, 22.8, 22.1, 21.9, 21.5, 19.7, 19.6, 19.5, 19.2, 18.9, 18.3, 18.1, 17.9, 17.7, 17.2, 15.2, 11.1.
HRMS (ESI) calculated for C152H239N34O553+ ([M + 3H]3+): m/z = 1140.2311, experimental 1140.2310, δ [ppm] = 0.1. Calculated for C152H240N34O554+ ([M + 4H]4+): m/z = 855.4252, experimental 855.4253, δ [ppm] = 0.1. Calculated for C152H241N34O555+ ([M + 5H]5+): m/z = 684.5416, experimental 684.5415, δ [ppm] = 0.1.
HasR 122–144 C-Terminal (PEG)7-MECAM-OH (30)
30 (31.0 mg, 8.1 μmol, 85%) was prepared following the general procedure for CuAAC with MECAM 4 (16.3 mg, 23.9 μmol, 2.5 equiv) and peptide 66 (30.0 mg, 9.6 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.67 (bs), 9.54 (bs), 9.43 (bs), 8.49 (bs), 8.32 (bs), 8.28 (s), 8.25–8.06 (m), 8.05 (d, J = 5.4 Hz), 8.03–7.85 (m), 7.84 (s), 7.82 (s), 7.81–7.50 (m), 7.47–7.28 (m), 7.26 (dd, J = 8.1 Hz, 1.2 Hz), 7.22 (s), 7.19 (dd, J = 8.2 Hz, 1.4 Hz), 6.98 (s), 6.87 (dd, J = 7.7 Hz, 0.8 Hz), 6.85 (dd, J = 7.7 Hz, 1.2 Hz), 6.75 (s), 6.71 (s), 6.60 (t, J = 7.9 Hz), 6.56 (t, J = 7.9 Hz), 5.05 (bs), 4.78 (d, J = 8.2 Hz), 4.54 (d, J = 6.6 Hz), 4.49 (d, J = 5.5 Hz), 4.46 (t, J = 5.3 Hz), 4.43 (d, J = 5.7 Hz), 4.41 (d, J = 6.0 Hz), 4.35 (d, J = 4.9 Hz), 4.33–4.10 (m), 4.07 (dd, J = 13.2 Hz, 8.7 Hz), 4.04–3.97 (m), 3.93 (s), 3.92 (s), 3.79 (t, J = 5.3 Hz), 3.72–3.53 (m), 3.52–3.42 (m), 3.29–3.27 (m), 3.17–3.01 (m), 2.78 (t, J = 2.7 Hz), 2.70 (t, J = 7.6 Hz), 2.64–2.58 (m), 2.54 (s), 2.48–2.33 (m), 2.29 (t, J = 7.4 Hz), 2.25–2.20 (m), 2.18 (td, J = 7.1 Hz, 2.7 Hz), 2.15–2.08 (m), 2.07–2.01 (m), 2.00 (s), 2.00–1.36 (m), 1.34–1.14 (m), 1.13–1.00 (m), 0.89–0.74 (m)
HRMS (ESI) calculated for C169H271N42O56S3+ ([M + 3H]3+): m/z = 1272.3118, experimental 1272.3127, δ [ppm] = 0.9. Calculated for C169H272N42O56S4+ ([M + 4H]4+): m/z = 954.4857, experimental 954.4871, δ [ppm] = 1.4. Calculated for C169H273N42O56S5+ ([M + 5H]5+): m/z = 763.7900, experimental 763.7907, δ [ppm] = 0.7.
