

Abstract
Immunotherapy with bispecific T cell engagers has shown efficacy in patients with hematologic malignancies and uveal melanoma. Antitumor effects of bispecific T cell engagers in most solid tumors are limited due to their short serum half-life and insufficient tumor concentration. We designed a novel serotype 5/3 oncolytic adenovirus encoding a human mucin1 antibody and the human CD3 receptor, Ad5/3-E2F-d24-aMUC1aCD3 (TILT-321). TILT-321 is engineered to replicate only in cancer cells, leading to a high concentration of the aMUC1aCD3 molecule in the tumor microenvironment. Infection and cell viability assays were performed to determine the oncolytic potential of the novel construct. The functionality of the virus-derived aMUC1aCD3 was evaluated in vitro. When TILT-321 was combined with allogeneic T cells, rapid tumor cell lysis was observed. TILT-321-infected cells secreted functional aMUC1aCD3, as shown by increased T cell activity and its binding to MUC1 and CD3. In vivo, TILT-321 treatment led to effective antitumor efficacy mediated by increased intratumoral T cell activity in an A549 and patient-derived ovarian cancer xenograft mouse model humanized with peripheral blood mononuclear cells (PBMC). This study provides a proof of concept for an effective strategy to overcome the key limitations of recombinant bispecific T cell engager delivery for solid tumor treatment.
Keywords: oncolytic virus, adenovirus, bispecific T cell engager, MUC1, immunotherapy
Graphical abstract
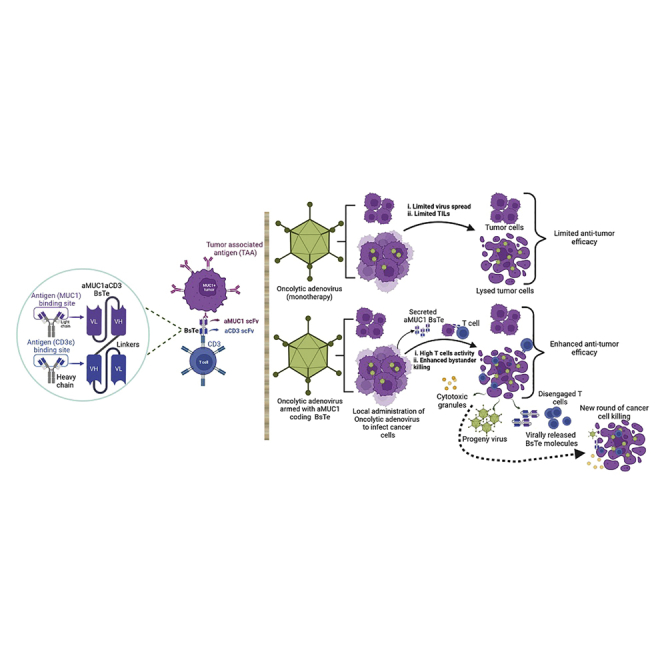
Bispecific T cell engagers (BsTes) and oncolytic adenovirus are promising immunotherapies. In clinical trials, both technologies demonstrated limited therapeutic effects when used as single agents. In this study, oncolytic adenovirus was armed with human mucin1 targeting BsTe to redirect and reactivate T cells against solid tumors more effectively.
Introduction
Immunotherapies that retarget T cells against tumors are promising tools to treat cancer and have demonstrated promising therapeutic potential in the last decade.1,2 The use of fusion recombinant proteins comprising two single-chain variable fragments with dual specificity for a tumor-associated antigen and T cell receptor (usually CD3ε), also known as bispecific T cell engagers (BsTe), is a potential strategy for T cell retargeting.3,4,5 Bispecific antibodies are composed of two distinct epitopes combined into a single molecule, which has the potential to improve tumor cell targeting and potency.5,6,7 The binding of BsTe to its targets causes T cell activation, facilitating an immunological synapse between the tumor cells and T cells, ultimately leading to apoptosis and lysis of the target tumor cell.4,8 Unlike many other T cell-based therapies, BsTe-mediated tumor cell death is independent of human leukocyte antigen expression and can occur in the absence of ex vivo stimulation or costimulatory signals.3,9 The first BsTe was synthesized in 199510 and, since its inception, numerous bispecific antibodies have been in preclinical and clinical development for the treatment of solid tumors, each with its molecular design and binding properties. Blinatumomab (Blincyto), a CD19xCD3 bispecific T cell engager (BiTE), was the first CD3 bispecific to be approved by the US Food and Drug Administration (FDA), and catumaxomab (EPCAMxCD3) has also received regulatory approval, although it was later withdrawn.10,11,12
Despite these promising initial results, BsTe treatment of non-melanoma solid tumors is frequently limited when delivered systemically because of its short serum half-life and insufficient tumor penetration, resulting in dose-limiting toxicity to organs before sufficient tumor concentrations are achieved.12,13 To compensate for their rapid elimination, BsTe proteins must be administered as a continuous infusion over several weeks, exposing patients to high systemic doses that can result in severe off-tumor toxicity.2 These shortcomings can be overcome by using vector delivery, such as oncolytic viruses. In 2014, the first study of a T cell engager-armed oncolytic vaccinia virus encoding EphA2 was reported.14 After this initial study, two studies proposed the use of oncolytic viruses encoding EGFR-targeting BsTe (ICO15K-cBiTE)15 and EpCAM/CD3 targeting BsTe.16
Oncolytic viruses selectively replicate in and lyse cancer cells, causing little or no harm to normal cells.17,18 This special feature has gained considerable attention as a novel immunotherapeutic agent for cancer treatment. Oncolytic adenoviruses are one of the most commonly used oncolytic virus backbones because of their capacity to induce tumor cell lysis and immune response stimulation.19 Modified oncolytic adenoviruses are tumor selective, highly immunogenic, can deliver transgenes, and express transgenes efficiently. Additionally, adenoviruses can produce high titers, making them attractive candidates for virotherapy. Oncolytic adenoviruses have demonstrated efficacy as monotherapy or in combination with other immunotherapies.20,21,22 Several clinical trials (NCT01147965, NCT00084316, NCT00583024, NCT03003676, and NCT04387461)23 have demonstrated its safety and potential, highlighting the importance of the immune system in cancer treatment. Of note, previous trials have demonstrated the safety of oncolytic adenoviruses but have also highlighted the importance of the immune response in achieving antitumor efficacy.24 To enhance immune system activation, many efforts to improve oncolytic adenoviruses rely on the insertion of immunostimulatory molecules (e.g., granulocyte-macrophage colony-stimulating factor, interleukin-2 [IL2], tumor necrosis factor alpha [TNF-α], or CD40L) into the viral genome, allowing for localized transgene expression in the tumor microenvironment after infection.21,25,26 These features described above render oncolytic adenoviruses a suitable platform for tumor-specific expression of molecules such as BsTe. Adenoviruses are strongly immunostimulatory through activation of danger signaling (pathogen-associated molecular patterns [PAMPs] and damage-associated molecular patterns [DAMPs]) in the tumor microenvironment20 and consequent recruitment of T cells, thus complementing the T cell-binding and -activating effects of BsTe.19,27,28 However, despite many promising preclinical results, currently, only one oncolytic adenovirus (H101; Oncorine) has been approved for therapeutic use, and that is only in China.
Considering the various benefits and challenges of BsTe and oncolytic adenoviruses, in this study, we engineered and characterized a novel serotype 5/3 oncolytic adenovirus armed with mucin 1 (MUC1) that targets the BsTe molecule. MUC1 is a promising immunotherapeutic target, as it is expressed at high levels, has altered glycosylation, and loses polarity in more than 80% of human malignancies.29,30,31 The truncated glycosylation of the MUC1 protein observed in cancer cells exposes cryptic peptide epitopes that can be recognized by antibodies or antibody fragments (e.g., BsTe molecules).32,33 These immunogenic areas are cancer specific, making them potential therapeutic targets. The cancer antigen 15-3 (CA15-3), also known as secretory MUC1 is a widely used tumor marker that can be measured in blood and is used to assess therapeutic efficacy in a variety of epithelial malignancies.34 MUC1/CA15-3 is a serological clinical marker of breast cancer that is used to track the treatment response and disease recurrence. Increased levels, on the other hand, may indicate disease progression. Breast, lung, gastric, pancreatic, liver, and gynecological cancers all had higher MUC1 protein expression, according to the human protein atlas The Cancer Genome Atlas (TCGA) dataset.35 Therefore, serum CA15-3 could also function as a useful biomarker for tumors that are potentially treatable with the virus characterized here. The previously observed low tumor specificity and efficacy of other glycan-based therapeutic strategies reveal the need for new functional therapies and combination therapies.36,37
We hypothesized that the new oncolytic adenovirus Ad5/3-E2F-d24-aMUC1aCD3 (TILT-321) replicates preferentially in cancer cells, lyses them, and ensures a sufficient local distribution of BsTe molecules to the tumor with minimal systemic exposure. We demonstrated that BsTe molecules armed with an oncolytic adenovirus can effectively redirect and activate T cells to fight solid cancer cells.
