

Abstract
With rapid advances in the development of metabolic pathways and synthetic biology toolkits, a persisting challenge in microbial bioproduction is how to optimally rewire metabolic fluxes and accelerate the concomitant high-throughput phenotype screening. Here we developed a biosensor-assisted titratable CRISPRi high-throughput (BATCH) screening approach that combines a titratable mismatch CRISPR interference and a biosensor mediated screening for high-production phenotypes in Escherichia coli. We first developed a programmable mismatch CRISPRi that could afford multiple levels of interference efficacy with a one-pot sgRNA pool (a total of 16 variants for each target gene) harboring two consecutive random mismatches in the seed region of sgRNA spacers. The mismatch CRISPRi was demonstrated to enable almost a full range of gene knockdown when targeting different positions on genes. As a proof-of-principle demonstration of the BATCH screening system, we designed doubly mismatched sgRNA pools targeting 20 relevant genes in E. coli and optimized a PadR-based p-coumaric acid biosensor with broad dynamic range for the eGFP fluorescence guided high-production screening. Using sgRNA variants for the combinatorial knockdown of pfkA and ptsI, the p-coumaric acid titer was increased by 40.6% to o 1308.6 mg/l from glycerol in shake flasks. To further demonstrate the general applicability of the BATCH screening system, we recruited a HpdR-based butyrate biosensor that facilitated the screening of E. coli strains achieving 19.0% and 25.2% increase of butyrate titer in shake flasks with sgRNA variants targeting sucA and ldhA, respectively. This work reported the establishment of a plug-and-play approach that enables multilevel modulation of metabolic fluxes and high-throughput screening of high-production phenotypes.
Keywords: Mismatch CRISPRi, Titratable repression, Biosensor, p-Coumaric acid, Butyrate
1. Introduction
Metabolic engineering harnesses the ability of living microorganisms to degrade renewable and low-cost carbon feedstocks in order to produce pharmaceutically or industrially important chemicals (Keasling, 2010; Khosla and Keasling, 2003; Lee et al., 2019; Nielsen and Keasling, 2011). However, the sophisticatedly regulated carbon metabolisms naturally coined in the genomes of most microbes always devote to cell survival and produce target chemicals non-efficiently (Montaño López et al., 2022; Zha et al., 2009; Zu et al., 2020). To obtain high-production strains that satisfy commercial relevance, traditional approaches rely on either domestication of microbes or deletion of specific non-essential competing genes (Gallone et al., 2016; Park et al., 2007; Steensels et al., 2019). With increasingly available genomic and transcriptomic data and better understanding of cellular metabolisms, increasing efforts have been made to systematically interrogate and rewire metabolic network in both “localized” and “global” scales (Andreozzi et al., 2016; Feist and Palsson, 2008; Lee et al., 2021; Park et al., 2009).
To that end, programmable synthetic biology toolkits have been developed to expedite genome-scale perturbation of metabolic fluxes, such as synthetic DNA oligonucleotides and CRISPR mediated multiplex genome engineering (Cong et al., 2013; Nyerges et al., 2016; Wang et al., 2009), retron-enabled genome editing (Lopez et al., 2022; Schubert et al., 2021; Simon et al., 2019), synthetic regulatory RNA mediated post-transcriptional interference (RNAi) (Na et al., 2013; Yang et al., 2015), and CRISPR-based transcriptional interference (CRISPRi) (Bikard et al., 2013; Gilbert et al., 2014; Hawkins et al., 2015). Among those, CRISPRi has been extensively used in systematic metabolic engineering due to its programmability and multiplexity, robust activity, and host flexibility (Banerjee et al., 2020; Liu et al., 2022; Schultenkämper et al., 2020; Wang et al., 2018b). However, CRISPRi mediated stringent gene repression may cause cell fitness defects especially when targeting growth-essential genes (Cui et al., 2018; de Bakker et al., 2022; Wang et al., 2021a), rendering this approach unable to thoroughly investigate the impact of expression levels of individual genes on desired high-production phenotypes.
To address this limitation, one ideal strategy would be to design massively pooled sgRNA libraries that guide dCas9 only to their respective gene targets while simultaneously affording parallel interrogation of individual genes on microbial production when repressed to different extents. CRISPRi repression efficiency could be modulated by engineered sgRNAs with random single mismatches (Jost et al., 2020; Qi et al., 2013), with interspersed double or multiple mismatches (Feng et al., 2021; Hawkins et al., 2020), with inducible hairpin or aptazyme-embedded structures (Ferry et al., 2017; Kocak et al., 2019; Siu and Chen, 2019; Tang et al., 2017), or with truncated spacers (Qi et al., 2013; Wang et al., 2021b). All these strategies require lab-intensive synthesis of large sgRNA libraries, empirically tested sgRNA structures, or specialized molecule inducers that have prevented their practical applications in metabolic engineering practices. Moreover, concomitant with large-scale sgRNA libraries, one recurring challenge would be the lack of efficient and cost-effective high-throughput CRISPRi screening methods for desired production phenotypes.
In this work, we developed a biosensor-assisted titratable CRISPRi high-throughput (BATCH) screening approach that combines a titratable mismatch CRISPR interference and a biosensor mediated screening for high-production phenotypes in Escherichia coli. Based on the fact that either mismatch position, type or combination could impair CRISPRi activity, we first defined the rule for a cost-effective one-pot mismatch sgRNA design that enabled a full range of CRISPRi repression efficiencies. Then, to use p-coumaric acid and butyrate as two proof-of-principle applications, we recruited their respective PadR and HpdR based biosensor-reporter system with broad dynamic properties. With mismatch sgRNA libraries targeting genes involved in central carbon metabolisms, the BATCH screening system identified multiple target genes that enhanced titers of both products. This work established a readily appliable approach of high-throughput and multilevel CRISPRi screen in metabolic engineering to obtaining desired high-production phenotypes with programmable and customizable mismatch sgRNA libraries.
2. Materials and Methods
2.1. Strains, medium and chemicals
All strains and plasmids used in this study are listed in Table 1. E. coli XL1-Blue (Stratagene, La Jolla, CA) was used for plasmid construction and storage. E. coli∷dCas9 was created by integrating the PLlacO1-dCas9 cassette into the genome at dkgB locus and was used as the host strain for CRISPRi test and butyrate production. Further deletion of pheA and tyrR in E. coli∷dCas9 resulted in strain E. coli∷dCas9 ΔpheA ΔtyrR, which was used as the host strain for p-coumaric acid production. Gene knockouts were conducted via P1 phage transduction method according to standard protocols (Thomason et al., 2007). Luria-Bertani (LB) medium (10 g/l tryptone, 5 g/l yeast extract and 10 g/l NaCl) was used for seed culture preparation. M9Y medium, prepared from M9 minimal medium (6 g/l Na2HPO4, 0.5 g/l NaCl, 3 g/l KH2PO4, 1 g/l NH4Cl, 1 mM MgSO4, 0.1 mM CaCl2) containing 5 g/l yeast extract and 20 g/l glycerol or 20 g/l glucose, was used for shake flask experiments for producing p-coumaric acid or butyrate, respectively. Antibiotics including ampicillin (100 mg/ml) and kanamycin (50 mg/ml) were added to the medium at a ratio of 0.1% when necessary. Butyrate and p-coumaric acid standards were purchased from Sigma-Aldrich and MP Biomedical LLC, respectively.
Table 1.