PfeA 33–51 C-Terminal (PEG)7-MECAM-OAc (31)
31 (16.0 mg (4.4 μmol, 80%) was prepared following the general procedure with MECAM 3 (12.8 mg, 13.7 μmol, 2.5 equiv) and 65 (15.0 mg, 5.5 μmol, 1.0 equiv).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.68 (s), 9.60 (s), 9.52 (d, J = 8.5 Hz), 8.98–8.91 (m), 8.84–8.80 (m), 8.78 (t, J = 5.8 Hz), 8.24 (d, J = 6.5 Hz), 8.21–8.15 (m), 8.10–8.01 (m), 7.99–7.86 (m), 7.86 (s), 7.84 (s), 7.83 (s), 7.82–7.78 (m), 7.77 (t, J = 5.3 Hz), 7.76–7.70 (m), 7.66 (d, J = 7.4 Hz), 7.54–7.51 (m), 7.48 (d, J = 1.4 Hz), 7.47 (t, J = 1.7 Hz), 7.46 (d, J = 1.4 Hz), 7.44 (d, J = 1.5 Hz), 7.43 (d, J = 1.5 Hz), 7.39–7.36 (m), 7.36 (d, J = 1.4 Hz), 7.35 (d, J = 1.5 Hz), 7.34 (t, J = 1.3 Hz), 7.33 (s), 7.32 (d, J = 1.4 Hz), 7.32 (s), 7.31 (d, J = 1.5 Hz), 7.31 (s), 7.30–7.26 (m), 7.25–7.14 (m), 7.01 (s), 6.88–6.72 (m), 4.51 (d, J = 5.4 Hz), 4.46 (t, J = 5.3 Hz), 4.42–4.37 (m), 4.36 (d, J = 6.5 Hz), 4.35–3.99 (m), 3.98 (t, J = 5.3 Hz), 3.93 (s), 3.91 (s), 3.79 (t, J = 5.3 Hz), 3.75 (d, J = 4.6 Hz), 3.70–3.64 (m), 3.55–3.40 (m), 3.27 (q, J = 5.9 Hz), 3.08 (dd, J = 13.3 Hz, 6.9 Hz), 2.99 (dd, J = 12.7 Hz, 6.7 Hz), 2.78 (t, J = 2.7 Hz), 2.75 (t, J = 6.8 Hz), 2.70 (t, J = 7.5 Hz), 2.62 (t, J = 7.6 Hz), 2.54 (s), 2.47–2.44 (m), 2.30 (t, J = 7.3 Hz), 2.28–2.19 (m), 2.18 (td, J = 7.1 Hz, 2.7 Hz), 2.18–2.15 (m), 2.15 (s), 2.15 (s), 2.14–2.07 (m), 2.03–1.88 (m), 1.86 (s), 1.85–1.79 (m), 1.78 (s), 1.77–1.68 (m), 1.65 (p, J = 7.2 Hz), 1.63–1.58 (m), 1.56–1.24 (m), 1.22 (t, J = 6.7 Hz), 1.10–0.98 (m), 0.89 (d, J = 6.8 Hz), 0.86 (d, J = 6.6 Hz), 0.85–0.77 (m).
13C NMR (176 MHz, DMSO-d6) δ [ppm] = 174.3, 174.1, 174.0, 173.9, 173.9, 173.6, 172.5, 172.4, 172.3, 171.8, 171.5, 171.4, 171.3, 171.2, 171.1, 171.1, 171.0, 171.0, 170.9, 170.9, 170.6, 170.0, 169.9, 169.8, 169.3, 169.3, 169.3, 169.2, 169.1, 168.8, 168.8, 168.7, 168.6, 168.5, 168.3, 168.3, 168.0, 167.9, 167.8, 164.8, 164.6, 153.0, 146.3, 146.1, 142.9, 142.8, 140.2, 140.1, 139.2, 139.2, 136.1, 135.8, 135.6, 135.6, 132.5, 130.8, 130.7, 130.6, 127.1, 127.0, 127.0, 126.2, 126.1, 126.1, 126.0, 125.6, 124.8, 124.8, 124.7, 124.7, 124.6, 124.6, 122.3, 122.3, 118.1, 117.8, 117.8, 117.8, 117.8, 116.1, 116.1, 115.9, 115.9, 83.9, 71.7, 70.3, 70.2, 69.8, 69.8, 69.7, 69.7, 69.6, 68.9, 68.8, 66.6, 66.5, 58.2, 58.1, 57.9, 57.7, 57.6, 56.9, 56.8, 52.8, 52.5, 52.4, 52.1, 52.0, 51.8, 51.7, 51.7, 51.1, 51.0, 49.2, 48.4, 48.3, 41.9, 41.0, 40.9, 40.4, 40.0, 38.4, 38.2, 38.1, 36.7, 36.5, 34.9, 33.0, 32.4, 31.7, 31.5, 31.5, 31.4, 31.4, 30.6, 30.4, 30.3, 30.2, 30.1, 30.1, 29.0, 28.9, 27.7, 27.7, 27.6, 27.6, 27.4, 27.3, 27.2, 25.2, 24.8, 24.4, 24.4, 24.1, 23.5, 23.1, 23.1, 22.7, 22.7, 22.5, 21.5, 21.5, 21.1, 20.4, 20.4, 20.2, 20.1, 19.6, 19.5, 19.5, 19.3, 19.2, 18.5, 18.2, 18.0, 18.0, 18.0, 17.5, 17.2, 15.2, 15.1, 11.0, 11.0.
HRMS (ESI) calculated for C164H251N34O613+ ([M + 3H]3+): m/z = 1224.2523, experimental 1224.2512, δ [ppm] = 1.1. Calculated for C164H252N34O614+ ([M + 4H]4+): m/z = 918.4410, experimental 918.4408, δ [ppm] = 0.2. Calculated for C164H253N34O615+ ([M + 5H]5+): m/z = 734.9543, experimental 734.9545, δ [ppm] = 0.2. Calculated for C164H254N34O616+ ([M + 6H]6+): m/z = 612.6298, experimental 612.6303, δ [ppm] = 0.5.