Results
Oncolytic adenovirus armed with human aMUC1-targeted BsTe exerts cytotoxic effects in cancer cell lines and exhibits synergy when combined with T cells
The oncolytic adenovirus coding for aMUC1aCD3 utilizes the Ad5/3-E2F-d24 virus backbone from adenovirus serotype 5, which carries the fiber knob from adenovirus serotype 3, as previously described.21,38 Briefly, the presence of an adenovirus serotype 3 knob increases cancer cell transduction via desmoglein 2, which is a receptor for Ad339,40 and is abundantly expressed in different cancer types.39,40,41,42,43,44,45,46,47,48 Furthermore, a 24-bp deletion (d24) in the Rb-binding domain of adenoviral E1A was combined with the E2F promoter to directly replicate the Rb/p16 pathway-deficient cancer cells.38 The transgene was introduced into the E3 region, resulting in a design in which viral replication was associated with aMUC1aCD3 expression (Figure 1A). This backbone has been reported as appealing because of its ability to achieve systemic transduction despite neutralizing antibodies, owing to its ability to use blood cells as stealth delivery vehicles.49
Figure 1.

In vitro characterization of Ad5/3-E2F-d24-aMUC1aCD3 virus
(A) Schematic representation of the genetic structure of Ad5/3-E2F-d24-aMUC1aCD3. VL and VH domains of aMUC1 and aCD3Ɛ are connected through glycine and serine (G3S) flexible linker. (B) Western blot analysis of 59-kDa aMUC1aCD3- BsTe transgene using CHO express host cells where lane M is a protein marker, lane P is a positive control (GenScript, catalog no. M0101), and lanes 1–2 are cell lysate supernatants from day 3 and day 6 post transfection of aMUC1aCD3, respectively. The analysis was performed under reduced conditions. (C) Comparison of the in vitro lytic effect of TILT-321 virus with its parental Ad5/3-E2F-d24 virus in A549 and (D) T47D tumor cells. The mean ± SEM of triplicate experiments is presented. The untreated control group is indicated by the dashed line. (E) Real-time xCELLigence-based cytotoxicity assay for assessment of lysis of MUC1-positive A549 (F) T47D and (G) MUC1-negative HEK-293 tumor cells in the presence of virus and T cells for up to 120 h. The mean ± SEM of duplicates is shown. Statistical significance is represented as ∗p = 0.0256, ∗∗p = 0.0041, and ∗∗∗p = 0.0009.
Western blotting detected the size of the aMUC1aCD3 transgene at a predicted molecular weight of 59 kDa (Figure 1B).9,50 The small size of BsTe (∼55 kDa) is advantageous for penetration and distribution throughout the tumor, as demonstrated by molecular imaging of radiolabeled BsTe.51
To select potential tumor target candidates for the testing of TILT-321, the expression of MUC1 in a panel of different cell lines was screened by flow cytometry (Figure S1). Compared with the unarmed virus, the aMUC1aCD3 armed virus exhibited a similar oncolytic capacity in different human cancer cells in the absence of immune cells (Figures 1C and 1D). These findings showed that insertion of the aMUC1aCD3 transgene did not restrict the lytic capabilities of the virus.
Interestingly, the addition of unstimulated T cells to cancer cells infected with aMUC1aCD3-armed virus resulted in tumor cell lysis at a slightly earlier time point (24–48 h post infection) in both MUC1+ cells (A549 and T47D) and a MUC1− cell line (HEK-293) compared with the controls (Figures 1E–1G). TILT-321 virus-mediated cell killing was statistically significant compared with tumor cells (A549 at 96 h, ∗∗∗p = 0.009; T47D at 144 h, ∗p = 0.0124; and HEK-293 at 96 h, ∗∗p = 0.0086) and tumor cells + T cell control group (A549 at 96 h, ∗p = 0.0256; T47D at 144 h, p = 0.0749; and HEK-293 at 96 h, ∗∗p = 0.0041).
Overall, we demonstrated that Ad5/3-E2F-d24-aMUC1aCD3 can lyse cancer cells both positive and negative for MUC1 expression in the presence of allogeneic T cells.
aMUC1aCD3-virus supernatant engages T cell and target cancer antigens
aMUC1aCD3-BsTe forms a bridge between tumor cells and T cells by simultaneously attaching to the target antigen (MUC1) and CD3 molecules on T cells.9 We observed very few T cell rosette structures (n = 9) in the uninfected control group (Figure 2A) and in cultures treated with the backbone virus supernatant (n = 12) group (Figure 2B). Most importantly, the co-cultures that included the supernatant of aMUC1aCD3-BsTe generated more T cell rosettes (n = 24) around the fluorescent cancer cells (Figure 2C). The frequent engagement of T cells around tumor cells was evident from the early time point (approximately 30 min) of incubation in quantified live-cell imaging. When photos were acquired after 4 h followed by washing with phosphate-buffered saline (PBS), rosettes (T cells around cancer cells) were visible in the same co-cultures, indicating aMUC1aCD3-BsTe-mediated attraction of T cells toward cancer cells (Figure 2D). The newly formed active T cell rosette is an early marker for T cell activation52 and provides additional evidence that aMUC1aCD3 present in the virus-derived supernatant mediates T cell activation.
Figure 2.

Ad5/3-E2F-d24-aMUC1aCD3-derived supernatants aggregate T cells around cancer cells
Co-cultures of T47D cells and T cells were treated with concentrated supernatants from (A) uninfected cells, (B) Ad5/3-E2F-d24-, or (C) Ad5/3-E2F-d24-aMUC1aCD3-infected cells. (D) Total count of quantified rosettes around tumor cells. A ratio of rosette-forming T cells to tumor cells equal to 4 was used for quantification. Four hours after co-culture, fluorescent and light microscopy images were taken and later quantified by using Fiji ImageJ.
Virus-secreted aMUC1aCD3 is functional and enhances T cell function
The functionality of virally released aMUC1aCD3 was investigated using a multistep process that included (1) binding to the target antigen MUC1, (2) binding to CD3, and (3) overall T cell activation. For the binding of aMUC1aCD3 to MUC1 antigen, a competitive binding study was performed with a commercially available anti-MUC1 antibody that binds to the same epitope. A lower signal from commercial anti-MUC1 antibodies was expected to envision the binding of aMUC1aCD3 to the target MUC1 antigen, because aMUC1aCD3-BsTe prevents binding of a labeled commercial anti-MUC1 antibody to MUC1 antigen. As expected, labeled anti-MUC1 antibody signal was statistically significantly lower in the aMUC1aCD3 supernatant-treated group than in the mock group, that is, uninfected cell supernatants at 1:1 dilution (∗∗∗p = 0.001) and Ad5/3-E2F-d24 supernatants (∗p = 0.05). Nevertheless, the binding was noticeably higher and dilution dependent at dilutions of 1:10 and 1:100, but not statistically significant (Figure 3A). This result revealed that aMUC1aCD3 released after virus infection and tumor cell lysis binds to target tumor cells and inhibits commercial anti-MUC1 antibodies from binding to MUC1 antigen on the T47D cell surface. Next, the binding of aMUC1aCD3-BsTe to the CD3Ɛ portion of the lymphocytes was studied by incubating three healthy donors of PBMC-derived unstimulated T cells with different supernatants. We confirmed that CD3 single-chain variable fragment can bind to CD3 T cell receptor complexes and activate CD4+CD69+CD8− and CD4+CD25+CD8− or CD8+CD69+CD4− and CD8+CD25+CD4− T cell populations when treated with aMUC1aCD3-virus supernatants (Figures S2A–S2D). T cell activation was further confirmed by checking the expression of cytokines in the supernatant of infected cells (Figures S2E and S2F).
Figure 3.

The functionality of virus-derived aMUC1aCD3 in co-cultures of T cells with MUC1+ tumor cells
(A) Ad5/3-E2F-d24-aMUC1aCD3-derived aMUC1 binding to MUC1 antigen. (B) Percentage of activated CD3+CD4+CD69+CD8− and CD3+CD8+CD69+CD4− and (C) CD3+CD4+CD25+CD8− and CD3+CD8+CD25+CD4− T cells assessed by flow cytometry in co-cultures with MUC1+ T47D tumor cells. T47D cells were infected with Ad5/3-E2F-d24 or Ad5/3-E2F-d24- aMUC1aCD3 virus using 1,000 pfu/mL and co-cultured with unstimulated T cells at ratio T:E = 5. After 72 h post infection, cells were collected, washed, stained with antibodies, and analyzed by flow cytometry. (D) Evaluation of cytokines GrzmB, (E) IL2, (F) TNF-α, and (G) IFN-γ in the supernatants harvested from infected cells for the validation of T cell activation using BD FACS Array bioanalyzer. (H) aMUC1aCD3 expressed by Ad5/3-E2F-d24-aMUC1aCD3-infected cells induces T cell proliferation. Carboxyfluorescein succinimidyl ester (CFSE)-labeled T cells were co-cultured with T47D cells (T:E = 5) in the presence of the indicated supernatants. Five days after co-culture, CFSE dilution (i.e., cell proliferation) in CD3+ T cells was evaluated by flow cytometry. (I) Ad5/3-E2F-d24-aMUC1aCD3-derived supernatant mediated T47D cell killing in the presence of an effector T cell (T:E = 5); 30 kDa concentrated supernatant was used and cell viability was measured on day 3 under Abs 450 nm. All experiments were run in quadruplicates, and the resulting data are presented as mean ± SEM. Statistical significance is represented as ∗p < 0.05, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Subsequently, we infected a MUC1+ cell monolayer (T47D) with armed and unarmed viruses followed by incubation with unstimulated T cells. Compared with the experimental setting mentioned in Figure S3, where virally released aMUC1aCD3 supernatants were added to the monolayer of tumor cells instead of the virus, this setting has more therapeutic relevance, as it mimics aMUC1aCD3-mediated activation of T cells regardless of their endogenous specificity. We found statistically higher levels of activated CD4+CD69+CD8− (∗p = 0.0225), CD4+CD25+ CD8− (∗∗∗∗p < 0.0001), and CD8+CD69+CD4− (∗∗∗∗p < 0.0001) T cells in the presence of aMUC1aCD3− virus than in the tumor + T cell group (Figures 3B and 3C). The co-cultures treated with aMUC1aCD3-virus had statistically significant levels of granzyme B (GrzmB) (∗p = 0.0253), IL2 (∗∗∗p = 0.0005), TNF-α (∗∗∗p = 0.0004), and interferon gamma (IFN-γ) (∗p = 0.0234) expression compared with T cell supernatants (Figures 3D–3G) and backbone supernatants (GrzmB, ∗p = 0.0210; IL2, ∗∗∗p = 0.0006; TNF-α, ∗∗∗p = 0.0004; IFN-γ, ∗p = 0.0173). A similar experimental setting was repeated using a co-culture of different supernatants and unstimulated T cells in a T47D monolayer (Figures S3A–S3F) with similar findings.
