

Abstract
Purpose:
The addition of immune checkpoint blockade (ICB) to platinum/etoposide chemotherapy changed the standard of care for small cell lung cancer (SCLC) treatment. However, ICB addition only modestly improved clinical outcomes, likely reflecting the high prevalence of an immunologically “cold” tumor microenvironment in SCLC, despite high mutational burden. Nevertheless, some patients clearly benefit from ICB and recent reports have associated clinical responses to ICB in SCLC with A) decreased neuroendocrine characteristics and B) activation of NOTCH signaling. We previously showed that inhibition of the LSD1 demethylase activates NOTCH and suppresses neuroendocrine features of SCLC, leading us to investigate whether LSD1 inhibition would enhance the response to PD1 inhibition in SCLC.
Experimental Design:
We employed a syngeneic immunocompetent model of SCLC, derived from a genetically engineered mouse model harboring Rb1/Trp53 inactivation, to investigate combining the LSD1 inhibitor bomedemstat with anti-PD1 therapy. In vivo experiments were complemented by cell-based studies in murine and human models.
Results:
Bomedemstat potentiated responses to PD1 inhibition in a syngeneic model of SCLC, resulting in increased CD8+ T cell infiltration and strong tumor growth inhibition. Bomedemstat increased MHC class I expression in mouse SCLC tumor cells in vivo and augmented MHC-I induction by interferon-ɣ and increased killing by tumor specific T cells in cell culture.
Conclusions:
LSD1 inhibition increased MHC-I expression and enhanced responses to PD1 inhibition in vivo, supporting a new clinical trial to combine bomedemstat with standard of care PD1 axis inhibition in SCLC.
Keywords: SCLC, small cell lung cancer, bomedemstat, IMG-7289, LSD1, KDM1A
Introduction
After decades with no notable change to the standard of care regimen for newly diagnosed extensive stage small cell lung cancer (ES-SCLC), the IMpower133(1) and CASPIAN(2) studies both demonstrated improved overall survival with the addition of an anti-PDL1 antibody to induction platinum doublet chemotherapy. These outcomes led to a new standard of care in ES-SCLC. However, the observed magnitude of survival improvement was modest, at approximately two months, and durable responses were rare. By comparison to other tumor types that also harbor a high tumor mutation burden, such as non-small cell lung cancer (NSCLC) or melanoma, these results suggested considerable remaining opportunity to improve the efficacy of immune checkpoint blockade (ICB) in SCLC. It is important to acknowledge, however, that a small subset of SCLC patients do seem to have durable responses and are therefore likely to strongly benefit from PD1/PDL1 axis inhibition(1,2).
Multiple lines of evidence offer possible explanations for the modest effects seen with immune checkpoint inhibitors in SCLC. SCLC typically expresses low levels of antigen presenting proteins(3–7) suggesting a mechanism by which mutation-induced neoepitopes may avoid recognition by surveilling immune cells. SCLC has also been associated with low levels of tumor infiltrating lymphocytes (TILs)(8,9) and PDL1 expression(10–12) raising the possibility that SCLC immune escape occurs prior to and/or independent of the PD1/PDL1 axis. Nevertheless, motivated, in part, by the observation of rare durable responses to ICB in SCLC, considerable effort has recently been directed to identifying biologically distinct subsets of SCLC, including subsets that benefit from ICB. Almost all SCLC harbors inactivating mutations or deletions in RB1 and TP53 while other frequent alterations in SCLC include amplification of MYCL, MYC and MYCN, mutations in the PIK3CA/PTEN pathway, deletions of epigenetic regulators such as KMT2D, CREBBP and EP300, and mutations in NOTCH family members(13–16). However, specific genetic alterations in SCLC have not yet been linked to differential responses to ICB. SCLC tumors also exhibit strongly differential activation of key transcription factors such as ASCL1, NEUROD1, and POU2F3 leading to the proposed transcription factor-defined SCLC subtypes (i.e. SCLC-A, -N, and -P)(17,18). Increased inflammation has also been identified in a subset of SCLC tumors associated with higher expression of MHC-I and lower expression of canonical neuroendocrine genes including ASCL1(19,20). Patients harboring tumors classified based on gene expression data as “inflamed” (termed SCLC-I) seemed to derive a greater clinical benefit from the addition of anti-PDL1 antibody to chemotherapy in IMpower133, the clinical trial that resulted in the approval of atezolizumab for SCLC(19). A second study strengthened the link between suppression of neuroendocrine features and ICB responses in SCLC(20), while a third study found increased NOTCH activation in patients who benefitted from ICB(21). Features of SCLC transcription may emerge as biomarkers for ICB activity in SCLC, although prospective validation and the development of rigorous and reproducible classification methods are needed. Furthermore, given the rarity of durable responses observed in IMpower133 and CASPIAN and the paucity of alternative treatment options, therapeutic methods to overcome intrinsic ICB resistance would likely have a much greater impact on clinical outcomes than diagnostic methods to predict sensitivity.
Lysine-specific Demethylase 1a (LSD1) encoded by KDM1A is a lysine demethylase active on mono- and dimethyl groups of histone H3 lysine 4 as well as non-histone substrates(22). LSD1 also interacts with other key chromatin regulators such as histone deacytelase (HDAC) 1, 2 or 3, DNA methyltransferase 1 (DNMT1), and the neuroendocrine regulators INSM1 and RCOR1 (also known as CoREST). In so doing, LSD1 participates in multi-protein complexes that generally act to repress gene expression. Mouse genetic studies have revealed that LSD1 is important for cell lineage determination and differentiation in the developing neuroendocrine pituitary gland(23), and LSD1 inhibitors can inhibit growth of SCLC cells and in vivo patient derived xenograft (PDX) models(24–26). We recently reported that inhibition of LSD1 using iadademstat (also known as ORY-1001) in patient derived xenograft (PDX) models of SCLC resulted in activation of NOTCH and suppression of ASCL1 and neuroendocrine transcriptional programs(24). Notch activation has been shown to suppress neuroendocrine programs during lung development(27,28), and in SCLC(29). LSD1 inhibition with GSK2879552 has been tested as a single agent in patients with relapsed SCLC but not in combination with ICB(30). It has also recently been shown that LSD1 genetic suppression or pharmacologic inhibition can enhance response to PD1 inhibitors in preclinical models of other tumor types(31–33). Given that NOTCH activation and suppression of neuroendocrine state was associated with clinical benefit from PDL1 inhibition in SCLC patients(19,21), we asked if LSD1 inhibition could potentiate immune responses against SCLC.
Materials and Methods
Mice
C57BL/6NJ mice were purchased from Jackson Laboratories (RRID:IMSR_JAX:005304). Non-obese Diabetic severe combined immunodeficient interleukin-2 receptor γ-deficient (NSG) mice were obtained from the Fred Hutchinson Cancer Research Center Co-Operative Center for Excellence in Hematology. Hemizygous OT-I mice on a CD45.1 background were generated in-house. Animal experiments were performed at the Fred Hutchinson Cancer Research Center with approval from the Institutional Animal Care and Use Committee (IACUC).
SCLC PDX model
Generation of the FHSC04 PDX model has been previously described(24).
mSCLC cell lines
RP-48 and RP-116 C57BL/6 mSCLC cell lines derived from Rb1lox/lox;Trp53lox/lox mice treated with intranasal Adeno-Cre have been previously described (negative for mycoplasma in 2/2022 by MycoProbe kit, R&D Systems) (6). The G8545 cell line was derived from a lung tumor that developed following intratracheal administration of Adeno-Cre virus to a Rb1lox/lox;Trp53lox/lox;Crebbplox/lox genotype mixed background (C57BL/6 and 129S) mouse as previously described (negative for mycoplasma in 4/2022 by MycoProbe kit, R&D Systems) (13). The FH-B6RP1 cell line was derived from a lung tumor that developed following intratracheal administration of Adeno-Cre virus to a Rb1lox/lox;Trp53lox/lox C57BL/6 mouse according to published methods (negative for mycoplasma in 4/2021 by MycoProbe kit, R&D Systems) (34).
Human cell lines
Human SCLC cell lines NCI-H510, NCI-H1048, NCI-H841, and NCI-H146 were purchased from ATCC and were negative for mycoplasma in 5/2021 by MycoProbe kit, R&D Systems.
Cell culture
Murine SCLC cell lines were grown in DMEM Complete media [DMEM 1x (Thermo Fisher Scientific 11965092), 15% FBS (Peak Serum PS-FB2), 1 mM sodium pyruvate (Thermo Fisher Scientific 11360-070), 100 μM β-mercaptoethanol (Fisher Scientific 501147851), 1% Penicillin-Streptomycin (10,000 U/mL, Thermo Fisher Scientific 15140122), 10 μg/mL recombinant human insulin zinc solution (Thermo Fisher Scientific 12585-014)]. Cell lines were incubated at 37°C in humidified 5% CO2. Human SCLC cell lines NCI-H510, NCI-H1048, and NCI-H841 were grown in DMEM/F-12 (Thermo Fisher Scientific 11320033) supplemented with insulin-transferrin-selenium (10, 5.5, and 0.0067 μg/ml, respectively, Thermo Fisher Scientific 41400045), 30 nM hydrocortisone (Sigma H0135), 10 nM β-estradiol (Sigma E2257), 1% Penicillin-Streptomycin (10,000 U/mL, Thermo Fisher Scientific 15140122), and 10% FBS (Peak Serum PS-FB2). Human cell line NCI-H146 was grown in RPMI 1640 (Thermo Fisher Scientific 11875093) supplemented with 1% Penicillin-Streptomycin (10,000 U/mL, Thermo Fisher Scientific 15140122), and 10% FBS (Peak Serum PS-FB2). Cell lines were incubated at 37°C in humidified 5% CO2.