PfeA 33–51 C-Terminal Disulfide-(PEG)5-MECAM-OH (32)
32 was prepared adapted from an established procedure reported by Song, Yang, Hall, Gurnani, and Perrier,67 which is a modified version of the procedures for disulfide bond formation by Bernatowicz, Matsueda and Matsueda.68
53 (6.1 mg, 5.7 μmol, 1.0 equiv) was dissolved in DMF (1 mL) and HEPES buffer (1.0 M, 1 mL). 70 (15 mg, 6.3 μmol, 1.1 equiv) dissolved in DMF (1 mL), DMSO (1 mL), and HEPES buffer (1.0 M, 1 mL) was added to the solution. The reaction mixture was stirred at 23 °C for 48 h, and the reaction progress was controlled by LCMS. The reaction mixture was concentrated by rotary evaporation and filtered after completeness. The remains in the filter were washed with ACN (20 mL), and the solvent was removed by rotary evaporation. The residue was taken up, diluted with ACN/Milli-Q H2O, and purified by RP-HPLC (C18 Phenomenex, 220 nm, collect all, 15–55% ACN/Milli-Q H2O, 0.1% HCOOH). The product-containing fractions were identified by LCMS and lyophilized to dryness to yield the product 32 as a white solid. Yield 15.0 mg (4.5 μmol, 78%).
1H NMR (700 MHz, DMSO-d6): δ [ppm] = 9.56 (s), 9.33 (t, J = 6.0 Hz), 9.17 (t, J = 5.8 Hz), 8.33 (d, J = 8.4 Hz), 8.22 (d, J = 7.8 Hz), 8.20–8.14 (m), 8.04 (t, J = 6.8 Hz), 8.01 (t, J = 5.6 Hz), 7.98–7.86 (m), 7.83 (s), 7.78 (d, J = 7.0 Hz), 7.74 (d, J = 7.7 Hz), 7.63 (d, J = 7.9 Hz), 7.30 (s), 7.28 (dd, J = 8.2 Hz, 1.3 Hz), 7.24 (s), 7.20 (s), 7.19 (dd, J = 8.3 Hz, 1.4 Hz), 7.03–6.99 (m), 6.91 (dd, J = 7.8 Hz, 1.3 Hz), 6.88 (dd, J = 7.8 Hz, 1.3 Hz), 6.78 (s), 6.77 (s), 6.66 (t, J = 7.9 Hz), 6.60 (t, J = 7.9 Hz), 4.46 (t, J = 5.3 Hz), 4.43 (s), 4.40 (d, J = 5.8 Hz), 4.38–4.33 (m), 4.29 (t, J = 6.8 Hz), 4.25 (t, J = 6.3 Hz), 4.23–4.07 (m), 4.04–3.94 (m), 3.79 (t, J = 5.4 Hz), 3.66 (d, J = 5.7 Hz), 3.64 (d, J = 5.5 Hz), 3.62–3.59 (m), 3.51 (s), 3.50–3.49 (m), 3.48 (s), 3.47 (s), 3.46–3.44 (m), 3.38 (t, J = 6.0 Hz), 3.19 (dd, J = 5.8 Hz), 3.00 (dd, J = 12.8 Hz, 6.9 Hz), 2.87 (t, J = 7.1 Hz), 2.76 (t, J = 7.3 Hz), 2.69 (t, J = 7.6 Hz), 2.48–2.42 (m), 2.33–2.08 (m), 2.06–1.83 (m), 1.81–1.17 (m), 1.11–0.99 (m), 0.94 (s), 0.90 (d, J = 6.9 Hz), 0.88 (d, J = 6.9 Hz), 0.86 (d, J = 6.6 Hz), 0.85–0.80 (m).
HRMS (ESI) calculated for C148H232N34O51S24+ ([M + 4H]4+): m/z = 841.4006, experimental 841.4019, δ [ppm] = 1.3. Calculated for C148H233N34O51S25+ ([M + 5H]5+): m/z = 673.3220, experimental 673.3218, δ [ppm] = 0.2.
Acknowledgments
We thank Ulrike Beutling formeasuring the HRMS samples and Christel Kakoschke and Kirsten Harmrolfs for measuring the NMR samples. We are thankful for helpful comments on the manuscript by Hazel Fuchs, Anna Vetter, and Vadim Korotkov.
Glossary
Abbreviations Used
- aa
amino acid
- DOTAM
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic amide
- MECAM
1,3,5-N,N′,N″-tris(2,3-dihydroxybenzoyl)-triaminomethylbenzene
- PMF
proton motive force
- PPI
protein–protein interactions
- TBDT
TonB-dependent transporter
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01489.
Author Contributions
% These authors contributed equally.
The presented work was supported by a “Kekulé-Stipendium” of the Fonds der chemischen Industrie (VCI)” as well as a grant from the Joint Programming Initiative on Antimicrobial Resistance (JPI AMR, Grant 01KI1825). We also acknowledge the Interdisciplinary Thematic Institute (ITI) InnoVec (Innovative Vectorization of Biomolecules, IdEx, ANR-10-IDEX-0002).
The authors declare no competing financial interest.
