

Abstract
Upon chronic antigen exposure, CD8+ T cells become exhausted, acquiring a dysfunctional state correlated with the inability to control infection or tumor progression. In contrast, stem-like CD8+ T progenitors maintain the ability to promote and sustain effective immunity. Adenovirus (Ad)–vectored vaccines encoding tumor neoantigens have been shown to eradicate large tumors when combined with anti–programmed cell death protein 1 (αPD-1) in murine models; however, the mechanisms and translational potential have not yet been elucidated. Here, we show that gorilla Ad vaccine targeting tumor neoepitopes enhances responses to αPD-1 therapy by improving immunogenicity and antitumor efficacy. Single-cell RNA sequencing demonstrated that the combination of Ad vaccine and αPD-1 increased the number of murine polyfunctional neoantigen-specific CD8+ T cells over αPD-1 monotherapy, with an accumulation of Tcf1+ stem-like progenitors in draining lymph nodes and effector CD8+ T cells in tumors. Combined T cell receptor (TCR) sequencing analysis highlighted a broader spectrum of neoantigen-specific CD8+ T cells upon vaccination compared to αPD-1 monotherapy. The translational relevance of these data is supported by results obtained in the first 12 patients with metastatic deficient mismatch repair (dMMR) tumors vaccinated with an Ad vaccine encoding shared neoantigens. Expansion and diversification of TCRs were observed in post-treatment biopsies of patients with clinical response, as well as an increase in tumor-infiltrating T cells with an effector memory signature. These findings indicate a promising mechanism to overcome resistance to PD-1 blockade by promoting immunogenicity and broadening the spectrum and magnitude of neoantigen-specific T cells infiltrating tumors.
INTRODUCTION
After chronic exposure to cancer or viral infection, CD8+ T cells acquire an exhausted phenotype, characterized by a poor ability to respond to antigenic stimulation (1). Tumor-reactive lymphocytes progressively become exhausted and rarely acquire memory features (2). Conversely to exhausted T cells, the Tcf1+ stem-like CD8+ T (TSTEM) progenitors of exhausted cells resemble self-renewal memory stem-like cells and proliferate after blockade of the PD-1 inhibitory pathway (3, 4) before differentiating into terminally exhausted CD8+ T cells (5). In line with their capacity to respond to immunotherapy, an increased percentage of Tcf1+ CD8+ T cells has been correlated with prolonged duration of responses to checkpoint blockade therapy in patients with cancer (6). As a consequence, understanding the functional programs to sustain Tcf1+ CD8+ T cell differentiation represents a critical step for the improvement of immunotherapy, aimed at increasing their number and maintaining their self-renewal capacity, thus overcoming resistance to checkpoint blockade treatment in cancer (7).
Several studies have shown that the clinical efficacy of PD-1 checkpoint blockade depends on the magnitude, quality, and tumor-infiltrating properties of CD8+ T cells targeting mutation-associated neoantigens (8, 9). As opposite to self-antigens, neoantigens are self-mutated peptides, without preexisting central tolerance and with the potential of inducing stronger immune response and effective antitumor activity. As a consequence, neoantigens have become of great interest as targets of cancer vaccines.
The development of a cancer vaccine driving both expansion and functional commitment of CD8+ T progenitors represents a critical determinant to achieve long-lasting antitumor efficacy. An effective cancer vaccine should induce, expand, and drive the differentiation of neoepitope-specific stem-like CD8+ T progenitors, with antitumor T cell responses and long-term memory protection against cancer recurrence (5, 10). However, major advances in the field have been hindered by the absence of vaccines able to promote expansion and tumor infiltration of neoepitope-specific T cells, to overcome the immunosuppressive effects of the tumor microenvironment.
Genetic vaccines based on adenoviruses (Ads) derived from nonhuman Great Apes (GAd) can elicit robust and effective T cell immunity in humans (11–14). Early vaccine studies in mice and clinical trials in humans for HIV, hepatitis C virus, Ebola, and emerging viruses such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have demonstrated the strong potential of GAd to induce potent T cell immunogenicity and protection (15–17). A key aspect of GAd is the capability to encode large gene inserts and therefore to target many neoantigens, offering the unique opportunity to overcome the issue of tumor heterogeneity and escape through immunoediting (18). In the context of cancer, we have previously shown in mice that the combination of neoantigen-based adenovector vaccine and anti–programmed cell death protein 1 (αPD-1) antibody acts synergistically to eradicate large established tumors (18). However, the mechanism underlying the antitumor effects and the synergy between the vaccine and αPD-1 have not yet been elucidated. We hypothesize that combining αPD-1 and a neoantigen-based adenovector vaccine would enhance the magnitude, quality, and breadth of CD8+ T cell responses. Here, we show that GAd vaccine combined with αPD-1 immunotherapy induced strong activation of CD8+ T cell clonotypes reactive against neoantigens, promoting the expansion of tumor-reactive Tcf1+ TSTEM progenitors in draining lymph nodes and accumulation of effector CD8+ T clonotypes in murine tumors. The impact of vaccination and αPD-1 on neoantigen-specific T cell responses was investigated in humans immunized with an experimental vaccine encoding shared neoantigens. Some colorectal [colorectal cancer (CRC)], gastric, and gastroesophageal junction (GEJ) tumors associated with mismatch repair deficiency (dMMR) represent a unique condition, in which tumors accumulate shared mutations, allowing the design of an “off-the-shelf” neoantigen-based vaccine (19). Here, we show expansion after treatment of vaccine-induced T cell clonotypes recognizing neoantigens in tumors of patients with dMMR. Overall, these results demonstrate an important role for adenoviral-based vectors to promote broadening, expansion, and differentiation of T cells to sustain an antitumor response.
RESULTS
GAd vaccine combined with αPD-1 treatment enhances immunogenicity and expands neoantigen-specific CD8 T cells in the tumor
To generate an Ad vaccine that targets tumor-specific neoantigens, we have chosen seven predicted immunogenic neoepitopes, derived from tumor-specific point mutations of the murine MC38 colorectal adenocarcinoma cell line. These peptides have been scored with the highest binding affinity for major histocompatibility complex class I (MHC-I) in comparison to the corresponding wild-type peptides and identified by mass spectrometry (MS) (20). First, we evaluated the stability and life span of neoepitope-MHC complexes, by measuring with flow cytometry the surface expression of the MHC-I molecules, in RMA-S cell line after loading with the neoantigen peptides (21). Results indicated higher and longer stability for Reps1, followed by Cpne1 and Adpgk peptides (fig. S1). The seven neoepitopes were then joined head to tail to generate a single synthetic antigenic molecule (Fig. 1A), whose corresponding gene was cloned into a GAd vector, as previously described (18). GAd vaccine and αPD-1 effectiveness was evaluated in mice with established subcutaneous MC38 tumors (Fig. 1A). Mice were subcutaneously injected with the adenocarcinoma colon cancer MC38 cell line, and 10 days later, once the tumor was established, the mice were immunized with GAd vaccine and treated with an antibody blocking PD-1 (αPD-1). In line with our previous findings (18), we found enhanced antitumor control in mice receiving combined GAd and αPD-1 treatment as compared to the αPD-1 monotherapy, with an increase in the overall survival (Fig. 1B).
Fig. 1. GAd vaccination combined with αPD-1 treatment reduces tumor growth by increasing the number of neoepitope-reactive CD8+ T cells in mice.
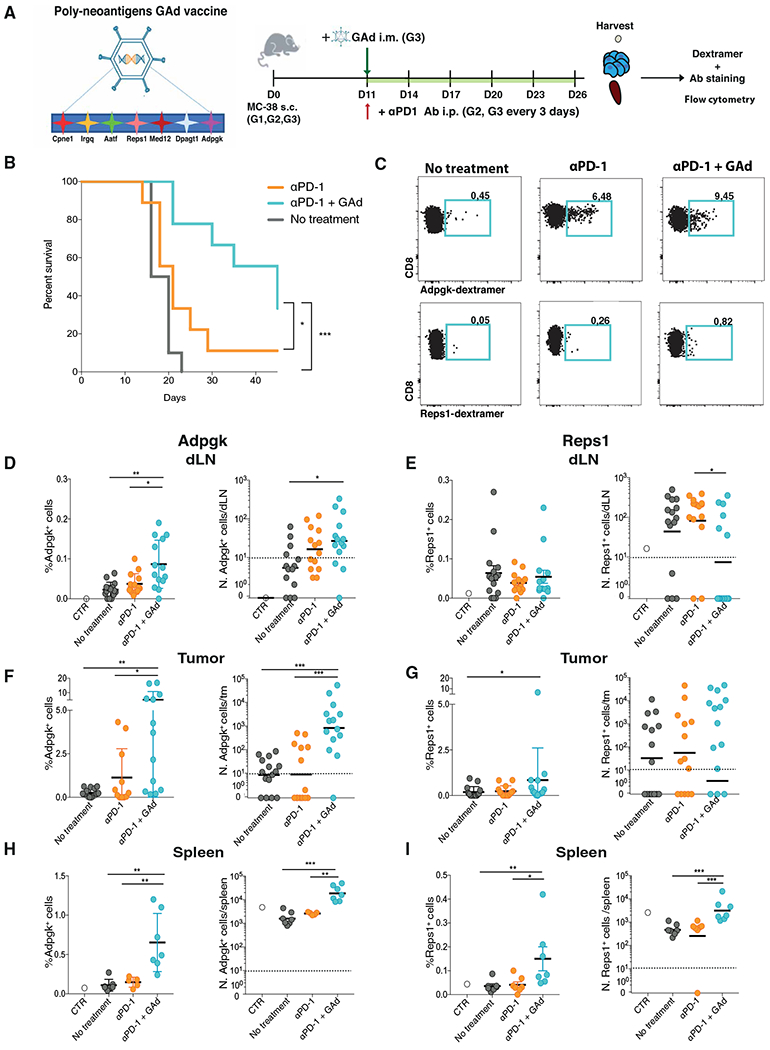
(A) Experimental design. C57BL/6J female mice (G1 to G3) were subcutaneously (s.c.) injected with MC38 adenocarcinoma colon cancer cell line. Eleven days later, G2 and G3 were intraperitoneally (i.p.) injected, every 3 days, with the monoclonal antibody (Ab) αPD-1. Eleven days after tumor implantation (tumor size between 70 and 100 mm3), G3 were also intramuscularly (i.m.) injected with poly-neoantigen Gad vaccine. Twenty-six days after tumor implantation, Db-Adpgk+ and Db-Reps1+CD8+ T cells were analyzed, in the draining lymph node, tumor, and spleen, by flow cytometry. (B) Overall survival of tumor-bearing mice was measured over time in G1, G2, and G3. (C) Representative dot plots of Db-Adpgk+CD8+ T cells and Db-Reps1+CD8+ T cells in the tumors; numbers represent percentages. (D, F, and H) Percentages and numbers (N.) of Db-Adpgk+CD8+ T cells were measured in draining lymph node (dLN) (D), tumor (tm) (F), and spleen (H). (E, G, and I) Percentages and numbers of Db-Reps1+CD8+ T cells were measured in draining lymph node (E), tumor (G), and spleen (I). Data are shown as mean with SEM (D to I, left) and geometric mean (D to I, right). *P < 0.05, **P < 0.01, and ***P < 0.001 (Mann-Whitney test). Graphs are representative of six experiments with seven mice per group.
Previous work was carried out in the same MC38 tumor model and identified the neoepitope Adpgk as the one conferring protection after peptide combined with adjuvant immunization and with the most prominent expansion in the tumor (20). In our model, neoantigen-specific CD8+ T cell responses in the draining lymph node, tumor, and spleen were characterized using the H-2Db-Adpgk peptide–MHC-I dextramer upon treatment with αPD-1 as compared to the combination GAd and αPD-1 (Fig. 1, C, D, F and H). We also analyzed CD8+ T cells specific for Reps1, a less effective neoepitope in tumor rejection. As expected, in the nontreated control tumor–bearing mice, the frequency and number of both Db-Adpgk+ and Db-Reps1+ CD8+ T cells were very low (Fig. 1, C to E) (18). The frequency of Db-Adpgk+CD8+ T cells increased in the draining lymph node, tumor, and spleen of tumor-bearing mice treated with GAd + αPD-1, as compared to control tumor–bearing animals (Fig. 1, D, F, and H). In mice treated with GAd immunization combined with αPD-1 antibody, we found about 100 and 10 times more Db-Adpgk+CD8+ T cells in tumor and spleen, respectively, as compared to mice receiving αPD-1 treatment alone. We saw no substantial differences in CD8+ T cell responses with GAd + αPD-1 treatment against Reps1 in draining lymph nodes and tumors (Fig. 1, E and G), but both frequency and number of Db-Reps1+CD8+ T cells were enhanced in the spleen (Fig. 1I). Thus, GAd vaccination promoted neoepitope immunogenicity, with expansion and accumulation of CD8+ T cells reactive against both Adpgk and Reps1 neoepitopes in the spleen of tumor-bearing mice. Conversely, Adpgk-reactive CD8+ T cells were also expanded in draining lymph nodes and tumors of these mice. These results indicate that two neoepitopes, Adpgk and Reps1, both identified by MS on tumor cells and with very similar features in terms of stability, predicted binding affinities, and in vivo immunogenicity, present different capabilities to infiltrate the tumor bed.