Strains and plasmids
| Strains | Description | Reference |
|---|---|---|
| E. coli XL1-Blue | recA1 endA1gyrA96thi-1hsdR17supE44relA1lac | Stratagene |
| E. coli BW25113(F′) | rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 F′[traD36 proAB lacIqZΔM15 Tn10(Tetr)] | (Atsumi et al., 2008) |
| E. coli∷dCas9 | E. coli BW25113(F′) with the integration of the PLlacO1-dCas9 cassette into the genome at dkgB locus | This study |
| E. coli∷dCas9 ΔpheA ΔtyrR | E. coli∷dCas9 with the deletion of pheA and tyrR | This study |
| Plasmids | Description | Reference |
| pZE12-luc | PLlacO1; luc; ColE1 ori; AmpR | (Lutz and Bujard, 1997) |
| pCS27 | PLlacO1; p15A ori; KanR | (Shen and Liao, 2008) |
| pZE-eGFP | pZE12luc harboring the PLlacO1-controlled eGFP expression cassette | (Wang et al., 2021b) |
| pCS-sgRNA | pCS27 harboring sgRNA scaffold targeting eGFP | (Wang et al., 2017) |
| pZE-sgRNA | pZE12-luc harboring the sgRNA scaffold amplified from pCS27-sgRNA and inserted in between XhoI and AvrII | This study |
| pHA-MCS | pZE12-luc with the luc gene replaced by a newly designed multi-cloning site | This study |
| pCS-sgegfp-cds1 | pCS27 harboring the PLlacO1-controlled transcriptional cassette of a fully matched sgRNA targeting the +39 bp of eGFP | This study |
| pCS-sgegfp-rbs | pCS-sgegfp targeting the RBS region of the eGFP | This study |
| pCS-sgegfp-cds2 | pCS-sgegfp targeting the coding sequence (starting from + 378 bp) | This study |
| pZE-Plpp0.2-PadR-P1-eGFP | pZE-Plpp0.2-PadR harboring the hybrid promoter P1-controlled eGFP expression cassette | This study |
| pZE-Plpp0.2-PadR-P2-eGFP | pZE-Plpp0.2-PadR harboring the hybrid promoter P2-controlled eGFP expression cassette | This study |
| pZE-Plpp0.2-PadR-P7-eGFP | pZE-Plpp0.2-PadR harboring the hybrid promoter P7-controlled eGFP expression cassette | This study |
| pZE-Plpp0.2-PadR-P9-eGFP | pZE-Plpp0.2-PadR harboring the hybrid promoter P9-controlled eGFP expression cassette | This study |
| pZE-CA | pZE-Plpp0.2-PadR-P9-eGFP harboring the codon-optimized RgTAL from Rhodotorula glutinis under control of the PLlacO1 promoter | This study |
| pCS-V2 | pCS27-Plpp1-HpdR-PhpdH-eGFP with deletion of a palindromic sequence (−209 to −118 bp) on the PhpdH promoter and HpdR under control of Plpp1.0 promoter | (Wang et al., 2021b) |
| pCS-THCTA | pCS27 harbors the butyrate synthetic pathway which includes the thl, hbd, crt, ter, and atoDA. | This study |
| pCS-HPE-THCTA | pCS-THCTA harboring Plpp1-HpdR-PhpdH-eGFP (HPE) biosensor cassette from pCS-V2 | This study |
2.2. Plasmid and strain construction
All plasmid constructions were carried out following standard molecular cloning protocols (Sambrook et al., 1989). Phusion high-fidelity DNA polymerase, restriction DNA endonucleases and Quick Ligation kit were purchased from New England Biolabs (Beverly, MA, USA). The high-copy plasmid pHA-MCS was derived from pZE12-luc by replacing luc gene with a multi-cloning site (MCS) and was applied as a negative control when needed. Fully matched sgRNA and rationally designed mismatch sgRNA variants targeting the eGFP coding sequence (starting from +39 bp) were constructed into the pCS-sgRNA scaffold plasmid using with ApaLI and BamHI, resulting in plasmids pCS-sgegfp-cds1, and pCS-sg-m7 to m16. Inverse PCR was applied to introduce random mismatches in sgRNA variants targeting genes of interest. To create mismatch sgRNA libraries targeting eGFP, the forward primer sgegfp-rbs-F (ApaLI) or sgegfp-cds2-F (ApaLI) was individually paired with the reverse primer sgRNA-R (ApaLI) (Table S1) to amplify the whole template plasmid pCS-sgRNA, resulting in linear plasmids containing sgRNA variants targeting the RBS region or the coding sequence (+378 bp) of eGFP, respectively. The resultant linear plasmids were then digested by ApaLI and DpnI, and subjected to ligation. These ligation products were co-transferred into E. coli∷dCas9 with the reporter plasmid pZE-eGFP. When plated on LB plates, transformants were randomly picked for eGFP fluorescence assay by cultivating in LB tubes and plasmids were extracted for gene sequencing analysis. To construct mismatch sgRNA variants targeting genes in E. coli genome for improved production of p-coumaric acid or butyrate, the inverse PCR templates were pCS-sgRNA and pZE-sgRNA, respectively. Selected targets and corresponding primers were listed in Table S1. Spacers of selected sgRNA variants that enabled high-producing phenotypes were later sequenced and included in Table S2.
For constructing p-coumaric acid responsive biosensor systems, the eGFP expression cassettes controlled by hybrid promoters (P1, P2, P7, and P9) were amplified from plasmids pZE-P1-eGFP, pZE-P2-eGFP, pZE-P7-eGFP, and pZE-P9-eGFP, respectively (Jiang et al., 2021). These cassettes were then inserted into plasmid pZE-Plpp0.2-PadR (Li et al., 2022) using AatII and XhoI, resulting in plasmids pZE-Plpp0.2-PadR-P1-eGFP, pZE-Plpp0.2-PadR-P2-eGFP, pZE-Plpp0.2-PadR-P7-eGFP, and pZE-Plpp0.2-PadR-P9-eGFP. The PLlacO1-controlled RgTAL expression cassette was amplified from plasmid pZE-RgTAL (Huang et al., 2013) and was constructed to plasmid pZE-Plpp0.2-PadR-P9-eGFP using BamHI and AvrII, yielding plasmid pZE-CA. The biosynthetic pathway of butyrate consisting of thl, hbd, crt, ter, and atoDA, were sequentially constructed to plasmid pCS27 to form plasmid pCS-THCTA using Acc65I, NdeI, SalI, EcoRI, BamHI, and MluI. The Plpp1-HpdR-PhpdH-eGFP (HPE) biosensor cassette was amplified from plasmid pCS-V2 (Wang et al., 2021b) and constructed to plasmid pCS-THCTA using XhoI and XbaI, yielding pCS-HPE-THCTA.
2.3. Biosensor dynamic range test and eGFP repression assay
To test the dynamic range or substrate scope of engineered biosensor systems, plasmids harboring the biosensor systems were transferred into E. coli BW25113(F′). Three independent colonies of each testing group were randomly picked and inoculated into 3.5 ml fresh LB medium (with appropriate antibiotics) as seed cultures. After overnight cultivation (10–12 h) in a 37°C rotatory shaker at a speed of 270 rpm, seed cultures were transferred into fresh 3.5 ml LB medium with a ratio of 5% (175 μl), and new cultures were placed in the shaker under 37°C with a speed of 270 rpm. After 1 h of cultivation, gradient concentrations of corresponding substrates (0–1000 mg/L for p-coumaric acid, and 0–2.5 g/L for butyrate) were fed into the cultures. After 12 h of cultivation post substrate induction, cultures were sampled and diluted by 10-folds before subjected to measurement of cell density (OD600) and green fluorescence intensities using the 96-well plate reader (Synergy HT, BioTek). The eGFP repression assay was conducted with E. coli∷dCas9 co-transformed with pZE-eGFP and pCS27 harboring sgRNAs. E. coli∷dCas9 co-transformed with pZE-eGFP and pCS27 empty plasmid was used as a control. The relative eGFP expression is calculated as the ratio of normalized eGFP fluorescence per OD600 (RFU/OD600) of cells with sgRNAs to those without sgRNAs. Green fluorescence intensities were determined using an excitation filter of 485/20 nm and an emission filter of 528/20 nm.
2.4. Screening high-production phenotypes in test tubes
For screening of p-coumaric acid high-production phenotypes, constructed mismatch sgRNA libraries on pCS27 for each target gene were separately co-transferred into E. coli∷dCas9 ΔpheA ΔtyrR with plasmid pZE-CA. For screening of butyrate high-production phenotypes, constructed mismatched sgRNA libraries on pZE12-luc for each target gene were separately co-transferred into E. coli∷dCas9 with plasmid pCS-HPE-THCTA. All transformants were plated on LB plates with appropriate antibiotics and 0.5 mM IPTG. Colonies with high green fluorescence intensities under blue light exposure were picked and inoculated in 3.5 ml M9Y medium containing 20 g/l glycerol (for p-coumaric acid production) or 20 g/l glucose (for butyrate production). IPTG was added in the medium to a final concentration of 0.5 mM at the beginning to induce expression of CRISPRi system and biosynthesis pathways. Samples were taken after 24 h of induction and were subjected to measurement of cell densities, green fluorescence intensities, and product accumulations. Cell density and green fluorescence intensity were quantitively analyzed using the 96-well plate reader (Synergy HT, BioTek) with the same protocol described earlier. Production of p-coumaric acid or butyrate was also analyzed by high-performance liquid chromatography (HPLC). Selected high-production strains were streaked in LB plates (with appropriate antibiotics) and plasmids were extracted for DNA sequencing of variant sgRNA spacers.
2.5. Validation of selected sgRNA variants via shake flasks fermentation
To validate the production performance of selected colonies from test tubes, three colonies of the strains streaked on LB plates were inoculated in 3.5 ml LB medium and grown at 37 °C for 8–10 h. Then, 1 ml of each seed culture was transferred to 20 ml fresh M9Y medium (containing 20 g/l glycerol or glucose) in 125-ml shake flasks (Chemglass Life Sciences LLC) and grown at 30 °C for 48 h. Shake flask experiments were performed in a 30 °C rotary shaker with a speed of 270 rpm. Glycerol was used as carbon source for p-coumaric acid production while glucose was used for butyrate production. IPTG was added in the medium with a final concentration of 0.5 mM. Samples were collected after 48 h post induction and were subjected for measurement of cell density (OD600) and product accumulation. Cell density (OD600) was measured using the spectrophotometer (VWR), and the products p-coumaric acid or butyrate were analyzed by HPLC.
2.6. Product analysis
Collected samples (1 ml) were centrifuged at 12,000 rpm for 15 minutes, and supernatants were filtered through 0.22 μm film before applied to HPLC analysis. Concentrations of p-coumaric acid were quantitatively analyzed by Agilent HPLC 1260 Infinity II (1260 Infinity II Diode Array Detector WR) with a reverse-phase ZORBAX SB-C18 column. A methanol-water (containing 0.1% trifluoroacetic acid) gradient system with a flow rate of 1 ml/min was used. The analyzing method, adopted from our previous study (Li et al., 2022), was set as follows: 5% methanol from 0 to 2 min, 5% to 80% methanol from 2 to 10 min, 80% methanol from 10 to 14 min, 80% to 5% methanol from 14 to 18 min, and 5% methanol from 18 to 20 min. A standard curve was drawn using gradient concentrations of p-coumaric acid solution prepared from p-coumaric acid standard (MP Biomedical LLC). The retention time of p-coumaric acid was 8.89 min and the detection wavelength was set at 280 nm.