Special Issue
Published as part of the Journal of Medicinal Chemistry virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
Supplementary Material
References
- Tacconelli E.; Carrara E.; Savoldi A.; Harbarth S.; Mendelson M.; Monnet D. L.; Pulcini C.; Kahlmeter G.; Kluytmans J.; Carmeli Y.; Ouellette M.; Outterson K.; Patel J.; Cavaleri M.; Cox E. M.; Houchens C. R.; Grayson M. L.; Hansen P.; Singh N.; Theuretzbacher U.; Magrini N.; Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18 (3), 318–327. 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- Murray C. J. L.; Ikuta K. S.; Sharara F.; Swetschinski L.; Robles Aguilar G.; Gray A.; Han C.; Bisignano C.; Rao P.; Wool E.; Johnson S. C.; Browne A. J.; Chipeta M. G.; Fell F.; Hackett S.; Haines-Woodhouse G.; Kashef Hamadani B. H.; Kumaran E. A. P.; McManigal B.; Agarwal R.; Akech S.; Albertson S.; Amuasi J.; Andrews J.; Aravkin A.; Ashley E.; Bailey F.; Baker S.; Basnyat B.; Bekker A.; Bender R.; Bethou A.; Bielicki J.; Boonkasidecha S.; Bukosia J.; Carvalheiro C.; Castañeda-Orjuela C.; Chansamouth V.; Chaurasia S.; Chiurchiù S.; Chowdhury F.; Cook A. J.; Cooper B.; Cressey T. R.; Criollo-Mora E.; Cunningham M.; Darboe S.; Day N. P. J.; De Luca M.; Dokova K.; Dramowski A.; Dunachie S. J.; Eckmanns T.; Eibach D.; Emami A.; Feasey N.; Fisher-Pearson N.; Forrest K.; Garrett D.; Gastmeier P.; Giref A. Z.; Greer R. C.; Gupta V.; Haller S.; Haselbeck A.; Hay S. I.; Holm M.; Hopkins S.; Iregbu K. C.; Jacobs J.; Jarovsky D.; Javanmardi F.; Khorana M.; Kissoon N.; Kobeissi E.; Kostyanev T.; Krapp F.; Krumkamp R.; Kumar A.; Kyu H. H.; Lim C.; Limmathurotsakul D.; Loftus M. J.; Lunn M.; Ma J.; Mturi N.; Munera-Huertas T.; Musicha P.; Mussi-Pinhata M. M.; Nakamura T.; Nanavati R.; Nangia S.; Newton P.; Ngoun C.; Novotney A.; Nwakanma D.; Obiero C. W.; Olivas-Martinez A.; Olliaro P.; Ooko E.; Ortiz-Brizuela E.; Peleg A. Y.; Perrone C.; Plakkal N.; Ponce-de-Leon A.; Raad M.; Ramdin T.; Riddell A.; Roberts T.; Robotham J. V.; Roca A.; Rudd K. E.; Russell N.; Schnall J.; Scott J. A. G.; Shivamallappa M.; Sifuentes-Osornio J.; Steenkeste N.; Stewardson A. J.; Stoeva T.; Tasak N.; Thaiprakong A.; Thwaites G.; Turner C.; Turner P.; van Doorn H. R.; Velaphi S.; Vongpradith A.; Vu H.; Walsh T.; Waner S.; Wangrangsimakul T.; Wozniak T.; Zheng P.; Sartorius B.; Lopez A. D.; Stergachis A.; Moore C.; Dolecek C.; Naghavi M. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399 (10325), 629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaden J. T.; Park L. P.; Maskarinec S. A.; Ruffin F.; Fowler V. G.; van Duin D. Results from a 13-Year prospective cohort study show increased mortality associated with bloodstream infections caused by Pseudomonas aeruginosa compared to other bacteria. Antimicrob. Agents Chemother. 2017, 61 (6), e02671-16. 10.1128/AAC.02671-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler M. S.; Gigante V.; Sati H.; Paulin S.; Al-Sulaiman L.; Rex J. H.; Fernandes P.; Arias C. A.; Paul M.; Thwaites G. E.; Czaplewski L.; Alm R. A.; Lienhardt C.; Spigelman M.; Silver L. L.; Ohmagari N.; Kozlov R.; Harbarth S.; Beyer P. Analysis of the clinical pipeline of treatments for drug resistant bacterial infections: Despite progress, more action is needed. Antimicrob. Agents Chemother. 2022, 66 (3), e01991-21. 10.1128/aac.01991-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theuretzbacher U.; Gottwalt S.; Beyer P.; Butler M.; Czaplewski L.; Lienhardt C.; Moja L.; Paul M.; Paulin S.; Rex J. H.; Silver L. L.; Spigelman M.; Thwaites G. E.; Paccaud J. P.; Harbarth S. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19 (2), e40–e50. 10.1016/S1473-3099(18)30513-9. [DOI] [PubMed] [Google Scholar]
- Breidenstein E. B. M.; de la Fuente-Núñez C.; Hancock R. E. W. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 2011, 19 (8), 419–426. 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Klahn P.; Bronstrup M. Bifunctional antimicrobial conjugates and hybrid antimicrobials. Nat. Prod. Rep. 2017, 34 (7), 832–885. 10.1039/C7NP00006E. [DOI] [PubMed] [Google Scholar]