GAd therapeutic vaccination increases the frequencies of neoepitope-specific memory precursors and effectors
We next characterized the phenotype of Adpgk-specific CD8+ T cells induced by GAd vaccination in tumor-bearing mice by measuring the frequency and number of CD127+ KLRG1− memory precursor, CD127+ CD62L+ KLRG1− central memory, CD127− KLRG1+ short-lived effector, or tissue-resident memory (TRM) CD103+ CD69+ CD8+ T cells, and also exhausted CD8+ T cells based on the expression of PD-1 and CD38 of Db-Adpgk+ CD8+ T cells (Fig. 2 and fig. S2, A to F) (22–25). Although a higher proportion of CD127+ KLRG1− memory precursors was observed in mice treated with αPD-1 alone as compared to the combined treatment with GAd in the draining lymph node (fig. S2A), increased numbers of CD127+ KLRG1− memory precursors were measured in tumor-bearing mice treated with GAd in draining lymph nodes, tumors, and spleens (Fig. 2, A and B to D, left panels). Combined treatment with GAd also associated with increased numbers of central memory CD127+ CD62L+ KLRG1− CD8+ T cells in the spleen (fig. S2F). A higher proportion of effector CD127− KLRG1+ Db-Adpgk–specific CD8+ T cells were also found in draining lymph nodes, tumors, and spleens of mice injected with GAd, with a 10-fold difference in tumors and spleens as compared to mice receiving αPD-1 alone (Fig. 2, A, and B to D, right panels, and fig. S2B). Furthermore, the analysis of neoepitope-specific Db-Reps1+ CD8+ T cells (fig. S3) revealed increased numbers of memory precursor CD127+ KLRG1− and CD127− KLRG1+ effector CD8+ T cells in spleens (fig. S3F).
Fig. 2. GAd combined with αPD-1 treatment induces neoepitope-reactive memory precursor CD8+ T cells in mice.
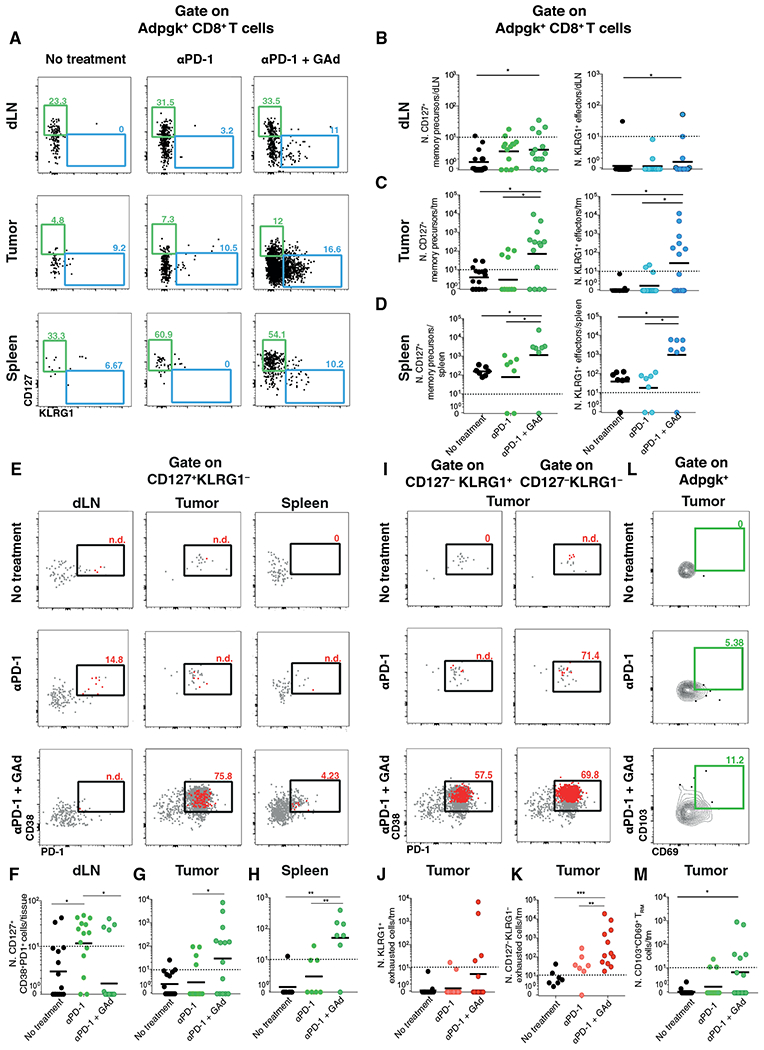
(A) Representative dot plots of CD127 and KLRG1 markers (CD127+KLRG1− memory precursors and CD127−KLRG1+ effectors); numbers represent percentages. (B to D) Number of Db-Adpgk+CD8+ T memory precursors and effectors were measured in draining lymph node (B), tumor (C), and spleen (D). (E) Representative dot plots of CD38 and PD-1 markers gated on CD127+KLRG1−Db-Adpgk+CD8+ T cells. (F to H) Numbers of CD38+PD-1+ cells gated on CD127+Db-Adpgk+CD8+ T cells were measured in draining lymph node (F), tumor (G), and spleen (H). (I) Representative dot plots of exhausted CD38+PD-1+cells analyzed on gated CD127−KLRG1+ (left) and on gated CD127−KLRG1− (right) Db-Adpgk+ CD8+ T cells in tumor; numbers represent percentages. In red, memory precursors gated on Db-Adpgk+ CD8+ T cells were displayed; in gray, the total number of Db-Adpgk+ CD8+ T cells was displayed. (J) Numbers of exhausted Db-Adpgk+ CD8+ T cells on gated CD127−KLRG1+ were measured in tumor. (K) Numbers of exhausted Db-Adpgk+ CD8+ T cells on gated CD127−KLRG1− were measured in tumor. (L) Representative dot plots of CD103 and CD69 markers (CD103+CD69+ TRM cells) in tumor; numbers represent percentages out of the total CD8+ T cells isolated from the tumor. (M) Numbers of Db-Adpgk+ CD8+ TRM cells were measured in tumors. Data are shown as geometric mean (B to D, F to H, J, K, and M). *P < 0.05, **P < 0.01, and ***P < 0.001 (Mann-Whitney test). Graphs are representative of six experiments with seven mice per group. Cells below the limit of detection were indicated as non-detectable (n.d.).
We next examined whether the memory precursor and effector subsets induced by GAd vaccination against neoepitopes could also express the exhaustion markers PD-1 and CD38 (Fig. 2E and figs. S2C and S4). Increased frequency and number of PD-1 and CD38 double-positive cells were found among the memory precursors in the tumors, whereas low frequency and numbers were detected in draining lymph nodes (Fig. 2, E to H). In mice receiving GAd vaccine, the frequency of CD127+ CD8+ T cells coexpressing PD-1 and CD38 was found reduced as compared to mice treated with αPD-1 alone (Fig. 2F). In GAd-treated mice, among Db-Adpgk+CD127− KLRG1+ effector and CD127− KLRG1− CD8+ T subsets in tumors (Fig. 2I), around 58 and 70% of the cells express both PD-1 and CD38. These results suggest that, although a fraction of cells acquired exhaustion markers after GAd vaccination (Fig. 2, J and K), an increase of exhausted T (TEX) cell number was measured only among CD127− KLRG1− CD8+ T cells. We did not observe a similar phenotype for Reps1-specific CD8+ T cells, and most of the cells were negative for these exhaustion markers (fig. S4). Last, the frequency of Db-Adpgk+ CD103+ CD69+ TRM CD8+ T cells was also increased in the tumors of αPD-1 + GAd–treated mice (Fig. 2, L and M).
Overall, these results suggest increased immunogenicity, and different paths of differentiation and expansion of neoantigen-specific CD8+ T cells generated after GAd immunization combined with αPD-1 in tumor-bearing mice. These responses are associated with a higher number of memory progenitors in both draining lymph nodes and spleens, and effector T cells in the tumors.
scRNA-seq analysis of neoantigen-specific CD8+ T cells demonstrates stem-like and effector cell accumulation after αPD-1 + GAd treatment
To further characterize the heterogeneity and gene expression programs of neoantigen-specific CD8+ T cell subsets induced after immunization with GAd and αPD-1 treatment, we performed single-cell RNA sequencing (scRNA-seq) analysis of CD8+ T cells from lymph nodes of control tumor–free mice and Db-Adpgk+ CD8+ T lymphocytes harvested from draining lymph nodes and tumors, fluorescence-activated cell sorting (FACS)–sorted 27 days after tumor implantation (Fig. 3A). Control and Db-Adpgk+ CD8+ T cells from individual mice were index-sorted into microwells and analyzed by scRNA-seq using Smart-Seq2 technology (26). After sequencing and quality control filtering, we obtained 57 naïve T cells from two control tumor–free mice and 1132 Db-Adpgk+ CD8+ T lymphocytes from nontreated (157 cells), αPD-1–treated (226 cells), and αPD-1 + GAd–treated (749 cells) tumor-bearing mice (Fig. 3B); for each condition, we processed nine, five, and eight biological replicates, respectively.
Fig. 3. scRNA-seq of CD8+ T cells identifies distinct subpopulations induced by αPD-1 + GAd vaccine in mice.
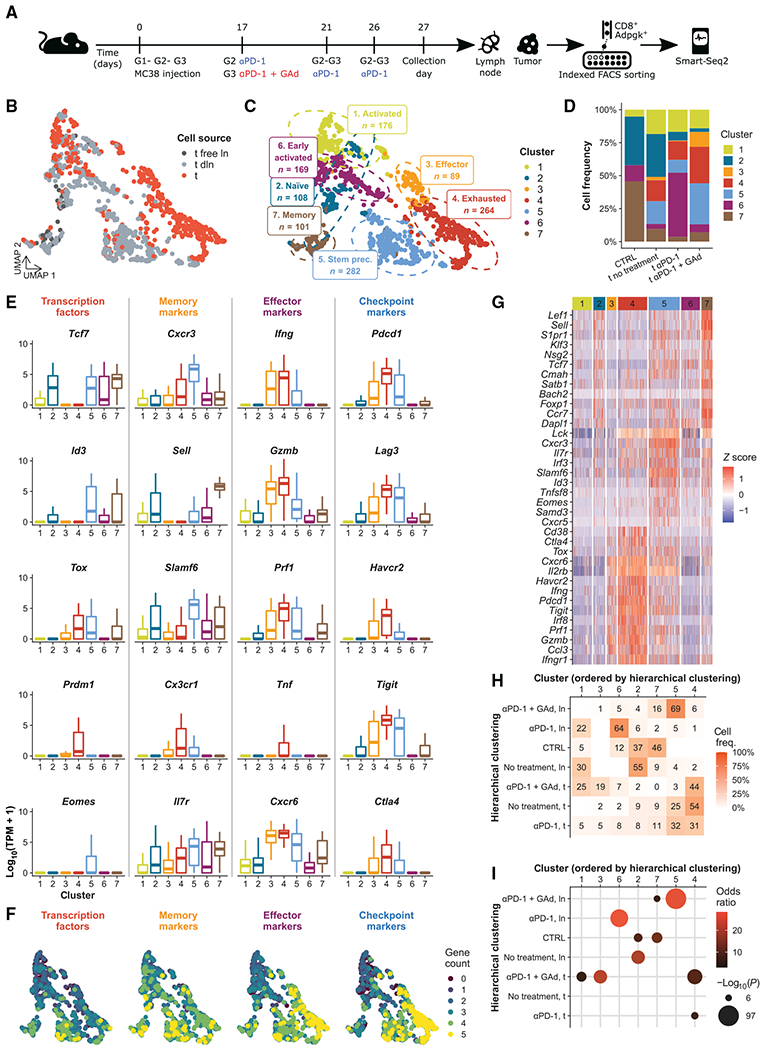
(A) Experimental design for scRNA-seq. Three groups of mice (G1 to G3) were treated as shown; CD8+ T cells isolated from untreated mice were used as the control (CTRL). (B) UMAP visualization of CD8+ T cells (n = 1189) color-coded by tissue of origin (t, tumor; ln, lymph node, dln, draining lymph node). (C) UMAP visualization of clustering results with phenotype annotations. (D) Distribution of Db-Adpgk+ CD8+ T cell frequencies in each experimental condition, color-coded by cluster. (E) Measurement of the expression of genes encoding transcription factors, memory, effector, and checkpoint markers is shown in boxplots. Kruskal-Wallis test, P ≤ 5.7−16. (F) Number of expressed marker genes from (E) projected onto UMAP. (G) Heatmap of selected top differentially expressed (DE) genes, TSTEM progenitor, and TEX CD8+ T cell signatures (29), color-coded by cluster. (H) Heatmap of cluster-wise cell frequencies for each treatment and cell type (ln, lymph node; t, tumor) and (I) results of Fisher’s exact test of those frequencies. The x- and y-axis orders were defined through hierarchical clustering. Frequencies were calculated from each combination of cell type and treatment (shown on rows) across all clusters; thus, the sum of the values of each row of the heatmap is 1 (=100%).