Butyrate was analyzed by a Dionex Ultimate 3000 HPLC equipped with a Coregel-64H column (Transgenomic, Omaha, NE). The collected samples (1 ml) were centrifuged at 12,000 rpm for 15 minutes, and the supernatants were filtered through 0.22 μm film before applied to HPLC analysis. The HPLC method was modified from our previous research (Wang et al., 2018a). Briefly, 4 mN sulfuric acid was used as the mobile phase with a flow rate of 0.4 ml/min. The column temperature was set at 45 °C. The retention time of butyrate was 38.897 min.
3. Results
3.1. Design of BATCH screening system
The BATCH screening system is composed of two critical parts: (1) a titratable CRISPRi for customizable and multilevel gene repression in microbial chassis, and (2) chemical inducible biosensors with broad dynamic ranges for phenotype screening (Fig. 1). The titratable CRISPRi represses all relevant gene targets within the metabolic network or competing genes of target biosynthetic pathways to different extents. Biosensors are committed to sensing intracellular product chemicals and inducing the reporter gene eGFP expression, thus linking the product concentration with fluorescence intensity. Implementation of the BATCH screening system involves two rounds of selection: the first round selection of fluorescent single colonies on plates for test tube cultivation and the second round selection of highly fluorescent transformants from test tube cultures for shake flask cultivation validation. A combination of selected beneficial repressions could be iteratively launched to screen hyperproduction phenotypes.
Figure 1. Schematic diagram of the BATCH screening workflow.
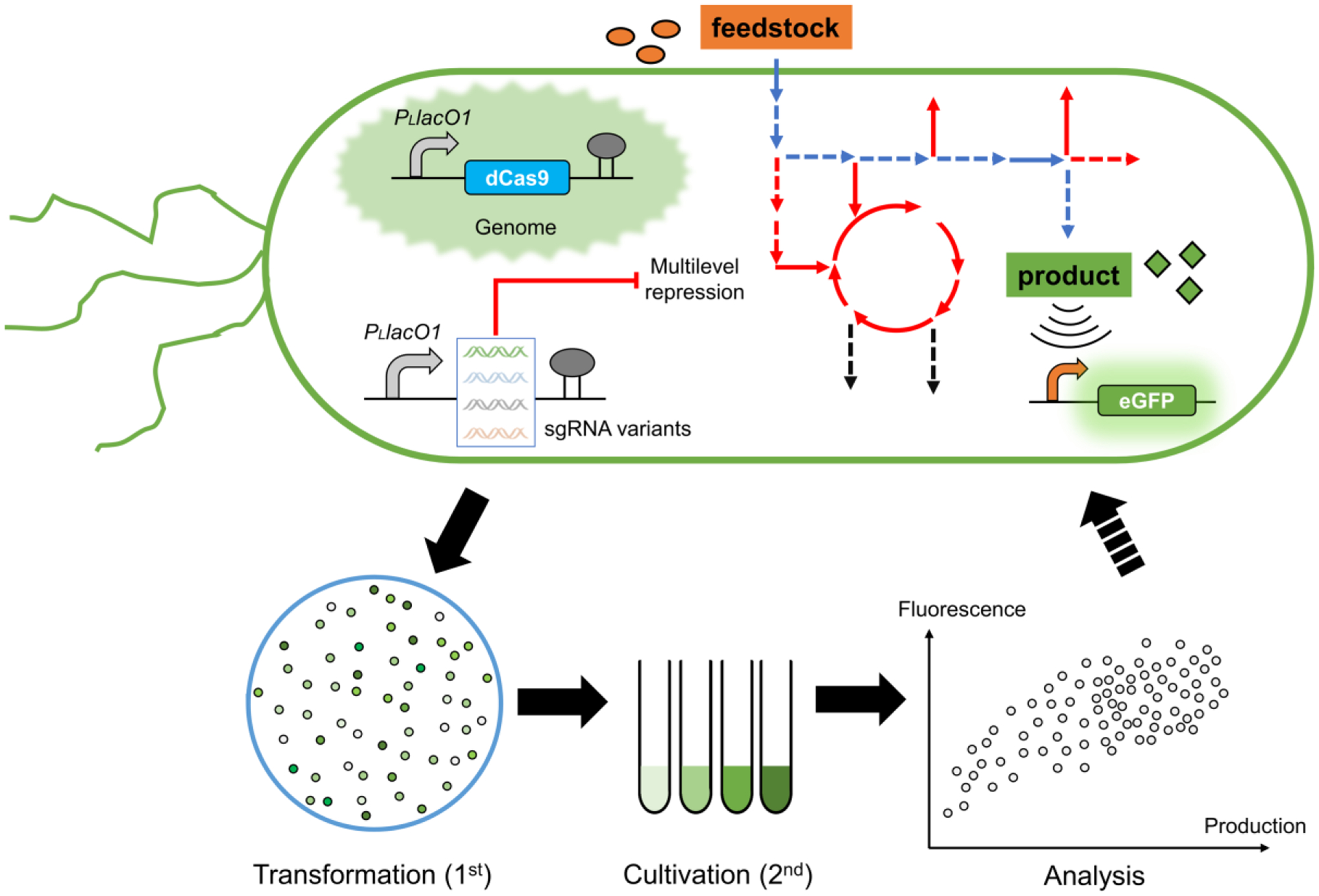
The BATCH screening involves sgRNA variants mediated multilevel CRISPRi of genes of interest within metabolic network to redirect carbon flux to target biosynthetic pathways. Improved production phenotypes will be reflected by the target chemical inducible reporter GFP fluorescence. The BATCH screening consists of a first round plate based selection and a second round test tube based selection followed by final shake flask based validation. Blue lines indicate biosynthetic pathways and red lines indicate relevant or potential competing pathways.
3.2. Establishing one-pot mismatch sgRNA libraries for titratable CRISPRi
First, in order to establish a titratable CRISPRi, we sought to alter the repression strengths of target genes simply by engineering customizable sgRNA libraries. Since CRISPRi activity is highly sensitive to mismatch positions, types and combinations between sgRNA and target DNA (Feng et al., 2021; Gilbert et al., 2014), we herein endeavored to establish a titratable CRISPRi method with a facile one-pot mismatch sgRNA library design.
To facilitate CRISPRi system construction and also avoid a potential toxicity of SpdCas9 overexpression in E. coli cells (Calvo-Villamañán et al., 2020; Zhang and Voigt, 2018), we constructed a strain E. coli∷dCas9 carrying SpdCas9 under the control of PLlacO1 promoter integrated into the genome at the low-expression dkgB locus (Bryant et al., 2014; Wang et al., 2021b). To generate mismatch sgRNAs, inverse PCR was applied for introducing mutations in sgRNA spacers with degenerate primers (Fig. 2A). We first tested the single mismatch sensitivity of sgRNA on eGFP repression, by transforming E. coli∷dCas9 with pCS-eGFP and pZE12-sgRNAs harboring mismatch sgRNAs targeting 5’ coding sequence (+39 bp) of eGFP (Fig. 2B and C). The eGFP repression assay indicated that single mismatches outside a 7bp PAM-proximal seed position are tolerated without significantly affecting CRISPRi repression efficiencies (Fig. 2C). This was consistent with previous findings that mismatches within the generally recognized ~8bp seed region greatly compromised or abolished the CRISPR/(d)Cas9 functionality, at least in E. coli (Datsenko et al., 2012; Gilbert et al., 2014; Qi et al., 2013; Sternberg et al., 2015). To further investigate the sensitivity of double mismatches, we narrowed down to construct sgRNA variants harboring saturated double substitutions (NN) in PAM-proximal 7–8th bp seed region. The repression assay showed that the 16 possible sgRNA variants almost enabled a broad repression profile of eGFP from 14.6% to 95.6%, suggesting that mismatching the 7–8th bp seed region could afford a multilevel gene repression (Fig. 2D). To examine the impact of target positions on the repression profile, we therefore designed double mismatch sgRNAs with random mutations of the 7–8th bp seed region that targeted both ribosome binding site (RBS) and alternate coding sequence (+378 bp) of eGFP with degenerate primers. In both cases, out of 32 randomly picked transformants, 12 different mismatch sgRNAs were obtained, accounting for a coverage of 75% of total variants. Both sgRNA variants enabled a full range of knockdown levels of eGFP (Fig. 2E and F), further confirming the generable applicability of one-pot mismatch sgRNA libraries for programmable and titratable CRISPRi.
Figure 2. Establishing titratable CRISPRi with mismatch sgRNA libraries.