Additionally, upon co-culture with aMUC1aCD3 supernatants, T cells presented a statistically significantly higher proliferative capacity (∗∗∗∗p < 0.0001), as demonstrated by the total count of CFSE-diluted CD3+ T cells present in co-culture with aMUC1aCD3 supernatants (Figure 3H). Cytokine analysis of the supernatants collected from such experiments confirmed T cell activation and proliferation, as shown in Figure S4.
When the concentration of aMUC1aCD3 supernatant was increased from 100 kDa (Figure S5) to 30 kDa filtration, cell viability decreased significantly from 50% to nearly complete lysis, demonstrating a dose-dependent response (Figure 3I).
Ad5/3-E2F-d24-aMUC1aCD3 virus improves tumor growth control in a human xenograft model of lung cancer
To study the therapeutic efficacy of Ad5/3-E2F-d24-aMUC1aCD3, we employed an in vivo human xenograft model of lung cancer. Intratumoral injections were administered according to the therapeutic scheme shown in Figure 4A. Human PBMC were injected intratumorally into mice to reconstitute the immunological dynamics of human tumors 48 h before virus administration. Overall, Ad5/3-E2F-d24-aMUC1aCD3 provided statistically superior tumor growth control by day 33 compared with the backbone and mock controls (Figures 4B–4D). After sample collection on day 35, the remaining mice (n = 5) from each group were assigned to a follow-up experiment. Mice from the PBMC and backbone + PBMC groups were euthanized on day 54 because of graft-versus-host disease (GVHD). All mice in the Ad5/3-E2F-d24-aMUC1aCD3+PBMC group remained alive until day 61, after which they developed similar symptoms recognized as GVHD. The pathologist confirmed the graft-versus-host reaction by identifying interstitial pneumonia (histiocytes, lymphocytes, and plasma cells) in the lungs. The graft-versus-host was moderate in the Ad5/3-E2F-d24-MUC1aCD3+PBMC and Ad5/3-E2F-d24+PBMC-treated groups, while the PBMC group showed a severe reaction. Furthermore, the Ad5/3-E2F-d24-MUC1aCD3+PBMC group had only mild hepatitis, whereas the Ad5/3-E2F-d24+PBMC and PBMC groups seemed to have moderate hepatitis. Presumably, the graft-versus-host was milder in Ad5/3-E2F-d24-MUC1aCD3+PBMC animals because more T cells were present in tumors than in normal organs. Other organs, such as the heart, spleen, and kidneys, showed no specific changes.
Figure 4.

In vivo antitumor efficacy of Ad5/3-E2F-d24-aMUC1aCD3 virus
(A) Schematic diagram of the in vivo experimental plan. Mice bearing subcutaneous A549 tumors were injected intratumorally with PBS or 1 × 1010 VP/tumor of Ad5/3-E2F-d24 or Ad5/3-E2F-d24-aMUC1aCD3 virus. Two days before virus administration, mice received an intratumoral injection of either 1 × 107 PBMC or PBS. Five animals from each group were euthanized on day 35 to collect their tumors and organs for biological assays (dashed line). (B) Overall tumor growth curve of all experimental groups receiving PBMC. (C) Quantification of E1A gene copy number as a measure of virus replication. Resulting data are presented as mean ± SEM (n = 10, per group). Statistical significance is represented as ∗∗p = 0.01, ∗∗∗∗p < 0.0001.
There was statistically better tumor growth control in the Ad5/3-E2F-d24-aMUC1aCD3+PBMC group than in the Ad5/3-E2F-d24+PBMC (∗∗∗∗p < 0.0001) and PBMC (∗∗∗∗p < 0.0001) groups when PBMC were present. The overall survival of Ad5/3-E2F-d24-aMUC1aCD3+PBMC-treated mice was statistically significant compared with that of Ad5/3-E2F-d24+PBMC (∗p = 0.0279) and PBMC-only groups (p = 0.0302) (Figure S6A) in the follow-up study. Quantitative analysis of viral DNA present in the tumors harvested on day 35 revealed the expression of the E1A gene in all groups treated with adenoviruses (Figure 4F). The levels of viral DNA in Ad5/3-E2F-d24-aMUC1aCD3 tumor samples were comparable with those in the unarmed virus, indicating replication of Ad5/3-E2F-d24-aMUC1aCD3 in an in vivo setting.
aMUC1aCD3-armed oncolytic virus therapy is associated with an increase in the intratumoral cytotoxic tumor microenvironment
The immunological mechanism of Ad5/3-E2F-d24-aMUC1aCD3 virus treatment was studied by analyzing different immune cell subsets in tumors collected on day 35. The total percentage of CD3+ T lymphocytes in the Ad5/3-E2F-d24-aMUC1aCD3 group was statistically significantly greater than that in the Ad5/3-E2F-d24 (∗∗p = 0.0011) and PBMC-only control groups (∗∗∗p = 0.0002) (Figure 5A), thus demonstrating the ability of aMUC1aCD3 to increase T cell activation and proliferation.
Figure 5.

Evaluation of immunological changes in the tumor microenvironment in Ad5/3-E2F-d24-aMUC1aCD3 virus-treated animals
(A–F) Tumors collected at day 35 were stained with fluorochrome-conjugated antibodies and analyzed by flow cytometry. The graphs show the total percentage of (A) CD3+ T cell in tumor, (B) tumor infiltrating CD3+CD4+ or CD3+CD8+ T cells, (C) CD3+CD4+CD69+ or CD3+CD8+CD69+ T cells, (D) TIM-3-expressing cells, and (E) CD3+CD8+TIM-3+ cells. (F) PD-1+ cells out of CD3+CD4+ or CD3+CD8+ T cell. Differences in cell percentages were analyzed with the one-way ANOVA test. Mean ± SEM are shown (n = 10). Statistical significance is represented as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Next, we investigated the percentage of CD3+CD4+ and CD3+CD8+ T lymphocytes. By day 35, the aMUC1aCD3-armed virus induced a statistically higher level of CD3+CD4+ (∗∗p = 0.0026) and CD3+CD8+ (∗∗∗∗p < 0.0001) T cells (Figure 5B) than the PBMC-only control group. Similarly, the total percentage of activated CD3+CD4+CD69+ and CD3+CD8+CD69+ T cells in tumors treated with aMUC1aCD3-armed adenovirus was statistically greater than that in the PBMC-only group (∗∗∗∗p < 0.0001) (Figure 5C). The presence of higher levels of cytotoxic T lymphocytes post viral infection in a tumor indicates a shift toward a pro-inflammatory tumor microenvironment.53,54 This finding confirmed that the virus modulates the T cell compartment by increasing the percentage of T cells, particularly activated T cells, when aMUC1aCD3-armed virus is used.
When analyzing the profile of exhausted immune cells in the tumors, groups treated with oncolytic adenoviruses showed a reduced fraction of T cell immunoglobulin and mucin domain-3 (TIM-3; a marker of immunosuppression)-positive cells, as shown in Figure 5D. The PBMC-only control group showed the statistically highest percentage of CD3+CD8+TIM-3 T cells (∗∗p = 0.0026, Figure 5E). In the armed virus group, the percentages of PD-1 cells out of CD3+CD4+ and CD3+CD8+ T cells increased noticeably, but not statistically significantly, compared with the unarmed virus and PBMC control groups (Figure 5F). PD-1 expression indicates T cell activation.
Oncolytic adenovirus armed with aMUC1aCD3 improves aMUC1aCD3-BsTe persistence in the tumor and antitumor effects in PDX-OvCa cells
To understand the immunological mechanism behind Ad5/3-E2F-d24-aMUC1aCD3 virus-mediated tumor growth control, we used an in vivo human xenograft model of PDX-OvCa tumors.55 All groups in this study received autologous PBMC. The therapeutic scheme for intratumoral injections is shown in Figure 6A. Overall, these findings suggest that the Ad5/3-E2F-d24-aMUC1aCD3 virus lysed tumor cells and released aMUC1aCD3-BsTe, resulting in greater T cell activity and enhanced antitumor efficacy in the PDX-OvCa model in vivo, ultimately extending the in vivo persistence of aMUC1aCD3-BsTe at tumor.