A principal components analysis (PCA) of single-cell transcriptome data is visualized as a set of Uniform Manifold Approximation and Projection (UMAP) plots in Fig. 3 (B and C). We observed a different distribution of Db-Adpgk+ CD8+ T cells isolated from the lymph nodes and tumors, underlining distinct gene expression profiles (Fig. 3B). To identify the distinct cell subsets, we clustered the PCA results. The analysis highlighted seven clusters (Fig. 3, C and D), which, on the basis of distinct transcription profiles (Fig. 3, E to G), were categorized as naïve (cluster 2), early activated (cluster 6), activated (cluster 1), TSTEM precursor (cluster 5), memory (cluster 7), effector (cluster 3), and TEX (cluster 4) Db-Adpgk+ CD8+ T cells. The memory population was mainly derived from lymph nodes, effector cells, and TEX cells belonging to tumors, whereas CD8+ T cells isolated from control tumor–free mice were found in the naïve and memory clusters (Fig. 3, B to D). Naïve cells were characterized by the expression of Tcf7 (Tcf1) and Sell (CD62L) (Fig. 3E). Although early activated cells displayed low expression of Tcf7 transcriptional factor and IL7r, both effector and TEX cell clusters showed high expression of the effector genes Ifng, Gzmb, Prf1, and Cxcr6 (Fig. 3, E and G). In addition, although these clusters shared Pdcd1, Lag3, and Tigit, these genes encoding checkpoints showed consistently higher expression in the exhausted subset (Fig. 3E). Conversely, the expression of Tcf7, Cxcr3, Il7r, and Slamf6 characterized the TSTEM precursor population. This cluster was the only one displaying the expression of Eomes. Memory cells were characterized by the expression of Sell and Il7r genes; in addition, the transcriptional factor Id3 was also transcribed.
To assess whether cells from specific clusters derived from lymph nodes or tumors or were related with a specific treatment, we examined the cell distribution among different clusters. Total CD8+ T cells from control tumor–free mice (Fig. 3D) were mainly localized in the memory and naïve clusters (46 and 37%, respectively), with lower frequencies in the early activated and activated clusters (12 and 5%). Db-Adpgk+ CD8+ T cells derived from nontreated tumor-bearing mice were found mainly in the naïve cluster (32%) and, to a lesser extent, in the activated, TSTEM precursor, TEX, and memory clusters (18, 17, 16, and 10%, respectively). Db-Adpgk+ CD8+ T cells from tumor-bearing mice treated with αPD-1 were predominantly found in the early activated cluster (49%), whereas some were found in the activated and TEX clusters (17 and 14%, respectively). αPD-1–treated mice also had smaller frequencies of the TSTEM precursor, naïve, memory, and effector clusters (10, 7, 3, and 0.4%, respectively). Db-Adpgk+ CD8+ T cells from tumor-bearing mice treated with αPD-1 + GAd were mainly distributed in the TSTEM precursor and TEX clusters (31 and 28%, respectively) and, in smaller proportions, in the activated, effector, memory, early activated, and naïve clusters (14, 11, 7, 6, and 3%, respectively). Db-Adpgk+ CD8+ T cells from tumors and lymph nodes did not distribute equally across clusters (Fig. 3, H and I). Db-Adpgk+ CD8+ T cells from lymph nodes of αPD-1 + GAd–treated mice were enriched in the TSTEM precursor cluster (69% of those cells were found in cluster 5); conversely, tumor-infiltrating Db-Adpgk+ CD8+ T cells from αPD-1 + GAd–treated mice were enriched in the TEX (44%) and in the effector clusters (19%). Conversely, cells from lymph nodes of αPD-1–treated mice were augmented in the early activated cluster (64%). Cells from lymph nodes of tumor-bearing mice without treatment were enriched in the naïve cluster (55%). Overall, these results suggest that the combined treatment αPD-1 + GAd promotes the differentiation of Db-Adpgk+ CD8+ T cells into TSTEM precursors in the lymph node and effector subsets in the tumor.
GAd combined with PD-1 blockade treatment shifts T cell differentiation and induces CD8+ TSTEM precursors
To better characterize the immune phenotype of each cluster, we performed differential expression analysis by comparing transcriptome data from each individual subset against all others. We identified 7313, 2664, 2167, 6723, 4256, 5907, and 2449 statistically significant genes (adjusted P ≤ 0.05) in the seven clusters, respectively (fig. S5B). Next, to determine the gene expression programs and the stages of differentiation corresponding to each cluster, we performed a gene set enrichment analysis (GSEA) with published immunological signatures from the Molecular Signatures Database (MSigDB) (27). GSEA by pairwise comparison of each cluster compared to all the other clusters revealed differential enrichments in the gene expression profiles (Fig. 4A). In the naïve population (cluster 2), the TCF7+ TSTEM precursor signatures were positively enriched (28) [Miller prog (29)] with 26% mean signature expression (Fig. 4B). Conversely, signatures for memory, exhausted, and effector cells were negatively enriched in the naïve population (30, 31). In the effector population (cluster 3), we observed positive enrichment for the effector versus memory signature (31) and negative enrichment for the exhaustion signature (6). TEX cells (cluster 4) showed positive enrichment for exhaustion signatures (6, 29) (with a mean number of 47% expressed genes) and TRM gene expression profile (44% expressed genes; Fig. 4, A and B). TSTEM precursors (cluster 5) were positively enriched for memory and effector signatures (30, 31). In this cluster, we also observed both TSTEM precursor (29) and T exhausted circulating cell (32) signatures (38 and 30% mean of expressed genes, respectively; Fig. 4, A and B). In contrast, the early activated subset (cluster 6) presented negative enrichments for the exhaustion (6), memory, and effector signatures (30, 31). In the memory population (cluster 7), we observed positive enrichments in TCF7+ cell tumor infiltration (28), TSTEM precursor, and circulating memory T cell signatures (32) but negative enrichment in the effector signature (31). Overall, these results confirmed that clusters 5 and 7 were enriched in Db-Adpgk+ CD8+ TSTEM precursor cells, with circulating and memory gene signatures, whereas cluster 4 identified the exhausted Db-Adpgk+ CD8+ T cell subset.
Fig. 4. Phenotype landscapes and trajectories of differentiation of Db-Adpgk+ CD8+ T cells from mice.
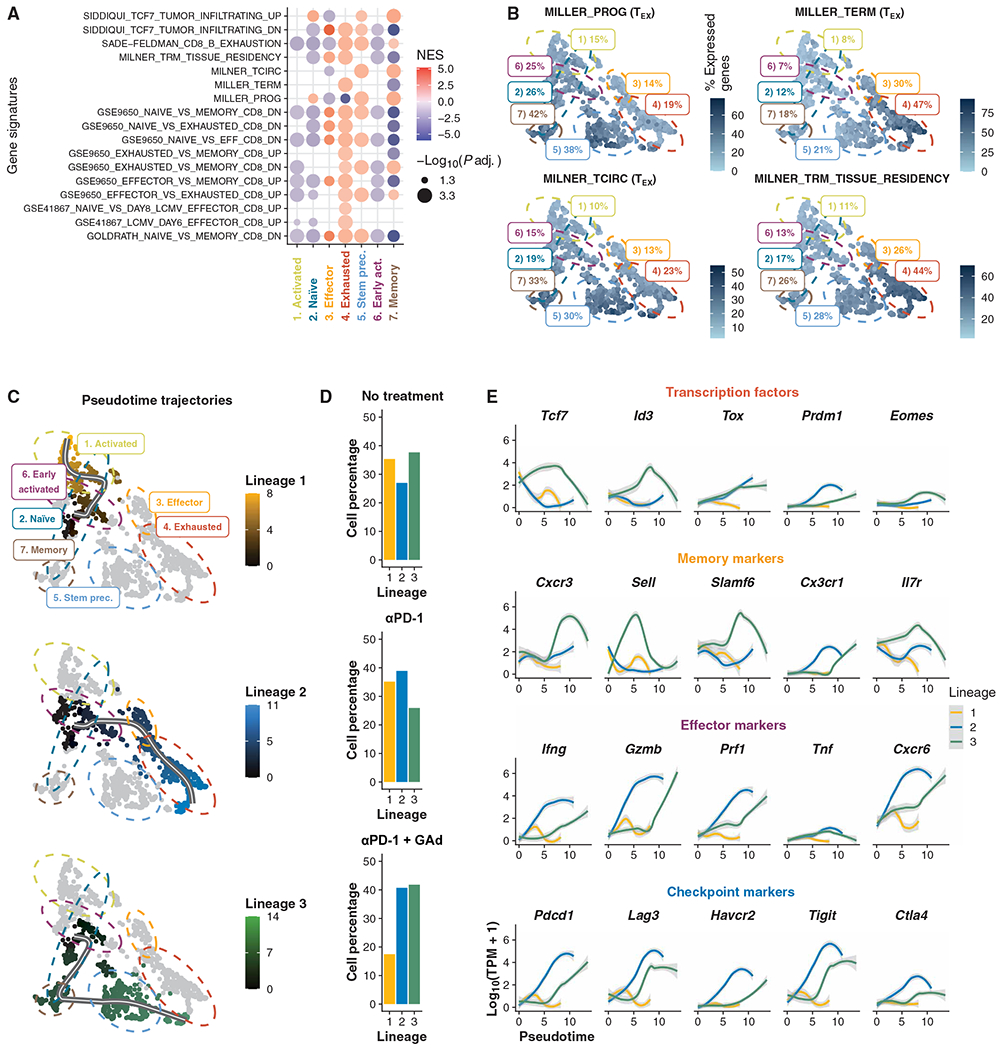
(A) GSEA of DE genes found in clusters (adjusted P ≤0.05). NES, normalized enrichment score. (B) Percentage of signature expression of TSTEM progenitors (MILLER_PROG.) and TEX CD8+ T cells (MILLER_TERM), core circulating (MILNER_TCIRC), and core tissue-resident memory T cell (MILNER_TRM_TISSUE_RESIDENCY) signatures (29, 32) in each cell. (C) Trajectory inference results shown as pseudotime projected onto UMAP. (D) Bar plots of cell percentages across experimental treatments for each lineage. (E) Locally weighted scatterplot smoothing of expression of transcription factors, memory, effector, and checkpoint markers along pseudotime trajectories.