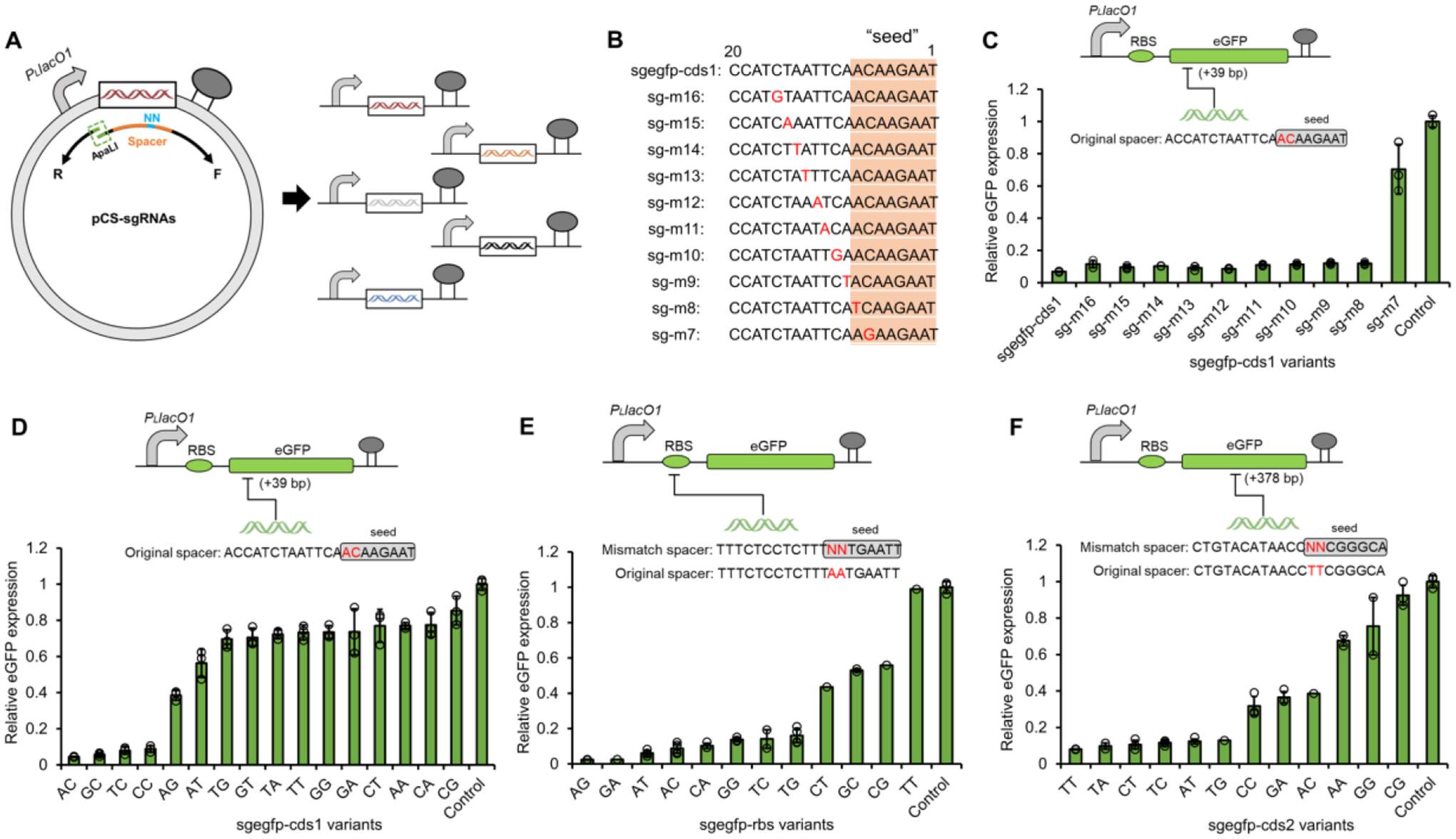
(A) The design of inverse PCR for one-pot construction of mismatch sgRNAs. (B) Spacer sequences (5’-3’) of single mismatch sgegfp-cds1 variants targeting the coding sequence (+39 bp) of eGFP. (C) The CRISPRi efficiencies of sgRNAs with single mismatch at different positions in the sgegfp-cds1 spacer (c), with rationally designed double mismatches in the 7–8th bp seed region of sgegfp-cds1 spacer (D), with randomly mutated double mismatches in the 7–8th bp seed region of sgegfp-rbs spacer (targeting the RBS region eGFP promoter) (E), and with randomly mutated double mismatches in the 7–8th bp seed region of sgegfp-cds2 spacer (targeting the coding sequence (+378 bp) of eGFP) (F). Control, E. coli∷dCas9 co-transformed with pZE-eGFP and pCS27 empty plasmid. In E and F, 32 single colonies were randomly picked from each plate and were subjected to both eGFP fluorescence assay and spacer determination via plasmid sequencing. All data in C and D represent the mean of three biologically independent samples and error bars show standard deviation.
3.3. Optimizing the p-coumaric acid biosensor PadR with broad dynamic behavior
For a proof-of-principle demonstration, we chose to target the p-coumaric acid biosynthetic pathway and optimized a PadR-based p-coumaric acid biosensor system for its high-production screening from glycerol. p-Coumaric acid has extensive applications in food, pharmaceutical and cosmetic industry, and also serves as an important precursor for more complex natural products such as flavonoids and polyphenols (Trantas et al., 2015; Wang et al., 2016).
To assist high-production phenotype screening, natural or engineered chemical-inducible biosensors with broad dynamic range would be critical. An engineered p-coumaric acid-responsive biosensor system PadR-PpadC from Bacillus subtilis was established in our previous study (Jiang et al., 2021). The transcriptional repressor PadR could repress the expression of downstream gene controlled by PpadC promoter, while the presence of p-coumaric acid could activate its transcription initiation (Jiang et al., 2021). To enable biosensor-assisted screening, four previously engineered hybrid PpadC promoters (P1, P2, P7, and P9) with optimized dynamic ranges and strong output strengths were selected to pair with PadR (Table S3). The hybrid promoters were constructed by inserting PadR binding boxes into the strong constitutive pL promoter (Jiang et al., 2021). PadR under control of the constitutive Plpp0.2 promoter and eGFP under control of the hybrid promoters were assembled into pZE12-luc, resulting pZE-Plpp0.2-PadR-P1-eGFP, pZE-Plpp0.2-PadR-P2-eGFP, pZE-Plpp0.2-PadR-P7-eGFP, and pZE-Plpp0.2-PadR-P9-eGFP (Fig. 3A). To test the dynamic performance, all plasmids harboring biosensor systems were transferred into E. coli BW25113(F′), and transformants were cultivated with supplementation of gradient concentrations of p-coumaric acid (0, 100, 200, 400, 600, and 1000 mg/l). The PadR-P9 biosensor system exhibited the best performance in the selected concentration range, resulting in a 14.2-fold eGFP induction with 1000 mg/l p-coumaric acid (Fig. 3B). Therefore, this combination was selected for the following BATCH screening of p-coumaric acid high-production phenotypes.
Figure 3. Optimizing the p-coumaric acid biosensor PadR with broad dynamic behavior.
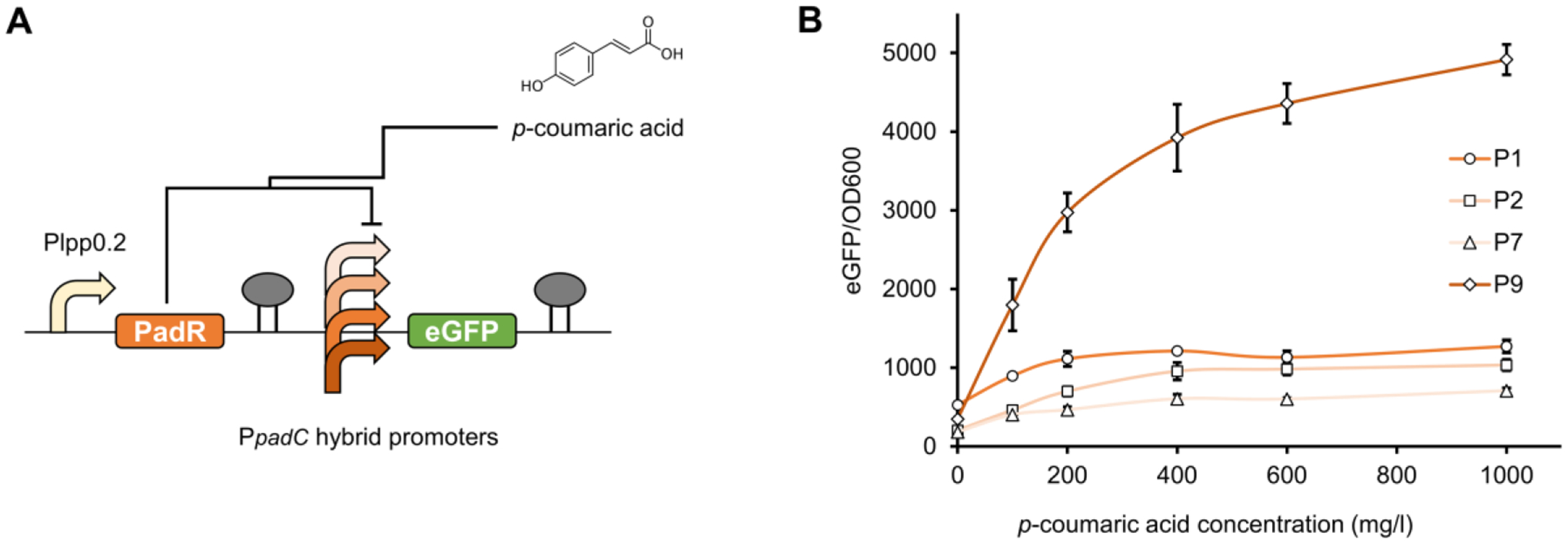
(A) The plasmid configuration for the PadR-based biosensor system. PadR was constitutively expressed under the control of the Plpp0.2 promoter and the reporter gene eGFP was controlled by the hybrid PpadC promoters (P1, P2, P7, and P9). (B) The dynamic range of the PadR-PpadC biosensor system harboring different hybrid promoters against different concentrations of p-coumaric acid (0, 100, 200, 400, 600, and 1000 mg/l). All data represent the mean of three biologically independent samples and error bars show standard deviation.