Figure 6.

Evaluation of in vivo persistence of aMUC1aCD3-BsTe and comparative antitumor efficacy of Ad5/3-E2F-d24-aMUC1aCD3 virus versus aMUC1aCD3-BsTe in PDX-OvCa xenograft mice model
(A) Schematic diagram of the in vivo experimental plan. (B) Tumor growth curve showing Ad5/3-E2F-d24- or Ad5/3-E2F-d24-aMUC1aCD3 virus-mediated antitumor efficacy in mice bearing subcutaneous PDX-OvCa tumors. (C) Detection of aMUC1aCD3-BsTe transgene in tumors versus serum samples collected at day 22 by In-Cell ELISA. (D) CD3+ T cell in tumor, (E) tumor-infiltrating CD3+CD4+ T cell, (F) tumor-infiltrating CD3+CD8+ T cell, (G) total percentage of CD3+CD4+CD69+ or CD3+CD8+CD69+ T cell in tumor. Differences in cell percentages were analyzed with the one-way ANOVA test. Mean ± SEM are shown (n = 7). Statistical significance is represented as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
First, the antitumor efficacy of Ad5/3-E2F-d24-aMUC1aCD3 virus was compared with that of aMUC1aCD3-BsTe alone. The Ad5/3-E2F-d24-aMUC1aCD3 virus-treated group statistically significantly outperformed all the other groups in terms of tumor growth control, including PBMC (∗∗∗∗p = <0.0001), Ad5/3-E2F-d24 (∗∗∗∗p = <0.0001), aMUC1aCD3-BsTe (∗∗∗∗p = <0.0001), and Ad5/3-E2F-d24 + aMUC1aCD3-BsTe (∗∗∗∗p = 0.0006) as shown in Figure 6B.
Then, the in vivo persistence of aMUC1aCD3-BsTe was investigated using tumor and blood serum samples. The prolonged in vivo persistence of aMUC1aCD3-BsTe in tumor versus serum samples was confirmed (Figure 6C). This finding suggests that the Ad5/3-E2F-d24-aMUC1aCD3 virus releases aMUC1aCD3-BsTe molecules at the tumor tissues, followed by tumor cell lysis after virus replication. Therefore, aMUC1aCD3-BsTe is more likely to persist in tumors in vivo. However, no aMUC1aCD3-BsTe was found in the blood serum samples collected at both times points: 48 h (Figure S7A) after the first treatment and on day 22.
T cell populations, such as CD3+, CD3+CD4, CD3+CD8, and those expressing CD69+, were assessed to further investigate the immunological events that occurred in the Ad5/3-E2F-d24-aMUC1aCD3+PBMC virus-treated group. The total percentage of CD3+ T cells (Figure 6D) was statistically significant in the Ad5/3-E2F-d24-aMUC1aCD3+PBMC virus-treated group compared with that in the PBMC (∗∗p = 0.0016), Ad5/3-E2F-d24 (∗p = 0.0160), aMUC1aCD3-BsTe+PBMC (∗∗p = 0.0013), and Ad5/3-E2F-d24+aMUC1aCD3-BsTe+PBMC (∗p = 0.0185) groups. When comparing CD3+CD4+ (Figure 6E) and CD3+CD8+ (Figure 6F) T cell populations in the Ad5/3-E2F-d24-aMUC1aCD3+PBMC virus-treated group with PBMC and Ad5/3-E2F-d24+PBMC groups, the results were similar, with a noticeably higher level but not statistically significant in the aMUC1aCD3-BsTe+PBMC and Ad5/3-E2F-d24 + aMUC1aCD3+PBMC groups, and the total percentage of CD3+CD4+CD69+ T cells (Figure 6G) was higher in the Ad5/3-E2F-d24-aMUC1aCD3 virus group, but not statistically significant compared with the other groups. However, the total percentage of CD3+CD8+CD69+ T cells was statistically significant compared with the aMUC1aCD3-BsTe+PBMC (∗p = 0.0165) group. In conclusion, a T cell profiling study in PDX-OvCa tumor samples confirmed that the Ad5/3-E2F-d24-aMUC1aCD3 virus increases T cell activity by engaging and activating T cells in tumor cell death.
Discussion
Adenoviruses are well-characterized, easily modified, and immunologically powerful tools for the production of oncolytic vectors that directly lyse tumor cells.18 They can also be armed with transgenes to promote tumor-specific lysis, antigen presentation, and immunostimulation.56 In this study, we constructed and performed in vitro and in vivo validations of a human MUC1 targeting T cell engager-armed oncolytic adenovirus. BsTe expressed by TILT-321 virus binds to the MUC1 antigen expressed by target tumor cells and CD3 receptors on the T cell surface. We found that the cytotoxicity of aMUC1aCD3-armed viruses in the presence of human T cells and MUC1-expressing cancer cells was higher than that of the controls, both in vitro and in vivo. However, the cell-killing potency of Ad5/3-E2F-d24-aMUC1aCD3 virus was not limited by the presence of MUC1 antigen in target cells.57 The virus also replicates and lyses MUC1-negative HEK-293 cells. While the virus replicates in tumor cells that are defective in the Rb/p16 pathway, the cell line produces adenoviral E1, which renders the Rb/p16 pathway not needed for virus replication.57 In vivo, Ad5/3-E2F-d24-aMUC1aCD3 virus-treated tumors demonstrated statistically superior tumor growth control and higher T cell activation in humanized A549 and PDX-OvCa xenograft models. These findings indicate that the virally encoded aMUC1aCD3-BsTe binds to its target tumor antigen and T cells simultaneously and mediates tumor cell killing, which is independent of major histocompatibility complex (MHC) presentation. However, the use of unmatched human leukocyte antigen PBMC in the A549 xenograft model resulted in a mild-to-moderate GVHD reaction, which was not observed in the PDX-OvCa xenograft with autologous PBMC. On day 47 of the A549 xenograft, the first mice developed visible GVHD symptoms, which were followed by tumor size suppression, skin rashes, and hair loss. Soon after, the experiment was terminated. While designing and carrying out the experiment, we attempted to minimize possible confounding factors for the antitumor efficacy readout (i.e., survival) of the in vivo experiments. For example, the same amount of virus and number of PBMC was administered to each group of mice and PBMC delivered intratumorally rather than intravenously. Given that visible signs of GVHD only appeared late in the study, this allows us to conclude that GVHD had little impact on tumor size or immunological (day 35) read-outs, and that antitumor efficacy could still be deduced from the various therapies tested. However, the phenomenon may have affected in vivo survival. The GVHD is a caveat of the humanized model using heterologous human PBMC in mice, and this is not expected to occur in humans, where the BsTe encoded by the virus recruits and activates autologous PBMC.
Engagement of tumor antigens and T cells by aMUC1aCD3-BsTe leads to the formation of immunological synapse structure and organization, with subsequent modulation of T cell function and persistence.12 Evidence of T cell activation was confirmed through the expression of cytokines such as GrzmB, IL2, TNF-α, and IFN-γ. Our in vitro data showing higher T cell function in the TILT-321 virus-treated group is in accordance with a study that combined an oncolytic adenovirus expressing EGFR-BiTE.15 Activated cytokine-induced killer cells armed with aMUC1/CD3 BsTe are currently being tested in phase II clinical trials (NCT03146637) for the treatment of advanced liver cancer.58 In this study, we utilized an approach in which aMUC1aCD3-BsTe is produced locally in the tumor by an oncolytic adenovirus. The advantage of our approach is that the virus self-amplifies at the tumor site, while T cells are recruited and activated to potentiate the effect of oncolysis. Oncolysis has immunological consequences that synergize with T cell activation, including pro-inflammatory signaling in the tumor microenvironment and induction of de novo antitumor immunity.19
MUC1 is a heavily glycosylated transmembrane protein of the mucin family, with a highly glycosylated extracellular domain.30 MUC1 is a highly appealing target for cancer immunotherapy due to three key characteristics. First, elevated levels are found in many tumors, most notably those of the breast and ovary (Figure S1), due to transcriptional upregulation. Second, although MUC1 is normally restricted to the luminal epithelium, its expression polarity is lost in response to stress and during transformation. Third, MUC1 glycosylation is severely dysregulated in cancer cells. Tumor-associated MUC1 contains a higher proportion of shorter glycans, such as Tn, sialyl Tn (STn), T (Thomsen-Friedenreich), and ST, owing to altered glycosyltransferase expression.33 MUC1 under glycosylation reveals cryptic epitopes within the variable-number tandem repeat, allowing several antibodies to bind the tumor specifically.33
We tested three sets of unstimulated T cells from healthy donors and examined the ability of virally released aMUC1aCD3 to activate T cells against tumor cells. Different donors provided slightly different responses, as expected. However, compared with the unarmed and control groups, the aMUC1aCD3-BsTe-treated group showed consistently greater T cell activity. This is also an expected outcome, as binding to CD3 is known to cause T cell activation. The competitive-binding In-Cell ELISA assay was used to demonstrate the binding of aMUC1aCD3-BsTe-containing supernatant samples from Ad5/3-E2F-d24-aMUC1aCD3-infected cells. Filtration of supernatants with 100-, 30-, and 10-kDa filters was an attempt to remove unwanted molecules to obtain pure aMUC1aCD3-BsTe. Nevertheless, other unwanted debris (such as cell proteins and cell debris) may still be present, which can ultimately affect the output of the assay results. The ideal scenario would include commercially available recombinant aMUC1aCD3-BsTe as a positive control. One of the major limitations of this study was the lack of a recombinant aMUC1aCD3-BsTe-positive control, which can be difficult to synthetize in adequate amounts to use in vitro and in vivo.