The scRNA-seq profiles revealed different immune phenotypes between lymph nodes and tumors, suggesting distinct stages of lineage commitments. On the basis of these considerations, we next asked whether CD8+ T cells reactive against Adpgk could follow distinct pathways of lineage differentiation after αPD-1 + GAd treatment. To determine Db-Adpgk+ CD8+ T cell transition to new states of differentiation, we traced the transcriptional trajectories corresponding to fate commitment (6, 33). Using supervised trajectory analysis, we identified three different paths originating from the naïve cell cluster, which could reflect likely trajectories of differentiation (Fig. 4, C and D). The first lineage trajectory included 360 cells and was related to T cell activation. The second lineage trajectory (548 cells) progressed through the early activated, effector, and TEX cell populations. Accordingly, we observed an increase in expression of effector and checkpoint markers along the second lineage (Fig. 4E). The third lineage (561 cells) progressed through the memory, TSTEM precursor, and TEX subsets. In the latter, we observed a transient increase in expression of genes encoding some memory markers, including Cxcr3, Sell, Slamf6, and Il7r, and transcription factors, such as Tcf7 and Id3, whereas checkpoint and effector markers displayed a progressive increase in expression from the TSTEM precursor toward the exhausted stage. Db-Adpgk+ CD8+ T cells from nontreated tumor-bearing mice were distributed roughly in equal proportions among the three possible lineages (38, 35, and 27% of lineages 3, 1, and 2, respectively; Fig. 4D). Likewise, Db-Adpgk+ CD8+ T cells from αPD-1–treated mice also encompassed similar proportions of cells in the three lineages (39, 35, and 26% in lineages 2, 1, and 3, respectively). In contrast, Db-Adpgk+ CD8+ T cells from mice treated with αPD-1 + GAd were distributed with the highest proportions in lineages 3 and 2 (42 and 41%, respectively), with a smaller fraction in lineage 1 (17%). The distinct stages of differentiation based on the gene expression patterns of each cluster were also validated by the analysis of surface protein expression obtained during the collection of single cells by FACS-indexed sorting (fig. S5C). Thus, the αPD-1 + GAd combined treatment drives T cell fate commitment toward two main possible lineages of differentiation: one through memory, TSTEM precursor, and TEX cells, and one through effectors and exhausted phenotypes.
Broader multifunctional CD8+ T clonotypes are induced by αPD-1 + GAd vaccination
To determine whether the increased percentage and number of TSTEM precursors and effectors observed in αPD-1– and αPD-1 + GAd–treated mice could be related to the clonal expansion of CD8+ T cells reactive against Adpgk, we leveraged the Smart-Seq2 sequencing data to analyze the full-length αβ T cell receptor (TCR) chain sequences at the single-cell level. This analysis led to the identification of 330 unique and 34 expanded TCR clonotypes (Fig. 5, A and B). Unique TCRs were identified mainly in naïve, memory, and early activated subsets, whereas expanded clonotypes (clone size > 30 cells) were primarily found in effector, TEX, and TSTEM precursor subsets (69, 74, and 56%, respectively; Fig. 5, A and B). The analysis of clonotype size through the naïve, early activated, effector, and exhausted subsets (Fig. 5B) underlined an increase in the proportion of expanded clonotypes throughout the progressive differentiation described in lineage 2 (Fig. 4C and fig. S5C). A similar behavior was found in lineage 3 encompassing memory, TSTEM precursor, and TEX cells (fig. S5C).
Fig. 5. Expanded TCR clonotypes are in memory progenitor stem-like and exhausted stages in lymph nodes and tumors of mice, respectively.
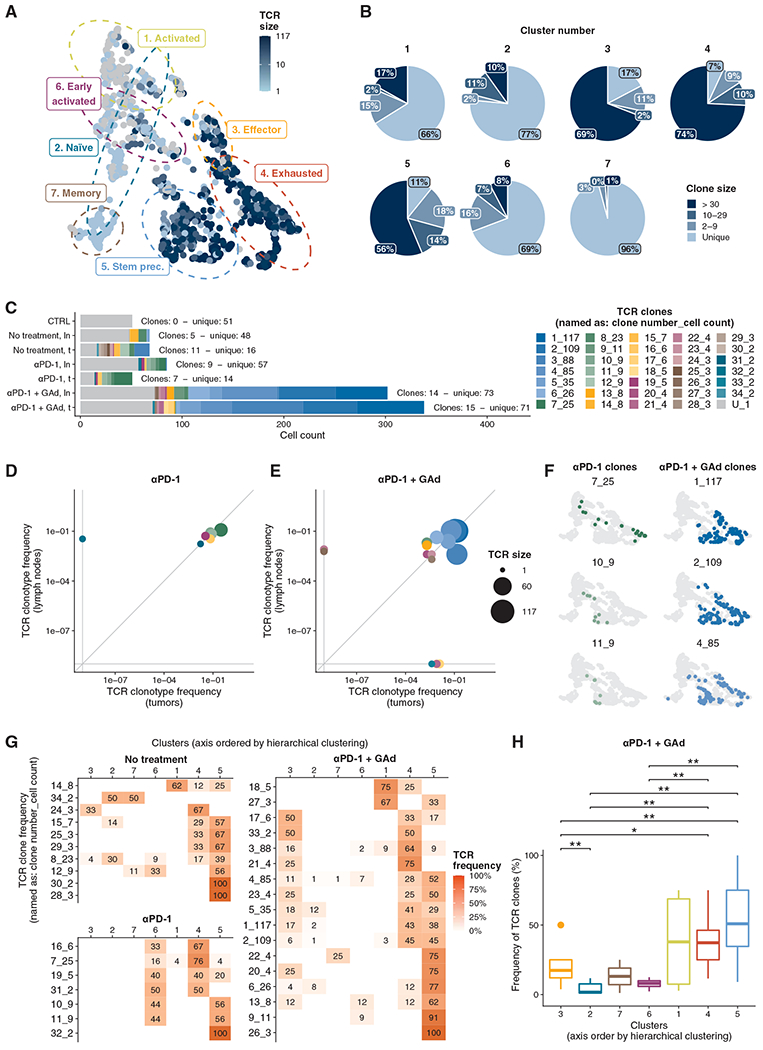
(A) T cell receptor (TCR) clonotype size of each Db-Adpgk+ CD8+ T cell projected onto UMAP. (B) Pie charts of TCR clonotype size for each cluster. (C) Distribution of TCR clonotype size in the different experimental groups, color-coded by TCR. The TCR label indicates the numeric ID of each clonotype and the number of cells belonging to the clonotypes before and after the underscore, respectively. “U” indicates unique TCRs. Numbers of clones and unique TCRs are annotated on the plot. (D and E) Scatterplot of cell frequencies for each TCR clonotype in lymph node and tumors isolated from (D) αPD-1–treated and (E) αPD-1 + GAd–treated mice, respectively. (F) Examples of TCR distributions projected onto UMAP for αPD-1– and αPD-1 + GAd–treated mice, respectively. (G) TCR-wise frequencies from cell counts of clonotypes across clusters. Axis order was defined through hierarchical clustering. (H) Distribution of TCR cell frequencies from αPD-1 + GAd–treated mice among clusters (Wilcoxon signed-rank test).
Furthermore, the results also highlighted that the different treatments generated distinct TCR patterns (Fig. 5C). Cells from lymph nodes of control tumor–free mice displayed exclusively unique TCR sequences (51 singletons). Nontreated tumor-bearing mice showed a distinct TCR distribution in lymph nodes and tumors: the former comprised Db-Adpgk+ CD8+ T cells predominantly with unique TCR (48 singletons and 5 clones), and the latter displayed a higher number of expanded clones of ~2 to 29 cells (16 singletons and 11 clones). In mice treated with αPD-1, tumor and lymph nodes comprised either expanded clones (7 and 9, respectively) or singletons (14 and 57, respectively). However, the highest number of expanded Db-Adpgk+ CD8+ T clones was measured in mice treated with the αPD-1 + GAd vaccine: 14 clones and 73 singletons in lymph nodes, and 15 clones and 71 singletons in tumors.
To better understand how the cells from each TCR clone were distributed between lymph nodes and tumors, we examined their frequency distribution in both compartments (Fig. 5, D and E). In mice treated with αPD-1, six of seven clones (86%) of Db-Adpgk+ CD8+ T cells were shared between the tumor and the lymph nodes, whereas one clone was found exclusively among Db-Adpgk+ CD8+ T cells from lymph nodes (Fig. 5D). In mice treated with αPD-1 + GAd, 11 of 17 clones (65%) were present in both tissues, whereas four and two clones (23 and 12%) were found exclusively in the tumor and lymph node microenvironments, respectively (Fig. 5E). Thus, these results indicate that the cells from the same TCR clone can be found in both lymph nodes and tumors, but in distinct differentiation stages. Examples of TCR clones derived from αPD-1 or αPD-1 + GAd treatment found in both tissues are shown in Fig. 5F. TCR clones found in αPD-1–treated mice were mostly enriched in the early activated, TEX, and T STEM precursors, whereas clones isolated from αPD-1 + GAd–treated mice were principally found in TSTEM precursor, effector, and TEX cell subsets (Fig. 5G). In addition, unique TCRs were mainly found in the memory, early activated, and naïve populations (28, 22, and 19% mean frequency, respectively; fig. S5D). More specifically, we identified 10 TCR clones in nontreated tumor-bearing mice, and only one clonotype with more than 10 cells distributed between TSTEM precursor and the naïve populations (39 and 30% mean frequency, respectively). Seven TCR clones were associated with αPD-1 treatment, but only one TCR clone had more than 10 cells. Most of the clonotypes associated with αPD-1 treatment belonged to the TEX, TSTEM precursor, and early activated populations. Conversely, we found 17 TCR clones in mice treated with αPD-1 + GAd, including seven clones with more than 10 cells and two clones with more than 100 cells. Whereas in nontreated tumor-bearing mice TSTEM precursor population (cluster 5) encompassed 8 TCR clones, in mice treated with αPD-1 + GAd both TCR clonotype number (14 clones) and expansion were increased. Whereas a limited number of TCR clones were observed in the effector population (cluster 3) of nontreated controls, αPD-1 + GAd–treated mice showed 12 clones in the same cluster, pointing to TCR repertoire broadening in the effector population due to the combined treatment.
In line with these results, we found differences in TCR clone frequencies in TSTEM cells (cluster 5) as compared to other subsets in αPD-1 + GAd–treated mice (Fig. 5H), also in accordance with the possible lineages of differentiation (Fig. 4C). Lineage 2, which progresses from naïve to TEX cluster, and lineage 3, which spans from naïve to TSTEM precursors, both displayed a significant increase in TCR clone frequency (fig. S5C; Wilcoxon test, P ≤ 0.01). Overall, these results indicate that combined treatment with αPD-1 + GAd is the strongest promoter of TCR clonotype expansion and diversification in both lymph nodes and tumors.
Neoantigen-based vaccine Nous-209 elicits strong and broad T cell responses in patients with dMMR tumors
Nous-209 vaccine is now being investigated in a phase 1 trial in patients with metastatic gastric, colorectal, and GEJ dMMR tumors combined with αPD-1 pembrolizumab (NCT04041310). This vaccine is based on heterologous prime boost with GAd vector followed by modified vaccinia Ankara (MVA) vector, both encoding 209 frame-shift peptides (FSPs), which are tumor-specific neoantigens shared across patients with dMMR cancer (19). Patients enrolled in this trial have received one GAd vaccination followed by three MVA immunizations, in a “prime/repeated boost” protocol, combined with treatment with αPD-1 every 3 weeks (Fig. 6A).
Fig. 6. Nous-209 vaccination elicits a strong and broad neoantigen-specific T cell response in patients with dMMR tumors.