3.4. BATCH screening of p-coumaric acid over-production phenotypes
The p-coumaric acid pathway initiates from the aromatic amino acid (AAA) pathway and ends up with deamination of tyrosine to p-coumaric acid via tyrosine ammonia-lyase (TAL) (Fig. 4A). Extending from the central carbon metabolism, the p-coumaric acid pathway suffers from multiple potential competing pathways that divert carbon fluxes. To redirect carbon flux to the p-coumaric acid pathway, we designed random double mismatch sgRNAs targeting 20 relevant genes in the glycolysis pathway (ptsI, pgi, pfkA, fbaA, phoA, gapA, pgk, gpmA, gpmM, eno, pykA, pykF, and csrA), the TCA cycle (aceE, gltA, and ppc), the fatty acid biosynthesis pathway (accA, and fabD), and the AAA pathway (ubiC, and trpE) (Fig. 4A). To minimize deleterious effects on cell growth and customize sgRNA design, sgRNAs targeted available NGG PAMs near the start codon for each gene target to block transcription elongation.
Figure 4. BATCH screening of p-coumaric acid over-production phenotypes.
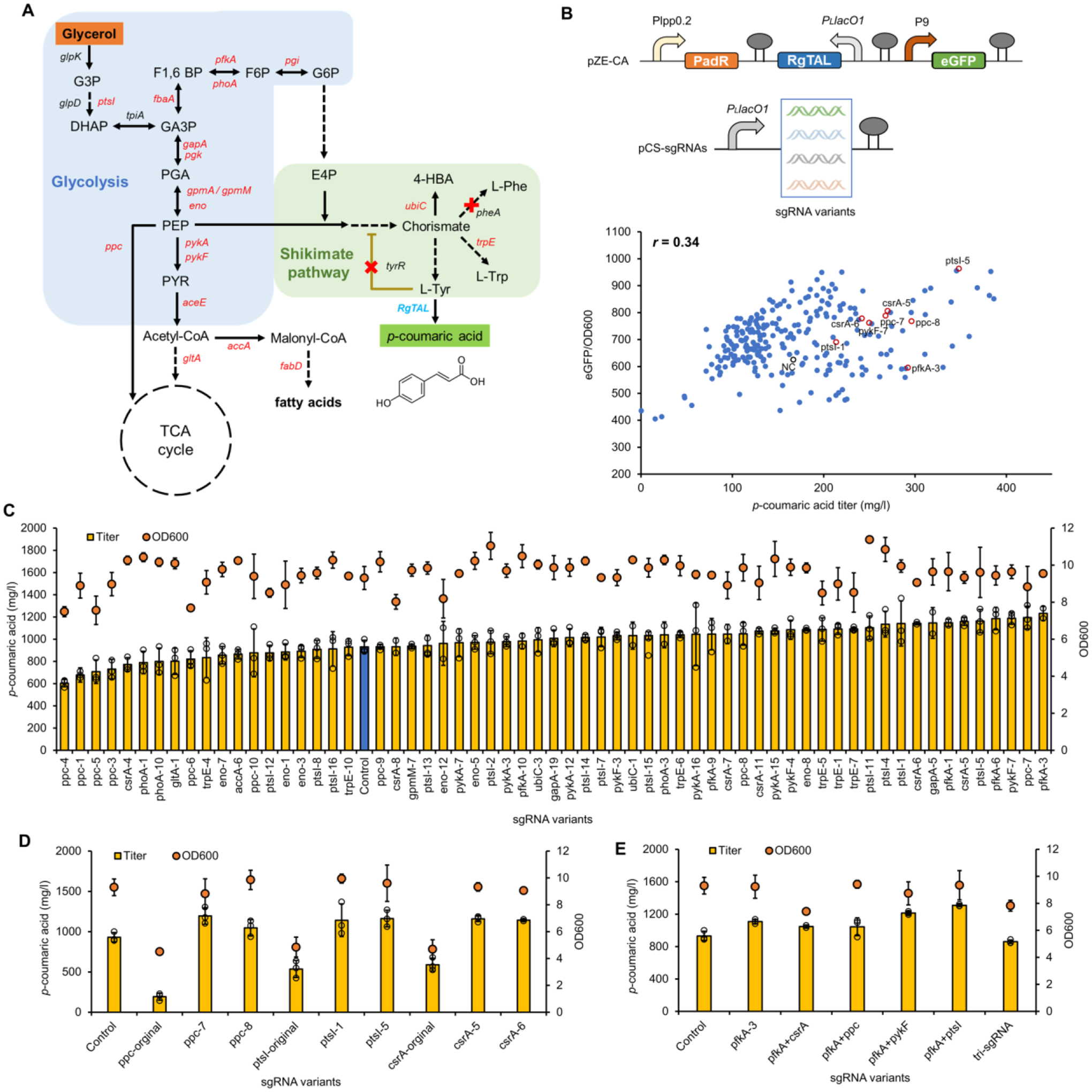
(A) The biosynthetic pathway of p-coumaric acid from glycerol. Genes marked in red are CRISPRi targets and red crosses are genes to be knocked out. (B) The correlation between p-coumaric acid titers (in test tubes) and green fluorescence intensities in E. coli∷dCas9 ΔpheA ΔtyrR with pZE-CA and pCS-sgRNA mismatch variants. p-Coumaric acid titers and normalized fluorescence intensities of the negative control strain with no sgRNA and strains with representative beneficial sgRNA variants were highlighted in black and red circles, respectively. (C) Titers of p-coumaric acid and cell densities (OD600) of p-coumaric acid producers with selected sgRNA variants in shake flasks. The blue bar represents the control strain without any sgRNA. (D) Comparison of p-coumaric acid production between authentic (fully matched) sgRNAs targeting ppc, ptsI and csrA, and their counterpart mismatch sgRNA variants in shake flasks. (E) The p-coumaric acid production test with combination of selected sgRNA variants in shake flasks. The number behind each gene name represented the order of selected colonies harboring counterpart mismatch sgRNA variants. Negative control (NC), E. coli∷dCas9 ΔpheA ΔtyrR with pZE-CA and pCS27. All data represent the mean of three biologically independent samples and error bars show standard deviation.
To implement the BATCH screening for p-coumaric acid over-production phenotype, E. coli∷dCas9 ΔtyrR ΔpheA with knockouts of the tyrosine repressor (tyrR) and the chorismate mutase/prephenate dehydratase (pheA) was applied as the host. E. coli∷dCas9 ΔtyrR ΔpheA was co-transformed with plasmids pZE-CA (harboring RgTAL and PadR biosensor) and pCS-sgRNAs harboring mismatch sgRNA libraries targeting each gene (Fig. 4B). After transformation and growing on LB plates supplemented with 20 g/l glycerol and 0.5 mM IPTG, a total of 284 colonies with notably high green fluorescence intensities were picked and cultivated in test tubes with M9 minimal medium containing 20 g/l glycerol (Fig. S1). Determination of eGFP fluorescence intensities and p-coumaric acid concentrations of all selected transformants exhibited a moderate positive correlation (Pearson correlation coefficient r = 0.34) (Fig. 4B). For each target gene, the difference of eGFP fluorescence intensities and p-coumaric acid concentration among mismatch sgRNA variants underlined the impact of gene repression level on p-coumaric acid production (Fig. S1). Among all targets, sgRNA variants towards csrA, fbaA, gapA, pfkA, phoA, ppc, ptsI, and pykF produced more hits with increased p-coumaric acid titers (Fig. S1). Shake flask based production tests with selected colonies from the test tube screening validated that mismatch sgRNAs towards csrA, ptsI, pykF, ppc, and pfkA afforded more than 25% increase of p-coumaric acid titers (Fig. 4C, and Fig. S1). The overall yields of p-coumaric acid per biomass were generally improved in accordance with p-coumaric acid titers (Fig. S2A). Especially, compared with the control strain (930.6 mg/l and an overall yield of 0.09 g/l/OD600), sgpfkA-3 permitted the highest production of p-coumaric acid, reaching a peak titer of 1232.0 mg/l and an overall yield of 0.13 g/l/OD600 (Fig. 4C and Fig. S2A).
One notable advantage of mismatch sgRNAs in BATCH screening over authentic sgRNAs in conventional CRISPRi is its non-deleterious effect to cell fitness especially when targeting essential genes. To corroborate that, we chose to compare the functionality of mismatch sgRNAs and authentic sgRNAs towards growth-essential genes such as ppc, ptsI and csrA. As expected, while authentic sgRNAs significantly impaired both cell growth (approximately 50% decrease) and p-coumaric acid titer, selected sgRNA variants improved p-coumaric acid production respectively by 28.4%, 25.0% and 24.6%, without sacrificing cell growth (Fig. 4D). This further underlined that the conditional and optimal gene repression with the BATCH screening system would expedite the rational high-production phenotype screening. One other advantage of BATCH screening is applying it for easy testing of combinatorial effects of beneficial repressions from initial rounds. Therefore, we combined selected mismatch sgRNAs to further boost p-coumaric acid production. The combination of sgpfkA-3 and sgptsI-5 increased p-coumaric acid titer to 1308.6 mg/l, accounting for a 40.6% increase over the control strain (Fig. 4E).
3.5. Application of the BATCH system for screening of butyrate over-production phenotypes
To explore the applicability of the BATCH screening system to other production scenarios, we then implemented it to the screening of butyrate over-production phenotypes. Butyrate is a C4 fatty acid and a commodity chemical with wide applications in chemical industry, food manufacturing, pharmaceuticals, perfume, and animal feed supplements (Guo et al., 2021; Jang et al., 2014). The butyrate pathway initiates from condensation of two acetyl-CoA to acetoacetyl-CoA followed by its reduction to 3-hydroxybutyryl-CoA with the enzyme thiolase (thl) and hydroxybutyryl-CoA dehydrogenase (hbd) (Fig. 5A). 3-Hydroxybutyryl-CoA was then reduced to butyryl-CoA by crotonase (crt) andtransenoyl-CoA reductase (ter), followed by release of butyrate from butyryl-CoA with acetyl-CoA:acetoacetyl-CoA synthase (atoDA) (Choi et al., 2012; Saini et al., 2014). As the direct precursor for butyrate and also a critical central metabolite, increasing acetyl-CoA supply or reducing its consumption could enhance butyrate titers.