Protein extraction and immunoblotting
Lysates from whole-cell extracts of cultured cell lines were prepared in diluted RIPA buffer (Cell Signaling 9806, 10x concentration) with added protease/phosphatase inhibitor. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific 23227). Immunoblotting was done using standard procedures. The following antibodies were used: anti-REST (1:1000; Fisher Scientific 07-579, RRID:AB_310728), anti-NOTCH1 (1:1000; Cell Signaling 3608, RRID:AB_2153354), anti-NOTCH2 (1:1000; Cell Signaling 5732, RRID:AB_10693319), anti-MASH1 (ASCL1; 1:1000; BD Pharmingen 556604, RRID:AB_396479), anti-HES1 (1:1000; Cell Signaling 11988, RRID:AB_2728766), anti-α-tubulin (1:1000; Santa Cruz Biotechnology sc-398103, RRID:AB_2832217), anti-RIG-1 (1:1000; Proteintech 20566-1-AP, RRID:AB_10700006), anti-PDL1 for mouse (1:1000; R&D Systems MAB90781-SP, RRID:AB_2921258), anti-PDL1 for human (1:1000; Cell Signaling 13684, RRID:AB_2687655), anti-beta-2-microglobulin (1:1000; Proteintech 13511-1-AP, RRID:AB_2062735), anti-MDA5 (1:1000; Proteintech 21775-1-AP, RRID:AB_10734593), anti-beta-ACTIN (1:1000; Proteintech 66009-1-lg, RRID:AB_2782959), anti-LSD1 (1:1000; Cell Signaling 2184, RRID:AB_2070132), anti-NOTCH1-ICD (1:1000, Cell Signaling 4147, RRID:AB_2153348), and anti-STING (1:1000, Cell Signaling 13647, RRID:AB_2732796).
In vitro cell viability and dose response
To assay the dose-response to bomedemstat, RP-116 and RP-48 cells were seeded in clear-bottom, opaque-walled 96-well culture plates with escalating dosages of bomedemstat. After 96-hours of co-culture, cell survival was evaluated using the CellTiter-Glo luminescent cell viability assay (Promega) with a BioTek plate reader to quantify luminescent signal.
Drug preparation for in vivo administration
Bomedemstat (Imago Biosciences) was resuspended in sterile water with 0.5% weight/volume Tween-80 and 0.5% weight/volume carboxymethylcellulose sodium salt. Bomedemstat vehicle was prepared as sterile H2O with 0.5% weight/volume Tween-80 and 0.5% weight/volume carboxymethylcellulose sodium salt. Anti-PD1 antibody (Bio X Cell BE0146) was diluted in cold PBS to a final concentration of 0.833 mg/mL.
Flank tumor generation and measurement
SCLC cells in the desired number were precipitated via gentle centrifugation and resuspended in DMEM complete media. Per mouse, 100 μL of cells in media were then mixed 1:1 with 100 μL of Matrigel (Sigma CLS354234). 200 μL of cells were implanted, unilaterally, into the left flanks of NSG or C57BL/6NJ mice using a 28-gauge fixed needle syringe. Flank tumor length and width were measured twice each using digital calipers and measurements were averaged. Estimated tumor volume was calculated using the modified ellipsoid formula [(LxW2)/2]. Animals were entered into a treatment group when estimated flank tumor volume was ~150 mm3. For the RP-48OVA-LLO190 syngeneic flank tumor experiments, in order to facilitate time-sensitive analyses such as multiparameter flow cytometry, treatment start was synchronized in batches, with starting tumor size balanced across treatment groups to the extent possible. Mice were sacrificed and tumors were collected at planned experiment conclusion or when tumors exceeded maximum allowed size (~2000 mm3) or if unacceptable ulceration or other impairment to animal health developed.
In vivo drug treatments
Vehicle was administered via oral gavage to match bomedemstat administration schedule across all in vivo experiments. Bomedemstat was administered via oral gavage throughout. NSG mice were treated as follows. For molecular analyses, bomedemstat was administered at 25 mg/kg daily. For tumor growth kinetics, bomedemstat was administered at 40 mg/kg on days 1-5 of a 7-day cycle as a 4 mg/mL solution. C57BL/6NJ mice were administered bomedemstat at 45 mg/kg/d. Anti-PD1 antibody was administered via intraperitoneal injection at a flat dose of 250 μg on days 1 and 4 of a 7-day cycle. Tumors were resected after euthanasia via cervical dislocation or carbon dioxide administration. Tumor fragments were snap-frozen and stored at −80°C.
Virus construction and transduction of mSCLC cells
To generate RP-48OVA-LLO190 cells, we used a retroviral plasmid based on the pMP71 backbone and encoding enhanced GFP fused to a peptide sequence from the listeriolysin O protein and a peptide sequence from chicken ovalbumin that was a gift from Dr. Stan Riddell (pJV119-pMP71-mGFP-LLO-SIINFEKL). Retrovirus was produced in 293T cells with Lipofectamine 2000 (ThermoFisher, 11668019) according to manufacturer’s directions and using accessory plasmids pUMVC (Addgene, #8449) and pCMV-VSV-G (Addgene, #8454). RP-48 cells were transduced and expanded for 1-2 weeks in culture. GFP-positive cells were then isolated via FACS. For the generation of CRISPR-mediated LSD1 knockout RP-48OVA-LLO190 cells, the following sgRNA sequences were cloned into the lentiCRISPRv2 vector (Addgene, #52961; a gift from Feng Zhang): Control-1 (5'-CGCTTCCGCGGCCCGTTCAA-3'), Control-2 (5'-CTAACAGCGCCGTTATCTAC-3'), Lsd1-1 (5'-ATATTCATCTTCTGAGAGGT-3'), Lsd1-2 (5'-TCTTCCTCAGGTGGGGCTTG-3'), and Lsd1-3 (5'-CCATGACCGAATGACCTCTC-3'). Lentivirus production with packaging plasmids psPAX2 (Addgene, #12260) and pMD2.G (Addgene, #12259) was performed as described above. RP-48OVA-LLO190 cells were transduced and selected for 72 hours with puromycin (1 μg/mL, InvivoGen ANTPR1).
Flow cytometry of RP-48OVA-LLO190 flank tumors
RP-48OVA-LLO190 flank tumors were generated and collected as described above. Two cohorts were analyzed: a 16-day cohort which focused on analysis of immune cell subsets, and a 10-day cohort which focused on analysis of SCLC cell MHC-I (H-2Kb) expression.
For the 16-day timepoint cohort, tumor tissue was prepared and analyzed as follows. Fresh tumor tissue was minced using a razor blade in 1-2 mm fragments. Tumor tissue was dissociated into a single cell suspension by incubating for 45 minutes at 37°C in RPMI with HEPES (ThermoFisher #22400089) with 80 U/mL DNase (Worthington #LS00206) and 300 U/mL Collagenase Type I (Worthington #CLS-1), filtration through a 70 μm strainer, and RBC lysis with ACK lysis buffer (Gibco) according to manufacturer’s instructions. Following RBC lysis, tumors were resuspended as single cell suspensions for viability and cell surface staining. Viability staining was performed with Fixable Viability Dye (eBioscience 65-0865-14) according to manufacturer’s instructions. For surface staining, cells were incubated at 4°C for 30 min in staining buffer (PBS, 2% FBS) with the following directly conjugated antibodies for murine proteins (from BioLegend unless otherwise specified): anti-CD45 (clone 30-F11, BD Biosciences, RRID:AB_2872789), -CD3 (145-2C11, RRID:AB_312670), -CD4 (RM4-5, RRID:AB_312714), -CD8 (53-6.7, BD, RRID:AB_2732919), -CD19 (6D5, RRID:AB_313646), -CD11b (M1/70, BD, RRID:AB_394491), -CD11c (N418, RRID:AB_11204262), -Ly6C (HK1.4, RRID:AB_10640119), -Ly6G (1A8, BD, RRID:AB_2737730), -F4/80 (BM8, RRID:AB_10901171), -NK1.1 (PK136, BD, RRID:AB_2738002). Data were acquired on Canto II or Fortessa flow cytometers (BD Biosciences) and analyzed using FlowJo software (Treestar).
For the 10-day timepoint cohort, tumor tissue was prepared and analyzed as follows. Fresh tumor tissue was minced using scissors into 1-2 mm fragments. Tumor tissue was dissociated into a single cell suspension by incubating for 45 minutes at 37°C in RPMI with HEPES (ThermoFisher #22400089) with 80 U/mL DNase (Worthington #LS00206) and 300 U/mL Collagenase Type I (Worthington #CLS-1), filtration through a 100 μm strainer, and RBC lysis with ACK lysis buffer (Gibco). Cells were stained using the Live/Dead Fixable Aqua Dead Cell stain kit (Invitrogen). For surface staining, cells were incubated at 4°C for 30 min in staining buffer (PBS, 2% FBS) with anti-CD45 (Biolegend 30-F11, RRID:AB_2872789) and anti-H-2Kb (Biolegend AF6-88.5, RRID:AB_313735) directly conjugated antibodies for murine proteins. Cells were fixed using Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and stored at 4°C in the dark until analysis. Data were acquired on Symphony flow cytometers (BD Biosciences) and analyzed using FlowJo software (Treestar).
RNAseq library construction and analysis
RNA was extracted from cell pellets or tumor samples using TRIzol reagent. RNAseq libraries were generated from 500ng of RNA using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina (New England Biolabs E7530L). Sequencing was performed using an Illumina HiSeq 2500 instrument in 50 bp single end read configuration. Fastq files were filtered to exclude reads that did not pass Illumina’s base call quality threshold. STAR v2.7.1(35) with 2-pass mapping was used to align reads to mouse genome build mm10 and GENCODE gene annotation M23 (https://www.gencodegenes.org/mouse/). FastQC 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and RSeQC 3.0.0(36) were used for QC including insert fragment size, read quality, read duplication rates, gene body coverage and read distribution in different genomic regions. FeatureCounts(37) in Subread 1.6.5(37) was used to quantify gene-level expression by unstranded read counting. Bioconductor package edgeR 3.26.8(38) was used to detect differential gene expression between sample groups. Genes with low expression were excluded using edgeR function filterByExpr with min.count = 5 and min.total.count = 15. The filtered expression matrix was normalized by TMM method(39) and subject to significance testing using GLM LRT method and cell line as blocking factor (if applicable).
Gene set enrichment and pathway analyses
RPKM or FPKM values generated as described above were analyzed using Gene Set Enrichment Analysis v4.2.2 software (Broad Institute and Regents of the University of California) with default settings except that mouse to human gene mapping was performed using the supplied “Mouse_ENSEMBL_Gene_ID_Human_Orthologs_MSigDB.v7.5.1.chip” file and “gene_set” permutation was used for generation of statistics. For Hallmark pathway analysis, the provided “ftp.broadinstitute.org://pub/gsea/gene_sets/h.all.v7.5.1.symbols.gmt” file was used. For enrichment of ASCL1 gene targets in mSCLC cells, we used a list of 141 ASCL1 target genes that were shared between mouse and human chromatin occupancy analyses(40).