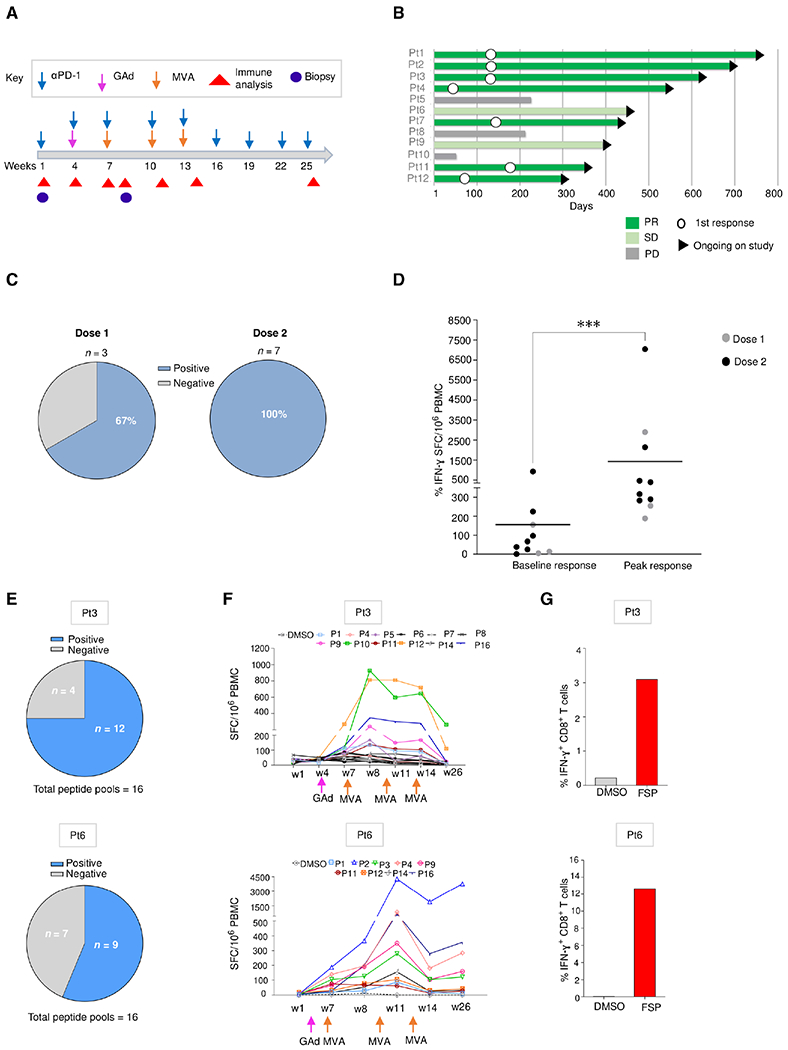
(A) Clinical event timeline for 12 vaccinated patients (Pt) from baseline to the latest time point; treatments include αPD-1 combined with GAd and MVA vaccine prime boost administrations. (B) Clinical responses after treatment as assessed by tumor imaging per RECIST v1.1: response duration shown as a swimmer plot for tumor response over time. White circles indicate time of first response. The arrowheads on the right indicate continuing study treatment. (C and D) Immune responses measured in patients (n = 10) after Nous-209 vaccine by ex vivo IFN-γ. Immunogenicity was assessed by ex vivo IFN-γ ELISpot on peripheral blood mononuclear cells (PBMCs) stimulated with 16 pools of overlapping peptides covering the entire vaccine sequence. (C) Frequency of patients showing a positive response after vaccination and number of SFCs/million PBMCs corresponding to the sum of the responses to the single pools. Dot plot in (D) represents peak responses for each individual subject, compared with baseline prevaccination responses after pembrolizumab. Lines represent the mean of immune response. (E to G) Breadth of immune responses: number of FSP-positive pools (E), kinetic T cell responses measured by ex vivo IFN-γ ELISpot (F), and IFN-γ+ FSP-specific CD8+ T cells measured by intracellular staining (ICS) and flow cytometry after vaccination (G) are reported for patients 3 (dose 1) and 6 (dose 2).
The study comprises two sequential cohorts: dose escalation and dose expansion cohorts testing two doses for both GAd-209-FSP and MVA-209-FSP. Here, we report data on the first 12 subjects evaluated as of 29 September 2021 (table S1). Clinical response according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 is an exploratory endpoint in the study. We observed durable partial response (PR) in the three patients enrolled in dose 1 (two CRC and one GEJ cancer). In dose 2 cohort (six CRC and three gastric cancers), four patients had PR, two had stable disease (SD), and three had progressive disease (PD) (Fig. 6B). The median follow-up for patients in dose 1 is 20.5 months (range, 18.1 to 22.5 months), and 9.4 months (range, 1.7 to 15.2 months) for subjects in dose 2 as of the above cutoff date. No dose-limiting toxicities (DLTs) were observed.
Vaccine immunogenicity was the secondary endpoint of the study and was evaluated by using an ex vivo enzyme-linked immunosorbent spot (ELISpot) assay against 16 peptide pools covering the entire sequence of the 209 FSPs encoded by the vaccine. To evaluate the impact of Nous-209 vaccination on the activation of specific T cell against the 209 FSPs, we collected peripheral blood mononuclear cells (PBMCs) at different time points before and after vaccination (Fig. 6A). Vaccine immunogenicity was demonstrated by ex vivo interferon-γ (IFN-γ) ELISpot assay in 67% of patients in dose 1 and in 100% of patients with evaluable samples in dose 2 (Fig. 6C). Responses at peak after vaccination reached a mean of ~1500 spot-forming cells (SFCs)/million PBMCs, whereas the baseline prevaccination responses measured after the first dose of pembrolizumab reached a mean of ~200 SFCs/million PBMCs (Fig. 6D). Responses were directed against several different peptide pools. The breadth and the kinetics of the response are shown for two patients (Pt3 and Pt6), one for each of the two dose groups (Fig. 6, E and F). Moreover, induction of high amounts of IFN-γ+ FSP-specific CD8+ T cells was measured in the two patients by intracellular staining followed by flow cytometry analysis (Fig. 6G). Overall, these results highlight the durable neoepitope-specific T cell responses in patients with cancer after vaccination with Nous-209 combined with PD-1 blockade.
Neoantigen-specific TCR clonotypes expand and diversify after Nous-209 vaccination in patients with dMMR tumors
To explore the variation of TCR repertoire upon treatment, we compared the intratumoral TCR repertoire from bulk RNA-seq in matched pre- and on-treatment tumors in three patients whose pre- and post-treatment biopsies were available (Pt1, Pt11, and Pt12). All three patients had a partial durable response according to RECIST v1.1 evaluation criteria (Fig. 6B). Consistent with the results in mice, the analysis of T cell infiltrates in all three patients showed increased number of TCR-β copies after Nous-209 treatment, highlighting expansion and diversification of clonotypes (Fig. 7A). The expansion of TCR clonotypes was associated with an enrichment in the estimate of the relative abundance of RNA belonging to effector memory T cells according to a recent pipeline for gene signature detection in tumors (Fig. 7B) (34).
Fig. 7. TCR neoantigen-specific T cell clonotype repertoire is expanded and diversified after Nous-209 vaccination.
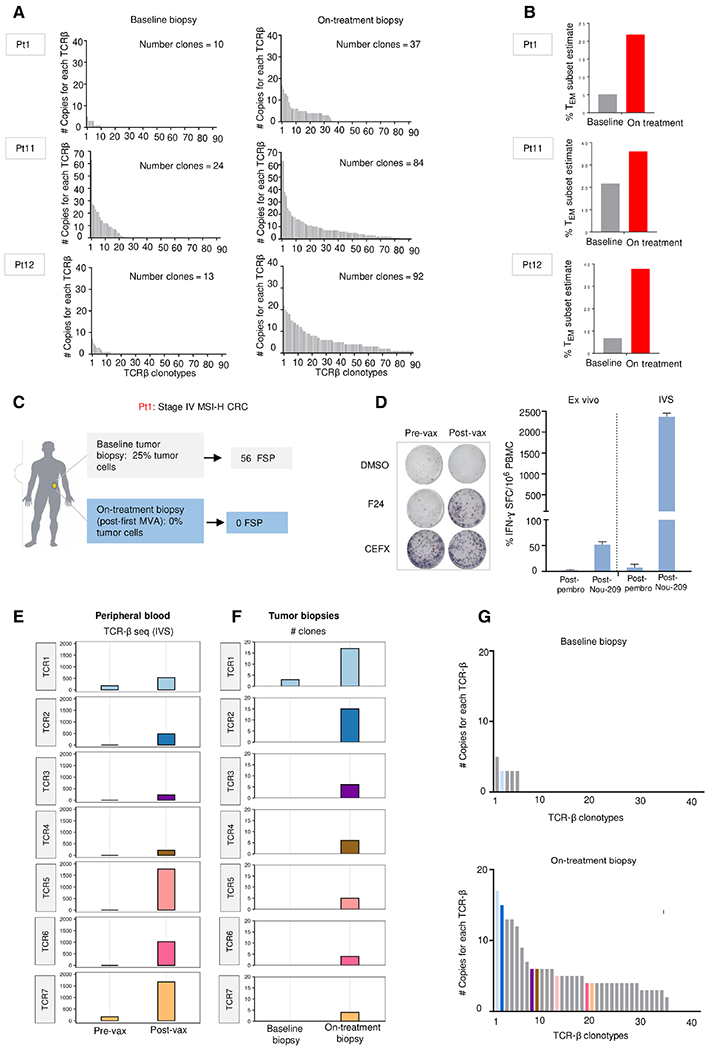
(A) Expansion and diversification of TCR-β repertoire in preand posttreatment tumor biopsies in three patients with clinical response (PR). (B) Estimated mRNA fraction of effector memory T (TEM) cells on tumor samples before and after treatment according to gene expression data (34). (C) Study of patient 1 (Pt1), a stage IV microsatellite instability–high (MSI-H) CRC, second line (2L) in PR. For this patient, baseline tumor biopsy and on-treatment biopsy were collected before the first pembrolizumab administration (week 1) and after first MVA (week 8), respectively. (D) T cell responses in Pt1 were measured by IFN-γ ELISpot assay performed ex vivo and after in vitro restimulation (IVS) with a peptide specific for one encoded FSP (FSP 24). Tested PBMC were collected after pembrolizumab (week 4) and after vaccination (week 7). DMSO and CEFX were used as negative and positive control, respectively. (E) TCR-β sequencing of PBMCs stimulated in vitro with the F24 peptide. The abundance of seven TCR-β clonotypes shared among PBMCs stimulated in vitro with F24 peptide and the clonotypes in the on-treatment tumor biopsy, detected by RNA-seq, are represented. (F) Bar plots represent the abundance of the seven clonotypes analyzed at baseline and after treatment on the tumor biopsies. Only one TCR (TCR1) was also found in the baseline biopsy as already present in the tumor and expanded after treatment, whereas the other six clones were exclusively detected in the posttreatment biopsy. (G) Expansion and diversification of TCR-β repertoire in pre- and posttreatment tumor biopsies. The clonotypes detected on the tumor biopsies of Pt1 are shown. Each bar is a TCR-β individual clone; colored bars indicated seven TCR clonotypes specific for F24 FSP identified in (F).
To investigate the migration of vaccine-induced CD8+ T cells from the peripheral blood to the tumor, we focused on patient 1, for which we identified a neoepitope specificity for CD8+ T cells induced by the vaccine. The peptide carrying the mutation eliciting the response, named F24, was one of the 56 FSPs in common between the vaccine and the baseline tumor biopsy, detected by next generation sequencing (NGS) data analysis (table S2) (Fig. 7C). The F24 mutation was found in about 40% of tumor cells according to the mutation clonality analysis. The F24 sequence includes an 8-mer peptide predicted as a strong binder of one of the patient haplotypes [IAKKRIKL; HLA-B*08:01; predicted median inhibitory concentration (IC50), 36 nM]. After ex vivo and after in vitro restimulation (IVS), although F24-specific CD8+ T cells were not detected in the PBMCs collected after pembrolizumab treatment (Fig. 7D), the F24-specific CD8+ T cells were found expanded after IVS of PBMCs collected after vaccination, as shown by the higher number of IFN-γ+ SFCs (Fig. 7D).
Expanded F24-specific CD8+ T cells were also subjected to TCR-β sequencing. Seven F24 FSP-reactive TCR-β clones expanded after IVS were also detected in the post-vaccine treatment tumor biopsy by bulk RNA analysis (week 8; Fig. 7, E and F). Of those, only one clonotype (TCR1) was found in the baseline biopsy and expanded after treatment, whereas the other six TCR clones were exclusively detected in the posttreatment biopsy (Fig. 7G). Together, these results indicate that neoantigen-specific CD8+ T cells induced by Nous-209 vaccine expand and diversify upon treatment, and successfully traffic and infiltrate into tumor bed.
DISCUSSION
The magnitude and the quality of neoepitope-specific CD8+ T cells are key determinants to drive effective antitumor responses. Conversely, for exhausted cells, TCF1+ CD8+ TSTEM cells can proliferate upon PD-1 blockade and act as a reservoir to continuously produce and replenish effector CD8+ T with cytolytic function (24). Recently, there is a growing interest in using CD8+ TSTEM cells for T cell–based therapies, such as adoptive T cell transfer or generation of chimeric antigenic receptor (CAR) T cells, where the use of a less differentiated CD8+ T cell subsets has been associated with improved therapeutic responses (35, 36). Because of these unique properties of plasticity and self-renewal, strategies aiming to expand T cells to have stem-like properties might provide opportunities for the development of more effective immunotherapies in clinical cancer care, especially in patients with resistance to checkpoint blockade therapies (7).