Figure 5. BATCH screening for butyrate over-production phenotypes.
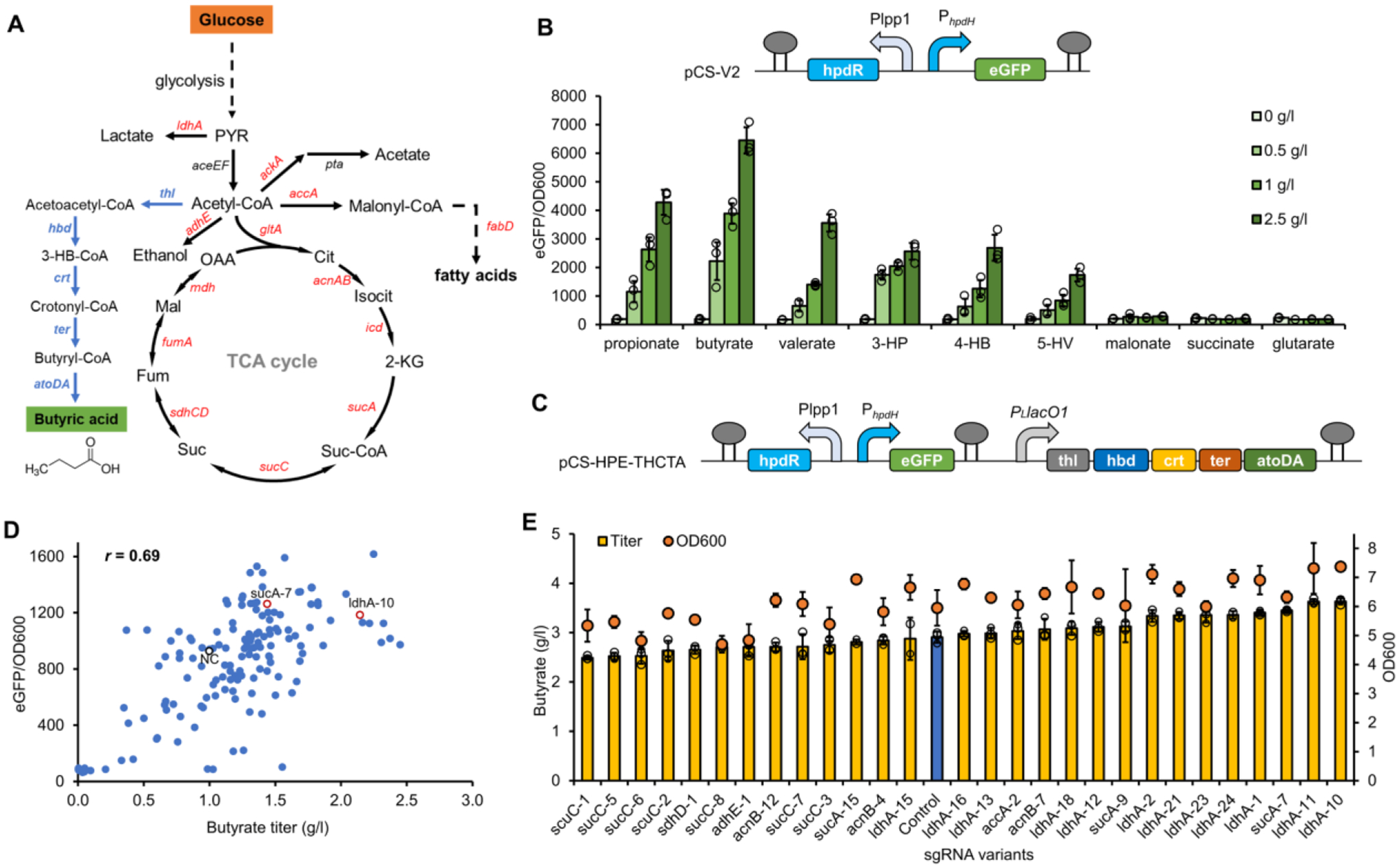
(A) The biosynthetic pathway for butyrate from glucose. Genes shown in blue are butyrate pathway genes and genes shown in red are CRISPRi targets. (B) The ligand profiling of the engineered HpdR-PhpdH biosensor against C3-C5 fatty acids (propionate, butyrate, and valerate), hydroxy acids (3-hydroxypropionate (3-HP), 4-hydroxybutyrate (4-HB), and 5-hydroxyvalerate (5-HV)), and diacids (malonate, succinate, and glutarate) with gradient concentrations of 0–2.5 g/l. (C) The plasmid scheme of pCS-HPE-HCTA for BATCH screening of butyrate high-production phenotypes. (D) The correlation between butyrate titers (in test tubes) and green fluorescence intensities in E. coli∷dCas9 with pCS-HPE-THCTA and pZE-sgRNA mismatch variants. Butyrate titers and normalized fluorescence intensities of negative control strain with no sgRNA and strains with representative beneficial sgRNA variants were highlighted in black and red circles, respectively. (E) Titers of butyrate and cell densities (OD600) of butyrate producers with selected sgRNA variants in shake flasks. The blue bar represents the control strain without any sgRNA. The number behind each gene name represented the order of selected colonies harboring counterpart mismatch sgRNA variants. Negative control (NC), E. coli∷dCas9 with pCS-HPE-THCTA and pHA-MCS. All data represent the mean of three biologically independent samples and error bars show standard deviation.
To deploy the BATCH screening, we first sought to identify and characterize a butyrate biosensor with broad dynamic range. We previously engineered a promiscuous 3-hydroxybutyrate responsive HpdR-PhpdH system on pCS-V2 plasmid, which harbors a deletion of a palindromic sequence (−209 to −118 bp) on the PhpdH promoter and expresses HpdR from Pseudomonas putida under control of Plpp1.0 promoter (Hanko et al., 2017; Wang et al., 2021b). To investigate the ligand scope of the engineered HpdR-PhpdH system, E. coli BW25113(F′) transformed with pCS-V2 was subjected to C3-C5 fatty acids, hydroxy acids, and diacids with gradient concentrations (0–2.5 g/l). The eGFP fluorescence assay showed that the engineered HpdR-PhpdH system exerted high sensitivity and broad dynamic range towards all C3-C5 fatty acids (propionate, butyrate, and valerate) and hydroxy acids (3-hydroxypropionate, 4-hydroxybutyrate, and 5-hydroxyvalerate) but not diacids (Fig. 5B). Among all ligands, butyrate afforded the highest eGFP induction fold (41-fold at 2.5 g/l) (Fig. 5B), indicating that the engineered HpdR-PhpdH system could serve as an ideal butyrate biosensor.
To experimentally implement the BATCH screening, the engineered HpdR-PhpdH system was assembled with the butyrate pathway, resulting in plasmid pCS-HPE-THCTA (Fig. 5C). To reserve acetyl-CoA for butyrate biosynthesis, 14 mismatch sgRNA libraries involved in pyruvate consumption (ldhA), acetyl-CoA consumption (ackA, and adhE), fatty acid pathway (accA and fabD), TCA cycle (gltA, acnA, acnB, icd, sucA, sucC, sdhC, sdhD, fumA and mdh) were constructed into pZE12-luc (resultant pZE-sgRNAs). All mismatch sgRNAs harboring 7–8th random mutations targeted available NGG PAMs near the start codon for each gene target to block transcription elongation. E. coli∷dCas9 was co-transformed with pCS-HPE-THCTA and pZE-sgRNAs and subjected to BATCH screening. For over 150 variants screened in test tubes, eGFP fluorescence intensities and butyrate titers were strongly correlated (Pearson correlation coefficient r = 0.69), confirming the applicability of biosensor-based screening of butyrate over-production phenotypes (Fig. 5D, and Fig. S3). The initial screening underlined that sgRNA variants targeting each gene enabled differential butyrate titers, and sgRNAs targeting acnB, ldhA, sucA and sucC generally afforded more sgRNA variants with enhanced butyrate titers (Fig. S3). Further validation of 27 selected over-production colonies in shake flask cultivations showed that the best sgRNA variants sgsucA-7 and sgldhA-10 increased butyrate titer by 19.0% and 25.2% over the control strain (2.90 g/l), reaching 3.45 and 3.63 g/l, respectively (Fig. 5E). Interestingly, the overall yields of butyrate per biomass were not necessarily improved, as we observed comparable overall yields per biomass between sgldhA-10 (0.49 g/l/OD600) and the control strain (0.48 g/l/OD600) (Fig. S2B). These results together demonstrated the general applicability of BATCH screening in different production scenarios.
4. Discussion
Identification of competing gene targets and optimal control of their expression are critical in achieving high production of desired biochemicals in metabolic engineering (Ko et al., 2020; Na et al., 2013). Due to the intricately regulated nature and the essentiality in cellular maintenance of metabolic network, conventional gene knockout strategies are often labor intensive and would miss identification of potential targets. RNAi or CRISPRi based screening has expedited large-scale profiling and repression of competing gene targets without modifying genomes. However, the static gene repression imposed by RNAi or CRISPRi is sometimes suboptimal and even detrimental to metabolic pathways in production scenarios (Crook et al., 2016; Rousset et al., 2018). This work established a superior BATCH screening system that renders (1) titratable CRISPRi using customizable mismatch sgRNA libraries for full-range gene repression, and (2) pinpointing the optimal gene repression with product biosensor based high-production screening.