- Miller M. J.; Liu R. Design and syntheses of new antibiotics inspired by nature’s quest for iron in an oxidative climate. Acc. Chem. Res. 2021, 54 (7), 1646–1661. 10.1021/acs.accounts.1c00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hider R. C.; Kong X. Chemistry and biology of siderophores. Nat. Prod. Rep. 2010, 27 (5), 637–657. 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- Schalk I. J.; Mislin G. L. A.; Brillet K. Chapter Two - Structure, function and binding selectivity and steoselectivity of sderophore–ion oter memane tansporters. Curr. Top. Membr. 2012, 69, 37–66. 10.1016/B978-0-12-394390-3.00002-1. [DOI] [PubMed] [Google Scholar]
- Schauer K.; Rodionov D. A.; de Reuse H. New substrates for TonB-dependent transport: do we only see the ‘tip of the iceberg’?. Trends Biochem. Sci. 2008, 33 (7), 330–338. 10.1016/j.tibs.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Hickman S. J.; Cooper R. E. M.; Bellucci L.; Paci E.; Brockwell D. J. Gating of TonB-dependent transporters by substrate-specific forced remodelling. Nat. Commun. 2017, 8, 14804. 10.1038/ncomms14804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalk I. J.; Mislin G. L. A. Bacterial Iron Uptake Pathways: Gates for the Import of Bactericide Compounds. J. Med. Chem. 2017, 60 (11), 4573–4576. 10.1021/acs.jmedchem.7b00554. [DOI] [PubMed] [Google Scholar]
- Bickel H.; Gäumann E.; Keller-Schierlein W.; Prelog V.; Vischer E.; Wettstein A.; Zähner H. Über eisenhaltige Wachstumsfaktoren, die Sideramine, und ihre Antagonisten, die eisenhaltigen Antibiotika Sideromycine. Experientia 1960, 16 (4), 129–133. 10.1007/BF02157712. [DOI] [PubMed] [Google Scholar]
- Braun V.; Pramanik A.; Gwinner T.; Köberle M.; Bohn E. Sideromycins: Tools and antibiotics. BioMetals 2009, 22, 3–13. 10.1007/s10534-008-9199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun V. Active transport of siderophore-mimicking antibacterials across the outer membrane. Drug Resist. Updat. 1999, 2 (6), 363–369. 10.1054/drup.1999.0107. [DOI] [PubMed] [Google Scholar]
- Braun V.; Herrmann C. Evolutionary relationship of uptake systems for biopolymers in Escherichia coli: cross-complementation between the TonB-ExbB-ExbD and the TolA-TolQ-TolR proteins. J. Mol. Microbiol. 1993, 8 (2), 261–268. 10.1111/j.1365-2958.1993.tb01570.x. [DOI] [PubMed] [Google Scholar]
- Braun V.; Günthner K.; Hantke K.; Zimmermann L. Intracellular activation of albomycin in Escherichia coli and Salmonella typhimurium. J. Bacteriol. 1983, 156 (1), 308–315. 10.1128/jb.156.1.308-315.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun V.; Patzer S. I.; Hantke K. Ton-dependent colicins and microcins: modular design and evolution. Biochimie 2002, 84 (5–6), 365–380. 10.1016/S0300-9084(02)01427-X. [DOI] [PubMed] [Google Scholar]
- Han S.; Zaniewski R. P.; Marr E. S.; Lacey B. M.; Tomaras A. P.; Evdokimov A.; Miller J. R.; Shanmugasundaram V. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc. Natl. Ac. Sci. 2010, 107 (51), 22002–22007. 10.1073/pnas.1013092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C.; Miller P. A.; Miller M. J. Iron transport-mediated drug delivery: Practical syntheses and in vitro antibacterial studies of tris-catecholate siderophore-aminopenicillin conjugates reveals selectively potent anti-pseudomonal activity. J. Am. Chem. Soc. 2012, 134 (24), 9898–9901. 10.1021/ja303446w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C.; Miller M. J. Chemical syntheses and in vitro antibacterial activity of two desferrioxamine B-ciprofloxacin conjugates with potential esterase and phosphatase triggered drug release linkers. Bioorg. Med. Chem. 2012, 20 (12), 3828–3836. 10.1016/j.bmc.2012.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C.; Miller M. J. Siderophore–fluoroquinolone conjugates containing potential reduction-triggered linkers for drug release: synthesis and antibacterial activity. Biometals 2015, 28 (3), 541–551. 10.1007/s10534-015-9830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M.; Miller P. A.; Möllmann U.; Claypool W. D.; Schroeder V. A.; Wolter W. R.; Suckow M.; Yu H.; Li S.; Huang W.; Zajicek J.; Miller M. J. Targeted Antibiotic Delivery: Selective Siderophore Conjugation with Daptomycin Confers Potent Activity against Multidrug Resistant Acinetobacter baumannii Both in Vitro and in Vivo. J. Med. Chem. 2017, 60 (11), 4577–4583. 10.1021/acs.jmedchem.7b00102. [DOI] [PubMed] [Google Scholar]