Here, we characterize neoepitope-specific CD8+ T cells in a mouse CRC model. We have selected the MC38 cell line, which is considered a valid tumor cell line for modeling hypermutated and microsatellite instability (MSI)–high CRC (37). We show that vaccination with GAd vector encoding tumor neoantigens in combination with αPD-1 enhances the magnitude of neoantigen-specific CD8+ T cells and promotes their differentiation into stem-like Tcf1+ CD8+ T cell progenitors.
scRNA-seq data of Adpgk-specific CD8+ T cells, induced by the vaccine in combination with αPD-1, highlighted a wide dynamic of CD8+ T cell expansion and differentiation with the accumulation of CD8+ TSTEM cells in the tumor-draining lymph nodes associated with memory T cell precursors in the spleen and CD8+ T effectors in tumor bed, suggesting that specific CD8+ T progenitors egress from the lymph nodes and further differentiate into effectors in the tumors. On the basis of these findings, it is likely that the superior antitumor efficacy achieved by GAd and αPD-1 combination compared to αPD-1 monotherapy is related with the magnitude and transcriptional quality of CD8+ T cell population induced by the vaccine.
Moreover, by using αβTCR sequences as a reliable molecular barcode to track the trajectories of Adpgk+-specific CD8+ T, we confirmed that the same CD8+ T cell clones can be found in both tissues in distinct differentiation stages. Our data support the role of CD8+ TSTEM cells as an active reservoir of progenitors of CD8+ T effectors and then exhausted cells, with the process of commitment for tumor-specific CD8+ T cells starting in the draining lymph nodes before migrating into the tumor (3). In the tumor microenvironment, as expected, chronic exposure to tumor antigens and the presence of multiple immunosuppressive signals lead to T cell exhaustion, a behavior confirmed by both trajectory inference and TCR clonal expansion. The TCR clonotype analyses have demonstrated an increased inter- and intraclonal heterogeneity of Adpgk+-specific CD8+ T cells in mice treated with GAd combined with αPD-1, as compared to mice receiving only αPD-1 monotherapy.
So far, the best example of vaccines generating long-lived memory T cells with stem-like features and mediating lifelong protection is the yellow fever vaccine, based on a highly effective attenuated virus (38). The synergistic mechanism of action of vaccination with checkpoint blockade in cancer is supportive of recent findings, showing that intravenous vaccination based on neoantigen peptides and Toll-like receptor 7/8 agonists (SNP-7/8a) linked to nanoparticles promotes increased numbers of CD8+ TSTEM cells, associated with better tumor rejection in a prophylactic cancer model (9).
Another key aspect of this study is related to the effect of the Ad vaccine in broadening the antigenic breadth and clonal diversity of CD8+ T cells in the context of cancer models (18). Our findings support the adenoviral vector platform as a promising approach promoting the generation of a diversified TSTEM cell reservoir for effective antitumor activity. In accordance with results obtained in the preclinical mouse model, vaccination targeting shared FSP neoantigens across patients with dMMR induced a potent and broad T cell response in the vast majority of patients. Broadening of T cell repertoire and an increase of T cells with an effector memory phenotype after the vaccination were shown in three patients responding clinically to the treatment. In one of these patients, we could track vaccine-induced neoantigen-specific TCR clonotypes in the tumor biopsy. Together, these results suggest that neoantigen-specific CD8+ T cells, induced by Nous-209, expand and diversify upon treatment, and successfully traffic and infiltrate the tumor microenvironment to exert active antitumor activity.
Our data support the development of viral vectored vaccines for neoantigen-based immunotherapies. Shared neoantigens are not common in tumor types other than MSI-high tumors, and a personalized approach is required. Related to the personalized approach, a major challenge for clinical application in patients with metastatic cancer is the need for fast manufacturing and timely delivery of individually tailored vaccines to patients. To this aim, we have developed a fast process for producing viral vectors encoding 60 unique patient-specific neoantigens and recently initiated a clinical trial in patients with metastatic melanoma and non–small cell lung cancer (NCT04990479).
This study has some limitations. First, there are some differences between the mouse model and the clinical trial in terms of treatment regimen and schedule. In mice, the vaccine was administered concomitant to αPD-1, whereas in the trial patients first received αPD-1 and then the vaccine. In patients, the vaccine was administered at the second injection of αPD-1 to establish a baseline immune response after αPD-1 treatment but before vaccination to evaluate vaccine-induced immune response. In contrast to the clinical trial, in the mouse model, vaccine schedule did not include a MVA boost after GAd priming. Tumor growth is much faster in the mouse model and does not allow for the assessment of the MVA boost. However, the ability of MVA to improve the magnitude and quality of adeno vaccine–induced T cells has been extensively demonstrated (15) and supports the use of the prime and boost regimen in patients.
Second, the effect of expansion and diversification of the intratumoral TCR-β is reported in three patients, and it was only possible in one to track vaccine-induced neoantigen-specific TCRs in the tumor biopsy after treatment. Unfortunately, only a limited number of requested samples from the clinical trial were available and were of good enough quality for proper analysis. Additional clinical samples will be analyzed in the future to extend our data. In addition, the patient population of this study is heterogeneous in terms of both line of treatment (first and second line of treatment) and tumor type, and the number of patients is too low to allow comparison with historical data of αPD-1 efficacy. Last, the TCR repertoire in the patient tumor biopsies was analyzed from bulk RNA-seq data and not by scRNA-seq, as were the samples from the mouse model. However, scRNA-seq of clinical tumor specimens poses several challenges both technically and logistically in multisite studies.
Overall, our findings indicate that Ad vector vaccines in combination with checkpoint inhibitor treatment can sustain the proliferation, expansion of neoepitope-specific T cell clones, and trafficking into the tumor both in mice and in patients with cancer. This vaccination platform encoding many different neoantigens is well suited to overcome αPD-1 therapy resistance by increasing polyclonal expansion and eliciting memory T cells against tumor rejection neoantigens, thus paving the way for more effective immunotherapy treatments.
MATERIALS AND METHODS
Study design
This is a multicenter, unrandomized, open-label phase 1 study designed to determine safety, immunogenicity, and preliminary efficacy of heterologous prime/boost GAd20-209-FSP/MVA-209-FSP vaccination schedule in combination with pembrolizumab, in subjects with second- or first-line unresectable or metastatic dMMR/MSI-H CRC, gastric, or GEJ tumors (NCT04041310). The study is composed of two sequential cohorts, i.e., dose escalation and expansion, testing two dose levels, i.e., dose level 1 [GAd20-209-FSP: 1.88 × 1010 viral particles (vp); MVA-209-FSP: 1.65 × 107 infectious unit (ifu)] and dose level 2 (GAd20-209-FSP: 1.88 × 1011 vp; MVA-209-FSP: 1.65 × 108 ifu). The study started at the first pembrolizumab infusion (day 1 of week 1); GAd20-209-FSP prime was administered on the day of the second pembrolizumab infusion (week 4); MVA-209-FSP boosts were given at third, fourth, and fifth pembrolizumab infusions (weeks 7, 10, and 13, respectively).
In the dose escalation portion of the study, a 3 + 3 + 3 dose escalation design was applied in up to two sequential cohorts. In the dose expansion portion of the study, the expansion cohort was determined by logistic feasibility rather than statistical considerations. No formal sample size calculation was carried out. Safety monitoring committee was established to assess safety of both dose levels, to make a recommendation for recommended phase 2 (RP2D) of the vaccine, as well as recommendations concerning the continuation, modification, and termination of the trial. The severity and relatedness with vaccination or adverse effects were assessed by the medical team in each center and graded according to Common Terminology Criteria for Adverse Events (CTCAE) v5.0. Objective response rate (ORR) was assessed using RECIST v1.1. Immunogenicity was assessed using ex vivo ELISpot. The full study protocol is provided in the Supplementary Materials (trial protocol no. NOUS-209-01-00). Subjects were expected to attend several visits after screening (up to 28 days) and in addition to vaccination visits. During the visits, the subjects underwent blood sampling and medical evaluation. Subjects also discussed with a doctor any potential adverse effects. Three subjects were vaccinated with dose level 1 of Nous-209 in combination with pembrolizumab treatment, not reporting any DLT. Subsequently, subjects were enrolled at dose level 2 of Nous-209 in combination with pembrolizumab. Nine subjects were vaccinated with dose level 2 of Nous-209 in combination with pembrolizumab treatment as of 29 September 2021, not reporting any DLT. Therefore, the high dose was declared as the RP2D.
Ethical statement
The study was approved by the Institutional Review Boards (IRBs) of MD Anderson University of Texas (protocol 2019-0651), Weill Cornell Medicine (protocol 19-08020684-02), John Hopkins (IRB 00220323), Roswell Park (MOD 00006973/P-457819), Karmanos Cancer (IRB 202000155), and City of Hope (IRB 19277/176947).
Vaccine vector generation
Production of GAd vaccine vector for preclinical studies has been performed as previously described (18). GAd-209-FSP and MVA-209-FSP vectors have been generated as previously described (19). For GAd, the transgenes were synthesized by GeneArt (Thermo Fisher Scientific) and subcloned via Eco R1–Not 1 restriction enzymes (New England Biolabs) into a shuttle plasmid containing the cytomegalovirus (CMV) promoter with two Tet Operator repeats and a bovine growth hormone (BGH) polyA. The expression cassettes were then transferred into the E1 deletion locus of pGAd20 plasmid by homologous recombination in BJ5183 cells. pGAd20 contains the genome of a gorilla Ad (serotype group C) deleted in E1 and E3 regions. For MVA-209, the transgenes were synthesized by GeneArt and subcloned via Bam H1–Asc 1 restriction enzymes (New England Biolabs) into a shuttle plasmid under the control of the P7.5 promoter. In addition, the shuttle plasmid carries an enhanced green fluorescent protein (eGFP) expression cassette and sequences homologous to the deletion III locus of MVA, to allow insertion of both expression cassettes (eGFP and transgene) in this locus. Recombinant MVA vectors were obtained by homologous recombination in chick embryo fibroblast (CEF) cells. GAd-209-FSP and MVA-209-FSP clinical lots have been manufactured by Reithera s.r.l. (Rome, Italy). For Good Manufacturing Practice (GMP) production of GAd-209-FSP, the four GAd-20 vectors have been amplified in suspension M9 cells [human embryonic kidney (HEK) 293 derivative] and purified by anion exchange chromatography. Vector titers were determined by quantitative polymerase chain reaction targeted on CMV promoter sequence, and the four vectors were mixed to generate the GAd-209-FSP vaccine. Vector infectious titers were determined by hexon immunostaining on infected M9 cells.
For GMP production of MVA-209-FSP, the four MVA vectors have been amplified in suspension AGE1.CR.pIX cells and, after purification, mixed to obtain the MVA-209-FSP vaccine. Infectious titer has been determined by immunostaining on infected Vero cells with anti–vaccinia virus antibodies.
Mice
C57BL/6J female mice were obtained from Envigo and used between 7 and 10 weeks of age. All the mice were housed in Molecular Biotechnology Center (MBC) (Turin University) specific pathogen free (SPF) Animal Facility. Live animal experiments were done in accordance with the guidelines of Italian and European Veterinary Department.
In vivo treatments
Vaccines were administered via intramuscular injections in the quadriceps by delivering a volume of 50 μl per side at 5 × 108 vp. For efficacy studies, anti–PD-1 antibody (αPD-1, BioXcell, clone RMP114, catalog number BE0146) was administered twice a week until day 16 after start of treatment. The antibody was administered intraperitoneally at a dosage of 200 μg. At the indicated time points, the draining inguinal lymph nodes, tumors, and spleens were harvested and analyzed by flow cytometry.
In vivo tumor progression
MC38 cells (2 × 105; C57BL/6 mouse colon adenocarcinoma, American Type Culture Collection) were subcutaneously injected as aforementioned. Before treatments start (day 0), animals were randomized (tumor size average per group, 70 to 100 mm3). Mice were sacrificed as soon as signs of distress or a tumor volume above 2000 mm3 occurred. Tumor growth was measured every 3 to 4 days. Tumor volume was calculated using the formula (tumor size W) × (tumor length L)2/2, where the length was the longer dimension.