The lack of aMUC1aCD3-BsTe in blood serum specifies tumor-specific lysis of the Ad5/3-E2F-d24-aMUC1aCD3 virus, resulting in minimal or no aMUC1aCD3-BsTe molecule leakage outside the tumor site. A review conducted by Goebeler and Bargou8 suggested that the short half-life of blinatumomab in the serum is 2–3 h. In this study, the detection of aMUC1aCD3-BsTe to target MUC1 antigen in PDX-OvCa tumors harvested 48 h after virus treatment suggested the extended persistence of aMUC1aCD3-BsTe in vivo and may also signify the oncolytic adenovirus mediated half-life extension. To further confirm this finding, aMUC1aCD3-BsTe concentrations will need to be precisely measured using techniques such as mass spectrometry. In addition, a biodistribution and tolerability study of TILT-321 should be studied under Good Laboratory Practice conditions before clinical trials in future.
Furthermore, we investigated whether T cells became exhausted over time after continuous stimulation with virally released aMUC1aCD3. We choose an exhaustion marker, TIM-3, and examined its expression in tumors treated with Ad5/3-E2F-d24-aMUC1aCD3+PBMC. Adenovirus (armed and backbone virus)-treated mice had a lower level of TIM-3, suggesting beneficial effects in the tumor microenvironment. More importantly, this finding supports the evidence presented previously59 that reported a direct inverse correlation between TIM-3+ infiltrating CD8+ T cells and survival in patients with cancer. We also observed an increase in PD-1+ cells from tumors treated with BsTe virus.
Arming an oncolytic adenovirus with aMUC1aCD3-BsTe is an appealing strategy for the treatment of solid tumors to overcome the limitations of the short serum half-life of BsTe molecules when used as recombinant molecules. This approach can also result in enhanced potency overuse of unarmed viruses. In addition to stimulating tumor-specific immunity, we can speculate that the aMUC1aCD3-armed virus induces an antiviral response at the local tumor site.
Conclusions
We developed a novel approach to treat cancer that overcomes the limitations of oncolytic viruses and BsTe when used as monotherapies. The proposed technology could be preferentially beneficial for the treatment of solid tumors expressing MUC1. Our results lay the groundwork for clinical exploration, the translation of which is currently ongoing.
Materials and methods
Cell lines
The human cancer cell lines A549 (lung adenocarcinoma), T47D (breast epithelial adenocarcinoma), and HEK-293 (human embryonic kidney) were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). All cell lines were cultured under the recommended conditions.
PDX-OvCa cells were generated as described previously.55
Preparation of PBMC and T cell isolation and expansion
Human PBMC were isolated from whole blood (Finnish Red Cross Blood, Finland) using lymphoprep (StemCell Technologies, USA) gradient density separation under sterile conditions. Cells were washed twice with PBS (Sigma, United Kingdom) and erythrocytes were lysed with ammonium-chloride-potassium (ACK) red blood cell lysis buffer (Sigma). T cells were negatively isolated using a human Pan T isolation kit (130-096-535, Miltenyi Biotec, Germany), following the manufacturer’s instructions.
Virus construction
The Ad5/3-E2F-d24-aMUC1aCD3/TILT-321 virus was constructed using a previously described technique.38 The genomic modifications, such as an E2F promoter and a 24-bp deletion in the constant region of E1A, were made for tumor-specific replication as described previously.38
The single-chain variable fragments of both aMUC1 and aCD3 were connected through a Gly-Gly-Gly-Ser flexible linker. The aMUC1aCD3-BsTe sequence was optimized for human codon usage and was synthesized using GenScript (GenScript, Germany). The resulting sequence was introduced into the E3 region by replacing the gp19k and 6.7k genes via a bacterial artificial chromosome (BAC) recombineering strategy38,60 and confirmed by next-generation sequencing.
The viruses were propagated in A549 cells and purified using cesium chloride gradient centrifugation. The tissue culture infectious dose (TCID50) assay was used to determine viral infectivity as described previously.38 The viral particle (VP)/mL titers of the purified viruses were determined by optical density 260 (OD260) readings.
Supernatant production and purification
HEK-293 (MUC1 negative) cells were infected with Ad5/3-E2F-d24 and Ad5/3-E2F-d24-aMUC1aCD3 viruses at a multiplicity of infection (MOI) of 100, and supernatants were harvested 48 h post infection. The 59-kDa aMUC1aCD3-BsTe molecules in the supernatants were filtered using Amicon Ultra-15 filter units (Merck Millipore, USA) with a molecular weight cutoff of 100 kDa to eliminate molecules >100 kDa. The supernatants were concentrated using 30- and 10-kDa filters to eliminate >30 and >10 kDa, respectively, when needed. Supernatants from uninfected cells were used as mock or negative controls (media only).
Competitive ELISA binding assays
A competitive binding assay was designed to detect the binding of aMUC1aCD3 to the target tumor antigen using an In-Cell ELISA Colorimetric Detection Kit (Invitrogen, 62200) and a commercially available anti-MUC1 antibody (clone HMFG2) that shared the same binding epitope as aMUC1aCD3-BsTe.
A 3-fold dilution (1:1, 1:10, and 1:100) of Ad5/3-E2F-d24-aMUC1aCD3 virus-infected 10-kDa (stock concentration 3.85 mg/mL) filtered supernatants was added and incubated with a T47D monolayer expressing MUC1 antigen for 1 h at 37°C to occupy the most available binding pockets, ensuring that no places were reserved for the commercial anti-MUC1 antibody. When evaluating aMUC1aCD3-BsTe persistence in vivo, PDX-OvCa tumor supernatants were harvested on day 22, and serum samples were collected at two time points (48 h after the first virus treatment and on day 22) and were incubated with a T47D cell monolayer to allow it to bind with MUC1 antigen expressed on the cell surface.
After incubation, the commercially available anti-MUC1 antibody (BioLegend, HMFG2) was incubated overnight at 4°C. Binding was detected by further incubating the target with horseradish peroxidase (HRP) conjugate, which detected immunoglobulin G (IgG) at absorbance (Abs) 450 nm and Abs 615 nm using a Hidex Sense (Turku, Finland).
Western blot analysis
GenScript (GenScript, Germany) performed western blot analysis to visualize the molecular weight of aMUC1aCD3-BsTe. CHO-expressing host cells were transfected with aMUC1aCD3, and cell lysate supernatants were collected on day 3 and day 6 post transfection under reducing conditions. Briefly, CHO cells were electrophoresed on denaturing 12% SDS-PAGE gels and the resulting gels were stained with Coomassie blue dye to assess purity.
T cell activation and proliferation assay
The functionality of the virally released BsTe was assessed through co-culture experiments. Ten-thousand target cells per well were seeded in 96-well plates in 100 μL of growth medium in quadruplicate, unless indicated otherwise. To assess T cell activation, target cells were mixed with 15 μL of 30-kDa concentrated supernatants and 50,000 effector cells (unstimulated T cells) per well at target to effector (T:E) cell ratio of 5 followed by 3 days of incubation. For the T cell proliferation assay, incubation was performed for 5 days. After incubation, the supernatants were collected and the cells were stained with antibodies targeting CD3, CD4, CD8, CD69, and CD25 surface receptors for flow cytometry. A detailed list of antibodies used is provided in Table S1.
Cell viability assay
Human cell lines A549 and T47D were plated in 96-well plates (flat bottom) in quadruplicates at 10,000 cells/well for 24 h and infected with 1, 10, 100, 1,000, or 10,000 VP/cell of either Ad5/3-E2F-d24 virus (also referred to in the text as a backbone or unarmed backbone) or Ad5/3-E2F-d24-aMUC1aCD3 (also referred to as an armed virus or TILT-321). Cell viability was measured after 3 days for A549 cells and 5 days for T47D cells by incubating wells for 1 h with 20% of CellTiter 96 Aqueous One Solution Proliferation Assay reagent (Promega, Wisconsin, USA). Absorbance was measured at 490 nm using a Hidex Sense spectrophotometer (Turku, Finland). Cell viability was determined by normalizing the absorbance data from treated wells to the absorbance data of negative controls.
Alternatively, to assess the cell viability of co-cultures treated with virally released aMUC1aCD3-BsTe in supernatants, 100 μL of 100-kDa filtered and 15 μL of 30-kDa filtered supernatants were mixed at T:E = 5. On day 3 post infection, the total supernatant was removed and the wells were gently washed twice with 100 μL of PBS. Cell viability was measured using 20% of CellTiter 96 AQueous One Solution Proliferation Assay reagent, as described above.
Oncolysis-enhanced cell-mediated cytotoxicity was assessed using an iCELLigence Real-Time Cell Analyzer (RTCA) iCELLigence system (Agilent Technologies, USA). A total of 50,000 T47D, A549, and HEK-293 cancer cells were seeded per well and incubated for 24 h at 37°C in a humidified chamber for overnight cell adherence. Cells were infected with Ad5/3-E2F-d24 or Ad5/3-E2F-d24-aMUC1aCD3 viruses at 1,000 plaque-forming units (pfu)/mL. Effector cells (unstimulated CD3+ T cells) were added at a ratio T:E = 5. Cell index (i.e., relative cell impedance) values were monitored every 15 min for 120 h and normalized to the cell index value immediately after treatment. The iCELLigence RTCA Agilent Technologies software (version 1.0) was used for data analysis.