- Wu J. Y.; Srinivas P.; Pogue J. M. Cefiderocol: A Novel Agent for the Management of Multidrug-Resistant Gram-Negative Organisms. Infect. Dis. Ther. 2020, 9 (1), 17–40. 10.1007/s40121-020-00286-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquesne S.; Destoumieux-Garzón D.; Peduzzi J.; Rebuffat S. Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat. Prod. Rep. 2007, 24 (4), 708–34. 10.1039/b516237h. [DOI] [PubMed] [Google Scholar]
- Neumann W.; Sassone-Corsi M.; Raffatellu M.; Nolan E. M. Esterase-catalyzed siderophore hydrolysis activates an enterobactin-ciprofloxacin conjugate and confers targeted antibacterial activity. J. Am. Chem. Soc. 2018, 140 (15), 5193–5201. 10.1021/jacs.8b01042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raines D. J.; Moroz O. V.; Blagova E. V.; Turkenburg J. P.; Wilson K. S.; Duhme-Klair A. K. Bacteria in an intense competition for iron: Key component of the Campylobacter jejuni iron uptake system scavenges enterobactin hydrolysis product. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (21), 5850–5855. 10.1073/pnas.1520829113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiach R.; Meijler M. M. Total synthesis of pyoverdin D. Org. Lett. 2013, 15 (7), 1702–1705. 10.1021/ol400490s. [DOI] [PubMed] [Google Scholar]
- Petrik M.; Umlaufova E.; Raclavsky V.; Palyzova A.; Havlicek V.; Haas H.; Novy Z.; Dolezal D.; Hajduch M.; Decristoforo C. Imaging of Pseudomonas aeruginosa infection with Ga-68 labelled pyoverdine for positron emission tomography. Sci. Rep. 2018, 8, 15698. 10.1038/s41598-018-33895-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perraud Q.; Cantero P.; Roche B.; Gasser V.; Normant V. P.; Kuhn L.; Hammann P.; Mislin G. L. A.; Ehret-Sabatier L.; Schalk I. J. Phenotypic Adaption of Pseudomonas aeruginosa by hacking siderophores produced by other microorganisms* Mol. Cell. Proteomics 2020, 19 (4), 589–607. 10.1074/mcp.RA119.001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira K.; Hu H.-Y.; Fetz V.; Prochnow H.; Rais B.; Müller P. P.; Brönstrup M. Multivalent siderophore–DOTAM conjugates as theranostics for imaging and treatment of cacterial infections. Angew. Chem., Int. Ed. 2017, 56 (28), 8272–8276. 10.1002/anie.201701358. [DOI] [PubMed] [Google Scholar]
- Pinkert L.; Lai Y.-H.; Peukert C.; Hotop S.-K.; Karge B.; Schulze L. M.; Grunenberg J.; Brönstrup M. Antibiotic conjugates with an crtificial MECAM-Based siderophore are potent agents against Gram-positive and Gram-negative bacterial pathogens. J. Med. Chem. 2021, 64 (20), 15440–15460. 10.1021/acs.jmedchem.1c01482. [DOI] [PubMed] [Google Scholar]
- Peukert C.; Langer L. N. B.; Wegener S. M.; Tutov A.; Bankstahl J. P.; Karge B.; Bengel F. M.; Ross T. L.; Brönstrup M. Optimization of artificial siderophores as 68Ga-complexed PET tracers for in vivo imaging of bacterial infections. J. Med. Chem. 2021, 64 (16), 12359–12378. 10.1021/acs.jmedchem.1c01054. [DOI] [PubMed] [Google Scholar]
- Brillet K.; Journet L.; Célia H.; Paulus L.; Stahl A.; Pattus F.; Cobessi D. A β Strand Lock Exchange for Signal Transduction in TonB-Dependent Transducers on the Basis of a Common Structural Motif. Structure 2007, 15 (11), 1383–1391. 10.1016/j.str.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Llamas M. A.; Mooij M. J.; Sparrius M.; Vandenbroucke-Grauls C. M. J. E.; Ratledge C.; Bitter W. Characterization of five novel Pseudomonas aeruginosa cell-surface signalling systems. Mol. Microbiol. 2008, 67 (2), 458–472. 10.1111/j.1365-2958.2007.06061.x. [DOI] [PubMed] [Google Scholar]