Flow cytometry of SCLC cultured cells
Cultured SCLC tumor cells were seeded with bomedemstat (1 μM) or equal volume of vehicle (DMSO) added to the culture media. Varying concentrations of recombinant murine IFN-ɣ (Peprotech 315-05 for RP-48 / RP-48OVA-LO190 or Abcam ab9922 for FH-B6RP1) or human IFN-ɣ (Sigma I17001) were added 24 or 48 hr before collecting cells. Cells were incubated at 37°C in humidified 5% CO2 for a total of 72 hr or 96 hr after plating. Cells were then harvested for flow cytometry analysis. Briefly, cells were stained using the Live/Dead Fixable Aqua Dead Cell stain kit (Invitrogen) according to the manufacturer’s protocol. For surface staining, cells were incubated at 4°C for 30 min in staining buffer (PBS, 2% FBS) with anti-H-2Kb (murine, Biolegend AF6-88.5, RRID:AB_313735) or anti-pan-MHC-I (human, Biolegend 311405, RRID:AB_314875) directly conjugated antibodies. Cells were fixed using Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and stored at 4C in the dark until analysis. Data were acquired on Symphony flow cytometers (BD Biosciences) and analyzed using FlowJo software (Treestar).
Gamma-secretase inhibitor and LSD1 inhibitor co-treatment
RP-48OVA-LLO190 tumor cells were seeded at a density of 4x105 cells in 3 mL of DMEM-complete media. On the day of tumor cell plating, bomedemstat (1 μM) or equal volume of DMSO with or without 0.5 μM gamma-secretase inhibitors dibenzazepine (DBZ, Selleckchem S2711) or RO4929097 (RO4, Selleckchem S1575) was added to the culture media. Cells were passaged on days 4 and 7. Recombinant murine IFN-ɣ (10 ng/mL, Peprotech 315-05) was added for the final 48h (days 8-10). Cells were then harvested for flow cytometry analysis as above. FH-B6RP1 cells were processed identically except that 2x106 cells were seeded, RO4 was omitted, cells were treated for a total of 96 hours, and IFN-ɣ was added for the final 24h (72-96h).
RP-48OVA-LLO190 and cytotoxic T cell co-culture
RP-48OVA-LLO190 tumor cells were seeded in 6-well tissue-culture-treated plates at a density of 1x105 cells in 3 mL of DMEM Complete media and treated with 1 μM bomedemstat or vehicle for 72 hr, with recombinant IFN-ɣ (10 ng/mL, Peprotech 315-05) added for the last 24 hr of culture. In parallel, CD8+ T cells were isolated by negative selection from spleens of OT-1 transgenic or wild-type B6 mice using a negative isolation kit (Stemcell 19853) and stimulated with 1 mg/mL each of plate-bound anti-CD3 and anti-CD28 (Biolegend 145-2C11 and 37.51, respectively) for 48hr in a 37°C, 5% CO2 incubator in complete RPMI (RPMI 1640, 10% heat inactivated FBS, 1 mM sodium pyruvate, 1 mM HEPES, 100 U/mL penicillin/streptomycin, 50 μM β-mercaptoethanol) supplemented with 50 U/mL recombinant murine IL-2 (Peprotech 212-12). 72 hr after tumor cell plating and 48 hr after T cell isolation, T cells and tumor cells were co-cultured in 96-well U-bottom plates at a ratio of 2:1 (50,000 tumor cells and 25,000 T cells) in complete RPMI. Co-cultures were incubated for 24 hr in a 37C, 5% CO2 incubator. Cells were then harvested for flow cytometry analysis. A defined number of counting beads (Polybead 18328-5) were spiked into each sample. Cells were stained with Live/Dead Fixable Aqua Dead Cell stain kit (Invitrogen) and surface stained with anti-CD8 (Biolegend 53-6.7, RRID:AB_2732919) and anti-CD45 (Biolegend 30-F11, RRID:AB_2872789) directly conjugated antibodies. Cells were fixed using Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and stored at 4°C in the dark until analysis. Data were acquired on Symphony flow cytometers (BD Biosciences) and analyzed using FlowJo software (Treestar). Percent survival was calculated as: 100-100*[[(# of live tumor cells in CD8 co-culture) − (# of live tumor cells in OT-1 co-culture)] / (# of live tumor cells in CD8 co-culture)].
CD107a degranulation assay
RP-48OVA-LLO190 tumor cells were seeded in 6-well tissue-culture-treated plates at a density of 1x105 cells in 3 mL of DMEM Complete media and treated with bomedemstat (1 μM) or vehicle for 72 hr, with recombinant IFNg (10ng/mL, Peprotech 315-05) added for the last 24 hr of culture. In parallel, CD8+ T cells were isolated as described for the co-culture assay above. 72 hr after tumor cell plating and 48 hr after T cell isolation, T cells and tumor cells were co-cultured in 96-well U-bottom (FB012931) plates at a ratio of 2:1 (100,000 T cells 50,000 tumor cells) in complete RPMI with PE conjugated anti-CD107a antibody (1 μg/mL, Biolegend 1D4B). For T cell independent stimulation PMA (50ng/mL) and ionomycin (1 μg/mL) were added in 0.2 mL of complete RPMI. Co-cultures were incubated for 24 hr in a 37C, 5% CO2 incubator. Cells were then harvested for flow cytometry analysis. Cells were stained with Live/Dead Fixable Aqua Dead Cell stain kit (Invitrogen) and surface stained with anti-CD8 (Biolegend 53-6.7, RRID:AB_2732919) and anti-CD45 (Biolegend 30-F11, RRID:AB_2872789) directly conjugated flow antibodies. Cells were fixed using Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and stored at 4C in the dark until analysis. Data were acquired on Symphony flow cytometers (BD Biosciences) and analyzed using FlowJo software (Treestar).
Statistical analyses
Protein expression in immunoblots was compared using unpaired t-tests. Flank tumor growth kinetics were compared using estimation of individual mixed effects models comparing bomedemstat+anti-PD1 to each other treatment group. RP-48OVA-LLO190 flank tumor immune cell subset frequencies, MHC-I positivity, and geometric mean fluorescence intensity (gMFI) were compared using one-way analysis of variance (ANOVA) with follow-up testing using Dunnett’s multiple comparison test, where follow-up tests were performed comparing the bomedemstat+anti-PD1 group to the other three treatment groups. SCLC cell line H-2Kb/pan-MHC-I positivity and gMFI were compared using multiple unpaired Welch’s t-tests. All statistical analysis was done in GraphPad Prism 9.
Graphics
Experimental design graphics were designed on biorender.com.
Data availability
The gene expression data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE208614.
Results
For these studies we employed bomedemstat (also known as IMG-7289), an orally available, irreversible LSD1 inhibitor that is currently being tested in clinical trials for the treatment of myeloproliferative neoplasms, but whose activity in SCLC has not been well studied. We first evaluated the activity of bomedemstat in a PDX model of SCLC previously shown to strongly respond to another LSD1 inhibitor, iadademstat (also known as ORY1001)(24). Similarly to iadademstat, bomedemstat at maximum tolerated dose induced complete regression of FHSC04 flank tumors (Supplementary Fig. S1), and intermediate dose treatment with bomedemstat led to NOTCH activation and suppressed neuroendocrine signaling, with reduced expression of ASCL1 (Fig. 1A, Supplementary Fig. S2).
Figure 1.
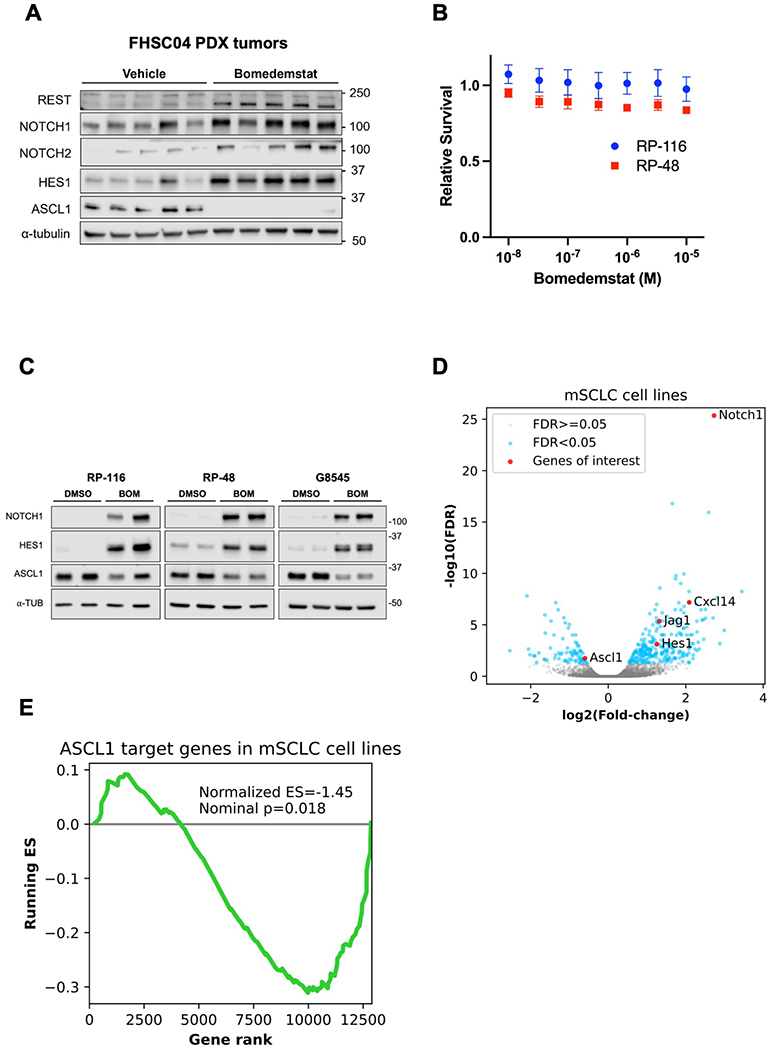
Bomedemstat treatment upregulates NOTCH and suppresses ASCL1 targets in human and murine SCLC models. A, Immunoblot analysis of FHSC04 PDX model tumors after treatment with vehicle or bomedemstat (25 mg per kg per day). B, Survival of murine SCLC (mSCLC) cell lines RP-116 and RP-48 treated with bomedemstat for 96 hours, relative to cells treated with DMSO (mean±SEM of three independent replicates), as determined by CellTiterGlo reagent. C, Immunoblot analysis of mSCLC cell lines after exposure to bomedemstat (1 μM) or DMSO for 96 hours. D, Differential gene expression analysis of RP-116, RP-48, and G8545 mSCLC cell lines after exposure to bomedemstat (1 μM) or DMSO for 96 hours, showing fold-change in bomedemstat-treated cells relative to DMSO. Selected genes of interest with respect to neuroendocrine differentiation and immune cell recruitment are highlighted. The −log10(FDR) is artificially capped at 25 for display purposes. E, Gene set enrichment analysis-derived running Enrichment Score (ES) of a custom gene set comprising 141 ASCL1 target genes in RP-116, RP-48, and G8545 mSCLC cell lines.