Cytokine analysis
Supernatants from the same virus-infected tumor cultures were collected and pooled together on day 3, and the presence of human GrzmB (560304), IL2 (558270), TNF-α (558273), and IFN-γ (558269) levels were determined using the BD Cytometric Bead Array (CBA) FlexSet kits following the manufacturer’s instructions. Data were acquired on a BD Accuri (BD Biosciences, USA) and analyzed using FCAP Array Software (catalog no. 641488).
Rosette microscopy
A total of 25,000 T47D cells per well were stained with CellTrace Far Red dye (Invitrogen, Massachusetts, United States) and seeded in a four-well μ-Slide (Ibidi, Martinsried, Germany) glass-bottom plate. After 24 h, unstimulated T cells were stained with CellTrace CFSE (Invitrogen, Massachusetts, United States) dye at a T:E ratio of 5, followed by the addition of Ad5/3-E2F-d24 or Ad5/3-E2F-d24-aMUC1aCD3 virus-infected cell supernatant. Live-cell imaging was performed for 4 h using Nikon Eclipse Ti-E. Each well was washed gently with PBS, and images of T cell rosette formation (i.e., circular or semi-circular shape of T cells [n = 4] around tumor cells) were taken after 4 h. The total number of T cell rosettes was quantified using Fiji macro software. After red tumor cells were gated, Fiji macro software was used to detect green T cells surrounding the red cells. The software generated a total count of T cells around tumor cells, which were then sorted to characterize the T cell rosettes. The formation of a rosette around the T cell was considered when the ratio of tumor cells to T cells was greater than or equal to 4. The minimum T cell threshold of 4 was set because it suggests the shape of a semi-circle, which is an early indication of rosette formation.
In vivo animal experiment
Four-week-old female immunodeficient NOD/SCID/IL2rg−/− (NSG) mice (Jackson Laboratory, USA) were used for in vivo studies.
For the Ad5/3-E2F-d24-aMUC1aCD3 virus-mediated antitumor efficacy and survival study, A549 tumors (5 × 106 cells per animal) were engrafted subcutaneously on their right lower back. When tumors reached 3–4 mm in the longest diameter, animals were randomized and treated intratumorally with 1 × 1010 VP per injection of Ad5/3-E2F-d24 or Ad5/3-E2F-d24-aMUC1aCD3 virus. The mock control group was injected intratumorally with PBS. Animals were treated every 3 days for a total of 12 rounds of injections. Two days before virus administration, the mice received an intratumoral injection of either 1 × 107 PBMC (n = 10 per group) or PBS (n = 10 per group).
To compare the activity of adenovirus-delivered BsTe therapy and BsTe therapy alone, 5 × 106 cells per animal of PDX-OvCa tumors were engrafted subcutaneously with 50% Matrigel (Corning, New York, USA) on their right lower back. After tumors reached 3–4 mm in diameter, the animals were randomized, and 1 × 107 autologous PBMC (n = 7 per group) were administered intratumorally. In addition, eight rounds of treatment with 1 × 1010 VP per Ad5/3-E2F-d24 or Ad5/3-E2F-d24-aMUC1aCD3 injection and 400 ng per tumor of virally released 10-kDa concentrated aMUC1aCD3-BsTe alone or in combination with Ad5/3-E2F-d24 virus was administered intratumorally. PBS was injected into the PBMC group. Blood was collected from the tails of mice after 48 h after the first virus administration to determine the persistence of aMUC1aCD3-BsTe in vivo in the serum sample. On day 22, tumors and blood were harvested from the mice.
A digital caliper was used to measure the tumor size, and tumor volumes were calculated as (length × width2)/2. Tumor volumes from the mechanism of action studies (n = 5 per group) were plotted, while the animals enrolled in the follow-up experiment were measured until the end of the experiment (day 53).
Histopathology analysis
Organs (liver, lung, heart, spleen, and kidney) from animals euthanized on day 35 were collected for histopathological analysis, stored in a 10% formalin solution for 24 h, and then transferred to 70% ethanol until paraffin embedding. Paraffin blocks were sectioned into 4- to 5-μm-thick slides and stained with hematoxylin and eosin. A veterinary pathologist evaluated the histological changes in the stained samples.
qPCR
Fragments of animal tumor samples were harvested on day 35, snap frozen, and stored at −80°C until further processing. DNA was extracted from these samples using the QIAamp DNA Mini Kit (51306, QIAGEN, Hilden, Germany) following the manufacturer's instructions. The purified DNA was quantified to check the relative expression of the E1A gene using Light Cycler Probes master mix (Roche, Basel, Switzerland), 50 μM forward (5′-TCCGGTTTCTATGCCAAACCT-3′) and reverse (5′-TCCTCCGGTGATAATGACAAGA-3′) primers with 10 μM probes (5′-FAM-TGATCGATCCACCCAGTGA-3′MGBNFQ), as described previously.61 The total gene expression was normalized to that of the hBactin housekeeping gene.62
Flow cytometry analysis
For all in vitro assays analyzed by flow cytometry, cells were stained with human aCD3 (PE-CF594, UCHT1, BioLegend), aCD4 (PE, RPA-T4, BioLegend), aCD8 (fluorescein isothiocyanate [FITC], RPA-T8, BioLegend), aCD69 (PE-Cy7, FN50, BioLegend), and aCD25 (PE-Cy5, M-A251, BioLegend) antibodies.
A panel of different tumor cell lines was stained with human aMUC1 (APC, 16A, BioLegend), and MUC1 expression was assessed using flow cytometry to select MUC1-positive and MUC1-negative cell lines.
Immunological analysis of tumors derived from the humanized xenograft mouse model, antibodies specific for human aCD3 (FITC, OKT3, BioLegend), aCD4 (AF700, A161A1, BioLegend), aCD8 (BV421, SK1, BioLegend), aCD69 (PE-Cy7, 17A2, BioLegend), aCD56 (APC, FN50, BioLegend), aTIM3 (PE, F38-2E2, BioLegend), and aPD-1 (PE-CF594, NAT105, BioLegend) were used in the NovoCyte Quanteon and NovoSampler Q System Bundle Flow Cytometry analyzer (Agilent, USA). Cell data processing and gating were performed using FlowJo v.10.6.1 (Ashland, USA). All antibodies used in this study are listed in Table S1.
Statistical analysis
GraphPad Prism v.9.2.0 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis and graphical representation of the data. For the comparison of two groups of in vitro assays, two-way ANOVA with Bonferroni tests was used, and one-way ANOVA with Tukey’s post hoc test was used to compare more than two groups of in vitro assays. Welch’s t test was used to compare the statistical difference among different group in the iCElligence assay. Tumor growth curves were compared using a mixed-model analysis. Survival curves were generated using the Kaplan-Meier method, and the differences in the two curves were compared using the log rank test. Results were considered statistically significant at p < 0.05. Data are presented as mean ± standard error of the mean (SEM).
Availability of data and material
All data are available upon request.
Acknowledgments
We thank Minna Oksanen for expert experimental and administrative assistance. We also thank the BIU Biomedicum Imaging Unit (University of Helsinki, Helsinki, Finland), the Laboratory Animal Center (LAC, University of Helsinki, Helsinki, Finland), and the Biomedicum Flow Cytometry Unit (University of Helsinki, Helsinki, Finland).
All animal experiments described in the paper were approved by the Provincial Government of Southern Finland and the Experimental Animal Committee of the University of Helsinki (license number ESAVI/12559/2021).
This study was supported by the Jane and Aatos Erkko Foundation, HUCH Research Funds (VTR), Finnish Cancer Organizations, University of Helsinki, TILT Biotherapeutics Ltd., Sigrid Juselius Foundation, Novo Nordisk Foundation, Päivikki, and Sakari Sohlberg Foundation. We thank the Orion Foundation, Ida Montin Foundation, and Finnish Cultural Foundation for grant support. This program was supported by an educational grant from the Gilead Nordic Fellowship Program. We thank Albert Ehrnrooth and Karl Fazer for their support.
Author contributions
S.B., J.S., S.V.K., J.C., and A.H. designed the experiments. S.B., D.Q., V.A., S.P., and T.V.K. conducted the experiments. S.B., J.S., and A.H. analyzed the results. All authors contributed to the writing and reviewing of the manuscript.
Declaration of interests
A.H. is a shareholder of Targovax ASA. (Norway). A.H., J.C., J.M.S., V.C.C., S.S., and R.H. are employees and shareholders of TILT Biotherapeutics, Ltd. Other co-authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.12.007.