- Celia H.; Noinaj N.; Zakharov S. D.; Bordignon E.; Botos I.; Santamaria M.; Barnard T. J.; Cramer W. A.; Lloubes R.; Buchanan S. K. Structural insight into the role of the Ton complex in energy transduction. Nature 2016, 538 (7623), 60–65. 10.1038/nature19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratliff A. C.; Buchanan S. K.; Celia H. Ton motor complexes. Curr. Opin. Struct. Biol. 2021, 67, 95–100. 10.1016/j.sbi.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takase H.; Nitanai H.; Hoshino K.; Otani T. Requirement of the Pseudomonas aeruginosa tonB gene for high-affinity iron acquisition and infection. Infect. Immun. 2000, 68 (8), 4498–4504. 10.1128/IAI.68.8.4498-4504.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs P. I.; Larsen R. A.; Postle K. Quantification of known components of the Escherichia coli TonB energy transduction system: TonB, ExbB, ExbD and FepA. Mol. Microbiol. 2002, 44, 271–281. 10.1046/j.1365-2958.2002.02880.x. [DOI] [PubMed] [Google Scholar]
- Tuckman M.; Osburne M. S. In vivo inhibition of TonB-dependent processes by a TonB box consensus pentapeptide. J. Bacteriol. 1992, 174 (1), 320–323. 10.1128/jb.174.1.320-323.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard S. P.; Herrmann C.; Stratilo C. W.; Braun V. In vivo synthesis of the periplasmic Domain of TonB inhibits transport through the FecA and FhuA iron siderophore transporters of Escherichia coli. J. Bacteriol. 2001, 183 (20), 5885–5895. 10.1128/JB.183.20.5885-5895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader M.; Journet L.; Meksem A.; Guillon L.; Schalk I. J. Mechanism of ferripyoverdine uptake by Pseudomonas aeruginosa outer membrane transporter FpvA: No diffusion channel formed at any time during ferrisiderophore uptake. Biochemistry 2011, 50 (13), 2530–2540. 10.1021/bi101821n. [DOI] [PubMed] [Google Scholar]
- Zimmermann L.; Stephens A.; Nam S.-Z.; Rau D.; Kübler J.; Lozajic M.; Gabler F.; Söding J.; Lupas A. N.; Alva V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430 (15), 2237–2243. 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Capasso S.; Mazzarella L.; Sica F.; Zagari A.; Salvadori S. Spontaneous cyclization of the aspartic acid side chain to the succinimide derivative. J. Chem. Soc. Chem. Comm. 1992, 12, 919–921. 10.1039/c39920000919. [DOI] [Google Scholar]
- Schilter D. Thiol oxidation: A slippery slope. Nat. Rev. Chem. 2017, 1, 0013 10.1038/s41570-016-0013. [DOI] [Google Scholar]
- Díaz-Mochón J. J.; Bialy L.; Bradley M. Full Orthogonality between Dde and Fmoc: The direct Synthesis of PNA–peptide conjugates. Org. Lett. 2004, 6 (7), 1127–1129. 10.1021/ol049905y. [DOI] [PubMed] [Google Scholar]
- Cassat J. E.; Skaar E. P. Iron in infection and immunity. Cell Host & Microbe 2013, 13 (5), 509–519. 10.1016/j.chom.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser V.; Baco E.; Cunrath O.; August P. S.; Perraud Q.; Zill N.; Schleberger C.; Schmidt A.; Paulen A.; Bumann D.; Mislin G. L. A.; Schalk I. J. Catechol siderophores repress the pyochelin pathway and activate the enterobactin pathway in Pseudomonas aeruginosa: an opportunity for siderophore–antibiotic conjugates development. Environ. Microbiol. 2016, 18 (3), 819–832. 10.1111/1462-2920.13199. [DOI] [PubMed] [Google Scholar]
- Cunrath O.; Geoffroy V. A.; Schalk I. J. Metallome of Pseudomonas aeruginosa: A role for siderophores. Environ. Microbiol. 2016, 18 (10), 3258–3267. 10.1111/1462-2920.12971. [DOI] [PubMed] [Google Scholar]
- Perraud Q.; Cantero P.; Munier M.; Hoegy F.; Zill N.; Gasser V.; Mislin G. L. A.; Ehret-Sabatier L.; Schalk I. J. Phenotypic adaptation of Pseudomonas aeruginosa in the presence of siderophore-antibiotic conjugates during epithelial cell infection. Microorganisms 2020, 8 (11), 1820. 10.3390/microorganisms8111820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culp E.; Wright G. D. Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 2017, 70 (4), 366–377. 10.1038/ja.2016.138. [DOI] [PubMed] [Google Scholar]
- Fritsch S.; Gasser V.; Peukert C.; Pinkert L.; Kuhn L.; Perraud Q.; Normant V.; Brönstrup M.; Schalk I. Uptake mechanisms and regulatory responses to MECAM- and DOTAM-based artificial siderophores and their antibiotic conjugates in Pseudomonas aeruginosa. ACS Infect. Dis. 2022, 8 (6), 1134–1146. 10.1021/acsinfecdis.2c00049. [DOI] [PubMed] [Google Scholar]