We then tested the activity of bomedemstat in cell lines derived from Rb1/Trp53-deleted murine SCLC (mSCLC) models. Bomedemstat did not substantially reduce cell growth at a dose of up to 10 micromolar (Fig. 1B). However, Western blot and RNAseq analyses revealed Notch pathway activation and suppression of a gene program driven by the neuroendocrine master regulator ASCL1 (Fig. 1C–D, Supplementary Fig. S3). Gene Set Enrichment Analysis(41,42) focusing on a set of 141 ASCL1 target genes(40) revealed broad suppression of ASCL1 targets with bomedemstat treatment (Fig. 1E). Thus, LSD1 inhibition with bomedemstat leads to Notch activation and suppression of neuroendocrine signaling in a PDX model of SCLC and in mSCLC cell lines.
We next asked whether bomedemstat would enhance responses to PD1 inhibition in vivo. Low mutational burden and neoantigen load in genetically engineered mouse models of SCLC(43) might obscure the effects of bomedemstat on anti-tumor immunity. Thus, we engineered an mSCLC cell line derived from a C57BL6 genetic background to express two model murine antigens, the SIINFEKL peptide derived from chicken ovalbumin (OVA) and the listeriolysin O 190-201 amino acid peptide (LLO190)(44), which we termed RP-48OVA-LLO190 (Fig. 2A). We then assessed whether LSD1 inhibition with bomedemstat would enhance anti-tumor immune responses to RP-48OVA-LLO190 flank tumors grown in syngeneic C57BL6 recipient animals. Treatment with bomedemstat or anti-PD1 antibody alone exerted minimal growth suppressive effect while treatment with bomedemstat in combination with anti-PD1 antibody significantly suppressed growth (Fig. 2B), suggesting the possibility that LSD1 inhibition was augmenting anti-tumor immunity. Thus, while PD1 inhibition alone did not suppress tumor growth in this model, strong tumor growth inhibition was seen when combined with LSD1 inhibition.
Figure 2.
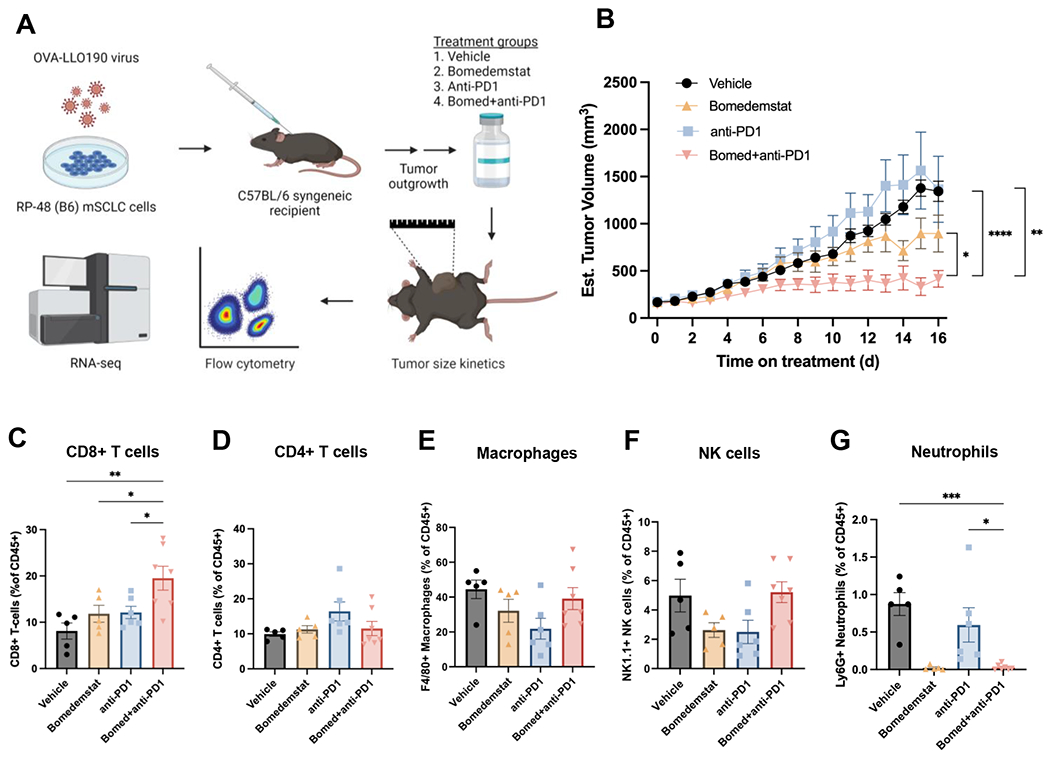
Bomedemstat and PD1 inhibition reduces tumor growth and increases cytotoxic T cell infiltration in an immunocompetent syngeneic murine model of SCLC. A, Schematic illustrating workflow of immunocompetent flank tumor experiments, including modification of mSCLC cells to express immunogenic model antigens, flank tumor engraftment, treatment with one of four listed regimens, and assessment of tumor growth kinetics, immune cell infiltration, and transcriptome. B, Growth kinetics of RP-48OVA-LLO190 flank tumors in C57BL/6 recipient mice treated with vehicle, bomedemstat (45 mg/kg daily), anti-PD1 (250 μg twice weekly), or bomedemstat and anti-PD1 (n=10 each, mean±SEM). Treatment was initiated once tumors reached ~150mm3. Mixed effects models comparing bomedemstat+PD1 to each other group, *P<0.05, **P<0.01, ***P<0.001. C, Flow cytometric quantification of CD8+ T cells, D, CD4+ T cells, E, F4/80+ macrophages, F, NK1.1+ Natural Killer (NK) cells, and, G, Ly6G+ neutrophils as a fraction of CD45+ cells in RP-48OVA-LLO190 flank tumors collected after 16 days of treatment (n=5-7 per group, mean±SEM). One-way ANOVA with Dunnett’s test comparing bomedemstat+anti-PD1 group to the other three groups, *P<0.05, **P<0.01, ***P<0.001. OVA-LLO190, Ovalbumin-Listeriolysin O 190-201 peptide; B6, C57BL/6 genetic background; bomed, bomedemstat; NK, natural killer.
To investigate whether bomedemstat alters the immune tumor microenvironment, we performed flow cytometric immunophenotyping of treated RP-48OVA-LLO190 flank tumors. We observed a selective increase in the prevalence of cytotoxic CD8+ T cells in tumors treated with bomedemstat and anti-PD1 antibody (Fig. 2C), without a clear change in all other immune cell subsets examined, with the exception of neutrophils (Fig. 2D–G, Supplementary Fig. S4). Although neutrophils were relatively infrequent across all groups, they were present at especially low frequencies in the bomedemstat-containing treatment groups (Fig. 2G). Of note, neutropenia is an expected side-effect of LSD1 inhibitor administration as LSD1 is required for terminal differentiation of myeloid lineage hematopoietic cells(45). Together, these data suggest that bomedemstat alters the composition of the immune tumor microenvironment in favor of cytotoxic CD8+ T cells.
We then performed RNAseq to further characterize the molecular features of treated RP-48OVA-LLO190 flank tumors. Bomedemstat and anti-PD1 combination therapy led to marked increase in inflammatory gene expression pathways as revealed by Gene Set Enrichment Analysis(41,42) when compared to bomedemstat alone, and both inflammatory and NOTCH pathways when compared to anti-PD1 antibody alone (Fig. 3A–B, Supplementary Fig. S5). Inspection of selected differentially expressed genes in pathways of interest in individual tumors confirmed systematic variation by treatment group (Fig. 3C). These data showed increased expression of antigen presentation machinery, including components of MHC-I, as well as chemokines such as CXC3L1 and CXCL14, which can mediate recruitment of T cells and dendritic cells, respectively. Activation of NOTCH pathway components and suppression of neuroendocrine transcripts was also observed when bomedemstat was combined with PD1 treatment (Fig. 3C). Growth kinetic, flow cytometric, and transcriptomic analyses therefore coordinately identified a signal of increased anti-tumor immunity when LSD1 inhibition with bomedemstat was combined with anti-PD1 antibody treatment.
Figure 3.