Supplemental information
References
- 1.Arnone C.M., Polito V.A., Mastronuzzi A., Carai A., Diomedi F.C., Antonucci L., Petrelli L.L., Vinci M., Ferrari F., Salviato E., et al. Oncolytic adenovirus and gene therapy BiTE for the treatment of with EphA2- grade gliomas. J. Immun. Can. 2021;9 doi: 10.1136/jitc-2020-001930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hao S., Inamdar V. v, Sigmund E.C., Zhang F., Stephan S.B., Watson C., Weaver S.J., Nielsen U.B., Stephan M.T. NC-ND license BiTE secretion from in situ-programmed myeloid cells results in tumor-retained pharmacology. J. Control Release. 2022;342:14–25. doi: 10.1016/j.jconrel.2021.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Einsele H., Borghaei H., Orlowski R.Z., Subklewe M., Roboz G.J., Zugmaier G., Kufer P., Iskander K., Kantarjian H.M. The BiTE (bispecific T-cell engager) platform: development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer. 2020;126:3192–3201. doi: 10.1002/cncr.32909. [DOI] [PubMed] [Google Scholar]
- 4.Suryadevara C.M., Gedeon P.C., Sanchez-Perez L., Verla T., Alvarez-Breckenridge C., Choi B.D., Fecci P.E., Sampson J.H. Are BiTEs the “missing link” in cancer therapy? Oncoimmunology. 2015;4:e1008339. doi: 10.1080/2162402X.2015.1008339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khalique H., Baugh R., Dyer A., Scott E.M., Seymour L.W., Frost S., Larkin S., Lei- J. Oncolytic herpesvirus expressing PD- BiTE for cancer therapy : exploiting tumor immune suppression as an opportunity for targeted immunotherapy. J. Immun. Can. 2021 doi: 10.1136/jitc-2020-001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kontermann R.E., Brinkmann U. Bispecific antibodies; different formats. Drug Discov. Today. 2015;20:838–847. doi: 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Nyakatura E.K., Soare A.Y., Lai J.R. Bispecific antibodies for viral immunotherapy. Hum. Vaccin. Immunother. 2017;13:836–842. doi: 10.1080/21645515.2016.1251536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goebeler M.E., Bargou R.C. T cell-engaging therapies — BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020;17:418–434. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 9.Vafa O., Trinklein N.D. Perspective: designing T-cell engagers with better therapeutic windows. Front. Oncol. 2020;10:446. doi: 10.3389/fonc.2020.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuraszeck T., Kasichayanula S., Benjamin J.E. Translation and clinical development of bispecific T-cell engaging antibodies for cancer treatment. Clin. Pharmacol. Ther. 2017;101:634–645. doi: 10.1002/cpt.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamperschroer C., Shenton J., Lebrec H., Leighton J.K., Moore P.A., Thomas O. Summary of a workshop on preclinical and translational safety assessment of CD3 bispecific. J. Immunotoxicol. 2020;17:67–85. doi: 10.1080/1547691X.2020.1729902. [DOI] [PubMed] [Google Scholar]
- 12.Klinger M., Benjamin J., Kischel R., Stienen S., Zugmaier G. Harnessing T cells to fight cancer with BiTE® antibody constructs - past developments and future directions. Immunol. Rev. 2016;270:193–208. doi: 10.1111/imr.12393. [DOI] [PubMed] [Google Scholar]
- 13.Dexter Posey A., Barber A., Sheng Guo Z., Suzuki M., Shaw R.A., Rosewell Shaw A. Oncolytic viruses partner with T-cell therapy for solid tumor treatment. Front. Immunol. 2018:2103. doi: 10.3389/fimmu.2018.02103. www.frontiersin.org 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu F., Wang X., Guo Z.S., Bartlett D.L., Gottschalk S.M., Song X.T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014;22:102–111. doi: 10.1038/mt.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fajardo C.A., Guedan S., Rojas L.A., Moreno R., Arias-Badia M., de Sostoa J., June C.H., Alemany R. Oncolytic adenoviral delivery of an EGFR-targeting t-cell engager improves antitumor efficacy. Cancer Res. 2017;77:2052–2063. doi: 10.1158/0008-5472.CAN-16-1708. [DOI] [PubMed] [Google Scholar]
- 16.Cioffi M., Dorado J., Baeuerle P.A., Heeschen C. EpCAM/CD3-bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin. Cancer Res. 2012;18:465–474. doi: 10.1158/1078-0432.CCR-11-1270. [DOI] [PubMed] [Google Scholar]
- 17.Biegert G.W.G., Rosewell Shaw A., Suzuki M. Current development in adenoviral vectors for cancer immunotherapy. Mol. Ther. Oncolytics. 2021;23:571–581. doi: 10.1016/j.omto.2021.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cervera-Carrascon V., Havunen R., Hemminki A. Oncolytic adenoviruses: a game changer approach in the battle between cancer and the immune system. Expert Opin. Biol. Ther. 2019;19:443–455. doi: 10.1080/14712598.2019.1595582. [DOI] [PubMed] [Google Scholar]
- 19.Cervera-Carrascon V., Quixabeira D.C.A., Havunen R., Santos J.M., Kutvonen E., Clubb J.H.A., Siurala M., Heiniö C., Zafar S., Koivula T., et al. Comparison of clinically relevant oncolytic virus platforms for enhancing T cell therapy of solid tumors. Mol. Ther. Oncolytics. 2020;17:47–60. doi: 10.1016/j.omto.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heiniö C., Havunen R., Santos J., de Lint K., Cervera-Carrascon V., Kanerva A., Hemminki A. TNFa and IL2 encoding oncolytic adenovirus activates pathogen and danger-associated immunological signaling. Cells. 2020;9:40798. doi: 10.3390/cells9040798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quixabeira D.C.A., Zafar S., Santos J.M., Cervera-Carrascon V., Havunen R., Kudling T.v., Basnet S., Anttila M., Kanerva A., Hemminki A. Oncolytic adenovirus coding for a variant interleukin 2 (vIL-2) cytokine Re-programs the tumor microenvironment and confers enhanced tumor control. Front. Immunol. 2021;12:674400. doi: 10.3389/fimmu.2021.674400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santos Apolonio J., Lima de Souza Gonçalves V., Cordeiro Santos M.L., Silva Luz M., Silva Souza J.V., Rocha Pinheiro S.L., de Souza W.R., Sande Loureiro M., de Melo F.F. Oncolytic virus therapy in cancer: a current review. World J. Virol. 2021;10:229–255. doi: 10.5501/wjv.v10.i5.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y., Liu Z., Li L., Wu J., Zhang H., Zhang H., Lei T., Xu B. Oncolytic adenovirus: prospects for cancer immunotherapy. Front. Microbiol. 2021;12:707290. doi: 10.3389/fmicb.2021.707290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemos de Matos A., Franco L.S., McFadden G. Oncolytic viruses and the immune system: the dynamic duo. Mol. Ther. Methods Clin. Dev. 2020;17:349–358. doi: 10.1016/j.omtm.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Havunen R., Kalliokoski R., Siurala M., Sorsa S., Santos J.M., Cervera-carrascon V., Anttila M., Hemminki A. Cytokine-coding oncolytic adenovirus TILT-123 is safe, selective, and effective as a single agent and in combination with immune checkpoint inhibitor anti-PD-1. Cells. 2021;10:246. doi: 10.3390/cells10020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zafar S., Sorsa S., Siurala M., Hemminki O., Havunen R., Cervera-Carrascon V., Santos J.M., Wang H., Lieber A., de Gruijl T., et al. CD40L coding oncolytic adenovirus allows long-term survival of humanized mice receiving dendritic cell therapy. Oncoimmunology. 2018;7:e1490856. doi: 10.1080/2162402X.2018.1490856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fajardo Calderón C. 2017. Arming oncolytic adenoviruses with bi-specific T-cell engagers to improve antitumor efficacy. PhD Thesis. [Google Scholar]
- 28.Porter C.E., Rosewell Shaw A., Jung Y., Yip T., Castro P.D., Sandulache V.C., Sikora A., Gottschalk S., Ittman M.M., Brenner M.K., Suzuki M. Oncolytic adenovirus armed with BiTE, cytokine and checkpoint inhibitor enables CAR T-cells to control growth of heterogeneous tumors. Mol. Ther. 2020;28:1251–1262. doi: 10.1016/j.ymthe.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acres B., Limacher J.M. MUC1 as a target antigen for cancer immunotherapy. Expert Rev. Vaccin. 2005;4:493–502. doi: 10.1586/14760584.4.4.493. [DOI] [PubMed] [Google Scholar]
- 30.Chen W., Zhang Z., Zhang S., Zhu P., Ko J.K.S., Yung K.K.L. Muc1: structure, function, and clinic application in epithelial cancers. Int. J. Mol. Sci. 2021;22:6567. doi: 10.3390/ijms22126567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coradini D., Casarsa C., Oriana S. Epithelial cell polarity and tumorigenesis: new perspectives for cancer detection and treatment. Acta Pharmacol. Sin. 2011;32:552–564. doi: 10.1038/aps.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor-Papadimitriou J., Burchell J.M., Graham R., Beatson R. Latest developments in MUC1 immunotherapy. Biochem. Soc. Trans. 2018;46:659–668. doi: 10.1042/BST20170400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilkie S., Picco G., Foster J., Davies D.M., Julien S., Cooper L., Arif S., Mather S.J., Taylor-Papadimitriou J., Burchell J.M., Maher J. Retargeting of human T cells to tumor-associated MUC1: the Evolution of a chimeric antigen receptor. J. Immunol. 2008;180:4901–4909. doi: 10.4049/jimmunol.180.7.4901. [DOI] [PubMed] [Google Scholar]