- Dean C. R.; Neshat S.; Poole K. PfeR, an enterobactin-responsive activator of ferric enterobactin receptor gene expression in Pseudomonas aeruginosa. J. Bacteriol. 1996, 178 (18), 5361–5369. 10.1128/jb.178.18.5361-5369.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser V.; Kuhn L.; Hubert T.; Aussel L.; Hammann P.; Schalk I. J. The esterase PfeE, the achilles’ Hheel in the battle for Iron between Pseudomonas aeruginosa and Escherichia coli. Int. J. Mol. Sci. 2021, 22 (6), 2814. 10.3390/ijms22062814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynié L.; Milenkovic S.; Mislin G. L. A.; Gasser V.; Malloci G.; Baco E.; McCaughan R. P.; Page M. G. P.; Schalk I. J.; Ceccarelli M.; Naismith J. H. The complex of ferric-enterobactin with its transporter from Pseudomonas aeruginosa suggests a two-site model. Nat. Commun. 2019, 10, 3673. 10.1038/s41467-019-11508-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghysels B.; Ochsner U.; Möllman U.; Heinisch L.; Vasil M.; Cornelis P.; Matthijs S. The Pseudomonas aeruginosa pirA gene encodes a second receptor for ferrienterobactin and synthetic catecholate analogues. FEMS Microbiol. Lett. 2005, 246 (2), 167–174. 10.1016/j.femsle.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Perraud Q.; Kuhn L.; Fritsch S.; Graulier G.; Gasser V.; Normant V.; Hammann P.; Schalk I. J. Opportunistic use of catecholamine neurotransmitters as siderophores to access iron by Pseudomonas aeruginosa. Environ. Microbiol. 2022, 24 (2), 878–893. 10.1111/1462-2920.15372. [DOI] [PubMed] [Google Scholar]
- Ankenbauer R. G.; Quan H. N. FptA, the Fe(III)-pyochelin receptor of Pseudomonas aeruginosa: A phenolate siderophore receptor homologous to hydroxamate siderophore receptors. J. Bacteriol. 1994, 176 (2), 307–319. 10.1128/jb.176.2.307-319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobessi D.; Celia H.; Folschweiller N.; Schalk I. J.; Abdallah M. A.; Pattus F. The crystal structure of the pyoverdine outer membrane receptor FpvA from Pseudomonas aeruginosa at 3.6Å resolution. J. Mol. Biol. 2005, 347 (1), 121–134. 10.1016/j.jmb.2005.01.021. [DOI] [PubMed] [Google Scholar]
- Cobessi D.; Celia H.; Pattus F. Crystal structure at high resolution of ferric-pyochelin and its membrane receptor FptA from Pseudomonas aeruginosa. J. Mol. Biol. 2005, 352 (4), 893–904. 10.1016/j.jmb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Dean C. R.; Poole K. Cloning and characterization of the ferric enterobactin receptor gene (pfeA) of Pseudomonas aeruginosa. J. Bacteriol. 1993, 175 (2), 317–324. 10.1128/jb.175.2.317-324.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perraud Q.; Cantero P.; Roche B.; Gasser V.; Normant V. P.; Kuhn L.; Hammann P.; Mislin G. L. A.; Ehret-Sabatier L.; Schalk I. J. Phenotypic adaption of Pseudomonas aeruginosa by hacking siderophores produced by other microorganisms*. Mol. Cell. Proteom. 2020, 19 (4), 589–607. 10.1074/mcp.RA119.001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira K.; Hu H.-Y.; Fetz V.; Prochnow H.; Rais B.; Müller P. P.; Brönstrup M. Multivalent siderophore–DOTAM conjugates as theranostics for imaging and treatment of bacterial infections. Angew. Chem., Int. Ed. 2017, 56 (28), 8272–8276. 10.1002/anie.201701358. [DOI] [PubMed] [Google Scholar]
- Svedhem S.; Hollander C.-Å.; Shi J.; Konradsson P.; Liedberg B.; Svensson S. C. T. Synthesis of a Series of Oligo(ethylene glycol)-Terminated Alkanethiol Amides Designed to Address Structure and Stability of Biosensing Interfaces. J. Org. Chem. 2001, 66 (13), 4494–4503. 10.1021/jo0012290. [DOI] [PubMed] [Google Scholar]
- Pinkert L.Synthese von Siderophor-Antibiotika-Konjugaten als neuartige antibakterielle Wirkstoffe. Doctoral Thesis, Gottfried Wilhelm Leibniz Universität Hannover, Hannover, Germany, 2021.
- Song Q.; Yang J.; Hall S. C. L.; Gurnani P.; Perrier S. Pyridyl Disulfide Reaction Chemistry: An Efficient Strategy toward Redox-Responsive Cyclic Peptide–Polymer Conjugates. ACS Macro Lett. 2019, 8 (10), 1347–1352. 10.1021/acsmacrolett.9b00538. [DOI] [PubMed] [Google Scholar]
- Bernatowicz M. S.; Matsueda R. E. I.; Matsueda G. R. Preparation of Boc-[S-(3-nitro-2-pyridinesulfenyl)]-cysteine and its use for unsymmetrical disulfide bond formation. Int. J. Pept. Protein Res. 1986, 28 (2), 107–112. 10.1111/j.1399-3011.1986.tb03236.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.