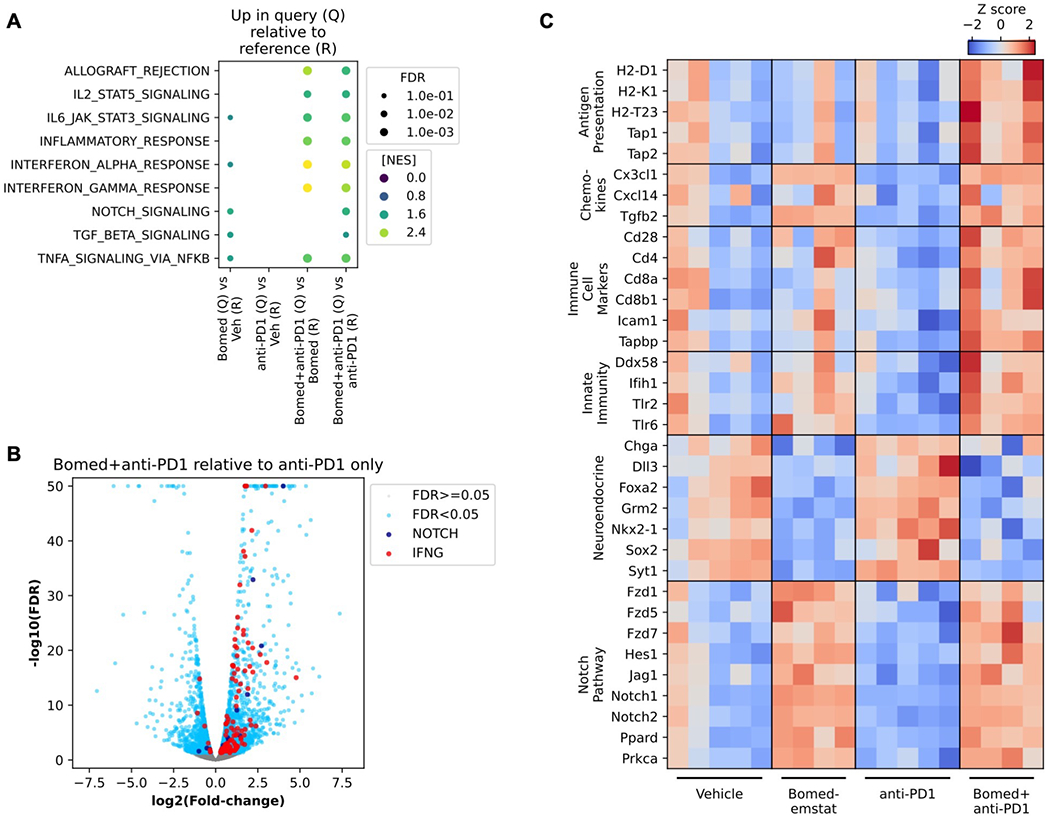
Increased NOTCH pathway and inflammatory gene expression signatures in murine SCLC flank tumors treated with bomedemstat and PD1 inhibition. A, Selected Molecular Signatures Database (MSigDB) Hallmark pathways that are upregulated in Query group relative to Reference group tumor transcriptomes, based on Gene Set Enrichment Analysis of RNA-seq data from RP-48OVA-LLO190 flank tumors in C57BL/6 recipient mice treated with vehicle, bomedemstat (45 mg/kg daily), anti-PD1 (250 μg twice weekly), or bomedemstat and anti-PD1, collected after 16 days on treatment (n=4-5 in each group). Points are shown if pathway enrichment was detected with false discovery rate (FDR) <0.1, point size is scaled to FDR, and point color is scaled to Normalized Enrichment Score (NES). B, Differential gene expression analysis comparing RP-48OVA-LLO190 flank tumors treated with bomedemstat and anti-PD1 vs anti-PD1 alone. Genes in MSigDB Hallmark NOTCH_SIGNALING (NOTCH) and INTERFERON_GAMMA_RESPONSE (IFNG) and FDR<0.05 are shown. The −log10(FDR) is artificially capped at 50 for display purposes. C, Z scores of log-transformed reads per kilobase per million (RPKM) values of selected genes in categories of interest in individual RP-48OVA-LLO190 flank tumors by treatment group. Bomed, bomedemstat.
A described mechanism of immune evasion in SCLC is epigenetic silencing of antigen presentation machinery gene expression leading to very low levels of surface MHC-I(7). Moreover, it has emerged recently that MHC-I re-expression can potentiate anti-tumor immunity in SCLC(6,20,21). To explore the possibility that LSD1 inhibition with bomedemstat induces MHC-I expression, we generated a second cohort of RP-48OVA-LLO190 flank tumors and again observed a clear trend of suppressed tumor growth in animals treated with the combination of bomedemstat and anti-PD1 antibody, although statistical significance was not reached at the early timepoint used for this cohort (Supplementary Fig. S6). Flow cytometric analysis of tumor cells revealed a higher fraction of MHC-I expressing cells in tumors treated with a bomedemstat-containing regimen relative to vehicle as well as increased surface MHC-I expression on MHC-I positive cells when treated with both agents (Fig. 4A–C, Supplementary Fig. S6).
Figure 4.
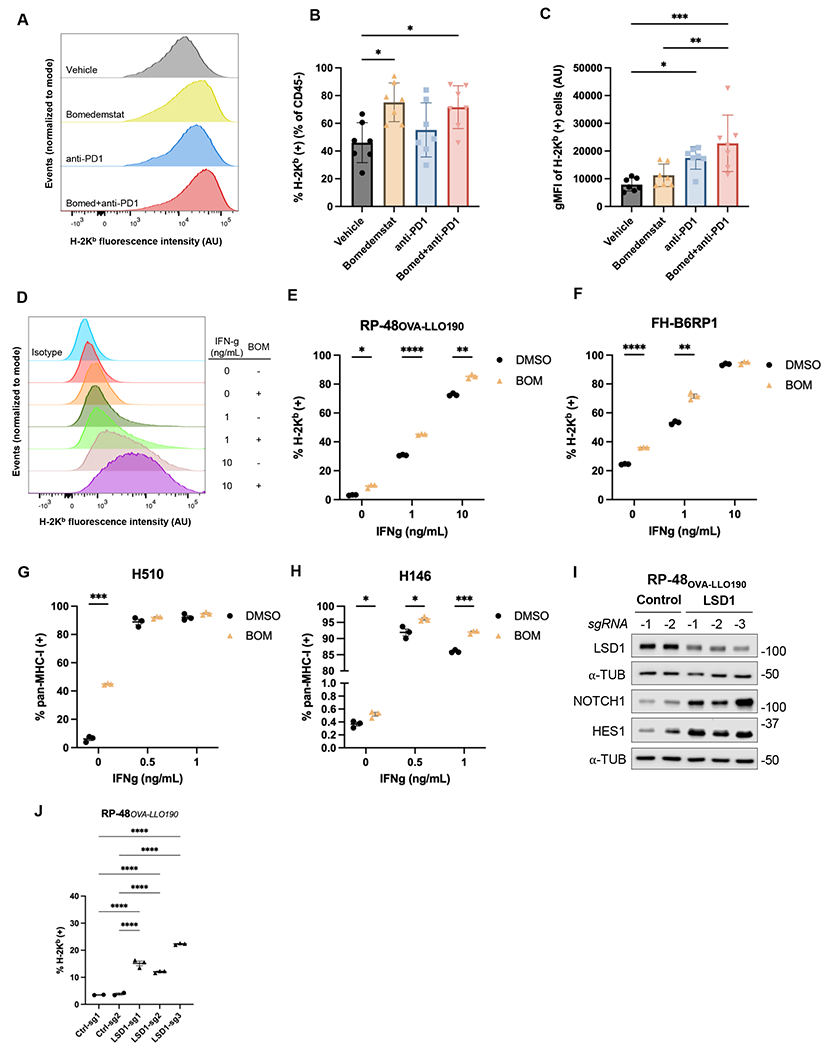
LSD1 inhibition drives MHC-I upregulation. A, Distributions of H-2Kb surface staining from representative RP-48OVA-LLO190 flank tumors collected after 10 days of treatment with vehicle, bomedemstat (45 mg/kg daily), anti-PD1 (250 μg twice weekly), or bomedemstat and anti-PD1 (normalized to modal value). B, Percent of H-2Kb positive cells and, C, geometric mean fluorescence intensity (gMFI) of cells in the positive gate in CD45- (tumor) cells from RP-48OVA-LLO190 flank tumors (n=7 in each group) (mean±SEM). One-way ANOVA with followup comparison of all groups, with Sidak’s correction for multiple comparisons, *P<0.05, **P<0.01, ***P<0.001. D, Distributions of H-2Kb surface staining from representative replicates and, E, percent of H-2Kb positive RP-48OVA-LLO190 cultured cells treated with bomedemstat (1 μM, BOM) or DMSO for a total of 72h, with addition of varying concentrations of interferon gamma (IFN-g) from 48-72h. Multiple unpaired t tests with unequal variance and Sidak’s correction for multiple testing, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. F, Percent of H-2Kb positive FH-B6RP-01 cells, G, pan-MHC-I positive H510 cells and, H, pan-MHC-I positive H146 cells treated with bomedemstat (1 μM) or DMSO for a total of 96h, with addition of varying concentrations of interferon gamma (IFN-g) from 72-96h. Multiple unpaired t tests with unequal variance and Sidak’s correction for multiple testing, *P<0.05, ***P<0.001. I, Immunoblot analysis and, J, percent of H-2Kb positive RP-48OVA-LLO190 cells transduced with a CRISPR knockout vector encoding a non-targeting (control) or LSD1-targeting sgRNA. One-way ANOVA with followup comparison of each control sgRNA with each LSD1 sgRNA, with Sidak’s correction for multiple comparisons, *P<0.05, **P<0.01, ***P<0.001, ***P<0.0001.
We then sought to confirm that this observation was a direct effect of bomedemstat on SCLC cells by using flow cytometry to determine surface MHC-I expression in cultured RP-48OVA-LLO190 cells. Echoing the effects observed in flank tumors, we found that bomedemstat alone or in combination with varying concentrations of IFN-ɣ led to an increase in the proportion of MHC-I positive cells as well as increased surface expression of MHC-I in MHC-I positive cells (Fig. 4D–E, Supplementary Fig. S7). To exclude the possibility that this was a cell line-specific effect or a consequence of expression of an exogenous class I model antigen, we then tested the effect of bomedemstat treatment on surface MHC-I expression in an additional Rb/p53-deleted murine cell line, FH-B6RP1. We again found that bomedemstat increased the proportion of cells expressing surface MHC-I as well as the intensity of surface expression in positive cells (Fig. 4F, Supplementary Fig. S7). These results confirm that bomedemstat reproducibly induces MHC-I across multiple murine models of SCLC and independently of exogenous expression of a model antigen.
We next tested whether upregulation of MHC-I also occurs with bomedemstat treatment in human SCLC cell lines. As LSD1 inhibition leads to loss of neuroendocrine features, we chose two human SCLC-A models, NCI-H510 and NCI-H146, that express high levels of ASCL1 and other neuroendocrine genes and relatively low levels of the MHC-I subunit B2M, according to publicly available RNAseq data(46). We again observed bomedemstat-induced MHC-I upregulation across a range of IFN-ɣ concentrations (Fig. 4G–H, Supplementary Fig. S7). We also treated NCI-H1048, a SCLC-P model, and NCI-H841, a SCLC-Y model, with bomedemstat. Unlike in the two SCLC-A models, MHC-I expression was observed in nearly all cells at baseline and bomedemstat exposure did not increase the percentage of MHC-I positive cells or the intensity of cell surface staining (Supplementary Fig. S8).
To support the notion that upregulation of MHC-I by bomedemstat is not an off-target effect of this compound, we used CRISPR to delete LSD1 in RP-48OVA-LLO190 cells. We first sought to confirm expected changes in LSD1 and NOTCH pathway levels in cells expressing one of three independent sgRNAs targeting LSD1 relative to cells expressing one of two non-targeting control sgRNAs. Transduction with LSD1-targeted sgRNAs led to modest but consistent reduction in LSD1 protein levels and strong upregulation of NOTCH pathway members (Fig. 4I). We then subjected these cells to flow cytometry to quantify surface MHC-I, finding that CRISPR-mediated LSD1 knockout increased the proportion of MHC-I positive cells (Fig. 4J) and increased surface MHC-I intensity in positive cells (Supplementary Fig. S7). Thus, decreased LSD1 activity, either via genetic or pharmacologic methods, induces MHC-I upregulation across a range of SCLC preclinical models.