The sensitivity of CRISPRi towards mismatches enabled fine-tuning of CRISPRi repression efficiency. In this work, we first located the mismatching window to the 7–8th bp within the seed region of sgRNA, whose random mutation almost afforded a full-scale of eGFP repression regardless of targeting positions (Fig. 2). With designable degenerate primers, mismatches could be easily created with one-pot PCR and incorporated into sgRNA plasmid libraries, as indicated by the 75% coverage from 32 randomly picked colonies (Fig. 2). These two features add greatly to the general applicability and manipulation simplicity of the BATCH screening system in metabolic engineering practices. Deployment of the BATCH system in p-coumaric acid production validated our hypothesis that repressing gene targets at different extents could dynamically affect cell fitness and product formation. As demonstrated by repressing growth-essential genes including ppc, ptsI and csrA, repression of all three genes with authentic sgRNAs significantly impaired cell growth and p-coumaric acid production, while mismatch sgRNAs marginally affected cell growth but significantly increased p-coumaric acid production (Fig. 4D). Thus, the BATCH screening system allows the production based screening of competing gene targets and their optimal repressions in production scenarios.
The BATCH screening system relies on the biosensors of target chemicals with broad dynamic ranges. Chemical-inducible biosensors have been widely used to accelerate high-production screening and implemented into dynamic control paradigms for carbon metabolism rewiring (Seok et al., 2021; Zhang et al., 2015). However, the lack of biosensors or poor dynamic performance always limited their wide applications. In this study, we recruited two engineered biosensors, the PadR-PpadC system for p-coumaric acid and the HpdR-PhpdH system for butyrate. When placed on high-copy plasmid, while the native PadR-PpadC (P1) showed low-induction fold (3.9-fold) and eGFP expression even in the presence of 1 g/l p-coumaric acid, the engineered PadR-PpadC (P9) with mutations in PPadC promoter showed markedly increased induction fold (14.2-fold) and elevated eGFP expression (Fig. 3). Biosensors for short-chain fatty acids (C3-C5) are rarely documented. We previously engineered a promiscuous 3-hydroxypropionate-responsive HpdR-PhpdH system with significantly improved sensitivity and dynamic range towards 3-hydroxybutyrate (Wang et al., 2021b). Ligand profiling substantiated that the engineered HpdR-PhpdH system could actually accept all C3-C5 fatty acids and counterpart hydroxy acids including butyrate (Fig. 5B). With broad dynamic ranges, both the engineered PadR-PpadC and HpdR-PhpdH system rendered modest to strong positive correlations between eGFP fluorescence and product titers, which successfully guided the screening of their high-production E. coli cells. These chemical-inducible biosensors demonstrated here in phenotype screening could also be readily translated into other applications like sensor-mediated dynamic gene regulations.
It is noteworthy that mismatch positions, types or numbers within sgRNA spacers would have an impact on CRISPRi repression efficiencies. Thus, it is feasible to create diverse mismatch sgRNAs to achieve more fine-tuned gene repression. For instance, introducing more mismatches would create larger sgRNA libraries (4n, n is the number of mismatches) for multilevel gene repression. Targeting different locations on genes (such as promoter, RBS, or coding sequence) by mismatched sgRNAs would also influence the CRISPRi repression range. Thus, it is possible to expand sgRNA libraries by targeting different locations on gene and potentially enable more beneficial variants. However, expanded sgRNA libraries would require screening of larger colony libraries to cover most of the potential sgRNA variants and would be labor-intensive without biosensor-based or other high-throughput screening technologies. To customize sgRNA design, we herein demonstrated the applicability of doubly mismatched sgRNA libraries targeting locations near start codons of gene targets to titrate gene expression and improve production performance.
Combination of multiple beneficial sgRNAs did not always result in the expected synergetic effect in production improvement. This underlined that individually modulated transcription level of each target gene may not be optimal in production scenario when combined. A potential solution to address this issue would be to screen multiple gene targets in a single round, which will be feasible with proper designs and appropriate DNA assembly methods. Given the multiplexity nature of CRISPRi, BATCH screening of multiple gene targets can be performed to expand the library size and potentially obtain combinatorial sgRNA variants with more desired high-producing phenotypes. We also noticed the variations of production improvement between test tube screenings and shake flask validations, which were mostly due to different cultivation conditions. This underscored that all obtained over-production strains from first round screening needed to be further confirmed in shake flask cultivations.
In conclusion, we developed a new BATCH screening system that could be readily applied for large-scale target identification and multilevel gene repression with one-pot mismatch sgRNA libraries. We demonstrated its applicability to identify multiple gene targets for production improvement of p-coumaric acid and butyrate within a single round of selection. Ideally, efficient assembly of sgRNA variants targeting multiple potential genes could also allow for the investigation of the combinatorial effects with the BATCH screening system. In summary, the screening system presented here would expedite the development of high-production microbial strains without genome intervention.
Supplementary Material
Highlights.
A biosensor-assisted titratable CRISPRi high-throughput screening was established.
One-pot mismatch sgRNA libraries could enable a full range of CRISPRi efficacy.
Biosensors with broad dynamic ranges allowed reporter-based phenotype screening.
Multiple gene targets were identified to enhance p-coumaric acid and butyrate titers.
Acknowledgements
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM128620. The authors also acknowledge the support from the College of Engineering, University of Georgia, Athens.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare that they have no conflicts of interest.
Data and materials availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data or materials related to this paper may be requested from the corresponding author.
Reference
- Andreozzi S, Chakrabarti A, Soh KC, Burgard A, Yang TH, Van Dien S, Miskovic L, Hatzimanikatis V, 2016. Identification of metabolic engineering targets for the enhancement of 1, 4-butanediol production in recombinant E. coli using large-scale kinetic models. Metabolic Engineering. 35, 148–159. [DOI] [PubMed] [Google Scholar]
- Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJY, Hanai T, Liao JC, 2008. Metabolic engineering of Escherichia coli for 1-butanol production. Metabolic Engineering. 10, 305–311. [DOI] [PubMed] [Google Scholar]
- Banerjee D, Eng T, Lau AK, Sasaki Y, Wang B, Chen Y, Prahl J-P, Singan VR, Herbert RA, Liu Y, 2020. Genome-scale metabolic rewiring improves titers rates and yields of the non-native product indigoidine at scale. Nature Communications. 11, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA, 2013. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Research. 41, 7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant JA, Sellars LE, Busby SJ, Lee DJ, 2014. Chromosome position effects on gene expression in Escherichia coli K-12. Nucleic acids research. 42, 11383–11392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo-Villamañán A, Ng JW, Planel R, Ménager H, Chen A, Cui L, Bikard D, 2020. On-target activity predictions enable improved CRISPR–dCas9 screens in bacteria. Nucleic Acids Research. 48, e64–e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Park JH, Kim TY, Lee SY, 2012. Metabolic engineering of Escherichia coli for the production of 1-propanol. Metabolic Engineering. 14, 477–486. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, 2013. Multiplex genome engineering using CRISPR/Cas systems. Science. 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook N, Sun J, Morse N, Schmitz A, Alper HS, 2016. Identification of gene knockdown targets conferring enhanced isobutanol and 1-butanol tolerance to Saccharomyces cerevisiae using a tunable RNAi screening approach. Applied Microbiology and Biotechnology. 100, 10005–10018. [DOI] [PubMed] [Google Scholar]
- Cui L, Vigouroux A, Rousset F, Varet H, Khanna V, Bikard D, 2018. A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nature Communications. 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, Semenova E, 2012. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nature Communications. 3, 1–7. [DOI] [PubMed] [Google Scholar]
- de Bakker V, Liu X, Bravo AM, Veening J-W, 2022. CRISPRi-seq for genome-wide fitness quantification in bacteria. Nature Protocols. 1–30. [DOI] [PubMed] [Google Scholar]
- Feist AM, Palsson BØ, 2008. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nature Biotechnology. 26, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Guo J, Wang T, Zhang C, Xing X. h., 2021. Guide-target mismatch effects on dCas9–sgRNA binding activity in living bacterial cells. Nucleic Acids Research. 49, 1263–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry QR, Lyutova R, Fulga TA, 2017. Rational design of inducible CRISPR guide RNAs for de novo assembly of transcriptional programs. Nature Communications. 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallone B, Steensels J, Prahl T, Soriaga L, Saels V, Herrera-Malaver B, Merlevede A, Roncoroni M, Voordeckers K, Miraglia L, 2016. Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell. 166, 1397–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, 2014. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Lu J, Gao C, Zhang L, Liu L, Chen X, 2021. Dynamic control of the distribution of carbon flux between cell growth and butyrate biosynthesis in Escherichia coli. Applied Microbiology and Biotechnology. 105, 5173–5187. [DOI] [PubMed] [Google Scholar]