Flow cytometry and index cell sorting
The cells were harvested at different time points after immunization. Single-cell suspensions were generated, red blood cells were lysed with a hypotonic buffer, and cell number was determined. Tumor-infiltrating lymphocytes were enriched by Percoll gradient before labeling. Cell suspensions were prepared in phosphate-buffered saline (PBS)–0.5% bovine serum albumin (BSA) and 2 mM EDTA, and cells were labeled according to the experiment with αCD8 (clone 53-6.7, reference 563332, BD), αTCRβ (clone H57-597, reference 563221, BD), αCD103 (clone M290, reference 562772, BD), αCD69 (clone H1.2F3, reference 552879, BD), αCD44 (clone IM-7, reference 565480, BD), αCD62L (clone MEL-14, reference 563252, BD), αCD127 (clone SB/199, reference 562419, BD), αKLRG1 (clone 2F1, reference 742199, BD), αH-2Kb (clone AF6-88.5, reference 116507, BioLegend), αH-2Db (clone REA619, reference 130-109-608, Miltenyi), αPD-1 (clone J43, reference 67-9985-82, Invitrogen), and αCD38 (clone 90, reference 11-0381-82, Invitrogen). Cells were then fixed with 1% paraformaldehyde. Fc receptors were blocked with the CD16/CD32 (2.4.G2) monoclonal antibody. Dead cells were stained with cell death dyes (eBioscience) according to the manufacturer’s instructions. Phenotypic characterization of lymphocytes was performed using BD LSRFortessa X-20 and sorted with BD FACSAria III (Becton Dickinson). The limit of detection was set above 10 recorded cells. The data were analyzed with FlowJo 10.4.2 software.
Peptide stability assay
RMA-S cells (5 × 105) were plated in a 24-well plate, and each peptide (Dpagt1, Reps1, Adpgk, Irgq, Aatf, and Cpne1) was added with a final concentration of 30 μM. The cells were incubated overnight at 37°C. Next, they were collected in a 50-ml tube, washed with 1× PBS, and plated in a 96-well plate. To monitor the relative stability of surface H2-Kb and H2-Db molecules, the cells were stained with αH-2Kb (AF6-88.5) and αH-2Db (clone REA619) antibody every 2 hours for 6 hours in total. Phenotypic characterization of lymphocytes was performed using BD LSRFortessa X-20 and sorted with BD FACSAria III (Becton Dickinson). The data were analyzed with FlowJo 10.4.2 software.
Single-cell RNA-seq
Dextramer-Adpgk+ CD8+ T cells were FACS-sorted from draining lymph nodes and tumors, harvested from nontreated tumor-bearing mice, and treated with αPD-1 and with αPD-1 combined with GAd. Neoepitope-specific CD8+ T cells were sorted into 384-well plates containing lysis buffer and immediately frozen. Single-cell library preparation was done as previously described (26). Libraries were pooled and sequenced [2 × 50–base pair (bp) and 2 × 100-bp paired-end reads], 106 reads per cell, on the NOVAseq 6000 instrument.
Single-cell RNA-seq processing
Single-cell sequencing files (base calls) were first demultiplexed using the bcl2fastq2 (v.2.20.0.422) software using default parameters for paired-end sequencing. The resulting FASTQ files were then aligned to the GRCm38 mouse (mm10) genome using STAR (v.2.7.3a) (39, 40), and transcript-per-million (TPM) counts were obtained by processing the alignments with RSEM (v.1.3.1) (41). After quality controls, 1189 cells were selected for further analysis with R (v.3.6.3) (R Core Team, 2020). Only genes detected in at least 5% of the cells with a log2TPM ≥ 2 were analyzed. Furthermore, mitochondrial and ribosomal genes were removed.
Data were regressed through linear modeling, then scaled, and centered along mean values of the gene expression matrix. PCA was run on the scaled data, and using the elbow plot approach, the first 14 principal components were selected for the UMAP (42) and clustering procedures. Clusters were identified using the Louvain algorithm (43) with the following parameters for k, resolution, and number of iterations: k = 35, r = 0.4, and i = 1000. Differentially expressed genes for each cluster and for each cluster subdivided by mouse response were obtained using MAST with default parameters (44).
TCR sequences were reconstructed using TraCeR (45) with default parameters. The outputs of TraCeR include the assembled nucleotide sequences for both α and β chains, the coding potential of the sequences, the estimation of TPM values, and the CDR3 sequences. Only α and β chains with TPM values higher than 10 and 15 TPM, respectively, were kept for analysis. Clones were defined as different cells that shared identical α-β pairs. In addition, cells that shared CDR3 and α sequence with other cells, but in which TraCeR failed to reconstruct the β sequences, were associated to the respective clones. In the rare event of matches with multiple clones, the TCR was disregarded and not considered for further analyses. Cells with missing α chains were not included in clones even if they shared a β sequence with other cells.
Supervised trajectory inference was done using the slingshot R package (46). We defined the start and end points of the lineages as the naïve and exhausted cell populations, respectively.
Patient PBMC preparation
Peripheral blood samples (PBMCs) from patients were isolated at different time points and cryopreserved at the clinical sites before shipment to the central laboratory for immunogenicity assessment. PBMC isolation and freezing procedures were performed within maximum 8 hours from blood collection to preserve PBMC functionality in immune-monitoring assays. Briefly, peripheral venous blood (80 ml) was collected in 8 × 10-ml lithium-heparin Vacutest blood collection tubes (Kima). PBMCs were isolated using Leucosep Bio-One Polypropylene Tube (prefilled) according to the instructions of the manufacturer. In brief, whole-blood samples were diluted 2:1 with Hanks’ balanced salt solution (HBSS) and transferred to the Leucosep tubes (20 ml of blood and 10 ml of HBSS for each tube). The tubes were then centrifuged at 800g for 15 min without braking at 20°C (±2°C). The cell suspension was collected, washed in HBSS for 10 min at 800g, and then washed two more times at 250g for 10 min at 20°C in HBSS + 5% fetal bovine serum (FBS) before counting. After counting, cells were thawed in freezing medium [10% dimethyl sulfoxide (DMSO) and 90% FBS] and placed at −80°C for 1 to 3 days before storage in liquid or vapor N2 tank. At the time of shipment, the samples were transferred from liquid N2 tank to MVE Vapor Shipper and then shipped to the central laboratory. PBMCs were stored in vapor-phase liquid nitrogen until time of the analysis. Median post-thaw PBMC viability was about 90.0%.
IFN-γ ELISpot assay
The frequency of IFN-γ–producing T cells was measured by ex vivo ELISpot-forming cell assay after antigen-specific stimulation. PBMCs were resuspended in R10, stimulated with a set of peptides designed to cover the 209 FSPs encoded by the vaccine, and arranged into 16 peptide pools (P1 to P16). Cells were plated at 2 × 105 cells per well in ELISpot plates (Human IFN-γ ELISpotPLUS kit, Mabtech) and incubated for 18 to 20 hours in the presence of the peptides in a 37°C (±1°C), humidified CO2 incubator. At the end of incubation, the ELISpot assay was developed according to the manufacturer’s instructions. Spontaneous cytokine production (background) was assessed by incubating PBMCs with the medium only plus the peptide diluent DMSO (negative control) (Sigma-Aldrich), whereas CEFX [a pool of known peptide epitopes for a range of human leukocyte antigen (HLA) subtypes and different infectious agents, namely, Clostridium tetani, coxsackievirus B4, Haemophilus influenzae, Helicobacter pylori, human Ad5, human herpesvirus 1, human herpesvirus 2, human herpesvirus3, human herpesvirus 4, human herpesvirus 5, human herpesvirus 6, human papillomavirus, JC polyomavirus, measles virus, rubella virus, Toxoplasma gondii, and vaccinia virus (JPT Peptide Technologies)] was used as positive control. Results are expressed as SFC/106 PBMCs in stimulating cultures after subtracting the DMSO background. The cutoff values for a positive response were calculated on 20 healthy donors. A response was considered positive if (i) the number of SFC/106 cells was greater than 48 and (ii) higher than three times the background DMSO value. A subject is defined as a responder if reactivity to at least 1 of the 16 FSP peptide pools is induced after vaccination. Induction of an immune response is defined as a change from negative at baseline (after pembrolizumab) to positive at any of the time points collected after vaccination. In case of detection of a positive reactivity against one of the peptide pools at baseline, the vaccine is expected to determine an increase (of at least 80%) of the preexisting reactivity to at least 1 of the 16 FSP peptide pools, and in this case, such patients will be considered as responders.
In vitro expansion of neoantigen-specific T cells
For in vitro expansion of antigen-specific T cells, PBMCs were cultured in RPMI 1640 medium supplemented with l-glutamine, Hepes, penicillin/streptomycin (Gibco), and 10% heat-inactivated human serum (Defined, HyClone). Cells (4 × 106 per well) in 1-ml volume were stimulated in a 24-well plate with individual (4 μg/ml) or peptide pool (each peptide at 4 μg/ml) in the presence of interleukin-7 (IL-7) (330 U/ml; PeproTech). On day 3, low-dose IL-2 (10 U/ml; PeproTech) was added. Half-medium change and supplementation of IL-2 were performed at days 3, 7, and 10. After 12 days, PBMCs were collected and resuspended in complete medium without cytokines and peptides for overnight resting at 37°C. The day after, T cell response against F24 peptide was tested by IFN-γ ELISpot assay.
Tumor biopsy RNA-seq
For all the patients, appropriate IRB approval and written informed consent were obtained. Formalin-fixed paraffin-embedded tissue specimens before and after treatment together with a blood sample were collected from patient 1. DNA and RNA were extracted from each collected sample and sequenced at CeGaT GmbH. Raw exome and RNA-seq reads were aligned on the GRCh37 human genome using Burrows-Wheeler Aligner (BWA) mem (PMC2705234) and HISAT2 2.0.4 (PMC7605509) software, respectively. Multimapping reads were filtered out using SAMtools 0.1.19 (PMC7931819). Optical duplicates were marked using Picard’s MarkDuplicates tool v1.14 (https://broadinstitute.github.io/picard/). DNA alignments were further optimized at regions around indels, and base scores were recalibrated after the optimization step using Genome Analysis Tool Kit (GATK) software v3.4.46 (PMC2928508). Somatic variant calling of small indels was performed using Mutect2 (PMC2928508), VarScan 2 (PMC3290792), and Scalpel (PMC5507611) with default parameters. All frameshift mutations detected by at least one variant caller were considered. Analysis of clonality of detected mutations was performed by using allele-specific copy number analysis of tumors (ASCAT) software (PubMed identifier PMID: 20837533) to detect copy number variation in tumor exome sequencing data, followed by PyClone (PMC4864026) to estimate the clonality of each somatic mutation. All the parameters were kept to default except for the input of ASCAT that was created with a custom tool that filters only the SNPs with at least the 30% of mutation allele frequency in blood. The list of filtered SNPs was then passed to the AlleleCounter tool to prepare the input for ASCAT.
MHC-I binding prediction
Patient HLA class I haplotypes were determined by using the OptiType software (PMC4441069) on the exome sequencing performed on blood sample. MHC-I binding predictions on the positive peptide were performed using the consensus method included in Immune Epitope Database 2.17 (PMID: 16767078). HLA binders (8- to 10-mer) with a predicted IC50 ≤ 500 nM were considered.
TCR-sequencing analysis
TCR sequencing (TCR-seq) of TCR-β chain was performed on IVS expanded T cells. Raw NGS data were analyzed with MiXCR 2.1.11 (PMID: 25924071) applying default parameters. The parameters were modified according to the manual to assess the TCR-β chain repertoire in RNA-seq performed on tumor biopsies at baseline and after vaccination. The derived CDR3 sequences were further analyzed by tracking the expression of clonotypes that were shared between post-vaccination tumor biopsy and in vitro sample at week 7 stimulated with F24 peptide.
Statistical analysis
Unpaired t test, or otherwise mentioned, was used for statistical analysis. SEM and P value were determined using Prism software (GraphPad Software Inc.). Symbols used: *P < 0.05, **P < 0.01, ***P < 0.001, and n.s. (not significant).
Supplementary Material
Acknowledgments:
We thank all the IIGM and CCI for helpful discussions and C. Parlato, K. Gizzi, S. Guarrera, G. Granato, F. Altruda and the MBC (University of Turin) animal facility for technical help. We thank Reithera for the GMP manufacturing of the vaccine clinical lots and S. Capone and M. Del Sorbo for support in performing and validating the ELISpot assay on the human samples.
Funding:
L. Pace received founding from IIGM/CSP, Armenise-Harvard foundation, AIRC IG 2020 ID 24463; FPO/Candiolo Advance Im-Immunity 5x1000. High-throughput sequencing of mouse data has been performed by the IIGM genomic platform of IIGM supported by CSP.