We then sought to determine mechanistic links between LSD1 inhibition and antigen presentation machinery upregulation. Given the central role of LSD1 in transcriptional regulation, we hypothesized that LSD1 inhibition might increase transcription of key antigen presentation machinery components. We therefore performed transcriptomic profiling of RP-48OVA-LLO190 cells that had been treated with bomedemstat for an extended duration (10 days), with or without IFN-ɣ for the last 48 hours. Regardless of IFN-ɣ exposure, we observed increased expression of antigen presentation machinery components B2m (a required MHC-I subunit), H2-D1 (a murine MHC-I locus), and Tapbp (a mediator of antigen loading onto newly formed MHC-I complexes) (Supplementary Fig. S9), supporting the hypothesis that transcriptional reprogramming is involved in upregulation of antigen presentation machinery.
We also queried whether PDL1 expression was altered in response to LSD1 inhibition in our system, as has previously been shown in other cancer types(31). Bomedemstat treatment increased PDL1 protein and transcript levels in the presence of IFN-ɣ sensitization in RP-48OVA-LLO190 cells (Fig. 5A, Supplementary Fig. S10), and CRISPR knockdown of LSD1 in RP-48OVA-LLO190 mSCLC cells was sufficient to induce PDL1 levels even in the absence of IFN-ɣ (Fig. 5B, Supplementary Fig. S10). In the human SCLC-A models tested, which exhibited widely varying PDL1 expression at baseline, we did not observe PDL1 increase, despite molecular evidence of induced antigen presentation machinery, i.e. increased B2M (Fig. 5C–D, Supplementary Fig. S10). We also searched for evidence of activation of cytoplasmic nucleic acid sensing pathways, which have previously been implicated in LSD1i-induced immunogenic signaling in other cancer models(31) and SCLC tumor cell intrinsic immunogenic signaling in response to other targeted therapies(47). In RP-48OVA-LLO190 cells, we observed increased transcript expression of the dsRNA sensors Ddx58 (Supplementary Fig. S9), which encodes RIG-I, and Ifih1 (Supplementary Fig. S9), which encodes MDA5, irrespective of IFN-ɣ treatment. Using immunoblotting, we also detected increases in STING, which is involved in cytoplasmic DNA sensing, in RP-48OVA-LLO190 cells harboring LSD1 knockout and H510 cells treated with bomedemstat (Fig. 5A–D, Supplementary Fig. S10). STING protein was not detected in H146 cells. Together, these data provide rationale for the potentiation of anti-PD1 treatment by bomedemstat.
Figure 5.

Molecular and functional immunogenicity induced by LSD1 inhibition. A, Immunoblot analysis of RP-48OVA-LLO190 cells treated with bomedemstat (1 μM, BOM) or DMSO for a total of 10 days, with or without interferon gamma (IFN-g, 10 ng/mL) for the final 48 hours. B, Immunoblot analysis of RP-48OVA-LLO190 cells transduced with a CRISPR knockout vector encoding a non-targeting (control) or LSD1-targeting sgRNA. C, Immunoblot analysis of H510 cells and, D, H146 cells treated with bomedemstat (1 μM) or DMSO for a total of 96 hours. E, Percent of CD8+ T cells positive for CD107a cytotoxic degranulation marker following 24h co-culture with RP-48OVA-LLO190 cells that were pre-treated with bomedemstat (1 μM) or DMSO for a total of 72h with or without IFN-ɣ (10 ng/mL) from 48-72h, shown for nonspecific bulk CD8 T cells (red) or antigen-specific OT-I CD8+ T cells (blue). PI, PMA+ionomycin. One-way ANOVA with Dunnett’s test performed on OT-I populations excluding PI and media, *P<0.05, **P<0.01, ****P<0.0001. F, Relative RP-48OVA-LLO190 cell survival following exposure to bomedemstat, IFN-g, and co-culture with OVA-specific OT-I or non-specific CD8+ T cells. Cells were treated with bomedemstat (1 μM) or DMSO for 72h, IFN-ɣ at 0 or 10 ng/mL for 24h prior to co-culture, and then co-cultured with OT-I or non-specific CD8+ T cells for 24h (2:1 effector:target ratio). Percent of cells surviving was calculated as the number of viable SCLC cells in co-culture with OT-I T cells relative to the number of viable SCLC cells in co-culture with non-specific CD8+ T cells multiplied by 100, as determined by flow cytometric quantification with counting bead spike-in (mean±SEM of 6 technical replicates performed in two groups of three at separate timepoints). Multiple unpaired t-tests with unequal variance and Sidak’s correction for multiple testing *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. G, Schematic of molecular targets and associated direct mechanisms, subsequent tumor cell-autonomous effects, and eventual multicellular phenotype of LSD1 inhibition in combination with PD1/PDL1 axis blockade. H, Schematic of an upcoming clinical trial of bomedemstat in combination with maintenance atezolizumab for patients with ES-SCLC who do not experience disease progression during four cycles of standard of care (SOC) induction platinum, etoposide, and anti-PDL1 antibody systemic therapy.
We also explored whether the increased chemokine transcript abundance detected in tumor tissue (Fig. 3C) was a tumor cell-intrinsic phenomenon using the transcriptomic profiling data from RP-48OVA-LLO190 cells treated with extended duration bomedemstat with or without IFN-g. Focusing on a curated consensus list of 133 immune system-related cytokine genes (48), we identified twelve genes that were differentially expressed, eight of which were significantly upregulated by bomedemstat irrespective of IFN-ɣ presence (Supplementary Fig. S9). We observed increased transcript abundance of the chemoattractive cytokines CX3CL1 (49) and CXCL14 (50) (Supplementary Fig. S9), which had also been observed in bomedemstat-treated flank tumors, as well as CXCL12 (51) and IL33 (52), among other genes.
LSD1 inhibition also strongly induces NOTCH signaling and NOTCH induction has been implicated in antigen presentation machinery upregulation in SCLC (21). We therefore tested whether suppression of NOTCH signaling was sufficient to alter the degree to which bomedemstat treatment increased MHC-I surface expression. Treatment with either of two gamma-secretase inhibitors led to substantial reduction in NOTCH pathway activation as evidenced by decreased induction of NOTCH1 intracellular domain and HES1 but did not reduce MHC-I upregulation in the two mSCLC models tested (Supplementary Fig. S11). These data suggest that LSD1 inhibition leads to MHC-I upregulation through one or more NOTCH-independent mechanisms.
Finally, we sought to determine whether bomedemstat treatment of mSCLC cells was sufficient to alter cytotoxic T cell function, as evidenced by cytotoxic degranulation and target cell survival. To test whether bomedemstat treatment of SCLC cells led to altered cytotoxic activation of antigen-specific T cells, we performed in vitro co-culture of RP-48OVA-LLO190 cells with OT-I (antigen-specific) or bulk CD8+ (antigen-nonspecific) T cells in the presence of the cytotoxic degranulation-marking antibody anti-CD107a. Pre-treatment of SCLC cells with bomedemstat and IFN-ɣ led to a modest but significant increase in the fraction of CD107a-positive OT-I antigen-specific T cells compared to IFN-ɣ alone, and a trend was also observed in the absence of IFN-ɣ although the difference was not statistically significant when corrected for multiple testing (Fig. 5E). In contrast, no effect on CD107a positivity was observed when RP-48OVA-LLO190 cells were co-cultured with antigen-nonspecific bulk CD8+ T cells, confirming the requirement for T cell recognition of a specific MHC-I-antigen complex (Fig. 5E). To establish that increased cytotoxic degranulation was associated with functional consequences, we employed the same experimental system and measured viable RP-48OVA-LLO190 cell numbers after bomedemstat exposure and co-culture with antigen-nonspecific CD8 T cells or OVA-specific OT-I T cells. We found that bomedemstat pre-treatment, with or without IFN-g, significantly reduced SCLC cell survival in the antigen-specific (“OT-I”) co-culture relative to the antigen-nonspecific (“CD8”) co-culture (Fig. 5F). These data establish that the molecular changes induced by bomedemstat in target SCLC cells are sufficient to increase cytotoxic CD8+ T cell activation in an antigen-dependent manner, and that increased antigen-specific CD8+ T cell activity is associated with decreased target cell survival.
We therefore constructed a framework for anti-tumor immune potentiation by the LSD1 inhibitor bomedemstat (Fig. 5G). Bomedemstat likely acts via both LSD1 demethylase activity inhibition as well as disruption of protein complexes involving LSD1, thereby altering chromatin organization and function and eventually resulting in more effective anti-tumor immunity and reduced tumor growth (Fig. 5G). To test the hypothesis that bomedemstat-mediated LSD1 inhibition can potentiate anti-tumor immunity, we have initiated a clinical trial for patients with extensive stage SCLC. This trial will test the combination of bomedemstat and standard-of-care maintenance atezolizumab in those patients with stable or reduced disease after four cycles of induction platinum, etoposide, and anti-PDL1 antibody (Fig. 5H). The primary outcomes of this single arm study are safety and tolerability of the combination of bomedemstat and atezolizumab and 6-month progression-free survival measured from the initiation of maintenance bomedemstat and atezolizumab relative to the historical control of IMpower133(1). Additional endpoints will include overall survival and correlative biomarker assessments of circulating cell-free DNA. Our in vitro studies and the results using an immunocompetent syngeneic mouse model taken together serve as proof-of-concept evidence that epigenetic reprogramming of SCLC cells with the LSD1 inhibitor bomedemstat can overcome SCLC-intrinsic immunosuppressive mechanisms including MHC-I downregulation and sensitize SCLC tumors to immune recognition and killing.