- Hanko EK, Minton NP, Malys N, 2017. Characterisation of a 3-hydroxypropionic acid inducible system from Pseudomonas putida for orthogonal gene expression control in Escherichia coli and Cupriavidus necator. Scientific Reports. 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins JS, Silvis MR, Koo B-M, Peters JM, Osadnik H, Jost M, Hearne CC, Weissman JS, Todor H, Gross CA, 2020. Mismatch-CRISPRi reveals the co-varying expression-fitness relationships of essential genes in Escherichia coli and Bacillus subtilis. Cell Systems. 11, 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins JS, Wong S, Peters JM, Almeida R, Qi LS, 2015. Targeted transcriptional repression in bacteria using CRISPR interference (CRISPRi). CRISPR. Springer, pp. 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Lin Y, Yan Y, 2013. Caffeic acid production enhancement by engineering a phenylalanine over-producing Escherichia coli strain. Biotechnology and Bioengineering. 110, 3188–96. [DOI] [PubMed] [Google Scholar]
- Jang Y-S, Im JA, Choi SY, Im Lee J, Lee SY, 2014. Metabolic engineering of Clostridium acetobutylicum for butyric acid production with high butyric acid selectivity. Metabolic Engineering. 23, 165–174. [DOI] [PubMed] [Google Scholar]
- Jiang T, Li C, Yan Y, 2021. Optimization of a p-Coumaric Acid Biosensor System for Versatile Dynamic Performance. ACS Synthetic Biology. 10, 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost M, Santos DA, Saunders RA, Horlbeck MA, Hawkins JS, Scaria SM, Norman TM, Hussmann JA, Liem CR, Gross CA, 2020. Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nature Biotechnology. 38, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasling JD, 2010. Manufacturing molecules through metabolic engineering. Science. 330, 1355–1358. [DOI] [PubMed] [Google Scholar]
- Khosla C, Keasling JD, 2003. Metabolic engineering for drug discovery and development. Nature Reviews Drug Discovery. 2, 1019–1025. [DOI] [PubMed] [Google Scholar]
- Ko Y-S, Kim JW, Lee JA, Han T, Kim GB, Park JE, Lee SY, 2020. Tools and strategies of systems metabolic engineering for the development of microbial cell factories for chemical production. Chemical Society Reviews. 49, 4615–4636. [DOI] [PubMed] [Google Scholar]
- Kocak DD, Josephs EA, Bhandarkar V, Adkar SS, Kwon JB, Gersbach CA, 2019. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nature Biotechnology. 37, 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Kim HU, Chae TU, Cho JS, Kim JW, Shin JH, Kim DI, Ko Y-S, Jang WD, Jang Y-S, 2019. A comprehensive metabolic map for production of bio-based chemicals. Nature Catalysis. 2, 18–33. [Google Scholar]
- Lee SY, Nielsen J, Stephanopoulos G, 2021. Metabolic Engineering: Concepts and Applications. John Wiley & Sons. [Google Scholar]
- Li C, Zou Y, Jiang T, Zhang J, Yan Y, 2022. Harnessing plasmid replication mechanism to enable dynamic control of gene copy in bacteria. Metabolic Engineering. 70, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liu M, Shi T, Sun G, Gao N, Zhao X, Guo X, Ni X, Yuan Q, Feng J, 2022. CRISPR-assisted rational flux-tuning and arrayed CRISPRi screening of an l-proline exporter for l-proline hyperproduction. Nature Communications. 13, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez SC, Crawford KD, Lear SK, Bhattarai-Kline S, Shipman SL, 2022. Precise genome editing across kingdoms of life using retron-derived DNA. Nature Chemical Biology. 18, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz R, Bujard H, 1997. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Research. 25, 1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaño López J, Duran L, Avalos JL, 2022. Physiological limitations and opportunities in microbial metabolic engineering. Nature Reviews Microbiology. 20, 35–48. [DOI] [PubMed] [Google Scholar]
- Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY, 2013. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nature Biotechnology. 31, 170–174. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Keasling JD, 2011. Synergies between synthetic biology and metabolic engineering. Nature Biotechnology. 29, 693–695. [DOI] [PubMed] [Google Scholar]
- Nyerges Á, Csörgő B, Nagy I, Bálint B, Bihari P, Lázár V, Apjok G, Umenhoffer K, Bogos B, Pósfai G, 2016. A highly precise and portable genome engineering method allows comparison of mutational effects across bacterial species. Proceedings of the National Academy of Sciences. 113, 2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Lee KH, Kim TY, Lee SY, 2007. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proceedings of the National Academy of Sciences. 104, 7797–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Kim TY, Lee SY, 2009. Constraints-based genome-scale metabolic simulation for systems metabolic engineering. Biotechnology Advances. 27, 979–988. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA, 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset F, Cui L, Siouve E, Becavin C, Depardieu F, Bikard D, 2018. Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLoS Genetics. 14, e1007749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini M, Wang ZW, Chiang C-J, Chao Y-P, 2014. Metabolic engineering of Escherichia coli for production of butyric acid. Journal of Agricultural and Food Chemistry. 62, 4342–4348. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T, 1989. Molecular cloning. Cold spring harbor laboratory press; New York. [Google Scholar]
- Schubert MG, Goodman DB, Wannier TM, Kaur D, Farzadfard F, Lu TK, Shipman SL, Church GM, 2021. High-throughput functional variant screens via in vivo production of single-stranded DNA. Proceedings of the National Academy of Sciences. 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultenkämper K, Brito LF, Wendisch VF, 2020. Impact of CRISPR interference on strain development in biotechnology. Biotechnology and Applied Biochemistry. 67, 7–21. [DOI] [PubMed] [Google Scholar]
- Seok JY, Han YH, Yang J-S, Yang J, Lim HG, Kim SG, Seo SW, Jung GY, 2021. Synthetic biosensor accelerates evolution by rewiring carbon metabolism toward a specific metabolite. Cell Reports. 36, 109589. [DOI] [PubMed] [Google Scholar]
- Shen CR, Liao JC, 2008. Metabolic engineering of Escherichia coli for 1-butanol and 1-propanol production via the keto-acid pathways. Metabolic Engineering. 10, 312–320. [DOI] [PubMed] [Google Scholar]
- Simon AJ, Ellington AD, Finkelstein IJ, 2019. Retrons and their applications in genome engineering. Nucleic Acids Research. 47, 11007–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K-H, Chen W, 2019. Riboregulated toehold-gated gRNA for programmable CRISPR–Cas9 function. Nature Chemical Biology. 15, 217–220. [DOI] [PubMed] [Google Scholar]
- Steensels J, Gallone B, Voordeckers K, Verstrepen KJ, 2019. Domestication of industrial microbes. Current Biology. 29, R381–R393. [DOI] [PubMed] [Google Scholar]
- Sternberg SH, LaFrance B, Kaplan M, Doudna JA, 2015. Conformational control of DNA target cleavage by CRISPR–Cas9. Nature. 527, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Hu JH, Liu DR, 2017. Aptazyme-embedded guide RNAs enable ligand-responsive genome editing and transcriptional activation. Nature Communications. 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL, 2007. E. coli genome manipulation by P1 transduction. Current Protocols in Molecular Biology. 1.17. 1–1.17. 8. [DOI] [PubMed] [Google Scholar]
- Trantas EA, Koffas MA, Xu P, Ververidis F, 2015. When plants produce not enough or at all: metabolic engineering of flavonoids in microbial hosts. Frontiers in Plant Science. 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM, 2009. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 460, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Guleria S, Koffas MA, Yan Y, 2016. Microbial production of value-added nutraceuticals. Current Opinion in Biotechnology. 37, 97–104. [DOI] [PubMed] [Google Scholar]
- Wang J, Teng Y, Zhang R, Wu Y, Lou L, Zou Y, Li M, Xie Z-R, Yan Y, 2021a. Engineering a PAM-flexible SpdCas9 variant as a universal gene repressor. Nature Communications. 12, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wu Y, Sun X, Yuan Q, Yan Y, 2017. De novo biosynthesis of glutarate via α-keto acid carbon chain extension and decarboxylation pathway in Escherichia coli. ACS Synthetic Biology. 6, 1922–1930. [DOI] [PubMed] [Google Scholar]
- Wang J, Yang Y, Zhang R, Shen X, Chen Z, Wang J, Yuan Q, Yan Y, 2018a. Microbial production of branched-chain dicarboxylate 2-methylsuccinic acid via enoate reductase-mediated bioreduction. Metabolic Engineering. 45, 1–10. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang R, Zhang J, Gong X, Jiang T, Sun X, Shen X, Wang J, Yuan Q, Yan Y, 2021b. Tunable hybrid carbon metabolism coordination for the carbon-efficient biosynthesis of 1, 3-butanediol in Escherichia coli. Green Chemistry. 23, 8694–8706. [Google Scholar]
- Wang T, Guan C, Guo J, Liu B, Wu Y, Xie Z, Zhang C, Xing X-H, 2018b. Pooled CRISPR interference screening enables genome-scale functional genomics study in bacteria with superior performance. Nature Communications. 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lin Y, Li L, Linhardt RJ, Yan Y, 2015. Regulating malonyl-CoA metabolism via synthetic antisense RNAs for enhanced biosynthesis of natural products. Metabolic Engineering. 29, 217–226. [DOI] [PubMed] [Google Scholar]
- Zha W, Rubin-Pitel SB, Shao Z, Zhao H, 2009. Improving cellular malonyl-CoA level in Escherichia coli via metabolic engineering. Metabolic Engineering. 11, 192–198. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jensen MK, Keasling JD, 2015. Development of biosensors and their application in metabolic engineering. Current Opinion in Chemical Biology. 28, 1–8. [DOI] [PubMed] [Google Scholar]
- Zhang S, Voigt CA, 2018. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Research. 46, 11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu Y, Prather KL, Stephanopoulos G, 2020. Metabolic engineering strategies to overcome precursor limitations in isoprenoid biosynthesis. Current Opinion in Biotechnology. 66, 171–178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data or materials related to this paper may be requested from the corresponding author.