- 34.Duffy M.J., Shering S., Sherry F., McDermott E., O’Higgins N. International Journal of Biological Markers. Wichtig Editore s.r.l.; 2000. CA 15-3: a prognostic marker in breast cancer; pp. 330–333. [DOI] [PubMed] [Google Scholar]
- 35.Tissue expression of MUC1 - Summary - The Human Protein Atlas. https://v15.proteinatlas.org/ENSG00000185499-MUC1/tissue
- 36.Deng, J., Wang, L., Chen, H., Li, L., Ma, Y., Ni, J., and Li, Y. The role of tumour-associated MUC1 in epithelial ovarian cancer metastasis and progression. 10.1007/s10555-013-9423-y. [DOI] [PubMed]
- 37.Mereiter S., Balmaña M., Campos D., Gomes J., Reis C.A. Glycosylation in the Era of cancer-targeted therapy: where are we heading? Cancer Cell. 2019;36:6–16. doi: 10.1016/j.ccell.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 38.Havunen R., Siurala M., Sorsa S., Grönberg-Vähä-Koskela S., Behr M., Tähtinen S., Santos J.M., Karell P., Rusanen J., Nettelbeck D.M., et al. Oncolytic adenoviruses armed with tumor necrosis factor alpha and interleukin-2 enable successful adoptive cell therapy. Mol. Ther. Oncolytics. 2017;4:77–86. doi: 10.1016/j.omto.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuryk L., Møller A.S.W. Chimeric oncolytic Ad5/3 virus replicates and lyses ovarian cancer cells through desmoglein-2 cell entry receptor. J. Med. Virol. 2020;92:1309–1315. doi: 10.1002/jmv.25677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hensen L.C.M., Hoeben R.C., Bots S.T.F. Adenovirus receptor expression in cancer and its multifaceted role in oncolytic adenovirus therapy. Int. J. Mol. Sci. 2020;21:6828. doi: 10.3390/ijms21186828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yashiro M., Nishioka N., Hirakawa K. Decreased expression of the adhesion molecule desmoglein-2 is associated with diffuse-type gastric carcinoma. Eur. J. Cancer. 2006;42:2397–2403. doi: 10.1016/j.ejca.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 42.Cai F., Zhu Q., Miao Y., Shen S., Su X., Shi Y. Desmoglein-2 is overexpressed in non-small cell lung cancer tissues and its knockdown suppresses NSCLC growth by regulation of p27 and CDK2. J. Cancer Res. Clin. Oncol. 2017;143:59–69. doi: 10.1007/s00432-016-2250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barber A.G., Castillo-Martin M., Bonal D.M., Rybicki B.A., Christiano A.M., Cordon-Cardo C. Characterization of desmoglein expression in the normal prostatic gland. Desmoglein 2 is an independent prognostic factor for aggressive prostate cancer. PLoS One. 2014;9:98786. doi: 10.1371/journal.pone.0098786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peitsch W.K., Doerflinger Y., Fischer-Colbrie R., Huck V., Bauer A.T., Utikal J., Goerdt S., Schneider S.W. Desmoglein 2 depletion leads to increased migration and upregulation of the chemoattractant secretoneurin in melanoma cells. PLoS One. 2014;9:89491. doi: 10.1371/journal.pone.0089491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanerva A., Zinn K.R., Chaudhuri T.R., Lam J.T., Suzuki K., Uil T.G., Hakkarainen T., Bauerschmitz G.J., Wang M., Liu B., et al. Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus. Mol. Ther. 2003;8:449–458. doi: 10.1016/S1525-0016(03)00200-4. [DOI] [PubMed] [Google Scholar]
- 46.Chang P.-H., Chen M.-C., Tsai Y.-P., Tan G.Y.T., Hsu P.-H., Jeng Y.-M., Tsai Y.-F., Yang M.-H., Hwang-Verslues W.W., Contributed M.-H.Y. Interplay between Desmoglein2 and Hypoxia Controls Metastasis in Breast Cancer. Proc. Natl. Acad. Sci. USA. 2021;118:2014408118. doi: 10.1073/pnas.2014408118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramani V.C., Hennings L., Haun R.S. 2008. Desmoglein 2 Is a Substrate of Kallikrein 7 in Pancreatic Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamekura R., Kolegraff K.N., Nava P., Hilgarth R.S., Feng M., Parkos C.A., Nusrat A. Loss of the desmosomal cadherin desmoglein-2 suppresses colon cancer cell proliferation through EGFR signaling. Oncogene. 2014;33:4531–4536. doi: 10.1038/onc.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zafar S., Quixabeira D.C.A., Kudling T.V., Cervera-Carrascon V., Santos J.M., Grönberg-Vähä-Koskela S., Zhao F., Aronen P., Heiniö C., Havunen R., et al. Ad5/3 is able to avoid neutralization by binding to erythrocytes and lymphocytes. Cancer Gene Ther. 2021;28:442–454. doi: 10.1038/s41417-020-00226-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeku O.O., Rao T.D., Laster I., Kononenko A., Purdon T.J., Wang P., Cui Z., Liu H., Brentjens R.J., Spriggs D. Bispecific T-cell engaging antibodies against MUC16 demonstrate efficacy against ovarian cancer in monotherapy and in combination with PD-1 and VEGF inhibition. Front. Immunol. 2021;12:663379. doi: 10.3389/fimmu.2021.663379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waaijer S.J.H., Warnders F.J., Stienen S., Friedrich M., Sternjak A., Cheung H.K., van Scheltinga A.G.T.T., Schröder C.P., de Vries E.G.E., Lub-De Hooge M.N. Molecular imaging of radiolabeled bispecific T-cell engager 89Zr-AMG211 targeting CEA-positive tumors. Clin. Cancer Res. 2018;24:4988–4996. doi: 10.1158/1078-0432.CCR-18-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wybran J., Dupont E. The active T rosette: an early marker for T-cell activation. Ann. Immunol. 1982;133D:211–218. [PubMed] [Google Scholar]
- 53.Cervera-Carrascon V., Quixabeira D.C.A., Santos J.M., Havunen R., Zafar S., Hemminki O., Heiniö C., Munaro E., Siurala M., Sorsa S., et al. Tumor microenvironment remodeling by an engineered oncolytic adenovirus results in improved outcome from PD-L1 inhibition. Oncoimmunology. 2020;9:1761229. doi: 10.1080/2162402X.2020.1761229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Santos J.M., Havunen R., Hemminki A. Methods in Enzymology. Academic Press Inc.; 2020. Modulation of the tumor microenvironment with an oncolytic adenovirus for effective T-cell therapy and checkpoint inhibition; pp. 205–230. [DOI] [PubMed] [Google Scholar]
- 55.Kudling T.v., Clubb J.H.A., Quixabeira D.C.A., Santos J.M., Havunen R., Kononov A., Heiniö C., Cervera-Carrascon V., Pakola S., Basnet S., et al. Local delivery of interleukin 7 with an oncolytic adenovirus activates tumor-infiltrating lymphocytes and causes tumor regression. Oncoimmunology. 2022;11:2096572. doi: 10.1080/2162402X.2022.2096572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mathis J.M., Stoff-Khalili M.A., Curiel D.T. Oncolytic adenoviruses - selective retargeting to tumor cells. Oncogene. 2005;24:7775–7791. doi: 10.1038/sj.onc.1209044. [DOI] [PubMed] [Google Scholar]
- 57.Rojas J.J., Cascallo M., Guedan S., Gros A., Martinez-Quintanilla J., Hemminki A., Alemany R. A modified E2F-1 promoter improves the efficacy to toxicity ratio of oncolytic adenoviruses. Gene Ther. 2009;16:1441–1451. doi: 10.1038/gt.2009.103. [DOI] [PubMed] [Google Scholar]
- 58.Lu Y.Y., Yu H., Tang Y. Efficacy and safety of MUC1 targeted CIK cells for the treatment of advanced liver cancer. J. Clin. Oncol. 2021;39:e16278. [Google Scholar]
- 59.Liikanen I., Basnet S., Quixabeira D.C.A., Taipale K., Hemminki O., Oksanen M., Kankainen M., Juhila J., Kanerva A., Joensuu T., et al. Oncolytic adenovirus decreases the proportion of TIM-3 + subset of tumor-infiltrating CD8 + T cells with correlation to improved survival in patients with cancer. J. Immunother. Cancer. 2022;10:e003490. doi: 10.1136/jitc-2021-003490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruzsics Z., Lemnitzer F., Thirion C. Engineering adenovirus genome by bacterial artificial chromosome (BAC) technology. Methods Mol. Biol. 2014;1089:143–158. doi: 10.1007/978-1-62703-679-5_11. [DOI] [PubMed] [Google Scholar]
- 61.Clubb J.H.A., Kudling T.v., Heiniö C., Basnet S., Pakola S., Cervera Carrascón V., Santos J.M., Quixabeira D.C.A., Havunen R., Sorsa S., et al. Adenovirus encoding tumor necrosis factor alpha and interleukin 2 induces a tertiary lymphoid structure signature in immune checkpoint inhibitor refractory head and neck cancer. Front. Immunol. 2022;13:794251. doi: 10.3389/fimmu.2022.794251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siurala M., Bramante S., Vassilev L., Hirvinen M., Parviainen S., Tähtinen S., Guse K., Cerullo V., Kanerva A., Kipar A., et al. Oncolytic adenovirus and doxorubicin-based chemotherapy results in synergistic antitumor activity against soft-tissue sarcoma. Int. J. Cancer. 2015;136:945–954. doi: 10.1002/IJC.29048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available upon request.