Discussion
Most SCLC tumors exhibit what is commonly referred to as an immunologically “cold” tumor and tumor microenvironment, with low expression of antigen processing and presentation machinery including MHC-I and low T cell infiltration. Recent studies, however, have identified a subset of human SCLC with high expression of antigen processing and presentation machinery genes associated with improved clinical responses to ICB(19–21). In one study, high MHC-I expression in clinical samples was associated with reduced ASCL1 and the neuroendocrine marker chromogranin, and MHC-I high status was strongly enriched in patients with more durable responses to ICB(20). In a second study, transcriptional analyses revealed a subset of SCLC that the authors termed SCLC-I (“Inflamed”), with low expression of neuroendocrine genes such as ASCL1 and high expression of the neuronal repressor REST(19). The SCLC-I subset experienced greater clinical benefit from the addition of ICB based on IMpower133 clinical outcomes data(19). In a third study, both low neuroendocrine differentiation and high antigen presentation machinery expression correlated with clinical responses to ICB in SCLC patient samples, but the most significant correlate of response to ICB was activation of NOTCH signaling(21).
Pharmacologic approaches that drive activation of key pathways associated with sensitivity to ICB could theoretically overcome intrinsic resistance to ICB in a substantial fraction of SCLC patients and thereby considerably improve the clinical benefit associated with ICB use. EZH2 inhibition, for example, was recently found to increase MHC-I expression in SCLC and may represent a promising approach to improving ICB efficacy(6,20). Our previous finding(24) that inhibition of another epigenetic regulator, LSD1, activated NOTCH, decreased expression of ASCL1 in SCLC, and reduced neuroendocrine characteristics, i.e., features associated with clinical responses to ICB, led us to test LSD1 inhibition as a potential mechanism to improve immune responses in SCLC. Indeed, we observed striking improvements in tumor growth inhibition when LSD1 inhibition was added to PD1 inhibition in an immunocompetent syngeneic mouse model of SCLC.
Our previous work with the LSD1 inhibitor iadademstat (ORY1001) showed that while most SCLC PDX models exhibit modest tumor growth inhibition with LSD1 suppression, one PDX model, FHSC04, derived from a SCLC patient that had relapsed following first line chemotherapy, exhibited a striking and durable regression with iadademstat monotherapy(24). Importantly, the activation of NOTCH and suppression of ASCL1 was a feature of all ASCL1-positive PDX models studied ex vivo(24). This finding is consistent with our current studies of bomedemstat, which did not exert substantial cytotoxic effect against the mSCLC cells tested but did induce NOTCH and suppress an ASCL1 gene program. Thus, while there may be potential for a rare exceptional response to LSD1 inhibition alone, the general ability of LSD1 inhibition to activate NOTCH and suppress neuroendocrine features of SCLC points to potential activity across a much broader subset of SCLC patients.
Potentiation of the anti-tumor efficacy of PD1 blockade by LSD1 in an immunocompetent model was associated with increased CD8 infiltration, suggesting a key role of antigen-specific cytotoxicity. Consistent with previous studies targeting the SCLC epigenome (6,20), tumors treated with LSD1i and PD1 blockade also displayed increased expression of antigen presentation machinery by both transcriptional and cell surface protein analysis, leading us to explore this phenomenon further using a panel of cell culture models. Bomedemstat treatment alone and in combination with IFN-ɣ increased cell surface MHC-I expression across a panel of murine and human SCLC-A models, and this effect was phenocopied by CRISPR knockout of LSD1, supporting LSD1’s central role. However, LSD1 inhibition did not induce MHC-I expression in either of the two ASCL1-low human models tested, which also expressed high MHC-I levels at baseline. Future investigations should determine whether subtype as defined by transcription factor expression, with or without other features, can reliably predict LSD1i-induced immunogenicity.
We also explored potential roles for NOTCH(21) and cytoplasmic nucleic acid sensor signaling(31,47) in LSD1i-induced MHC-I upregulation. Substantial reduction of NOTCH activation did not attenuate MHC-I upregulation in our studies, suggesting that LSD1i may retain immunogenic activity in the considerable fraction of SCLC tumors that harbor inactivating NOTCH mutations(15). We did however detect cytoplasmic nucleic acid sensor upregulation in multiple models, raising the question of whether LSD1i treatment will lead to similar immunogenicity in NOTCH-high tumors, where it has recently been shown that STING signaling is suppressed by NOTCH activity(53).
Bomedemstat treatment of SCLC cells in culture also increased markers of antigen-specific T cell activation and sensitivity to antigen-specific T cell killing. Our studies focused on MHC-I derepression as a likely mechanism by which LSD1 inhibition sensitizes SCLC to PD1 blockade, but the complete mechanism in the fully immunocompetent system we utilized is likely to be more complex. Bomedemstat treatment of tumors was associated with lower neutrophil levels, and while myelosuppression is an expected effect of LSD1 inhibitor therapy(45), neutrophils have previously been implicated in promoting an immunosuppressive immune microenvironment in lung cancer(54). Whether neutrophil depletion from the TME plays a role in the anti-tumor immunity induced by bomedemstat remains to be determined. We also observed increased mRNA expression of potentially immunostimulatory chemokines such as CX3CL1 and CXCL14 in LSD1i+anti-PD1-treated tumors, and these transcripts were again upregulated by LSD1i in cultured mSCLC cells along with other potential chemoattractants such as IL33 and CXCL12. The role of LSD1i-induced chemokine secretion will also be important to delineate. Moreover, while our studies have been tumor cell focused, it has recently been shown that LSD1 can promote the terminal differentiation of T cells, and that LSD1 suppression can increase the population of progenitor exhausted CD8+ T cells, which is the CD8+ T cell population most responsive to PD1 inhibition(32).
Based on our observations that LSD1 inhibition can potentiate responses to PD1 axis inhibition in vivo, we reasoned that the addition of an LSD1 inhibitor to standard maintenance anti-PDL1 antibody in first line treatment of ES-SCLC might overcome intrinsic ICB resistance in a meaningful fraction of patients, leading to improved clinical outcomes. Importantly, however, a Phase 1 clinical trial of the LSD1 inhibitor GSK2879552 administered as monotherapy in patients with relapsed SCLC was terminated(30). Bomedemstat has been administered to more than 170 patients with hematologic malignancies at doses of up to 6 mg/kg/d (55–57), i.e. doses up to ten times higher than those planned in the SCLC clinical study. We therefore expect that bomedemstat will be generally well-tolerated in patients with SCLC and that combination therapy with bomedemstat and ICB merits clinical exploration. To this end, a clinical trial has been initiated in which bomedemstat is added to maintenance atezolizumab in extensive stage SCLC following induction treatment with platinum/etoposide chemotherapy and PDL1 inhibitor (Clinicaltrials.gov identifier: NCT05191797). We hypothesize that bomedemstat will sensitize intrinsically ICB-resistant SCLC and therefore substantially improve progression-free survival. Additional, exploratory endpoints will include analysis of tumor tissue and circulating cell-free DNA for biomarkers of sensitivity to bomedemstat and atezolizumab.
After decades of incremental progress in SCLC research, a recent proliferation of promising discoveries has occurred (3,58). Prominent among these are (A) a new standard of care for patients with untreated ES-SCLC that clearly albeit modestly improves overall survival for all comers (i.e. chemotherapy with ICB), (B) proposal of an epigenetically driven taxonomy of SCLC subtypes, (C) the association of NOTCH pathway activation and neuroendocrine feature loss with benefit from ICB, and (D) the identification of targeted therapies that can induce SCLC epigenetic reprogramming (6,20). A critical next step will be to determine how to best integrate epigenetic markers and therapies into routine SCLC care, including how to achieve optimal benefit with ICB. Since only about 15% of patients appeared to derive more durable benefit from the addition of ICB to platinum doublet (1,2), it is less likely that epigenetic features will be employed as clinical biomarkers to select patients for ICB use. Instead, therapeutic manipulation of the SCLC epigenome to sensitize intrinsically resistant SCLC tumors to ICB treatment represents an alternative and potentially more broadly beneficial approach. We have found that LSD1 inhibition in SCLC drives epigenetic reprogramming including NOTCH pathway activation and neuroendocrine gene suppression, increases MHC-I presentation and T cell killing and reduces tumor growth when combined with PD1 axis blockade. Of note, our findings are highly consistent with a very recently published study also describing the immunostimulatory role of LSD1 blockade in SCLC(59). LSD1 inhibition in combination with ICB is therefore an especially promising example of clinical translation of the emerging paradigm of epigenetic heterogeneity and plasticity in SCLC biology, with the potential to substantially improve on the outcomes that ICB can achieve for patients with SCLC.
Supplementary Material
Translational Relevance.
Although now part of standard-of-care, most patients with small cell lung cancer (SCLC) do not derive durable benefit from immune checkpoint blockade (ICB), and strategies to improve the activity of ICB in SCLC are therefore needed. Recent studies have shown that SCLC tumors that have low neuroendocrine (NE) features and exhibit NOTCH pathway activation may be more likely to benefit from ICB. We recently showed that blocking the chromatin modifying enzyme Lysine-specific Demethylase 1a (LSD1) can suppress NE features and activate NOTCH in SCLC models. Here, using immunocompetent murine models of SCLC, we report that inhibiting LSD1 increases the anti-tumor activity of ICB, augments CD8+ T cell infiltration into SCLC tumors, increases expression of antigen presentation machinery on tumor cells, and potentiates killing of SCLC cells by CD8+ T cells in vitro. Based on these results, we recently opened a clinical trial testing the addition of LSD1 inhibition to standard-of-care maintenance ICB in SCLC.
Acknowledgements
This work was supported by the National Institutes of Health (Fred Hutch NIH Lung Spore award P50CA228944 to DM and AMH, training grant awards T32HL007093 and K12CA076930 to JH, University of Washington/Fred Hutch Cancer Center Support Grant P30CA015704); a Conquer Cancer Foundation Young Investigator Award to JH. We acknowledge support from the Fred Hutch Flow Cytometry, Genomics and Bioinformatics and Comparative Medicine Shared Resources.
Conflict of interest statement:
Dr. Friesen reports personal fees from Alpine Immune Sciences, outside the submitted work; Dr. Srivastava reports grants from Lyell Immunopharma, personal fees from Lyell Immunopharma, outside the submitted work; In addition, Dr. Srivastava has a patent PCT/US2018/056796 with royalties paid to Bristol Myers Squibb, and a patent PCT/US2018/049812 with royalties paid to Lyell Immunopharma. Hugh Rienhoff is an employee of Imago BioSciences, an equity holder and an inventor on mulitple patents for the composition of matter and method of use of bomedemstat. Dr. MacPherson reports grants from Janssen, grants from Roche, outside the submitted work. Dr. Martins reports grants from Merck, grants from NIH/NCI, grants from Pfizer, grants from Genentech Inc., grants from Calithera Biosciences, Inc., grants from AstraZeneca Pharmaceuticals, all outside the submitted work. Prof Sutherland reports grants from the National Health and Medical Research Council (NHMRC; APP1159955; APP1138275) and holds a Victorian Cancer Agency Mid-Career Fellowship (MCRF18003) and a Peter and Julie Alston WEHI Centenary Fellowship and direct research support from AGIOS Pharmaceuticals and Pfizer Pharmaceuticals, outside the submitted work. The remaining authors have no disclosures to report.