Competing interests:
E.S. is the founder of Nouscom. A.M.D., G.L., F.L., E.M., T.F., and P.D. are employees of Nouscom; E.S. and G.L. are inventors of the patent application related to the manuscript “A universal vaccine based on shared tumor neoantigens for prevention and treatment of micro satellite instable (MSI) cancers,” WO2019/012082. D.T.L. serves on advisory boards for Merck, Bristol Myers Squibb, Nouscom, and Janssen and has received research funding from Merck, Bristol Myers Squibb, Aduro Biotech, Curegenix, Medivir, Nouscom, and Abbvie. D.T.L. has received speaking honoraria from Merck and is an inventor of licensed intellectual property related to technology for dMMR for diagnosis and therapy (WO2016077553A1) from Johns Hopkins University. The terms of these arrangements are being managed by Johns Hopkins. M.F. serves on advisory boards for Nouscom, Amgen Inc., Array BioPharma, Bayer Corporation, GlaxoSmithKline, and Seattle Genetics Inc. and is a consultant of Pfizer Inc., Zhuhai Yufan Biotechnology Co. Ltd., Taiho Oncology, HalioDx, Speaker Bureau of Guardant360, Grant to Institution from Amgen Inc., AstraZeneca Pharmaceuticals, Novartis Oncology, and Bristol Myers Squibb Verastem. M.J.O. is consultant of Phanes Therapeutics, Takeda Pharmaceuticals, Ipsen Biopharmaceuticals, Pfizer, Merck, GlaxoSmithKline, Promega, 3D Medicine, Acrotech Biopharma, Nouscom, and Gritstone and received research funding from Takeda, Merck, BMS, AstraZeneca, Nouscom, and Gritstone. A.F.S. received research funding from Nouscom. M.A.S. received research funding from Merck, Bristol Meyers Squibb, and Oncolys Biopharma and is in advisory board of Daiichi and Lilly.
Footnotes
SUPPLEMENTARY MATERIALS
www.science.org/doi/10.1126/scitranslmed.abo7604
MDAR Reproducibility Checklist
View/request a protocol for this paper from Bio-protocol.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Viral vectors (GAd and MVA) are available from Nouscom with a material transfer agreement upon reasonable request to E.S. The bioinformatics data are available in the Gene Expression Omnibus (GEO) database with accession number GSE206811.
REFERENCES AND NOTES
- 1.Pace L, Temporal and epigenetic control of plasticity and fate decision during CD8+ T-cell memory differentiation. Cold Spring Harb. Perspect. Biol 13, a037754 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, Forman J, Huang T, Li S, Zhang W, Xu Q, Cieri N, Clauser KR, Shukla SA, Neuberg D, Justesen S, MacBeath G, Carr SA, Fritsch EF, Hacohen N, Sade-Feldman M, Livak KJ, Boland GM, Ott PA, Keskin DB, Wu CJ, Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature 596, 119–125 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue HH, Ahmed R, Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, Xu L, Chen X, Hao Y, Wang P, Zhu C, Ou J, Liang H, Ni T, Zhang X, Zhou X, Deng K, Chen Y, Luo Y, Xu J, Qi H, Wu Y, Ye L, Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–416 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin JX, Konieczny BT, Im SJ, Freeman GJ, Leonard WJ, Kissick HT, Ahmed R, Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1+ stem-like CD8+ T cells during chronic infection. Immunity 51, 1043–1058.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, Freeman SS, Reuben A, Hoover PJ, Villani AC, Ivanova E, Portell A, Lizotte PH, Aref AR, Eliane JP, Hammond MR, Vitzthum H, Blackmon SM, Li B, Gopalakrishnan V, Reddy SM, Cooper ZA, Paweletz CP, Barbie DA, Stemmer-Rachamimov A, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, Hacohen N, Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Restifo NP, Smyth MJ, Snyder A, Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 16, 121–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Z, Leet DE, Allesoe RL, Oliveira G, Li S, Luoma AM, Liu J, Forman J, Huang T, Iorgulescu JB, Holden R, Sarkizova S, Gohil SH, Redd RA, Sun J, Elagina L, Giobbie-Hurder A, Zhang W, Peter L, Ciantra Z, Rodig S, Olive O, Shetty K, Pyrdol J, Uduman M, Lee PC, Bachireddy P, Buchbinder EI, Yoon CH, Neuberg D, Pentelute BL, Hacohen N, Livak KJ, Shukla SA, Olsen LR, Barouch DH, Wucherpfennig KW, Fritsch EF, Keskin DB, Wu CJ, Ott PA, Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med 27, 515–525 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baharom F, Ramirez-Valdez RA, Tobin KKS, Yamane H, Dutertre CA, Khalilnezhad A, Reynoso GV, Coble VL, Lynn GM, Mule MP, Martins AJ, Finnigan JP, Zhang XM, Hamerman JA, Bhardwaj N, Tsang JS, Hickman HD, Ginhoux F, Ishizuka AS, Seder RA, Intravenous nanoparticle vaccination generates stem-like TCF1+ neoantigen-specific CD8+ T cells. Nat. Immunol 22, 41–52 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, Yu S, Kurachi M, Herati RS, Vella LA, Baxter AE, Wu JE, Khan O, Beltra JC, Giles JR, Stelekati E, McLane LM, Lau CW, Yang X, Berger SL, Vahedi G, Ji H, Wherry EJ, TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 51, 840–855.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, Hayton EJ, Black A, Bridgeman A, Rosario M, Hill AV, Berrie E, Moyle S, Frahm N, Cox J, Colloca S, Nicosia A, Gilmour J, McMichael AJ, Dorrell L, Hanke T, Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol. Ther 22, 464–475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A, Siani L, Naddeo M, Grazioli F, Esposito ML, Ambrosio M, Sparacino A, Bartiromo M, Meola A, Smith K, Kurioka A, O’Hara GA, Ewer KJ, Anagnostou N, Bliss C, Hill AV, Traboni C, Klenerman P, Cortese R, Nicosia A, Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci. Transl. Med 4, 115ra2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Hara GA, Duncan CJ, Ewer KJ, Collins KA, Elias SC, Halstead FD, Goodman AL, Edwards NJ, Reyes-Sandoval A, Bird P, Rowland R, Sheehy SH, Poulton ID, Hutchings C, Todryk S, Andrews L, Folgori A, Berrie E, Moyle S, Nicosia A, Colloca S, Cortese R, Siani L, Lawrie AM, Gilbert SC, Hill AV, Clinical assessment of a recombinant simian adenovirus ChAd63: A potent new vaccine vector. J. Infect. Dis 205, 772–781 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasso E, D’Alise AM, Zambrano N, Scarselli E, Folgori A, Nicosia A, New viral vectors for infectious diseases and cancer. Semin. Immunol 50, 101430 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, Halliday J, Kelly C, Bowen D, Fergusson J, Kurioka A, Ammendola V, Del Sorbo M, Grazioli F, Esposito ML, Siani L, Traboni C, Hill A, Colloca S, Davis M, Nicosia A, Cortese R, Folgori A, Klenerman P, Barnes E, A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci. Transl. Med 6, 261ra153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanini S, Capone S, Antinori A, Milleri S, Nicastri E, Camerini R, Agrati C, Castilletti C, Mori F, Sacchi A, Matusali G, Gagliardini R, Ammendola V, Cimini E, Grazioli F, Scorzolini L, Napolitano F, Plazzi MM, Soriani M, De Luca A, Battella S, Sommella A, Contino AM, Barra F, Gentile M, Raggioli A, Shi Y, Girardi E, Maeurer M, Capobianchi MR, Vaia F, Piacentini M, Kroemer G, Vitelli A, Colloca S, Folgori A, Ippolito G, Ottou S, Vita S, Vergori A, D’Abramo A, Petrecchia A, Montaldo C, Scalise E, Grassi G, Casetti R, Bordoni V, Notari S, Colavita F, Meschi S, Lapa D, Bordi L, Murachelli S, Tambasco T, Grillo A, Masone E, Marchioni E, Bardhi D, Porzio O, Cocca F, Murachelli S, Turrini I, Malescio F, Ziviani L, Lawlor R, Poli F, Martire F, Zamboni D, Mazzaferri F, GRAd-COV2, a gorilla adenovirus-based candidate vaccine against COVID-19, is safe and immunogenic in younger and older adults. Sci. Transl. Med 14, eabj1996 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Capone S, Raggioli A, Gentile M, Battella S, Lahm A, Sommella A, Contino AM, Urbanowicz RA, Scala R, Barra F, Leuzzi A, Lilli E, Miselli G, Noto A, Ferraiuolo M, Talotta F, Tsoleridis T, Castilletti C, Matusali G, Colavita F, Lapa D, Meschi S, Capobianchi M, Soriani M, Folgori A, Ball JK, Colloca S, Vitelli A, Immunogenicity of a new gorilla adenovirus vaccine candidate for COVID-19. Mol. Ther 29, 2412–2423 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Alise AM, Leoni G, Cotugno G, Troise F, Langone F, Fichera I, De Lucia M, Avalle L, Vitale R, Leuzzi A, Bignone V, Di Matteo E, Tucci FG, Poli V, Lahm A, Catanese MT, Folgori A, Colloca S, Nicosia A, Scarselli E, Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat. Commun 10, 2688 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leoni G, D’Alise AM, Cotugno G, Langone F, Garzia I, De Lucia M, Fichera I, Vitale R, Bignone V, Tucci FG, Mori F, Leuzzi A, Di Matteo E, Troise F, Abbate A, Merone R, Ruzza V, Diodoro MG, Yadav M, Gordon-Alonso M, Vanhaver C, Panigada M, Soprana E, Siccardi A, Folgori A, Colloca S, van der Bruggen P, Nicosia A, Lahm A, Catanese MT, Scarselli E, A genetic vaccine encoding shared cancer neoantigens to treat tumors with microsatellite instability. Cancer Res. 80, 3972–3982 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L, Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Townsend A, Ohlen C, Bastin J, Ljunggren HG, Foster L, Karre K, Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature 340, 443–448 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, Quivy JP, Almouzni G, Amigorena S, The epigenetic control of stemness in CD8+ T cell fate commitment. Science 359, 177–186 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Pace L, Tempez A, Arnold-Schrauf C, Lemaitre F, Bousso P, Fetler L, Sparwasser T, Amigorena S, Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science 338, 532–536 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, Araki K, Ahmed R, CD8 T cell exhaustion in chronic infection and cancer: Opportunities for interventions. Annu. Rev. Med 69, 301–318 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, Schietinger A, Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R, Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W, Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Miller BC, Sen DR, Abosy RA, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, Haining WN, Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luckey CJ, Bhattacharya D, Goldrath AW, Weissman IL, Benoist C, Mathis D, Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc. Natl. Acad. Sci. U.S.A 103, 3304–3309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R, Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, Wang W, Pipkin ME, Goldrath AW, Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Speiser DE, Ho P-C, Verdeil G, Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol 16, 599–611 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Schillebeeckx I, Earls J, Flanagan KC, Hiken J, Bode A, Armstrong JR, Messina DN, Adkins D, Ley J, Alborelli I, Jermann P, Glasscock JI, T cell subtype profiling measures exhaustion and predicts anti-PD-1 response. Sci. Rep 12, 1342 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montacchiesi G, Pace L, Epigenetics and CD8+ T cell memory. Immunol. Rev 305, 77–89 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Krishna S, Lowery FJ, Copeland AR, Bahadiroglu E, Mukherjee R, Jia L, Anibal JT, Sachs A, Adebola SO, Gurusamy D, Yu Z, Hill V, Gartner JJ, Li YF, Parkhurst M, Paria B, Kvistborg P, Kelly MC, Goff SL, Altan-Bonnet G, Robbins PF, Rosenberg SA, Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 370, 1328–1334 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, Hermann-Kleiter N, Lower M, Baier G, Krogsdam A, Trajanoski Z, Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat. Commun 9, 32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, McGuire DJ, Cohen KW, Alexe G, Nagar S, McCausland MM, Gupta S, Tata P, Haining WN, McElrath MJ, Zhang D, Hu B, Greenleaf WJ, Goronzy JJ, Mulligan MJ, Hellerstein M, Ahmed R, Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobin A, Gingeras TR, Optimizing RNA-seq mapping with STAR. Methods Mol. Biol 1415, 245–262 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Li B, Dewey CN, RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McInnes L, Healy J, Melville J, UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv:1802.03426 [stat.ML] (9 February 2018). [Google Scholar]
- 43.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E, Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008 (2008). [Google Scholar]
- 44.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, Linsley PS, Gottardo R, MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stubbington MJT, Lönnberg T, Proserpio V, Clare S, Speak AO, Dougan G, Teichmann SA, T cell fate and clonality inference from single-cell transcriptomes. Nat. Methods 13, 329–332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S, Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Viral vectors (GAd and MVA) are available from Nouscom with a material transfer agreement upon reasonable request to E.S. The bioinformatics data are available in the Gene Expression Omnibus (GEO) database with accession number GSE206811.