Abbreviations:
- SCLC
small cell lung cancer
- PD1
Programmed cell death-protein 1
- MHC
major histocompatibility complex
- IFN
interferon
- ICB
immune checkpoint blockade
References
- 1.Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 2018;379(23):2220–9 doi 10.1056/NEJMoa1809064. [DOI] [PubMed] [Google Scholar]
- 2.Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet 2019;394(10212):1929–39 doi 10.1016/S0140-6736(19)32222-6. [DOI] [PubMed] [Google Scholar]
- 3.Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nature reviews Cancer 2017;17(12):725–37 doi 10.1038/nrc.2017.87. [DOI] [PubMed] [Google Scholar]
- 4.Yazawa T, Kamma H, Fujiwara M, Matsui M, Horiguchi H, Satoh H, et al. Lack of class II transactivator causes severe deficiency of HLA-DR expression in small cell lung cancer. J Pathol 1999;187(2):191–9 doi . [DOI] [PubMed] [Google Scholar]
- 5.Funa K, Gazdar AF, Minna JD, Linnoila RI. Paucity of beta 2-microglobulin expression on small cell lung cancer, bronchial carcinoids and certain other neuroendocrine tumors. Lab Invest 1986;55(2):186–93. [PubMed] [Google Scholar]
- 6.Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019;36(4):385–401 e8 doi 10.1016/j.ccell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doyle A, Martin WJ, Funa K, Gazdar A, Carney D, Martin SE, et al. Markedly decreased expression of class I histocompatibility antigens, protein, and mRNA in human small-cell lung cancer. J Exp Med 1985;161(5):1135–51 doi 10.1084/jem.161.5.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iams WT, Shiuan E, Meador CB, Roth M, Bordeaux J, Vaupel C, et al. Improved Prognosis and Increased Tumor-Infiltrating Lymphocytes in Patients Who Have SCLC With Neurologic Paraneoplastic Syndromes. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2019;14(11):1970–81 doi 10.1016/j.jtho.2019.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonanno L, Pavan A, Dieci MV, Di Liso E, Schiavon M, Comacchio G, et al. The role of immune microenvironment in small-cell lung cancer: Distribution of PD-L1 expression and prognostic role of FOXP3-positive tumour infiltrating lymphocytes. Eur J Cancer 2018;101:191–200 doi 10.1016/j.ejca.2018.06.023. [DOI] [PubMed] [Google Scholar]
- 10.Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018;33(5):853–61 e4 doi 10.1016/j.ccell.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17(7):883–95 doi 10.1016/S1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- 12.Schultheis AM, Scheel AH, Ozretic L, George J, Thomas RK, Hagemann T, et al. PD-L1 expression in small cell neuroendocrine carcinomas. Eur J Cancer 2015;51(3):421–6 doi 10.1016/j.ejca.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Jia D, Augert A, Kim DW, Eastwood E, Wu N, Ibrahim AH, et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer discovery 2018;8(11):1422–37 doi 10.1158/2159-8290.CD-18-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Augert A, Zhang Q, Bates B, Cui M, Wang X, Wildey G, et al. Small Cell Lung Cancer Exhibits Frequent Inactivating Mutations in the Histone Methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J Thorac Oncol 2017;12(4):704–13 doi 10.1016/j.jtho.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524(7563):47–53 doi 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nature genetics 2012. doi 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019;19(5):289–97 doi 10.1038/s41568-019-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang YH, Klingbeil O, He XY, Wu XS, Arun G, Lu B, et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev 2018;32(13-14):915–28 doi 10.1101/gad.314815.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021;39(3):346–60 e7 doi 10.1016/j.ccell.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigi M, Campisi M, et al. Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov 2021;11(8):1952–69 doi 10.1158/2159-8290.CD-20-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roper N, Velez MJ, Chiappori A, Kim YS, Wei JS, Sindiri S, et al. Notch signaling and efficacy of PD-1/PD-L1 blockade in relapsed small cell lung cancer. Nat Commun 2021;12(1):3880 doi 10.1038/s41467-021-24164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004;119(7):941–53 doi 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007;446(7138):882–7 doi 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- 24.Augert A, Eastwood E, Ibrahim AH, Wu N, Grunblatt E, Basom R, et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci Signal 2019;12(567) doi 10.1126/scisignal.aau2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takagi S, Ishikawa Y, Mizutani A, Iwasaki S, Matsumoto S, Kamada Y, et al. LSD1 Inhibitor T-3775440 Inhibits SCLC Cell Proliferation by Disrupting LSD1 Interactions with SNAG Domain Proteins INSM1 and GFI1B. Cancer Res 2017;77(17):4652–62 doi 10.1158/0008-5472.CAN-16-3502. [DOI] [PubMed] [Google Scholar]
- 26.Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015;28(1):57–69 doi 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Ito T, Udaka N, Yazawa T, Okudela K, Hayashi H, Sudo T, et al. Basic helix-loop-helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development 2000;127(18):3913–21. [DOI] [PubMed] [Google Scholar]
- 28.Morimoto M, Nishinakamura R, Saga Y, Kopan R. Different assemblies of Notch receptors coordinate the distribution of the major bronchial Clara, ciliated and neuroendocrine cells. Development 2012;139(23):4365–73 doi 10.1242/dev.083840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim JS, Ibaseta A, Fischer MM, Cancilla B, O’Young G, Cristea S, et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017;545(7654):360–4 doi 10.1038/nature22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauer TM, Besse B, Martinez-Marti A, Trigo JM, Moreno V, Garrido P, et al. Phase I, Open-Label, Dose-Escalation Study of the Safety, Pharmacokinetics, Pharmacodynamics, and Efficacy of GSK2879552 in Relapsed/Refractory SCLC. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2019;14(10):1828–38 doi 10.1016/j.jtho.2019.06.021. [DOI] [PubMed] [Google Scholar]
- 31.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018;174(3):549–63 e19 doi 10.1016/j.cell.2018.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Debo B, Li M, Shi Z, Sheng W, Shi Y. LSD1 inhibition sustains T cell invigoration with a durable response to PD-1 blockade. Nature communications 2021;12(1):6831 doi 10.1038/s41467-021-27179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2019;38(3):390–405 doi 10.1038/s41388-018-0451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 2009;4(7):1064–72 doi 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 2012;28(16):2184–5 doi 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 37.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30(7):923–30 doi 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 38.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26(1):139–40 doi 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010;11(3):R25 doi 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell reports 2016. doi 10.1016/j.celrep.2016.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34(3):267–73 doi 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 43.McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014;156(6):1298–311 doi 10.1016/j.cell.2014.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veatch JR, Singhi N, Srivastava S, Szeto JL, Jesernig B, Stull SM, et al. A therapeutic cancer vaccine delivers antigens and adjuvants to lymphoid tissues using genetically modified T cells. J Clin Invest 2021;131(16) doi 10.1172/JCI144195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sprussel A, Schulte JH, Weber S, Necke M, Handschke K, Thor T, et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012;26(9):2039–51 doi 10.1038/leu.2012.157. [DOI] [PubMed] [Google Scholar]
- 46.Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569(7757):503–8 doi 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov 2019;9(5):646–61 doi 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carrasco Pro S, Dafonte Imedio A, Santoso CS, Gan KA, Sewell JA, Martinez M, et al. Global landscape of mouse and human cytokine transcriptional regulation. Nucleic Acids Res 2018;46(18):9321–37 doi 10.1093/nar/gky787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conroy MJ, Lysaght J. CX3CL1 Signaling in the Tumor Microenvironment. Adv Exp Med Biol 2020;1231:1–12 doi 10.1007/978-3-030-36667-4_1. [DOI] [PubMed] [Google Scholar]
- 50.Hara T, Tanegashima K. Pleiotropic functions of the CXC-type chemokine CXCL14 in mammals. J Biochem 2012;151(5):469–76 doi 10.1093/jb/mvs030. [DOI] [PubMed] [Google Scholar]
- 51.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res 2010;16(11):2927–31 doi 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 52.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol 2016;16(11):676–89 doi 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 53.Hong D, Knelson EH, Li Y, Durmaz YT, Gao W, Walton E, et al. Plasticity in the Absence of NOTCH Uncovers a RUNX2-Dependent Pathway in Small Cell Lung Cancer. Cancer Res 2022;82(2):248–63 doi 10.1158/0008-5472.CAN-21-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kargl J, Zhu X, Zhang H, Yang GHY, Friesen TJ, Shipley M, et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight 2019;4(24) doi 10.1172/jci.insight.130850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palandri F, Vianelli N, Ross DM, Cochrane T, Lane SW, Larsen SR, et al. A Phase 2 Study of the LSD1 Inhibitor Img-7289 (bomedemstat) for the Treatment of Essential Thrombocythemia (ET). Blood 2021;138(Supplement 1):386- doi 10.1182/blood-2021-148210. [DOI] [Google Scholar]
- 56.Gill H, Yacoub A, Pettit KM, Bradley T, Gerds AT, Tatarczuch M, et al. A Phase 2 Study of the LSD1 Inhibitor Img-7289 (bomedemstat) for the Treatment of Advanced Myelofibrosis. Blood 2021;138(Supplement 1):139- doi 10.1182/blood-2021-148682. [DOI] [Google Scholar]
- 57.Palandri F, Ross D, Cochrane T, Tate C, Lane S, Larsen S, et al. P1033: A PHASE 2 STUDY OF THE LSD1 INHIBITOR IMG-7289 (BOMEDEMSTAT) FOR THE TREATMENT OF ESSENTIAL THROMBOCYTHEMIA (ET). HemaSphere 2022;6:923–4 doi 10.1097/01.HS9.0000847000.38667.91. [DOI] [Google Scholar]
- 58.Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers 2021;7(1):3 doi 10.1038/s41572-020-00235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen EM, Taniguchi H, Chan JM, Zhan YA, Chen X, Qiu J, et al. Targeting LSD1 rescues MHC class I antigen presentation and overcomes PD-L1 blockade resistance in small cell lung cancer. J Thorac Oncol 2022. doi 10.1016/j.jtho.2022.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gene expression data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE208614.